
Home » Uncategorized (Page 63)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pidotimod, 匹多莫德 , пидотимод , بيدوتيمود ,
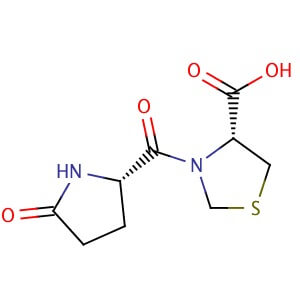

Pidotimod
H-Pyr-Thz-OH
(4R)-3-[(2S)-5-oxopyrrolidine-2-carbonyl]-1,3-thiazolidine-4-carboxylic acid
CAS 121808-62-6
Thymodolic acid, Pidotimod, Timodolic acid, PGT/1A, Axil, Onaka, Pigitil, Polimod
(4R)-3-(5-oxo-L-prolyl)-l ,3-thiazolidine-4-carboxylic acid, ITI 231723.
3-(L-pyroglutamyl)-L-thiazolidine-4-carboxylic acid
- 4-Thiazolidinecarboxylic acid, 3-[(5-oxo-2-pyrrolidinyl)carbonyl]-, [R-(R*,S*)]-
- (4R)-3-[[(2S)-5-Oxo-2-pyrrolidinyl]carbonyl]-4-thiazolidinecarboxylic acid
- Adimod
- Axil (pharmaceutical)
- Pigitil
| Pidotimod; 121808-62-6; (R)-3-((S)-5-Oxopyrrolidine-2-carbonyl)thiazolidine-4-carboxylic acid; Pidotomod; PGT/1A; Pidotimod [INN]; | |
| Molecular Formula: | C9H12N2O4S |
|---|---|
| Molecular Weight: | 244.26758 g/mol |
Stefano Poli, Corona Lucio Del
Pidotimod is an immunostimulant.[1]
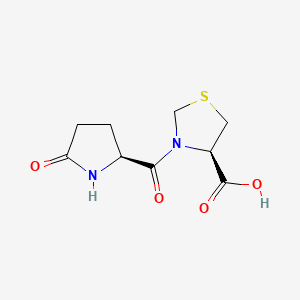

Pidotimod, whose chemical name is (4R)-3-(5-oxo-L-prolyl)-l ,3-thiazolidine-4-carboxylic acid, was first disclosed in ITI 231723. It is a synthetic peptide-like molecule provided with an in vitro and in vivo immunomodulating action (Giagulli et al., International Immunopharmacology, 9, 2009, 1366-1373). The immune system assists in maintaining a homeostatic balance between the human body and all foreign substances. An abnormality in this balance may cause a defective or aberrant response towards non-self substances, as well as loss of tolerance toward self-antigens, in such cases, the immune system imbalance exhibits clinically as signs of disease.
Pidotimod has been shown to induce dendritic cell maturation and up-regulate the expression of HLA-DR and co-stimulatory molecules CD83 and CD86, which are integral to communication with adaptive immunity cells. Pidotimod has also been shown to stimulate dendritic cells to release pro-inflammatory molecules such as MCP-1 and TNF-a cytokines, and to inhibit thymocyte apoptosis caused by a variety of apoptosis-inducing molecules. Pidotimod exerts a protective action against infectious processes, although not through direct antimicrobial or antiviral action. Rather, pidotimod stimulates both innate and acquired immunity by enhancing humoral and cell-mediated immunity mechanisms.
Pidotimod, which may be administered as solid or liquid forms, for example, via an oral route, has been shown to increase natural resistance to viral or bacterial infections in animal models. Efficacy demonstrated in patients includes respiratory, urinary and genital infections, in particular recurrent respiratory infections in pediatric patients, respiratory infections in asthmatic patients and chronic obstructive pulmonary disease in adults and elderly patients.
Besides exhibiting activity to illnesses characterized by immune defects, pidotimod has been reported to be of benefit in to patients with other kinds of diseases, not directly related to immune defects, including gastroenterology diseases such as ulcerative colitis and irritable bowel syndrome, and dermatological diseases such as psoriasis and atopic dermatitis where symptoms relating to these diseases have been attenuated. In gastroenterology diseases pidotimod may be administered either by oral or by rectal route. Oral route or topical application, for example in creams or gels containing pidotimod, may be used to treat dermal conditions.
Further use of pidotimod includes treatment of inflammatory diseases, in particular those characterized by an aberrant activation of the non-canonical NF-kB pathway. Diseases implicated by such activation include allergic diseases, autoimmune diseases, and numerous other inflammatory diseases. Allergic diseases include allergic rhinitis, allergic conjunctivitis, contact dermatitis, eczema and allergic vasculitis.
Autoimmune diseases include alopecia areata, ankylosing spondylitis, autoimmune cardiomyopathy, autoimmune connective tissue diseases, autoimmune enteropathy, autoimmune hepatitis, autoimmune peripheral neuropathy, autoimmune pancreatitis, autoimmune polyendocrine syndrome, autoimmune thrombocytopenic purpura, autoimmune urticaria, autoimmune uveitis, celiac disease, chronic fatigue syndrome, cystic fibrosis, hashimoto’s thyroiditis, idiopathic pulmonary fibrosis, idiopathic thrombocytopenic purpura, IGA nephropathy, juvenile idiopathic arthritis for juvenile rheumatoid arthritis, or Still’s disease) Kawasaki’s disease, lichen planus, lupus erythematosus, rheumatoid arthritis, rheumatic fever, Sj5gren’s syndrome, spondyloarthropathy, temporal arteritis (or giant cell arteritis), urticarial vasculitis, and vitiligo.
Other inflammatory diseases include Alzheimer’s disease, atherosclerosis, chronic liver diseases, chronic nephropathy, gastritis, glomerulonephritis, hydradenitis suppurativa, hypogammaglobulinemia, interstitial cystitis, lichen sclerosus, liver steatosis, metabolic syndrome, obesity, Parkinson’s disease, pemphigus vulgaris, post-ischemic inflammation, raynaud phenomenon, restless leg syndrome, retroperitoneal fibrosis, and thrombocytopenia.

PATENT
Synthesis pidotimod
A method for producing pidotimod, characterized in that: comprising the steps of: a) L- thiazolidine-4-carboxylic acid: L- cysteine formaldehyde solution was added dropwise, stirred at room temperature, filtered to give L- thiazolidine-4-carboxylic acid; (2) metal ion load type cation exchange resin preparation: strongly acidic with hydrochloric acid cation exchange resin is converted to the hydrogen form, the hydrogen form strong acid cation exchange resin was added a solution of a metal ion compound In, 40 ~ 80 ° C for 1 to 6 hours, cooled to room temperature, and dried to obtain a supported metal ion cation exchange resin; (3) Synthesis of pidotimod: the step (1) of L- thiazolidine – 4- carboxylic acid, in step (2) of the load as a catalyst metal ion type cation exchange resin, L- pyroglutamic acid and N, N- dimethylformamide mixed, 40 ~ 80 ° C for 1 to 4 hours, filtered to give a white solid, the white solid was acidified with hydrochloric acid, to give the finished pidotimod.

In four flask IOg L- thiazolidine-4-carboxylic acid, 11. 3g g L- pyroglutamic acid, 320mL N, N- dimethylformamide, 12g modified resin, 70 ° C the reaction 2 hours. Filtration, the reaction mixture by rotary evaporation, after removal of part of the solvent, placed in an ice bath to cool, the precipitated solid was suction filtered to give a white solid, this white solid was acidified with 37% hydrochloric acid, was allowed to stand at KTC, crystallization, filtration, a white product 14. 4g, a yield of 78.3%. Measured melting point 192 ~ 194 ° C, [a] 25D = – 150 ° (literature values mp: 192 ~ 194 ° C, [a] 25D = – 150 °).The whole preparation reaction pidotimod total yield of 64%. By HPLC, pidotimod content of 98.5%.
PAPER
http://europepmc.org/abstract/med/19604731
10.1016/j.jchromb.2009.06.038
PATENT
Example 14 – Preparation of Pidotimod
Pidotimod was prepared following Example 1 of EP0422566 Al .
PATENT
WO2015036009,
https://www.google.com/patents/WO2015036009A1?cl=en
PATENT
EP276752,
PATENT
http://google.com/patents/EP0422566B1?cl=en
EXAMPLE 1
A solution of 16.78 g (0.084 mole) of ethyl L-thiazolidine-4-carboxylate hydrochloride in 33 ml of water is treated with 16.78 g of potassium carbonate and extracted with 40 ml of ethyl acetate. The organic phase is dried over sodium sulfate, filtered and diluted to 85 ml with ethyl acetate. The solution is stirred and cooled to 0-5°C, then 19.2 g (0.093 mole) of dicyclohexylcarbodiimide dissolved in 20 ml of ethyl acetate and 12 g (0.093 mole) of L-pyroglutamic acid are added thereto. The reaction mixture is stirred for 1 hour at 0-5°C, then 12 hours at room temperature, dicyclohexylurea is filtered, the filtrate is evaporated under vacuum and the oily residue, consisting in ethyl 3-(L-pyroglutamyl)-L-thiazolidine-4-carboxylate is taken up into 25 ml of water. 3.73 g of sodium hydroxide dissolved in 13.3 ml of water are dropped into the resulting solution. After 30 minutes, the reaction mixture is acidified with concentrated hydrochloric acid at 0-5°C, kept for 2 hours at 5°C, then filtered washing with little cool water and dried to obtain 17.8 g (87.6%) of 3-(L-pyroglutamyl)-L-thiazolidine-4-carboxylic acid, m.p. 193-194°C.
EXAMPLE 2
23 g (0.1 mol) of L-N-t-butoxycarbonylpyroglutamic acid (E. Schröder and E. Klinger, Ann. Chem., 673, 1964, 202) and 16.1 g (0.1 mol) of ethyl L-thiazolidine-4-carboxylate are dissolved in 150 ml of THF, to the solution stirred at 0-5°C, 21 g (0.105 mol) of dicyclohexylcarbodiimide are added and the slurry is stirred for 15 hours at room temperature. The dicyclohexylurea is filtered, the wear filtrate is evaporated u.v. and the oily residue is kept in 40 ml of water. In the solution 6.6 g of potassium hydroxyde in a little water are dropped in 30′ at 15-20°C, the pH is adjusted to 2 with hydrochloric acid at 0-5°C and after 2 hours the precipitated L-pyroglutamyl-L-thiazolidine-4-carboxylic acid is filtered and dried, giving 88%, mp. 193-4°.
CLIP
Drugs Fut 1991,16(12),1096
Liebigs Ann Chem 1964,673

The synthesis of pidotimod has been carried out using N-tert-butoxycarbonyl-L-pyroglutamic acid as starting material, in order to avoid the formation of diketopiperazine derivatives. L-Glutamic acid (I) was condensed with di-tert-butyl dicarbonate by means of triethylamine in DMF to give N-(tert-butoxycarbonyl)-L-glutamic acid (II), which is dissolved in THF and treated with dicyclohexylcarbodiimide (DCC) to obtain N-(tert-butoxycarbonyl)-L-glutamic anhydride (III). The treatment of anhydride (III) with dicyclohexylamine in THF-ethyl ether affords the dicyclohexylamine salt of N-(tert-butoxycarbonyl)-L-pyroglutamic acid (IV), which by acidification with aqueous citric acid yields the corresponding free acid (V). The condensation of equimolecular amounts of N-(tert-butoxycarbonyl)-L-pyroglutamic acid (V) with L-thiazolidine-4-carboxylic acid ethyl ester (VIII) by means of DCC in methylene chloride gives the coupled ester (IX), which is hydrolyzed with aqueous NaOH, and the corresponding sodium salt acidified to yield the N-tert-butoxycarbonyl derivative (X). Finally, this compound is deprotected with trifluoroacetic acid to obtain crystalline pidotimod (XI). The intermediate thiazolidine (VIII) has been obtained as follows: Esterification of L-thiazolidine-4-carboxylic acid (VI) with ethanol by means of SOCl2 gives the corresponding ethyl ester hydrochloride (VII), which by treatment with K2CO3 in water yields the free ester (VIII).
CLIP
Arzneim-Forsch Drug Res 1994,44(12a),1402

Two new related routes for the synthesis of pidotimod have been reported: 1) The condensation of L-pyroglutamic acid (I) with L-thiazolidine-4-carboxylic acid ethyl ester (II) by means of dicyclohexylcarbodiimide (DCC) in methylene chloride gives the corresponding dipeptide ethyl ester (III), which is saponified with aqueous 1N NaOH. 2) By condensation of the activated ester L-pyroglutamic acid pentachlorophenyl ester (IV) with L-thiazolidine-4-carboxylic acid (V) by means of triethylamine in DMF.
PATENT
Novel crystalline, amorphous and solid forms of di-pidotimod benzathine (designated as Forms M and H), their hydrates, processes for their preparation and compositions comprising them are claimed. Also claimed is their use for treating viral or bacterial infections, respiratory, urinary and/or genital infections, ulcerative colitis, irritable bowel syndrome, psoriasis and atopic dermatitis
Example 14 – Preparation of Pidotimod
Pidotimod was prepared following Example 1 of EP0422566 Al .
NMR
Figure 17 is a Ή solution-state NMR spectrum of Form H
SEE
CN 104447947
Indian Pat. Appl. (2014), IN 2013MU00181 A
WO 2014111957
CN 103897025
| CN1557303A * | Jan 16, 2004 | Dec 29, 2004 | 太阳石(唐山)药业有限公司 | Use of Pidotimod in preparation of hepatitis B treating medicine |
| EP0382180A2 * | Feb 7, 1990 | Aug 16, 1990 | POLI INDUSTRIA CHIMICA S.p.A. | Derivatives of thiazolidine-4-carboxylic acid, its preparation and pharmaceutical compositions containing it |
| IT1231723B | Title not available |
| Reference | ||
|---|---|---|
| 1 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; DUAN, RUOZHU ET AL: “Application and prospects of immunostimulants“, XP002722997, retrieved from STN Database accession no. 2006:478774 |
| 2 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LI, YIPING ET AL: “Effects of pidotimod on immune function of patients with chronic hepatitis C“, XP002722996, retrieved from STN Database accession no. 2007:598452 |
| 3 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; WU, RONGRONG ET AL: “Application of immunomodulatory drugs in treatment of chronic hepatitis B“, XP002722995, retrieved from STN Database accession no. 2010:125278 |
| 4 | * | DATABASE MEDLINE [Online] US NATIONAL LIBRARY OF MEDICINE (NLM), BETHESDA, MD, US; March 2002 (2002-03), VARGAS CORREA JORGE B ET AL: “[Pidotimod in recurring respiratory infection in children with allergic rhinitis, asthma, or both conditions].“, XP002722994, Database accession no. NLM12092522 & VARGAS CORREA JORGE B ET AL: REVISTA ALERGIA MEXICO (TECAMACHALCO, PUEBLA, MEXICO : 1993) 2002 MAR-APR, vol. 49, no. 2, March 2002 (2002-03), pages 27-32, XP8168769, ISSN: 0002-5151 |
| 5 | * | GOURGIOTIS DIMITRIOS ET AL: “Immune modulator pidotimod decreases the in vitro expression of CD30 in peripheral blood mononuclear cells of atopic asthmatic and normal children“, JOURNAL OF ASTHMA, ASTHMA PUBLICATIONS SOCIETY, OSSINING, NY, US, vol. 41, no. 3, 1 January 2004 (2004-01-01), pages 285-287, XP008164025, ISSN: 0277-0903, DOI: 10.1081/JAS-120026085 |
| 6 | * | XIN JIN ET AL: “Sublingual Surprise: A New Variant of Oral Lichen Planus“, THE AMERICAN JOURNAL OF MEDICINE, vol. 127, no. 1, 1 January 2014 (2014-01-01), pages 28-30, XP055112640, ISSN: 0002-9343, DOI: 10.1016/j.amjmed.2013.10.002 |
References
- Du XF, Jiang CZ, Wu CF, Won EK, Choung SY (September 2008). “Synergistic immunostimulating activity of pidotimod and red ginseng acidic polysaccharide against cyclophosphamide-induced immunosuppression”. Archives of pharmacal research 31 (9): 1153–9.doi:10.1007/s12272-001-1282-6. PMID 18806958.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(4R)-3-(5-oxo-L-prolyl)-1,3-thiazolidine-4-carboxylic acid
|
|
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Identifiers | |
| ATC code | L03AX05 (WHO) |
| PubChem | CID 65944 |
| ChemSpider | 59348 |
| UNII | 785363R681 |
| KEGG | D07261 |
| ChEMBL | CHEMBL1488165 |
| Synonyms | (4R)-3-[(2S)-5-oxopyrrolidine-2-carbonyl]-1,3-thiazolidine-4-carboxylic acid |
| Chemical data | |
| Formula | C9H12N2O4S |
| Molar mass | 244.26758 g/mol |
//////////////Pidotimod, Thymodolic acid, Pidotimod, Timodolic acid, PGT/1A, Axil, Onaka, Pigitil, Polimod, H-Pyr-Thz-OH, 121808-62-6, ITI 231723, peptide, QA-7522, SMR000466390, Thymodolic acid, Timodolic acid, UNII:785363R681, 匹多莫德 , пидотимод , بيدوتيمود ,
O=C(O)[C@H]2N(C(=O)[C@H]1NC(=O)CC1)CSC2
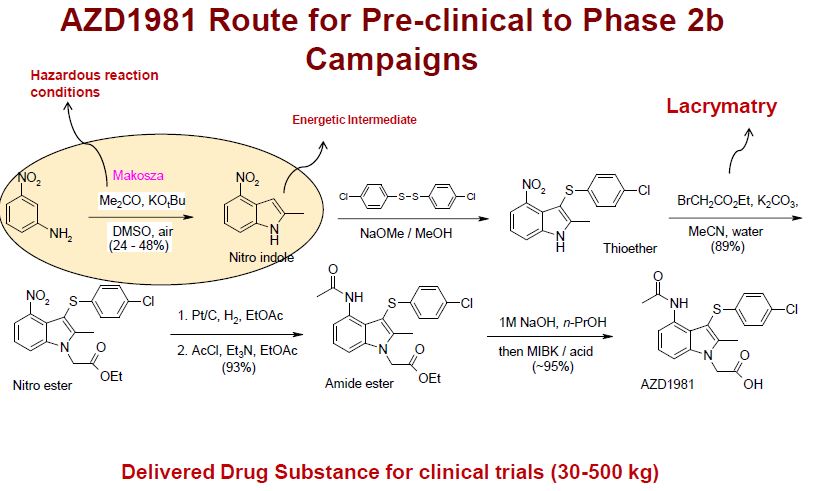
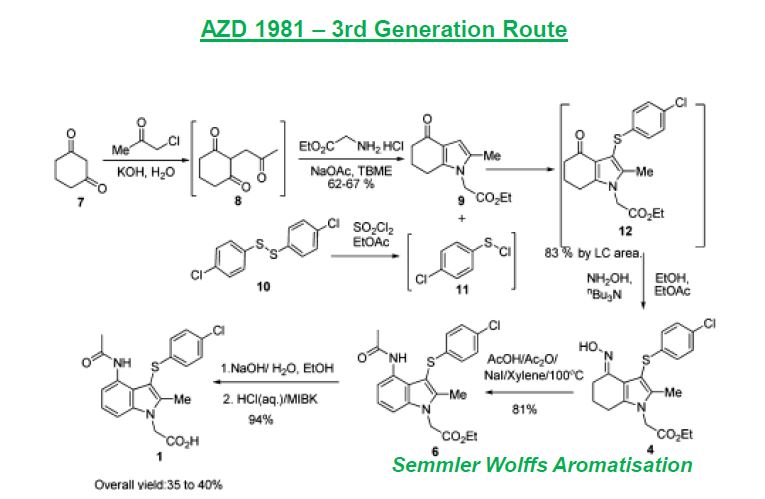
AZD 1981
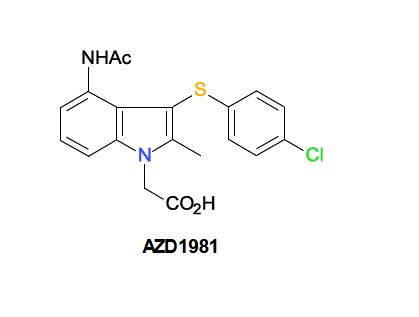
| AZD1981; AZD-1981; 802904-66-1; UNII-2AD53WQ2CX; ; AZD 1981; | |
| Molecular Formula: | C19H17ClN2O3S |
|---|---|
| Molecular Weight: | 388.86788 g/mol |
- 1H-Indole-1-acetic acid, 4-(acetylamino)-3-[(4-chlorophenyl)thio]-2-methyl-
- 2-[4-acetamido-3-(4-chlorophenyl)sulfanyl-2-methylindol-1-yl]acetic acid
- Originator AstraZeneca
- Developer AstraZeneca; Johns Hopkins University
- Class Antiasthmatics
- Mechanism of Action Prostaglandin D2 receptor antagonists
-
- Phase II Urticaria
- Discontinued Asthma; Chronic obstructive pulmonary disease
Most Recent Events
- 09 Mar 2016 AZD 1981 is still in phase II trials for Urticaria in USA (PO)
- 07 Mar 2016 Johns Hopkins University in collaboration with AstraZeneca completes a phase II trial in Urticaria in USA (PO) (NCT02031679)
- 04 Mar 2016 Efficacy and safety data from a phase II trial in Urticaria presented at the Annual Meeting of the American Academy of Allergy, Asthma and Immunology (AAAAI-2016)
https://ncats.nih.gov/files/AZD1981.pdf
SEE
AZD1981 is a potent, selective CRTh2 (DP2) receptor antagonist with IC50 of 4 nM, showing >1000-fold selectivity over more than 340 other enzymes and receptors, including DP1. Phase 2.
118 patients were randomised to treatment (AZD1981 n = 61; placebo n = 57); 83% of patients were male and the mean age was 63 years (range 43-83). There were no significant differences in the mean difference in change from baseline to end of treatment between AZD1981 and placebo for the co-primary endpoints of pre-bronchodilator FEV1 (AZD1981-placebo: -0.015, 95% CI: -0.10 to 0.070; p = 0.72) and CCQ total score (difference: 0.042, 95% CI: -0.21 to 0.30; p = 0.75). Similarly, no differences were observed between treatments for the other outcomes of lung function, COPD symptom score, 6-MWT, BODE index, and use of reliever medication. AZD1981 was well tolerated.
CONCLUSION:
There was no beneficial clinical effect of AZD1981, at a dose of 1000 mg twice daily for 4 weeks, in patients with moderate to severe COPD. AZD1981 was well tolerated and no safety concerns were identified.

Biological Activity
| Description | AZD1981 is a potent, selective CRTh2 (DP2) receptor antagonist with IC50 of 4 nM, showing >1000-fold selectivity over more than 340 other enzymes and receptors, including DP1. Phase 2. | |||||
|---|---|---|---|---|---|---|
| Targets | CRTh2 (DP2) receptor [1] | |||||
| IC50 | 4 nM | |||||
| In vitro | AZD1981, as a potent antagonist in a disease relevant cell system, inhibits DK-PGD2-induced CD11b expression in human eosinophils with IC50 of 10 nM. [1] AZD1981 blocks DP2-mediated shape change in human eosinophils and basophils in blood, as well as DP2-mediated chemotaxis of human Th2 cells and eosinophils. Moreover, AZD1981 also blocks the binding of [3H]PGD2 to mouse, rat, guinea pig, rabbit and dog recombinant DP2. [2] | |||||
| In vivo | AZD1981 has high oral bioavailability in male sprague dawley rats. [1] In guinea pig hind limb model, AZD1981 (100 nM) completely inhibits DK-PGD2-induced eosinophil mobilization. [2] | |||||
| Features | An orally available selective DP2(CRTh2) receptor antagonist in clinical development for asthma. | |||||
Protocol(Only for Reference)
Kinase Assay: [2]
| DP2 binding studies | A scintillation proximity assay (SPA) following [3H]PGD2 binding to membranes of HEK cells expressing recombinant DP2 is used. The potency of AZD1981 as an antagonist is determined by quantifying its ability to displace specific radio-ligand binding. Briefly, membranes from HEK293 expressing recombinant human DP2 are pre-bound to Wheat Germ Agglutinin-coated PVT-SPA beads for 18 h at 4°C. Assays were started by the addition of 25 μL of membrane-coated beads (10 mg/mL of beads) to an assay buffer (50 mm HEPES pH 7.4 containing 5 mm MgCl2) containing 2.5 nM [3H]PGD2 in the absence or the presence of increasing concentrations of the tested compounds (50 μL final volume). Non-specific binding is determined in the same conditions but in the presence of 10 μM DK-PGD2. Plates are incubated for 2 h at room temperature, and bead-associated radioactivity is measured using a Wallac Microbeta counter. The concentration of the compounds causing 50% inhibition of binding of [3H]PGD2 to the receptor is calculated (IC50). Ki values have not been derived from IC50, as there is no evidence of a simple competitive interaction with PGD2. The same methodology is used for recombinant human, murine, rat, guinea pig, dog and rabbit DP2. Reversibility of binding to the human receptor was assessed by recovery of [3H]PGD2 binding after removal of AZD1981 by washing of the membrane-coated SPA beads. HEK-membrane-coated beads are incubated in the presence of AZD1981 for 2 h at room temperature to bind the compound to DP2. To remove the bound AZD1981, beads are centrifuged (1 min at 1300× g), and the pellet resuspended in 1 mL of assay buffer. This is repeated four times. Aliquots (30 μL) are transferred to 96-well plates, and [3H]PGD2 binding is evaluated as above. Parallel samples containing (i) 10 μM DK-PGD2 during the 2 h incubation and in the wash buffer; (ii) AZD1981 at 2 μM in the wash buffer; and (iii) vehicle are processed alongside to determine non-specific binding and the ‘no wash’ condition whilst controlling for loss of beads during the washing process. The time from first wash to end of first reading is approximately 13 min. |
|---|
Animal Study: [1]
| Animal Models | Male sprague dawley rats. |
|---|---|
| Formulation | |
| Dosages | 1 mg/kg(i.v.), 4 mg/kg(oral) |
| Administration | i.v. or oral administration |
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
| Rat dose (mg/kg) = mouse dose (22.4 mg/kg) × | mouse Km(3) | = 11.2 mg/kg |
| rat Km(6) |
References
[1] Luker T, et al. Bioorg Med Chem Lett. 2011, 21(21), 6288-6292.
[2] Schmidt JA, et al. Br J Pharmacol. 2013, 168(7), 1626-1638.
Clinical Trial Information( data from http://clinicaltrials.gov, updated on 2016-07-09)
| NCT Number | Recruitment | Conditions | Sponsor /Collaborators |
Start Date | Phases |
|---|---|---|---|---|---|
| NCT02031679 | Recruiting | Chronic Idiopathic Urticaria | Johns Hopkins University|AstraZeneca | January 2014 | Phase 2 |
| NCT01311635 | Completed | Healthy | AstraZeneca | April 2011 | Phase 1 |
| NCT01254461 | Completed | Drug Interaction | AstraZeneca | February 2011 | Phase 1 |
| NCT01265641 | Completed | Asthma | AstraZeneca | January 2011 | Phase 1 |
| NCT01199341 | Completed | Pharmakokinetic | AstraZeneca | October 2010 | Phase 1 |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015210655 | 2015-07-30 | CERTAIN (2S)-N-[(1S)-1-CYANO-2-PHENYLETHYL]-1,4-OXAZEPANE-2-CARBOXAMIDES AS DIPEPTIDYL PEPTIDASE 1 INHIBITORS |
| US2015072963 | 2015-03-12 | COMPOSITIONS AND METHODS FOR REGULATING HAIR GROWTH |
| US2014328861 | 2014-11-06 | Combination of CRTH2 Antagonist and a Proton Pump Inhibitor for the Treatment of Eosinophilic Esophagitis |
| US8772305 | 2014-07-08 | Substituted pyridinyl-pyrimidines and their use as medicaments |
| US8227622 | 2012-07-24 | Pharmaceutical Process and Intermediates 714 |
| US2012178764 | 2012-07-12 | Novel Compounds |
| US2011263614 | 2011-10-27 | Novel compounds |
| US7781598 | 2010-08-24 | Process for the preparation of substituted indoles |
| US7687535 | 2010-03-30 | Substituted 3-sulfur indoles |
| US2009163518 | 2009-06-25 | Novel Compounds |
///////////
CC1=C(C2=C(N1CC(=O)O)C=CC=C2NC(=O)C)SC3=CC=C(C=C3)Cl
AZD 3514 MALEATE
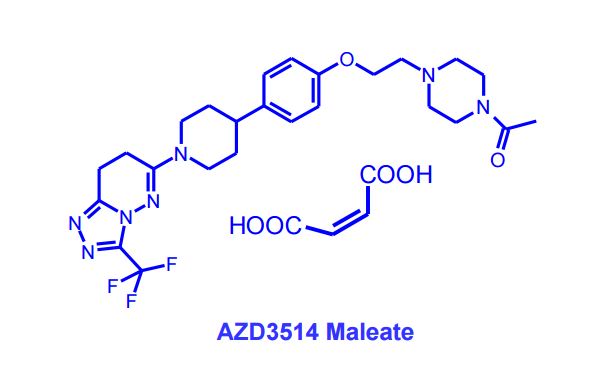
AZD3514; AZD 3514; AZD-3514.
CAS 1240299-33-5
Chemical Formula: C25H32F3N7O2
Exact Mass: 519.25696
1-(4-(2-(4-(1-(3-(trifluoromethyl)-7,8-dihydro-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)piperidin-4-yl)phenoxy)ethyl)piperazin-1-yl)ethanone
Ethanone, 1-[4-[2-[4-[1-[7,8-dihydro-3-(trifluoromethyl)-1,2,4-triazolo[4,3-b]pyridazin-6-yl]-4-piperidinyl]phenoxy]ethyl]-1-piperazinyl]
6-f4-{4-[2-f4-acetylpiperazin-l-yl)ethoxylphenyl}piperidin-l-yl)-3-( trifluoromethyr)-7,8-dihvdro [ 1 ,2,41 triazolo [4,3-bl pyridazine
6-(4-{4-[2-(4-acetylpiperazin-l- vDethoxyl phenyllpiperidin- l-vD-3-f trifluoromethyl)-7.,8-(iihv(iro [ 1 ,2,41 triazolo [4,3- blpyridazine
- 1-[4-[2-[4-[1-[7,8-Dihydro-3-(trifluoromethyl)-1,2,4-triazolo[4,3-b]pyridazin-6-yl]-4-piperidinyl]phenoxy]ethyl]-1-piperazinyl]ethanone
- Originator AstraZeneca
- Class Antineoplastics
- Mechanism of Action Androgen receptor antagonists
AZD-3514 is a potent androgen receptor downregulator with potential anticancer cancer activity. AZD3514 is being evaluated in a Phase I clinical trial in patients with castrate-resistant prostate cancer.
AZD3514 is currently in Phase I trail. This trial is looking at a new drug called AZD3514 for men who have prostate cancer that has spread to other parts of the body and is no longer responding to hormone therapy. Doctors often use hormone therapy to treat prostate cancer. This may keep it under control for long periods of time. But researchers are looking for treatments that will help men who have prostate cancer that stops responding to hormone therapy. Prostate cancer needs the hormone testosterone to grow. The testosterone locks into receptors on the cancer cells. AZD3514 works by breaking down these receptors so that testosterone canÂ’t tell the prostate cancer cells to grow.

6-(4-{4-[2-(4-Acetylpiperazin-1-yl)ethoxy]phenyl}piperidin-1-yl)-3-(trifluoromethyl)-7,8-ihydro[1,2,4]triazolo[4,3-b]pyridazine
as a white, free flowing solid.
1H NMR (400 MHz, CDCl3): δ 1.62 (2H, m), 1.88 (2H, m), 2.02 (3H, s), 2.49 (4H, m), 2.65 – 2.78 (5H, m), 2.94 (2H, m), 3.15 (2H, t), 3.42 (2H, m), 3.57 (2H, m), 4.03 (2H, t), 4.24 (2H, m), 6.80 (2H, d), 7.06 (2H, d);
m/z = 520 [M+H]+. RT = 0.87: 99% purity.
HRMS found 520.26373,
Prostate cancer is the second leading cause of death from cancer among men in developed countries, and was projected to account for 25% of newly-diagnosed cases and 9% of deaths due to cancer in the USA in 2010. The androgen receptor (AR), a ligand binding transcription factor in the nuclear hormone receptor super family, is a key molecular target in the etiology and progression of prostate cancer.Binding of the endogenous AR ligand dihydrotestosterone stabilizes and protects the AR from rapid proteolytic degradation. The early stages of prostate cancer tumor growth are androgen dependent and respond well to androgen ablation, either via surgical castration or by chemical castration with a luteinizing hormone releasing hormone agonist in combination with an AR antagonist, such as bicalutamide.
Although introduction of androgen deprivation therapy represented a major advance in prostate cancer treatment, recurrence within 1–2 years typically marks transition to the so-called castrate-resistant state, in which the tumor continues to grow in the presence of low circulating endogenous ligand and is no longer responsive to classical AR antagonists. Castrate-resistant prostate cancer (CRPC) is a largely unmet medical need with a 5-year survival rate of less than 15%. Antimitotic agents docetaxel and cabazitaxel, testosterone biosynthesis inhibitor abiraterone acetate and second generation AR antagonist enzalutamide (MDV3100) are the currently approved small-molecule drugs that have been shown to provide survival benefit.
Recent evidence from both pre-clinical and clinical studies is consistent with the importance of re-activation of AR signaling in a majority of castrate-resistant prostate tumors. It is also well established that the functional AR in castrate-resistant tumors is frequently mutated or amplified, and that over-expression can convert hormone-responsive cell lines to hormone refractory. Recent second-generation AR antagonists have been designed that retain antagonism in over-expressing cell lines, and among these agents enzalutamide has recently successfully met efficacy criteria in a large Phase III clinical trial.
By analogy with fulvestrant, an estrogen receptor (ER) downregulator approved by the FDA in 2002 for treatment of advanced breast cancer and initially characterized as a pure ER antagonist, a ligand which downregulates the AR represents one of a number of potential approaches to treatment of CRPC via a sustained reduction in tumor AR content. We recently described derivation from a novel 3-(trifluoromethyl)-[1,2,4]triazolo[4,3-b]pyridazine ligand of AR inhibitor 1 The compound also causes AR downregulation15 and high plasma levels following oral administration in pre-clinical models compensate for moderate cellular potency
Figure 1.

Structures of lead AR downregulator 1 and chemotype 2.
Scheme 3.

Synthesis of compounds 10, 11a–b, 12. Reagents and conditions: (a) 2-(1-Methyl-1H-pyrazol-5-yl)ethanol,27 Ph3P, diisopropyl azodicarboxylate, THF, 20 °C; (b) 2-(4-acetylpiperazine-1-yl)ethanol,28 Ph3P, diisopropyl azodicarboxylate, THF, 20 °C; (c) H2, 10% Pd-C, MeOH, 50 °C.
PATENT
| Robert Hugh Bradbury, Gregory Richard Carr,Alfred Arthur Rabow, Korupoju Srinivasa Rao,Harikrishna Tumma, | |
| Applicant | Astrazeneca Ab, Astrazeneca Uk Limited |
Preparation of 6-f4-{4-[2-f4-acetylpiperazin-l-yl)ethoxylphenyl}piperidin-l-yl)-3-
( trifluoromethyr)-7,8-dihvdro [ 1 ,2,41 triazolo [4,3-bl pyridazine

A solution of acetyl chloride (0.027 mL, 0.38 mmol) in DCM (0.5 mL) was added dropwise to 6-[4- [4- [2-(piperazin- 1 -yl)ethoxy]phenyl]piperidin- 1 -yl] -3 -(trifluoromethyl)- 7,8-dihydro-[l,2,4]triazolo[4,3-b]pyridazine (150 mg, 0.31 mmol) and triethylamine (0.088 mL, 0.63 mmol) in DCM (1 mL) cooled to 00C under nitrogen. The resulting solution was stirred at 00C for 5 minutes then allowed to warm to room temperature and stirred for 15 minutes. The reaction mixture was diluted with water (2 mL), passed through a phase separating cartridge and then the organic layer was evaporated to afford crude product. The crude product was purified by preparative HPLC (Waters XBridge Prep Cl 8 OBD column, 5μ silica, 19 mm diameter, 100 mm length), using decreasingly polar mixtures of water (containing 1% ammonia) and MeCN as eluents. Fractions containing the desired compound were evaporated to dryness to give 6-(4-{4-[2-(4-acetylpiperazin-l- yl)ethoxy]phenyl}piperidin-l-yl)-3-(trifluoromethyl)-7,8-dihydro[l,2,4]triazolo[4,3- b]pyridazine (80 mg, 49%) as a gum.
IH NMR (399.9 MHz, CDC13) δ 1.69 (2H, m), 1.95 (2H, m), 2.08 (3H, s), 2.56 (4H, m), 2.71 – 2.84 (5H, m), 3.00 (2H, m), 3.22 (2H, t), 3.48 (2H, m), 3.63 (2H, m), 4.10 (2H, t), 4.31 (2H, m), 6.86 (2H, d), 7.12 (2H, d); m/z = 520 [M+H]+.
The 6-[4-[4-[2-(piperazin- 1 -yl)ethoxy]phenyl]piperidin- 1 -yl]-3-(trifluoromethyl)-7,8- dihydro-[l,2,4]triazolo[4,3-b]pyridazine used as starting material was prepared as follows :-
Preparation of tert-butyl 4-[2-[4-(l-(benzyloxycarbonyl)-l,2,3,6-tetrahydropyridin-4- yl)phenoxy]ethyl]piperazine-l-carboxylate DIAD (12.60 mL, 64.00 mmol) was added dropwise to benzyl 4-(4-hydroxyphenyl)-5,6- dihydropyridine-l(2H)-carboxylate (obtained as described in Example 4.1, preparation of starting materials) (16.5 g, 53.34 mmol), tert-butyl 4-(2-hydroxyethyl)piperazine-l- carboxylate (CAS 77279-24-4) (14.74 g, 64.00 mmol) and triphenylphosphine (16.79 g, 64.00 mmol) in THF (150 mL) under nitrogen. The resulting solution was stirred at ambient temperature for 16 hours. The reaction mixture was evaporated to dryness then the residue was stirred in ether (200 mL) for 10 minutes at room temperature. The resulting precipitate was removed by filtration and discarded. The ether filtrate was washed with water (100 mL) followed by saturated brine (100 mL), then dried over MgSO4, filtered and evaporated to give crude product. The crude product was purified by flash silica chromatography, elution gradient 20 to 60% EtOAc in isohexane. Fractions containing the desired product were evaporated to dryness to afford tert-butyl 4-[2-[4-(l- (benzyloxycarbonyl)- 1,2,3, 6-tetrahydropyridin-4-yl)phenoxy]ethyl]piperazine-l- carboxylate (34.6 g, 82%) as a gum which was contaminated with 34% by weight triphenylphosphine oxide.
IH NMR (399.9 MHz, DMSO-d6) δ 1.40 (9H, s), 2.42 – 2.47 (6H, m), 2.71 (2H, m), 3.32 (4H, m), 3.62 (2H, m), 4.03 – 4.10 (4H, m), 5.12 (2H, s), 6.06 (IH, m), 6.92 (2H, d), 7.31 – 7.40 (7H, m); m/z = 522 [M+H]+.
Preparation of tert-butyl 4-[2-[4-(piperidin-4-yl)phenoxy]ethyl]piperazine-l- carboxylate tert-Butyl 4-[2-[4-(l-(benzyloxycarbonyl)-l,2,3,6-tetrahydropyridin-4- yl)phenoxy]ethyl]piperazine-l-carboxylate (66% pure by weight) (34.62 g, 43.80 mmol) and 5% palladium on carbon (50% wet) (4.47 g, 1.05 mmol) in MeOH (250 mL) were stirred under an atmosphere of hydrogen at 5 bar and 600C for 4 hours. The catalyst was removed by filtration and the solvents evaporated to give crude product. The crude product was purified by flash silica chromatography, eluting with 60% EtOAc in isohexane then 15% 2M ammonia/MeOH in DCM. Pure fractions were evaporated to dryness to afford tert-butyl 4-[2-[4-(piperidin-4-yl)phenoxy]ethyl]piperazine-l-carboxylate (15.42 g, 90%) as a solid. IH NMR (399.9 MHz, CDC13) δ 1.46 (9H, s), 1.62 (2H, m), 1.81 (2H, m), 2.50 – 2.59 (5H, m), 2.73 (2H, m), 2.80 (2H, t), 3.18 (2H, m), 3.44 (4H, m), 4.09 (2H, t), 6.85 (2H, d), 7.13 (2H, d); m/z = 390 [M+H]+.
Preparation of tert-butyl 4-[2-[4-[l-(3-(trifluoromethyl)-[l,2,4]triazolo[4,3- b]pyridazin-6-yl]piperidin-4-yl]phenoxy]ethyl]piperazine-l-carboxylate
DIPEA (2.348 mL, 13.48 mmol) was added to 6-chloro-3-(trifluoromethyl)- [l,2,4]triazolo[4,3-b]pyridazine (obtained as described in Monatsh. Chem. 1972, 103, 1591) (2 g, 8.99 mmol) and tert-butyl 4-[2-[4-(piperidin-4-yl)phenoxy]ethyl]piperazine-l- carboxylate (3.68 g, 9.44 mmol) in DMF (30 mL). The resulting solution was stirred at 800C for 2 hours. The reaction mixture was cooled to room temperature and the solvents evaporated to dryness. The resulting solid was triturated with water then collected by filtration, washed with ether and dried to afford tert-butyl 4-[2-[4-[l-(3-(trifluoromethyl)- [l,2,4]triazolo[4,3-b]pyridazin-6-yl]piperidin-4-yl]phenoxy]ethyl]piperazine-l -carboxylate (5.02 g, 97%) as a solid.
IH NMR (399.9 MHz, CDC13) δ 1.46 (9H, s), 1.76 (2H, m), 2.00 (2H, m), 2.54 (4H, m), 2.75 – 2.86 (3H, m), 3.11 (2H, m), 3.46 (4H, m), 4.11 (2H, m), 4.37 (2H, m), 6.87 (2H, d), 7.13 (3H, m), 7.92 (IH, d); m/z = 576 [M+H]+.
Preparation of tert-butyl 4-[2-[4-[l-[3-(trifluoromethyl)-7,8-dihydro-
[1 ,2,4] triazolo [4,3-b] pyridazin-6-yl)piperidin-4-yl] phenoxy] ethyl] piperazine- 1- carboxylate
10% Palladium on carbon (0.924 g, 0.87 mmol) was added to tert-butyl 4-[2-[4-[l-(3- (trifluoromethyl)-[l,2,4]triazolo[4,3-b]pyridazin-6-yl]piperidin-4- yl]phenoxy]ethyl]piperazine-l -carboxylate (2.5 g, 4.34 mmol) and ammonium formate (2.74 g, 43.43 mmol) in ethanol (100 mL). The resulting mixture was stirred at 78°C, with further portions of ammonium formate being added every 5 hours until the reaction was complete. The reaction mixture was cooled to room temperature and the catalyst was removed by filtration. The filtrate was evaporated to dryness, redissolved in DCM (100 mL) and the solution was washed with water (100 mL) followed by brine (50 mL), then the solvents were evaporated to afford tert-butyl 4-[2-[4-[l-[3-(trifluoromethyl)-7,8-dihydro- [l,2,4]triazolo[4,3-b]pyπdazin-6-yl)pipeπdin-4-yl]phenoxy]ethyl]piperazine-l-carboxylate (2.02O g, 81%) as a solid.
IH NMR (399.9 MHz, CDC13) δ 1.46 (9H, s), 1.69 (2H, m), 1.95 (2H, m), 2.52 (4H, m), 2.71 – 2.82 (5H, m), 3.00 (2H, m), 3.22 (2H, t), 3.45 (4H, m), 4.09 (2H, m), 4.31 (2H, m), 6.86 (2H, d), 7.12 (2H, d); m/z = 578 [M+H]+.
Preparation of 6- [4-[4- [2-(piperazin-l-yl)ethoxy] phenyl] piperidin-1-yl] -3- (trifluor omethyl)-7,8-dihydr o- [ 1 ,2,4] triazolo [4,3-b] pyridazine
TFA (10 mL) was added to tert-butyl 4-[2-[4-[l-[3-(trifluoromethyl)-7,8-dihydro- [l,2,4]triazolo[4,3-b]pyπdazin-6-yl)pipeπdin-4-yl]phenoxy]ethyl]piperazine-l-carboxylate (2.02 g, 3.50 mmol) in DCM (10 mL). The resulting solution was stirred at ambient temperature for 1 hour then added to an SCX column. The desired product was eluted from the column using 2M ammonia/MeOH and the solvents were evaporated to afford 6-[4-[4- [2-(piperazin-l-yl)ethoxy]phenyl]piperidin-l-yl]-3-(trifluoromethyl)-7,8-dihydro- [l,2,4]triazolo[4,3-b]pyridazine (1.660 g, 99%) as a solid.
IH NMR (399.9 MHz, CDC13) δ 1.68 (2H, m), 1.95 (2H, m), 2.55 (4H, m), 2.70 – 2.80 (5H, m), 2.91 (4H, m), 3.00 (2H, m), 3.22 (2H, t), 4.09 (2H, t), 4.30 (2H, m), 6.87 (2H, d), 7.11 (2H, d); m/z = 478 [M+H]+.
Example 5.2
Larger scale preparation of 6-(4-{4-[2-(4-acetylpiperazin-l- vDethoxyl phenyllpiperidin- l-vD-3-f trifluoromethyl)-7.,8-dihvdro [ 1 ,2,41 triazolo [4,3- blpyridazine
Ammonium formate (99 g, 1568.94 mmol) was added to 6-[4-[4-[2-(4-acetylpiperazin-l- yl)ethoxy]phenyl]piperidin- 1 -yl]-3-(trifluoromethyl)[ 1 ,2,4]triazolo[4,3-b]pyridazine (81.2 g, 156.89 mmol) and 10% palladium on carbon (8.35 g, 7.84 mmol) in EtOH (810 mL) under nitrogen. The resulting mixture was stirred at 700C for 6 hours, then ammonium formate (50 g) was added. The mixture was stirred at 700C for 2 hours then further portions of 10% palladium on carbon (8.35 g, 7.84 mmol) and ammonium formate (50 g) were added and stirring continued at 700C for a further 10 hours. Ammonium formate (50 g) was added and the reaction mixture was stirred at 700C for 24 hours then cooled to room temperature. The catalyst was removed by filtration and the reaction charged with further 10% palladium on carbon (8.35 g, 7.84 mmol) and stirred at 700C for 16 hours. Further ammonium formate (50 g) was added and the stirring continued for 5 hours. The reaction mixture was cooled to room temperature and a further portion of 10% palladium on carbon (8.35 g, 7.84 mmol) was added. The mixture was heated to 700C for a 30 hours, cooled to room temperature and the catalyst removed by filtration and washed with EtOH. The solvent was evaporated and the residue dissolved in DCM (500 mL) and the solution washed with water (500 mL). The aqueous layer was re-extracted with DCM (500 mL), then EtOAc (500 mL x 2). The combined extracts were dried over MgSO4, filtered and evaporated to give crude product. The crude product was purified by flash silica chromatography, elution gradient 0 to 5% MeOH in DCM. Pure fractions were evaporated to dryness to afford a gum, which was slurried with ether (300 mL) and re-evaporated. Methyl tert-butyl ether (250 mL) was added and the mixture was stirred vigorously for 3 days. The solid was collected by filtration and dried to afford 6-(4-{4-[2-(4- acetylpiperazin- 1 -yl)ethoxy]phenyl}piperidin- 1 -yl)-3-(trifluoromethyl)-7,8- dihydro[l,2,4]triazolo[4,3-b]pyridazine (60.8 g, 75%) as a solid.
IH NMR (399.9 MHz, CDC13) δ 1.62 (2H, m), 1.88 (2H, m), 2.02 (3H, s), 2.49 (4H, m), 2.65 – 2.78 (5H, m), 2.94 (2H, m), 3.15 (2H, t), 3.42 (2H, m), 3.57 (2H, m), 4.03 (2H, t), 4.24 (2H, m), 6.80 (2H, d), 7.06 (2H, d); m/z = 520 [M+H]+.
The 6-[4-[4-[2-(4-acetylpiperazin-l-yl)ethoxy]phenyl]piperidin-l-yl]-3-
(trifluoromethyl)[l,2,4]triazolo[4,3-b]pyridazine used as starting material was prepared as follows :-
Preparation of 4-(piperidin-4-yl)phenol Benzyl 4-(4-hydroxyphenyl)-5,6-dihydropyridine-l(2H)-carboxylate (obtained as described in Example 4.1, preparation of starting materials) (37.7 g, 121.86 mmol) and 5% palladium on carbon (7.6 g, 3.57 mmol) in methanol (380 mL) were stirred under an atmosphere of hydrogen at 5 bar and 25°C for 2 hours. The catalyst was removed by filtration, washed with MeOH and the solvents evaporated. The crude material was triturated with diethyl ether, then the desired product collected by filtration and dried under vacuum to afford 4-(piperidin-4-yl)phenol (20.36 g, 94%) as a solid. IH NMR (399.9 MHz, DMSO-d6) δ 1.46 (2H, m), 1.65 (2H, m), 2.45 (IH, m), 2.58 (2H, m), 3.02 (2H, m), 6.68 (2H, d), 7.00 (2H, d), 9.15 (IH, s); m/z = 178 [M+H]+.
Preparation of 4- { 1- [3-(trifluor omethyl) [1 ,2,4] triazolo [4,3-b] pyridazin-6-yl] piperidin- 4-yl}phenol
DIPEA (48.2 mL, 276.86 mmol) was added to 6-chloro-3-(trifluoromethyl)- [l,2,4]triazolo[4,3-b]pyridazine (obtained as described in Monatsh. Chem. 1972, 103, 1591) (24.65 g, 110.74 mmol) and 4-(piperidin-4-yl)phenol (20.61 g, 116.28 mmol) in DMF (200 mL). The resulting solution was stirred at 800C for 1 hour. The reaction mixture was cooled to room temperature, then evaporated to dryness and re-dissolved in DCM (1 L) and washed with water (2 x 1 L). The organic layer was washed with saturated brine (500 mL), then dried over MgSO4, filtered and evaporated to afford crude product. The crude product was triturated with ether to afford 4-{l-[3- (trifluoromethyl)[l,2,4]triazolo[4,3-b]pyridazin-6-yl]piperidin-4-yl}phenol (36.6 g, 91%) as a solid.
IH NMR (399.9 MHz, DMSO-d6) δ 1.64 (2H, m), 1.87 (2H, m), 2.75 (IH, m), 3.09 (2H, m), 4.40 (2H, m), 6.69 (2H, d), 7.05 (2H, d), 7.65 (IH, d), 8.24 (IH, d), 9.15 (IH, s); m/z = 364 [M+H]+.
Preparation of 2-(4-{l-[3-(trifluoromethyl)[l,2,4]triazolo[4,3-b]pyridazin-6- yl]piperidin-4-yl}phenoxy)ethanol
A solution of ethylene carbonate (121 g, 1376.13 mmol) in DMF (200 mL) was added dropwise to a stirred suspension of 4-{l-[3-(trifluoromethyl)[l,2,4]triazolo[4,3- b]pyridazin-6-yl]piperidin-4-yl}phenol (100 g, 275.23 mmol) and potassium carbonate (76 g, 550.45 mmol) in DMF (200 mL) at 800C over a period of 15 minutes under nitrogen.
The resulting mixture was stirred at 800C for 20 hours. The reaction mixture was cooled to room temperature, then concentrated and diluted with DCM (2 L), and washed sequentially with water (1 L) and saturated brine (500 mL). The organic layer was dried over MgSO4, filtered and evaporated to afford crude product. The crude product was purified by flash silica chromatography, elution gradient 70 to 100% EtOAc in isohexane. Fractions containing the desired product were evaporated to dryness then triturated with EtOAc (150 mL). The resulting solid was washed with further EtOAc (50 mL) and ether then dried to give 2-(4- { 1 -[3-(trifluoromethyl)[ 1 ,2,4]triazolo[4,3-b]pyridazin-6-yl]piperidin-4- yl}phenoxy)ethanol. The filtrate was evaporated and further purified by flash silica chromatography, elution gradient 70 to 100% EtOAc in isohexane. Fractions containing the desired product were evaporated to dryness then triturated with ether, dried and combined with the material previously collected to afford 2-(4- { 1 -[3-
(trifluoromethyl)[ 1 ,2,4]triazolo[4,3-b]pyridazin-6-yl]piperidin-4-yl}phenoxy)ethanol (89 g, 79%) as a solid.
IH NMR (399.9 MHz, DMSO-d6) δ 1.66 (2H, m), 1.88 (2H, m), 2.80 (IH, m), 3.10 (2H, m), 3.70 (2H, m), 3.95 (2H, t), 4.41 (2H, m), 4.85 (IH, t), 6.87 (2H, d), 7.18 (2H, d), 7.67 (IH, d), 8.25 (IH, d); m/z = 408 [M+H]+.
Preparation of 2-(4-{ 1- [3-(trifluoromethyl) [ 1 ,2,4] triazolo [4,3-b] pyridazin-6- yl] piperidin-4-yl}phenoxy)ethyl methanesulfonate
A solution of methanesulfonyl chloride (20.37 mL, 262.16 mmol) in DCM (300 mL) was added to 2-(4- { 1 -[3-(trifluoromethyl)[ 1 ,2,4]triazolo[4,3-b]pyridazin-6-yl]piperidin-4- yl}phenoxy)ethanol (89 g, 218.46 mmol) and triethylamine (60.9 mL, 436.93 mmol) in DCM (900 mL) at 00C over a period of 30 minutes under nitrogen. The resulting solution was stirred at 00C for 1 hour. The reaction mixture was diluted with DCM (1 L), and washed with water (2 L). The organic layer was dried over MgSO4, filtered and evaporated to afford 2-(4- { 1 -[3-(trifluoromethyl)[ 1 ,2,4]triazolo[4,3-b]pyridazin-6-yl]piperidin-4- yl}phenoxy)ethyl methanesulfonate (104 g, 98%) as a solid.
IH NMR (399.9 MHz, DMSO-d6) δ 1.67 (2H, m), 1.89 (2H, m), 2.83 (IH, m), 3.11 (2H, m), 3.23 (3H, s), 4.23 (2H, t), 4.41 (2H, m), 4.52 (2H, t), 6.91 (2H, d), 7.21 (2H, d), 7.66 (IH, d), 8.24 (IH, d); m/z = 486 [M+H]+. Preparation of 6-[4-[4-[2-(4-acetylpiperazin-l-yl)ethoxy]phenyl]piperidin-l-yl]-3- (trifluor omethyl) [ 1 ,2,4] triazolo [4,3-b] pyridazine DIPEA (107 mL, 613.00 mmol) was added to 2-(4-{l-[3-
(trifluoromethyl)[l,2,4]triazolo[4,3-b]pyridazin-6-yl]piperidin-4-yl}phenoxy)ethyl methanesulfonate (99 g, 204.33 mmol) and N-acetylpiperazine (28.8 g, 224.77 mmol) in DMA (500 mL). The resulting solution was stirred at 1100C for 1 hour. The reaction mixture was cooled to room temperature and the solvents were evaporated. The residue was dissolved in ethyl acetate (1 L) and the solution was washed with water (1 L). The aqueous was re-extracted with ethyl acetate (1 L) and the combined organics were washed with brine (1 L), dried over MgSO4, filtered and evaporated to give crude product. The aqueous layer was basifϊed to pH 12 with 2M NaOH, then extracted with ethyl acetate (1 L), washed with brine (IL), dried over MgSO4, filtered and evaporated to give further crude product. The crude product was purified by flash silica chromatography, elution gradient 0 to 3% MeOH in DCM then 5% MeOH in DCM. Pure fractions were evaporated to give 6-[4-[4-[2-(4-acetylpiperazin-l-yl)ethoxy]phenyl]piperidin-l-yl]-3- (trifluoromethyl)[l,2,4]triazolo[4,3-b]pyridazine (81 g, 77%) as a solid. IH NMR (399.9 MHz, DMS0-d6) δ 1.59-1.73 (2H, m), 1.87 (2H, d), 1.99 (3H, s), 2.42 (2H, t), 2.71 (2H, t), 2.76-2.86 (IH, t), 3.08 (2H, t), 3.38-3.47 (4H, m), 4.08 (2H, t), 4.41 (2H, d), 6.88 (2H, d), 7.18 (2H, d), 7.62 (IH, d), 8.26 (IH, d); m/z = 518 [M+H]+.
Example 5.5
Alternative route for the preparation of 6-(4-{4-[2-(4-acetylpiperazin-l- vDethoxyl phenyllpiperidin- l-vD-3-f trifluoromethyl)-7.,8-(iihv(iro [ 1 ,2,41 triazolo [4,3- blpyridazine Form A
Methanol (375.0 mL) was added to 6-[4-[4-[2-(4-acetylpiperazin-l- yl)ethoxy]phenyl]piperidin-l-yl]-3-(trifluoromethyl)[ 1,2,4] triazolo[4,3-b]pyridazine (25.0 g, 48 m mol) in a 2.0 L autoclave reactor and to this was added 10% Pd/C (12.5 g, 50% w/w) paste at 22-25°C under nitrogen gas atmosphere. The reaction was performed under hydrogen pressure (5.0 bar) at 500C temperature for 10.0 h. The reaction mass was cooled to room temperature and the catalyst removed by filtration. Filtered cake was washed with methanol. The solvent was evaporated and the residue was azeotropically distilled by ethylacetate (2 x 125.0 mL) at 400C under reduced pressure to 3.0 rel vol (75.0 mL). Drop wise addition of tert-butylmethylether (MTBE, 375.0 mL) to the reaction mass resulted in solid material, which was collected by filtration and washed with MTBE (50.0 mL). The material was dried under reduced pressure with nitrogen gas bleed at 500C to afford the desired product 6-(4-{4-[2-(4-acetylpiperazin-l-yl)ethoxy]phenyl}piperidin-l-yl)-3- (trifluoromethyl)-7,8-dihydro[l,2,4]triazolo [4,3-b]pyridazine (22.3 g, 88%) as a white color free flowing solid. The isolated material was confirmed by XRPD as Form A. IH NMR (400.13 MHz, CDC13): δ 1.62 (2H, m), 1.88 (2H, m), 2.02 (3H, s), 2.49 (4H, m), 2.65 – 2.78 (5H, m), 2.94 (2H, m), 3.15 (2H, t), 3.42 (2H, m), 3.57 (2H, m), 4.03 (2H, t), 4.24 (2H, m), 6.80 (2H, d), 7.06 (2H, d); m/z = 520 [M+H]+.
The 6-[4-[4-[2-(4-acetylpiperazin-l-yl)ethoxy]phenyl]piperidin-l-yl]-3- (trifluoromethyl)[ 1,2,4] triazolo[4,3-b]pyridazine used as starting material was prepared as follows :-
Preparation of 4- { 1- [3-(trifluor omethyl) [1 ,2,4] triazolo [4,3-b] pyridazin-6-yl] piperidin- 4-yl}phenol: Dimethylacetamide (250.0 mL) was added to 6-chloro-3-(trifluoromethyl)- [l,2,4]triazolo[4,3-b]pyridazine [CAS: 40971-95-7] (50.0 g, 225 m mol) at 22-25°C in a suitable round bottom flask followed by 4-(piperidin-4-yl)phenol [CAS: 62614-84-0] (60.9 g, 236 m mol) at 22-25°C. The reaction mass was stirred to obtain a clear solution. Triethylamine (79.1 mL, 561 m mol) was slowly added to the reaction mass by drop wise addition over a period of 60 min at 25-300C. Temperature was raised to 400C and the reaction mass stirred for 1.0 h. After completion of reaction, water (500.0 mL) was added to the reaction mass by drop wise addition over a period of 30 min at 40-430C. The slurry mass was stirred for 30 min at 400C and then filtered under reduced pressure. The wet material was slurry washed using water (500.0 mL) for 30 min at 400C. The solid was collected by filtration and the material washed with water (125.0 mL). The material was dried under reduced pressure with nitrogen gas bleed at 500C to afford the desired product 4-{l-[3-(trifluoromethyl)[l,2,4]triazolo[4,3-b]pyridazin-6-yl]piperidin-4-yl}phenol (75.1 g, 89.9%) as a free flowing solid. IH NMR (400.13 MHz, DMSO-d6): δ 1.64 (2H, m), 1.87 (2H, m), 2.75 (IH, m), 3.09 (2H, m), 4.40 (2H, m), 6.69 (2H, d), 7.05 (2H, d), 7.65 (IH, d), 8.24 (IH, d), 9.15 (IH, s); m/z = 364 [M+H]+.
Preparation of 6-[4-[4-[2-(4-acetylpiperazin-l-yl)ethoxy]phenyl]piperidin-l-yl]-3- (trifluor omethyl) [ 1 ,2,4] triazolo [4,3-b] pyridazine:
Dichloromethane (225.0 mL) and 4-{l-[3-(trifluoromethyl)[l,2,4]triazolo[4,3-b]pyridazin- 6-yl]piperidin-4-yl} phenol (50.0 g, 138 m mol) were charged to a suitable round bottom flask at 22-25°C. Triphenylphosphine (72.2 g, 275 m mol) and l-[4-(2-hydroxy- ethyl)piperazin-l-yl]ethanone [CAS: 83502-55-0] (47.4 g, 275 m mol) were added successively to the reaction mass and stirred for 10 min at 22-25°C. Di-isopropyl azodicarboxylate (55.65 g, 275 m mol) in dichloromethane (75.0 mL) was added to the reaction mass slowly drop wise at 25-300C over a period of 60-90 min. The resulting reaction mass was stirred for 1.0 h at 25-300C to complete the reaction. n-Heptane (600.0 mL) was introduced to the reaction mass by drop wise addition over a period of 15-30 min at 22-25°C and stirred for 30 min at the same temperature. Thus precipitated solid was filtered and washed with n-heptane (150.0 mL). The material was then suck dried for 30 min under reduced pressure. The crude material was purified by slurry washing in methanol (325.0 mL) at 22-25°C. The solid was then collected by filtration and washed with methanol (50.0 mL). The material was dired under reduced pressure with nitrogen gas bleed at 500C to afford the desired product 6-[4-[4-[2-(4-acetylpiperazin-l- yl)ethoxy]phenyl]piperidin- 1 -yl]-3-(trifluoromethyl)[ 1 ,2,4] triazolo[4,3-b]pyridazine (61.2 g, 84%) as a free flowing solid.
IH NMR (400.13 MHz, DMSO-d6): δ 1.59-1.73 (2H, m), 1.87 (2H, d), 1.99 (3H, s), 2.42 (2H, t), 2.71 (2H, t), 2.76-2.86 (IH, t), 3.08 (2H, t), 3.38-3.47 (4H, m), 4.08 (2H, t), 4.41 (2H, d), 6.88 (2H, d), 7.18 (2H, d), 7.62 (IH, d), 8.26 (IH, d); m/z = 518 [M+H]+.
Example 5.8
Preparation of 6-(4-{4-[2-(4-acetylpiperazin-l-yl)ethoxy]phenyl}piperidin-l-yl)-3-(trifluor omethyl)-7,8-dihydr 0 [1 ,2,4] triazolo [4,3-b] pyridazine maleate

A clear solution of maleic acid (0.445 g, 3.84 m mol) in methanol (1.0 mL) was added to a clear solution of 6-(4-{4-[2-(4-acetylpiperazin-l-yl)ethoxy]phenyl}piperidin-l-yl)-3- (trifluoromethyl)-7,8-dihydro[l,2,4]triazolo[4,3-b]pyridazine, obtained as described in Example 5.5, (2.0 g, 3.84 m mol) in methanol (2.0 mL) at 22-25°C and the resulting clear solution heated to 500C for 30 min. The reaction mass was cooled to 22-25°C and ethylacetate (16.0 mL) added drop wise to the reaction mass at 22-25°C. The reaction mass was then stirred for 60 min at 22-25°C. The resulting white color material was collected by filtration and washed with ethylacetate (5.0 mL). The material was dried under reduced pressure with nitrogen gas bleed at 500C to afford the desired product 6-(4- {4-[2-(4-acetylpiperazin-l-yl)ethoxy]phenyl}piperidin-l-yl)-3-(trifluoromethyl)-7,8- dihydro[l,2,4]triazolo[4,3-b]pyridazine maleate (2.21 g, 90.0%) as free flowing white color material.
IH NMR (400.13 MHz, DMSO-d6): δ 1.62 (2H, m), 1.77 (2H, m), 2.02 (3H, s), 2.75 (IH, m), 2.77 (2H, m), 2.80 (2H, m), 2.95 (4H, m), 3.16 (2H, t), 3.36 (6H, m), 4.22 (4H, m), 6.08 (2H, s), 6.91 (2H, d), 7.17 (2H, d).
PAPER
Bioorg Med Chem Lett. 2013 Apr 1;23(7):1945-8
Volume 23, Issue 7, 1 April 2013, Pages 1945–1948

Discovery of AZD3514, a small-molecule androgen receptor downregulator for treatment of advanced prostate cancer
- Oncology iMed, AstraZeneca, Mereside, Alderley Park, Macclesfield SK10 4TG, UK
Removal of the basic piperazine nitrogen atom, introduction of a solubilising end group and partial reduction of the triazolopyridazine moiety in the previously-described lead androgen receptor downregulator 6-[4-(4-cyanobenzyl)piperazin-1-yl]-3-(trifluoromethyl)[1,2,4]triazolo[4,3-b]pyridazine (1) addressed hERG and physical property issues, and led to clinical candidate 6-(4-{4-[2-(4-acetylpiperazin-1-yl)ethoxy]phenyl}piperidin-1-yl)-3-(trifluoromethyl)-7,8-dihydro[1,2,4]triazolo[4,3-b]pyridazine (12), designated AZD3514, that is being evaluated in a Phase I clinical trial in patients with castrate-resistant prostate cancer.

http://www.sciencedirect.com/science/article/pii/S0960894X13002321
SYNTHESIS
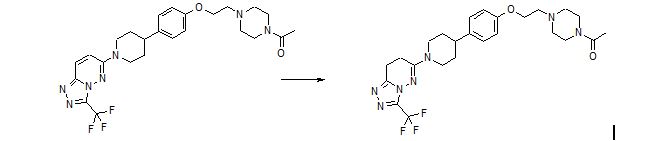 AZD 3514
AZD 3514
6-(4-{4-[2-(4-Acetylpiperazin-1-yl)ethoxy]phenyl}piperidin-1-yl)-3-(trifluoromethyl)-7,8-dihydro[1,2,4]triazolo[4,3-b]pyridazine AZD 3514
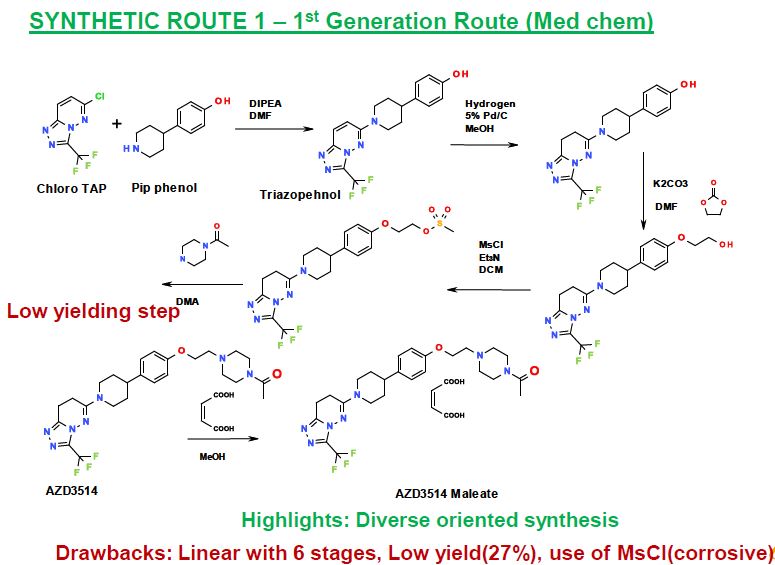
SYNTHETIC ROUTE 2ND GENERATION
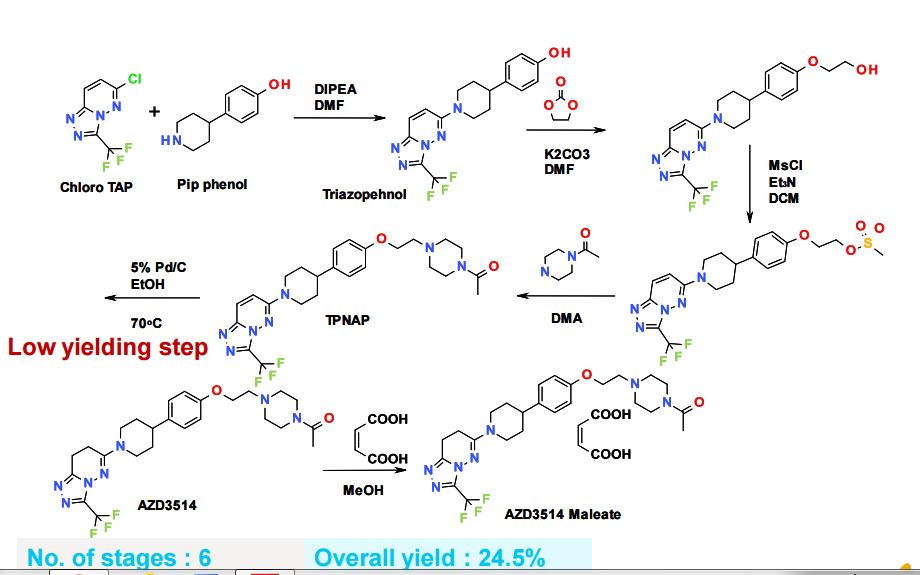
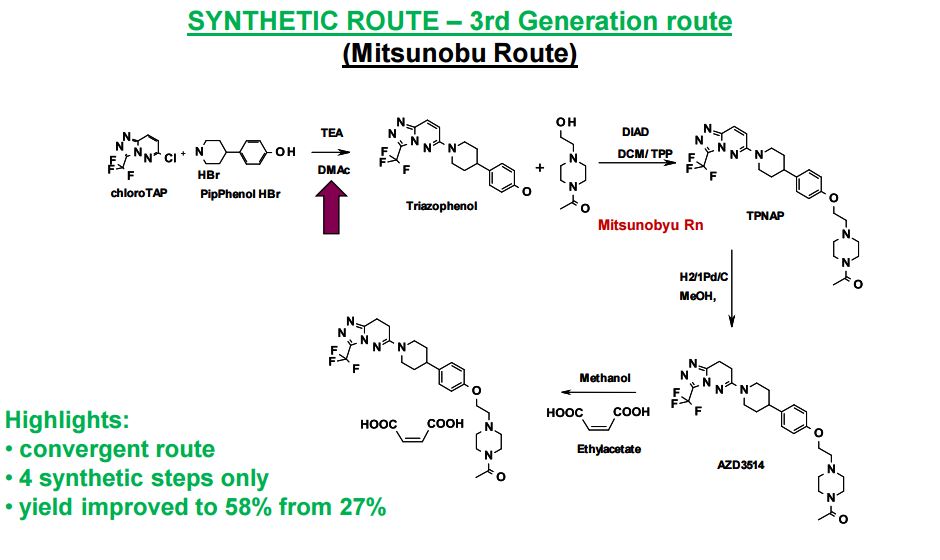
SYNTHETIC ROUTE 4TH GENERATION
REFERENCES
1: Bradbury RH, Acton DG, Broadbent NL, Brooks AN, Carr GR, Hatter G, Hayter BR, Hill KJ, Howe NJ, Jones RD, Jude D, Lamont SG, Loddick SA, McFarland HL, Parveen Z, Rabow AA, Sharma-Singh G, Stratton NC, Thomason AG, Trueman D, Walker GE, Wells SL, Wilson J, Wood JM. Discovery of AZD3514, a small-molecule androgen receptor downregulator for treatment of advanced prostate cancer. Bioorg Med Chem Lett. 2013 Apr 1;23(7):1945-8. doi: 10.1016/j.bmcl.2013.02.056. Epub 2013 Feb 21. PubMed PMID: 23466225.
Some pics, Team at Astrazeneca , Bangalore, INDIA

Vijaykumar Sengodan Chellappan

Jagannath V, PMP®

Associate Research Scientist II at AstraZeneca India Pvt Ltd

Rifahath Mon
Associate Research Scientist at AstraZeneca

Route Scouting, Process Design, Technology Transfer, Trouble shooting, QbD, Green Chemistry

Srinivasa Rao Korupoju

Harikrishna Tumma Ph. D.

Rashmi HV

Anandan Muthusamy

Partha Pratim Bishi,




///////////////AZD 3514 MALEATE, AZD 3514 , AZD-3514, Prostate cancer, Androgen receptor downregulator, AZD3514, 1240299-33-5
Lobeglitazone sulfate (Duvie)
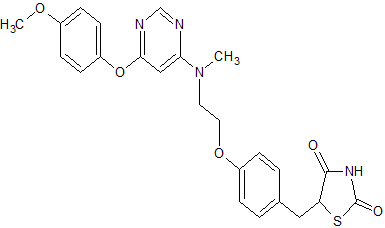
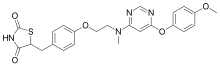
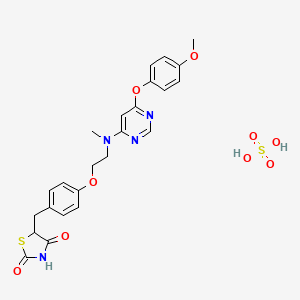
Lobeglitazone Sulfate, CKD-501, IDR-105
(Duvie®)Approved KOREA

Chong Kun Dang (Originator)
Adjunct to diet and exercise to improve glycemic control in adults with type 2 Diabetes mellitus
A dual PPARα and PPARγ agonist used to treat type 2 diabetes.
![]()
Trade Name:Duvie®MOA:Dual PPARα and PPARγ agonistIndication:Type 2 diabetes
CAS No. 607723-33-1(FREE)
CAS 763108-62-9(Lobeglitazone Sulfate)
2,4-Thiazolidinedione, 5-((4-(2-((6-(4-methoxyphenoxy)-4- pyrimidinyl)methylamino)ethoxy)phenyl)methyl)-, sulfate (1:1);



- Developer Chong Kun Dang; EQUIS & ZAROO
- Class Antihyperglycaemics; Pyrimidines; Small molecules; Thiazolidinediones
- Mechanism of Action Peroxisome proliferator-activated receptor alpha agonists; Peroxisome proliferator-activated receptor gamma agonists
- MarketedType 2 diabetes mellitus
-
Most Recent Events
- 01 May 2016Chong Kun Dang Pharmaceutical completes two phase I drug-interaction trials in Healthy volunteers in South Korea (PO) (NCT02824874; NCT02827890)
- 01 Apr 2016Chong Kun Dang Pharmaceutical initiates two phase I drug-interaction trials in Healthy volunteers in South Korea (PO) (NCT02824874; NCT02827890)
- 01 Mar 2016Chong Kun Dang completes a phase I pharmacokinetic trial in Impaired hepatic function in Healthy volunteers in South Korea, NCT02007941)
- Lobeglitazone sulfate was approved by the Ministry of Food and Drug Safety (Korea) on July 4, 2013. It was developed and marketed as Duvie® by Chong Kun Dang Corporation.Lobeglitazone is an agonist for both PPARα and PPARγ, and it works as an insulin sensitizer by binding to the PPAR receptors in fat cells and making the cells more responsive to insulin. It is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes.Duvie® is available as tablet for oral use, containing 0.5 mg of free Lobeglitazone. The recommended dose is 0.5 mg once daily.


Lobeglitazone (trade name Duvie, Chong Kun Dang) is an antidiabetic drug in the thiazolidinedione class of drugs. As an agonistfor both PPARα and PPARγ, it works as an insulin sensitizer by binding to the PPAR receptors in fat cells and making the cells more responsive to insulin.[3]
![]()
Chong Kun Dang
Lobeglitazone sulfate was approved by the Ministry of Food and Drug Safety (Korea) on July 4, 2013. It was developed and marketed as Duvie® by Chong Kun Dang Corporation.
Lobeglitazone is an agonist for both PPARα and PPARγ, and it works as an insulin sensitizer by binding to the PPAR receptors in fat cells and making the cells more responsive to insulin. It is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes.
Duvie® is available as tablet for oral use, containing 0.5 mg of free Lobeglitazone. The recommended dose is 0.5 mg once daily.
Lobeglitazone which was reported in our previous works belongs to the class of potent PPARα/γ dual agonists (PPARα EC50: 0.02 μM, PPARγ EC50: 0.018 μM, rosiglitazone; PPARα EC50: >10 μM, PPARγ EC50: 0.02 μM, pioglitazone PPARα EC50: >10 μM, PPARγ EC50: 0.30 μM). Lobeglitazone has excellent pharmacokinetic properties and was shown to have more efficacious in vivo effects in KKAy mice than rosiglitazone and pioglitazone.17 Due to its outstanding pharmacokinetic profile, lobeglitazone was chosen as a promising antidiabetes drug candidate.
Medical uses
Lobeglitazone is used to assist regulation of blood glucose level of diabetes mellitus type 2 patients. It can be used alone or in combination with metformin.[4]
Lobeglitazone was approved by the Ministry of Food and Drug Safety (Korea) in 2013, and the postmarketing surveillance is on progress until 2019.[4][5]
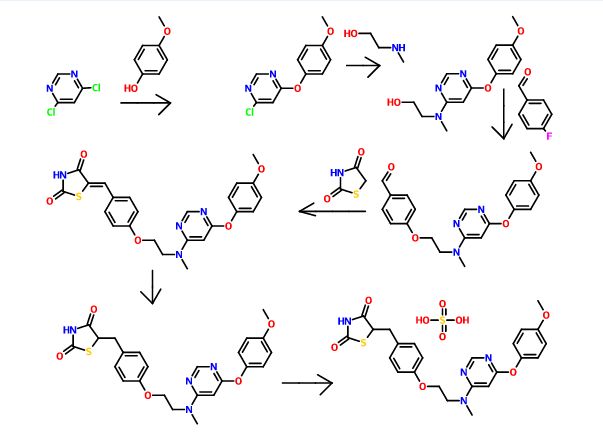
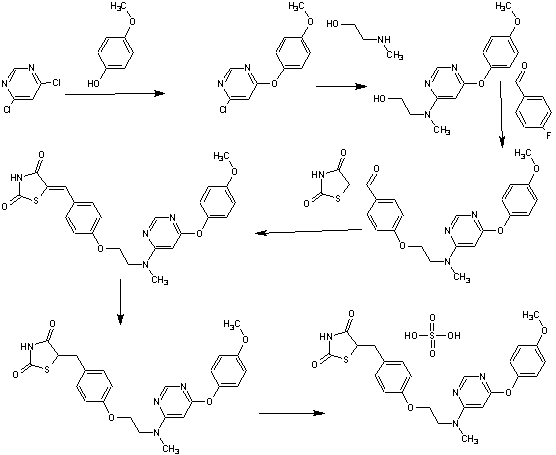
SYNTHESIS

Chong Kun Dang’s Modcol Flu Dry Syrup is released in four different versions: All-Day, Night, Nose and Cough. [CHONG KUN DANG]


PAPER
Org. Process Res. Dev. 2007, 11, 190-199.
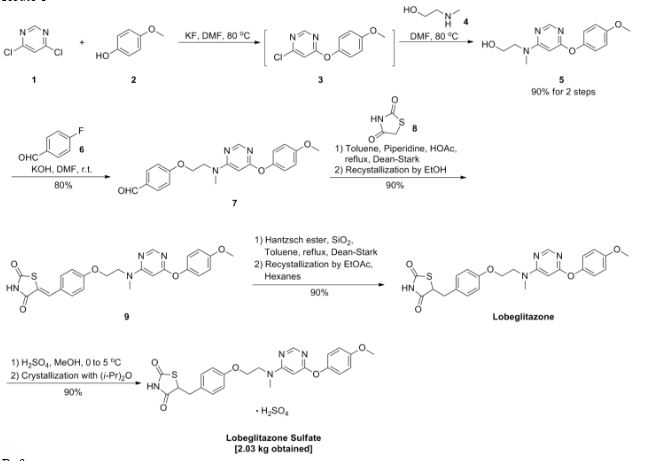
Process Development and Scale-Up of PPAR α/γ Dual Agonist Lobeglitazone Sulfate (CKD-501)
http://pubs.acs.org/doi/abs/10.1021/op060087u

A scaleable synthetic route to the potent PPARα/γ dual agonistic agent, lobeglitazone (1), used for the treatment of type-2 diabetes was developed. The synthetic pathway comprises an effective five-step synthesis. This process involves a consecutive synthesis of the intermediate, pyrimidinyl aminoalcohol (6), from the commercially available 4,6-dichloropyrimidine (3) without the isolation of pyrimidinyl phenoxy ether (4). Significant improvements were also made in the regioselective 1,4-reduction of the intermediate, benzylidene-2,4-thiazolidinedione (10), using Hantzsch dihydropyridine ester (HEH) with silica gel as an acid catalyst. The sulfate salt form of lobeglitazone was selected as a candidate compound for further preclinical and clinical study. More than 2 kg of lobeglitazone sulfate (CKD-501, 2) was prepared in 98.5% purity after the GMP batch. Overall yield of 2 was improved to 52% from 17% of the original medicinal chemistry route.
Silica gel TLC Rf = 0.35 (detection: iodine char chamber, ninhydrin solution, developing solvents: CH2Cl2/MeOH, 20:1); mp 111.4 °C; IR (KBr) ν 3437, 3037, 2937, 2775, 1751, 1698, 1648, 1610, 1503, 1439, 1301, 1246, 1215, 1183 cm-1;
1H NMR (400 MHz, CDCl3) δ 3.09 (m, 4H), 3.29 (m, 1H), 3.76 (s, 3H), 3.97 (m, 2H), 4.14 (m, 2H), 4.86 (m, 1H), 6.06 (bs, 1H), 6.86 (m, 2H), 7.00 (m, 2H), 7.13 (m, 4H), 8.30 (s, 1H), 11.99 (s, NH);
13C NMR (100 MHz, CDCl3) δ 37.1, 38.2, 53.7, 53.8, 56.3, 62.2, 65.8, 86.0, 115.1, 116.0, 123.0, 129.8, 131.2, 145.7, 153.4, 157.9, 158.1, 161.1, 166.5, 172.4, 172.5, 176.3, 176.5;
MS (ESI)m/z (M + 1) 481.5; Anal. Calcd for C24H26N4O9S2: C, 49.82; H, 4.53; N, 9.68; S, 11.08. Found: C, 49.85; H, 4.57; N, 9.75; S, 11.15.
PATENT
Clip
Lobeglitazone sulfate (Duvie) Lobeglitazone sulfate, an oral peroxisome proliferator-activated receptor (PPARa/c) dual agonist with IC50 = 20 and 18 nM respectively, was developed by Chong Kun Dang Pharmaceutical in Korea for the treatment of diabetes.135 This drug is differentiated from two other PPAR agonists available—pioglitazone and rosiglitazone —which lack PPARa activity.135 The most likely processscale preparation of lobeglitazone sulfate follows the route described in a process communication from Chong Kun Dang Pharmaceutical.136
Commercially available 4,6-dichloropyrimidine (152) was treated with a stoichiometric equivalent of p-methoxyphenol (153) in the presence of KF in warm DMF (Scheme 24). Upon completion of this reaction, 2-methylaminoethanol was added to the mixture to provide pyrimidine 154 in high yield.137
Next, alcohol 154 underwent a substitution reaction with p-fluorobenzaldehyde (155) under basic conditions to provide alkoxy benzaldehyde 156 which was converted to the benzylidene thiazolidindione 158 upon subjection to Knoevenagel conditions with 2,4-thiazolidinedione (157) in 90% yield.
Finally, reduction of olefin 158 was facilitated by treatment with the Hantzsch ester (159) in the presence of silica gel followed by treatment with methanolic sulfuric acid (96%) at low temperature to ultimately furnish lobeglitazone sulfate in 90% yield.
135. Jin, S. M.; Park, C. Y.; Cho, Y. M.; Ku, B. J.; Ahn, C. W.; Cha, B.-S.; Min, K. W.;Sung, Y. A.; Baik, S. H.; Lee, K. W.; Yoon, K.-H.; Lee, M.-K.; Park, S. W. Diab.Obes. Metab. 2015, 17, 599.
136. Lee, H. W.; Ahn, J. B.; Kang, S. K.; Ahn, S. K.; Ha, D.-C. Org. Process Res. Dev.2007, 11, 190.
137. Lee, H. W.; Kim, B. Y.; Ahn, J. B.; Kang, S. K.; Lee, J. H.; Shin, J. S.; Ahn, S. K.; Lee,S. J.; Yoon, S. S. Eur. J. Med. Chem. 2005, 40, 862.
References
- Lee JH, Noh CK, Yim CS, Jeong YS, Ahn SH, Lee W, Kim DD, Chung SJ. (2015). “Kinetics of the Absorption, Distribution, Metabolism, and Excretion of Lobeglitazone, a Novel Activator of Peroxisome Proliferator-Activated Receptor Gamma in Rats.”.Journal of Pharmaceutical sciences 104 (9): 3049–3059.doi:10.1002/jps.24378. PMID 25648999.
- Kim JW, Kim JR, Yi S, Shin KH, Shin HS, Yoon SH, Cho JY, Kim DH, Shin SG, Jang IJ, Yu KS. (2011). “Tolerability and pharmacokinetics of lobeglitazone (CKD-501), a peroxisome proliferator-activated receptor-γ agonist: a single- and multiple-dose, double-blind, randomized control study in healthy male Korean subjects.”. Clinical therapeutics 33 (11): 1819–1830.doi:10.1016/j.clinthera.2011.09.023. PMID 22047812.
- Lee JH, Woo YA, Hwang IC, Kim CY, Kim DD, Shim CK, Chung SJ. (2009). “Quantification of CKD-501, lobeglitazone, in rat plasma using a liquid-chromatography/tandem mass spectrometry method and its applications to pharmacokinetic studies.”. Journal of Pharmaceutical and Biomedical Analysis 50 (5): 872–877.doi:10.1016/j.jpba.2009.06.003. PMID 19577404.
- “MFDS permission information of Duvie Tablet 0.5mg”(Release of Information). Ministry of Food and Drug Safety. Retrieved2014-10-23.
- “국내개발 20번째 신약‘듀비에정’허가(20th new drug developed in Korea ‘Duvie Tablet’ was approved)”. Chong Kun Dang press release. 2013-07-04. Retrieved 2014-10-23.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-[(4-[2-([6-(4-Methoxyphenoxy)pyrimidin-4-yl]-methylamino)ethoxy]phenyl)methyl]-1,3-thiazolidine-2,4-dione
|
|
| Clinical data | |
| Trade names | Duvie |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | >99%[1] |
| Metabolism | liver (CYP2C9, 2C19, and 1A2)[1] |
| Biological half-life | 7.8–9.8 hours[2] |
| Identifiers | |
| CAS Number | 607723-33-1 |
| PubChem | CID 9826451 |
| DrugBank | DB09198 |
| ChemSpider | 8002194 |
| Synonyms | CKD-501 |
| Chemical data | |
| Formula | C24H24N4O5S |
| Molar mass | 480.53616 g/mol |
Identifications:
| 1H NMR (Estimated) for Lobeglitazone |
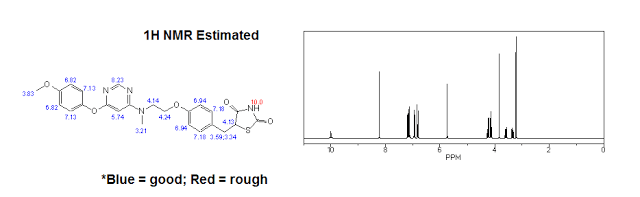
Experimental: 1H NMR (400 MHz, CDCl3) δ 3.12 (m, 4H), 3.45 (m, 1H), 3.83 (s, 3H), 4.00 (m, 2H), 4.16 (m, 2H), 4.50 (m, 1H), 5.84 (bs, 1H), 6.83 (m, 2H), 7.06 (m, 2H), 7.15 (m, 2H), 8.31 (s, 1H), 8.89 (bs, NH).
///Lobeglitazone Sulfate, CKD-501, Duvie®, Approved KOREA, Chong Kun Dang, A dual PPARα and PPARγ agonist , type 2 diabetes, CKD 501, 763108-62-9, 607723-33-1, IDR-105
CN(CCOC1=CC=C(C=C1)CC2C(=O)NC(=O)S2)C3=CC(=NC=N3)OC4=CC=C(C=C4)OC.OS(=O)(=O)O
TOFOGLIFLOZIN 托格列净

TOFOGLIFLOZIN
托格列净
CSG-452, R-7201, RG-7201
903565-83-3 (anhydrous)
(1S,3′R,4′S,5′S,6′R)-6-(4-Ethylbenzyl)-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2′-pyran]-3′,4′,5′-triol hydrate (1:1)
PMDA Pharmaceuticals and Medical Devices Agency, Japan Approved mar24, 2014
THERAPEUTIC CLAIM Treatment of diabetes mellitus
CHEMICAL NAMES
1. Spiro[isobenzofuran-1(3H),2′-[2H]pyran]-3′,4′,5′-triol, 6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-, hydrate (1:1), (1S,3’R,4’S,5’S,6’R)-
2. (1S,3’R,4’S,5’S,6’R)-6-[(4-ethylphenyl)methyl]-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2′-pyran]-3′,4′,5′-triol monohydrate
3. (1S,3’R,4’S,5’S,6’R)-6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-
spiro[isobenzofuran-1(3H),2′-[2H]pyran]-3′,4′,5′-triol monohydrate
(3S,3’R,4’S,5’S,6’R)-5-[(4-ethylphenyl)methyl]-6′-(hydroxymethyl)spiro[1H-2-benzofuran-3,2′-oxane]-3′,4′,5′-triol;hydrate
MW404.5, MF C22H26O6
INNOVATOR Chugai Pharmaceuticals
Sanofi, kowa
Deberza®………..KOWA/Apleway®……………SANOFI
CODE DESIGNATION CSG 452
Tofogliflozin (USAN, codenamed CSG452) is an experimental drug for the treatment of diabetes mellitus and is being developed byChugai Pharma in collaboration with Kowa and Sanofi.[1] It is an inhibitor of subtype 2 sodium-glucose transport protein (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. As of September 2012, the drug is in Phase III clinical trials.[2][3]
Tofogliflozin is an SGLT-2 inhibitor first launched in 2014 in Japan by Sanofi and Kowa for the oral treatment of type II diabetes.
The product was discovered by Chugai and was licensed to Roche in 2007. In 2011, this license agreement was terminated. In 2012, the product was licensed to Kowa and Sanofi by Chugai Pharmaceutical in Japan for the treatment of diabetes type 2. In 2015, the license between Kowa and Chugai was expanded for developments and marketing of the agent in the U.S. and the E.U.
Chemistry
The active moiety or anhydrous form (ChemSpider ID: 28530778, CHEMBL2110731) has the chemical formula C22H26O6 and amolecular mass of 386.44 g/mol.
The United States Adopted Name tofogliflozin applies to the monohydrate, which is the form used as a drug.[4] The International Nonproprietary Name tofogliflozin applies to the anhydrous compound[5] and the drug form is referred to as tofogliflozin hydrate.

Several drugs are available for the treatment of type 2 diabetes mellitus (T2DM), but few patients achieve and maintain glycaemic control without weight gain and hypoglycaemias. Sodium glucose co-transporter 2 (SGLT-2) inhibitors are an emerging class of drugs with an original mechanism of action involving inhibition of renal glucose reabsorption. Two agents of this class, dapagliflozin and canagliflozin, have already been approved, although we need more data on cardiovascular outcomes along with bladder and breast cancer. Tofogliflozin is a further SGLT-2 inhibitor, which exhibits the highest selectivity for SGLT-2, the most potent antidiabetic action and a reduced risk of hypoglycaemia. Recently, a 52-week, multicentre, open-label, randomised controlled trial in Japanese T2DM patients has shown that tofogliflozin exhibits adequate safety and efficacy as monotherapy or as add-on treatment in patients suboptimally controlled with oral agents. Despite the very promising characteristics of this new drug, important questions remain to be answered, mainly additional data on safety outcomes and potential beneficial effects of tofogliflozin, for instance in prediabetes and diabetic nephropathy. Moreover, it would be welcome to examine the utility of its therapeutic use in combination with insulin and metformin.
Tofogliflozin has recently demonstrated safety and efficacy as monotherapy or add-on treatment . This is very important, granted our expectations of SGLT-2 inhibitors as useful alternative oral hypoglycaemic agents. Although important questions remain to be answered, the results of the new trial add to the importance of SGLT-2 inhibitors as a useful new class of oral hypoglycaemic agents.
CLIP
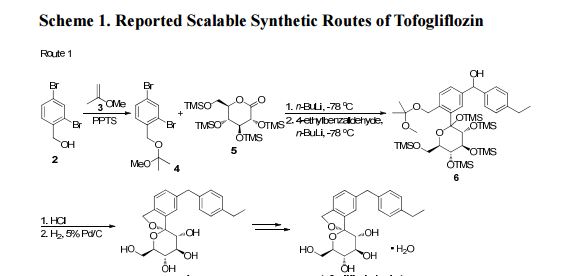
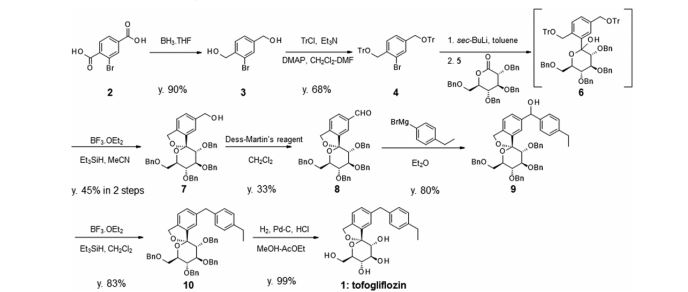
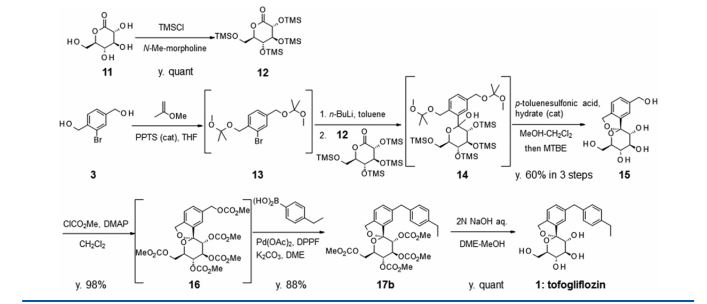
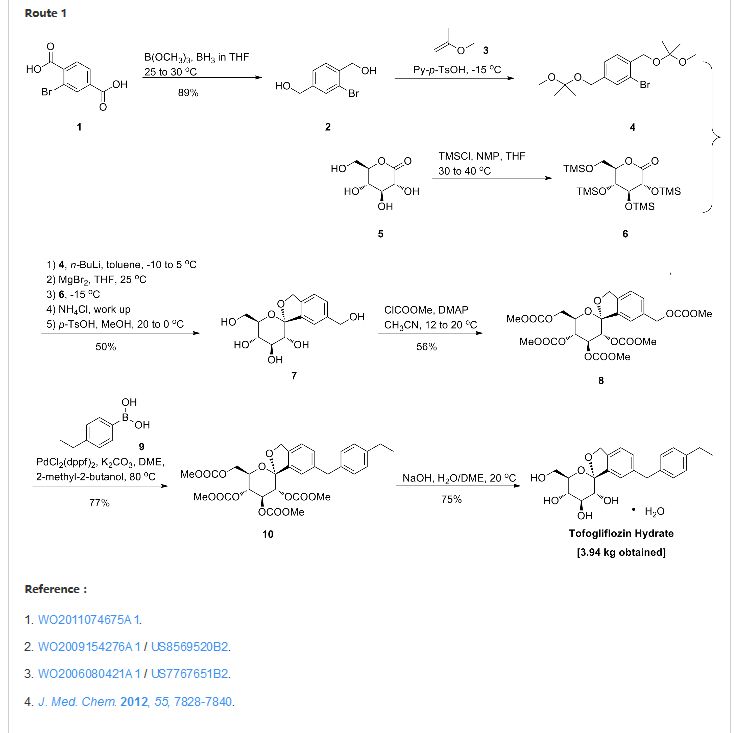
There are two scalable synthetic routes reported to prepare tofogliflozin.2 An efficient production synthesis of tofogliflozin hydrate from alcohol 2 was first described by Murakata et al. (Scheme 1, route 1).2a In 2016, Ohtake et al. reported an improved synthetic route, which achieved in just 7 linear steps (Scheme 1, route 2).2b They selected the optimal protecting groups for the purpose of chemoselective activation and crystalline purification, and obtained the pure tofogliflozin in a good overall yield. However, these methods suffer from several drawbacks. Firstly, some reagents, such as BH3 (Scheme 1, route 2) and 2-Methoxyproene (3, Scheme 1), are toxic or highly volatile. Meanwhile, the use of Palladium reagents may lead to an excess of residual heavy metal in the final product. Secondly, manufacturing costs in these methods are high due to the application of expensive raw materials and reagents. Last but not least, the key tactical stages that involve Br/Li exchange of aryl bromide followed by addition to gluconolactone 5 need the cryogenic conditions (< -60 oC), and this method is not suitable for industrial production. Herein, we report a newly developed synthetic method for tofogliflozin hydrate starting from readily available raw materials and affording good overall yield.
SCHEME 2 FOR

2. (a) Murakata, M.; Ikeda, T.; Kimura, N.; Kawase, A.; Nagase, M.; Yamamoto, K.; Takata, N.; Yoshizaki, S.; Takano, K. Crystal of spiroketal derivative, and process for production thereof. European Appl. EP 2308886 A1, April 13, 2011. (b) Ohtake, Y.; Emura, T.; Nishimoto, M.; Takano, K.; Yamamoto, K.; Tsuchiya, S.; Yeu, S.; Kito, Y.; Kimura, N.; Takeda, S.; Tsukazaki, M.; Murakata, M.; Sato, T. J. Org. Chem. 2016, 81, 2148.

CLIP
Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S.-H.; Ahn, K.-H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. J. Med. Chem. 2012, 55, 7828−7840
DOI: 10.1021/acs.joc.5b02734 J. Org. Chem. 2016, 81, 2148−2153
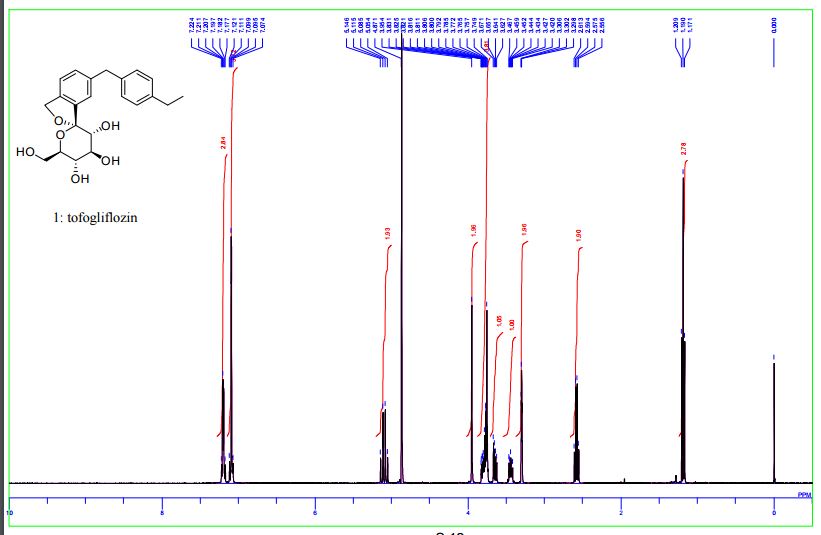
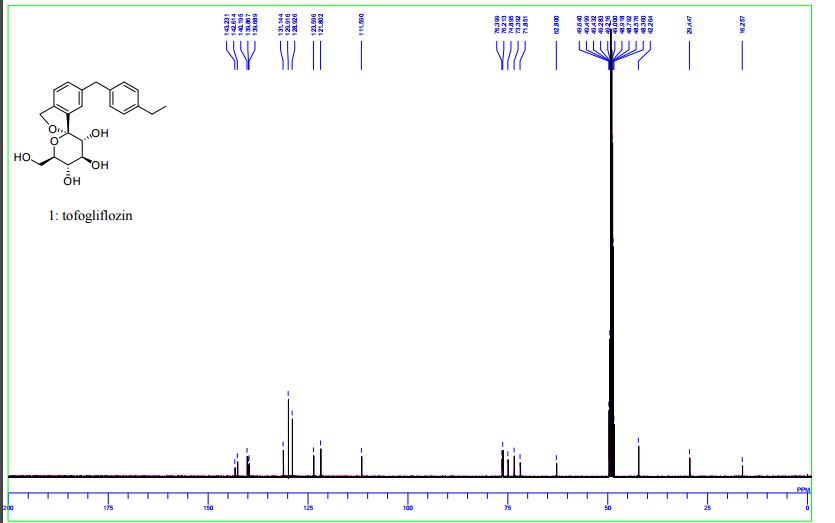
(1S,3′R,4′S,5′S,6′R)-6-[(4-Ethylphenyl)methyl]-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2′- pyran]-3′,4′,5′-triol (1, tofogliflozin).
To a solution of 17b (89.9 g, 145 mmol) in DME (653 mL) and MeOH (73.0 mL), 2 N NaOH aq. solution (726 mL, 1.45 mol) was added dropwise for 1 h at waterbath temperature. After stirring at rt for 1 h, 2 N H2SO4 aq. solution (436 mL) was added slowly to the mixture. Water (700 mL) was added to the mixture, and the resultant mixture was extracted with AcOEt (500 mL × 2). The resultant organic layer was washed with brine (1.00 L) and then dried over anhydrous Na2SO4 (250 g). The mixture was concentrated in vacuo to obtain 1 (57.3 g, quant) as a colorless amorphous solid;
[α]D 26 +24.2° (c 1.02, MeOH);
1 H NMR (400 MHz, CD3OD) δ: 1.19 (3H, t, J = 7.6 Hz), 2.58 (2H, q, J = 7.6 Hz), 3.42−3.47 (1H, m), 3.63−3.67 (1H, m), 3.75−3.88 (4H, m), 3.95 (2H, s), 5.06 (1H, d, J = 12.5 Hz), 5.12 (1H, d, J = 12.5 Hz), 7.07−7.14 (4H, m), 7.17−7.23 (3H, m);
13C NMR (100 MHz, CD3OD) δ: 16.3, 29.4, 42.3, 62.8, 71.9, 73.4, 74.9, 76.2, 76.4, 111.6, 121.8, 123.6, 128.9, 129.9, 131.1, 139.7, 139.9, 140.2, 142.6, 143.2;
MS (ESI) m/z: 387 [M + H]+ ; HRMS (ESI) calcd for C22H27O6 [M + H]+ 387.1802, found 387.1801
DOI: 10.1021/acs.joc.5b02734 J. Org. Chem. 2016, 81, 2148−2153
Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S.-H.; Ahn, K.-H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. J. Med. Chem. 2012, 55, 7828−7840
SGLT2 inhibitors inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with type 2 diabetes. Glucopyranosyl-substituted benzene derivative are described in the prior art as SGLT2 inhibitors, for example in
WO 01/27128, WO 03/099836, WO 2005/092877, WO 2006/034489,
WO 2006/064033, WO 2006/117359, WO 2006/117360,
WO 2007/025943, WO 2007/028814, WO 2007/031548,
WO 2007/093610, WO 2007/128749, WO 2008/049923, WO 2008/055870, WO 2008/055940.
PATENTS
Papers
Chinese Chemical Letters, 2013 , vol. 24, 2 pg. 131 – 133
Journal of Medicinal Chemistry, 2012 , vol. 55, 17 pg. 7828 – 7840
NMR

1 H-NMR (CD 3 OD) δ: 1.19 (3H, t, J = 7.5Hz), 2.59 (2H, q, J = 7.5Hz) ,3.42-3 .46 (1H , m), 3.65 (1H, dd, J = 5.5,12.0 Hz) ,3.74-3 .82 (4H, m), 3.96 (2H, s), 5.07 (1H , d, J = 12.8Hz), 5.13 (1H, d, J = 12.8Hz) ,7.08-7 .12 (4H, m) ,7.18-7 .23 (3H, m) .
MS (ESI +): 387 [M +1] +.
Second set
http://pubs.acs.org/doi/full/10.1021/jm300884k
J. Med. Chem., 2012, 55 (17), pp 7828–7840
1H NMR (400 MHz, CD3OD) δ: 1.20 (3H, t, J = 7.6 Hz), 2.58 (2H, q, J = 7.6 Hz), 3.42–3.47 (1H, m), 3.63–3.67 (1H, m), 3.75–3.88 (4H, m), 3.95 (2H, s), 5.06 (1H, d, J = 12.3 Hz), 5.12 (1H, d, J = 12.5 Hz), 7.07–7.14 (4H, m), 7.17–7.23 (3H, m).
13C NMR (100 MHz, CD3OD) δ: 16.3, 29.4, 42.3, 62.8, 71.9, 73.4, 74.9, 76.2, 76.4, 111.6, 121.8, 123.6, 128.9, 129.9, 131.1, 139.7, 139.9, 140.2, 142.6, 143.2.
MS (ESI): 387 [M + H]+. HRMS (ESI), m/z calcd for C22H27O6 [M + H]+ 387.1802, found 387.1801.
THIRD SET
(1S,3′R,4′S,5′S,6′R)-6-[(4-Ethylphenyl)methyl]-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2′- pyran]-3′,4′,5′-triol (1, tofogliflozin).
To a solution of 17b (89.9 g, 145 mmol) in DME (653 mL) and MeOH (73.0 mL), 2 N NaOH aq. solution (726 mL, 1.45 mol) was added dropwise for 1 h at waterbath temperature. After stirring at rt for 1 h, 2 N H2SO4 aq. solution (436 mL) was added slowly to the mixture. Water (700 mL) was added to the mixture, and the resultant mixture was extracted with AcOEt (500 mL × 2). The resultant organic layer was washed with brine (1.00 L) and then dried over anhydrous Na2SO4 (250 g). The mixture was concentrated in vacuo to obtain 1 (57.3 g, quant) as a colorless amorphous solid;
[α]D 26 +24.2° (c 1.02, MeOH);
1 H NMR (400 MHz, CD3OD) δ: 1.19 (3H, t, J = 7.6 Hz), 2.58 (2H, q, J = 7.6 Hz), 3.42−3.47 (1H, m), 3.63−3.67 (1H, m), 3.75−3.88 (4H, m), 3.95 (2H, s), 5.06 (1H, d, J = 12.5 Hz), 5.12 (1H, d, J = 12.5 Hz), 7.07−7.14 (4H, m), 7.17−7.23 (3H, m);
13C NMR (100 MHz, CD3OD) δ: 16.3, 29.4, 42.3, 62.8, 71.9, 73.4, 74.9, 76.2, 76.4, 111.6, 121.8, 123.6, 128.9, 129.9, 131.1, 139.7, 139.9, 140.2, 142.6, 143.2;
MS (ESI) m/z: 387 [M + H]+ ; HRMS (ESI) calcd for C22H27O6 [M + H]+ 387.1802, found 387.1801
DOI: 10.1021/acs.joc.5b02734 J. Org. Chem. 2016, 81, 2148−2153
Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S.-H.; Ahn, K.-H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. J. Med. Chem. 2012, 55, 7828−7840
PATENT
Prepn
[Example 1] (1S, 3’R, 4’S, 5’S, 6’R) -6 – [(4 – ethyl-phenyl) methyl] -3 ‘, 4’, 5 ‘, 6′-tetrahydro- -6′-(hydroxymethyl) – spiro [isobenzofuran -1 (3H), 2’-[2H] pyran] -3 ‘, 4′, one of the preparation step [compound of formula (IX)] 5’-triol Preparation of methanol (2 – hydroxymethyl-phenyl – bromo-4)

To the mixing solution (1mol / L, 78.9kg, 88.4mol) of borane-tetrahydrofuran complex in tetrahydrofuran (6.34kg, 61.0mol) and, trimethoxyborane, two tetrahydrofuran (33.1kg) in – bromoterephthalic was added at below 30 ℃ solution (7.5kg, 30.6mol) of the acid, and the mixture was stirred for 1 hour at 25 ℃. Then cooled to 19 ℃ The reaction mixture was stirred for 30 minutes and added a mixed solution of tetrahydrofuran and methanol (3.0kg) of (5.6kg). In addition to methanol (15.0kg) in the mixture was kept for a while.
Again, to the mixing solution (1mol / L, 78.9kg, 88.4mol) of borane-tetrahydrofuran complex in tetrahydrofuran (6.34kg, 61.0mol) and, trimethoxyborane, two tetrahydrofuran (33.0kg) in – was added at below 30 ℃ solution (7.5kg, 30.6mol) of bromo terephthalic acid, and the reaction was carried out for 1 hour at 25 ℃. Then cooled to 18 ℃ The reaction mixture was stirred for 30 minutes and added a mixed solution of tetrahydrofuran and methanol (3.0kg) of (5.6kg). After addition of methanol (15.0kg) in the mixture is combined with the reaction mixture obtained in the previous reaction, and then the solvent was distilled off under reduced pressure. After addition of methanol (36kg) residue was obtained, and the solvent was evaporated under reduced pressure. Furthermore, (54 ℃ dissolved upon confirmation) which was dissolved by warming was added to methanol (36kg) to the residue. After cooling to room temperature the solution was stirred for 30 minutes added water (60kg). After addition of water (165kg) In addition to this mixture was cooled to 0 ℃, and the mixture was stirred for one hour. Centrifuge the obtained crystals were washed twice with water (45kg), and dried for 2 hours under reduced pressure to give (11.8kg, 54.4mol, 89% yield) of the title compound.
1 H-NMR (DMSO-d 6) δ: 4.49 (4H, t, J = 5.8Hz), 5.27 (1H, t, J = 5.8Hz), 5.38 (1H, t, J = 5.8Hz), 7.31 (1H, d, J = 7.5Hz), 7.47 (1H, d, J = 7.5Hz), 7.50 (1H, s).
Preparation of benzene (ethoxy methyl – methyl – – methoxy-1 1) – bromo-1 ,4 – 2:2 process bis

(- Bromo-4 – 2-hydroxyethyl methyl phenyl) in tetrahydrofuran (57kg) in the solution (8.0kg, 36.9mol) of methanol, I added (185.12g, 0.74mol) of pyridinium p-toluenesulfonate. After cooling to -15 ℃ below the mixture, 2 – was added at -15 ℃ or less (7.70kg, 106.8mol) methoxy propene, and the mixture was stirred 1 h at -15 ~ 0 ℃. Was added aqueous potassium carbonate (25 wt%, 40kg) and the reaction mixture was warmed to room temperature and separate the organic layer was added toluene (35kg). After washing with water (40kg) The organic layer was evaporated under reduced pressure. Was dissolved in toluene (28kg) and the residue obtained was obtained as a toluene solution of the title compound.
1 H-NMR (CDCl 3) δ: 1.42 (6H, s), 1.45 (6H, s), 3.24 (3H, s), 3.25 (3H, s), 4.45 ( 2H, s), 4.53 (2H, s), 7.28 (1H, dd, J = 1.5,8.0 Hz), 7.50 (1H, d, J = 8.0Hz), 7. 54 (1H, d, J = 1.5Hz).
MS (ESI +): 362 [M +2] +.
Preparation of on – (3R, 4S, 5R, 6R) -3,4,5 – tris (trimethylsilyloxy)-6 – trimethylsilyloxy methyl – tetrahydropyran-2: Step 3

Glucono -1,5 – – D-(+) in tetrahydrofuran (70kg) in the solution (35.8kg, 353.9mol) of N-methylmorpholine (7.88kg, 44.23mol) and lactone, chlorotrimethylsilane ( was added at 40 ℃ less 29.1kg, and 267.9mol), and the mixture was stirred for 2 hours at 30 ~ 40 ℃ resulting mixture. Was cooled to 0 ℃ the reaction mixture was added toluene (34kg) water (39kg), and the organic layer was separated. Twice sodium dihydrogen phosphate aqueous solution (5 wt%, 39.56kg) in, washed once with water (39kg) the organic layer the solvent was evaporated under reduced pressure. Was dissolved in toluene (34.6kg) and the residue obtained was obtained as a toluene solution of the title compound.
1 H-NMR (CDCl 3) δ: 0.13 (9H, s), 0.17 (9H, s), 0.18 (9H, s), 0.20 (9H, s), 3.74- 3.83 (3H, m), 3.90 (1H, t, J = 8.0Hz), 3.99 (1H, d, J = 8.0Hz), 4.17 (1H, dt, J = 2 .5,8.0 Hz).
Step 4: (1S, 3’R, 4’S, 5’S, 6’R) -3 ‘, 4’, 5 ‘, 6′-tetrahydro -6,6′ – bis (hydroxymethyl) – spiro [ (3H), 2’-[2H] pyran] -3 ‘, 4′, 5’-Preparation of triol isobenzofuran-1

(Methyl – – – methoxy 1-ethoxy-methyl) – bromo-1 ,4 – 2 prepared in step 2 bis cooled to below -10 ℃ toluene solution of benzene, hexane solution to (15 wt% n-butyl lithium , was added at below 0 ℃ 18.2kg, and 42.61mol), and the mixture was stirred 1.5 h at 5 ℃ resulting mixture. (10.5kg, 40.7mol), was added tetrahydrofuran (33.4kg) then magnesium bromide diethyl ether complex in the mixture, and the mixture was stirred for 1 hour at 25 ℃. Was added at below -10 ℃ toluene solution of the on – tris (trimethylsilyloxy) -6 – – 3,4,5 cooled to -15 ℃ below the mixture prepared in step 3 trimethylsilyloxy methyl – tetrahydropyran-2 was. After stirring 0.5 h at -15 ℃ or less, poured into 20% aqueous ammonium chloride solution to (80kg) of this solution, and the organic layer was separated. After washing with water (80kg) and the organic layer obtained, and the solvent was evaporated under reduced pressure. I was dissolved in methanol (43kg) residue was obtained. Was stirred for 1 hour at 20 ℃ was added (1.4kg, 7.4mol) and p-toluenesulfonic acid monohydrate in the mixture. Thereafter, it was stirred for another hour and cooled to 0 ℃, centrifuged crystals obtained was washed with methanol (25kg), and dried for 8 hours at reduced pressure under 40 ℃, (5.47kg, yield the title compound I got 50%) rate.
1 H-NMR (DMSO-d 6) δ :3.20-3 .25 (1H, m) ,3.41-3 .45 (1H, m) ,3.51-3 .62 (4H, m) , 4.39 (1H, t, J = 6.0Hz) ,4.52-4 .54 (3H, m), 4.86 (1H, d, J = 4.5Hz), 4.93 (1H, d, J = 5.5Hz), 4.99 (1H, d, J = 12.5Hz), 5.03 (1H, d, J = 12.5Hz), 5.23 (1H, t, J = 5 .8 Hz) ,7.24-7 .25 (2H, m), 7.29 (1H, dd, J = 1.5,8.0 Hz).
Step 5: (1S, 3’R, 4’S, 5’S, 6’R) -6 – [(methoxycarbonyl) methyl] -3 ‘, 4’, 5 ‘, 6′-tetrahydro-3’ , 4 ‘, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – Preparation of [(3H), 2’-[2H] pyran isobenzofuran] spiro

(1S, 3’R, 4’S, 5’S, 6’R) – tetrahydro -6,6 ‘- bis (hydroxymethyl) – spiro [isobenzofuran -1 (3H), 2’-[2H] pyran ] -3 ‘, 4′, 5’-triol 4 (5.3kg, 17.8mol) and – dissolved in acetonitrile (35kg) (13.7kg, 112.1mol) a chloroformate, in the solution of dimethylaminopyridine I was added at 12 ℃ or less (10.01kg, 105.9mol) methyl. Heated to 20 ℃, After stirring for 1 h, was added ethyl acetate (40kg) and water (45kg), and the organic layer was separated and the mixture. Once (45.4kg) aqueous solution consisting of (9.01kg) sodium chloride and potassium hydrogen sulfate (1.35kg), sodium chloride aqueous solution (weight 10%, 44.5kg), sodium chloride aqueous solution (the organic layer was washed successively 20% by weight, in 45.0kg), and the solvent was evaporated under reduced pressure. Was dissolved in ethylene glycol dimethyl ether (18kg) and the residue obtained was then evaporated under reduced pressure. Was dissolved in ethylene glycol dimethyl ether (13.2kg) again and the residue obtained was obtained as ethylene glycol dimethyl ether solution of the title compound. I was used as it was in the six step.
1 H-NMR (CDCl 3) δ: 3.54 (3H, s), 3.77 (6H, s), 3.811 (3H, s), 3.812 (3H, s), 4.23 ( 1H, dd, J = 2.8,11.9 Hz), 4.32 (1H, dd, J = 4.0,11.9 Hz) ,4.36-4 .40 (1H, m), 5.11 -5.24 (5H, m), 5.41 (1H, d, J = 9.8Hz), 5.51 (1H, t, J = 9.8Hz), 7.25 (1H, d, J = 7.5Hz), 7.42 (1H, d, J = 7.5Hz), 7.44 (1H, s).
MS (ESI +): 589 [M +1] +, 606 [M +18] +.
Step 6: (1S, 3’R, 4’S, 5’S, 6’R) -6 – [(4 – ethyl-phenyl) methyl] -3 ‘, 4’, 5 ‘, 6’-tetrahydro-3 ‘4’, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – Preparation of [(3H), 2′-[2H] pyran isobenzofuran] spiro

[(Methoxycarbonyl) methyl] -3 ‘, 4’, 5 ‘, 6’-tetrahydro – (1S, 3’R, 4’S, 5’S, 6’R) -6 which had been prepared in Step 5 – 3 ‘, 4′, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – spiro [isobenzofuran -1 (3H), 2’-[2H] pyran] Ethylene glycol dimethyl ether in solution, 2 – (2.46kg, 17.8mol), 4 butanol (25kg), anhydrous potassium carbonate – – methyl-2 were sequentially added (3.73kg, 24.9mol) ethyl phenyl boronic acid, in the reaction vessel was replaced with argon atmosphere, was bubbled with argon mixture. To the mixture – after the addition (0.72kg, 0.88mol) and palladium (II) chloride dichloromethane adduct [1,1 ‘-bis (diphenylphosphino) ferrocene], it was replaced with argon again inside of the vessel, one at 80 ℃ I was stirring time. After cooling, I added sequentially (0.859kg, 5.3mol) of ethylene glycol dimethyl ether (9.85kg), ethyl acetate (19kg), N-acetyl-L-cysteine in the mixture. After stirring for 2.5 h the mixture was filtered and added Celite (5.22kg), and washed with ethyl acetate (78kg) and the filter residue. The combined washings and filtrate, and the solvent is evaporated off under reduced pressure, and in addition (0.58kg, 3.6mol) and ethanol (74kg), N-acetyl-L-cysteine residue was obtained, which is heated to 70 ℃ or I was dissolved residue is then. After addition of water (9.4kg) in the solution, cooled to 60 ℃, and the mixture was stirred for 1 h. After confirming solid precipitated, cooled to 0 ℃ from 60 ℃ over 2.5 hours or more The mixture was stirred for 1 hour or more at 5 ℃ less. Centrifuge the resulting solid was washed twice with a mixture of water (35kg) and ethanol (55kg). Was dissolved at 70 ℃ ethanol (77kg) again, wet powder was obtained (10.21kg), cooled to 60 ℃ added water (9.7kg), and the mixture was stirred for 1 h. After confirming solid precipitated, cooled to 0 ℃ from 60 ℃ over 2.5 hours or more, and the mixture was stirred for 1 hour or more at 5 ℃ less. (9.45kg, dry powder rate 8.47kg, 13.7mol which was centrifuged obtained crystals were washed with a mixture of water (32kg) and ethanol (51kg), was obtained as a moist powder the title compound, 77% overall yield from the previous step).
1 H-NMR (CDCl 3) δ: 1.20 (3H, t, J = 7.5Hz), 2.60 (2H, q, J = 7.5Hz), 3.50 (3H, s), 3 .76 (3H, s), 3.77 (3H, s), 3.81 (3H, s), 3.96 (2H, s), 4.23 (1H, dd, J = 2.8,11 .9 Hz), 4.33 (1H, dd, J = 4.5,11.9 Hz) ,4.36-4 .40 (1H, m) ,5.11-5 .20 (3H, m), 5 .41 (1H, d, J = 10.0Hz), 5.51 (1H, t, J = 10.0Hz) ,7.07-7 .11 (4H, m), 7.14 (1H, d, J = 7.8Hz), 7.19 (1H, dd, J = 1.5,7.8 Hz), 7.31 (1H, d, J = 1.5Hz).
MS (ESI +): 619 [M +1] +, 636 [M +18] +.
Step 7: (1S, 3’R, 4’S, 5’S, 6’R) -6 – [(4 – ethyl-phenyl) methyl] -3 ‘, 4’, 5 ‘, 6’-tetrahydro-6 , 4 ‘, 5′-Preparation of triol’ – -3 [(3H), 2′-[2H] pyran isobenzofuran] spiro – (hydroxymethyl) ‘

(1S, 3’R, 4’S, 5’S, 6’R) -6 – [(4 – ethyl-phenyl) methyl] -3 ‘, 4’, 5 ‘, 6′-tetrahydro-3’, 4 ‘, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – wet powder spiro [(3H), 2’-[2H] pyran isobenzofuran -1] (8.92kg, In addition at 20 ℃ (4mol / L, 30.02kg, the 104.2mol) aqueous solution of sodium hydroxide, 1 hour the reaction mixture to a solution of (28kg) ethylene glycol dimethyl ether dry end conversion 8.00kg, of 12.9mol) the mixture was stirred. And the organic layer was separated by addition of water (8.0kg) in the mixture. The ethyl acetate aqueous sodium chloride solution (25 wt%, 40kg) and a (36kg) in the organic layer and the aqueous layer was removed after washing. The washed again aqueous sodium chloride solution (25 wt%, 40kg) in the organic layer was evaporated under reduced pressure. Were added and acetone (32.0kg) water (0.8kg) residue was obtained. After the solvent was evaporated under reduced pressure, dissolved in acetone (11.7kg) in water (15.8kg) and the residue obtained was cooled to below 5 ℃. Was added below 10 ℃ water (64kg) to the mixture, and the mixture was stirred for 1 hour at below 10 ℃. Centrifuge the resulting crystals were washed with a mixture of water (8.0kg) and (1.3kg) acetone. For 8 hours through-flow drying 13 ~ 16 ℃ temperature ventilation, under the conditions of 24-33% relative humidity the wet powder, the monohydrate crystal (3.94kg, 9.7mol, 75% yield) of the title compound I was obtained as: (4.502 wt% water content).
Method of measuring the amount of water:
Analysis: coulometric KF titration analyzer: trace moisture measurement device manufactured by Mitsubishi Chemical Corporation Model KF-100
Anolyte: Aqua micron AX (manufactured by Mitsubishi Chemical Corporation)
Catholyte: Aqua micron CXU (manufactured by Mitsubishi Chemical Corporation)
1 H-NMR (CD 3 OD) δ: 1.19 (3H, t, J = 7.5Hz), 2.59 (2H, q, J = 7.5Hz) ,3.42-3 .46 (1H , m), 3.65 (1H, dd, J = 5.5,12.0 Hz) ,3.74-3 .82 (4H, m), 3.96 (2H, s), 5.07 (1H , d, J = 12.8Hz), 5.13 (1H, d, J = 12.8Hz) ,7.08-7 .12 (4H, m) ,7.18-7 .23 (3H, m) .
MS (ESI +): 387 [M +1] +.
PATENT
Example 1 Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose Step 1: Synthesis of 3,4,5-tris(trimethylsilyloxy)-6-trimethylsilyloxymethyl-tetrahydropyran-2-one

To a solution of D-(+)-glucono-1,5-lactone (7.88 kg) and N-methylmorpholine (35.8 kg) in tetrahydrofuran (70 kg) was added trimethylsilyl chloride (29.1 kg) at 40° C. or below, and then the mixture was stirred at a temperature from 30° C. to 40° C. for 2 hours. After the mixture was cooled to 0° C., toluene (34 kg) and water (39 kg) were added thereto. The organic layer was separated and washed with an aqueous solution of 5% sodium dihydrogen phosphate (39.56 kg×2) and water (39 kg×1). The solvent was evaporated under reduced pressure to give the titled compound as an oil. The product was used in the next step without further purification.
1H-NMR (CDCl3) δ: 0.13 (9H, s), 0.17 (9H, s), 0.18 (9H, s), 0.20 (9H, s), 3.74-3.83 (3H, m), 3.90 (1H, t, J=8.0 Hz), 3.99 (1H, d, J=8.0 Hz), 4.17 (1H, dt, J=2.5, 8.0 Hz).
Step 2: Synthesis of 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene

Under a nitrogen atmosphere, to a solution of 2,4-dibromobenzyl alcohol (40 g, 0.15 mol) in tetrahydrofuran (300 ml) was added 2-methoxypropene (144 ml, 1.5 mol) at room temperature, and then the mixture was cooled to 0° C. At the same temperature, pyridinium p-toluenesulfonic acid (75 mg, 0.30 mmol) was added and the mixture was stirred for 1 hour. The reaction mixture was poured into a saturated aqueous solution of sodium hydrogen carbonate cooled to 0° C., and extracted with toluene. The organic layer was washed with a saturated aqueous solution of sodium chloride, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure to give the titled compound as an oil in quantitative yield. The product was used in the next step without further purification.
1H-NMR (CDCl3) δ: 1.44 (6H, s), 3.22 (3H, 4.48 (2H, s), 7.42 (1H, d, J=8.0 Hz), 7.44 (1H, dd, J=1.5, 8.0 Hz), 7.68 (1H, d, J=1.5 Hz).
Step 3: Synthesis of 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-(4-ethylphenyl)hydroxymethyl-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran

Under a nitrogen atmosphere, 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene (70 g, 207 mmol), which was obtained in the previous step, was dissolved in toluene (700 mL) and t-butylmethyl ether (70 ml), and n-butyllithium in hexane (1.65 M, 138 ml, 227 mmol) was added dropwise at 0° C. over 30 minutes. After the mixture was stirred for 1.5 hours at 0° C., the mixture was added dropwise to a solution of 3,4,5-tris(trimethylsilyloxy)-6-trimethylsilyloxymethyl-tetrahydropyran-2-one (Example 1, 108 g, 217 mol) in tetrahydrofuran (507 ml) at −78° C., and the reaction mixture was stirred for 2 hours at the same temperature. Triethylamine (5.8 ml, 41 mmol) and trimethylsilyl chloride (29.6 ml, 232 mmol) were added thereto, and the mixture was warmed to 0° C. and stirred for 1 hour to give a solution containing 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-bromo-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran.
The resulting solution was cooled to −78° C., and n-butyllithium in hexane (1.65 M, 263 ml, 434 mmol) was added dropwise thereto at the same temperature. After the mixture was stirred at −78° C. for 30 minutes, 4-ethylbenzaldehyde (62 ml, 455 mmol) was added dropwise at −78° C., and the mixture was stirred at the same temperature for 2 hours. A saturated aqueous solution of ammonium chloride was added to the reaction mixture, and the organic layer was separated, and washed with water. The solvent was evaporated under reduced pressure to give a product containing the titled compound as an oil (238 g). The product was used in the next step without further purification.
A portion of the oil was purified by HPLC (column: Inertsil ODS-3, 20 mm I.D.×250 mm; acetonitrile, 30 mL/min) to give four diastereomers of the titled compound (two mixtures each containing two diastereomers).
Mixture of Diastereomers 1 and 2:
1H-NMR (500 MHz, CDCl3) δ: −0.47 (4.8H, s), −0.40 (4.2H, s), −0.003-0.004 (5H, m), 0.07-0.08 (1314, m), 0.15-0.17 (18H, m), 1.200 and 1.202 (3H, each t, J=8.0 Hz), 1.393 and 1.399 (3H, each s), 1.44 (3H, s), 2.61 (2H, q, J=8.0 Hz), 3.221 and 3.223 (3H, each s), 3.43 (1H, t, J=8.5 Hz), 3.54 (1H, dd, J=8.5, 3.0 Hz), 3.61-3.66 (1H, m), 3.80-3.85 (3H, m), 4.56 and 4.58 (1H, each d, J=12.4 Hz), 4.92 and 4.93 (1H, each d, J=12.4 Hz), 5.80 and 5.82 (1H, each d, J=3.0 Hz), 7.14 (2H, d, J=8.0 Hz), 7.28-7.35 (3H, m), 7.50-7.57 (2H, m).
MS (ESI+): 875 [M+Na]+.
Mixture of Diastereomers 3 and 4:
1H-NMR (500 MHz, toluene-d8, 80° C.) δ: −0.25 (4H, s), −0.22 (5H, s), 0.13 (5H, s), 0.16 (4H, s), 0.211 and 0.214 (9H, each s), 0.25 (9H, s), 0.29 (9H, s), 1.21 (3H, t, J=7.5 Hz), 1.43 (3H, s), 1.45 (3H, s), 2.49 (2H, q, J=7.5 Hz), 3.192 and 3.194 (3H, each s), 3.91-4.04 (4H, m), 4.33-4.39 (2H, m), 4.93 (1H, d, J=14.5 Hz), 5.10-5.17 (1H, m), 5.64 and 5.66 (1H, each s), 7.03 (2H, d, J=8.0 Hz), 7.28-7.35 (3H, m), 7.59-7.64 (1H, m), 7.87-7.89 (1H, m).
MS (ESI+): 875 [M+Na]+.
Step 4: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)hydroxymethyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose

Under a nitrogen atmosphere, the oil containing 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-(4-ethylphenyl)hydroxymethyl-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran (238 g), which was obtained in the previous step, was dissolved in acetonitrile (693 ml). Water (37 ml) and 1N HCl aq (2.0 ml) were added and the mixture was stirred at room temperature for 5.5 hours. Water (693 ml) and n-heptane (693 ml) were added to the reaction mixture and the aqueous layer was separated. The aqueous layer was washed with n-heptane (693 ml×2), and water was evaporated under reduced pressure to give a product containing water and the titled compound (a diastereomer mixture) as an oil (187 g). The product was used in the next step without further purification.
1H-NMR (500 MHz, CD3OD) δ: 1.200 (3H, t, J=7.7 Hz), 1.201 (3H, t, J=7.7 Hz), 2.61 (2H, q, J=7.7 Hz), 3.44-3.48 (1H, m), 3.63-3.68 (111, m), 3.76-3.84 (4H, m), 5.09 (1H, d, J=12.8 Hz), 5.15 (1H, d, J=12.8 Hz), 5.79 (1H, s), 7.15 (2H, d, J=7.7 Hz), 7.24 and 7.25 (1H, each d, J=8.4 Hz), 7.28 (2H, d, J=7.7 Hz), 7.36 (1H, dd, J=8.4, 1.5 Hz), 7.40-7.42 (114, m).
MS (ESI+): 425 [M+Na]+.
Step 5: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (crude product)

To a solution of the oil containing 1,1-anhydro-1-C-[5-(4-ethylphenyl)hydroxymethyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (187 g), which was obtained in the previous step, in 1,2-dimethoxyethane (693 ml) was added 5% Pd/C (26 g, 6.2 mmol, water content ratio: 53%), and the mixture was stirred in the atmosphere of hydrogen gas at room temperature for 4 hours. After filtration, the filtrate was evaporated under reduced pressure to give an oil containing the titled compound (59 g). The purity of the resulting product was 85.7%, which was calculated based on the area ratio measured by HPLC. The product was used in the next step without further purification.
1H-NMR (CD3OD) δ: 1.19 (3H, t, J=7.5 Hz), 2.59 (2H, q, J=7.5 Hz), 3.42-3.46 (1H, m), 3.65 (1H, dd, J=5.5, 12.0 Hz), 3.74-3.82 (4H, m), 3.96 (2H, s), 5.07 (1H, d, J=12.8 Hz), 5.13 (1H, d, J=12.8 Hz), 7.08-7.12 (4H, m), 7.18-7.23 (3H, m).
MS (ESI+): 387 [M+1]+.
Measurement Condition of HPLC:
Column: Cadenza CD-C18 50 mm P/NCD032
Mobile phase: Eluent A: H2O, Eluent B: MeCN
Gradient operation: Eluent B: 5% to 100% (6 min), 100% (2 min)
Flow rate: 1.0 mL/min
Temperature: 35.0° C.
Detection wavelength: 210 nm
Step 6: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-2,3,4,6-tetra-O-methoxycarbonyl-β-D-glucopyranose
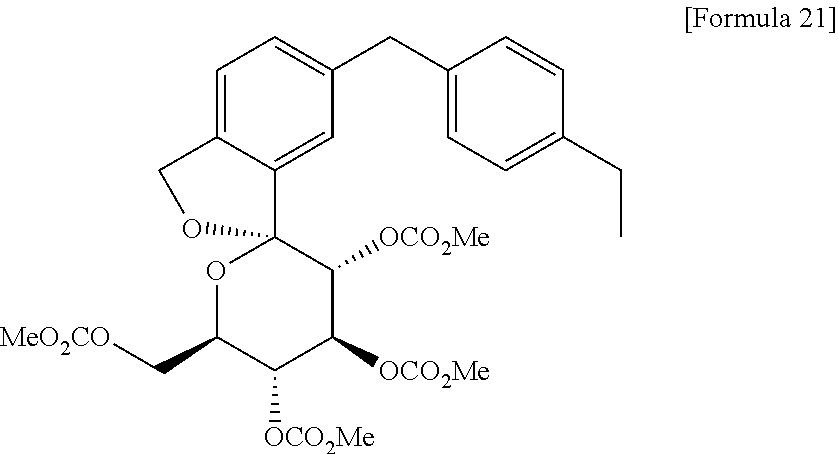
Under a nitrogen atmosphere, to a solution of the oil containing 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (59 g) and 4-(dimethylamino)pyridine (175 g, 1436 mmol) in acetonitrile (1040 ml) was added dropwose methyl chloroformate (95 ml, 1231 mmol) at 0° C. The mixture was allowed to warm to room temperature while stirred for 3 hours. After addition of water, the mixture was extracted with isopropyl acetate. The organic layer was washed with an aqueous solution of 3% potassium hydrogensulfate and 20% sodium chloride (three times) and an aqueous solution of 20% sodium chloride, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. To the resulting residue was added ethanol (943 mL) and the mixture was heated to 75° C. to dissolve the residue. The mixture was cooled to 60° C. and a seed crystal of the titled compound was added thereto. The mixture was cooled to room temperature and stirred for 1 hour. After precipitation of solid was observed, water (472 ml) was added thereto, and the mixture was stirred at room temperature for 2 hours. The resulting crystal was collected by filtration, washed with a mixture of water and ethanol (1:1), and dried under reduced pressure to give the titled compound (94 g). To the product (91 g) was added ethanol (1092 ml), and the product was dissolved by heating to 75° C. The solution was cooled to 60° C. and a seed crystal of the titled compound was added thereto. The mixture was cooled to room temperature and stirred for 1 hour. After precipitation of solid was observed, water (360 ml) was added thereto, and the mixture was stirred at room temperature for 2 hours. The resulting crystal was collected by filtration, washed with a mixture of water and ethanol (1:1), and dried under reduced pressure to give the titled compound [83 g, total yield from 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene used in Step 3: 68%].
1H-NMR (CDCl3) δ: 1.20 (3H, t, J=7.5 Hz), 2.60 (2H, q, J=7.5 Hz), 3.50 (3H, s), 3.76 (3H, s), 3.77 (3H, s), 3.81 (3H, s), 3.96 (2H, s), 4.23 (1H, dd, J=2.5, 11.8 Hz), 4.33 (1H, dd, J=4.5, 12.0 Hz), 4.36-4.40 (1H, m), 5.11-5.20 (3H, m), 5.41 (1H, d, J=10.0 Hz), 5.51 (1H, t, J=10.0 Hz), 7.07-7.11 (4H, m), 7.14 (1H, d, J=7.5 Hz), 7.19 (1H, dd, J=1.5, 7.8 Hz), 7.31 (1H, d, J=1.5 Hz).
MS (ESI+): 619 [M+1]+, 636 [M+18]+.
Another preparation was carried out in the same manner as Step 6, except that a seed crystal was not used, to give the titled compound as a crystal.
Step 7: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose

To a solution of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-2,3,4,6-tetra-O-methoxycarbonyl-β-D-glucopyranose (8.92 kg as wet powder, corresponding to 8.00 kg of dry powder) in 1,2-dimethoxyethane (28 kg) was added a solution of sodium hydroxide (4 mol/L, 30.02 kg) at 20° C., and the mixture was stirred for 1 hour. Water (8.0 kg) was added to the mixture and the layers were separated. To the organic layer were added an aqueous solution of 25% sodium chloride (40 kg) and ethyl acetate (36 kg). The organic layer was separated, washed with an aqueous solution of 25% sodium chloride (40 kg), and the solvent was evaporated under reduced pressure. The purity of the resulting residue was 98.7%, which was calculated based on the area ratio measured by HPLC. To the resulting residue were added acetone (32.0 kg) and water (0.8 kg), and the solvent was evaporated under reduced pressure. To the resulting residue were added acetone (11.7 kg) and water (15.8 kg), and the solution was cooled to 5° C. or below. Water (64 kg) was added to the solution at 10° C. or below, and the mixture was stirred at the same temperature for 1 hour. The resulting crystal was collected by centrifugation, and washed with a mixture of acetone (1.3 kg) and water (8.0 kg). The resulting wet powder was dried by ventilation drying under a condition at air temperature of 13 to 16° C. and relative humidity of 24% to 33% for 8 hours, to give a monohydrate crystal (water content: 4.502%) of the titled compound (3.94 kg). The purity of the resulting compound was 99.1%, which was calculated based on the area ratio measured by HPLC.
1H-NMR (CD3OD) δ: 1.19 (3H, t, J=7.5 Hz), 2.59 (2H, q, J=7.5 Hz), 3.42-3.46 (1H, m), 3.65 (1H, dd, J=5.5, 12.0 Hz), 3.74-3.82 (4H, m), 3.96 (2H, s), 5.07 (1H, d, J=12.8 Hz), 5.13 (1H, d, J=12.8 Hz), 7.08-7.12 (4H, m), 7.18-7.23 (311, m).
MS (ESI+): 387 [M+1]+.
Measurement Condition of HPLC:
Column: Capcell pack ODS UG-120 (4.6 mm I.D.×150 mm, 3 μm, manufactured by Shiseido Co., Ltd.)
Mobile phase: Eluent A: H2O, Eluent B: MeCN
Mobile phase sending: Concentration gradient was controlled by mixing Eluent A and Eluent B as indicated in the following table.
| TABLE 1 | ||||
| Time from | ||||
| injection (min) | Eluent A (%) | Eluent B (%) | ||
| 0 to 15 | 90→10 | 10→90 | ||
| 15 to 17.5 | 10 | 90 | ||
| 17.5 to 25 | 90 | 10 | ||
Flow rate: 1.0 mL/min
Temperature: 25.0° C.
Detection wavelength: 220 nm
Method for Measurement of Water Content:
Analysis method: coulometric titration method
KF analysis apparatus: Type KF-100 (trace moisture measuring apparatus manufactured by Mitsubishi Chemical Corporation)
Anode solution: Aquamicron AX (manufactured by Mitsubishi Chemical Corporation)
Cathode solution: Aquamicron CXU (manufactured by Mitsubishi Chemical Corporation)
PATENT
The compound of the present invention can be synthesized as shown in Scheme 1:

wherein R11 and R12 have the same meaning as defined above for substituents on Ar1, A is as defined above, and P represents a protecting group for a hydroxyl group.
CLIP
Tofogliflozin hydrate (Deberza)
Tofogliflozin hydrate, which is a sodium-glucose co-transporter 2 inhibitor, was approved in Japan for the treatment of type 2 diabetes
at the same time as luseogliflozin hydrate (XIX). The drug was discovered by Chugai Pharmaceutical and jointly developed
with Sanofi-Aventis and Kowa.263
Tofogliflozin hydrate reduces glucose levels by inhibiting the reuptake of glucose by selectively
inhibiting SGLT2, and plays a key role in the reuptake of glucose in the proximal tubule of the kidneys.264–266 The synthetic
approach described in Scheme 48 represents the largest scale reported to date in a patent application.263,266–268
Reduction of commercially available 2-bromoterephtalic acid (268, Scheme 48) through the use of trimethoxyborane and borane-THF proceeded in 89% yield to afford diol 269.
Subjection of this compound to 2-methoxypropene (270) under acidic conditions generated bis-acetonide 271. This bromide then underwent lithium–halogen exchange followed by exposure to magnesium bromide and treatment with lactone 272 (which was prepared by persilylation of commercially available (3R,4S,5S,6R)-3,4,5-trihydroxy-6-hydroxymethyl)tetrahydro-2Hpyran-2-one (277, Scheme 49).
This mixture was worked up with aqueous ammonium chloride and upon treatment with p-TsOH in methanol resulted in spiroacetal 273. Next, global protection of all alcohol functionalities within 273 was affected by reaction with methylchloroformate and DMAP in acetonitrile.
The benzyl carbonate within 274 was selectively exchanged via Suzuki coupling with 4-ethylphenylboronic acid (275) to afford methylene dibenzyl system 276. Subsequent treatment with aqueous sodium hydroxide in methanol followed by crystallization from 1:6 acetone and water furnished the desired product tofogliflozin hydrate (XXXIV) in 75% yield.
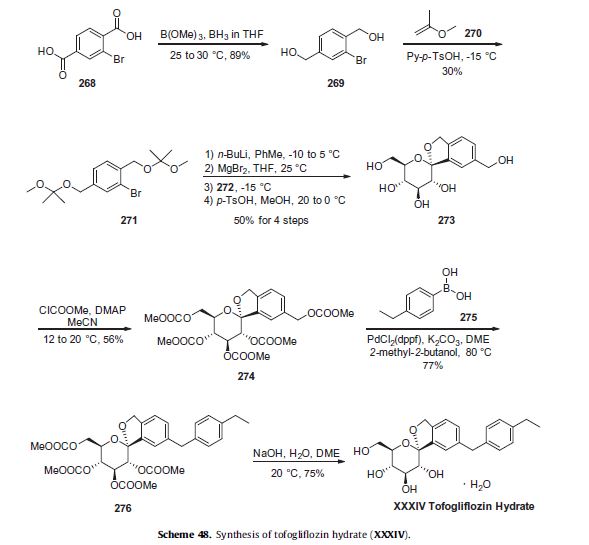
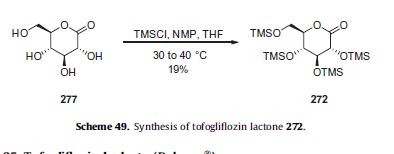
263 Takamitsu, K.; Tsutomu, S.; Masahiro, N. WO Patent 2006080421A1, 2006.
264. http://www.info.pmda.go.jp/shinyaku/P201400036/index.html.
265. Pafili, K.; Papanas, N. Expert Opin. Pharmacother. 2014, 15, 1197.
266. Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.;Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S. Y.; Ahn, K. H.;Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.;Kato, M.; Ikeda, S. J. Med. Chem. 2012, 55, 7828.
267. Murakata, M.; Ikeda, T.; Kawase, A.; Nagase, M.; Kimura, N.; Takeda, S.;Yamamoto, K.; Takano, K.; Nishimoto, M.; Ohtake, Y.; Emura, T.; Kito, Y. WOPatent 2011074675A1, 2011.
268. Murakata, M.; Takuma, I.; Nobuaki, K.; Masahiro, N.; Kawase, A.; Nagase, M.;Yamamoto, K.; Takata, N.; Yoshizaki, S. WO Patent 2009154276A1, 2009.
Paper
A Scalable Synthesis of Tofogliflozin Hydrate

A newly process for the synthesis of tofogliflozin hydrate, a sodium-glucose cotransporter type 2 (SGLT2) inhibitor, was described. Three improvements were achieved, including the development of a regioselective Friedel–Crafts reaction, a high-yield reduction, and a mild metal–halogen exchange. These improvements ultimately resulted in the isolation of tofogliflozin hydrate as a white solid in >99% purity (HPLC area) and 23% overall yield after 12 steps without column chromatography.

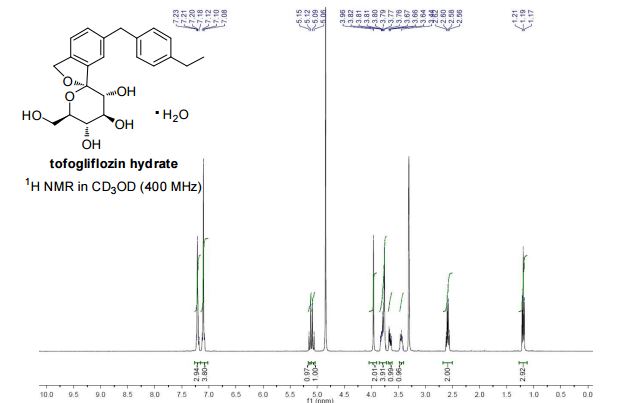
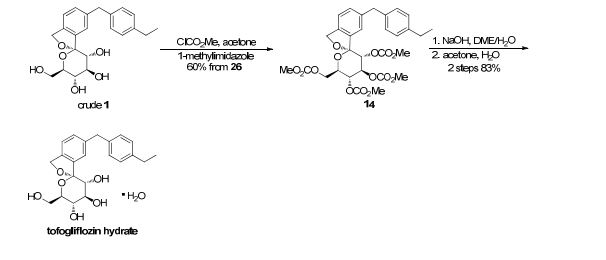
Tofogliflozin hydrate white solid with 99.56% purity by HPLC. Water content: 4.47%.
Mp: 71−80 oC. [α]20 D = +23.9 (c = 1.0, CH3OH).
1H NMR (400 MHz, CD3OD) δ 7.23-7.18 (m, 3H), 7.12-7.08(m, 4H), 5.13 (d, J = 12.4 Hz, 1H), 5.07 (d, J = 12.4 Hz, 1H), 3.96 (s, 2H), 3.83-3.73 (m, 4H), 3.65 (dd, J = 11.9, 5.5 Hz, 1H), 3.41-3.47 (m, 1H), 2.59 (q, J = 7.6 Hz, 2H), 1.19 (t, J = 7.6 Hz, 3H).
13C NMR (100 MHz, CD3OD) δ 143.2, 142.6, 140.2, 139.9, 139.7, 131.2, 129.9, 128.9, 123.6, 121.8, 111.6, 76.4, 76.2, 74.9, 73.4, 71.9, 62.8, 42.3, 29.5, 16.3.
HRMS (ESI) m/z: [M+H]+ Calcd for C22H27O6 387.1802; Found 387.1805.
IR (KBr, cm-1) ν: 3362, 2962, 2927, 1637, 1513, 1429, 1095, 1034, 808, 770. Spectroscopic data were identical with those reported.1b, 2
1. (a) Suzuki, M.; Honda, K.; Fukazawa, M.; Ozawa, K.; Hagita, H.; Kawai, T.; Takeda, M.; Yata, T.; Kawai, M.; Fukuzawa, T.; Kobayashi, T.; Sato, T.; Kawabe, Y.; Ikeda, S. J. Pharmacol. Exp. Ther. 2012, 341, 692.
(b) Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S.-H.; Ahn. K.-H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. J. Med. Chem. 2012, 55, 7828.
(c) Ikeda, S.; Takano, Y.; Cynshi, O.; Tanaka, R.; Christ, A. D.; Boerlin, V.; Beyer, U.; Beck, A.; Ciorciaro, C.; Meyer, M.; Kadowaki, T. Diabetes, Obesity and Metabolism 2015, 17, 984.
2. (a) Murakata, M.; Ikeda, T.; Kimura, N.; Kawase, A.; Nagase, M.; Yamamoto, K.; Takata, N.; Yoshizaki, S.; Takano, K. Crystal of spiroketal derivative, and process for production thereof. European Appl. EP 2308886 A1, April 13, 2011.
(b) Ohtake, Y.; Emura, T.; Nishimoto, M.; Takano, K.; Yamamoto, K.; Tsuchiya, S.; Yeu, S.; Kito, Y.; Kimura, N.; Takeda, S.; Tsukazaki, M.; Murakata, M.; Sato, T. J. Org. Chem. 2016, 81, 2148.
References
- Chugai Pharmaceutical: Development Pipeline
- Nagata, T.; Fukazawa, M.; Honda, K.; Yata, T.; Kawai, M.; Yamane, M.; Murao, N.; Yamaguchi, K.; Kato, M.; Mitsui, T.; Suzuki, Y.; Ikeda, S.; Kawabe, Y. (2012). “Selective SGLT2 inhibition by tofogliflozin reduces renal glucose reabsorption under hyperglycemic but not under hypo- or euglycemic conditions in rats”. AJP: Endocrinology and Metabolism 304 (4): E414–E423. doi:10.1152/ajpendo.00545.2012.PMID 23249697.
- Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S. Y.; Ahn, K. H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. (2012). “Discovery of Tofogliflozin, a NovelC-Arylglucoside with anO-Spiroketal Ring System, as a Highly Selective Sodium Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes”. Journal of Medicinal Chemistry 55 (17): 7828–7840. doi:10.1021/jm300884k.PMID 22889351.
- Statement on a nonproprietary name adopted by the USAN council: Tofogliflozin.
- http://www.who.int/entity/medicines/publications/druginformation/innlists/RL65.pdf
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(1S,3′R,4′S,5′S,6′R)-6-(4-Ethylbenzyl)-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2′-pyran]-3′,4′,5′-triol hydrate (1:1)
|
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 1201913-82-7 903565-83-3 (anhydrous) |
| ATC code | None |
| PubChem | CID 46908928 |
| ChemSpider | 28527871 |
| KEGG | D09978 |
| ChEMBL | CHEMBL2105711 |
| Synonyms | CSG452 |
| Chemical data | |
| Formula | C22H28O7 |
| Molar mass | 404.45 g/mol |
//////////TOFOGLIFLOZIN, 托格列净 , CSG-452, R-7201, RG-7201, 1201913-82-7 , 903565-83-3, oral hypoglycaemic agents, SGLT-2 inhibitors, type 2 diabetes mellitus, Deberza
CCc1ccc(cc1)Cc2ccc3c(c2)[C@]4([C@@H]([C@H]([C@@H]([C@H](O4)CO)O)O)O)OC3.O
The glucopyranosyl-substituted benzene derivatives are proposed as inducers of urinary sugar excretion and as medicaments in the treatment of diabetes.
The term “canagliflozin” as employed herein refers to canagliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2005/012326 and WO 2009/035969 for example. Preferred hydrates, solvates and crystalline forms are described in the patent applications WO 2008/069327 for example.
atigliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2004/007517 for example.
ipragliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2004/080990, WO 2005/012326 and WO 2007/114475 for example.
tofogliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2007/140191 and WO 2008/013280 for example.
remogliflozin and prodrugs of remogliflozin, in particular remogliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods of its synthesis are described in the patent applications EP 1213296 and EP 1354888 for example.
sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
luseoghflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

ertugliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

and is described for example in WO 2010/023594.
The compound of the formula

is described for example in WO 2008/042688 or WO 2009/014970.
Dapagliflozin

The compound is described for example in WO 03/099836. Crystalline forms are described for example in WO 2008/002824.
Remogliflozin and Remogliflozin Etabonate

The compound is described for example in EP 1354888 A1.
Sergliflozin and Sergliflozin Etabonate

The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
1-Chloro-4-(β-D-glucopyranos-1-yl)-2-(4-ethyl-benzyl)-benzene

The compound is described in WO 2006/034489.
(1S)-1,5-anhydro-1-[5-(azulen-2-ylmethyl)-2-hydroxyphenyl]-D-glucitol

The compound (4-(Azulen-2-ylmethyl)-2-(β-D-glucopyranos-1-yl)-1-hydroxy-benzene) is described in WO 2004/013118 and WO 2006/006496. The crystalline choline salt thereof is described in WO 2007/007628.
(1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-D-glucitol

The compound is described in WO 2004/080990 and WO 2005/012326. A cocrystal with L-proline is described in WO 2007/114475.
Thiophen Derivatives of the Formula (7-1)

wherein R denotes methoxy or trifluoromethoxy. Such compounds and their method of production are described in WO 2004/007517, DE 102004063099 and WO 2006/072334.
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene

The compound is described in WO 2005/012326. A crystalline hemihydrate is described in WO 2008/069327.
Spiroketal Derivatives of the Formula (9-1)

wherein R denotes methoxy, trifluoromethoxy, ethoxy, ethyl, isopropyl or tert. butyl. Such compounds are described in WO 2007/140191 and WO 2008/013280.
EDQM announces revision of general chapter Monocyte Activation Test (2.6.30)
DRUG REGULATORY AFFAIRS INTERNATIONAL

On 23 June, the EDQM in Strasbourg announced the revision of the pharmacopoeial general chapter 2.6.30 on Monocyte Activation Test.
During the last two years, the chapters of the European Pharmacopoeia relating to the detection of Endotoxins and Pyrogens were successively updated or revised, e.g. 5.1.10. “Guidelines for Using the Test for Bacterial Endotoxins” or 2.6.8.” Pyrogens” (see Pharmeuropa – Comments concerning revised texts about Bacterial Endotoxins). There, amongst others, the EDQM announced that the chapter 2.6.8. now includes a reference to 2.6.30. “Monocyte Activation Test” as a potential replacement for the test for pyrogens.
Last week, the EDQM published the information that during its 155th Session held in Strasbourg on 21-22 June 2016, the European Pharmacopoeia (Ph. Eur.) Commission adopted a revision of the general chapter Monocyte Activation Test (2.6.30).
It has been a goal of the Ph. Eur. Commission since nearly 30 years to consider the…
View original post 526 more words
Drafts of revised USP plastic packaging chapters and : removal of the biological reactivity test for oral and topical dosage forms
DRUG REGULATORY AFFAIRS INTERNATIONAL

In a recent Pharmacopeial Forum two revised USP general chapters have been published for comment. With these drafts, the USP expert committee is removing the requirement for <87> Biological Reactivity Tests, In Vitro testing for packaging materials and systems for oral and topical dosage forms. Read more about the draft chapters of <661.1> Plastic Materials of Construction and <661.2> Plastic Packaging Systems for Pharmaceutical Use.testing for packaging materials and systems for oral and topical dosage forms. Read more about the draft chapters of <661.1> Plastic Materials of Construction and <661.2> Plastic Packaging Systems for Pharmaceutical Use.
read
In Pharmacopeial Forum 42(4) [Jun-Jul 2016] drafts of two revised USP general chapters <661.1> Plastic Materials of Construction and <661.2> Plastic Packaging Systems for Pharmaceutical Use have been published for comment. Deadline for comments is September 30, 2016. With these drafts, the USP General Chapters – Packaging and Distribution Expert Committee…
View original post 2,996 more words
EMA reviews Medicines manufactured at U.S. Company
DRUG REGULATORY AFFAIRS INTERNATIONAL

Following the issuance of two Non-Compliance Reports for two sites of the US based company, EMA has started a review of medicines manufactured by Pharmaceutics International Inc., USA.
The European Medicines Agency (EMA) has started a review of medicines manufactured by Pharmaceutics International Inc., USA. This follows the issuance of two Non-Compliance Reports for two sites of the US based company after an inspection in February 2016 conducted by the MHRA (the medicines regulatory agency in the United Kingdom) which highlighted several shortcomings in relation to good manufacturing practice (GMP).
Pharmaceutics International Inc. manufactures the centrally authorised medicine Ammonaps (sodium phenylbutyrate) and is also the registered manufacturing site for some other medicines that have been authorised through national procedures in the European Union (EU).
This inspection which was a follow-up to an inspection in June 2015 aimed to assess whether corrective measures agreed previously had been appropriately implemented. It found…
View original post 201 more words
New bicalutamide/enzalutamide derivatives as antiproliferative agents for the treatment of prostate cancer
3,3,3-trifluoro-2-hydroxy-N-(4-nitro-3-(trifluoromethyl)phenyl)-2-(((2-(trifluoromethyl)phenyl)thio)methyl)propanamide
Cas 1929605-82-2

Dr Marcella Bassetto
Post Doctoral Research Associate
- bassettom@cardiff.ac.uk
- https://www.researchgate.net/profile/Marcella_Bassetto
- http://marcellabassetto.blogspot.in/
SYNTHESIS

Scheme .
Synthetic strategy used in the synthesis of 52. Reagents and conditions: (a) NaH (1 equiv.), THF, 0 °C to RT, 3 h; (b) KCN (1.2 equiv.), 25% H2SO4, 0 °C to RT, 20 h; c) HCl, AcOH, reflux, 24 h; (d) 8, SOCl2(1.3 equiv.), DMA, RT, 72 h.
3-Bromo-1,1,1-trifluoroacetone (48) was coupled with thiophenol 47 to afford 49, which was then converted into cyano derivative 50 using potassium cyanide and 25% sulfuric acid [16]. Intermediate 51 was obtained after refluxing 50 in concentrated HCl and glacial acetic acid. Coupling of 51 with commercially available 4-nitro-3-(trifluoromethyl)aniline 8yielded the desired amide 52.
Synthesis of 1,1,1-rifluoro-3-((2-(trifluoromethyl)phenyl)thio)propan-2-one (49)
To a mixture of NaH (10.47 mmol) in 10 mL anhydrous THF was added a solution of 2-(trifluoromethyl)benzenethiol (10.47 mmol) in 2mL anhydrous THF at 0 °C. This mixture was stirred for 20 min. 3-Bromo-1,1,1-trifluoropropan-2-one was then added dropwise to the mixture at 0 °C, the reaction was warmed to r.t. and stirred for 12 h. The mixture was filtered trough celite, the filtered pad was washed with THF, and the filtrate was evaporated to dryness. The residue was purified by flash column chromatography eluting with n-hexane/EtOAc 100:0 v/v increasing to n-hexane/EtOAc 85:15 v/v to give a pale yellow oil in 93% yield. 1H-NMR (CDCl3): d 7.76-7.69 (m, 2H), 7.60-7.53 (m, 1H), 7.42-7.38 (m, 1H), 3.44 (s, 2H). 19F-NMR (CDCl3): d -59.91 (s, 3F), -85.26 (s, 3F). 13C-NMR (CDCl3): d 189.6, 137.7, 135.9, 134.5, 133.2, 130.6, 129.6 (q, J= 26.3 Hz), 127.0 (q, J= 3.8 Hz), 124.3 (q, J= 4.1 Hz), 124.0 (q, J= 3.7 Hz), 94.4 (q, J= 30.4 Hz), 40.4.
Synthesis of 3,3,3-trifluoro-2-hydroxy-2-(((2-(trifluoromethyl)phenyl)thio)methyl)propanenitrile (50)
A 20% aqueous solution of H2SO4 (3.4 mL) was added dropwise to a mixture of 49 (11.03 mmol) and KCN (13.24 mmol) in 5 mL H2O at 0 °C. The reaction mixture was warmed to r.t. and stirred for 20 h. The mixture was then diluted with water (50 mL) and extracted with Et2O (3 x 150 mL). The organic extracts were washed with sat. aq. NaHCO3 and brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography eluting with n-hexane/EtOAc 100:0 v/v increasing to n-hexane/EtOAc 95:5 v/v to give a pale yellow oil in 86% yield. 1H-NMR (CDCl3): d 7.80 (d, J= 7.8 Hz, 1H), 7.77-7.76 (m, 1H), 7.72-7.59 (m, 1H), 7.52-7.49 (m, 1H), 4.36 (bs, 1H), 3.58 (d, J= 14.6 Hz, 1H), 3.44 (d, J= 14.6 Hz, 1H). 19F-NMR (CDCl3): d -57.08 (s, 3F), -79.51 (s, 3F). 13C-NMR (CDCl3): d 135.4, 132.8, 132.5 (q, J= 30.1 Hz), 129.1, 128.7 (q, J= 5.5 Hz), 126.7, 124.9, 124.6, 122.6, 122.4, 120.4, 114.0, 71.4 (q, J= 32.9), 40.75.
1.1.1 Synthesis of 3,3,3-trifluoro-2-hydroxy-2-(((2-(trifluoromethyl)phenyl)thio)methyl)propanoic acid (51)
A mixture of 51 (6.89 mmol), concentrated HCl (23.4 mL) and AcOH (4.1 mL) was refluxed o.n. with vigorous stirring. The mixture was then diluted with water (100 mL) and extracted with Et2O (4 x 100 mL), which was in turn washed with sat. aq. NaHCO3 (4 x 100 mL). The water solution was acidified with concentrated HCl to pH 1 and extracted with Et2O (4x 150 mL). The Et2O extracts were dried over Na2SO4, filtered and concentrated to dryness to give a pale yellow waxy solid in 41% yield. 1H-NMR (CDCl3): d 9.57 (bs, 1H), 7.70 (d, J= 7.7 Hz, 1H), 7.67 (d, J= 7.7 Hz, 1H), 7.54-7.51 (m, 1H), 7.39-7.36 (m, 1H), 3.60 (s, 2H). 19F-NMR (CDCl3): d -60.10 (s, 3F), -77.7 (s, 3F). 13C-NMR (CDCl3): d 172.0, 134.1, 134.0, 131.2 (q, J= 30.1 Hz), 127.5, 126.7 (q, J= 5.6 Hz), 124.2 (q, J= 121.9 Hz), 121.9 (q, J= 126.7 Hz), 78.2 (q, J= 28.7 Hz), 37.7.
Synthesis of 3,3,3-trifluoro-2-hydroxy-N-(4-nitro-3-(trifluoromethyl)phenyl)-2-(((2-(trifluoromethyl)phenyl)thio)methyl)propanamide (52)
Thionyl chloride (1.16 mmol) was added dropwise to a stirring solution of 51 in anhydrous DMA at -10 °C under Ar atmosphere. The reaction mixture was stirred for 1 h, then a solution of 8 in 2 mL anhydrous DMA was added dropwise. The reaction mixture was warmed to r.t. and stirred for 72 h. The mixture was then diluted with sat. aq. NaHCO3 (40 mL) and extracted with Et2O (3 x 40 mL). The organic extracts were filtered trough celite, dried over Na2SO4 and evaporated to dryness. The residue was purified by flash column chromatography eluting with n-hexane/EtOAc 100:0 v/v increasing to n-hexane/EtOAc 80:20 v/v to give a pale yellow solid in 13% yield.
1H-NMR (CDCl3): d 8.93 (bs, 1H), 7.94 (d, J= 8.8 Hz, 1H), 7.87 (d, J= 2.2 Hz, 1H), 7.72 (d, J= 8.1 Hz, 1H), 7.69 (dd, J= 8.8 Hz, 2.2 Hz, 1H), 7.50-7.47 (m, 2H), 7.26-7.23 (m, 1H), 4.41 (s, 1H), 4.19 (d, 14.7 Hz, 1H), 3.45 (d, J= 14.7 Hz, 1H).
19F-NMR (CDCl3): d -59.7 (s, 3F), -60.12 (s, 3F), -77.4 (s, 3F).
13C-NMR (CDCl3): d 164.6, 143.8, 140.0, 134.7, 132.6, 131.1 (q, J= 29.8 Hz), 130.5, 128.3, 126.8 (q, J= 5.5 Hz), 126.7, 125.2 (q, J= 36.3 Hz), 124.5, 123.9, 122.6, 122.4, 122.2, 121.7, 120.4, 118.2 (q, J= 5.8 Hz), 76.3 (q, J= 27.8 Hz), 38.5.
MS [ESI, m/z]: 523.0 [M+H]+.
EI-HMRS (M-H)– found 521.0215, calculated for C18H0N2O4F9S 521.0218.
HPLC (method 1): retention time = 23.84 min.
clips
Prostate cancer (PC) is a leading cause of male death worldwide and it is the most frequently diagnosed cancer among men aged 65–74 [1]. The prognosis varies greatly, being highly dependent on a number of factors such as stage of diagnosis, race and age. Currently, PC treatment includes androgen deprivation, surgery, radiation, endocrine therapy and radical prostatectomy.
PC cell growth is strongly dependent on androgens, therefore blocking their effect can be beneficial to the patient’s health. Such outcomes can be achieved by antagonism of the androgen receptor (AR) using anti-androgen drugs, which have been extensively explored either alone or in combination with castration [2]. Flutamide (Eulexin®) (1) (in its active form as hydroxyflutamide (2)), bicalutamide (Casodex®) (3), nilutamide (Niladron®) (4) and enzalutamide (previously called MDV3100) (Xtandi®) (5) are all non-steroidal androgen receptor antagonists approved for the treatment of PC (Fig. 1). In many cases, after extended treatment over several years, these anti-androgens become ineffective and the disease may progress to a more aggressive and lethal form, known as castration resistant prostate cancer (CRPC). The major cause of this progressive disease is the emergence of different mutations on the AR, which cause the anti-androgen compounds to function as agonists, making them tumour-stimulating agents[3].

-
Fig. 1.
Structure of anti-androgen small molecules approved by FDA or in clinical development for the treatment of PC.
Among the drugs used for the treatment of PC, bicalutamide and enzalutamide selectively block the action of androgens while presenting fewer side effects in comparison with other AR antagonists [4], [5] and [6]. The structure of these molecules is characterised by the presence of a trifluoromethyl substituted anilide, which appears to be critical for biological activity (Fig. 1). As a means to improve the anti-proliferative activity of these compounds, and in order to exploit the well established potential of the fluorine atom in enhancing the pharmacological properties and drug-like physicochemical characteristics of candidate compounds [7], [8] and [9], a wide array of diverse new structures has been rationally designed and synthesised, through the introduction of fluoro-, trifluoromethyl- and trifluoromethoxy groups in diverse positions of both aromatic rings of the parent scaffolds. Our modifications resulted in a marked improvement of in vitro anti-proliferative activities on a range of human PC cell lines (VCap, LNCaP, DU-145 and 22RV1). In addition, we probed full versus partial AR antagonism for our new compounds.
Paper

Volume 118, 8 August 2016, Pages 230–243

Design and synthesis of novel bicalutamide and enzalutamide derivatives as antiproliferative agents for the treatment of prostate cancer
School of Pharmacy and Pharmaceutical Sciences, Redwood Building, King Edward VII Avenue, CF10 3NB, Cardiff, Wales, UK
- doi:10.1016/j.ejmech.2016.04.052
- E-mail address: Ferlas1@cardiff.ac.uk (Dr. S. Ferla)
Highlights
- •Synthesis of novel fluorinated bicalutamide and enzalutamide analogs.
- •Anti-proliferative activity in four human prostate cancer cell lines improved up to 50 folds.
- •Full AR antagonist effect exhibited by the new compounds.
- •Activity switch from partial agonist to full AR antagonist for enobosarm scaffold.
- •AR open conformation homology model and molecular modeling studies.
Abstract
Prostate cancer (PC) is one of the major causes of male death worldwide and the development of new and more potent anti-PC compounds is a constant requirement. Among the current treatments, (R)-bicalutamide and enzalutamide are non-steroidal androgen receptor antagonist drugs approved also in the case of castration-resistant forms. Both these drugs present a moderate antiproliferative activity and their use is limited due to the development of resistant mutants of their biological target.
Insertion of fluorinated and perfluorinated groups in biologically active compounds is a current trend in medicinal chemistry, applied to improve their efficacy and stability profiles. As a means to obtain such effects, different modifications with perfluoro groups were rationally designed on the bicalutamide and enzalutamide structures, leading to the synthesis of a series of new antiproliferative compounds. Several new analogues displayed improved in vitro activity towards four different prostate cancer cell lines, while maintaining full AR antagonism and therefore representing promising leads for further development.
Furthermore, a series of molecular modelling studies were performed on the AR antagonist conformation, providing useful insights on potential protein-ligand interactions.
http://www.sciencedirect.com/science/article/pii/S0223523416303452
Top cancer scientist dies of the disease he spent his life trying to cure
Professor Chris McGuigan, 57, of Cardiff University, was trying to invent new drugs to use in the fight against the disease

Professor Chris McGuigan, 57, was trying to invent new drugs to use in the fight against the disease.
But the tragic scientist, who was head of medicinal chemistry at Cardiff University’s School of Pharmacy and Pharmaceutical Sciences, died after his own fight with cancer.
A spokesman for Cardiff University said: “Professor McGuigan had been at the heart of scientific research for more than 30 years. He was an exceptionally gifted inventor and chemist.
“His loss will be felt cross the university and the wider scientific community.

“He had a strong drive to use his scientific ideas for social good, working tirelessly to address medical needs where they were unmet.
“Our thoughts are with his family, friends and close colleagues at this very sad time.”
Prof McGuigan’s research led him to try and develop new drugs for cancer, HIV, hepatitis B and C, shingles, measles, influenza and central nervous system (CNS) disease.
He also invented four new experimental drugs that were used in human clinical trials.
Prof McGuigan, who lived in Cardiff, is survived by wife Maria, 50, and his two young daughters Phoebe and Grace.
References
-
- Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008
- Int. J. Cancer, 127 (2010), pp. 2893–2917
-
- Antiandrogen monotherapy: a new form of treatment for patients with prostate cancer
- Urology, 58 (2001), pp. 16–22
-
- Flutamide withdrawal syndrome: its impact on clinical trials in hormone-refractory prostate cancer
- J. Clin. Oncol., 11 (1993), pp. 1566–1572
-
- Antiandrogens in prostate cancer
- Investig. New Drugs, 17 (1999), pp. 271–284
-
- The role of antiandrogen monotherapy in the treatment of prostate cancer
- BJU Int., 91 (2003), pp. 455–461
-
- Antiandrogens in the treatment of prostate cancer
- Eur. Urol., 51 (2007), pp. 306–313
-
- Fluorine-containing natural products
- J. Fluor. Chem., 100 (1999), pp. 127–133
-
- Fluorine substituent effects on bioactivity
- J. Fluor. Chem., 109 (2001), pp. 3–11
-
- Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade 2001–2011
- Chem. Rev., 114 (2014), pp. 2432–2506
-
- A two-step synthesis of the anti-cancer drug (R,S)-Bicalutamide
- Synthesis, 7 (2002), pp. 850–852
-
- Nucleohilic aromatic substitution of methacrylamide anion and its application to the synthesis of the anticancer drug bicalutamide
- J. Org. Chem., 26 (2003), pp. 10181–10182
-
-
- Bicalutamide: clinical pharmacokinetics and metabolism
- Clin. Pharmacokinet., 13 (2004), pp. 855–878
-
-
- Resolution of the nonsteroidal antiandrogen 4′-cyano-3-[(4-fluorophenyl)sulfonyl]-2-hydroxy-2-methyl-3′-(trifluoromethyl)-propionanilide and the determination of the absolute configuration of the active enantiomer
- J. Med. Chem., 31 (1988), pp. 885–887
-
- Novel nonsteroidal ligands with binding affinity and potent functional activity for the androgen receptor
- Eur. J. Med. Chem., 37 (2002), pp. 619–634
///////////1929605-82-2, bicalutamide and enzalutamide derivatives, antiproliferative agents, treatment of prostate cancer, School of Pharmacy and Pharmaceutical Sciences, Redwood Building, King Edward VII Avenue, CF10 3NB, Cardiff, Wales, UK
FC(F)(F)c1cc(ccc1[N+]([O-])=O)NC(=O)C(O)(CSc2ccccc2C(F)(F)F)C(F)(F)F
Eldecalcitol, an active vitamin D3 analog used to treat osteoporosis
Eldecalcitol
(1S,2S,3S,5Z)-5-[(2E)-2-[(1R,3aS,7aR)-1-[(2R)-6-hydroxy-6-methylheptan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1H-inden-4-ylidene]ethylidene]-2-(3-hydroxypropoxy)-4-methylidenecyclohexane-1,3-diol
(1R,2R,3R,5Z,7E)-2-(3-Hydroxypropyloxy)-9,10-secocholesta-5,7,10(19)-triene-1,3,25-triol
| AC1O5QQ2; CAS 104121-92-8; AN-3697; ED 71, Edirol® | |
| Molecular Formula: | C30H50O5 |
|---|---|
| Molecular Weight: | 490.715 g/mol |
APPROVED JAPAN , 2011-01-21, Chugai (Originator) , Roche,Taisho Toyama
Eldecalcitol was approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on January 21, 2011. It was developed by Chugai Pharmaceutical (a member of Roche) and marketed as Edirol® by Chugai Pharmaceutical and Taisho.
Eldecalcitol is an orally active vitamin D analogue leading to greater absorption of bind calcium. It is usually used to treat osteoporosis.
Edirol® is available as capsule for oral use, containing 0.5 μg or 0.75 μg of free Eldecalcitol, and the recommended dose is 0.75 μg once daily.
ED-71, a vitamin D analog, is a more potent inhibitor of bone resorption than alfacalcidol in an estrogen-deficient rat model of osteoporosis. ED-71, effectively and safely increased lumbar and hip bone mineral density (BMD) in osteoporotic patients who also received vitamin D3 supplementation.
Eldecalcitol is a drug used in Japan for the treatment of osteoporosis.[1] It is an analog of vitamin D.[2] Osteoporosis is a common bone disease among the older generation, with an estimated prevalence of over 200 million people.[1] This condition often results in bone fractures due to abnormally low bone mass density, and is a leading cause of disability, especially among developed countries with longer average life spans. Osteoporosis is more common in women than with men.
Discovery
Chugai Pharmaceutical/Roche are the originators of the medicinal drug eldecalcitol through Taisho Pharmaceutical Holdings and Chugai Pharmaceutical. The trade name of eldecalcitol is Edirol, and its Chemical Abstracts Service (CAS) registry number is 104121-92-8. Eldecalcitol was approved for use in Japan on January 2011. The approval came from the Japanese Ministry of Health, Labor, and Welfare for the objective of a treatment for osteoporosis.[3]
Effects
Clinical trials have suggested that eldecalcitol, a vitamin D analog, has strong effects to reduce calcium reabsorption into the body from bones, therefore increasing bone mineral density, and to increase calcium absorption in intestines.[4] In animals, eldecalcitol inhibits the activity of osteoclasts for the function to reduce bone degradation for calcium, while still able to maintain osteoblast function so as to not hinder bone formation.[5] Unlike other vitamin D analogs, eldecalcitol does not significantly suppress parathyroid hormone levels, promising a better treatment for osteoporosis in comparison to other medications.[6] Bone mineral density increases with eldecalcitol use, in addition to strengthening bone structure. This occurs due to the function of the eldecalcitol drug, which decreases bone reabsorption as observed through a bone reabsorption marker. Bone geometry assessments show that eldecalcitol increases cortical bone area in patients with osteoporosis more so than other vitamin D analogs, such as alfacalcidol. There was also the maintenance of thickness of cortical bone mass, strongly indicating that eldecalcitol improves the strength and mass of bone, specifically cortical bone structure.[7] Adverse effects of eldecalcitol include an increase in blood and urinary calcium levels. Abnormally high levels of calcium can lead to problems associated with hypercalcemia.
Treatment for Osteoporosis
Eldecalcitol can be used for the treatment of hypocalcaemia or osteoporosis. Calcium absorption increases with the presence of eldecalcitol by the body, occurring in the intestines, which is useful for those who have low calcium levels. Eldecalcitol is more often used due to its effects to treat osteoporosis. In the aging population, the bone matrix becomes weakened through untreated osteoporosis. This leads to an increased risk of severe fractures that include spinal and hip fractures in addition to vertebral and wrist fractures. This creates a burden on the health care system due to a decline in the quality of life for the individuals that suffer from this condition. Some risk factors leading to the predisposition of developing osteoporosis are previous incidents of bone fractures and a reduction in bone mineral density.[1] These factors expectantly increase as age increases. Bone health is reliant on maintaining physiologically needed levels of calcium, where the body constantly maintains this calcium homeostasis through osteoblast and osteoclast activity. Osteoblast activity serves this function of maintaining appropriate calcium levels by depositing calcium in bones when blood calcium levels are above normal. In contrast, osteoclasts break down bone tissue to increase blood calcium levels if they are low.[8] This activity is performed after absorption of calcium by the body, which requires the actions of vitamin D. The active metabolite of vitamin D, calcitriol, performs its function through interactions with the calcitriol receptor. This nuclear hormone receptor is responsible for calcium absorption which, in turn, is involving in bone depletion and formation. The new analogs of vitamin D, such as eldecalcitol, are observed to have stronger effects in preventing bone loss, fractures, and falls in comparison to calcitriol.[9] Eldecalcitol is even more effective than its counterpart alfacalcidol, another vitamin D analog. Studies have shown eldecalcitol is more effective than alfacalcidol in preventing vertebral and wrist fractures, and even falls, with osteoporotic patients with vitamin D insufficiencies.[10] Eldecalcitol is also more effective at preventing fractures than vitamin D and calcium supplements.[1] Eldecalcitol increases calcium absorption for vitamin D deficient patients, and therefore could be used for osteoporosis treatment for all age groups.
Pharmacology
Analogs of vitamin D are being explored intensely for their regulatory effects on calcium metabolism with the purpose of treating osteoporosis, a skeletal disease associated with low bone mass and deterioration of bone tissue. Vitamin D is imperative for absorption of calcium to maintain bone strength.
Mechanism of Action
Eldecalcitol is an orally administered drug to patients, which binds to vitamin D receptors and binding protein for the goal of achieving greater specificity to bind calcium for its absorption. This greater affinity is 2.7-fold that of the active vitamin D form of calcitriol. Eldecalcitol is readily absorbed into the body, with a long elimination half-life of over eight hours, reaching maximum absorption in 3.4 hours.[1]
Dosage
Eldecalcitol is present in the form of pills for oral administration. In preclinical models with healthy male volunteers, oral doses of eldecalcitol ranged from 0.1 to 1.0 micrograms once daily to show an increase in bone mineral density.[11] Preclinical trials show improvements for doses at 0.5 and 0.75 micrograms, which are the recommended dosage amounts for the Edirol product as approved by the Japanese Ministry of Health, Labor, and Welfare for treating osteoporosis.[3]
Chemistry
The class of eldecalcitol is a vitamin D3 derivative. This molecule has a molecular weight of 490.71 grams per mole. The eldecalcitol analog of calcitriol, contains a hydroxypropyl group in the lower cyclohexane ring. The synthesis of eldecalcitol incorporates two units assembled together. The IUPAC names include (3S, 4S, 5R)-oct-1-en-7-yne-3,4,5-triol that is fused to a bicyclic system, (R)-6-((1R, 3aR, 7aR, E)-4-(bromomethylene)-7a-methyloctahydro-1H-inden-1-yl)-2-methylheptan-2-ol. The assembly process includes a Diels-Alder reaction to give the fully protected eldecalcitol. In order to get the parent molecule, the hydroxyl groups have to be deprotected. The chemistry of eldecalcitol allows for its binding 2.7-fold more potently than calcitriol. In addition, some vitamin D derivatives have been known to inhibit the serum parathyroid hormone. Eldecalcitol only weakly inhibits the serum parathyroid hormone, making it an even more appealing medicinal drug for its physiological uses in the treatment of osteoporosis.[3] Animal studies of eldecalcitol, in ovariectomized rats, show improvements in bone mass while lowering bone reabsorption to demonstrate its effectiveness in osteoporosis treatment.[5]
PAPER
Heterocycles, Vol 92, No. 6, 2016, pp.1013-1029
Published online, 22nd March, 2016
■ Diverse and Important Contributions by Medicinal Chemists to the Development of Pharmaceuticals: An Example of Active Vitamin D3 Analog, Eldecalcitol
Noboru Kubodera*
*International Institute of Active Vitamin D Analogs, 35-6, Sankeidai, Mishima, Shizuoka 411-0017, Japan
Abstract
Presented herein are diverse and important contributions by medicinal chemists to different stages of pharmaceutical development. The conceptual elements reviewed, which are intended for young chemists who engage in drug discovery research, draw upon the author’s experience in developing eldecalcitol, an active vitamin D3 analog used to treat osteoporosis. The review covers exploratory research for a lead candidate compound; process development for practical manufacturing; and synthesis of other compounds relevant to the program, such as tritiated compounds, postulated metabolites, and miscellaneous analogs for mode of action studies.
PAPER
Eldecalcitol [1α,25-dihydroxy-2β-(3-hydroxypropoxy)vitamin D3], an analog of calcitriol (1α,25-dihydroxyvitamin D3), possesses a hydroxypropoxy substituent at the 2β-position of calcitriol. Eldecalcitol has potent biological effects on bone disease such as osteoporosis. The marketing of eldecalcitol has very recently started in Japan. In consideration of this, we have been investigating practical synthesis of eldecalcitol for industrial-scale production. Eldecalcitol was initially synthesized in a linear manner. The 27-step linear sequence was, however, suboptimal due to its lengthiness and low overall yield (ca. 0.03%). Next, we developed a convergent approach based on the Trost coupling reaction, in which the A-ring fragment (ene-yne part obtained in 10.4% overall yield) and the C/D-ring fragment (bromomethylene part obtained in 27.1% overall yield) are coupled to produce the triene system of eldecalcitol (15.6%). Although the overall yield of the convergent synthesis was better than that of the linear synthesis, significant improvements were still necessary. Therefore, additional biomimetic studies were investigated. Process development for the practical production of eldecalcitol is described herein.
http://ar.iiarjournals.org/content/32/1/303/F3.expansion.html
Convergent synthesis of eldecalcitol (5) by coupling A-ring fragment 37 with C/D-ring fragment 40. Reagents and conditions: a: HO(CH2)3OH/t-BuOK, 120°C. b: t-BuCOCl/pyridine/CH2Cl2, rt. c: H2/Pd(OH)2/MeOH, rt. d: Me2C(OMe)2/TsOH/acetone, rt. e: DMSO/(COCl)2/CH2Cl2, −60°C. f: CH2=CHMgBr/THF, −60°C. g: t-BuCOCl/Et3N/DMAP/CH2Cl2, rt. h: 1 M HCl/MeOH, rt. i: Ph3P/DEAD/benzene, reflux. j: LiC ≡ CTMS/BF3-OEt2, −78°C. k: 10 N NaOH/MeOH, rt. l: TBSOTf/Et3N/CH2Cl2, 0°C. m: TESOTf/Et3N/CH2Cl2, 0°C. n: O3/CH2Cl2/MeOH, −78°C then NaBH4/MeOH, −78°C. o: NMO/TPAP/4Ams/CH2Cl2, rt. p: Ph3P+CH2BrBr−/NaHMDS/ THF, −60°C to rt. q: (dba)3Pd2-CHCl3/PPh3/Et3N/toluene, reflux. r: TBAF/THF/toluene, reflux.

Industrial synthesis of alfacalcidol (4) and biomimetic synthesis of eldecalcitol (5) from cholesterol (42). Reagents and conditions: a: [Al(Oi-Pr)3]/cyclohexanone. b: DDQ/AcOEt. c: NaOEt/EtOH. d: NaBH4/MeOH/THF. e: Ac2O/DMPA/pyridine, rt. f: NBS/AIBN/n-hexane, reflux. g: γ-collidine/toluene, reflux. h: KOH/MeOH, rt. i: PTAD/CH2Cl2, rt. j: TBSCl/imidazole. k: MCPBA/CH2Cl2. l: DMI, 140°C. m: TBAF/THF. n: NaBH4/EtOH. o: 400 W high pressure mercury lamp/THF, 0°C then reflux without mercury lamp. p: HO(CH2)3OH/t-BuOK, 110°C. q: Microbial 25-hydroxylation.

ROUTE1
Reference:1. US4666634A / EP184206A2.

Reference:1. Anticancer. Res. 2012, 32, 303-310.
2. Drugs. Fut. 2005, 30, 450-461.

Reference:1. J. Am. Chem. Soc. 2001, 123, 3716-3722.

Reference:1. Bioorg. Med. Chem. Lett. 1997, 7, 2871-2874.
2. Anticance. Res. 2009, 29, 3571-3578.

Reference:1. EP0503630A1.
2. Drugs Fut. 2005, 30, 450-461.

Reference:1. Bioorg. Med. Chem. 1998, 6, 2517-2523.
References
- Sanford, M; McCormack, PL (2011). “Eldecalcitol: A review of its use in the treatment of osteoporosis”. Drugs 71 (13): 1755–70. doi:10.2165/11206790-000000000-00000. PMID 21902297.
- Hatakeyama, S; Yoshino, M (2010). “Synthesis and preliminary biological evaluation of 20-epieldecalcitol [20-epi-1α,25-dihydroxy-2β-(3-hydroxypropoxy)vitamin D3: 20-epi-ED-71]”. The Journal of Steroid Biochemistry and Molecular Biology 121 (1–2): 25–28.doi:10.1016/j.jsbmb.2010.03.041. PMID 20304058.
- Robichaud; Stamford; Weinstein; McAlpine; Primeau; Lowe; Bernstein; Bronson; Manoj, Desai (2012). Annual Reports in Medicinal Chemistry 47 (1st ed.). San Diego: Elsevier Inc. pp. 529–531. ISBN 9780123964922.
- Nogachi, Y; Kawate, H; Nomura, M; Takayanagi, R (2013). “Eldecalcitol for the treatment of osteoporosis”. Europe PubMed Central 8: 1313–1321. doi:10.2147/CIA.S49825.
- Smith, S; Doyle, N; Boyer, M; Chouinard, L; Saito, H (2013). “Eldecalcitol, a vitamin D analog, reduces bone turnover and increases trabecular an cortical bone mass, density, and strength in ovariectomized cynomolgus monkeys”. Bone 57 (1): 116–122.doi:10.1016/j.bone.2013.06.005. PMID 23774444.
- Harada, S; Uno, S; Takahashi, F; Saito, H (2010). “Eldecalcitol is less effective in suppressing parathyroid hormone compared to calcitriol in vivo“. The Journal of Steroid Biochemistry and Molecular Biology 121 (1–2): 281–283.doi:10.1016/j.jsbmb.2010.04.001. PMID 20398764.
- Nakamura, T; Takano, T; Fukunaga, M; Shiraki, M; Matsumoto, T (2013). “Eldecalcitol is more effective for the prevention of osteoporotic fractures than alfacalcidol”. Journal of Bone and Mineral Metabolism 31 (4): 417–422. doi:10.1007/s00774-012-0418-5.PMC 3709079. PMID 23575909.
- Matsuo, K; Irie, N (2008). “Osteoclast-osteoblast communication”. Archives of Biochemistry and Biophysics 473 (2): 201–209. doi:10.1016/j.abb.2008.03.027.PMID 18406338.
- Saito, H; Takeda, S; Amizuka, N (2013). “Eldecalcitol and calcitriol stimulates ‘bone minimodeling,’ focal bone formation without prior bone resorption, in rat trabecular bone”.The Journal of Steroid Biochemistry and Molecular Biology 136 (1): 178–182.doi:10.1016/j.jsbmb.2012.10.004.
- Matsumoto, T; Ito, M; Hayashi, Y; Hirota, T; Tanigawara, Y; Sone, T; Fukunaga, M; Shiraki, M; Nakamura, T (2011). “A new active vitamin D3 analog, eldecalcitol, prevents the risk of osteoporotic fractures—A randomized, active comparator, double-blind study”. Bone49 (4): 605–612. doi:10.1016/j.bone.2011.07.011. PMID 21784190.
- Harada, S; Mizoguchi, T; Kobayashi, Y; Nakamichi, Y; Takeda, S; Sakai, S; Takahashi, F; Saito, H; Yasuda, H; Udagawa, N; Suda, T; Takahashi, N (2012). “Daily administration of eldecalcitol (ED-71), an active vitamin D analog, increases bone mineral density by suppressing RANKL expression in mouse trabecular bone”. Journal of Bone and Mineral Research 27 (1): 461–473. doi:10.1002/jbmr.555.
| No. | Major Technical Classification | Publication No. | Patent No. | Legal Status | Filling Date | Estimated Expiry Date |
| 1 | Preparation | CN85108857A | CN1008368B | Granted/expired | 1985/12/4 | 2005/12/4 |
| 2 | Crystal | CN1223639A | CN1216861C | Granted | 1997/6/16 | 2017/6/16 |
| 3 | Preparation | CN1637017A | CN1276927C |
| Patent ID | Date | Patent Title |
|---|---|---|
| US7927613 | 2011-04-19 | Pharmaceutical co-crystal compositions |
| US7323580 | 2008-01-29 | CRYSTALS OF A VITAMIN D DERIVATIVE AND A METHOD FOR THE PREPARATION THEREOF |
| US7235679 | 2007-06-26 | Crystals of a vitamin D derivative and a method for the preparation thereof |
| EP0924199 | 2006-05-10 | CRYSTALS OF VITAMIN D DERIVATIVES AND PROCESS FOR THE PREPARATION THEREOF |
| US2005009794 | 2005-01-13 | Crystals of a vitamin D derivative and a method for the preparation thereof |
| US6831183 | 2004-12-14 | Crystals of a vitamin D derivative and a method for the preparation thereof |
| US6448421 | 2002-09-10 | CRYSTALS OF VITAMIN D DERIVATIVES AND PROCESS FOR THE PREPARATION THEREOF |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(1S,2S,3S,5Z,7E)-2-(3-Hydroxypropoxy)-9,10-secocholesta-5,7,10-triene-1,3,25-triol
|
|
| Clinical data | |
| Trade names | Edirol |
| Identifiers | |
| CAS Number | 104121-92-8 |
| ATC code | None |
| PubChem | CID 6438982 |
| ChemSpider | 4943418 |
| Chemical data | |
| Formula | C30H50O5 |
| Molar mass | 490.715 g/mol |
///////////eldecalcitol, active vitamin D3 analog, treat osteoporosis, AC1O5QQ2, 104121-92-8, AN-3697, ED 71, ED-71, Edirol®, PMDA, JAPAN
O[C@H]1CC(\C(=C)[C@H](O)[C@H]1OCCCO)=C\C=C2/CCC[C@]3([C@H]2CC[C@@H]3[C@H](C)CCCC(O)(C)C)C
OR
CC(CCCC(C)(C)O)C1CCC2C1(CCCC2=CC=C3CC(C(C(C3=C)O)OCCCO)O)C