
Home » Posts tagged 'Antibiotics'
Tag Archives: Antibiotics
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
CEFOPERAZONE
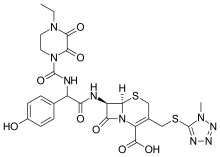
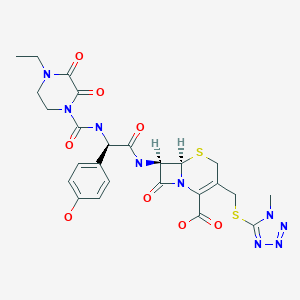
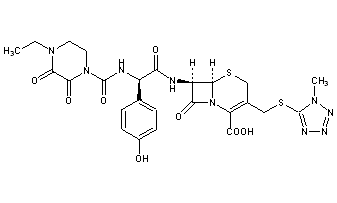
Cefoperazone
- Molecular FormulaC25H27N9O8S2
- Average mass645.667 Da
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Cefoperazone sodium | 5FQG9774WD | 62893-20-3 | NCFTXMQPRQZFMZ-WERGMSTESA-M |
(6R,7R)-7-{[(2R)-2-{[(4-ethyl-2,3-dioxopiperazin-1-yl)carbonyl]amino}-2-(4-hydroxyphenyl)acetyl]amino}-3-{[(1-methyl-1H-tetrazol-5-yl)thio]methyl}-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
(6R,7R)-7-[(2R)-2-[(4-ethyl-2,3-dioxopiperazine-1-carbonyl)amino]-2-(4-hydroxyphenyl)acetamido]-3-{[(1-methyl-1H-1,2,3,4-tetrazol-5-yl)sulfanyl]methyl}-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
263-749-4[EINECS], 4742
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[(2R)-2-[[(4-ethyl-2,3-dioxo-1-piperazinyl)carbonyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo- , (6R,7R)- [ACD/Index Name]
5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[(2R)-2-[[(4-ethyl-2,3-dioxo-1-piperazinyl)carbonyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-, (6R,7R)-
62893-19-0[RN]
7-[D-(-)-a-(4-Ethyl-2,3-dioxo-1-piperazinecarboxamido)-a-(4-hydroxyphenyl)acetamido]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-3-cephem-4-carboxylic Acid
7U75I1278D
Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 188-190 | Saikawa, I., Takano, S., Yoshida, C., Takashima, 0..Momonoi, K., Kuroda, S., Komatsu, M., Yasuda, T.and Kodama, Y.; British Patent 1,508,071; April 19,1978; assigned to Toyama Chemical Co., Ltd. and U.S. Patent 4,110,327; August 29,1978; also assigned to Toyama Chemical Co., Ltd. |
| logP | -0.74 | HANSCH,C ET AL. (1995) |
Cefoperazone
CAS Registry Number: 62893-19-0
CAS Name: (6R,7R)-7-[[(2R)-[[(4-Ethyl-2,3-dioxo-1-piperazinyl)carbonyl]amino](4-hydroxyphenyl)acetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acidAdditional Names: 7-[D-(-)-a-(4-ethyl-2,3-dioxo-1-piperazinecarboxamido)-a-(4-hydroxyphenyl)acetamido]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-3-cephem-4-carboxylic acid
Molecular Formula: C25H27N9O8S2
Molecular Weight: 645.67
Percent Composition: C 46.50%, H 4.21%, N 19.52%, O 19.82%, S 9.93%
Literature References: Broad spectrum third generation cephalosporin antibiotic. Prepn: I. Saikawa et al.,BE837682; eidem,US4410522 (1976, 1983 both to Toyama); eidem,Yakugaku Zasshi99, 929 (1979). Stability in aq soln: eidem,ibid. 1207. In vitro activity: M. V. Borobio et al.,Antimicrob. Agents Chemother.17, 129 (1980). Kinetics in rats: J. Fabre et al.,Schweiz. Med. Wochenschr.110, 264 (1980); in humans: A. F. Allaz, ibid.109, 1999 (1979). Review of pharmacology and therapeutic efficacy: R. N. Brogden et al.,Drugs22, 423-460 (1981). Symposium on clinical studies: ibid. Suppl. 1, 1-124.
Properties: Crystals from acetonitrile/water, mp 169-171° (hydrated). Stable at pH 4.0-7.0; slightly unstable in acid; highly unstable in alkaline soln.
Melting point: mp 169-171° (hydrated)
Derivative Type: Sodium salt
CAS Registry Number: 62893-20-3
Manufacturers’ Codes: CP-52640-2; T-1551
Trademarks: Bioperazone (Biopharma); Cefazone (Firma); Cefobid (Pfizer); Cefobine (Pfizer); Cefobis (Pfizer); Cefogram (Metapharma); Cefoneg (Tosi); Cefosint (Proter); Dardum (Lisapharma); Farecef (Lafare); Kefazon (Esseti); Novobiocyl (Francia); Pathozone (Pfizer); Peracef (Pfizer); Perocef (Pulitzer); Tomabef (Aandersen)
Molecular Formula: C25H26N9NaO8S2
Molecular Weight: 667.65
Percent Composition: C 44.97%, H 3.93%, N 18.88%, Na 3.44%, O 19.17%, S 9.61%
Therap-Cat: Antibacterial., Therap-Cat-Vet: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Cephalosporins.
Cefoperazone is a third-generation cephalosporin antibiotic, marketed by Pfizer under the name Cefobid. It is one of few cephalosporin antibiotics effective in treating Pseudomonas bacterial infections which are otherwise resistant to these antibiotics.
It was patented in 1974 and approved for medical use in 1981.[1] Cefoperazone/sulbactam (Sulperazon) is a co-formulation with sulbactam.
Cefoperazone is a broad-spectrum cephalosporin antibiotic used for the treatment of bacterial infections in various locations, including the respiratory tract, abdomen, skin, and female genital tracts.
Cefoperazone is a semisynthetic broad-spectrum cephalosporin proposed to be effective against Pseudomonas infections. It is a third-generation antiobiotic agent and it is used in the treatment of various bacterial infections caused by susceptible organisms in the body, including respiratory tract infections, peritonitis, skin infections, endometritis, and bacterial septicemia. While its clinical use has been discontinued in the U.S., cefoperazone is available in several European countries most commonly under the product name, Sulperazon.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

English: I. Saikawa, S. Takano, Y. Shuntaro, C. Yoshida, 0.
Takashima, K. Momonoi, S. Kuroda, M. Komatsu, T. Yasuda, and Y. Kodama, German Offen., DE 2,600,880 (1977); Chem.
Abstr., 87_, 184533b (1977).
SYN
Following is one of the synthesis routes:
alpha-(4-Ethyl-2,3-dioxo-1-piperazinocarbonylamino)-p-hydroxyphenylacetic acid (I) is condensed with 7-amino-3-[(1-methyl-1H-tetrazol-5-yl)thiomethyl]-3-cephem-4-carboxylic acid (II) in the presence of ethyl chlorocarbonate and N,O-bis(trimethylsilyl)acetamide in acetonitrile to produce Cefoperazone sodium.

SYN
Antibiotics
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Cefoperazone
Cefoperazone, (6R,7R)-7-[(R)-2-(4-ethyl-2,3-dioxo-1-piperazincarboxamido)-2-(p-hydroxyphenyl)acetamido]-3-[[(1-methyl-1 H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid (32.1.2.84), is synthesized by acylating 7-amino-3-(1-methyl-1,2,3,4-tetrazol-5-yl)-thiomethyl-3-cefem-4-carboxylic acid (32.1.2.24) with a mixed anhydride synthesized from ethyl chloroformate and α-(4-ethylpiperazin-2, 3-dion-1-carbonylamino)-4-hydroxyphenylacetic acid (32.1.2.83), which in turn is synthesized from 4-ethylpiperazin-2,3-dion-1-carboxylic acid (32.1.1.29) and the sodium salt of 4-hydroxyphenylglycine [163–168].


Cefoperazone also has a broad spectrum of antimicrobial action, including most clinically significant microorganisms: Gram-positive, Gram-negative, aerobic, and anaerobic. It is stable with respect to most beta-lactamases of Gram-positive and Gram-negative bacteria.
Cefoperazone is used for bacterial infections of the lower respiratory tract, urinary and sexual tracts, bones, joints, skin, soft tissues, abdominal, and gynecological infections. Synonyms of this drug are cefazon, cefobid, cefobis, and many others.
Spectrum of bacterial susceptibility
Cefoperazone has a broad spectrum of activity and has been used to target bacteria responsible for causing infections of the respiratory and urinary tract, skin, and the female genital tract. The following represents MIC susceptibility data for a few medically significant microorganisms.
- Haemophilus influenzae: 0.12 – 0.25 µg/ml
- Staphylococcus aureus: 0.125 – 32 µg/ml
- Streptococcus pneumoniae: ≤0.007 – 1 µg/ml[2]
Adverse effects
Cefoperazone contains an N-methylthiotetrazole (NMTT or 1-MTT) side chain. As the antibiotic is broken down in the body, it releases free NMTT, which can cause hypoprothrombinemia (likely due to inhibition of the enzyme vitamin K epoxide reductase) and a reaction with ethanol similar to that produced by disulfiram (Antabuse), due to inhibition of aldehyde dehydrogenase.[3]
Mechanism of action
Cefoperazone exerts its bactericidal effect by inhibiting the bacterial cell wall synthesis, and sulbactam acts as a beta-lactamase inhibitor, to increase the antibacterial activity of cefoperazone against beta-lactamase-producing organisms.
References
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 494. ISBN 9783527607495.
- ^ “Cefoperazone (Cefobid) – The Antimicrobial Index Knowledgebase – TOKU-E”. antibiotics.toku-e.com.
- ^ Stork CM (2006). “Antibiotics, antifungals, and antivirals”. In Nelson LH, Flomenbaum N, Goldfrank LR, Hoffman RL, Howland MD, Lewin NA (eds.). Goldfrank’s toxicologic emergencies. New York: McGraw-Hill. p. 847. ISBN 0-07-143763-0. Retrieved 2009-07-03.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a601206 |
| ATC code | J01DD12 (WHO) QJ51DD12 (WHO) |
| Pharmacokinetic data | |
| Excretion | Hepatic |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 62893-19-0 |
| PubChem CID | 44185 |
| DrugBank | DB01329 |
| ChemSpider | 40206 |
| UNII | 7U75I1278D |
| KEGG | D07645 |
| ChEMBL | ChEMBL507674 |
| CompTox Dashboard (EPA) | DTXSID2022759 |
| ECHA InfoCard | 100.057.936 |
| Chemical and physical data | |
| Formula | C25H27N9O8S2 |
| Molar mass | 645.67 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
//////////cefoperazone, Antibacterial, Antibiotics, Lactams, Cephalosporins, CP-52640-2, T-1551, CP 52640-2, T 1551
[H][C@]12SCC(CSC3=NN=NN3C)=C(N1C(=O)[C@@]2([H])NC(=O)[C@H](NC(=O)N1CCN(CC)C(=O)C1=O)C1=CC=C(O)C=C1)C(O)=O

NEWDRUG APPROVALS
one time
$10.00
BIAPENEM

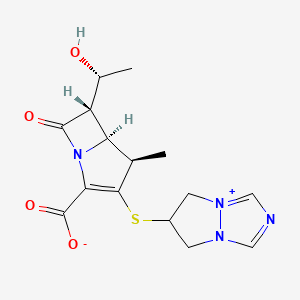
Biapenem
RPX7009
- Molecular FormulaC15H18N4O4S
- Average mass350.393 Da
Biapenern
CL 186-815LJ
C10,627LJ
C10627LJC 10627
omegacin
YR5U3L9ZH1
(4R,5S,6S)-3-((6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-yl)thio)-6-((R)-1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
[4R-[4a,5b,6b(R*)]]-6-[[2-Carboxy-6-(1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5 H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
120410-24-4[RN]
5H-Pyrazolo[1,2-a][1,2,4]triazol-4-ium, 6-[[(4R,5S,6S)-2-carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-, inner salt [ACD/Index Name]
6-[[(4R,5S,6S)-2-Carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
7074
(4R,5S,6S)-3-(6,7-Dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-ylsulfanyl)-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
TL8000539UNII:YR5U3L9ZH1UNII-YR5U3L9ZH1биапенем
بيابينام比
阿培南
INDIA CDSCO APPROVED 25 SEPT 2021, BDR PHARMA, 
https://www.cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadCTApprovals/BDR.pdfhttps://medicaldialogues.in/news/industry/pharma/bdr-pharma-gets-dcgi-nod-for-generic-antibiotic-drug-biapenem-82384
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
Biapenem (INN) is a carbapenem antibiotic. It has in vitro activity against anaerobes.[1] 1-β-methyl-carbapenem antibiotic. Approved in Japan in 2001.
PATENT
EP 168707
EP 289801
JP 02088578
ZA 9100014
EP 533149
CN 1995040
IN 2006DE01555
CN 101121716
IN 2008CH00177
CN 101805359
CN 101851206
CN 101935321
CN 111875622
WO 2018074916
WO 2016059622
US 20150328323
WO 2015151081
WO 2015155753
WO 2015151078
US 20150284416
WO 2015151080
US 20150038726
WO 2014104488
IN 2013MU00181
WO 2014111957
CN 103570750
WO 2014097221
IN 2012CH01371
WO 2013150550
PAPERS
Journal of Organic Chemistry (1992), 57(15), 4243-9.
Heterocycles (1993), 36(8), 1729-34.
Journal of Antibiotics (1993), 46(12), 1866-82.
e-EROS Encyclopedia of Reagents for Organic Synthesis (2008), 1-3.
Bioorganic & medicinal chemistry letters (2009), 19(17), 5162-5.
IP.com Journal (2014), 14(12A), 1-3
IP.com Journal (2014), 14(10A), 1-2.
Bioorganic & medicinal chemistry (2013), 21(18), 5841-50.

NEW DRUG APPROVALS
one time
$10.00
PATENT
https://patents.google.com/patent/WO2014097221A1/esBiapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)- hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3 -yljthio] 6,7-dihydro-5H- pyrazolo[l,2-a][l,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.

Formula 1U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.EXAMPLESExample 1 : Purification of BiapenemBiapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.Yield: 84%HPLC Purity: 99.87% Example 2: Purification of BiapenemBiapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.Yield: 81.77%HPLC Purity: 99.80%
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013150550
The present invention relates to an improved process for the preparation of carbapenem antibiotic; more particularly relates to the preparation of Ertapenem monosodium salt of formula (I) having purity greater than 98.5% and having pharmaceutically acceptable level of residual solvent and palladium content.
The US patents namely US 5,478,820 and US 5,856,321 disclose various processes for preparing Ertapenem and its sodium salt. Example 12 of US 5,478,820 discloses a process in which the Ertapenem was isolated using column purification followed by freeze-drying technique. According to Example-4 of this patent disodium salt of Ertapenem was prepared by dissolving crude product in water using NaHCO3, followed by purification using column chromatography and subsequent lyophilization.
US 6,504,027 provides a process for preparing Ertapenem in crystalline form which comprises deprotecting and extracting a polar organic solution containing a crude mono-protected Ertapenem of formula
wherein P represents protecting group and X represents charge balancing group like sodium
with C4.10 alcohol in the presence of ion-pairing reagent followed by adjusting the pH of the aqueous layer to 5.5 and crystallizing using methanol and 1-propanol to produce a crystalline compound; this patent process involves operations like
multiple extractions which is cumbersome in plant and said operation affects the overall yield.
US 7,145,002 provides a process for producing Ertapenem or its sodium salt and/or its solvate in crystalline form. This patent states (refer para 3, lines 31-41) that contact of Ertapenem sodium with water and alcoholic solvents results in the formation of crystalline solvates. The processes reported in examples- 1 & 2 provide crystalline Ertapenem monosodium which is isolated from a mixture of methanol, 1-propanol and water followed by washing with aqueous isopropyl alcohol which results in the formation of crystalline solvate of Ertapenem sodium. Applicant found the Ertapenem monosodium obtained according to this process contain higher amount of residual solvent and palladium content.
US 7,022,841 provide a process for reducing the levels of organic solvents in Ertapenem to pharmaceutically acceptable levels. This patent discloses (Refer para 1, lines 52-60) that Ertapenem sodium obtained from water/alcohol mixture according to US 7, 145,002 becomes amorphous when water content of the solid is reduced and further the organic solvent present in the solid is not readily removed. In view of this drawback, this patent provides a process wherein the water content of Ertapenem sodium is maintained between 13-25% during the washing and drying process. This patent further discloses that (Refer para 9, lines 6-14) the washing of Ertapenem sodium can be carried out using anhydrous solvents which results in the formation of amorphous solid, which is then dried using hydrated nitrogen by increasing the water content of the solid. Due to the hygroscopic and unstable nature of Ertapenem sodium when in contact with water, the above processes result in more degradation of Ertapenem. The patent further discloses in example 5 that the degradation of Ertapenem sodium is more when it takes more time for drying.
Further this patent requires repetitive washing and control of moisture content to get the desired results.
For isolation of Ertapenem sodium from the reaction mass, all the above discussed prior art patents utilize methanol and 1-propanol as crystallization solvent. The filtration of Ertapenem sodium formed by using these solvents or their mixture takes longer time duration and subsequent drying for the removal of residual solvent also takes several hours due to occlusion of solvent into Ertapenem sodium. During these operations the Ertapenem sodium degrades an results in the formation of many impurities such as several dimers, methanolysis impurity etc., and hence the reported processes is not suitable to manufacture Ertapenem sodium on commercial scale with purity greater than 98.5% and with pharmaceutically acceptable level of residual solvent content.
Methanolysis impurity Dimer-I
Dimer-II
Further the applicant found that Ertapenem monosodium isolated by following the process reported in prior art was having palladium content above the pharmaceutically acceptable level. Hence the process reported in prior art is not suitable on manufacturing scale where maintaining stringent technological condition is cumbersome and involves higher operating cost.
Thus all the reported processes suffer in terms of one or more of the following facts:
■ Filtration time of Ertapenem sodium takes several hours.
■ Drying time takes several hours due to occlusion of solvent and nature of the solid.
■ Stringent technological condition is required for maintenance of moisture content during washing & drying operation.
■ Palladium content is found to be higher (greater than 25 ppm) which is not acceptable for pharmaceutical products.
■ The isolated Ertapenem sodium is having higher amount of residual solvents.
■ The purity is reduced over to several hours of filtration & drying.
With our continued research for developing a process for the preparation of Ertapenem monosodium of formula (I) to overcome the above mentioned drawbacks, we surprisingly found that when esters of organic acid were used as solvents in place of 1-propanol, the solid obtained was easily filterable with less cycle time. Further the washing with hydrocarbon solvents containing 0-75% alcoholic solvent followed by drying results in Ertapenem having residual solvent content well below the pharmaceutically acceptable levels. The use of thiourea, thiosemicarbazide or their N-substituted derivatives in the presence of organic solvents during isolation brings down the palladium content to pharmaceutically acceptable level.
The Ertapenem or its sodium salt can be prepared according the processes provi
(I)
P’ and P” represent carboxylic protecting groups and X is H or Na
Scheme-1
The present invention is illustrated with the following examples, which should not be construed to limit the scope of the invention.
Example- I
Preparation of Ertapenem monosodium of formula (I)
Step-I:
To a stirred solution of p-nitrobenzyl (4R,5S,6S)-3-(diphenyloxy)phosphoryloxy-6-[(lR)-l-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound II) (100 g) and (2S,4S)-2-[[(3-carboxyphenyl) amino]carbonyl]-4-mercapto-l-(4-nitrobenzyl)pyrrolidinecarboxylate (compound III) (75 g) in N,N-dimethylformamide was added Ν,Ν-diisopropylethylamine at -30 to -40° C and stirred. The reaction mass, after completion of the reaction, was quenched with a mixture of phosphate buffer solution-ethyl acetate and the pH was adjusted to 5 – 6 with phosphoric acid. The organic layer was separated, washed with water and subjected to carbon treatment. To the organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl), a solution of sodium 2-ethylhexanoate (42 g in 500 mL methanol) was added and taken to next step as such. (If required the compound of formula (IV) is isolated either as sodium salt or as free acid by following the process reported in prior art and taken further)
Step-II:
To the Step-I organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl & X is Na), 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8- 10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered to remove palladium on carbon. To the filtrate, thiourea (5 g) and tetrahydrofuran were added and stirred. The aqueous layer was separated and treated with carbon and neutral alumina at 10-15° C while degassing and filtered. The filtrate was added to methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 46 g; Purity by HPLC: 98.93%; Palladium content: 1.8 ppm by ICP MS
The HPLC purity of Ertapenem monosodium was checked using the following parameters
Column : Zorbax Eclipse plus C8, (50 mm x 4.6 mm), 1.8μ).
Mobile phase : Ammoniam acetate buffer: Acetonitile: water
Detector : UV at 250 nm
Flow rate : 0.5 mL/min
Run time : 45 min.
Example- II
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon & neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with cyclohexane (200 ml) and
dried under vacuum. Yield: 44 g; Purity by HPLC: 98.84%; Palladium content: 0.93 ppm by ICP MS
The term ICP MS method refers to the inductively coupled plasma mass spectrometry. The following parameter was used to determine the content of palladium.
The carbapenem was digested in a closed vessel system in presence of reagents Nitric acid, Hydrogen peroxide and Hydrochloric acid by using Microwave reaction system with microwave radiation power 1200 Watts. The digested sample was introduced into inductively coupled plasma mass spectrometer by help of Peltier cooled spray chamber. The sample aerosol is getting atomized then ionized in the argon plasma. The ionized Palladium was estimated by using Quadrupole mass detector. The sample was quantified against NIST traceable reference standards at mass number ! 05.
Example- III
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was separated and treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of toluene: ethanol (200 ml) and dried under vacuum. Yield: 42 g; Purity by HPLC: 99.03%
Example- IV
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the filtrate was treated with thiosemicarbazide and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C followed by the addition of ethyl acetate and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 41 g; Purity by HPLC: 99.13%; Palladium content: 1.71 ppm by ICP MS
Example- V
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, a mixture of ethyl acetate containing 10% methyl acetate was added and stirred. The solid obtained was
filtered, washed with cyclohexane:ethanol and dried under vacuum. Yield: 40.5 g; Purity by HPLC: 98.77%; Palladium content: 1.43 ppm by ICP MS
Example-VI
(V ) (V I )
The diprotected Meropenem of formula (V) (where P and P’ were p-nitrobenzyl) was dissolved in tetrahydrofuran and 3-(N-morpholino)propanesulfonic acid buffer and hydrogenated using palladium on carbon at 9-10 kg hydrogen pressure. The mass was filtered and the filtrate was washed with ethyl acetate. The aqueous layer was treated with thiourea and 2-methyltetrahydrofuran. The aqueous layer was separated, treated with carbon and degassed. The carbon was filtered off and acetone was added to the filtrate to crystallize Meropenem trihydrate of formula (VI). The product was filtered and washed with aq. acetone and dried under vacuum to get Meropenem trihydrate. Purity: 99.8%; Pd content: 0.08 ppm
Reference example-I:
Preparation of Ertapenem monosodium of formula (I)
To Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered. The filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass ethyl acetate was added and stirred. The solid obtained was filtered, washed with ethanol (5 * 100 ml) and dried under vacuum. Yield: 31 g; Purity by HPLC: 96.76%
Reference example-II:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass , as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol and dried under vacuum. Yield: 43 g; Purity by HPLC: 98.6%; Palladium content: 35.8 ppm by ICP MS.
Reference example-HI:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with 1-propanol at -5° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass methanol and 1-propanol were added and stirred. The solid obtained was filtered, washed with ethanol and dried under nitrogen atmosphere in vacuum. Yield: 25 g; Purity by HPLC: 97 %.: palladium content: 38.2 ppm
The following tables illustrate the advantages of the present invention over prior art process:
Table-I: Comparison of present process with prior art process
The crystallization and washing method disclosed in US 7,022,841 was followed.
The above table indicates that the use of ethyl acetate as crystallization solvent results with improved yield and high purity with less filtration and drying time thereby increasing the productivity significantly on manufacturing scale. Further the use of thiourea or thiosemicarbazide as reagents in the present process results in the pharmaceutically acceptable level of palladium content.
Table-II: Comparison of solvents for washing Ertapenem monosodium
The above table indicates that the use of hydrocarbon solvents containing 0-75% of alcoholic solvent helps in washing to remove the residual solvent content in shorter duration and with single run wash. On the other hands the use of ethanol alone results in Ertapenem monosodium having less yield and purity requiring repetitive washing.
Table-IH: Effect of different reagent in reduction of palladium content
Reagent : thiourea, thiosemicarbazide or its N-substituted derivatives
Advantages of the process of the present invention:
> The use of ester of an organic acid for the crystallization of Ertapenem sodium results in fast filtration and reduced cycle time, thereby increasing the productivity.
> Washing of Ertapenem sodium with hydrocarbon solvent optionally containing alcohol results in improved physical nature of Ertapenem sodium resulting in reduced washing and drying time thereby avoid the degradation of Ertapenem and providing Ertapenem sodium with purity greater than 98.5% by HPLC.
Use of thiourea, thiosemicarbazide or their N-substituted derivatives in the process results in Ertapenem sodium having pharmaceutically acceptable level of palladium content.
PATENT
https://patents.google.com/patent/WO2002057266A1/enEXAMPLE

PNB = p-nitrobenzyl

Ia’A hydrogenator is charged with 63 g of 10% Pd on carbon catalyst (dry weight) in 1.8 L of water. The vessel is placed under hydrogen then vented and placed under nitrogen. Sodium hydroxide (68 g, 50%) is charged adjusting the pH to about 7.5 with carbon dioxide.The enol phosphate (170 g) and the thiol (86 g) are dissolved in 1.3‘L of N- ethylpyrrolidinone (NEP). The mixture is cooled to below -40°C and 1,1,3,3- tetramethylguanidine (109 g) is added. After 3 hours, the reaction mixture is quenched into the hydrogenator at below 15°C adjusting the pH to about 8 with carbon dioxide. The vessel is placed under hydrogen. When the reaction is complete, the hydrogen is vented and the reaction mixture is treated with activated carbon and filtered. The filtrate is extracted with iso-amyl alcohol containing diphenylphosphoric acid (240 g) and 50% NaOH (44 g). The resulting aqueous solution is further extracted with iso-amyl alcohol to give an aqueous solution containing at least 90 mg/mL of the product. Both extractions are performed using two CINC centrifugal separators set in series for countercurrent extraction. The pH is adjusted to 5.5 with acetic acid. The product is crystallized by adding equal volumes of methanol and 1- propanol at below -5°C and isolated by filtration. The solid is washed with a mixture of 2-propanol and water (85: 15 v/v) then dried to yield a compound of formula la’.While certain preferred embodiments of the invention have been described herein in detail, numerous alternative embodiments are contemplated as falling within the scope of the appended claims. Consequently the invention is not to be limited thereby.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleUS4866171A1987-04-111989-09-12Lederle (Japan), Ltd.(1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazolium-6-yl)]thio-6-[R-1-hydroxyethyl]-1-methyl-carbapenum-3-carboxylateUS5241073A1990-10-121993-08-31Lederle (Japan)Process for preparing (1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo [1,2-a][1,2,4]triazolium-6-yl)]thio-6-[(R)-1-hydroxyethyl]-1-methyl-carbapenem-3-carboxylate and starting materials thereofUS5286856A1991-09-201994-02-15Takeda Chemical Industries, Ltd.Production of crystalline penemWO2002057266A1 *2001-01-162002-07-25Merck & Co., Inc.Improved process for carbapenem synthesisWO2009047604A1 *2007-10-082009-04-16Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of carbapenem antibioticCN102268025A *2011-07-152011-12-07海南美兰史克制药有限公司一种比阿培南化合物及其制法
References
- ^ Aldridge KE, Morice N, Schiro DD (April 1994). “In vitro activity of biapenem (L-627), a new carbapenem, against anaerobes”. Antimicrob. Agents Chemother. 38 (4): 889–93. doi:10.1128/aac.38.4.889. PMC 284564. PMID 8031067.
External links
- (in Japanese) Omegacin
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | IV |
| ATC code | J01DH05 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 120410-24-4 |
| PubChem CID | 71339 |
| ChemSpider | 64442 |
| UNII | YR5U3L9ZH1 |
| ChEBI | CHEBI:3089 |
| ChEMBL | ChEMBL285347 |
| CompTox Dashboard (EPA) | DTXSID5046435 |
| Chemical and physical data | |
| Formula | C15H18N4O4S |
| Molar mass | 350.39 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04552444 | Clinical Efficacy of Combination Therapy Based on High-dose Biapenem in CRKP Infections | Recruiting | 2020-09-17 | |
| NCT01772836 | Safety Study of Intravenous Biapenem (RPX2003) and RPX7009 Given Alone and in Combination | Phase 1 | Completed | 2013-07-11 |
| NCT01702649 | Safety, Tolerability, Pharmacokinetics of Intravenous RPX2003 (Biapenem) in Healthy Adult Subjects | Phase 1 | Completed | 2012-12-03 |
NIPH Clinical Trials Search of Japan
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| UMIN000017219 | Feasibility and efficacy of the de-escalation therapy by Biapenem for postoperative bacterial pneumonia. | None | Recruiting | 2015-04-22 |
| UMIN000003964 | Clinical evaluation of Biapenem 0.3g, three times daily dosing in eldery patients with pneumonia (moderate and severe infection) | Not applicable | Complete: follow-up complete | 2010-07-29 |
/////////BIAPENEM, TL8000539, UNII:YR5U3L9ZH1, UNII-YR5U3L9ZH1, биапенем, بيابينام ,比阿培南 , Biapenern, CL 186-815, CL 186815, L 627, LJC 10627, Omegacin, Antibacterial, Antibiotics, Lactams, Carbapenems, ind 2021, india 2021, approvals 2021
CC1C2C(C(=O)N2C(=C1SC3CN4C=NC=[N+]4C3)C(=O)[O-])C(C)O
https://clinicaltrials.gov/search/intervention=Biapenem

updated
Biapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)-hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3-yljthio] 6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.
Formula 1
U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.
U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.
U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.
The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.
The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.
EXAMPLES
Example 1 : Purification of Biapenem
Biapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.
Yield: 84%
HPLC Purity: 99.87%
Example 2: Purification of Biapenem
Biapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.
Yield: 81.77%
HPLC Purity: 99.80%
PATENT
Background of the Invention Biapenem is a synthetic broad-spectrum carbapenem antibiotic which suppresses bacterial growth by inhibiting the enzymes responsible for bacterial cell wall synthesis, and shows broad-spectrum antibacterial activity both against gram-positive bacteria and gram-negative bacteria. Biapenem is chemically known as (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][ 1,2,4] triazol-8-ium-6-ylsulfanyl)-6-( 1 -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo [3.2.0]hept-2-ene-2-carboxylate and marketed in Japan as OMEGACIN®.Various methods are reported in the prior art for the preparation of Biapenem of formula (I) which includes the condensation of compound of formula (II) with compound of formula (III) and subsequent deprotection of the protecting group as shown in scheme-1. wherein R1 is hydrogen or hydroxy protecting group such as tert-butyl dimethyl silyl and the like, R2 is hydrogen or carboxyl protecting group such as p-nitrobenzyl, p-methoxy benzyl, allyl and the like, A is an activating group such as P(0)(OR)2, SO2R and the like wherein R is selected from substituted or unsubstituted C1-6 alkyl, aralkyl or aryl to form the compound of formula (II). The X” in compound of formula (III) is halogen selected from Br or CI.Biapenem was first disclosed in US 4,866,171 and the said patent also discloses a process for the preparation of the same. US 5,241,073 disclosed the method for the preparation of compound of formula (III) followed by condensation with compound of general formula (II) using base such as N-ethyldiisopropylamine and subsequent deprotection yields Biapenem which was isolated by column chromatography followed by crystallization from ethanol.EP 0289801 discloses a process for the preparation of crystalline Biapenem wherein Biapenem was dissolved in water and lyophilized to get amorphous compound. The amorphous compound was dissolved in water at 40° C followed by cooling to get crystalline product. This patent further provides the PXRD values of the crystalline Biapenem. The Biapenem obtained according to the process provided in this patent takes longer time for reconstitution and hence not suitable.US 5,286,856 and US 5,424,069 provide a process for the crystallization of Biapenem which utilizes freeze-drying technique and vial lyophillisation method respectively. These patents disclose (refer para 1, lines 10-33 of US’ 856) that the process provided in EP 0289801 results with Biapenem crystals which take relatively longer time for dissolution during use. To overcome the above issues, these patents utilize the freeze-drying and vial lyophillisation methods. The said methods involve freezing of the solution containing Biapenem followed by raising the temperature and repeating the cooling and heating process followed by lyophillisation to get the crystalline product. Lyophillisation and related process are capital intensive techniques and uneconomical in commercial scale operations.All the above said prior arts utilize either the lyophillisation technique or preparing the amorphous material and crystallizing it from water to get crystalline Biapenem.Biapenem is available as powder for injection which needs to be reconstituted with water or saline solution before injection. The process of preparing a solution having an appropriate concentration of an active ingredient for the administration is called “reconstitution”. The reconstitution time (RCT) plays a critical role in injectable powders. Short reconstitution time is preferable for both a member of medical center and patients. If the reconstitution time is too long, it will increase the preparation time thus making it difficult to administrate it to many patients at the same, which will eventually lower the competitiveness of the drug. The problem before the applicants is to find economic and robust process for the preparation of Biapenem with high purity and yield which should dissolve in water in less than 25 seconds (reconstitution time). With our continued intensive and diligent research for developing a process for the preparation of Biapenem having high purity and yield with reconstitution time of less than 25 seconds, we have identified an improved process which is commercially viable and eliminates the issues associated with reconstitution time. The process of this invention is simple and obviates the use of freeze crystallization. Further the present invention fulfils the need for a process for the manufacture of Biapenem which is convenient to operate in commercial scale
Objectives of the inventionThe main objective of the present invention is to provide a simple and commercially viable, industrially scalable process for the crystallization of Biapenem of formula (I) with high purity and good yield.Yet another objective of the present invention is to provide a simple and commercially suitable process for the preparation of Biapenem of formula (I) with reconstitution time less than 25 seconds. The reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Summary of the inventionAccordingly the primary aspect of the present invention is to provide an improved process for the preparation of Biapenem of formula (I) the said process comprises;(i) obtaining a solution of Biapenem in water containing co-solvent; and(ii) adding anti-solvent in to the solution of step (i) or vice-versa to crystallize Biapenem followed by filtration. Detailed Description In an embodiment of the present invention, the co-solvent used in step (i) is selected from alcoholic solvents consisting of methanol, ethanol, isopropyl alcohol, n-propanol, n-butanol and iso-butanol or mixtures thereof; preferably methanol, ethanol and isopropyl alcohol; more preferably methanol.In another embodiment of the present invention the anti-solvent used in step (ii) is selected from acetone, methyl ethyl ketone, methyl isobutyl ketone, ethyl acetate, methyl acetate, butyl acetate, tetrahydrofuran or mixtures thereof; preferably acetone. In yet another embodiment of the present invention, the solution of Biapenem in step (i) can be obtained by (a) dissolving Biapenem in water followed by addition of co-solvent (b) dissolving Biapenem in water containing the co-solvent (c) the aqueous solution containing Biapenem can be obtained directly from the reaction mass followed by addition of co-solvent (d) the aqueous solution of Biapenem containing co-solvent can be obtained directly from the reaction mass. The said solutions, if necessary can be subjected to sterile filtration before the addition of anti-solvent. Thus the present invention provided a process for the preparation of sterile Biapenem having reconstitution time less than 25 seconds, more preferably less than 15 seconds.The prior art lyophillisation process for the preparation of Biapenem requires capital investment and high operating cost due to the involvement of repetitive heating and cooling process which is tedious technology in commercial scale operations. The reported prior art process for the crystallization of Biapenem of formula (I) from water results in the formation of crystalline powder which takes longer time for dissolution in water or saline solution (reconstitution time). Surprisingly, applicant found that the use of co-solvents during the crystallization of Biapenem results with Biapenem having reconstitution time of less than 25 seconds. This constitutes the novelty of the present invention.In this present invention the Biapenem of formula (I) is obtained as crystalline solid with purity above 99.0 % by HPLC with good stability and further can be easily filled in vials.
The following examples are provided by way of illustration only and should not be construed to limit the scope of the invention.
Crystallization of (4R,5S,6S)-3-(6,7-dihvdro-5H-pyrazolo[l,2-al 11,2,41 triazol-8-ium-6-vlsulfanvl)-6-(l-hydroxvethvl)-4-methvl-7-oxo-l-azabicyclo [3.2.01hept-2-ene-2-carboxvlate [Biapenem of formula (1)1:Example -1:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. Activated carbon and EDTA were added to the clear solution and filtered through hi-flow bed, washed with water followed by filtration through micron filters in sterile area. To the filtrate, methanol (600 mL) was added followed by acetone under stirring. To the reaction mass, Biapenem seed material was added and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 85 g Purity by HPLC: 99.5% Reconstitution time (RCT): < 15 seconds
Example -2:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. To the filtrate, isopropyl alcohol (500 ml) was added followed by acetone under stirring. The mass was cooled and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 83 g Purity by HPLC: 99.6% Reconstitution time: < 15 seconds
Example -3;To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through micron filters. To the filtrate, ethanol (600 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 84 g Purity by HPLC: 99.5% Reconstitution time : < 15 seconds
Example -4:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through hi-flow bed, washed with water followed by filtration through micron filters. To the filtrate, methanol (450 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem. Yield: 87 g Purity by HPLC: 99.4% Reconstitution time (RCT): < 15 seconds
Reference example -1:Preparation of Biapenem (Non-Sterile)Step-I: Preparation of p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pvrazolofl,2-al[l,2,41triazol-8-ium-6-vlsulfanvn-6-(l-hvdroxvethyl)-4-methvI-7-oxo-l-azabicvclo[3.2.01hept-2-ene-2-carboxylate [Compound of formula (IV)1To a mixture of acetonitrile and DMF, P-Nitrobenzyl (4R,5S,6S)-3-(dipheny loxy)phosphory loxy-6- [(1R)-1 -hydroxyethy 1] -4-methy 1-7-oxo-1 -azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound of formula II) and 6,7-dihydro-6-mercapto-5H-pyrazolo[l,2-a] [1,2,4] triazole chloride (compound of formula III) were added and cooled to 0-5° C. To this mixture, N-ethyldiisopropyl amine was added and stirred till the completion of the reaction, followed by the addition of dichloromethane to crystallize the p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo[3.2.0] hept-2-ene-2-carboxylate which was filtered and dried under nitrogen.
Step-II: Preparation of BiapenemTo a solution of MOPS buffer and THF, p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l-hydroxy ethyl)-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (Compound of formula-IV) was added at pH 7-8 and cooled to 5-10° C. The mixture was hydrogenated using palladium on carbon as catalyst. The catalyst was filtered and the filtrate was treated with activated carbon and filtered. The filtrate was extracted with dichloromethane and the layers separated. The aqueous layer was degassed. To the aqueous layer, acetone was added to crystallize Biapenem at 20-25° C. The product was filtered, washed with aqueous acetone and dried under vacuum to get Biapenem (Non-Sterile).
Reference example -2: Crystallization of Biapenem
Example -1 was repeated without the addition of methanol.Yield: 84 g Purity by HPLC: 99.5%Reconstitution time : > 90 secondsThe reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Table-1: Comparative Data:The comparative data provided in the table-1 clearly indicates that the addition of co-solvent during crystallization provides Biapenem with reconstitution time less than 25 seconds.
CLARITHROMYCIN


Clarithromycin
Synonyms:A-56268, TE-031, 6-O-methylerythromycin, ATC:J01FA09Use:macrolide antibioticChemical name:6-O-methylerythromycinFormula:C38H69NO13
- MW:747.96 g/mol
- CAS-RN:81103-11-9
- 81103-11-9
klacid XL / Klaricid XL / Macladin / Naxy / Veclam / Zeclar
(3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-6-{[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy}-14-ethyl-12,13-dihydroxy-4-{[(2R,4R,5S,6S)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy}-7-methoxy-3,5,7,9,11,13-hexamethyl-1-oxacyclotetradecane-2,10-dione
Synthesis Reference
Jih-Hua Liu, David A. Riley, “Preparation of crystal form II of clarithromycin.” U.S. Patent US5844105, issued May, 1997. US5844105
NEW DRUG APPROVALS
ONE TIME
$10.00
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Clarithromycin citrate | 16K08R7NG0 | 848130-51-8 | MDRWXDRMSKEMRE-AZFLODHXSA-N |
ClarithromycinCAS Registry Number: 81103-11-9CAS Name: 6-O-MethylerythromycinManufacturers’ Codes: A-56268; TE-031Trademarks: Biaxin (Abbott); Clarosip (Grñenthal); Clathromycin (Taisho); Cyllind (Abbott); Klacid (Abbott); Klaricid (Abbott); Macladin (Guidotti); Naxy (Sanofi Winthrop); Veclam (Zambon); Zeclar (Abbott)Molecular Formula: C38H69NO13Molecular Weight: 747.95Percent Composition: C 61.02%, H 9.30%, N 1.87%, O 27.81%Literature References: Semisynthetic macrolide antibiotic; derivative of erythromycin, q.v. Prepn: Y. Watanabe et al.,EP41355; eidem,US4331803 (1981, 1982 both to Taisho); and in vitro antibacterial activity: S. Morimoto et al.,J. Antibiot.37, 187 (1984). In vitro and in vivo antibacterial activity: P. B. Fernandes et al.,Antimicrob. Agents Chemother.30, 865 (1986). Comparative antibacterial spectrum in vitro: C. Benson et al.,Eur. J. Clin. Microbiol.6, 173 (1987); H. M. Wexler, S. M. Finegold, ibid. 492. HPLC determn in biological fluids: D. Croteau et al.,J. Chromatogr.419, 205 (1987); in plasma: H. Amini, A. Ahmadiani, J. Chromatogr. B817, 193 (2005). Acute toxicity study: S. Abe et al.,Chemotherapy (Tokyo)36, Suppl. 3, 274 (1988). Symposium on pharmacology and comparative clinical studies: J. Antimicrob. Chemother.27, Suppl. A, 1-124 (1991). Comprehensive description: I. I. Salem, Anal. Profiles Drug Subs. Excip.24, 45-85, (1996).Properties: Colorless needles from chloroform + diisopropyl ether (1:2), mp 217-220° (dec). Also reported as crystals from ethanol, mp 222-225° (Morimoto). uv max (CHCl3): 288 nm (e 27.9). uv max (CHCl3): 240, 288 nm; (methanol): 211, 288 nm. [a]D24 -90.4° (c = 1 in CHCl3). Stable at acidic pH. LD50 in male, female mice, male, female rats (mg/kg): 2740, 2700, 3470, 2700 orally, 1030, 850, 669, 753 i.p., >5000 all s.c. (Abe).Melting point: mp 217-220° (dec); mp 222-225° (Morimoto)Optical Rotation: [a]D24 -90.4° (c = 1 in CHCl3)Absorption maximum: uv max (CHCl3): 288 nm (e 27.9). uv max (CHCl3): 240, 288 nmToxicity data: LD50 in male, female mice, male, female rats (mg/kg): 2740, 2700, 3470, 2700 orally, 1030, 850, 669, 753 i.p., >5000 all s.c. (Abe)Therap-Cat: Antibacterial.Keywords: Antibacterial (Antibiotics); Macrolides.
Clarithromycin, a semisynthetic macrolide antibiotic derived from erythromycin, inhibits bacterial protein synthesis by binding to the bacterial 50S ribosomal subunit. Binding inhibits peptidyl transferase activity and interferes with amino acid translocation during the translation and protein assembly process. Clarithromycin may be bacteriostatic or bactericidal depending on the organism and drug concentration.
Clarithromycin, sold under the brand name Biaxin among others, is an antibiotic used to treat various bacterial infections.[2] This includes strep throat, pneumonia, skin infections, H. pylori infection, and Lyme disease, among others.[2] Clarithromycin can be taken by mouth as a pill or liquid.[2]
Common side effects include nausea, vomiting, headaches, and diarrhea.[2] Severe allergic reactions are rare.[2] Liver problems have been reported.[2] It may cause harm if taken during pregnancy.[2] It is in the macrolide class and works by decreasing protein production of some bacteria.[2]
Clarithromycin was developed in 1980 and approved for medical use in 1990.[3][4] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[5] Clarithromycin is available as a generic medication.[2] It is made from erythromycin and is chemically known as 6-O-methylerythromycin.[6]
Medical uses
Clarithromycin is primarily used to treat a number of bacterial infections including pneumonia, Helicobacter pylori, and as an alternative to penicillin in strep throat.[2] Other uses include cat scratch disease and other infections due to bartonella, cryptosporidiosis, as a second line agent in Lyme disease and toxoplasmosis.[2] It may also be used to prevent bacterial endocarditis in those who cannot take penicillin.[2] It is effective against upper and lower respiratory tract infections, skin and soft tissue infections and helicobacter pylori infections associated with duodenal ulcers.
Spectrum of bacterial susceptibility
Staphylococcus aureusAerobic Gram-positive bacteria
Aerobic Gram-negative bacteria
- Haemophilus parainfluenzae
- Haemophilus influenzae
- Moraxella catarrhalis
Helicobacter
Mycobacteria
Mycobacterium avium complex consisting of:
Other bacteria
Safety and effectiveness of clarithromycin in treating clinical infections due to the following bacteria have not been established in adequate and well-controlled clinical trials:[7]
Aerobic Gram-positive bacteria
- Streptococcus agalactiae
- Streptococcus (Groups C, F, G)
- Viridans group streptococci
Aerobic Gram-negative bacteria
Anaerobic Gram-positive bacteria
- Clostridium perfringens
- Peptococcus Niger
- Cutibacterium acnes
Anaerobic Gram-negative bacteria
- Prevotella melaninogenica (formerly Bacteroides melaninogenicus)
Contraindications
- Clarithromycin should not be taken by people who are allergic to other macrolides or inactive ingredients in the tablets, including microcrystalline cellulose, croscarmelose sodium, magnesium stearate, and povidone
- Clarithromycin should not be used by people with a history of cholestatic jaundice and/or liver dysfunction associated with prior clarithromycin use.[7]
- Clarithromycin should not be used in the setting of hypokalaemia (low blood potassium)
- Use of clarithromycin with the following medications: cisapride, pimozide, astemizole, terfenadine, ergotamine, ticagrelor, ranolazine or dihydroergotamine is not recommended.[7]
- It should not be used with colchicine in people with kidney or liver impairment.[7]
- Concomitant use with cholesterol medications such as lovastatin or simvastatin.[7]
- Hypersensitivity to clarithromycin or any component of the product, erythromycin, or any macrolide antibiotics.[7]
- QT prolongation or ventricular cardiac arrhythmias, including torsade de pointes.[7]
Side effects
The most common side effects are gastrointestinal: diarrhea (3%), nausea (3%), abdominal pain (3%), and vomiting (6%). It also can cause headaches, insomnia, and abnormal liver function tests. Allergic reactions include rashes and anaphylaxis. Less common side effects (<1%) include extreme irritability, hallucinations (auditory and visual), dizziness/motion sickness, and alteration in senses of smell and taste, including a metallic taste. Dry mouth, panic attacks, and nightmares have also been reported, albeit less frequently.[8]
Cardiac
In February 2018, the FDA issued a Safety Communication warning with respect to an increased risk for heart problems or death with the use of clarithromycin, and has recommended that alternative antibiotics be considered in those with heart disease.[9]
Clarithromycin can lead to a prolonged QT interval. In patients with long QT syndrome, cardiac disease, or patients taking other QT-prolonging medications, this can increase risk for life-threatening arrhythmias.[10]
In one trial, the use of short-term clarithromycin treatment was correlated with an increased incidence of deaths classified as sudden cardiac deaths in stable coronary heart disease patients not using statins.[11] Some case reports suspect it of causing liver disease.[12]
Liver and kidney
Clarithromycin has been known to cause jaundice, cirrhosis, and kidney problems, including kidney failure.[citation needed]
Central nervous system
Common adverse effects of clarithromycin in the central nervous system include dizziness, headaches. Rarely, it can cause ototoxicity, delirium and mania.
Infection
A risk of oral candidiasis and vaginal candidiasis, due to the elimination of the yeast’s natural bacterial competitors by the antibiotic, has also been noted.
Pregnancy and breastfeeding
Clarithromycin should not be used in pregnant women except in situations where no alternative therapy is appropriate.[7] Clarithromycin can cause potential hazard to the fetus hence should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.[7] For lactating mothers it is not known whether clarithromycin is excreted in human milk.[7]
Interactions
Clarithromycin inhibits a liver enzyme, CYP3A4, involved in the metabolism of many other commonly prescribed drugs. Taking clarithromycin with other medications that are metabolized by CYP3A4 may lead to unexpected increases or decreases in drug levels.
A few of the common interactions are listed below.
Colchicine
Clarithromycin has been observed to have a dangerous interaction with colchicine as the result of inhibition of CYP3A4 metabolism and P-glycoprotein transport. Combining these two drugs may lead to fatal colchicine toxicity, particularly in people with chronic kidney disease.[7]
Statins
Taking clarithromycin concurrently with certain statins (a class of drugs used to reduce blood serum cholesterol levels) increases the risk of side effects, such as muscle aches and muscle break down (rhabdomyolysis).[13]
Calcium channel blockers
Concurrent therapy with calcium channel blocker may increase risk of low blood pressure, kidney failure, and death, compared to pairing calcium channel blockers with azithromycin, a drug similar to clarithromycin but without CYP3A4 inhibition.[14] Administration of clarithromycin in combination with verapamil have been observed to cause low blood pressure, low heart rate, and lactic acidosis.[7]
Carbamazepine
Clarithromycin may double the level of carbamazepine in the body by reducing its clearance, which may lead to toxic symptoms of carbamazepine, such as double vision, loss of voluntary body movement, nausea, as well as hyponatremia.[15]
HIV medications
Depending on the combination of medications, clarithromycin therapy could be contraindicated, require changing doses of some medications, or be acceptable without dose adjustments.[16] For example, clarithromycin may lead to decreased zidovudine concentrations.[17]
Mechanism of action
Clarithromycin prevents bacteria from multiplying by acting as a protein synthesis inhibitor. It binds to 23S rRNA, a component of the 50S subunit of the bacterial ribosome, thus inhibiting the translation of peptides.[citation needed]
Pharmacokinetics
MetabolismUnlike erythromycin, clarithromycin is acid-stable, so can be taken orally without having to be protected from gastric acids. It is readily absorbed, and diffuses into most tissues and phagocytes. Due to the high concentration in phagocytes, clarithromycin is actively transported to the site of infection. During active phagocytosis, large concentrations of clarithromycin are released; its concentration in the tissues can be over 10 times higher than in plasma. Highest concentrations are found in liver, lung tissue, and stool.
Clarithromycin has a fairly rapid first-pass metabolism in the liver. Its major metabolites include an inactive metabolite, N-desmethylclarithromycin, and an active metabolite, 14-(R)-hydroxyclarithromycin. Compared to clarithromycin, 14-(R)-hydroxyclarithromycin is less potent against mycobacterial tuberculosis and the Mycobacterium avium complex. Clarithromycin (20%-40%) and its active metabolite (10%-15%) are excreted in urine. Of all the drugs in its class, clarithromycin has the best bioavailability at 50%, which makes it amenable to oral administration. Its elimination half-life is about 3 to 4 hours with 250 mg administered every 12 h, but increased to 5 to 7 h with 500 mg administered every 8 to 12 h. With any of these dosing regimens, the steady-state concentration of this metabolite is generally attained within 3 to 4 days.[18]
History
Clarithromycin was invented by researchers at the Japanese drug company Taisho Pharmaceutical in 1980.[3] The product emerged through efforts to develop a version of the antibiotic erythromycin that did not experience acid instability in the digestive tract, causing side effects, such as nausea and stomachache. Taisho filed for patent protection for the drug around 1980 and subsequently introduced a branded version of its drug, called Clarith, to the Japanese market in 1991. In 1985, Taisho partnered with the American company Abbott Laboratories for the international rights, and Abbott also gained FDA approval for Biaxin in October 1991. The drug went generic in Europe in 2004 and in the US in mid-2005.
Society and culture

A pack of Clarithromycin tablets manufactured by Taisho Pharmaceutical
Available forms
Clarithromycin is available as a generic medication.[2] In the United States, clarithromycin is available as immediate release tablets, extended release tablets, and granules for oral suspension.[2]
Brand names
Clarithromycin is available under several brand names in many different countries, for example Biaxin, Crixan, Claritron, Clarihexal, Clacid, Claritt, Clacee, Clarac, Clariwin, Claripen, Clarem, Claridar, Cloff, Fromilid, Infex, Kalixocin, Karicin, Klaricid, Klaridex, Klacid, Klaram, Klabax, MegaKlar, Monoclar, Resclar, Rithmo, Truclar, Vikrol and Zeclar.
Manufacturers
In the UK the drug product is manufactured in generic form by a number of manufacturers including Somex Pharma, Ranbaxy, Aptil and Sandoz.
SYN
CN 109705180

SYN
Indian Pat. Appl., 2014DE00731, 31 Aug 2016

SYN
Heterocycles, 31(12), 2121-4; 1990

SYN
https://patents.google.com/patent/WO2006064299A1/enErythromycin A is known to be a useful macrolide antibiotic having a strong activity against Gram-positive bacteria, this compound has an undesirable property that it loses rapidly the antibacterial activity by the acid in stomach when administered orally, where- upon its blood concentration remains at a low level. 6-0-Alkyl derivatives of Erythromycin- A are well known as an useful antibacterial agents. 6-O-Methyl-Erythromycin-A (Clarithromycin) and a pharmaceutically acceptable salt is a potent macrolide antibiotic as reported in US Patent No. 4,331 ,803. Clarithromycin is stable in acidic medium and also remarkable in vivo activity and has a strong antibacterial property against Gram-positive bacteria compared to Erythromycin- A. This compound shows excellent effect for the treatment of infections by oral administration.A number of synthetic processes have been reported for preparing 6-O-alkyl erythromycin. US Patent No. 4,331 ,803 discloses a method for the preparation of Clarithromycin by methylating 6-OH group of 2′-O-3′-N-benzyloxycarbonyl erythromycinFormula (III)

21,3′-O-Protected ErythromycinMethylation of 6-OH group of the 2′,3′-benzyloxycarbonyl erythromycin was carried out using methyl iodide in the presence of a suitable base in a solvent. Clarithromycin was obtained from the compound after removing benzyloxycarbonyl group by hydrogenolysis and then subjecting to the reductive methylation in the presence of excess amount of farmaldehyde. Clarithromycin can also be synthesized by the methylation of 6-OH position of Erythromycin-A-9-OximeFormula (II)

Erythromycin-9-OximeSynthesis of Clarithromycin using 9-oxime or its derivatives are well reported in US Patent Nos. 5,274,085; 4,680,386; 4,668,776; 4,670,549 and 4,672,109. In case of Erythromycin-9-Oxime derivatives, the oxime is protected before methylation step with 2- alkenyl group (US Patent Nos. 4,670,549; 4,668,776) or benzyl group (US Patent Nos. 4,680,386 and 4,670,549). However, it has been reported (Ref. Journal of Antibiotics 46, No. 6, Page No. 647, year 1993) that when the Erythromycin-A-9-Oxime is protected by trimethylsilyl group, which is very unstable under basic condition pose potential impurities formation during methylation. There are some methods reported in US Patent Nos., e.g. , 4,680,386; 4,670,549 and US Patent No. 4,311,803 for the synthesis of Clarithromycin by using chlorobenzyloxycarbonyl group for protection at 2′ and 3′ function of of Erythromycin-A-9-Oxime derivatives.For the protection of 2′-OH group (US Patent No. 4,311 ,803) requires large amounts of benzyl chloroformate which poses problems in handling because of its severe irritating and toxic properties. This protection step also leads to the formation of 3′ -N- demethylation, which requires an additional re-methylation step. The de-protection of chlorobenzyloxy carbonyl group leads to the formation of undesired side products. In earlier reported processes, e.g. , US Patent No. 4,990,602; EP 0,272,110 Al where the methylation has been done on Erythromycin-A-9-Oxime derivatives by the protection of 2′ and 4″ hydroxyl groups using DMSO and THF as a solvent at 0° to 50C or at room temperature, smooth methylation takes place with less side product formation. However, by using the above methylation processes the formation of 6, 11-O-dimethyl erythromycin- A (Compound- A) is always more than 1.0 % in Clarithromycin. Hence, there is a need for an efficient methylation process for the production of Clarithromycin with lesser amount of 6,11-O-dimethyl erythromycin-A than reported previously.




EXAMPLE 1Erythromycin-A-9-OximeTo a solution of 201 Ltr water in 561 Kg isopropyl alcohol is added 282 Kg (4057 mol) of hydroxyl amine hydrochloride under stirring and the reaction mixture is brought to 10 to 200C. Caustic flakes (162 Kg, 4050 mol) is added slowly to the reaction mixture by keeping temperature between 10° to 200C. After 15 minutes of completion of addition, pH of reaction mixture is adjusted to 6.5 to 7.0 by the slow addition of glacial acetic acid (96 Ltr, 100.8 Kg, 1678.6 mole). To the stirred reaction mass is added 300 Kg (408.8 mole) of Erythromycin-A base and reaction mixture is stirred at 55° C for 28 hours. After completion of the reaction, mixture is brought to ambient temperature and to it a mixture of ammonia solution (270 Kg) and water (600 Ltr) is added within 1 hour followed by 3000 Ltr of fresh water in next two hours and stirred the reaction mass for further 1 hour. White solid product obtained is centrifuged, wet cake is washed with water and dried at 6O0C for 12 hours to give 270 Kg of erythromycin-A Oxime. Melting point = 156° to 158°C.EXAMPLE 22′,4″-O-Bis(trimethylsilyl)-erythro?nycin-A-9[O-(l-methoxy-l-methyl ethyl)oximeTo a solution of 80 Kg (106.8 mole) of Erythromycin-A-9-Oxime in 400 Ltr of dichloromethane is added 38.50 Kg (534 mole) of 2-methoxy propene at 100C temperature 19.25 Kg (166.6 mole) of pyridine hydrochloride is added under stirring and the reaction mixture is stirred at 8 to 12° C for 6 hours then to it is added 19.30 Kg (119.5 mole) of HMDS and stirring is continued for 12 to 15 hours at 15° to 18°C temperature. After completion of reaction, 400 Ltr of saturated aqueous sodium carbonate solution is added and the mixture is stirred thoroughly at room temperature. Aqueous layer is further extracted with fresh DCM (100 Ltr). Both DCM extracts are mixed together and washed with water (200 Ltr) followed by brine solution (200 Ltr). The solvent is evaporated under reduced pressure. To the obtained crude solid mass is charged isopropyl alcohol (240 Ltr) and distilled out 80 Ltr of isopropyl alcohol. To the reaction mixture 160 Ltr of water is charged and stirring continued at room temperature for 1 hour. Solid crystalline product obtained is centrifuged and dried at 60° to 650C for 8 hours under vacuum to give 85 Kg of title compound. Melting point = 125° to 126°C. HPLC Purity = More than 90 % .EXAMPLE 3Clarithromycin-9- OximeTo a solution of 80 Kg (82.98 mole) of 2′,4″-O-bis(trimethylsilyl)-erythromycin-A- 9-[O-(l-methoxy methyl ethyl)Oxime] in 1200 Ltr of a mixture of dimethyl sulfoxide and diethylether (1 : 1) are added methyl iodide (20.62 Kg, 145.2 mole) and 6.48 Kg (98.35 mole) of 85 % potassium hydroxide powder and the reaction mixture is stirred for 90 minutes at room temperature. To the reaction mass is added 53 Kg of 40 % dimethylamine solution and stirring is continued for 1 hour diethylether layer is separated and DMSO layer is further extracted with fresh diethylether (200 Ltr). Combined ether layer is washed with water and concentrated in vacuum. To the obtained semi solid mass 330 Ltr of isopropyl alcohol is charged and then distilled out 165 Ltr of isopropyl alcohol. To the obtained slurry 165 Ltr of water and 21.71 Kg formic acid (99%) are added and the mixture is stirred at room temperature for 30 minutes. 622 Ltr of water is added to the reaction mixture and pH is adjusted between 10.5 and 11.5 with 25 % aqueous sodium hydroxide solution. Solid compound obtained is centrifuged and wet cake is kept as such for further reaction on the basis of moisture content. Wet weight = 95 Kg, Moisture Content = 33 %, Dried weight = 62 KgEXAMPLE 46-O-Methyl erythromycin- A (Clarithromycin)62 Kg of 6-O-Methyl erythromycin-9-Oxime is charged into a mixture of 434 Ltr of isopropyl alcohol and water (1: 1) and to it is added 38.80 Kg of sodium metabisulphite (203 mole) and then the mixture is heated to reflux for 6 to 8 hours. To the reaction mixture is charged water (620 Ltr) at ambient temperature and then the mixture is adjusted to pH about 10.5 to 11.5 by adding 25% aqueous sodium hydroxide solution and stirred for further 1 hour. White solid crude product is centrifuged, washed with water (300 Ltr), dried at 65° to 750C for 8 hours to give 40 Kg of crude Clarithromycin which on re- crystallization with chloroform isopropyl alcohol mixture provided 20 Kg of Clarithromycin (Form II).
SYNEP 0041355; US 4331803J Antibiot 1984,37(2),187-189
| EP 0147062 |
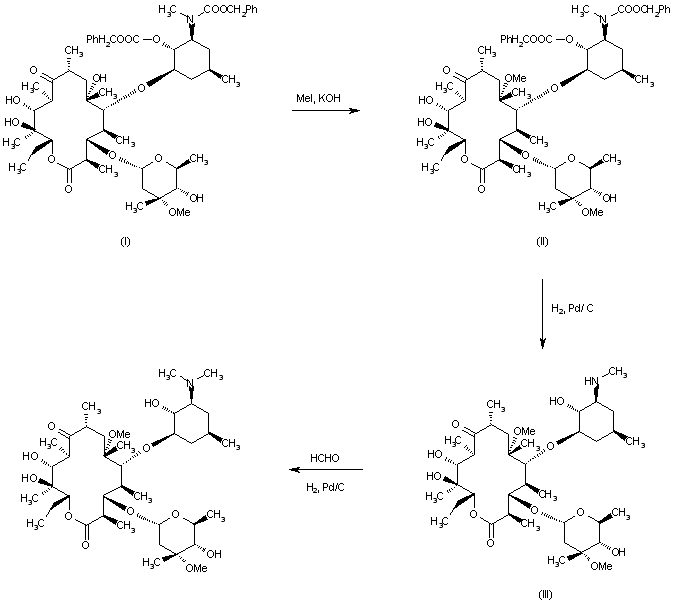
The methylation of 2′-O,N-bis(benzyloxycarbonyl)-N-demethylerythromycin A (I) with methyl iodide and KOH or NaHI in DMSO-dimethoxyethane gives the 6-O-methyl derivative (II), which is deprotected by hydrogenation with H2 over Pd/C in ethanol acetic acid affording 6-O-methyl-N-demethylerythromycin A (III). Finally, this compound is methylated with formaldehyde under reductive conditions (H2-Pd/C) in ethanol/acetic acid.
CLIP


2 Clarithromycin. Initial attempts of making clarithromycin (2) from erythromycin (1) by methylation of 8 gave approximately equal amounts of 2 and 10 by methylation at O-6 and O-11, respectively (Scheme 2, route A).[28–30] This allowed 2 to be obtained in approximately 39% yield, but it contained a small impurity of di-O-methylated 9. To improve the yields and obtain 2 in pure form, other alternatives were explored. During methylation of analogues of 8 it was observed that the conformation of the macrocyclic core plays an important role for the regioselectivity of the O-methylation.[31] As oximes are readilyhydrolysed and may have different conformations than ketone 8, oximes 11 and 13 were subjected to methylation. Interestingly, methylation of 13, but not of 11, proved to be highly selective for O-6 and provided 14 in 86% yield (Scheme2 route B); an observation which supports that 13 populates different conformations compared to 8 and 11 under the methylation conditions.[31] Compound 14 was then hydrogenated with Pd/C to deprotect the two benzyloxycarbonyl groups and the 2-chlorobenzyl group. The N-methylamine was then methylated by reductive amination and the oxime was deprotected by hydrolysis to provide clarithromycin (2). This procedure was further modified for process-scale synthesis so that clarithromycin (2) could be obtained in 70% yield starting from oxime 11 without the isolation of any intermediate.[32][28] M. Shigeo, T. Yoko, W. Yoshiaki, O. Sadafumi, J. Antibiot. 1984, 37, 187 – 189. [29] Y. Watanabe, T. Adachi, T. Asaka, M. Kashimura, S. Morimoto, Heterocycles 1990, 31, 2121 – 2124. [30] E. H. Flynn, H. W. Murphy, R. E. McMahon, J. Am. Chem. Soc. 1955, 77, 3104 – 3106. [31] Y. Watanabe, S. Morimoto, T. Adachi, M. Kashimura, T. Asaka, J. Antibiot. 1993, 46, 647 – 660.32] R. A. Dominguez, M. D. C. C. Rodriguez, L. . D. Tejo, R. N. Rib, J. S. Cebrin, J. I. B. Bilbao, 2003, US6642364B2.
References
- ^ https://www.ema.europa.eu/documents/psusa/clarithromycin-list-nationally-authorised-medicinal-products-psusa/00000788/202004_en.pdf
- ^ Jump up to:a b c d e f g h i j k l m n “Clarithromycin”. The American Society of Health-System Pharmacists. Archivedfrom the original on September 3, 2015. Retrieved September 4, 2015.
- ^ Jump up to:a b Greenwood D (2008). Antimicrobial drugs : chronicle of a twentieth century medical triumph (1 ed.). Oxford: Oxford University Press. p. 239. ISBN 9780199534845. Archived from the original on 2016-03-05.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 498. ISBN 9783527607495.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Kirst HA (2012). Macrolide Antibiotics (2 ed.). Basel: Birkhäuser Basel. p. 53. ISBN 9783034881050. Archived from the original on 2016-03-05.
- ^ Jump up to:a b c d e f g h i j k l “BIAXIN® Filmtab® (clarithromycin tablets, USP) BIAXIN® XL Filmtab® (clarithromycin extended-release tablets) BIAXIN® Granules (clarithromycin for oral suspension, USP)” (PDF). November 2, 2015. Archived (PDF) from the original on August 24, 2015. Retrieved November 2, 2015.
- ^ “Clarithromycin Side Effects in Detail – Drugs.com”. Drugs.com. Archived from the original on 2017-08-19. Retrieved 2017-08-18.
- ^ “Safety Alerts for Human Medical Products – Clarithromycin (Biaxin): Drug Safety Communication – Potential Increased Risk of Heart Problems or Death in Patients With Heart Disease”. FDA. Retrieved 24 February 2018.
- ^ Yamaguchi S, Kaneko Y, Yamagishi T, et al. [Clarithromycin-induced torsades de pointes]. Nippon Naika Gakkai Zasshi. 2003;92(1):143–5.
- ^ Winkel P, Hilden J, Fischer Hansen J, Hildebrandt P, Kastrup J, Kolmos HJ, et al. (2011). “Excess sudden cardiac deaths after short-term clarithromycin administration in the CLARICOR trial: why is this so, and why are statins protective?”. Cardiology. 118 (1): 63–7. doi:10.1159/000324533. PMID 21447948. S2CID 11873791.
- ^ Tietz A, Heim MH, Eriksson U, Marsch S, Terracciano L, Krähenbühl S (January 2003). “Fulminant liver failure associated with clarithromycin”. The Annals of Pharmacotherapy. 37 (1): 57–60. doi:10.1345/1542-6270(2003)037<0057:flfawc>2.0.co;2. PMID 12503933.
- ^ Patel AM, Shariff S, Bailey DG, Juurlink DN, Gandhi S, Mamdani M, et al. (June 2013). “Statin toxicity from macrolide antibiotic coprescription: a population-based cohort study”. Annals of Internal Medicine. 158 (12): 869–76. doi:10.7326/0003-4819-158-12-201306180-00004. PMID 23778904. S2CID 21222679.
- ^ Gandhi S, Fleet JL, Bailey DG, McArthur E, Wald R, Rehman F, Garg AX (December 2013). “Calcium-channel blocker-clarithromycin drug interactions and acute kidney injury”. JAMA. 310 (23): 2544–53. doi:10.1001/jama.2013.282426. PMID 24346990.
- ^ Gélisse P, Hillaire-Buys D, Halaili E, Jean-Pastor MJ, Vespignan H, Coubes P, Crespel A (November 2007). “[Carbamazepine and clarithromycin: a clinically relevant drug interaction]”. Revue Neurologique. 163 (11): 1096–9. doi:10.1016/s0035-3787(07)74183-8. PMID 18033049.
- ^ Sekar VJ, Spinosa-Guzman S, De Paepe E, De Pauw M, Vangeneugden T, Lefebvre E, Hoetelmans RM (January 2008). “Darunavir/ritonavir pharmacokinetics following coadministration with clarithromycin in healthy volunteers”. Journal of Clinical Pharmacology. 48 (1): 60–5. doi:10.1177/0091270007309706. PMID 18094220. S2CID 38368595.
- ^ Polis MA, Piscitelli SC, Vogel S, Witebsky FG, Conville PS, Petty B, et al. (August 1997). “Clarithromycin lowers plasma zidovudine levels in persons with human immunodeficiency virus infection”. Antimicrobial Agents and Chemotherapy. 41 (8): 1709–14. doi:10.1128/AAC.41.8.1709. PMC 163990. PMID 9257746.
- ^ Ferrero JL, Bopp BA, Marsh KC, Quigley SC, Johnson MJ, Anderson DJ, et al. (1990). “Metabolism and disposition of clarithromycin in man”. Drug Metabolism and Disposition. 18 (4): 441–6. PMID 1976065.
- ReferencesAllevi, P. et al.: Bioorg. Med. Chem. (BMECEP) 7, 12, 2749 (1999)Watanabe, Y. et al.: Heterocycles (HTCYAM) 31, 12, 2121 (1990).EP 158 467 (Taisho Pharmaceutical Co.; 22.3.1985; J-prior. 6.4.1984).EP 272 110 (Taisho Pharmaceutical Co.; 16.12.1987; J-prior. 17.12.1986).US 2 001 037 015 (Teva Pharm.; 15.12.2000; USA-prior. 29.2.2000).KR 2 000 043 839 (Hanmi Pharm.; ROK-prior. 29.12.1998).EP 1 150 990 (Hanmi Pharm.; 7.11.2001; ROK-prior. 29.12.1998)EP 41 355 (Taisho Pharmaceutical Co.; 27.5.1981; J-prior. 4.6.1980).Preparation of O,N-dicarbobenzoxy-N-demethylerythromycin:Flynn, E. H. et al.: J. Am. Chem. Soc. (JACSAT) 77, 3104 (1955).Process for preparation of erythromycin A oxime:US 5 808 017 (Abbott; 15.9.1998; USA-prior. 10.4.1996).Alternative synthesis of clarithromycin:Liao, G.; Zhang, G.; He, T.: Zhongguo Kangshengsu Zazhi (ZKZAEY) 27, 3, 148 (2002) (in Chinese).EP 1 134 229 (Hanmi Pharmac. Co.; 19.9.2001; ROK-prior. 15.3.2000).Crystal form 0 of clarithromycin:The Merck Index, 13th Ed., 2362, p. 408.US 5 945 405 (Abbott; 31.8.1999; USA-prior. 17.1.1997).
External links
- “Clarithromycin”. Drug Information Portal. U.S. National Library of Medicine.
- US patent 4331803, Watanabe Y, Morimoto S, Omura S, “Novel erythromycin compounds”, issued 1981-05-19, assigned to Taisho Pharmaceutical
- “FDA review finds additional data supports the potential for increased long-term risks with antibiotic clarithromycin (Biaxin) in patients with heart disease” (PDF). FDA Drug Safety Communication.
| Clinical data | |
|---|---|
| Trade names | Biaxin, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a692005 |
| License data | EU EMA: by INNUS DailyMed: Clarithromycin |
| Pregnancy category | AU: B3 |
| Routes of administration | By mouth, intravenous |
| Drug class | Macrolides |
| ATC code | J01FA09 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)US: ℞-onlyEU: Rx-only [1]In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 50% |
| Protein binding | low binding |
| Metabolism | hepatic |
| Elimination half-life | 3–4 h |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 81103-11-9 |
| PubChem CID | 84029 |
| DrugBank | DB01211 |
| ChemSpider | 10342604 |
| UNII | H1250JIK0A |
| KEGG | D00276 |
| ChEMBL | ChEMBL1741 |
| CompTox Dashboard (EPA) | DTXSID3022829 |
| ECHA InfoCard | 100.119.644 |
| Chemical and physical data | |
| Formula | C38H69NO13 |
| Molar mass | 747.964 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCC[C@@H]1[C@@]([C@@H]([C@H](C(=O)[C@@H](C[C@@]([C@@H]([C@H]([C@@H]([C@H](C(=O)O1)C)O[C@H]2C[C@@]([C@H]([C@@H](O2)C)O)(C)OC)C)O[C@H]3[C@@H]([C@H](C[C@H](O3)C)N(C)C)O)(C)OC)C)C)O)(C)O | |
| hideInChIInChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21(4)28(40)19(2)17-37(9,47-14)33(52-35-29(41)25(39(11)12)16-20(3)48-35)22(5)30(23(6)34(44)50-26)51-27-18-36(8,46-13)32(43)24(7)49-27/h19-27,29-33,35,41-43,45H,15-18H2,1-14H3/t19-,20-,21+,22+,23-,24+,25+,26-,27+,29-,30+,31-,32+,33-,35+,36-,37-,38-/m1/s1 Key:AGOYDEPGAOXOCK-KCBOHYOISA-N | |
| (what is this?) (verify) |
////////////////////CLARITHROMYCIN, Antibacterial, Antibiotics, Macrolides, A-56268, TE-031,
#CLARITHROMYCIN, #Antibacterial, #Antibiotics, #Macrolides, #A-56268, #TE-031,
AZITHROMYCIN, アジスロマイシン;
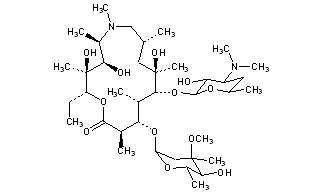
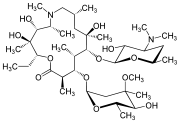
AZITHROMYCIN
C38H72N2O12,
748.9845
アジスロマイシン;
| CAS: | 83905-01-5 |
| PubChem: | 51091811 |
| ChEBI: | 2955 |
| ChEMBL: | CHEMBL529 |
| DrugBank: | DB00207 |
| PDB-CCD: | ZIT[PDBj] |
| LigandBox: | D07486 |
| NIKKAJI: | J134.080H |
Azithromycin is an antibiotic used for the treatment of a number of bacterial infections.[3] This includes middle ear infections, strep throat, pneumonia, traveler’s diarrhea, and certain other intestinal infections.[3] It can also be used for a number of sexually transmitted infections, including chlamydia and gonorrhea infections.[3] Along with other medications, it may also be used for malaria.[3] It can be taken by mouth or intravenously with doses once per day.[3]
Common side effects include nausea, vomiting, diarrhea and upset stomach.[3] An allergic reaction, such as anaphylaxis, QT prolongation, or a type of diarrhea caused by Clostridium difficile is possible.[3] No harm has been found with its use during pregnancy.[3] Its safety during breastfeeding is not confirmed, but it is likely safe.[4] Azithromycin is an azalide, a type of macrolide antibiotic.[3] It works by decreasing the production of protein, thereby stopping bacterial growth.[3]
Azithromycin was discovered 1980 by Pliva, and approved for medical use in 1988.[5][6] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[7] The World Health Organization classifies it as critically important for human medicine.[8] It is available as a generic medication[9] and is sold under many trade names worldwide.[2] The wholesale cost in the developing world is about US$0.18 to US$2.98 per dose.[10] In the United States, it is about US$4 for a course of treatment as of 2018.[11] In 2016, it was the 49th most prescribed medication in the United States with more than 15 million prescriptions.[12]
Medical uses
Azithromycin is used to treat many different infections, including:
- Prevention and treatment of acute bacterial exacerbations of chronic obstructive pulmonary disease due to H. influenzae, M. catarrhalis, or S. pneumoniae. The benefits of long-term prophylaxis must be weighed on a patient-by-patient basis against the risk of cardiovascular and other adverse effects.[13]
- Community-acquired pneumonia due to C. pneumoniae, H. influenzae, M. pneumoniae, or S. pneumoniae[14]
- Uncomplicated skin infections due to S. aureus, S. pyogenes, or S. agalactiae
- Urethritis and cervicitis due to C. trachomatis or N. gonorrhoeae. In combination with ceftriaxone, azithromycin is part of the United States Centers for Disease Control-recommended regimen for the treatment of gonorrhea. Azithromycin is active as monotherapy in most cases, but the combination with ceftriaxone is recommended based on the relatively low barrier to resistance development in gonococci and due to frequent co-infection with C. trachomatis and N. gonorrhoeae.[15]
- Trachoma due to C. trachomatis[16]
- Genital ulcer disease (chancroid) in men due to H. ducrey
- Acute bacterial sinusitis due to H. influenzae, M. catarrhalis, or S. pneumoniae. Other agents, such as amoxicillin/clavulanate are generally preferred, however.[17][18]
- Acute otitis media caused by H. influenzae, M. catarrhalis or S. pneumoniae. Azithromycin is not, however, a first-line agent for this condition. Amoxicillin or another beta lactam antibiotic is generally preferred.[19]
- Pharyngitis or tonsillitis caused by S. pyogenes as an alternative to first-line therapy in individuals who cannot use first-line therapy[20]
Bacterial susceptibility
Azithromycin has relatively broad but shallow antibacterial activity. It inhibits some Gram-positive bacteria, some Gram-negative bacteria, and many atypical bacteria.
A strain of gonorrhea reported to be highly resistant to azithromycin was found in the population in 2015. Neisseria gonorrhoeae is normally susceptible to azithromycin,[21] but the drug is not widely used as monotherapy due to a low barrier to resistance development.[15] Extensive use of azithromycin has resulted in growing Streptococcus pneumoniae resistance.[22]
Aerobic and facultative Gram-positive microorganisms
- Staphylococcus aureus (Methicillin-sensitive only)
- Streptococcus agalactiae
- Streptococcus pneumoniae
- Streptococcus pyogenes
Aerobic and facultative Gram-negative microorganisms
- Haemophilus ducreyi
- Haemophilus influenzae
- Moraxella catarrhalis
- Neisseria gonorrhoeae
- Bordetella pertussis
- Legionella pneumophila
Anaerobic microorganisms
- Peptostreptococcus species
- Prevotella bivia
Other microorganisms
- Chlamydophila pneumoniae
- Chlamydia trachomatis
- Mycoplasma genitalium
- Mycoplasma pneumoniae
- Ureaplasma urealyticum
Pregnancy and breastfeeding
No harm has been found with use during pregnancy.[3] However, there are no adequate well-controlled studies in pregnant women.[23]
Safety of the medication during breastfeeding is unclear. It was reported that because only low levels are found in breast milk and the medication has also been used in young children, it is unlikely that breastfed infants would suffer adverse effects.[4] Nevertheless, it is recommended that the drug be used with caution during breastfeeding.[3]
Airway diseases
Azithromycin appears to be effective in the treatment of COPD through its suppression of inflammatory processes.[24] And potentially useful in asthma and sinusitis via this mechanism.[25] Azithromycin is believed to produce its effects through suppressing certain immune responses that may contribute to inflammation of the airways.[26][27]
Adverse effects
Most common adverse effects are diarrhea (5%), nausea (3%), abdominal pain (3%), and vomiting. Fewer than 1% of people stop taking the drug due to side effects. Nervousness, skin reactions, and anaphylaxis have been reported.[28] Clostridium difficile infection has been reported with use of azithromycin.[3] Azithromycin does not affect the efficacy of birth control unlike some other antibiotics such as rifampin. Hearing loss has been reported.[29]
Occasionally, people have developed cholestatic hepatitis or delirium. Accidental intravenous overdose in an infant caused severe heart block, resulting in residual encephalopathy.[30][31]
In 2013 the FDA issued a warning that azithromycin “can cause abnormal changes in the electrical activity of the heart that may lead to a potentially fatal irregular heart rhythm.” The FDA noted in the warning a 2012 study that found the drug may increase the risk of death, especially in those with heart problems, compared with those on other antibiotics such as amoxicillin or no antibiotic. The warning indicated people with preexisting conditions are at particular risk, such as those with QT interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or those who use certain drugs to treat abnormal heart rhythms.[32][33][34]
Pharmacology
Mechanism of action
Azithromycin prevents bacteria from growing by interfering with their protein synthesis. It binds to the 50S subunit of the bacterial ribosome, thus inhibiting translation of mRNA. Nucleic acid synthesis is not affected.[23]
Pharmacokinetics
Azithromycin is an acid-stable antibiotic, so it can be taken orally with no need of protection from gastric acids. It is readily absorbed, but absorption is greater on an empty stomach. Time to peak concentration (Tmax) in adults is 2.1 to 3.2 hours for oral dosage forms. Due to its high concentration in phagocytes, azithromycin is actively transported to the site of infection. During active phagocytosis, large concentrations are released. The concentration of azithromycin in the tissues can be over 50 times higher than in plasma due to ion trapping and its high lipid solubility.[citation needed] Azithromycin’s half-life allows a large single dose to be administered and yet maintain bacteriostatic levels in the infected tissue for several days.[35]
Following a single dose of 500 mg, the apparent terminal elimination half-life of azithromycin is 68 hours.[35] Biliary excretion of azithromycin, predominantly unchanged, is a major route of elimination. Over the course of a week, about 6% of the administered dose appears as unchanged drug in urine.
History
A team of researchers at the pharmaceutical company Pliva in Zagreb, SR Croatia, Yugoslavia, — Gabrijela Kobrehel, Gorjana Radobolja-Lazarevski, and Zrinka Tamburašev, led by Dr. Slobodan Đokić — discovered azithromycin in 1980.[6] It was patented in 1981. In 1986, Pliva and Pfizer signed a licensing agreement, which gave Pfizer exclusive rights for the sale of azithromycin in Western Europe and the United States. Pliva put its azithromycin on the market in Central and Eastern Europe under the brand name Sumamed in 1988. Pfizer launched azithromycin under Pliva’s license in other markets under the brand name Zithromax in 1991.[36] Patent protection ended in 2005.[37]
Society and culture
_tablets.jpg)
Zithromax (azithromycin) 250 mg tablets (CA)
Cost
It is available as a generic medication.[9] The wholesale cost is about US$0.18 to US$2.98 per dose.[10] In the United States it is about US$4 for a course of treatment as of 2018.[11] In India, it is about US$1.70 for a course of treatment.[citation needed]
Available forms
Azithromycin is commonly administered in film-coated tablet, capsule, oral suspension, intravenous injection, granules for suspension in sachet, and ophthalmic solution.[2]
Usage
In 2010, azithromycin was the most prescribed antibiotic for outpatients in the US,[38] whereas in Sweden, where outpatient antibiotic use is a third as prevalent, macrolides are only on 3% of prescriptions.[39]
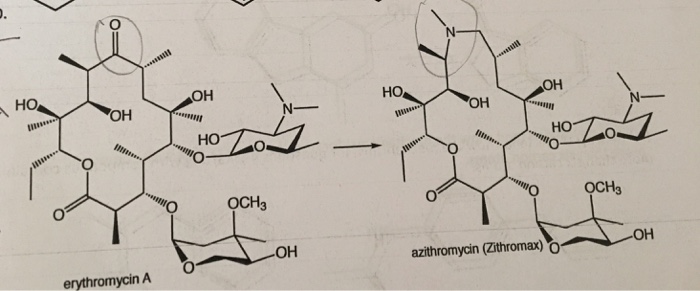
READ
References
- ^ Jump up to:ab “Azithromycin Use During Pregnancy”. Drugs.com. 2 May 2019. Retrieved 24 December 2019.
- ^ Jump up to:abcdef “Azithromycin International Brands”. Drugs.com. Archived from the original on 28 February 2017. Retrieved 27 February 2017.
- ^ Jump up to:abcdefghijklm “Azithromycin”. The American Society of Health-System Pharmacists. Archived from the original on 5 September 2015. Retrieved 1 August 2015.
- ^ Jump up to:ab “Azithromycin use while Breastfeeding”. Archived from the original on 5 September 2015. Retrieved 4 September 2015.
- ^ Greenwood, David (2008). Antimicrobial drugs : chronicle of a twentieth century medical triumph (1. publ. ed.). Oxford: Oxford University Press. p. 239. ISBN9780199534845. Archived from the original on 5 March 2016.
- ^ Jump up to:ab Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 498. ISBN9783527607495.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ World Health Organization (2019). Critically important antimicrobials for human medicine (6th revision ed.). Geneva: World Health Organization. hdl:10665/312266. ISBN9789241515528. License: CC BY-NC-SA 3.0 IGO.
- ^ Jump up to:ab Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. ISBN9781284057560.
- ^ Jump up to:ab “Azithromycin”. International Drug Price Indicator Guide. Retrieved 4 September 2015.
- ^ Jump up to:ab “NADAC as of 2018-05-23”. Centers for Medicare and Medicaid Services. Retrieved 24 May 2018.
- ^ “The Top 300 of 2019”. clincalc.com. Retrieved 22 December2018.
- ^ Taylor SP, Sellers E, Taylor BT (2015). “Azithromycin for the Prevention of COPD Exacerbations: The Good, Bad, and Ugly”. Am. J. Med. 128 (12): 1362.e1–6. doi:10.1016/j.amjmed.2015.07.032. PMID26291905.
- ^ Mandell LA, Wunderink RG, Anzueto A, Bartlett JG, Campbell GD, Dean NC, Dowell SF, File TM, Musher DM, Niederman MS, Torres A, Whitney CG (2007). “Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults”. Clin. Infect. Dis. 44 Suppl 2: S27–72. doi:10.1086/511159. PMID17278083.
- ^ Jump up to:ab “Gonococcal Infections – 2015 STD Treatment Guidelines”. Archived from the original on 1 March 2016.
- ^ Burton M, Habtamu E, Ho D, Gower EW (2015). “Interventions for trachoma trichiasis”. Cochrane Database Syst Rev. 11 (11): CD004008. doi:10.1002/14651858.CD004008.pub3. PMC4661324. PMID26568232.
- ^ Rosenfeld RM, Piccirillo JF, Chandrasekhar SS, Brook I, Ashok Kumar K, Kramper M, Orlandi RR, Palmer JN, Patel ZM, Peters A, Walsh SA, Corrigan MD (2015). “Clinical practice guideline (update): adult sinusitis”. Otolaryngol Head Neck Surg. 152 (2 Suppl): S1–S39. doi:10.1177/0194599815572097. PMID25832968.
- ^ Hauk L (2014). “AAP releases guideline on diagnosis and management of acute bacterial sinusitis in children one to 18 years of age”. Am Fam Physician. 89 (8): 676–81. PMID24784128.
- ^ Neff MJ (2004). “AAP, AAFP release guideline on diagnosis and management of acute otitis media”. Am Fam Physician. 69 (11): 2713–5. PMID15202704.
- ^ Randel A (2013). “IDSA Updates Guideline for Managing Group A Streptococcal Pharyngitis”. Am Fam Physician. 88 (5): 338–40. PMID24010402.
- ^ The Guardian newspaper: ‘Super-gonorrhoea’ outbreak in Leeds, 18 September 2015Archived 18 September 2015 at the Wayback Machine
- ^ Lippincott Illustrated Reviews : Pharmacology Sixth Edition. p. 506.
- ^ Jump up to:ab “US azithromycin label”(PDF). FDA. February 2016. Archived(PDF) from the original on 23 November 2016.
- ^ Simoens, Steven; Laekeman, Gert; Decramer, Marc (May 2013). “Preventing COPD exacerbations with macrolides: A review and budget impact analysis”. Respiratory Medicine. 107 (5): 637–648. doi:10.1016/j.rmed.2012.12.019. PMID23352223.
- ^ Gotfried, Mark H. (February 2004). “Macrolides for the Treatment of Chronic Sinusitis, Asthma, and COPD”. CHEST. 125 (2): 52S–61S. doi:10.1378/chest.125.2_suppl.52S. ISSN0012-3692. PMID14872001.
- ^ Zarogoulidis, P.; Papanas, N.; Kioumis, I.; Chatzaki, E.; Maltezos, E.; Zarogoulidis, K. (May 2012). “Macrolides: from in vitro anti-inflammatory and immunomodulatory properties to clinical practice in respiratory diseases”. European Journal of Clinical Pharmacology. 68 (5): 479–503. doi:10.1007/s00228-011-1161-x. ISSN1432-1041. PMID22105373.
- ^ Steel, Helen C.; Theron, Annette J.; Cockeran, Riana; Anderson, Ronald; Feldman, Charles (2012). “Pathogen- and Host-Directed Anti-Inflammatory Activities of Macrolide Antibiotics”. Mediators of Inflammation. 2012: 584262. doi:10.1155/2012/584262. PMC3388425. PMID22778497.
- ^ Mori F, Pecorari L, Pantano S, Rossi M, Pucci N, De Martino M, Novembre E (2014). “Azithromycin anaphylaxis in children”. Int J Immunopathol Pharmacol. 27 (1): 121–6. doi:10.1177/039463201402700116. PMID24674687.
- ^ Dart, Richard C. (2004). Medical Toxology. Lippincott Williams & Wilkins. p. 23.
- ^ Tilelli, John A.; Smith, Kathleen M.; Pettignano, Robert (2006). “Life-Threatening Bradyarrhythmia After Massive Azithromycin Overdose”. Pharmacotherapy. 26 (1): 147–50. doi:10.1592/phco.2006.26.1.147. PMID16506357.
- ^ Baselt, R. (2008). Disposition of Toxic Drugs and Chemicals in Man (8th ed.). Foster City, CA: Biomedical Publications. pp. 132–133.
- ^ Denise Grady (16 May 2012). “Popular Antibiotic May Raise Risk of Sudden Death”. The New York Times. Archived from the original on 17 May 2012. Retrieved 18 May 2012.
- ^ Ray, Wayne A.; Murray, Katherine T.; Hall, Kathi; Arbogast, Patrick G.; Stein, C. Michael (2012). “Azithromycin and the Risk of Cardiovascular Death”. New England Journal of Medicine. 366(20): 1881–90. doi:10.1056/NEJMoa1003833. PMC3374857. PMID22591294.
- ^ “FDA Drug Safety Communication: Azithromycin (Zithromax or Zmax) and the risk of potentially fatal heart rhythms”. FDA. 12 March 2013. Archived from the original on 27 October 2016.
- ^ Jump up to:ab “Archived copy”. Archived from the original on 14 October 2014. Retrieved 10 October 2014.
- ^ Banić Tomišić, Z. (2011). “The Story of Azithromycin”. Kemija U Industriji. 60 (12): 603–617. ISSN0022-9830. Archived from the original on 8 September 2017.
- ^ “Azithromycin: A world best-selling Antibiotic”. http://www.wipo.int. World Intellectual Property Organization. Retrieved 18 June 2019.
- ^ Hicks, LA; Taylor TH, Jr; Hunkler, RJ (April 2013). “U.S. outpatient antibiotic prescribing, 2010”. The New England Journal of Medicine. 368 (15): 1461–1462. doi:10.1056/NEJMc1212055. PMID23574140.
- ^ Hicks, LA; Taylor TH, Jr; Hunkler, RJ (September 2013). “More on U.S. outpatient antibiotic prescribing, 2010”. The New England Journal of Medicine. 369 (12): 1175–1176. doi:10.1056/NEJMc1306863. PMID24047077.
External links
Keywords: Antibacterial (Antibiotics); Macrolides.
- “Azithromycin”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Zithromax, Azithrocin, others[2] |
| Other names | 9-deoxy-9α-aza-9α-methyl-9α-homoerythromycin A |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a697037 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth (capsule, tablet or suspension), intravenous, eye drop |
| Drug class | Macrolide antibiotic |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 38% for 250 mg capsules |
| Metabolism | Liver |
| Elimination half-life | 11–14 h (single dose) 68 h (multiple dosing) |
| Excretion | Biliary, kidney (4.5%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| NIAID ChemDB | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.126.551 |
| Chemical and physical data | |
| Formula | C38H72N2O12 |
| Molar mass | 748.984 g·mol−1 g·mol−1 |
| 3D model (JSmol) | |
/////////AZITHROMYCIN, Antibacterial, Antibiotics, Macrolides, CORONA VIRUS, COVID 19, アジスロマイシン ,
Substances Referenced in Synthesis Path
CAS-RN Formula Chemical Name CAS Index Name
76801-85-9 C37H70N2O12 2-deoxo-9a-aza-9a-homoerythromycin A 1-Oxa-6-azacyclopentadecan-15-one,
13-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-2-eth- yl-3,4,10-trihydroxy-3,5,8,10,12,14-hexamethyl-11-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]-, [2R-(2
R*,3S*,4R*,5R*,8R*,10R*,11R*,12S*,13S*,1
4R*)]-
90503-04-1 C37H70N2O14 [2R-(2R*,3S*,4R*,5R*,8R*,10R*,11R*,12S*,
13S*,14R*)]-13-[(2,6-dideoxy-3-C-methyl3-O-methyl-α-L-ribo-hexopyranosyl)
oxy]-2-ethyl-3,4,6,10-tetrahydroxy3,5,8,10,12,14-hexamethyl-13-[[3,4,6-
trideoxy-3-(dimethyloxidoamino)-
β-D-xylo-hexopyranosyl] oxy]-1-oxa-6-azacyclopentadecan-15-one
1-Oxa-6-azacyclopentadecan-15-one,
13-[(2,6-dideoxy-3-C-methyl-3-Omethyl-α-L-ribo-hexopyranosyl)
oxy]-2-ethyl-3,4,6,10-tetrahydroxy3,5,8,10,12,14-hexamethyl-13-[[3,4,6-
trideoxy-3-(dimethyloxidoamino)-β-Dxylo-hexopyranosyl]oxy]-, [2R-(2R*,3S*,4R
*,5R*,8R*,10R*,11R*,12S*,13S*,14R*)]-
90503-05-2 C38H72N2O14 [2R-(2R*,3S*,4R*,5R*,8R*,10R*,11R*,12S*,
13S*,14R*)]-13-[(2,6-dideoxy-3-C-methyl3-O-methyl-α-L-ribo-hexopyranosyl) oxy]-2-ethyl-3,4,10-trihydroxy3,5,6,8,10,12,14-heptamethyl-11-[[3,4,6-
trideoxy-3-(dimethyloxidoamino)-
β-D-xylo-hexopyranosyl]
oxy]-1-oxa-6-azacyclopentadecan-15-one
6-oxide
1-Oxa-6-azacyclopentadecan-15-one,
13-[(2,6-dideoxy-3-C-methyl-3-Omethyl-α-L-ribo-hexopyranosyl)
oxy]-2-ethyl-3,4,10-trihydroxy3,5,6,8,10,12,14-heptamethyl-11-[[3,4,6-
trideoxy-3-(dimethyloxidoamino)-βD-xylo-hexopyranosyl]oxy]-, 6-oxide,
[2R-(2R*,3S*,4R*,5R*,8R*,10R*,11R*,12S*,1
3S*,14R*)]-
50-00-0 CH2O formaldehyde Formaldehyde
74-88-4 CH3I methyl iodide Methane, iodoTrade Names
Country Trade Name Vendor Annotation
D Ultreon Pfizer
Zithromax Pfizer Pharma/Gödecke/Parke-Davis
numerous generic preparations
F Azadose Pfizer
Monodose Pfizer
Zithromax Pfizer
GB Zithromax Pfizer
I Azitrocin Bioindustria
Ribotrex Pierre Fabre
Trocozina Sigma-Tau
Zithromax Pfizer
J Zithromac Pfizer
USA Azasite InSite Vision
Zithromax Pfizer as dihydrate
Formulations
cps. 100 mg, 250 mg; Gran. 10%; susp. 200 mg (as dihydrate); tabl. 250 mg
References
Djokic, S. et al.: J. Antibiot. (JANTAJ) 40, 1006 (1987).
a DOS 3 140 449 (Pliva; appl. 12.10.1981; YU-prior. 6.3.1981).
US 4 517 359 (Pliva; 14.5.1985; appl. 22.9.1981; YU-prior. 6.3.1981).
b EP 101 186 (Pliva; appl. 14.7.1983; USA-prior. 19.7.1982, 15.11.1982).
US 4 474 768 (Pfizer; 2.10.1984; prior. 19.7.1982, 15.11.1982).
educt by ring expansion of erythromycin A oxime by Beckmann rearrangement:
Djokic, S. et al.: J. Chem. Soc., Perkin Trans. 1 (JCPRB4) 1986, 1881-1890.
Bright, G.M. et al.: J. Antibiot. (JANTAJ) 41, 1029 (1988). US 4 328 334 (Pliva; 4.5.1982; YU-prior. 2.4.1979).
stable, non-hygroscopic dihydrate: EP 298 650 (Pfizer; appl. 28.6.1988).
medical use for treatment of protozoal infections:
US 4 963 531 (Pfizer; 16.10.1990; prior. 16.8.1988, 10.9.1987).
When Antibiotics Fail, Are Herbs the Answer?


Complex Compounds in Natural Herbs:
Now looking at the rate of adaptation and mutability of bacteria, it is inevitable that they will form resistance to most forms of simplistic human made antibiotic compounds. And when everything fails we will fall back to the old biblical medicinal herbs such as Ginger, Garlic, Black Pepper, Ashwagandha, etc. These herbs not only contain dozens of mild antibiotic compounds, they are also widely available in abundance.

{kind=link}
{kind=link}
{kind=link}
{kind=link}