
Home » Posts tagged 'ANTIMALARIAL'
Tag Archives: ANTIMALARIAL
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
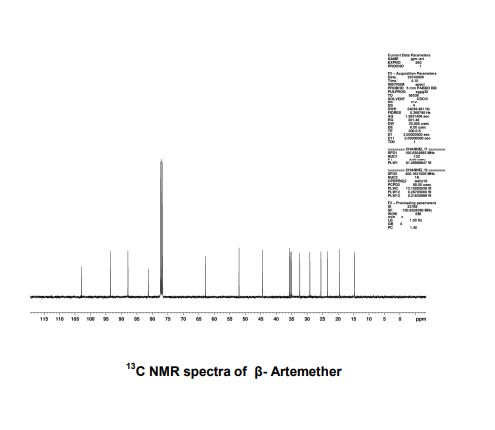
ARTEMETHER
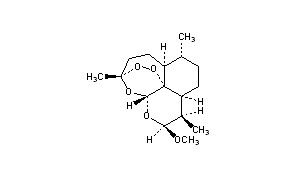
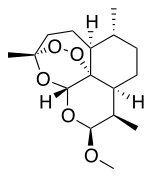
ARTEMETHER
- Molecular FormulaC16H26O5
- Average mass298.375 Da
(3R,5aS,6R,8aS,9R,10S,12R,12aR)-10-methoxy-3,6,9-trimethyldecahydro-3,12-epoxy[1,2]dioxepino[4,3-i]isochromene
(4S,5R,8S,9R,10S,12R,13R)-10-Methoxy-1,5,9-trimethyl-11,14,15,16-tetraoxatetracyclo[10.3.1.04,13.08,13]hexadecane[
3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, decahydro-10-methoxy-3,6,9-trimethyl-, (3R,5aS,6R,8aS,9R,10S,12R,12aR)-
3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, decahydro-10-methoxy-3,6,9-trimethyl-, (5aS,6R,8aS,9R,10S,12R,12aR)-
71963-77-4[RN]
dihydroartemisinin methyl ether
Dihydroqinghaosu Methyl Ether
KD4165000
PALUTHER
- SM 224
- SM-224
- 3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, decahydro-10-methoxy-3,6,9-trimethyl-, [3R-(3α,5aβ,6β,8aβ,9α,10α,12β,12aR*)]-
- (3R,5aS,6R,8aS,9R,10S,12R,12aR)-Decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin
- (+)-Artemether
Artemether
CAS Registry Number: 71963-77-4
CAS Name: (3R,5aS,6R,8aS,9R,10S,12R,12aR)-Decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin
Additional Names: dihydroartemisinin methyl ether; dihydroqinghaosu methyl ether; o-methyldihydroartemisinin
Manufacturers’ Codes: SM-224
Trademarks: Paluther (RPR)
Molecular Formula: C16H26O5, Molecular Weight: 298.37,
Percent Composition: C 64.41%, H 8.78%, O 26.81%
Literature References: Derivative of artemisinin, q.v. Prepn: Y. Li et al.,K’o Hsueh T’ung Pao24, 667 (1979), C.A.91, 211376u (1979); eidem,Acta Pharm. Sin.16, 429 (1981). Absolute configuration: X.-D. Luo et al.,Helv. Chim. Acta67, 1515 (1984). NMR spectral study: F. S. El-Feraly et al.,Spectrosc. Lett.18, 843 (1985). Inhibition of protein synthesis: H. M. Gu et al.,Biochem. Pharmacol.32, 2463 (1983). Antimalarial activity: S. Thaithong, G. H. Beale, Bull. WHO63, 617 (1985). Series of articles on chemistry, pharmacology and antimalarial efficacy: China Cooperative Research Group on Qinghaosu, J. Tradit. Chin. Med.2, 3-50 (1982). Toxicity data: eidem,ibid. 31. Clinical trial in cerebral malaria in children: M. B. van Hensbroek et al.,N. Engl. J. Med.335, 69 (1996). Review: R. N. Price, Expert Opin. Invest. Drugs9, 1815-1827 (2000).
Properties: Crystals, mp 86-88°. [a]D19.5 +171° (c = 2.59 in CHCl3). LD50 i.m. in mice: 263 mg/kg (China Cooperative Research Group on Qinghaosu).
Melting point: mp 86-88°
Optical Rotation: [a]D19.5 +171° (c = 2.59 in CHCl3)
Toxicity data: LD50 i.m. in mice: 263 mg/kg (China Cooperative Research Group on Qinghaosu)
Therap-Cat: Antimalarial.
Keywords: Antimalarial.
Artemether is an antimalarial agent used in combination with lumefantrine for the treatment of acute uncomplicated malaria caused by Plasmodium falciparum.
Artemether is an antimalarial agent used to treat acute uncomplicated malaria. It is administered in combination with lumefantrine for improved efficacy. This combination therapy exerts its effects against the erythrocytic stages of Plasmodium spp. and may be used to treat infections caused by P. falciparum and unidentified Plasmodium species, including infections acquired in chloroquine-resistant areas.
Artemether is a natural product which effectively kills both malarial parasites P. falciparum and P. vivax. Artemether is usually used in combination with Lumefantrine for the treatment of malaria. Arthemether also kills trematodes of the species Schistosoma, providing protection against schistosomiasis. Sesquiterpene lactones like artemether, artesunate, and artemisinin have potential applications in certain types of cancer and inflammatory conditions.
Artemether is a medication used for the treatment of malaria.[2] The injectable form is specifically used for severe malaria rather than quinine.[2] In adults, it may not be as effective as artesunate.[2] It is given by injection in a muscle.[2] It is also available by mouth in combination with lumefantrine, known as artemether/lumefantrine.[3][4]
Artemether causes relatively few side effects.[5] An irregular heartbeat may rarely occur.[5] While there is evidence that use during pregnancy may be harmful in animals, there is no evidence of concern in humans.[5] The World Health Organization (WHO) therefore recommends its use during pregnancy.[5] It is in the artemisinin class of medication.[5]
Artemether has been studied since at least 1981, and been in medical use since 1987.[6] It is on the World Health Organization’s List of Essential Medicines.[7]
Synthesis Reference
Haynes RK, Vonwiller SC: Extraction of artemisinin and artemisinic acid: preparation of artemether and new analogues. Trans R Soc Trop Med Hyg. 1994 Jun;88 Suppl 1:S23-6. Pubmed.
REF
ChemMedChem (2007), 2, (10), 1448-1463
PAT
| Malaria is a serious parasitic disease caused by Plasmodium parasites in the human body. Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malaria and Plasmodium knowlesi are the parasites that live in humans, of which P. vivax and P. falciparum are the most common. |
| Traditional anti-malarial drugs mainly include quinine, chloroquine, primaquine, and pyrimethamine. In 1972, the antimalarial active ingredient artemisinin extracted from the Compositae plant Artemisia annuaL by Chinese scientists is the most popular antimalarial effect after chloroquine, pyrimethamine, primary amine and sulfonamide. Drugs, especially for the treatment of cerebral malaria and anti-chloroquine malaria. |
| At present, a large number of artemisinin derivatives have been synthesized and screened for antimalarial activity. Artemether is a compound with excellent curative effect. In addition to the advantages of artemisinin’s quick effect and low toxicity, its solubility in oil is also higher than that of artemisinin. Artemisinin is large, which is especially beneficial for the preparation of preparations. Since artemether has two products, α and β epimers, and the antimalarial activity of artemether is mainly isomer β, so the industrial automation and intelligent production of β-artemether and the improvement of the process are realized. , reducing the impurities produced by the reaction, improving the quality of the product, and improving the purity of the product are the problems that need to be solved in today’s scientific research. |
| Patent CN104557965B discloses a preparation process of β-artemether, which mainly includes adding dihydroartemisinin and etherification reagent to alcohol to form a reaction system, and then adding acid to the reaction system for reaction. Water or non-alkaline aqueous solution is added to the reaction system to crystallize, namely β-artemether. The preparation process claims to effectively inhibit the production of isomer α-artemether in the reaction, and can make the etherification reaction proceed mildly, with simple post-treatment and high purity; although the purity of the product has been improved, the yield and Purity needs to be further improved. |
| Patent CN102731523B discloses a method for preparing β-artemether, which mainly includes the reaction of artemisinin under the action of a reducing agent to generate dihydroartemisinin, and the reaction of dihydroartemisinin with p-toluenesulfonic acid to generate β-artemisinin. The crude artemether is crystallized with methanol, ethanol, ethylene glycol or isopropanol, filtered, washed and dried. The method for preparing B-artemether of the invention has mild conditions, is environmentally friendly, is suitable for industrial production, and has a product yield of over 90 percent and a purity of 99.2 percent. The crystallization step of the invention adopts organic reagents, which adversely affects the quality control of subsequent products. |
| Patent CN103180325B discloses a method for preparing β-artemether, which uses dihydroartemisinin as a raw material and undergoes etherification reaction with trimethyl orthoformate in organic solvents including esters and alkanes to obtain β-artemether. The method of the invention is easy to control in process operation, high in yield, low in cost and high in product quality, and is suitable for industrial production. The method requires vacuum distillation, the obtained crude product needs to be redissolved with methanol, decolorized with activated carbon, etc., new impurities are easily introduced, the operation is not simple enough, and the efficiency is low. |
| Patent CN107793428A discloses a preparation method of artemether, hydrogenating artemisinin to obtain dihydroartemisinin, adding trimethyl orthoformate, reacting with boron trifluoride ether solution, slowly adding saturated sodium bicarbonate solution dropwise, The system was adjusted to neutrality, the liquids were separated, the aqueous phase was extracted with dichloromethane, the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure to obtain a solid; the obtained solid was dissolved in methanol, and an appropriate amount of activated carbon was added to obtain a solid. Reflux and decolorize, filter, add pure water dropwise to the filtrate, crystallize, wash with water, and dry to obtain artemether. However, this method requires steps such as extraction with an organic reagent dichloromethane and decolorization with activated carbon, which is cumbersome to handle. |
| Therefore, the following problems generally exist in the process of preparing β-artemether at present: |
| (1) when preparing β-artemether, the reaction time is longer, the impurities are large, and the purity and yield of the product are not high enough; |
| (2) The use of organic reagents in the subsequent purification process has a certain impact on the quality control of the product; |
| (3) The batch production equipment is adopted, the subsequent process steps are many, the degree of industrialization is low, the production efficiency is low, and it does not meet the requirements of GMP. |
| Example 1 |
| This embodiment includes the following steps: |
| (1) at room temperature, add methanol 2400L in the 3000L stirred tank (1), then add 600kg of dihydroartemisinin through the solid feed pump, and circulate and disperse evenly; |
| (2) add etherification agent trimethyl orthoformate and acid catalyst acetyl chloride through three-way automatic feeding mixing reactor again, the volume ratio is 500:100:3, the mixing reactor control temperature is 5 ℃, and the flow rate of control feeding is 5L /min; |
| (3) in the continuous flow pipeline, enter the second mixer and add 5% sodium bicarbonate solution to neutralize, and the adding speed is 1.0L/min, and is filtered through the fine filter; |
| (4) Then directly enter the 2000L crystallization reaction kettle 11 with 300L of water added in advance and keep the temperature at 10°C. At the same time, purified water was added to the reaction kettle at a rate of 12L/min, and the crystallization was continued for 1.5h; the jacket of the crystallization kettle was fed with -10°C chilled water for 30min, and the temperature of the system was controlled to 5°C. |
| (5) centrifugal washing, obtaining crude artemether 704.5kg, drying to obtain artemether fine product 608.6kg, β-artemether purity 99.83%, α-artemether impurity 0.12%, and other single impurities less than 0.1%, The content is 99.8%, the mass yield is 96.1%, and the molar yield is 91.42%. |
| Example 2 |
| This embodiment includes the following steps: |
| (1) at room temperature, 2400L of methanol was pumped into the 3000L reactor 1, and then 800kg of dihydroartemisinin was added by the solid feed pump, and the circulation was uniformly dispersed; |
| (2) add etherifying agent dimethyl phosphate and acid catalyst boron trifluoride ether through the three-way automatic feeding mixing reactor again, the volume ratio is 500:105:3.5, the mixing reactor control temperature is 3 ℃, and the control feeding flow rate is 3L/min; |
| (3) in the continuous flow pipeline, enter the second mixer and add 3% sodium bicarbonate solution to neutralize, and the speed of addition is 1.8L/min, through the fine filter; |
| (4) Directly enter the 2000L crystallization reaction kettles 11 and 12 with 300L of water added in advance and the temperature kept at 10°C. At the same time, purified water was added to the reaction kettle at 9 L/min, and the crystallization was continued for 2.5 hours; the jacket of the crystallization kettle was fed with -10 °C chilled water for 30 minutes, and the temperature of the system was controlled to 10 °C |
| (5) centrifugal washing, obtain crude artemether 939.3kg, oven dry to obtain artemether fine product 809.7kg, β-artemether purity 99.81%, α-artemether impurity 0.11%, other single impurities are less than 0.1%, The content is 99.8%, the mass yield is 96.2%, and the molar yield is 91.6%. |
| Example 3 |
| This embodiment includes the following steps: |
| (1) 2400L of methanol was pumped into the 3000L reactor F1 at room temperature, and then 400kg of dihydroartemisinin was added through the solid feed pump, and the circulation was uniformly dispersed; |
| (2) Add etherification agent dimethyl phosphate and acid catalyst trimethylchlorosilane through the three-way automatic feeding mixing reactor, the volume ratio is 500:95:2.5, the mixing reactor is controlled at a temperature of 8 °C, and the feeding liquid is controlled to be added. The flow rate is 7L/min, and the reaction time is; |
| (3) in the continuous flow pipeline, enter the second mixer and add 8% sodium bicarbonate solution for neutralization, and the rate of addition is 0.6L/min, passing through the fine filter; |
| (4) Directly enter into the 2000L crystallization reactor J2 with 300L water added in advance and keeping the temperature at 10°C. At the same time, purified water was added to the reaction kettle at 15 L/min, and the crystallization was continued for 1 hour; the jacket of the crystallization kettle was fed with -10 °C chilled water for 30 minutes, and the temperature of the system was controlled to 0 °C |
| (5) centrifugal washing, obtain crude artemether 939.3kg, oven dry to obtain artemether fine product 809.7kg, β-artemether purity 99.81%, α-artemether impurity 0.11%, other single impurities are less than 0.1%, The content is 99.8%, the mass yield is 95.5%, and the molar yield is 90.9%. |
| Comparative Example 1 |
| The difference between this embodiment and Example 1 is that hydrochloric acid is used instead of the acidic catalyst. Finally, 633.6kg of crude artemether was obtained, and 550.3kg of fine artemether was obtained by drying. The purity of β-artemether was 94.20%, and the impurities of α-artemether were 3.66%. %, and the molar yield was 80.6%. |
| Comparative Example 2 |
| The difference between this embodiment and Example 1 is that the step of adding water in advance in the crystallization kettle is removed. Finally, 645.1kg of crude artemether was obtained, and 562.2kg of fine artemether was obtained by drying. The purity of β-artemether was 99.68%, the impurity of α-artemether was 0.22%, and the average of single and impurity was less than 0.1%. The mass yield was 88.7%. %, and the molar yield was 84.4%. |
| In Comparative Example 2, the step of adding water in advance in the crystallization was removed, the purity of β-artemether was 99.68%, and the yield was 88.7%. The yield dropped by 7.6%. |
| The above detailed description is a specific description of one of the feasible embodiments of the present invention, and this embodiment is not intended to limit the patent scope of the present invention. Any equivalent implementation or modification that does not depart from the present invention shall be included in the present invention. within the scope of the technical solution. |
SYN1
Synthetic Reference
Continuous synthesis of artemisinin-derived medicines; Gilmore, Kerry; Kopetzki, Daniel; Lee, Ju Weon; Horvath, Zoltan; McQuade, D. Tyler; Seidel-Morgenstern, Andreas; Seeberger, Peter H. Chemical Communications (Cambridge, United Kingdom); Volume 50; Issue 84; Pages 12652-12655; Journal; 2014
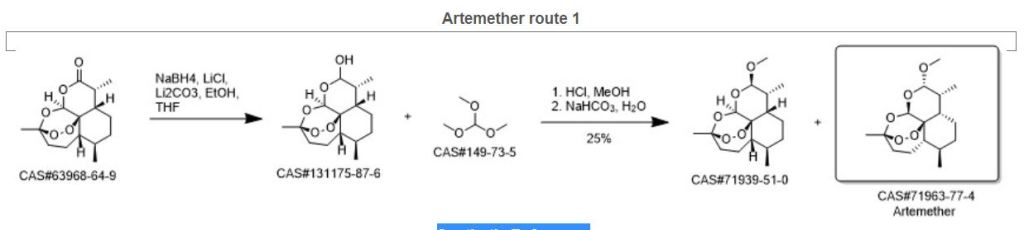
SYN2
Synthetic Reference
An Improved Manufacturing Process for the Antimalaria Drug Coartem. Part I; Boehm, Matthias; Fuenfschilling, Peter C.; Krieger, Matthias; Kuesters, Ernst; Struber, Fritz; Organic Process Research & Development; Volume 11; Issue 3; Pages 336-340; Journal; 2007
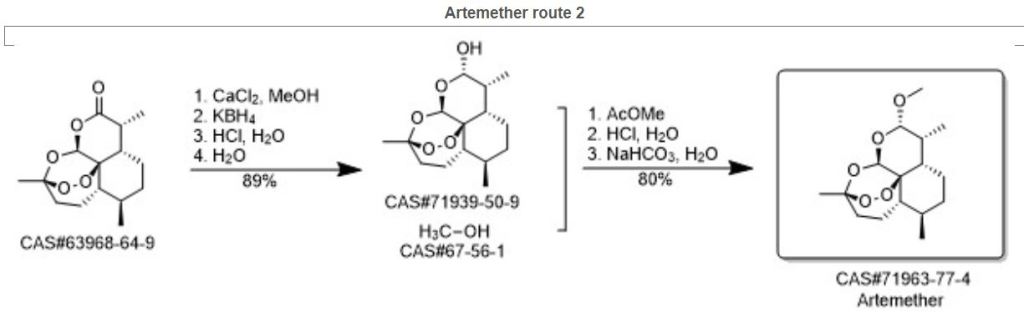
SYN3
Synthetic Reference
Some transition metal complexes bearing artemisinin derivatives and (N-N-O) tridentate chromium (III) complexes ligated by 2-benzolmidazo-yl-6-acetyl-pyridines for catalytic behaviour towards ethylene; Obaleye, Joshua Ayoola; Amolegbe, Saliu Alao; Adewuyi, Sheriff; Sun, Wenhua; Oshodi, Margaret Damilola; Journal of Chemistry and Chemical Engineering; Volume 4; Issue 12; Pages 23-32; Journal; 2010
SYN4
Synthetic Reference
Method and apparatus for the synthesis of dihydroartemisinin and artemisinin derivatives; Kopetzki, Daniel; McQuade, David Tyler; Seeberger, Peter H.; Gilmore, Kerry; Assignee Max-Planck-Gesellschaft zur Foerderung der Wissenschaften e.V., Germany; 2015; Patent Information; Jan 21, 2015; EP 2826779 A1
PAPER
https://pubs.rsc.org/en/content/articlehtml/2014/ra/c4ra05531d
An efficient one pot green synthesis of β-artemether/arteether from artemisinin has been developed using a sodium borohydride-cellulose sulfuric acid (CellSA) catalyst system. The green methodology is high yielding and the catalyst has good recyclability.
Experimental section
Representative procedure for catalyst preparation
Preparation of cellulose sulfuric acid.To a magnetically stirred mixture of 5.00 g of cellulose (DEAE for column chromatography, Merck) in 20 ml of n-hexane, 1.0 g of chlorosulfonic acid (9 mmol) was added dropwise at 0 °C over 2 h. HCl gas was removed from the reaction vessel immediately. After the addition was complete, the mixture was stirred for 2 h. Then, the mixture was filtered, washed with 30 ml of acetonitrile, and dried at room temperature to obtain 5.47 g cellulose sulfuric acid as a white powder.17
General procedure for the arteether from artemisinin in one-pot
To a solution of artemisinin (200 mg, 0.71 mmol) in ethanol (15 ml) and trimethyl orthoacetate (0.5 ml) was added NaBH4 (67 mg, 1.77 mmol, 2.5 equ.) and cellulose sulfuric acid (0.015 g). Reaction mixture was carried out at −5 to 0 °C for 60 min, and then stirred at room temperature for 1.5 h. Then we added a solution of sodium bicarbonate to quenched the reaction. The slurry was stirred in an below 20 °C for 1 h and allowed to settle for 30 min. Solid crude arteether was collected by filtration, and the cake was washed with of ethanol. The reaction mass was heated to 40 ± 5 °C in water. The reaction mass was seeded with pure β-arteether. Then it was filtered, washed with chilled 50% solution of ethanol in water and dried.
General procedure for the artemether from artemisinin in one-pot
Artemisinin (200 mg, 0.71 mmol) in methanol (15 ml) and trimethylorthoformate (0.5 ml), cellulose sulfuric acid (0.015 g), was carried out at −5 to 0 °C for 60 min, and then stirred at room temperature for 1.5 h. The reaction was monitored by TLC and HPLC to check completion of the reaction. The cellulose sulfuric acid was removed by filtration, the filtrate was concentrated. Then we added a solution of sodium bicarbonate to terminate the reaction. Then, follow above recrystallization method.
Preparation of cellulose sulfuric acid. To a magnetically stirred mixture of 5.00 g of cellulose (DEAE for column chromatography, Merck) in 20 ml of n-hexane, 1.0 g of chlorosulfonic acid (9 mmol) was added dropwise at 0 0 C over 2 h. HCl gas was removed from the reaction vessel immediately. After the addition was complete, the mixture was stirred for 2 h. Then, the mixture was filtered, washed with 30 ml of acetonitrile, and dried at room temperature to obtain 5.47 g cellulose sulfuric acid as a white powder. K General procedure for the arteether from artemisinin in one-pot. To a solution of artemisinin (200 mg, 0.71 mmol) in ethanol (15 mL) and trimethyl orthoacetate (0.5 mL) was added NaBH4 (67 mg, 1.77 mmol, 2.5 equ.) and cellulose sulfuric acid (0.015 g). Reaction mixture was was carried out at -5 to 0°C for 60 min, and then stirred at room temperature for 1.5 h. Then we added a solution of sodium bicarbonate to quenched the reaction. The slurry was stirred in an below 20 0 C for 1 h and allowed to settle for 30 min. Solid crude arteether was collected by filtration, and the cake was washed with of ethanol. The reaction mass was heated to 40± 5 0 C in water. The reaction mass was seeded with pure β–arteether. Then it was filtered, washed with chilled 50% solution of ethanol in water and dried. General procedure for the artemether from artemisinin in one-pot. Artemisinin (200 mg, 0.71 mmol) in methanol (15 ml) and trimethylorthoformate (0.5 ml), cellulose sulfuric acid (0.015 g), was carried out at -5 to 0°C for 60 min, and then stirred at room temperature for 1.5 h. The reaction was monitored by TLC and HPLC to check completion of the reaction. The cellulose sulfuric acid was removed by filtration, the filtrate was concentrated. Then we added a solution of sodium bicarbonate to terminate the reaction. Then, follow above recrystallization method.
PATENT
https://patents.google.com/patent/US6683193B2/en
Approximately, out of the 4 billion people suffering from malaria, 1-3 million, mostly children die every year worldwide. The rapidly spreading multidrug resistant parasite to standard quinoline based antimalarial drugs such as chloroquine and mefloquine based antimalarial complicate chemotherapy treatment of malaria patients.
Artemether is a methyl ether derivative of dihydroartemisinin. Dihydroartemisinin is derived from arternisinin, a novel sesquiterpene endoperoxide isolated from the plant Artemisia annua. Artemisinin and its derivative artemether, arteether, artelinate and artesunate a novel class of antimalarials derived from Artemisia annua are now proving their promising activity and being used for the treatment; of uncomplicated severe complicated/cerebral and multi drug resistant malaria.
Artemether, developed in France and China has undergone extensive preclinical, animal, toxicological studies as well as clinical studies. Artemether is more potential as compared to artemisinin and an antimalarial drug especially for treating multi drug resistant and complicated strains of Plasmodium falciparum.
Artemether shows rapid shizonticidal action with quicker parasite clearance rate, short half life less side effect and low recrudence rate. Brossi, et al (Brossi, A; Venugopalan, B, Domingueg, G L; Yeh, H. J. C; Flippend-Anderson, J. L.; Buchs, P; Luo, X. D.; Milhous,W and peters, W; J. Med. Chem. 31, 646-649, 1988) reported the preparation of arteether, the ethyl ether derivative of dihydroartemisinin in two steps: First artemisinin was reduced with an excess of sodium borohydride in methanol at 0 to −5 degree C. in 3 hours to dihydroartemisinin in 79% yield. In the second step arteether is prepared by dissolving the dihydroartemisinin in the solvent mixture of benzene and ethanol at 45 degree C. followed by addition of BF3 etherate and refluxing the reaction mixture at 70 degree C. for one hour. After completion of the reaction it was worked up, dried over anhydrous sodium sulphate with removal of the solvent dichloromethane. The reaction yielded arteether along with some impurities. Column chromatography of the reaction mixture over silica gel, 1:20 ratio yielded pure alpha and beta arteether in nearly qualitative yield.
EL-Feraly etal. (E L Feraly, F. S; Al-Yahya M A; Orabi, K. Y; Mc-Phail D R and Me Phail A. T. J.Nat.Prod. 55, 878-883 1992) reported the preparation of arteether by a process in which anhydrodihydroartemisinin, prepared from artemisinin was dissolved in absolute alcohol. The reaction mixture was stirred in the presence of p-toluene sulphonic acid used as a catalyst. On workup it yielded a mixture of beta arteether and C-11 epimer in the ratio of 3:1. In this process only beta arteether, is obtained and separation of C-11 epimer is difficult and preparation of anhydrodihydroartemisinin is a tedious process. The reaction took 22 hours to complete. The lewis acid catalyst used in this reaction is required in large amount (60 mg. acid catalyst by 100 mg. anhydrodihydroartemisinin).
In another method Bhakuni etal (Bhakuni, R. S.; Jain D. C and Sharma R. P. Indian. J. Chemistry, 34B, 529-30, 1995) arteether, artemether and other ether derivatives were prepared from dihydroartemisinin in different alcohol and benzene in the presence of chlorotrimethylsilane catalyst in 2-4 hours at room temperature. After workup of the reaction mixture and removal of the solvent, the impure reaction products were purified over silica gel column to obtained the pure mixture of alpha, beta ethers.
Another method is reported by Lin et al. (Lin, A. J. and Miller, R. E, J.Med Chero. 38,764-770, 1995) In this method the new ether derivatives were prepared by dissolving dihydroarternisinin in anhydrous ether and appropriate alcohol followed by BF3-etherate. The reaction mixture was stirred at room temperature for 24 hours. The yield of the purified products ranged from 40-90%. Purification was achieved by the use of silica gel chromatography.
Another method described by Jain et al (Jain D. C, Bhakuni R. S, Saxena S, kumar, S and Vishwakarma, R. A.) the preparation of arteether from artemisinin comprises: Reduction of artemisinin into dihydroartemisinin. Isolation of dihydroartemisinin. Acylation of dihydroartemisinin by dissolving it in alcohol and adding trialkylorthoformate in the reaction mixture, which produce ethers in quantitative yield in 10 hours at 40 degree C.
The above mentioned methods carry some disadvantages being less cost effective and more time consuming as compared to the present invention. Moreover, benzene, a carcinogenic solvent, used in the previous methods is not acceptable according to the health standard. Further, all the above methods require at least two separate steps to convert artemisinin into ethers i.e. reduction of the artemisinin into dihydroartemisinin in the first pot followed by isolation of dihydroartemisinin and then comes the second step of conversion of dihydroartemisinin into different ethers in the second pot. However, the present invention provide an efficient method for conversion of artemisinin into artemether
EXAMPLE 1
Artemisinin (3 g.) was dissolved in dry methanol (40 ml) at room temperature. It was cooled to −5 degree C. Now sodium borohydride (700 mg) was added slowly for 30 minutes and the reaction mixture was stirred for about 1.5 hours. The reaction was monitored by TLC to check completion of the reduction step. Now cation exchange resin (8 g) was added slowly at cooling temperature and the reaction mixture was further stirred at room temperature for about 2 hours. Cooled water was added to the reaction mixture and the resin was filtered.
The filtrate was neutralized with 5% sodium bicarbonate solution followed by extracting with dichloromethane (3×50 ml). The dichloromethane extract was dried over anhydrous sodium sulphate and evaporation of the solvent yielded 3.21 g, of artemether along with some impurities. The impure artemether was purified over silica gel column (1:5 ratio) in hexane:ethyl acetate (96:4) furnished pure alpha and beta artemether 2.43 g (81% w/w). Small portion of artemether was separated by prep TLC into alpha and beta isomers and characterized by the analysis of their IR, Mass and 1H NMR data.
EXAMPLE 2
The experiment was carried out following example 1 except in place of solid acid catalyst in the second reaction. Liquid acid catalyst chlorotrimethylsilane was added at cooling temperature for methylation reaction. The overall yield of pure alpha, beta artemether after column chromatography was 2.46 gm (82% w/w).
EXAMPLE 3
Artemisinin (100 g.) was dissolved in dry methanol (3 ml). Added sodium borohydride (30 mg.) at −5° C. The reaction mixture was stirred for 2 hours. After completion of the reaction, trifluroacetic acid (0.5 ml) was added and the reaction mixture was stirred for 5 hours. The methylation was incompleted and after workup the artemether was purified by prep TLC to yield 46 mg (46%) pure alpha, beta artemether.
EXAMPLE 4
The experiment was carried following example 1 except before column chromatography, the beta isomer (40%) was recrystallized in hexane from impure artemether and remaining mother liquor was purified over silica gel column in 1:5 ratio to yield alpha and beta artemether in 80% w/w.

PAPER
https://www.sciencedirect.com/science/article/abs/pii/S0920586114003307
The earlier developed flow protocol for stoichiometric reduction of an important biologically derived pharmaceutical precursor, artemisinin, to dihydroartemisinin was extended to a sequential reaction to produce one of the final APIs, artemether. A highly active heterogeneous catalyst was found for the etherification reaction. The use of QuadraSil catalyst allows to eliminate one step of reaction workup. A comparative Life Cycle Assessment of both reactions has shown advantages of the flow process over the optimized literature batch protocols. Results of LCA highlight the significance of solvents in pharmaceuticals manufacture and the advantage of flow technology, enabling small solvent inventories to be used.
Graphical abstract

PAPER
http://chem.vander-lingen.nl/articles/Target:_Artemether/id/126/itemid/663
In a previous episode chemical company Sanofi was granted exclusive access to certain yeast cells that produce a precursor to anti-malarial drug artemisinin. One of the charities making this all possible is the Bill and Melinda Gates Foundation. Another charity that has apparently entered into the drug business is the Clinton Health Access Initiative. Bill together with Rodger Stringham and David Teager report on an improved process for the conversion of artemisinin to artemether in Organic Process Research & Development (DOI).
Does the Clinton Health Access Initiative have a pilot-plant facility or even an organic lab? Unless it is all cramped in suite 400 on Dorchester Avenue in Boston, the article is not very explicit. The acknowledgements mention Mangalam Drugs and Organics.
Case at hand: artemether has the carbonyl group replaced by a methoxy group in a two-step reduction – methylation. So far so good. The point is that principal supplier Novartis reports up to 68% overall yields but that many Indian and Chinese suppliers working with the procedure generously supplied by same Novartis, report considerably lower figures (58-62%). But Why? And how can the process be improved?
Any organic chemist knows reported yields in the literature should be considered with caution. Chemists tend to be over-optimistic / self-delusionional but this scenario was not considered. No bottlenecks were encountered in step 1, the reduction with sodium borohydride. Only the beta form was isolated due to its poor solubility in the quench. Drying the product without heat prevented formation of one byproduct. Moving on to step two, the methylation with HCl in methanol was more troublesome. The byproducts lurking around the corner are the anomer and the elimination product. Co-solvent (co-reagent?) trimethyl orthoformate made all the difference. The critical element in the workup was first adding more methanol before adding the base quench otherwise you end up with a nasty gum. The new record yield for the improved synthesis is 72%.
But what have all these suppliers been doing wrong with the existing Novartis procedure? The answer to that question, remains unclear. The Novartis yield for step two with co-solvent methylacetate (not the formate) was confirmed so no surprise there.
///////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Artemether is an antimalarial drug for uncomplicated malaria caused by P. falciparum (and chloroquine-resistant P. falciparum) or chloroquine-resistant P. vivax parasites.[8] Artemether can also be used to treat severe malaria.[2]
The World Health Organization (WHO) recommends the treatment of uncomplicated P. falciparum with artemisinin-based combination therapy.[9] Given in combination with lumefantrine, it may be followed by a 14-day regimen of primaquine to prevent relapse of P. vivax or P. ovale malarial parasites and provide a complete cure.[10]
Artemether can also be used in treating and preventing trematode infections of schistosomiasis when used in combination with praziquantel.[11]
Artemether is rated category C by the FDA based on animal studies where artemisinin derivatives have shown an association with fetal loss and deformity. Some studies, however, do not show evidence of harm.[12][13]
Side effects
Possible side effects include cardiac effects such as bradycardia and QT interval prolongation.[14] Also, possible central nervous system toxicity has been shown in animal studies.[15][16]
Drug interactions
Plasma artemether level was found to be lower when the combination product was used with lopinavir/ritonavir.[16] There is also decreased drug exposure associated with concurrent use with efavirenz or nevirapine.[17][18]
Artemether/lumefantrine should not be used with drugs that inhibit CYP3A4.[19]
Hormonal contraceptives may not be as efficacious when used with artemether/lumefantrine.[19]
Pharmacology
Mechanism of action
Artemether is an artemisinin derivative and the mechanism of action for artemisinins is.[medical citation needed]
Artemether interact with ferriprotoporphyrin IX (heme) or ferrous ions in the acidic parasite food vacuole, and generates cytotoxic radical species
The accepted mode of action of the peroxide containing drug involve its interaction with heme (byproduct of hemoglobin degradation), derived from proteolysis of haemoglobin. This interaction results in the formation of toxic oxygen and carbon centered radicals.
One of the proposed mechanisms is that through inhibiting anti-oxidant and metabolic enzymes, artemisinin derivatives inflict oxidative and metabolic stress on the cell. Some pathways affected may concern glutathione and glucose metabolism. As a consequence, lesions and reduced growth of the parasite may result.[20]
Another possible mechanism of action suggests that arteristinin drugs exert their cidal action through inhibiting PfATP6. Since PfATP6 is an enzyme regulating cellular calcium concentration, its malfunctioning will lead to intracellular calcium accumulation, which in turns causes cell death.[21]
Pharmacokinetics
Absorption of artemether is improved 2- to 3-fold with food. It is highly bound to protein (95.4%). Peak concentrations of artemether are seen 2 hours after administration.[4]
Artemether is metabolized in the human body to the active metabolite, dihydroartemisinin, primarily by hepatic enzymes CYP3A4/5.[4] Both the parent drug and active metabolite are eliminated with a half-life of about 2 hours.[4]
Chemistry
Artemether is a methyl ether derivative of artemisinin, which is a peroxide-containing lactone isolated from the antimalarial plant Artemisia annua. It is also known as dihydroartemisinin methyl ether, but its correct chemical nomenclature is (+)-(3-alpha,5a-beta,6-beta,8a-beta, 9-alpha,12-beta,12aR)-decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano(4,3-j)-1,2-benzodioxepin. It is a relatively lipophilic and unstable drug,[22] which acts by creating reactive free radicals in addition to affecting the membrane transport system of the plasmodium organism.[14]
References
- ^ “Artemether – Drugs.com”. http://www.drugs.com. Archived from the original on 20 December 2016. Retrieved 7 December 2016.
- ^ Jump up to:a b c d e f Esu, Ekpereonne B.; Effa, Emmanuel E.; Opie, Oko N.; Meremikwu, Martin M. (18 June 2019). “Artemether for severe malaria”. The Cochrane Database of Systematic Reviews. 6: CD010678. doi:10.1002/14651858.CD010678.pub3. ISSN 1469-493X. PMC 6580442. PMID 31210357.
- ^ “Artemether and Lumefantrine”. The American Society of Health-System Pharmacists. Archived from the original on 20 December 2016. Retrieved 28 November 2016.
- ^ Jump up to:a b c d “Coartem- artemether and lumefantrine tablet”. DailyMed. 5 August 2019. Retrieved 26 April 2020.
- ^ Jump up to:a b c d e Kovacs, SD; Rijken, MJ; Stergachis, A (February 2015). “Treating severe malaria in pregnancy: a review of the evidence”. Drug Safety. 38 (2): 165–81. doi:10.1007/s40264-014-0261-9. PMC 4328128. PMID 25556421.
- ^ Rao, Yi; Zhang, Daqing; Li, Runhong (2016). Tu Youyou and the Discovery of Artemisinin: 2015 Nobel Laureate in Physiology or Medicine. World Scientific. p. 162. ISBN 9789813109919. Archived from the original on 2017-09-10.
- ^ World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. 2019. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Makanga, Michael; Krudsood, Srivicha (2009-10-12). “The clinical efficacy of artemether/lumefantrine (Coartem)”. Malaria Journal. 8 (Suppl 1): S5. doi:10.1186/1475-2875-8-S1-S5. ISSN 1475-2875. PMC 2760240. PMID 19818172.
- ^ Treatment of Uncomplicated Plasmodium falciparum Malaria. World Health Organization. 2015-01-01. Archived from the original on 2017-09-10.
- ^ Treatment Of Uncomplicated Malaria Caused By P. vivax, P. ovale, P. malariae or P. knowlesi. World Health Organization. 2015-01-01. Archived from the original on 2017-09-10.
- ^ Pérez del Villar, Luis; Burguillo, Francisco J.; López-Abán, Julio; Muro, Antonio (2012-01-01). “Systematic review and meta-analysis of artemisinin based therapies for the treatment and prevention of schistosomiasis”. PLOS ONE. 7 (9): e45867. Bibcode:2012PLoSO…745867P. doi:10.1371/journal.pone.0045867. ISSN 1932-6203. PMC 3448694. PMID 23029285.
- ^ Dellicour S, Hall S, Chandramohan D, Greenwood B (2007). “The safety of artemisinins during pregnancy: a pressing question”. Malaria Journal. 6: 15. doi:10.1186/1475-2875-6-15. PMC 1802871. PMID 17300719.
- ^ Piola P, Nabasumba C, Turyakira E, et al. (2010). “Efficacy and safety of artemether—lumefantrine compared with quinine in pregnant women with uncomplicated Plasmodium falciparum malaria: an open-label, randomised, non-inferiority trial”. Lancet Infect Dis. 10 (11): 762–769. doi:10.1016/S1473-3099(10)70202-4. hdl:10144/116337. PMID 20932805.
- ^ Jump up to:a b “Artemether”. http://www.antimicrobe.org. Archived from the original on 2017-02-23. Retrieved 2016-11-09.
- ^ “WHO Model Prescribing Information: Drugs Used in Parasitic Diseases – Second Edition: Protozoa: Malaria: Artemether”. apps.who.int. Archived from the original on 2016-11-10. Retrieved 2016-11-09.
- ^ Jump up to:a b Askling, Helena H.; Bruneel, Fabrice; Burchard, Gerd; Castelli, Francesco; Chiodini, Peter L.; Grobusch, Martin P.; Lopez-Vélez, Rogelio; Paul, Margaret; Petersen, Eskild (2012-01-01). “Management of imported malaria in Europe”. Malaria Journal. 11: 328. doi:10.1186/1475-2875-11-328. ISSN 1475-2875. PMC 3489857. PMID 22985344.
- ^ van Geertruyden, J.-P. (2014). “Interactions between malaria and human immunodeficiency virus anno 2014”. Clinical Microbiology and Infection. 20 (4): 278–285. doi:10.1111/1469-0691.12597. PMC 4368411. PMID 24528518.
- ^ Kiang, Tony K. L.; Wilby, Kyle J.; Ensom, Mary H. H. (2013-10-26). “Clinical Pharmacokinetic Drug Interactions Associated with Artemisinin Derivatives and HIV-Antivirals”. Clinical Pharmacokinetics. 53 (2): 141–153. doi:10.1007/s40262-013-0110-5. ISSN 0312-5963. PMID 24158666. S2CID 1281113.
- ^ Jump up to:a b Stover, Kayla R.; King, S. Travis; Robinson, Jessica (2012-04-01). “Artemether-Lumefantrine: An Option for Malaria”. Annals of Pharmacotherapy. 46 (4): 567–577. doi:10.1345/aph.1Q539. ISSN 1060-0280. PMID 22496476. S2CID 7678606.
- ^ Saeed, ME; Krishna, S; Greten, HJ; Kremsner, PG; Efferth, T (August 2016). “Antischistosomal activity of artemisinin derivatives in vivo and in patients”. Pharmacological Research. 110: 216–26. doi:10.1016/j.phrs.2016.02.017. PMID 26902577.
- ^ Guo, Zongru (2016-03-01). “Artemisinin anti-malarial drugs in China”. Acta Pharmaceutica Sinica B. 6 (2): 115–124. doi:10.1016/j.apsb.2016.01.008. PMC 4788711. PMID 27006895.
- ^ De Spiegeleer, B.M.J.; D’Hondt, M.; Vangheluwe, E.; Vandercruyssen, K.; De Spiegeleer, B.G.I.; Jansen, H.; Koijen, I.; Van Gompel, J. (2012). “Relative response factor determination of artemether degradants with a dry heat stress approach”. Journal of Pharmaceutical and Biomedical Analysis. 70: 111–116. doi:10.1016/j.jpba.2012.06.002. hdl:1854/LU-2938963. PMID 22770733.
| Clinical data | |
|---|---|
| Trade names | Many[1] |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Intramuscular[2] |
| ATC code | P01BE02 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 71963-77-4 |
| PubChem CID | 68911 |
| DrugBank | DB06697 |
| ChemSpider | 62138 |
| UNII | C7D6T3H22J |
| KEGG | D02483 |
| ChEBI | CHEBI:195280 |
| ChEMBL | ChEMBL1237051 |
| PDB ligand | D8Z (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID7040651 |
| ECHA InfoCard | 100.189.847 |
| Chemical and physical data | |
| Formula | C16H26O5 |
| Molar mass | 298.379 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 86 to 88 °C (187 to 190 °F) |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////ARTEMETHER, ANTIMALARIAL, SM 224, SM-224
[H][C@@]12CC[C@@H](C)[C@]3([H])CC[C@@]4(C)OO[C@@]13[C@]([H])(O[C@H](OC)[C@@H]2C)O4

NEW DRUG APPROVALS
ONE TIME
$10.00
ZY 19489, MMV 253
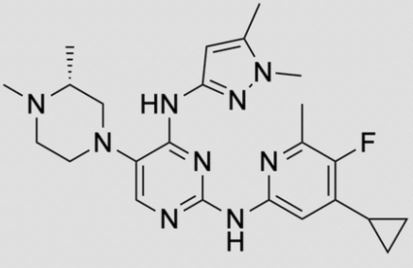

![2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=92045019&t=l)
ZY 19489, MMV 253
C24 H32 FN9, 465.5
CAS 1821293-40-6
MMV253, GTPL10024, MMV674253
N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-((3R)-2-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-3,4-dimethylpiperazin-1-yl)pyrimidin-2-amine
2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine
- N2-(4-Cyclopropyl-5-fluoro-6-methyl-2-pyridinyl)-5-[(3R)-3,4-dimethyl-1-piperazinyl]-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-2,4-pyrimidinediamine
- (R)-N2-(4-Cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine

SYN
IN 201721031453
The invention relates to triaminopyrimidine compd. of formula I, pharmaceutically acceptable salts thereof, hydrates, solvates, polymorphs, optically active forms thereof, in solid state forms useful for preventing or treating malaria. The invention also relates to a process for prepn. of triaminopyrimidine compd. and intermediates thereof. Compd. I was prepd. by condensation of 5-bromouracil with tert-Bu (R)-2-methylpiperazine-1-carboxylate to give tert-Bu (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine-1-carboxylate, which underwent chlorination followed by condensation with 1,5-dimethyl-1H-pyrazol-3-amine followed by condensation with 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride to give (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine, which underwent Boc-deprotection followed by methylation to give I.
SYN
WO 2019049021
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019049021
Malaria is caused by protozoan parasites of the genus Plasmodium that infect and destroy red blood cells, leading to fever, severe anemia, cerebral malaria and, if untreated, death.
International (PCT) Publication No. WO 2015/165660 (the WO ‘660) discloses triaminopyrimidine compounds, intermediates, pharmaceutical compositions and methods for use for preventing or treating malaria. The WO ‘660 discloses a process for preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine (compound 5) as depicted in scheme-1.
Scheme 1
WO ‘660 discloses a process for preparation of triaminopyrimidine compounds depicted in scheme-2.

WO ‘660 discloses the preparation of compounds 8 and 4 by using microwave technique using Biotage microwave vial. WO ‘660 in example- 13, discloses the isolation of compound 1 by concentration of reaction mixture to obtain crude product, which was purified through reverse phase HPLC GILSON instrument to obtain pure solid compound 1 in 40.8% yield, without providing the purity of the solid compound 1. The process disclosed in WO ‘660 is not industrially advantageous as it requires microwave conditions as well as chromatographic purification and provides compound 1 with lower yields. The compound 1 prepared may not be suitable for pharmaceutical preparations based on various regulatory requirements.
Polymorphism, the occurrence of different crystalline forms, is a property of some molecules. A single molecule can exist in different crystalline forms having distinct physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis – TGA, or different scanning calorimetry – DSC, Powder x-ray diffraction pattern – PXRD, infrared absorption – IR). One or more these techniques may be used to distinguish different polymorphic forms of a compound.
Different salts and solid states (e.g. solvates, hydrates) of an active pharmaceutical ingredient may possess different physio-chemical properties. Such variation in the properties of different salts and solid states forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (both chemical and polymorph) and shelf-life. These variations in the properties of different salts and solid states forms may offer improvements to the final dosage form for example, to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms or amorphous form, which may in turn provide additional opportunities to assess variations in the properties and characteristics of an active pharmaceutical ingredient.
In view of the above, the present invention provides a process for the preparation of triaminopyrimidine compound 1 or pharmaceutically acceptable salts thereof or hydrates or solvates or polymorphs or optically active forms thereof, which is industrially scalable, environment friendly and efficient so as to obtain compounds of the invention in higher yields and purity.
The process for the preparation of triaminopyrimidine compound 1 or intermediates thereof of the present invention, takes the advantage by using appropriate solvent systems and isolation techniques as well as purification techniques, thereby to overcome problems of lower yields, chromatography purifications and microwave reactions of the prior art.
SUMMARY OF THE INVENTION
The present invention provides solid state forms of triaminopyrimidine compound
1,
1
Examples: Preparation of Intermediates
Example-1: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a 250 mL 4N round bottom flask, process water (30 ml) and cyclopropanecarboxylic acid (14.19 g, 164.88 mmol) were added at 25 to 35°C and started stirring. Sulphuric acid (4.4 ml, 82.44 mmol) was charged to the reaction mixture. Silver nitrate (4.18 g, 24.73 mmol), 6-Chloro-3-fluoro-2-methylpyridine (6 g, 41.22 mmol) were charged to the reaction mixture. Aqueous solution of ammonium persulphate (65.85 g, 288.54 mmol in 90 mL water) was added to the reaction mixture in 30 to 60 min at temperature NMT 60 °C. After the completion of the reaction as monitored by HPLC, toluene (30 ml) was added to the reaction mixture and stirred for 15 min. The reaction mixture filtered, separated layers from filtrate and extracted aqueous layer using toluene (30 mL). The organic layer was washed with aqueous sodium carbonate solution (30 mL) and water. The organic layer was distilled completely under vacuum at 60 °C to obtain 3.37 g syrupy mass as titled compound.
Example-2: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a suitable glass assembly, process water (7.5 L) and cyclopropanecarboxylic acid (3.55 Kg, 41.24 mol) were added at 25 to 35 °C and stirred. Sulphuric acid (2.02 Kg, 20.59 mol), silver nitrate (1.05 Kg, 6.21 mol), 6-chloro-3-fluoro-2-methylpyridine (1.5 Kg, 10.3 mol) were added to the reaction mixture. Aqueous solution of ammonium persulphate (16.46 g, 72.13 mmol in 22.5 L water) was added to the reaction mixture at 55 to 60 °C and maintained. After the completion of the reaction as monitored by HPLC, toluene (7.5 L) was added to the reaction mixture and stirred for 15 min. The reaction mixture was filtered, organic layer was separated and aqueous layer was extracted using toluene (6 L), filtered the reaction mixture and washed the solid with toluene (1.5 L). The combined organic layer was washed with 20% sodium carbonate solution (9 L) and water. The organic layer was concentrated completely under vacuum at 60 °C to obtain 880 g (86.50%) syrupy mass of titled compound.
Example-3: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a 100 mL 3N round bottom flask, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (2.69 g, 14.48 mmol) and toluene (30 mL) were added at 25 to 35 °C. Diphenylmethanimine (3.15 g, 17.38 mmol) was charged to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (270 mg, 0.43 mmol) and palladium acetate (98 mg, 0.43 mmol) were added to the reaction mixture. Sodium-ie/ -butoxide (2.78 g, 28.96 mmol) was added to the reaction mixture and heated to 100 to 110° C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C and filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-4: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a suitable assembly, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (880) and toluene (7.5 L) were added at 25 to 35 °C. Diphenylmethanimine (787 g, 4.34 mmol) and BOC anhydride (237 g, 1.086 mol) was added to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (67.6 g, 0.108 mmol) and palladium acetate (24.4 g, 0.108 mol) were added to the reaction mixture. S odium- ieri-butoxide (870 g, 9.05 mol) was added to the reaction mixture and heated to 100 to 110 °C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C, water (6 L) was added. The reaction mixture was filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-5: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a 100 mL 3N round bottom flask, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-3 was added water (25 mL) at 25 to 35° C. The cone. HCl (3 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride, charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (9 mL) and ethyl acetate (9 mL) was added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 1.62 g title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity.
Example-6: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a suitable glass assembly, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-4 was added water (6 L) at 25 to 35° C. The cone. HCl (750 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride (3 L) and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride (3 L), charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (1.5 L) and ethyl acetate (1.5 L) were added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 489 g (96.80%) title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.5), Differential scanning calorimetry (FIG.6) and Thermogravimetric analysis (FIG.7).
Example 7: Preparation of 2,3-dibromobutanenitrile
In a 2 L round bottom flask, dichloromethane (550 mL) and 2-butenenitrile 110 g
(1.64 mol) were cooled to 20 to 25 °C. A solution of bromine 275 g (1.72 mol) in dichloromethane (220 mL) was dropwise added at 20 to 25 °C. Hydrobromic acid 1.43 ml (0.0082 mol) in acetic acid (33%) solution was added into the reaction mixture and stirred for 4 hours. After the completion of reaction, Na2S203 (550 mL) 4% aqueous solution was added and the reaction mixture was stirred for 15 min. The separated organic layer was distilled under vacuum completely to obtain 364.2 g (97.9%) of title compound as an oil.
Example 8: Preparation of l,5-dimethyl-lH-pyrazol-3-amine
In a 5 L round bottom flask, water (1. 36 L), sodium hydroxide 340 g (8.99 mol) were added and the reaction mixture was cooled to 0 to 5°C. A solution of methyl hydrazine sulphate 237.8 g (1.65 mol) in 680 mL water was added dropwise to the reaction mixture and stirred below 10 °C. 2,3-dibromobutanenitrile 340 g (1.5 mol) prepared in example-7 was added and the reaction mixture was stirred below 10 °C for 2 hours. After the completion of reaction, toluene (630 mL) was added and the reaction mixture was stirred for 15 min. The aqueous layer was separated and the organic layer was removed. The aqueous layer was extracted with dichloromethane (5.1 L). The combined organic layer was distilled completely under vacuum to obtain residue. Diisopropyl ether (680 mL) was added and the reaction mixture was stirred at 0 to 5 °C for 1 hour. The reaction mixture was filtered, washed with diisopropyl ether and dried to obtained 121.5 g (72.93%) of title compound having 95.63% purity.
Examples: Preparation of triaminopyrimidine compounds
Example-9: Preparation of tert-butyl (R)-4-(2,4-dioxo-l,2,3,4-tetrahydro- pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 2 L four neck round bottom flask, 1.25 Kg (6.545 mol) 5-bromouracil, 1.87 Kg (9.360 mol) tert-butyl (R)-2-methylpiperazine-l-carboxylate and 5L pyridine were added at 25 to 35° C. The reaction mass was stirred for 15 hours at 115 to 120°C. After completion, the reaction mass was cooled to 25 to 35°C. 12.5 L water was added and stirred for 1 hour. The reaction mass was filtered, washed with 2.5 L water and dried to obtain 1.37 Kg (67.4%) of title compound.
Example-10: Preparation of tert-butyl (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine- 1 -carboxylate
In 20 L four neck round bottom flask, 1.36 Kg (4.382 mmol) tert-butyl (R)-4-(2,4-dioxo-1, 2,3, 4-tetrahydropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate and 6.8 L phosphorus oxychloride were added at 25 to 35° C. 26.5 mL pyridine (0.329 mol) was added and the reaction mass was heated to 105 to 110 °C and stirred for 4 hours. After the completion of the reaction, phosphorus oxychloride was distilled completely at atmospheric pressure. 2.72 L acetone was added and the reaction mixture was quenched into 4.08 L water. Acetone was removed by distillation under vacuum. 20% sodium carbonate solution was added to adjust pH 7.5-8.5 of the reaction mixture. 1.14 Kg (5.258 mol) di-tert-butyl dicarbonate and 9.52 L ethyl acetate were added and stirred for 2 hours at 25 to 35 °C. After the completion of the reaction, the organic layer was separated and aqueous layer was extracted with 6.8 L ethyl acetate. The combined ethyl layers were distilled to remove ethyl acetate completely under vacuum to obtain residue. 1.36 L isopropyl alcohol was added to the residue and isopropyl alcohol was removed completely. 4.08 L isopropyl alcohol and 6.8 L water were added to the residue and stirred for 1 hour. The reaction mass was filtered, washed with water and dried to obtain 1.25 Kg of title compound.
Example-11: Preparation of tert-butyl (R)-4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 640 g (1.843 mol) tert-butyl (R)-4-(2, 4-dichloropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate, 225.3 g (2.027 g) 1,5-dimethyl-lH-pyrazol-3-amine and 9.6L toluene were added at 25 to 35°C. 1.2 Kg (3.686 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 12.41 g (0.0553 mol) palladium acetate and 34.43 g (0.0553 mol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added and the reaction mass was maintained for 16 hours at 110 to 115 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed the bed with 1.28 L toluene. Toluene was distilled completely and 2.56 L dichlromethane was added. The compound was adsorbed by 1.92 Kg silica gel (60-120 mesh). The dichloromethane was distilled completely under vacuum and 12.8 L mixture of ethyl acetate and hexane was added to the residue and stirred for 2 hours. The silica gel was filtered and the filtrate was distilled completely under vacuum to obtain 595 g title compound.
Example-12: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 595 g (1.40 mol) tert-butyl (R)- 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 305 g (1.38 mol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride and 11.5 L toluene were added at 25 to 35°C. 1.08 Kg (3.32 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 17.21 g (27.6 mmol) palladium acetate and 6.21 g (27.6 mmol) racemic 2,2′-bis(diphenylphosphino)-l, -binaphthyl were added. The reaction mass was stirred for 6 hours at 110 tol l5 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
Example-13: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 500 mL four neck round bottom flask, 7.5 g (17.77 mmol) (R)-tert-butyl 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 3.92 g (17.77 mmol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride compound and 150 mL toluene were added at 25 to 35 °C. 20 g (61.3 mmol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. Then, 130 mg (0.58 mmol) palladium acetate and 360 mg (0.58 mmol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added. The reaction mass was stirred for 18 hours at 110 to 115° C under nitrogen. After completion, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
2 4
Example-14: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1, 5-dimethyl-lH-pyrazol-3-yl)-5-(3-methylpiperazin-l-yl)pyrimidine-2,4-diamine
In 50 L glass assembly, the filtrate containing tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate from example 13 was taken. 11.5 L water and 1.28 L Cone. HC1 were added at 25 to 35 °C. The reaction mass was stirred for 2 hours at 50 to 55 °C. After the completion of the reaction, reaction mixture was cooled to room temperature and filtered over celite bed and washed with water. The separated the aqueous layer from filtrate was basified by using 20% sodium carbonate solution and extracted with 12.8 L methylene dichloride. The organic layer was distilled completely under vacuum to obtain residue. 9.6 L acetonitrile was added to the residue and heated to reflux for 30 min. The reaction mixture was cooled and stirred at 25 to 35 °C for 1 hour. The reaction mixture was filtered, washed with 640 mL acetonitrile and dried to obtain 360 g titled compound.
2 4
Example-15: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 250 mL four neck round bottom flask, 4.7 g (10.4 mmol) (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine was dissolved in 56 mL ethanol. 1.89 g (23.32 mmol) formaldehyde and 1.44 g (22.90 mmol) sodium cyanoborohydride were added. Adjusted pH 5-6 using acetic acid and stirred the reaction mass at 25 to 35 °C for 2 hours. After completion, ethanol was distilled completely under vacuum. 47 mL water was added to the residue. The reaction mass was basified by 20% sodium carbonate solution and extracted with methylene dichloride. Both the organic layers were combined and distilled completely under vacuum. 94 mL acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mass was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 5 mL acetonitrile and dried to obtain 3.7 g title compound as crystalline solid, having HPLC purity of about 99.61%.
2 4
Example-16: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 20 L round bottom flask, 725 g (1.60 mol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazine-l-yl)pyrimidine-2,4-diamine was dissolved in 6.52 L dichloromethane. 261.5 g (3.2 mol) formaldehyde and 510.4 g (2.4 mol) sodium triacetoxyborohydride were added and stirred the reaction mixture at 25 to 35 °C for 2 hours. After the completion of the reaction, 3.63 L water was added into the reaction mixture. The reaction mixture was basified by 20% sodium carbonate solution and the organic layer was separated. The aqueous layer was extracted with 1.45 L methylene dichloride. The combined organic layers were distilled completely under vacuum. 14.5 L acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 1.45 L acetonitrile and dried to obtain 632 g of title compound as crystalline solid having 99.01% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.l) and Differential Scanning Calorimetry (FIG.2).
2 4
Example-17: Preparation of (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine In a 10 mL round bottom flask, 300 mg (0.644 mmol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine, 2.7 mL acetonitrile and 0.3 mL water were added and the reaction mixture was heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35 °C and stirred for 1 hour. The reaction mass was filtered, washed with acetonitrile and dried to obtain 201 mg (67%) title compound as crystalline solid. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.3) and Differential Scanning Calorimetry (FIG.4).
SYN
WO 2015165660
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015165660
Example 13
Synthetic scheme 1
Synthetic scheme 2
(R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine
In a 50 mL round-bottomed flask (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (190 mg, 0.42 mmol, Example 2) was taken in DCM (2 mL) to give a yellow suspension. To this Hunig’s Base (0.184 mL, 1.05 mmol) was added and the suspension turned clear. After 10 minutes, it turned into a white suspension. After another 10 minutes, the mixture was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 mL) and formaldehyde (0.042 mL, 0.63 mmol) was added and stirred for 10 minutes. White suspension slowly cleared to yellow solution. To this clear solution sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get white suspension. After 30 minutes LCMS showed completion of reaction. The reaction mixture was concentrated and the crude was purified through reverse phase HPLC GILSON instrument to get the pure solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8 %).1H NMR (300
MHz, DMSO-d6) δ ppm 0.67 – 0.78 (m, 2 H) 1.00 (d, J=6.22 Hz, 3 H) 1.02 – 1.08 (m, 2 H) 1.96 – 2.10 (m, 1 H) 2.23 (s, 7 H) 2.30 – 2.38 (m, 4 H) 2.73 – 2.96 (m, 4 H) 3.33 (s, 3 H) 6.83 (s, 1 H) 7.67 (d, J=5.09 Hz, 1 H) 8.00 (s, 1 H) 8.03 (s, 1 H) 9.26 (s,1 H) MS (ES+), (M+H)+ = 466.45 for C21H32FN9.
SYN
Nature Communications (2015), 6, 6715.
https://www.nature.com/articles/ncomms7715
Hameed P., S., Solapure, S., Patil, V. et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat Commun 6, 6715 (2015). https://doi.org/10.1038/ncomms7715
The widespread emergence of Plasmodium falciparum (Pf) strains resistant to frontline agents has fuelled the search for fast-acting agents with novel mechanism of action. Here, we report the discovery and optimization of novel antimalarial compounds, the triaminopyrimidines (TAPs), which emerged from a phenotypic screen against the blood stages of Pf. The clinical candidate (compound 12) is efficacious in a mouse model of Pf malaria with an ED99 <30 mg kg−1 and displays good in vivo safety margins in guinea pigs and rats. With a predicted half-life of 36 h in humans, a single dose of 260 mg might be sufficient to maintain therapeutic blood concentration for 4–5 days. Whole-genome sequencing of resistant mutants implicates the vacuolar ATP synthase as a genetic determinant of resistance to TAPs. Our studies highlight the potential of TAPs for single-dose treatment of Pf malaria in combination with other agents in clinical development.

(A) Pyridine, microwave, 150 °C, 45 min. (B) (i) POCl3, reflux, 6 h (ii) sodium carbonate, di-tert-butyl dicarbonate, room temperature, 16 h. (C) N,N-Diisopropylethylamine (DIPEA), ethanol, microwave, 110 °C, 1 h. (D) (i) Potassium tert-butoxide, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), pd2(dba)3, toluene, reflux, 12 h. (E) HCl (4 N) in dioxane, 15–30 min. (F) Compound 9, DIPEA, dichloromethane, formaldehyde (HCHO), sodium cyanoborohydride, 15 min.
Synthesis of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3, 4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (12). (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (compound 9, 190 mg, 0.42 mmol) was taken in dichloromethane (2 ml) to give a yellow suspension. To this Hunig’s Base (0.184 ml, 1.05 mmol) was added and the suspension turned clear. After 10 min of stirring, reaction mixture turned into a white suspension and then it was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 ml), and formaldehyde (0.042 ml, 0.63 mmol) was added and stirred for 10 min. To this clear solution, sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get a white suspension. The reaction mixture was concentrated and the crude product was purified through reverse-phase chromatography to get the pure off-white solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8%). Yield: 40.8%, purity: >95% by HPLC (ultraviolet at 220 and 254 nm). 1H NMR (300 MHz, DMSO-d6) δ 9.26 (s,1H), 8.03 (s, 1H) 8.00 (s, 1H) 7.67 (d, J=5.1 Hz, 1H) 6.83 (s, 1H) 3.33 (s, 3H) 2.96–2.73 (m, 4H) 2.75–2.50 (m, 1H) 2.38–2.30 (m, 4H) 2.23 (s, 7H) 2.10–1.96 (m, 1H),1.08–1.02 (m, 2H) 1.00 (d, J=6.2 Hz, 3H) 0.78–0.67 (m, 2H). 13C-NMR (126 MHz, DMO-d6) δ 155.30, 154.67, 152.10, 150.93, 148.98, 146.81. 145.29, 141.95, 140.31, 138.81, 124.91, 106.20, 97.07, 58.78, 51.87, 42.16, 35.28, 17.23. 10.99 and 8.77, HRMS (ESI): m/z calculated for C24H32FN9+H [M+H]: 466.2765. Found: 466. 2838. Traces of LC-MS, HRMS, 1H NMR and 13C-NMR of compound 12 are shown in Supplementary Figs 1–3.


| Product vision |
|
| MoA |
|
| Key features |
|
| Challenges |
|
| Status |
|
| Next milestone |
|
| Previously |
|
Zydus receives Orphan Drug Designation from USFDA for ZY-19489, a novel compound to treat malaria;
ZY19489 is a novel antimalarial compound active against all current clinical strains of P. falciparum and P. vivax, including drug-resistant strains.
Zydus Cadila listed as Cadila Healthcare Limited announced that its antimalarial compound ZY19489 (MMV253), currently in development together with Medicines for Malaria Venture (MMV), a leading product development partnership (PDP) in antimalarial drug research, has received Orphan Drug Designation from the USFDA.
Orphan drug designation provides eligibility for certain development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions, and seven-year marketing exclusivity upon FDA approval.
The company said that the Phase I study of ZY19489 has demonstrated a long half-life and potential for a single-dose cure for malaria. In a separate malaria challenge trial, potent antimalarial activity has been demonstrated following single-dose oral administration of ZY19489.
“As a global community facing threats from rapidly mutating malaria strains and the rise in artemisinin resistance cases, we have to be prepared with novel therapeutic drugs. ZY-19489 is a potential single dose radical cure for P. falciparum and P. vivax malaria which is a major global health risk today,” Pankaj R. Patel, Chairman, Zydus Group, said.
“ZY19489 is a potent, first in class molecule, originally discovered and elaborated in India” said Dr. Timothy Wells, Chief Scientific Officer, MMV. “It has tremendous potential as part of a new generation of treatments and is fully active against drug resistant strains of malaria which are increasingly a concern.”
Artemisinin resistance is seen as a mounting challenge to the global fight against malaria. ZY19489 is being developed to provide an effective alternative to the current front-line antimalarial drugs for the treatment of P. falciparum and P. vivax malaria, as artemisinin-based combination therapies (ACTs) are under threat of resistance.
As per the World Malaria Report 2021, there were an estimated 241 million cases of malaria worldwide and the estimated number of malaria deaths stood at 627,000 in 2020. A major health concern, it is estimated that a child dies from malaria every minute. About 96% of malaria deaths globally were in 29 countries. India accounted for about 82% of all malaria deaths in the WHO South-East Asia Region.
////////////ZY 19489, MMV 253, Orphan Drug Designation, PHASE 1, ZYDUS CADILA, ANTIMALARIAL
Cn1nc(Nc2nc(Nc3cc(C4CC4)c(F)c(C)n3)ncc2N2C[C@@H](C)N(C)CC2)cc1C
CC1CN(CCN1C)C2=CN=C(N=C2NC3=NN(C(=C3)C)C)NC4=NC(=C(C(=C4)C5CC5)F)C
CHLOROQUINE, クロロキン;Хлорохин , クロロキン , كلوروكين
CHLOROQUINE
| Formula |
C18H26ClN3
|
|---|---|
| CAS |
54-05-7
|
| Mol weight |
319.8721
|
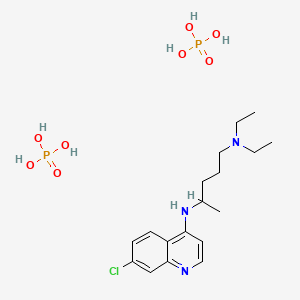
Chloroquine is a medication used primarily to prevent and to treat malaria in areas where that parasitic disease is known to remain sensitive to its effects.[1] A benefit of its use in therapy, when situations allow, is that it can be taken by mouth (versus by injection).[1] Controlled studies of cases involving human pregnancy are lacking, but the drug may be safe for use for such patients.[verification needed][1][2] However, the agent is not without the possibility of serious side effects at standard doses,[1][3] and complicated cases, including infections of certain types or caused by resistant strains, typically require different or additional medication.[1] Chloroquine is also used as a medication for rheumatoid arthritis, lupus erythematosus, and other parasitic infections (e.g., amebiasis occurring outside of the intestines).[1] Beginning in 2020, studies have proceeded on its use as a coronavirus antiviral, in possible treatment of COVID-19.[4]
Chloroquine, otherwise known as chloroquine phosphate, is in the 4-aminoquinoline class of drugs.[1] As an antimalarial, it works against the asexual form of the malaria parasite in the stage of its life cycle within the red blood cell.[1] In its use against rheumatoid arthritis and lupus erythematosus, its activity as a mild immunosuppressive underlies its mechanism.[1] Antiviral activities, established and putative, are attributed to chloroquines inhibition of glycosylation pathways (of host receptor sialylation or virus protein post-translational modification), or to inhibition of virus endocytosis (e.g., via alkalisation of endosomes), or other possible mechanisms.[5] Common side effects resulting from these therapeutic uses, at common doses, include muscle problems,[clarification needed] loss of appetite, diarrhea, and skin rash.[clarification needed][1] Serious side effects include problems with vision (retinopathy), muscle damage, seizures, and certain anemias.[1][6]
Chloroquine was discovered in 1934 by Hans Andersag.[7][8] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[9] It is available as a generic medication.[1] The wholesale cost in the developing world is about US$0.04.[10] In the United States, it costs about US$5.30 per dose.[1]
Medical uses
Malaria

Distribution of malaria in the world:[11]
♦ Elevated occurrence of chloroquine- or multi-resistant malaria
♦ Occurrence of chloroquine-resistant malaria
♦ No Plasmodium falciparum or chloroquine-resistance
♦ No malaria
Chloroquine has been used in the treatment and prevention of malaria from Plasmodium vivax, P. ovale, and P. malariae. It is generally not used for Plasmodium falciparum as there is widespread resistance to it.[12][13]
Chloroquine has been extensively used in mass drug administrations, which may have contributed to the emergence and spread of resistance. It is recommended to check if chloroquine is still effective in the region prior to using it.[14] In areas where resistance is present, other antimalarials, such as mefloquine or atovaquone, may be used instead. The Centers for Disease Control and Prevention recommend against treatment of malaria with chloroquine alone due to more effective combinations.[15]
Amebiasis
In treatment of amoebic liver abscess, chloroquine may be used instead of or in addition to other medications in the event of failure of improvement with metronidazole or another nitroimidazole within 5 days or intolerance to metronidazole or a nitroimidazole.[16]
Rheumatic disease
As it mildly suppresses the immune system, chloroquine is used in some autoimmune disorders, such as rheumatoid arthritis and lupus erythematosus.[1]
Side effects
Side effects include blurred vision, nausea, vomiting, abdominal cramps, headache, diarrhea, swelling legs/ankles, shortness of breath, pale lips/nails/skin, muscle weakness, easy bruising/bleeding, hearing and mental problems.[17][18]
- Unwanted/uncontrolled movements (including tongue and face twitching) [17]
- Deafness or tinnitus.[17]
- Nausea, vomiting, diarrhea, abdominal cramps[18]
- Headache.[17]
- Mental/mood changes (such as confusion, personality changes, unusual thoughts/behavior, depression, feeling being watched, hallucinating)[17][18]
- Signs of serious infection (such as high fever, severe chills, persistent sore throat)[17]
- Skin itchiness, skin color changes, hair loss, and skin rashes.[18][19]
- Chloroquine-induced itching is very common among black Africans (70%), but much less common in other races. It increases with age, and is so severe as to stop compliance with drug therapy. It is increased during malaria fever; its severity is correlated to the malaria parasite load in blood. Some evidence indicates it has a genetic basis and is related to chloroquine action with opiate receptors centrally or peripherally.[20]
- Unpleasant metallic taste
- This could be avoided by “taste-masked and controlled release” formulations such as multiple emulsions.[21]
- Chloroquine retinopathy
- Electrocardiographic changes[22]
- This manifests itself as either conduction disturbances (bundle-branch block, atrioventricular block) or Cardiomyopathy – often with hypertrophy, restrictive physiology, and congestive heart failure. The changes may be irreversible. Only two cases have been reported requiring heart transplantation, suggesting this particular risk is very low. Electron microscopy of cardiac biopsies show pathognomonic cytoplasmic inclusion bodies.
- Pancytopenia, aplastic anemia, reversible agranulocytosis, low blood platelets, neutropenia.[23]
Pregnancy
Chloroquine has not been shown to have any harmful effects on the fetus when used for malarial prophylaxis.[24] Small amounts of chloroquine are excreted in the breast milk of lactating women. However, this drug can be safely prescribed to infants, the effects are not harmful. Studies with mice show that radioactively tagged chloroquine passed through the placenta rapidly and accumulated in the fetal eyes which remained present five months after the drug was cleared from the rest of the body.[23][25] Women who are pregnant or planning on getting pregnant are still advised against traveling to malaria-risk regions.[24]
Elderly
There is not enough evidence to determine whether chloroquine is safe to be given to people aged 65 and older. Since it is cleared by the kidneys, toxicity should be monitored carefully in people with poor kidney functions.[23]
Drug interactions
Chloroquine has a number of drug-drug interactions that might be of clinical concern:[citation needed]
- Ampicillin– levels may be reduced by chloroquine;[23]
- Antacids– may reduce absorption of chloroquine;[23]
- Cimetidine– may inhibit metabolism of chloroquine; increasing levels of chloroquine in the body;[23]
- Cyclosporine– levels may be increased by chloroquine;[23] and
- Mefloquine– may increase risk of convulsions.[23]
Overdose
Chloroquine is very dangerous in overdose. It is rapidly absorbed from the gut. In 1961, a published compilation of case reports contained accounts of three children who took overdoses and died within 2.5 hours of taking the drug. While the amount of the overdose was not stated, the therapeutic index for chloroquine is known to be small.[26] One of the children died after taking 0.75 or 1 gram, or twice a single therapeutic amount for children. Symptoms of overdose include headache, drowsiness, visual disturbances, nausea and vomiting, cardiovascular collapse, seizures, and sudden respiratory and cardiac arrest.[23]
An analog of chloroquine – hydroxychloroquine – has a long half-life (32–56 days) in blood and a large volume of distribution (580–815 L/kg).[27] The therapeutic, toxic and lethal ranges are usually considered to be 0.03 to 15 mg/l, 3.0 to 26 mg/l and 20 to 104 mg/l, respectively. However, nontoxic cases have been reported up to 39 mg/l, suggesting individual tolerance to this agent may be more variable than previously recognised.[27]
Pharmacology
Chloroquine’s absorption of the drug is rapid. It is widely distributed in body tissues. It’s protein binding is 55%.[ It’s metabolism is partially hepatic, giving rise to its main metabolite, desethylchloroquine. It’s excretion os ≥50% as unchanged drug in urine, where acidification of urine increases its elimination It has a very high volume of distribution, as it diffuses into the body’s adipose tissue.
Accumulation of the drug may result in deposits that can lead to blurred vision and blindness. It and related quinines have been associated with cases of retinal toxicity, particularly when provided at higher doses for longer times. With long-term doses, routine visits to an ophthalmologist are recommended.
Chloroquine is also a lysosomotropic agent, meaning it accumulates preferentially in the lysosomes of cells in the body. The pKa for the quinoline nitrogen of chloroquine is 8.5, meaning—in simplified terms, considering only this basic site—it is about 10% deprotonated at physiological pH (per the Henderson-Hasselbalch equation) This decreases to about 0.2% at a lysosomal pH of 4.6.Because the deprotonated form is more membrane-permeable than the protonated form, a quantitative “trapping” of the compound in lysosomes results.
Mechanism of action

Medical quinolines
Malaria

Hemozoin formation in P. falciparum: many antimalarials are strong inhibitors of hemozoin crystal growth.
The lysosomotropic character of chloroquine is believed to account for much of its antimalarial activity; the drug concentrates in the acidic food vacuole of the parasite and interferes with essential processes. Its lysosomotropic properties further allow for its use for in vitro experiments pertaining to intracellular lipid related diseases,[28][29] autophagy, and apoptosis.[30]
Inside red blood cells, the malarial parasite, which is then in its asexual lifecycle stage, must degrade hemoglobin to acquire essential amino acids, which the parasite requires to construct its own protein and for energy metabolism. Digestion is carried out in a vacuole of the parasitic cell.[citation needed]
Hemoglobin is composed of a protein unit (digested by the parasite) and a heme unit (not used by the parasite). During this process, the parasite releases the toxic and soluble molecule heme. The heme moiety consists of a porphyrin ring called Fe(II)-protoporphyrin IX (FP). To avoid destruction by this molecule, the parasite biocrystallizes heme to form hemozoin, a nontoxic molecule. Hemozoin collects in the digestive vacuole as insoluble crystals.[citation needed]
Chloroquine enters the red blood cell by simple diffusion, inhibiting the parasite cell and digestive vacuole. Chloroquine then becomes protonated (to CQ2+), as the digestive vacuole is known to be acidic (pH 4.7); chloroquine then cannot leave by diffusion. Chloroquine caps hemozoin molecules to prevent further biocrystallization of heme, thus leading to heme buildup. Chloroquine binds to heme (or FP) to form the FP-chloroquine complex; this complex is highly toxic to the cell and disrupts membrane function. Action of the toxic FP-chloroquine and FP results in cell lysis and ultimately parasite cell autodigestion. [31] Parasites that do not form hemozoin are therefore resistant to chloroquine.[32]
Resistance in malaria[edit source]
Since the first documentation of P. falciparum chloroquine resistance in the 1950s, resistant strains have appeared throughout East and West Africa, Southeast Asia, and South America. The effectiveness of chloroquine against P. falciparum has declined as resistant strains of the parasite evolved. They effectively neutralize the drug via a mechanism that drains chloroquine away from the digestive vacuole. Chloroquine-resistant cells efflux chloroquine at 40 times the rate of chloroquine-sensitive cells; the related mutations trace back to transmembrane proteins of the digestive vacuole, including sets of critical mutations in the P. falciparum chloroquine resistance transporter (PfCRT) gene. The mutated protein, but not the wild-type transporter, transports chloroquine when expressed in Xenopus oocytes (frog’s eggs) and is thought to mediate chloroquine leak from its site of action in the digestive vacuole.[33] Resistant parasites also frequently have mutated products of the ABC transporter P. falciparum multidrug resistance (PfMDR1) gene, although these mutations are thought to be of secondary importance compared to Pfcrt. Verapamil, a Ca2+ channel blocker, has been found to restore both the chloroquine concentration ability and sensitivity to this drug. Recently, an altered chloroquine-transporter protein CG2 of the parasite has been related to chloroquine resistance, but other mechanisms of resistance also appear to be involved.[34] Research on the mechanism of chloroquine and how the parasite has acquired chloroquine resistance is still ongoing, as other mechanisms of resistance are likely.[citation needed]
Other agents which have been shown to reverse chloroquine resistance in malaria are chlorpheniramine, gefitinib, imatinib, tariquidar and zosuquidar.[35]
Chloroquine has antiviral effects.[36] It increases late endosomal or lysosomal pH, resulting in impaired release of the virus from the endosome or lysosome – release requires a low pH. The virus is therefore unable to release its genetic material into the cell and replicate.[37][38]
Chloroquine also seems to act as a zinc ionophore, that allows extracellular zinc to enter the cell and inhibit viral RNA-dependent RNA polymerase.[39][40]
Other
Chloroquine inhibits thiamine uptake.[41] It acts specifically on the transporter SLC19A3.
Against rheumatoid arthritis, it operates by inhibiting lymphocyte proliferation, phospholipase A2, antigen presentation in dendritic cells, release of enzymes from lysosomes, release of reactive oxygen species from macrophages, and production of IL-1.
History
In Peru the indigenous people extracted the bark of the Cinchona plant[42] trees and used the extract (Chinchona officinalis) to fight chills and fever in the seventeenth century. In 1633 this herbal medicine was introduced in Europe, where it was given the same use and also began to be used against malaria.[43] The quinoline antimalarial drug quinine was isolated from the extract in 1820, and chloroquine is an analogue of this.
Chloroquine was discovered in 1934, by Hans Andersag and coworkers at the Bayer laboratories, who named it “Resochin”.[44] It was ignored for a decade, because it was considered too toxic for human use. During World War II, United States government-sponsored clinical trials for antimalarial drug development showed unequivocally that chloroquine has a significant therapeutic value as an antimalarial drug. It was introduced into clinical practice in 1947 for the prophylactic treatment of malaria.[45]
Society and culture
.jpg)
Resochin tablet package
Formulations
Chloroquine comes in tablet form as the phosphate, sulfate, and hydrochloride salts. Chloroquine is usually dispensed as the phosphate.[46]
Names
Brand names include Chloroquine FNA, Resochin, Dawaquin, and Lariago.[47]
Other animals
Chloroquine is used to control the aquarium fish parasite Amyloodinium ocellatum.[48]
Research
COVID-19
In late January 2020 during the 2019–20 coronavirus outbreak, Chinese medical researchers stated that exploratory research into chloroquine and two other medications, remdesivir and lopinavir/ritonavir, seemed to have “fairly good inhibitory effects” on the SARS-CoV-2 virus, which is the virus that causes COVID-19. Requests to start clinical testing were submitted.[49] Chloroquine had been also proposed as a treatment for SARS, with in vitro tests inhibiting the SARS-CoV virus.[50][51]
Chloroquine has been recommended by Chinese, South Korean and Italian health authorities for the treatment of COVID-19.[52][53] These agencies noted contraindications for people with heart disease or diabetes.[54] Both chloroquine and hydroxychloroquine were shown to inhibit SARS-CoV-2 in vitro, but a further study concluded that hydroxychloroquine was more potent than chloroquine, with a more tolerable safety profile.[55] Preliminary results from a trial suggested that chloroquine is effective and safe in COVID-19 pneumonia, “improving lung imaging findings, promoting a virus-negative conversion, and shortening the disease course.”[56] Self-medication with chloroquine has caused one known fatality.[57]
On 24 March 2020, NBC News reported[58] a fatality due to misuse of a chloroquine product used to control fish parasites.[59]
Other viruses
In October 2004, a group of researchers at the Rega Institute for Medical Research published a report on chloroquine, stating that chloroquine acts as an effective inhibitor of the replication of the severe acute respiratory syndrome coronavirus (SARS-CoV) in vitro.[60]
Chloroquine was being considered in 2003, in pre-clinical models as a potential agent against chikungunya fever.[61]
Other
The radiosensitizing and chemosensitizing properties of chloroquine are beginning to be exploited in anticancer strategies in humans.[62][63] In biomedicinal science, chloroquine is used for in vitro experiments to inhibit lysosomal degradation of protein products.
SYN


CLIP
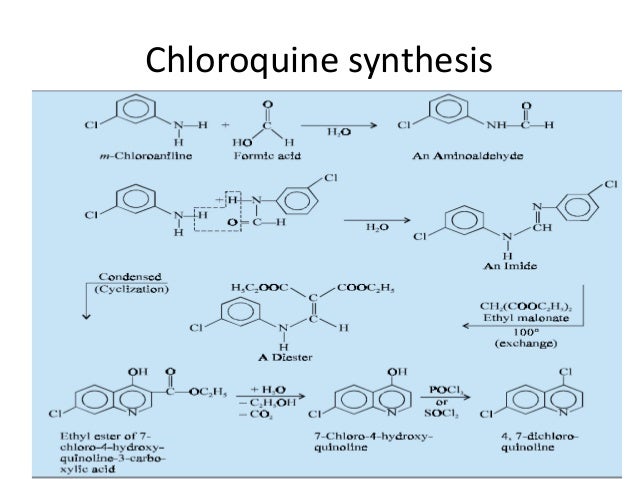
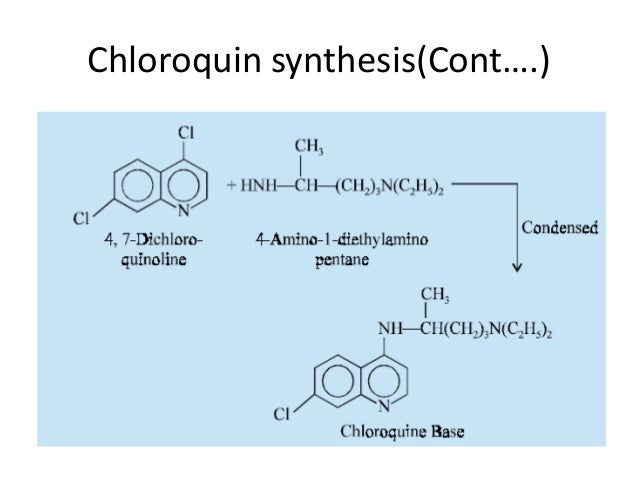
CLIP

CLIP
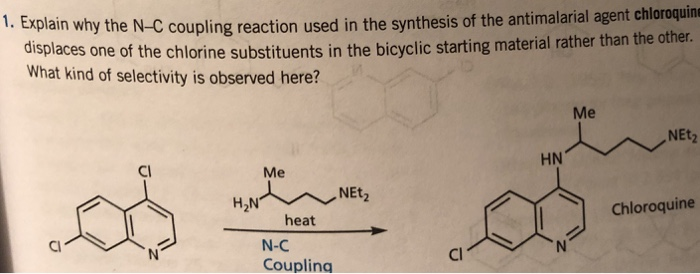
CLIP

References
- ^ Jump up to:a b c d e f g h i j k l m n “Aralen Phosphate”. The American Society of Health-System Pharmacists. Archived from the original on 8 December 2015. Retrieved 2 December 2015.
- ^ “Chloroquine Use During Pregnancy”. Drugs.com. Archivedfrom the original on 16 April 2019. Retrieved 16 April 2019.
There are no controlled data in human pregnancies.
- ^ Mittra, Robert A.; Mieler, William F. (1 January 2013). Ryan, Stephen J.; Sadda, SriniVas R.; Hinton, David R.; Schachat, Andrew P.; Sadda, SriniVas R.; Wilkinson, C. P.; Wiedemann, Peter; Schachat, Andrew P. (eds.). Retina (Fifth Edition). W.B. Saunders. pp. 1532–1554 – via ScienceDirect.
- ^ Cortegiani A, Ingoglia G, Ippolito M, Giarratano A, Einav S (March 2020). “A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19”. Journal of Critical Care. doi:10.1016/j.jcrc.2020.03.005. PMID 32173110.
- ^https://www.sciencedirect.com/science/article/pii/S0924857920300881
- ^https://www.sciencedirect.com/science/article/pii/B9781455707379000898
- ^ Manson P, Cooke G, Zumla A, eds. (2009). Manson’s tropical diseases (22nd ed.). [Edinburgh]: Saunders. p. 1240. ISBN 9781416044703. Archived from the original on 2 November 2018. Retrieved 9 September 2017.
- ^ Bhattacharjee M (2016). Chemistry of Antibiotics and Related Drugs. Springer. p. 184. ISBN 9783319407463. Archived from the original on 1 November 2018. Retrieved 9 September 2017.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “Chloroquine (Base)”. International Drug Price Indicator Guide. Archived from the original on 27 August 2018. Retrieved 4 December 2015.
- ^ “Frequently Asked Questions (FAQs): If I get malaria, will I have it for the rest of my life?”. US Centers for Disease Control and Prevention. 8 February 2010. Archived from the original on 13 May 2012. Retrieved 14 May 2012.
- ^ Plowe CV (2005). “Antimalarial drug resistance in Africa: strategies for monitoring and deterrence”. Malaria: Drugs, Disease and Post-genomic Biology. Current Topics in Microbiology and Immunology. 295. pp. 55–79. doi:10.1007/3-540-29088-5_3. ISBN 3-540-25363-7. PMID 16265887.
- ^ Uhlemann AC, Krishna S (2005). “Antimalarial multi-drug resistance in Asia: mechanisms and assessment”. Malaria: Drugs, Disease and Post-genomic Biology. Current Topics in Microbiology and Immunology. 295. pp. 39–53. doi:10.1007/3-540-29088-5_2. ISBN 3-540-25363-7. PMID 16265886.
- ^ “Chloroquine phosphate tablet – chloroquine phosphate tablet, coated”. dailymed.nlm.nih.gov. Archived from the original on 8 December 2015. Retrieved 4 November 2015.
- ^ CDC. Health information for international travel 2001–2002. Atlanta, Georgia: U.S. Department of Health and Human Services, Public Health Service, 2001.
- ^ Amebic Hepatic Abscesses~treatment at eMedicine
- ^ Jump up to:a b c d e f “Drugs & Medications”. http://www.webmd.com. Retrieved 22 March 2020.
- ^ Jump up to:a b c d “Chloroquine Side Effects: Common, Severe, Long Term”. Drugs.com. Retrieved 22 March 2020.
- ^ “Chloroquine: MedlinePlus Drug Information”. medlineplus.gov. Retrieved 22 March 2020.
- ^ Ajayi AA (September 2000). “Mechanisms of chloroquine-induced pruritus”. Clinical Pharmacology and Therapeutics. 68 (3): 336. PMID 11014416.
- ^ Vaziri A, Warburton B (1994). “Slow release of chloroquine phosphate from multiple taste-masked W/O/W multiple emulsions”. Journal of Microencapsulation. 11 (6): 641–8. doi:10.3109/02652049409051114. PMID 7884629.
- ^ Tönnesmann E, Kandolf R, Lewalter T (June 2013). “Chloroquine cardiomyopathy – a review of the literature”. Immunopharmacology and Immunotoxicology. 35 (3): 434–42. doi:10.3109/08923973.2013.780078. PMID 23635029.
- ^ Jump up to:a b c d e f g h i “Aralen Chloroquine Phosphate, USP” (PDF). Archived (PDF) from the original on 25 March 2020. Retrieved 24 March 2020.
- ^ Jump up to:a b “Malaria – Chapter 3 – 2016 Yellow Book”. wwwnc.cdc.gov. Archived from the original on 14 January 2016. Retrieved 11 November 2015.
- ^ Ullberg S, Lindquist NG, Sjòstrand SE (September 1970). “Accumulation of chorio-retinotoxic drugs in the foetal eye”. Nature. 227 (5264): 1257–8. Bibcode:1970Natur.227.1257U. doi:10.1038/2271257a0. PMID 5452818.
- ^ Cann HM, Verhulst HL (January 1961). “Fatal acute chloroquine poisoning in children”. Pediatrics. 27: 95–102. PMID 13690445.
- ^ Jump up to:a b Molina DK (March 2012). “Postmortem hydroxychloroquine concentrations in nontoxic cases”. The American Journal of Forensic Medicine and Pathology. 33 (1): 41–2. doi:10.1097/PAF.0b013e3182186f99. PMID 21464694.
- ^ Chen PM, Gombart ZJ, Chen JW (March 2011). “Chloroquine treatment of ARPE-19 cells leads to lysosome dilation and intracellular lipid accumulation: possible implications of lysosomal dysfunction in macular degeneration”. Cell & Bioscience. 1 (1): 10. doi:10.1186/2045-3701-1-10. PMC 3125200. PMID 21711726.
- ^ Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, et al. (April 2010). “Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61”. The Journal of Neuroscience. 30(17): 5948–57. doi:10.1523/JNEUROSCI.0157-10.2010. PMC 2868326. PMID 20427654.
- ^ Kim EL, Wüstenberg R, Rübsam A, Schmitz-Salue C, Warnecke G, Bücker EM, et al. (April 2010). “Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells”. Neuro-Oncology. 12 (4): 389–400. doi:10.1093/neuonc/nop046. PMC 2940600. PMID 20308316.
- ^ Hempelmann E (March 2007). “Hemozoin biocrystallization in Plasmodium falciparum and the antimalarial activity of crystallization inhibitors”. Parasitology Research. 100 (4): 671–6. doi:10.1007/s00436-006-0313-x. PMID 17111179.
- ^ Lin JW, Spaccapelo R, Schwarzer E, Sajid M, Annoura T, Deroost K, et al. (June 2015). “Replication of Plasmodium in reticulocytes can occur without hemozoin formation, resulting in chloroquine resistance” (PDF). The Journal of Experimental Medicine. 212(6): 893–903. doi:10.1084/jem.20141731. PMC 4451122. PMID 25941254. Archived (PDF) from the original on 22 September 2017. Retrieved 4 November 2018.
- ^ Martin RE, Marchetti RV, Cowan AI, Howitt SM, Bröer S, Kirk K (September 2009). “Chloroquine transport via the malaria parasite’s chloroquine resistance transporter”. Science. 325 (5948): 1680–2. Bibcode:2009Sci…325.1680M. doi:10.1126/science.1175667. PMID 19779197.
- ^ Essentials of medical pharmacology fifth edition 2003, reprint 2004, published by-Jaypee Brothers Medical Publisher Ltd, 2003, KD Tripathi, pages 739,740.
- ^ Alcantara LM, Kim J, Moraes CB, Franco CH, Franzoi KD, Lee S, et al. (June 2013). “Chemosensitization potential of P-glycoprotein inhibitors in malaria parasites”. Experimental Parasitology. 134 (2): 235–43. doi:10.1016/j.exppara.2013.03.022. PMID 23541983.
- ^ Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R (November 2003). “Effects of chloroquine on viral infections: an old drug against today’s diseases?”. The Lancet. Infectious Diseases. 3(11): 722–7. doi:10.1016/s1473-3099(03)00806-5. PMID 14592603.
- ^ Al-Bari MA (February 2017). “Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases”. Pharmacology Research & Perspectives. 5 (1): e00293. doi:10.1002/prp2.293. PMC 5461643. PMID 28596841.
- ^ Fredericksen BL, Wei BL, Yao J, Luo T, Garcia JV (November 2002). “Inhibition of endosomal/lysosomal degradation increases the infectivity of human immunodeficiency virus”. Journal of Virology. 76 (22): 11440–6. doi:10.1128/JVI.76.22.11440-11446.2002. PMC 136743. PMID 12388705.
- ^ Xue J, Moyer A, Peng B, Wu J, Hannafon BN, Ding WQ (1 October 2014). “Chloroquine is a zinc ionophore”. PloS One. 9(10): e109180. doi:10.1371/journal.pone.0109180. PMC 4182877. PMID 25271834.
- ^ te Velthuis AJ, van den Worm SH, Sims AC, Baric RS, Snijder EJ, van Hemert MJ (November 2010). “Zn(2+) inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture”. PLoS Pathogens. 6 (11): e1001176. doi:10.1371/journal.ppat.1001176. PMC 2973827. PMID 21079686.
- ^ Huang Z, Srinivasan S, Zhang J, Chen K, Li Y, Li W, et al. (2012). “Discovering thiamine transporters as targets of chloroquine using a novel functional genomics strategy”. PLOS Genetics. 8 (11): e1003083. doi:10.1371/journal.pgen.1003083. PMC 3510038. PMID 23209439.
- ^ Fern, Ken (2010–2020). “Cinchona officinalis – L.” Plans for a Future. Archived from the original on 25 August 2017. Retrieved 2 February 2020.
- ^ V. Kouznetsov, Vladímir (2008). “Antimalarials: construction of molecular hybrids based on chloroquine” (PDF). Universitas Scientiarum: 1. Archived (PDF) from the original on 22 February 2020. Retrieved 22 February 2020 – via scielo.
- ^ Krafts K, Hempelmann E, Skórska-Stania A (July 2012). “From methylene blue to chloroquine: a brief review of the development of an antimalarial therapy”. Parasitology Research. 111 (1): 1–6. doi:10.1007/s00436-012-2886-x. PMID 22411634.
- ^ “The History of Malaria, an Ancient Disease”. Centers for Disease Control. 29 July 2019. Archived from the original on 28 August 2010.
- ^ “Chloroquine”. nih.gov. National Institutes of Health. Retrieved 24 March 2020.
- ^ “Ipca Laboratories: Formulations – Branded”. Archived from the original on 6 April 2019. Retrieved 14 March 2020.
- ^ Francis-Floyd, Ruth; Floyd, Maxine R. “Amyloodinium ocellatum, an Important Parasite of Cultured Marine Fish” (PDF). agrilife.org.
- ^ “Could an old malaria drug help fight the new coronavirus?”. asbmb.org. Archived from the original on 6 February 2020. Retrieved 6 February 2020.
- ^ Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M (October 2004). “In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine”. Biochemical and Biophysical Research Communications. 323 (1): 264–8. doi:10.1016/j.bbrc.2004.08.085. PMID 15351731.
- ^ Devaux CA, Rolain JM, Colson P, Raoult D. New insights on the antiviral effects of chloroquine against coronavirus: what to expect for COVID-19? Int J Antimicrob Agents. 2020 Mar 11:105938. doi:10.1016/j.ijantimicag.2020.105938 PMID 32171740
- ^ “Physicians work out treatment guidelines for coronavirus”. m.koreabiomed.com (in Korean). 13 February 2020. Archivedfrom the original on 17 March 2020. Retrieved 18 March 2020.
- ^ “Azioni intraprese per favorire la ricerca e l’accesso ai nuovi farmaci per il trattamento del COVID-19”. aifa.gov.it (in Italian). Retrieved 18 March 2020.
- ^ “Plaquenil (hydroxychloroquine sulfate) dose, indications, adverse effects, interactions… from PDR.net”. http://www.pdr.net. Archivedfrom the original on 18 March 2020. Retrieved 19 March 2020.
- ^ Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, et al. (March 2020). “In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)”. Clinical Infectious Diseases. doi:10.1093/cid/ciaa237. PMID 32150618.
- ^ Gao J, Tian Z, Yang X (February 2020). “Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies”. Bioscience Trends. 14: 72–73. doi:10.5582/bst.2020.01047. PMID 32074550. Archived from the original on 19 March 2020. Retrieved 19 March 2020.
- ^ Edwards, Erika; Hillyard, Vaughn (23 March 2020). “Man dies after ingesting chloroquine in an attempt to prevent coronavirus”. NBC News. Retrieved 24 March 2020.
- ^ “A man died after ingesting a substance he thought would protect him from coronavirus”. NBC News. Retrieved 25 March 2020.
- ^ “Banner Health experts warn against self-medicating to prevent or treat COVID-19”. Banner Health (Press release). 23 March 2020. Retrieved 25 March 2020.
- ^ Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M (October 2004). “In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine”. Biochemical and Biophysical Research Communications. 323 (1): 264–8. doi:10.1016/j.bbrc.2004.08.085. PMID 15351731.
- ^ Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R (November 2003). “Effects of chloroquine on viral infections: an old drug against today’s diseases?”. The Lancet. Infectious Diseases. 3(11): 722–7. doi:10.1016/S1473-3099(03)00806-5. PMID 14592603.
- ^ Savarino A, Lucia MB, Giordano F, Cauda R (October 2006). “Risks and benefits of chloroquine use in anticancer strategies”. The Lancet. Oncology. 7 (10): 792–3. doi:10.1016/S1470-2045(06)70875-0. PMID 17012039.
- ^ Sotelo J, Briceño E, López-González MA (March 2006). “Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial”. Annals of Internal Medicine. 144 (5): 337–43. doi:10.7326/0003-4819-144-5-200603070-00008. PMID 16520474.
“Summaries for patients. Adding chloroquine to conventional chemotherapy and radiotherapy for glioblastoma multiforme”. Annals of Internal Medicine. 144 (5): I31. March 2006. doi:10.7326/0003-4819-144-5-200603070-00004. PMID 16520470.
External links
“Chloroquine”. Drug Information Portal. U.S. National Library of Medicine.
- “Medicines for the Prevention of Malaria While Traveling – Chloroquine (Aralen)” (PDF) (Fact sheet). U.S. Centers for Disease Control and Prevention (CDC).
 The dictionary definition of chloroquine at Wiktionary
The dictionary definition of chloroquine at Wiktionary
 |
|
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ˈklɔːrəkwɪn/ |
| Trade names | Aralen, other |
| Other names | Chloroquine phosphate |
| AHFS/Drugs.com | Monograph |
| License data |
|
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Liver |
| Elimination half-life | 1-2 months |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| NIAID ChemDB | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.000.175 |
| Chemical and physical data | |
| Formula | C18H26ClN3 |
| Molar mass | 319.872 g·mol−1 |
| 3D model (JSmol) | |
//////////////CHLOROQUINE,, クロロキン, ANTIMALARIAL, COVID 19, CORONA VIRUS, Хлорохин , クロロキン , كلوروكين
Hydroxychloroquine, ヒドロキシクロロキン, гидроксихлорохин , هيدروكسيكلوروكين , 羟氯喹 ,
|
Hydroxychloroquine
ヒドロキシクロロキン;
|
|
| Formula |
C18H26ClN3O
|
|---|---|
| cas |
118-42-3
sulphate 747-36-4
|
| Mol weight |
335.8715
|
Hydroxychloroquine (HCQ), sold under the brand name Plaquenil among others, is a medication used for the prevention and treatment of certain types of malaria.[2] Specifically it is used for chloroquine-sensitive malaria.[3] Other uses include treatment of rheumatoid arthritis, lupus, and porphyria cutanea tarda.[2] It is taken by mouth.[2] It is also being used as an experimental treatment for coronavirus disease 2019 (COVID-19).[4]
Common side effects include vomiting, headache, changes in vision and muscle weakness.[2] Severe side effects may include allergic reactions.[2] Although all risk cannot be excluded it remains a treatment for rheumatic disease during pregnancy.[5] Hydroxychloroquine is in the antimalarial and 4-aminoquinoline families of medication.[2]
Hydroxychloroquine was approved for medical use in the United States in 1955.[2] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[6] The wholesale cost in the developing world is about US$4.65 per month as of 2015, when used for rheumatoid arthritis or lupus.[7] In the United States the wholesale cost of a month of treatment is about US$25 as of 2020.[8] In the United Kingdom this dose costs the NHS about £ 5.15.[9] In 2017, it was the 128th most prescribed medication in the United States with more than five million prescriptions.[10]
Medical use
Hydroxychloroquine treats malaria, systemic lupus erythematosus, rheumatic disorders like rheumatoid arthritis, porphyria cutanea tarda, and Q fever.[2]
In 2014, its efficacy to treat Sjögren syndrome was questioned in a double-blind study involving 120 patients over a 48-week period.[11]
Hydroxychloroquine is widely used in the treatment of post-Lyme arthritis. It may have both an anti-spirochaete activity and an anti-inflammatory activity, similar to the treatment of rheumatoid arthritis.[12]
Contraindications
The drug label advises that hydroxychloroquine should not be prescribed to individuals with known hypersensitivity to 4-Aminoquinoline compounds.[13] There are a range of other contraindications[14] [15] and caution is required if patients have certain heart conditions, diabetes, psoriasis etc.
Side effects[
The most common adverse effects are a mild nausea and occasional stomach cramps with mild diarrhea. The most serious adverse effects affect the eye, with dose-related retinopathy as a concern even after hydroxychloroquine use is discontinued.[2] For short-term treatment of acute malaria, adverse effects can include abdominal cramps, diarrhea, heart problems, reduced appetite, headache, nausea and vomiting.[2]
For prolonged treatment of lupus or rheumatoid arthritis, adverse effects include the acute symptoms, plus altered eye pigmentation, acne, anemia, bleaching of hair, blisters in mouth and eyes, blood disorders, convulsions, vision difficulties, diminished reflexes, emotional changes, excessive coloring of the skin, hearing loss, hives, itching, liver problems or liver failure, loss of hair, muscle paralysis, weakness or atrophy, nightmares, psoriasis, reading difficulties, tinnitus, skin inflammation and scaling, skin rash, vertigo, weight loss, and occasionally urinary incontinence.[2] Hydroxychloroquine can worsen existing cases of both psoriasis and porphyria.[2]
Children may be especially vulnerable to developing adverse effects from hydroxychloroquine.[2]
Eyes
One of the most serious side effects is retinopathy (generally with chronic use).[2][16] People taking 400 mg of hydroxychloroquine or less per day generally have a negligible risk of macular toxicity, whereas the risk begins to go up when a person takes the medication over 5 years or has a cumulative dose of more than 1000 grams. The daily safe maximum dose for eye toxicity can be computed from one’s height and weight using this calculator. Cumulative doses can also be calculated from this calculator. Macular toxicity is related to the total cumulative dose rather than the daily dose. Regular eye screening, even in the absence of visual symptoms, is recommended to begin when either of these risk factors occurs.[17]
Toxicity from hydroxychloroquine may be seen in two distinct areas of the eye: the cornea and the macula. The cornea may become affected (relatively commonly) by an innocuous cornea verticillata or vortex keratopathy and is characterized by whorl-like corneal epithelial deposits. These changes bear no relationship to dosage and are usually reversible on cessation of hydroxychloroquine.
The macular changes are potentially serious. Advanced retinopathy is characterized by reduction of visual acuity and a “bull’s eye” macular lesion which is absent in early involvement.
Overdose
Due to rapid absorption, symptoms of overdose can occur within a half an hour after ingestion. Overdose symptoms include convulsions, drowsiness, headache, heart problems or heart failure, difficulty breathing and vision problems.
Hydroxychloroquine overdoses are rarely reported, with 7 previous cases found in the English medical literature. In one such case, a 16-year-old girl who had ingested a handful of hydroxychloroquine 200mg presented with tachycardia (heart rate 110 beats/min), hypotension (systolic blood pressure 63 mm Hg), central nervous system depression, conduction defects (ORS = 0.14 msec), and hypokalemia (K = 2.1 meq/L). Treatment consisted of fluid boluses and dopamine, oxygen, and potassium supplementation. The presence of hydroxychloroquine was confirmed through toxicologic tests. The patient’s hypotension resolved within 4.5 hours, serum potassium stabilized in 24 hours, and tachycardia gradually decreased over 3 days.[18]
Interactions
The drug transfers into breast milk and should be used with care by pregnant or nursing mothers.[citation needed]
Care should be taken if combined with medication altering liver function as well as aurothioglucose (Solganal), cimetidine (Tagamet) or digoxin (Lanoxin). HCQ can increase plasma concentrations of penicillamine which may contribute to the development of severe side effects. It enhances hypoglycemic effects of insulin and oral hypoglycemic agents. Dose altering is recommended to prevent profound hypoglycemia. Antacids may decrease the absorption of HCQ. Both neostigmine and pyridostigmine antagonize the action of hydroxychloroquine.[19]
While there may be a link between hydroxychloroquine and hemolytic anemia in those with glucose-6-phosphate dehydrogenase deficiency, this risk may be low in those of African descent.[20]
Specifically, the FDA drug label for hydroxychloroquine lists the following drug interactions [13]:
- Digoxin (wherein it may result in increased serum digoxin levels)
- Insulin or antidiabetic drugs (wherein it may enhance the effects of a hypoglycemic treatment)
- Drugs that prolong QT interval and other arrhythmogenic drugs (as Hydroxychloroquine prolongs the QT interval and may increase the risk of inducing ventricular arrhythmias if used concurrently)
- Mefloquine and other drugs known to lower the convulsive threshold (co-administration with other antimalarials known to lower the convulsion threshold may increase risk of convulsions)
- Antiepileptics (concurrent use may impair the antiepileptic activity)
- Methotrexate (combined use is unstudied and may increase the frequency of side effects)
- Cyclosporin (wherein an increased plasma cylcosporin level was reported when used together).
Pharmacology[
Pharmacokinetics
Hydroxychloroquine has similar pharmacokinetics to chloroquine, with rapid gastrointestinal absorption and elimination by the kidneys. Cytochrome P450 enzymes (CYP2D6, 2C8, 3A4 and 3A5) metabolize hydroxychloroquine to N-desethylhydroxychloroquine.[21]
Pharmacodynamics
Antimalarials are lipophilic weak bases and easily pass plasma membranes. The free base form accumulates in lysosomes (acidic cytoplasmic vesicles) and is then protonated,[22] resulting in concentrations within lysosomes up to 1000 times higher than in culture media. This increases the pH of the lysosome from 4 to 6.[23] Alteration in pH causes inhibition of lysosomal acidic proteases causing a diminished proteolysis effect.[24] Higher pH within lysosomes causes decreased intracellular processing, glycosylation and secretion of proteins with many immunologic and nonimmunologic consequences.[25] These effects are believed to be the cause of a decreased immune cell functioning such as chemotaxis, phagocytosis and superoxide production by neutrophils.[26] HCQ is a weak diprotic base that can pass through the lipid cell membrane and preferentially concentrate in acidic cytoplasmic vesicles. The higher pH of these vesicles in macrophages or other antigen-presenting cells limits the association of autoantigenic (any) peptides with class II MHC molecules in the compartment for peptide loading and/or the subsequent processing and transport of the peptide-MHC complex to the cell membrane.[27]
Mechanism of action
Hydroxychloroquine increases[28] lysosomal pH in antigen-presenting cells. In inflammatory conditions, it blocks toll-like receptors on plasmacytoid dendritic cells (PDCs).[citation needed] Hydroxychloroquine, by decreasing TLR signaling, reduces the activation of dendritic cells and the inflammatory process. Toll-like receptor 9 (TLR 9) recognizes DNA-containing immune complexes and leads to the production of interferon and causes the dendritic cells to mature and present antigen to T cells, therefore reducing anti-DNA auto-inflammatory process.
In 2003, a novel mechanism was described wherein hydroxychloroquine inhibits stimulation of the toll-like receptor (TLR) 9 family receptors. TLRs are cellular receptors for microbial products that induce inflammatory responses through activation of the innate immune system.[29]
As with other quinoline antimalarial drugs, the mechanism of action of quinine has not been fully resolved. The most accepted model is based on hydrochloroquinine and involves the inhibition of hemozoin biocrystallization, which facilitates the aggregation of cytotoxic heme. Free cytotoxic heme accumulates in the parasites, causing their deaths.[citation needed]
Brand names
It is frequently sold as a sulfate salt known as hydroxychloroquine sulfate.[2] 200 mg of the sulfate salt is equal to 155 mg of the base.[2]
Brand names of hydroxychloroquine include Plaquenil, Hydroquin, Axemal (in India), Dolquine, Quensyl, Quinoric.[30]
Research
COVID-19
Hydroxychloroquine and chloroquine have been recommended by Chinese and South Korean health authorities for the experimental treatment of COVID-19.[31][32] In vitro studies in cell cultures demonstrated that hydroxychloroquine was more potent than chloroquine against SARS-CoV-2.[33]
On 17 March 2020, the AIFA Scientific Technical Commission of the Italian Medicines Agency expressed a favorable opinion on including the off-label use of chloroquine and hydroxychloroquine for the treatment of SARS-CoV-2 infection.[34]
clip

clip
https://d-nb.info/1166863441/34
white solid (0.263 g, 78%). 1H NMR
(600 MHz, CDCl3
) δ 8.48 (d, J = 5.4 Hz, 1H), 7.93 (d, J = 5.4 Hz, 1H), 7.70 (d, J = 9.2 Hz, 1H), 7.34 (dd, J = 8.8, 7.3 Hz, 1H), 6.39 (d, J = 5.4 Hz, 1H), 4.96 (d, J = 7.5 Hz, 1H), 3.70 (sx,J = 6.8 Hz, 1H), 3.55 (m, 2H), 2.57 (m, 5H), 2.49 (m, 2H),
1.74–1.62 (m, 1H), 1.65–1.53 (m, 3H), 1.31 (d, J = 6.9 Hz, 3H),
1.24 (d, J = 7.2 Hz, 2H);
13C NMR (125 MHz, CDCl3) δ 152.2,
149.5, 149.2, 135.0, 129.0, 125.4, 121.2, 117.4, 99.4, 58.6, 54.9,
53.18, 48.5, 47.9, 34.5, 24.1, 20.6, 11.9. Spectra were obtained
in accordance with those previously reported [38,39].
38. Cornish, C. A.; Warren, S. J. Chem. Soc., Perkin Trans. 1 1985,
2585–2598. doi:10.1039/P19850002585
39. Münstedt, R.; Wannagat, U.; Wrobel, D. J. Organomet. Chem. 1984,
264, 135–148. doi:10.1016/0022-328X(84)85139-6
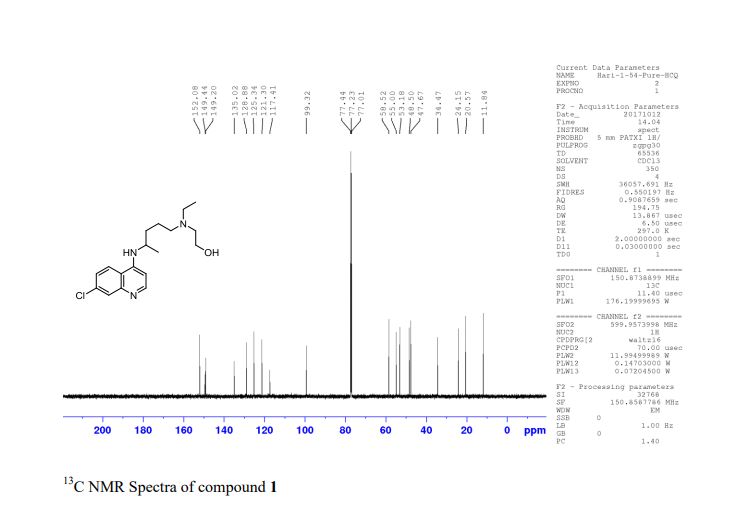
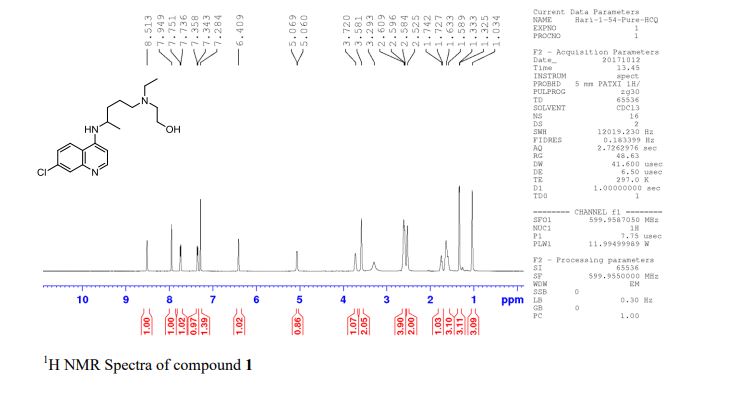
References
- ^ Jump up to:a b “Hydroxychloroquine Use During Pregnancy”. Drugs.com. 28 February 2020. Retrieved 21 March 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p “Hydroxychloroquine Sulfate Monograph for Professionals”. The American Society of Health-System Pharmacists. 20 March 2020. Archived from the original on 20 March 2020. Retrieved 20 March 2020.
- ^ Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia. Jones & Bartlett Learning. p. 463. ISBN 9781284057560.
- ^ Cortegiani, Andrea; Ingoglia, Giulia; Ippolito, Mariachiara; Giarratano, Antonino; Einav, Sharon (10 March 2020). “A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19”. Journal of Critical Care. doi:10.1016/j.jcrc.2020.03.005. ISSN 0883-9441.
- ^ Flint, Julia; Panchal, Sonia; Hurrell, Alice; van de Venne, Maud; Gayed, Mary; Schreiber, Karen; Arthanari, Subha; Cunningham, Joel; Flanders, Lucy; Moore, Louise; Crossley, Amy (1 September 2016). “BSR and BHPR guideline on prescribing drugs in pregnancy and breastfeeding – Part I: standard and biologic disease modifying anti-rheumatic drugs and corticosteroids”. Rheumatology. 55 (9): 1693–1697. doi:10.1093/rheumatology/kev404. ISSN 1462-0324.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “Single Drug Information | International Medical Products Price Guide”. Retrieved 31 December 2019.[dead link]
- ^ “NADAC as of 2019-08-07”. Centers for Medicare and Medicaid Services. Retrieved 19 March 2020.
Typical dose is 600mg per day. Costs 0.28157 per dose. Month has about 30 days.
- ^ British national formulary: BNF 69 (69 ed.). British Medical Association. 2015. p. 730. ISBN 9780857111562.
- ^ “The Top 300 of 2020”. ClinCalc. Retrieved 18 March 2020.
- ^ Effects of Hydroxychloroquine on Symptomatic Improvement in Primary Sjögren Syndrome, Gottenberg, et al. (2014) “Archived copy”. Archived from the original on 11 July 2015. Retrieved 10 July 2015.
- ^ Steere, AC; Angelis, SM (October 2006). “Therapy for Lyme Arthritis: Strategies for the Treatment of Antibiotic-refractory Arthritis”. Arthritis and Rheumatism. 54 (10): 3079–86. doi:10.1002/art.22131. PMID 17009226.
- ^ Jump up to:a b “Plaquenil- hydroxychloroquine sulfate tablet”. DailyMed. 3 January 2020. Retrieved 20 March 2020.
- ^ “Plaquenil (hydroxychloroquine sulfate) dose, indications, adverse effects, interactions”. pdr.net. Retrieved 19 March 2020.
- ^ “Drugs & Medications”. webmd.com. Retrieved 19 March 2020.
- ^ Flach, AJ (2007). “Improving the Risk-benefit Relationship and Informed Consent for Patients Treated with Hydroxychloroquine”. Transactions of the American Ophthalmological Society. 105: 191–94, discussion 195–97. PMC 2258132. PMID 18427609.
- ^ Marmor, MF; Kellner, U; Lai, TYY; Lyons, JS; Mieler, WF (February 2011). “Revised Recommendations on Screening for Chloroquine and Hydroxychloroquine Retinopathy”. Ophthalmology. 118 (2): 415–22. doi:10.1016/j.ophtha.2010.11.017. PMID 21292109.
- ^ Marquardt, Kathy; Albertson, Timothy E. (1 September 2001). “Treatment of hydroxychloroquine overdose”. The American Journal of Emergency Medicine. 19 (5): 420–424. doi:10.1053/ajem.2001.25774. ISSN 0735-6757. PMID 11555803.
- ^ “Russian Register of Medicines: Plaquenil (hydroxychloroquine) Film-coated Tablets for Oral Use. Prescribing Information” (in Russian). Sanofi-Synthelabo. Archived from the original on 16 August 2016. Retrieved 14 July 2016.
- ^ Mohammad, Samya; Clowse, Megan E. B.; Eudy, Amanda M.; Criscione-Schreiber, Lisa G. (March 2018). “Examination of Hydroxychloroquine Use and Hemolytic Anemia in G6PDH-Deficient Patients”. Arthritis Care & Research. 70 (3): 481–485. doi:10.1002/acr.23296. ISSN 2151-4658. PMID 28556555.
- ^ Kalia, S; Dutz, JP (2007). “New Concepts in Antimalarial Use and Mode of Action in Dermatology”. Dermatologic Therapy. 20 (4): 160–74. doi:10.1111/j.1529-8019.2007.00131.x. PMID 17970883.
- ^ Kaufmann, AM; Krise, JP (2007). “Lysosomal Sequestration of Amine-containing Drugs: Analysis and Therapeutic Implications”. Journal of Pharmaceutical Sciences. 96 (4): 729–46. doi:10.1002/jps.20792. PMID 17117426.
- ^ Ohkuma, S; Poole, B (1978). “Fluorescence Probe Measurement of the Intralysosomal pH in Living Cells and the Perturbation of pH by Various Agents”. Proceedings of the National Academy of Sciences of the United States of America. 75 (7): 3327–31. doi:10.1073/pnas.75.7.3327. PMC 392768. PMID 28524.
- ^ Ohkuma, S; Chudzik, J; Poole, B (1986). “The Effects of Basic Substances and Acidic Ionophores on the Digestion of Exogenous and Endogenous Proteins in Mouse Peritoneal Macrophages”. The Journal of Cell Biology. 102 (3): 959–66. doi:10.1083/jcb.102.3.959. PMC 2114118. PMID 3949884.
- ^ Oda, K; Koriyama, Y; Yamada, E; Ikehara, Y (1986). “Effects of Weakly Basic Amines on Proteolytic Processing and Terminal Glycosylation of Secretory Proteins in Cultured Rat Hepatocytes”. The Biochemical Journal. 240 (3): 739–45. doi:10.1042/bj2400739. PMC 1147481. PMID 3493770.
- ^ Hurst, NP; French, JK; Gorjatschko, L; Betts, WH (1988). “Chloroquine and Hydroxychloroquine Inhibit Multiple Sites in Metabolic Pathways Leading to Neutrophil Superoxide Release”. The Journal of Rheumatology. 15 (1): 23–27. PMID 2832600.
- ^ Fox, R (1996). “Anti-malarial Drugs: Possible Mechanisms of Action in Autoimmune Disease and Prospects for Drug Development”. Lupus. 5: S4–10. doi:10.1177/096120339600500103. PMID 8803903.
- ^ Waller; et al. Medical Pharmacology and Therapeutics (2nd ed.). p. 370.
- ^ Takeda, K; Kaisho, T; Akira, S (2003). “Toll-Like Receptors”. Annual Review of Immunology. 21: 335–76. doi:10.1146/annurev.immunol.21.120601.141126. PMID 12524386.
- ^ “Hydroxychloroquine trade names”. Drugs-About.com. Retrieved 18 June 2019.
- ^ “Diagnosis and Treatment Protocol for Novel Coronavirus Pneumonia”. China Law Translate. 3 March 2020. Retrieved 18 March 2020.
- ^ “Physicians work out treatment guidelines for coronavirus”. Korea Biomedical Review. 13 February 2020. Retrieved 18 March2020.
- ^ Yao, Xueting; Ye, Fei; Zhang, Miao; Cui, Cheng; Huang, Baoying; Niu, Peihua; Liu, Xu; Zhao, Li; Dong, Erdan; Song, Chunli; Zhan, Siyan (9 March 2020). “In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)”. Clinical Infectious Diseases. doi:10.1093/cid/ciaa237. ISSN 1537-6591. PMID 32150618.
- ^ “Azioni intraprese per favorire la ricerca e l’accesso ai nuovi farmaci per il trattamento del COVID-19”. Italian Medicines Agency (AIFA) (in Italian). 17 March 2020. Retrieved 18 March2020.
External links
- “Hydroxychloroquine”. Drug Information Portal. U.S. National Library of Medicine.
 |
|

Hydroxychloroquine freebase molecule
|
|
| Clinical data | |
|---|---|
| Trade names | Plaquenil, others |
| Other names | Hydroxychloroquine sulfate |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a601240 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth (tablets) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Variable (74% on average); Tmax = 2–4.5 hours |
| Protein binding | 45% |
| Metabolism | Liver |
| Elimination half-life | 32–50 days |
| Excretion | Mostly Kidney (23–25% as unchanged drug), also biliary (<10%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.003.864 |
| Chemical and physical data | |
| Formula | C18H26ClN3O |
| Molar mass | 335.872 g/mol g·mol−1 |
| 3D model (JSmol) | |
///////////Hydroxychloroquine, Hydroxy chloroquine, HCQ, ヒドロキシクロロキン , covid 19, coronavirus, antimalarial, гидроксихлорохин , هيدروكسيكلوروكين , 羟氯喹 , Oxychlorochin, Plaquenil , Plaquenil®,
Lumefantrine
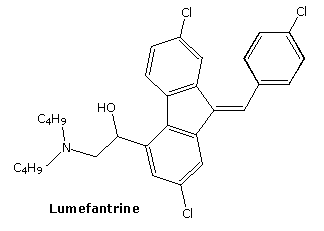
lumefantrine
- Molecular FormulaC30H32Cl3NO
- Average mass528.940 Da
- Benflumetol
- dl-Benflumelol
UNIIF38R0JR742
CAS number82186-77-4
2-(dibutylamino)-1-[(9Z)-2,7-dichloro-9-[(4-chlorophenyl)methylidene]-9H-fluoren-4-yl]ethan-1-ol
| (±)-2,7-Dichloro-9-((Z)-p-chlorobenzylidene)-α-((dibutylamino)methyl)fluorene-4-methanol |
| 2-Dibutylamino-1-[2,7-dichloro-9-(4-chloro-benzylidene)-9H-fluoren-4-yl]-ethanol |
| 2-Dibutylamino-1-{2,7-dichloro-9-[1-(4-chloro-phenyl)-meth-(Z)-ylidene]-9H-fluoren-4-yl}-ethanol |
| Benflumetol |
| dl-Benflumelol |
UNII F38R0JR742
CAS number 82186-77-4
Weight Average: 528.94
Monoisotopic: 527.154947772
Chemical Formula C30H32Cl3NO
Lumefantrine (or benflumetol) is an antimalarial drug. It is only used in combination with artemether. The term “co-artemether” is sometimes used to describe this combination.[1] Lumefantrine has a much longer half-life compared to artemether and so is therefore thought to clear any residual parasites that remain after combination treatment.[2]
Lumefantrine, along with pyronaridine and naphtoquine, were synthesized in course of the Project 523 antimalaria drug research initiated in 1967; these compounds are all used in combination antimalaria therapies.[3][4][5]

Lumefantrine is an antimalarial drug chemically known as 2-(dibutylamino)-1-[(9Z)-2, 7-dichloro-9-(4- chlorobenzylidene)-9H-floren-4-yl] ethanol, which is used in the prevention and treatment of Malaria in worm blooded animals. Lumefantrine is using the combination of β-Artemether in the treatment of Malaria
SYN
Synthetic Reference
Beutler, Ulrich; Fuenfschilling, Peter C.; Steinkemper, Andreas. An Improved Manufacturing Process for the Antimalaria Drug Coartem. Part II. Organic Process Research & Development. Volume 11. Issue 3. Pages 341-345. 2007.

SYN 2
Synthetic Reference
Rao, Dharmaraj Ramachandra; Kankan, Rajendra Narayanrao; Phull, Manjinder Singh. Process for preparation of lumefantrine as antimalarial agent with improved method. Assignee Cipla Co., Ltd., India. CN 1865227. (2006).

SYN 3
Synthetic Reference
Sethi, Madhuresh Kumar; Gonuguntla, Anantavena Rani; Arikatla, Siva Lakshmi Devi; Mulukutla, Suryanarayana; Yerramalla, Rajakrishna; Bontalakoti, Jaganmohanarao; Vemula, Lakshminarayana; Thirunavukarasu, Jayaprakash. Synthesis and characterization of novel related substances of Lumefantrine, an anti-malarial drug. Pharma Chemica. Volume 8. Issue 3. Pages 91-100. 2016.

SYN4
Synthetic Reference
Mathur, Prafull; Mathur, Suvigya; Vishwanath, Kannan; Mishra, Anand Kumar. Preparation of lumefantrine. Assignee Aanjaneya Lifecare Limited, India. IN 2013MU00611. (2015).

SYN 5
Synthetic Reference
Wu, Guang-liang; Dai, Ying-jie; Kang, Cong-min; Zi, Yan. A new synthetic technology of anti-malarial drug lumefantrine. Zhongguo Xinyao Zazhi. Volume 21. Issue 24. Pages 2944-2947. 2012.

SYN 6
Synthetic Reference
Krishna, Bettadapura Gundappa; Verma, Sudhakar; Krishna, Sujatha; Naik, Gajanan; Arulmoli, Thangavel. A process for preparation of lumefantrine. Assignee SeQuent Scientific Limited, India. IN 2012CH00470. (2012).

SYN 7
Synthetic Reference
Bansi, Lal; Genbhau, Gund Vitthal; Prabhakar, Bapat Chintamani; Popat, Bochiya Pravin; Banshi, Punde Dnyanadeo; Venkata, Reddy Prabhakar Gorla. Improved one pot process for the synthesis of lumefantrine. Assignee Calyx Chemicals and Pharmaceuticals Ltd., India. IN 2009MU01437. (2010).

SYN 8
Synthetic Reference
Shailesh, Singh; Dhaval, Vashi; Vinod, Gaikwad; Sanjay, Chowkekar; Sanjay, Bute. Process for the preparation of lumefantrine. Assignee Ajanta Pharma Ltd., India. IN 2008MU01677. (2010).

SYN 9
Synthetic Reference
Rawalnath, Sakhardande Rajiv; Kanji, Khatri Navin; Nilkanth, Firake Pandharinath; Vasant, Panchal Rajesh; Nagesh, Babrekar Chandan; Madhukar, Mohite Dhanaji. Preparation of lumefantrine. Assignee Saxena, Alok, India. IN 2006MU00260. (2007).

///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
https://patents.google.com/patent/CN107501316A/en
(19) (PDF) Process for high purity lumefantrine. Available from: https://www.researchgate.net/publication/221933417_Process_for_high_purity_lumefantrine [accessed Feb 06 2022].
(19) (PDF) Process for high purity lumefantrine. Available from: https://www.researchgate.net/publication/221933417_Process_for_high_purity_lumefantrine [accessed Feb 06 2022].
(19) (PDF) Process for high purity lumefantrine. Available from: https://www.researchgate.net/publication/221933417_Process_for_high_purity_lumefantrine [accessed Feb 06 2022].
PAPER
Tropical Journal of Pharmaceutical Research October 2013; 12 (5): 791-798 ISSN: 1596-5996 (print); 1596-9827 (electronic) © Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. All rights reserved. Available online at http://www.tjpr.org
CLIP
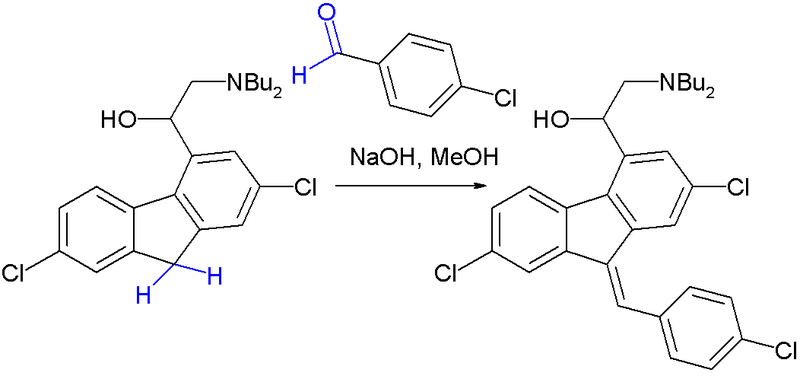
NMR

1H NMR

COSY

NOESY

HSQC

DEPT

13C NMR

DSC

CLIP
CLIP
Click to access DPC-2016-8-3-91-100.pdf
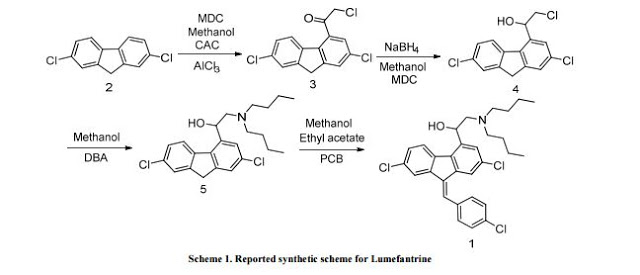
REFERENCES
[1] Ulrich Beutler, C Peter.; Fuenfschilling.; and Andreas, Steinkemper.; Novartis Pharma AG; Chemical and Analytical Development: CH-4002 Basel, Switzerland, Organic Process Research & Development 2007, 11, 341- 345.
[2] Boehm, M. Fuenfschilling.; Krieger, P. C.; Kuesters, E. M.; Struber, F.; Org. Process Res. DeV. 2007, 11, 336- 340.
[3] (a) Rao, D. R.; Kankan, R. N.; Phull, M. S.; Patent Application CN 1009-3724 20060424, 2005. (b) Deng, R.; Zhong, J.; Zhao, D.; Wang, J.; Yaoxue, X. 2000, 35 (1), 22. (c) Allmendinger, Th.; Wernsdorfer, W. H. PCT WO 99/67197.
[4] Perrumattam, J.; Shao, Ch.; Confer, W. L. Synthesis 1994, 1181.
[5] Fuenfschilling, P. C.; Hoehn P.; Mutz J.-P. Organic Process Res. Dev. 2007, 11, 13.
[6] Di Nunno, L.; Scilimati, A. Tetrahedron 1988, 44, 3639.
[7] Pharmacopeial Forum, Vol. 36(2) [Mar.-Apr. 2010]
Preparation of 2-(dibutylamino)-1-[(9Z)-2, 7-dichloro-9-(4-chlorobenzylidene)-9H-floren-4-yl] ethanol (Lumefantrine) 1.
To a stirred solution of NaOH (1.97 g 0.0492 mol) in methanol (100 ml) there was added 1-(2, 7- dichloro-9 H-fluren-4-yl)-2-(dibutyl amino) ethanol (10 g, 0.0246 mol) and para chloro benzaldehyde (5.24 g 0.0372). The suspension obtained was stirred at reflux temperature till the absence of starting material by TLC. After confirming the product formation reaction mixture was cooled to room temperature and further stirred at same temperature for overnight. The precipitated solids were filtered and washed with methanol and dried under vacuum at 50°C to get desired compound. (Purity by HPLC: 99%).
IR (cm-1): 3408, 3092, 2953, 2928, 2870, 2840, 1634, 1589, 1487, 1465, 1443, 1400, 1365, 1308, 1268, 1241, 1207, 1173, 1156, 1085, 1071, 1014, 980, 933, 874, 839, 815, 806, 770;
1H NMR (CDCl3, δ ppm): 7.75 (d, 1H, CH, J 1.5 Hz), 7.68 (d, 1H, CH, J 1.5 Hz), 7.60-7.63 (m, 1H, CH), 7.32-7.35 (dd, 1H, CH, J 1.7,8.3 Hz), 7.45-7.50 (m, 1H, CH), 5.35-5.39 (dd, 1H, CH, J 3.0,9.9 Hz), 2.41-2.74 (m, 1H, CH2Ha), 2.86-2.92 (m, 1H, CH2Hb), 2.41-2.74 (m, 4H, CH2), 1.25-1.56 (m, 8H, CH2), 0.97 (t, 1H, CH, J 7.2 Hz), 7.60-7.63 (m, 1H, CH), 7.45-7.50 (m, 4H, CH), 4.54 (broad, 1H, OH),
13C NMR (CDCl3, δ ppm): 138.2, 141.5, 120.6, 133.2, 126.3, 135.0, 135.0, 136.4, 123.9, 128.3, 132.8, 123.0, 139.8, 65.5, 60.0, 53.5, 29.1, 20.6, 14.0, 127.6, 134.7, 130.5, 129.1, 133.2;
MS: m/z: 528 [M+H]+ ; Analysis calcd. for C30H32Cl3NO: C, 68.12; H, 6.10; N, 2.65% Found: C, 68.38; H, 6.14; N, 2.63 %.
CLIP
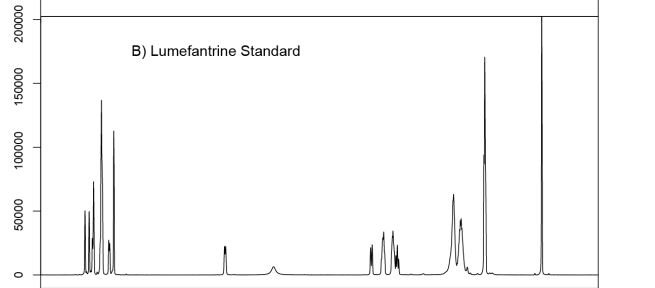
One-dimensional 1H NMR spectrum of B) a lumefantrine standard,
A CLIP
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-50532012000100010&lng=en&nrm=iso




CLIP
A simple and precise method for quantitative analysis of lumefantrine …
[2–4] Thus, today lumefantrine is a drug of choice in antimalarial treatment against P. …. The NMRspectra observed triplet at 0.943-0.989 (methyl protons of alkyl …
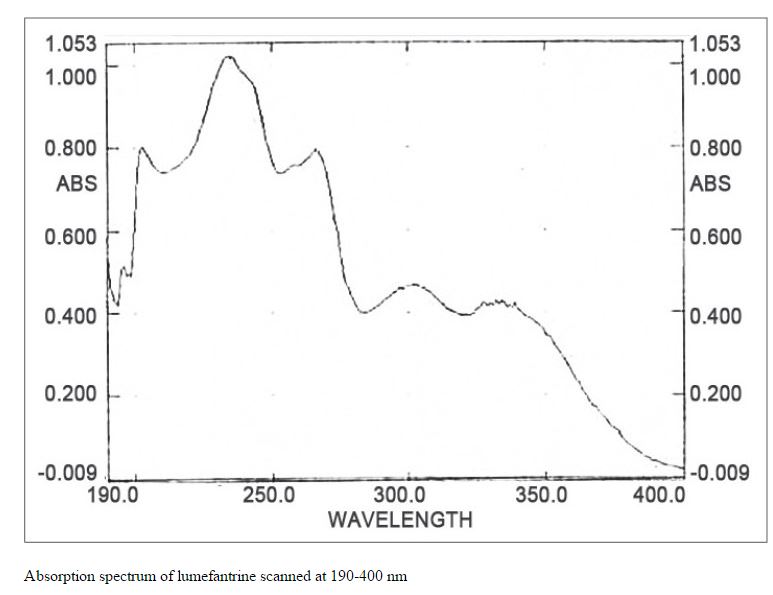
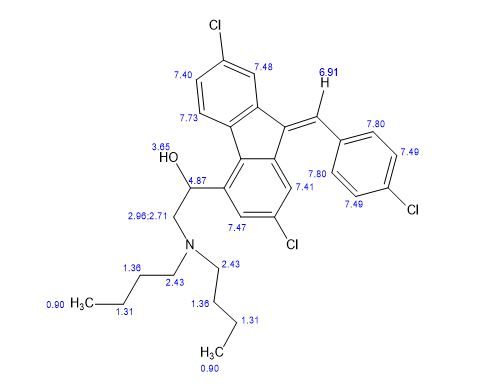


References
- Jump up^ Toovey S, Jamieson A, Nettleton G (2003). “Successful co-artemether (artemether-lumefantrine) clearance of falciparum malaria in a patient with severe cholera in Mozambique”. Travel medicine and infectious disease. 1 (3): 177–9. doi:10.1016/j.tmaid.2003.09.002. PMID 17291911.
- Jump up^ White, Nicholas J.; van Vugt, Michele; Ezzet, Farkad (1999). “Clinical Pharmacokinetics and Pharmacodynamics of Artemether-Lumefantrine”. Clinical Pharmacokinetics. 37 (2): 105–125. doi:10.2165/00003088-199937020-00002. ISSN 0312-5963.
- Jump up^ Cui, Liwang; Su, Xin-zhuan (2009). “Discovery, mechanisms of action and combination therapy of artemisinin”. Expert Review of Anti-infective Therapy. 7 (8): 999–1013. doi:10.1586/eri.09.68. PMC 2778258
 . PMID 19803708.
. PMID 19803708. - Jump up^ http://aac.asm.org/content/56/5/2465.full
- Jump up^ Laman, M; Moore, BR; Benjamin, JM; Yadi, G; Bona, C; Warrel, J; Kattenberg, JH; Koleala, T; Manning, L; Kasian, B; Robinson, LJ; Sambale, N; Lorry, L; Karl, S; Davis, WA; Rosanas-Urgell, A; Mueller, I; Siba, PM; Betuela, I; Davis, TM (2014). “Artemisinin-naphthoquine versus artemether-lumefantrine for uncomplicated malaria in Papua New Guinean children: an open-label randomized trial”. PLoS Med. 11: e1001773. doi:10.1371/journal.pmed.1001773. PMC 4280121. PMID 25549086.
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a609024 |
| Routes of administration |
Oral |
| ATC code | P01BF01 (WHO) (combination with artemether) |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 82186-77-4 |
| PubChem (CID) | 6437380 |
| DrugBank | DB06708 |
| ChemSpider | 4941944 |
| UNII | F38R0JR742 |
| KEGG | D03821 |
| ChEBI | CHEBI:156095 |
| ChEMBL | CHEMBL38827 |
| Chemical and physical data | |
| Formula | C30H32Cl3NO |
| Molar mass | 528.939 g/mol |
| 3D model (Jmol) | Interactive image |
///////////lumefantrine, lumefantrene, Antimalarial, CPG-56695, CPG 56695,
CCCCN(CCCC)CC(O)C1=C2C(=CC(Cl)=C1)\C(=C/C1=CC=C(Cl)C=C1)C1=C2C=CC(Cl)=C1
Arteflene





Alternative solid-state forms of a potent antimalarial aminopyridine: X-ray crystallographic, thermal and solubility aspects

Graphical abstract: Alternative solid-state forms of a potent antimalarial aminopyridine: X-ray crystallographic, thermal and solubility aspects
Alternative solid-state forms of a potent antimalarial aminopyridine: X-ray crystallographic, thermal and solubility aspects
Dyanne L. Cruickshank, Yassir Younis, Nicholas M. Njuguna, Dennis S. B. Ongarora, Kelly Chibale and Mino R. Caira
CrystEngComm, 2014, 16, 5781 DOI:10.1039/C3CE41798K
3-(6-Methoxypyridin-3-yl)-5-(4-methylsulfonyl phenyl)-pyridin-2-amine (MMP) is a member of a novel class of orally active antimalarial drugs. This aminopyridine molecule has shown potent in vitro antiplasmodial activity and in vivo antimalarial activity in Plasmodium berghei-infected mice. The aqueous solubility of this molecule is, however, limited.
Thus investigations aimed at improving the physicochemical properties, including solubility, of MMP were accordingly conducted. Five salts of MMP were formed with co-former molecules saccharin, salicylic acid, fumaric acid, oxalic acid and suberic acid, but a cocrystal was obtained when the co-former adipic acid was employed.
All these new multi-component systems have been fully characterised using X-ray diffraction and thermal methods. Semi-quantitative, turbidimetric solubility tests in a phosphate-buffered saline solution at a pH of 7.4 were performed on the salts and the cocrystal of MMP. The saccharinate salt, fumarate salt and the cocrystal of MMP proved to have greater solubility than MMP itself. This work illustrates the importance of screening and modifying candidate drug compounds in their preliminary stages of development.
Alternative solid-state forms of a potent antimalarial aminopyridine: X-ray crystallographic, thermal and solubility aspects
Dyanne L. Cruickshank,a Yassir Younis,a Nicholas M. Njuguna,a Dennis S. B. Ongarora,a Kelly Chibalea and Mino R. Caira*a
*corresponding authors
aDepartment of Chemistry, University of Cape Town, Rondebosch 7701, South Africa
E-mail: mino.caira@uct.ac.za;
Fax: +27 21 650 5195 ;
Tel: +27 21 650 3071
CrystEngComm, 2014,16, 5781-5792
DOI: 10.1039/C3CE41798K
Bulaquine a CDRI India Antimalarial

Bulaquine
CAS NO.: 79781-00-3
2(3H)-Furanone, dihydro-3-(1-((4-((6-methoxy-8-quinolinyl)amino)pentyl)amino)ethylidene)-,
3-[l-[[4-[(6-methoxy-8-quinolinyl)amino]pentyl]amino]- ethyMene]-dihydro-2(3H)furanone
N1– (3-ethylidinotetrahydrofuran-2-one)-N4– (6-methoxy-8-quinolinyl)-1,4-pentanediamine

BULAQUINE
https://www.ncbi.nlm.nih.gov/pubmed?cmd=search&term=%22bulaquine%22%5BNM%5D
………………….

http://www.cdriindia.org/Bulaquin.htm
|
|
|
It is being increasingly felt that the eroding efficacy of commonly used antimalarials has contributed substantially to the resurgence of malaria during last three decades. Although new antimalarials have appeared in the market during this time, none has yet supplemented chloroquine. There are no drugs in the market or in advanced stages of development that appear to be as well tolerated as chloroquine. Combinations of existing antimalarials especially those now available in rural clinics and market hold great potential for effective, self-administered therapies for uncomplicated malaria, particularly where relapses are frequently encountered. Applying combined therapies to the problem should demand a high standard of proof of safety and efficacy in randomised double blind, placebo controlled trials. Bulaquin is without any side effects that have been observed with primaquine. A comparative data analysis on initial (0 day pre-drug) and final (+7 day post-drug) values of haemoglobin, methaemoglobin, prothrombin time, partial thromboplastin time and fibrinogen in healthy human subjects treated with primaquine (15 mg OD x7 days) and Bulaquin (25 mg OD x7 days) have been carried out. The study has shown that one week primaquine treatment leads to rise in methaemoglobin levels from 3.97% to 16.32%, which is highly significant in comparison to the 2.29% and 3.02% levels of methaemoglobin before and after 7 days treatment with Bulaquin respectively. Thus, it is evident that primaquine treatment produces rise in methaemoglobin contrary to Bulaquine does not produce rise in methaemoglobin levels. This result manifests a clear superiority of Bulaquin over Primaquine. Bulaquin has been licenced to Nicholas Piramal India Ltd., Mumbai for marketing. Nicholas Piramal has introduced Bulaquin alongwith chloroquine into the market as a combination pack under the trade name Aablaquine. The objective of the combined therapy is to control P.vivax malaria more effectively by providing initial cure and thereafter preventing relapses by use of this combination pack. It is hoped that the introduction of this combination pack of Bulaquin should contribute substantially to the ongoing National Malaria action programme advocated by Government of India. |
|
http://www.google.com/patents/EP1292306A2?cl=en
Malaria, caused by a parasitic protozoan called Plasmodium, is one of the most serious and complex tropical parasitic diseases. Generally human malaria is caused by four species of malarial parasites which are Plasmodium falciparwn, Plasmodium vivax, Plasmodium ovale and Plasmodium malariae. Of these P. falciparum and P. vivαx are most widespread and cause most of the mortality and morbidity associated with these types of infections.
It is known that the malarial parasites undergo complex life cycle in humans, which is initiated through the bite of an infected female Anopheles mosquito. When the mosquito bites a host, some of the sporozoites are injected into the bloodstream of the host and through the circulation they reach the liver where they multiply and liberate merozoites into the bloodstream which then invade the erythrocytes. In case of infections caused by P. vivαx, most of the time the parasites remain dormant in the liver which stage is termed hypnozoites. Hypnozoites are reactivated and reinitiate blood stage parasitemias causing relapse. It has often been observed that people infected with P. vivax do not experience any symptoms for a very long period after their initial illness but become symptomatic after certain period (Korean J. Intern Med, 1999 Juk 14(2): 86-9).
A number of drugs ranging from those of natural origin to synthetic ones have been developed for the treatment of malaria. Quinine and artemisinin are the commonly known drugs of natural origin, which are mostly used for the treatment of malaria. A number of synthetic anti- malarial drugs such as chloroquine, mefloquine, primaquine, halofantrine, ainodiaquine, proguanil, maloprim are known in the literature. Of all the synthetic anti-malarial agents chloroquine has been the most widely prescribed drug for the treatment of malaria of all the types, for more than last 60 years.
Chloroquine has been the effective treatment so far for the P. vivax malarial infections, however, some strains of P. vivax have shown resistance to this well known drug {Ann. Trop. Med. ParasitoL, 1999 Apr; 93(3): 225-230). In recent years drug resistant malaria has become one of the most serious problems in malaria control. Drug resistance necessitates the use of drugs which are more expensive and may have dangerous side effects. To overcome the problems associated with drug resistance, treatments comprising combinations of anti-malarial agents are on the rise. A number of anti-malarial combinations are already known in the malarial chemotherapy. For example, a combination of amodiaquine and tetracycline, a combination of sulfadoxine and pyrimethamine known as fansidar, are known therapies for the treatment of P. falάparum. Also fansimef, a combination of mefloquine with sulfadoxine and pyrimetha min e is used against multidrug resistant strains of P. faldparum.
United States Patent No. 5 998 449 describes a method for the treatment of malaria wherein combination of atovaquone and proguanil is used for the treatment of malaria. In US Patent No. 5 834 505, combination of fenozan with another anti-malarial agent selected from artemisinin, sodium artesunate, chloroquine, mefloquine is described for the prophylactic and curative treatment of malaria.
All the aforementioned anti-malarial combinations reported heretofore are generally used for the treatment of P. faldparum. None of the standard anti-malarial combination treatment regimens have been found to be favourable for the treatment of P. vivax malaria which is the most relapsing type of malaria. For a very long time chloroquine was used for the treatment of infections caused by P. vivax, however, chloroquine eradicates only the asexual erythrocytic stages of P. vivax and does not eliminate the hypnozoites. Until recently primaquine has been the drug of choice for the treatment of malarial relapse. Generally the standard therapy for the P. vivax malarial infection comprises of a sequential chloroquine-primaquine combination treatment regimen wherein primaquine is administered for 14 days following the 3 days course of chloroquine. WHO (World Health Organisation) also recommends a 14 days primaquine treatment for P. vivax malarial infection. A shorter duration of cMoroquine-primaquine treatment regimen was also tried out wherein primaquine was administered only for 5 days following the chloroquine course. However, the outcome of the treatment was not encouraging, since the percentage relapse was more than the standard 14 days primaquine treatment regimen (Trans. R. Soc. Trop. Med. Hyg., 93(6), 641-643). Also primaquine is known to cause hemolytic anemia in persons deficient in the enzyme glucose-6-phosphate dehydrogenase (G6PD) (Pharmacol Rev. 21: 73-103 (1969); Rev. Cubana Med trop, 1997; 49 (2): 136-8 ). Moreover, methemoglobin toxicity is another predictable dose-related adverse effect associated with primaquine. Needless to say that in the case of sequential combination therapy the patient may not complete the course once the symptoms of malaria are diminished, hence this may increase the chances of relapse. Thus, the chloroquine- primaquine treatment regimen is not safe with respect to toxicity of primaquine and has a further limitation from the standpoint of patient compliance due to longer duration of treatment.
Another anti-relapse agent namely tafenoquine is disclosed in United States Patent 4 617 394. Though more effective than primaquine, the drug was found to cause methemoglobin toxicity almost three times more than that of primaqu ie (Fundam. Appl. Toxicol. 1988, 10(2), 270-275), hence has drawbacks in terms of safety.
The compound, 3-[l-[[4-[(6-nιethoxy-8-quinolinyl)aιnino]pentyl)am.ino]- ethylidene]-dihydro-2(3H)furanone is a derivative of primaquine. It was described in Indian Patent Specification No. 158111 as 6-methoxy-8-(4-
N-(3′-aceto-4^5′-dihydro-2-furanylamino)- l-methylbutylamino)quinoline , the structure of which was revised to that represented by the following formula I. As per the revised structure, the compound is named 3-[l-[[4-
[(6-metJhoxy-8-quinolmyl)amino]pentyl]amino]ethylidene]-dihydro-2(3H)- furanone (hereinafter referred to as compound I). The revised structure is described in WHO Drug Information Vol. 13, No. 4, pg. 268 (1999).

The compound of formula (I) has been found to be safer and less toxic than the parent compound primaquine (Am. J. Trop. Med. Hyg, 1989 Dec; 41(6): 635-637). Its anti-relapse activity has been found to be comparable to primaquine.
Over the years primaquine was the only drug used for the radical cure of malaria caused by P. vivax. Primaquine is associated with a number of severe adverse effects, therefore there is a need to develop agents which are more effective and/ or less toxic than primaquine. The compound I has been found to exhibit anti-relapse activity comparable to Primaquine (Am. J. Trop. Med. Hyg., 41(6): 633-637 (1989)). However, this compound has been shown to cause less methemoglobin formation (Am. J. Trop Hyg., 41(6): 638-642 (1989) ) and also has less effect on anti-oxidant defence enzymes than primaquine (Biochem Pharmacol. 46(10): 1859- 1860 (1993) ). Thus, this primaquine derivative (I) is found to be less toxic as compared to the parent drug, primaquine.
Therefore, there is a longfelt need for a more practical, effective, patient compliant and safe remedy for the radical cure of P. vivax malarial infection.
The inventors have found that the longfelt need may be fulfilled by providing a treatment regimen consisting of regulated use of chloroquine and 3-[l-[[4-[(6-methoxy-8-quinolinyl)aιnino]pentyl]amino]ethylidene]- dihydro-2(3H)furanone of formula I over a period of between 5 to 8 days.
It has also been found that the treatment regimen may be executed most effectively and in a user friendly manner by providing a combination kit which comprises two anti-malarial agents, namely chloroquine and 3-[l- [[4-[(6-meth.oxy-8-qumolmyl)am^
…………………

The title enamine derivative is prepared by condensation of primaquine (I) with acetyl butyrolactone (II) by means of piperidine.
……………….
http://www.google.com/patents/EP1055427B1?cl=en
-
manufacture fo a medicament for the treatment of malaria of primaquine derivative N1-(3-ethylidinotetrahydrofuran-2-one)-N4-(6-methoxy-8-quinolinyl)-1,4-pentanediamine as a gametocytocidal agent. More particularly, this invention relates to the use of primaquine derivative N1– (3-ethylidinotetrahydrofuran-2-one)-N4– (6-methoxy-8-quinolinyl)-1,4-pentanediamine of formula 1 shown below useful for controlling the spread of malaria by virtue of its high therapeutic value as a gametocytocidal agent.

-
The primaquine derivative of the present invention does not damage either normal or G-6PD deficient erythrocytes to the extent it is observed with the use of primaquine.
-
scheme:

-

The following example illustrates the details of the process of this invention:
N1– (3-ethylidinotetrahydrofuran-2-one)-N4– (6-methoxy-8-quinolinyl)-1,4-pentanediamine
-
A mixture of primaquine base (0.97g, 3.7 mmole) freshly distilled 3-acetyl-r-butyrolactone (1.0g, 7.8 mmole) and a base like piperidine (2-3 drops) were stirred under magnetic stirrer at room temperature. In an hour or so the reaction mixture solidified. The product was titrated in ether and filtered to get the product. It was crystallised from alcoholic solvent like propanol. Yield 0.89g, m.p. 118-120°C.
…………..
BELL A.: “Recent developments in the chemotherapy of malaria.” CURRENT OPINION IN ANTI-INFECTIVE INVESTIGATIONAL DRUGS, (2000) 2/1 (63-70). , XP001038054 |
||
| 2 | * | DUTTA, G. P. ET AL: “Radical curative activity of a new 8-aminoquinoline derivative ( CDRI 80/53) against Plasmodium cynomolgi B in monkeys” AM. J. TROP. MED. HYG. (1989), 41(6), 635-7 , 1989, XP001037488 cited in the application |
| 3 | * | KAR, K. ET AL: “Pharmacology of compound CDRI 80/53;a potential new antirelapse antimalarial agent” INDIAN J. PARASITOL. (1988), 12, 259-62 , 1988, XP001034143 |
| 4 | * | NEWTON P. ET AL: “Malaria: New developments in treatment and prevention.” ANNUAL REVIEW OF MEDICINE, (1999) 50/- (179-192). , XP001036946 |
| 5 | * | PALIWAL, JYOTI KUMAR ET AL: “Simultaneous determination of a new antimalarial agent, CDRI compound 80/53, and its metabolite primaquine in serum by high-performance liquid chromatography” J. CHROMATOGR., BIOMED. APPL. (1993), 616(1), 155-60 , 1993, XP000955186 |
| 6 | * | PURI, S. K. ET AL: “Methemoglobin toxicity and hematological studies on malaria anti-relapse compound CDRI 80/53 in dogs” AM. J. TROP. MED. HYG. (1989), 41(6), 638-42 , 1989, XP001037486 cited in the application |
| 7 | * | SETHI, N. ET AL: “Long term toxicity studies with a synthetic anti-relapse antimalarial compound 80/53 in rats and monkeys” INDIAN J. PARASITOL. (1993), 17(1), 15-26 , 1993, XP001034142 |
| 8 | * | VALECHA, NEENA ET AL: “Comparative antirelapse efficacy of CDRI compound 80/53 (Bulaquine) vs. primaquine in double blind clinical trial” CURR. SCI. (2001), 80(4), 561-563 , 2001, XP001037095 |

{kind=link}
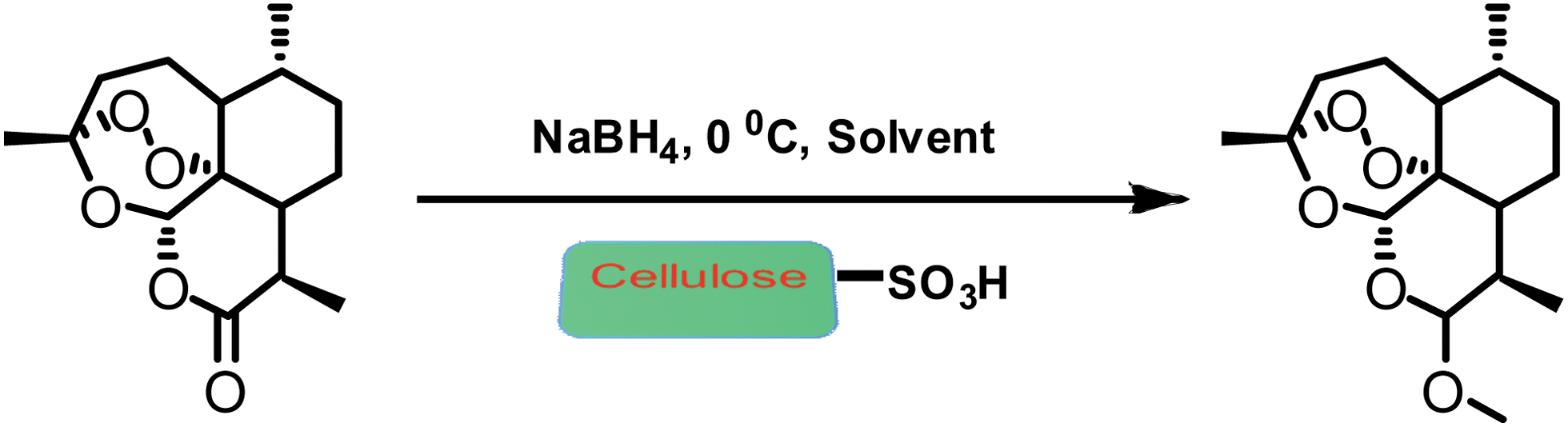{kind=link}
{kind=link}
{kind=link}