
Home » Uncategorized (Page 4)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Gintemetostat
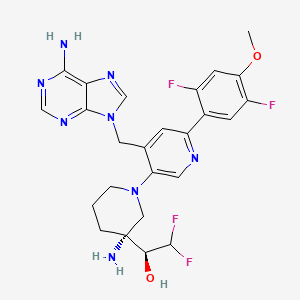
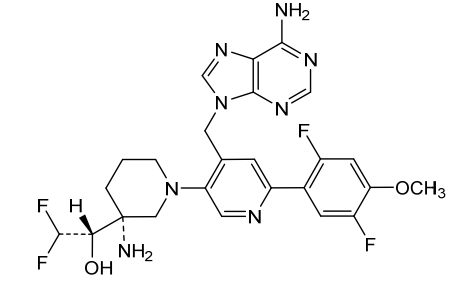
Gintemetostat
(1S)-1-[(3R)-3-amino-4′-[(6-amino-9H-purin-9-yl)methyl]-6′-(2,5-difluoro-4-methoxyphenyl)-3,4,5,6-tetrahydro-2H-[1,3′-bipyridin]-3-yl]-2,2-difluoroethan1-ol
antineoplastic, KTX 1001, NSD2 inhibitor 161, A48CGJ5UQM
CAS 2604513-16-6
MF C25H26F4N8O2 MW 546.5 g/mol
(S)-1-((R)-3-Amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6-(2,5-difluoro-4-methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol
Gintemetostat (also known as KTX-1001) is a first-in-class, orally administered small molecule being developed to treat relapsed and refractory multiple myeloma. It works as a selective inhibitor of NSD2 (also known as MMSET), targeting the epigenetic drivers of high-risk cancers.
How it Works
- Mechanism: Gintemetostat selectively binds to the catalytic SET domain of the NSD2 enzyme.
- Effect: By blocking this enzyme, it downregulates oncogenic signaling, decreases cancer cell growth, and can enhance T-cell activation against the tumor.
Target Patient Population
- High-Risk Myeloma: The drug focuses heavily on patients harboring the t(4;14) translocation, a genetic alteration found in 10-15% of patients that often causes aggressive relapses.
- Refractory Cases: It has shown notable single-agent activity in heavily pretreated patients who have exhausted standard-of-care, triple-class refractory treatment options.
Current Clinical Status
- Phase 1 Trial: Early data from phase 1 trials (such as NCT05651932) showed the drug has manageable safety profiles and offers clinical benefit (ranging from stable disease to very good partial response) in patients with aggressive, hard-to-treat multiple myeloma.
- Future Developments: Researchers are expanding studies to pair gintemetostat with other standard myeloma treatments, such as proteasome inhibitors and CELMoDs, to create stronger synergistic anti-cancer effects.
Gintemetostat is an orally available small molecule inhibitor of the histone-lysine N-methyltransferase nuclear receptor-binding SET domain protein 2 (NSD2; MMSET; WHSC1), with potential antineoplastic activity. Upon oral administration, gintemetostat selectively targets and binds to NSD2, and inhibits its catalytic activity and the mono- and di-methylation of histone H3 lysine 36 (H3K36). This modulates the expression of genes involved in cellular processes including cellular proliferation, which may lead to decreased growth of cancer cells. NSD2, a member of the NSD family of histone lysine methyltransferase enzymes that catalyzes the mono- and di-methylation of H3K36, is overexpressed and dysregulated in many types of cancers.
SYN
Discovery of a Highly Potent and Selective Inhibitor Targeting Protein Lysine Methyltransferase NSD2
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2024-09-04
PMID: 39230932
DOI: 10.1021/acs.jmedchem.4c00639
SYN
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021028854&_cid=P12-MQ0AZT-13511-1
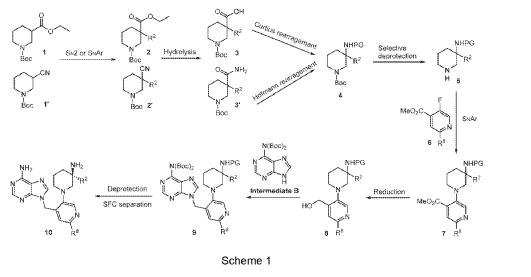
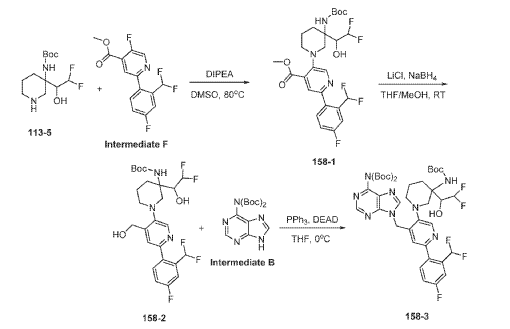
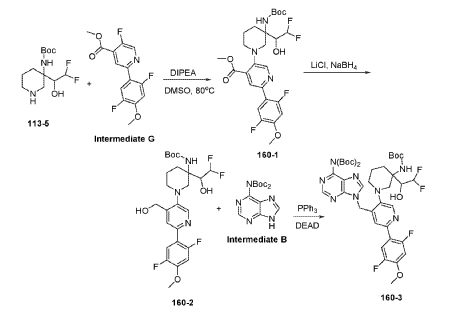
Example 160 and Example 161: (R)-1-((R)-3-amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6- (2,5-difluoro-4-methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol and (S)-1-((R)-3- amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6-(2,5-difluoro-4-methoxyphenyl)pyridin-3- yl)piperidin-3-yl)-2,2-difluoroethan-1-ol
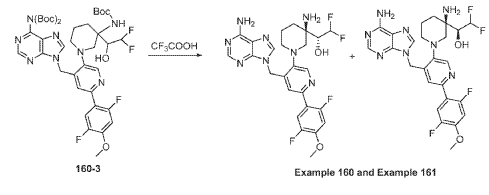
To a solution of tert-butyl (tert-butoxycarbonyl)(9-((5-(3-((tert-butoxycarbonyl)amino)-3-(2,2- difluoro-1-hydroxyethyl)piperidin-1-yl)-2-(2,5-difluoro-4-methoxyphenyl)pyridin-4-yl)methyl)-9H- purin-6-yl)carbamate (Intermediate 160-3) (200 mg, 0.237 mmol) in DCM (18 mL), was added TFA (36 mL), and the reaction mixrture was stirred at rt for 30 min under N2 atmosphere. The reaction mixture was concentrated in vacuo to give the crude product. The crude product was purifed by Pre-HPLC and SFC to afford (R)-1-((R)-3-amino-1-(4-((6-amino-9H-purin-9- yl)methyl)-6-(2,5-difluoro-4-methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol (Example 160) and (S)-1-((R)-3-amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6-(2,5-difluoro-4- methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol (Example 161).
Example 160: 1H NMR (400 MHz, CD3OD) d ppm 8.48 (s, 1H), 8.20 (d, J = 1.6 Hz, 2H), 7.58 (dd, J = 12.2, 7.3 Hz, 1H), 7.11 (d, J = 1.3 Hz, 1H), 6.90 (dd, J = 12.6, 7.1 Hz, 1H), 6.06 (td, J = 55.1, 3.9 Hz, 1H), 5.67 (s, 2H), 3.87 (s, 3H), 3.75 – 3.58 (m, 1H), 3.25 – 2.75 (m, 4H), 2.26 – 1.60 (m, 4H). LC-MS: [M+H]+ = 547.2, 548.2.
Example 161: 1H NMR (400MHz, CD3OD) d = 8.51 – 8.44 (m, 1H), 8.24 – 8.16 (m, 2H), 7.62 – 7.48 (m, 1H), 7.03 (s, 1H), 6.93 – 6.79 (m, 1H), 6.25 – 5.86 (m, 1H), 5.71 – 5.59 (m, 2H), 4.00 (m, 1H), 3.88 – 3.80 (m, 3H), 3.28 – 2.87 (m, 4H), 1.99 – 1.56 (m, 4H). LC-MS: [M+H]+ =547.4.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
{kind=link}
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: EP-4559915-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: EP-4013755-B1Priority Date: 2019-08-14Grant Date: 2025-01-08
- Piperidinyl-methyl-purinamines as NSD2 inhibitors and anticancer agentsPublication Number: CN-114585622-APriority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as NSD2 inhibitors and anti-cancer agentsPublication Number: US-12312353-B2Priority Date: 2019-08-14Grant Date: 2025-05-27
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: WO-2021026803-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: EP-4013755-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: WO-2021028854-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamine D-tartrate, crystalline forms, and use thereof in the treatment of medical diseases and conditionsPublication Number: CN-119744262-APriority Date: 2022-05-18
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: EP-4526305-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: US-2023002388-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purinamines as NSD2 inhibitors and anticancer agentsPublication Number: CN-114585622-BPriority Date: 2019-08-14Grant Date: 2024-08-09
- Piperidinyl-methyl-purineamines as NSD2 inhibitors and anti-cancer agentsPublication Number: US-11420970-B1
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: AU-2023273656-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225144-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine fumaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225150-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: US-2025326752-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purinic amine D-tartrate salt, crystalline form and their use in the treatment of medical diseases and conditionsPublication Number: KR-20250012083-APriority Date: 2022-05-18
- Pharmaceutical compositions containing a piperidinyl-methyl-purine amine and their use in treating diseases and conditionsPublication Number: US-2024207192-A1Priority Date: 2022-12-12
- Pharmaceutical compositions containing a piperidinyl-methyl-purine amine and their use in treating diseases and conditionsPublication Number: WO-2024129670-A1Priority Date: 2022-12-12
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: US-2024002385-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225154-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225141-A1Priority Date: 2022-05-18
////////////gintemetostat, ANAX LABS, antineoplastic, KTX 1001, NSD2 inhibitor 161, A48CGJ5UQM
Zidebactam
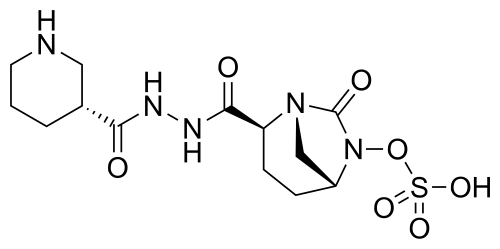
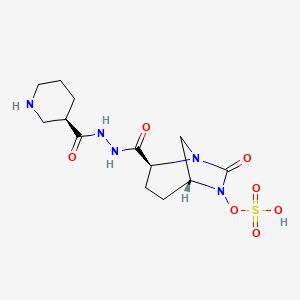
Zidebactam
FDA 2026, APPROVALS 2026
To treat complicated urinary tract infections, including pyelonephritis, caused by designated susceptible microorganisms
CAS 1436861-97-0, UNII: YPM97423DB, Wockhardt Biopharm, WCK-5107, WCK5107
Molecular Formula, C13-H21-N5-O7-S
Molecular Weight, 391.4029
Disclosed in PCT International Patent Application No. PCT/IB2012/054290D
- 01 Aug 2015 Phase-I clinical trials in Bacterial infections (In volunteers, Combination therapy) in USA (IV) (NCT02532140)
trans- sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(2S, 5R)-sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(1R,2S,5R)-l,6-Diazabicyclo [3.2.1] octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-[2-[(3R)-3-piperidinylcarbonyl]hydrazide]
trans- sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(2S, 5R)-sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(lR,2S,5R)-l,6-Diazabicyclo [3.2.1] octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-[2-[(3R)-3 -piperidinylcarbonyl] hydrazide]
1,6-Diazabicyclo(3.2.1)octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-(2-((3R)-3-piperidinylcarbonyl)hydrazide), (1R,2S,5R)-
Zidebactam potassium
cas is 1706777-49-2
Zidebactam (WCK-5107) is an antibiotic adjuvant drug which acts as a beta-lactamase inhibitor, preventing the breakdown of other antibiotic drugs.[1]
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019016393&_cid=P20-MPYVFE-00532-1
PATENT
http://www.google.com/patents/WO2013030733A1?cl=en

Scheme-1

Example-2
trans-sulfuric acid mono-r2-(N,-r(R)-piperidin-3-carbonyll-hvdrazinocarbonyl)-7-oxo-l,6- diaza-bicyclo Γ3.2.11 oct-6-νΠ ester
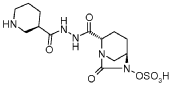
Step-1: Preparation of trans-3-[N’-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester:
By using the procedure described in Step-1 of Example- 1 above, and by using trans-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylic acid (25 gm, 0.084 mol), N,N-dimethyl formamide (625 ml), EDC hydrochloride (24 gm, 0.126 mol), HOBt (16.96 gm, 0.126 mol), (R)-N-tert-butoxycarbonyl-piperidin-3-carboxylic acid hydrazide (21.40 gm , 0.088 mol) to provide the title compound in 17.0 gm quantity, 41% yield as a white solid.
Analysis: MS (ES+) CzsHasNsOe = 502.1 (M+l);
I^NMR (CDCI3) = 8.40 (br s, IH), 7.34-7.44 (m, 5H), 5.05 (d, IH), 4.90 (d, IH), 4.00 (br d, IH), 3.82 (br s, IH), 3.30 (br s, IH), 3.16-3.21 (m, IH), 3.06 (br d, IH), 2.42 (br s, IH), 2.29-2.34 (m, IH), 1.18-2.02 (m, 4H), 1.60-1.75 (m, 4H), 1.45-1.55 (m, 2H),1.44 (s, 9H).
Step-2: Preparation of trans-3-[N’-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester:
By using the procedure described in Step-2 of Example- 1 above, and by using trans-3-[N ‘ -(6-benzyloxy-7-oxo- 1 ,6-diaza-bicyclo [3.2.1 ]octane-2-carbonyl)-hydrazinocarbonyl] -(R)-piperidin-l-carboxylic acid tert-butyl ester (16.5 gm , 0.033 mol), methanol (170 ml) and 10% palladium on carbon (3.5 gm) to provide the title compound in 13.5 gm quantity as a pale pink solid and it was used for the next reaction immediately.
Analysis: MS (ES+) CiglfeNsOe = 411.1 (M+l);
Step-3: Preparation of tetrabutylammonium salt of trans-3-[N’-(6-sulfooxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane-2-carbonyl)-hydrazinocarbonyl] -(R)-piperidin- 1 -carboxylic acid tert-butyl ester:
By using the procedure described in Step-3 of Example- 1 above, and by using trans-3-[N’-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)-piperidin-1 -carboxylic acid tert-butyl ester (13.5 gm , 0.033 mol), pyridine (70 ml) and pyridine sulfur trioxide complex (26.11 gm, 0.164 mol), 0.5 N aqueous potassium dihydrogen
phosphate solution (400 ml) and tetrabutylammonium sulphate (9.74 gm, 0.033 mol) to provide the title compound in 25 gm quantity as a yellowish solid, in quantitative yield.
Analysis: MS (ES-)
as a salt = 490.0 (M-l) as a free sulfonic acid;
Step-4: trans-sulfuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl]ester:
By using the procedure described in Step-4 of Example- 1 above, and by using tetrabutylammonium salt of trans-3-[N’-(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester (24 gm , 0.032 mmol), dichloromethane (60 ml) and trifluoroacetic acid (60 ml) to provide the title compound in 10 gm quantity as a white solid, in 79% yield.
Analysis: MS (ES-)= C13H21N5O7S = 390.2 (M-l) as a free sulfonic acid;
HXNMR (DMSO-d6) = 9.97 (d, 2H), 8.32 (br s, 2H), 4.00 (br s, IH), 3.81 (d, IH), 3.10-3.22 (m, 3H), 2.97-3.02 (m, 2H), 2.86-2.91 (m, IH), 2.65-2.66 (m, IH), 1.97-2.03 (m, IH), 1.57-1.88 (m, 7H).
-32.6°, (c 0.5, water).
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015110885
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014135931
| Clinical data | |
|---|---|
| License data | US DailyMed: Zidebactam |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1436861-97-0 |
| PubChem CID | 77846445 |
| DrugBank | DB13090 |
| ChemSpider | 44209501 |
| UNII | YPM97423DB |
| ChEMBL | ChEMBL4533605 |
| Chemical and physical data | |
| Formula | C13H21N5O7S |
| Molar mass | 391.40 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
References
- Karvouniaris M, Almyroudi MP, Abdul-Aziz MH, Blot S, Paramythiotou E, Tsigou E, et al. (April 2023). “Novel Antimicrobial Agents for Gram-Negative Pathogens”. Antibiotics. 12 (4). Basel, Switzerland: 761. doi:10.3390/antibiotics12040761. PMC 10135111. PMID 37107124.
ERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
///////ZIDEBACTAM, ANAX LABS, FDA 2026, APPROVALS 2026, Cypsedo, WCK-5107, WCK 5107, YPM97423DB
see………http://apisynthesisint.blogspot.in/2015/11/wck-5107-in-phase-1-from-wockhardt.html
SEE BACTAM SERIES…………..http://apisynthesisint.blogspot.in/p/bactam-series.html
C1C[C@H](CNC1)C(=O)NNC(=O)[C@@H]2CC[C@@H]3C[N@]2C(=O)N3OS(=O)(=O)O
or
O=C(NNC(=O)[C@@H]2CC[C@@H]1CN2C(=O)N1OS(=O)(=O)O)[C@@H]3CCCNC3
C1CC(CNC1)C(=O)NNC(=O)C2CCC3CN2C(=O)N3OS(=O)(=O)[O-].[Na+]
Gadosircoclamide
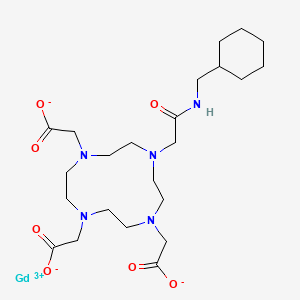
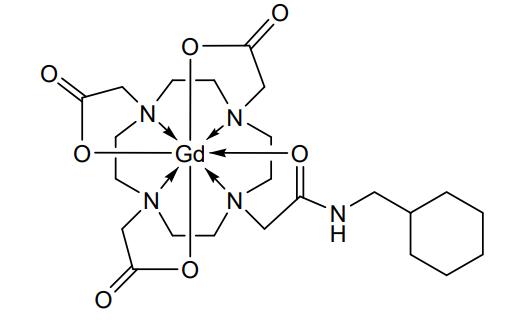
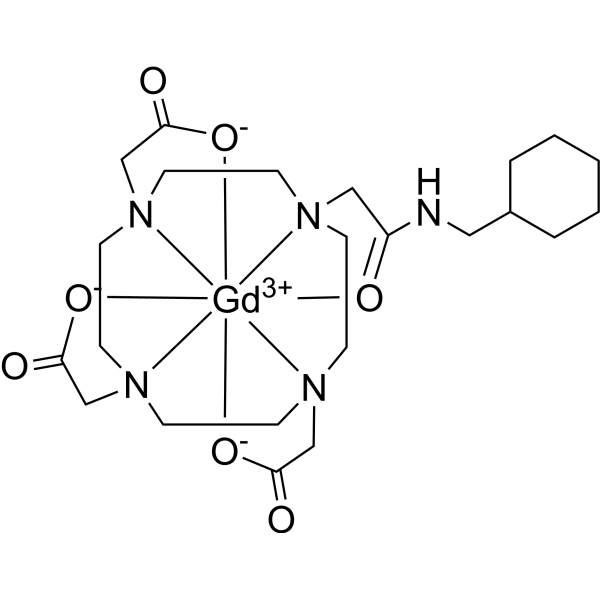
Gadosircoclamide
CAS 1801159-68-1
MF C23H38GdN5O7. MW653.8 g/mol
2-[4,7-bis(carboxylatomethyl)-10-[2-(cyclohexylmethylamino)-2-oxoethyl]-1,4,7,10-tetrazacyclododec-1-yl]acetate;gadolinium(3+)
- [10-[2-[(Cyclohexylmethyl)amino]-2-(oxo-kappaO)ethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetato(3-)-kappaN1,kappaN4,kappaN7,kappaN10,kappaO1,kappaO4,kappaO7]gadolinium
- [2,2′,2”-(10-{2-[(cyclohexylmethyl)amino]-2-oxo-kappaOethyl}-1,4,7,10-tetraazacyclododecane-1,4,7-triyl-kappa4N1,N4,N7,N10)tri(acetato-kappaO)]gadolinium
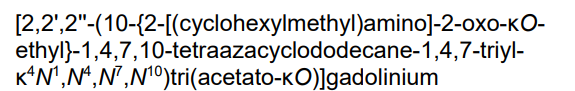
radiodiagnostic agent, 7V6P6PCM4U
Gadosircoclamide (CAS # 1801159-68-1) is a specialized gadolinium-based coordination complex used primarily as a magnetic resonance imaging (MRI) contrast agent. It is designed to enhance image contrast, help visualize lesions, and accurately track abnormalities during diagnostic scans.
SYN
R=H
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015105352&_cid=P11-MPXG49-92326-1
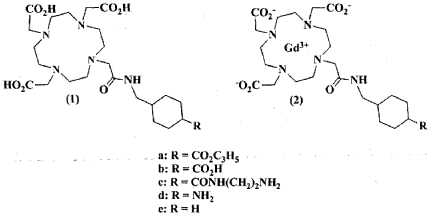
4) Synthesis of le.
DG3A-(f BuO)3 (3.0 g, 5.8 mmol) was added to a solution of 2-chlorocyclohexylmethylacetamid (1.2 g, 6.4 mmol) in acetanitrile (30 mL) prepared according to the conventional literature method (Cho, SD; Song, SY; Kim, . H.; Zhao, BX; Ahn, C; J oo, WH; Yoon, YJ; Falck, JR; Shin, DS B / (or. Chem. St. 2004, 25, 415) . The solution was stirred at room temperature for 24 hours. Solid impurities were removed by filtration, and the filtrate was evaporated under vacuum to obtain an oil phase residue. Subsequently, column chromatography on a silica phase (gradient elution: CH₂C1₂ to 10 % MeOH -CH₂Cl₂ , R f = 0.4 ( MeOH/ CH₂Cl₂ = A 1:9 mixture was performed and evaporated under reduced pressure to obtain a yellowish-white solid. As described in the preparation of the above Id, deprotection with TFA was performed to obtain a yellowish-white solid as a product. Yield: 2.4 g (82%). 1H R ( O): δ = 3.74/3.57 (m, 8H, -NCH₂CO₂- ) , 3.30 (m, 10H , overlapped -NCH₂CH₂N- ( 8H) & -CONHCH₂- ( 2H )), 3.10 (m, 8H, -NCH₂CH₂N- ) , 1.98/1.44/1.27 (in, 4H, -CH₂- , cyclohexyl ) , 1.88 (m, 1H, -NHCH₂CH- ) . Anal . Calculated for C₂₂H₃₅N₅₀ 0 7 · 3CF 3 C00H ■ 3H 2 0 : C, 38.14; H, 5.49; N, 7.94. Found: C, 37.83; H, 5.76; N, 8.44. MALDI-T0F MS (m/z): Calcd for C22H39N5O7,: 485.28, Found: 486.42 ([MH] + ), 508.44 ([MNa] + ).
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
[1].
Kim T, et al., Gadolinium complex comprising do3a-tranexamic acid conjugate. WO2015105352
////////////gadosircoclamide. anax labs, radiodiagnostic agent, 7V6P6PCM4U
Cipepofol
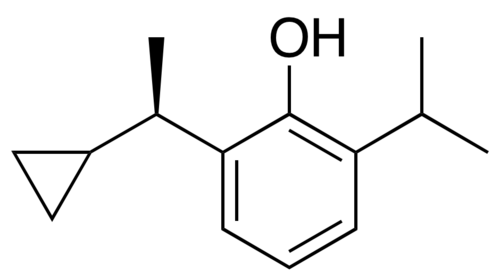
Cipepofol
CAS1637741-58-2
MW 204.31 g/mol MF C14H20O
2-[(1R)-1-cyclopropylethyl]-6-propan-2-ylphenol
FDA 2026, APPROVALS 2026, Cypsedo, HSK 3486, CS-0064163, GTPL 10812, HSK-3486, HY-116152, M3WGS532VY
- OriginatorSichuan Haisco Pharmaceutical
- ClassCyclopropanes; General anaesthetics; Phenols; Small molecules
- Mechanism of ActionGABA A receptor agonists
- RegisteredAnaesthesia; Sedation
- 10 Apr 2026Sichuan Haisco Pharmaceutical plans a phase III trial for Anesthesia (In Children, In adolescents) (IV) in May 2026 (NCT07510945)
- 28 Aug 2024No recent reports of development identified for preclinical development in Sedation in USA (IV, Infusion)
- 01 Aug 2024Zhongda Hospital plans a clinical trial for Sedation (IV) in August 2024 (NCT06538883)
To induce general anesthesia in adults undergoing surgery
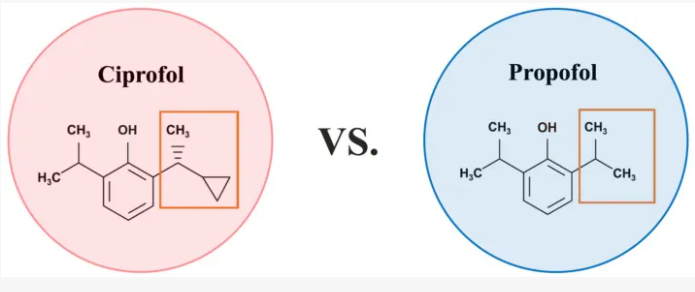
Cipepofol (also known as ciprofol or HSK3486) is a novel, short-acting intravenous anesthetic and sedative. As a structural analog of propofol, it targets \(GABA_{A}\) receptors but is 4 to 6 times more potent. It offers faster recovery, improved cardiovascular stability, and significantly less injection pain than propofol.
Key Clinical Advantages
- Superior Efficacy: Requires a lower dose to achieve the same sedative depth as propofol.
- Better Safety Profile: Associated with a lower incidence of injection pain, reduced respiratory depression, and better hemodynamic (blood pressure) stability.
- Fast Acting: Characterized by rapid onset and quick recovery times, making it ideal for procedures like gastrointestinal endoscopy, bronchoscopy, and general anesthesia induction.
Recent Developments
- FDA Approval: Cipepofol (sold under the brand name CYPSEDO) officially received U.S. FDA marketing approval, becoming the first China-originated innovative intravenous anesthetic to enter the global market.
- Ongoing Trials: Clinical trials and post-marketing studies are actively evaluating its safety in specific populations, such as elderly patients and children.
Cipepofol (INNTooltip International Nonproprietary Name, USANTooltip United States Adopted Name), also known as ciprofol or by its developmental code name HSK3486, is a general anesthetic related to propofol which is used for anesthesia and sedation.[1][2][3][4] The drug is used by intravenous infusion.[1] A short-acting and highly selective γ-aminobutyric acid positive allosteric modulator,[5] ciprofol is 4 to 6 times more potent than other phenol derivatives such as propofol or fospropofol.[6]
In May 2026, cipepofol was approved by the US FDA.[7] Manufactured by Haisco Pharmaceutical Group of Chengdu, Sichuan, China, ciprofol underwentphase I and II trials in Australia and China.[8][9][10] In these early studies, ciprofol was comparable in efficacy to propofol and was associated with fewer adverse events.[4][6][11][12][13][14][15][16][17][18]
Physical properties
Ciprofol is an optically active 2,6-disubstituted alkylphenol with a cyclopropylethyl group incorporated at the second carbon atom. This cyclopropyl group increases the steric effects and introduces stereoselective effects over its anesthetic properties. These properties appear to increase the anesthetic potency of ciprofol, when compared with propofol.[9]
Medical use
Ciprofol is used for the intravenous induction of general anesthesia.[3][4] Studies published in 2022 and 2023 found it was efficacious as a general anesthetic in patients undergoing gynecological surgery[6][11] and kidney transplantation,[19] as well as for endoscopic procedures such as bronchoscopy,[15][20] esophagogastroduodenoscopy and colonoscopy.[21][22]
Ciprofol has also been used for sedation of critically ill patients undergoing mechanical ventilation in the intensive care unit,[23] as well as for the treatment of agitation and delirium in that patient population.[24] When combined with mild therapeutic hypothermia, ciprofol may also be useful as a cerebral protective agent in the setting of cerebral ischemia-reperfusion injury.[25]
Experimental use
In experimental models of isoproterenol-induced myocardial infarction (using mice as subjects), ciprofol appears to protect the heart against oxidative damage, inflammation and apoptosis of cardiac muscle cells.[26]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US428011434&_cid=P12-MPW0XO-91017-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014180305&_cid=P12-MPW0R4-87054-1
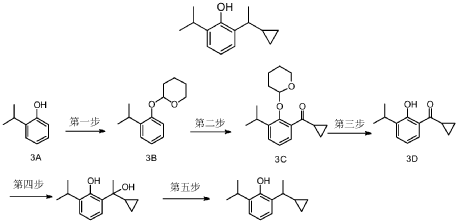
Example 16
[-cyclopropylethyl] -6 -isopropylphenol (compound 16)
2- [(lR)-l-cyclopropylethyl]-6-isopropyl -phenol
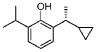
Preparation methods of Examples 16-17:
2-(1-Cyclopropylethyl-6-isopropylphenol (compound 3) 600 mg was used for resolution. Preparation conditions: (Instrument: Agilent 1260/CH-Y-J0404; Column: CHIRALPAK OJ-H (4.6 mm < 250 mm, 5 μm) No.: OJ-H-27; Mobile phase: A: isopropanol, B: n-hexane; Flow rate: 1.0 mL/min; Back pressure: 100 bar; Column temperature: 35°C; Wavelength: 210 nm; Period: 10 min)
Two optical isomers were obtained after separation: peak 1 (retention time: 10.72 min, 280 mg, pale yellow liquid, ee%=99%) and peak 2 (retention time: 13.58 min, 280 mg, pale yellow liquid, ee%=99%).
峰 1 : MS m/z(ESI): 203.1(Ml).
toMR (400 MHz,CDCl3 ) : δ 7.14(dd, 1H), δ 7.08(dd, 1H), 6.91 (t, 1H), 4.93 (s, 1H), 3.22-3.14(m, 1H), 2.55-2.48 (m, 1H), 1.33 (d, 6H), 1.28 (d, 3H), 1.10-1.05 (m, 1H), 0.60-0.58 (m, 1H), 0.49-0.46 (m, 1H), 0.25-0.18 (m, 2H).
峰 2: MS m/z(ESI): 203.1(Ml).
iHNMR (400 MHz,CDCl3) : 57.14(dd, 1H), δ 7.08(dd, 1H), 6.93 (t, 1H), 4.93 (s 1H), 3.22-3.15(m, 1H), 2.55-2.48 (m, 1H), 1.32 (d, 6H), 1.28 (d, 3H), 1.10-1.04 (m, 1H), 0.60-0.58 (m, 1H), 0.49-0.46 (m, 1H), 0.25-0.18 (m, 2H).
PAT
- Phenol derivative compound, methods of preparing said compound, pharmaceutical composition comprising said compound and use thereofPublication Number: BR-112015028212-B1Priority Date: 2013-05-09
- Phenol derivative and its preparation method and application in medicinePublication Number: CN-104507899-APriority Date: 2013-05-09
- Phenol derivative and preparation method and use in medicine thereofPublication Number: US-9517988-B2Priority Date: 2013-05-09Grant Date: 2016-12-13
- Phenol derivative and preparation method and use in medicine thereofPublication Number: EP-2995604-B1Priority Date: 2013-05-09Grant Date: 2019-07-10
- Phenol derivative, its production method and medicinal applicationPublication Number: JP-6431155-B2Priority Date: 2013-05-09Grant Date: 2018-11-28
- Phenol derivative, preparation method and medical application thereofPublication Number: CN-104507899-BPriority Date: 2013-05-09Grant Date: 2016-11-30
- Phenol derivative and method of preparation and medical use thereofPublication Number: ES-2746987-T3Priority Date: 2013-05-09Grant Date: 2020-03-09
- Phenol derivative and preparation method and use in medicine thereofPublication Number: US-2016060197-A1Priority Date: 2013-05-09
- Isopropyl phenol derivative and preparation method thereofPublication Number: WO-2016026459-A1Priority Date: 2014-08-22
- Phenol derivative and preparation method and use in medicine thereofPublication Number: AU-2014264103-B2Priority Date: 2013-05-09Grant Date: 2018-03-22
- Phenol derivative and preparation method and use in medicine thereofPublication Number: AU-2014264103-A1Priority Date: 2013-05-09
- Phenol derivative and preparation method and use in medicine thereofPublication Number: AU-2014264103-C1Priority Date: 2013-05-09Grant Date: 2018-08-02
- Phenol derivative and preparation method and use in medicine thereofPublication Number: EP-2995604-A1Priority Date: 2013-05-09
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- “Sichuan Haisco Pharmaceutical”. AdisInsight. 28 August 2024. Retrieved 1 October 2025.
- “Ciprofol (Cipepofol): A γ-Aminobutyric Acid Receptor Agonist for Induction of Anesthesia”. Chemistry and Pharmacology of Drug Discovery. Wiley. 2024. pp. 251–274. doi:10.1002/9781394225156.ch12. ISBN 978-1-394-22512-5. Retrieved 1 October 2025.
- Wang X, Wang X, Liu J, Zuo YX, Zhu QM, Wei XC, et al. (March 2022). “Effects of ciprofol for the induction of general anesthesia in patients scheduled for elective surgery compared to propofol: a phase 3, multicenter, randomized, double-blind, comparative study”. European Review for Medical and Pharmacological Sciences. 26 (5): 1607–1617. PMID 35302207.
- Zeng Y, Wang DX, Lin ZM, Liu J, Wei XC, Deng J, et al. (February 2022). “Efficacy and safety of HSK3486 for the induction and maintenance of general anesthesia in elective surgical patients: a multicenter, randomized, open-label, propofol-controlled phase 2 clinical trial”. European Review for Medical and Pharmacological Sciences. 26 (4): 1114–1124. PMID 35253166.
- Liao J, Li M, Huang C, Yu Y, Chen Y, Gan J, et al. (2022). “Pharmacodynamics and Pharmacokinetics of HSK3486, a Novel 2,6-Disubstituted Phenol Derivative as a General Anesthetic”. Frontiers in Pharmacology. 13 830791. doi:10.3389/fphar.2022.830791. PMC 8851058. PMID 35185584.
- Chen BZ, Yin XY, Jiang LH, Liu JH, Shi YY, Yuan BY (August 2022). “The efficacy and safety of ciprofol use for the induction of general anesthesia in patients undergoing gynecological surgery: a prospective randomized controlled study”. BMC Anesthesiology. 22 (1) 245. doi:10.1186/s12871-022-01782-7. PMC 9347095. PMID 35922771.
- “Novel Drug Approvals for 2026”. U.S. Food and Drug Administration. 29 May 2026. Retrieved 31 May 2026.
- Lu M, Liu J, Wu X, Zhang Z (2023). “Ciprofol: A Novel Alternative to Propofol in Clinical Intravenous Anesthesia?”. BioMed Research International. 2023 7443226. doi:10.1155/2023/7443226. PMC 9879693. PMID 36714027.
- Qin L, Ren L, Wan S, Liu G, Luo X, Liu Z, et al. (May 2017). “Design, Synthesis, and Evaluation of Novel 2,6-Disubstituted Phenol Derivatives as General Anesthetics”. Journal of Medicinal Chemistry. 60 (9): 3606–3617. doi:10.1021/acs.jmedchem.7b00254. PMID 28430430.
- Nair A, Seelam S (2022). “Ciprofol- a game changing intravenous anesthetic or another experimental drug!”. Saudi Journal of Anaesthesia. 16 (2): 258–259. doi:10.4103/sja.sja_898_21. PMC 9009555. PMID 35431734.
- Man Y, Xiao H, Zhu T, Ji F (March 2023). “Study on the effectiveness and safety of ciprofol in anesthesia in gynecological day surgery: a randomized double-blind controlled study”. BMC Anesthesiology. 23 (1) 92. doi:10.1186/s12871-023-02051-x. PMC 10039513. PMID 36964501.
- Chen X, Guo P, Yang L, Liu Z, Yu D (2022). “Comparison and Clinical Value of Ciprofol and Propofol in Intraoperative Adverse Reactions, Operation, Resuscitation, and Satisfaction of Patients under Painless Gastroenteroscopy Anesthesia”. Contrast Media & Molecular Imaging. 2022 9541060. doi:10.1155/2022/9541060. PMC 9314164. PMID 35935320.
- Zhong J, Zhang J, Fan Y, Zhu M, Zhao X, Zuo Z, et al. (May 2023). “Efficacy and safety of Ciprofol for procedural sedation and anesthesia in non-operating room settings”. Journal of Clinical Anesthesia. 85 111047. doi:10.1016/j.jclinane.2022.111047. PMID 36599219. S2CID 255468218.
- Liang P, Dai M, Wang X, Wang D, Yang M, Lin X, et al. (June 2023). “Efficacy and safety of ciprofol vs. propofol for the induction and maintenance of general anaesthesia: A multicentre, single-blind, randomised, parallel-group, phase 3 clinical trial”. European Journal of Anaesthesiology. 40 (6): 399–406. doi:10.1097/EJA.0000000000001799. PMC 10155686. PMID 36647565.
- Luo Z, Tu H, Zhang X, Wang X, Ouyang W, Wei X, et al. (March 2022). “Efficacy and Safety of HSK3486 for Anesthesia/Sedation in Patients Undergoing Fiberoptic Bronchoscopy: A Multicenter, Double-Blind, Propofol-Controlled, Randomized, Phase 3 Study”. CNS Drugs. 36 (3): 301–313. doi:10.1007/s40263-021-00890-1. PMC 8927014. PMID 35157236.
- Hu C, Ou X, Teng Y, Shu S, Wang Y, Zhu X, et al. (November 2021). “Sedation Effects Produced by a Ciprofol Initial Infusion or Bolus Dose Followed by Continuous Maintenance Infusion in Healthy Subjects: A Phase 1 Trial”. Advances in Therapy. 38 (11): 5484–5500. doi:10.1007/s12325-021-01914-4. PMC 8523013. PMID 34559359.
- Teng Y, Ou M, Wang X, Zhang W, Liu X, Liang Y, et al. (September 2021). “Efficacy and safety of ciprofol for the sedation/anesthesia in patients undergoing colonoscopy: Phase IIa and IIb multi-center clinical trials”. European Journal of Pharmaceutical Sciences. 164 105904. doi:10.1016/j.ejps.2021.105904. PMID 34116176.
- Zhu Q, Luo Z, Wang X, Wang D, Li J, Wei X, et al. (April 2023). “Efficacy and safety of ciprofol versus propofol for the induction of anesthesia in adult patients: a multicenter phase 2a clinical trial”. International Journal of Clinical Pharmacy. 45 (2): 473–482. doi:10.1007/s11096-022-01529-x. PMC 10147789. PMID 36680620.
- Qin K, Qin WY, Ming SP, Ma XF, Du XK (July 2022). “Effect of ciprofol on induction and maintenance of general anesthesia in patients undergoing kidney transplantation”. European Review for Medical and Pharmacological Sciences. 26 (14): 5063–5071. PMID 35916802.
- Wu B, Zhu W, Wang Q, Ren C, Wang L, Xie G (2022). “Efficacy and safety of ciprofol-remifentanil versus propofol-remifentanil during fiberoptic bronchoscopy: A prospective, randomized, double-blind, non-inferiority trial”. Frontiers in Pharmacology. 13 1091579. doi:10.3389/fphar.2022.1091579. PMC 9812563. PMID 36618929.
- Li J, Wang X, Liu J, Wang X, Li X, Wang Y, et al. (August 2022). “Comparison of ciprofol (HSK3486) versus propofol for the induction of deep sedation during gastroscopy and colonoscopy procedures: A multi-centre, non-inferiority, randomized, controlled phase 3 clinical trial”. Basic & Clinical Pharmacology & Toxicology. 131 (2): 138–148. doi:10.1111/bcpt.13761. PMC 9543620. PMID 35653554.
- Long YQ, Feng CD, Ding YY, Feng XM, Liu H, Ji FH, et al. (2022). “Esketamine as an Adjuvant to Ciprofol or Propofol Sedation for Same-Day Bidirectional Endoscopy: Protocol for a Randomized, Double-Blind, Controlled Trial With Factorial Design”. Frontiers in Pharmacology. 13 821691. doi:10.3389/fphar.2022.821691. PMC 8975265. PMID 35370640.
- Liu Y, Yu X, Zhu D, Zeng J, Lin Q, Zang B, et al. (May 2022). “Safety and efficacy of ciprofol vs. propofol for sedation in intensive care unit patients with mechanical ventilation: a multi-center, open label, randomized, phase 2 trial”. Chinese Medical Journal. 135 (9): 1043–1051. doi:10.1097/CM9.0000000000001912. PMC 9276409. PMID 34924506.
- Liu GL, Wu GZ, Ge D, Zhou HJ, Cui S, Gao K, et al. (2023). “Efficacy and safety of ciprofol for agitation and delirium in the ICU: A multicenter, single-blind, 3-arm parallel randomized controlled trial study protocol”. Frontiers in Medicine. 9 1024762. doi:10.3389/fmed.2022.1024762. PMC 9868613. PMID 36698817.
- Wang YC, Wu MJ, Zhou SL, Li ZH (January 2023). “Protective effects of combined treatment with ciprofol and mild therapeutic hypothermia during cerebral ischemia-reperfusion injury”. World Journal of Clinical Cases. 11 (3): 487–492. doi:10.12998/wjcc.v11.i3.487. PMC 9923870. PMID 36793629.
- Yang Y, Xia Z, Xu C, Zhai C, Yu X, Li S (2022). “Ciprofol attenuates the isoproterenol-induced oxidative damage, inflammatory response and cardiomyocyte apoptosis”. Frontiers in Pharmacology. 13 1037151. doi:10.3389/fphar.2022.1037151. PMC 9723392. PMID 36483733.
- Vittori A, Di Fabio C, Cascella M, Marinangeli F, Francia E, Mascilini I, et al. (January 2026). “Advantages of Ciprofol with Special Consideration of Pediatric Anesthesia”. Children (Basel, Switzerland). 13 (2). doi:10.3390/children13020188. PMC 12939459. PMID 41749542.
- Liu SB, Yao X, Tao J, Yang JJ, Zhao YY, Liu DW, et al. (March 2023). “Population total and unbound pharmacokinetics and pharmacodynamics of ciprofol and M4 in subjects with various renal functions”. British Journal of Clinical Pharmacology. 89 (3): 1139–1151. doi:10.1111/bcp.15561. PMID 36217805. S2CID 252818288.
- Hu Y, Li X, Liu J, Chen H, Zheng W, Zhang H, et al. (December 2022). “Safety, pharmacokinetics and pharmacodynamics of a novel γ-aminobutyric acid (GABA) receptor potentiator, HSK3486, in Chinese patients with hepatic impairment”. Annals of Medicine. 54 (1): 2769–2780. doi:10.1080/07853890.2022.2129433. PMC 9559057. PMID 36217101.
- Li X, Yang D, Li Q, Wang H, Wang M, Yan P, et al. (2021). “Safety, Pharmacokinetics, and Pharmacodynamics of a Single Bolus of the γ-aminobutyric Acid (GABA) Receptor Potentiator HSK3486 in Healthy Chinese Elderly and Non-elderly”. Frontiers in Pharmacology. 12 735700. doi:10.3389/fphar.2021.735700. PMC 8430033. PMID 34512361.
- Ding YY, Long YQ, Yang HT, Zhuang K, Ji FH, Peng K (December 2022). “Efficacy and safety of ciprofol for general anaesthesia induction in elderly patients undergoing major noncardiac surgery: A randomised controlled pilot trial”. European Journal of Anaesthesiology. 39 (12): 960–963. doi:10.1097/EJA.0000000000001759. PMID 36214498. S2CID 252779399.
- Duan G, Lan H, Shan W, Wu Y, Xu Q, Dong X, et al. (April 2023). “Clinical effect of different doses of ciprofol for induction of general anesthesia in elderly patients: A randomized, controlled trial”. Pharmacology Research & Perspectives. 11 (2) e01066. doi:10.1002/prp2.1066. PMC 9944862. PMID 36811327. S2CID 257098376.
- Yang Y, Xia Z, Xu C, Zhai C, Yu X, Li S (2022). “Ciprofol attenuates the isoproterenol-induced oxidative damage, inflammatory response and cardiomyocyte apoptosis”. Frontiers in Pharmacology. 13 1037151: 1037151. doi:10.3389/fphar.2022.1037151. PMC 9723392. PMID 36483733.
- Bian Y, Zhang H, Ma S, Jiao Y, Yan P, Liu X, et al. (January 2021). “Mass balance, pharmacokinetics and pharmacodynamics of intravenous HSK3486, a novel anaesthetic, administered to healthy subjects”. British Journal of Clinical Pharmacology. 87 (1): 93–105. doi:10.1111/bcp.14363. PMID 32415708. S2CID 218658207.
Further reading
- Bajwa SJ, Vinayagam S, Shinde S, Dalal S, Vennel J, Nanda S (January 2023). “Recent advancements in total intravenous anaesthesia and anaesthetic pharmacology”. Indian Journal of Anaesthesia. 67 (1): 56–62. doi:10.4103/ija.ija_1022_22. PMC 10034929. PMID 36970470.
- Skiljic S, Budrovac D, Cicvaric A, Neskovic N, Kvolik S (February 2023). “Advances in Analgosedation and Periprocedural Care for Gastrointestinal Endoscopy”. Life. 13 (2): 473. Bibcode:2023Life…13..473S. doi:10.3390/life13020473. PMC 9962362. PMID 36836830.
- Wei A, Yang L, Ma S, Jin G, Yang M, Zhou J (November 2022). “A case report of ciprofol overdose during anesthesia/analgesia and literature review: clinical presentation, blood pressure, and management”. The Journal of International Medical Research. 50 (11) 3000605221132466. doi:10.1177/03000605221132466. PMC 9659933. PMID 36366740.
| Clinical data | |
|---|---|
| Other names | Ciprofol; CS-0064163; CS0064163; GTPL10812; GTPL-10812; HSK-3486; HSK3486; HY-116152; HY116152; (R)-2-(1-Cyclopropylethyl)-6-isopropylphenol |
| Routes of administration | Intravenous infusion[1] |
| Drug class | GABAA receptor positive allosteric modulator |
| Pharmacokinetic data | |
| Metabolism | Liver glucuronidation |
| Excretion | Kidney |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1637741-58-2 |
| PubChem CID | 86301664 |
| DrugBank | DB16295 |
| ChemSpider | 76794458 |
| UNII | M3WGS532VY |
| KEGG | D12449 |
| ChEMBL | ChEMBL4094894 |
| Chemical and physical data | |
| Formula | C14H20O |
| Molar mass | 204.313 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
////////cipepofol, FDA 2026, APPROVALS 2026, Cypsedo, HSK 3486, CS-0064163, GTPL 10812, HSK-3486, HY-116152, M3WGS532VY, ANAESTHETIC
Fudapirine
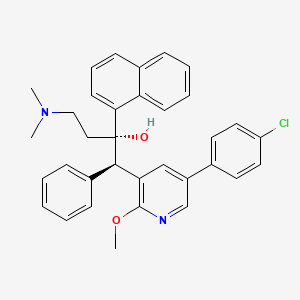
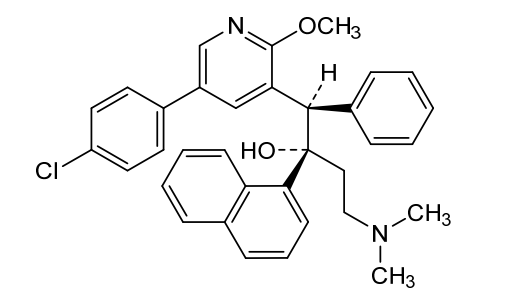
Fudapirine
CAS 1859978-72-5
MFC34H33ClN2O2 MW537.1 g/mol
- (alphaS,betaR)-5-(4-Chlorophenyl)-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-pyridineethanol
- 3-Pyridineethanol, 5-(4-chlorophenyl)-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-, (alphaS,betaR)-
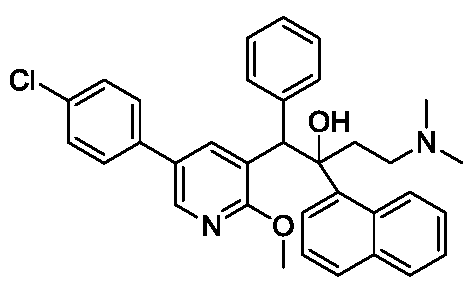
(1R,2S)-1-[5-(4-chlorophenyl)-2-methoxy-3-pyridinyl]-4-(dimethylamino)-2-naphthalen-1-yl-1-phenylbutan-2-ol
(1R,2S)-1-[5-(4-chlorophenyl)-2-methoxypyridin-3-yl]-4-(dimethylamino)-2-(naphthalen-1-yl)-1-phenylbutan-2-ol
antibacterial, Sudapyridine, WX-081, WX 081, 7X86XPE5TG,
- A Phase III Study of Oral Sudapyridine (WX-081) Tablets in Rifampicin-Resistant Pulmonary Tuberculosis PatientsCTID: NCT05824871Phase: Phase 3Status: RecruitingDate: 2025-06-29
- Drug-Drug Interaction and Food Effect of Sudapyridine(WX-081) With Itraconazole and Rifampin in Healthy Chinese AdultsCTID: NCT06701136Phase: Phase 1Status: Not yet recruitingDate: 2025-02-12
- Sudapyridine (WX-081) in Healthy VolunteersCTID: NCT06117514Phase: Phase 1Status: CompletedDate: 2023-11-07
- Evaluation of Early Bactericidal Activity and Safety in Pulmonary Tuberculosis With WX-081CTID: NCT04608955Phase: Phase 2Status: CompletedDate: 2023-09-11
Fudapirine (also known as sudapyridine or WX-081) is a novel, next-generation antimycobacterial drug primarily being developed to treat tuberculosis (TB). It belongs to a chemical class called diarylquinolines, making it a close analogue of bedaquiline, an already established drug used for drug-resistant tuberculosis.Key Facts About FudapirinePrimary Function: It displays powerful anti-mycobacterial activity against Mycobacterium tuberculosis strains.
Mechanism: As a diarylquinoline, it selectively inhibits bacterial ATP synthase, effectively cutting off the energy supply that the tuberculosis bacteria need to survive and replicate.Development Stage: According to pharmacology databases like the IUPHAR/BPS Guide to PHARMACOLOGY, the drug has advanced into Phase III clinical evaluation.Official Naming: While initially designated as WX-081, the World Health Organization (WHO) assigned it the International Nonproprietary Name (INN) fudapirine in early 2025.Other Applications: Beyond standard TB, researchers are investigating its therapeutic potential against non-tuberculous mycobacterial (NTM) infections.
PAT
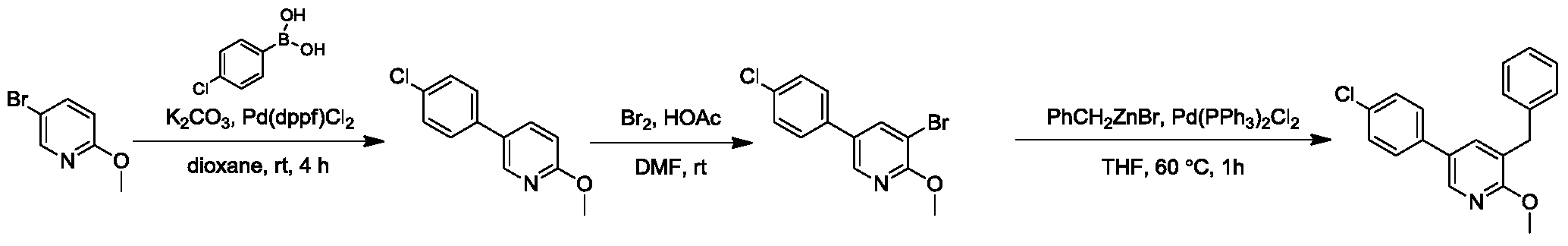
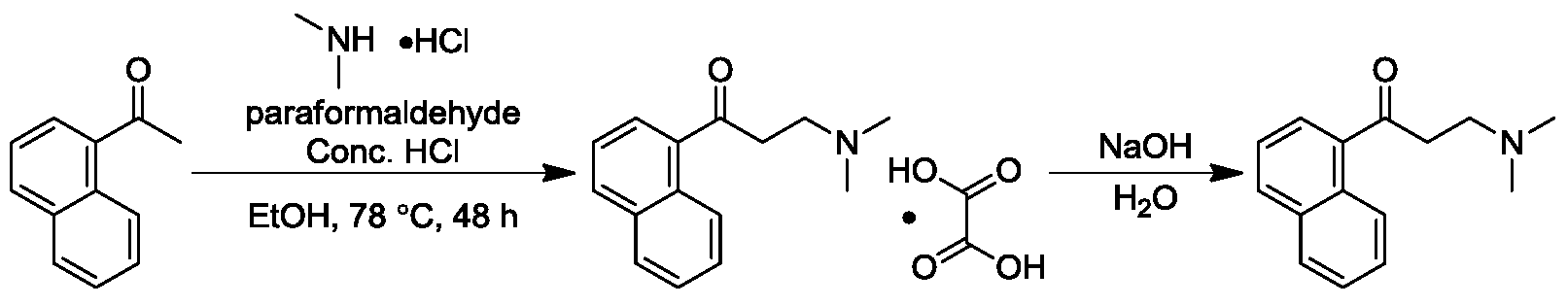
Step 5:
Synthesis of
1-(5-(4-chlorophenyl)-2-methoxypyridin-3-yl)-4-(dimethylamino)-2-(naphth-1-yl)-1-phenylbut-2-ol
Titration of n-butyllithium : Under nitrogen protection, 1.00 g
of diphenylacetic acid (Alfa, 4.71 mmol) was added to 10 mL of tetrahydrofuran to form a colorless and transparent solution. A hexane solution of
n-butyllithium was slowly added dropwise to this solution using a syringe. The solution was observed to turn yellow locally during the addition, but the yellow color quickly disappeared. The endpoint was reached when a yellow solution formed after one drop and did not fade within half a minute. The volumes of
n-butyllithium were recorded (1.927 mL and 1.985 mL, with an average volume of 1.95 mL). Therefore, the concentration of
the n-butyllithium hexane solution used was 2.42 mol/L.
[0171]TMP (2.74 kg, 19.3 mol) was dissolved in anhydrous tetrahydrofuran (12 L). The reaction temperature was cooled to -65°C using a dry ice-acetone bath, and then
n-butyllithium (8 L, 19.3 mol, 2.42 mol/L n-hexane solution) was added dropwise. The temperature was controlled between -20°C and -78°C. The reaction system was observed to gradually change color from light yellow to red to deep red, eventually forming a yellow suspension. Stirring was continued at this temperature for 30 minutes. Then, the reaction temperature was lowered to -75°C to -80°C, and over 4–6 hours, a solution of
3-benzyl-5-(4-chlorophenyl)-2-methoxypyridine (4.08 kg, 12.9 mol) in anhydrous tetrahydrofuran (6 L) was slowly added dropwise. The temperature was maintained between -65°C and -78°C, and a mild exothermic reaction with a deep red color was observed. After the initial addition was complete, a solution of 3-(dimethylamino)-1-(naphth-1-yl)propyl-1-one (3.26 kg, 12.9 mol, 90% purity) in anhydrous tetrahydrofuran (2.0 L) was slowly added dropwise over 2–4 hours. The system exhibited significant exothermic activity, and the flow rate was controlled to maintain the temperature at -65°C to -78°C. After the addition was complete, the temperature was maintained at -65°C to -78°C, and stirring was continued for another half hour. HPLC analysis showed that the content of
3-benzyl-5-(4-chlorophenyl)-2-methoxypyridine was less than 10%. The reaction solution was slowly added to a saturated
ammonium chloride solution (40 L) for quenching, and the mixture was separated. The aqueous phase was extracted with ethyl acetate (30 L). The combined organic phases were washed and separated with saturated brine (30 L). The organic phases were concentrated under reduced pressure at 40–50 °C to obtain a yellow oily crude product (13.5 kg). The crude product was stirred in a mixed solvent of ethyl acetate/n-heptane (4 L, 1/4) at 5–15 °C for 16 hours to precipitate a white solid. The product was filtered, and the filter cake was slurried with ethanol (4 L × 2). After filtration, the filter cake was vacuum dried to constant weight (50 °C, 24–48 hours) to obtain the target compound 1-(5-(4-chlorophenyl)-2-methoxypyridin-3-yl)-4-(dimethylamino)-2-(naphth-1-yl)-1-phenylbut-2-ol (1.83 kg, yield 23.23%), a white solid. HPLC identification showed that isomer A accounted for 88.3% and isomer B accounted for 4.8%.
1 H NMR (400MHz, CDCl 3 )δ: 8.85(d,J=2.3Hz,1H),8.64(d,J=8.7Hz,1H),8.32(d,J=2.4Hz,1H),7. 98-7.86(m,2H),7.72-7.61(m,2H),7.57(d,J=8.4Hz,2H),7.54-7.43(m,3 H),7.33(t,J=7.8Hz,1H),7.20-7.17(m,2H),6.95-6.87(m,3H),5.85(s,1 H),4.17(s,3H),2.60-2.51(m,1H),2.19-2.04(m,2H),2.01-1.97(m,7H).
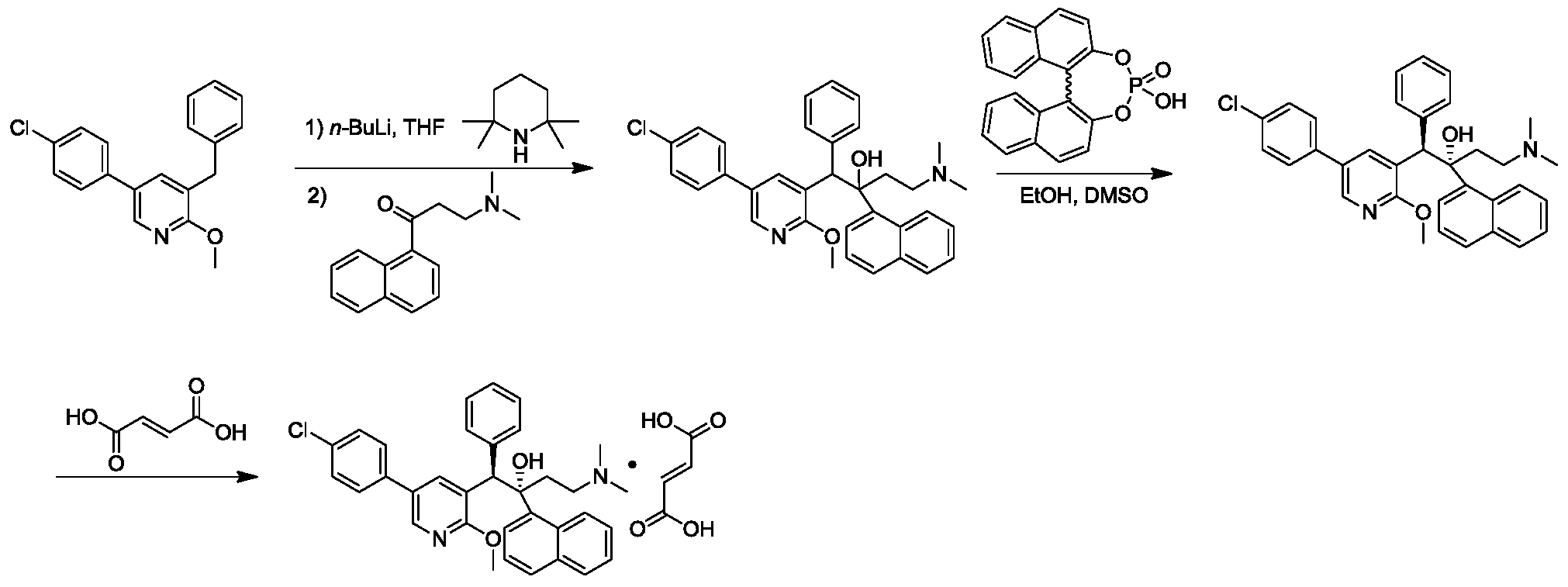
[0172]Step 6: Synthesis of (1R,2S)-1-(5-(4-chlorophenyl)-2-methoxypyridin-3-yl)-4-(dimethylamino)-2-(naphth-1-yl)-1-phenylbut-2-ol compound I-2
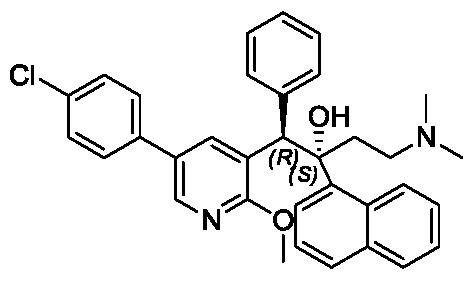
[0175]Two parallel batches were prepared: R-(-)-binaphthol phosphate (519.3 g, 1.49 mol) was suspended in DMSO (1.0 L) and heated to 50 °C with stirring until dissolved and clear. 1-(5-(4-chlorophenyl)-2-methoxypyridin-3-yl)-4-(dimethylamino)-2-(naphthaleneethanol-1-yl)-1-phenylbutyl-2-ol (910 g, 1.49 mol, isomer A content 88.3%) was added to an ethanol (24 L) solution, and the DMSO (1.0 L) solution of R-(-)-binaphthol phosphate prepared above was added dropwise over 1–2 hours with stirring (196 rpm). Undissolved particulate compounds began to dissolve, but a more viscous emulsion formed. After the addition was complete, the reaction mixture was stirred at 15–35 °C for 16 hours. The reaction solution was heated to reflux in an oil bath and refluxed for 1 hour. Heating was then stopped, and the reaction solution was cooled to 15–35°C and stirred for 16 hours. The reaction solution was filtered (two batches were combined for processing). Due to the high viscosity of the solids, filtration was slow. The filter cake was slurried three times with ethanol (20 liters). The combined organic phases were concentrated to constant weight to obtain a yellow oily substance (5 kg). Water (10 liters) and ethyl acetate (5 liters) were added to this crude product. The pH of the system was adjusted to 11 with a 10% cold
sodium hydroxide aqueous solution, and stirring was continued for 1 hour. Then, the mixture was separated. A large amount of solid precipitated from the system. The solid obtained by filtration was isomer A (350 g, purity 97%, white solid). The filtrate was concentrated under reduced pressure at 50°C to constant weight, and ethanol (1.0 L) was added. The mixture was stirred at 15-35°C for 16 hours, filtered, and the filter cake was washed three times with ethanol (400 mL) to obtain a white solid. The solid was dried under vacuum to constant weight (50°C, 24-48 hours) to obtain compound I-2 (400 g, purity 95%, ee value greater than 99.5%, yield 24%), a white solid.
1 H NMR (400MHz, CDCl
3 )δ: 8.85(d,J=2.3Hz,1H),8.64(d,J=8.7Hz,1H),8.32(d,J=2.4Hz,1H),7. 98-7.86(m,2H),7.72-7.61(m,2H),7.57(d,J=8.4Hz,2H),7.54-7.43(m,3 H),7.33(t,J=7.8Hz,1H),7.20-7.17(m,2H),6.95-6.87(m,3H),5.85(s,1 H),4.17(s,3H),2.60-2.51(m,1H),2.19-2.04(m,2H),2.01-1.97(m,7H).
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- In vitro and Intracellular Antibacterial Activity of Sudapyridine (WX-081) Against TuberculosisPublication Name: Infection and Drug ResistancePublication Date: 2023-01PMCID: PMC9840375PMID: 36647451DOI: 10.2147/idr.s390187
- Discovery and preclinical profile of sudapyridine (WX-081), a novel anti-tuberculosis agentPublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2022-09-01PMID: 35636648DOI: 10.1016/j.bmcl.2022.128824
- Sudapyridine (WX-081), a Novel Compound against Mycobacterium tuberculosisPublication Name: Microbiology SpectrumPublication Date: 2022-02-23PMCID: PMC8849072PMID: 35170994DOI: 10.1128/spectrum.02477-21
PAT
- Pharmaceutical composition for treating mycobacterium tuberculosis infection and application thereofPublication Number: CN-117243956-APriority Date: 2023-09-08
- Pharmaceutical composition containing clarithromycin and Shu Da pyridinePublication Number: CN-116763807-APriority Date: 2023-08-08
- Pharmaceutical composition containing clarithromycin and Shu Da pyridinePublication Number: CN-116763807-BPriority Date: 2023-08-08Grant Date: 2025-04-11
- A kind of preparation method of pyridine derivative compound and its intermediate and crystal formPublication Number: CN-108473428-BPriority Date: 2016-01-13Grant Date: 2021-07-23
/////////fudapirine, anax labs, antibacterial, Sudapyridine, WX-081, WX 081, 7X86XPE5TG,
Fosrugocrixan
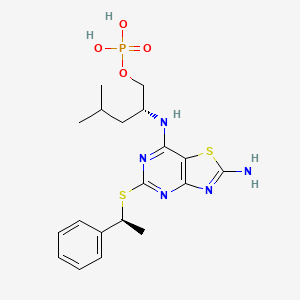
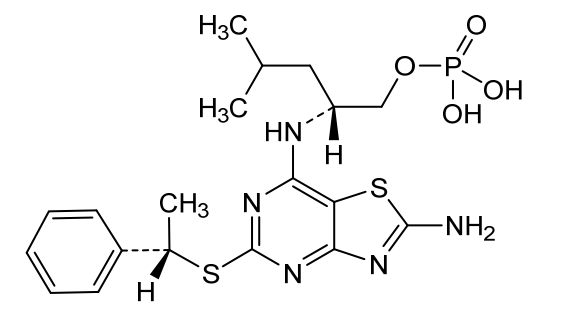
Fosrugocrixan
CAS 2408145-38-8
MF C19H26N5O4PS2, MW483.5 g/mol
[(2R)-2-[[2-amino-5-[(1S)-1-phenylethyl]sulfanyl-[1,3]thiazolo[4,5-d]pyrimidin-7-yl]amino]-4-methylpentyl] dihydrogen phosphate
- (2R)-2-[(2-amino-5-{[(1S)-1-phenylethyl]sulfanyl}[1,3]thiazolo[4,5-d]pyrimidin-7-yl)amino]-4-methylpentyl dihydrogen phosphate
- 1-Pentanol, 2-[[2-amino-5-[[(1S)-1-phenylethyl]thio]thiazolo[4,5-d]pyrimidin-7-yl]amino]-4-methyl-, 1-(dihydrogen phosphate), (2R)-
(2R)-2-[(2-amino-5-{[(1S)-1-phenylethyl]sulfanyl}[1,3]thiazolo[4,5-d]pyrimidin-7-yl)amino]-4-methylpentyl dihydrogen phosphate
CX3C chemokine receptor 1 (CX3CR1) antagonist, antiinflammatory, 4ZXD25SC4S, KAND-145, KAND 145
- OriginatorKancera
- DeveloperNovakand Pharma
- ClassAnti-inflammatories; Antineoplastics; Small molecules
- Mechanism of ActionChemokine CXCL13 inhibitors
- Phase IOvarian cancer
- PreclinicalChronic lymphocytic leukaemia
- No development reportedInflammation
- 22 Sep 2025Kancera is now called Novakand Pharma
- 28 Apr 2025No recent reports of development identified for preclinical development in Ovarian-cancer in Sweden (IV)
- 03 May 2024Efficacy and adverse event data from a phase I trials in healthy volunteers released by Kancera
Fosrugocrixan (also known by its developmental code KAND145) is a novel, small-molecule drug candidate acting as a selective antagonist for CX3C chemokine receptor 1 (CX3CR1), commonly known as the fractalkine receptor.
Key Characteristics and Mechanism
- Drug Class: It represents a first-in-class small molecule immune modulator.
- Phosphate Prodrug: Fosrugocrixan is designed as a soluble phosphate prodrug. Once inside the body (in vivo), it converts into its active drug form, rugocrixan (formerly KAND567).
- Mechanism of Action: By blocking the CX3CR1 fractalkine pathway, it controls and prevents the trafficking of disease-promoting immune cells. This blockage provides potent anti-inflammatory activity.
Clinical Development and Targets
The drug is being actively developed by Novakand Pharma (a company formerly known as Kancera). Its primary therapeutic targets span several conditions driven by runaway inflammation and immune responses:
- Cardiovascular Diseases: Specifically targeted to manage conditions where hyper-inflammation damages tissue (such as post-myocardial infarction or heart conditions).
- Autoimmune & Inflammatory Diseases: Evaluated for broad anti-inflammatory potential.
- Oncology: Investigated for its ability to regulate the tumor microenvironment.
SYN
WO 2020008064
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020008064&_cid=P21-MPQB4Y-95682-1
SYN
Karlström et al. J. Med. Chem., 2013, 56, 3177-3190
https://pubs.acs.org/doi/10.1021/jm3012273
PAT
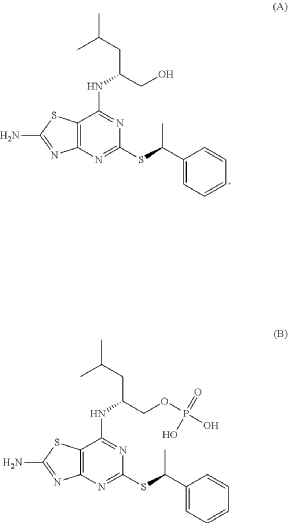
(2R)-2-[(2-Amino-5-{[(1S)-1-phenylethyl]sulfanyl}[1,3]thiazolo[4,5-d]pyrimidin-7-yl)amino]-4-methylpentyl dihydrogen phosphate (B), are known to act as antagonists of the fractalkine receptor (CX3CR1) (Karlström et al. J. Med. Chem., 2013, 56, 3177-3190; WO 2020/008064)
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US336567291&_cid=P21-MPQAYT-90868-1
Example 1
Preparation of (2R)-2-[(2-Amino-5-{[(1S)-1-phenylethyl]sulfanyl}[1,3]thiazolo[4,5-d]pyrimidin-7-yl)amino]-4-methylpentyl dihydrogen phosphate
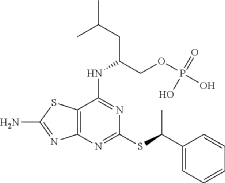
Phosphorus oxychloride (337 mg, 2.2 mmol) was dissolved in THE (0.75 mL) and water (25 mg, 1.4 mmol) was added. The mixture was cooled in an ice-bath and pyridine (111 mg, 113 μL, 1.4 mmol) was added followed by (2R)-2-[(2-amino-5-{[(1S)-1-phenylethyl]sulfanyl}-[1,3] thiazolo[4,5-d]pyrimidin-7-yl)amino]-4-methylpentan-1-ol hydrochloride (110 mg, 0.25 mmol) (Karlstr6m S., et al., J. Med. Chem., 2013, 56, 3177-3190; WO 2006/107258). The reaction mixture was stirred at ice-bath temperature for 1 h. To a mixture of phosphorus oxychloride (337 mg, 2.2 mmol) and water (25 mg, 1.4 mmol) in THE was added, at ice-bath temperature pyridine (111 mg, 113 μL, 1.4 mmol). Half of this mixture was added to the reaction mixture described above. The reaction mixture was stirred at ice-bath temperature for another 1 h. Water (3 mL) was added and the reaction mixture was stirred for 15 min at ice-bath temperature and 20 min at room temperature. DCM (3 mL) was added and the phases were separated. The aqueous phase was extracted with another portion of DCM (3 mL) and the organic phases were combined. At this point the product started to precipitate as a pale-yellow gum in the organic phase. MeOH was added and the now homogeneous solution was transferred to a round-bottomed flask and was evaporated to yield 120 mg of crude product, which according to HPLC was ca. 93% pure. The crude material was dissolved in a MeOH/water mixture and the pH was adjusted to about 6-7 with 1 M NaOH. The material was purified by preparative HPLC (basic method). The pure fractions were pooled, evaporated, and dried in vacuum. The product was assumed to be the diammonium salt after purification. 1H NMR (600 MHz, CD 3OD) δ H ppm 7.43-7.47 (m, 2H) 7.30-7.35 (m, 2H) 7.20-7.24 (m, 1H) 5.08 (q, J=7.03 Hz, 1H) 4.59-4.68 (m, 1H) 3.92 (ddd, J=10.12, 5.67, 4.30 Hz, 1H) 3.88 (dt, J=10.12, 4.94 Hz, 1H) 1.74 (d, J=7.03 Hz, 3H) 1.71-1.79 (m, 1H) 1.68 (ddd, J=13.87, 9.54, 5.67 Hz, 1H) 1.57 (ddd, J=13.87, 8.54, 5.33 Hz, 1H) 0.98 (d, J=6.71 Hz, 3H) 0.96 (d, J=6.56 Hz, 3H). MS (ESI +) m/z 484 [M+H] +.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidines and their use in treating conditions associated with elevated levels of cx3cr1 and/or cx3cl1Publication Number: EP-3818065-B1Priority Date: 2018-07-06Grant Date: 2023-03-15
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidine and their use in treating conditions associated with elevated levels of CX3CR1 and/or CX3CL1Publication Number: KR-102736870-B1Priority Date: 2018-07-06Grant Date: 2024-12-03
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidines and their use in treating conditions associated with elevated levels of cx3cr1 and/or cx3cl1Publication Number: US-2021340167-A1Priority Date: 2018-07-06
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-D]pyrimidine and their use in the treatment of conditions associated with elevated levels of cx3cr1 and/or cx3cl1 – Patent Application 20070123333Publication Number: JP-7506653-B2Priority Date: 2018-07-06Grant Date: 2024-06-26
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidines and their use in treating conditions associated with elevated levels of CX3CR1 and/or CX3CL1Publication Number: US-11339183-B2Priority Date: 2018-07-06Grant Date: 2022-05-24
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo-[4,5-D]-pyrimidines and their uses in the treatment of conditions associated with high levels of CX3CR1 and/or CX3CL1Publication Number: IL-279818-B1Priority Date: 2018-07-06
- PHOSPHATE AND PHOSPHONATE DERIVATIVES OF 7-AMINO-5-THIO-THIAZOLO[4,5-D]PYRIMIDINE AND THEIR USE IN THE TREATMENT OF CONDITIONS ASSOCIATED WITH INCREASED LEVELS OF CX3CR1 AND/OR CX3CL1Publication Number: HR-P20230532-T1Priority Date: 2018-07-06
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidines and their use in treating conditions associated with elevated levels of CX3CR1 and/or CX3CL1Publication Number: US-12060380-B2Priority Date: 2018-07-06Grant Date: 2024-08-13
- 7-Amino-5-thio-thiazolo [4,5-D] Pyrimidine phosphate and phosphonate derivatives and their use in therapeutic conditions associated with elevated levels of CX3CR1 and / or CX3CL1Publication Number: JP-2021530474-APriority Date: 2018-07-06
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo-[4,5-D]-pyrimidines and their uses in the treatment of conditions associated with high levels of CX3CR1 and/or CX3CL1Publication Number: IL-279818-B2Priority Date: 2018-07-06
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo[4,5-d]pyrimidines and their use in treating conditions associated with elevated levels of cx3cr1 and/or cx3cl1Publication Number: US-2021292349-A1Priority Date: 2018-07-06
- Phosphate and phosphonate derivatives of 7-amino-5-thio-thiazolo [4,5-D ] pyrimidine modulators of CX3CR1 receptor and medical uses thereofPublication Number: CN-112867725-BPriority Date: 2018-07-06Grant Date: 2024-09-27
- New usePublication Number: US-2025195548-A1Priority Date: 2023-12-19
- Fractalkine receptor antagonists for use in the prevention of thrombus formation and/or growthPublication Number: EP-4593833-A1Priority Date: 2023-12-19
- Fractalkine receptor antagonists for use in the prevention of thrombus formation and/or growthPublication Number: WO-2025133022-A1Priority Date: 2023-12-19
- New usePublication Number: US-2025195549-A1Priority Date: 2023-12-19
- New treatments of viral infectionsPublication Number: WO-2021224494-A1Priority Date: 2020-05-08
////////fosrugocrixan, anax labs, CX3C chemokine receptor 1 (CX3CR1) antagonist, antiinflammatory, 4ZXD25SC4S, KAND-145, KAND 145
Fosizensertib
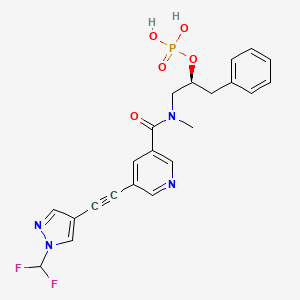
Fosizensertib
CAS 2905377-00-4
MF C22H21F2N4O5P MW490.4 g/mol
[(2S)-1-[[5-[2-[1-(difluoromethyl)pyrazol-4-yl]ethynyl]pyridine-3-carbonyl]-methylamino]-3-phenylpropan-2-yl] dihydrogen phosphate
(2S)-1-(5-{[1-(difluoromethyl)-1H-pyrazol-4-yl]ethynyl}-Nmethylpyridine-3-carboxamido)-3-phenylpropan-2-yl dihydrogen
phosphate
receptor-interacting serine/threonine protein (RIP-1) kinase inhibitor, ABBV-668, ABBV 668, 6GA6XSX5SL
Fosizensertib (also known by the developmental code ABBV-668) is an investigational small molecule drug being evaluated for the treatment of ulcerative colitis and other chronic autoimmune or inflammatory conditions.
Mechanism of Action
- Target: It acts as a selective inhibitor of receptor-interacting serine/threonine-protein kinase 1 (RIPK1), an enzyme that plays a critical role in regulating cellular inflammation and necroptosis (programmed cell death).
- Prodrug Design: Fosizensertib functions as a phosphate prodrug. When administered, it is essentially inactive in vitro (inhibiting RIPK1 by less than 10%).
- Bioactivation: Once inside the body, it undergoes in vivo dephosphorylation to convert into its active metabolite (Compound 2), which strongly inhibits RIPK1 activity to suppress inflammatory pathways.
According to resources like the IUPHAR/BPS Guide to Pharmacology and PubChem, its core chemical metrics include:
Fosizensertib was assigned its International Nonproprietary Name (INN) by the World Health Organization (WHO). Developed by the pharmaceutical company AbbVie, it is classified as a clinical candidate intended for oral administration. It is currently restricted strictly to laboratory research and clinical evaluation settings and is not approved for general prescription or veterinary use.
PAT
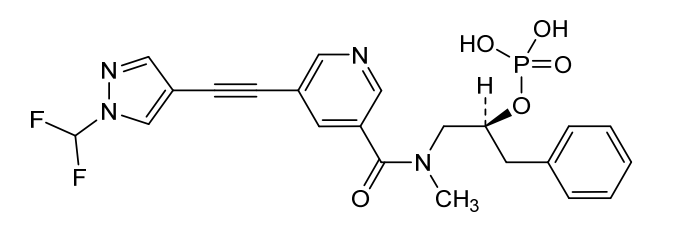
(S)-1-(5-((1-(difluoromethyl)-1H-pyrazol-4-yl)ethynyl)-N-methylnicotinamido)-3-phenylpropan-2-yl dihydrogen phosphate;
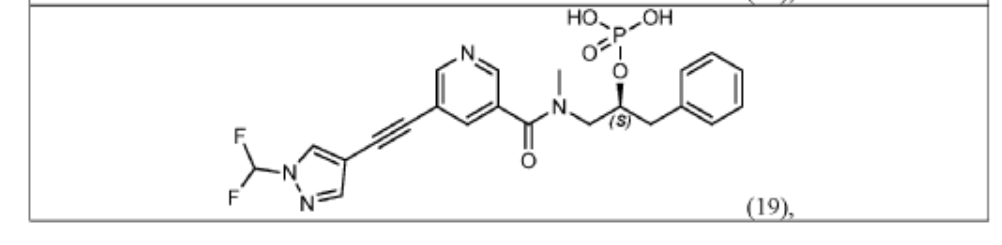
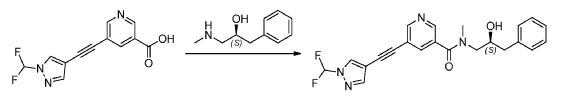
Examples #18 and 19: (S)–di–tert–butyl (1–(5–((1–(difluoromethyl)–1H–pyrazol–4– yl)ethynyl)–N–methylnicotinamido)–3–phenylpropan–2–yl) phosphate (Example #18) and (S)–1–(5–((1–(difluoromethyl)–1H–pyrazol–4–yl)ethynyl)–N–methylnicotinamido)–3– phenylpropan–2–yl dihydrogen phosphate (Example #19)
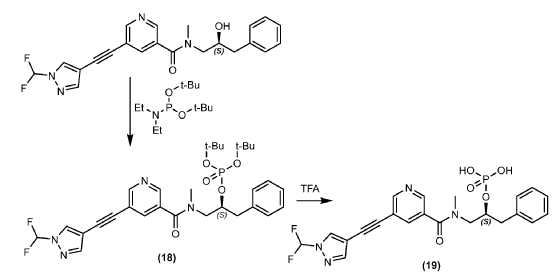
[0173] To a solution of (S)-5-((1-(difluoromethyl)-1H-pyrazol-4-yl)ethynyl)-N-(2-hydroxy- 3-phenylpropyl)-N-methylnicotinamide (Example #2) (500 mg, 1.22 mmol) in N-Methyl-2- pyrrolidinone (1000 mL) was added di-tert-butyl diethylphosphoramidite (304 mg, 1.22 mmol) and 1H-tetrazole (10.8 mL, 4.87 mmol) in one portion at 20 °C under N2. The mixture was stirred at 40 °C for 3 hours. Hydrogen peroxide (5.0 mL, 49 mmol) was added to the solution at 0 °C, and the mixture was stirred for an additional 2 hours. The mixture was poured into saturated Na2SO3 (75 mL) and extracted with ethyl acetate (EtOAc) (3 × 100 mL). The organic phase was washed with brine (100 mL), dried over Na2SO4, concentrated under reduced pressure to give the crude t-butyl phosphate ester, which was chromatographed on silica gel (petroleum ether: ethyl acetate=1:1-1:4) to provide (S)-di-tert-butyl (1-(5-((1-(difluoromethyl)-1H-pyrazol-4- yl)ethynyl)-N-methylnicotinamido)-3-phenylpropan-2-yl) phosphate (Example #18) (384 mg, 0.64 mmol, 52% yield). LC/MS (Table B, Method aa) Rt = 1,73 min; MS m/z: 545.20 (M-tBu)+; 1H NMR (400 MHz, DMSO-d6) δ 8.76 – 8.40 (m, 3H), 8.15 – 7.60 (m, 3H), 7.27-7.01 (m, 5H), 4.78-4.46 (br m, 1H), 3.75-2.72 (m, 7H), 1.50-1.18 (m, 18H). tBu = tert–butyl; Et = ethyl.
[0174] A flask was charged with (S)-di-tert-butyl (1-(5-((1-(difluoromethyl)-1H-pyrazol-4- yl)ethynyl)-N-methylnicotinamido)-3-phenylpropan-2-yl) phosphate (Example #18) (381 mg, .632 mmol), dichloromethane (DCM) (5 mL) and trifluoroacetic acid (TFA) (0.61 mL, 7.9 mmol) and stirred at room temperature for approximately 19 hours. The mixture was concentrated under reduced pressure, then purified via reverse phase liquid chromatography (Atlantis® Prep T3 Phenomenex 5 μm 19 x 50 mm column, 5 to 95 acetonitrile (MeCN):water (formic acid buffer) at 1 mL/minute) to provide the title compound, Example #19 (230 mg, 0.47 mmol, 74% yield). LC/MS (Table B, Method ff) Rt = 1.96 min; MS m/z: 491.0 (M+H)+; 1H NMR (400 MHz,
DMSO-d6) δ 8.78 – 8.69 (m, 1H), 8.65 – 8.57 (m, 1H), 8.44 (d, J = 1.0 Hz, 1H), 8.14 – 8.08 (m, 1H), 8.03 – 7.99 (m, 1H), 7.97 (s, 1H), 7.88 – 7.84 (m, 1H), 7.76 (s, 1H), 7.73 – 7.69 (m, 1H), 7.35 – 7.28 (m, 2H), 7.27 – 7.21 (m, 1H), 7.19 – 7.12 (m, 1H), 7.03 (br d, J = 7.5 Hz, 1H), 4.80 – 4.73 (m, 1H), 4.52 – 4.45 (m, 1H), 3.84 – 3.76 (m, 1H), 3.66 (br d, J = 13.5 Hz, 1H), 3.33 (br dd, J = 9.5, 13.5 Hz, 1H), 3.27 – 3.11 (m, 1H), 3.08 – 3.00 (m, 1H), 2.97 (s, 1H), 2.95 (br s, 1H), 2.92 (s, 2H), 2.90 – 2.85 (m, 1H), 2.79 – 2.69 (m, 1H), 2.07 (s, 1H), 1.78 (s, 1H), 1.74 (s, 1H).
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Nicotinamide ripk1 inhibitorsPublication Number: WO-2023018643-A1Priority Date: 2021-08-10
- Nicotinamide ripk1 inhibitorsPublication Number: EP-4301744-A1Priority Date: 2021-08-10
- Nicotinamide RIPK1 inhibitorsPublication Number: US-11767310-B2Priority Date: 2021-08-10Grant Date: 2023-09-26
- Nicotinamide ripk1 inhibitorsPublication Number: US-2023127127-A1Priority Date: 2021-08-10
/////////fosizensertib, anax labs, ABBV-668, ABBV 668, 6GA6XSX5SL
Bulevirtide-gmod

Bulevirtide-gmod
CAS 2012558-47-1.
MF C248H355N65O72 MW 5399 g/mol
FDA 2026, APPROVALS 2026, 5/22/2026, Hepcludex, WKM56H3TLB
To treat chronic hepatitis delta virus infection in adults without cirrhosis or with compensated cirrhosis
N-myristoyl-glycyl-L-threonyl-L-asparagyl-L-leucyl-L-seryl-L-valyl-L-prolyl-L-asparagyl-L-prolyl-L-leucyl-glycyl-L-phenylalanyl-L-phenylalanyl-L-prolyl-L-alpha-aspartyl-L-histidyl-L-glutaminyl-L-leucyl-L-alpha-aspartyl-L-prolyl-L-alanyl-L-phenylalanyl-glycyl-L-alanyl-L-asparagyl-L-seryl-L-asparagyl-L-asparagyl-L-prolyl-L-alpha-aspartyl-L-tryptophyl-L-alpha-aspartyl-L-phenylalanyl-L-asparagyl-L-prolyl-L-asparagyl-L-lysyl-L-alpha-aspartyl-L-histidyl-L-tryptophyl-L-prolyl-L-alpha-glutamyl-L-alanyl-L-asparagyl-L-lysyl-L-valyl-glycinamide
(4S)-4-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-4-amino-2-[[(2S)-1-[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-4-amino-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-1-[(2S)-4-amino-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S,3R)-3-hydroxy-2-[[2-(tetradecanoylamino)acetyl]amino]butanoyl]amino]-4-oxobutanoyl]amino]-4-methylpentanoyl]amino]-3-hydroxypropanoyl]amino]-3-methylbutanoyl]pyrrolidine-2-carbonyl]amino]-4-oxobutanoyl]pyrrolidine-2-carbonyl]amino]-4-methylpentanoyl]amino]acetyl]amino]-3-phenylpropanoyl]amino]-3-phenylpropanoyl]pyrrolidine-2-carbonyl]amino]-3-carboxypropanoyl]amino]-3-(1H-imidazol-4-yl)propanoyl]amino]-5-oxopentanoyl]amino]-4-methylpentanoyl]amino]-3-carboxypropanoyl]pyrrolidine-2-carbonyl]amino]propanoyl]amino]-3-phenylpropanoyl]amino]acetyl]amino]propanoyl]amino]-4-oxobutanoyl]amino]-3-hydroxypropanoyl]amino]-4-oxobutanoyl]amino]-4-oxobutanoyl]pyrrolidine-2-carbonyl]amino]-3-carboxypropanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-3-carboxypropanoyl]amino]-3-phenylpropanoyl]amino]-4-oxobutanoyl]pyrrolidine-2-carbonyl]amino]-4-oxobutanoyl]amino]hexanoyl]amino]-3-carboxypropanoyl]amino]-3-(1H-imidazol-4-yl)propanoyl]amino]-3-(1H-indol-3-yl)propanoyl]pyrrolidine-2-carbonyl]amino]-5-[[(2S)-1-[[(2S)-4-amino-1-[[(2S)-6-amino-1-[[(2S)-1-[(2-amino-2-oxoethyl)amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxohexan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-1-oxopropan-2-yl]amino]-5-oxopentanoic acid
Bulevirtide-gmod, sold under the brand name Hepcludex, is the first and only FDA-approved medication for treating chronic hepatitis delta virus (HDV) infection in adults. Developed by Gilead Sciences, it received accelerated approval from the U.S. Food and Drug Administration (FDA) on May 22, 2026, filling a critical gap for patients with this severe viral liver disease.
Indication and Clinical Use
- Target Patient Profile: Approved for adults with chronic HDV who have compensated cirrhosis or no cirrhosis.
- The Clinical Need: HDV only occurs as a co-infection in individuals who already have Hepatitis B (HBV). It is considered the most aggressive form of viral hepatitis, often accelerating liver scarring (fibrosis), liver failure, and liver cancer.
- Basis of Approval: The FDA granted accelerated approval based on Phase 3 MYR301 study data, which demonstrated a significant reduction in viral HDV RNA and the normalization of alanine aminotransferase (ALT) liver enzymes.
Mechanism of Action
Bulevirtide-gmod is a first-in-class entry inhibitor. It works by binding to and blocking the sodium taurocholate co-transporting polypeptide (NTCP) receptor on liver cells. Because HDV and HBV rely on this specific receptor to enter hepatocytes, the drug successfully disrupts the viral life cycle and prevents the virus from spreading to healthy liver cells.
Dosage and Administration
- Form: Supplied as a lyophilized powder for injection.
- Dose: The recommended dose is 8.5 mg once daily.
- Administration: Delivered via subcutaneous injection (under the skin).
Safety and Side Effects
- Boxed Warning: The drug carries a prominent warning regarding the risk of severe acute exacerbations of hepatitis D and B if treatment is discontinued. Stopping the medication can cause severe, life-threatening viral flares, requiring close medical monitoring for at least 6 months post-treatment.
- Common Side Effects: The most frequent adverse reactions of patients) include:
- Injection site reactions
- Headache
- Abdominal pain
- Fatigue
- Pruritus (itching)
Bulevirtide, sold under the brand name Hepcludex, is an antiviral medication used for the treatment of chronic hepatitis D (in the presence of hepatitis B).[8]
The most common side effects include raised levels of bile salts in the blood and reactions at the site of injection.[8]
Bulevirtide works by attaching to and blocking a receptor (target) through which the hepatitis delta and hepatitis B viruses enter liver cells.[8] By blocking the entry of the virus into the cells, it limits the ability of HDV to replicate and its effects in the body, reducing symptoms of the disease.[8]
Bulevirtide was approved for medical use in the European Union in July 2020,[8] and in Canada in August 2025.[5]
Medical uses
Bulevirtide is indicated for the treatment of chronic hepatitis delta virus (HDV) infection in plasma (or serum) HDV-RNA positive adult patients with compensated liver disease.[8][10]
Pharmacology
Mechanism of action
Bulevirtide binds and inactivates the sodium/bile acid cotransporter, blocking both hepatitis B and hepatitis D viruses from entering hepatocytes.[11]
The hepatitis B virus uses its surface lipopeptide pre-S1 for docking to mature liver cells via their sodium/bile acid cotransporter (NTCP) and subsequently entering the cells. Myrcludex B is a synthetic N-acylated pre-S1[12][13] that can also dock to NTCP, blocking the virus’s entry mechanism.[14]
Bulevirtide is also effective against hepatitis D because the hepatitis D virus uses the same entry receptor as the hepatitis B virus and is only effective in the presence of a hepatitis B virus infection.[14]
Pre-clinical data in mice suggests that pharmacological inhibition of NTCP-mediated bile salt uptake may also be effective to lower hepatic bile salt accumulation in cholestatic conditions. This reduces hepatocellular damage.[15] An increased ratio of phospholipid to bile salts seen in bile upon NTCP inhibition may further contribute to the protective effect as bile salts are less toxic in presence of phospholipids.[16]
Structural formula
Bulevirtide is a 47-amino acid peptide with the following sequence:[17]
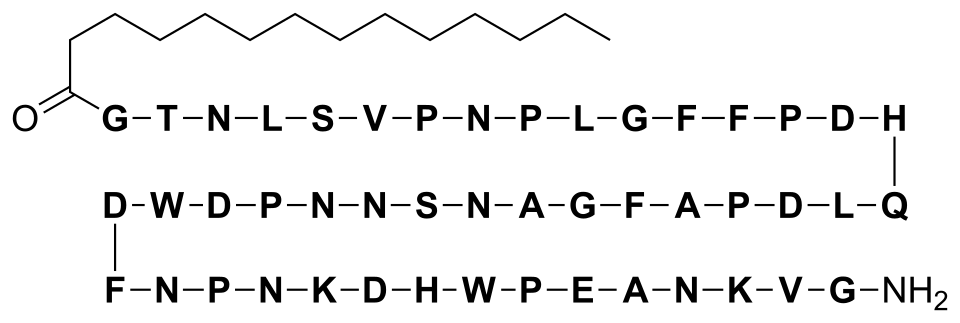
CH3(CH2)12CO–Gly–Thr–Asn–Leu–Ser–Val–Pro-Asn-Pro-Leu-Gly-Phe-Phe-Pro-Asp–His–Gln-Leu-Asp-Pro-Ala-Phe-Gly-Ala-Asn-Ser-Asn-Asn-Pro-Asp-Trp-Asp-Phe-Asn-Pro-Asn-Lys-Asp-His-Trp-Pro-Glu-Ala-Asn-Lys-Val-Gly-NH2 (C13H27CO-GTNLSVPNPLGFFPDHQLDPAFGANSNNPDWDFNPNKDHWPEANKVG-NH2)
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024073572&_cid=P11-MPNG4J-82875-1
PATENTS
- Therapy of atherosclerosis, primary biliary cirrhosis and nrlp3 inflammasome-associated disease by htcp inhibitorsPublication Number: EP-3392267-A1Priority Date: 2017-04-18
- Therapy of atherosclerosis, primary biliary cirrhosis and nrlp3 inflammasome-associated disease by htcp inhibitorsPublication Number: US-2018296634-A1Priority Date: 2017-04-18
- Therapy of atherosclerosis, primary biliary cirrhosis and nrlp3 inflammasome-associated disease by htcp inhibitorsPublication Number: US-2021196786-A1Priority Date: 2017-04-18
- Therapy of atherosclerosis, primary biliary cirrhosis and NRLP3 inflammasome-associated disease by HTCP inhibitorsPublication Number: US-10925925-B2Priority Date: 2017-04-18Grant Date: 2021-02-23
- Combination therapy of hbv and hdv infectionPublication Number: EP-3204030-B1Priority Date: 2014-10-07Grant Date: 2022-04-27
- Combination therapy of hbv and hdv infectionPublication Number: EP-4098273-A1Priority Date: 2014-10-07
- Combination therapy of hbv and hdv infectionPublication Number: US-2022040178-A1Priority Date: 2014-10-07
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- Deterding K, Wedemeyer H (2019). “Beyond Pegylated Interferon-Alpha: New Treatments for Hepatitis Delta”. AIDS Reviews. 21 (3): 126–134. doi:10.24875/AIDSRev.19000080. PMID 31532397. S2CID 202674681.
- “Hepcludex (bulevirtide acetate)”. Therapeutic Goods Administration (TGA). 12 August 2024. Retrieved 12 October 2024.
- “Therapeutic Goods (Poisons Standard—June 2024) Instrument 2024”. Federal Register of Legislation. 30 May 2024. Retrieved 10 June 2024.
- “Hepcludex (Gilead Sciences Pty Ltd)”. Therapeutic Goods Administration (TGA). 13 September 2024. Retrieved 15 September 2024.
- “Hepcludex Product information”. Health Canada. 8 August 2025. Retrieved 20 August 2025.
- “Summary Basis of Decision for Hepcludex”. Drug and Health Products Portal. 29 September 2025. Retrieved 12 October 2025.
- “Hepcludex 2 mg powder for solution for injection – Summary of Product Characteristics (SmPC)”. (emc). 30 March 2022. Retrieved 1 July 2022.
- “Hepcludex EPAR”. European Medicines Agency (EMA). 26 May 2020. Retrieved 12 August 2020. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Hepcludex Product information”. Union Register of medicinal products. Retrieved 3 March 2023.
- “Summary of opinion: Hepcludex” (PDF). European Medicines Agency (EMA). 28 May 2020.
- Francisco EM (29 May 2020). “Hepcludex”. European Medicines Agency (EMA). Archived from the original on 15 June 2020. Retrieved 6 August 2020.
- Volz T, Allweiss L, Ben MBarek M, Warlich M, Lohse AW, Pollok JM, et al. (May 2013). “The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus”. Journal of Hepatology. 58 (5): 861–867. doi:10.1016/j.jhep.2012.12.008. PMID 23246506.
- Abbas Z, Abbas M (August 2015). “Management of hepatitis delta: Need for novel therapeutic options”. World Journal of Gastroenterology. 21 (32): 9461–9465. doi:10.3748/wjg.v21.i32.9461. PMC 4548107. PMID 26327754.
- Spreitzer H (14 September 2015). “Neue Wirkstoffe – Myrcludex B”. Österreichische Apothekerzeitung (in German) (19/2015): 12.
- Na+ -taurocholate cotransporting polypeptide inhibition has hepatoprotective effects in cholestasis in mice. Slijepcevic D, Roscam Abbing RLP, Fuchs CD, Haazen LCM, Beuers U, Trauner M, Oude Elferink RPJ, van de Graaf SFJ. Hepatology. 2018 Sep;68(3):1057-1069. doi: 10.1002/hep.29888
- Roscam Abbing RL, Slijepcevic D, Donkers JM, Havinga R, Duijst S, Paulusma CC, et al. (January 2020). “Blocking Sodium-Taurocholate Cotransporting Polypeptide Stimulates Biliary Cholesterol and Phospholipid Secretion in Mice”. Hepatology. 71 (1): 247–258. doi:10.1002/hep.30792. PMC 7003915. PMID 31136002.
- Sauter M, Blank A, Stoll F, Lutz N, Haefeli WE, Burhenne J (September 2021). “Intact plasma quantification of the large therapeutic lipopeptide bulevirtide”. Analytical and Bioanalytical Chemistry. 413 (22): 5645–5654. doi:10.1007/s00216-021-03384-7. PMC 8410713. PMID 34018034.
| Clinical data | |
|---|---|
| Pronunciation | /bjuːˈlɛvɪrtaɪd/ byoo-LEH-vir-tyde |
| Trade names | Hepcludex |
| Other names | MyrB, Myrcludex-B[1] |
| License data | US DailyMed: Bulevirtide |
| Pregnancy category | AU: B1[2] |
| Routes of administration | Subcutaneous |
| ATC code | J05AX28 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)[3][4][2]CA: ℞-only[5][6]UK: POM (Prescription only)[7]EU: Rx-only[8][9] |
| Identifiers | |
| CAS Number | 2012558-47-1 |
| DrugBank | DB15248 |
| ChemSpider | 129157549 |
| UNII | WKM56H3TLB |
| KEGG | D11877as salt: D11878 |
| ChEMBL | ChEMBL4297711 |
| Chemical and physical data | |
| Formula | C248H355N65O72 |
| Molar mass | 5398.951 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
/////////Bulevirtide-gmod, ANAX LABS, FDA 2026, APPROVALS 2026, Hepcludex, WKM56H3TLB, ANTIVIRALS
Flormotridazum (18F)
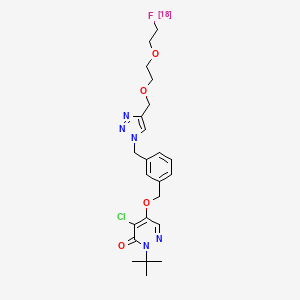
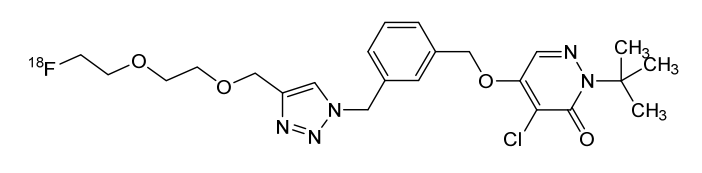
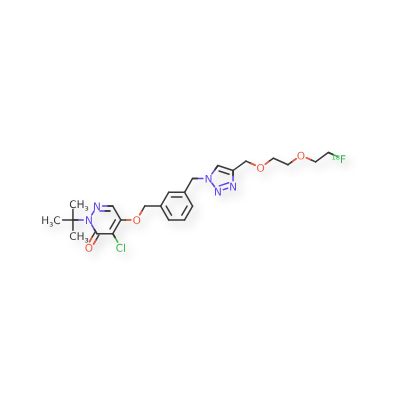
Flormotridazum (18F)
CAS 2798832-03-6
MF C23H29Cl18FN5O4 MW492.961
2-tert-butyl-4-chloro-5-[(3-{[4-({2-[2-(18F)fluoroethoxy]ethoxy}methyl)-1H-1,2,3-triazol-1-yl]methyl}phenyl)methoxy]pyridazin-3(2H)-one
| 3(2H)-Pyridazinone, 4-chloro-2-(1,1-dimethylethyl)-5-[[3-[[4-[[2-[2-(fluoro-18F)ethoxy]ethoxy]methyl]-1H-1,2,3-triazol-1-yl]methyl]phenyl]methoxy]- |
2-tert-butyl-4-chloro-5-[(3-{[4-({2-[2-(18F)fluoroethoxy]ethoxy}methyl)-1H-1,2,3-triazol-1-yl]methyl}phenyl)methoxy]pyridazin-3(2H)-one
imaging agent, 7AR6ZH8YUU
Flormotridaz (18F) (also referred to by its International Nonproprietary Name, flormotridazum) is an advanced radiopharmaceutical compound utilized in nuclear medicine. It is specifically engineered as a radioactive diagnostic tracer containing the fluorine-18 positron-emitting isotope.
Core Characteristics & Chemical Profile
- Substance Classification: Radioactive Diagnostic Agent / Small Molecule.
- Mechanism Basis: It shares core structural similarities and structural lineage with pyridazinone-based mitochondrial complex 1 (MC-1) inhibitors, heavily linking its functionality to target-specific tissues with high metabolic or mitochondrial activity.
Mechanism and Clinical Application
Like related fluorine-18 labeled pyridazinone analogues, this agent is designed for Positron Emission Tomography (PET) imaging workflows. [1]
- Administration: The agent is administered intravenously as a sterile unit dose before scanning.
- Cellular Targeting: It binds selectively to specific intracellular molecular targets (such as mitochondrial pathways) within highly active tissues.
- PET Imaging: As the Fluorine-18 radioisotope decays, it emits positrons. These positrons encounter electrons to produce gamma rays, which the PET scanner captures to map high-resolution, three-dimensional metabolic layouts of internal organ systems.
Contextual Comparison
In clinical nuclear medicine, molecular tracers tagged with Fluorine-18 offer significant clinical benefits over older Single-Photon Emission Computed Tomography (SPECT) agents. Their 110-minute half-life allows them to be manufactured at centralized cyclotron facilities and distributed directly to regional medical centres as ready-to-use unit doses, eliminating the need for an on-site cyclotron
Flormotridaz (\(^{18}\text{F}\)):
- CN112807276B: “Preparation method and application of a pyridazinone myocardial perfusion PET radiopharmaceutical” (Covers the definitive radiosynthesis scheme).
- CN115947775A: “Method for preparing compound (I), compound (I), and uses thereof”.
- WO2024008073A1 / CN114832118B: “Compound I liquid composition, preparation method and use thereof” (Covers final formulation stabilization utilizing vitamin C and gentisic acid)
PAT
https://patents.google.com/patent/WO2024008073A1/zh
Compound I, chemically named 2-tert-butyl-4-chloro-5-((3-((4-((2-(2-fluoro[ 18F ]ethoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)benzyl)oxy)pyridazine-3(2H)-one. Chemical structural formula:Molecular formula : C₂₃H₂₉Cl₁₈FN₅O₄
Molecular weight: 492.97The mechanism of action of compound I as a myocardial perfusion PET imaging agent: Once compound I enters cardiomyocytes, it can rapidly interact with respiratory chain complex I (MC-I) in mitochondria and remain in the myocardium for a long time. Preliminary animal studies showed that it has high cardiac uptake and low hepatic uptake 15 minutes after injection, and maintains a good heart-liver ratio 60 minutes after injection, showing good potential for myocardial perfusion imaging.In this application, Compound I liquid composition or Compound I is used as a myocardial perfusion PET imaging agent.Precursor of Compound I: Chemical name is methyl 2-(2-((1-(3-(((1-(tert-butyl)-5-chloro-6-oxo-1,6-dihydropyridazin-4-yl)oxy)methyl)benzyl)-1H-1,2,3-triazol-4-yl)methoxy)ethoxy)ethyl-4-methylbenzenesulfonate, chemical structural formula is:Molecular formula : C30H36ClN5O7S
Molecular weight: 646.16Amino polyethers (K222 ) are tribridged crown ether molecules with cavitary structures, and are typical nitrogen-containing cavitary ethers, belonging to the category of cavitary ethers. Due to their unique coordination properties, nitrogen-containing cavitary ethers can effectively and selectively complex transition metal and heavy metal cations, forming more stable complexes. Furthermore, they possess both lipophilic and hydrophilic properties, thus showing promising research potential.In existing technologies, the classic synthetic method for amino polyether (K
222 ) is the highly diluted method proposed by Lehn et al., which is a typical non-template ion synthesis method. The specific steps involve dissolving the starting materials 1,8-diamino-3,6-dioxane and 1,8-diacyl chloride-3,6-dioxane in a large amount of benzene solvent and heating the reaction for 8 hours. Then, a reduction reaction with lithium aluminum hydride is performed for 24 hours, followed by column chromatography separation and recrystallization to obtain amino polyether (K
222 ). This method requires a large amount of solvent, such as benzene, has a long synthetic route, is complex, has a low yield, and is not economically efficient. Besides the highly diluted method, another classic synthetic method for amino polyether (K222 ) is proposed by Kulstad and Malmsten, which uses Na 2CO 3
as a template to obtain a sodium iodide complex of amino polyether (K 222 ) in acetonitrile , and then decomplexes it using a resin to obtain amino polyether (K 222 ). The specific steps are as follows: 1,2-bis(2-iodoethoxy)ethane and benzylamine are refluxed in acetonitrile solution for 3 days. An intermediate is then obtained through post-processing. This intermediate is recrystallized from acetone and filtered to obtain a NaI complex. This complex is then decomplexed under acidic conditions using cation exchange resins and anion exchange resins to prepare amino polyether (K222 ) . This method uses simple equipment, requires little solvent, and has relatively mild reaction conditions. However, the applicant has found that the decomplexing method using ion exchange resins fails to proceed when the sodium ion content decreases to a certain level, resulting in a low yield.
PAT
https://patents.google.com/patent/CN114773179B/en
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
//////////flormotridazum (18F), anax labs, imaging agent, 7AR6ZH8YUU
Florensocatib
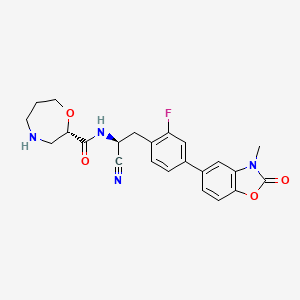
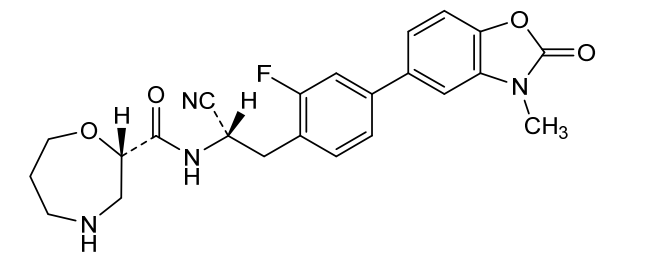
Florensocatib
CAS 2762114-61-2
MF C23H23FN4O4 MW438.5 g/mol
(2S)-N-[(1S)-1-cyano-2-[2-fluoro-4-(3-methyl-2-oxo-1,3-benzoxazol-5-yl)phenyl]ethyl]-1,4-oxazepane-2-carboxamide
(2S)-N-{(1S)-1-cyano-2-[2-fluoro-4-(3-methyl-2-oxo-2,3-dihydro1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-
carboxamide
cathepsin inhibitor, HSK 31858, CHF 10196, DPP1-IN-1, RWC743JRK7
Florensocatib (originally designated as HSK31858 or CHF10196) is an investigative, highly potent, oral reversible inhibitor of dipeptidyl peptidase 1 (DPP1). It is being actively researched for its ability to reduce the frequency of pulmonary exacerbations in adults suffering from inflammatory respiratory diseases like bronchiectasis
Mechanism of Action
DPP1 (also known as cathepsin C) is a lysosomal protease enzyme responsible for activating neutrophil serine proteases (NSPs). In conditions like non-cystic fibrosis bronchiectasis, hyperactive neutrophils accumulate in the airways, causing severe tissue damage, chronic inflammation, and airway widening.
By inhibiting DPP1, florensocatib prevents the activation of these damaging enzymes, effectively targeting the primary driver of neutrophilic inflammation in the lungs.
Clinical Development & Trial Progress
Florensocatib is undergoing global evaluation across multiple advanced clinical trials:
- The SAVE-BE Trial: An earlier clinical phase where the drug demonstrated high potency and favorable efficacy profiles in treating inflammatory lung conditions.
- The HOPE-BE Trial: A definitive Phase III protocol launched to evaluate the long-term safety and overall reduction of pulmonary exacerbation frequencies specifically among Chinese adults.
- Global Phase III Status: According to records on ClinicalTrials.gov, randomised, double-blind trials are evaluating the drug against a placebo in participants aged 12 to 85 for treatment windows stretching up to 78 weeks.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US395653715&_cid=P21-MPKKTA-33002-1
Example 1: (S)—N—((S)-1-cyano-2-(2-fluoro-4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide (Compound 1)
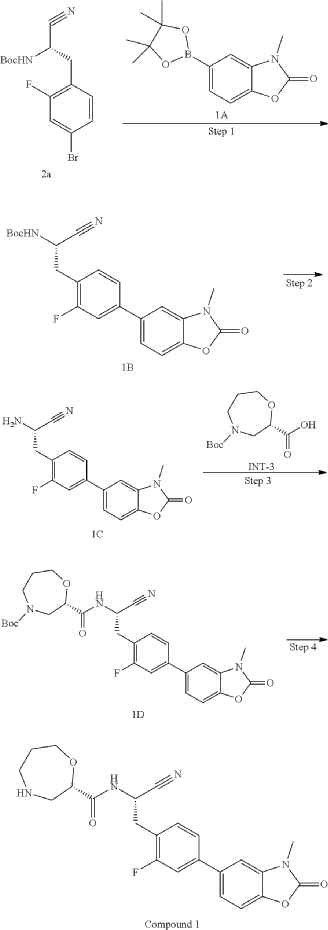
Step 4: (S)—N—((S)-1-cyano-2-(2-fluoro-4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide (Compound 1)
| 1H NMR (400 MHz, CDCl 3) δ 7.43-7.22 (m, 5H), 7.12 (d, 1H), 5.19 (dd, 1H), 4.18-4.04 (m, 1H), 4.05-3.95 (m, 1H), 3.78 (m, 1H), 3.46 (s, 3H), 3.41-3.17 (m, 3H), 3.03-2.87 (m, 3H), 1.88 (m, 2H). |
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US395653715&_cid=P21-MPKKYT-35548-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022042591&_cid=P21-MPKKS9-32509-1
(S)-N-((S)-1-cyano-2-(2-fluoro-4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide(compound 1)
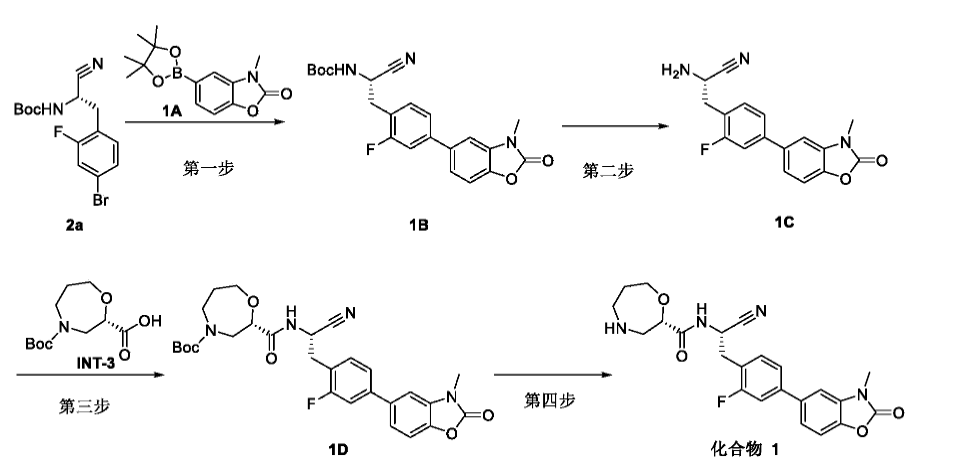
Step 4: (S)-N-((S)-1-cyano-2-(2-fluoro-4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazacycloheptane-2-carboxamide (Compound 1)
[0360]
(S)-N-((S)-1-cyano-2-(2-fluoro-4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide(compound 1)
[0361]1D (0.32 g, 0.59 mmol) was dissolved in formic acid (2.5 mL), and the mixture was reacted at 50 °C for 10 min after the addition was complete. The solution was concentrated to dryness, and ethyl acetate (20 mL) was added. The pH was adjusted to approximately 8 by dropwise addition of saturated sodium bicarbonate solution. The organic layer was separated and extracted with ethyl acetate (25 mL × 3). The organic phases were combined, dried over anhydrous sodium sulfate, filtered, concentrated, and the residue was purified by silica gel column chromatography (dichloromethane:methanol (v/v) = 20:1) to give title compound 1 (0.15 g, 58.0%). LC-MS (ESI): m/z = 439.1 [M+H] + .
[0362]
1H NMR(400MHz,CDCl 3)δ7.43–7.22(m,5H),7.12(d,1H),5.19(dd,1H),4.18–4.04(m,1H),4.05–3.95(m,1H),3.78(m,1H),3.46(s,3H),3.41–3.17(m,3H),3.03–2.87(m,3H),1.88(m,2H).
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Nitrile derivative that acts as inhibitor of dipeptidyl peptidase 1 and use thereofPublication Number: US-2023121807-A1Priority Date: 2020-08-26
- Nitrile derivative that acts as inhibitor of dipeptidyl peptidase 1 and use thereofPublication Number: WO-2022042591-A1Priority Date: 2020-08-26
- Nitrile derivative as dipeptidyl peptidase 1 inhibitor and application thereofPublication Number: CN-115996923-APriority Date: 2020-08-26
- Nitrile derivative that acts as inhibitor of dipeptidyl peptidase 1 and use thereofPublication Number: EP-4129989-A1Priority Date: 2020-08-26
- Dipeptidyl peptidase 1 inhibitor polymorph and preparation method and application thereofPublication Number: CN-118696040-APriority Date: 2022-02-22
- Salt and crystal form of dipeptidyl peptidase inhibitor compoundPublication Number: EP-4484414-A1Priority Date: 2022-02-22
- Nitrile derivative that acts as inhibitor of dipeptidyl peptidase 1 and use thereofPublication Number: US-11807635-B2Priority Date: 2020-08-26Grant Date: 2023-11-07
- A kind of nitrile derivative as dipeptidyl peptidase 1 inhibitor and use thereofPublication Number: TW-202214616-APriority Date: 2020-08-26
- Nitrile derivatives acting as inhibitors of dipeptidyl peptidase 1 and uses thereofPublication Number: KR-20230084134-APriority Date: 2020-08-26
- Salt and crystal form of dipeptidyl peptidase inhibitor compoundPublication Number: WO-2023160542-A1Priority Date: 2022-02-22
- Dipeptidyl peptidase 1 inhibitor polymorph, preparation method and use thereforPublication Number: WO-2023160579-A1Priority Date: 2022-02-22
- Salts and crystalline forms of dipeptidyl peptidase inhibitor compoundsPublication Number: CN-118660873-APriority Date: 2022-02-22
- Preparation method of nitrogen-containing heterocyclic compoundPublication Number: WO-2023160541-A1Priority Date: 2022-02-22
- Preparation method of nitrogen-containing heterocyclic compoundPublication Number: CN-118742547-APriority Date: 2022-02-22
- Dipeptidyl peptidase 1 inhibitors and uses thereofPublication Number: WO-2025059526-A1Priority Date: 2023-09-15
- Pharmaceutical composition containing dipeptidyl peptidase small molecule inhibitorPublication Number: WO-2024193695-A1Priority Date: 2023-03-23
- Uses of certain 1,4-oxazepane-2-carboxamides as dpp1 inhibitorsPublication Number: WO-2024173556-A2Priority Date: 2023-02-15
- Novel dpp1 inhibitors and uses thereofPublication Number: WO-2024026433-A2Priority Date: 2022-07-28
- Novel dpp1 inhibitors and uses thereofPublication Number: EP-4561565-A2Priority Date: 2022-07-28
/////////florensocatib, anax labs, cathepsin inhibitor, HSK 31858, CHF 10196, DPP1-IN-1, RWC743JRK7