
Home » Posts tagged 'Anthony crasto' (Page 18)
Tag Archives: Anthony crasto
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
OCRELIZUMAB

Ocrelizumab is a humanized anti-CD20 monoclonal antibody. It targets mature B lymphocytes[1] and hence is an immunosuppressive drug candidate. It is under development by Hoffmann–La Roche‘s subsidiary Genentech, and Biogen Idec.
It had reached Phase III clinical trials for rheumatoid arthritis[2] and lupus erythematosus,[3]and Phase II for multiple sclerosis[4] and hematological cancer.[5]
In March 2010, Roche announced the suspension of clinical trials in rheumatoid arthritis and lupus erythematosus. This step followed excess deaths due to opportunistic infections. Development for multiple sclerosis continues.[6]
In October 2010 Roche announced 24 week results from the PhII study in relapse remittingMS. The drug demonstrated a statistically significant reduction in disease activity as measured by brain lesions (measured by MRI scans) and relapse rate compared to placebo. Both doses (200 mg & 600 mg) were well tolerated.Anti-B cell therapy with rituximab has been shown to be safe and beneficial for RA treatment. Rituximab is approved and marketed for the treatment of RA in patients who have failed other therapies. Ocrelizumab, a fully human monoclonal antibody against CD20, may have less immunogenicity and less complement activation than rituximab which, theoretically, may reduce the development of drug neutralizing antibodies and infusion reactions. Here, Genovese at al report the results of a Phase I/II dose finding study of Ocrelizumab in RA patients who have failed other DMARDs (including prior TNF inhibitors).
- K. John Morrow Jr (2008-06-15). “Methods for Maximizing Antibody Yields”. Genetic Engineering & Biotechnology News (Mary Ann Liebert, Inc.). p. 36. Retrieved 2008-07-06. (Note: information included in this article only found in table present in print version of article.)
- Kausar, F; Mustafa, K; Sweis, G; Sawaqed, R; Alawneh, K; Salloum, R; Badaracco, M; Niewold, TB et al. (2009). “Ocrelizumab: a step forward in the evolution of B-cell therapy”. Expert opinion on biological therapy 9 (7): 889–95. doi:10.1517/14712590903018837.PMID 19463076.
- http://www.clinicaltrials.gov/ct2/show/NCT00539838 A Study to Evaluate Two Doses of Ocrelizumab in Patients With Active Systemic Lupus Erythematosus (BEGIN)
- http://www.clinicaltrials.gov/ct2/show/NCT00676715 A Study of the Efficacy and Safety of Ocrelizumab in Patients With Relapsing-Remitting Multiple Sclerosis.
- Hutas, G (2008). “Ocrelizumab, a humanized monoclonal antibody against CD20 for inflammatory disorders and B-cell malignancies”.Current opinion in investigational drugs (London, England : 2000) 9 (11): 1206–15. PMID 18951300.
- Katie Reid (2010-03-08). Update 2. Roche suspends arthritis treatment after deaths. Reuters. Retrieved 2010-03-08
GSK and Genmab seek alternative approval for leukaemia drug Arzerra

Arzerra
GlaxoSmithKline and Genmab A/S have announced the submission of leukaemia drug Arzerra to the European Medicines Agency (EMA) for a variation in marketing authorisation.
The companies are seeking authorisation for the drug to be used in combination with an alkylator-based therapy for treatment of Chronic Lymphocytic Leukemia (CLL) patients who have not received prior treatment and are inappropriate for fludarabine-based therapy.
READ ALL AT
Ofatumumab(trade name Arzerra, also known as HuMax-CD20) is a human monoclonal antibody (for the CD20 protein) which appears to inhibit early-stage B lymphocyte activation. It is FDA approved for treating chronic lymphocytic leukemia that is refractory to fludarabine and alemtuzumab (Campath) and has also shown potential in treating Follicular non-Hodgkin’s lymphoma, Diffuse large B cell lymphoma, rheumatoid arthritis and relapsing remitting multiple sclerosis. Ofatumumab has also received conditional approval in Europe for the treatment of refractory chronic lymphocytic leukemia. This makes ofatumumab the first marketing application for an antibody produced by Genmab, as well as the first human monoclonal antibody which targets the CD20 molecule that will be available for patients with refractory CLL.
Chronic lymphocytic leukemia (CLL) is a slowly progressing cancer of the blood and bone marrow. Arzerra (ofatumumab) has been approved by the FDA for treating CLL.
Patients with CLL whose cancer is no longer being controlled by other forms of chemotherapy can be prescribed Arzerra.
People older than fifty are mainly affected by CLL. A group of white blood cells known as B-cells that are part of the body’s immune system is the source of CLL. Every year, about ¼ of people diagnosed with CLL die from the disease.

Arzerra is an anti-CD20 monoclonal antibody that targets a membrane-proximal (which means close to the cell surface), small loop epitope, which is a portion of a molecule to which an antibody binds, on the CD20 molecule on B-cells. This epitope isn’t similar to binding sites that are targeted by other CD20 antibodies that are currently available. The CD20 molecule is highly expressed in most B-cell malignancies, making it a key target in CLL therapy.

MECHANISM OF ACTION:
Binding specifically to both the small and large extracellular loops of the CD20 molecule, Arzerra is an anti-CD20 monoclonal antibody. The CD20 molecule is expressed on normal B lymphocytes (pre-B- to mature B-lymphocyte) and on B-cell CLL. The CD20 molecule isn’t internalized following antibody binding and it isn’t shed from the cell surface. The Fc domain of ofatumumab mediates immune effector functions to result in B-cell lysis in vitro, while the Fab domain binds to the CD20 molecule. Complement-dependent cytotoxicity and antibody-dependent, cell-mediated cytotoxicity has been suggested as the possible mechanisms of cell lysis.
Products receive accelerated approval based on a surrogate endpoint, such as a reduction in the size of the tumor or decrease in the number of cancerous white cells or in an enlarged spleen or lymph nodes. These indirect measures for clinical outcomes are considered reasonably likely to predict that the drug will allow patients to live with fewer side effects of a disease or to live longer. Arzerra was approved under the FDA’s accelerated approval process, which allows earlier approval of drugs that meet unmet medical needs.

To confirm that the addition of Arzerra to standard chemotherapy delays the progression of the disease, the manufacturer of this medication is currently conducting a clinical trial in CLL patients. This is because the accelerated approval process requires further study of the drug.
Epratuzumab
Epratuzumab
Epratuzumab is a humanised anti-CD22 monoclonal antibody under investigation (clinical development phase III) for its efficacy in SLE. CD22 is a B cell specific surface protein that is considered to be involved in B cell function.
| Expected indication | Systemic lupus erythematosus |
| R&D stage | Phase 3 ongoing (started in December 2010) |
| Next milestone | Phase 3 results (H1 2014) |
| Quick facts |
|
Epratuzumab is a humanized monoclonal antibody. Potential uses may be found inoncology and in treatment of inflammatory autoimmune disorders, such as lupus (SLE).[1][2] The manufacturers in August 2009 announced success in early trials against SLE.[3]
Epratuzumab binds to the glycoprotein CD22 of mature and malignant B-cells.
- Epratuzumab, a humanized monoclonal antibody targeting CD22: characterization of in vitro properties Clinical Cancer Research Vol. 9, September 1, 2003 free full text
- Dose-Fractionated Radioimmunotherapy in Non-Hodgkin’s Lymphoma Using DOTA-Conjugated, 90Y-Radiolabeled, Humanized Anti-CD22 Monoclonal Antibody, Epratuzumab Clinical Cancer Research Vol. 11, July 15, 2005 free full text
- Reuters: UCB and Immunomedics Announce Positive Results for Epratuzumab Phase IIb Study in Systemic Lupus Erythematosus (SLE)
Epratuzumab is a humanized IgG1 antibody that acts as an antagonist of the CD22 receptor present on B cells. UCB is currently enrolling patients for the 2 Phase III trials, EMBODY-1 and EMBODY-2. The primary objective of both studies is to measure the percent of subjects meeting treatment response criteria at week 48 among those patients with moderate to severe SLE. Epratuzumab is dosed at either 600 mg per week or 1200 mg every other week administered over four 12-week treatment cycles.
The cumulative dose for both treatment arms is 2400 mg for each of the 4-week dosing periods. The estimated primary completion date is January 2014 for both EMBODY-1 and EMBODY-2. –
UCB pipeline. UCB Web site. www.ucb.com/rd/pipeline/new-development/epratuzumab. Published July 10, 2010. Accessed June 18, 2011
Brussels (Belgium), June 13th 2013, 0700 CEST – UCB today announced new data from an open-label extension (SL0008) of the EMBLEM™ phase 2b study evaluating the long-term effects of epratuzumab treatment in adult patients with moderate-to-severe systemic lupus erythematosus (SLE). The primary outcome of the open-label extension was to assess the safety of epratuzumab in patients with SLE.4
Relative to the 12 week, double-blind, placebo-controlled EMBLEM™ study, data from the open-label, long-term extension identified no new safety or tolerability signals.1 In addition, relative to EMBLEM™ baseline values, secondary outcome data indicated that the efficacy of epratuzumab as measured by reduction in disease activity was maintained over two years.2 Secondary outcome data also indicated that relative to EMBLEM™ baseline values, treatment over two years with epratuzumab was associated with decreases in corticosteroid use in patients receiving >7.5 mg/day.1 These data were presented this week at the European League Against Rheumatism 2013 Congress in Madrid, Spain.
Epratuzumab, licensed from Immunomedics Inc. (NASDAQ: IMMU), is an investigational medicine and the first CD-22/B-Cell receptor (BCR) targeted monoclonal antibody to be evaluated in clinical studies for the treatment of SLE. Also known as lupus, SLE is a complex, systemic autoimmune disease that affects many different organ systems, including the skin, joints, lungs, kidneys and blood.3,5
“In EMBLEM™, a dose-ranging, phase 2b study, reduction in disease activity was observed in patients treated with epratuzumab,” said Professor Daniel J Wallace MD, Clinical Professor of Medicine, Cedars-Sinai Medical Center, California, US. “This double-blind study had a relatively short 12-week, placebo-controlled, treatment period and it was important to accumulate long-term data on epratuzumab in the treatment of SLE. The phase 2b extension study adds new two year open-label data on epratuzumab to that already available from the 12-week, randomized, controlled study.”
EMBLEM™ was designed to identify a suitable dosing regimen for epratuzumab.6 A total of 227 patients with moderate-to-severe SLE received either: placebo, epratuzumab cumulative dose of 200 mg (100 mg every other week), 800 mg (400 mg every other week), 2400 mg (600 mg weekly), 2400 mg (1200 mg every other week) or 3600 mg (1800 mg every other week).3,6 In the open-label extension 203 patients from any arm of the EMBELM™ study received 1200 mg epratuzumab at weeks 0 and 2 of 12-week cycles.1,2,7
Data on epratuzumab presented at EULAR 2013
Evaluation of the safety profile of long-term epratuzumab treatment in patients with moderate-to-severe SLE1
Safety variables were primary outcome measures in SL0008 and included duration of exposure, adverse events, infusion reactions and infections.
Exposure to epratuzumab was a median 845 days over a median 10 treatment cycles. Adverse events (AEs) caused discontinuation in 29 (14.3%) patients. The most common serious AEs were SLE flare (3.4%), lupus nephritis (2%) and symptomatic cholelithiasis (1.5%). The most common infections/infestations were urinary tract infection (24.6%) and upper respiratory tract infection (23.2%). There were no opportunistic infections and no patterns of specific serious or severe infections.
Evaluation of long-term efficacy of epratuzumab as measured by reduction in disease activity in patients with moderate-to-severe SLE2
Secondary outcome measures in SL0008 included efficacy as measured by reduction in disease activity, and assessed by: British Isles Lupus Assessment Group (BILAG) improvement, SLE disease activity index (SLEDAI) score, Physician Global Assessment (PGA) score and combined treatment response defined as BILAG improvement without worsening, no SLEDAI worsening and no PGA worsening, relative to EMBLEM™ baseline.
The median BILAG total score was 25.0 at EMBLEM™ baseline and 9.0 at week 108. The score was 14.0 at SL0008 screening. Median SLEDAI score was 12.0 at EMBLEM™ baseline and 4.0 at week 108. The score was 10.0 at SL0008 screening. The median PGA score was 50.0 at EMBLEM™ baseline and 17.5 at week 108 with a score of 31.0 at SL0008 screening.
The proportion of patients achieving the combined treatment response was 32.5% at SL0008 screening (n=203) and 60.3% at week 108 (n=116).
Effect of corticosteroid use of long-term epratuzumab treatment in patients with moderate-to-severe SLE1
Corticosteroid doses were monitored throughout SL0008 and was a secondary outcome measure.
Median corticosteroid dose at EMBLEM™ baseline and SL0008 screening was 10.0 mg/day. At week 116, this was 5 mg/day (n=112). Data indicated that treatment over two years with epratuzumab was associated with decreases in corticosteroid use in patients receiving >7.5 mg/day with a corresponding increase in the proportion of patients receiving lower doses or no longer receiving corticosteroids.
The proportion of patients requiring 7.5-20 mg/day and >20 mg/day decreased (49.8% and 10.8% at baseline and 33.9% and 8.0% respectively, at week 116) and the proportion of patients receiving >0–7.5mg/day or no longer receiving corticosteroids increased (33.5% and 5.9% at baseline and 45.5% and 12.5% respectively, at week 116).
Orexigen files obesity drug Contrave for approval in Europe

Orexigen files obesity drug Contrave for approval in Europe
Orexigen Therapeutics has submitted the Marketing Authorization Application (MAA) for Contrave, an investigational weight-loss drug to the European Medicines Agency (EMA).
 The La Jolla, CA-based drug firm is using the EMA’s centralised procedure to seek approval for Contrave Orexigen (32 mg naltrexone sustained release (SR) / 360 mg bupropion SR) for the management of obesity, including weight loss and maintenance of weight loss, in conjunction with lifestyle modification. The company filed the application after meeting with the European agency to discuss the filing strategy and “both were supportive” of the company’s plan to file in advance of an eagerly-anticipated interim analysis of a cardiovascular outcomes trial called the Light study. Orexigen and the European regulator have also agreed upon an investigation plan in children and adolescents.
The La Jolla, CA-based drug firm is using the EMA’s centralised procedure to seek approval for Contrave Orexigen (32 mg naltrexone sustained release (SR) / 360 mg bupropion SR) for the management of obesity, including weight loss and maintenance of weight loss, in conjunction with lifestyle modification. The company filed the application after meeting with the European agency to discuss the filing strategy and “both were supportive” of the company’s plan to file in advance of an eagerly-anticipated interim analysis of a cardiovascular outcomes trial called the Light study. Orexigen and the European regulator have also agreed upon an investigation plan in children and adolescents.
read all at
http://www.pharmatopics.com/2013/10/orexigen-files-obesity-drug-contrave-approval-europe/
Tetraphase to Present New Data at IDWeek on Eravacycline’s Potential Activity in Treating Serious Respiratory Infections

eravacycline
http://www.ama-assn.org/resources/doc/usan/eravacycline.pdf
1-Pyrrolidineacetamide, N-[(5aR,6aS,7S,10aS)-9-(aminocarbonyl)-7-(dimethylamino)-
4-fluoro-5,5a,6,6a,7,10,10a,12-octahydro-1,8,10a,11-tetrahydroxy-10,12-dioxo-2-
naphthacenyl]-
(4S,4aS,5aR,12aS)-4-(dimethylamino)-7-fluoro-3,10,12,12a-tetrahydroxy-1,11-dioxo-9-
[(pyrrolidin-1-ylacetyl)amino]-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide
MOLECULAR FORMULA C27H31FN4O8
MOLECULAR WEIGHT 558.6
SPONSOR Tetraphase Pharmaceuticals, Inc.
CODE DESIGNATION TP-434
CAS REGISTRY NUMBER 1207283-85-9
WHO NUMBER 9702
Tetraphase Pharmaceuticals Inc. (NASDAQ:TTPH) today announced that it will present two posters at IDWeek 2013 that examine the potential of its lead antibiotic candidate eravacycline to treat serious multi-drug resistant (MDR) infections. The first will highlight positive results of a Phase 1 study assessing the bronchopulmonary disposition safety and tolerability of eravacycline in healthy men and women; this study represents the first clinical assessment of eravacycline for potential use in treating pneumonia. The second poster will provide the results of a study that examined the activity of eravacycline in vitro against multiple Gram-negative and Gram-positive pathogens to set quality-control limits for monitoring eravacycline activity in future testing programs.
http://www.pharmiweb.com/pressreleases/pressrel.asp?ROW_ID=79335#.Uk6MOoanonU
More: http://www.pharmiweb.com/pressreleases/pressrel.asp?ROW_ID=79335#.Uk6MOoanonU#ixzz2gkEMTtQm
Eravacycline (TP-434) is a synthetic fluorocycline antibiotic in development.
Eravacycline (TP-434 or 7-fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline) is a novel fluorocycline that was evaluated for antimicrobial activity against panels of recent aerobic and anaerobic Gram-negative and Gram-positive bacteria. Eravacycline showed potent broad spectrum activity against 90% of the isolates (MIC90) in each panel at concentrations ranging from ≤0.008 to 2 μg/mL for all species panels except Pseudomonas aeruginosa and Burkholderia cenocepacia (MIC90 values of 32 μg/mL for both organisms). The antibacterial activity of eravacycline was minimally affected by expression of tetracycline-specific efflux and ribosomal protection mechanisms in clinical isolates. Further, eravacycline was active against multidrug-resistant bacteria, including those expressing extended spectrum β-lactamases and mechanisms conferring resistance to other classes of antibiotics, including carbapenem resistance. Eravacycline has the potential to be a promising new IV/oral antibiotic for the empiric treatment of complicated hospital/healthcare infections and moderate-to-severe community-acquired infections.
Tetraphase’s lead product candidate, eravacycline, has also received an award from Biomedical Advanced Research and Development Authority (BARDA) that provides for funding to develop eravacycline as a potential counter-measure to certain biothreat pathogens. It is worth up to USD 67 million.
Process R&D of Eravacycline: The First Fully Synthetic Fluorocycline in Clinical Development
Eravacycline (TP-434)
We are developing our lead product candidate, eravacycline, as a broad-spectrum intravenous and oral antibiotic for use as a first-line empiric monotherapy for the treatment of multi-drug resistant (MDR) infections, including MDR Gram-negative bacteria. We developed eravacycline using our proprietary chemistry technology. We completed a successful Phase 2 clinical trial of eravacycline with intravenous administration for the treatment of patients with complicated intra-abdominal infections (cIAI) and have initiated the Phase 3 clinical program.
Eravacycline is a novel, fully synthetic tetracycline antibiotic. We selected eravacycline for development from tetracycline derivatives that we generated using our proprietary chemistry technology on the basis of the following characteristics of the compound that we observed in in vitro studies of the compound:
- potent antibacterial activity against a broad spectrum of susceptible and multi-drug resistant bacteria, including Gram-negative, Gram-positive, atypical and anaerobic bacteria;
- potential to treat the majority of patients as a first-line empiric monotherapy with convenient dosing; and
- potential for intravenous-to-oral step-down therapy.
In in vitro studies, eravacycline has been highly active against emerging multi-drug resistant pathogens like Acinetobacter baumannii as well as clinically important species ofEnterobacteriaceae, including those isolates that produce ESBLs or are resistant to the carbapenem class of antibiotics, and anaerobes.
Based on in vitro studies we have completed, eravacycline shares a similar potency profile with carbapenems except that it more broadly covers Gram-positive pathogens like MRSA and enterococci, is active against carbapenem-resistant Gram-negative bacteria and unlike carbapenems like Primaxin and Merrem is not active against Pseudomanas aeruginosa. Eravacycline has demonstrated strong activity in vitro against Gram-positive pathogens, including both nosocomial and community-acquired methicillin susceptible or resistantStaphylococcus aureus strains, vancomycin susceptible or resistant Enterococcus faecium andEnterococcus faecalis, and penicillin susceptible or resistant strains of Streptococcus pneumoniae. In in vitro studies for cIAI, eravacycline consistently exhibited strong activity against enterococci and streptococci. One of the most frequently isolated anaerobic pathogens in cIAI, either as the sole pathogen or often in conjunction with another Gram-negative bacterium, is Bacteroides fragilis. In these studies eravacycline demonstrated activity against Bacteroides fragilis and a wide range of Gram-positive and Gram-negative anaerobes.
Key Differentiating Attributes of Eravacycline
The following key attributes of eravacycline, observed in clinical trials and preclinical studies of eravacycline, differentiate eravacycline from other antibiotics targeting multi-drug resistant infections, including multi-drug resistant Gram-negative infections. These attributes will make eravacycline a safe and effective treatment for cIAI, cUTI and other serious and life-threatening infections for which we may develop eravacycline, such as ABSSSI and acute bacterial pneumonias.
- Broad-spectrum activity against a wide variety of multi-drug resistant Gram-negative, Gram-positive and anaerobic bacteria. In our recently completed Phase 2 clinical trial of the intravenous formulation of eravacycline, eravacycline demonstrated a high cure rate against a wide variety of multi-drug resistant Gram-negative, Gram-positive and anaerobic bacteria. In addition, in in vitro studies eravacycline demonstrated potent antibacterial activity against Gram-negative bacteria, including E. coli; ESBL-producing Klebsiella pneumoniae; Acinetobacter baumannii; Gram-positive bacteria, including MSRA and vancomycin-resistant enterococcus, or VRE; and anaerobic pathogens. As a result of this broad-spectrum coverage, eravacycline has the potential to be used as a first-line empiric monotherapy for the treatment of cIAI, cUTI, ABSSSI, acute bacterial pneumonias and other serious and life-threatening infections.
- Favorable safety and tolerability profile. Eravacycline has been evaluated in more than 250 subjects in the Phase 1 and Phase 2 clinical trials that we have conducted. In these trials, eravacycline demonstrated a favorable safety and tolerability profile. In our recent Phase 2 clinical trial of eravacycline, no patients suffered any serious adverse events, and safety and tolerability were comparable to ertapenem, the control therapy in the trial. In addition, in the Phase 2 clinical trial, the rate at which gastrointestinal adverse events such as nausea and vomiting that occurred in the eravacycline arms was comparable to the rate of such events in the ertapenem arm of the trial.
- Convenient dosing regimen. In our recently completed Phase 2 clinical trial we dosed eravacycline once or twice a day as a monotherapy. Eravacycline will be able to be administered as a first-line empiric monotherapy with once- or twice-daily dosing, avoiding the need for complicated dosing regimens typical of multi-drug cocktails and the increased risk of negative drug-drug interactions inherent to multi-drug cocktails.
- Potential for convenient intravenous-to-oral step-down. In addition to the intravenous formulation of eravacycline, we are also developing an oral formulation of eravacycline. If successful, this oral formulation would enable patients who begin intravenous treatment with eravacycline in the hospital setting to transition to oral dosing of eravacycline either in hospital or upon patient discharge for convenient home-based care. The availability of both intravenous and oral administration and the oral step-down will reduce the length of a patient’s hospital stay and the overall cost of care.
Additionally, in February 2012, Tetraphase announced a contract award from the Biomedical Advanced Research and Development Authority (BARDA) worth up to $67 million for the development of eravacycline, from which Tetraphase may receive up to approximately $40 million in funding. The contract includes pre-clinical efficacy and toxicology studies; clinical studies; manufacturing activities; and associated regulatory activities to position the broad-spectrum antibiotic eravacycline as a potential empiric countermeasure for the treatment of inhalational disease caused by Bacillus anthracis, Francisella tularensis and Yersinia pestis.\
……………………………………………………………………………………….

Process research and development of the first fully synthetic broad spectrum 7-fluorotetracycline in clinical development is described. The process utilizes two key intermediates in a convergent approach. The key transformation is a Michael–Dieckmann reaction between a suitable substituted aromatic moiety and a key cyclohexenone derivative. Subsequent deprotection and acylation provide the desired active pharmaceutical ingredient in good overall yield.
Process R&D of Eravacycline: The First Fully Synthetic Fluorocycline in Clinical Development
FDA Grants QIDP Designation to Eravacycline, Tetraphase’s Lead Antibiotic Product Candidate
– Eravacycline designated as a QIDP for complicated intra-abdominal infection (cIAI) and complicated urinary tract infection (cUTI) indications –
July 15, 2013 08:30 AM Eastern Daylight Time
WATERTOWN, Mass.–(BUSINESS WIRE)–Tetraphase Pharmaceuticals, Inc. (NASDAQ: TTPH) today announced that the U.S. Food and Drug Administration (FDA) has designated the company’s lead antibiotic product candidate, eravacycline, as a Qualified Infectious Disease Product (QIDP). The QIDP designation, granted for complicated intra-abdominal infection (cIAI) and complicated urinary tract infection (cUTI) indications, will make eravacycline eligible to benefit from certain incentives for the development of new antibiotics provided under the Generating Antibiotic Incentives Now Act (GAIN Act). These incentives include priority review and eligibility for fast-track status. Further, if ultimately approved by the FDA, eravacycline is eligible for an additional five-year extension of Hatch-Waxman exclusivity.
Aerial Biopharma announce positive Phase II results for narcolepsy drug
ADX-N05, ARL-N05, SKL-N05
Aerial Biopharma announce positive Phase II results for narcolepsy drug
A new drug to treat excessive daytime sleepiness associated with narcolepsy has shown positive results from a phase 2b clinical trial, US-based Aerial Biopharma has announced this week.
read all at
Secukinumab
Secukinumab is an anti-IL17A drug being investigated for a number of inflammatory conditions. For plaque psoriasis, Novartis is planning to evaluate a dose of 150 mg subcutaneously compared with placebo.
The primary outcome measure of the planned Phase III trial named ERASURE is to evaluate the efficacy in patients with moderate to severe chronic plaque-type psoriasis. Novartis is also planning to evaluate secukinumab dosed at either 150 or 300 mg versus Enbrel (enterecept) 50 mg in a Phase III trial entitled FIXTURE.
Final data collection for the primary outcome measures in both ERASURE and FIXTURE are anticipated in March 2013.
Secukinumab is a human monoclonal antibody designed for the treatments of uveitis,rheumatoid arthritis, and psoriasis. It targets member A from the cytokine family ofinterleukin 17.[1][2]
Secukinumab was developed by Novartis Pharma AG and has completed Phase II clinical trials for plaque psoriasis in 2011.[3]
CAS registry numbers
- 875356-43-7 (heavy chain)
- 875356-44-8 (light chain)
- ^ “Statement On A Nonproprietary Name Adopted By The USAN Council: Secukinumab”. American Medical Association.
- ^ Hueber, W.; Patel, D. D.; Dryja, T.; Wright, A. M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M. H.; Psoriasis Study, P.; Durez, P. P.; Tak, J. J.; Gomez-Reino, C. S.; Rheumatoid Arthritis Study, R. Y.; Foster, C. M.; Kim, N. S.; Samson, D. S.; Falk, D.; Chu, Q. D.; Callanan, K.; Nguyen, A.; Uveitis Study, F.; Rose, K.; Haider, A.; Di Padova, F. (2010). “Effects of AIN457, a Fully Human Antibody to Interleukin-17A, on Psoriasis, Rheumatoid Arthritis, and Uveitis”. Science Translational Medicine 2 (52): 52ra72.doi:10.1126/scitranslmed.3001107. PMID 20926833. edit
- ^ Papp K.A. et al. ‘Secukinumab efficacy and safety preliminary results from a phase II subcutaneous dose-ranging study in the treatment of moderate-to-severe plaque psoriasis.’ Presented at: 20th Congress of the European Academy of Dermatology and Venereology; 20-24 October, 2011; Lisbon, Portugal.

Dimeric Thymosin beta-4……..accelerates the rate of wound healing

Structure of a Longitudinal Actin Dimer Assembled by Tandem W Domains
Thymosin beta 4 (Tβ4) is a peptide with 43 amino acids that is critical for repair and remodeling tissues on the skin, eye, heart, and neural system following injury
Thymosin beta-4 is a protein that in humans is encoded by the TMSB4X gene.
The protein consists (in humans) of 43 amino acids (msdkpdmaei ekfdksklkk tetqeknplp sketieqekq ages) molWt 4921

NMR structure of a β-thymosin. Both thymosin α1 and β-thymosins areintrinsically unstructured proteins, i.e. they lack a stable fold when free in aqueous solution. This structure, mostly alpha helix, was artificially stabilised by an organic solvent. The thymosin illustrated, originally named β9 is the cow orthologue of human β10
It has been studied in a number of clinical trials.
The thymosin beta-4 peptide, if used after a heart attack, might reactivate cardiacprogenitor cells to repair damaged heart tissue.
Doping in Sports
Thymosin beta-4 was allegedly used by some players in various Australian football codes and is under investigation by the Australian Sports Anti-Doping Authority for anti-doping violations (Feb/Mar 2013):
https://theconversation.edu.au/cronulla-sharks-and-thymosin-beta-4-is-it-doping-12694
FDA, EMA Accept Omeros Ophthalmology Product NDA
OMS302
US and European Regulators Accept for Review OMS302 Marketing Applications
— OMS302 Remains on Track for Planned 2014 Commercial Launch —
SEATTLE, Oct. 2, 2013 /PRNewswire/ — Omeros Corporation (NASDAQ: OMER) announced today that the New Drug Application (NDA) for its ophthalmology product, OMS302, has been confirmed for filing by the U.S. Food and Drug Administration (FDA), which means that the application, submitted in July of this year, is sufficiently complete to permit a substantive review. The company also announced that its Marketing Authorization Application (MAA) for OMS302, submitted last month, has been validated by the European Medicines Agency (EMA). Validation of the MAA confirms that the submission package is administratively complete and is ready for formal review by Europe’s Committee for Medicinal Products for Human Use (CHMP).
read all at
http://www.pharmalive.com/fda-ema-accept-omeros-opthamology-product-nda
Isavuconazole – Basilea reports positive results from study

This post is updated in sept 2015……..
Isavuconazole (BAL4815; trade name Cresemba) is a triazole antifungal drug. Its prodrug, isavuconazonium sulfate (BAL8557), was granted approval by the U.S. Food and Drug Administration (FDA) on March 6, 2015[1]
During its Phase III drug trials, Astellas partnered with Basilea Pharmaceutica, the developer of the drug, for rights to co-development and marketing of isavuconazole. [2]
On May 28, 2013, Basilea Pharmaceutica announced it had been granted orphan drug status by the FDA for treatment of aspergillosis.[3] Since then, it has also been granted orphan drug status for the treatment of invasive candidiasis.[4]

Isavuconazonium sulfate (BAL8557)—a prodrug of isavuconazole.
CLINICAL TRIALS…LINK
PATENTS
|
6-27-2012
|
Process for the manufacture of enantiomerically pure antifungal azoles as ravuconazole and isavuconazole
|
|
|
11-18-2011
|
Antifungal Composition
|
|
|
9-29-2010
|
PROCESS FOR PREPARATION OF WATER-SOLUBLE AZOLE PRODRUGS
|
|
|
12-3-2008
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
3-14-2007
|
N-phenyl substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
11-3-2004
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
10-10-2001
|
Azoles for treatment of fungal infections
|
Several azoles are currently used for systemic mycoses. However, none of them fulfills the needs of clinical requirement in full extent, particularly with regard 0 to broad antifungal spectrum including aspergillus fumigatus, less drug-drug interaction, and appropriate plasma half-life for once a day treatment. Other clinical requirements which are not fulfilled by the azoles currently used, are efficacy against major systemic mycoses including disseminated aspergillosis, safety, and oral or parenteral formulations. Particularly, demand of a 5 parenteral administration of the azoles is increasing for the treatment of serious systemic mycoses. Most of the azoles on the market as well as under development are highly lipophilic molecules that make the parenteral formulation difficult.

Isavuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol; formula I, R1 and R3 represent fluorine and R2 represents hydrogen] as well as Ravuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol; formula I, R1 and R2 represent fluorine and R3 represents hydrogen] are useful antifungal drugs as reported in U.S. Pat. No. 5,648,372 from Feb. 1, 1995 or in U.S. Pat. No. 5,792,781 from Sep. 18, 1996 or in U.S. Pat. No. 6,300,353 from Oct. 9, 2001 (WO99/45008).
Since compounds of general formula I contain two adjacent chiral centers, synthesis of enantiomerically pure compound is complex and until now, all patented syntheses are not efficient enough and do not allow cost effective manufacturing on a technical scale:
Thus, U.S. Pat. Nos. 5,648,372 or 5,792,781 describe enantioselective synthesis of compounds of formula I (specifically Ravuconazole) from chiral 3-hydroxy-2-methyl propionic acid in 12 steps with overall yield lower than 5%. In another approach including 13 steps and low overall yield, (R)-lactic acid was used as the starting material (Chem. Pharm. Bull. 46(4), 623 (1998) and ibid. 46(7), 1125 (1998)).
Because both starting materials contain only one chiral center, in a number of inefficient steps, the second, adjacent chiral center has to be created by a diastereoselective reaction (using either Corey or Sharpless epoxidation method) which is not sufficiently selective leading mostly to a mixture of two diastereomers which have to be separated.
The second approach, based on (R)-methyl lactate, was recently very thoroughly optimized by BMS on a multi kilogram scale but it still does not fulfill requirements for cost effective manufacturing process (Organic Process Research & Development 13, 716 (2009)). The overall yield of this optimized 11 steps process is still only 16% (Scheme 1).

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (U.S. Pat. No. 6,300,353 from Oct. 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to U.S. Pat. No. 6,300,353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec. 1, 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in U.S. Pat. No. 6,133,485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid.
This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (U.S. Pat. No. 6,300,353). It is obvious that this process using less expensive (S)-lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from Jun. 26, 2001) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(II)-catalyst and diethyl zinc (Scheme 2).

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 1611 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
All known processes for enantiomerically pure compounds of formula I have definitely too many operation steps and specifically very low overall yield. The chiral starting materials used, either 3-hydroxy-2-methyl propionic acid or (S)- or (R)-methyl lactate, contain only one chiral center and consequently, in number of steps, the second adjacent chiral center has to be ineffectively generated which makes the entire process long and expensive. The only known process, which generates both chiral centers simultaneously, requires again expensive chiral starting material (R)-2-butynol.
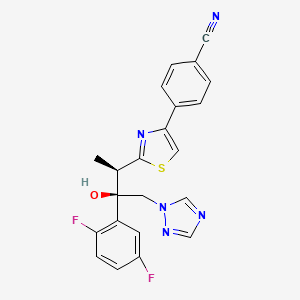
ISAVUCONAZOLE
…………………………………………….
synthetic scheme A, starting from 4-[(2R)-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propionyl]morpholine [which can be prepared by a same procedure as described in Chem. Pharm. Bull. 41, 1035, 1993.]. This synthesis route has been described for example in European Patent Application No. 99101360.8.


(a)
………………………………………………………………………
Example 1 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1R)-10-camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41% yield, 99.1% ee);
m.p. 164-166° C.; [α]=−30° (c=1, methanol, 25° C.);
NMR (CDCl3): 1.23 (3H, d, J=8 Hz), 4.09 (1H, q, J=8 Hz), 4.26 (1H, d, J=14 Hz), 4.92 (1H, d, J=14 Hz), 5.75 (1H, s), 6.75-6.85 (2H, m), 7.45-7.54 (2H, m), 7.62 (1H, s), 7.69 (1H, s), 7.75 (1H, d, J=8 Hz), 7.86 (1H, s), 8.03 (1H, d, J=8 Hz).
The analytical data were identical with published (U.S. Pat. No. 5,648,372 and Chem. Pharm. Bull. 1998, 46, 623-630).
Example 2 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
Racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (44 g) and (1R)-10-camphorsulfonic acid (20 g) were suspended in methanol (ca. 300 ml), the slurry was stirred intensively, warmed up to ca. 70° C. and a small addition of acetic acid was added to obtain a clear solution. After cooling of the solution to rt and then to 0° C., the mixture was seeded with enantiomerically pure salt and stirred for another 2 hrs. The crystalline solid was collected by filtration, washed with cooled methanol and dried under reduced pressure. The crystals were partitioned between methylenechloride (300 ml) and saturated aqueous sodium bicarbonate solution (200 ml). The organic layer was washed twice with water (50 ml), dried with magnesium sulphate, filtrated and evaporated under reduced pressure to give the title compound (16.9 g, 38% yield, 95% ee). The analytical data were identical with published (U.S. Pat. No. 5,648,372 or Chem. Pharm. Bull. 1998, 46, 623).
Example 3 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (10 g) in acetone (ca. 200 ml) a solution of (1R)-10-camphorsulfonic acid (3.9 g) in methanol (50 ml) was added and the mixture was heated shortly under reflux until a clear solution was obtained. The solution was then slowly cooled to rt, seeded with crystals of the desired enantiomeric salt and let overnight. The solid precipitate was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This salt was then taken up in methylenechloride and water and basified with aqueous sodium bicarbonate solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride. The organic phases were combined, dried with sodium sulphate, filtrated and the solvent removed under reduced pressure. The crude product was then dissolved in ethanol, the slurry heated for 20 min, small amount of water was added, the solution slowly cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with cold ethanol and dried under reduced pressure to provide the title enantiomerically pure compound (3.9 g, 39% yield, 96% ee). The analytical date were identical with published in U.S. Pat. No. 6,300,353 B1 and WO 99/45008.
Example 4 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (100 g) in acetone (1000 ml) a solution of (1R)-10-camphorsulfonic acid (47 g) in methanol (500 ml) was added at rt, then slurry was heated under stirring to almost reflux for ca. 30 min, then cooled slowly to rt, seeded with the pure enantiomeric salt and stirred over night. The solid was collected by filtration, washed with methanol/acetone mixture, dried under reduced pressure. The residue was taken up with a solvent mixture of methylenechloride/water and after addition of saturated aqueous sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated, the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 39 g (39% yield, 92% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
Example 5 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
A solution of the racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (4.4 g) and (1R)-10-camphorsulfonic acid (2 g) in toluene (40 ml) containing glacial acetic acid (0.6 ml) was warmed up to approximately 70° C., then allowed to cool slowly to 20° C., seeded with the pure enantiomeric salt whereupon the pure enantiomeric salt start to crystallize out. After ca. 2 hrs at this temperature the solid was collected, washed with cold toluene and dried. The crystals were taken with a solvent mixture of methylenechloride/water and after addition of aqueous saturated sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated and the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 2 g (45% yield, 99% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
…………………………………..
WO 1999045008
The following synthetic scheme 1 illustrates the manufacture of one of the compounds of formula I′:



……………………………….
Bioorganic and medicinal chemistry letters, 2003 , vol. 13, 2 p. 191 – 196
http://www.sciencedirect.com/science/article/pii/S0960894X02008922
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.

-

-
Scheme 1.
Chemistry
Scheme 1.We synthesized a series of new triazolium derivatives of Figure 1, Figure 3 and Scheme 1. CompoundsScheme 1 and Scheme 2, 6, 9, 10 and 11 were first prepared as outlined in Scheme 2 in order to analyze their stability and ability to release Figure 1, Figure 3 and Scheme 1. Next, aromatic analogues 18, 19, 20,21 and Figure 1, Figure 3 and Scheme 3 were synthesized for optimization of 11 to increase its water solubility and conversion rate. Compounds in the second series had sarcosine esters6 to make them water soluble, and they were also designed to generate acetaldehyde7 instead of formaldehyde for a better safety profile. The synthetic procedures for the second series of the derivatives are outlined in Scheme 3.

-
Scheme 2.
(a) ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt (quant); (b) Figure 1, Figure 3 and Scheme 1, CH3CN, 80 °C (60%); (c) (1) ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (30%, two steps); (d) (1) NaI, CH3CN, 50 °C ; (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 50 °C (88%, two steps); Synthesis of Scheme 1 and Scheme 2: (1) N-3-hydroxypropyl-N-methylamine, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) AcCl, Et3N, CH2Cl2, rt (20%, two steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (82%); Synthesis of 10: (1) l-prolinol, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (<10%, 2 steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (92%); Synthesis of 11: (1) 2-hydroxymethyl-N-methylaniline, ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt; (2) Ac2O, diisopropylethylamine, rt (20%, two steps); (3)Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux (63%).
-
Figure options

-
Scheme 3.
(a) (1) oxalyl chloride, DMF, 0 °C; (2) KOtBu, THF, −5 °C (97%, two steps); (b) CH3NH2, MeOH, rt (90%); (c) LiAlH4, THF, 0 °C (80%); (d) (1) ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (2) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (84%, two steps); (e) (1) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C; (2) DOWEX-1 Cl− form, aqueous MeOH, rt (65%, two steps); (f) (1) HCl, EtOAc, rt; (2) lyophilization (69%, two steps); Synthesis of 18: (1) (i) (4,5-difluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (quant, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 80 °C; (50%,); (3) HCl, EtOAc, rt (90%); Synthesis of 19: (1) (i) 2-fluoro-6-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (74%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux; (3) HCl, EtOAc, rt (29%, two steps); Synthesis of 20: (1) (i) (5-fluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (91%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 70 °C (72%); (3) HCl, EtOAc, rt (88%); Synthesis of 21: (1) (i) (4-chloro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (71%, two steps); (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 65 °C; (3) HCl, EtOAc, rt (65%, two steps).

read more at
Boyd, B.; Castaner, J. BAL-4815/BAL-8557
Drugs Fut 2006, 31(3): 187
Antimicrobial Agents and Chemotherapy, 2008 , vol. 52, 4 p. 1396 – 1400
Ohwada, J.; Tsukazaki, M.; Hayase, T.; Oikawa, N.; Isshiki, Y.; Umeda, I.; Yamazaki, T.; Ichihara, S.; Shimma, N.Development of novel water antifungal, RO0098557
21st Med Chem Symp (November 28-30, Kyoto) 2001, Abst 1P-06
Ohwada, J.; Tsukazaki, M.; Hayase, T.; et al.
RO0098557, a novel water soluble azole prodrug for parenteral and oral administration (I). Design, synthesis, physicochemical properties and bioconversion42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-820
Tasaka et al., Chem. Pharm. Bull. 41(6) pp. 1035-1042 (1993).
Clinical trials
There have been three phase III clinical trials of isavuconazole, ACTIVE, VITAL and SECURE. As of June 2015, SECURE and VITAL have been presented in abstract form and results from ACTIVE have not been released.[9]
The SECURE trial compared voriconazole and isavuconazole in invasive fungal infections due to aspergillus. Isuvaconazole was found to be non-inferior to voriconazole, anothertriazole antifungal, with all cause mortality at 18.6%, compared to 20.2% in the voriconazole group. It additionally demonstrated a similar side effect profile.[10]
Data from the VITAL study showed that isavuconazole could be used in treatment of invasive mucormycosis, but did not evaluate its clinical efficacy for this indication.[11]
The ACTIVE trial is a comparison of isuvaconazole and caspofungin for invasive candida infections and results are anticipated in the second half of 2015.[12][13]
References
- [1]
- Saboo, Alok. “Basilea Announces Global Partnership With Astellas for Its Antifungal Isavuconazole.” FierceBiotech. N.p., 24 Feb. 2010. Web.
- “Basilea reports isavuconazole orphan drug designation by U.S. FDA.” Market Wired. 28 May 2013.
- “FDA Grants Orphan Drug Designation to Astellas for Isavuconazole for the Treatment of Invasive Candidiasis.” News Releases. Astellas. 3 Nov 2014.
- Cresemba (isovuconazole sulfate) [prescribing information]. Astella Pharma US, Inc. Revised March 2015.
- Jump up^ “Aspergillosis.” Centers for Disease Control and Prevention. Centers for Disease Control and Prevention, 08 Sept. 2014.
- Jump up^ “Astellas Receives FDA Approval for CRESEMBA® (isavuconazonium Sulfate) for the Treatment of Invasive Aspergillosis and Invasive Mucormycosis.” PR Newswire. N.p., 6 Mar. 2015.
- Jump up^ “Isavuconazonium.” Micromedex Solutions. Truven Health Analytics, n.d. Web. <www.micromedexsolutions.com>.
- Jump up^ Pettit, Natasha N.; Carver, Peggy L. (2015-07-01). “Isavuconazole A New Option for the Management of Invasive Fungal Infections”. Annals of Pharmacotherapy 49 (7): 825–842.doi:10.1177/1060028015581679. ISSN 1060-0280. PMID 25940222.
- Mujais, A. “2014: M-1756. A Phase 3 Randomized, Double-Blind, Non-Inferiority Trial Evaluating Isavuconazole (ISA) vs. Voriconazole (VRC) for the Primary Treatment of Invasive Fungal Disease (IFD) Caused by Aspergillus spp. or other Filamentous Fungi (SECURE): Outcomes by Malignancy Status”. http://www.icaaconline.com. Retrieved 2015-06-19.
- “Abstract: An Open-Label Phase 3 Study of Isavuconazole (VITAL): Focus on Mucormycosis (IDWeek 2014)”. idsa.confex.com. Retrieved 2015-06-19.
- Ltd., Basilea. “Basilea Pharmaceutica – Portfolio – Isavuconazole”. http://www.basilea.com. Retrieved 2015-06-19.
- “Isavuconazole (BAL8557) in the Treatment of Candidemia and Other Invasive Candida Infections – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2015-06-19.
| US4861879 | Feb 9, 1988 | Aug 29, 1989 | Janssen Pharmaceutica N.V. | [[4-[4-Phenyl-1-piperazinyl)phenoxymethyl]-1-3-dioxolan-2-yl]-methyl]-1H-imidazoles and 1H-1,2,4-triazoles |
| US5900486 | Sep 9, 1997 | May 4, 1999 | Hoffmann-La Roche Inc. | N-benzylazolium derivatives |
| AU4536497A | Title not available | |||
| EP0667346A2 | Feb 3, 1995 | Aug 16, 1995 | Eisai Co., Ltd. | Azole antifungal agents, process for the preparation there of and intermediates |
| WO1992017474A1 | Mar 26, 1992 | Oct 15, 1992 | Pfizer | Triazole antifungal agents |
| US5648372 | Feb 1, 1995 | Jul 15, 1997 | Eisai Co., Ltd. | Antifungal agents, and compositions |
| US5686646 * | May 23, 1995 | Nov 11, 1997 | Schering-Plough Corporation | Chiral hydrazine derivatives |
| US5746840 * | Mar 28, 1997 | May 5, 1998 | Janssen Pharmaceutica, N.V. | Process for preparing enantiomerically pure 6-{4-chlorophenyl) (1 H-1,2,4-triazol-1-YL) methyl}-1-methyl-1 H-benzotriazole |
| US5792781 | Sep 18, 1996 | Aug 11, 1998 | Eisai Co., Ltd. | Antifungal agents, processes for the preparation thereof, and intermediates |
| US6020497 | Oct 9, 1998 | Feb 1, 2000 | Merck & Co., Inc. | 3-substitutes isoxazolidines as chiral auxiliary agents |
| US6133485 | Apr 15, 1998 | Oct 17, 2000 | Synphar Laboratories, Inc. | Asymmetric synthesis of 2-(2,4-difluorophenyl)-1-heterocycl-1-yl butan-2,3-diols |
| US6300353 | Mar 5, 1999 | Oct 9, 2001 | Basilea Pharmaceutica Ag, A Swiss Company | Azoles for treatment of fungal infections |
| US6383233 | Mar 7, 1997 | May 7, 2002 | Reuter Chemicscher Apparatebau Kg | Separation process |
| US6812238 * | Oct 31, 2000 | Nov 2, 2004 | Basilea Pharmaceutica Ag | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7151182 * | Sep 3, 2004 | Dec 19, 2006 | Basilea Pharmaceutica Ag | Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7803949 * | Dec 20, 2006 | Sep 28, 2010 | Eisai R&D Management Co., Ltd. | Process for preparation of water-soluble azole prodrugs |
| US20030236419 | Dec 31, 2002 | Dec 25, 2003 | Sumika Fine Chemicals Co., Ltd. | Production methods of epoxytriazole derivative and intermediate therefor |
| US20040176432 | Jun 17, 2002 | Sep 9, 2004 | Milan Soukup | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
| WO2003002498A1 * | Jun 17, 2002 | Jan 9, 2003 | Basilea Pharmaceutica Ag | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{2-[(1R,2R)-(2,5-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-1,3-thiazol-4-yl}benzonitrile
|
|
| Clinical data | |
| Trade names | Cresemba (prodrug form) |
| AHFS/Drugs.com | entry |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, intravenous |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 6918485 |
| ChemSpider | 5293682 |
| UNII | 60UTO373KE |
| ChEBI | CHEBI:85979 |
| ChEMBL | CHEMBL409153 |
| NIAID ChemDB | 416566 |
| Chemical data | |
| Formula | C22H17F2N5OS |
| Molecular mass | 437.47 g/mol |


/////
