
Home » Posts tagged 'Antineoplastic' (Page 2)
Tag Archives: Antineoplastic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Luvometinib
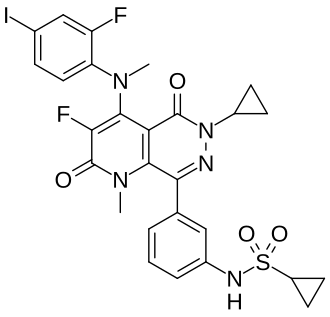
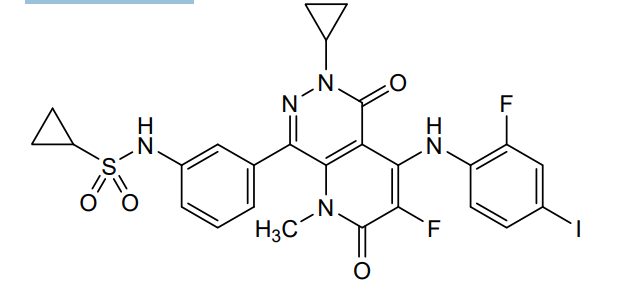
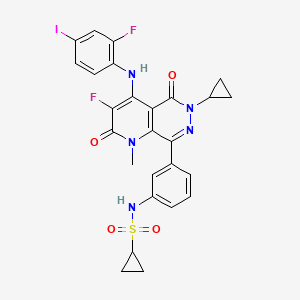
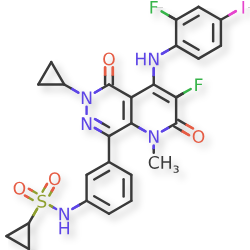
Luvometinib
CAS 2739690-43-6
MF C26H22F2IN5O4S MW665.5 g/mol
CHINA 2025, APPROVALS 2025
N-[3-[6-cyclopropyl-3-fluoro-4-(2-fluoro-4-iodoanilino)-1-methyl-2,5-dioxopyrido[2,3-d]pyridazin-8-yl]phenyl]cyclopropanesulfonamide
N-{3-[6-cyclopropyl-3-fluoro-4-(2-fluoro-4-iodoanilino)-1-methyl-2,5-dioxo-1,2,5,6-tetrahydropyrido[2,3-
d]pyridazin-8-yl]phenyl}cyclopropanesulfonamide
mitogen-activated protein kinase (MEK) inhibitor, antineoplastic, FCN 159, FCN-159, B2DYT4V89X
Luvometinib is a drug for the treatment of various types of cancer. It is a selective, orally administered inhibitor of mitogen-activated protein kinase kinases 1 and 2 (MEK1/MEK2), developed by Fosun Pharma for the treatment of rare malignancies, especially those driven by abnormal abnormal mitogen-activated protein kinase (MAPK) activation.[1][2]
In May 2025, it was approved in China for the treatment of histiocytic neoplasms such as Langerhans cell histiocytosis (LCH) and the genetic disease neurofibromatosis type 1 (NF1).[2]
Luvometinib is an orally bioavailable inhibitor of mitogen-activated protein kinase kinase (MAP2K, MAPK/ERK kinase, or MEK) 1 and 2, with potential antineoplastic activity. Upon administration, luvometinib selectively binds to and inhibits the activity of MEK1 and MEK2, preventing the activation of MEK1/2-dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling and tumor cell proliferation. MEK1/2 are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway that regulates cell growth. This pathway is often dysregulated in a variety of tumor cell types through BRAF, KRAS and NRAS mutations.
Luvometinib is a small molecule drug. The usage of the INN stem ‘-tinib’ in the name indicates that Luvometinib is a tyrosine kinase inhibitor. Luvometinib is under investigation in clinical trial NCT07004075 (FCN-159 Monotherapy Versus Chemotherapy by Investigator’s Choice in Pediatric Low-grade Glioma Patients With BRAF Alteration). Luvometinib has a monoisotopic molecular weight of 665.04 Da.
SYN
Example 8
N-(3-(6-allyl-3-ƒluoro-4-(2-ƒluoro-4-iodophenylamino)-1-methyl-2,5-dioxo-1,2,5,6- tetrahydropyrido[2,3-d]pyridazin-8-yl)phenyl)cyclopropanesulƒonamide (8)
The title compound 8 was prepared following the same procedure as described for Example 5 by substituting methanesulfonyl chloride with cyclopropanesulfonyl chloride. MS-ESI (m/z): 666 [M + 1]+.
PAT
Example 8
N-(3-(6-cyclopropyl-3-fluoro-4-(2-fluoro-4-iodophenylamino)-1-methyl-2,5-dioxo-1,2,5,6-tetrahydropyrido[2,3-d]pyridazin-8-yl)phenyl)cyclopropanesulfonamide (8)
[0136] The title compound 8 was prepared following the same procedure as described for Example 5 by substituting methanesulfonyl chloride with cyclopropanesulfonyl chloride. MS-ESI (m/z): 666 [M + 1] +.
PAT
- Protein kinase inhibitorsPublication Number: CN-106905316-BPriority Date: 2013-04-18Grant Date: 2021-06-01
- Certain protein kinase inhibitorsPublication Number: EP-2986611-B1Priority Date: 2013-04-18Grant Date: 2019-02-06
- Certain protein kinase inhibitorsPublication Number: US-10022374-B2Priority Date: 2013-04-18Grant Date: 2018-07-17



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Cheng Y, Tian H (2017). “Current Development Status of MEK Inhibitors”. Molecules. 22 (10). Basel, Switzerland: 1551. doi:10.3390/molecules22101551. PMC 6151813. PMID 28954413.
- Keam SJ (2025). “Luvometinib: First Approval”. Drugs. 85 (9): 1177–1183. doi:10.1007/s40265-025-02217-6. PMID 40751881.
| Clinical data | |
|---|---|
| Trade names | 复迈宁 (Fu Mainin) |
| Other names | FCN-159 |
| Routes of administration | Oral |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2739690-43-6 |
| PubChem CID | 135210935 |
| IUPHAR/BPS | 13495 |
| UNII | B2DYT4V89X |
| Chemical and physical data | |
| Formula | C26H22F2IN5O4S |
| Molar mass | 665.45 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////luvometinib, CHINA 2025, APPROVALS 2025, antineoplastic, FCN 159, FCN-159, B2DYT4V89X, ANAX, PTFEON, ADVECT, BLUE JET
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
ADVERTISEMENT
Advect Process Systems Ltd.
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
ADVERTISEMENT
BLUE JET HEALTHCARE LTD, https://bluejethealthcare.com
Looking for a Reliable SNAC Manufacturer? Let’s Talk.
At Blue Jet Healthcare Ltd, we specialize in the scalable, high-purity production of SNAC—a critical excipient powering the next generation of oral peptide therapeutics.
With increasing demand for SNAC across global pharma pipelines, choosing the right manufacturing partner is essential. Quality, timelines, and consistency matter.

Phone No. +91 (22) 22075307 / +91 (22) 22071691
Business Development/ Contract Manufacturing: marketing1@bluejethealthcare.com, madhu.gautam71@gmail.com
Lutetium (177Lu) tezuvotide tetraxetan
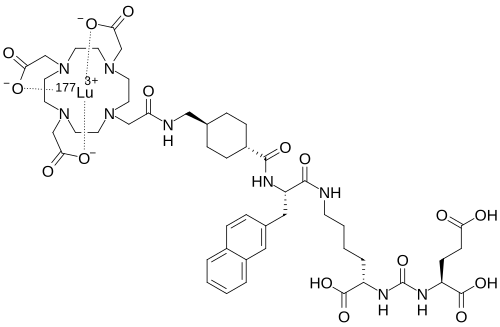
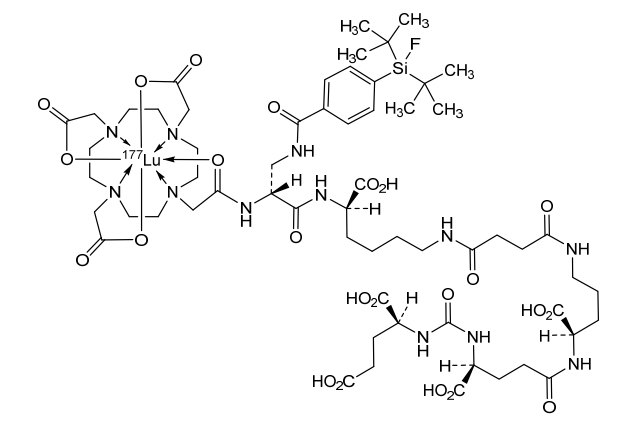
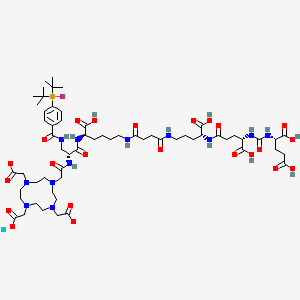
Lutetium (177Lu) tezuvotide tetraxetan
CAS2613239-73-7
MF C60H92F177LuN12O23Si , 1573.5 g/mol
2-[4-[2-[[(2R)-1-[[(1R)-1-carboxy-5-[[4-[[(4R)-4-carboxy-4-[[(4S)-4-carboxy-4-[[(1S)-1,3-dicarboxypropyl]carbamoylamino]butanoyl]amino]butyl]amino]-4-oxobutanoyl]amino]pentyl]amino]-3-[[4-[ditert-butyl(fluoro)silyl]benzoyl]amino]-1-oxopropan-2-yl]amino]-2-oxoethyl]-7,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;lutetium-177(3+)

antineoplastic, 177LU-RHPSMA-10.1, RHPSMA-10.1 LUTETIUM LU-177, FJ9Z7Y8MRW
Lutetium (177Lu) tezuvotide tetraxetan ($^{177}$Lu-rhPSMA-10.1) is an experimental radioligand therapy, developed by Bracco, that targets prostate-specific membrane antigen (PSMA) to treat metastatic castration-resistant prostate cancer. It uses a radiohybrid (rh) PSMA molecule, designed to have high binding affinity to PSMA-positive cancer cells and deliver targeted beta-minus radiation.
Key details:
- Mechanism: It binds to PSMA-expressing cells, leading to DNA damage and tumor cell death.
- Target: Prostate-specific membrane antigen (PSMA)-positive metastatic castration-resistant prostate cancer.
- Distinction: It is distinct from the FDA-approved ${177}$Lu-vipivotide tetraxetan (Pluvicto), though it is part of the same class of PSMA-targeted radiopharmaceutical agents.
- Status: It has been tested in clinical trials as a potential therapy for advanced prostate cancer, including studies evaluating its efficacy and safety.
It is important to distinguish between the various PSMA-targeted agents, such as vipivotide tetraxetan, which is approved for use.
Lutetium Lu 177 Tezuvotide Tetraxetan is a radioconjugate composed of PSMA-10.1, a prostate-specific membrane antigen (PSMA)-targeting ligand and radiolabeled with the beta-emitting radioisotope lutetium Lu 177 (177Lu), with potential antineoplastic activity against PSMA-expressing tumor cells. Upon intravenous administration, lutetium Lu 177 tezuvotide tetraxetan targets and binds to PSMA-expressing tumor cells. Upon binding, PSMA-expressing tumor cells are destroyed by 177Lu through the specific delivery of beta particle radiation. PSMA, a tumor-associated antigen (TAA) and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells as well as a variety of other solid tumors.
An open-label, multicentre, integrated Phase 1 & 2 study to evaluate the safety, tolerability, radiation dosimetry and anti-tumour activity of Lutetium (177Lu) rhPSMA-10.1 injection in men with metastatic castrate-resistant prostate cancer
EudraCT: 2022-002407-37
Phase: Phase 2
Status: Trial now transitioned
Date: 2023-05-22
PAT
https://patents.google.com/patent/WO2024121722A1/en
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////////lutetium (177Lu) tezuvotide tetraxetan, antineoplastic, 177LU-RHPSMA-10.1, RHPSMA-10.1 LUTETIUM LU-177, FJ9Z7Y8MRW
Istisociclib
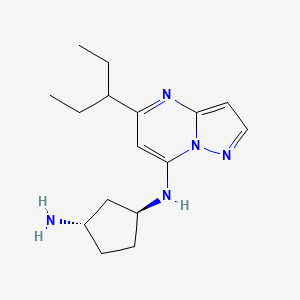
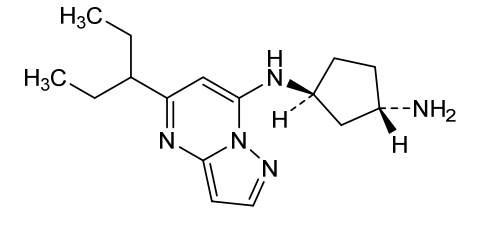
Istisociclib
KB 130742
CAS 2416873-83-9
MF C16H25N5, 287.40 g/mol
trans-(1S,3S)-3-N-(5-pentan-3-ylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine
- (1S,3S)-N1-[5-(1-Ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]-1,3-cyclopentanediamine
- 1,3-Cyclopentanediamine, N1-[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]-, (1S,3S)-
(1S,3S)-N1-[5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine
cyclin-dependent kinase (CDK) inhibitor, antineoplastic, KB-0742, 2416873-83-9, KB 0742, F7J6KSY5I8, UB-18422, KB-130742, KB 00130742
Istisociclib is a small molecule drug. The usage of the INN stem ‘-ciclib’ in the name indicates that Istisociclib is a cyclin dependant kinase inhibitor. Istisociclib is under investigation in clinical trial NCT04718675 (A Study of KB-0742 in Participants With Relapsed or Refractory Solid Tumors Including Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC)). Istisociclib has a monoisotopic molecular weight of 287.21 Da.
Istisociclib is an orally bioavailable, selective inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon oral administration, istisociclib targets, binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins and oncogenic transcription factors including MYC and androgen receptor (AR). This induces cell cycle arrest and apoptosis and prevents tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA Pol II at serine 2 (p-Ser2-RNAPII), and is an important cofactor for various oncogenic transcription factors. It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
ISTISOCICLIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
A Study of KB-0742 in Participants With Relapsed or Refractory Solid Tumors Including Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC)
CTID: NCT04718675
Phase: Phase 1/Phase 2
Status: Terminated
Date: 2025-02-17
REF
- Discovery of KB-0742, a Potent, Selective, Orally Bioavailable Small Molecule Inhibitor of CDK9 for MYC-Dependent CancersPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-11-15PMCID: PMC10726352PMID: 37967851DOI: 10.1021/acs.jmedchem.3c01233
- CDK9 inhibitors in cancer researchPublication Name: RSC Medicinal ChemistryPublication Date: 2022-06-22PMCID: PMC9215160PMID: 35814933DOI: 10.1039/d2md00040g
- From Structure Modification to Drug Launch: A Systematic Review of the Ongoing Development of Cyclin-Dependent Kinase Inhibitors for Multiple Cancer TherapyPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-04-29PMID: 35485642DOI: 10.1021/acs.jmedchem.1c02064
- Lessons Learned from Past Cyclin-Dependent Kinase Drug Discovery EffortsPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-02PMID: 35235745DOI: 10.1021/acs.jmedchem.1c02190
PAT
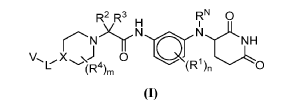
(lS,3S)-N1-(5-(pentan-3-yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diamine is a pharmaceutically active compound that has been studied for various uses, such as for the treatment of cancer. As used herein, the term “Compound A” is used to refer to both the free base and salt forms of (lS,3S)-N1-(5-(pentan-3-yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diamine. The free base of Compound A has the CAS number of 2416873-83-9 and structure of formula (I):
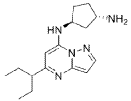
SYN
Example 35: (1S,3S)-N3-[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine (35)
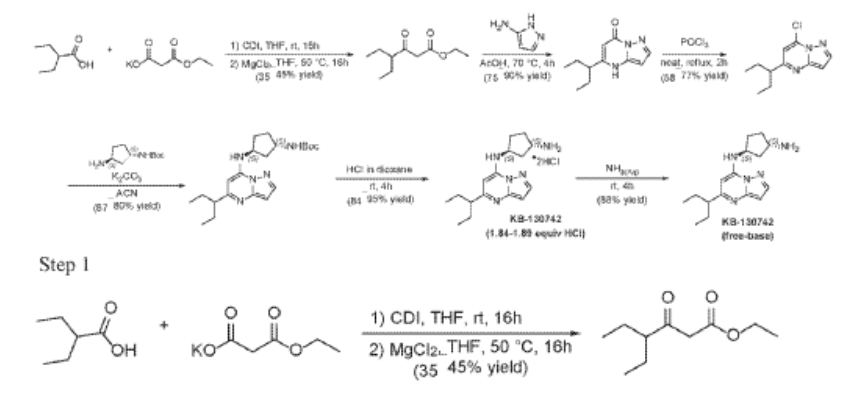
2-Ethylbutanoic acid (7.5 g, 64.57 mmol) was dissolved in THF (150 mL) and cooled to 0 °C. Within 20 min CDI (16.23 g, 100.08 mmol) was added portion-wise. The reaction warmed to room temp (rt) and the mixture was stirred at rt overnight (Solution A). In another flask MgCl2 (6.14 g, 64.57 mmol) and potassium 3-ethoxy-3-oxo-propanoate (17 g, 100.1 mmol) were mixed with THF (150 mL) and stirred under argon overnight at 50 °C. The resultant white suspension was cooled to rt and solution A was added dropwise over 10 min and the reaction mixture (RM) was stirred for 16h at room temperature. After several minutes a sticky, amorphous solid appeared whereupon after several hours the reaction mixture became homogenous in appearance. The RM was concentrated to about a third, taken up in half sat. potassium bisulphate solution and extracted twice with ethyl acetate. The organic layers were subsequently washed with a sat. sodium bicarbonate solution, combined, dried over anhydrous sodium sulfate, filtered and evaporated. Purification by column chromatography gave ethyl 4-ethyl-3-oxo-hexanoate (4.3 g, 23.087 mmol, 35.8% yield) as a transparent liquid. The RM was monitored by TLC (10% EA in Hex, Product Rf=0.6, SM Rf=0.1).
Step 2
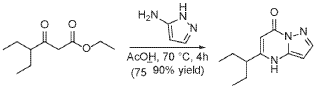
To a suspension of ethyl 4-ethyl-3-oxo-hexanoate (4.4 g, 23.62 mmol) in acetic acid (11 mL) at 70 °C was added 1H-pyrazol-5-amine (4.71 g, 56.7 mmol) in two portions (the second portion was added after 2 hours of stirring the first portion) over a 4 hour period. Upon consumption of SM as indicated by TLC, the reaction was cooled to rt and the solvent was evaporated in a rotary evaporator. The residue was treated with ethyl acetate and filtered to give 5-(1-ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (3.7 g, 17.7 mmol, 74.9% yield) as an off-white solid. The reaction mixture was monitored by TLC (5% MeOH in DCM, Product Rf=0.3, SM Rf=0.8).
Step 3

A stirred solution of 5-(1-ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (3.7 g, 18.03 mmol) in POCl3 (33.7 mL, 360.52 mmol) was heated to reflux for 4 hours. The reaction mixture was cooled to room temperature, excess reagent was evaporated in a rotary evaporator, and the residue was treated with ice-water. The chlorinated product was extracted from aqueous mixture by DCM. The organic layer was separated, dried over anhydrous Na2SO4, filtered and purified by column chromatography to give 7- chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (3.1 g, 13.9 mmol, 76.9% yield) as a light yellow liquid. The reaction mixture was monitored by TLC (20% EA in Hex, Product Rf=0.6, SM Rf=0.1).
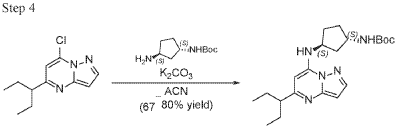
To a stirred solution 7-chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (2.3 g, 10.28 mmol), tert-Butyl ((1S,3S)-3-aminocyclopentyl)carbamate (2.27 g, 11.31 mmol) and K2CO3 (4.26 g, 30.84 mmol) in MeCN (20 mL) were heated to reflux for 16 hours. The reaction mixture was filtered, concentrated under reduced pressure and purified by column chromatography, eluent 30% EA in hexane to give tert-butyl N-[(1S,3S)-3-[[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (4.5 g, 11.6 mmol, 112.8% yield) as an off-white solid. The reaction mixture was monitored by TLC (40% EA in Hex, Product Rf=0.5, SM Rf=0.7).
Step 5
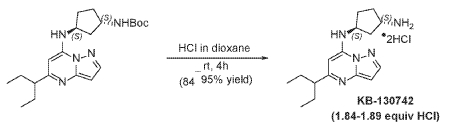
To tert-butyl N-[(1 S,3S)-3-[[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (1.0 g, 2.58 mmol) in l,4-Dioxane (0.2 mL), 4 M HC1 in Dioxane (3.22 mL, 12.9 mmol) was added and stirred at room temperature for 4 hours. The reaction mixture was evaporated in vacuo, triturated with pentane and lyophilized from MeCN:H20 to give [(lS,3S)-3-[[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium dichloride (0.9 g, 2.5 mmol, 96.8% yield) as a pale-yellow sticky solid. The reaction mixture was monitored by TLC ( 100% EA, Product Rf=0.l, SM Rf=0.8). 1H NMR (400 MHz, DMSO-d6) d 15.00 (s, 1H), 9.93-9.86 (m, 1H), 8.51 (s, 3H), 8.30 (s, 1H), 6.84 (s, 1H), 6.58 (s, 1H), 4.95 (q, J = 7.8 Hz, 1H), 3.77- 3.66 (m, 1H), 2.84-2.71 (m, 1H), 2.29-2.05 (m, 4H), 1.94-1.63 (m, 6H), 0.81 (t, J = 7.4 Hz, 6H). LC-MS (m/z 287.21, found 288.0 [M+H+])
Step 6
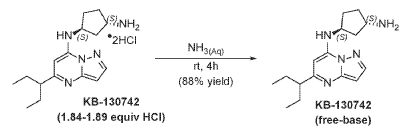
To [(1S,3S)-3-[[5-(l-ethylpropyl)pyrazolo[I,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium-di chloride (0.2 g, 0.5600 mmol) in aq. NH3 (4.0 mL, 0.56 mmol) was added and stirred at room temperature for 4 hours. The reaction mixture was evaporated in vacuo, triturated with pentane and lyophilized from MeCN:H20 to give (lS,3S)-N3-[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-7-yl]cyclopentane-l,3-diamine (140 mg, 0.49 mmol, 87.8% yield) as a pale-yellow sticky solid. The reaction mixture was monitored by TLC (100% EA, Product Rf=0.1, SM Rf=0.8). 1H NMR (400 MHz, DMSO-d6) d 7.95 (d, J = 2.2 Hz, 1H), 6.86 (s, 1H), 6.29 (d, J = 2.2 Hz, 1H), 5.95 (s, 1H), 4.31-4.19 (m, 1H), 3.57-3.44 (m, 1H), 2.52-2.44 (m, 1H), 2.36-2.22 (m, 1H),
2.09–1.79 (m, 3H), 1.80–1.59 (m, 5H), 1.58–1.24 (m, 3H), 0.83 (t, J = 7.4 Hz, 6H). LC-MS (m/z 287.21, found 288.5 [M+H+]).
PAT
- Compounds, compositions and methods for modulating CDK9 activityPublication Number: CN-112996790-BPriority Date: 2018-10-30Grant Date: 2023-11-03
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: US-2024132506-A1Priority Date: 2018-10-30
- Compounds, Compositions, and Methods for Modulating CDK9 ActivityPublication Number: US-2020131189-A1Priority Date: 2018-10-30
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: US-2022002305-A1Priority Date: 2018-10-30
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: EP-3873911-A1Priority Date: 2018-10-30
- Substituted pyrazolo[1,5-a]pyrimidines for modulating CDK9 activityPublication Number: US-11845754-B2Priority Date: 2018-10-30Grant Date: 2023-12-19
- Substituted pyrazolo[1,5-a]pyrimidines for modulating CDK9 activityPublication Number: US-11155560-B2Priority Date: 2018-10-30Grant Date: 2021-10-26
- Compounds and methods for modulating cdk9 activityPublication Number: EP-4240422-A1Priority Date: 2020-11-05
- Compounds and methods for modulating cdk9 activityPublication Number: WO-2022098843-A1Priority Date: 2020-11-05
- Compounds and methods for modulating cdk9 activityPublication Number: US-2025188084-A1Priority Date: 2020-11-05
- Chimeric degraders of cyclin-dependent kinase 9 and uses thereofPublication Number: WO-2021216828-A1Priority Date: 2020-04-24
- Chimeric degraders of cyclin-dependent kinase 9 and uses thereofPublication Number: US-2023158159-A1Priority Date: 2020-04-24
- Polymorphic and salt forms of (ls,3s)-n-(5-(pentan-3- yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diaminePublication Number: EP-4436569-A1Priority Date: 2021-11-24
- Polymorphs and salt forms of (1S,3S)-N1-(5-(pentan-3-yl)pyrazolo[1,5-A]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: KR-20240110634-APriority Date: 2021-11-24
- Compositions and methods for enhanced protein productionPublication Number: EP-4412621-A2Priority Date: 2021-09-22
- Compositions and methods for enhanced protein productionPublication Number: US-2024252688-A1Priority Date: 2021-09-22
- Compositions and methods for enhanced protein productionPublication Number: WO-2023056202-A2Priority Date: 2021-09-22
- A cdk9 inhibitor for use in the treatment of cancer in a subject having an asxl1 mutationPublication Number: WO-2025217597-A1Priority Date: 2024-04-12
- Polymorphic and salt forms of (1s,3s)-n1-(5-(pentan-3- yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: WO-2023096922-A8Priority Date: 2021-11-24
- Polymorphic and salt forms of (ls,3s)-n1-(5-(pentan-3- yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diaminePublication Number: WO-2023096922-A1Priority Date: 2021-11-24
- Polymorphic and salt forms of (1s,3s)-n1-(5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl) cyclopentane-1,3-diaminePublication Number: US-2025059193-A1Priority Date: 2021-11-24
- Polymorphic forms and salt forms of (1S,3S)-N1-(5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: CN-118678952-APriority Date: 2021-11-24
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////istisociclib, cyclin-dependent kinase (CDK) inhibitor, antineoplastic, KB-0742, 2416873-83-9, KB 0742, F7J6KSY5I8, UB-18422, KB-130742, KB 00130742
Istisociclib
Istisociclib
KB 130742
CAS 2416873-83-9
MF C16H25N5, 287.40 g/mol
trans-(1S,3S)-3-N-(5-pentan-3-ylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine
- (1S,3S)-N1-[5-(1-Ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]-1,3-cyclopentanediamine
- 1,3-Cyclopentanediamine, N1-[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]-, (1S,3S)-
(1S,3S)-N1-[5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine
cyclin-dependent kinase (CDK) inhibitor, antineoplastic, KB-0742, 2416873-83-9, KB 0742, F7J6KSY5I8, UB-18422, KB-130742, KB 00130742
Istisociclib is a small molecule drug. The usage of the INN stem ‘-ciclib’ in the name indicates that Istisociclib is a cyclin dependant kinase inhibitor. Istisociclib is under investigation in clinical trial NCT04718675 (A Study of KB-0742 in Participants With Relapsed or Refractory Solid Tumors Including Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC)). Istisociclib has a monoisotopic molecular weight of 287.21 Da.
Istisociclib is an orally bioavailable, selective inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon oral administration, istisociclib targets, binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins and oncogenic transcription factors including MYC and androgen receptor (AR). This induces cell cycle arrest and apoptosis and prevents tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA Pol II at serine 2 (p-Ser2-RNAPII), and is an important cofactor for various oncogenic transcription factors. It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
ISTISOCICLIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
A Study of KB-0742 in Participants With Relapsed or Refractory Solid Tumors Including Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC)
CTID: NCT04718675
Phase: Phase 1/Phase 2
Status: Terminated
Date: 2025-02-17
REF
- Discovery of KB-0742, a Potent, Selective, Orally Bioavailable Small Molecule Inhibitor of CDK9 for MYC-Dependent CancersPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-11-15PMCID: PMC10726352PMID: 37967851DOI: 10.1021/acs.jmedchem.3c01233
- CDK9 inhibitors in cancer researchPublication Name: RSC Medicinal ChemistryPublication Date: 2022-06-22PMCID: PMC9215160PMID: 35814933DOI: 10.1039/d2md00040g
- From Structure Modification to Drug Launch: A Systematic Review of the Ongoing Development of Cyclin-Dependent Kinase Inhibitors for Multiple Cancer TherapyPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-04-29PMID: 35485642DOI: 10.1021/acs.jmedchem.1c02064
- Lessons Learned from Past Cyclin-Dependent Kinase Drug Discovery EffortsPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-02PMID: 35235745DOI: 10.1021/acs.jmedchem.1c02190
PAT
(lS,3S)-N1-(5-(pentan-3-yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diamine is a pharmaceutically active compound that has been studied for various uses, such as for the treatment of cancer. As used herein, the term “Compound A” is used to refer to both the free base and salt forms of (lS,3S)-N1-(5-(pentan-3-yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diamine. The free base of Compound A has the CAS number of 2416873-83-9 and structure of formula (I):
SYN
Example 35: (1S,3S)-N3-[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine (35)
2-Ethylbutanoic acid (7.5 g, 64.57 mmol) was dissolved in THF (150 mL) and cooled to 0 °C. Within 20 min CDI (16.23 g, 100.08 mmol) was added portion-wise. The reaction warmed to room temp (rt) and the mixture was stirred at rt overnight (Solution A). In another flask MgCl2 (6.14 g, 64.57 mmol) and potassium 3-ethoxy-3-oxo-propanoate (17 g, 100.1 mmol) were mixed with THF (150 mL) and stirred under argon overnight at 50 °C. The resultant white suspension was cooled to rt and solution A was added dropwise over 10 min and the reaction mixture (RM) was stirred for 16h at room temperature. After several minutes a sticky, amorphous solid appeared whereupon after several hours the reaction mixture became homogenous in appearance. The RM was concentrated to about a third, taken up in half sat. potassium bisulphate solution and extracted twice with ethyl acetate. The organic layers were subsequently washed with a sat. sodium bicarbonate solution, combined, dried over anhydrous sodium sulfate, filtered and evaporated. Purification by column chromatography gave ethyl 4-ethyl-3-oxo-hexanoate (4.3 g, 23.087 mmol, 35.8% yield) as a transparent liquid. The RM was monitored by TLC (10% EA in Hex, Product Rf=0.6, SM Rf=0.1).
Step 2
To a suspension of ethyl 4-ethyl-3-oxo-hexanoate (4.4 g, 23.62 mmol) in acetic acid (11 mL) at 70 °C was added 1H-pyrazol-5-amine (4.71 g, 56.7 mmol) in two portions (the second portion was added after 2 hours of stirring the first portion) over a 4 hour period. Upon consumption of SM as indicated by TLC, the reaction was cooled to rt and the solvent was evaporated in a rotary evaporator. The residue was treated with ethyl acetate and filtered to give 5-(1-ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (3.7 g, 17.7 mmol, 74.9% yield) as an off-white solid. The reaction mixture was monitored by TLC (5% MeOH in DCM, Product Rf=0.3, SM Rf=0.8).
Step 3
A stirred solution of 5-(1-ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (3.7 g, 18.03 mmol) in POCl3 (33.7 mL, 360.52 mmol) was heated to reflux for 4 hours. The reaction mixture was cooled to room temperature, excess reagent was evaporated in a rotary evaporator, and the residue was treated with ice-water. The chlorinated product was extracted from aqueous mixture by DCM. The organic layer was separated, dried over anhydrous Na2SO4, filtered and purified by column chromatography to give 7- chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (3.1 g, 13.9 mmol, 76.9% yield) as a light yellow liquid. The reaction mixture was monitored by TLC (20% EA in Hex, Product Rf=0.6, SM Rf=0.1).
To a stirred solution 7-chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (2.3 g, 10.28 mmol), tert-Butyl ((1S,3S)-3-aminocyclopentyl)carbamate (2.27 g, 11.31 mmol) and K2CO3 (4.26 g, 30.84 mmol) in MeCN (20 mL) were heated to reflux for 16 hours. The reaction mixture was filtered, concentrated under reduced pressure and purified by column chromatography, eluent 30% EA in hexane to give tert-butyl N-[(1S,3S)-3-[[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (4.5 g, 11.6 mmol, 112.8% yield) as an off-white solid. The reaction mixture was monitored by TLC (40% EA in Hex, Product Rf=0.5, SM Rf=0.7).
Step 5
To tert-butyl N-[(1 S,3S)-3-[[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (1.0 g, 2.58 mmol) in l,4-Dioxane (0.2 mL), 4 M HC1 in Dioxane (3.22 mL, 12.9 mmol) was added and stirred at room temperature for 4 hours. The reaction mixture was evaporated in vacuo, triturated with pentane and lyophilized from MeCN:H20 to give [(lS,3S)-3-[[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium dichloride (0.9 g, 2.5 mmol, 96.8% yield) as a pale-yellow sticky solid. The reaction mixture was monitored by TLC ( 100% EA, Product Rf=0.l, SM Rf=0.8). 1H NMR (400 MHz, DMSO-d6) d 15.00 (s, 1H), 9.93-9.86 (m, 1H), 8.51 (s, 3H), 8.30 (s, 1H), 6.84 (s, 1H), 6.58 (s, 1H), 4.95 (q, J = 7.8 Hz, 1H), 3.77- 3.66 (m, 1H), 2.84-2.71 (m, 1H), 2.29-2.05 (m, 4H), 1.94-1.63 (m, 6H), 0.81 (t, J = 7.4 Hz, 6H). LC-MS (m/z 287.21, found 288.0 [M+H+])
Step 6
To [(1S,3S)-3-[[5-(l-ethylpropyl)pyrazolo[I,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium-di chloride (0.2 g, 0.5600 mmol) in aq. NH3 (4.0 mL, 0.56 mmol) was added and stirred at room temperature for 4 hours. The reaction mixture was evaporated in vacuo, triturated with pentane and lyophilized from MeCN:H20 to give (lS,3S)-N3-[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-7-yl]cyclopentane-l,3-diamine (140 mg, 0.49 mmol, 87.8% yield) as a pale-yellow sticky solid. The reaction mixture was monitored by TLC (100% EA, Product Rf=0.1, SM Rf=0.8). 1H NMR (400 MHz, DMSO-d6) d 7.95 (d, J = 2.2 Hz, 1H), 6.86 (s, 1H), 6.29 (d, J = 2.2 Hz, 1H), 5.95 (s, 1H), 4.31-4.19 (m, 1H), 3.57-3.44 (m, 1H), 2.52-2.44 (m, 1H), 2.36-2.22 (m, 1H),
2.09–1.79 (m, 3H), 1.80–1.59 (m, 5H), 1.58–1.24 (m, 3H), 0.83 (t, J = 7.4 Hz, 6H). LC-MS (m/z 287.21, found 288.5 [M+H+]).
PAT
- Compounds, compositions and methods for modulating CDK9 activityPublication Number: CN-112996790-BPriority Date: 2018-10-30Grant Date: 2023-11-03
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: US-2024132506-A1Priority Date: 2018-10-30
- Compounds, Compositions, and Methods for Modulating CDK9 ActivityPublication Number: US-2020131189-A1Priority Date: 2018-10-30
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: US-2022002305-A1Priority Date: 2018-10-30
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: EP-3873911-A1Priority Date: 2018-10-30
- Substituted pyrazolo[1,5-a]pyrimidines for modulating CDK9 activityPublication Number: US-11845754-B2Priority Date: 2018-10-30Grant Date: 2023-12-19
- Substituted pyrazolo[1,5-a]pyrimidines for modulating CDK9 activityPublication Number: US-11155560-B2Priority Date: 2018-10-30Grant Date: 2021-10-26
- Compounds and methods for modulating cdk9 activityPublication Number: EP-4240422-A1Priority Date: 2020-11-05
- Compounds and methods for modulating cdk9 activityPublication Number: WO-2022098843-A1Priority Date: 2020-11-05
- Compounds and methods for modulating cdk9 activityPublication Number: US-2025188084-A1Priority Date: 2020-11-05
- Chimeric degraders of cyclin-dependent kinase 9 and uses thereofPublication Number: WO-2021216828-A1Priority Date: 2020-04-24
- Chimeric degraders of cyclin-dependent kinase 9 and uses thereofPublication Number: US-2023158159-A1Priority Date: 2020-04-24
- Polymorphic and salt forms of (ls,3s)-n-(5-(pentan-3- yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diaminePublication Number: EP-4436569-A1Priority Date: 2021-11-24
- Polymorphs and salt forms of (1S,3S)-N1-(5-(pentan-3-yl)pyrazolo[1,5-A]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: KR-20240110634-APriority Date: 2021-11-24
- Compositions and methods for enhanced protein productionPublication Number: EP-4412621-A2Priority Date: 2021-09-22
- Compositions and methods for enhanced protein productionPublication Number: US-2024252688-A1Priority Date: 2021-09-22
- Compositions and methods for enhanced protein productionPublication Number: WO-2023056202-A2Priority Date: 2021-09-22
- A cdk9 inhibitor for use in the treatment of cancer in a subject having an asxl1 mutationPublication Number: WO-2025217597-A1Priority Date: 2024-04-12
- Polymorphic and salt forms of (1s,3s)-n1-(5-(pentan-3- yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: WO-2023096922-A8Priority Date: 2021-11-24
- Polymorphic and salt forms of (ls,3s)-n1-(5-(pentan-3- yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diaminePublication Number: WO-2023096922-A1Priority Date: 2021-11-24
- Polymorphic and salt forms of (1s,3s)-n1-(5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl) cyclopentane-1,3-diaminePublication Number: US-2025059193-A1Priority Date: 2021-11-24
- Polymorphic forms and salt forms of (1S,3S)-N1-(5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: CN-118678952-APriority Date: 2021-11-24
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////istisociclib, cyclin-dependent kinase (CDK) inhibitor, antineoplastic, KB-0742, 2416873-83-9, KB 0742, F7J6KSY5I8, UB-18422, KB-130742, KB 00130742
Gridegalutamide
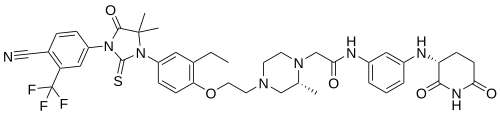
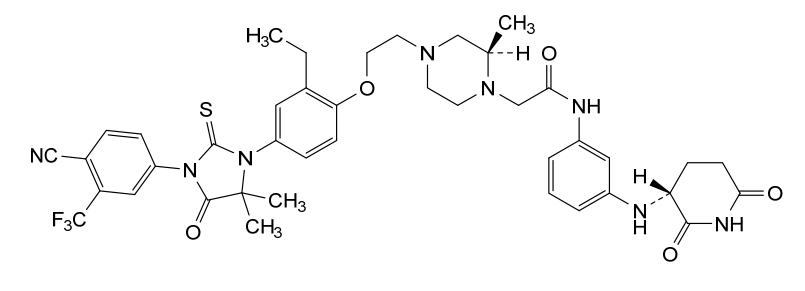
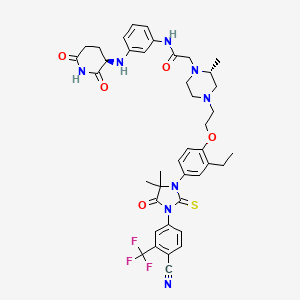
Gridegalutamide
CAS 2446929-86-6
MF C41H45F3N8O5S MW818.9 g/mol
2-[(2R)-4-[2-[4-[3-[4-cyano-3-(trifluoromethyl)phenyl]-5,5-dimethyl-4-oxo-2-sulfanylideneimidazolidin-1-yl]-2-ethylphenoxy]ethyl]-2-methylpiperazin-1-yl]-N-[3-[[(3R)-2,6-dioxopiperidin-3-yl]amino]phenyl]acetamide

antiandrogen, antineoplastic, BMS 986365, CC 94676, BMS-986365, CC-94676, CEL 010355,
VA228VR2DI,
Gridegalutamide is an investigational oral androgen receptor (AR) degrader being developed for the treatment of metastatic castration-resistant prostate cancer (mCRPC). It belongs to a class of drugs called proteolysis targeting chimeras (PROTACs), which are designed to selectively degrade specific proteins by hijacking the ubiquitin-proteasome system.[1][2] CC-94676 employs a unique dual mechanism of action, combining AR degradation with AR antagonism, potentially offering advantages over traditional AR inhibitors in overcoming resistance mechanisms.[3] Initially developed by Celgene and now under Bristol Myers Squibb, CC-94676 has demonstrated AR protein degradation and suppression of tumor growth in CRPC mouse models.[2] As of 2024, CC-94676 is being evaluated in phase I clinical trials for patients with mCRPC who have progressed on androgen deprivation therapy and at least one prior secondary hormonal therapy.[1][2]
Gridegalutamide is a small molecule drug. The usage of the INN stem ‘-lutamide’ in the name indicates that Gridegalutamide is a non-steroid antiandrogen. Gridegalutamide is under investigation in clinical trial NCT04428788 (Study to Evaluate the Safety and Tolerability of CC-94676 in Participants With Metastatic Castration-Resistant Prostate Cancer). Gridegalutamide has a monoisotopic molecular weight of 818.32 Da.
GRIDEGALUTAMIDE is a small molecule drug with a maximum clinical trial phase of II (across all indications) and has 3 investigational indications.
Gridegalutamide is an orally bioavailable androgen receptor (AR) degrader, with potential antineoplastic activity. Upon administration, gridegalutamide causes degradation of AR, prevents AR-mediated signaling and inhibits the proliferation of AR-overexpressing tumor cells. AR plays a key role in tumor cell proliferation in castration-resistant prostate cancer (CRPC).
- A Study to Evaluate the Drug Levels, Metabolism and Excretion, and Absolute Bioavailability of BMS-986365 in Healthy Male ParticipantsCTID: NCT06433505Phase: Phase 1Status: CompletedDate: 2025-03-26
- Study to Evaluate the Safety and Tolerability of CC-94676 in Participants With Metastatic Castration-Resistant Prostate CancerCTID: NCT04428788Phase: Phase 1Status: CompletedDate: 2025-12-22
SYN
DRUGHUNTER
https://drughunter.com/molecule/gridegalutamide-bms-986365-cc-94676
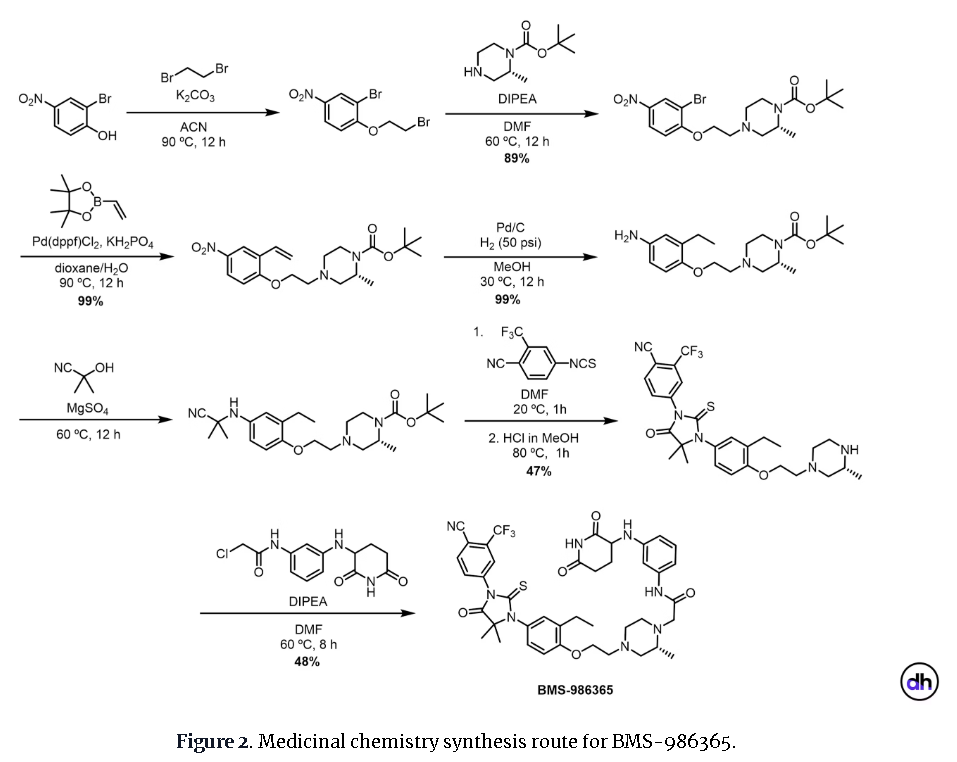
PAT
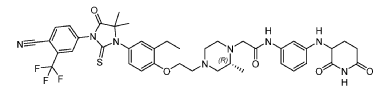
Example 17: 2-((R)-4-(2-(4-(3-(4-Cyano-3-(trifluoromethyl)phenyl)-5,5-dimethyl-4-oxo-2- thioxoimidazolidin-1-yl)-2-ethylphenoxy)ethyl)-2-methylpiperazin-1-yl)-N-(3-((2,6- dioxopiperidin-3-yl)amino)phenyl)acetamide hydrochloride



PAT
- Combination therapy with substituted 3- ((3-aminophenyl) amino) piperidine-2, 6-dione compoundsPublication Number: CN-120152718-APriority Date: 2022-11-09
- Combination therapy with substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compoundsPublication Number: WO-2024102706-A1Priority Date: 2022-11-09
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-2020199073-A1Priority Date: 2018-12-19
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-11149007-B2Priority Date: 2018-12-19Grant Date: 2021-10-19
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-11873283-B2Priority Date: 2018-12-19Grant Date: 2024-01-16
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-2024368083-A1Priority Date: 2018-12-19
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-12404241-B2Priority Date: 2018-12-19Grant Date: 2025-09-02
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-2023002321-A1Priority Date: 2018-12-19
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
ADVERTISEMENT
Advect Process Systems Ltd. https://advectprocess.com

ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
References
- Salama AK, Trkulja MV, Casanova E, Uras IZ (December 2022). “Targeted Protein Degradation: Clinical Advances in the Field of Oncology”. International Journal of Molecular Sciences. 23 (23) 15440. doi:10.3390/ijms232315440. PMC 9741350. PMID 36499765.
- Xie H, Liu J, Alem Glison DM, Fleming JB (2021). “The clinical advances of proteolysis targeting chimeras in oncology”. Exploration of Targeted Anti-Tumor Therapy. 2 (6): 511–521. doi:10.37349/etat.2021.00061. PMC 9400722. PMID 36046114.
- Rathkopf DE, Patel MR, Choudhury AD, Rasco D, Lakhani N, Hawley JE, et al. (September 2024). “Safety and clinical activity of BMS-986365 (CC-94676), a dual androgen receptor ligand-directed degrader and antagonist, in heavily pretreated patients with metastatic castration-resistant prostate cancer”. Annals of Oncology. 36 (1): 76–88. doi:10.1016/j.annonc.2024.09.005. PMC 12094577. PMID 39293515.
| Clinical data | |
|---|---|
| Other names | BMS-986365; CC-94676 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2446929-86-6 |
| PubChem CID | 153513643 |
| ChemSpider | 133326102 |
| UNII | VA228VR2DI |
| KEGG | D12866 |
| ChEMBL | ChEMBL6068413 |
| Chemical and physical data | |
| Formula | C41H45F3N8O5S |
| Molar mass | 818.92 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////gridegalutamide, ANAX, ADVECT, antiandrogen, antineoplastic, BMS 986365, CC 94676, BMS-986365, CC-94676, CEL 010355, VA228VR2DI,
Fanregratinib


Fanregratinib
CAS 1628537-44-9
MF C27H33ClN6O2, 509.0 g/mol
4-chloro-3-[2-[2-[4-[(3S,5R)-3,5-dimethylpiperazin-1-yl]anilino]pyrimidin-5-yl]ethyl]-5-methoxy-N-methylbenzamide
- 4-Chloro-3-(2-(2-((4-((3s,5r)-3,5-dimethylpiperazin-1-yl)phenyl)amino)pyrimidin-5-yl)ethyl)-5-methoxy-N-methylbenzamide
- 4-chloro-3-(2-(2-(4-((3S,5R)-3,5-dimethylpiperazin-1-yl)phenylamino)pyrimidin-5-yl)ethyl)-5-methoxy-N-methylbenzamide
- 4-chloro-3-[2-[2-[4-[(3S,5R)-3,5-dimethylpiperazin-1-yl]anilino]pyrimidin-5-yl]ethyl]-5-methoxy-N-methylbenzamide
- Benzamide, 4-chloro-3-[2-[2-[[4-[(3R,5S)-3,5-dimethyl-1-piperazinyl]phenyl]amino]-5-pyrimidinyl]ethyl]-5-methoxy-N-methyl-, cis-
- Benzamide, 4-chloro-3-[2-[2-[[4-[(3R,5S)-3,5-dimethyl-1-piperazinyl]phenyl]amino]-5-pyrimidinyl]ethyl]-5-methoxy-N-methyl-, rel-
- cis-4-Chloro-3-[2-[2-[[4-[(3R,5S)-3,5-dimethyl-1-piperazinyl]phenyl]amino]-5-pyrimidinyl]ethyl]-5-methoxy-N-methylbenzamide
- rel-4-Chloro-3-[2-[2-[[4-[(3R,5S)-3,5-dimethyl-1-piperazinyl]phenyl]amino]-5-pyrimidinyl]ethyl]-5-methoxy-N-methylbenzamide

fibroblast growth factor receptor tyrosine kinase inhibitor, antineoplastic, 8RWL2B2CLS
- OriginatorHutchison MediPharma
- DeveloperHutchison MediPharma; HUTCHMED
- ClassAntineoplastics; Small molecules
- Mechanism of ActionType 1 fibroblast growth factor receptor antagonists; Type 3 fibroblast growth factor receptor antagonists; Type-2 fibroblast growth factor receptor antagonists
- PreregistrationCholangiocarcinoma
- Phase IIMesothelioma
- Phase I/IISolid tumours
- 29 Dec 2025Preregistration for Cholangiocarcinoma (Late-stage disease, Metastatic disease, Second-line therapy or greater, Inoperable/Unresectable) in China (PO)
- 29 Dec 2025Updated efficacy data from a phase II trial in Cholangiocarcinoma released by HUTCHMED
- 03 Nov 2025HUTCHMED announces intention to submit new drug application to NMPA for Cholangiocarcinoma in first half of 2026
FANREGRATINIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
Fanregratinib is a small molecule drug. The usage of the INN stem ‘-gratinib’ in the name indicates that Fanregratinib is a fibroblast growth factor receptor (FGFR) inhibitor. Fanregratinib has a monoisotopic molecular weight of 508.24 Da.
Fanregratinib is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) types 1, 2, and 3 (FGFR1/2/3), with potential antineoplastic activity. Upon administration, fanregratinib binds to and inhibits FGFR1/2/3, which may result in the inhibition of FGFR1/2/3-related signal transduction pathways. This inhibits proliferation in FGFR1/2/3-overexpressing tumor cells. FGFR, a family of receptor tyrosine kinases (RTKs) upregulated in many tumor cell types, plays a key role in cellular proliferation, migration and survival.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US159751913&_cid=P10-MKQ98D-55657-1
Example 9
Synthesis of Compounds 79-91, 146-155
Compound 79
4-chloro-3-(2-(2-((4-((3S,5R)-3,5-dimethylpiperazin-1-yl)phenyl)amino)pyrimidin-5-yl)ethyl)-5-methoxy-N-methylbenzamide

(C) 4-chloro-3-(2-(2-(4-((3S,5R)-3,5-dimethylpiperazin-1-yl)phenylamino)pyrimidin-5-yl)ethyl)-5-methoxy-N-methylbenzamide
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014139465&_cid=P10-MKQ9F3-62190-1
Example 9: Synthesis of Compounds 78-103
Compound 78
4-chloro-3-(2-(2-((4-((3S,5/f)-3,5-dimethylpiperazin-l-yl)phenyl)amino)pyrimidin-5
-yl)ethyl)-5-methoxy-N-methylbenzamide

(A) Methyl 4-chloro-3-((JE)-2-(2-(4-((3S,5R)-3,5-dimethylpiperazin-l-yl)
phenylamino)pyrimidin-5-yl)vinyl)-5-methoxybenzoate
A mixture of (E)-methyl 4-chloro-3-(2-(2-chloropyrimidin-5-yl)vinyl)-5-methoxy benzoate (150mg, 0.442 mmol), 4-((35′,5i?)-3,5-dimethylpiperazin-l-yl)aniline (109 mg, 0.531 mmol) and TFA (0.1 mL, 1.326 mmol) in propan~2-oi (5 mL) was stirred at 150 °C for 1 h under microwave. The resulting mixture was concentrated, basified with ammonia water, purified via ISCO (DCM/MeOH) to afford the title compound as a
yellow solid (130 mg, 57.9% yield). MS (m/z): 508.2(M+H)+.
(B) 4-chloro-3-((£)-2-(2-(4-((3S,5R)-3,5-dimethylpiperazin-l-yl)phenylamino)pyrim idin-5-yl)vinyl)-5-methoxy-N-methylbenzamide
A mixture of methyl 4-chloro-3-((E)-2-(2-(4-((35′,5i?)-3,5-dimethylpiperazin-l-yl) phenylamino)pyrimidin-5-yl)vinyl)-5-methoxybenzoate (250 mg, 0.492 mmol) and methylamine (6 mL, 35% solution in ethanol) was stirred at 145 °C for 22 min under microwave. The resulting mixture was concentrated, purified via ISCO (DCM/MeOH) to afford the title compound as a yellow solid (145 mg, 58.1%> yield). MS (m/z):
506.9(M+H)+.
(C) 4-chloro-3-(2-(2-(4-((3S,5R)-3,5-dimethylpiperazin-l-yl)phenylamino)pyrimidin -5-yl)ethyl)-5-methoxy-N-methylbenzamide
A mixture of 4-chloro-3-((E)-2-(2-(4-((35*,5i?)-3,5-dimethylpiperazin-l-yl)
phenylamino)pyrimidin-5-yl)vinyl)-5-methoxy-N-methylbenzamide (120 mg, 0.237 mmol), 4-methylbenzenesulfonohydrazide (528 mg, 2.84 mmol) and sodium acetate (233 mg, 2.84 mmol) in THF (6mL) and water (6mL) was stirred overnight at 100 °C under nitrogen atmosphere. The resulting mixture was concentrated. The residue was partitioned between 2N HC1 (15 mL) and EA (15 mL). The aqueous layer was then adjusted to pH=8 with 30% NaOH and extracted with DCM (2* 15 mL). The combined extracts were concentrated and the residue was purified via ISCO (eluted with MeOH in H20 0-100%) to afford the title compound as a yellow solid (50 mg, 41.5% yield). MS (m/z): 509.0(M+H)+. 1H NM (400 MHz, CD3OD) δ 8.1 1 (s, 2H), 7.44 (d, J = 9.1 Hz, 2H), 7.37 (d, J = 2.0 Hz, 1H), 7.30 (d, J = 2.0 Hz, 1H), 6.95 (d, J = 9.1 Hz, 2H), 3.93 (s, 3H), 3.53 – 3.44 (m, 2H), 3.10 – 2.99 (m, 4H), 2.90 (s, 3H), 2.82 (t, J = 7.6 Hz, 2H), 2.25 (t, J = 7.5 Hz, 2H), 1.16 (d, J = 6.4 Hz, 6H).
PAT
- Novel pyrimidine and pyridine compounds and their usagePublication Number: WO-2014139465-A1Priority Date: 2013-03-15
- Novel pyrimidine and pyridine compounds and usage thereofPublication Number: WO-2014139145-A1Priority Date: 2013-03-15
- Pyrimidine and pyridine compounds and their usePublication Number: BR-112015020772-B1Priority Date: 2013-03-15
- Pyrimidine and pyridine compounds and their usagePublication Number: US-9701680-B2Priority Date: 2013-03-15Grant Date: 2017-07-11
- Novel pyrimidine and pyridine compounds and usage thereofPublication Number: US-2016052926-A1Priority Date: 2013-03-15
- Novel pyrimidine and pyridine compounds and their usagePublication Number: EP-2970120-A1Priority Date: 2013-03-15
- Novel pyrimidine and pyridine compounds and their usagePublication Number: EP-2970120-B1Priority Date: 2013-03-15Grant Date: 2018-09-12
- Novel pyrimidine and pyridine compounds and their usagePublication Number: US-2016024021-A1Priority Date: 2013-03-15
- The salts of a compound and the crystalline forms thereofPublication Number: WO-2021073494-A1Priority Date: 2019-10-14
- The salts of a compound and the crystalline forms thereofPublication Number: US-2023121346-A1Priority Date: 2019-10-14
- The salts of a compound and the crystalline forms thereofPublication Number: TW-202128669-APriority Date: 2019-10-14
- Salts of compounds and crystalline forms thereofPublication Number: CN-114555558-APriority Date: 2019-10-14
- SALTS OF THE COMPOUND 4-CHLORO-3-(2-(2-((4-((3S,5R)-3,5-DIMETHYLPIPERAZIN-1-YL)PHENYL)AMINO)PYRIMIDIN-5-YL)ETHYL)-5 -METHOXY-N-METHYLBENZAMIDE AND CRYSTALLINE FORMS THEREOFPublication Number: AR-120202-A1Priority Date: 2019-10-14
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////fanregratinib, fibroblast growth factor receptor tyrosine kinase inhibitor, antineoplastic, 8RWL2B2CLS
Enrupatinib



Enrupatinib
CAS 2222689-47-4
MF C27H26N6O3 MW 482.5 g/mol
6-[3-methoxy-4-[(6-methyl-3-pyridinyl)methoxy]anilino]-3-morpholin-4-ylquinoxaline-5-carbonitrile

colony-stimulating factor 1 receptor (CSF1R) inhibitor, antineoplastic, EI 1071, EI-1071, 9L35RVQ9J6
ENRUPATINIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
EI-1071 is a selective Colony Stimulating Factor-1 Receptor kinase inhibitor.
- A Study to Evaluate the Safety, Tolerability and Amount of EI-1071 in Blood in Healthy VolunteersCTID: NCT04238364Phase: Phase 1Status: CompletedDate: 2025-02-25
- A Phase 2 Study to Assess the Safety of EI-1071 and the Effects of EI-1071 on Neuroinflammation in Alzheimer’s Disease PatientsCTID: NCT06745583Phase: Phase 2Status: RecruitingDate: 2025-07-28
- OriginatorElixiron Immunotherapeutics
- Developer4B Technologies; Elixiron Immunotherapeutics
- ClassAntidementias; Antineoplastics; Small molecules
- Mechanism of ActionMacrophage colony-stimulating factor receptor antagonists
- Phase IIAlzheimer’s disease
- Phase IAmyotrophic lateral sclerosis; Giant cell tumour of tendon sheath
- No development reportedBreast cancer; Colorectal cancer
- 27 Jul 2025Pharmacodynamics data from preclinical studies in Alzheimer’s disease presented at the Alzheimer’s Association International Conference 2025 (AAIC-2025)
- 20 Dec 2024Phase-II clinical trials in Alzheimer’s disease (Treatment-experienced) in Taiwan (PO) (NCT06745583)
- 28 Jul 2024Adverse event data from a phase I trial in Alzheimer’s disease presented at the Alzheimer’s Association International Conference 2024 (AAIC-2024)
SYN
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018071348&_cid=P12-MKLZ1L-03304-1

PAT
- Solid dispersion, pharmaceutical composition, and preparation method and use thereforPublication Number: WO-2025067450-A1Priority Date: 2023-09-28
- Method of enhanced absorption of quinoxaline type iii receptor tyrosine kinase inhibitorsPublication Number: WO-2025019622-A2Priority Date: 2023-07-18
- Quinoxaline compounds as type iii receptor tyrosine kinase inhibitorsPublication Number: US-2019308949-A1Priority Date: 2016-10-10
- Quinoxaline Compounds as Type III Receptor Tyrosine Kinase InhibitorsPublication Number: CN-110325515-APriority Date: 2016-10-10
- Quinoxaline compounds as type III receptor tyrosine kinase inhibitorsPublication Number: JP-7206188-B2Priority Date: 2016-10-10Grant Date: 2023-01-17
- quinoxaline compounds as type III tyrosine kinase receptor inhibitorsPublication Number: BR-112019007271-A2Priority Date: 2016-10-10
- Quinoxaline compounds as type III receptor tyrosine kinase inhibitorsPublication Number: AU-2017342928-A1Priority Date: 2016-10-10
- QUINOXALINE COMPOUNDS AS TYPE III RECEPTOR TYROSINKINASE INHIBITORSPublication Number: RU-2019113764-APriority Date: 2016-10-10
- Quinoxaline compounds as type iii receptor tyrosine receptor inhibitorsPublication Number: IL-265829-APriority Date: 2016-10-10
- Quinoxaline compounds as type iii receptor tyrosine kinase inhibitorsPublication Number: CA-3039919-A1Priority Date: 2016-10-10
- Quinoxaline compounds as type iii receptor tyrosine kinase inhibitorsPublication Number: WO-2018071348-A1Priority Date: 2016-10-10
- Quinoxaline compounds as inhibitors of type III receptor tyrosine kinasePublication Number: CN-110325515-BPriority Date: 2016-10-10Grant Date: 2023-06-20
- Quinoxaline compounds as type III receptor tyrosine kinase inhibitorsPublication Number: US-10689362-B2Priority Date: 2016-10-10Grant Date: 2020-06-23
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
////////enrupatinib, ANAX, colony-stimulating factor 1 receptor (CSF1R) inhibitor, antineoplastic, EI 1071, EI-1071, 9L35RVQ9J6
Deulorlatinib


Deulorlatinib
CAS 2131126-33-3
MFC21H162H3FN6O2, MW 409.4 g/mol

- (10R)-7-Amino-12-fluoro-10,15,16,17-tetrahydro-10,16-dimethyl-2-(methyl-d3)-15-oxo-2H-4,8-methenopyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile
- 2H-4,8-Methenopyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile, 7-amino-12-fluoro-10,15,16,17-tetrahydro-10,16-dimethyl-2-(methyl-d3)-15-oxo-, (10R)-
(10R)-7-amino-12-fluoro-2-(2H3)methyl-10,16-dimethyl15-oxo-10,15,16,17-tetrahydro-2H-8,4-
(metheno)pyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile
tyrosine kinase inhibitor, antineoplastic, 7PW3UT8C9B, TGRX 326, TGRX-326
Deulorlatinib is an orally bioavailable inhibitor of the receptor tyrosine kinases anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1), with potential antineoplastic activity. Upon oral administration, deulorlatinib targets, binds to and inhibits the activity of ALK and ROS1, which leads to the disruption of ALK- and ROS1-mediated signaling and the inhibition of cell growth in ALK- and ROS1-expressing tumor cells. ALK belongs to the insulin receptor superfamily and plays an important role in nervous system development. ALK is not expressed in healthy adult human tissue but ALK dysregulation and gene rearrangements are associated with a variety of tumor cell types. ROS1, overexpressed in certain cancer cells, plays a key role in cell growth and survival of cancer cells.
- TGRX-326 Chinese Phase III for Advanced Non-small Cell Lung Cancer (NSCLC)CTID: NCT06082635Phase: Phase 3Status: Active, not recruitingDate: 2025-05-18
- TGRX-326 Pharmacokinetic Drug InteractionCTID: NCT06294561Phase: Phase 1Status: CompletedDate: 2024-06-27
- TGRX-326 Chinese Phase I for Advanced Non-small Cell Lung Cancer (NSCLC)CTID: NCT05441956Phase: Phase 1Status: Active, not recruitingDate: 2025-05-18
- TGRX-326 Chinese Phase II for Advanced Non-small Cell Lung Cancer (NSCLC)CTID: NCT05955391Phase: Phase 2Status: Active, not recruitingDate: 2025-05-18
SYN
WO 2017/148325 A1
syn
https://patentscope.wipo.int/search/en/detail.jsf?docId=US348430040&_cid=P11-MKG9AH-82468-1



Example 6: Synthesis of (10R)-7-amino-12-fluoro-2-(methyl-d3)-10,16-dimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile (the Compound of Formula (A))



| To a 250 mL three-necked flask equipped with magnetic stirring were added the compound of formula (J) (7.0 g, 42.2 mmol) and anhydrous dichloromethane (120 mL), and stirred until the solution became clear. The compound of formula (H) (8.77 g, 46.4 mmol) and then triethylamine (4.69 g, 46.4 mmol) were successively added. The mixture was stirred at room temperature under nitrogen atmosphere for 30 minutes to give a pale yellow clear solution for further use. |
| Alkylation of the Compound of Formula (E-a) with the Compound of Formula (F) to Form the Compound of Formula (D-a): |
| To another 250 mL three-necked flask equipped with magnetic stirring were added the compound of formula (E-a) (11.2 g, 59.3 mmol) and acetonitrile (200 mL), and cesium carbonate (25.7 g, 79.0 mmol) was added with stirring. The mixture was heated to 50° C. under nitrogen atmosphere, and stirred at this temperature for 30 min. The above-mentioned solution of the compound of formula (F) in acetonitrile was slowly added dropwise at 50° C. over 10 minutes. After the dropwise addition was completed, the mixture was reacted with stirring at this temperature for 2 hours. By TLC (DCM:MeOH=20:1) and HPLC monitoring, the reaction was completed. After cooling to room temperature, the reaction was quenched by adding water (200 mL). The reaction solution was diluted with ethyl acetate (300 mL), stirred for 5 minutes, and then filtered through Celite to remove insoluble solids. The filter cake was washed with ethyl acetate (50 mL). The organic layer was separated from the filtrate, and the aqueous phase was extracted with ethyl acetate (60 mL×2). The organic phases were combined, washed with a saturated aqueous solution of sodium carbonate (100 mL×3) and then saturated brine (60 mL), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to dryness under reduced pressure to give 17.5 g of a brown solid in a yield of 90.1% and a purity (HPLC) of >85% (ee>95%). LC-MS (APCI): m/z=390.1 (M+1) +. |
| Introduction of Boc Protecting Group into the Compound of Formula (D-a) to Form the Compound of Formula (C): |
| To a 250 mL single-necked flask equipped with magnetic stirring were added the compound of formula (D-a) (17.5 g, 35.8 mmol) and dichloromethane (200 mL), and stirred until the solution became clear. Triethylamine (14.5 g, 143.2 mmol) and then DMAP (850 mg, 7.2 mmol) were successively added. Boc2O (23.4 g, 107.4 mmol) was slowly added dropwise, and the mixture was reacted with stirring at room temperature under nitrogen atmosphere overnight. By TLC (DCM:MeOH=20:1) and HPLC monitoring, the reaction was completed. The reaction solution was evaporated under reduced pressure to remove the solvent, and the residue was purified by silica gel column chromatography (EA/PE=0-35%) to give 15.4 g of a white solid in a yield of 62.4% and a purity (HPLC) of >95% (ee>95%). LC-MS (APCI): m/z=590.1 (M+1−100) +. 1H NMR (300 MHz, CDCl 3) (δ/ppm): 8.06 (d, J=1.8 Hz, 1H), 7.53-7.48 (m, 1H), 7.24-7.20 (m, 2H), 7.04-6.98 (m, 1H), 6.81 (s, 1H), 5.66-5.59 (m, 1H), 4.89-4.69 (m, 2H), 2.97 (s, 3H), 1.58 (d, J=6.0 Hz, 3H), 1.47 (s, 18H). |
| Cyclization of the Compound of Formula (C) Using Palladium Catalyst to Form the Compound of Formula (B): |
| To a 500 mL single-necked flask equipped with magnetic stirring were added the compound of formula (C) (15.4 g, 22.3 mmol) and 2-methyl-2-butanol (300 mL), and stirred until the solution became clear. Potassium acetate (6.56 g, 66.9 mmol) was added. The system was evacuated with suction and purged with nitrogen gas three times. Palladium acetate (0.75 g, 3.35 mmol) and n-butylbis(1-adamantyl)phosphine (1.60 g, 4.46 mmol) were quickly added. The system was evacuated with suction and purged with nitrogen gas three times. The reaction solution was heated to 110° C. under nitrogen atmosphere, and reacted with stirring at this temperature overnight. By TLC (PE:EA=1:1) and HPLC monitoring, the reaction was completed. The reaction solution was cooled to room temperature, diluted with dichloromethane (300 mL), and filtered through Celite to remove insoluble solids. The filter cake was washed with dichloromethane (50 mL). The filtrates were combined, and concentrated to dryness under reduced pressure. To the residue was added acetonitrile (150 mL), and the mixture was heated to reflux for 1 hour. The oil bath was removed, and the mixture was allowed to slowly cool to room temperature. A large amount of a white solid precipitated out, and the precipitated solid was filtered. The filter cake was washed with acetonitrile (10 mL), and dried to give 8.2 g of a white solids in a yield of 60.4% and a purity (HPLC) of >99.5% (ee>99.9%). LC-MS (APCI): m/z=510.1 (M+1−100) +. 1H NMR (300 MHz, CDCl 3) (δ/ppm): 8.22 (d, J=1.8 Hz, 1H), 7.29-7.25 (m, 1H), 7.22-7.16 (m, 2H), 7.03-6.96 (m, 1H), 5.76-5.70 (m, 1H), 4.42 (q, J=14.1 Hz, 2H), 3.15 (s, 3H), 1.76 (d, J=6.0 Hz, 3H), 1.44 (s, 18H). |
| Removal of the Boc from the Compound of Formula (B) Using an Acid to Form the Compound of Formula (A): |

To a 250 mL single-necked flask equipped with magnetic stirring were added the compound of formula (B) (8.2 g, 13.5 mmol) and dichloromethane (100 mL), and stirred until the solution became clear. The mixture was cooled in an ice-water bath, and trifluoroacetic acid (20 mL) was slowly added dropwise. After the addition was completed, the ice bath was removed, and the mixture was reacted with stirring at room temperature for 2 hours. By TLC (DCM:MeOH=20:1) and HPLC monitoring, the reaction was completed. The reaction solution was evaporated under reduced pressure to remove the organic solvent. Dichloromethane (100 mL) and a saturated aqueous solution of sodium bicarbonate (60 mL) were added under cooling, and the mixture was stirred for 10 minutes. The organic phase was separated, and the aqueous layer was extracted with dichloromethane (50 mL×2). The organic phases were combined, washed successively with water (30 mL) and then saturated brine (500 mL), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to give 5.1 g of an amorphous white solid in a yield of 92.6% and a purity (HPLC) of >99.5% (ee>99.9%). LC-MS (APCI): m/z=410.2 (M+1) +. 1H NMR (300 MHz, CDCl 3) (δ) ppm 7.79 (d, J=1.8 Hz, 1H), 7.31-7.27 (m, 1H), 7.23-7.19 (m, 1H), 7.06-6.97 (m, 1H), 6.87 (d, J=1.8 Hz, 1H), 5.75-5.70 (m, 1H), 5.09 (br s, 2H), 4.40 (q, J=14.1 Hz, 2H), 3.12 (s, 3H), 1.78 (d, J=6.6 Hz, 3H).
PAT
Preparation method for deuterated macrocyclic compound
Publication Number: US-2022024908-A1
Priority Date: 2018-11-28
ADVERTISEMENT
Advect Process Systems Ltd.
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////deulorlatinib, tyrosine kinase inhibitor, antineoplastic, 7PW3UT8C9B, TGRX 326, TGRX-326
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
NEXT
ADVERTISEMENT
Advect Process Systems Ltd.
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1
Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
Casdatifan


Casdatifan
CAS 2709069-30-5
MF C21H17F4NO3S, 439.4 g/mol
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-(methanesulfonyl)-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methylsulfonyl-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methylsulfonyl-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
(5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methanesulfonyl-2, 3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
hypoxia-inducible factor (HIF) inhibitor, antineoplastic, AB 521, DP73UWL6LE
Casdatifan is an orally bioavailable allosteric inhibitor of hypoxia inducible factor (HIF)-2alpha, with potential antineoplastic activity. Upon oral administration, casdatifan targets and allosterically binds to a hydrophobic pocket on HIF-2alpha leading to a confirmational change that prevents HIF-2alpha heterodimerization with HIF-1beta and binding to the hypoxia response element (HRE) binding site on DNA. This results in decreased transcription and expression of HIF-2alpha downstream target genes, many of which regulate tumor cell growth and survival. Blocking HIF-2alpha reduces the proliferation of HIF-2alpha-expressing tumor cells. HIF-2alpha, a heterodimeric transcription factor overexpressed under hypoxic conditions in many cancer cell types, promotes proliferation, progression and metastasis of tumors.
- A Phase 1 Study of AB521 Monotherapy and Combination Therapies in Renal Cell Carcinoma and Other Solid TumorsCTID: NCT05536141Phase: Phase 1Status: RecruitingDate: 2026-01-02
- A Relative Bioavailability Study and Food Effect Study of AB521 in Healthy Adult VolunteersCTID: NCT05999513Phase: Phase 1Status: CompletedDate: 2024-10-17
- A Study to Investigate the Efficacy and Safety of Volrustomig ± Casdatifan vs Nivolumab + Ipilimumab as 1L Treatment for Advanced ccRCCCTID: NCT07000149Phase: Phase 3Status: Active, not recruitingDate: 2025-11-14
- Study of Zanzalintinib (XL092) + AB521 and Zanzalintinib + AB521 + Nivolumab in Participants With Advanced Clear Cell Renal Cell Carcinoma (ccRCC) or Other Advanced Solid Tumors (STELLAR-009)CTID: NCT06191796Phase: Phase 1Status: TerminatedDate: 2025-06-12
- Drug-Drug Interaction Study of Casdatifan in Healthy Adult Participants (ARC-29)CTID: NCT06919991Phase: Phase 1Status: CompletedDate: 2025-11-13
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.4c00497




PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021188769&_cid=P12-MKDEE0-87371-1
Example 215: (5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methanesulfonyl-2, 3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile



ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735
Email : info@anaxlab.com
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US442743749&_cid=P12-MKDEE0-87371-1
Example 2: Synthesis of (5R,6S,8R)-3,5,6-trifluoro-8-[(1S,2R)-2-fluoro-1-hydroxy-7-methanesulfonyl-2,3-dihydro-1H-inden-4-yl]-5,6,7,8-tetrahydronaphthalene-1-carbonitrile

Step i: Synthesis of Compound 11

Product 10 of step h (37.85 g, 78.28 mmol, 1.0 equiv.) was dissolved in THF (400 mL) at 23° C. A solution of hydrochloric acid (320 mL, 6M) was added dropwise over 20 min, and the mixture was stirred at 30° C. for 4 h. After this time, the reaction reached completion, as shown by LC/MS (MeCN/H 2O—20%→100%, 6 min). The reaction mixture was diluted with water (1 L) and EtOAc (0.6 L), back-extracted twice with EtOAc, and washed with water, sat. sol. NaHCO 3, and brine. The organic layer was dried over Na 2SO 4, filtered, and concentrated. The material (32.25 g, 94%) was triturated with CH 2Cl 2 (45 mL) at 45° C., filtered, and washed with a minimum of cold CH 2Cl 2 and cold hexanes to afford 11 as a white crystalline solid (26.15 g, 76%, 12:1 dr). 1H NMR (400 MHZ, DMSO-d 6) δ 7.96 (ddd, J=8.3, 2.7, 1.3 Hz, 1H), 7.89 (dd, J=8.9, 2.7 Hz, 1H), 7.57 (d, J=8.1 Hz, 1H), 6.66 (d, J=8.1 Hz, 1H), 5.95 (ddd, J=51.2, 13.5, 2.2 Hz, 1H), 5.89 (d, J=5.6 Hz, 1H), 5.47 (ddd, J=10.0, 6.2, 4.9 Hz, 1H), 5.26 (qd, J=52.5, 5.4 Hz, 1H), 5.12 (tddd, J=47.4, 18.7, 10.3, 2.7 Hz, 1H), 4.83 (t, J=5.4 Hz, 1H), 3.30 (s, 3H), 3.28-3.13 (m, 2H), 2.71-2.60 (m, 1H), 2.02-1.85 (m, 1H). 19F NMR (376 MHZ, DMSO-d 6) δ −112.3, −179.6, −196.7, −199.4. ESI MS [M+Na] + for C 21H 17F 4NO 3SNa, calcd 462.0, found 461.9.
PAT
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2023021476-A1Priority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of HIF-2αPublication Number: US-11407712-B2Priority Date: 2020-03-19Grant Date: 2022-08-09
- Tetralin and tetrahydroquinoline compounds as inhibitors of HIF-2alphaPublication Number: US-12103907-B2Priority Date: 2020-03-19Grant Date: 2024-10-01
- Tetralin and tetrahydroquinoline compounds as HIF-2α inhibitorsPublication Number: CN-115298165-APriority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2021317079-A1Priority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: WO-2021188769-A1Priority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2024254079-A1Priority Date: 2020-03-19
- Process for preparing tetralin compoundsPublication Number: US-12145901-B1Priority Date: 2021-09-17Grant Date: 2024-11-19
- Tetralin and tetrahydroquinoline compounds as inhibitors of HIF-2alphaPublication Number: US-11787762-B2Priority Date: 2020-03-19Grant Date: 2023-10-17
- Tetrahydronaphthalene and tetrahydroquinoline compounds as HIF-2 alpha inhibitorsPublication Number: CN-119118872-APriority Date: 2020-03-19
- Tetralin and tetrahydroquinoline compounds as HIF-2α inhibitorsPublication Number: CN-115298165-BPriority Date: 2020-03-19Grant Date: 2024-09-17
- Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alphaPublication Number: US-2025214930-A1Priority Date: 2020-03-19
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735
Email : info@anaxlab.com
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////Casdatifan, hypoxia-inducible factor (HIF) inhibitor, antineoplastic, AB 521, DP73UWL6LE
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions,, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
Cambritaxestat


Cambritaxestat
CAS 1979939-16-6
MFC25H22ClF3N4O2 MW502.9 g/mol
N-[(1S)-1-(4-chlorophenyl)ethyl]-3-[3-[[4-(trifluoromethoxy)phenyl]methyl]imidazo[4,5-b]pyridin-2-yl]propanamide
N-[(1S)-1-(4-chlorophenyl)ethyl]-3-(3-{[4-(trifluoromethoxy)phenyl]methyl}-3H-imidazo[4,5-b]pyridin-2-yl)propanamide
autotaxin inhibitor, antineoplastic, Orphan Drug, IOA 289, IOA-289, IOA289, LYY3P2KA27, CRT 0273750
- OriginatorCancer Research Technology; Merck & Co
- DeveloperiOnctura
- ClassAntifibrotics; Antineoplastics; Small molecules
- Mechanism of ActionAngiogenesis inhibitors; Cell proliferation inhibitors; ENPP2 protein inhibitors
- Orphan Drug StatusYes – Pancreatic cancer
- Phase I/IIPancreatic cancer
- Phase ISolid tumours
- PreclinicalNon-alcoholic steatohepatitis
- 14 Oct 2025Efficacy and adverse event data from a phase I/II trial in Pancreatic cancer released by iOnctura
- 04 Oct 2024Cambritaxestat is still in phase-I development in Solid-tumours (In volunteers) in Italy (PO, Capsule) (NCT05027568)
- 31 May 2024Efficacy and adverse event data from a phase I/II trial in Pancreatic cancer presented at the 60th Annual Meeting of the American Society of Clinical Oncology (ASCO-2024)
Cambritaxestat is an autotaxin inhibitor.
Cambritaxestat is an orally bioavailable small molecule inhibitor of autotaxin (ATX; ectonucleotide pyrophosphatase/phosphodiesterase family member 2; ENPP2), with potential antifibrotic and antineoplastic activities. Upon oral administration, cambritaxestat targets and binds to both the substrate pocket and the lysophosphatidic acid (LPA) carrier channel of ATX, thereby inhibiting the activity of ATX. This both directly inhibits the proliferation of tumor cells and reduces fibrosis in the tumor microenvironment (TME), allowing lymphocytes to infiltrate into the tumor and enhancing immune responses against tumor cells. ATX, a secreted glycoprotein with lysophospholipase D activity, hydrolyzes lysophosphatidylcholine (LPC) to LPA. LPA-mediated signaling plays an important role in cellular migration, proliferation and survival in fibrotic response. ATX and LPA are overexpressed in many tumors.
- A Study to Assess an ATX Inhibitor (IOA-289) in Healthy VolunteersCTID: NCT05027568Phase: Phase 1Status: CompletedDate: 2025-03-20
- A Study to Assess an ATX Inhibitor (IOA-289) in Patients with Metastatic Pancreatic CancerCTID: NCT05586516Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-03-20
SYN
WO2016/124939
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016124939&_cid=P22-MKBYYZ-98558-1


SYN

WO2016/124939 describes various ATX inhibitor compounds and their use in the treatment of proliferative disorders in which ATX activity is implicated, including Compound 1.
Compound 1 is example 40 in WO2016/124939, which document is incorporated herein by reference in its entirety. WO2016/124939 describes over 200 examples. Compound 1’s structure is according to Formula I.

PAT
- Autotaxin inhibitory compoundsPublication Number: US-10654846-B2Priority Date: 2015-02-06Grant Date: 2020-05-19
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-B3Priority Date: 2015-02-06Grant Date: 2024-05-29
- Autotaxin inhibitor compoundsPublication Number: ES-2778898-T7Priority Date: 2015-02-06Grant Date: 2024-11-15
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-A1Priority Date: 2015-02-06
- Home chemokine inhibiting compoundsPublication Number: CN-107428752-BPriority Date: 2015-02-06Grant Date: 2021-06-29
- Autotaxin inhibitory compoundsPublication Number: US-11453666-B2Priority Date: 2015-02-06Grant Date: 2022-09-27
- Autotaxin Inhibitory CompundsPublication Number: US-2020283435-A1Priority Date: 2015-02-06
- Autotaxin inhibitory compoundsPublication Number: WO-2016124939-A1Priority Date: 2015-02-06
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: WO-2022207648-A1Priority Date: 2021-03-29
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: EP-4313059-A1Priority Date: 2021-03-29
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: US-2024216385-A1Priority Date: 2021-03-29
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-B1Priority Date: 2015-02-06Grant Date: 2020-01-08
- Autotaxin inhibitory compoundsPublication Number: US-2018016274-A1Priority Date: 2015-02-06
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: WO-2022258693-A1Priority Date: 2021-06-09
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: US-2025057820-A1Priority Date: 2021-06-09
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: EP-4351563-A1Priority Date: 2021-06-09
- Autotaxin (ATX) inhibitors for the treatment of pancreatic cancerPublication Number: CN-117295496-APriority Date: 2021-06-09
- PI3K-δ inhibitors for the treatment of pancreatic cancerPublication Number: CN-116997340-APriority Date: 2021-03-29
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- Characterization and translational development of IOA-289, a novel autotaxin inhibitor for the treatment of solid tumorsPublication Name: Immuno-Oncology and TechnologyPublication Date: 2023-06PMCID: PMC10205783PMID: 37234285DOI: 10.1016/j.iotech.2023.100384
- The IUPHAR Guide to Immunopharmacology: connecting immunology and pharmacologyPublication Name: ImmunologyPublication Date: 2020-03-02PMCID: PMC7160657PMID: 32020584DOI: 10.1111/imm.13175
- Discovery of potent inhibitors of the lysophospholipase autotaxinPublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2016-11-15PMID: 27780639DOI: 10.1016/j.bmcl.2016.10.036
///////Cambritaxestat, autotaxin inhibitor, antineoplastic, Orphan Drug, IOA 289, IOA-289, IOA289, LYY3P2KA27, CRT 0273750

{kind=link}
{kind=link}