
Home » Posts tagged 'ANAX LAB'
Tag Archives: ANAX LAB
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
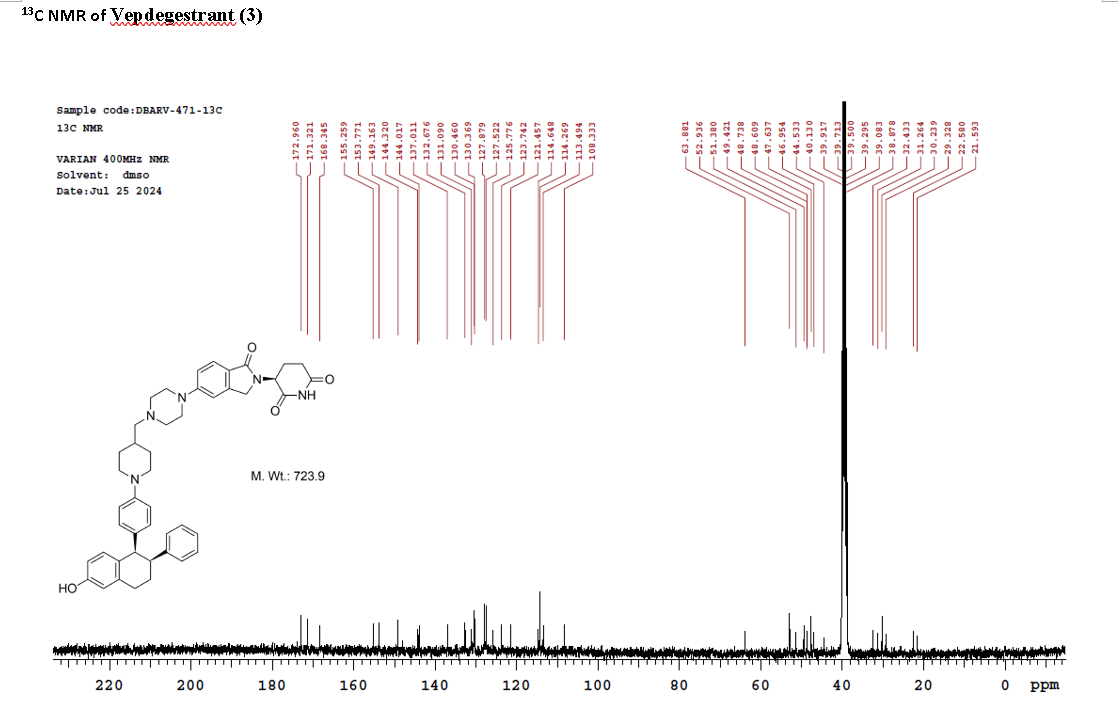
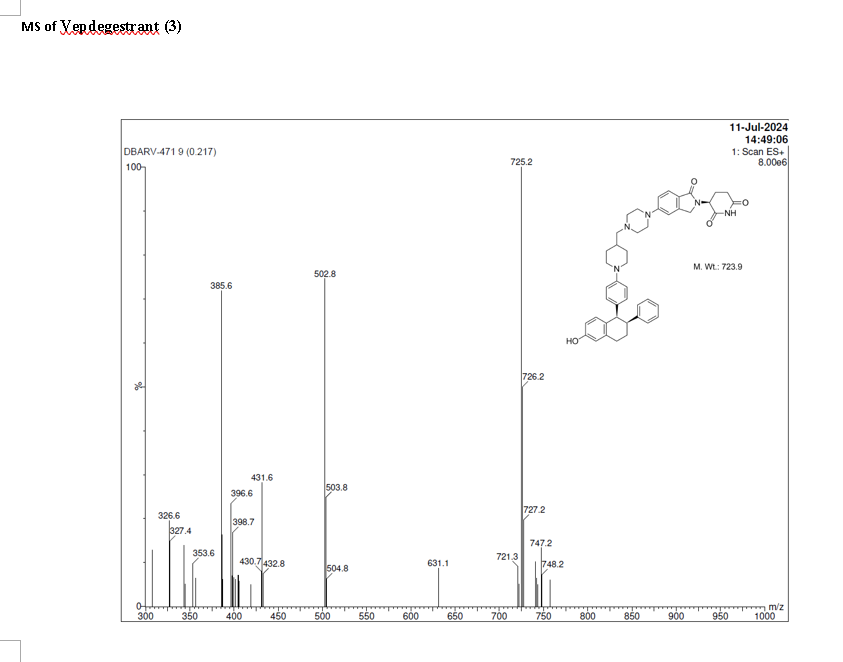
Vepdegestrant
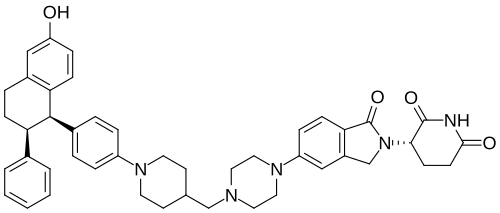
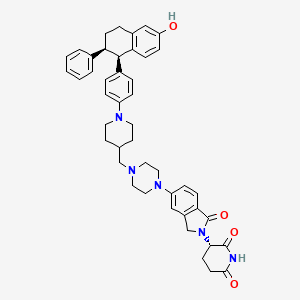
Vepdegestrant
CAS 2229711-08-2
MW 723.9 g/mol, C45H49N5O4
- (S)-3-(5-(4-((1-(4-((1R,2S)-6-Hydroxy-2-phenyl-1,2,3,4-tetrahydronaphthalen-1-yl)phenyl)piperidin-4-yl)methyl)piperazin-1-yl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione
- (3S)-3-[5-[4-[[1-[4-[(1R,2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione
- (2(1)R,2(2)S,8(3)S)-2-hydroxy-2(1),2(2),2(3),2-tetrahydro-7(5,2)-isoindola-6(1,4)-piperazina-4(1,4),8(3)-dipiperidina-2(2,1)-naphthalena-1(1),3(1,4)-dibenzenaoctaphane-7(1),8(2),8(7(3)H)-trione
- 2,6-Piperidinedione, 3-(1,3-dihydro-1-oxo-5-(4-((1-(4-((1R,2S)-1,2,3,4-tetrahydro-6-hydroxy-2-phenyl-1-naphthalenyl)phenyl)-4-piperidinyl)methyl)-1-piperazinyl)-2H-isoindol-2-yl)-, (3S)-
(3S)-3-[6-[4-[[1-[4-[(1R,2S)-6-hydroxy-2-phenyl-1,2,3,4-tetrahydronaphthalen-1-yl]phenyl]piperidin-4-yl]methyl]piperazin-1-yl]-3-oxo-1H-isoindol-2-yl]piperidine-2,6-dione
5/1/2026, FDA 2026, APROVALS 2026, Veppanu, ARV 471, WC1U3R1YMI, PF 07850327
To treat estrogen receptor-positive, human epidermal growth factor receptor 2-negative, ESR1-mutated advanced or metastatic breast cancer with disease progression following at least one line of endocrine therapy
On May 1, 2026, the FDA approved vepdegestrant (Veppanu), a first-in-class oral PROTAC estrogen receptor (ER) degrader developed by Arvinas and Pfizer, for adults with ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer who have progressed on endocrine therapy. It demonstrated significant progression-free survival (PFS) improvements compared to fulvestrant.
Key Details About Vepdegestrant (Veppanu):
- Mechanism of Action: As an oral PROTAC (Proteolysis-Targeting Chimera), vepdegestrant targets the estrogen receptor for degradation, designed to be more effective than traditional endocrine therapies, particularly in ESR1-mutated tumors.
- Approved Indication: For treating adults with ER+/HER2-, ESR1-mutated advanced/metastatic breast cancer (detected by Guardant360 CDx) after at least one line of endocrine therapy.
- Dosage: The recommended dose is 200 mg taken orally once daily with food.
- Clinical Efficacy (VERITAC-2): In trials, vepdegestrant showed a significantly longer PFS compared to intramuscular fulvestrant.
- Side Effects & Risks: Common side effects include decreased white blood cell counts, increased liver function tests, muscle/bone pain, fatigue, and nausea. Warnings include embryo-fetal toxicity and QTc interval prolongation (heart rhythm issues).
- Companion Diagnostic: Guardant360 CDx was approved alongside the drug to identify patients with ESR1 mutations
Vepdegestrant (developmental code name ARV-471) is an investigational oral proteolysis-targeting chimera (PROTAC) compound that targets the estrogen receptor for protein degradation. It is being developed for the treatment of estrogen receptor-positive, HER2-negative (ER+/HER2-) breast cancer by Arvinas and Pfizer.[1][2][3]
Mechanism of action
Vepdegestrant is designed as a PROTAC that recruits the ubiquitin-proteasome system to target the estrogen receptor for degradation.[4] The compound contains both an E3 ubiquitin ligase-binding moiety and an estrogen receptor-binding domain, intended to bring these proteins into proximity to trigger ubiquitination and subsequent proteasomal degradation of the ER protein.[5] In laboratory studies, vepdegestrant demonstrated ER degradation in ER-positive breast cancer cell lines with reported DC50 values of approximately 1-2 nM.[6]
Vepdegestrant is an orally available hetero-bifunctional molecule and selective estrogen receptor (ER) alpha-targeted protein degrader, using the proteolysis targeting chimera (PROTAC) technology, with potential antineoplastic activity. Vepdegestrant is composed of an ER alpha ligand attached to an E3 ligase recognition moiety. Upon oral administration,vepdegestrant targets and binds to the ER ligand binding domain on ER alpha. E3 ligase is recruited to the ER by the E3 ligase recognition moiety and ER alpha is tagged by ubiquitin. This causes ubiquitination and degradation of ER alpha by the proteasome. This decreases ER alpha protein levels, decreases the expression of ER alpha-target genes and halts ER-mediated signaling. This results in an inhibition of proliferation in ER alpha-overexpressing tumor cells. In addition, the degradation of the ER alpha protein releases the ARV-471 and can bind to additional ER alpha target proteins. ER alpha is overexpressed in a variety of cancers and plays a key role in cancer cell proliferation.
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/slct.202405939
PAT
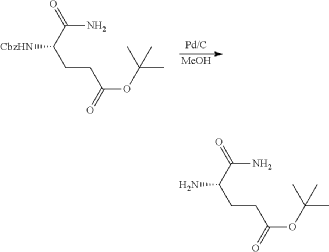
Step 11: Preparation of 3-[5-[4-[[1-[4-[(1R, 2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione (Compound (I-b))
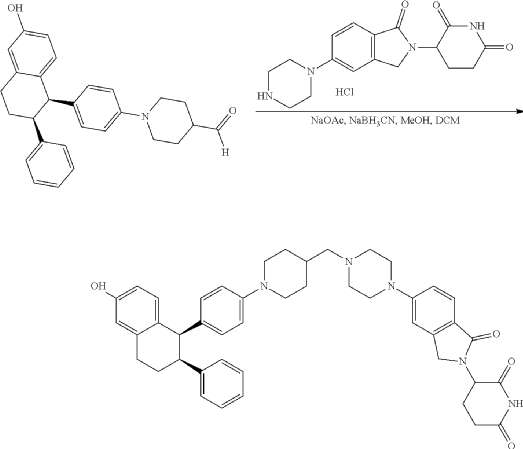
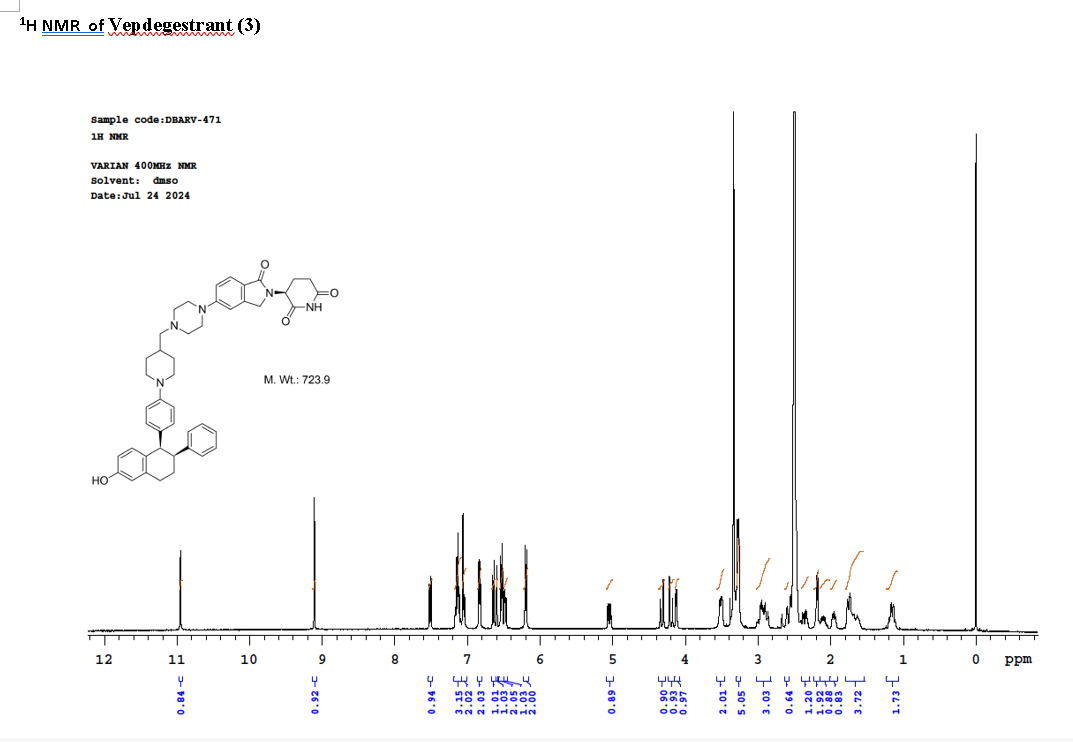
To a solution of 3-(1-oxo-5-piperazin-1-yl-isoindolin-2-yl)piperidine-2,6-dione hydrochloride (319 mg, 0.87 mmol, prepared in Step 17 described for Exemplary Compound 62) in methanol (4 mL) and dichloromethane (4 mL) was added sodium acetate (120 mg, 1.46 mmol, 2 eq). The mixture was stirred at 20° C. for 0.5 h, then to the mixture was added 1-[4-[(1R,2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]piperidine-4-carbaldehyde (300 mg, 0.73 mmol, 1 eq) and sodium cyanoborohydride (137 mg, 2.19 mmol, 3 eq). The mixture was stirred at 20° C. for 12 h. LC-MS showed the starting material was consumed completely and one main peak with desired MW was detected. The reaction mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex luna C 18 column, 250×50 mm, 10 um; mobile phase: [water (0.05% HCl)-acetonitrile]; B %: acetonitrile 10%-40% in 30 min). The desired compound 3-[5-[4-[[1-[4-[(1R, 2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione (288.4 mg, 0.37 mmol, 51% yield) was obtained as a white solid of hydrochloride salt. LC-MS (ESI) m/z: 724.4 [M+1] +; 1H NMR (400 MHz, DMSO-d 6) δ 10.97 (s, 1H), 10.83 (s, 0.9H, HCl), 7.60 (d, J=8.5 Hz, 1H), 7.40 (br s, 2H), 7.22-7.11 (m, 5H), 6.83 (d, J=6.0 Hz, 2H), 6.69-6.63 (m, 2H), 6.58-6.47 (m, 3H), 5.07 (dd, J=5.2, 13.2 Hz, 1H), 4.41-4.30 (m, 2H), 4.28-4.21 (m, 1H), 4.00 (d, J=12.7 Hz, 2H), 3.61 (d, J=11.0 Hz, 2H), 3.54-3.36 (m, 6H), 3.16 (br s, 4H), 3.06-2.84 (m, 3H), 2.76-2.53 (m, 1H), 2.43-2.33 (m, 1H), 2.27 (br s, 1H), 2.16-2.04 (m, 3H), 2.02-1.69 (m, 5H).
Synthesis of (3S)-3-[5-[4-[[1-[4-[(1R, 2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione (Compound (I-c))
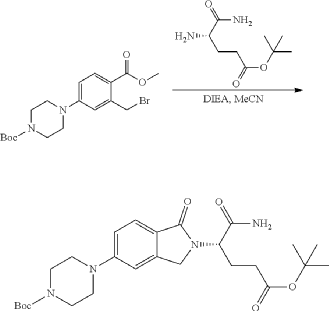
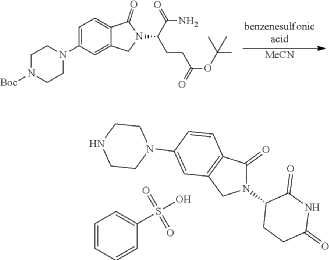
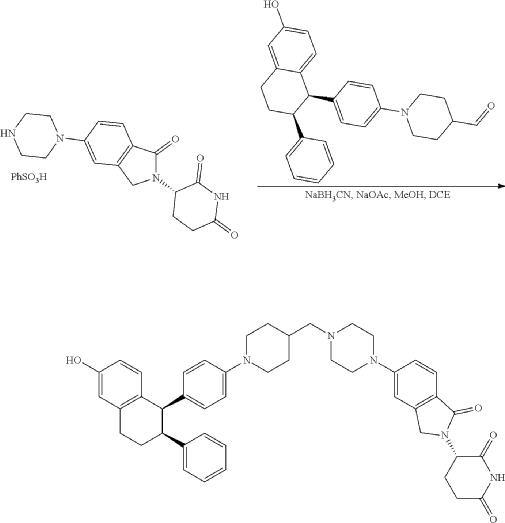
PAT
- Tetralin and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: CN-118834201-APriority Date: 2016-12-01
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: EP-3689868-B1Priority Date: 2016-12-01Grant Date: 2023-09-27
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: US-10647698-B2Priority Date: 2016-12-01Grant Date: 2020-05-12
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: US-2025320195-A1Priority Date: 2016-12-01
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: US-10899742-B1Priority Date: 2016-12-01Grant Date: 2021-01-26
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: US-11104666-B2Priority Date: 2016-12-01Grant Date: 2021-08-31
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: US-12172981-B2Priority Date: 2016-12-01Grant Date: 2024-12-24



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
References
- Iwata, H.; Naito, Y.; Hattori, M.; Yoshimura, A.; Yonemori, K.; Aizawa, M.; et al. (November 2023). “58P Safety and pharmacokinetics (PK) of vepdegestrant in Japanese patients with estrogen receptor (ER)+/human epidermal growth factor receptor 2 (HER2)- advanced breast cancer: Results from a Japanese phase I study”. Annals of Oncology. 34: S1488–S1489. doi:10.1016/j.annonc.2023.10.193. S2CID 265657144.
- Iwata, H.; Hamilton, E.P.; Ma, C.X.; De Laurentiis, M.; Hurvitz, S.A.; Wander, S.A.; et al. (November 2023). “73TiP Global phase III studies evaluating vepdegestrant in estrogen receptor (ER)+/human epidermal growth factor receptor 2 (HER2)- advanced breast cancer: VERITAC-2 and VERITAC-3”. Annals of Oncology. 34: S1493. doi:10.1016/j.annonc.2023.10.207. S2CID 265654990.
- “Arvinas, Pfizer reworking partnership on ‘Protac’ cancer drug | BioPharma Dive”. http://www.biopharmadive.com. Retrieved 17 September 2025.
- “Estrogen Receptor”. Arvinas. Retrieved 17 September 2025.
- Sakamoto, Kathryn M.; Kim, Kwon B.; Kumagai, Ayumu; Mercurio, Frank; Crews, Craig M.; Deshaies, Raymond J. (18 January 2022). “PROTAC targeted protein degraders: the past is prologue”. Nature Reviews Drug Discovery. 21 (3): 181–200. doi:10.1038/s41573-021-00371-6. PMC 8765495. PMID 35046570.
- “Vepdegestrant (ARV-471) PROTAC ER Degrader”. MedChemExpress. Retrieved 17 September 2025.
- Hamilton, Erika P.; Ma, Cynthia; De Laurentiis, Michelino; Iwata, Hiroji; Hurvitz, Sara A.; Wander, Seth A.; et al. (2024). “VERITAC-2: a Phase III study of vepdegestrant, a PROTAC ER degrader, versus fulvestrant in ER+/HER2- advanced breast cancer”. Future Oncology (London, England). 20 (32): 2447–2455. doi:10.1080/14796694.2024.2377530. ISSN 1744-8301. PMC 11524203. PMID 39072356.
- “A Study to Compare the Efficacy and Safety of Vepdegestrant (ARV-471) Versus Fulvestrant in Participants With Estrogen Receptor-positive, HER2-negative Advanced Breast Cancer (VERITAC-2)”. ClinicalTrials.gov. 30 June 2025. Retrieved 17 September 2025.
- “Arvinas and Pfizer Announce Positive Topline Results from Phase 3 VERITAC-2 Clinical Trial”. Arvinas. Retrieved 17 September 2025.
- “VERITAC-2 Trial Shows Vepdegestrant Significantly Improves Survival in ESR1-Mutant Breast Cancer”. Applied Clinical Trials Online. 24 March 2025. Retrieved 17 September 2025.
- “Arvinas Announces Results from the VERITAC-2 Trial Selected as Late-Breaking Oral Presentation at the 2025 ASCO Annual Meeting”. Arvinas. 23 April 2025. Retrieved 17 September 2025.
- Gough, Sheryl M.; Flanagan, John J.; Teh, Jimmy (15 August 2024). “Oral Estrogen Receptor PROTAC Vepdegestrant (ARV-471) Is Highly Efficacious as Monotherapy and in Combination with CDK4/6 or PI3K/mTOR Pathway Inhibitors in Preclinical ER+ Breast Cancer Models”. Clinical Cancer Research. 30 (16): 3549–3562. doi:10.1158/1078-0432.CCR-23-3465. PMC 11325148. PMID 38819400.
- “FDA Grants Fast Track Status to Vepdegestrant for ER+/HER2– Metastatic Breast Cancer”. Oncology Live. 6 February 2024. Retrieved 17 September 2025.
- “Vepdegestrant Gains FDA Fast Track Designation in ER+/HER2- Breast Cancer”. Targeted Oncology. 6 February 2024. Retrieved 17 September 2025.
- “Arvinas Announces Submission of New Drug Application to U.S. FDA for Vepdegestrant for Patients with ESR1-Mutated ER+/HER2- Advanced or Metastatic Breast Cancer” (Press release). Arvinas. 24 June 2025. Retrieved 17 September 2025.
External links
| Clinical data | |
|---|---|
| Pronunciation | /ˌvɛpdəˈdʒɛstrənt/ VEP-də-JES-trənt |
| Other names | ARV-471 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2229711-68-4 |
| PubChem CID | 134562533 |
| ChemSpider | 114935295 |
| UNII | WC1U3R1YMI |
| ChEMBL | ChEMBL5095210 |
| Chemical and physical data | |
| Formula | C45H49N5O4 |
| Molar mass | 723.918 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- Targeting the Estrogen Receptor for the Treatment of Breast Cancer: Recent Advances and ChallengesPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-06-28PMID: 37377342DOI: 10.1021/acs.jmedchem.3c00136
- Emerging targeted protein degradation tools for innovative drug discovery: From classical PROTACs to the novel and beyondPublication Name: European Journal of Medicinal ChemistryPublication Date: 2022-03-05PMID: 35092900DOI: 10.1016/j.ejmech.2022.114142
- Structural and Physicochemical Features of Oral PROTACsPublication Name: Journal of Medicinal ChemistryPublication Date: 2024-07-30PMID: 39078401DOI: 10.1021/acs.jmedchem.4c01017
- Discovery of the cereblon-recruiting tubulin PROTACs effective in overcoming Taxol resistance in vitro and in vivoPublication Name: European Journal of Medicinal ChemistryPublication Date: 2024-02-05PMID: 38171146DOI: 10.1016/j.ejmech.2023.116067
- Current advances and development strategies of orally bioavailable PROTACsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2023-12-05PMID: 37708797DOI: 10.1016/j.ejmech.2023.115793
- Discovery of ERD-3111 as a Potent and Orally Efficacious Estrogen Receptor PROTAC Degrader with Strong Antitumor ActivityPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-08-30PMID: 37647546DOI: 10.1021/acs.jmedchem.3c01186
- Expanding Chemical Probe Space: Quality Criteria for Covalent and Degrader ProbesPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-07-05PMCID: PMC10388296PMID: 37403870DOI: 10.1021/acs.jmedchem.3c00550
////////////vepdegestrant, anax lab, approvals 2026, fda 2026, Veppanu, FDA 2026, APROVALS 2026, Veppanu, ARV 471, WC1U3R1YMI, PF 07850327
Dencatistat
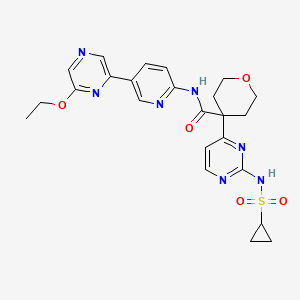
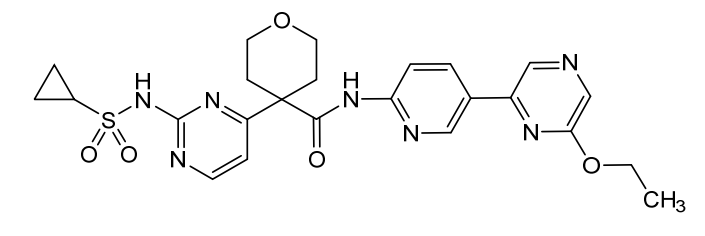
Dencatistat
CAS 2377000-84-3
MFC24H27N7O5S MW 525.6 g/mol
4-[2-(cyclopropylsulfonylamino)pyrimidin-4-yl]-N-[5-(6-ethoxypyrazin-2-yl)-2-pyridinyl]oxane-4-carboxamide
4-[2-(cyclopropanesulfonamido)pyrimidin-4-yl]-N-[5-(6-ethoxypyrazin-2-yl)pyridin-2-yl]oxane-4-carboxamide
CTP synthase 1 inhibitor, antineoplastic, STP 938, CTPS1-IN-2, QG9C9SZZ3T
Dencatistat (formerly known as STP938) is a first-in-class, orally bioavailable cancer drug designed to target specific blood cancers and solid tumours
Dencatistat is an orally bioavailable, small molecule inhibitor of cytidine triphosphate synthase 1 (CTPS1), with potential antineoplastic activity. Upon oral administration, dencatistat targets, binds to and inhibits the activity of CTPS1, thereby decreasing the production of cytidine triphosphate (CTP), an essential building block of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). This may disrupt DNA and RNA synthesis and trigger apoptosis. CTPS1, an enzyme that catalyzes the rate-limiting step in pyrimidine synthesis, plays an important and nonredundant role in B-cell and T-cell proliferation. CTPS1 is required for rapid cell division in certain types of cancers that arise from blood cells.
Mechanism of Action
It works by inhibiting CTPS1 (Cytidine Triphosphate Synthase 1), a key enzyme that cancer cells “addicted” to for DNA synthesis.
- Targeted approach: It aims to kill cancer cells while leaving healthy cells unharmed by exploiting a “synthetic lethal” dependency in certain tumours.
- Precision medicine: It is particularly being tested in patients whose tumours lack CTPS2, a backup enzyme, which makes them highly vulnerable to dencatistat.
🏥 Clinical Status
Developed by Step Pharma, the drug is currently in several clinical trials:
- Lymphoma: Phase 1/2 trials for relapsed or refractory T-cell and B-cell lymphomas.
- Solid Tumours: Phase 1 studies for patients with solid tumours, specifically ovarian and endometrial cancers.
- Essential Thrombocythaemia: A Phase 1b trial for this blood disorder was initiated in 2025.
- Orphan Drug Status: Received FDA Orphan Drug Designation for T-cell lymphoma in May 2025.
- OriginatorStep Pharma
- ClassAnti-inflammatories; Antineoplastics; Antirheumatics; Antithrombotics; Small molecules
- Mechanism of ActionCTPS1 protein inhibitors
- Orphan Drug StatusYes – T-cell lymphoma
- Phase I/IIB-cell lymphoma; T-cell lymphoma
- Phase ISolid tumours; Thrombocytosis
- PreclinicalGraft-versus-host disease; Inflammation
- No development reportedRheumatoid arthritis
- 23 Feb 2026Step Pharma plans phase II trials for Gynaecological cancer
- 10 Feb 2026Preclinical development in Inflammation is till ongoing in France (PO) (Step Pharma pipeline, February 2026)
- 15 Oct 2025Adverse event data from a phase I/II trial in T-cell lymphoma/B-cell lymphoma released by Step Pharma
SYN
US20250177394, Compound CTPS1-IA
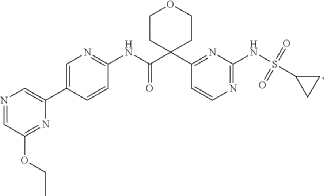
PAT
PAT
A. Preparation of Active Ingredient
20 Example A1 – Preparation of crude 4-(2-(cyclopropanesulfonamido)pyrimidin-4-yl)-N-(5- (6-ethoxypyrazin-2-yl)pyridin-2-yl)tetrahydro-2H-pyran-4-carboxamide
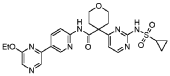

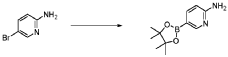
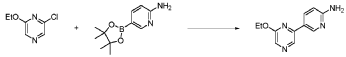
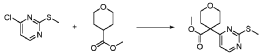
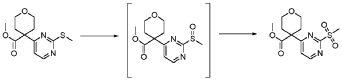
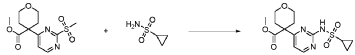
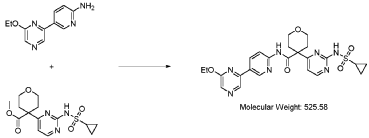
Step 4 – Preparation of crude 4-(2-(cyclopropanesulfonamido)pyrimidin-4-yl)-N-(5-(6- ethoxypyrazin-2-yl)pyridin-2-yl)tetrahydro-2H-pyran-4-carboxamide
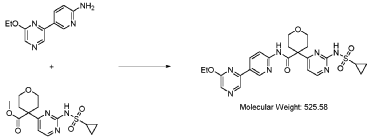
4-(2-(cyclopropanesulfonamido)pyrimidin-4-yl)tetrahydro-2H-pyran-4-carboxylate (1.76 kg, 5.15 mol, 1.00 equiv.) and 5-(6-ethoxypyrazin-2-yl)pyridin-2-amine (1.22 kg, 5.65 mol, 1.10 equiv.) were suspended in a mixture of THF (27.1 L, 15.5 rel. vol.) and DMSO (2.63 L, 1.50 rel. vol.) and stirred until the solids were evenly dispersed. The mixture was concentrated by
STP-P3718PCT
102
distillation at atmospheric pressure and approximately 70 oC to a volume of 15 L. The temperature was adjusted to 20 ± 5 oC, potassium tert-butoxide (6.92 kg 20 wt% solution in THF, 12.3 mol, 2.40 equiv.) was added over 1 h and the reaction mixture stirred at 20 ± 5 oC for 70 minutes until completion. THF (880 mL, 0.500 rel vol.) was charged, followed by acetic acid (780 5 mL, 820 g, 13.6 mol, 2.64 equiv.) over 10 minutes, followed by methanol (4.40 L, 2.50 rel. vol.), followed by water (13.2 L, 7.50 rel. vol.) over 35 minutes. The mixture was stirred at 20 ± 5 oC for 15 minutes and then 16 h at 0 ± 5 oC. The resulting suspension was filtered and washed with water (2 × 8.80 L, 2 × 5.00 rel. vol.), followed by methanol (4.40 L, 2.50 rel. vol.) The filter cake was dried at 35 oC under a flow of nitrogen for 20 h to afford crude 4-(2-10 (cyclopropanesulfonamido)pyrimidin-4-yl)-N-(5-(6-ethoxypyrazin-2-yl)pyridin-2-yl)tetrahydro- 2H-pyran-4-carboxamide (“CTPS1-IA”).
PAT
- Aminopyrimidine derivatives as CTPS1 inhibitorsPublication Number: JP-7428692-B2Priority Date: 2018-03-23Grant Date: 2024-02-06
- Aminopyrimidine derivatives as ctps1 inhibitorsPublication Number: EP-3768674-A1Priority Date: 2018-03-23
- Aminopyrimidine derivative as a CTPS1 inhibitorPublication Number: JP-2021518436-APriority Date: 2018-03-23
- Aminopyrimidine derivatives as ctps1 inhibitorsPublication Number: EP-3768674-B1Priority Date: 2018-03-23Grant Date: 2024-01-03
- Aminopyrimidine derivatives as CTPS1 inhibitorsPublication Number: CN-111868051-APriority Date: 2018-03-23
- Aminopyrimidine derivatives as CTPS1 inhibitorsPublication Number: ES-2974445-T3Priority Date: 2018-03-23Grant Date: 2024-06-27
- CompoundsPublication Number: US-2021024507-A1Priority Date: 2018-03-23
- CompoundsPublication Number: US-2021387965-A1Priority Date: 2018-10-23
- CompoundsPublication Number: US-2023192673-A1Priority Date: 2018-06-04
- Aminopyrimidine derivatives as CTPS1 inhibitorsPublication Number: CN-111868051-BPriority Date: 2018-03-23Grant Date: 2024-04-09
- Aminopyrimidine derivatives as ctps1 inhibitorsPublication Number: WO-2019180244-A1Priority Date: 2018-03-23
- Aminopyrimidine derivatives as ctps1 inhibitorsPublication Number: WO-2019179652-A1Priority Date: 2018-03-23
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
////////dencatistat, anax lab, CTP synthase 1 inhibitor, antineoplastic, STP 938, CTPS1-IN-2, QG9C9SZZ3T
Delocamten
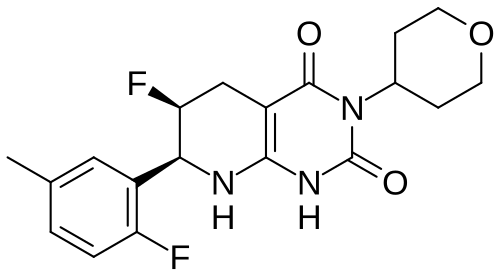
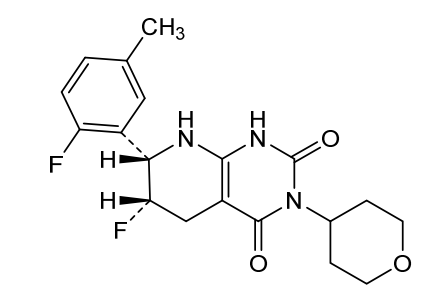
Delocamten
CAS 2417411-02-8
MFC19H21F2N3O3 MW377.4 g/mol
(6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(oxan-4-yl)-5,6,7,8-tetrahydro-1H-pyrido[2,3-d]pyrimidine-2,4-dione
(6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(oxan-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione
cardiac myosin inhibitor, MYK-224; BMS-986435, MYK 224, BMS 986435, IE5886BN8T
Delocamten (development code MYK-224) is a small-molecule cardiac myosin inhibitor developed by Bristol Myers Squibb for hypertrophic cardiomyopathy.[1][2][3]
Delocamten is a small molecule drug. Delocamten is under investigation in clinical trial NCT06122779 (Study to Evaluate Safety, Tolerability and Drug Levels of BMS-986435/MYK-224 in Participants With Heart Failure With Preserved Ejection Fraction (HFpEF)). Delocamten has a monoisotopic molecular weight of 377.16 Da.
SYN
Example 1-3: Preparation of (6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(tetrahydro-2H-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2, 4 (1H, 3H)-dione (3)
Step 7. Synthesis of (6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(tetrahydro-2H-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2, 4 (1H, 3H)-dione (3). A mixture of crude 3G (1.0 g, 2.53 mmol) in CH 3CN (15 mL) was put into a microwave reactor with stirring at 120° C. for 30 min. Subsequently, the mixture was concentrated and the residue was purified by preparative HPLC (column: C18 silica gel; mobile phase: CH3CN:H 2O=20:80 (v v) increasing to CH3CN:H 2O=80:20 (v v) within 40 min; detector: UV 254 nm) to give compound 3 (302 mg, 32%), as a white solid, which was identified as Form 1 polymorph (see Example 2). LC-MS (ES, m/z): 378 [M+H] +; 1H NMR (300 MHz, d-DMSO): δ 10.20 (s, 1H), 7.38-7.05 (m, 3H), 6.45 (s, 1H), 5.11-4.81 (m, 3H), 3.89 (dd, J=10.8, 3.9 Hz, 2H), 3.34-3.27 (m, 3H), 2.76-2.48 (m, 4H), 2.28 (s, 3H), 1.39-1.36 (m, 2H); 19F NMR (376 MHz, d 6-DMSO): δ −123.51 (t, J=86.5 Hz), −191.57 (d, J=129.34 Hz).
PAT
Example 1-3: Preparation of (6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3- (tetrahydro-2H-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2, 4 (1H, 3H)-dione (3).
Scheme 3
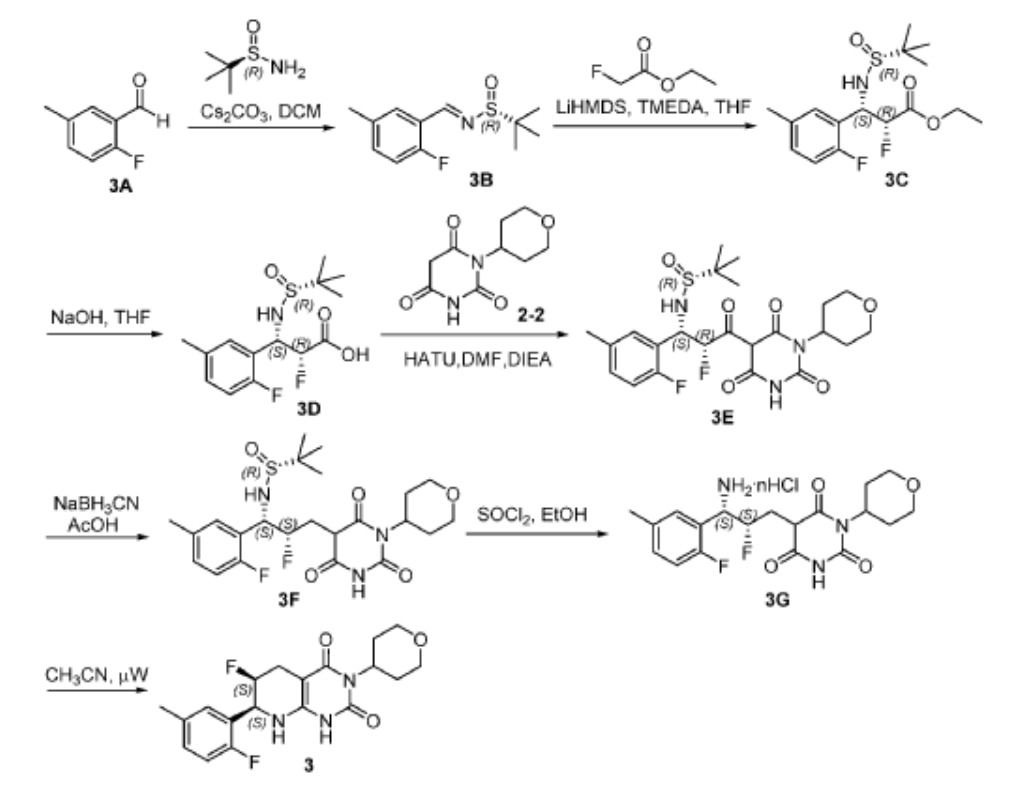
[0165] Step 1. Synthesis of (R,E)-N-(2-fluoro-5-methylbenzylidene)-2-methylpropane-2-sulfinamide (3B). The reaction mixture was filtered and the filtrate was diluted with ether (150 mL). Subsequently, the resulting suspension was filtered. The filtrate was concentrated and the residue was dried in vacuo to give 3B (8.7 g, 97%) as a yellow oil. LC-MS (ES, m/z): 242 [M+H] + ; 1 H NMR (400 MHz, d 6 -DMSO): d 8.87 (s, 1H), 7.76 (m, 1H), 7.29 (m, 1H), 7.03 (m, 1H), 2.37 (d, J = 1.0 Hz, 3H), 1.27 (s, 9H).
[0166] Step 2. Synthesis of ethyl (2R,3S)-3-(((R)-tert-butylsulfinyl)amino)-2-fluoro-3-(2-fluoro-5-methylphenyl)propanoate (3C). To a solution of 3B (4 g, 16.6 mmol), ethyl 2- fluoroacetate (2.6 g, 24.6 mmol), and TMEDA (4.8 mL) in anhydrous THF (40 mL) was added LiHMDS (1 M in THF, 24.6 mL, 24.6 mmol) dropwise at -78 o C over 30 min under an atmosphere of Ar. After stirring at -78 o C for 1 h, the reaction was quenched by adding 1 N aq.
HCl (50 mL), while maintaining the inner temperature of the mixture at < -20 o C. Subsequently, the mixture was concentrated to remove most of the organic solvent and then extracted with EtOAc (100 mL x 3). The combined organic extracts were washed with brine (100 mL) and dried over anhydrous Na2SO4. The solvent was removed and the residue was dried in vacuo to give crude 3C (6.0 g) as a yellow oil, which was used for the next step without further purification. LC-MS (ES, m/z): 348 [M+H] + .
[0167] Step 3. Synthesis of (2R,3S)-3-(((R)-tert-butylsulfinyl)amino)-2-fluoro-3-(2-fluoro-5-methylphenyl)propanoic acid (3D). To a solution of 3C (6.0 g, 17.3 mmol) in THF (40 mL) was added 1N aq. NaOH (34.6 mL, 34.6 mmol) at rt. After stirring at rt for 1 h, the reaction mixture was added ice water (50 mL). The resulting mixture was extracted with EtOAc (100 mL x 2). The aqueous layer was adjusted to pH 5 with sat. aq. citric acid, followed by extraction with EtOAc (100 mL x 3). Subsequently, the combined organic extracts were washed with brine (100 mL) and dried over anhydrous Na 2 SO 4 . The solvent was removed and the residue was purified by preparative HPLC (Column: LC-MS (ES, m/z): 320 [M+H] + ; 1 H NMR (400 MHz, d 6 -DMSO): d 13.57 (br, 1H), 7.55 (dd, J = 7.5, 2.2 Hz, 1H), 7.23– 6.94 (m, 2H), 6.04 (d, J = 10.8 Hz, 1H), 5.37– 4.86 (m, 2H), 2.29 (s, 3H), 1.12 (s, 9H).
[0168] Step 4. Synthesis of (R)-N-((1S,2R)-2-fluoro-1-(2-fluoro-5-methylphenyl)-3-oxo-3- (2,4,6-trioxo-1-(tetrahydro-2H-pyran-4-yl)hexahydropyrimidin-5-yl)propyl)-2-methylpropane-2-sulfinamide (3E). A solution of 3D (700 mg, 2.19 mmol), 2-2 (698 mg, 3.29 mmol), and HATU (1.25 g, 3.29 mmol) in DMF (10 mL) was added DIEA (849 mg, 6.57 mmol) at 0 o C under an atmosphere of Ar. aq. sodium bicarbonate (30 mL) and the resulting solution was extracted with ethyl acetate (50 mL x3). The combined organic extracts were washed with brine (50 mL x 2) and dried over anhydrous Na 2 SO 4 . The solvent was removed and the residue was dried in vacuo to give crude 3E (1.3 g) as a white solid, which was used for the next step without further purification. LC-MS (ES, m/z): 514 [M+H] + ; 1 H NMR (400 MHz, d 6 -DMSO): d 12.16 (br, 1H), 7.66– 7.45 (m, 1H), 7.23– 6.98 (m, 2H), 6.37 (m, 1H), 6.13 (d, J = 10.7 Hz, 1H), 5.22 (m, 1H), 4.79 (m, 1H), 3.94 (m, 2H), 3.35 (t, J = 11.7 Hz, 2H), 2.52– 2.39 (m, 2H), 2.29 (s, 3H), 1.49 (d, J = 12.2 Hz, 2H), 1.04 (s, 9H).
[0169] Step 5. Synthesis of (R)-N-((1S,2S)-2-fluoro-1-(2-fluoro-5-methylphenyl)-3-(2,4,6- trioxo-1-(tetrahydro-2H-pyran-4-yl)hexahydropyrimidin-5-yl)propyl)-2-methylpropane-2-sulfinamide (3F). A solution of crude 3E (1.3 g, 2.53 mmol) in AcOH (10 mL) was added NaBH3CN (398 mg, 6.33 mmol) at 0 o C under an atmosphere of Ar. After stirring at rt for 1 h, the reaction mixture was added ice water (20 mL) and the resulting solution was extracted with EtOAc (50 mL x 3). Next, the combined organic extracts were washed with brine (50 mL) and
dried over anhydrous Na2SO4. The solvent was removed and the residue was dried in vacuo to give crude 3F (1.3 g) as a white solid, which was used for the next step without further purification. LC-MS (ES, m/z): 500 [M+H] + ; 1 H NMR (400 MHz, d 6 -DMSO): d 11.31 (d, J = 28.1 Hz, 1H), 7.41 (d, J = 7.4 Hz, 1H), 7.27– 6.84 (m, 2H), 6.11– 5.78 (m, 2H), 5.08– 4.43 (m, 3H), 3.87 (m, 3H), 2.29 (s, 6H), 1.99 (s, 1H), 1.53– 1.28 (m, 2H), 1.10 (d, J = 2.1 Hz, 10H).
[0170] Step 6. Synthesis of 5-((2S,3S)-3-amino-2-fluoro-3-(2-fluoro-5-methylphenyl)propyl)-1-(tetrahydro-2H-pyran-4-yl)pyrimidine-2, 4, 6 (1H, 3H, 5H)-trione (3G). A solution of crude 3F (1.3 g, 2.60 mmol) in ethanol (10 mL) was added thionyl chloride (334 mg) at 0 o C. After stirring at rt for 1 h, the reaction mixture was concentrated and the residue was dried in vacuo to give crude 3G (1.0 g) as a white solid, which was used for the next step without further purification. LC-MS (ES, m/z): 396 [M+H] + .
[0171] Step 7. Synthesis of (6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(tetrahydro-2H-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2, 4 (1H, 3H)-dione (3). A mixture of crude 3G (1.0 g, 2.53 mmol) in CH 3 CN (15 mL) was put into a microwave reactor with stirring at 120 o C for 30 min. Subsequently, the mixture was concentrated and the residue was purified by preparative HPLC (column: C18 silica gel; mobile phase: CH3CN:H2O = 20:80 (v/v) increasing to CH3CN:H2O = 80:20 (v/v) within 40 min; detector: UV 254 nm) to give compound 3 (302 mg, 32%), as a white solid, which was identified as Form 1 polymorph (see Example 2). LC-MS (ES, m/z): 378 [M+H] + ; 1 H NMR (300 MHz, d 6 -DMSO): d 10.20 (s, 1H), 7.38– 7.05 (m, 3H), 6.45 (s,1H), 5.11– 4.81 (m, 3H), 3.89 (dd, J = 10.8, 3.9 Hz, 2H), 3.34– 3.27 (m, 3H), 2.76–2.48 (m, 4H), 2.28 (s, 3H), 1.39–1.36 (m, 2H); 19 F NMR (376 MHz, d 6 -DMSO): d -123.51 (t, J = 86.5 Hz), -191.57 (d, J = 129.34 Hz).
PAT
- Tetrahydropyran (thp)-substituted bicyclic-pyrimidinedione compoundsPublication Number: EP-4464321-A2Priority Date: 2018-10-29
- Tetrahydropyran-substituted bicyclic pyrimidinedione compounds (THP)Publication Number: ES-2986923-T3Priority Date: 2018-10-29Grant Date: 2024-11-13
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-2024025894-A1Priority Date: 2018-10-29
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-12344607-B2Priority Date: 2018-10-29Grant Date: 2025-07-01
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-2025282779-A1Priority Date: 2018-10-29
- Tetrahydropyran (THP) Substituted Bicyclic Pyrimidinedione CompoundsPublication Number: CN-113056465-APriority Date: 2018-10-29
- Tetrahydropyrane (THP) -substituted bicyclic pyrimidinedione compoundsPublication Number: CN-119977963-APriority Date: 2018-10-29
- Tetrahydropyran (thp)-substituted bicyclic-pyrimidinedione compoundsPublication Number: EP-3873904-B1Priority Date: 2018-10-29Grant Date: 2024-07-10
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-11034693-B2Priority Date: 2018-10-29Grant Date: 2021-06-15
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-2022106314-A1Priority Date: 2018-10-29
- Tetrahydropyran (THP)-Substituted Bicyclic-Pyrimidinedione CompoundsPublication Number: JP-2024063091-APriority Date: 2018-10-29
- Tetrahydropyran (thp)-substituted bicyclic-pyrimidinedione compoundsPublication Number: TW-202426449-APriority Date: 2018-10-29
- Tetrahydropyrane (THP) -substituted bicyclic pyrimidinedione compoundsPublication Number: CN-113056465-BPriority Date: 2018-10-29Grant Date: 2025-01-28
- Tetrahydropyran (thp)-substituted bicyclic-pyrimidinedione compoundsPublication Number: US-2020165247-A1Priority Date: 2018-10-29
- BICYCLIC PYRIMIDINODIONA COMPOUNDS REPLACED WITH TETRAHYDROPYRAN, POLYMORPHIC FORM OF THE SAME AND THE USE OF THE SAME FOR THE TREATMENT OF HCMPublication Number: AR-116880-A1Priority Date: 2018-10-29
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-2022340569-A1Priority Date: 2018-10-29
- Tetrahydropyran (thp)-substituted bicyclic-pyrimidinedione compoundsPublication Number: EP-3873904-A1Priority Date: 2018-10-29
- CRYSTALLINE FORMS OF (6S,7S)-6-FLUORO-7-(2-FLUORO-5-METHYLPHENYL)- 3-(TETRAHYDRO-2H-PYRAN-4-YL)-5,6,7,8-TETRAHYDROPYRIDO[2,3- d]PYRIMIDINE-2,4(1H,3H)-DIONEPublication Number: WO-2024026058-A8Priority Date: 2022-07-29
- Crystalline forms of (6s,7s)-6-fluoro-7-(2-fluoro-5-methylphenyl)- 3-(tetrahydro-2h-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3- d]pyrimidine-2,4(1h,3h)-dionePublication Number: EP-4561697-A1Priority Date: 2022-07-29
- Methods of Administering Myosin InhibitorsPublication Number: US-2023338378-A1Priority Date: 2022-04-26
- Methods of treatment with myosin modulatorPublication Number: US-2023158027-A1Priority Date: 2019-11-10
- tetrahydropyran-substituted bicyclic pyrimidinedione compounds (thp)Publication Number: BR-112021008077-A2Priority Date: 2018-10-29
- CRYSTALLINE FORMS OF (6S,7S)-6-FLUORO-7-(2-FLUORO-5-METHYLPHENYL)-3-(TETRAHYDRO-2H-PYRAN-4-YL)-5,6,7,8-TETRAHYDROPYRIDO[2,3-d]PYRIMIDINE-2,4(1H,3H)-DIONEPublication Number: US-2025034129-A1Priority Date: 2023-07-28
- CRYSTALLINE FORMS OF (6S,7S)-6-FLUORO-7-(2-FLUORO-5-METHYLPHENYL)-3-(TETRAHYDRO-2H-PYRANO-4-IL)-5,6,7,8-TETRAHYDROPYRIDE[2 ,3-D]PYRIMIDINE-2,4(1H,3H)-DIONEPublication Number: AR-130058-A1Priority Date: 2022-07-29
- Crystalline forms of (6s,7s)-6-fluoro-7-(2-fluoro-5-methylphenyl)- 3-(tetrahydro-2h-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3- d]pyrimidine-2,4(lh,3h)-dionePublication Number: WO-2024026058-A1Priority Date: 2022-07-29
- Crystalline form of (6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(tetrahydro-2H-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4(1H,3H)-dionePublication Number: CN-119855816-APriority Date: 2022-07-29
- Crystalline forms of (6s,7s)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(tetrahydro-2h-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4(1h,3h)-dionePublication Number: TW-202412788-APriority Date: 2022-07-29
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- Lehman, Sarah J.; Crocini, Claudia; Leinwand, Leslie A. (June 2022). “Targeting the sarcomere in inherited cardiomyopathies”. Nature Reviews Cardiology. 19 (6): 353–363. doi:10.1038/s41569-022-00682-0. ISSN 1759-5010. PMC 9119933. PMID 35304599.
- Sebastian, Sneha Annie; Padda, Inderbir; Lehr, Eric J.; Johal, Gurpreet (September 2023). “Aficamten: A Breakthrough Therapy for Symptomatic Obstructive Hypertrophic Cardiomyopathy”. American Journal of Cardiovascular Drugs. 23 (5): 519–532. doi:10.1007/s40256-023-00599-0. PMID 37526885. S2CID 260348901.
- Packard, Elizabeth; de Feria, Alejandro; Peshin, Supriya; Reza, Nosheen; Owens, Anjali Tiku (December 2022). “Contemporary Therapies and Future Directions in the Management of Hypertrophic Cardiomyopathy”. Cardiology and Therapy. 11 (4): 491–507. doi:10.1007/s40119-022-00283-5. PMC 9652179. PMID 36243823.
////////delocamten, ANAX LAB, cardiac myosin inhibitor, MYK-224; BMS-986435, MYK 224, BMS 986435, IE5886BN8T
Darlifarnib
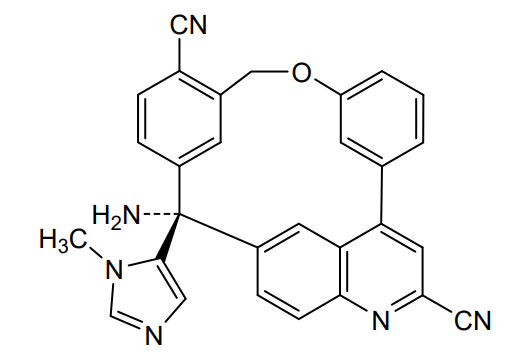
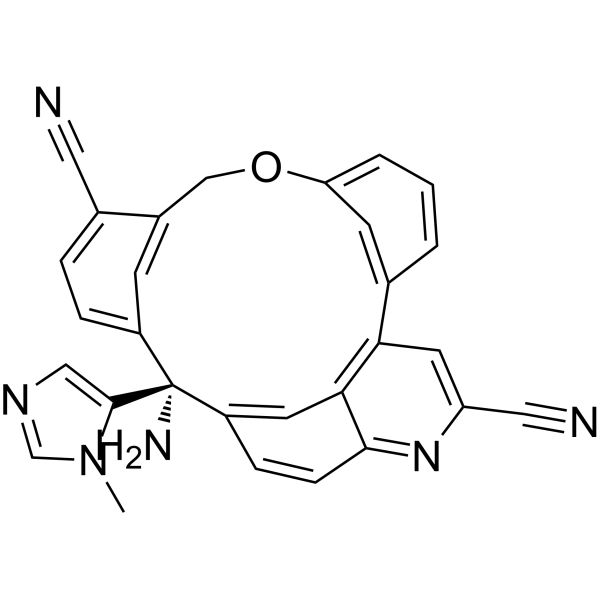
Darlifarnib
CAS 2939824-30-1
MF C29H20N6O MW 468.51
14-amino-14-(3-methylimidazol-4-yl)-7-oxa-19-azapentacyclo[13.6.2.12,6.19,13.018,22]pentacosa-1(22),2(25),3,5,9,11,13(24),15(23),16,18,20-undecaene-10,20-dicarbonitrile

farnesyl transferase inhibitor, antineoplastic, KO-2806, KO 2806, T206317
Darlifarnib (KO-2806) is an investigational, orally active next-generation farnesyl transferase inhibitor (FTI) being developed by Kura Oncology to treat solid tumors, such as clear cell renal cell carcinoma (ccRCC). It inhibits the enzyme farnesyl transferase, blocking KRAS and mTORC1 signaling to induce tumor regression. It is often combined with other agents to overcome resistance.
Key Details About Darlifarnib
- Mechanism of Action: As a FTI, darlifarnib binds to and inhibits farnesyl transferase, which prevents the activation of RAS oncogenes and inhibits downstream mTORC1 signaling, leading to tumor cell death.
- Target Indications: Preclinical and early clinical data show potential in treating KRAS-mutant cancers, including non-small cell lung cancer (NSCLC), colorectal cancer (CRC), and clear cell renal cell carcinoma (ccRCC).
- Combination Therapy: Data from the Phase 1 FIT-001 trial (presented in April 2026) showed that combining darlifarnib with the TKI cabozantinib demonstrated robust activity in patients with pretreated, advanced ccRCC.
- Overcoming Resistance: Darlifarnib is designed to re-sensitize tumors that have become resistant to prior therapies, such as RAS inhibitors and tyrosine kinase inhibitors (TKIs).
- Status: It is an investigational drug and not yet FDA-approved.
- OriginatorKura Oncology
- ClassAntineoplastics; Small molecules
- Mechanism of ActionFarnesyltranstransferase inhibitors
- Phase IAdenocarcinoma; Colorectal cancer; Non-small cell lung cancer; Renal cell carcinoma; Solid tumours
- 12 Jan 2026Kura Oncology plans the one or more expansion cohorts of KO 2806 and cabozantinib in patients with advanced renal cell carcinoma in the first half of 2026
- 22 Oct 2025Pharmacodynamics data from a preclinical trial in Cancer presented at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics 2025 (AACR-NCI-EORTC-2025)
- 18 Oct 2025Adverse events and efficacy data from a phase I trial in Non-small cell lung cancer, Renal cell carcinoma, Adenocarcinoma released by Kura Oncology
PAT
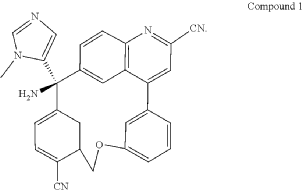
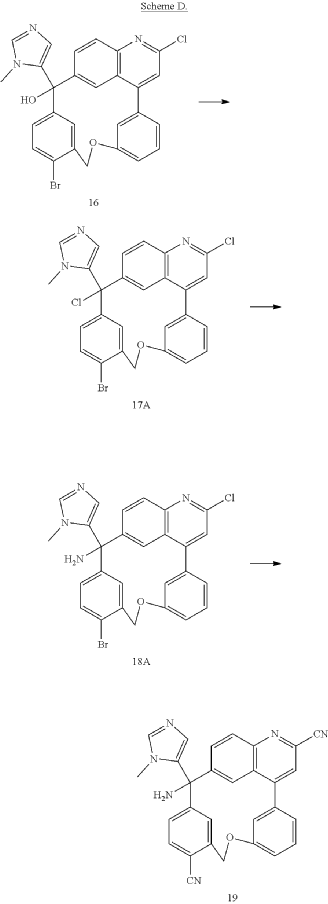
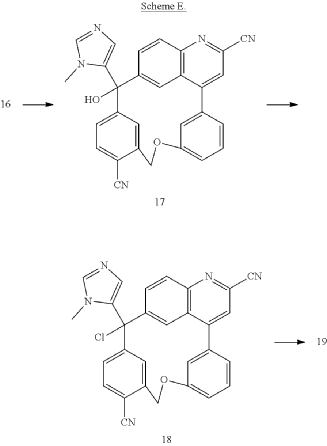
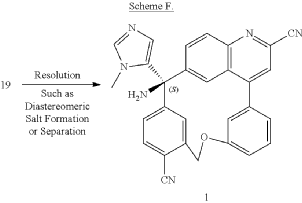
PAT
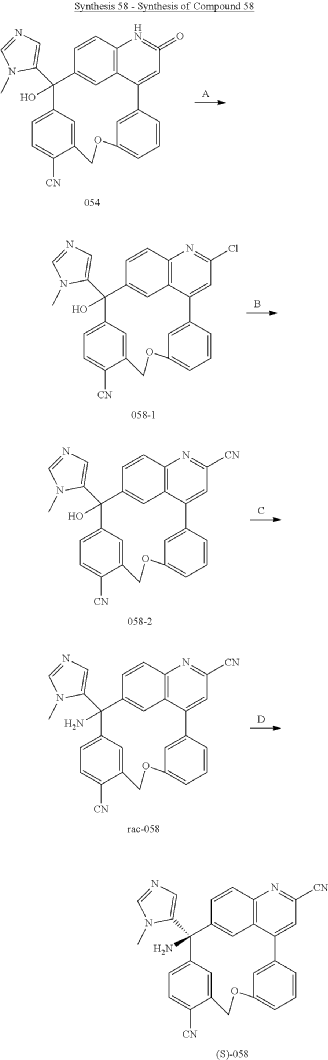
Step A: Preparation of (058-1)
Step B: Preparation of (058-2)
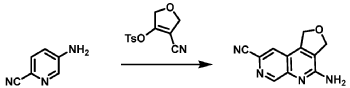
Step C: Preparation of (rac)-3-amino-3-(1-methyl-1H-imidazol-5-yl)-6-oxa-2(4,6)-quinolina-1,4(1,3)-dibenzenacyclohexaphane-22,44-dicarbonitrile (rac-058)
Step D: Preparation of (S)-3-amino-3-(1-methyl-1H-imidazol-5-yl)-6-oxa-2(4,6)-quinolina-1,4(1,3)-dibenzenacyclohexaphane-22,44-dicarbonitrile ((S)-058)
PAT
- Macrocyclic compounds and compositions, and methods of preparing and using the samePublication Number: US-2023322711-A1Priority Date: 2021-11-30
- Macrocyclic compounds and compositions, and methods of preparing and using the samePublication Number: US-12018011-B2Priority Date: 2021-11-30Grant Date: 2024-06-25
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
/////////////darlifarnib, ANAX LAB, farnesyl transferase inhibitor, antineoplastic, KO-2806, KO 2806, T206317
Danifexor
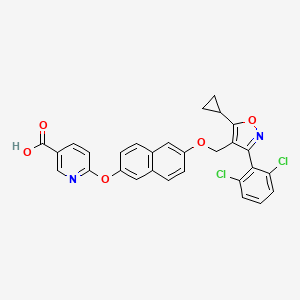
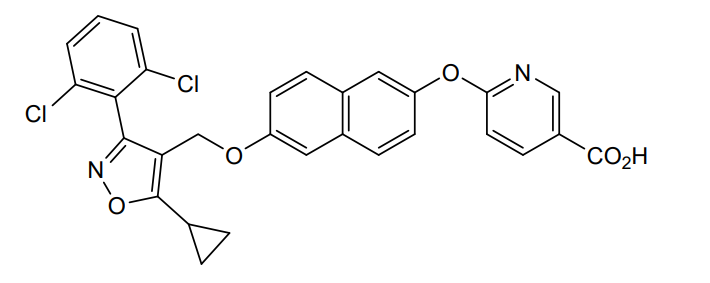
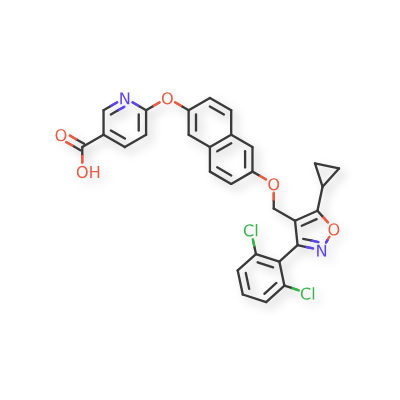
Danifexor
CAS 2648738-68-3
MF C29H20Cl2N2O5 MW547.386
6-[6-[[5-cyclopropyl-3-(2,6-dichlorophenyl)-1,2-oxazol-4-yl]methoxy]naphthalen-2-yl]oxypyridine-3-carboxylic acid
| 3-Pyridinecarboxylic acid, 6-[[6-[[5-cyclopropyl-3-(2,6-dichlorophenyl)-4-isoxazolyl]methoxy]-2-naphthalenyl]oxy]- |

farnesoid X receptor agonist, TUU8G1CX9O, HEC 96719, ASC42
Danifexor is an investigational drug that acts as a potent and selective agonist for the farnesoid X receptor (FXR). It was primarily being developed for the treatment of liver diseases such as Primary Biliary Cholangitis (PBC). ProbeChem +1
However, recent reports from April 2024 indicate that development for Danifexor has been discontinued because it was deemed non-competitive against other emerging therapies for PBC.
Key Properties and Identifiers
Danifexor is a non-steroidal molecule with specific chemical markers used in laboratory research:
- Target: Farnesoid X receptor (FXR).
Therapeutic Context
The drug was designed to target the FXR pathway, which regulates bile acid, lipid, and glucose metabolism.
- Primary Goal: Treatment of Primary Biliary Cholangitis (PBC), a chronic liver disease.
- Mechanism: As an agonist, it binds to and activates FXR to help reduce the toxic buildup of bile acids in the liver.
SYN
Example 1
Preparation of 6-((6-((5-cyclopropyl-3-(2,6-dichlorophenyl)isoxazol-4-yl) methoxy)naphthalen-2-yl)oxy)nicotinic acid (Compound 1)
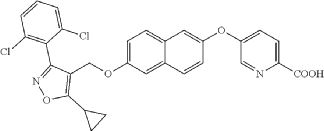
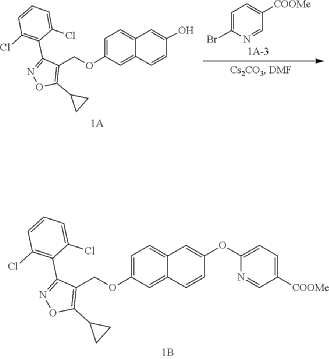
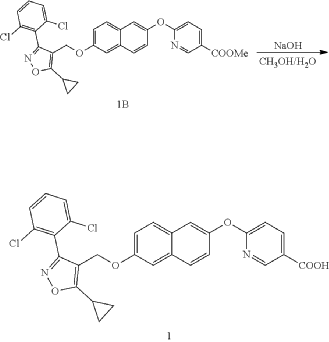
(a) Referring to the following reaction equation (Route A), Compound 1A-1 (1.0 g, 2.88 mmol, 1 eq.), Compound 1A-2 (0.46 g, 2.88 mmol, 1 eq.) and cesium carbonate (1.88 g, 5.76 mmol, 2 eq.) were dissolved in DMF (10 ml). The reaction was carried out at 65° C. for 2 h. After cooling, 10 ml water and 10 ml EA (ethyl acetate) were added for extraction, and the organic phase was washed with water and concentrated to dryness to give Compound 1A, 6-((5-cyclopropyl-3-(2,6-dichlorophenyl)isoxazol-4-yl)methoxy)naphthalen-2-ol, 0.8 g, yield: 65.0%. LCMS (ESI): calculated for C 23H 17C 12NO 3; [M+H] +: 426.1, found: 426.1.
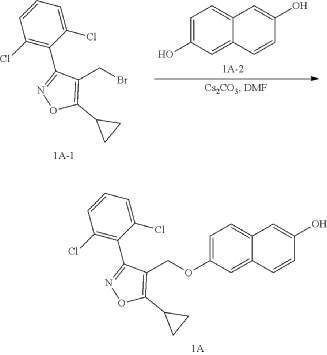
b) Referring to the following reaction equation, Compound 1A (0.2 g, 0.47 mmol, 1 eq.), 6-bromonicotinic acid methyl ester (0.1 g, 0.47 mmol, 1 eq.) and cesium carbonate (0.306 g, 0.94 mmol, 2 eq.) were dissolved in DMF (10 ml). The reaction was carried out at 65° C. for 2 h. After cooling, 10 ml water and 10 ml EA were added for extraction, and the organic phase was washed with water and concentrated to dryness to give Compound 1B, methyl 6((6((5-cyclopropyl-3-(2,6-dichlorophenyl)isoxazol-4-yl)methoxy)naphthalene-2-yl)oxy)nicotinate, 0.21 g, yield: 80.0%. LCMS (ESI): calculated for C 30H 22C 12N 2O 5; [M+H] +: 561.1, found: 561.1.
PAT
PAT
- Use and pharmaceutical composition of phenylisoxazolyl methylene-naphthalene-ether derivativesPublication Number: WO-2021109713-A1Priority Date: 2019-12-03
- Compounds for modulating activity of fxr and uses thereofPublication Number: WO-2021109712-A1Priority Date: 2019-12-03
- Uses and pharmaceutical compositions of phenylisoxazolylmethylene-naphthalene-ether derivativesPublication Number: CN-112891348-APriority Date: 2019-12-03
- Compounds that modulate FXR activity and their applicationsPublication Number: CN-112898289-APriority Date: 2019-12-03
- Compounds that modulate FXR activity and their applicationsPublication Number: CN-112898289-BPriority Date: 2019-12-03Grant Date: 2022-11-04
- Uses and pharmaceutical compositions of phenylisoxazolyl methylene-naphthalene-ether derivativesPublication Number: KR-20220101697-APriority Date: 2019-12-03
- Compounds for modulating activity of fxr and uses thereofPublication Number: EP-4073070-A1Priority Date: 2019-12-03
- Use and pharmaceutical composition of phenylisoxazolyl methylene-naphthalene-ether derivativesPublication Number: EP-4073071-A1Priority Date: 2019-12-03
- Compounds for modulating activity of fxr and uses thereofPublication Number: WO-2021108974-A1Priority Date: 2019-12-03
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
////////danifexor, ANAX LAB, farnesoid X receptor agonist, TUU8G1CX9O, HEC 96719, ASC42
Dabogratinib
Dabogratinib
CAS 2800223-30-5
MF C25H24Cl2N6O3S, 559.5 g/mol
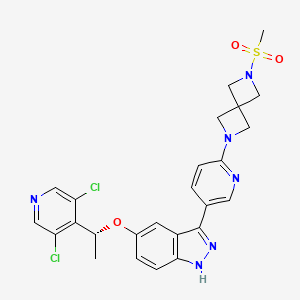
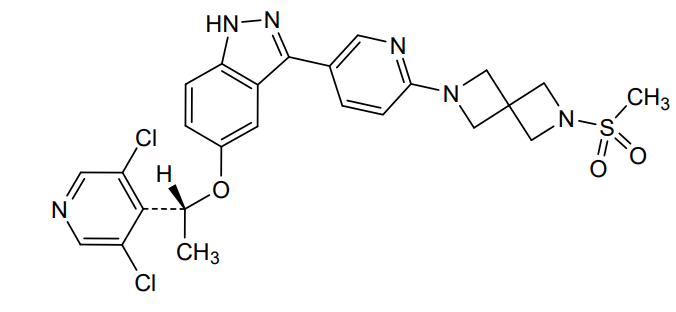
5-[(1R)-1-(3,5-dichloro-4-pyridinyl)ethoxy]-3-[6-(2-methylsulfonyl-2,6-diazaspiro[3.3]heptan-6-yl)-3-pyridinyl]-1H-indazole
(R)-5-(1-(3,5-Dichloropyridin-4-yl)ethoxy)-3-(6-(6-(methylsulfonyl)-2,6-diazaspiro[3.3]heptan-2-yl)pyridin-3-yl)-1H-indazole
[6-(5-{5-[(1R)-1-(3,5-dichloropyridin-4-yl)ethoxy]-1H-indazol-3-yl}pyridin-2-yl)-2,6-diazaspiro[3.3]heptan-2-yl](methyl)-λ6sulfanedioneTYRA-300
fibroblast growth factor receptor inhibitor, antineoplastic, TYRA-300, TYRA 300, A1AV2, FH245S2JZJ
Dabogratinib (TYRA-300) is an orally active, highly selective inhibitor of fibroblast growth factor receptor 3 (FGFR3), designed to treat cancers with FGFR3 alterations and genetic diseases like achondroplasia. It shows potent tumor growth inhibition in preclinical studies and early phase I/II (SURF301) clinical activity against advanced bladder cancer and metastatic urothelial carcinoma.
Key Aspects of Dabogratinib (TYRA-300)
- Mechanism: It acts as a selective inhibitor of FGFR3 with a high selectivity over other isoforms (FGFR1/2/4), which helps minimize toxicity.
- Target Indications: It is being developed for FGFR3-mutant cancers, including non-muscle invasive bladder cancer (NMIBC) and metastatic urothelial carcinoma, as well as pediatric achondroplasia.
- Preclinical Performance: Studies showed that it reduces tumor growth and drives tumor regression, especially in xenograft models with FGFR3-activating mutations (e.g., S249C).
- Clinical Trials:
- SURF301 (Phase I/II): Ongoing study, Tyra Biosciences reported early efficacy in patients with advanced metastatic urothelial carcinoma (mUC) harboring FGFR3 mutations/fusions.
- SURF302 (Phase II): Evaluating the drug in patients with FGFR3-altered, low-grade, intermediate-risk non–muscle invasive bladder cancer (NMIBC).
- BEACH301 (Phase II): Studying the drug in children with achondroplasia, as it is designed to increase long-bone growth.
- Properties: It is an orally bioavailable molecule with an IC50 of
for FGFR3.

Dabogratinib is an orally bioavailable, selective inhibitor of human fibroblast growth factor receptor 3 (FGFR3), with potential antineoplastic activity. Upon oral administration, dabogratinib specifically targets and binds to certain FGFR3 activating gene alterations, and specifically the gatekeeper mutants V555L/M. This blocks FGFR3-mediated signaling and leads to an inhibition of tumor cell proliferation in FGFR3-overexpressing cells. FGFR3, a receptor tyrosine kinase, is involved in angiogenesis and in the proliferation, differentiation, and survival of tumor cells. FGFR3 expression is associated with poor prognosis. It is overexpressed by certain tumor cell types.
- Efficacy and Safety of TYRA-300 in Participants With FGFR3 Altered Low Grade, Intermediate Risk Non-Muscle Invasive Bladder CancerCTID: NCT06995677Phase: Phase 2Status: RecruitingDate: 2026-04-09
- A Study of TYRA-300 in Children With Achondroplasia: BEACH301CTID: NCT06842355Phase: Phase 2Status: RecruitingDate: 2026-03-06
- Safety and Preliminary Anti-Tumor Activity of TYRA-300 in Advanced Urothelial Carcinoma and Other Solid Tumors With FGFR3 Gene AlterationsCTID: NCT05544552Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2026-01-12
PAT
Example 46. 5-[(1R)-1-(3,5-dichloro-4-pyridyl)ethoxy]-3-[6-(2-methylsulfonyl-2,6-diazaspiro[3.3]heptan-6-yl)-3-pyridyl]-1H-indazole
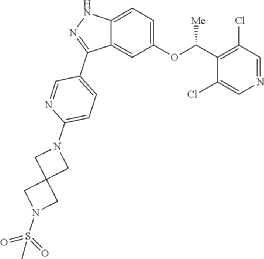
(5-[(1R)-1-(3,5-dichloro-4-pyridyl)ethoxy]-3-[6-(2-methylsulfonyl-2,6-diazaspiro[3.3]heptan-6-yl)-3-pyridyl]-1H-indazole. Triethylamine (20.5 uL, 0.148 mmol, 1.2 equiv) and methylsulfonyl chloride (9.5 uL, 0.123 mmol, 1.0 equiv) were sequentially added at room temperature to a solution of example 45 (59.0 mg, 0.123 mmol, 1 equiv) in anhydrous THE (3 mL). After stirring for 2 hours, the reaction mixture was concentrated under reduced pressure and diluted with saturated brine (30 mL) and dichloromethane (30 mL). The layers were separated. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure on to Celite (1 g). The product was purified on an Interchim automated chromatography system (RediSep Rf Gold HP C18, 15.5 g cartridge), eluting with a gradient of 0 to 100% acetonitrile in water. The fractions containing product were collected and lyophilized to give a white solid (45.0 mg, 65% yield). Analysis: LCMS: m/z=559.2 (M+H); 1H NMR (400 MHz, DMSO-d6) δ 13.02 (br s, 1H), 8.59 (s, 2H), 8.52 (dd, J=0.6, 2.2 Hz, 1H), 7.87 (dd, J=2.4, 8.6 Hz, 1H), 7.46 (d, J=8.9 Hz, 1H), 7.16 (d, J=2.1 Hz, 1H), 7.09 (dd, J=2.3, 9.0 Hz, 1H), 6.54 (dd, J=0.4, 8.6 Hz, 1H), 6.10 (q, J=6.6 Hz, 1H), 4.17 (s, 4H), 4.12 (s, 4H), 3.03 (s, 3H), 1.76 (d, J=6.6 Hz, 3H).
PAT
- Indazole compoundsPublication Number: TW-202241906-APriority Date: 2020-12-30
- Indazole compounds as kinase inhibitorsPublication Number: EP-4271673-A1Priority Date: 2020-12-30
- Indazole compounds as kinase inhibitorsPublication Number: WO-2022147246-A1Priority Date: 2020-12-30
- Indazole compounds as kinase inhibitorsPublication Number: US-12264149-B2Priority Date: 2020-12-30Grant Date: 2025-04-01
- Polymorphic compounds and uses thereofPublication Number: EP-4547670-A1Priority Date: 2022-06-29
- Indazole compounds as kinase inhibitorsPublication Number: US-12071428-B2Priority Date: 2020-12-30Grant Date: 2024-08-27
- Indazole Compounds as Kinase InhibitorsPublication Number: KR-20230152654-APriority Date: 2020-12-30
- Indazole compounds as kinase inhibitorsPublication Number: US-2024109865-A1Priority Date: 2020-12-30
- Indazole compounds as kinase inhibitorsPublication Number: US-2024208941-A1Priority Date: 2020-12-30
- Tyra-300 (5-[(1r)-1-(3,5-dichloro-4-pyridyl)ethoxy]-3-[6-(2-methylsulfonyl-2,6-diazaspiro[3.3]heptan-6-yl)-3-pyridyl]-1h-indazole ) in combination with a pd-1 or pd-l1 antagonist for use in the treatment of cancerPublication Number: WO-2025064744-A1Priority Date: 2023-09-22
- Fgfr inhibitors and methods of use thereofPublication Number: WO-2025061029-A1Priority Date: 2023-09-18
- Polymorphic compounds and uses thereofPublication Number: AU-2023300357-A1Priority Date: 2022-06-29
- Polymorphic compounds and uses thereofPublication Number: WO-2024006883-A1Priority Date: 2022-06-29
- Polymorphic compounds and uses thereofPublication Number: TW-202408493-APriority Date: 2022-06-29
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2024-09-11
PMID: 39258897
DOI: 10.1021/acs.jmedchem.4c01531
////////dabogratinib, anax lab, fibroblast growth factor receptor inhibitor, antineoplastic, TYRA-300, TYRA 300, A1AV2, FH245S2JZJ
Colfosceril miristate
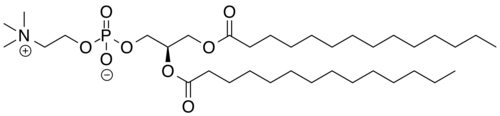
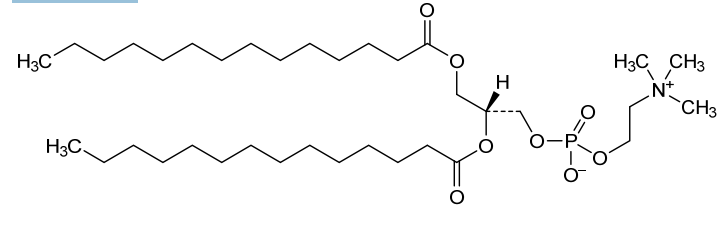
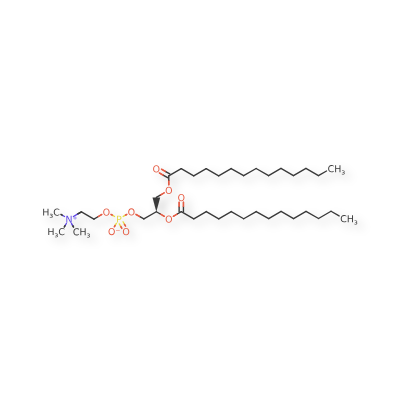
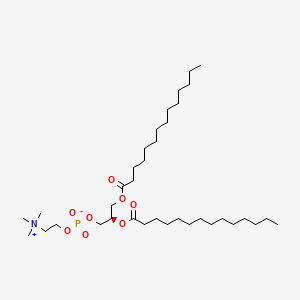
Colfosceril miristate
CAS 18194-24-6
MF C36H72NO8P MW677.9325
1,2 Dimyristoyl glycero 3 phosphorylcholine
(2R)-2,3-bis(tetradecanoyloxy)propyl 2-(trimethylazaniumyl)ethyl phosphate
surfactant replacement, DIMYRISTOYL LECITHIN, Dimyristoyllecithin, DMCP, DMPC
Colfosceril miristate (1,2-dimyristoyl-sn-glycero-3-phosphocholine or DMPC) is a synthetic phospholipid commonly used in research to study lipid bilayers, liposomes, and drug delivery systems. It serves as a model membrane system due to its phase transition properties and has shown potential in enhancing nanoparticle uptake and acting as a drug stabilizer.
Key Aspects of Colfosceril Miristate (DMPC):
- Scientific Application: Primarily used in laboratory research for studying lipid monolayers and bilayers.
- Drug Delivery: Employed in the creation of liposomes for drug delivery applications.
- Biological Activity: Exhibits antiproliferative effects on various tumor cell lines and can increase the cellular uptake of nanoparticles.
- Characteristics: It is a synthetic phospholipid, frequently studied for its phase transition temperature (approx.
).
- Storage: Should be stored at
or
to maintain stability.
Important Distinction:
It is crucial not to confuse Colfosceril miristate (DMPC) with Colfosceril palmitate (DPPC). Colfosceril palmitate is a different synthetic surfactant historically used in medicine to treat neonatal respiratory distress syndrome
1,2-DIMYRISTOYL-SN-GLYCERO-3-PHOSPHOCHOLINE (dimyristoyl phosphatidylcholine, DMPC) is a synthetic phospholipid used in liposomes and lipid bilayers for the study of biological membranes. DMPC is a frequently studied artificial lipid because it undergoes a phase transition at a convenient temperature. Upon cooling below 23.6°C it undergoes a transition from the liquid crystalline phase to the solid rippled phase, characterized by periodic corrugations of the bilayer.
Dimyristoylphosphatidylcholine is a phosphatidylcholine, a kind of phospholipid. Along with other lipids, it can be used to prepare liposomes.[1]
PAT
PAT
Compositions of Phosphorylated Tau Peptides and Uses Thereof
Publication Number: US-2025326806-A1
- Antigen Binding Polypeptides, Polypeptide Complexes and Methods of Use ThereofPublication Number: US-2025326823-A1
- Variant rna-guided cas12f4 nucleases and dna binding proteinsPublication Number: US-2025327095-A1
- Modulation of novel immune checkpoint targetsPublication Number: US-12447213-B2Grant Date: 2025-10-21
- Therapeutic peptidesPublication Number: US-12448425-B2Grant Date: 2025-10-21
- Cytokine conjugates for the treatment of proliferative and infectious diseasesPublication Number: US-2025325632-A1
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
| Names | |
|---|---|
| Systematic IUPAC name(2R)-2,3-Bis(tetradecanoyloxy)propyl 2-(trimethylazaniumyl)ethyl phosphate | |
| Other names1,2-dimyristoylphosphatidylcholine, 1,2-dimyristoyl-sn-glycero-3-phosphocholine, 1,2-ditetradecanoyl-sn-glycero-3-phosphocholine, DMPC, 14:0 PC | |
| Identifiers | |
| CAS Number | 18194-24-6 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:45240 |
| ChEMBL | ChEMBL1235508 |
| ChemSpider | 4573168 |
| ECHA InfoCard | 100.038.245 |
| EC Number | 242-085-9 |
| PubChem CID | 5459377 |
| UNII | 52QK2NZ2T0 |
| CompTox Dashboard (EPA) | DTXSID00860227 |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C36H72NO8P |
| Molar mass | 677.945 g·mol−1 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).Infobox references | |
References
- Liposomal drug delivery system from laboratory to clinic, N. A. Kshirsagar, S. K. Pandya, G. B. Kirodian, S. Sanath, Journal of Postgraduate Medicine, 51 (#5) (2005), pp. 5-15, PMID 16519249.
//////////colfosceril miristate, ANAX LAB, surfactant replacement, DIMYRISTOYL LECITHIN, Dimyristoyllecithin, DMCP, DMPC
Anvumetostat
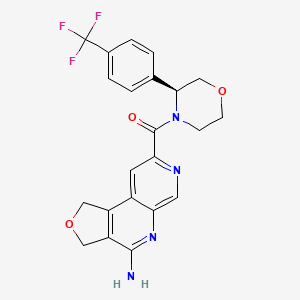
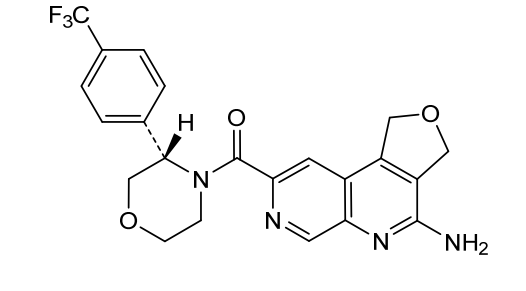
Anvumetostat
CAS 2790567-82-5
MF C22H19F3N4O3 MW444.4 g/mol
(4-amino-1,3-dihydrofuro[3,4-c][1,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone
(4-amino-1,3-dihydrofuro[3,4-c][1,7]naphthyridin-8-yl){(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl}methanone
antineoplastic, AMG 193, QAT649EJ5E, PRMT5-IN-27,
Anvumetostat (also known as AMG 193) is an orally available, small-molecule inhibitor of protein arginine methyltransferase 5 (PRMT5), primarily being developed for the treatment of advanced solid tumours with MTAP-null (methylthioadenosine phosphorylase-deficient) mutations.
Mechanism of Action
- Targeting PRMT5: It is a potent and selective MTA-cooperative inhibitor of PRMT5.
- Synthetic Lethality: In cells where the MTAP gene is deleted (a common occurrence in various cancers), a metabolite called MTA (methylthioadenosine) accumulates. Anvumetostat selectively binds to the PRMT5-MTA complex, inhibiting its methyltransferase activity.
- Cellular Impact: By blocking PRMT5, the drug reduces the methylation of arginine residues in histones (H2A, H3, and H4), which can lead to decreased growth or death of cancer cells.
Clinical Development
Anvumetostat was initially developed by Amgen, Inc. and is currently in clinical trials. Institute (.gov) +1
- Current Status: As of early 2026, it is in Phase 2 of global research and development.
- Study Focus: Trials are evaluating its efficacy both as a monotherapy and in combination with other treatments for adult patients with metastatic or locally advanced MTAP-null cancers.
Key Identifiers
- Alternate Names: AMG 193, AMG-193.
- Chemical Class: Orally bioavailable small molecule.
- Genetic Biomarker: Specifically targets cancers with MTAP-null status
Anvumetostat is an orally available small molecule inhibitor of protein arginine methyltransferase 5 (PRMT5), with potential antiproliferative and antineoplastic activities. Upon oral administration, anvumetostat selectively binds to PRMT5 and inhibits its function. By inhibiting its methyltransferase activity, levels of both monomethylated and dimethylated arginine residues in histones H2A, H3 and H4 are decreased. This modulates the expression of genes involved in several cellular processes, including cellular proliferation. This may increase the expression of antiproliferative genes and/or decrease the expression of genes that promote cell proliferation, which may lead to decreased growth of rapidly proliferating cells, including cancer cells. PRMT5, a type II methyltransferase that catalyzes the formation of both omega-N monomethylarginine (MMA) and symmetric dimethylarginine (sDMA) on histones and a variety of other protein substrates involved in signal transduction and cellular transcription, is overexpressed in several neoplasms. Elevated levels are associated with decreased patient survival. Methylthioadenosine phosphorylase (MTAP) is deleted in certain cancer cells leading to an accumulation of methylthioadenosine (MTA). As MTA inhibits PRMT5, MTAP-null cancer cells are specifically sensitive to PRMT5 inhibitors.
SYN
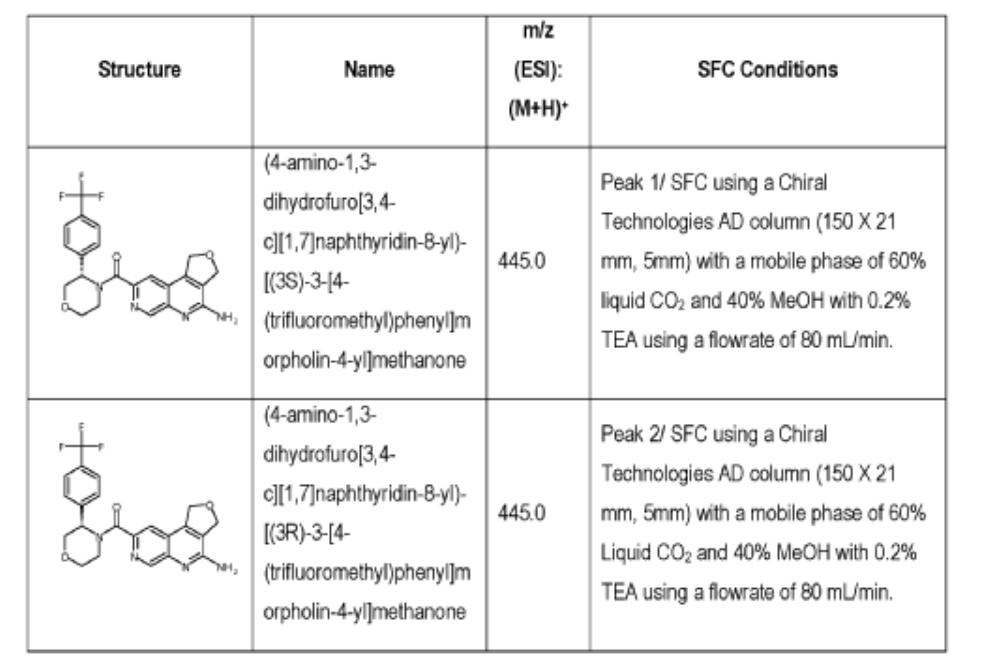
[0163] Examples 481 and 482: (4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4- (trifluoromethyl)phenyl)morpholino)methanone
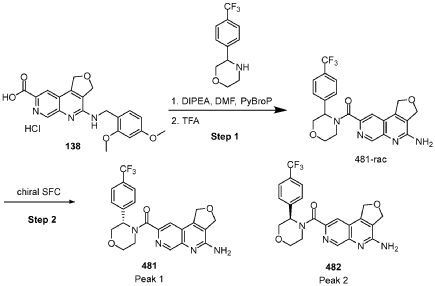
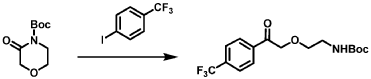
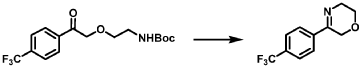
[0164] Step 1: To a solution of 3-(4-(trifluoromethyl)phenyl)morpholine (0.100 g, 0.432 mmol, Enamine), 4-((2,4-dimethoxybenzyl)amino)-l,3-dihydrofuro[3,4-c][l,7]naphthyridine-8-carboxylic acid hydrochloride (138) (0.271 g, 0.649 mmol) and l,l’-dimethyltriethylamine (0.559 g, 0.755 mL, 4.32 mmol, Sigma- Aldrich Corporation) in DMF (4 mL) was added bromotripyrrolidinophosphonium hexafluorophosphate (0.202 g, 0.432 mmol, Sigma-Aldrich Corporation) and the resulting mixture was heated at 50 °C for 30 min. The reaction was brought to rt, diluted with water, sat.NaHCCh and extracted with EtOAc (3x). The combined organics were dried over Na2SO4, filtered and concentrated. The residue was then chromatographed on silica gel using 0-50% 3:1 EtOAc/EtOH in heptane to afford (4-((2,4-dimethoxybenzyl)amino)- 1 ,3 -dihy drofuro [3 ,4-c] [ 1 ,7]naphthyridin-8-y 1) (3 – (4 -(trifluoromethyl)phenyl)morpholino)methanone (0.160 g, 0.269 mmol, 62.2% yield) as a light yellow solid, m/z (ESI): 595 (M+H)+.
[0165] To a solution of (4-((2,4-dimethoxybenzyl)amino)-l,3-dihydrofuro[3,4-c] [l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone (0.160 g, 0.269 mmol, 62.2 % yield) in DCM (2 mL) was added TFA (14.80 g, 10 mL, 130 mmol, Aldrich) and the resulting mixture was heated at 50 °C for 1 h. The reaction was concentrated, washed with 10% Na2CO3 and extracted with DCM. The combined organics were concentrated and chromatographed on silica gel using 0-50% 3:1 EtOAc/EtOH in heptane to afford (4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone as the TFA salt (0.078 g, 0.140 mmol, 32.3% yield) as an off-white solid, m/z (ESI): 445 (M+H)+.
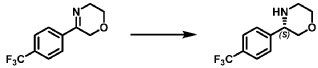
[0166] Step 2: (S)-(4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4- (trifluoromethy l)phenyl)morpholino)methanone and (R)-(4-amino- 1 ,3 -dihy drofuro [3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone
(4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone 2,2,2-trifluoroacetate were separated via preparative SFC using a Chiral Technologies AD column (150 x 21 mm, 5mm) with a mobile phase of 60% Liquid CO2 and 40% MeOH with 0.2% TEA using a flowrate of 80 mL/min to generate peak 1, (S)-(4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone with an ee of >99%, and peak 2, (R)-(4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone with an ee of 99.28%. Peak assignment determined by
SFC with AD column with 60% Liquid CO2 and 40% MeOH with 0.2% TEA and absolute
stereochemistry was arbitrarily assigned.
Peak 1: (S)-(4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone (481) as a white solid . m/z (ESI): 445 (M+H)+. NMR 1H (400 MHz, DMSO-d6) 5 ppm 8.67 – 9.03 (m, 1 H), 7.85 (s, 1 H), 7.77 (br s, 4 H), 7.07 (br s, 2 H), 5.75 (s, 1 H), 5.37 (br s, 2 H), 5.04 (br s, 2 H), 4.46 – 4.61 (m, 1 H), 3.89 (br dd, J=12.2, 3.3 Hz, 4 H), 3.58 (br d, ./=5,8 Hz, 1 H). 19F NMR (377 MHz, DMSO-d6 ) 5 ppm -60.90 (br s, 3 F).
Peak 2: (R)-(4-amino- 1 ,3 -dihy drofuro [3 ,4-c] [ 1 ,7]naphthyridin-8-yl)(3 -(4-(trifluoromethyl)phenyl)morpholino)methanone (482) as a white solid, m/z (ESI): 445 (M+H)+. NMR 1H (400 MHz, DMSO-d6) 5 ppm 8.88 (br s, 1 H), 7.85 (s, 1 H), 7.77 (br d, J=1.7 Hz, 4 H), 7.07 (br s, 2 H),
5.69 – 5.78 (m, 1 H), 5.37 (br s, 2 H), 5.04 (br s, 2 H), 4.45 – 4.61 (m, 1 H), 3.89 (br dd, J=12.4, 3.3 Hz, 4 H), 3.51 – 3.64 (m, 1 H). 19F NMR (DMSO-d6, 377 MHz) 5 -60.90 (s, 3 F).
SYN
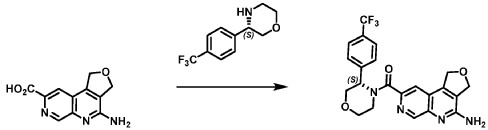
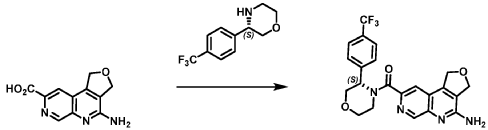
Example 4. Synthesis of Compound I – (4-amino-1 ,3-di hydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone
Reaction Scale 1
[0137] 4-Amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridine-8-carboxylic acid (1.0 kg, 4.3 mol, 1.0 equiv), (3S)-3-[4-(trifluoromethyl)phenyl]morpholine (1.2 kg, 5.2 mmol, 1.2- equiv), and DMF, (6.6 kg, 7.0 V) were charged to a clean, dry reactor. To the mixture was added triethylamine (1.1 Kg, 13.8 mol, 2.6 equiv). The mixture was cooled to 10 ± 5 °C and O-(benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate (TBTU) (1.67 kg, 5.2 mol, 1.2 equiv) was added slowly. Next, an additional amount of DMF (0.94 Kg, 1 V) was added. The reaction mixture was warmed to 25 ± 5 °C and stirred over 18 hours. Water (1 .0 kg, 1 V) was charged followed by MeCN (1 .6 kg, 2 V) and the reaction mass was warmed to 45 °C. Next, water (7.0 Kg, 7 V) was added over 30 min. A seed lot of 4-amino-1 ,3-dihydrofuro[3,4-c][1,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone (10 g, 22 mmol, 0.01 equiv), was charged and the mixture was stirred at 45 °C for over 2 hours before being cooled to 20 °C over 10 hours. Water (12.0 kg, 12 V) was added over 2 hours at 20 °C and further stirred for over 4 hours before being filtered. The reactor was rinsed with a mixture of 10% DMF in water (9.83 kg, 10 V) and the resulting rinse mixture was used to wash the cake. The reactor was rinsed with a mixture of water (10.0k kg, 10 V) and the resulting rinse mixture was used to wash the cake. This rinsing and washing protocol was repeated once more with water (10.0k kg, 10V). The cake was dried under vacuum with a stream of nitrogen to afford (4-amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-
(trifluoromethyl)phenyl]morpholin-4-yl]methanone. LCMS: 445.20 1H NMR (400 MHz, DMS0-d6 at 130 °C): 8.87 (s, 1 H), 7.80 (s, 1 H), 7.73 (d, 0=8.7 Hz, 2H), 7.71 (d, 0=8.7 Hz, 2H), 6.58 (br s, 2H), 5.72 (br s, 1 H), 5.38 (m, 2H), 5.09 (t, 0=3.5 Hz, 2H), 4.44 (br d, 0=12.3 Hz, 1 H), 4.08 (br d, 0=13.4 Hz, 1 H), 3.96 (dd, 0=12.3, 3.7 Hz, 1 H), 3.86 (br dd, 0=11 .4, 3.0 Hz, 1 H), 3.66 (td, 0=11 .4, 3.0 Hz, 1 H), 3.28 (m, 1 H).
Reaction Scale 2
[0138] 4-Amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridine-8-carboxylic acid (85.0 g, 352.2 mmol, 1.0 equiv), (3S)-3-[4-(trifluoromethyl)phenyl]morpholine (99.6 g, 422.6 mmol, 1.2- equiv), and DMF, (674 mL, 8.7 mol, 7.9 V) were charged to a clean, dry 5 L reactor. To the mixture was added 1 -methylimidazole (75.2 g, 916.2 mmol, 2.6 equiv). The mixture was cooled to 0 °C and N,N,N’,N’-tetramethylchloroformamidinium hexafluorophosphate (TCFH) (118.6 g, 422.6 mmol, 1.2 equiv) was added slowly. Next, an additional amount of DMF (170 mL, 2 V) was added at 0 °C. The reaction mixture was warmed to 25 °C and stirred overnight. Next, the reaction mass was warmed to 45 °C and 2-methyltetrahydrofuran, (169.2 mL, 2 V) was added followed by slow addition of water (850 mL, 10 V) over 30 min by addition funnel. A seed lot of 4-amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone (1.6 g, 3.5 mmol, 0.1 equiv), was charged as a slurry in a 1 :1 v/v of DMF and water (31 .3 mL) and the mixture was stirred at 45 °C for approximately 12 hrs. Water (510 mL, 6 V) was added over 1 h 10 min by addition funnel and the mixture was further stirred at 45°C for 30 min before being filtered. The reactor was rinsed with water (340 mL, 4 V) and the resulting rinse mixture was used to wash the cake. This rinsing and washing protocol was repeated twice more. The cake was dried under vacuum with a stream of nitrogen to afford (4-amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone. LCMS: 445.20 1H NMR (400 MHz, DMSO-d6 at 130 °C): 8.87 (s, 1 H), 7.80 (s, 1 H), 7.73 (d, J=8.7 Hz, 2H), 7.71 (d, J=8.7 Hz, 2H), 6.58 (br s, 2H), 5.72 (br s, 1 H), 5.38 (m, 2H), 5.09 (t, J=3.5 Hz, 2H), 4.44 (br d, J=12.3 Hz, 1 H), 4.08 (br d, J=13.4 Hz, 1 H), 3.96 (dd, J=12.3, 3.7 Hz, 1 H), 3.86 (br dd, J=11.4, 3.0 Hz, 1 H), 3.66 (td, J=11.4, 3.0 Hz, 1 H), 3.28 (m, 1 H).
Reaction Scale 3:
[0139] 4-Amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridine-8-carboxylic acid (Compound A’) (20.0 g, 86.5 mmol, 1.0 equiv) was added to dimethylsulfoxide (400 mL) at 20 °C. To the mixture was added 1 ,T-carbonyldiimidazole (15.4 g, 95.2 mmol, 1.1 equiv) and the mixture was heated to 60 °C for 1 hour. A solution of (S)-3-(4-(trifluoromethyl)phenyl)morpholin-4-ium chloride (25.5 g, 95.2 mmol, 1.1 equiv) and dimethylsulfoxide (40 mL) was added, and the mixture was heated to 80 °C for 11 hours. The reaction mixture was cooled to 35 °C, then water (265 mL) was added, then the batch was cooled to 20 °C. The reaction was filtered, washed with 40% water:DMSO (80 mL), then washed with water (100 mL). The cake was dried under vacuum with a stream of nitrogen to afford (4-amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone (Compound I). LCMS: 445.20 1H NMR (400 MHz, DMSO-d6 at 130 °C): 8.87 (s, 1 H), 7.80 (s, 1 H), 7.73 (d, J=8.7 Hz, 2H), 7.71 (d, J=8.7 Hz, 2H), 6.58 (br s, 2H), 5.72 (br s, 1 H), 5.38 (m, 2H), 5.09 (t, >3.5 Hz, 2H), 4.44 (br d, >12.3 Hz, 1H), 4.08 (br d, >13.4 Hz, 1 H), 3.96 (dd, >12.3, 3.7 Hz, 1 H), 3.86 (br dd, >11 .4, 3.0 Hz, 1 H), 3.66 (td, >11 .4, 3.0 Hz, 1 H), 3.28 (m, 1 H).
Recrystallization of Compound I
[0140] A clean, dry 5 L reactor was charged with (4-amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone (279.7 g, 0.6 mol, 1.0 equiv) followed by acetone (6.2 L,
22 V). The mixture was stirred at 40 °C for 15 minutes before cooling to 25 °C. The reactor was discharged into a flask and the reactor was rinsed with acetone and the process stream was polish-filtered back into the reactor.
The reactor jacket was set to 65 °C and the reaction volume was reduced to approximately 6 V by distillation at atmospheric pressure, crystallization was observed. The reaction temperature was set to cool to 20 °C over two hours. Heptane (2.8 L, 10 V) was added over two hours. The slurry was filtered and the cake was washed twice with a 4:1 Heptane/acetone mix (750 mL, 3 V each) and dried under vacuum with a nitrogen purge to afford (4-amino-1,3-dihydrofuro[3,4-c][1,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl] methanone.

ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Process for synthesizing naphthyridine derivatives and intermediates thereofPublication Number: EP-4396170-A1Priority Date: 2021-08-30
- PRMTS inhibitorsPublication Number: CN-116888120-APriority Date: 2020-12-16
- Prmts inhibitorsPublication Number: WO-2022132914-A1Priority Date: 2020-12-16
- Prmts inhibitorsPublication Number: EP-4263545-A1Priority Date: 2020-12-16
- Mta-cooperative prmt5 inhibitors for use in the treatment of cancerPublication Number: EP-4572760-A1Priority Date: 2022-08-15
- Cancer treatments using mta-cooperative prmt5 inhibitorsPublication Number: WO-2023196545-A1Priority Date: 2022-04-08
- Process for the synthesis of naphthyridine derivatives and intermediates thereofPublication Number: CN-117897379-APriority Date: 2021-08-30
- Process for synthesizing naphthyridine derivatives and intermediates thereofPublication Number: WO-2023034786-A1Priority Date: 2021-08-30
- Process for Synthesizing Naphthyridine Derivatives and Intermediates ThereofPublication Number: US-2024360147-A1Priority Date: 2021-08-30
- Prmt5 inhibitor for use in cancer therapyPublication Number: WO-2024170488-A1Priority Date: 2023-02-13
- Cancer treatments using a prmt5 inhibitor and a mat2a inhibitorPublication Number: WO-2024118897-A1Priority Date: 2022-11-30
- Cancer treatments using a prmt5 inhibitor and a mat2a inhibitorPublication Number: EP-4626435-A1Priority Date: 2022-11-30
- MTA synergizes with PRMT5 inhibitors for cancer treatmentPublication Number: CN-119730853-APriority Date: 2022-08-15
- Mta-cooperative prmt5 inhibitors for use in the treatment of cancerPublication Number: WO-2024038004-A1Priority Date: 2022-08-15
////////anvumetostat, ANAX LAB, antineoplastic, AMG 193, QAT649EJ5E, PRMT5-IN-27,
Amsulostat
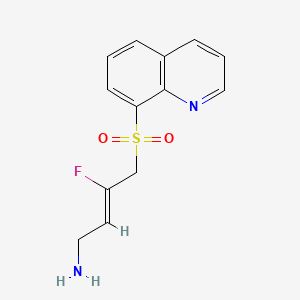
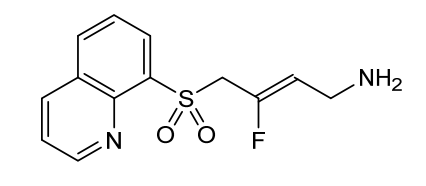
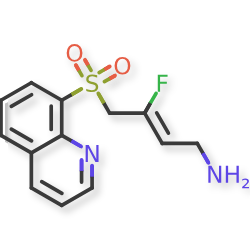
Amsulostat
CAS 2409963-83-1
MF C13H13FN2O2S MW280.32
2-Buten-1-amine, 3-fluoro-4-(8-quinolinylsulfonyl)-, (2Z)-
(2Z)-3-fluoro-4-(quinolin-8-ylsulfonyl)but-2-en-1-amine
(Z)-3-fluoro-4-quinolin-8-ylsulfonylbut-2-en-1-amine
(2Z)-3-fluoro-4-(quinolin-8-ylsulfonyl)but-2-en-1-amine
protein-lysine-oxidase (LOX) inhibitor, antifibrotic, PXS 5505, LOX inhibitor PXS-5505, LOX-IN-3, pan-LOX inhibitor PXS-5505, DO94E28WYW, SNT 5505
Amsulostat is an orally available, small-molecule, irreversible inhibitor of all lysyl oxidases (LOX) family members, with potential antifibrotic activity. Upon oral administration, amsulostat targets, binds to and inhibits the activity of all enzymes in the LOX family. This prevents the post-translational oxidative deamination of lysine residues on target proteins, including collagen and elastin, and reduces the formation of deaminated lysine (allysine), the formation of inter- and intramolecular cross-linkages and may prevent remodeling of the extracellular matrix (ECM), thereby reducing fibrotic tissue formation in certain chronic fibrotic diseases. LOX is often upregulated in fibrotic tissue and plays a key role in fibrosis.
Amsulostat (formerly PXS-5505) is an orally available, investigational, pan-lysyl oxidase (pan-LOX) inhibitor designed by Syntara to treat fibrotic diseases and solid tumors. It works by preventing collagen cross-linking and remodeling of the extracellular matrix, effectively reducing fibrosis. The drug is currently in Phase 2 clinical trials for myelofibrosis, showing promise in reducing symptom burden and spleen volume, and is also being studied for myelodysplastic syndrome (MDS) and pancreatic cancer.
Key Aspects of Amsulostat:
- Mechanism of Action: Irreversibly inhibits all LOX family members (LOX, LOXL1-4), reducing fibrotic tissue.
- Clinical Status (Myelofibrosis): Phase 2a data showed 73% of patients (who were suboptimal responders to ruxolitinib) achieved
reduction in total symptom score, with significant spleen volume reduction.
- Clinical Status (Other Cancers): Phase 2 trials (AZALOX) are evaluating its use in myelodysplastic syndrome (MDS) and chronic myelomonocytic leukemia (CMML). It is also being tested in combination with chemotherapy for pancreatic cancer to improve drug delivery to tumors.
- Regulatory Status: Has received Orphan Drug Designation for primary myelofibrosis from the FDA (USA) and EMA (Europe).
- Safety Profile: Clinical trials have reported it is well-tolerated with no treatment-related serious adverse events in early findings.

Amsulostat’s ability to target the stiff, fibrotic environment surrounding tumors makes it a promising “add-on” therapy to increase the effectiveness of existing cancer treatments, including chemotherapy and immunotherapy.
An orally available, small-molecule, irreversible inhibitor of all lysyl oxidases (LOX) family members, with potential antifibrotic activity. Upon oral administration, amsulostat targets, binds to and inhibits the activity of all enzymes in the LOX family. This prevents the post-translational oxidative deamination of lysine residues on target proteins, including collagen and elastin, and reduces the formation of deaminated lysine (allysine), the formation of inter- and intramolecular cross-linkages and may prevent remodeling of the extracellular matrix (ECM), thereby reducing fibrotic tissue formation in certain chronic fibrotic diseases. LOX is often upregulated in fibrotic tissue and plays a key role in fibrosis.
SYN
syn

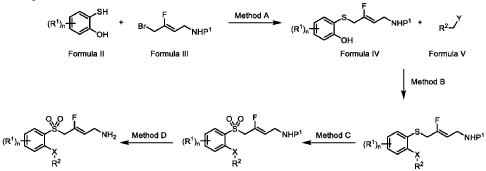
(Z)-3-fluoro-4-(quinolin-8-ylsulfonyl)but-2-en-1-amine dihydrochloride (Compound 33)
[0282] White solid; m.p.150-153 °C; 1H NMR (300 MHz, CD3OD) d ppm: 9.18 (d, J = 4.7 Hz, 1H), 8.70 (dd, J = 8.4, 2.6 Hz, 1H), 8.57 (d, J = 7.4 Hz, 1H), 8.45 (d, J = 8.5 Hz, 1H), 7.99– 7.68 (m, 2H), 5.22 (dt, J = 32.9, 7.4 Hz, 1H), 5.00 (d, J = 19.4 Hz, 2H), 3.60 (d, J = 7.7 Hz, 2H); LCMS: for C13H13FN2O2S calculated 280.1, found 281.1 [M+1]+.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Haloallylamine sulfone derivative inhibitors of lysyl oxidases and uses thereofPublication Number: EP-3829520-B1Priority Date: 2018-08-03Grant Date: 2024-11-27
- Haloallylamine sulfone derivative inhibitors of lysyl oxidases and uses thereofPublication Number: US-2025082593-A1Priority Date: 2018-08-03
- Haloallylamine sulfone derivative inhibitors of lysyl oxidases and uses thereofPublication Number: US-2021353571-A1Priority Date: 2018-08-03
- Lox enzyme inhibiting methods and compositionsPublication Number: EP-4138824-A1Priority Date: 2020-04-21
- Haloallylamine sulfone derivative inhibitors of lysyl oxidases and uses thereofPublication Number: WO-2020024017-A1Priority Date: 2018-08-03
- Inhibitors of haloallylamine sulfone derivatives of lysyl oxidase and uses thereofPublication Number: KR-20210045984-APriority Date: 2018-08-03
- Halide allylamine derivate inhibitor for amine oxidase and its usePublication Number: TW-202019877-APriority Date: 2018-08-03
- Haloallylamine sulfone derivative inhibitors of lysyl oxidases and uses thereofPublication Number: US-12178791-B2Priority Date: 2018-08-03Grant Date: 2024-12-31
- Bithiazol deratives as inhibitors of lysyl oxidasesPublication Number: EP-4333843-A1Priority Date: 2021-05-13
- Inhibitors of lysyl oxidasesPublication Number: US-11712437-B2Priority Date: 2021-05-13Grant Date: 2023-08-01
- Method for selecting cancer patients for whom combination therapy of retinoid with cancer therapeutic agent is effective, and combination drug of retinoid with cancer therapeutic agentPublication Number: CN-115997122-APriority Date: 2020-06-26
- Method for selecting cancer patient for which combination therapy of retinoid and cancer treatment agent will be effective, and combined drug of retinoid and cancer treatment agentPublication Number: EP-4173639-A1Priority Date: 2020-06-26
- Method for selecting cancer patients for whom combination therapy with retinoid and cancer therapeutic agent is effective, and combination medicament with retinoid and cancer therapeutic agentPublication Number: US-2023301950-A1Priority Date: 2020-06-26
////////////amsulostat, ANAX LAB, protein-lysine-oxidase (LOX) inhibitor, antifibrotic, PXS 5505, LOX inhibitor PXS-5505, LOX-IN-3, pan-LOX inhibitor PXS-5505, DO94E28WYW, SNT 5505
Amezalpat
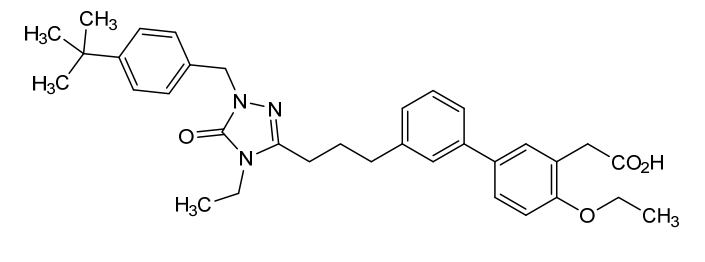
Amezalpat
CAS 1616372-41-8
MF C34H41N3O4 MW555.7 g/mol
- [1,1′-Biphenyl]-3-acetic acid, 3′-[3-[1-[[4-(1,1-dimethylethyl)phenyl]methyl]-4-ethyl-4,5-dihydro-5-oxo-1H-1,2,4-triazol-3-yl]propyl]-4-ethoxy-
- 2-(3′-(3-(1-(4-(tert-Butyl)benzyl)-4-ethyl-5-oxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)propyl)-4-ethoxy-[1,1′-biphenyl]-3-yl)acetic acid
- 3′-(3-(1-((4-(1,1-DIMETHYLETHYL)PHENYL)METHYL)-4-ETHYL-4,5-DIHYDRO-5-OXO-1H-1,2,4-TRIAZOL-3-YL)PROPYL)-4-ETHOXY(1,1′-BIPHENYL)-3-ACETIC ACID
2-[5-[3-[3-[1-[(4-tert-butylphenyl)methyl]-4-ethyl-5-oxo-1,2,4-triazol-3-yl]propyl]phenyl]-2-ethoxyphenyl]acetic acid
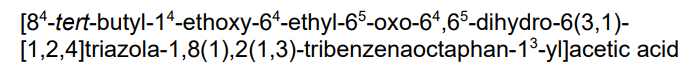
peroxisome proliferator-activated receptor alpha (PPARα) antagonist, antineoplastic, TPST 1120, FDA Fast Track, Orphan Drug, 1EQ4LQN9N3
Amezalpat (formerly TPST-1120) is an investigational, oral, small-molecule inhibitor targeting peroxisome proliferator-activated receptor alpha (PPAR being developed by Tempest Therapeutics. It works by directly targeting tumor cells and reducing immune suppression in the tumor microenvironment. In combination with atezolizumab and bevacizumab, it has shown improved survival in hepatocellular carcinoma (HCC) patients, receiving FDA Fast Track and Orphan Drug designations.
Key Details on Amezalpat
- Indication: Primarily being studied for unresectable or metastatic hepatocellular carcinoma (liver cancer).
- Mechanism: A selective, competitive antagonist of PPAR
, which plays a role in fatty acid metabolism in cancer cells.
- Clinical Efficacy: A phase 1b/2 study indicated that adding amezalpat to standard-of-care (atezolizumab + bevacizumab) improved median overall survival to 21 months compared to 15 months for the control, according to Tempest Therapeutics.
- Trial Status: A pivotal Phase 3 study (NCT06680258) to evaluate this combination as a first-line treatment is planned for 2025.
- Other Potential Uses: Preclinical data suggests potential activity in other advanced solid tumors, including renal cell carcinoma.
Disclaimer: Amezalpat is an investigational agent and is not yet approved by the FDA for widespread clinical use.
Amezalpat is an orally bioavailable, small molecule, selective and competitive antagonist of peroxisome proliferator activated receptor alpha (PPARa), with potential immunomodulating and antineoplastic activities. Upon oral administration, amezalpat targets, binds to and blocks the activity of PPARa, thereby blocking transcription of PPARa target genes leading to an intracellular metabolism shift from fatty acid oxidation (FAO) to glycolysis in FAO-dependent tumors and reducing the production of fatty acids in the tumor microenvironment (TME). As fatty acids are essential for tumor cell growth in FAO-dependent tumor cells and are needed for the metabolism of suppressive immune cells in the TME, including regulatory T-cells (Tregs), reducing the amount of fatty acids leads to a direct killing of FAO-dependent tumor cells. It also skews macrophages from the immune suppressive M2 phenotype to an effector M1 phenotype and facilitates the cytotoxicity of immune effector cells, thereby stimulating an anti-tumor immune response and further killing tumor cells. Amezalpat also restores the natural inhibitor of angiogenesis thrombospondin-1 (TSP-1) and stimulator of interferon genes (STING) in the TME. PPARa, a ligand-activated nuclear transcription factor and metabolic checkpoint, regulates the expression of FAO genes and lipid metabolism. It plays a key role in immunosuppression in the TME. FAO is a metabolic pathway essential to tumor growth, survival and immunosuppression.
SYN
Example 6: 2-(3′-(3-(1-(4-(tert-Butyl)benzyl)-4-ethyl-5-oxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)propyl)-4-ethoxy-[1,1′-biphenyl]-3-yl)acetic acid
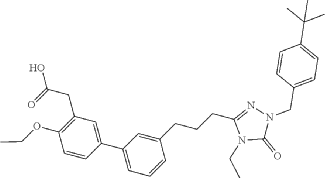
SYN
SYN
WO2014099503 TRIAZOLONE COMPOUNDS AND USES THEREOF
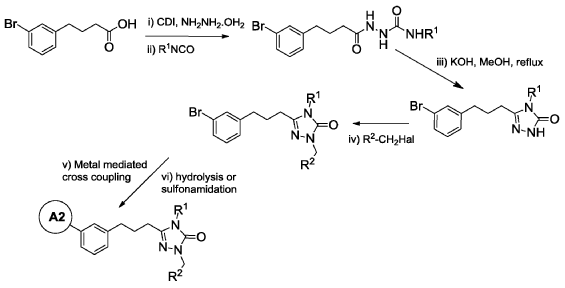
Example 6: 2-(3′-(3-(1-(4-(tert-Butyl)benzyl)-4-ethyl-5-oxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)propyl)-4-ethoxy-[1, 1′-biphenyl]-3-yl)acetic acid
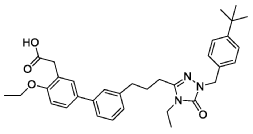
Pat
WO2025235527 CRYSTALLINE FORMS OF A PPAR ALPHA ANTAGONIST
2-(3′-(3-(l-(4-(tertbutyl)benzyl)-4-ethyl-5-oxo-4,5-dihydro-lH-l,2,4-triazol-3-yl)propyl)-4-ethoxy-[1,T-biphenyl]-3-yl)acetic acid, depicted below as Compound A
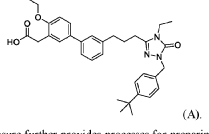
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Triazolone compounds and uses thereofPublication Number: US-2017239223-A1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: WO-2014099503-A1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: US-10568871-B2Priority Date: 2012-12-20Grant Date: 2020-02-25
- Triazolone compounds and uses thereofPublication Number: US-2024041837-A1Priority Date: 2012-12-20
- Compound or pharmaceutically acceptable salt thereof, pharmaceutical composition and uses thereofPublication Number: BR-112015013350-B1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: US-2015344446-A1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: US-11666557-B2Priority Date: 2012-12-20Grant Date: 2023-06-06
- Triazolone compounds and uses thereofPublication Number: US-2020138790-A1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: US-9676754-B2Priority Date: 2012-12-20Grant Date: 2017-06-13
- Triazolone compounds and uses thereofPublication Number: CA-2894281-CPriority Date: 2012-12-20Grant Date: 2021-04-20
- Triazolone compounds and uses thereofPublication Number: WO-2024102620-A2Priority Date: 2022-11-09
- Triazolone compounds and uses thereofPublication Number: AU-2013363398-B2Priority Date: 2012-12-20Grant Date: 2017-06-01
- Triazolone compounds and uses thereofPublication Number: EP-2935228-B9Priority Date: 2012-12-20Grant Date: 2017-12-06
- Triazolone compounds and uses thereofPublication Number: CA-2894281-A1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: EP-2935228-B1Priority Date: 2012-12-20Grant Date: 2017-08-02
/////////////amezalpat, ANAX LAB, antineoplastic, TPST 1120, FDA Fast Track, Orphan Drug, 1EQ4LQN9N3

{kind=link}
{kind=link}
{kind=link}