
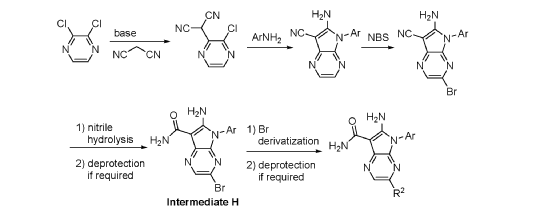
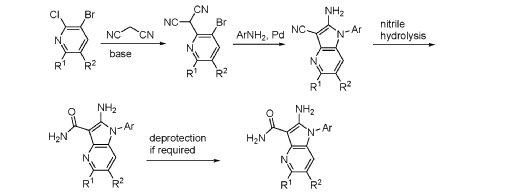
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Mangaciclanol
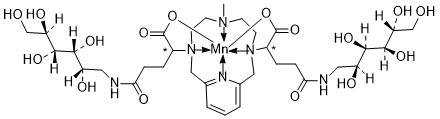
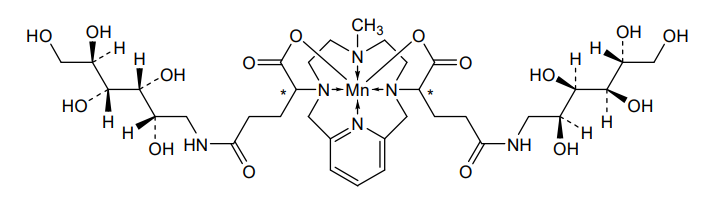
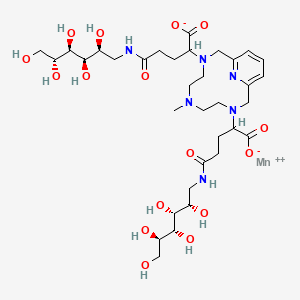
Mangaciclanol
Cas 2169771-05-3
MF C34H56MnN6O16 MW859.8 g/mol
- ANU6AE7NAP
- [[1,1′-[(6-Methyl-3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-1(15),11,13-triene-3,9-diyl-kappaN3,kappaN6,kappaN9,kappaN15)bis[[4-(carboxy-kappaO)-1-oxo-4,1-butanediyl]imino]]bis[1-deoxy-D-glucitolato]](2-)]manganese
2-[9-[1-carboxylato-4-oxo-4-[[(2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl]amino]butyl]-6-methyl-3,6,9,15-tetrazabicyclo[9.3.1]pentadeca-1(15),11,13-trien-3-yl]-5-oxo-5-[[(2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl]amino]pentanoate;manganese(2+)

diagnostic imaging agent, ANU6AE7NAP



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
/////////mangaciclanol, diagnostic imaging agent, ANU6AE7NAP
Elinzanetant
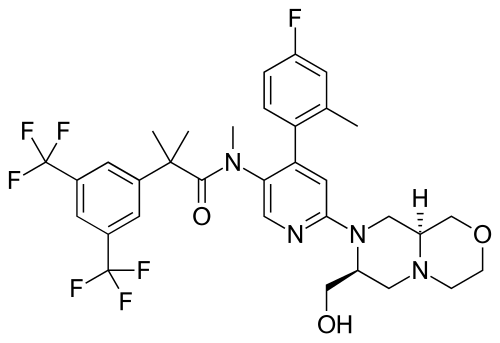
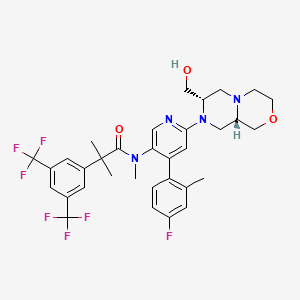
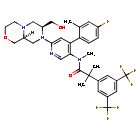
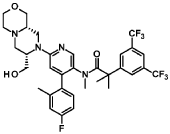
Elinzanetant
CAS 929046-33-3
MW 668.6 g/mol MF C33H35F7N4O3
N-[6-[(7S,9aS)-7-(hydroxymethyl)-3,4,6,7,9,9a-hexahydro-1H-pyrazino[2,1-c][1,4]oxazin-8-yl]-4-(4-fluoro-2-methylphenyl)-3-pyridinyl]-2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethylpropanamide
FDA 10/24/2025, Lynkuet, To treat moderate-to-severe vasomotor symptoms due to menopause
BAY-3427080; GSK-1144814; NT-814, UNII-NZW2BOW35N
Elinzanetant, sold under the brand name Lynkuet, is a medication used for the treatment of moderate to severe vasomotor symptoms due to menopause.[4] It is an neurokinin 1 and neurokinin 3 receptor antagonist.[4] It was developed by Bayer Healthcare.[4] It is taken by mouth.[4]
Elinzanetant is a non-hormonal, selective, neurokinin 1 (NK-1) and neurokinin 3 (NK-3) receptor antagonist.[5] By blocking NK-1 and NK-3 receptors signaling, elinzanetant is postulated to normalize neuronal activity involved in thermo- and sleep regulation in the hypothalamus.[5]
Elinzanetant is an orally bioavailable neurokinin/tachykinin 1 receptor (NK1-receptor; NK1R; NK-1R) and NK3 receptor (NK-3R; NK3R) antagonist, that may be used to treat vasomotor symptoms in menopausal woman. Upon oral administration, elinzanetant targets, competitively binds to and blocks the activity of the NK1R and NK3R in the central nervous system (CNS), thereby inhibiting the binding of the endogenous ligands and neuropeptides substance P (SP; neurokinin-1; NK1) and neurokinin B (NKB). This inhibits NK1R/NK3R-mediated signal transduction and may prevent certain menopausal symptoms such as hot flashes. Neurokinin-mediated signaling may increase during hormone deficiency and may cause hot flashes.
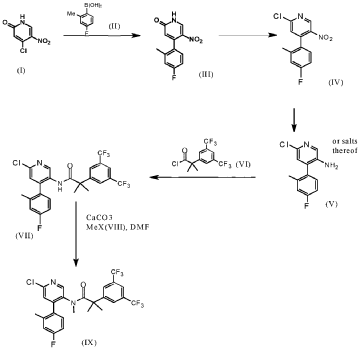
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021094247&_cid=P20-MHLSZY-53200-1
“Compound A” refers to 2-[3,5-Bis(trifluoromethyl)phenyl]-N-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hydroxymethyl)hexahydropyrazino[2,l-c][l,4]oxazin-8(lH)-yl]-3-pyridi-nyl}-N,2-dimethylpropanamide, and has the chemical structure depicted below.
(Compound A).
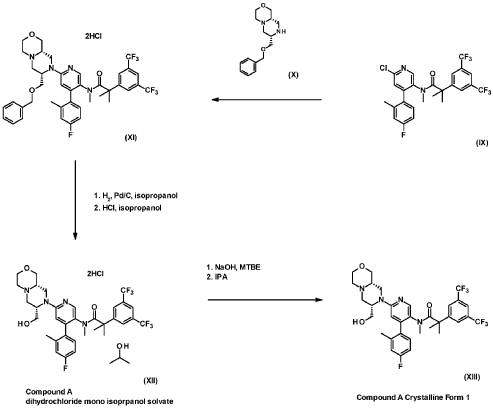
Example 8
2-[3.5-Bis(trifluoromethyl¾phenyl1-N-{4-(4-fluoro-2-methylphenyl¾-6-[(7S.9aS¾-7-(hvdroxymethyl¾hexa-hvdropyrazino[2,l-c1[l,41oxazin-8(lH)-yl1-3-pyridinyl}-N, 2-dimethyl propanamide as anhydrous crys talline form (Compound A)
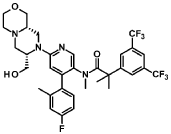
Example 7 (2-[3,5-Bis(trifluoromethyl)phenyl]-N-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hy-droxymethyl)hexahydropyrazino[2,l-c][l,4]oxazin-8(lH)-yl]-3-pyridinyl}-N,2-dimethylpropanamide dihydrochloride salt mono-isopropanol solvate (Compound XII)) (3.4 kg), methyl-f-butyl ether (from now on, MTBE) (15.0 L/kg of Example 7) and NaOH 2.5N (4.9 L/kg of Example 7) were loaded, heated to 40QC and stirred for 10 to 30 min. The layers were settled for not less than 30 min at 40QC and the bottom aqueous layer discarded.
An aqueous solution of L-cysteine 9 wt% (5.0 L water per kg of Example 7+ 0.5 w/w L-cysteine per Ex ample 7) was added over the organic layer and stirred at 40QC for not less than 60 min. The layers were settled for not less than 30 min at 40QC and the bottom aqueous layer discarded.
Water (5.0 L/kg of Example 7) was added over the organic layer and stirred at 40QC for not less than 15 min. The layers were settled for not less than 60 min at 40QC and the bottom aqueous layer dis carded.
Water (5.0 L/kg of Example 7) was added over the organic layer and stirred at 40QC for not less than 15 min. The layers were settled for not less than 60 min at 40QC and the bottom aqueous layer dis carded.
The organic layer was concentrated at atmospheric pressure to 2.5 L/kg of Example 7. Iso-octane (8.3 L/kg of Example 7) was added at 50/55QC in not less than lh and the solution distilled under light vac uum to 4.0 L/kg of Example 7. A sample was taken for controlling the water and MTBE removal.
Isopropanol (0.8 L/kg of Example 7) was added and stirred at 65/75QC until total dissolution. The solu tion was cooled down to 45/55QC and filtered to remove any foreign matters. Iso-octane (4.5 L/kg of Example 7) was added and the batch heated to 70QC for not less than 30 min. The solution was cooled down to 50QC and seeded with a slurry of 2-[3,5-Bis(trifluoromethyl)phenyl]-N-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hydroxymethyl)hexahydropyrazino[2,l-c][l,4]oxazin-8(lH)-yl]-3-pyridi-nyl}-N,2-dimethylpropanamide(0.008% w/w of Example 7) in iso-octane (0.07 L/kg of Example 7) and isopropanol (0.01 L/kg of Example 7). The seeds were aged at 50QC for not less than 3h and additional iso-octane (4.2 L/kg of Example 7) was added in not less than 3h keeping the temperature at 50/55QC. The slurry was held at 50QC for not less than 8h, cooled down to 0QC in not less than 5h and aged for not less than 3h before proceeding with the centrifugation step.
The slurry was centrifuged and the cake washed with iso-octane (2 x 3.3 L/kg of Example 7).
The wet product was dried under vacuum at 50QC to obtain 2.34 kg of the title compound (yield = 82.7%). This product was sieved for delumping to obtain 2.26kg of the title compound with a 99.8% purity as a white powder.
NMR spectrometer: Varian Agilent Mercury Vx 400 (16 scans, sw 6400 Hz, 25 °C).
*H NMR (400 MHz, DMSO-ds): d 8.02 (s, 1 H), 7.85 (s, 1 H), 7.74 (bd, 2 H), 7.22-6.92 (m, 3 H), 6.61 (s, 1 H), 4.70 (m, 1 H), 4.21 (bd, 1 H), 4.09 (bd, 1 H), 3.75 (m, 3 H), 3.55 (td, 11.3 Hz, 2.2 Hz, 1 H), 3.40 (bd, 1 H), 3.15 (t, 10.5 Hz, 1 H), 3.02 (d, 11.3 Hz, 1 H), 2.63 (d, 11.3 Hz, 1 H), ca. 2.5 (bd, 2 H), 2.31-2.00 (m, 7 H), 1.58-1.10 (m, 6 H).
SYN
- WO2007028654
- https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007028654&_cid=P20-MHLT4M-58180-1
Example 34
2-[3,5-Bis(trifluoromethyl)phenyl]-yV-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hydroxymethyl)hexahydropyrazino[2,1 -c][1 ,4]oxazin-8(1 H)-yl]-3-pyridinyl}-A/,2-dimethylpropanamide (E34)
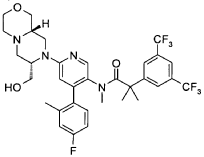
2-[3,5-bis(trifluoromethyl)phenyl]-Λ/-[6-[(7S,9aS)-7-({[(1 , 1 -dimethylethyl)(dimethyl)silyl]oxy}methyl)hexahydropyrazino[2,1-c][1 ,4]oxazin-8(1H)-yl]-4-(4-fluoro-2-methylphenyl)-3-pyridinyl]-Λ/,2-dimethylpropanamide (D24) (390 mg, 0.498 mmol) was dissolved 17 ml. of methanol. To this solution was added concentrated HCI (0.9 mL) at 00C, and stirring was continued at room temperature for 3h (complete conversion). The reaction mixture was loaded on a SCX cartridge and washed with MeOH. The product was eluted with 0.5 M methanolic ammonia. The product-containing fractions were evaporated, leaving the target compound as a white solid: 310 mg, 0.464 mmol, 93%.
UPLC/MS: m/z= 669 (M+1 ).
1H-NMR (DMSO-d6): δ (ppm) 8.07-7.97 (s, 1 H), 7.88-7.81 (s, 1 H), 7.79-7.69 (br. s, 2H), 7.19-7.11 (d, 1 H), 7.14-7.06 (br. s, 2H) 6.64-6.56 (s, 1 H), 4.75-4.65 (m, 1 H), 4.31-4.13 (br. S, 1 H), 4.15-4.01 (br. s, 1 H), 3.80-3.68 (m, 3H), 3.58-3.49 (t, 1 H); 3.43-3.34 (m, 1 H); 3.18-3.09 (t, 1 H); 3.04-2.98 (d, 1 H); 2.68-2.58 (d, 1 H); 2.51-2.45 (s, 3H); 2.20-2.13 (s, 3H); 2.29-2.00 (m, 4H); 1.54-1.39 (s, 3H); 1.39-1.28 (s, 3H).
SYN
Crystalline forms of a pyridine derivative
Publication Number: WO-2010015626-A1
Priority Date: 2008-08-05
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010015626&_cid=P20-MHLT79-60873-1
ntermediate 7
2-r3.5-bis(trifluoromethyl)phenvn-Λ/-f4-(4-fluoro-2-methylphenyl)-6-r(7S,9aS)-7-(hvdroxymethyl)hexahvdro-pyrazinoF2,1-c1f1 ,41oxazin-8(1/-/)-vn-3-pyridinyl}-Λ/.2-dimethylpropanamide
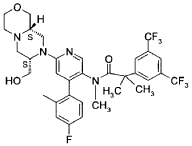
16.90 g of bis(trifluoromethyl)phenyl]-Λ/-[6-chloro-4-(4-fluoro-2-methylphenyl)-3-pyridinyl]- Λ/,2-dimethylpropanamide (WO 2005/002577) 4.58 g sodium tert-butoxide and 2.1O g Bis-(tri-terf-butylphposphine-palladium(O) catalyst was loaded into the vessel under nitrogen.
10.00 g intermediate 6 dissolved in 338 mL toluene was charged to afford a dark brown solution. The solution was heated to 800C and stirred for at least 16 h (thin suspension obtained).
The reaction mixture was cooled down to 20-25°C and 16.90 g celite was added to give a brown suspension. The suspension was filtered over 16.90 g celite and washed with 33.8 mL toluene. 338 mL sat. sodium bicarbonate solution was added and the biphasic system was stirred for 5 min. at 20-250C. After phase separation, the aqueous layer was extracted twice with 118 mL toluene. The combined organic layers were treated with 90 mL of a 10% aqueous cysteine solution and stirred for 1 h at 25°C. After phase separation the organic layer was treated again with 90 mL of a 10% cysteine-solution and stirred for a further 1 h at 25°C. After phase separation , the organic layer was washed with 85 mL half saturated sodium bicarbonate solution then solvent exchanged to dioxane. The dioxane solution was cooled down to 10-150C. 63.5 mL of 4M hydrogen chloride in dioxane was added at 10-150C over at least 10 min. The solution was warmed to 20-25°C and stirred for 2 h.
Dioxane was concentrated down to 85 mL at 45°C under reduced pressure. 85 mL water and 254 mL dichloromethane were added to the residue to give a thin suspension. The biphasic system was stirred for 5 min. at 20-250C. The layers were separated and the organic phase was washed with 33.8 mL saturated sodium bicarbonate solution at 20-25°C (pH adjusted to 7-8). The biphasic system was stirred for 5 min. at 20-250C and the organic layer separated and concentrated under reduced pressure at 500C to afford crude title compound as a pale brown solid. 8.00 g of the title compound (78.8% a/a HPLC) was dissolved in 16 mL ethyl acetate. The filter was loaded with 80 to 104 g silica gel and conditioned with ethyl acetate. The product solution was loaded on top of the column and chromatography was started using ethyl acetate as solvent. The product fractions were combined and partially concentrated at 45-50°C under reduced pressure. To the mixture was added 2.64 g to 4.00 g silicycle (Si-Thiol, 1.2 mmol/g) at rt and stirred for 2 h. Filtration over 8.00 g celite and washing with 32 mL ethyl acetate gave the filtrate which was concentrated to dryness at 45°C afford the title compound as a light brown solid. ( Weight yield 72%) 1H-NMR [ppm, CDCI3]: 8.04-7.91 , (m, 1 H); 7.77, (s, 1 H); 7.72-7.60, (m, 2H); 7.59-7.16, (m, 1H); 7.06-6.74, (m, 2H); 6.44, (s, 1 H); 4.64-4.43, (m, 1 H); 4.38-4.18, (m, 1 H); 4.07-3.96, (m, 2H); 3.95-3.76, (m, 3H); 3.76-3.61 , (m, 1 H); 3.37-3.27, (m, 1 H); 3.16-2.98, (m, 2H); 2.84-2.70, (m, 1 H); 2.67-2.51 , (m, 2H); 2.49-2.22, (m, 5H); 2.19-2.06, (m, 2H); 1.64-1.31 , (m, 5H), OH broad and not observed
LIT
- Elinzanetant: a phase III therapy for postmenopausal patients with vasomotor symptomsPublication Name: Expert Opinion on Investigational DrugsPublication Date: 2024-01-02PMID: 38224099DOI: 10.1080/13543784.2024.2305122
- Elinzanetant (NT-814), a Neurokinin 1,3 Receptor Antagonist, Reduces Estradiol and Progesterone in Healthy WomenPublication Name: The Journal of clinical endocrinology and metabolismPublication Date: 2021-02-24PMCID: PMC8277204PMID: 33624806DOI: 10.1210/clinem/dgab108
- Pleiotypic responses of regenerating liverPublication Name: Advances in Enzyme RegulationPublication Date: 1976PMID: 9791DOI: 10.1016/0065-2571(76)90023-6
PAT
Pyridine Derivatives and Their Use in the Treatment of Psychotic Disorders
Publication Number: US-2008269208-A1
Priority Date: 2005-06-06
- Pyridine Derivatives And Their Use In The Treatment Of Psychotic DisordersPublication Number: US-2011190276-A1Priority Date: 2005-09-09
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: US-7683056-B2Priority Date: 2005-09-09Grant Date: 2010-03-23
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: US-7919491-B2Priority Date: 2005-09-09Grant Date: 2011-04-05
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: US-8097618-B2Priority Date: 2005-09-09Grant Date: 2012-01-17
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: WO-2007028654-A1Priority Date: 2005-09-09
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Elinzanetant is indicated for the treatment of moderate to severe vasomotor symptoms associated with menopause.[4]
Society and culture
Legal status
In September 2025, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Lynkuet, intended for the treatment of moderate to severe vasomotor symptoms (hot flushes).[5] The applicant for this medicinal product is Bayer AG.[5]
Lynkuet was approved for medical use in the United States in October 2025.[6]
Names
Elinzanetant is the international nonproprietary name.[7]
Elinzanetant is sold under the brand name Lynkuet.[8]
References
- “Details for: Lynkuet”. Drug and Health Products Portal. 23 July 2025. Retrieved 28 September 2025.
- “Lynkuet product information”. Lynkuet. 23 July 2025. Retrieved 28 September 2025.
- “MHRA approves elinzanetant to treat moderate to severe vasomotor symptoms (hot flushes) caused by menopause”. Medicines and Healthcare products Regulatory Agency (Press release). 8 July 2025. Retrieved 28 September 2025.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219469s000lbl.pdf
- “Lynkuet EPAR”. European Medicines Agency (EMA). 19 September 2025. Retrieved 27 September 2025. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 24 October 2025. Retrieved 29 October 2025.
- World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 84”. WHO Drug Information. 34 (3). hdl:10665/340680.
- “Bayer’s Lynkuet (elinzanetant), the First and Only Neurokinin 1 and Neurokinin 3 Receptor Antagonist, Receives FDA Approval for Moderate to Severe Hot Flashes Due to Menopause” (Press release). Bayer. 24 October 2025. Retrieved 29 October 2025 – via Business Wire.
External links
- Clinical trial number NCT05042362 for “A Study to Learn More About How Well Elinzanetant Works and How Safe it is for the Treatment of Vasomotor Symptoms (Hot Flashes) That Are Caused by Hormonal Changes Over 26 Weeks in Women Who Have Been Through the Menopause (OASIS-1)” at ClinicalTrials.gov
- Clinical trial number NCT05099159 for “A Study to Learn More About How Well Elinzanetant Works and How Safe it is for the Treatment of Vasomotor Symptoms (Hot Flashes) That Are Caused by Hormonal Changes Over 26 Weeks in Women Who Have Been Through the Menopause (OASIS-2)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lynkuet |
| Other names | BAY-3427080; GSK-1144814; NT-814 |
| License data | US DailyMed: Elinzanetant |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | CA: ℞-only[1][2]UK: POM (Prescription only)[3]US: ℞-only[4] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 929046-33-3 |
| PubChem CID | 16063568 |
| ChemSpider | 17223178 |
| UNII | NZW2BOW35N |
| KEGG | D12123 |
| CompTox Dashboard (EPA) | DTXSID101337049 |
| Chemical and physical data | |
| Formula | C33H35F7N4O3 |
| Molar mass | 668.657 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////elinzanetant, Lynkuet, FDA 2025, APPROVALS 2025, BAY 3427080, GSK 1144814, NT 814
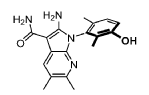
Lunresertib
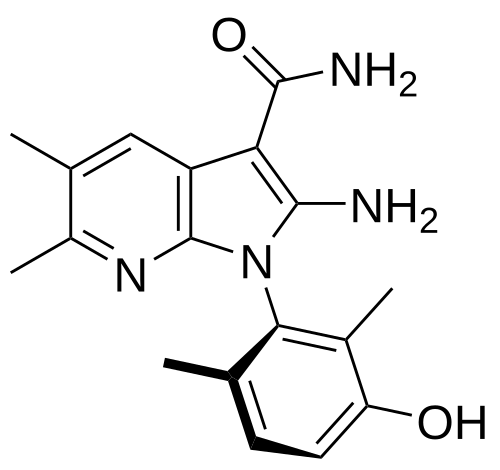
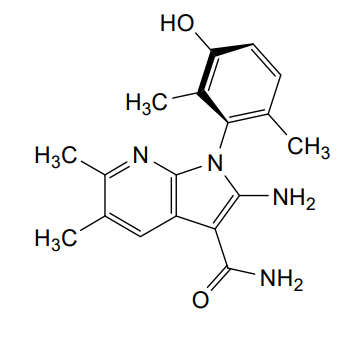
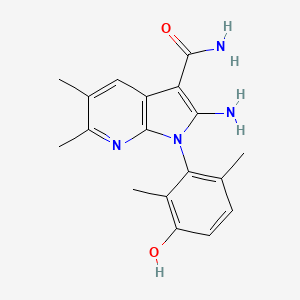
Lunresertib
CAS 2719793-90-3
MF C18H20N4O2 MW 324.4 g/mol
(1P)-2-amino-1-(3-hydroxy-2,6-dimethylphenyl)-5,6-dimethyl1H-pyrrolo[2,3-b]pyridine-3-carboxamide
serine/ threonine kinase inhibitor, antineoplastic, N95U3A7N57, RP-6306, RP 6306
2-Amino-1-(3-hydroxy-2,6-dimethylphenyl)-5,6-dimethylpyrrolo[2,3-b]pyridine-3-carboxamide
Lunresertib is an investigational new drug that is being evaluated for the treatment of cancer. It is an oral small molecule inhibitor of PKMYT1, developed by Repare Therapeutics.[1] This drug targets cell cycle regulation in tumors with specific genetic alterations, including CCNE1 amplifications or FBXW7 and PPP2R1A loss of function mutations. It is currently in phase 1/2 clinical trials, both as monotherapy or in combination with camonsertib, an ATR inhibitor.[2]
Lunresertib is an orally bioavailable inhibitor of the human membrane-associated tyrosine– and threonine-specific cdc2-inhibitory kinase (PKMYT1), with potential antineoplastic activity. Upon oral administration, lunresertib targets, binds to and inhibits the activity of PKMYT1. This results in the inhibition of CDK1 phosphorylation, which may promote both premature mitosis and a prolonged mitotic arrest, and lead to the accumulation of unrepaired DNA damage and apoptosis in susceptible tumor cells, such as CCNE1-overexpressing tumor cells. PKMYT1 phosphorylates CDK1 specifically when CDK1 is complexed to cyclins, which blocks progression from G2 into mitosis.NCI Thesaurus (NCIt)
- Study of RP-6306 With FOLFIRI in Advanced Solid TumorsCTID: NCT05147350Phase: Phase 1Status: TerminatedDate: 2025-08-20
- Study of RP-6306 Alone or in Combination With RP-3500 or Debio 0123 in Patients With Advanced Solid TumorsCTID: NCT04855656Phase: Phase 1Status: RecruitingDate: 2025-08-06
- RP-6306 in Patients With Advanced CancerCTID: NCT05605509Phase: Phase 2Status: Active, not recruitingDate: 2025-07-14
- Study of RP-6306 With Gemcitabine in Advanced Solid TumorsCTID: NCT05147272Phase: Phase 1Status: TerminatedDate: 2025-06-17
- Liquid-biopsy Informed Platform Trial to Evaluate CDK4/6-inhibitor Resistant ER+/HER2- Metastatic Breast CancerCTID: NCT05601440Phase: Phase 2Status: RecruitingDate: 2025-01-14
- Phase 1 Study of RP-6306 With Carboplatin and Paclitaxel in TP53 Ovarian and Uterine Cancer
- CTID: NCT06107868
- Phase: Phase 1
- Status: Active, not recruiting
- Date: 2024-03-22
PAT
- Compounds, Pharmaceutical Compositions, and Methods of Preparing and Using CompoundsPublication Number: JP-2023521633-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: US-2023151014-A1Priority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: US-2023158022-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: EP-4126879-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: IL-296934-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of making the compounds and methods of using themPublication Number: KR-20230011279-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions and methods of making compounds and methods of their usePublication Number: CN-115916783-APriority Date: 2020-04-01
- Methods of using MYT1 inhibitorsPublication Number: JP-2023519430-APriority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: WO-2021195782-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: AU-2021250744-A1Priority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: CA-3173955-A1Priority Date: 2020-04-01
- Methods of using MYT1 inhibitorsPublication Number: CN-115811976-APriority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: EP-4125907-A1Priority Date: 2020-04-01
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021195781&_cid=P20-MHLE6P-37080-1
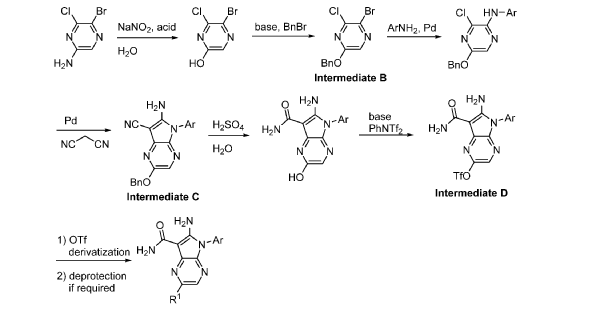
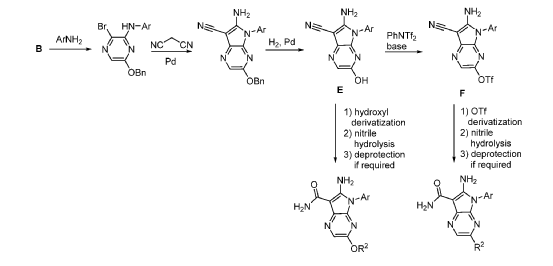
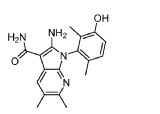
Step 9. To a suspension of 2-amino-1-(3-methoxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (2.22 g, 6.56 mmol, 77% purity) in DCM (25 mL) was added tribromoborane in DCM (1 M, 26 mmol, 26 mL) dropwise. The reaction mixture was stirred at RT for 45 min, then concentrated to dryness. The crude product was taken in DCM and placed in an ice bath and MeOH was added carefully (exotherm). The mixture was concentrated to dryness then co-evaporated twice with MeOH. The residue was triturated with saturated aqueous NaHCO3. The solids were collected by filtration on a Buchner funnel, washed with H2O and air-dried. The still wet solid was dissolved in DCM/MeOH, concentrated to dryness and triturated in 20% MeOH/DCM (50 mL). The solid was collected by filtration, washed with 20% MeOH/DCM, air-dried then dried in vacuo to afford 2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (1.60g, 75% yield) as a light beige solid. MS: [M+1]: 325.1. A different batch was purified by preparative HPLC to yield 2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (63% yield) as an off-white fluffy solid.
1H NMR (400 MHz, DMSO-d6) δ 9.51 (s, 1H), 7.82 (s, 1H), 7.05 (d, J = 8.3 Hz, 1H), 6.90 (d, J =
8.2 Hz, 1H), 6.71 (br s, 2H), 6.64 (br s, 2H), 2.26 (s, 3H), 2.23 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
Chiral SFC separation of Compound 181 (1.60g, 4.93 mmol) (Instrument: Waters Prep 100 SFC-MS; Column: Phenomenex Lux Cellulose-2, 30 x 250 mm, 5 μm; Conditions: isocratic at 55% IPA + 10mM Ammonium Formate with 45% CO2 ; Flow Rate: 70 mL/min) provided
Compound 182 and Compound 183.
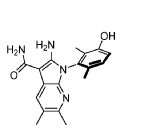
Compound 182 from SFC separation of 181. Peak 1 (retention time 3.94 min, 99.86%): (S)-2- amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (381 mg) was obtained as an off white fluffy solid. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 7.83 (s, 1 H), 7.05 (d, J = 8.3 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H), 6.72 (s, 2H), 6.65 (s, 2H), 2.26 (s, 3H), 2.24 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
Compound 183 from SFC separation of 181. Peak 2 (retention time 4.35 min, 98.09%): (R)-2- amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (495 mg) was obtained as an off white fluffy solid. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 7.83 (s, 1 H), 7.05 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.72 (s, 2H), 6.66 (s, 2H), 2.26 (s, 3H), 2.24 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
SYN
https://pubs.acs.org/doi/full/10.1021/acs.oprd.4c00493
REF
- The Science and Art of Structure-Based Virtual ScreeningPublication Name: ACS Medicinal Chemistry LettersPublication Date: 2024-03-25PMCID: PMC11017385PMID: 38628791DOI: 10.1021/acsmedchemlett.4c00093
- Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306Publication Name: Journal of Medicinal ChemistryPublication Date: 2022-07-26PMCID: PMC9837800PMID: 35880755DOI: 10.1021/acs.jmedchem.2c00552
- CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibitionPublication Name: NaturePublication Date: 2022-04-20PMCID: PMC9046089PMID: 35444283DOI: 10.1038/s41586-022-04638-9
- Contributions in the domain of cancer research: Review¶Negative regulators of cyclin-dependent kinases and their roles in cancersPublication Name: Cellular and molecular life sciences : CMLSPublication Date: 2001-11PMCID: PMC11337304PMID: 11766887DOI: 10.1007/pl00000826
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | RP-6306 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2719793-90-3 |
| PubChem CID | 156869388 |
| ChemSpider | 115008046 |
| UNII | N95U3A7N57 |
| KEGG | D12736 |
| ChEMBL | ChEMBL5199076 |
| Chemical and physical data | |
| Formula | C18H20N4O2 |
| Molar mass | 324.384 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Szychowski J, Papp R, Dietrich E, Liu B, Vallée F, Leclaire ME, et al. (August 2022). “Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306”. Journal of Medicinal Chemistry. 65 (15): 10251–10284. doi:10.1021/acs.jmedchem.2c00552. PMC 9837800. PMID 35880755.
- Previtali V, Bagnolini G, Ciamarone A, Ferrandi G, Rinaldi F, Myers SH, et al. (July 2024). “New Horizons of Synthetic Lethality in Cancer: Current Development and Future Perspectives”. Journal of Medicinal Chemistry. 67 (14): 11488–11521. doi:10.1021/acs.jmedchem.4c00113. PMC 11284803. PMID 38955347.
///////lunresertib, Serine/ threonine kinase inhibitor, antineoplastic, N95U3A7N57, RP-6306, RP 6306
Lunbotinib
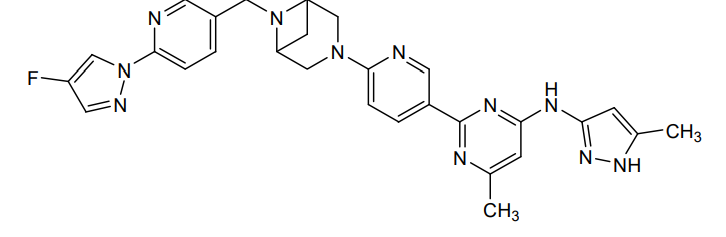
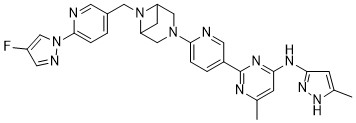
Lunbotinib
CAS 2479961-46-9
MF C28H28FN11 MW537.6 g/mol
2-[6-(6-{[6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl]methyl}-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl]-6-methyl-N-(5-methyl1H-pyrazol-3-yl)pyrimidin-4-amine
tyrosine kinase inhibitor, antineoplastic, KL3T9ZU6HQ
- 2-(6-(6-((6-(4-fluoropyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo(3.1.1)heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine
- 2-[6-[6-[[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine
Lunbotinib is an orally bioavailable selective inhibitor of the proto-oncogene receptor tyrosine kinase rearranged during transfection (RET), with potential antineoplastic activity. Upon oral administration, lunbotinib selectively binds to various RET fusions and mutations, including solvent front resistance mutations, and inhibits the activity of RET. This results in an inhibition of cell growth of tumors that exhibit increased RET activity due to these fusions and mutations. RET overexpression, activating mutations, and fusions result in the upregulation and/or overactivation of RET tyrosine kinase activity in various cancer cell types. Dysregulated RET activity plays a key role in the development and progression of certain cancers. Lunbotinib is able to penetrate the blood-brain barrier (BBB) and may also be able to overcome resistance mechanisms to first generation selective RET inhibitors (SRIs).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020168939&_cid=P12-MHKH7H-14851-1
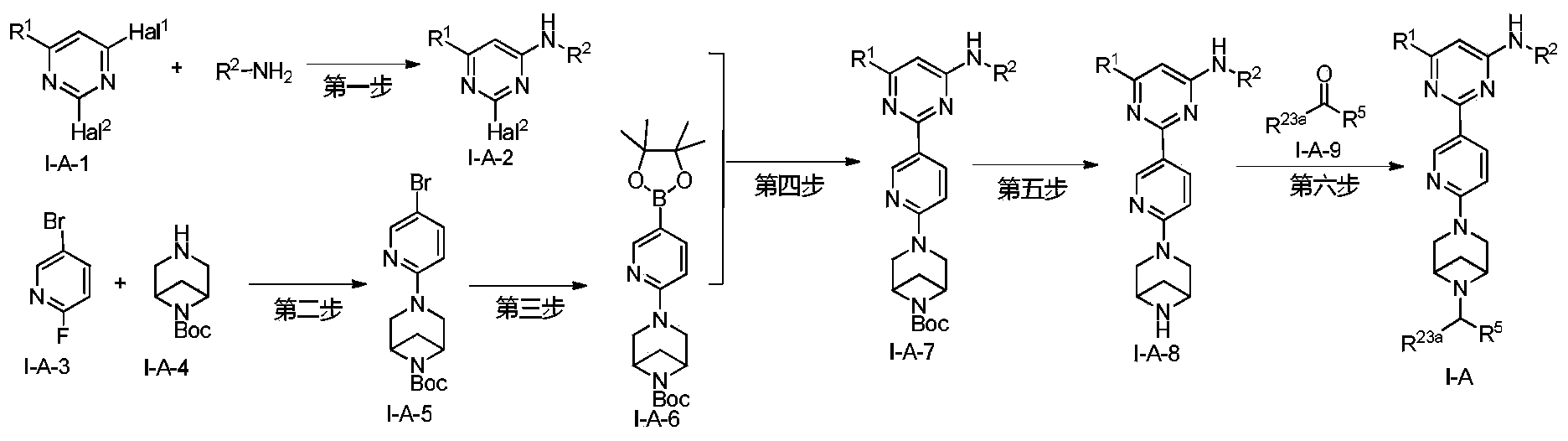
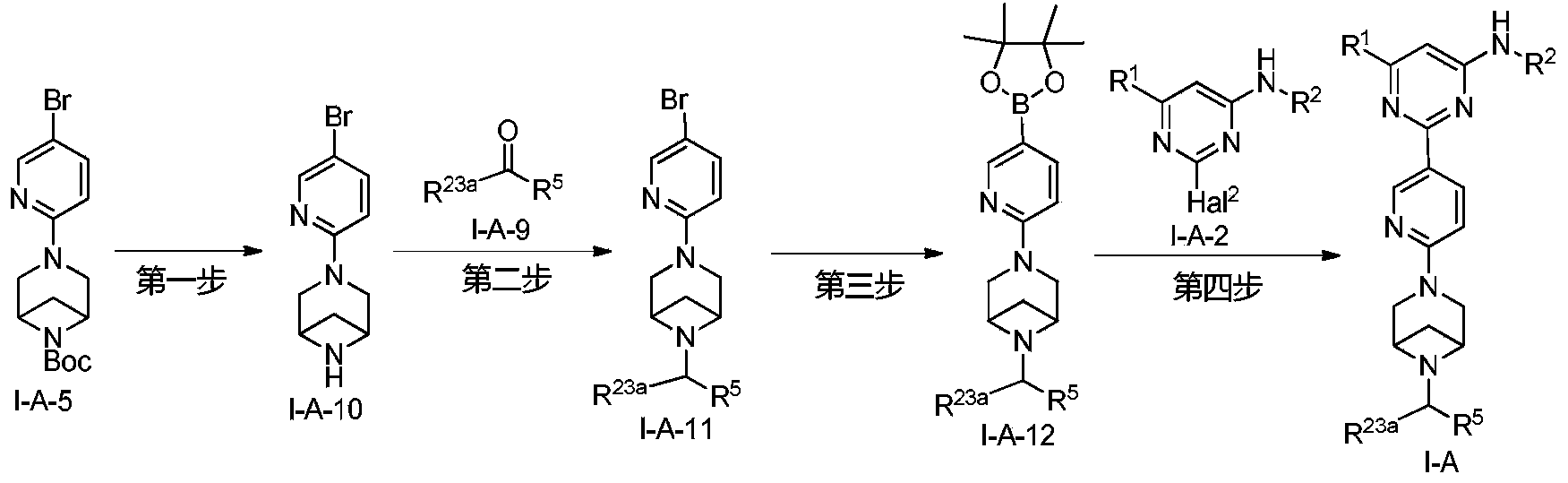
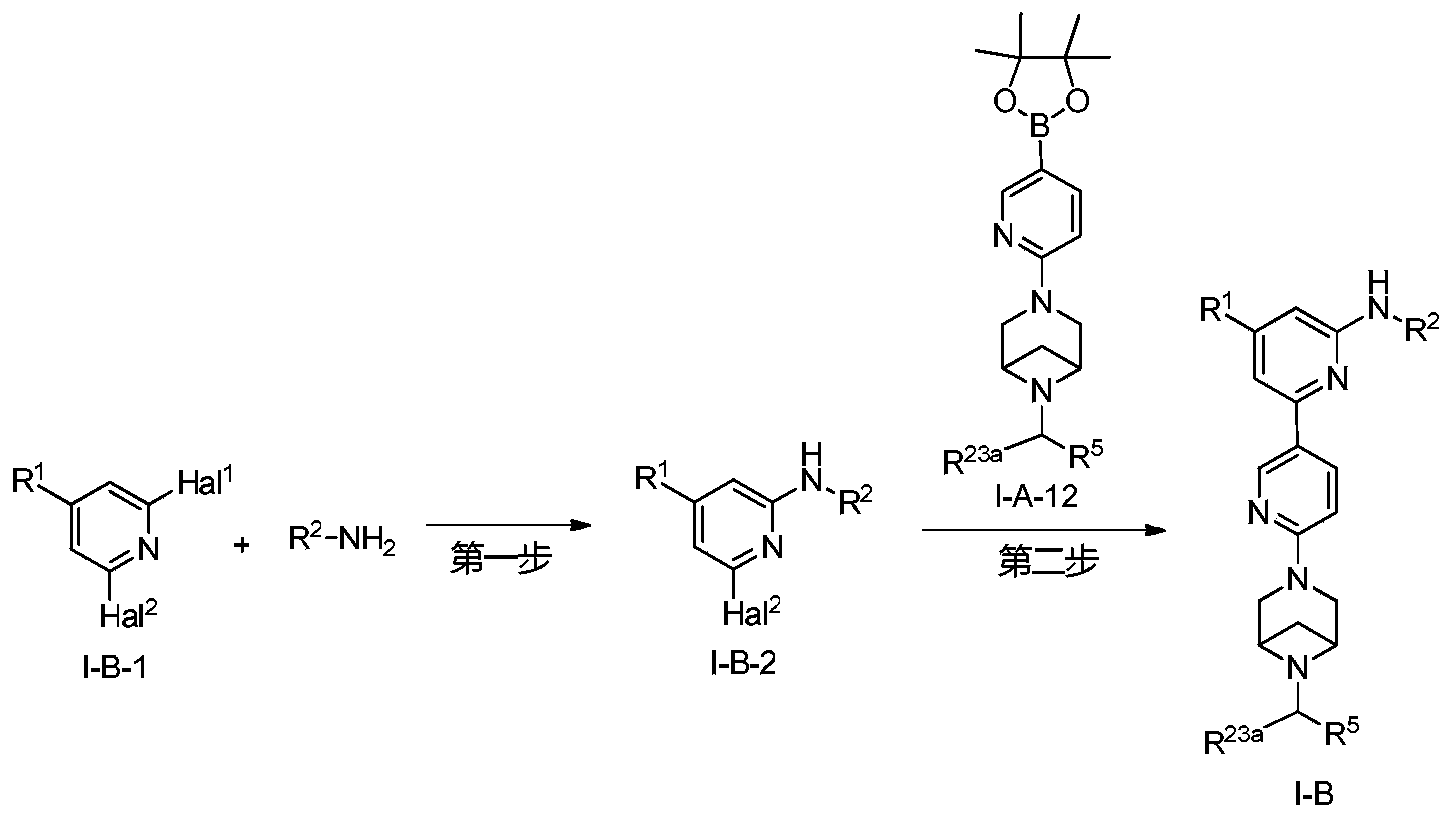
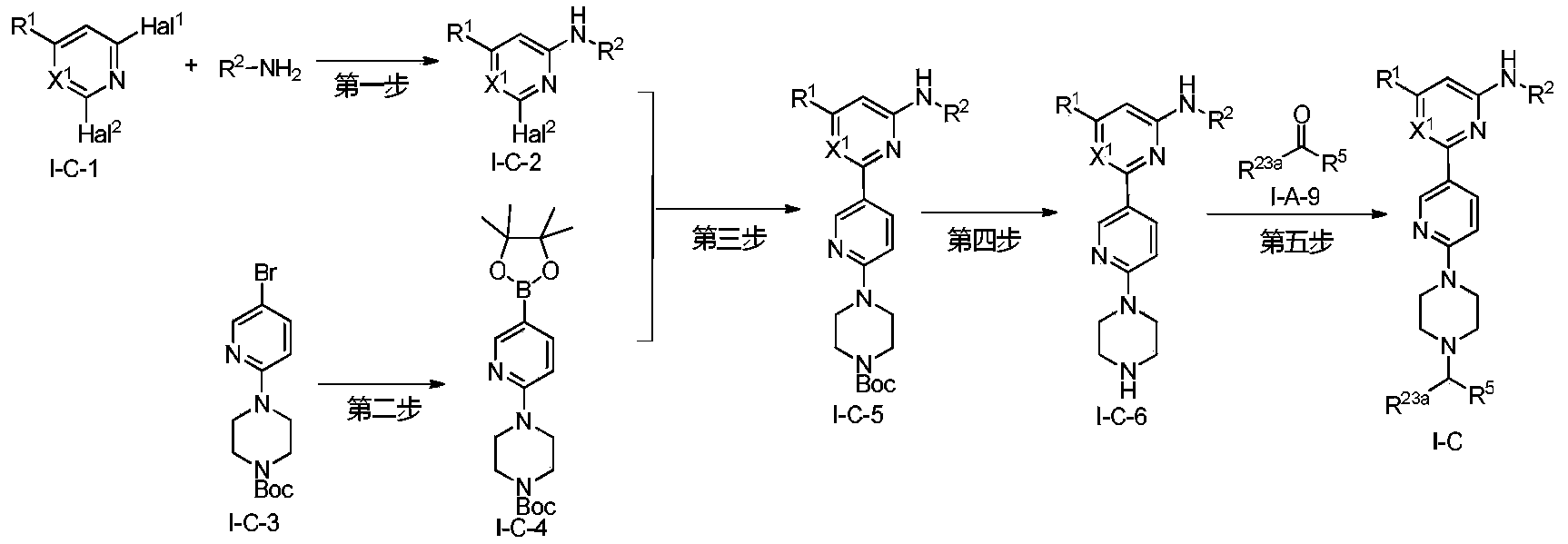
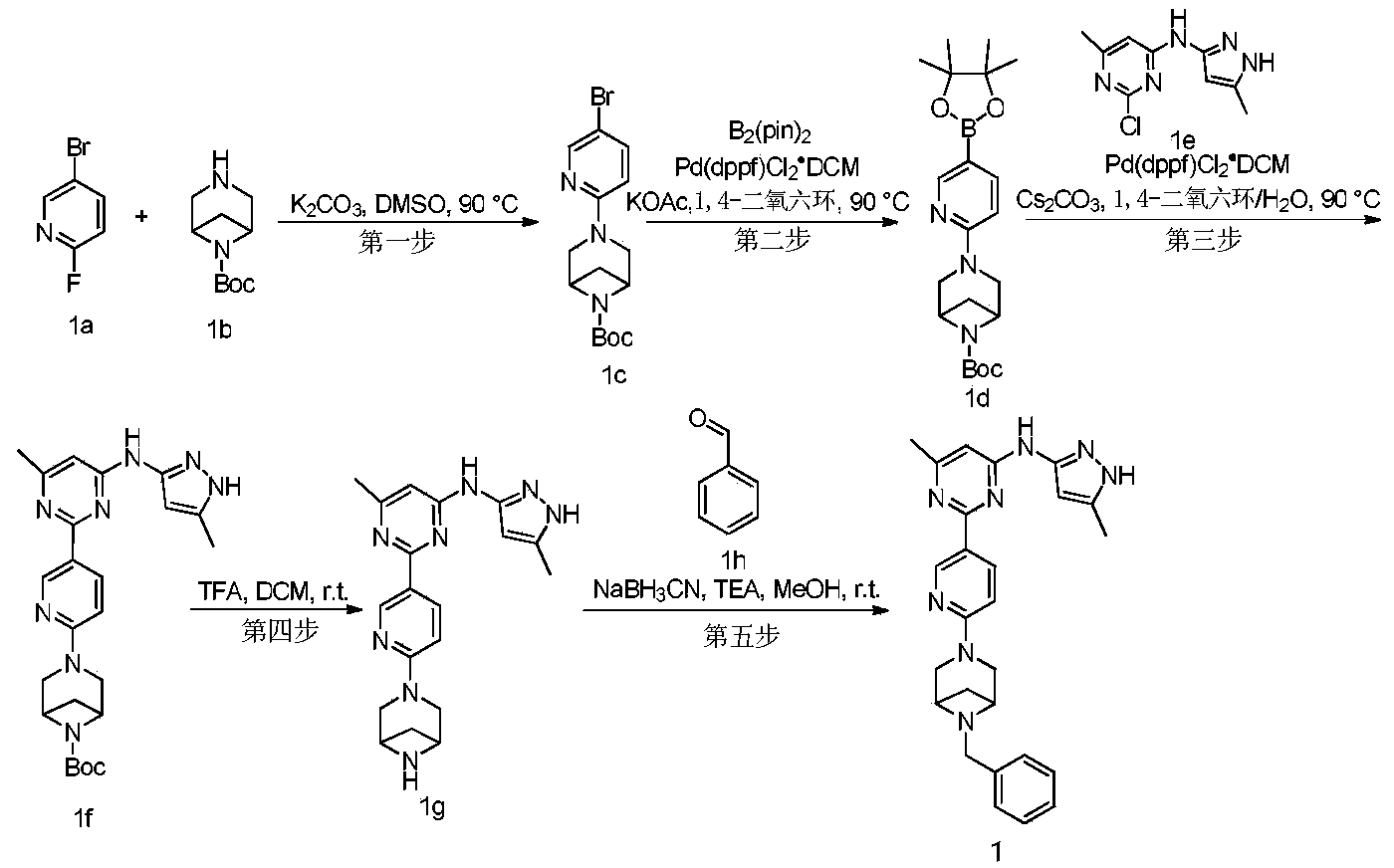
Example 6: 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 17)
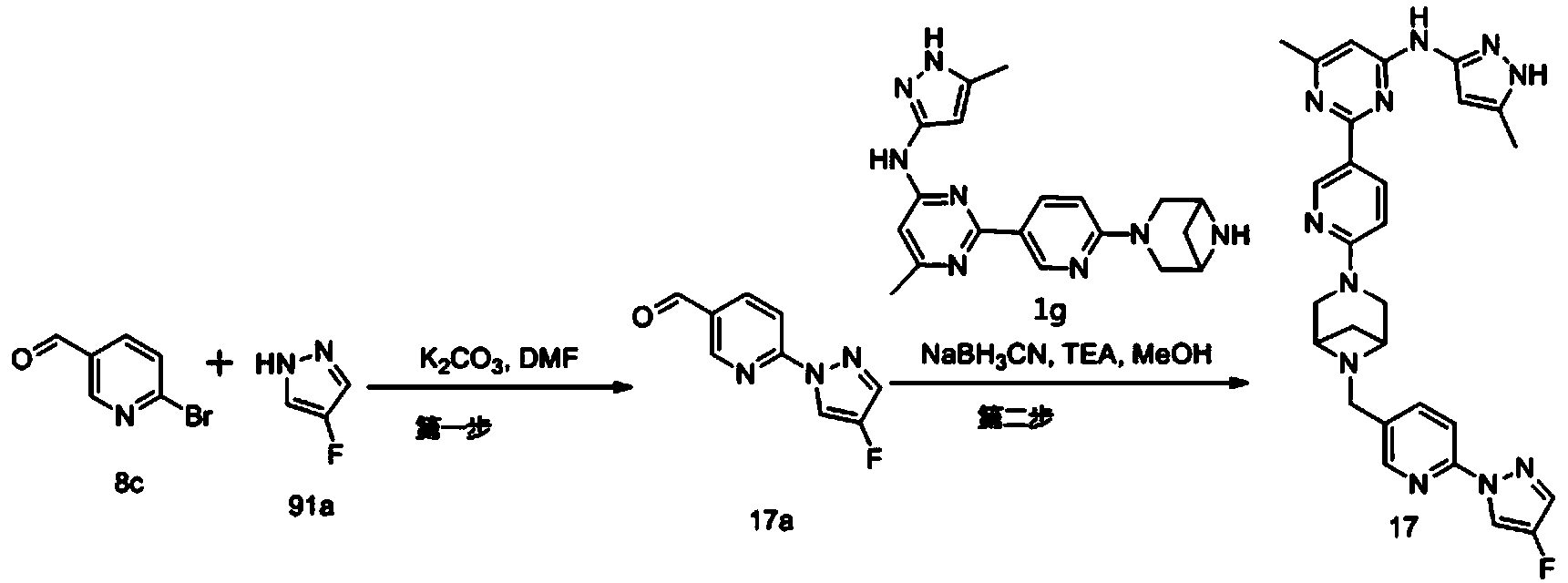
Step 1: Preparation of 6-(4-fluoro-1H-pyrazol-1-yl)nicotinaldehyde (compound 17a)
[0396]Compound 8c (2.0 g), 91a hydrochloride (1.58 g), and potassium carbonate (4.45 g) were sequentially added to DMF (15 mL), and the mixture was heated to 80 °C and stirred for 14 h. The reaction mixture was cooled to room temperature, diluted with water (100 mL), and extracted with DCM (50 mL x 2). The organic phases were combined, washed with water and saturated brine, dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography (PE:EA = 10:1) to give compound 17a (0.81 g). MS m/z (ESI): 192.1 [M+H]
[0397]Step 2: Preparation of 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 17)
[0398]1 g of trifluoroacetate (22.82 mg) and compound 17a (27.47 mg) were added to methanol (1.0 mL), followed by the sequential addition of triethylamine (4.45 mg) and sodium cyanoborohydride (13.86 mg), and the reaction was carried out at room temperature for 14 h. After the reaction was completed, the reaction solution was concentrated to dryness under reduced pressure and purified by Prep-HPLC to obtain compound 17 (7.0 mg). MS m/z (ESI): 538.3 [M+H]
[0399]
1H NMR(400MHz,DMSO-d 6)δ11.98(s,1H),9.66(s,1H),9.12(d,J=2.16Hz,1H),8.67(dd,J=4.54,0.64Hz,1H),8.43(dd,J=8.94,2.28Hz,1H),8.41(d,J=1.68,1H),7.98(dd,J=8.48Hz,2.12 1H),7.92(d,J=4.28,1H),7.87(d,J=8.4,1H),6.78(d,J=9.0Hz,2H),6.31(br,1H),3.78-3.71(m,4H),3.68-3.52(m,4H),2.59-2.52(m,1H),2.33(s,3H),2.25(s,3H),1.60(d,J=8.36Hz,1H).
PAT
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: US-2022144847-A1Priority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-113316578-BPriority Date: 2019-02-19Grant Date: 2023-10-31
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-117263945-APriority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-117327078-APriority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing same, methods for their preparation and usePublication Number: JP-7615056-B2Priority Date: 2019-02-19Grant Date: 2025-01-16
- Salt and crystal form of pyrimidine compound, and preparation methods thereforPublication Number: US-2023295174-A1Priority Date: 2020-07-28
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: WO-2020168939-A1Priority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same, and preparation methods and uses thereofPublication Number: CN-113316578-APriority Date: 2019-02-19
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: EP-3929198-A1Priority Date: 2019-02-19
- Heterocyclic compounds, drug compositions containing them, methods of their manufacture and usePublication Number: JP-2022521859-APriority Date: 2019-02-19
- Use of heterocyclic compound for treating diseases related to ret genetic change and method thereforPublication Number: WO-2024240017-A1Priority Date: 2023-05-19
- Uses and methods of heterocyclic compounds for treating diseases associated with kinase resistance mutationsPublication Number: CN-116801882-APriority Date: 2021-03-24
- Use of heterocyclic compound in treating diseases related to kinase drug-resistant mutation and method thereforPublication Number: EP-4316490-A1Priority Date: 2021-03-24
- Salt and crystal form of pyrimidine compound, and preparation methods thereforPublication Number: EP-4190781-A1Priority Date: 2020-07-28
- Salts, crystal forms of pyrimidine compounds and methods for their preparationPublication Number: JP-2023535361-APriority Date: 2020-07-28
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Lunbotinib, tyrosine kinase inhibitor, antineoplastic, KL3T9ZU6HQ
Lomedeucitinib
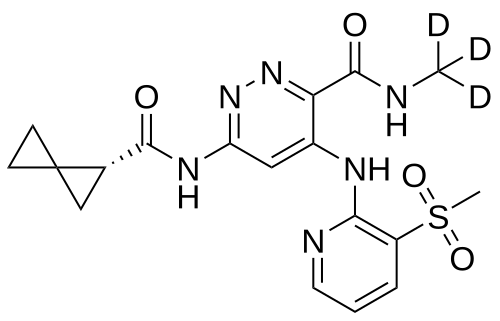
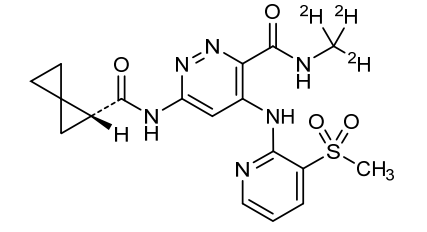
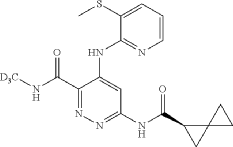
Lomedeucitinib
CAS 2328068-29-5
MF C18H172H3N6O4S
MW 419.5 g/mol

4-{[3-(methanesulfonyl)pyridin-2-yl]amino}-N-(2H3)methyl-6-[(1R)-spiro[2.2]pentane-1-carboxamido]pyridazine-3-carboxamide
4-[(3-methylsulfonyl-2-pyridinyl)amino]-6-[[(2R)-spiro[2.2]pentane-2-carbonyl]amino]-N-(trideuteriomethyl)pyridazine-3-carboxamide
Janus kinase inhibitor, anti-inflammatory, BMS-986322, BMS 986322, EYQ7KA55XA
Lomedeucitinib is an investigational new drug that is being evaluated for the treatment of psoriasis and psoriatic arthritis. It is a tyrosine kinase 2 (TYK2) inhibitor.[1]
- A Study to Evaluate Effectiveness and Safety of BMS-986322 in Participants With Moderate-to-Severe PsoriasisCTID: NCT05730725Phase: Phase 2Status: CompletedDate: 2024-09-19
- A Study to Evaluate the Drug Levels, Metabolism, and Removal of BMS-986322 in Healthy Adult Male ParticipantsCTID: NCT06088264Phase: Phase 1Status: CompletedDate: 2024-03-29
- A Study Investigating Interactions Between BMS-986322 and Rosuvastatin, Metformin and Methotrexate in Healthy ParticipantsCTID: NCT05615012Phase: Phase 1Status: CompletedDate: 2024-03-27
- A Study to Investigate the Interaction of BMS-986322 and a Combined Oral Hormonal Contraceptive (Ethinyl Estradiol [EE]/Norethindrone [NET]) in Healthy Female ParticipantsCTID: NCT05579574Phase: Phase 1Status: CompletedDate: 2023-08-18
- A Study to Assess the Safety and Tolerability of BMS-986322 in Healthy Participants of Japanese DescentCTID: NCT05546151Phase: Phase 1Status: CompletedDate: 2023-06-22
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US333829535&_cid=P10-MHIXWK-98212-1
General Scheme for Examples 252 and 253:
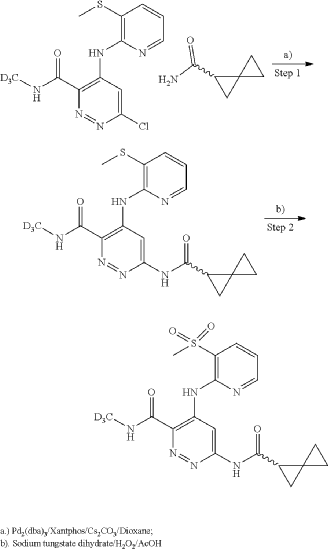
Example 252
Step 1
| A mixture of cesium carbonate (149 mg, 0.457 mmol), Xantphos (14.43 mg, 0.025 mmol), Pd 2(dba) 3 (11.42 mg, 0.012 mmol), 6-chloro-N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)pyridazine-3-carboxamide (65 mg, 0.208 mmol), and (R)-spiro[2.2]pentane-1-carboxamide (50.8 mg, 0.457 mmol) in dioxane (3 mL) was degassed using a vacuum/N2 fill cycle three times. The reaction was heated at 110° C. for 16 hours. The reaction was diluted with water and DCM. The DCM layer was separated and washed two more times with water and then dried (Na 2SO 4), filtered and concentrated. Purification via automated flash chromatography, eluting with methanol in DCM from 0 to 10%, gave the title compound (R)—N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (54 mg, 67% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ 12.15 (br s, 1H), 9.88 (s, 1H), 8.68 (br s, 1H), 8.36 (br d, J=3.5 Hz, 1H), 8.25 (br s, 1H), 7.72 (br d, J=7.4 Hz, 1H), 6.97 (br dd, J=7.0, 5.1 Hz, 1H), 2.51 (s, 3H), 2.21-2.09 (m, 1H), 1.58-1.10 (m, 6H), 1.08-0.93 (m, 5H). |
| LCMS (ESI) m/e 388.1 [(M+H) +, calc’d C 18H 18D 3N 6O 2S 1, 388.1]; LC/MS retention time (method D): t R=0.80 min. |
Step 2
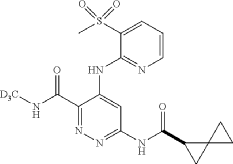
To a suspension of hydrogen peroxide (30% solution in water, 0.258 mL, 2.52 mmol) and (R)—N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (0.0489 g, 0.126 mmol) in AcOH (1 mL) was added sodium tungstate dihydrate (0.042 g, 0.126 mmol) at room temperature. After stirring at room temperature for 1 hour, the reaction was diluted with water, basified with Na 2CO 3 powder and extracted three times with DCM. The DCM layers were combined, washed with Na 2S 2O 3 (5% solution), dried (Na 2SO 4), filtered and concentrated. The crude product was purified using reverse phase prepHPLC to give the title compound (R)—N-(methyl-d3)-4-((3-(methylsulfonyl)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (16.2 mg, 31%) as a colorless solid. 1H NMR (500 MHz, DMSO-d 6) δ 12.07 (s, 1H), 11.22 (s, 1H), 9.49 (s, 1H), 9.16 (s, 1H), 8.63 (dd, J=4.6, 1.5 Hz, 1H), 8.29 (dd, 0.1=7.8, 1.4 Hz, 1H), 7.34 (dd, 0.1=7.8, 4.7 Hz, 1H), 2.48-2.43 (m, 1H), 1.46-1.41 (m, 1H), 1.42-1.36 (m, 1H), 0.95-0.82 (m, 3H), 0.80-0.73 (m, 1H). (3H methyl sulfone was buried under DMSO peak). LCMS (ESI) m/e 420.0 [(M+H) +, calc’d C 18H 18D 3N 6O 4S, 420.1]; LC/MS retention time (method E): t R=1.38 min; OR: −205.39 (20° C.).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US242383764&_cid=P10-MHIXVD-97150-1
PAT
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-11787779-B2Priority Date: 2017-11-21Grant Date: 2023-10-17
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2024002364-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: KR-102702228-B1Priority Date: 2017-11-21Grant Date: 2024-09-02
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: NZ-805343-APriority Date: 2017-11-21
- Sulfonepyridine alkylamide-substituted heteroaryl compoundsPublication Number: JP-2023098942-APriority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2023255024-A1Priority Date: 2017-11-21
- Heteroaryl compounds substituted with sulfone pyridinylalkylamidesPublication Number: CN-111315737-BPriority Date: 2017-11-21Grant Date: 2024-06-18
- The heteroaryl compounds are substituted with sulfone-pyridine alkyl amidesPublication Number: IL-274816-B2Priority Date: 2017-11-21
- Sulfonepyridine alkylamide substituted heteroaryl compoundsPublication Number: JP-7490107-B2Priority Date: 2017-11-21Grant Date: 2024-05-24
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2022228101-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: TW-I776994-BPriority Date: 2017-11-21Grant Date: 2022-09-11
- Sulfonepyridine alkylamide-substituted heteroaryl compoundsPublication Number: JP-7258903-B2Priority Date: 2017-11-21Grant Date: 2023-04-17
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2022228101-B2Priority Date: 2017-11-21Grant Date: 2023-08-03
- The heteroaryl compounds are substituted with sulfone-pyridine alkyl amidesPublication Number: IL-274816-B1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2019152948-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: CA-3083122-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: KR-20200089706-APriority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-11021462-B2Priority Date: 2017-11-21Grant Date: 2021-06-01
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2021253554-A1Priority Date: 2017-11-21
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | BMS-986322 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2328068-29-5 |
| PubChem CID | 138620496 |
| IUPHAR/BPS | 13210 |
| UNII | EYQ7KA55XA |
| KEGG | D12725 |
| ChEMBL | ChEMBL5314608 |
| Chemical and physical data | |
| Formula | C18H17D3N6O4S |
| Molar mass | 419.47 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Ahsan S, Degener R, Schlamp M (2024). “Non-Invasive Treatments Invade the Psoriasis Pipeline”. Drugs in Context. 13: 2024–5–6. doi:10.7573/dic.2024-5-6. PMC 11313207. PMID 39131603.
////////lomedeucitinib, Janus kinase inhibitor, anti-inflammatory, BMS-986322, BMS 986322, EYQ7KA55XA
Lirodegimod
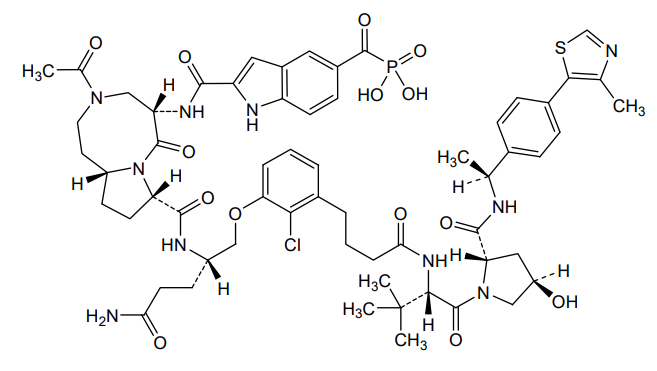
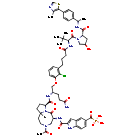
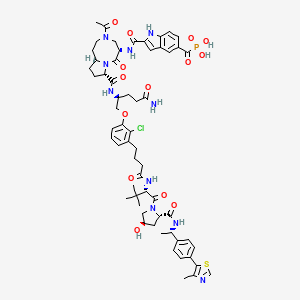
Lirodegimod
CAS 2502186-79-8
MF C60H74ClN10O14PS, MW 1257.79

[2-[[(5S,8S,10aR)-3-acetyl-8-[[(2S)-5-amino-1-[2-chloro-3-[4-[[(2S)-1-[(2S,4R)-4-hydroxy-2-[[(1S)-1-[4-(4-methyl-1,3-thiazol-5-yl)phenyl]ethyl]carbamoyl]pyrrolidin-1-yl]-3,3-dimethyl-1-oxobutan-2-yl]amino]-4-oxobutyl]phenoxy]-5-oxopentan-2-yl]carbamoyl]-6-oxo-1,2,4,5,8,9,10,10a-octahydropyrrolo[1,2-a][1,5]diazocin-5-yl]carbamoyl]-1H-indole-5-carbonyl]phosphonic acid
KT 333, KT333, ANTINEOPLASTIC, Fast Track (United States), Orphan Drug (United States), 4Q6ZHJ2MNA
Lirodegimod is a small molecule drug. The usage of the INN stem ‘-imod’ in the name indicates that Lirodegimod is a immunomodulator, both stimulant/suppressive and stimulant. Lirodegimod has a monoisotopic molecular weight of 1256.45 Da.
Safety, PK, PD, Clinical Activity of KT-333 in Adult Patients With Refractory Lymphoma, Large Granular Lymphocytic Leukemia, Solid Tumors
CTID: NCT05225584
Phase: Phase 1
Status: Completed
Date: 2025-03-19
PAT
- Stat3 degraders and uses thereofPublication Number: US-2023212201-A1Priority Date: 2021-12-11
- Stat3 degraders and uses thereofPublication Number: US-2025019388-A1Priority Date: 2021-12-11
- Stat degraders and uses thereofPublication Number: US-2024016942-A1Priority Date: 2020-03-17
- Stat degraders and uses thereofPublication Number: WO-2020206424-A1Priority Date: 2019-04-05
- Stat degraders and uses thereofPublication Number: US-11746120-B2Priority Date: 2019-04-05Grant Date: 2023-09-05
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Lirodegimod, KT 333, KT333, ANTINEOPLASTIC, Fast Track, Orphan Drug, 4Q6ZHJ2MNA
Linustedastat
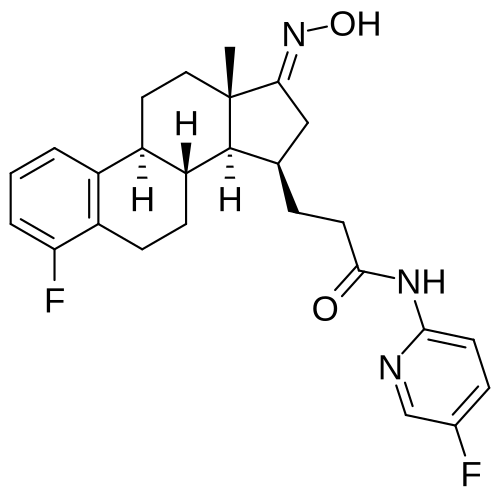
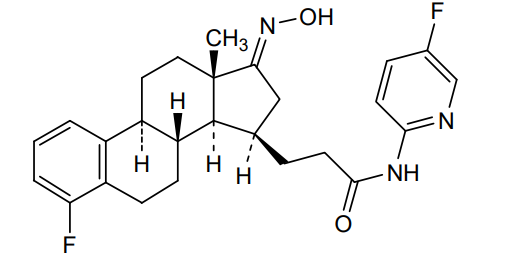
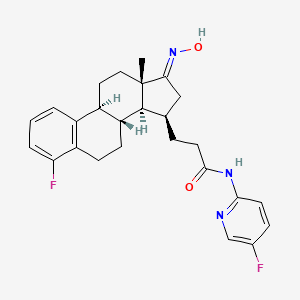
Linustedastat
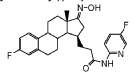
CAS 2254299-48-2
MFC26H29F2N3O2 MW 453.5 g/mol
FOR-6219, OG-6219, FOR 6219, OG 6219, PP3PLL7GZY, Phase 2, Endometriosis
3-[(8R,9S,13S,14S,15R,17E)-4-fluoro-17-hydroxyimino-13-methyl-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthren-15-yl]-N-(5-fluoro-2-pyridinyl)propanamide
- (15beta,17E)-4-Fluoro-N-(5-fluoro-2-pyridinyl)-17-(hydroxyimino)estra-1,3,5(10)-triene-15-propanamide
- 3-[(17E)-4-fluoro-17-(hydroxyimino)estra-1,3,5(10)-trien-15beta-yl]-N-(5-fluoropyridin-2-yl)propanamide
- Estra-1,3,5(10)-triene-15-propanamide, 4-fluoro-N-(5-fluoro-2-pyridinyl)-17-(hydroxyimino)-, (15beta,17E)-
3-[(17E)-4-fluoro-17-(hidroxiimino)estra-1,3,5(10)-trien-15β-il]-N-(5-fluoropiridin-2-il)propanamida
inhibidor de la hidroxiesteroide 17-beta deshidrogenasa 1(HSD17B1)
- OriginatorHormos Medical; Solvay Pharmaceuticals B.V.; University of Turku
- DeveloperOrganon
- ClassSmall molecules
- Mechanism of ActionEstradiol dehydrogenase inhibitors
- Phase IIEndometriosis
- 02 Jul 2025Efficacy data from the phase II ELENA trial in Endometriosis released by Organon
- 28 May 2025Organon completes a phase-II clinical trials in Endometriosis (In adults) in Latvia, Sweden, Poland, Italy, France, Hungary, Germany, Czech Republic, Czech Republic, Bulgaria, Belgium, USA (PO) (NCT05560646)
- 28 Nov 2023No recent reports of development identified for phase-I development in Endometriosis(In volunteers) in United Kingdom (PO)
Linustedastat (developmental code names FOR-6219 and OG-6219) is a 17β-hydroxysteroid dehydrogenase 1 (17β-HSD1; HSD17B1) inhibitor which is under development for the treatment of endometriosis.[1][2][3][4][5] It is a steroidal compound derived from estrone and works by preventing the formation of the more potent estrogen estradiol from the minimally active precursor estrone.[1][2][5] This in turn results in antiestrogenic effects that may be useful in the treatment of estrogen-dependent conditions.[1][2][5] As of November 2023, the drug is in phase 2 clinical trials for endometriosis.[1][2] It is also under preclinical investigation for treatment of breast cancer and endometrial cancer.[5]
A Study to Investigate Efficacy and Safety of OG-6219 BID in 3 Dose Levels Compared With Placebo in Participants Aged 18 to 49 With Moderate to Severe Endometriosis-related Pain
CTID: NCT05560646
Phase: Phase 2
Status: Completed
Date: 2025-05-29
Pat
WO2018224736
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018224736&_cid=P21-MHFVBM-49409-1
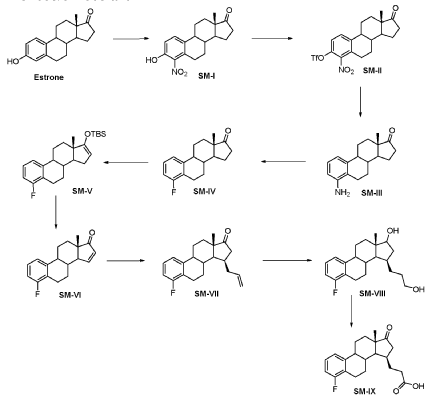
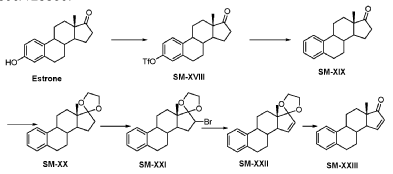
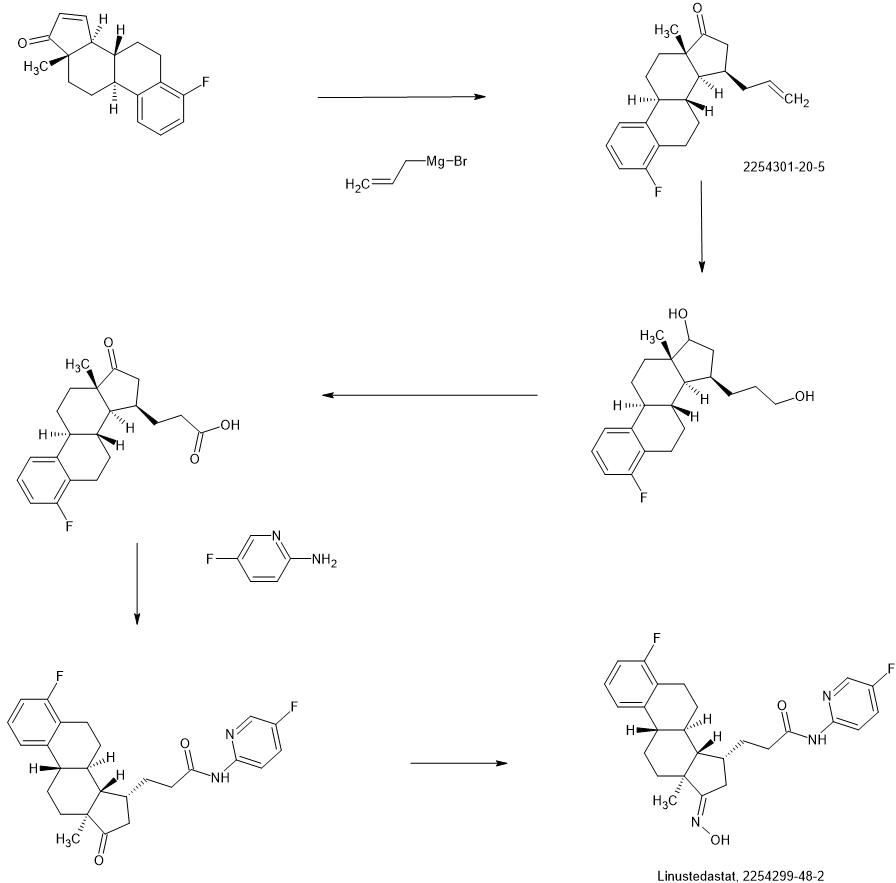
Compound 26
3-((13S,15R,E)-3-fluoro-17-(hydroxyimino)-13-methyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-15-yl)-N-(5-fluoropyridin-2-yl)propanamide
Example 26 was prepared in 94% yield from the compound 25 by the same method as with Example 2 in three hours reaction time.
1H NMR (200 MHz, DMSO-d6): 1.03 (s, 3 H), 1.12 – 2.48 (m, 15 H), 2.57 – 2.78 (m, 1 H), 2.80 – 2.95 (m, 2 H), 6.79 – 7.01 (m, 2 H), 7.18 – 7.38 (m, 1 H), 7.72 (td, 1 H), 8.15 (dd, 1 H), 8.31 (d, 1 H), 10.18 (s, 1 H), 10.64 (s, 1 H). MS m/z (TOF ES+): 454 (M+1).
SYNTHESIS
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | FOR-6219; OG-6219; 3-[(17E)-4-Fluoro-17-(hydroxyimino)estra-1,3,5(10)-trien-15β-yl]-N-(5-fluoropyridin-2-yl)propanamide |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2254299-48-2 |
| PubChem CID | 171390018 |
| UNII | PP3PLL7GZY |
| KEGG | D13078 |
| Chemical and physical data | |
| Formula | C26H29F2N3O2 |
| Molar mass | 453.534 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “FOR 6219”. AdisInsight. 28 November 2023. Retrieved 15 August 2024.
- “Delving into the Latest Updates on Linustedastat with Synapse”. Synapse. 3 August 2024. Retrieved 15 August 2024.
- Barra F, Romano A, Grandi G, Facchinetti F, Ferrero S (June 2019). “Future directions in endometriosis treatment: discovery and development of novel inhibitors of estrogen biosynthesis”. Expert Opin Investig Drugs. 28 (6): 501–504. doi:10.1080/13543784.2019.1618269. hdl:11380/1201688. PMID 31072144.
- Perrone U, Evangelisti G, Laganà AS, Bogliolo S, Ceccaroni M, Izzotti A, Gustavino C, Ferrero S, Barra F (December 2023). “A review of phase II and III drugs for the treatment and management of endometriosis”. Expert Opin Emerg Drugs. 28 (4): 333–351. doi:10.1080/14728214.2023.2296080. PMID 38099328.
- Rižner TL, Romano A (2023). “Targeting the formation of estrogens for treatment of hormone dependent diseases-current status”. Front Pharmacol. 14 1155558. doi:10.3389/fphar.2023.1155558. PMC 10175629. PMID 37188267.
Several compounds with inhibitory action on the enzyme HSD17B1 have been developed and one steroidal compound, a competitive HSD17B1 inhibitor (OG-6219) recently entered the clinical phase for endometriosis […] and it is in the preclinical phase for endometrial and breast cancer (Husen et al., 2006a; Husen et al., 2006b; Konings et al., 2018b; Jarvensivu et al., 2018; Xanthoulea et al., 2021). […] Only the C15 estrone derivative developed by Organon Finland, former Forendo pharma (compound FOR-6219/OR-6219) reached the clinical phase for endometriosis with three clinical trials registered in the database Clinical Trails (Table 2). Phase 1 and 1b trials NCT04686669 and NCT03709420 determined the bio-availability of the compound administered orally as gelatine capsule in 12 subjects (NCT04686669) and then the safety, tolerability, food interactions, the pharmacokinetics and pharmacodynamics of escalating doses of the drug in 87 subjects (NCT03709420). The phase 2 randomized, double-blind, Elena study (NCT05560646) is currently recruiting patients and aims at evaluating the efficacy and safety of OG-6219 in women with moderate to severe endometriosis […]
External links
//////////Linustedastat, FOR-6219, OG-6219, FOR 6219, OG 6219, PP3PLL7GZY, Phase 2, Endometriosis
Iodofalan (131I)
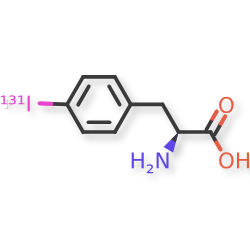
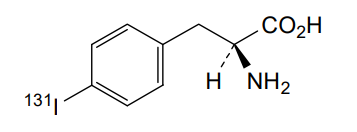
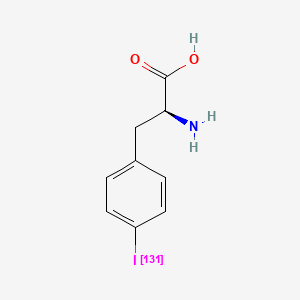
Iodofalan (131I)
CAS 76641-05-9
MFC9H10131INO2
Molecular FormulaC9H10INO2
Molecular Weight295.09
4-(131I)iodo-L-phenylalanine
(2S)-2-amino-3-(4-iodophenyl)propanoic acid
radiopharmaceutical, antineoplastic, Phase 2, Glioblastoma, 606VTF676Y, 131I-TLX-101, ACD 101
- 4-Iodophenylalanine I-131
- 4-(131I)Iodo-L-phenylalanine
- 4-Iodo-L-phenylalanine-131I
- ACD-101
- L-Phenylalanine, 4-(iodo-131I)-
- OriginatorTherapeia
- DeveloperTelix Pharmaceuticals; Therapeia
- ClassAmino acids; Antineoplastics; Radioisotopes; Radiopharmaceutical diagnostics; Radiopharmaceuticals; Small molecules
- Mechanism of ActionApoptosis stimulants; Positron-emission tomography enhancers
- Orphan Drug StatusYes – Glioblastoma
- Phase IIGlioblastoma
- 14 Oct 2025Telix Pharmaceuticals receives IND approval for TLX 101 in Glioblastoma
- 27 Jul 2025Telix Pharmaceuticals plans a phase III IPAX BrIGHT trial for Glioblastoma (Monotherapy, Combination therapy, Recurrent, Second-line therapy or greater) in Australia(IV) (NCT07100730)(EudraCT2025-521785-10) in September 2025
- 16 Apr 2025Telix has submitted for ethics approval a registration-enabling study of TLX101 in recurrent glioblastoma.
Iodofalan (131I) is a radiopharmaceutical that has garnered significant attention in oncological research due to its targeted therapeutic potential. This compound, which includes the radioactive isotope Iodine-131, has been explored for its efficacy in treating certain types of cancers, particularly those associated with the thyroid. Various research institutions worldwide have been studying Iodofalan (131I) to better understand its clinical benefits, optimize its usage, and minimize potential side effects. As a drug type, Iodofalan (131I) is categorized as a targeted radiopharmaceutical therapy, which leverages the properties of radioactive isotopes to destroy cancer cells with precision. Currently, its primary indications include differentiated thyroid cancer and non-resectable metastatic thyroid cancer, among other investigational uses.
Iodofalan (131I) Mechanism of Action
The mechanism of action for Iodofalan (131I) centers on the properties of Iodine-131, a beta-emitting isotope. When administered, Iodofalan (131I) is selectively absorbed by thyroid cells. This selectivity is due to the thyroid gland’s natural ability to uptake iodine, a key element required for the production of thyroid hormones. Cancerous thyroid tissues retain this ability, making them ideal targets for Iodofalan (131I) therapy.
Once absorbed by the thyroid cancer cells, the radioactive decay of Iodine-131 begins. This decay process emits beta particles, which possess sufficient energy to destroy nearby cells. The radiation from these beta particles causes direct DNA damage, leading to cell death. Additionally, the gamma radiation emitted by Iodine-131 can be used diagnostically to track the distribution and uptake of the compound in the body via imaging techniques such as SPECT (Single Photon Emission Computed Tomography).
The dual role of Iodofalan (131I) in both treatment and diagnostic contexts underscores its importance in managing thyroid cancers. By delivering a localized radiation dose to thyroid cancer cells, Iodofalan (131I) minimizes damage to surrounding healthy tissues, which is a significant advantage over traditional external beam radiotherapy.
What is the indication of Iodofalan (131I)?
The primary indication for Iodofalan (131I) is the treatment of differentiated thyroid cancer, a category that includes papillary and follicular thyroid cancers. These subtypes are characterized by their ability to absorb iodine, making them particularly amenable to radioiodine therapy. Iodofalan (131I) is typically used in cases where the thyroid cancer is not amenable to surgical removal or has metastasized to other parts of the body. In such scenarios, the radiopharmaceutical offers a non-invasive therapeutic option that can target and destroy cancer cells even in distant metastatic sites.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42129729&_cid=P21-MHE8B5-15309-1
EXAMPLE 1
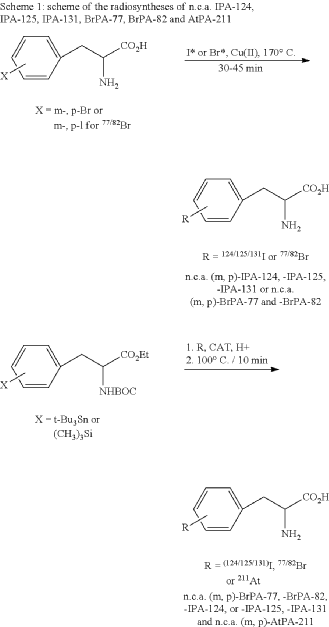
EXAMPLE 2
General synthesis of 3,4-[124I]iodo-L-phenylalanine (m, p-IPA-124), 3,4-[125I]iodo-L-phenylalanine (m,p-IPA-125) and 3,4-[131I]iodo-L-phenylalanine (m,p-IPA-131) by non-isotopic radioiodo-debromination
PAT
- Pharmaceutical combinations and uses thereofPublication Number: US-2024197715-A1Priority Date: 2022-11-18
- Pharmaceutical combinations and uses thereofPublication Number: WO-2024105610-A1Priority Date: 2022-11-18
- Iodine-labeled homoglutamic acid and glutamic acid derivativesPublication Number: US-2013034497-A1Priority Date: 2009-11-17
- MALIGNAS NEOPLASIAS THERAPY.Publication Number: ES-2341575-T3Priority Date: 2005-11-25Grant Date: 2010-06-22
- Therapy of malignant neoplasiasPublication Number: US-2007128108-A1Priority Date: 2005-11-18
- Therapy of malignant neoplasias
- Publication Number: US-9682158-B2
- Priority Date: 2005-11-18
- Grant Date: 2017-06-20
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Iodofalan (131I), radiopharmaceutical, antineoplastic, Phase 2, Glioblastoma, 606VTF676Y, 131I-TLX-101, ACD 101
Inlexisertib
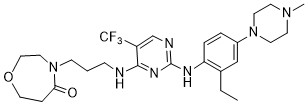
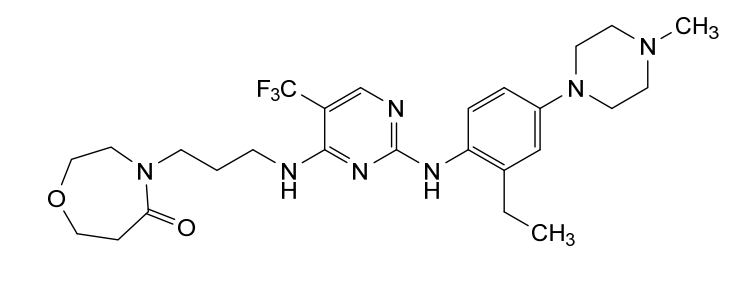
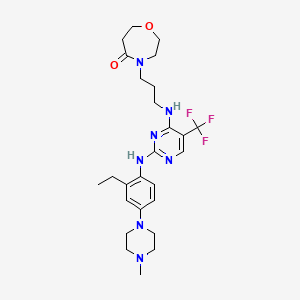
Inlexisertib
CAS 2543673-19-2
MF C26H36F3N7O2, 535.62
4-(3-((2-((2-ethyl-4-(4-methylpiperazin-1-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4-yl)amino)propyl)-1,4-oxazepan-5-one
4-[3-[[2-[2-ethyl-4-(4-methylpiperazin-1-yl)anilino]-5-(trifluoromethyl)pyrimidin-4-yl]amino]propyl]-1,4-oxazepan-5-one

serine/ threonine kinase inhibitor, antineoplastic, DCC 3116, JM2ZTM8S7S
Inlexisertib is an orally bioavailable inhibitor of the serine/threonine-protein kinase ULK 1 and 2, with potential antineoplastic activity. Upon oral administration, inlexisertib targets and binds to ULK1/2. This inhibits cancer autophagy, which mutant RAS cancer cells use for their survival, and results in tumor cell death. ULK1/2 mediates the autophagocytotic process and is often upregulated in cancers, especially in mutant RAS cancers. Autophagy plays a key role in a tumor cell proliferation and survival, and mediates tumor cell resistance.
- A Study of Inlexisertib (DCC-3116) in Combination With Anticancer Therapies in Participants With Advanced MalignanciesCTID: NCT05957367Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-06-05
- A Phase 1/2 Study of Inlexisertib (DCC-3116) in Patients With RAS/MAPK Pathway Mutant Solid TumorsCTID: NCT04892017Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-05-06
SYN
https://patents.google.com/patent/US11530206B2/en
PAT
Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereof
Publication Number: JP-7593947-B2
Priority Date: 2019-05-10
Grant Date: 2024-12-03
- PHENYLAMINOPYRIMIDINE AMIDE INHIBITORS OF AUTOPHAGY AND METHODS OF THEIR APPLICATIONPublication Number: HR-P20231730-T1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-12071432-B2Priority Date: 2019-05-10Grant Date: 2024-08-27
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118878519-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118878520-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118930524-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: AU-2020275392-B2Priority Date: 2019-05-10Grant Date: 2023-09-14
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-114127057-BPriority Date: 2019-05-10Grant Date: 2024-07-12
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-3966207-B1Priority Date: 2019-05-10Grant Date: 2023-11-01
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-4342469-A2Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: ES-2966807-T3Priority Date: 2019-05-10Grant Date: 2024-04-24
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-3966207-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: KR-20220008873-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitor and how to use itPublication Number: JP-2022531801-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-11530206-B2Priority Date: 2019-05-10Grant Date: 2022-12-20
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-2023039712-A1Priority Date: 2019-05-10
- Combination of dcc-3116 and mapkap pathway inhibitors for use in the treatment of cancerPublication Number: WO-2024050351-A1Priority Date: 2022-09-02
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-2020354352-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: WO-2020231806-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: AU-2020275392-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitor and method of usePublication Number: CN-114127057-APriority Date: 2019-05-10
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020231806&_cid=P12-MHCSWS-98394-1

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Inlexisertib, serine/ threonine kinase inhibitor, antineoplastic, DCC 3116, JM2ZTM8S7S
Imocitrelvir
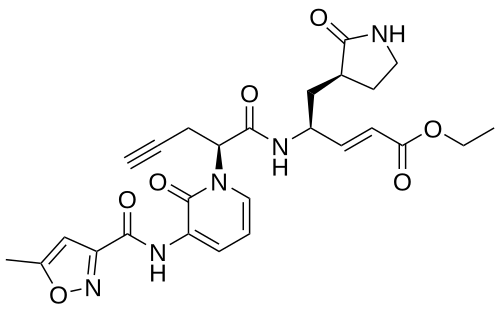
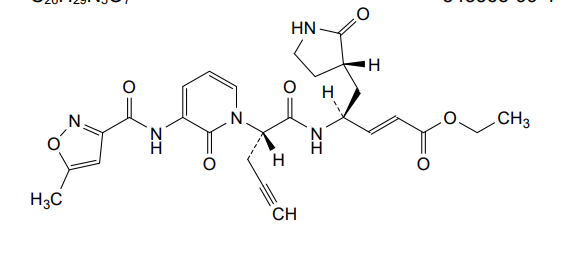
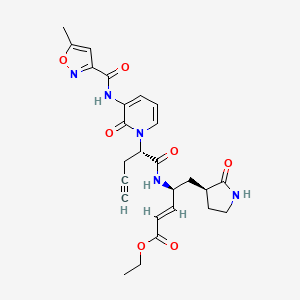
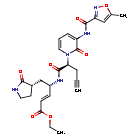
Imocitrelvir
CAS 343565-99-1
MFC26H29N5O7 MW523.5 g/mol
ethyl (2E,4S)-4-{(2S)-2-[3-(5-methyl-1,2-oxazole-3-carboxamido)-2-oxopyridin-1(2H)-yl]pent-4-ynamido}-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate
ethyl (E,4S)-4-[[(2S)-2-[3-[(5-methyl-1,2-oxazole-3-carbonyl)amino]-2-oxo-1-pyridinyl]pent-4-ynoyl]amino]-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate
protease inhibitor, antiviral, AG-7404, V-7404, AG 7404, V 7404, VQ1AN3OO42
Imocitrelvir is an investigational new drug that is being evaluated for the treatment of viral infections. It is a 3C protease inhibitor in picornaviruses. Originally developed by Pfizer for treating human rhinovirus infections,[1] this small molecule has shown promise against a broader range of viruses, including polioviruses.[2][3]
SYN
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2003-09-17
PMID: 14521419
DOI: 10.1021/jm030166l
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016044656&_cid=P21-MHBDH2-20719-1
PAT
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001040189&_cid=P21-MHBDI9-21481-1
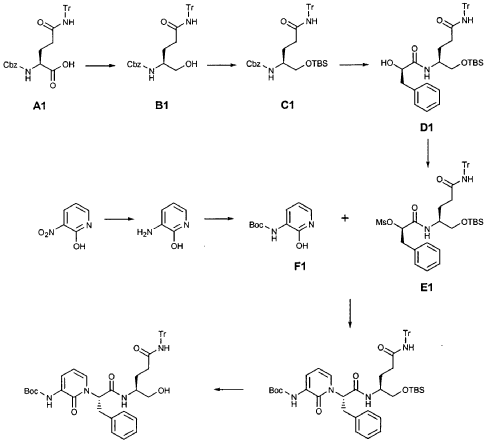
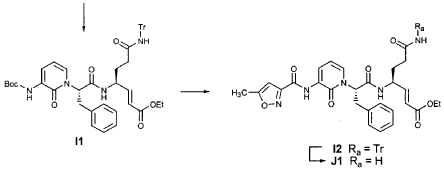
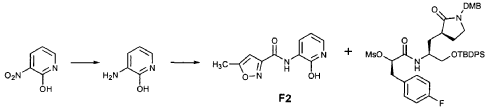
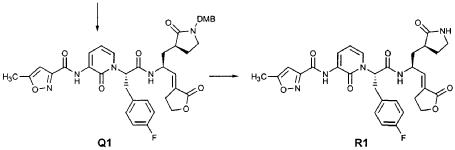
EXAMPLE 21
Preparation of Compound 22: tra«5-(4S,3″”S)-4-(2′-{3″-[(5′”-Methylisoxazole-3′”-carbonyl)amino]-2″-oxo-2″H-pyridin- 1 “-yl} acetylamino)-5-(2″”-oxopyrrilidin-3″”-yl)pent-2-enoic Acid Ethyl Ester
Preparation of Intermediate {3-[(5′-Methylisoxazole-3′-carbonyl)amino]-2-oxo-2H-pyridin-l-yl} acetic Acid tert-Butyl Ester
To a solution of 5-methylisoxazole-3-carboxylic acid (2′-hydroxy-4′-methylpyridin-3′-yl)amide (F2, Example 19) (0.520 g, 2.37 mmol, 1 equiv) in TΗF (20 mL) at 0 °C was added NaΗ (0.095 g, 2.37 mmol, 1.0 equiv). The resulting mixture was stirred at 0 °C for 20 min, and then t-butyl bromoacetate (0.385 mL, 2.61 mmol, 1.1 equiv) was added. The reaction mixture was stirred and warmed to room temperature for 30 min, then was partitioned between 0.5 N ΗC1 (100 mL) and EtOAc (2 x 100 mL). The combined organic layers were dried over Na2SO and were concentrated. Purification of the residue by flash column chromatography (30% EtOAc in hexanes) provided the title intermediate (0.628 g, 79%) as a white solid: IR (cm-1) 3343, 1743, 1651, 1581, 1156; Η NMR (CDC13) δ 1.52 (s, 9H), 2.53 (s, 3H), 4.65 (s, 2H), 6.32 (t, 1H, 7= 7.2), 6.51 (s, IH), 7.01 (dd, 1H, 7= 6.9, 1.8), 8.50 (dd, 1H, 7= 7.5, 1.8), 9.63 (s, br. IH); Anal. C16H19N3O5: C, H, N.
Preparation of Compound 22
The preceding intermediate was transformed into Compound 22 by a process that was analogous to that described in Example 25 for the transformation of V3 to product R3: mp = 102-106 °C; IR (cm”1) 3336, 1684, 1534, 1457; JH NMR (CDCI3) δ 1.27 (t, 3H, 7= 7.2), 1.67-1.75 (m, IH), 1.98-2.09 (m, IH), 2.37-2.49 (m, IH), 2.53 (s, 3H), 2.55-2.61 (m, IH), 3.34-3.46 (m, 2H), 3.51-3.52 (m, IH), 4.17 (q, 2H, 7= 7.2), 4.61-4.78 (m, 3H), 5.98 (dd, IH, 7 = 15.6, 1.5), 6.20 (s, br. IH), 6.35 (t, 1H, 7= 7.8), 6.51 (s, IH), 6.85 (dd, IH, 7= 15.6, 5.1), 7.17 (d, IH, 7= 7.2), 8.33 (d, IH, 7= 7.2), 8.49 (d, IH, 7= 7.5), 9.57 (s, br. IH); Anal.
C23H27N5O7: C, H, N.
EXAMPLE 24
Preparation of Compound 25: trans-(2’S,3″”‘S,4S)-4-(3,-(4″-Fluorophenyl)-2′-{3″‘-[(5″”-methylisoxazole-3″”-carbonyl)amino]-2′”-oxo-2′”H-pyridin- “-yl}propionylamino)-5-(2″ oxopyrrolidin-3′””-yl)pent-2-enoic Acid Ethyl Ester
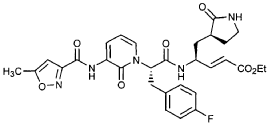
The title compound was prepared from F2 (Example 19) in a manner analogous to that described for the conversion of U2 to 13 in Example 23 utilizing intermediate Y2 (Example 25) where appropriate: IR (cm-1) 3331, 1690, 1590, 1531, 1455; !H NMR (CDCI3) δ 1.30 (t, 3H, 7= 7.0), 1.45-1.55 (m, IH), 1.64-1.75 (m, IH), 2.03-2.31 (m, 3H), 2.49 (s, 3H), 3.10 (dd, IH, 7= 13.7, 7.9), 3.20-3.46 (m, 3H), 4.20 (q, 2H, 7= 7.0), 4.36-4.47 (m, IH), 5.67 (dd, IH, 7 = 15.7, 1.4), 5.85-5.92 (m, IH), 6.29 (t, 1H, 7= 7.2), 6.45 (s, IH), 6.70 (dd, IH, 7= 15.7, 5.7), 6.86 (s, IH), 6.90-6.97 (m, 2H), 7.10-7.16 (m, 2H), 7.60 (dd, IH, 7= 7.2, 1.6), 8.37 (dd, IH, 7 = 7.2, 1.6), 8.51 (d, IH, 7= 6.6), 9.47 (s, IH).
EXAMPLE 25
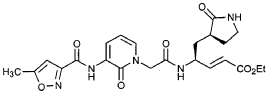
Preparation of Compound 26: tr_.«5-(2’S,3″”S,4S)-4-(2′-{3″-[(5″‘-Methyl-isoxazole-3′”-carbonyl)amino]-2″-oxo-2″H-pyridin-l”-yl}butyrylamino)-5-(2″”-oxopyrrolidin-3″”-yl)pent-2-enoic Acid Ethyl Ester (R3)
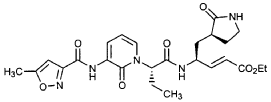
Preparation of Intermediate (2R)-2-Trifluoromethanesulfonyl-oxybutyric acid tert-butyl ester (U3)
Commercially available T3 (0.575 g, 3.59 mmol, 1 equiv) was dissolved in CH2CI2 (25 mL) and cooled in an ice bath. 2,6-Lutidine (0.836 mL, 7.18 mmol, 2 equiv) and trifluoromethanesulfonic anhydride (1.15 mL, 6.84 mmol, 1.9 equiv) were added and the reaction mixture was stirred 30 min. It was then diluted with MTBE (400 mL), washed with a mixture of brine and 1 N HCl (2:1, 100 mL) and brine (100 mL), dried over Na2SO4 and evaporated to provide the title intermediate which was used without further purification.
Preparation of Intermediate (2S)-2- {3′-[(5″-Methylisoxazole-3″-carbonyl)amino]-2′-oxo-2’H-pyri din- l’-yl} butyric Acid tert-Butyl Ester (V3)
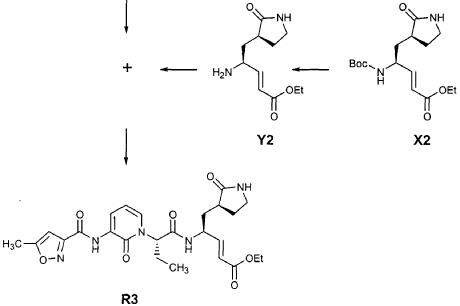
Intermediate F2 from above (0.200 g, 0.912 mmol, 1.1 equiv) was suspended in TΗF (6 mL). Sodium hydride (60% dispersion in mineral oil, 0.0332 g, 0.830 mmol, 1 equiv) was added in one portion. After stirring 30 min, a solution of intermediate U3 (0.830 mmol, 1 equiv, based on T3) in TΗF (7 mL) was added dropwise. The resulting mixture was stirred 2 hours, then diluted with EtOAc (200 mL) and washed with brine (2 x 50 mL). The organic phase was dried over MgSO4 and evaporated. The residue was purified by flash column chromatography (25% EtOAc in hexanes) to provide the title intermediate (0.178 g, 59%) as an oil: R/= 0.30 (25% EtOAc in hexanes); IR (cm”1) 3331, 1731, 1690, 1649, 1602, 1531 ; *Η NMR (CDCI3) δ 0.93 (t, 3H, 7= 7.3), 1.45 (s, 9H), 1.83-2.01 (m, IH), 2.17-2.31 (m, IH), 2.50 (s, 3H), 5.44-5.51 (m, IH), 6.32 (t, IH, 7= 7.2), 6.48 (s, IH), 7.10 (dd, IH, 7= 7.2, 1.8), 8.45 (dd, 1H, 7= 7.2, 1.8), 9.64 (s, IH); Anal. C18H23N3O5: C, H, N.
Preparation of Intermediate (2S)-2- {3′-[(5″-Methylisoxazole-3″-carbonyl)amino]-2′-oxo-2’H-pyridin-l’-yl}butyric Acid (W3)
Intermediate V3 from above (0.143 g, 0.397 mmol, 1 equiv) was stirred for 1 h in a solution of TFA (2 mL) in CΗ2CI2 (3 mL). The volatiles were evaporated. The residue was suspended in toluene (10 mL) and concentrated to dryness, providing the title intermediate which was used without further purification.
Preparation of Intermediate trα«5-(3’S,4S)-4-Amino-5-(2′-oxopyrrolidin-3′-yl)pent-2-enoic Acid Ethyl Ester (Y2)
Intermediate X2, prepared according to the method disclosed in the co-pending application, U.S. Provisional Patent Application No. 60/150,358, filed August 24, 1999(0.130 g, 0.398 mmol, 1 equiv), was stirred for 30 min in a solution of TFA (2 mL) in CH2CI2 (3 mL). The volatiles were evaporated. The residue was suspended in toluene (10 mL) and concentrated to dryness, providing the title intermediate which was used without further purification.
Preparation of Product R3 (Compound 26)
Intermediates W3 and Y2 (as prepared above) were combined in CH2CI2 (7 mL) and cooled in an ice bath. HOBt (0.064 g, 0.47 mmol, 1.2 equiv), iP^NEt (0.484 mL, 2.78 mmol, 7 equiv) and EDC (0.084 g, 0.44 mmol, 1.1 equiv) were added sequentially. The reaction mixture was allowed to warm to 23 °C overnight, then diluted with EtOAc (500 mL) and washed with 5% KHSO4 , half saturated NaHCO3, and brine (100 mL each). The organic phase was dried over MgSO4 and evaporated. The residue was purified by flash column chromatography (gradient elution, 2→3% CH3OH in CH2CI2) to provide the title intermediate (0.119 g, 58%) as a white foam: IR (cm”1) 3331, 1684, 1649, 1590, 1531; JH NMR (CDCI3) δ 0.92 (t, 3H, J = 7.3), 1.29 (t, 3H, J = 7.1), 1.47-1.58 (m, IH), 1.62-1.77 (m, IH), 1.85-2.00 (m, IH), 2.08-2.33 (m, 4H), 2.49 (s, 3H), 3.25-3.42 (m, 2H), 4.19 (q, 2H, J = 7.1), 4.39-4.50 (m, IH), 5.73 (dd, IH, J = 8.8, 6.8), 5.97 (dd, IH, J = 15.7, 1.4), 6.34 (t, IH, J = 7.2), 6.46 (s, IH), 6.86 (dd, IH, J = 15.7, 5.9), 7.18 (s, IH), 7.59 (dd, IH, J = 7.2, 1.8), 8.42 (dd, IH, J = 7.2, 1.8), 8.58-8.62 (m, IH), 9.56 (s, 1); Anal. C25H31N5O7O.5OH2O: C, H, N.
PAT
- Treatment of infection by human enterovirus d68Publication Number: US-2020016243-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus d68Publication Number: WO-2016044656-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus d68Publication Number: US-2021052708-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus D68Publication Number: US-11191817-B2Priority Date: 2014-09-17Grant Date: 2021-12-07
- Therapeutic compounds and methodsPublication Number: US-2025051283-A1
- Protease Inhibitors for Treatment or Prevention of Coronavirus DiseasePublication Number: US-2023192660-A1Priority Date: 2020-05-08
- Composition and combined medication method for treating enterovirus infectionPublication Number: US-2019030027-A1Priority Date: 2016-01-29
- Composition and combined medication method for treating enterovirus infectionPublication Number: US-10864210-B2Priority Date: 2016-01-29Grant Date: 2020-12-15
- Treatment of infection by human enterovirus D68Publication Number: US-10328128-B2Priority Date: 2014-09-17Grant Date: 2019-06-25
- Treatment of infection by human enterovirus d68Publication Number: US-2017290893-A1Priority Date: 2014-09-17
- Nucleotide and nucleoside therapeutic compositions, combinations and related uses thereofPublication Number: CN-117881402-APriority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: EP-4333859-A1Priority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations, and related usesPublication Number: JP-2024517807-APriority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: WO-2022235874-A1Priority Date: 2021-05-05
- Protease inhibitors for treatment or prevention of coronavirus diseasePublication Number: EP-4146267-A1Priority Date: 2020-05-08
- 4′-substituted nucleosides and nucleotides as antiviral agentsPublication Number: WO-2024227159-A2Priority Date: 2023-04-28
- Therapeutic compoundsPublication Number: WO-2024206284-A2Priority Date: 2023-03-27
- Antibody molecules binding to sars-cov-2Publication Number: WO-2024168061-A2Priority Date: 2023-02-07
- Predictive model for variants associated with drug resistance and theranostic applications thereofPublication Number: WO-2023172635-A1Priority Date: 2022-03-08
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: CA-3216679-A1Priority Date: 2021-05-05
LIT
- Structure and inhibition of SARS-CoV-1 and SARS-CoV-2 main proteases by oral antiviral compound AG7404Publication Name: Antiviral ResearchPublication Date: 2022-12PMCID: PMC9632241PMID: 36336176DOI: 10.1016/j.antiviral.2022.105458
- Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug DesignPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-09-30PMID: 34591488DOI: 10.1021/acs.jmedchem.1c01215
- In Vitro Antiviral Activity of New Oxazoline Derivatives as Potent Poliovirus InhibitorsPublication Name: Journal of Medicinal ChemistryPublication Date: 2018-12-04PMCID: PMC9169555PMID: 30512950DOI: 10.1021/acs.jmedchem.8b01482
- A Novel Series of Highly Potent Small Molecule Inhibitors of Rhinovirus ReplicationPublication Name: Journal of Medicinal ChemistryPublication Date: 2017-06-15PMID: 28581749DOI: 10.1021/acs.jmedchem.7b00175
- Anti-poliovirus activity of protease inhibitor AG-7404, and assessment of in vitro activity in combination with antiviral capsid inhibitor compoundsPublication Name: Antiviral ResearchPublication Date: 2013-05PMID: 23499651DOI: 10.1016/j.antiviral.2013.03.003
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | AG-7404, V-7404 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 343565-99-1 |
| PubChem CID | 5280053 |
| IUPHAR/BPS | 13223 |
| UNII | VQ1AN3OO42 |
| ChEMBL | ChEMBL141157 |
| Chemical and physical data | |
| Formula | C26H29N5O7 |
| Molar mass | 523.546 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Imocitrelvir”. PatSnap.
- Xie H, Rhoden EE, Liu HM, Ogunsemowo F, Mainou BA, Burke RM, et al. (November 2024). “Antiviral Development for the Polio Endgame: Current Progress and Future Directions”. Pathogens. 13 (11). Basel, Switzerland: 969. doi:10.3390/pathogens13110969. PMC 11597170. PMID 39599522.
- Bandyopadhyay AS, Burke RM, Hawes KM (June 2024). “Polio Eradication: Status, Struggles and Strategies”. The Pediatric Infectious Disease Journal. 43 (6): e207-211. doi:10.1097/INF.0000000000004330. PMID 38564755.
////////Imocitrelvir, protease inhibitor, antiviral, AG-7404, V-7404, AG 7404, V 7404, VQ1AN3OO42
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}