
Home » Uncategorized (Page 68)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ICH M8 “Specification for Submission Formats for eCTD”
DRUG REGULATORY AFFAIRS INTERNATIONAL

This additional specification describes the way files should be constructed for inclusion in the eCTD.
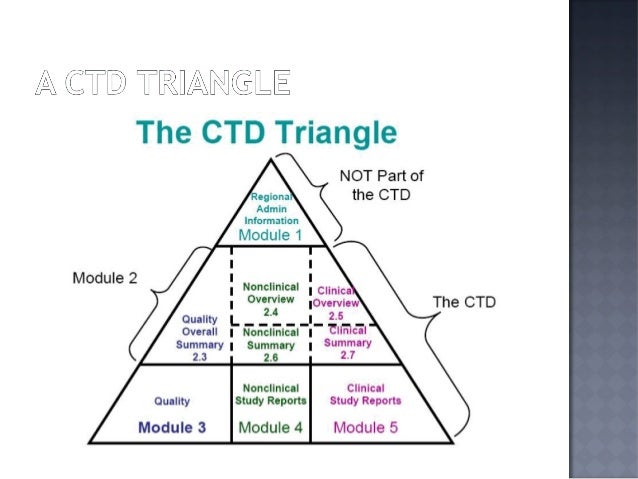 Key Points:
Key Points:
- It is not necessary to use a product from Adobe or from any specific company to produce PDF documents.
- All ICH regional regulatory authorities are able to read and accept PDF files saved as PDF version 1.4 through 1.7, PDF/A-1, or PDF/A-2 compliant to ISO 32000-1:2008.
- The size of a PDF file should not exceed 500MB.
- Regulatory authorities cannot guarantee the availability of any fonts except Times New Roman, Arial, and Courier and fonts supported in the Acrobat product set itself. Therefore, all additional fonts used in the PDF files should be embedded to ensure that those fonts would always be available to the reviewer.
- Times New Roman, 12-point font, is adequate in size for narrative text and should be used whenever possible. Times New Roman font sizes 9-10 or an equivalent size…
View original post 496 more words
CDRI 830

CDRI 830
CDRI S006-830
S 006-830
CAS 1550975-42-2
N-[2-[4-[(4-methoxyphenyl)-thiophen-2-ylmethyl]phenoxy]ethyl]-N-propan-2-ylpropan-2-amine
| Molecular Formula: | C26H33NO2S |
|---|---|
| Molecular Weight: | 423.61072 g/mol |


CDRI-830 of thiophene containing trisubstituted methane (TRSM) class was identified as an anti-tubercular lead with MIC value of 1.33 mg/L against Mycobacterium tuberculosis H37Rv strain, non-toxicity against Vero C-1008 cell line (selectivity index >10), ex vivo efficacy (in mouse and human macrophages) equivalent to first line TB drugs, lung CFU count (2.2×107) comparable to pyrazinamide (1.9×107) and ethambutol (1.27×107). CDRI-830 has exhibited potent bactericidal activity against single and multi-drug resistant clinical isolates of M. tuberculosis. Furthermore, CDRI-830 has demonstrated good pharmacokinetic properties with fast intestinal absorption, peak plasma concentration one hour post oral dose, optimum elimination half-life (9-13 h), plasma protein binding (~60%), favorable bioavailability (45-50%) and mean residence time (18-20 h).

CDRI S006-830 is a potent triethylamine containing thiophene antitubercular compound of the Central Drug Research Institute, India. The present study aimed to conduct comprehensive metabolic investigations of CDRI S006-830 to corroborate its preclinical investigations. Preliminary metabolic investigations were performed to assess the metabolic stability, enzyme kinetics, reaction phenotyping, and metabolite identification of CDRI S006-830 in rat, rabbit, dog, and human liver microsomes using liquid chromatography with mass spectrometry. The observed in vitro t1/2 and Clint values were 9.9 ± 1.29, 4.5 ± 0.52, 4.5 ± 0.86, 17 ± 5.21 min and 69.60 ± 8.37, 152.0 ± 17.26, 152.34 ± 27.63, 33.62 ± 21.04 μL/min/mg in rat, rabbit, dog and human liver microsomes respectively. These observations suggested that CDRI S006-830 rapidly metabolized in the presence of NADPH in liver microsomes of rat, rabbit and dog while moderately metabolized in human liver microsomes. It was observed that CDRI S006-830 exhibited monophasic Michaelis–Menten kinetics. The metabolism of CDRI S006-830 was primarily mediated by CYP3A4 and was deduced by CYP reaction phenotyping with known potent inhibitors. CYP3A4 involvement was also confirmed by cDNA-expressed recombinant human isozyme activity with different CYPs. Four major phase-I metabolites of S006-830, (M-1 to M-4) were detected in rat, rabbit, dog (except M4) and human liver microsomes……..http://onlinelibrary.wiley.com/doi/10.1002/dta.1802/abstract?systemMessage=Wiley+Online+Library+will+be+unavailable+on+Saturday+14th+May+11%3A00-14%3A00+BST+%2F+06%3A00-09%3A00+EDT+%2F+18%3A00-21%3A00+SGT+for+essential+maintenance.Apologies+for+the+inconvenience.

NMR
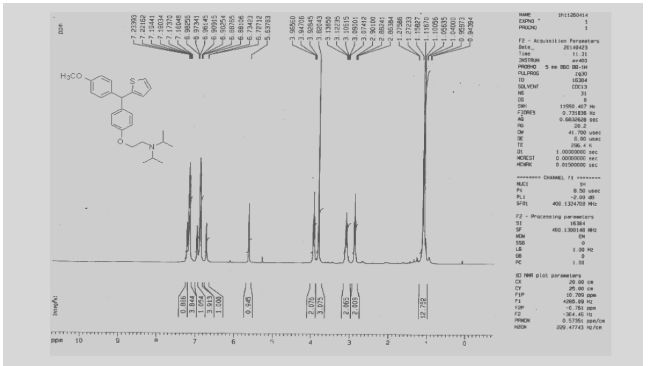
13C NMR

SYNTHESIS

Gautam Panda

| Associate Professor AcSIR ( Academy of Scientific and Innovative Research, New Delhi, India) Principal Scientist and Group Leader Medicinal and Process Chemistry Division CSIR-CDRI ( Central Drug Research Institute ) Sector-10, Jankipuram Extension, Sitapur Road, Lucknow-226031 Phone (Office) : 0522-2772450, 2772550, Ext. 4661, 4662 Phone (Res.) : 0522-2746635 Fax : 0522-2771941 Email : gautam.panda@gmail.com, gautam_panda@cdri.res.in Webpage: http://www.cdriindia.org/gautampanda.htm |
PATENT
Indian Pat. Appl. (2012), IN 2010DE00685
Abstract:
The invention relates to Thiophene containing Trisubstituted Methanes (TRSMs) and a process for the preparation thereof. The invention particularly relates to a process for the preparation of substituted secondary and tertiary amino alkoxy diary! thiophenyl methanes and their use as potential antimycobacterial agents. Novel diaryl thiophenyl methanes of formula I have been prepared. The present invention provides novel diaryl thiophenyl methanes substituted with a secondary or tertiary amino alkoxy group and a process for the preparation of the said compounds of general formula I comprising general formula la and lb useful in antimycobacterial activity wherein R1 is selected from an aryl group or thophene moiety wherein the aryl group is selected from a group consisting of substituted phenyl groups such as methoxy phenyl, thiomethoxy phenyl, phenyl, p-chlorophenyl, p-fluorophenyl; R2 is selected from a group consisting of aminoalkoxyl, alkyl/dialkyl aminoalkoxy, cyclic alkyl aminoalkoxy. R3 is selected from a group consisting of H, lower alkyl, lower alkoxy group such as methyl, ethyl, n-propyl, isopropyl, n-butyl, secondary butyl, tertiary butyl, n-amyl, n-hexyl, 2-ethyl butyl; R4 is selected from a group consisting of H,OH,methyl.
The invention relates to thiophene containing Trisubstituted Methanes (TRSMs) and a process for the preparation thereof. The invention particularly relates to a process for the preparation of substituted secondary and tertiary amino alkoxy diaryl thiophenyl methanes and their use as potential antimycobacterial agents. Novel diaryl thiophenyl methanes of formula I have been prepared.
The present invention provides novel diaryl thiophenyl methanes substituted with a secondary or tertiary amino alkoxy group and a process for the preparation of the said compounds of general formula I comprising formula la and lb useful in antimycobacterial activity wherein R1 is selected from an aryl group or thophene moiety wherein the aryl group is selected from a group consisting of substituted phenyl groups such as methoxy phenyl, thiomethoxy phenyl, phenyl, p-chlorophenyl, p-fluorophenyl; R2 is selected from a group consisting of aminoalkoxyl, alkyl/dialkyl aminoalkoxy, cyclic alkyl aminoalkoxy. R3 is selected from a group consisting of H, lower alkyl, lower alkoxy group such as methyl, ethyl, n-propyl, isopropyl, n-butyl, secondary butyl, tertiary butyl, n-amyl, n-hexyl, 2-ethyl butyl ; R4 is selected from a group consisting of H,OH,methyl etc.
(Formula Removed)
Background of the Invention
Tuberculosis is a growing international health concern; it is the leading infectious cause of death in the world today (Dolin, P.J. et al Bull. WHO 1994, 72, 213; Daffe, M. et al Adv. Microb. Physiol 1998, 39, 131). It is estimated that worldwide 100 million people are infected annually.
Approximately ten million develop the disease, with five million of these progressing to the infectious stage and ultimately three million dying. Even though improved methods of prevention, detection, diagnosis and modern treatment have greatly reduced the number of people getting infected and dying from it, the emergence of multi-drug-resistant (MDR) strains and the global human immunodeficiency virus (HIV) augments the risk of developing TB many fold. Resistance has been described for all first-line drugs (isoniazid, rifampin, pyrazinamide, ethambutol and streptomycin) and for several second-line and newer drugs (ethionamide, fluoroquinolones, macrolides, nitroimidazopyrans). Because MDR strains are the result of cumulative mutations, growth of Mycobacterium tuberculosis (MT) can successfully be controlled in the host by concomitant treatment with more than one drug. This has resulted in the development of new agents (Panda, G. et al Indian Journal of Chemistry, 2009, 48B, 1121-1127; Parai, M. K. et al Bioorganic & Medicinal Chemistry Letters, 2008, 18, 289-292) for the preparation of Disseminated Mycobacterium avium complex (DMAC) infection as well as combinations of both new and standard agents for its treatment. The search for more effective agents against Mycobacterium tuberculosis (MT) and Mycobacterium avium complex (MAC) is ongoing in an attempt to enhance survival and reduce morbidity, as proven by the high number of publications (Jing-Ping Lu et al J. Med. Chem., 2010, 53, 3, 1329-1337; Liqiang Chen et al J. Med. Chem., 2010, 53 (12), 4768-4778; Jiyoung A et al J. Med. Chem., 2009, 52 (17), 5485-5495; Maria-Teresa Gutierrez-Lugo et al J. Med. Chem., 2008, 51 (9), 2606-2612; Li Liu et al J. Med. Chem., 2010, 53 (7), 2882-2891 and references cited therein) and patents of new antituberculous drugs recently published. Preclinical data, such as in vitro measures of drug activity and pharmacokinetics, are used in the design of new treatment regimens. Assessment of pharmacodynamic activity from standard in vitro minimum inhibitory concentrations (MICs) alone is insufficient to predict in vivo potency. Achievable serum and tissue concentrations as well as pharmacokinetic characteristics must be considered.
Because of this, there is an urgent need for anti-TB drugs with improved properties such as enhanced activity against MDR strains, reduced toxicity, shortened duration of therapy, rapid mycobactericidal mechanism of action and the ability to penetrate host cells and exert anti-mycobacterial effects in the intracellular environment.
Following is the description of thiophene containing trisubstituted methanes having antimycobacterial activity.



PAPER
European Journal of Medicinal Chemistry (2015), 95, 357-368
http://www.sciencedirect.com/science/article/pii/S0223523415002032
Thiophene containing trisubstituted methanes [TRSMs] as identified lead against Mycobacterium tuberculosis
- a Medicinal and Process Chemistry Division, CSIR-Central Drug Research Institute, B.S. 10/1, Jankipuram Extension, Sitapur Road, Lucknow-226031, UP, India
- b Biochemistry Division, CSIR-Central Drug Research Institute, B.S. 10/1, Jankipuram Extension, Sitapur Road, Lucknow-226031, UP, India
Triarylmethanes (TRAMs) and thiophene containing trisubstituted methanes (TRSMs) have been reported by us, having potential against Mycobacterium tuberculosis andMycobacterium fortuitum strains, respectively. Further, extension through synthesis and biological evaluation of novel TRSMs resulted into an identified lead 36 (S006-830) [(diisopropyl-(2-{4-[(4-methoxy-phenyl)- thiophen-2-yl-methyl]-phenoxy}-ethyl)-amine)] with MIC: 1.33 mg/L, non-toxic against Vero C-1008 cell line with selectivity index >10,ex vivo efficacy equivalent to first line TB drugs-isoniazid (INH), rifampicin (RFM) and pyrazinamide (PZA) in the mouse and human macrophages, and lung CFU count of 2.2 × 107 (approximately 15 fold lesser than untreated mice, 31 × 107) with efficacies comparable to ethambutol (EMB) (1.27 × 107) and PZA (1.9 × 107). Further, S006-830 also showed potent bactericidal activity against multi-drug resistant and single-drug resistant clinical isolates of M. tuberculosis

.
PAPER
Synthetic Communications (2014), 44(23), 3408-3413
Abstract


Total Synthesis of an Experimental Antitubercular DrugDOI:
10.1080/00397911.2014.942745
Uma Reddy Paillaab, Veera Reddy Aravaa* & L. K. Ravindranathb
pages 3408-3413
http://www.tandfonline.com/doi/abs/10.1080/00397911.2014.942745
REFERENCES
http://www.ingentaconnect.com/content/ben/cpa/2015/00000011/00000001/art00008?crawler=true
S006-830 against H37RV, single, multi-drug resistant M. tuberculosis; CFU in the lungs with S006-830, EMB, PZA (European Journal of Medicinal Chemistry 2015, 95, 357-368, J Antimicrob Chemother. 2012; 67(5):1188-97, Bioorg Med Chem Lett, 2008, 18, 289-292)
| 1. DiaryloxyMethanoPhenanthrenes: A New Class of Antituberculosis Agents, G. Panda,Shagufta, Jitendra Kumar Mishra, Vinita Chaturvedi, Anil K. Srivastava, Manju, RanjanaSrivastava and Brahm S. Srivastava, 1178DEL2004 Filing date 24/06/04 | |
| 2. Thiophene containing Trisubstituted Methanes (TRSMs) as antitubercular agents, Gautam Panda, Maloy Kumar Parai, Priyanka Singh, Sudhir Sinha, Vinita Chaturvedi, Anil Gaikwad, PCT in process (685/DEL/2010) dt 20-2-2010 |
/////////S 006-830, CDRI 830, 1550975-42-2
c1c(ccc(c1)OC)C(c2ccc(cc2)OCCN(C(C)C)C(C)C)c3sccc3
7th Annual Clinical Trials Summit 2016, 24th May 2016, The Lalit Hotel, Mumbai, India


successfully conducting clinical trials”
Insight,
and your colleagues to be a sponsor/ delegate for our upcoming “7th
Annual Clinical Trials Summit 2016” The conference will Be held on 14th
May 2016, The Lalit Hotel, Mumbai, India.
Development, Cadila, Sanofi Aventis, Johnson & Johnson, GNH India, Clintech
India, Boehringer Ingelheim, Reliance Life Sciences, Abbott, Glenmark
Pharmaceuticals, Sanofi, Nishith Desai Associates, Novartis, Tata Consultancy
Services, Janssen India (Pharmaceutical companies of Johnson & Johnson),
SIRO Clinpharm, and few more..
Standard
Price:- 1 or 2 Delegates – (INR 7,000 + Tax (14.5%) per delegate)
Group
Discounts – 3 or 4 Delegates – (INR 6,500 + Tax (14.5%) per delegate)
Group
Discounts – 5 and above Delegates – (INR 5,500 + Tax (14.5%) per delegate)
Conference
Sponsor & Exhibition Stall – Should you wish to Sponsor, or purchase a
Exhibition Stall (Booth) or a paid Speaker Slot, you can simply email your
interest and queries to TEL: + 91
9171350244
or deepak@virtueinsight.co.in, deepakrajvirtueinsight@gmail.com
consideration. I look forward to hearing from you.
friends or colleagues by forwarding this email to anyone you think may benefit
from it.

trials”“A critical guide for successfully conducting clinical trials”
APIs from Legitimate and Reliable Sources
DRUG REGULATORY AFFAIRS INTERNATIONAL

APIs from Legitimate and Reliable Sources
1. Introduction
Counterfeit and sub-standard APIs are increasingly present. Not only are they a fact of non-compliance but also they form a serious and increasing risk for patient safety. Various initiatives have been taken such as the founding of the FDA Counterfeit Drug Task Force, the European Commission’s current “Public consultation in preparation of a legal proposal to combat counterfeit medicines for human use” and the WHO Program “IMPACT” (International Medical Products Anti-Counterfeiting Taskforce).
API =Active pharmaceutical ingredient (synonym: drug substance)
Counterfeit API =Active pharmaceutical ingredient for which source and/or quality are falsely represented on the label, on the certificate of analysis or otherwise
Rogue API =API that is counterfeit or severely, deliberately non-compliant.

This writeup focuses on the interaction between the API manufacturer and the medicinal product manufacturer and provides possible measures that may be taken by both partners in order to ensure only…
View original post 2,586 more words
USP publishes draft of a new general chapter for plastic components used in manufacturing
DRUG REGULATORY AFFAIRS INTERNATIONAL
In the Pharmacopoeial Forum (PF) 42(3) (May-June 2016) the USP General Chapters – Packaging and Distribution Expert Committee proposes a new general chapter <661.3> Plastic Components and Systems Used in Pharmaceutical Manufacturing and a revised version of general chapter <1661> Evaluation of Plastic Packaging and Manufacturing Systems and Their Materials of construction with Respect to Their User Safety Impact. Read more about USPs Proposal on Plastic Components and Systems Used in Pharmaceutical Manufacturing.
<1661> Evaluation of Plastic Packaging and Manufacturing Systems and Their Materials of construction with Respect to Their User Safety Impact. Read more about USPs Proposal on Plastic Components and Systems Used in Pharmaceutical Manufacturing.
see
In the Pharmacopoeial Forum (PF) 42(3) (May-June 2016) the USP General Chapters – Packaging and Distribution Expert Committee proposes a new chapter to address the qualification of plastic components used in the manufacture of APIs (pharmaceutical and…
View original post 754 more words
EMA’s new Draft Guideline on the Sterilisation of Medicinal Products, APIs, Excipients and Primary Containers
DRUG REGULATORY AFFAIRS INTERNATIONAL

For medicinal products administrated in sterile form, the process to reduce the microbial level is a critical manufacturing step with regard to quality. The EMA has recently published the draft of a guideline on that topic which contains a range of clarifications. Read more about the coming requirements on sterilisation of medicinal products, APIs, excipients and final containers
see
As referred to in the European Pharmacopoeia, the procedure for terminal sterilisation of a medicinal product, an API, or an excipient is generally the method of choice. Yet, this might be difficult in many cases for product stability reasons. That’s why other microbial reduction processes can be used like sterilising filtration or aseptic processing. So far, there has been some uncertainty about these methods and their acceptance in a marketing authorisation procedure or a variation application, and about which data have to be submitted.
EMA’s new draft guideline entitled “Guideline…
View original post 781 more words
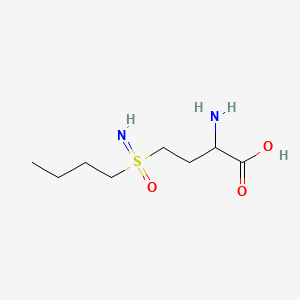
Buthionine Sulphoximine

Buthionine Sulphoximine
NDA Filed in china
A gamma-glutamylcysteine synthetase inhibitor potentially for the treatment of solid tumors.
![]()
NSC-326231; BSO
CAS No. 5072-26-4
BUTHIONINE SULFOXIMINE; DL-Buthionine-[S,R]-sulfoximine; 5072-26-4; Buthionine sulfoxamine; Buthionine-S,R-sulfoximine; Buthione sulfoximine;
| Molecular Formula: | C8H18N2O3S |
|---|---|
| Molecular Weight: | 222.30512 g/mol |
Buthionine sulfoximine (BSO) is a sulfoximine which reduces levels of glutathione and is being investigated as an adjunct withchemotherapy in the treatment of cancer.[1] The compound inhibits gamma-glutamylcysteine synthetase, the enzyme required in the first step of glutathione synthesis. Buthionine sulfoximine may also be used to increase the sensitivity of parasites to oxidativeantiparasitic drugs.[2]
Buthionine sulphoximine is an oncolytic agent in early clinical development at the National Cancer Institute (NCI) for the treatment of neuroblastoma in pediatric patients in combination with melphalan and bone marrow or peripheral stem cell transplantation.
DATA

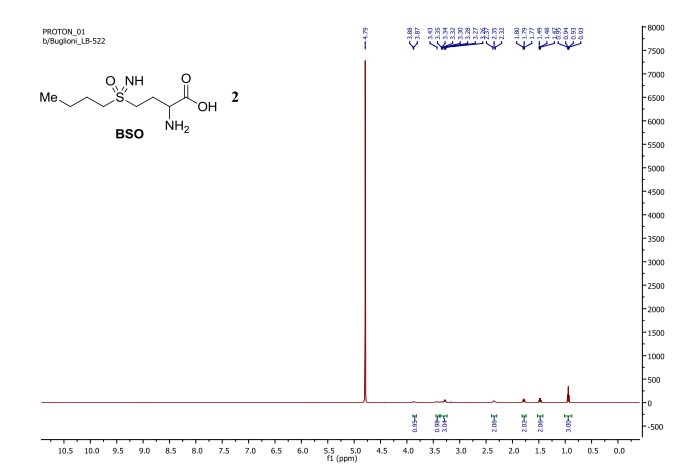
1H NMR
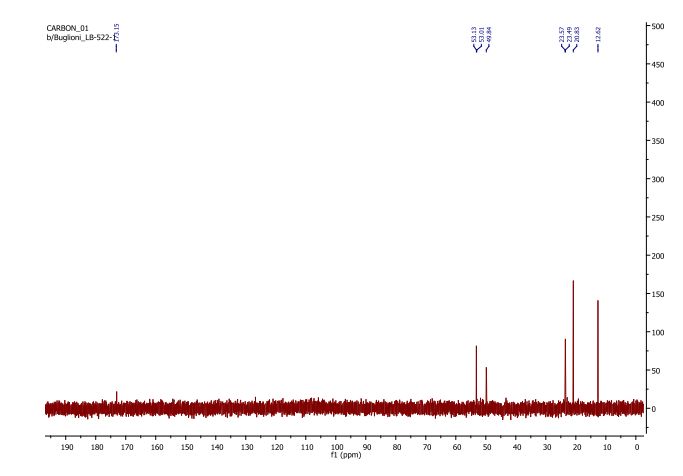
13C NMR
Synthesis
Methionine and buthionine sulfoximines: Syntheses under mild and safe imidation/oxidation conditions
Advanced Synthesis&Catalysis (2014), 356, (10), 2209-2213
Abstract

Methionine and buthionine sulfoximines (MSO and BSO) are non-natural amino acids known to inhibit the biosynthesis of glutathione (GSH). The current syntheses of these biologically active molecules involve harsh reaction conditions and the use of hazardous reagents for the sulfur imidation. Here, improved syntheses of MSO and BSO are presented including safe and mild one-pot imidation/oxidation sequences and single-step deprotections of three different functionalities.
Methionine and Buthionine Sulfoximines: Syntheses under Mild and Safe Imidation/Oxidation Conditions
DOI: 10.1002/adsc.201400354
http://onlinelibrary.wiley.com/doi/10.1002/adsc.201400354/abstract
References
- Defty, CL; Marsden, JR (2012). “Melphalan in regional chemotherapy for locally recurrent metastatic melanoma.”. Current topics in medicinal chemistry 12 (1): 53–60. PMID 22196271.
- “Definition of buthionine sulfoximine – National Cancer Institute Drug Dictionary”.
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
2-amino-4-(butylsulfonimidoyl)butanoic acid
|
|
| Other names
BSO
|
|
| Identifiers | |
| 5072-26-4 |
|
| ChEBI | CHEBI:28714 |
| ChemSpider | 19896 |
| Jmol 3D model | Interactive image |
| MeSH | Buthionine+sulfoximine |
| PubChem | 21157 |
| Properties | |
| C8H18N2O3S | |
| Molar mass | 222.305 g/mol |
| Density | 1.29 g/mL |
| Melting point | 215 °C (419 °F; 488 K) |
| Boiling point | 382.3 °C (720.1 °F; 655.5 K) |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
////NSC-326231, BSO, 5072-26-4, Butionine sulfoximine, Neuroblastoma
CCCCS(=N)(=O)CCC(C(=O)O)N
Processes for Constructing Homogeneous Antibody Drug Conjugates

Antibody drug conjugates (ADCs) are synthesized by conjugating a cytotoxic drug or “payload” to a monoclonal antibody. The payloads are conjugated using amino or sulfhydryl specific linkers that react with lysines or cysteines on the antibody surface. A typical antibody contains over 60 lysines and up to 12 cysteines as potential conjugation sites. The desired DAR (drugs/antibody ratio) depends on a number of different factors and ranges from two to eight drugs/antibody. The discrepancy between the number of potential conjugation sites and the desired DAR, combined with use of conventional conjugation methods that are not site-specific, results in heterogeneous ADCs that vary in both DAR and conjugation sites. Heterogeneous ADCs contain significant fractions with suboptimal DARs that are known to possess undesired pharmacological properties. As a result, new methods for synthesizing homogeneous ADCs have been developed in order to increase their potential as therapeutic agents. This article will review recently reported processes for preparing ADCs with improved homogeneity. The advantages and potential limitations of each process are discussed, with emphasis on efficiency, quality, and in vivo efficacy relative to similar heterogeneous ADCs.
| ADC | Sponsor | Indications | Status | Payload | Linked to | Target |
|---|---|---|---|---|---|---|
| Adcetris | Seattle Genetics | HL and ALCL | approved | MMAE | cysteine | CD30 |
| Kadcyla | Genentech/Roche | breast cancer | approved | DM1 | lysine | Her2 |
| inotuzumab ozogamicin | Pfizer | NHL and ALL | Phase III | calicheamicin | lysine | CD22 |
| lorvotuzumab mertansine | Immunogen | SCLC | Phase II | DM1 | lysine | CD56 |
| glembatumumab vedotin | Celldex | BC, melanoma | Phase II | MMAE | cysteine | GPNMB |
| PSMA-ADC | Progenics | prostate | Phase II | MMAE | cysteine | FOLH1 |
| SAR-3419 | Sanofi | DLBCL, ALL | Phase II | DM4 | lysine | CD19 |
| ABT-414 | Abbvie | glioblastoma | Phase II | MMAE | cysteine | EGFR |
| BT-062 | Biotest | mult. myeloma | Phase II | DM4 | lysine | CD138 |
| HLL1-Dox | Immunomedics | CLL, MM, NHL | Phase II | doxorubicin | cysteine | CD74 |
| Immu-130 | Immunomedics | CRC | Phase II | SN-38 | cysteine | CEACAM5 |
| Immu-132 | Immunomedics | solid tumors | Phase II | SN-38 | cysteine | EGP1 |
| SYD985 | Synthon | breast cancer | Phase II | duocarmycin | cysteine | Her2 |
| SAR-3419 | Sanofi | DLBCL, ALL | Phase II | DM4 | lysine | CD19 |
| IMGN853 | ImmunoGen | solid tumors | Phase I | DM4 | lysine | FOLR1 |
| IMGN529 | ImmunoGen | BCL,CLL, NHL | Phase I | DM1 | lysine | CD37 |
| ASG-22M6E | Astellas | solid tumors | Phase I | MMAE | cysteine | nectin-4 |
| AGS-16M8F | Astellas | RCC | Phase I | MMAF | cysteine | AGS16 |
| AMG 172 | Amgen | RCC | Phase I | DM1 | lysine | CD27L |
| AMG 595 | Amgen | glioblastoma | Phase I | DM1 | lysine | EGFR8 |
| BAY94-9343 | Bayer | solid tumors | Phase I | DM4 | lysine | mesothelin |
a
Source: www.clinicaltrials.gov.
Processes for Constructing Homogeneous Antibody Drug Conjugates
//////Processes, Constructing, Homogeneous, Antibody Drug Conjugates
EMA publishes finalised Process Validation Guideline for Biotech Products
DRUG REGULATORY AFFAIRS INTERNATIONAL
.jpg?n=3254)
Approximately two years ago the EMA published a draft guideline on process validation for the manufacture of biotech products. Now the final guideline has been published under the title “Guideline on process validation for the manufacture of biotechnology-derived active substances and data to be provided in the regulatory submission“.
READ
Approximately two years ago the EMA published a draft guideline on process validation for the manufacture of biotech products. Now the final guideline has been published under the title “Guideline on process validation for the manufacture of biotechnology-derived active substances and data to be provided in the regulatory submission”.
The scope of the guideline is to provide guidance on the data to be included in a regulatory submission to demonstrate that the active substance manufacturing process is in a validated state. The guideline focuses on recombinant proteins and polypeptides, their derivates, and products of which they are components (e.g…
View original post 300 more words
Quality Documentation of API mix in the Marketing Authorisation Procedure
DRUG REGULATORY AFFAIRS INTERNATIONAL
For different reasons, the manufacture of APIs may sometimes require adding excipients. In the context of an authorisation procedure, this practice reveals to be problematic. Read more here about the data required for the quality documentation of a API mix in an ASMF or a CEP.
The manufacture of APIs sometimes requires adding of one or several excipients like for example an antioxidant or an inert matrix for stabilisation purposes. Occasionally, corresponding mixtures can be manufactured to optimize workability for further processing or filling (e.g. improvement of flowability). Yet, within a marketing authorisation procedure, such an API mix can possibly be accepted differently than the pure API.
To clarify the questions around this topic, EMA’s QWP has published a document entitled “Quality Working Party questions and answers on API mix“. Please find hereinafter a summary of the questions addressed in the document:
What is an API mix?
View original post 476 more words