
Home » Posts tagged 'Antiallergic'
Tag Archives: Antiallergic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TRANILAST
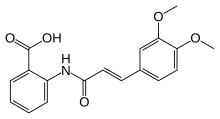
Tranilast
- Molecular FormulaC18H17NO5
- Average mass327.331 Da
Tranilast (INN, brand name Rizaben) is an antiallergic drug. It was developed by Kissei Pharmaceuticals and was approved in 1982 for use in Japan and South Korea for bronchial asthma. Indications for keloid and hypertrophic scar were added in the 1980s.
Kissei has developed and launched tranilast in Japan and South Korea for the treatment of allergic rhinitis, asthma and atopic dermatitis. Kissei, in collaboration with GlaxoSmithKline was additionally developing tranilast for the prevention of restenosis following percutaneous transluminal coronary angioplasty.
Medical uses
It is used Japan, South Korea, and China to treat asthma, keloid scars, and hypertrophic scars, and as an ophthalmic solution for allergic pink eye.[1]
It should not be taken women who are or might become pregnant, and it is secreted in breast milk.[1]
Interactions
People who are taking warfarin should not also take tranilast, as they interact.[1] It appears to inhibit UGT1A1 so will interfere with metabolism of drugs that are affected by that enzyme.[1]
Adverse effects
When given systemically, tranilast appears to cause liver damage; in a large well-conducted clinical trial it caused elevated transaminases three times the upper limit of normal in 11 percent of patients, as well as anemia, kidney failure, rash, and problems urinating.[1]
Given systemically it inhibits blood formation, causing leukopenia, thrombocytopenia, and anemia.[1]
Society and culture
As of March 2018 it was marketed in Japan, China, and South Korea under the brand names Ao Te Min, Arenist, Brecrus, Garesirol, Hustigen, Krix, Lumios, Rizaben, Tramelas, Tranilast and it was marketed as a combination drug with salbutamol under the brand name Shun Qi.[2]
In 2016 the FDA proposed that tranilast be excluded from the list of active pharmaceutical ingredients that compounding pharmacies in the US could formulate with a prescription.[1]
Pharmacology
It appears to work by inhibiting the release of histamine from mast cells; it has been found to inhibit proliferation of fibroblasts but its biological target is not known.[3] It has been shown to inhibit the release of many cytokines in various cell types, in in vitro studies.[3] It has also been shown to inhibit NALP3 inflammasome activation and is being studied as a treatment for NALP3-driven inflammatory diseases.[4]
Chemistry
Tranilast is an analog of a metabolite of tryptophan, and its chemical name is 3′,4′-dimethoxycinnamoyl) anthranilic acid (N-5′).[3]
It is almost insoluble in water, easily soluble in dimethylsulfoxide, soluble in dioxane, and very slightly soluble in ether. It is photochemically unstable in solution.[3]
Orally active anti-allergic agent. Prepn: K. Harita et al., DE 2402398; idem, US 3940422 (1974, 1976 both to Kissei).
Y. Kamijo, M. Kobayashi, and A. Ajisawa, Jpn. Kokai, 77/83,428 (1977) via Chem. Abstr.,
88:6,569f (1978).
Research
After promising results in three small clinical trials, tranilast was studied in a major clinical trial (the PRESTO trial) by SmithKline Beecham in partnership with Kissei for prevention of restenosis after percutaneous transluminal coronary revascularization,[5] but was not found effective for that application.[1][6]
As of 2016, Altacor was developing a formulation of tranilast to prevent of scarring following glaucoma surgery and had obtained an orphan designation from the EMA for this use.[7][8]
History
It was developed by Kissei and first approved in Japan and South Korea for asthma in 1982, and approved uses for keloid and hypertrophic scars were added later in the 1980s.[3]
PATENT
tranilast product case US03940422 , expired in all the regional territories.
PATENT
WO2013144916 claiming tranilast complexes and cocrystals with nicotinamide, saccharin, gentisic acid, salicylic acid, urea, 4-aminobenzoic acid and 2,4-dihydroxybenzoic acid
Patent
WO-2020035546
Nuformix Ltd
Novel crystalline forms of tranilast or its salts as histamine H1 receptor antagonist useful for treating allergy, allergic rhinitis and atopic dermatitis.
Tranilast, (2-[[3-(3,4-dimethoxyphenyl)-l-oxo-2-propenyl]amino] benzoic acid, shown below), was originally developed as an anti-allergy drug due to its ability to inhibit the release of inflammatory mediators, such as histamine, from mast cells and basophils (P. Zampini. IntJ Immunopharmacol. 1983;
Tranilast
Tranilast has been marketed in Japan, China and South Korea by Kissei Pharmaceutical Co. Ltd, for allergic conditions such as allergic conjunctivitis, bronchial asthma, allergic rhinitis and atopic dermatitis, under the Rizaben® brand name for more than thirty years. More recently tranilast has also been shown to have anti-proliferative properties. Tranilast was shown to inhibit the proliferation of fibroblasts and suppress collagen synthesis (M. Isaji. Biochem Pharmacol. 1987; 36: 469-474) and also to inhibit the transformation of fibroblasts to myofibroblasts and their subsequent contraction (M. Isaji. Life Sci. 1994; 55: 287-292). This additional behaviour led to tranilast gaining additional approval for the treatment of keloids and hypertrophic scars.
[004] Over recent years many researchers have explored the anti-proliferative effects of tranilast to assess its potential in fibrotic and cancerous conditions. Its anti-proliferative action is believed to be due to its ability to inhibit transforming growth factor beta (TGF-b) (H. Suzawa. Jpn J Pharmacol. 1992 Oct; 60(2): 91-96). Fibrosis is a condition that can affect most organs of the body and fibroblast proliferation, differentiation and collagen synthesis are known to be key factors in the progression of most types of fibrosis. Tranilast has been shown in-vivo to have potential beneficial effects in
numerous fibrotic conditions. Tranilast has been shown in-vivo to have potential in lung fibrosis (M. Kato. Eur RespirJ. 2013; 42(57): 2330), kidney fibrosis (DJ Kelly, J Am Soc Nephrol. 2004; 15(10): 2619-29), cardiac fibrosis (J Martin, Cardiovasc Res. 2005; 65(3): 694-701), ocular fibrosis (M J Moon, BMC Opthalmol. 2016; 16: 166) and liver fibrosis (M Uno, Hepatology. 2008; 48(1): 109-18.
[005] Tranilast’s anti-tumor action has also recently been demonstrated, in-vitro and in-vivo. Tranilast has been shown to inhibit the proliferation, apoptosis and migration of several cell lines including breast cancer (R. Chakrabarti. Anticancer Drugs. 2009 Jun; 20(5): 334-45) and prostate cancer (S. Sato. Prostate. 2010 Feb; 70(3): 229-38) cell lines. In a study of mammary carcinoma in mice tranilast was found to produce a significant reduction in metastasis (R. Chakrabarti. Anticancer Drugs. 2009 Jun; 20(5): 334-45). In a pilot study in humans, tranilast was shown to have the potential to improve the prognosis of patients with advanced castration-resistant prostate cancer (K. Izumi. Anticancer Research. 2010 Jul; 30: 73077-81). In-vitro studies also showed the therapeutic potential of tranilast in glioma (M Platten. IntJ Cancer. 2001; 93:53-61), pancreatic cancer (M Hiroi, J Nippon Med Sch. 2002; 69: 224-234) and gastric carcinoma (M Yashiro, Anticancer Res. 2003; 23: 3899-3904).
[006] Given the wide range of fibrotic conditions and cancers for which tranilast could have a potential therapeutic benefit, as well as the different patient types and specific areas of the body requiring treatment, it is anticipated that patients would benefit from having multiple delivery methods for the administration of tranilast so as to best suit the patient’s needs. The pharmaceutical compositions could include, for example, a solid oral dosage, a liquid oral dosage, an injectable composition, an inhalable composition, a topical composition or a transdermal composition.
[007] Kissei Pharmaceutical Co. Ltd explored the anti-proliferative effect of tranilast in the prevention of restenosis associated with coronary intervention. In a Phase II clinical study Kissei found that the current approved dose of tranilast (300 mg/day) was insufficient to prevent restenosis and that a higher dose of 600 mg/day was needed to achieve a decrease in restenosis rates (H. Tamai, Am Heart J.1999; 138(5): 968-75). However, it was found that a 600 mg daily dosage can result in a ten-fold inter-patient variation in plasma concentrations of the drug (30-300 pmol/L) (H Kusa ma. Atherosclerosis. 1999; 143: 307-313) and in the Phase III study of tranilast for the prevention of restenosis the dose was further increased to 900mg daily (D Holmes, Circulation. 2002; 106(10): 1243-1250).
[008] The marketed oral form of tranilast (Rizaben®) contains tranilast in its pure crystalline form. Crystalline tranilast has extremely low aqueous solubility (solubility of 14.5 pg/ml in water and 0.7 pg/ml in pH 1.2 buffer solution (Society of Japanese Pharmacopoeia. 2002)). Whilst, high energy amorphous forms are often used as a means of improving the solubility of poorly soluble drug
compounds, literature shows that an amorphous form of tranilast is not completely photostable in the solid state and that it undergoes photodegradation on storage when exposed to light (S. Onoue. EurJ Pharm Sci. 2010; 39: 256-262).
[009] It is expected that the very low solubility of tranilast is a limiting factor in the oral bioavailability of the drug. Given the limited time any drug has to firstly dissolve in the
gastrointestinal tract and then be absorbed into the bloodstream, this issue will become even more limiting as the oral dose of tranilast is increased. The poor solubility of tranilast is also possibly a key factor in the high inter-patient variability reported for higher dose tranilast pharmacokinetics. As a BCS class II drug (low solubility/high permeability) it is expected that absorption from the gastrointestinal tract is hampered by the dissolution rate of the drug in gastrointestinal media as well as its overall solubility. For treatment of chronic proliferative diseases such as fibrosis and cancer it is vital for the delivery method of a drug to produce consistent, predictable plasma levels that are maintained above the minimum effective concentration. To achieve efficacious oral delivery of tranilast at higher doses there is a need for new solid forms of the drug with both high solubility and rapid dissolution rates.
[010] Given the severity of conditions involving cancer or fibrosis there is also a need for systemic treatment options by which tranilast can be delivered by healthcare specialists that do not require the patient to swallow solid oral dosage forms. Alternative dosage forms suitable for these needs could include, for example, injectable compositions, liquid oral formulations or nebulized inhaled formulations. These would require a liquid formulation of tranilast suitable for systemic delivery. [Oil] Given the potential of tranilast to treat ocular diseases, such as allergic conjunctivitis, Kissei Pharmaceutical Co. Ltd recognised the need to develop an eye drop formulation of tranilast for localised treatment. However, as well as having very low aqueous solubility, tranilast is also photochemically unstable when stored in solution, resulting in significant degradation (N Hori, Chem. Pharm. Bull. 1999; 47(12): 1713-1716). Therefore, the only way Kissei were able to achieve an eye drop liquid composition of tranilast was to use both solubilising and stabilising agents in the formulation (US Patent 5356620). The resulting 0.5% (w/v) eye drop formulation is currently also marketed under the Rizaben® brand name. However, the focus of this formulation and of the subsequent research that has attempted to produce alternative solution formulations of tranilast has always been solely on external delivery of tranilast using compositions such as eye drops and skin ointments etc. None of the liquid formulations of tranilast previously described have been produced for systemic delivery such as for oral or IV delivery. Excipients used in the previously reported external preparations are not suitable for systemic delivery. Also, despite the successful
development of an eye drop formulation of tranilast, the package insert of the marketed Rizaben® eye drops states that the product should not be stored in a refrigerator as crystals may precipitate.
[012] Thus, there remains a need for aqueous pharmaceutical compositions of tranilast suitable for systemic delivery. Given the potential photochemical degradation issue of long term storage of tranilast in solution and also the disadvantage of the larger storage facilities needed to store bulkier solution based formulations it would also be advantageous to develop a stable highly soluble solid form of tranilast that can be quickly dissolved at the time of treatment by the patient or healthcare provider to produce the required liquid formulation.
[013] Following efforts to make a liquid formulation of tranilast, Kissei made the statement that tranilast and pharmaceutically acceptable salts thereof are too insoluble in water to prepare an aqueous solution (US Patent 5356620). Since that US patent the only crystalline pharmaceutically acceptable salt to have been published is the sodium salt (N Geng, Cryst. Growth Des. 2013; 13: 3546-3553). In line with the findings of Kissei the authors of this paper stated that the apparent solubility of the crystalline tranilast sodium salt is even less than that of pure tranilast. Also, when they performed a dissolution study of tranilast in a sodium containing media they found that as the tranilast dissolved it gradually precipitated out of solution as its sodium salt indicating that the sodium salt has a lower thermodynamic solubility than the pure drug. The authors of this paper also successfully prepared the non-pharmaceutically acceptable crystalline cytosine salt of tranilast. Despite this crystalline cytosine salt showing approximately a two-fold solubility improvement over pure crystalline tranilast, not only would this crystalline cytosine salt not be suitable for systemic delivery to a patient due to cytosine not having FDA acceptability but this improvement in solubility would not be great enough to produce high dose tranilast liquid formulations such as an injectable formulation.
[014] Patent application EP1946753 discloses an attempt to prepare an external preparation of tranilast and claims the preparation of ionic liquid salts of tranilast with organic amines. The inventors claim that blending tranilast with the organic amine results in a liquid form. This application does not disclose the formation of any solid state, crystalline tranilast salts with organic amines. They demonstrate that these ionic liquid forms of tranilast have higher solubility in solvents suitable for external application to the skin and that these preparations have higher photostability than pure tranilast in the same formulation. However, this improved photostability still results in a significant proportion of the tranilast being photo-degraded and would not be suitable for long term storage. Also, the solvents used for preparation of these ionic liquid salt formulations are not suitable for internal delivery of tranilast. Moreover, there is no mention in EP1946753 of improved solubility in aqueous or bio-relevant media.
PATENT
https://patents.google.com/patent/US20150119428
-
Tranilast, (2-[[3-(3,4-dimethoxyphenyl)-1-oxo-2-propenyl]amino]benzoic acid), shown below, is a therapeutic agent that exhibits an anti-allergic effect. It has been shown to inhibit the release of inflammatory mediators, such as histamine, from mast cells and basophils (P. Zampini. Int J Immunopharmacol. 1983; 5(5): 431-5). Tranilast has been used as an anti-allergic treatment, for several years in Japan and South Korea, for conditions such as allergic conjunctivitis, bronchial asthma, allergic rhinitis and atopic dermatitis.
-

- [0004]
Tranilast is currently marketed in Japan and South Korea by Kissei Pharmaceutical Co. Ltd under the Rizaben® brand name. As well as displaying an anti-allergic effect tranilast has been shown to possess anti-proliferative properties. Tranilast was found to inhibit the proliferation of fibroblasts and suppress collagen synthesis (M. Isaji. Biochem Pharmacol. 1987; 36: 469-474) and also to inhibit the transformation of fibroblasts to myofibroblasts and their subsequent contraction (M. Isaji. Life Sci. 1994; 55: 287-292). On the basis of these effects tranilast is now also indicated for the treatment of keloids and hypertrophic scars. Its anti-fibrotic action is believed to be due to its ability to inhibit transforming growth factor beta (TGF-β) (H. Suzawa. Jpn J Pharmacol. 1992 October; 60(2): 91-96). TGF-β induced fibroblast proliferation, differentiation and collagen synthesis are known to be key factors in the progression of idiopathic pulmonary fibrosis and tranilast has been shown in-viva to have potential in the treatment of this chronic lung disease (T. Jiang. Afr J Pharm Pharmaco. 2011; 5(10): 1315-1320). Tranilast has also been shown in-vivo to be have potential beneficial effects in the treatment of airway remodelling associated with chronic asthma (S. C. Kim. J Asthma 2009; 46(9): 884-894.
- [0005]
It has been reported that tranilast also has activity as an angiogenesis inhibitor (M. Isaji. Br. J Pharmacol. 1997; 122(6): 1061-1066). The results of this study suggested that tranilast may be beneficial for the treatment of angiogenic diseases such as diabetic retinopathy and age related macular degeneration. As well as showing inhibitory effects on mast cells and fibroblasts, tranilast has also demonstrated an ability to diminish tumor necrosis factor-alpha (TNF-α) from cultured macrophages (H. O. Pae. Biochem Biophys Res Commun. 371: 361-365) and T-cells (M. Platten. Science. 310: 850-855), and inhibited NF-kB-dependent transcriptional activation in endothelial cells (M. Spieker. Mol Pharmacol. 62: 856-863). Recent studies have revealed that tranilast attenuates inflammation and inhibits bone destruction in collagen induced arthritis in mice suggesting the possible usefulness of tranilast in the treatment of inflammatory conditions such as arthritis (N. Shiota. Br. Pharmacol. 2010; 159 (3): 626-635).
- [0006]
As has recently been demonstrated, in-vitro and in-vivo, tranilast also possesses an anti-tumor action. Tranilast has been shown to inhibit the proliferation, apoptosis and migration of several cell lines including breast cancer (R. Chakrabarti. Anticancer Drugs. 2009 June; 20(5): 334-45) and prostate cancer (S. Sato. Prostate. 2010 February; 70(3): 229-38) cell lines. In a study of mammary carcinoma in mice tranilast was found to produce a significant reduction in metastasis (R. Chakrabarti. Anticancer Drugs. 2009 June; 20(5): 334-45). In a pilot study in humans, tranilast was shown to have the potential to improve the prognosis of patients with advanced castration-resistant prostate cancer (K. Izurni. Anticancer Research. 2010 July; 30: 73077-81).
- [0007]
It has been reported that tranilast has the ability to induce or enhance neurogenesis and, therefore, could be used as an agent to treat neuronal conditions such as cerebral ischernia, glaucoma, multiple sclerosis, amyotrophic lateral sclerosis, Alzheimer’s disease, neurodegenerative trinucleotide repeat disorders, neurodegenerative lyosomal storage diseases, spinal cord injury and trauma, dementia, schizophrenia and peripheral neuropathy (A. Schneider. EP2030617).
- [0008]
Tranilast’s beneficial properties have been reported to have utility in several ocular conditions. Tranilast is currently approved in Japan and Korea far the treatment of allergic conjunctivitis. WO2010137681 claims the use of tranilast as a prophylactic or therapeutic agent for the treatment of retinal diseases. The anti-fibrotic properties of tranilast have been reported to be of benefit in maintaining the filtering blob during glaucoma surgery and this has been demonstrated in a pilot study in humans (E. Chihara.J Glaucoma. 1999; 11(2): 127-133). There have also been several reported cases of the beneficial use of tranilast in the prevention of postoperative recurrence of pterygium (C. Fukui. Jap J Opthalmol. 1999; 12: 547-549). Tsuji recently reported that tranilast may be beneficial not only in the prevention of ptergium recurrence, but also for the inhibition of symblepharon and granuloma formation (A. Tsuji. Tokai J Exp Clin Med. 2011; 36(4): 120-123). Collectively it has been demonstrated that tranilast possesses anti-allergic, anti-fibrotic, anti-inflammatory, anti-tumor, neurogenesis enhancing end angiogenesis inhibitory properties and as such may be useful for the treatment of diseases associated with such properties.
- [0009]
Tranilast occurs as a yellow crystalline powder that is identified by CAS Registry Number: 53902-12-8. As is typical of cinnamic acid derivatives (G. M. J. Schmidt J Chem. Soc. 1964: 2000) tranilast is photochemically unstable when in solution, tranforming into cis-isomer and dimer forms on exposure to light (N. Hori. Cehm Pharm Bull. 1999; 47: 1713-1716). Although pure crystalline tranilast is photochemically stable in the solid state it is practically insoluble in water (14.5 μg/ml) and acidic media (0.7 μg/ml in pH 1.2 buffer solution) (Society of Japanese Pharmacopoeia. 2002). Although tranilast has shown activity in various indications, it is possible that the therapeutic potential of the drug is currently limited by its poor solubility and photostability. High energy amorphous forms are often used as a means of improving the solubility of poorly soluble APIs, however, literature shows that amorphous solid dispersions of tranilast are not completely photostable in the solid state and that they undergo photodegradation on storage when exposed to light (S. Onoue. Eur J Pharm Sci. 2010; 39: 256-262). US20110136835 describes a combination of tranilast and allopurinol and its use in the treatment of hyperuricemia associated with gout and has one mention of a “co-crystal form”, but lacks any further description or characterization.
Patent
References
- ^ Jump up to:a b c d e f g h “FDA Proposed Rules” (PDF). Federal Register. 81 (242): 91071–91082. December 16, 2016. Another version of same published at here
- ^ “International brands for Tranilast”. Drugs.com. Retrieved 10 March 2018.
- ^ Jump up to:a b c d e Darakhshan, S; Pour, AB (January 2015). “Tranilast: a review of its therapeutic applications”. Pharmacological Research. 91: 15–28. doi:10.1016/j.phrs.2014.10.009. PMID 25447595.
- ^ Y. Huang et al, “Tranilast directly targets NLRP3 to treat inflammasome-driven diseases.”, EMBO Mol Med., 10(4), 2018
- ^ “Kissei’s existing business flat but R&D pipeline should lead to growth”. The Pharma Letter. 8 September 2000.
- ^ Holmes, D. R; Savage, M; Lablanche, J. M; Grip, L; Serruys, P. W; Fitzgerald, P; Fischman, D; Goldberg, S; Brinker, J. A; Zeiher, A. M; Shapiro, L. M; Willerson, J; Davis, B. R; Ferguson, J. J; Popma, J; King Sb, 3rd; Lincoff, A. M; Tcheng, J. E; Chan, R; Granett, J. R; Poland, M (2002). “Results of Prevention of REStenosis with Tranilast and its Outcomes (PRESTO) Trial”. Circulation. 106 (10): 1243–50. doi:10.1161/01.CIR.0000028335.31300.DA. PMID 12208800.
- ^ “Tranilast – Altacor: ALT-401”. AdisInsight. Retrieved 10 March 2018.
- ^ “EU/3/10/756 Orphan Designation”. European Medicines Agency. 6 August 2010. Retrieved 10 March 2018.
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.150.125 |
| Chemical and physical data | |
| Formula | C18H17NO5 |
| Molar mass | 327.336 g·mol−1 |
| 3D model (JSmol) | |
///////////////Tranilast, Rizaben, antiallergic, Kissei Pharmaceuticals, Japan, South Korea, bronchial asthma, keloid, hypertrophic scar
Ibudilast

IBUDILAST, MN 166
AV-411
KC-404
MN-166
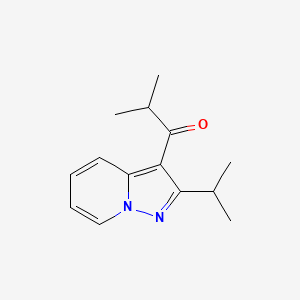
2-methyl-1-(2-propan-2-ylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one
1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)-2-methylpropan-1-one
KYORIN Kyorin Seiyaku Kk……….INNOVATOR

Ibudilast is an anti-inflammatory and neuroprotective oral agent which shows an excellent safety profile at 60 mg/day and provides significantly prolonged time-to-first relapse and attenuated brain volume shrinkage in patients with relapsing-remitting (RR) and/or secondary progressive (SP) multiple sclerosis (MS). Ibudilast is currently in development in the U.S. (codes: AV-411 or MN-166), but is approved for use as an antiinflammatory in Japan.



Ibudilast (development codes: AV-411 or MN-166) is an antiinflammatory drug used mainly in Japan, which acts as aphosphodiesterase inhibitor, inhibiting the PDE-4 subtype to the greatest extent,[1] but also showing significant inhibition of other PDE subtypes.[2][3]
Ibudilast has bronchodilator, vasodilator [4] and neuroprotective effects,[5][6] and is mainly used in the treatment of asthma andstroke.[7] It inhibits platelet aggregation,[8] and may also be useful in the treatment of multiple sclerosis.[9]
Ibudilast crosses the blood–brain barrier and suppresses glial cell activation. This activity has been shown to make ibudilast useful in the treatment of neuropathic pain and it not only enhances analgesia produced by opioid drugs, but also reduces the development oftolerance.[10]
It may have some use reducing methamphetamine[11] and alcohol[12] addiction.
It may have some use reducing methamphetamine addiction.[11]
Avigen has identified the potential of ibudilast (AV-411) for the treatment of neuropathic pain and other neurological indications, including opiate withdrawal. As an inhibitor of glial cells, ibudilast can deactivate these cells which produce various chemicals, including proinflammatory cytokines, in response to nerve damage or viral infection to amplify and maintain pain. Preclinical evaluation to date indicates that it reverses the painful sensory abnormality allodynia in chemotherapy- and trauma-induced neuropathic pain models.
Originator Kyorin and Banyu Pharmaceutical (now MSD KK following the merger of Banyu and Schering-Plough KK in 2010) have been developing ibudilast under a collaborative agreement. MediciNova obtained exclusive, worldwide rights outside of Japan, China, Taiwan and South Korea from Kyorin in October 2004 to develop and commercialize the compound for MS. In 2012, a codevelopment agreement was signed between MediciNova and the University of Colorado for the treatment of post-traumatic brain injury.

Sixteenth revised Japanese Pharmacopoeia chemicals, etc. IBUDILAST Ibudilast C14H18N2O: 230.31 [ 50847-11-5 ] that this product was dried when to quantify, including ibudilast (C14H18N2O) 98.5 ~ 101.0%.


http://www.google.co.in/patents/US3850941PATENT
EXAMPLE 1 Synthesis of 2-isopropyl-3-is0butyrylpyrazolo[1,5-a] pyridine (KC404) A mixture of 1-amino-Z-methylpyridinium iodide g.), isobutyric anhydride (500 g.) and K CO (81 g.) was refluxed for 8 hr. After cooling, the precipitated crystals were filtered off and water was added to the filtrate, The solution was made basic to pH 11 with K CO’ and extracted with ethyl acetate (1000 ml.). The extract’was washed with water (400 ml.), dried over Na SO and concentrated under reduced pressure. The residue was distilled to give 58 g. of colorless crystalline product, hp, 110- 175 (7.5 mm. Hg). Recrystallization from hexane gave colorless prisms, melting point 53.554.
Analysis- Calcd.: C, 73.01; H, 7.88; N, 12.17 Found: C, 72.86; H, 7.94; N, 12.09
CLIP

http://www.customsynthesis.com/ibud.html
PATENT
http://www.google.com/patents/US8119657
FIG. 6 is a synthetic reaction scheme illustrating one approach for preparing (S)-AV1013; the approach employs chiral chromatography of an N-protected form of the racemate as described in detail in Example 1.
FIG. 7 demonstrates additional reaction schemes for synthesizing (S)-AV1013.


Example 1Synthesis of (S)-2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one hydrochloride
(S)-2-Amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one hydrochloride (also referred to herein as S-AV1013.HCl) was prepared on a preparative scale using two different routes to obtain the intermediate isopropylpyrazolo[1,5-a]pyridine (IPPP). In the first approach (method 1), ibudilast was employed as the starting material to obtain IPPP; an alternate synthetic approach (method 2) employed ibudilast acid as the starting material.
Step 1Method 1Preparation of Isopropylpyrazolo[1,5-a]pyridine (IPPP) from ibudilast

A 5 L 3-neck round-bottom flask was equipped with a mechanical stirrer, thermocouple, heating mantle and a Y-adapter with a nitrogen inlet. The flask was charged with water (350 mL, USP), concentrated sulfuric acid (350 mL) and ibudilast (3-isobutyryl-2-isopropylpyrazolo[1,5-a]pyridine) (140 g, 0.608 mol). The flask was purged with nitrogen, and the mixture was stirred while it was heated to 135° C. An aliquot was removed for HPLC analysis, which showed that all starting material was consumed after 5 hours at 135° C., so the mixture was allowed to cool to room temperature overnight. The mixture was cooled in an ice bath, and water (1400 mL, USP) was added over 10 min, with the temperature maintained below 25° C. With continuous cooling in an ice bath, the mixture was neutralized by adding sodium hydroxide (50% w/w aq., 1150 mL) dropwise, with the temperature maintained below 25° C. Ethyl acetate (250 mL) was added, and the layers were separated. The aqueous layer was washed with ethyl acetate (2×300 mL). The combined ethyl acetate extracts were washed sequentially with 250 mL portions of saturated aqueous sodium bicarbonate and saturated aqueous sodium chloride, then dried over anhydrous sodium sulfate for 30 minutes. Activated carbon (20 g) and silica (60 g) were added and stirred before filtering over a pad of Celite. The filtrate was concentrated under reduced pressure to obtain 96.5 g of IPPP (2-isopropyl-pyrazolo[1,5-a]pyridine, 99% crude yield, 99.6 area % pure by HPLC) as an amber oil.
1H-NMR (CDCl3) δ 1.4 (d, 6H), 3.2 (m, 1H), 6.3 (s, 1H), 6.6 (t, 1H), 7.0 (m, 1H), 7.4 (d, 1H), 8.4 (d, 1H). HPLC: RT=9.1 min (99.6 area %).
CLIP
Ibudilast (3-isobutyryl-2-isopropylpyrazolo[l,5-α]pyridine) is a small molecule drug that has been used for many years in Japan and Korea for the treatment of bronchial asthma as well as for treatment of cerebrovascular disorders such as post-stroke dizziness. It is sold in these countries under the tradename, Ketas®. Marketed indications for ibudilast in Japan include its use as a vasodilator, for treating allergy, eye tissue regeneration, ocular disease, and treatment of allergic ophthalmic disease (Thompson Current Drug Reports). Its use in the treatment of both chronic brain infarction (ClinicalTrials.gov) and multiple sclerosis (News.Medical.Net; Pharmaceutical News, 2 Aug 2005) is currently being explored in separate, ongoing clinical trials.
The mechanisms of action of ibudilast have been widely explored. Its role as a non-selective inhibitor of cyclic nucleotide phosphodiesterase (PDE) has been described
(Fujimoto, T., et al., J. of Neuroimmunology, 95 (1999) 35-92). Additionally, ibudilast has been reported to act as an LTD4 antagonist, an anti-inflammatory, a PAF antagonist, and a vasodilatator agent (Thompson Current Drug Reports). Ibudilast is also thought to exert a neuroprotective role in the central nervous system of mammals, presumably via suppression of the activation of glial cells (Mizuno et al. (2004) Neuropharmacology 46: 404-411). New uses for ibudilast continue to be explored.http://www.google.com/patents/WO2007146087A2?cl=en
PATENT
http://www.google.com/patents/WO2007142924A1?cl=en
IBUDILAST
Ibudilast is a small molecule drug (molecular weight of 230.3) having the structure shown below.

Ibudilast is also found under ChemBank ID 3227, CAS # 50847-1 1-5, and Beilstein Handbook Reference No. 5-24-03-00396. Its molecular formula corresponds to [Ci4HIgN2O]. Ibudilast is also known by various chemical names which include 2- methyl-l-(2-(l-methylethyI)pyrazolo(l,5-a)pyridin-3-yl)l-propanone; 3-isobutyryl-2- isopropylpyrazolo(l,5-a)pyridine]; and l-(2-isopropyl-pyrazolo[l,5-a]pyridin-3-yl)-2- methyl-propan-1-one. Other synonyms for ibudilast include Ibudilastum (Latin), BRN 0656579, KC-404, and the brand name Ketas®. Ibudilast, as referred to herein, is meant to include any and all pharmaceutically acceptable salt forms thereof, prodrug forms (e.g., the corresponding ketal), and the like, as appropriate for use in its intended formulation for administration.
Ibudilast is a non-selective nucleotide phosphodiesterase (PDE) inhibitor (most active against PDE-3 and PDE-4), and has also been reported to have LTD4 and PAF antagonistic activities. Its profile appears effectively anti-inflammatory and unique in comparison to other PDE inhibitors and anti-inflammatory agents. PDEs catalyze the hydrolysis of the phosphoester bond on the 3 ‘-carbon to yield the corresponding 5′- nucleotide monophosphate. Thus, they regulate the cellular concentrations of cyclic nucleotides. Since extracellular receptors for many hormones and neurotransmitters utilize cyclic nucleotides as second messengers, the PDEs also regulate cellular responses to these extracellular signals. There are at least eight classes of PDEs: Ca2+/calmodul in-dependent PDEs (PDEl); cGMP-stimulated PDEs (PDE2); cGMP- inhibited PDEs (PDE3); cAMP-specific PDEs (PDE4); cGMP-binding PDEs (PDE5); photoreceptor PDEs (PDE6); high affinity, cAMP-specific PDEs (PDE7); and high affinity cGMP-specific PDEs (PDE9).
SYNTHESIS

DE 2315801; FR 2182914; JP 7714799, WO 0196278
By condensation of 1-amino-2-methylpyridinium iodide (I) with isobutyric anhydride (II) by means of K2CO3 at reflux temperature.
Patent
PATENT
2-methyl -l- [2- (l- methylethyl) – pyrazolo [l, 5_a] pyrimidine _3_ yl] _1_ acetone (ibudilast, generic drug name: IBUDILAST ) is an anti-allergic asthma drugs, anti-leukotrienes can twist and platelet-activating factor, promote the secretion of mucus in the respiratory tract, respiratory cilia function, enhance the role of prostacyclin, increase cerebral blood flow, improve brain metabolism. For the treatment of bronchial asthma, sequelae of cerebral embolism, cerebral arteriosclerosis.
ibudilast preparation methods are mainly the following two:
Method a: (The Jourrtal of Organic Chemistry, 1968, 33, 3766 ~3770) Synthesis Road
Lines are as follows:

The route to 2-picoline as starting material to give amino-2-methyl-pyridine iodide I-, after pyrimidine, the role of isobutyryl chloride to give the title compound. The final product obtained by this route need be purified by column chromatography, thereby increasing the difficulty of the operation, in addition to column chromatography, eluent used larger benzene toxicity, is not suitable for industrial production.
Method II: (Journal of the American Chemical Society, 2005,127, 751-760) co
A route is as follows:

The route to 2-picoline as starting material to obtain the sulfamic acid, potassium iodide I- amino-2-picoline under the action of potassium carbonate, then with isobutyric anhydride to give the title compound effect. This route of the first-stage reaction process locked, the yield is low, is not suitable for industrial production.
so there ibudilast conventional method for preparing the operational difficulties or low yield, making it impossible to achieve industrial production problems.
DETAILED DESCRIPTION IX: with a specific embodiment of the present embodiment is one of one to eight different points: in the second step of the recrystallization specific operation is as follows: First, the collected fractions was cooled to 10 ° c~25 ° C, to give a pale yellow solid, and then n-hexane was added to the pale yellow solid, and the temperature was raised to 50 ° C~68 ° C, at a temperature of 50 ° C~68 ° C incubation 5min~IOmin, then cooled to 10 ° C~ 25 ° C, and at a temperature of 10 ° C~25 ° C incubated O. 5h~Ih, and finally filtered to obtain ibudilast; the volume of the pale yellow solid quality and hexane ratio of Ig: (ImL~2mL), to obtain ibudilast.
PAPER
Bioorganic & medicinal chemistry letters (2011), 21(11), 3307-12.
Ibudilast [1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)-2-methylpropan-1-one] is a nonselective phosphodiesterase inhibitor used clinically to treat asthma. Efforts to selectively develop the PDE3- and PDE4-inhibitory activity of ibudilast led to replacement of the isopropyl ketone by a pyridazinone heterocycle. Structure–activity relationship exploration in the resulting 6-(pyrazolo[1,5-a]pyridin-3-yl)pyridazin-3(2H)-ones revealed that the pyridazinone lactam functionality is a critical determinant for PDE3-inhibitory activity, with the nitrogen preferably unsubstituted. PDE4 inhibition is strongly promoted by introduction of a hydrophobic substituent at the pyridazinone N(2) centre and a methoxy group at C-7′ in the pyrazolopyridine. Migration of the pyridazinone ring connection from the pyrazolopyridine 3′-centre to C-4′ strongly enhances PDE4 inhibition. These studies establish a basis for development of potent PDE4-selective and dual PDE3/4-selective inhibitors derived from ibudilast.

UPDATE AS ON JAN 2016
…………..MediciNova’s ibudilast gets FDA rare paediatric disease status to treat Krabbe disease
MediciNova has received rare paediatric disease status from the US Food and Drug Administration (FDA) for its MN-166 (ibudilast) to treat Type 1 Early Infantile Krabbe disease.
![]()
References
- Huang Z, Liu S, Zhang L, Salem M, Greig GM, Chan CC, Natsumeda Y, Noguchi K. Preferential inhibition of human phosphodiesterase 4 by ibudilast. Life Sciences. 2006 May 1;78(23):2663-8.
- Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Research. 1999 Aug 7;837(1-2):203-12.
- Gibson LC, Hastings SF, McPhee I, Clayton RA, Darroch CE, Mackenzie A, Mackenzie FL, Nagasawa M, Stevens PA, Mackenzie SJ. The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. European Journal of Pharmacology. 2006 May 24;538(1-3):39-42.
- Kishi Y, Ohta S, Kasuya N, Sakita S, Ashikaga T, Isobe M. Ibudilast: a non-selective PDE inhibitor with multiple actions on blood cells and the vascular wall. Cardiovascular Drug Reviews. 2001 Fall;19(3):215-25.
- Mizuno T, Kurotani T, Komatsu Y, Kawanokuchi J, Kato H, Mitsuma N, Suzumura A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004 Mar;46(3):404-11.
- Yoshioka M, Suda N, Mori K, Ueno K, Itoh Y, Togashi H, Matsumoto M. Effects of ibudilast on hippocampal long-term potentiation and passive avoidance responses in rats with transient cerebral ischemia. Pharmacological Research. 2002 Apr;45(4):305-11.
- Wakita H, Tomimoto H, Akiguchi I, Lin JX, Ihara M, Ohtani R, Shibata M. Ibudilast, a phosphodiesterase inhibitor, protects against white matter damage under chronic cerebral hypoperfusion in the rat. Brain Research. 2003 Nov 28;992(1):53-9.
- Rile G, Yatomi Y, Qi R, Satoh K, Ozaki Y. Potentiation of ibudilast inhibition of platelet aggregation in the presence of endothelial cells. Thrombosis Research. 2001 May 1;102(3):239-46.
- Feng J, Misu T, Fujihara K, Sakoda S, Nakatsuji Y, Fukaura H, Kikuchi S, Tashiro K, Suzumura A, Ishii N, Sugamura K, Nakashima I, Itoyama Y. Ibudilast, a nonselective phosphodiesterase inhibitor, regulates Th1/Th2 balance and NKT cell subset in multiple sclerosis. Multiple Sclerosis. 2004 Oct;10(5):494-8.
- Ledeboer A, Hutchinson MR, Watkins LR, Johnson KW. Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opinion on Investigational Drugs. 2007 Jul;16(7):935-50.
- http://www.huffingtonpost.com/2013/04/03/meth-addiction-cure-ucla-ibudilast_n_2863126.html?utm_hp_ref=mostpopular#slide=more268305
- http://onlinelibrary.wiley.com/doi/10.1111/adb.12106/abstrac
SEE……Synthesis technology of ibudilast
Shandong Huagong (2014), 43, (8), 29-30. Publisher: (Shandong Huagong Bianjibu, ) CODEN:SHHUA4 ISSN:1008-021X.
Literature References:
Leukotriene D4 antagonist. Prepn: T. Irikura et al., DE 2315801; eidem, US 3850941 (1973, 1974 both to Kyorin).
Pharmacology and antiallergic activity: K. Nishino et al., Jpn. J. Pharmacol. 33, 267 (1983); H. Nagai et al., ibid. 1215.
In vitro cerebral vasodilating activity: M. Ohashi et al., Arch. Int. Pharmacodyn. 280, 216 (1986);
in vivo activity: W. M. Armstead et al., J. Pharmacol. Exp. Ther. 244, 138 (1988).
Bronchodilating activity in animals: S. Mue et al., Arch. Int. Pharmacodyn. 283,153 (1986).
Antiplatelet activity in animals: M. Ohashi et al., ibid. 321; M. Ohashi et al., Gen. Pharmacol. 17, 385 (1986).

| Patent | Submitted | Granted |
|---|---|---|
| Method for treating neuropathic pain and associated syndromes [US7534806] | 2006-07-20 | 2009-05-19 |
| Synergistic combination [US2006205806] | 2006-09-14 | |
| Pharmaceutical composition of a pde4 or pde 3/4 inhibitor and histamine receptor antagonist [US2005112069] | 2005-05-26 | |
| Methods and reagents for the treatment of immunoinflammatory disorders [US2005119160] | 2005-06-02 | |
| Methods of inducing ovulation using a non-polypeptide camp level modulator [US7507707] | 2005-07-07 | 2009-03-24 |
| Methods and reagents for the treatment of immunoinflammatory disorders [US2005192261] | 2005-09-01 | |
| Synergistic combination [US7056936] | 2004-02-19 | 2006-06-06 |
| Methods for the treatment of respiratory diseases and conditions with a selective iNOS inhibitor and a PDE inhibitor and compositions therefor [US2004087653] | 2004-05-06 | |
| Remedies for multiple sclerosis [US6395747] | 2002-05-28 | |
| Combination [US2005014762] | 2005-01-20 |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4097483 | Aug 31, 1976 | Jun 27, 1978 | Kyorin Pharmaceutical Co., Ltd. | Pyrazolo 1,5-a!pyridines |
| US7585875 * | Jun 6, 2007 | Sep 8, 2009 | Avigen, Inc. | Phosphodiesterase inhibitors; neuropathic pain, inflammation, opioid dependence or withdrawal; 2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one for example |
| US20070015924 | Jun 15, 2006 | Jan 18, 2007 | Cardiome Pharma Corp. | stereoselective preparation of aminocyclohexyl ether compounds such as trans-(1R,2R)-aminocyclohexyl ether compounds and/or trans-(1S,2S)-aminocyclohexyl ether compounds; useful in treating arrhythmias |
| US20080070912 | Jun 6, 2007 | Mar 20, 2008 | Avigen, Inc. | Phosphodiesterase inhibitors; neuropathic pain, inflammation, opioid dependence or withdrawal; 2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one for example |
| US20090062330 | Jul 8, 2008 | Mar 5, 2009 | Medicinova, Inc. | Treatment of progressive neurodegenerative disease with ibudilast |
| US20090318437 * | Jun 10, 2009 | Dec 24, 2009 | Gaeta Federico C A | SUBSTITUTED PYRAZOLO[1,5-a] PYRIDINE COMPOUNDS AND THEIR METHODS OF USE |
| WO2007142924A1 | May 29, 2007 | Dec 13, 2007 | Avigen Inc | Ibudilast for inhibiting macrophage migration inhibitory factor (mif) activity |
| WO2007146087A2 * | Jun 6, 2007 | Dec 21, 2007 | Avigen Inc | SUBSTITUTED PYRAZOLO [1,5-α] PYRIDINE COMPOUNDS AND THEIR METHODS OF USE |
| WO2010151551A1 * | Jun 22, 2010 | Dec 29, 2010 | Medicinova, Inc. | ENANTIOMERIC COMPOSITIONS OF 2-AMINO-1-(2-ISOPROPYLPYRAZOLO[1,5-a]PYRIDIN-3-YL)PROPAN-1-ONE AND RELATED METHODS |
| US8119657 | Jun 22, 2010 | Feb 21, 2012 | Medicinova, Inc. | Enantiomeric compositions of 2-amino-1-(2-isopropylpyrazolo[1,5-α]pyridin-3-yl)propan-1-one and related methods |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003104178A1 * | Jun 6, 2003 | Dec 18, 2003 | Cortical Pty Ltd | Napththalene derivatives which inhibit the cytokine or biological activity of macrophage migration inhibitory factor (mif) |
| WO2003104203A1 * | Jun 6, 2003 | Dec 18, 2003 | Cortical Pty Ltd | Therapeutic molecules and methods-1 |
| WO2004058713A1 * | Dec 18, 2003 | Jul 15, 2004 | Jason Chyba | Differential tumor cytotoxocity compounds and compositions |
| WO2005058304A1 * | Dec 17, 2004 | Jun 30, 2005 | Cortical Pty Ltd | Implantable device containing inhibitor of macrophage migration inhibitory factor |
| WO2006045505A1 * | Oct 19, 2005 | May 4, 2006 | Novartis Ag | Mif-inhibitors |
| WO2006108671A1 * | Apr 13, 2006 | Oct 19, 2006 | Novartis Ag | 3,4-dihydro-benzo[e][1,3]oxazin-2-ones |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-methyl-1-(2-propan-2-ylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one
|
|
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Identifiers | |
| CAS Number | 50847-11-5 |
| ATC code | R03DC04 |
| PubChem | CID: 3671 |
| IUPHAR/BPS | 7399 |
| DrugBank | DB05266 |
| ChemSpider | 3543 |
| UNII | M0TTH61XC5 |
| KEGG | D01385 |
| ChEMBL | CHEMBL19449 |
| Chemical data | |
| Formula | C14H18N2O |
| Molecular mass | 230.31 g/mol |
Keywords: Antiallergic; Antiasthmatic (Nonbronchodilator); Leukotriene Antagonist; Vasodilator (Cerebral).

SEE………http://apisynthesisint.blogspot.in/2016/01/ibudilast.html
////////////////////
CC(C)C1=NN2C=CC=CC2=C1C(=O)C(C)C
