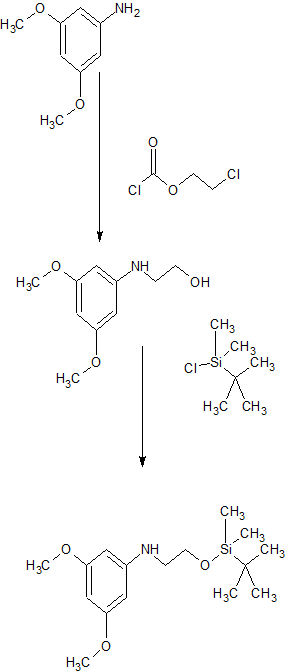
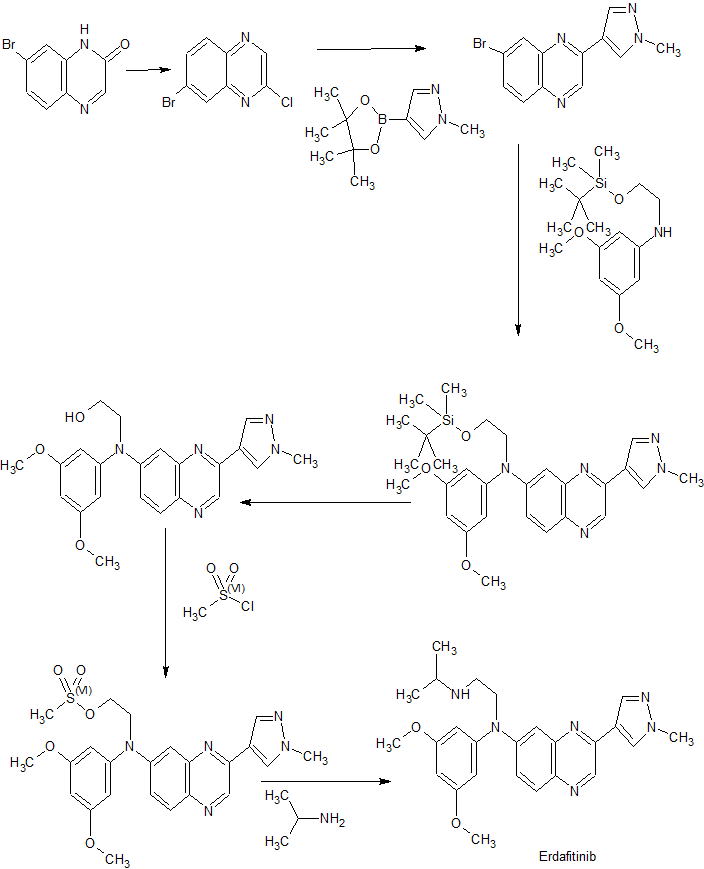
VX-659, BAMOCAFTOR
N-(Benzenesulfonyl)-6-[3-[2-[1-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide
3-Pyridinecarboxamide, N-(phenylsulfonyl)-6-[3-[2-[1-(trifluoromethyl)cyclopropyl]ethoxy]-1H-pyrazol-1-yl]-2-[(4S)-2,2,4-trimethyl-1-pyrrolidinyl]-
N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide
| CAS Number |
2204245-48-5 |
|
|
BAMOCAFTOR |
| M. Wt |
591.65 |
| Formula |
C28H32F3N5O4S |


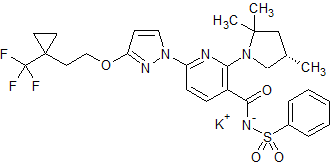
Bamocaftor potassium
CAS 2204245-47-4
| Molecular Formula |
C28 H31 F3 N5 O4 S . K |
| Molecular Weight |
629.735 |
VX-659
VX-659 potassium salt
VY7D8MTV72 (UNII code)
WHO 11167
3-Pyridinecarboxamide, N-(phenylsulfonyl)-6-[3-[2-[1-(trifluoromethyl)cyclopropyl]ethoxy]-1H-pyrazol-1-yl]-2-[(4S)-2,2,4-trimethyl-1-pyrrolidinyl]-, potassium salt (1:1)
Potassium (benzenesulfonyl)[6-(3-[2-[1-(trifluoromethyl)cyclopropyl]ethoxy]-1H-pyrazol-1-yl)-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carbonyl]azanide
PHASE 2 CYSTIC FIBRIOSIS , VERTEX
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) (DeltaF508 Mutant) Correctors

Bamocaftor potassium is a CFTR channel (DeltaF508-CFTR Mutant) corrector in phase II clinical trials at Vertex, in patients with CF who are homozygous for the F508del mutation of the CF transmembrane conductance regulator (CFTR) gene, or who are heterozygous for the F508del mutation and a minimal function (MF) CFTR mutation not likely to respond to tezacaftor, ivacaftor, or tezacaftor/ivacaftor and also in combination with tezacaftor and VX-561 in F508del/MF in patients with cystic fibrosis.
The compound is also developed by the company as a fixed-dose combination of VX-659, tezacaftor and ivacaftor.
Vertex Pharmaceuticals is developing a combination regimen comprising VX-659, a next-generation cystic fibrosis transmembrane conductance regulator (CFTR) corrector, with tezacaftor and ivacaftor, as a triple fixed-dose combination tablet. In March 2019, Vertex planned to file an NDA in the US in 3Q19 concurrently in patients aged 12 years or older with one F508del mutation and one minimal function mutation and in patients with two F508del mutations for either the VX-659 or VX-445 triple combination regimen; the regimen selected for a regulatory filing would be based on final 24-week data.
PATENT
WO 2018064632
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018064632
Example 4: Synthesis of Compounds 1-65
[00229] Synthetic Example 1: Synthesis of N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (Compound 1)
[00230] Part A: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride

[00231] Step 1: Synthesis of methyl-2,4-dimethyl-4-nitro-pentanoate

[00232] Tetrahydrofuran (THF, 4.5 L) was added to a 20 L glass reactor and stirred under N2 at room temperature. 2-Nitropropane (1.5 kg. 16.83 mol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.282 kg, 8.42 mol) were then charged to the reactor, and the jacket temperature was increased to 50 °C. Once the reactor contents were close to 50 °C, methyl methacrylate (1.854 kg, 18.52 mol) was added slowly over 100 minutes. The reaction temperature was maintained at or close to 50 °C for 21 hours. The reaction mixture was concentrated in vacuo then transferred back to the reactor and diluted with methyl tert-butyl ether (MTBE) (14 L). 2 M HC1 (7.5 L) was added, and this mixture was stirred for 5 minutes then allowed to settle. Two clear layers were visible – a lower yellow aqueous phase and an upper green organic phase. The aqueous layer was removed, and the organic layer was stirred again with 2 M HC1 (3 L). After separation, the HC1 washes were recombined and stirred with MTBE (3 L) for 5 minutes. The aqueous layer was removed, and all of the organic layers were combined in the reactor and stirred with water (3 L) for 5 minutes. After separation, the organic layers were concentrated in vacuo to afford a cloudy green oil. This was dried with MgSC and filtered to afford methyl-2,4-dimethyl-4-mtro-pentanoate as a clear green oil (3.16 kg, 99% yield). 1H NMR (400 MHz, Chloroform-d) δ 3.68 (s, 3H), 2.56 – 2.35 (m, 2H), 2.11 – 2.00 (m, 1H), 1.57 (s, 3H), 1.55 (s, 3H), 1.19 (d, J= 6.8 Hz, 3H). [00233] Step 2: Synthesis of methyl (2S)-2,4-dimethyl-4-nitro-pentanoate

[00234] A reactor was charged with purified water (2090 L; 10 vol) and then potassium phosphate monobasic (27 kg, 198.4 moles; 13 g/L for water charge). The pH of the reactor contents was adjusted to pH 6.5 (± 0.2) with 20% (w/v) potassium carbonate solution. The reactor was charged with racemic methyl-2,4-dimethyl-4-nitro-pentanoate (209 kg; 1104.6 moles), and Palatase 20000L lipase (13 L, 15.8 kg; 0.06 vol).
[00235] The reaction mixture was adjusted to 32 ± 2 °C and stirred for 15-21 hours, and pH 6.5 was maintained using a pH stat with the automatic addition of 20% potassium carbonate solution. When the racemic starting material was converted to >98% ee of the S-enantiomer, as determined by chiral GC, external heating was switched off. The reactor was then charged with MTBE (35 L; 5 vol), and the aqueous layer was extracted with MTBE (3 times, 400-1000L). The combined organic extracts were washed with aqueous Na2CO3 (4 times, 522 L, 18 % w/w 2.5 vol), water (523 L; 2.5 vol), and 10% aqueous NaCl (314 L, 1.5 vol). The organic layer was concentrated in vacuo to afford methyl (2S)-2,4-dimethyl-4-nitro-pentanoate as a mobile yellow oil (>98% ee, 94.4 kg; 45 % yield).
[00236] Step 3: Synthesis of (3S)-3,5,5-trimethylpyrrolidin-2-one

[00237] A 20 L reactor was purged with N2. The vessel was charged sequentially with DI water-rinsed, damp Raney® Ni (2800 grade, 250 g), methyl (2S)-2,4-dimethyl-4-nitro-pentanoate (1741g, 9.2 mol), and ethanol (13.9 L, 8 vol). The reaction was stirred at 900 rpm, and the reactor was flushed with H2 and maintained at -2.5 bar. The reaction mixture was then warmed to 60 °C for 5 hours. The reaction mixture was cooled and filtered to remove Raney nickel, and the solid cake was rinsed with ethanol (3.5 L, 2 vol). The ethanolic solution of the product was combined with a second equal sized batch and concentrated in vacuo to reduce to a minimum volume of ethanol (-1.5 volumes). Heptane (2.5 L) was added, and the suspension was concentrated again to -1.5 volumes. This was repeated 3 times; the resulting suspension was cooled to 0-5 °C, filtered under suction, and washed with heptane (2.5 L). The product was dried under vacuum for 20 minutes then transferred to drying trays and dried in a vacuum oven at 40 °C overnight to afford (3S)-3,5,5-trimethylpyrrolidin-2-one as a white crystalline solid (2.042 kg, 16.1 mol, 87 %). 1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (dd, J = 12.4, 8.6 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
[00238] Step 4: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride

[00239] A glass lined 120 L reactor was charged with lithium aluminium hydride pellets (2.5 kg, 66 mol) and dry THF (60 L) and warmed to 30 °C. The resulting suspension was charged with (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C, then cautiously quenched with the addition of ethyl acetate (EtOAc) (1.0 L, 10 moles), followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq), and then a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 equiv water with 1.4 equiv sodium hydroxide relative to aluminum), followed by 7.5 L water. After the addition was complete, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by vacuum distillation to a slurry. Isopropanol (8 L) was added and the solution was concentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added, and 1he product was slurried by warming to about 50 °C. MTBE (6 L) was added, and the
slurry was cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L MTBE and dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (4S)-2,2,4- trimethylpyrrolidine’HCl as a white, crystalline solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (br d, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, / = 11.4, 8.6 Hz, 1H), 2.50 – 2.39 (m, 1H), 1.97 (dd, J= 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J= 6.6 Hz, 3H).
[00240] Part B: Synthesis of N-(benzenesulfonyl)-6-[3-[2-[l- (trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4- trimethylpyrrolidin-l-yl]pyridine-3-carboxamide
[00241] Synthesis of starting materials:
[00242] Synthesis of tert-Butyl 2,6-dichloropyridine-3-carboxylate

[00243] A solution of 2,6-dichloropyridine-3-carboxylic acid (10 g, 52.08 mmol) in THF (210 mL) was treated successively with di-tert-butyl dicarbonate (17 g, 77.89 mmol) and 4-(dimethylamino)pyridine (3.2 g, 26.19 mmol) and stirred overnight at room temperature. At this point, HC1 IN (400 mL) was added, and the mixture was stirred vigorously for about 10 minutes. The product was extracted with ethyl acetate (2x300mL), and the combined organic layers were washed with water (300 mL) and brine (150 mL) and dried over sodium sulfate and concentrated under reduced pressure to give 12.94 g (96% yield) of tert- butyl 2,6-dichloropyndine-3-carboxylate as a colorless oil. ESI-MS m/z calc. 247.02, found 248.1 (M+l) +; Retention time: 2.27 minutes. 1H NMR (300 MHz, CDC13) ppm 1.60 (s, 9H), 7.30 (d, .7=7.9 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H).
[00244] Synthesis of tert-Butyl 3-oxo-2,3-dihydro-lH-pyrazole-l-carboxylate

[00245] A 50L reactor was started, and the jacket was set to 20 °C, with stirring at 150 rpm, reflux condenser (10 °C) and nitrogen purge. MeOH (2.860 L) and methyl (E)-3-methoxyprop-2-enoate (2.643 kg, 22.76 mol) were added, and the reactor was capped. The reaction was heated to an internal temperature of 40 °C, and the system was set to hold jacket temperature at 40 °C. Hydrazine hydrate (1300 g of 55 %w/w, 22.31 mol) was added portion wise via addition funnel over 30 min. The reaction was heated to 60 °C for 1 h. The reaction mixture was cooled to 20 °C and triethyamine (2.483 kg, 3.420 L, 24.54 mol) was added portion-wise, maintaining reaction temperature <30 °C. A solution of Boc anhydride (di-tert-butyl dicarbonate) (4.967 kg, 5.228 L. 22.76 mol) in MeOH (2.860 L) was added portion-wise maintaining temperature <45 °C. The reaction mixture was stirred at 20 °C for 16 h. The reaction solution was partially concentrated to remove MeOH, resulting in a clear, light amber oil. The resulting oil was transferred to the 50L reactor, stirred and water (7.150 L) and heptane (7.150 L) were added. The additions caused a small amount of the product to precipitate. The aqueous layer was drained into a clean container, and the interface and heptane layer were filtered to separate the solid (product). The aqueous layer was transferred back to the reactor, and the collected solid was placed back into the reactor and mixed with the aqueous layer. A dropping funnel was added to the reactor and loaded with acetic acid (1.474 kg, 1.396 L, 24.54 mol) and added dropwise. The jacket was set to 0 °C to absorb the quench exotherm. After the addition was complete (pH=5), the reaction mixture was stirred for 1 h. The solid was collected by filtration and washed with water (7.150 L), and washed a second time with water (3.575 L). The crystalline solid was transferred into a 20L rotovap bulb, and heptane (7.150 L) was added. The mixture was slurried at 45 °C for 30 mins, and 1-2 volumes of solvent were distilled off The slurry in the rotovap flask was filtered, and the solids were washed with heptane (3.575 L). The solid was further dried in vacuo (50 °C, 15 mbar) to give tert-butyl 5-oxo-lH-pyrazole-2-carboxylate (2921 g, 71%) as a coarse, crystalline solid. 1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.98 (d, J= 2.9 Hz, 1H), 5.90 (d, J= 2.9 Hz, 1H), 1.54 (s, 9H).
[00246] Synthesis of 2-[l-(trifluoromethyl)cyclopropyl]ethanol

[00247] To a solution of lithium aluminum hydride (293 mg, 7.732 mmol) in THF (10.00 mL) in an ice-bath, 2-[l-(trifluoromethyl)cyclopropyl]acetic acid (1.002 g, 5.948 mmol) in THF (3.0 mL) was added dropwise over a period of 30 minutes keeping the reaction temperature below 20 ° C. The mixture was allowed to gradually warm to ambient temperature and was stirred for 18 h. The mixture was cooled with an ice-bath and sequentially quenched with water (294 mg, 295 μL, 16.36 mmol), NaOH (297 μL of 6 M, 1.784 mmol), and then water (884.0 μL, 49.07 mmol) to afford a granular solid in the mixture. The solid was filtered off using celite, and the precipitate was washed with ether. The filtrate was further dried with MgSO4 and filtered and concentrated in vacuo to afford the product with residual THF and ether. The mixture was taken directly into the next step without further purification.
[00248] Step 1: tert-Butyl 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazole-1-carboxylate

[00249] rerf-Butyl 5-oxo-lH-pyrazole-2-carboxylate (1.043 g, 5.660 mmol), 2-[l-(trifluoromethyl)cyclopropyl]ethanol (916 mg, 5.943 mmol), and triphenyl phosphine (1.637 g, 6.243 mmol) were combined in THF (10.48 mL) and the reaction was cooled in an ice-bath. Diisopropyl azodicarboxylate (1.288 g, 1.254 mL, 6.368 mmol) was added dropwise to the reaction mixture, and the reaction was allowed to warm to room temperature for 16 hours. The mixture was evaporated, and the resulting material was partitioned between ethyl acetate (30 mL) and IN sodium hydroxide (30 mL). The organic layer was separated, washed with brine (30 mL), dried over sodium sulfate, and concentrated. The crude material was purified by silica gel chromatography eluting with a gradient of ethyl acetate in hexanes (0- 30%) to give tert-butyl 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazole-l-carboxylate (1.03 g, 57%). ESI-MS m/z calc. 320.13, found 321.1 (M+l) +; Retention time: 0.72 minutes.
[00250] Step 2: 3-[2-[l-(Trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole

[00251] terr-Butyl-3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazole-l-carboxylate (1.03 g, 3.216 mmol) was dissolved in dichloromethane (10.30 mL) with trifluoroacetic acid (2.478 mL, 32.16 mmol), and the reaction was stirred at room temperature for 2 hours. The reaction was evaporated, and the resulting oil was partitioned between ethyl acetate (10 mL) and a saturated sodium bicarbonate solution.
The organic layer was separated, washed with brine, dried over sodium sulfate, and evaporated to give 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (612 mg, 86%). ESI-MS m/z calc. 220.08, found 221.0 (M+1) +; Retention time: 0.5 minutes. ¾ NMR (400 MHz, DMSO-d6) δ 11.86 (s, 1H), 7.50 (t, J = 2.1 Hz, 1H), 5.63 (t, J= 2.3 Hz, 1H), 4.14 (t, J= 7.1 Hz, 2H), 2.01 (t, J= 7.1 Hz, 2H), 0.96 – 0.88 (m, 2H), 0.88 -0.81 (m, 2H).
[00252] Step 3: tert- Butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxylate

[00253] tert-Butyl 2,6-dichloropyridine-3-carboxylate (687 mg, 2.770 mmol), 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (610 mg, 2.770 mmol), and freshly ground potassium carbonate (459 mg, 3.324 mmol) were combined in anhydrous DMSO (13.75 mL). l,4-diazabicyclo[2.2.2]octane (DABCO (1,4-diazabicyclo[2.2.2]octane), 62 mg, 0.5540 mmol) was added, and the mixture was stirred at room temperature under nitrogen for 16 hours. The reaction mixture was diluted with water (20 mL) and stirred for 15 minutes. The resulting solid was collected and washed with water. The solid was dissolved in dichloromethane and dried over magnesium sulfate. The mixture was filtered and concentrated to give ferf-butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (1.01 g, 84%). ESI-MS m/z calc. 431.12, found 432.1 (M+1) +; Retention time: 0.88 minutes.
[00254] Step 4: 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid

[00255] tert-Butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (1.01 g, 2.339 mmol) and trifluoroacetic acid (1.8 mL, 23.39 mmol) were combined in dichloromethane (10 mL) and heated at 40 °C for 3 h. The reaction was concentrated. Hexanes were added, and the mixture was concentrated again to give 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (873 mg, 99%) ESI-MS m/z calc. 375.06, found 376.1 (M+l)+; Retention time: 0.69 minutes.
[00256] Step 5: N-(Benzenesulfonyl)-2-chloro-6-[3- [2- [1-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxamide

[00257] A solution of 2-chloro-6-[3-[2-[l- (trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (0.15 g, 0.3992 mmol) and carbonyl diimidazole (77 mg, 0,4790 mmol) in THF (2.0 mL) was stirred for one hour, and benzenesulfonamide (81 mg, 0.5190 mmol) and DBU (72 μL, 0.4790 mmol) were added. The reaction was stirred for 16 hours, acidified with 1 M aqueous citric acid, and extracted with ethyl acetate. The combined extracts were dried over sodium sulfate and evaporated. The residue was purified by silica gel chromatography eluting with a gradient of methanol in dichloromethane (0-5%) to give N-(benzenesulfonyl)-2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyndine-3-carboxamide (160 mg, 78%). ESI-MS m/z calc. 514.07, found 515.1 (M+l)+; Retention time: 0.74 minutes.
[00258] Step 6: N-(Benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy] pyrazol-l-yl] -2- [(4S)-2,2,4-trimethylpyrrolidin-l-yl] pyridine-3-carboxamide

[00259] A mixture of N-(benzenesulfonyl)-2-chloro-6-[3-[2-[l -(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxamide (160 mg, 0.3107 mmol), (4S)-2,2,4-trimethylpyrrolidine hydrochloride salt (139 mg, 0.9321 mmol), and potassium carbonate (258 mg, 1.864 mmol) in DMSO (1.5 mL) was stirred at 130 °C for 17 hours. The reaction mixture was acidified with 1 M aqueous citric acid and extracted with ethyl acetate. The combined extracts were dried over sodium sulfate and evaporated to yield a crude product that was purified by reverse-phase HPLC utilizing a gradient of 10-99% acetonitrile in 5 mM aqueous HCI to yield N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (87 mg, 47%). ESI-MS mJz calc. 591.21, found 592.3 (M+l) +; Retention time: 2.21 minutes. 1H NMR (400 MHz, DMSO-d6) δ 12.48 (s, 1H), 8.19 (d, J = 2.8 Hz, 1H), 8.04 – 7.96 (m, 2H), 7.81 (d, J= 8.2 Hz, 1H), 7.77 – 7.70 (m, 1H), 7.70 – 7.62 (m, 2H), 6.92 (d, J= 8.2 Hz, 1H), 6.10 (d, J= 2.8 Hz, 1H), 4.31 (t, J= 7.0 Hz, 2H), 2.42 (t, J = 10.5 Hz, 1H), 2.28 (dd, J = 10.2, 7.0 Hz, 1H), 2.17 – 2.01 (m, 3H), 1.82 (dd, J= 11.9, 5.5 Hz, 1H), 1.52 (d, .7= 9.4 Hz, 6H), 1.36 (t, J= 12.1 Hz, 1H), 1.01 – 0.92 (m, 2H), 0.92 – 0.85 (m, 2H), 0.65 (d, J = 6.3 Hz, 3H). pKa: 4.95±0.06.
Alternate synthesis of 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid
[00263] Step 1: ethyl 3-hydroxy-lH-pyrazole-4-carboxylate

[00264] A mixture of EtOH (20.00 L, 10 vol) and diethyl 2-(ethoxymethylene)propanedioate (2000 g, 9.249 mol, 1.0 equiv) was added under nitrogen purge a to a 50 L reactor equipped with a reflux condenser (10 °C) and the jacket set to 40 °C. The mixture was stirred, and then hydrazine hydrate (538.9 g of 55 %w/w, 523.7 mL of 55 %w/w, 9.249 mol, 1.00 equiv) was added in portions via an addition funnel. Once the addition was complete, the reaction was heated to 75 °C for 22 h to afford a solution of ethy l 3-hydroxy-lH-pyrazole-4-carboxylate that was used directly in the next step.
[00265] Step 2: l-(tert-butyl) 4-ethyl 3-hydroxy-lH-pyrazole-l,4-dicarboxylate

[00266] The solution of ethyl 3-hydroxy-lH-pyrazole-4-carboxylate was cooled from 75 °C to 40 °C, then triethylamine (TEA) (46.80 g, 64.46 mL, 462.5 mmol, 0.05 eq.) was added. A solution of Boc anhydride (2.119 kg, 9.711 mol 1.05 equiv) in EtOH (2.000 L, 1 equiv) was added to the reactor over 35 min. The mixture was stirred for 4 hours to complete the reaction; then water (10.00 L, 5.0 vol) was added over 15 mins. The resulting mixture was cooled to 20 °C to complete crystallization of the product. The crystals were allowed to age for 1 hour, then the mixture was filtered. The solid was washed with a mixture of EtOH (4.000 L, 2.0 vol) and water (2.000 L, 1 0 vol) The solid was then dried in vacuo to afford l-(tert-butyl)-4-ethyl-3-hydroxy-lH-pyrazole-1,4-dicarboxylate (1530 g, 65%) as colorless, fine needle, crystalline solid. ‘H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1H), 8.40 (s, 1H), 4.20 (q, J = 7.1 Hz, 2H), 1.56 (s, 9H), 1.25 (t, J = 7.1 Hz, 3H).
[00267] Step 3: l-(tert-butyl) 4-ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-l,4-dicarboxylate

[00268] A 5L reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temperature and nitrogen purge. The vessel was charged with toluene (1.0L, 10.0 vol), 2-[l-(tnfluoromethyl)cyclopropyl]ethanol (lOO.Og, 648.8 mmol, 1.0 equiv), and l-(tert-butyl) 4-ethyl 3-hydroxy-lH-pyrazole-l,4-dicarboxylate (166.3 g, 648.8 mmol), and the mixture was stirred. The reaction mixture was charged with triphenyl phosphine (195.7 g, 746.1 mmol, 1.15 equiv), then the reactor was set to maintain an internal temperature of 40 °C. Diisopropyl azoldicarboxylate (150.9 g, 746.1 mmol, 1.15 equiv) was added into an addition funnel and was added to the
reaction while maintaining the reaction temperature between 40 and 50 °C (addition was exothermic, exotherm addition controlled), and stirred for a total of 2.5 hours. Once the reaction was deemed complete by HPLC, heptane was added (400 mL, 4 vol), the solution was cooled to 20 °C over 60 minutes, and the bulk of tnphenylphosphine oxide-DIAD complex (TPPO-DIAD) crystallized out. Once at room temp, the mixture was filtered, and the solid was washed with heptane (400 mL, 4.0 vol) and pulled dry. The filtrate was used in the next step as a solution in toluene-heptane without further purification.
[00269] Step 4: ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylate

[00270] A 500mL reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temp, and nitrogen purge. The vessel was charged with a toluene solution consisting of approximately 160 mmol, 65.0 g of 1 -(tert-buty 1) 4-ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-l,4-dicarboxylate in 3 vol of toluene (prepared by concentrating a 25% portion of filtrate from previous reaction down to 4 volumes in a rotovap). The reaction was set to maintain an internal temperature at 40 °C and KOH (33.1 g, 1.5 eq. of aqueous 45 % KOH solution) was added in one portion, resulting in a mild exothermic addition, while CO2 was generated upon removal of the protecting group. The reaction proceeded for 1.5 hr, monitored by HPLC, with the product partially crystallizing during the reaction. Heptane (160 mL, 2.5 vol) was added to the reaction mixture and the reaction was cooled to room temperature over 30 minutes. The resulting mixture was filtered, and the solid was washed with heptane (80.00 mL, 1.25 vol), pulled dry, then dried in vacuo (55 °C, vacuum). 52.3 g of ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylate was obtained as a crude, colorless solid that was used without further purification.
[00271] Step 5: 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid

[00272] A 500mL reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temp, and nitrogen purge. The vessel was charged with methanol (150.0 mL, 3.0 vol), a solution of ethyl 3-(2-(l-(triiluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylate (50.0 g, 171.1 mmol, 1.0 equiv), and the reaction was stirred to suspend the solids. The reactor was set to maintain internal temperature at 40 °C. To the mixture was added KOH (96 g of aqueous 45 % KOH, 1.71 mol, 10.0 equiv) in portions maintaining the internal temperature <50 °C. Once addition was complete, the reaction was set to maintain temperature at 50 °C, and the reaction proceeded for 23 hours, monitored by HPLC. Once complete the reaction was cooled to 10 °C then partially concentrated on a rotary evaporator to remove most of the MeOH. The resulting solution was diluted with water (250 mL, 5.0 vol) and 2-Me-THF (150 mL, 3.0 vol), and transferred to the reactor, stirred at room temp, then stopped, and layers were allowed to separate. The layers were tested, with remaining TPPO-DIAD complex in the organic layer and product in the aqueous layer. The aqueous layer was washed again with 2-Me-THF (100 mL, 2.0 vol), the layers separated, and the aqueous layer returned to the reactor vessel. The stirrer was started and set to 450 rpm, and the reactor jacket was set to 0 °C. The pH was adjusted to pH acidic by addition of 6M aqueous HC1 (427mL, 15 equiv) portion wise, maintaining the internal temperature between 10 and 30 °C. The product began to crystallize close to pH neutral and was accompanied with strong off-gassing, and so the acid was added slowly, and then further added to reach pH 1 once the off-gassing had ended. To the resulting suspension was added 2-Me-THF (400 mL, 8.0 vol), and the product was allowed to dissolve into the organic layer. Stirring was stopped, the layers were separated, and the aqueous layer was returned to the reactor, stirred and re-extracted with 2-Me-THF (100 mL, 2.0 vol). The organic lay ers were combined in the reactor and stirred at room temperature, washed with brine (lOOmL, 2 vols), dried over Na2S04, filtered through celite, and the solid was washed with 2-Me-THF (50 mL, 1.0 vol). The filtrate was transferred to a clean rotovap flask, stirred, warmed to 50 °C and heptane (200 mL, 4.0 vol) added, and then partially concentrated with the addition of heptane (300 mL, 6.0 vol) and then seeded with 50mg of 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid), and the product crystallized during solvent removal. The distillation was stopped when the bulk of the 2-Me-THF had distilled off. The bath heater was turned off, the vacuum removed, and the mixture was allowed to stir and cool to room temperature. The mixture was filtered (slow speed) and the solid was washed with heptane (100 mL, 2.0 vol), and the solid was collected and dried in vacuo (50 °C, rotovap). 22.47 g of 3-(2-(l-(triiluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid was obtained as an off-white solid. 1H NMR (400 MHz, DMSO-d) δ 12.45 (s, 2H), 8.01 (s, 1H), 4.26 (t, J = 7.0 Hz, 2H), 2.05 (t, J= 7.0 Hz, 2H), 0.92 (m, 4H).
[00273] Step 6: 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole

[00274] A mixture of toluene (490.0 mL), 3-(2-(l- (triiluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid (70.0 g, 264.9 mmol), and DMSO (70.00 mL) was placed in a reactor and heated to 100 °C with stirring. DBU (approximately 20.16 g, 19.80 mL, 132.4 mmol) was added to the reactor over 15 min. The mixture was stirred for 20 h to complete the reaction and then cooled to 20 °C. The mixture was washed with water (350.0 mL), then 0.5N aq HC1 (280.0 mL), then water (2 x 140.0 mL), and lastly with bnne (210.0 mL). The organic layer was dried with Na2S04, and then activated charcoal (5 g, Darco 100 mesh) was added to the stirred slurry. The dried mixture was filtered through celite, and the solid was washed with toluene (140.0 mL) and then pulled dry. The filtrate was concentrated in a rotovap (50 °C, vac) to afford 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-
pyrazole (30.89 g, 53%) as an amber oil. 1H NMR (400 MHz, DMSO-4,) δ 11.87 (s, 1H), 7.50 (d, J= 2.4 Hz, 1H), 5.63 (d, 7= 2.4 Hz, 1H), 4.23 – 4.06 (m, 2H), 2.01 (t, J= 7.1 Hz, 2H), 1.00 – 0.77 (m, 4H).
[00275] Step 7: ethyl 2-chloro-6-[3-[2-[l- (trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate

[00276] A mixture of DMF (180.0 mL), ethyl 2,6-dichloropyridine-3-carboxylate (approximately 29.97 g, 136.2 mmol), 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (30.0 g, 136.2 mmol), and K2CO3, (325 mesh, approximately 24.48 g, 177.1 mmol) was added to a stirred reactor at 20 °C. DABCO (approximately 2.292 g, 20.43 mmol) was then added to the reactor, and the mixture was stirred at 20 °C for 1 hour, and then the temperature was increased to 30 °C, and the mixture stirred for 24 hours to complete the reaction. The mixture was cooled to 20 °C; then water (360 mL) was added slowly. The mixture was then drained from the reactor and the solid was isolated by filtration. The solid was then washed with water (2 x 150 mL), and then the solid was dried under vacuum at 55 °C to afford ethyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (51.37 g, 93%) as a fine, beige colored solid. 1H NMR (400 MHz, DMSO-c4) δ 8.44 (d, J= 2.9 Hz, 1H), 8.41 (d, J= 8.5 Hz, 1H), 7.75 (d, J= 8.5 Hz, 1H), 6.21 (d, J= 2.9 Hz, 1H), 4.34 (m, 4H), 2.09 (t, J= 7.1 Hz, 2H), 1.34 (t, J= 7.1 Hz, 3H), 1.00 – 0.84 (m, 4H).
[00277] Step 8: 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid

[00278] A solution of ethyl 2-chloro-6-[3-[2-[l- (trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (50.0 g, 123.8 mmol) in THF (300.0 mL) was prepared in a reactor at 20 °C. EtOH (150.0 mL) was added, followed by aqueous NaOH (approximately 59.44 g of 10 %w/w, 148.6 mmol). The mixture was stirred for 1 hour to complete the reaction; then aq IN HCl (750.0 mL) was slowly added. The resulting suspension was stirred for 30 mm at 10 °C, and then the solid was isolated by filtration. The solid was washed with water (150 mL then 2 x 100 mL) and then pulled dry by vacuum. The solid was then further dried under vacuum with heating to afford 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (42.29 g, 91%). 1H NMR (400 MHz, DMSO-d 6) 5 13.63 (s, 1H), 8.48 – 8.35 (m, 2H), 7.73 (d, J= 8.4 Hz, 1H), 6.20 (d, J= 2.9 Hz, 1H), 4.35 (t, J = 7.1 Hz, 2H), 2.09 (t, J= 7.1 Hz, 2H), 1.01 – 0.82 (m, 4H).
PATENT
WO2018227049
Follows on from WO2018227049 , claiming a composition comprising this compound and at least one of tezacaftor, ivacaftor, deutivacaftor or lumacaftor, useful for treating CF.
PATENT
WO-2019079760
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019079760&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline forms of the compound, the potassium salt of which is presumed to be VX-659 , Such as Forms A, B, C, D, E, H and M , processes for their preparation and compositions comprising them are claimed. Also claimed are their use for treating cystic fibrosis, and compositions comprising VX-659, ivacaftoR, lumacaftor and tezacaftor .
This application claims priority to U.S. Provisional Application No.
62/574,677, filed October 19, 2017; U.S. Provisional Application No. 62/574,670, filed October 19, 2017; and U.S. Provisional Application No. 62/650,057, filed March 29, 2018, the entire contents of each of which are expressly incorporated herein by reference in their respective entireties.
[0002] Disclosed herein are crystalline forms of Compound I and pharmaceutically acceptable salts thereof, which are modulators of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), compositions comprising the same, methods of using the same, and processes for making the same.
[0003] Cystic fibrosis (CF) is a recessive genetic disease that affects approximately 70,000 children and adults worldwide. Despite progress in the treatment of CF, there is no cure.
[0004] In patients with CF, mutations in CFTR endogenously expressed in respiratory epithelia lead to reduced apical anion secretion causing an imbalance in ion and fluid transport. The resulting decrease in anion transport contributes to enhanced mucus accumulation in the lung and accompanying microbial infections that ultimately cause death in CF patients. In addition to respiratory disease, CF patients typically suffer from gastrointestinal problems and pancreatic insufficiency that, if left untreated, result in death. In addition, the majority of males with cystic fibrosis are infertile, and fertility is reduced among females with cystic fibrosis.
[0005] Sequence analysis of the CFTR gene has revealed a variety of disease-causing mutations (Cutting, G. R. et al. (1990) Nature 346:366-369; Dean, M. et al. (1990) Cell 61 :863 :870; and Kerem, B-S. et al. (1989) Science 245: 1073-1080; Kerem, B-S et al. (1990) Proc. Natl. Acad. Sci. USA 87:8447-8451). To date, greater than 2000 mutations in the CF gene have been identified; currently, the CFTR2 database contains information on only 322 of these identified mutations, with sufficient evidence to define 281 mutations as disease causing. The most prevalent disease-causing mutation is a deletion of phenylalanine at position 508 of the CFTR amino acid sequence and is
commonly referred to as the F508del mutation. This mutation occurs in approximately 70% of the cases of cystic fibrosis and is associated with severe disease.
[0006] The deletion of residue 508 in CFTR prevents the nascent protein from folding correctly. This results in the inability of the mutant protein to exit the endoplasmic reticulum (ER) and traffic to the plasma membrane. As a result, the number of CFTR channels for anion transport present in the membrane is far less than observed in cells expressing wild-type CFTR, i.e., CFTR having no mutations. In addition to impaired trafficking, the mutation results in defective channel gating.
Together, the reduced number of channels in the membrane and the defective gating lead to reduced anion and fluid transport across epithelia. (Quinton, P. M. (1990), FASEB J. 4: 2709-2727). The channels that are defective because of the F508del mutation are still functional, albeit less functional than wild-type CFTR channels. (Dalemans et al. (1991), Nature Lond. 354: 526-528; Pasyk and Foskett (1995), J. Cell. Biochem. 270: 12347-50). In addition to F508del, other disease-causing mutations in CFTR that result in defective trafficking, synthesis, and/or channel gating could be up-or down-regulated to alter anion secretion and modify disease progression and/or severity.
[0007] CFTR is a cAMP/ATP-mediated anion channel that is expressed in a variety of cell types, including absorptive and secretory epithelia cells, where it regulates anion flux across the membrane, as well as the activity of other ion channels and proteins. In epithelial cells, normal functioning of CFTR is critical for the maintenance of electrolyte transport throughout the body, including respiratory and digestive tissue. CFTR is composed of approximately 1480 amino acids that encode a protein which is made up of a tandem repeat of transmembrane domains, each containing six
transmembrane helices and a nucleotide binding domain. The two transmembrane domains are linked by a large, polar, regulatory (R)-domain with multiple
phosphorylation sites that regulate channel activity and cellular trafficking.
[0008] Chloride transport takes place by the coordinated activity of ENaC and CFTR present on the apical membrane and the Na+-K+-ATPase pump and CI- channels expressed on the basolateral surface of the cell. Secondary active transport of chloride from the luminal side leads to the accumulation of intracellular chloride, which can then passively leave the cell via CI“ channels, resulting in a vectorial transport. Arrangement of Na+/2C17K+ co-transporter, Na+-K+– ATPase pump and the basolateral membrane K+ channels on the basolateral surface and CFTR on the luminal side coordinate the secretion of chloride via CFTR on the luminal side. Because water is probably never actively transported itself, its flow across epithelia depends on tiny transepithelial osmotic gradients generated by the bulk flow of sodium and chloride.
[0009] Compound I and pharmaceutically acceptable salts thereof are potent CFTR modulators. Compound I is N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl) cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide, and has the following structure:

Example 1: Synthesis of N-(benzenesulfonyl)-6-[3-[2-[l- (trifluoromethyl)cyclopropyl] ethoxy] pyrazol-l-yl]-2- [(4S)-2,2,4- trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (Compound I)
Part A: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride

° THF, Base
N02 1 “* N02 | -k/ B) HC‘
Step 1: Synthesis of methyl-2,4-dimethyl-4-nitro-pentanoate

[00381] Tetrahydrofuran (THF, 4.5 L) was added to a 20 L glass reactor and stirred under N2 at room temperature. 2-Nitropropane (1.5 kg, 16.83 mol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.282 kg, 8.42 mol) were then charged to the reactor, and the jacket temperature was increased to 50 °C. Once the reactor contents were close to 50 °C, methyl methacrylate (1.854 kg, 18.52 mol) was added slowly over 100 minutes. The reaction temperature was maintained at or close to 50 °C for 21 hours. The reaction mixture was concentrated in vacuo then transferred back to the reactor and diluted with methyl fert-butyl ether (MTBE) (14 L). 2 M HC1 (7.5 L) was added, and this mixture was stirred for 5 minutes then allowed to settle. Two clear layers were visible – a lower yellow aqueous phase and an upper green organic phase. The aqueous layer was removed, and the organic layer was stirred again with 2 M HC1 (3 L). After separation, the HC1 washes were recombined and stirred with MTBE (3 L) for 5 minutes. The aqueous layer was removed, and all of the organic layers were combined in the reactor and stirred with water (3 L) for 5 minutes. After separation, the organic layers were concentrated in vacuo to afford a cloudy green oil. This was dried with MgS04 and filtered to afford methyl-2,4-dimethyl-4-nitro-pentanoate as a clear green oil (3.16 kg, 99% yield). ¾ MR (400 MHz, Chloroform-i ) δ 3.68 (s, 3H), 2.56 – 2.35 (m, 2H), 2.11 – 2.00 (m, 1H), 1.57 (s, 3H), 1.55 (s, 3H), 1.19 (d, J= 6.8 Hz, 3H).
Step 2: Synthesis of methyl (2S)-2,4-dimethyl-4-nitro-pentanoate

[00382] A reactor was charged with purified water (2090 L; 10 vol) and then potassium phosphate monobasic (27 kg, 198.4 moles; 13 g/L for water charge). The pH of the reactor contents was adjusted to pH 6.5 (± 0.2) with 20% (w/v) potassium carbonate solution. The reactor was charged with racemic methyl-2,4-dimethyl-4-nitro-pentanoate (209 kg; 1104.6 moles), and Palatase 20000L lipase (13 L, 15.8 kg; 0.06 vol).
[00383] The reaction mixture was adjusted to 32 ± 2 °C and stirred for 15-21 hours, and pH 6.5 was maintained using a pH stat with the automatic addition of 20% potassium carbonate solution. When the racemic starting material was converted to >98% ee of the S-enantiomer, as determined by chiral GC, external heating was
switched off. The reactor was then charged with MTBE (35 L; 5 vol), and the aqueous layer was extracted with MTBE (3 times, 400-1000L). The combined organic extracts were washed with aqueous Na2CCb (4 times, 522 L, 18 % w/w 2.5 vol), water (523 L; 2.5 vol), and 10% aqueous NaCl (314 L, 1.5 vol). The organic layer was concentrated in vacuo to afford methyl (2,S)-2,4-dimethyl-4-nitro-pentanoate as a mobile yellow oil (>98% ee, 94.4 kg; 45 % yield).
Step 3: Synthesis of (3S)-3,5,5-trimethylpyrrolidin-2-one

[00384] A 20 L reactor was purged with N2. The vessel was charged sequentially with DI water-rinsed, damp Raney® Ni (2800 grade, 250 g), methyl (2S)-2,4-dimethyl-4-nitro-pentanoate (1741g, 9.2 mol), and ethanol (13.9 L, 8 vol). The reaction was stirred at 900 rpm, and the reactor was flushed with H2 and maintained at -2.5 bar. The reaction mixture was then warmed to 60 °C for 5 hours. The reaction mixture was cooled and filtered to remove Raney nickel, and the solid cake was rinsed with ethanol (3.5 L, 2 vol). The ethanolic solution of the product was combined with a second equal sized batch and concentrated in vacuo to reduce to a minimum volume of ethanol (-1.5 volumes). Heptane (2.5 L) was added, and the suspension was concentrated again to -1.5 volumes. This was repeated 3 times; the resulting suspension was cooled to 0-5 °C, filtered under suction, and washed with heptane (2.5 L). The product was dried under vacuum for 20 minutes then transferred to drying trays and dried in a vacuum oven at 40 °C overnight to afford (3S)-3,5,5-trimethylpyrrolidin-2-one as a white crystalline solid (2.042 kg, 16.1 mol, 87 %). ¾ MR (400 MHz, Chloroform-i ) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (dd, J = 12.4, 8.6 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
Step 4: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride

[00385] A glass lined 120 L reactor was charged with lithium aluminium hydride pellets (2.5 kg, 66 mol) and dry THF (60 L) and warmed to 30 °C. The resulting suspension was charged with (¾)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C, then cautiously quenched with the addition of ethyl acetate (EtOAc) (1.0 L, 10 moles), followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq), and then a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 equiv water with 1.4 equiv sodium hydroxide relative to aluminum), followed by 7.5 L water. After the addition was complete, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HC1 (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by vacuum distillation to a slurry. Isopropanol (8 L) was added and the solution was concentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added, and the product was slurried by warming to about 50 °C. MTBE (6 L) was added, and the slurry was cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L MTBE and dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (4S)-2,2,4-trimethylpyrrolidine»HCl as a white, crystalline solid (6.21 kg, 75% yield). ¾ NMR (400 MHz, DMSO-^6) δ 9.34 (br d, 2H), 3.33 (dd, J= 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50 – 2.39 (m, 1H), 1.97 (dd, J= 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J= 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J= 6.6 Hz, 3H).
Part B: Synthesis of N-(benzenesulfonyl)-6-[3-[2-[l- (trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4- trimethylpyrrolidin-l-yl]pyridine-3-carboxamide
HO‘ CF,

Synthesis of starting materials:
Synthesis of terf-Butyl 2,6-dichloropyridine-3-carboxylate

[00386] A solution of 2,6-dichloropyridine-3-carboxylic acid (10 g, 52.08 mmol) in THF (210 mL) was treated successively with di-tert-butyl dicarbonate (17 g, 77.89 mmol) and 4-(dimethylamino)pyridine (3.2 g, 26.19 mmol) and stirred overnight at room temperature. At this point, HCI IN (400 mL) was added, and the mixture was stirred vigorously for about 10 minutes. The product was extracted with ethyl acetate (2x300mL), and the combined organic layers were washed with water (300 mL) and brine (150 mL) and dried over sodium sulfate and concentrated under reduced pressure to give 12.94 g (96% yield) of tert-butyl 2,6-dichloropyridine-3-carboxylate as a colorless oil. ESI-MS m/z calc. 247.02, found 248.1 (M+1) +; Retention time: 2.27 minutes. ¾ NMR (300 MHz, CDCh) ppm 1.60 (s, 9H), 7.30 (d, J=7.9 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H).
Synthesis of terf-Butyl 3-oxo-2,3-dihydro-lH-pyrazole-l-carboxylate

[00387] A 50L reactor was started, and the jacket was set to 20 °C, with stirring at 150 rpm, reflux condenser (10 °C) and nitrogen purge. MeOH (2.860 L) and methyl (E)-3-methoxyprop-2-enoate (2.643 kg, 22.76 mol) were added, and the reactor was capped. The reaction was heated to an internal temperature of 40 °C, and the system was set to hold jacket temperature at 40 °C. Hydrazine hydrate (1300 g of 55 %w/w, 22.31 mol) was added portion wise via addition funnel over 30 min. The reaction was heated to 60 °C for 1 h. The reaction mixture was cooled to 20 °C and triethyamine (2.483 kg, 3.420 L, 24.54 mol) was added portion-wise, maintaining reaction
temperature <30 °C. A solution of Boc anhydride (di-tert-butyl dicarbonate) (4.967 kg, 5.228 L, 22.76 mol) in MeOH (2.860 L) was added portion-wise maintaining temperature <45 °C. The reaction mixture was stirred at 20 °C for 16 h. The reaction solution was partially concentrated to remove MeOH, resulting in a clear, light amber oil. The resulting oil was transferred to the 50L reactor, stirred and water (7.150 L) and heptane (7.150 L) were added. The additions caused a small amount of the product to precipitate. The aqueous layer was drained into a clean container, and the interface and heptane layer were filtered to separate the solid (product). The aqueous layer was transferred back to the reactor, and the collected solid was placed back into the reactor and mixed with the aqueous layer. A dropping funnel was added to the reactor and loaded with acetic acid (1.474 kg, 1.396 L, 24.54 mol) and added dropwise. The jacket was set to 0 °C to absorb the quench exotherm. After the addition was complete (pH=5), the reaction mixture was stirred for 1 h. The solid was collected by filtration and washed with water (7.150 L) and washed a second time with water (3.575 L). The crystalline solid was transferred into a 20L rotovap bulb, and heptane (7.150 L) was added. The mixture was slurried at 45 °C for 30 mins, and 1-2 volumes of solvent were distilled off. The slurry in the rotovap flask was filtered, and the solids were washed with heptane (3.575 L). The solid was further dried in vacuo (50 °C, 15 mbar) to give tert-butyl 5-oxo-lH-pyrazole-2-carboxylate (2921 g, 71%) as a coarse, crystalline solid. ¾ MR
(400 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.98 (d, J= 2.9 Hz, 1H), 5.90 (d, J
1H), 1.54 (s, 9H).
Synthesis of 2-[l-(trifluoromethyl)cyclopropyl]ethanol

[00388] To a solution of lithium aluminum hydride (293 mg, 7.732 mmol) in THF (10.00 mL) in an ice-bath, 2-[l-(trifluoromethyl)cyclopropyl]acetic acid (1.002 g, 5.948 mmol) in THF (3.0 mL) was added dropwise over a period of 30 minutes keeping the reaction temperature below 20 0 C. The mixture was allowed to gradually warm to ambient temperature and was stirred for 18 h. The mixture was cooled with an ice-bath and sequentially quenched with water (294 mg, 295 μΐ., 16.36 mmol), NaOH (297 μΐ. of 6 M, 1.784 mmol), and then water (884.0 μΐ., 49.07 mmol) to afford a granular solid in the mixture. The solid was filtered off using celite, and the precipitate was washed with ether. The filtrate was further dried with MgS04 and filtered and concentrated in vacuo to afford the product with residual THF and ether. The mixture was taken directly into the next step without further purification.
Step 1: tert-Butyl 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazole-l-carboxylate

[00389] tert-Butyl 5-oxo-lH-pyrazole-2-carboxylate (1.043 g, 5.660 mmol), 2-[l-(trifluoromethyl)cyclopropyl]ethanol (916 mg, 5.943 mmol), and triphenyl phosphine (1.637 g, 6.243 mmol) were combined in THF (10.48 mL) and the reaction was cooled in an ice-bath. Diisopropyl azodicarboxylate (1.288 g, 1.254 mL, 6.368 mmol) was added dropwise to the reaction mixture, and the reaction was allowed to warm to room temperature for 16 hours. The mixture was evaporated, and the resulting material was partitioned between ethyl acetate (30 mL) and IN sodium hydroxide (30 mL). The organic layer was separated, washed with brine (30 mL), dried over sodium sulfate, and concentrated. The crude material was purified by silica gel chromatography eluting with a gradient of ethyl acetate in hexanes (0- 30%) to give tert-butyl 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazole-l-carboxylate (1.03 g, 57%). ESI-MS m/z calc. 320.13, found 321.1 (M+1) +; Retention time: 0.72 minutes.
Step 2: 3-[2-[l-(Trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole

[00390] tert-Butyl-3 -[2-[ 1 -(trifluoromethyl)cyclopropyl]ethoxy]pyrazole- 1 -carboxylate (1.03 g, 3.216 mmol) was dissolved in dichloromethane (10.30 mL) with trifluoroacetic acid (2.478 mL, 32.16 mmol), and the reaction was stirred at room temperature for 2 hours. The reaction was evaporated, and the resulting oil was partitioned between ethyl acetate (10 mL) and a saturated sodium bicarbonate solution. The organic layer was separated, washed with brine, dried over sodium sulfate, and evaporated to give 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (612 mg, 86%). ESI-MS m/z calc. 220.08, found 221.0 (M+1) +; Retention time: 0.5 minutes. ¾ MR (400 MHz, DMSO-d6) δ 11.86 (s, 1H), 7.50 (t, J= 2.1 Hz, 1H), 5.63 (t, J= 2.3 Hz, 1H), 4.14 (t, J= 7.1 Hz, 2H), 2.01 (t, J= 7.1 Hz, 2H), 0.96 – 0.88 (m, 2H), 0.88 -0.81 (m, 2H).
Step 3: tert-Butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxylate

[00391] tert-Butyl 2,6-dichloropyridine-3-carboxylate (687 mg, 2.770 mmol), 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (610 mg, 2.770 mmol), and freshly ground potassium carbonate (459 mg, 3.324 mmol) were combined in anhydrous DMSO (13.75 mL). l,4-diazabicyclo[2.2.2]octane (DAB CO (1,4-diazabicyclo[2.2.2]octane), 62 mg, 0.5540 mmol) was added, and the mixture was
stirred at room temperature under nitrogen for 16 hours. The reaction mixture was diluted with water (20 mL) and stirred for 15 minutes. The resulting solid was collected and washed with water. The solid was dissolved in dichloromethane and dried over magnesium sulfate. The mixture was filtered and concentrated to give tert-butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (1.01 g, 84%). ESI-MS m/z calc. 431.12, found 432.1 (M+l) +; Retention time: 0.88 minutes.
Step 4: 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid

[00392] tert-Butyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (1.01 g, 2.339 mmol) and trifluoroacetic acid (1.8 mL, 23.39 mmol) were combined in dichloromethane (10 mL) and heated at 40 °C for 3 h. The reaction was concentrated. Hexanes were added, and the mixture was concentrated again to give 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (873 mg, 99%) ESI-MS m/z calc. 375.06, found 376.1 (M+l)+; Retention time: 0.69 minutes.
Step 5: N-(Benzenesulfonyl)-2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxamide

[00393] A solution of 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]
ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (0.15 g, 0.3992 mmol) and carbonyl diimidazole (77 mg, 0.4790 mmol) in THF (2.0 mL) was stirred for one hour, and
benzenesulfonamide (81 mg, 0.5190 mmol) and DBU (72 μΐ^, 0.4790 mmol) were added. The reaction was stirred for 16 hours, acidified with 1 M aqueous citric acid, and extracted with ethyl acetate. The combined extracts were dried over sodium sulfate and evaporated. The residue was purified by silica gel chromatography eluting with a gradient of methanol in dichloromethane (0-5%) to give N-(benzenesulfonyl)-2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxamide (160 mg, 78%). ESI-MS m/z calc. 514.07, found 515.1 (M+l)+; Retention time: 0.74 minutes.
Step 6: N-(Benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide

[00394] A mixture of N-(benzenesulfonyl)-2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxamide (160 mg, 0.3107 mmol), (4S)-2,2,4-trimethylpyrrolidine hydrochloride salt (139 mg, 0.9321 mmol), and potassium carbonate (258 mg, 1.864 mmol) in DMSO (1.5 mL) was stirred at 130 °C for 17 hours. The reaction mixture was acidified with 1 M aqueous citric acid and extracted with ethyl acetate. The combined extracts were dried over sodium sulfate and evaporated to yield a crude product that was purified by reverse-phase HPLC utilizing a gradient of 10-99%) acetonitrile in 5 mM aqueous HC1 to yield N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (87 mg, 47%). ESI-MS m/z calc. 591.21, found 592.3 (M+l) +; Retention time: 2.21 minutes. 1H MR (400 MHz, DMSO-d6) δ 12.48 (s, 1H), 8.19 (d, J= 2.8 Hz, 1H), 8.04 – 7.96 (m, 2H), 7.81 (d, J= 8.2 Hz, 1H), 7.77 – 7.70 (m, 1H), 7.70 – 7.62 (m, 2H), 6.92 (d, J= 8.2 Hz, 1H), 6.10 (d, J= 2.8 Hz, 1H), 4.31 (t, J= 7.0 Hz, 2H), 2.42 (t, J= 10.5 Hz, 1H), 2.28 (dd, J = 10.2, 7.0 Hz, 1H), 2.17 – 2.01 (m, 3H), 1.82 (dd, J= 11.9, 5.5 Hz, 1H), 1.52 (d, J = 9.4 Hz, 6H), 1.36 (t, J= 12.1 Hz, 1H), 1.01 – 0.92 (m, 2H), 0.92 – 0.85 (m, 2H), 0.65 (d, J = 6.3 Hz, 3H). pKa: 4.95±0.06.
Synthesis of sodium salt of N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl) cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (sodium salt of Compound I)
[00395] N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide (1000 mg, 1.679 mmol) was dissolved in ethanol (19.87 ml) under warming, filtered clear through a syringe filter (0.2 μπι), washed with warm ethanol (10 ml) and the warm solution was treated with 1M NaOH (1.679 ml, 1.679 mmol). The solution was evaporated at 30-35 °C, co-evaporated 3 times with ethanol (-20 ml), to give a solid, which was dried overnight under vacuum in a drying cabinet at 45 °C with a nitrogen bleed to give 951 mg of a cream colored solid. The solid was further dried under vacuum in a drying cabinet at 45 °C with a nitrogen bleed over the weekend. 930 mg (89%) of the sodium salt of N-(benzenesulfonyl)-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-l-yl]pyridine-3-carboxamide was obtained as an off-white amorphous solid. ¾ NMR (400 MHz, DMSO-d) δ 8.15 (d, J= 2.7 Hz, 1H), 7.81 (dd, J= 6.7, 3.1 Hz, 2H), 7.61 (d, J= 7.9 Hz, 1H), 7.39 (dd, J= 4.9, 2.0 Hz, 3H), 6.74 (d, J= 7.9 Hz, 1H), 6.01 (d, J= 2.6 Hz, 1H), 4.29 (t, J= 7.0 Hz, 2H), 2.93 – 2.78 (m, 2H), 2.07 (t, J= 7.1 Hz, 3H), 1.78 (dd, J= 11.8, 5.6 Hz, 1H), 1.52 (d, J= 13.6 Hz, 6H), 1.33 (t, J= 12.0 Hz, 1H), 1.00 – 0.92 (m, 2H), 0.89 (q, J= 5.3, 4.6 Hz, 2H), 0.71 (d, J= 6.3 Hz, 3H). EST-MS m/z calc. 591.2127, found 592.0 (M+l)+; Retention time: 3.28 minutes. XRPD (see FIG. 16).
Alternate synthesis of 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy] pyrazol-l-yl] pyridine-3-carboxylic acid
Step 1: ethyl 3-hydroxy-lH-pyrazole-4-carboxylate

[00396] A mixture of EtOH (20.00 L, 10 vol) and diethyl 2-(ethoxymethylene) propanedioate (2000 g, 9.249 mol, 1.0 equiv) was added under nitrogen purge a to a 50 L reactor equipped with a reflux condenser (10 °C) and the jacket set to 40 °C. The mixture was stirred, and then hydrazine hydrate (538.9 g of 55 %w/w, 523.7 mL of 55 %w/w, 9.249 mol, 1.00 equiv) was added in portions via an addition funnel. Once the addition was complete, the reaction was heated to 75 °C for 22 h to afford a solution of ethyl 3-hydroxy-lH-pyrazole-4-carboxylate that was used directly in the next step.
Step 2: l-(tert-butyl) 4-ethyl 3-hydroxy-lH-pyrazole-l,4-dicarboxylate

[00397] The solution of ethyl 3 -hydroxy- lH-pyrazole-4-carboxylate was cooled from 75 °C to 40 °C, then triethylamine (TEA) (46.80 g, 64.46 mL, 462.5 mmol, 0.05 eq.) was added. A solution of Boc anhydride (2.119 kg, 9.711 moll .05 equiv) in EtOH (2.000 L, 1 equiv) was added to the reactor over 35 min. The mixture was stirred for 4 hours to complete the reaction; then water (10.00 L, 5.0 vol) was added over 15 mins. The resulting mixture was cooled to 20 °C to complete crystallization of the product. The crystals were allowed to age for 1 hour, then the mixture was filtered. The solid was washed with a mixture of EtOH (4.000 L, 2.0 vol) and water (2.000 L, 1.0 vol). The solid was then dried in vacuo to afford l-(tert-butyl)-4-ethyl-3-hydroxy-lH-pyrazole-1,4-dicarboxylate (1530 g, 65%) as colorless, fine needle, crystalline solid. ¾ NMR (400 MHz, DMSO-de) δ 11.61 (s, 1H), 8.40 (s, 1H), 4.20 (q, J = 7.1 Hz, 2H), 1.56 (s, 9H), 1.25 (t, J = 7.1 Hz, 3H).
Step 3: l-(tert-butyl) 4-ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-ΙΗ-pyr azole- 1 ,4-dicarboxylate

[00398] A 5L reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temperature and nitrogen purge. The vessel was charged with toluene (1.0L, 10.0 vol), 2-[l-(trifluoromethyl)cyclopropyl]ethanol (lOO.Og, 648.8 mmol, 1.0 equiv), and l-(tert-butyl) 4-ethyl 3-hydroxy-lH-pyrazole-l,4-dicarboxylate (166.3 g, 648.8 mmol), and the mixture was stirred. The reaction mixture was charged with triphenyl phosphine (195.7 g, 746.1 mmol, 1.15 equiv), then the reactor was set to maintain an internal temperature of 40 °C. Diisopropyl azoldicarboxylate (150.9 g, 746.1 mmol, 1.15 equiv) was added into an addition funnel and was added to the reaction while maintaining the reaction temperature between 40 and 50 °C (addition was exothermic, exotherm addition controlled), and stirred for a total of 2.5 hours. Once the reaction was deemed complete by HPLC, heptane was added (400 mL, 4 vol), the solution was cooled to 20 °C over 60 minutes, and the bulk of triphenylphosphine oxide-DIAD complex (TPPO-DIAD) crystallized out. Once at room temp, the mixture was filtered, and the solid was washed with heptane (400 mL, 4.0 vol) and pulled dry. The filtrate was used in the next step as a solution in toluene-heptane without further purification.
Step 4: ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylate

[00399] A 500mL reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temp, and nitrogen purge. The vessel was charged with a toluene solution consisting of approximately 160 mmol, 65.0 g of l-(tert-butyl) 4-ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-l,4-dicarboxylate in 3 vol of toluene (prepared by concentrating a 25% portion of filtrate from previous reaction down to 4 volumes in a rotovap). The reaction was set to maintain an internal temperature at 40 °C and KOH (33.1 g, 1.5 eq. of aqueous 45 % KOH solution) was added in one portion, resulting in a mild exothermic addition, while CO2 was generated upon removal of the protecting group. The reaction proceeded for 1.5 hr, monitored by HPLC, with the product partially crystallizing during the reaction. Heptane (160 mL, 2.5 vol) was added to the reaction mixture and the reaction was cooled to room temperature over 30 minutes. The resulting mixture was filtered, and the solid was
washed with heptane (80.00 mL, 1.25 vol), pulled dry, then dried in vacuo (55 °C, vacuum). 52.3 g of ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylate was obtained as a crude, colorless solid that was used without further purification.
Step 5: 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid

[00400] A 500mL reactor was started with the jacket set to 40 °C, stirring at 450 rpm, reflux condenser at room temp, and nitrogen purge. The vessel was charged with methanol (150.0 mL, 3.0 vol), a solution of ethyl 3-(2-(l-(trifluoromethyl)cyclopropyl) ethoxy)-lH-pyrazole-4-carboxylate (50.0 g, 171.1 mmol, 1.0 equiv), and the reaction was stirred to suspend the solids. The reactor was set to maintain internal temperature at 40 °C. To the mixture was added KOH (96 g of aqueous 45 % KOH, 1.71 mol, 10.0 equiv) in portions maintaining the internal temperature <50 °C. Once addition was complete, the reaction was set to maintain temperature at 50 °C, and the reaction proceeded for 23 hours, monitored by HPLC. Once complete the reaction was cooled to 10 °C then partially concentrated on a rotary evaporator to remove most of the MeOH. The resulting solution was diluted with water (250 mL, 5.0 vol) and 2-Me-THF (150 mL, 3.0 vol), and transferred to the reactor, stirred at room temp, then stopped, and layers were allowed to separate. The layers were tested, with remaining TPPO-DIAD complex in the organic layer and product in the aqueous layer. The aqueous layer was washed again with 2-Me-THF (100 mL, 2.0 vol), the layers separated, and the aqueous layer returned to the reactor vessel. The stirrer was started and set to 450 rpm, and the reactor jacket was set to 0 °C. The pH was adjusted to pH acidic by addition of 6M aqueous HC1 (427mL, 15 equiv) portion wise, maintaining the internal temperature between 10 and 30 °C. The product began to crystallize close to pH neutral and was accompanied with strong off-gassing, and so the acid was added slowly, and then further added to reach pH 1 once the off-gassing had ended. To the resulting suspension was added 2-Me-THF (400 mL, 8.0 vol), and the product was allowed to dissolve into
the organic layer. Stirring was stopped, the layers were separated, and the aqueous layer was returned to the reactor, stirred and re-extracted with 2-Me-THF (100 mL, 2.0 vol). The organic layers were combined in the reactor and stirred at room temperature, washed with brine (lOOmL, 2 vols), dried over Na2S04, filtered through celite, and the solid was washed with 2-Me-THF (50 mL, 1.0 vol). The filtrate was transferred to a clean rotovap flask, stirred, warmed to 50 °C and heptane (200 mL, 4.0 vol) added, and then partially concentrated with the addition of heptane (300 mL, 6.0 vol) and then seeded with 50mg of 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid), and the product crystallized during solvent removal. The distillation was stopped when the bulk of the 2-Me-THF had distilled off. The bath heater was turned off, the vacuum removed, and the mixture was allowed to stir and cool to room temperature. The mixture was filtered (slow speed) and the solid was washed with heptane (100 mL, 2.0 vol), and the solid was collected and dried in vacuo (50 °C, rotovap). 22.47 g of 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole-4-carboxylic acid was obtained as an off-white solid. ¾ MR (400 MHz, DMSO-de) δ
12.45 (s, 2H), 8.01 (s, 1H), 4.26 (t, J= 7.0 Hz, 2H), 2.05 (t, J= 7.0 Hz, 2H), 0.92 (m,
4H).
Step 6: 3-(2-(l-(trifluoromethyl)cyclopropyl)ethoxy)-lH-pyrazole

[00401] A mixture of toluene (490.0 mL), 3-(2-(l-(trifluoromethyl)cyclopropyl) ethoxy)-lH-pyrazole-4-carboxylic acid (70.0 g, 264.9 mmol), and DMSO (70.00 mL) was placed in a reactor and heated to 100 °C with stirring. DBU (approximately 20.16 g, 19.80 mL, 132.4 mmol) was added to the reactor over 15 min. The mixture was stirred for 20 h to complete the reaction and then cooled to 20 °C. The mixture was washed with water (350.0 mL), then 0.5N aq HC1 (280.0 mL), then water (2 x 140.0 mL), and lastly with brine (210.0 mL). The organic layer was dried with Na2S04, and then activated charcoal (5 g, Darco 100 mesh) was added to the stirred slurry. The dried mixture was filtered through celite, and the solid was washed with toluene (140.0 mL) and then pulled dry. The filtrate was concentrated in a rotovap (50 °C, vac) to afford 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (30.89 g, 53%) as an amber oil. 1H MR (400 MHz, DMSO-d) δ 11.87 (s, 1H), 7.50 (d, J= 2.4 Hz, 1H), 5.63 (d, J = 2.4 Hz, 1H), 4.23 – 4.06 (m, 2H), 2.01 (t, J= 7.1 Hz, 2H), 1.00 – 0.77 (m, 4H).
Step 7: ethyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy] pyrazol-l-yl]pyridine-3-carboxylate

[00402] A mixture of DMF (180.0 mL), ethyl 2,6-dichloropyridine-3-carboxylate (approximately 29.97 g, 136.2 mmol), 3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]-lH-pyrazole (30.0 g, 136.2 mmol), and K2CO3, (325 mesh, approximately 24.48 g, 177.1 mmol) was added to a stirred reactor at 20 °C. DABCO (approximately 2.292 g, 20.43 mmol) was then added to the reactor, and the mixture was stirred at 20 °C for 1 hour, and then the temperature was increased to 30 °C, and the mixture stirred for 24 hours to complete the reaction. The mixture was cooled to 20 °C; then water (360 mL) was added slowly. The mixture was then drained from the reactor and the solid was isolated by filtration. The solid was then washed with water (2 x 150 mL), and then the solid was dried under vacuum at 55 °C to afford ethyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (51.37 g, 93%) as a fine, beige colored solid. ¾ MR (400 MHz, DMSO-^e) δ 8.44 (d, J= 2.9 Hz, 1H), 8.41 (d, J= 8.5 Hz, 1H), 7.75 (d, J= 8.5 Hz, 1H), 6.21 (d, J= 2.9 Hz, 1H), 4.34 (m, 4H), 2.09 (t, J= 7.1 Hz, 2H), 1.34 (t, J= 7.1 Hz, 3H), 1.00 – 0.84 (m, 4H).
Step 8: 2-Chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid

[00403] A solution of ethyl 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl] ethoxy]pyrazol-l-yl]pyridine-3-carboxylate (50.0 g, 123.8 mmol) in THF (300.0 mL) was prepared in a reactor at 20 °C. EtOH (150.0 mL) was added, followed by aqueous NaOH (approximately 59.44 g of 10 %w/w, 148.6 mmol). The mixture was stirred for 1 hour to complete the reaction; then aq IN HC1 (750.0 mL) was slowly added. The resulting suspension was stirred for 30 min at 10 °C, and then the solid was isolated by filtration. The solid was washed with water (150 mL then 2 x 100 mL) and then pulled dry by vacuum. The solid was then further dried under vacuum with heating to afford 2-chloro-6-[3-[2-[l-(trifluoromethyl)cyclopropyl]ethoxy]pyrazol-l-yl]pyridine-3-carboxylic acid (42.29 g, 91%). ¾ NMR (400 MHz, DMSO-i¾) δ 13.63 (s, 1H), 8.48 -8.35 (m, 2H), 7.73 (d, J= 8.4 Hz, 1H), 6.20 (d, J= 2.9 Hz, 1H), 4.35 (t, J= 7.1 Hz, 2H), 2.09 (t, J= 7.1 Hz, 2H), 1.01 – 0.82 (m, 4H).
Example 2: Preparation of a Spray Dried Dispersion (SDD) of Compound I
[00404] A spray dried dispersion of Compound I (free form) was prepared using Buchi Mini Spray Dryer B290. HPMCAS-HG (6.0 grams) was dissolved in 200 mL of MeOH/DCM (1/1), and Compound I (6.0 grams) was added and stirred for 30 minutes forming a clear solution. The resulting solution was spray dried under the following conditions resulting in a 50 wt% Compound 1/50 wt% HPMCAS- HG spray dried dispersion (Yield: 80%, Solid load: 6%). FIG. 14 shows the XRPD spectrum of a SDD of 50% Compound I in HPMCAS-HG. FIG. 15 is spectrum showing modulated differential scanning calorimetry (MDSC) spectrum of a spray dried dispersion (SDD) of 50% Compound I in HPMCAS-HG.
Table 64 SDD of Compound I

Example 3: Synthesis of Compound II: (R)-l-(2,2-Difluorobenzo[d][l,3]dioxol-5- yl)-N-(l-(2,3-dihydroxypropyl)-6-fluoro-2-(l-hydroxy-2- -2-yl)-lH-indol-5-yl)cyclopropanecarboxamide

Step 1: (R)-Benzyl 2-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropanoate and ((S)-2,2-Dimethyl-l,3-dioxolan-4-yl)methyl 2-(l-(((R)-2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropanoate
[00405] Cesium carbonate (8.23 g, 25.3 mmol) was added to a mixture of benzyl 2-(6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropanoate (3.0 g, 8.4 mmol) and (S)-(2,2-dimethyl-l,3-dioxolan-4-yl)methyl 4-methylbenzenesulfonate (7.23 g, 25.3 mmol) in DMF (N,N-dimethylformamide) (17 mL). The reaction was stirred at 80 °C for 46 hours under a nitrogen atmosphere. The mixture was then partitioned between ethyl acetate and water. The aqueous layer was extracted with ethyl acetate. The combined ethyl acetate layers were washed with brine, dried over MgS04, filtered and concentrated. The crude product, a viscous brown oil which contains both of the products shown above, was taken directly to the next step without further purification. (R)-Benzyl 2-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropanoate, ESI-MS m/z calc. 470.2, found 471.5 (M+l)+. Retention time 2.20 minutes. ((S)-2,2-Dimethyl-l,3-dioxolan-4-yl)methyl 2-(l-(((R)-2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropanoate, ESI-MS m/z calc. 494.5, found 495.7 (M+l)+. Retention time 2.01 minutes.
Step 2: (R)-2-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropan-l-ol
[00406] The crude reaction mixture obtained in step (A) was dissolved in THF (tetrahydrofuran) (42 mL) and cooled in an ice-water bath. LiAlH4 (16.8 mL of 1 M solution, 16.8 mmol) was added drop-wise. After the addition was complete, the
mixture was stirred for an additional 5 minutes. The reaction was quenched by adding water (1 mL), 15% NaOH solution (1 mL) and then water (3 mL). The mixture was filtered over Celite, and the solids were washed with THF and ethyl acetate. The filtrate was concentrated and purified by column chromatography (30-60% ethyl acetate-hexanes) to obtain (R)-2-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropan-l-ol as a brown oil (2.68g, 87 % over 2 steps). ESI-MS m/z calc. 366.4, found 367.3 (M+l)+. Retention time 1.68 minutes. 1H MR (400 MHz, DMSO-^6) δ 8.34 (d, J = 7.6 Hz, 1H), 7.65 (d, J = 13.4 Hz, 1H), 6.57 (s, 1H), 4.94 (t, J = 5.4 Hz, 1H), 4.64 – 4.60 (m, 1H), 4.52 – 4.42(m, 2H), 4.16 – 4.14 (m, 1H), 3.76 – 3.74 (m, 1H), 3.63 – 3.53 (m, 2H), 1.42 (s, 3H), 1.38 – 1.36 (m, 6H) and 1.19 (s, 3H) ppm. (DMSO is dimethylsulfoxide).
Step 3: (R)-2-(5-amino-l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-lH-indol-2-yl)-2-methylpropan-l-ol
[00407] (R)-2-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-5-nitro-lH-indol-2-yl)-2-methylpropan-l-ol (2.5 g, 6.82 mmol) was dissolved in ethanol (70 mL) and the reaction was flushed with N2. Then Pd-C (250 mg, 5% wt) was added. The reaction was flushed with nitrogen again and then stirred under H2 (atm). After 2.5 hours only partial conversion to the product was observed by LCMS. The reaction was filtered through Celite and concentrated. The residue was re-subjected to the conditions above. After 2 hours LCMS indicated complete conversion to product. The reaction mixture was filtered through Celite. The filtrate was concentrated to yield the product (1.82 g, 79 %). ESI-MS m/z calc. 336.2, found 337.5 (M+l)+. Retention time 0.86 minutes. ¾ NMR (400 MHz, DMSO-^6) δ 7.17 (d, J = 12.6 Hz, 1H), 6.76 (d, J = 9.0 Hz, 1H), 6.03 (s, 1H), 4.79 – 4.76 (m, 1H), 4.46 (s, 2H), 4.37 – 4.31 (m, 3H),4.06 (dd, J = 6.1, 8.3 Hz, 1H), 3.70 – 3.67 (m, 1H), 3.55 – 3.52 (m, 2H), 1.41 (s, 3H), 1.32 (s, 6H) and 1.21 (s, 3H) ppm.
Step 4: (R)-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-N-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-2-(l-hydroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide
[00408] DMF (3 drops) was added to a stirring mixture of l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)cyclopropanecarboxylic acid (1.87 g, 7.7 mmol) and thionyl chloride (1.30 mL, 17.9 mmol). After 1 hour a clear solution had formed. The
solution was concentrated under vacuum and then toluene (3 mL) was added and the mixture was concentrated again. The toluene step was repeated once more and the residue was placed on high vacuum for 10 minutes. The acid chloride was then dissolved in dichloromethane (10 mL) and added to a mixture of (R)-2-(5 -amino- 1-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-lH-indol-2-yl)-2-methylpropan-l-ol (1.8 g, 5.4 mmol) and triethylamine (2.24 mL, 16.1 mmol) in dichloromethane (45 mL). The reaction was stirred at room temperature for 1 hour. The reaction was washed with IN HC1 solution, saturated NaHCCb solution and brine, dried over MgSCb and concentrated to yield the product (3g, 100%). ESI-MS m/z calc. 560.6, found 561.7 (M+l)+. Retention time 2.05 minutes. ¾ NMR (400 MHz, DMSO-^6) δ 8.31 (s, 1H), 7.53 (s, 1H), 7.42 – 7.40 (m, 2H), 7.34 – 7.30 (m, 3H), 6.24 (s, 1H), 4.51 – 4.48 (m, 1H), 4.39 – 4.34 (m,2H), 4.08 (dd, J = 6.0, 8.3 Hz, 1H), 3.69 (t, J = 7.6 Hz, 1H), 3.58 – 3.51 (m, 2H), 1.48 – 1.45 (m, 2H), 1.39 (s, 3H), 1.34 – 1.33 (m, 6H), 1.18 (s, 3H) and 1.14 -1.12 (m, 2H) ppm
Step 5: (R)-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-N-(l-(2,3-dihydroxypropyl)-6-fluoro-2-(l-hydroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide
[00409] (R)-l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)-N-(l-((2,2-dimethyl-l,3-dioxolan-4-yl)methyl)-6-fluoro-2-(l -hydroxy -2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide (3.0 g, 5.4 mmol) was dissolved in methanol (52 mL). Water (5.2 mL) was added followed by p-TsOH.H20 (p-toluenesulfonic acid hydrate) (204 mg, 1.1 mmol). The reaction was heated at 80 °C for 45 minutes. The solution was concentrated and then partitioned between ethyl acetate and saturated NaHCCb solution. The ethyl acetate layer was dried over MgS04 and concentrated. The residue was purified by column chromatography (50-100 % ethyl acetate – hexanes) to yield the product. (1.3 g, 47 %, ee >98% by SFC). ESI-MS m/z calc. 520.5, found 521.7 (M+l)+. Retention time 1.69 minutes. ¾ NMR (400 MHz, DMSC 6) δ 8.31 (s, 1H), 7.53 (s, 1H), 7.42 – 7.38 (m, 2H), 7.33 – 7.30 (m, 2H), 6.22 (s, 1H), 5.01 (d, J = 5.2 Hz, 1H), 4.90 (t, J = 5.5 Hz, 1H), 4.75 (t, J = 5.8 Hz, 1H), 4.40 (dd, J = 2.6, 15.1 Hz, 1H), 4.10 (dd, J = 8.7, 15.1 Hz, 1H), 3.90 (s, 1H), 3.65 – 3.54 (m, 2H), 3.48 – 3.33 (m, 2H), 1.48 -1.45 (m, 2H), 1.35 (s, 3H), 1.32 (s, 3H) and 1.14 – 1.11 (m, 2H) ppm.
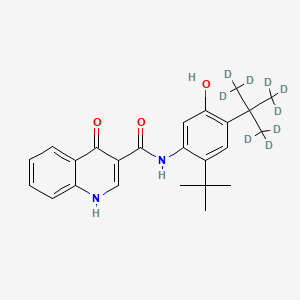
Example 4: Synthesis of Compound III: N-(2,4-di-terf-butyl-5-hydroxyphi oxo-l,4-dihydroquinoline-3-carboxamide
Part A: Synthesis of 4-oxo-l,4-dihydroquinoline-3-carboxylic acid

Step 1: 2-Phenylaminomethylene-malonic acid diethyl ester
[00410] A mixture of aniline (25.6 g, 0.275 mol) and diethyl 2-(ethoxymethylene)malonate (62.4 g, 0.288 mol) was heated at 140-150 °C for 2 h. The mixture was cooled to room temperature and dried under reduced pressure to afford 2-phenylaminomethylene-malonic acid diethyl ester as a solid, which was used in the next step without further purification. ¾ MR (OMSO-de) δ 1 1.00 (d, 1H), 8.54 (d, J = 13.6 Hz, 1H), 7.36-7.39 (m, 2H), 7.13-7.17 (m, 3H), 4.17-4.33 (m, 4H), 1.18-1.40 (m, 6H).
Step 2: 4-Hydroxyquinoline-3-carboxylic acid ethyl ester
[00411] A I L three-necked flask fitted with a mechanical stirrer was charged with 2-phenylaminomethylene-malonic acid diethyl ester (26.3 g, 0.100 mol), polyphosphoric acid (270 g) and phosphoryl chloride (750 g). The mixture was heated to 70 °C and stirred for 4 h. The mixture was cooled to room temperature and filtered. The residue was treated with aqueous Na2CCb solution, filtered, washed with water and dried. 4-Hydroxyquinoline-3-carboxylic acid ethyl ester was obtained as a pale brown solid (15.2 g, 70%). The crude product was used in next step without further purification.
Step 3: 4-Oxo-l,4-dihydroquinoline-3-carboxylic acid
[00412] 4-Hydroxyquinoline-3-carboxylic acid ethyl ester (15 g, 69 mmol) was suspended in sodium hydroxide solution (2N, 150 mL) and stirred for 2 h at reflux. After cooling, the mixture was filtered, and the filtrate was acidified to pH 4 with 2N HCl. The resulting precipitate was collected via filtration, washed with water and dried under vacuum to give 4-oxo-l,4-dihydroquinoline-3-carboxylic acid as a pale white solid (10.5 g, 92 %). ¾ MR (DMSO-^e) δ 15.34 (s, 1 H), 13.42 (s, 1 H), 8.89 (s, 8.28 (d, J = 8.0 Hz, 1H), 7.88 (m, 1H), 7.81 (d, J = 8.4 Hz, 1H), 7.60 (m, 1H).
Part B: Synthesis of N-(2,4-di-terf-butyl-5-hydroxyphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide

Step 1: Carbonic acid 2,4-di-ferf-butyl-phenyl ester methyl ester
[00413] Methyl chloroformate (58 mL, 750 mmol) was added dropwise to a solution of 2,4-di-fert-butyl-phenol (103.2 g, 500 mmol), Et3N (139 mL, 1000 mmol) and DMAP (3.05 g, 25 mmol) in dichloromethane (400 mL) cooled in an ice-water bath to 0 °C. The mixture was allowed to warm to room temperature while stirring overnight, then filtered through silica gel (approx. 1L) using 10% ethyl acetate – hexanes (~ 4 L) as the eluent. The combined filtrates were concentrated to yield carbonic acid 2,4-di-tert-butyl-phenyl ester methyl ester as a yellow oil (132 g, quant.). ¾ MR (400 MHz, DMSO-i¾) δ 7.35 (d, J = 2.4 Hz, 1H), 7.29 (dd, J = 8.5, 2.4 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 3.85 (s, 3H), 1.30 (s, 9H), 1.29 (s, 9H).
Step 2: Carbonic acid 2,4-di-ferf-butyl-5-nitro-phenyl ester methyl ester and Carbonic acid 2,4-di-terf-butyl-6-nitro-phenyl ester methyl ester
[00414] To a stirring mixture of carbonic acid 2,4-di-tert-butyl-phenyl ester methyl ester (4.76 g, 180 mmol) in cone, sulfuric acid (2 mL), cooled in an ice-water bath, was added a cooled mixture of sulfuric acid (2 mL) and nitric acid (2 mL). The addition was done slowly so that the reaction temperature did not exceed 50 °C. The reaction was allowed to stir for 2 h while warming to room temperature. The reaction mixture was then added to ice-water and extracted into diethyl ether. The ether layer was dried (MgS04), concentrated and purified by column chromatography (0 – 10% ethyl acetate – hexanes) to yield a mixture of carbonic acid 2,4-di-tert-butyl-5-nitro-phenyl ester methyl ester and carbonic acid 2,4-di-tert-butyl-6-nitro-phenyl ester methyl ester as a pale yellow solid (4.28 g), which was used directly in the next step.
Step 3: 2,4-Di-terf-butyl-5-nitro-phenol and 2,4-Di-terf-butyl-6-nitro-phenol
[00415] The mixture of carbonic acid 2,4-di-tert-butyl-5-nitro-phenyl ester methyl ester and carbonic acid 2,4-di-tert-butyl-6-nitro-phenyl ester methyl ester (4.2 g, 14.0 mmol) was dissolved in MeOH (65 mL) before KOH (2.0 g, 36 mmol) was added. The mixture was stirred at room temperature for 2 h. The reaction mixture was then made acidic (pH 2-3) by adding cone. HC1 and partitioned between water and diethyl ether. The ether layer was dried (MgS04), concentrated and purified by column
chromatography (0 – 5 % ethyl acetate – hexanes) to provide 2,4-di-tert-butyl-5-nitro-phenol (1.31 g, 29% over 2 steps) and 2,4-di-tert-butyl-6-nitro-phenol. 2,4-Oi-tert-butyl-5-nitro-phenol: ¾ MR (400 MHz, DMSO-i¾) δ 10.14 (s, 1H, OH), 7.34 (s, 1H), 6.83 (s, 1H), 1.36 (s, 9H), 1.30 (s, 9H). 2,4-Di-tert-butyl-6-nitro-phenol: ¾ MR (400 MHz, CDCh) δ 11.48 (s, 1H), 7.98 (d, J = 2.5 Hz, 1H), 7.66 (d, J = 2.4 Hz, 1H), 1.47 (s, 9H), 1.34 (s, 9H).
Step 4: 5-Amino-2,4-di-terf-butyl-phenol
[00416] To a refluxing solution of 2,4-di-tert-butyl-5-nitro-phenol (1.86 g, 7.40 mmol) and ammonium formate (1.86 g) in ethanol (75 mL) was added Pd-5% wt. on activated carbon (900 mg). The reaction mixture was stirred at reflux for 2 h, cooled to room temperature and filtered through Celite. The Celite was washed with methanol and the combined filtrates were concentrated to yield 5-amino-2,4-di-tert-butyl-phenol as a grey solid (1.66 g, quant.). ¾ MR (400 MHz, DMSO-^e) δ 8.64 (s, 1H, OH), 6.84 (s, 1H), 6.08 (s, 1H), 4.39 (s, 2H, H2), 1.27 (m, 18H); HPLC ret. time 2.72 min, 10-99 % CftCN, 5 min run; ESI-MS 222.4 m/z [M+H]+.
Step 5: N-(5-hydroxy-2,4-di-ieri-butyl-phenyl)-4-oxo-lH-quinoline-3-carboxamide

[00417] To a suspension of 4-oxo-l,4-dihydroquinolin-3-carboxylic acid (35.5 g, 188 mmol) and HBTU (85.7 g, 226 mmol) in DMF (280 mL) was added Et3N (63.0 mL, 451 mmol) at ambient temperature. The mixture became homogeneous and was allowed to stir for 10 min before 5-amino-2,4-di-tert-butyl-phenol (50.0 g, 226 mmol) was added in small portions. The mixture was allowed to stir overnight at ambient temperature. The mixture became heterogeneous over the course of the reaction. After all of the acid was consumed (LC-MS analysis, MH+ 190, 1.71 min), the solvent was removed in vacuo. EtOH (ethyl alcohol) was added to the orange solid material to produce a slurry. The mixture was stirred on a rotovap (bath temperature 65 °C) for 15 min without placing the system under vacuum. The mixture was filtered and the captured solid was washed with hexanes to provide a white solid that was the EtOH crystalate. Et20
(diethyl ether) was added to the solid obtained above until a slurry was formed. The mixture was stirred on a rotovapor (bath temperature 25 °C) for 15 min without placing the system under vacuum. The mixture was filtered and the solid captured. This procedure was performed a total of five times. The solid obtained after the fifth precipitation was placed under vacuum overnight to provide N-(5-hydroxy-2,4-di-tert-butyl-phenyl)-4-oxo-lH-quinoline-3-carboxamide (38 g, 52%). HPLC ret. time 3.45 min, 10-99% CftCN, 5 min run; 1H MR (400 MHz, DMSO-i¾) δ 12.88 (s, 1H), 11.83 (s, 1H), 9.20 (s, 1H), 8.87 (s, 1H), 8.33 (dd, J = 8.2, 1.0 Hz, 1H), 7.83-7.79 (m, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.54-7.50 (m, 1H), 7.17 (s, 1H), 7.10 (s, 1H), 1.38 (s, 9H), 1.37 (s, 9H); ESI-MS m/z calc’d 392.21; found 393.3 [M+H]+.
PAPER
The New England journal of medicine (2018), 379(17), 1599-1611
https://www.nejm.org/doi/10.1056/NEJMoa1807119
////////////VX-659, VX 659, VX659, PHASE 2, CYSTIC FIBRIOSIS , VERTEX, Bamocaftor potassium
[K+].C[C@@H]1CN(c2nc(ccc2C(=O)[N-]S(=O)(=O)c3ccccc3)n4ccc(OCCC5(CC5)C(F)(F)F)n4)C(C)(C)C1
C[C@@H]1CN(c2nc(ccc2C(=O)NS(=O)(=O)c3ccccc3)n4ccc(OCCC5(CC5)C(F)(F)F)n4)C(C)(C)C1

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO






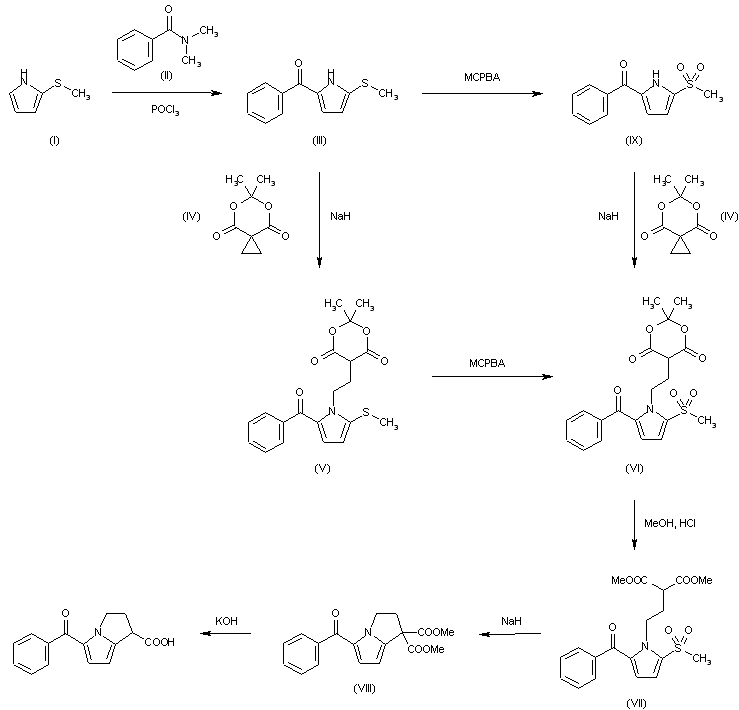



 Cavosonstat (N91115) was an experimental therapy being developed by Nivalis Therapeutics. Its primary mechanism of action was to inhibit the S-nitrosoglutathione reductase (GSNOR) enzyme and to stabilize cystic fibrosis transmembrane regulator (CFTR) protein activity. A press release published in February announced the end of research for this therapy in cystic fibrosis (CF) patients with F508del mutations. The drug, which did not meet primary endpoints in a Phase 2 trial, had been referred to as the first of a new class of compounds that stabilizes the CFTR activity.
Cavosonstat (N91115) was an experimental therapy being developed by Nivalis Therapeutics. Its primary mechanism of action was to inhibit the S-nitrosoglutathione reductase (GSNOR) enzyme and to stabilize cystic fibrosis transmembrane regulator (CFTR) protein activity. A press release published in February announced the end of research for this therapy in cystic fibrosis (CF) patients with F508del mutations. The drug, which did not meet primary endpoints in a Phase 2 trial, had been referred to as the first of a new class of compounds that stabilizes the CFTR activity.