
Home » Uncategorized (Page 39)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Alatrofloxacin Mesylate
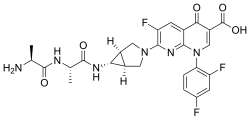
Alatrofloxacin Mesylate
| Chemical Names: | Alatrofloxacin mesylate; UNII-2IXX802851; 146961-77-5; Alatrofloxacin mesylate [USAN]; 157605-25-9; 2IXX802851 |
|---|---|
| Molecular Formula: | C27H29F3N6O8S |
| Molecular Weight: | 654.618 g/mol |
| CAS No. | 146961-76-4 (Alatrofloxacin ); 157605-25-9 (Alatrofloxacin Mesylate); |
| Chemical Name | (1α, 5α, 6α)-L-alanyl-N-[3-[6-carboxy-8-(2,4-difluorophenyl)-3-fluoro-5,8-dihydro-5-oxo-1,8-naphthyridine-2-yl]-3-azabicyclo[3.1.0]hex-6-yl]-L-alaninamide, monomethanesulfonate |
Research Code:CP-116517-27; CP-116517, Trade Name:Trovan I.V.® MOA:Quinolone antibiotic Indication:Life- or limb-threatening infections caused by susceptible strains Status:Withdrawn Company:Pfizer (Originator)
Alatrofloxacin (Trovan IV) is a fluoroquinolone antibiotic developed by Pfizer, delivered as a mesylate salt.[1]
Trovafloxacin and alatrofloxacin were both withdrawn from the U.S. market in 2001
Alatrofloxacin mesylate was first approved by the U.S. Food and Drug Administration (FDA) on Dec 18, 1997. It was developed and marketed as Trovan I.V. ® by Pfizer in the US.
Alatrofloxacin mesylate is a fluoronaphthyridone related to the fluoroquinolones with in vitro activity against a wide range of gram-negative and gram-positive aerobic and anaerobic microorganisms. The bactericidal action of alatrofloxacin results from inhibition of DNA gyrase and topoisomerase IV. Trovan I.V.® is indicated for the treatment of patients initiating therapy in in-patient health care facilities (i.e., hospitals and long term nursing care facilities) with serious, life- or limb-threatening infections caused by susceptible strains of the designated microorganisms in the conditions listed below.
Trovan I.V.® is available as injection solution for intravenous use, containing 7.86 mg/ml of Alatrofloxacin mesylate. The recommended starting dose is 200 mg or 300 mg administered intravenously.
Alatrofloxacin mesylate was withdrawn from the U.S. market in 2001.


Alatrofloxacin mesilate
-
- Synonyms:CP 116517, CP 116517-27
- ATC:J01MA
- Use:antibiotic, prodrug of trovafloxacin
- Chemical name:l-Alanyl-N-[(1α,5α,6α)-3-[6-carboxy-8-(2,4-difluorophenyl)-3-fluoro-5,8-dihydro-5-oxo-1,8-naphthyridin-2-yl]-3-azabicyclo[3.1.0]hex-6-yl]-l-alaninamide monomethanesulfonate
- Formula:C26H25F3N6O5 • CH4O3S
- MW:654.62 g/mol
- CAS-RN:146961-77-5
Derivatives
base
- Formula:C26H25F3N6O5
- MW:558.52 g/mol
- CAS-RN:146961-76-4
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 27317-69-7 | C11H20N2O5 | N–tert-butoxycarbonyl-l-alanyl-l-alanine | L-Alanine, N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl- |
| 186772-86-1 | C33H37F3N6O7 | N-[(1,1-dimethylethoxy)carbonyl]-l-alanyl-N-[(1α,5α,6α)-3-[8-(2,4-difluorophenyl)-6-(ethoxycarbonyl)-3-fluoro-5,8-dihydro-5-oxo-1,8-naphthyridin-2-yl]-3-azabicyclo[3.1.0]hex-6-yl]-l-alaninamide | L-Alaninamide, N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl-N-[(1α,5α,6α)-3-[8-(2,4-difluorophenyl)-6-(ethoxycarbonyl)-3-fluoro-5,8-dihydro-5-oxo-1,8-naphthyridin-2-yl]-3-azabicyclo[3.1.0]hex-6-yl]- |
| 171176-56-0 | C22H19F3N4O3 | ethyl (1α,5α,6α)-7-(6-amino-3-azabicyclo[3.1.0]hex-3-yl)-1-(2,4-difluorophenyl)-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylate | 1,8-Naphthyridine-3-carboxylic acid, 7-(6-amino-3-azabicyclo[3.1.0]hex-3-yl)-1-(2,4-difluorophenyl)-6-fluoro-1,4-dihydro-4-oxo-, ethyl ester, (1α,5α,6α)- |
| 134575-66-9 | C27H27F3N4O5 | ethyl (1α,5α,6α)-1-(2,4-difluorophenyl)-7-[6-[[(1,1-dimethylethoxy)carbonyl]amino]-3-azabicyclo[3.1.0]hex-3-yl]-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylate | 1,8-Naphthyridine-3-carboxylic acid, 1-(2,4-difluorophenyl)-7-[6-[[(1,1-dimethylethoxy)carbonyl]amino]-3-azabicyclo[3.1.0]hex-3-yl]-6-fluoro-1,4-dihydro-4-oxo-, ethyl ester, (1α,5α,6α)- |
| 75-75-2 | CH4O3S | methanesulfonic acid | Methanesulfonic acid |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | TROVAN | Pfizer | wfm |
| F | Turvel | Pfizer | wfm |
| GB | Turvel | Pfizer | wfm |
| I | Turvel | Pfizer | wfm |
| USA | Trovan | Pfizer | wfm |
(wfm = withdrawn from market)
Formulations
- vial 200 mg/40 ml, 300 mg/60 ml (5 mg/ml) (as mesilate)
References
-
- US 5 164 402 (Pfizer; 17.11.1992; appl. 4.2.1991; WO-prior. 16.8.1989).
- US 5 229 396 (Pfizer; 20.7.1993; appl. 24.7.1992).
- WO 9 700 268 (Pfizer; appl. 27.3.1996; USA-prior. 15.6.1995).
- US 5 763 454 (Pfizer; 9.6.1998; appl. 21.5.1997; WO-prior. 6.6.1995).
-
polymorphs:
- US 6 080 756 (Pfizer; 27.6.2000; appl. 30.1.1998; WO-prior. 5.7.1996).
References
“Center for Drug Evaluation and Research – Application Number: 020759/020760 – Chemistry Review(s)” (PDF). Food and Drug Administration. Retrieved 29 August 2014.
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a605016 |
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | N/A |
| Protein binding | 76% (trovafloxacin) |
| Metabolism | Quickly hydrolyzed to trovafloxacin |
| Elimination half-life | 9 to 12 hours (trovafloxacin) |
| Excretion | Fecal and renal(trovafloxacin) |
| Identifiers | |
| CAS Number | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C26H25F3N6O5 |
| Molar mass | 558.509 g/mol |
| 3D model (JSmol) | |
/////////////////
Chenodeoxycholic acid, ケノデオキシコール酸

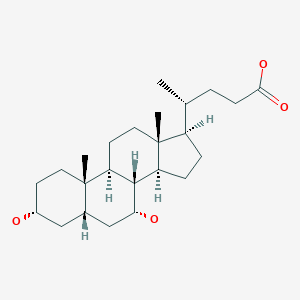
Chenodeoxycholic acid
Chenodiol
- Molecular FormulaC24H40O4
- Average mass392.572
|
Chenodeoxycholate;
Chenodeoxycholic acid; 3alpha,7alpha-Dihydroxy-5beta-cholanic acid; Chenodiol |
Synthesis ReferenceHenry Francis Frost, Fritz Fabian, Christopher James Sharpe, William Arthur Jones, “Process for preparing chenodeoxycholic acid.” U.S. Patent US4022806, issued October, 1974. US4022806
First ref
- By Windaus, A.; Bohne, A.; Schwarzkopf, E.
- From Z. physiol. Chem. (1924), 140, 177-85
- By Wieland, Heinrich; Reverey, Gustav
- From Z. physiol. Chem. (1924), 140, 186-202.

Chenodeoxycholic acid (also known as chenodesoxycholic acid, chenocholic acid and 3α,7α-dihydroxy-5β-cholan-24-oic acid) is a bile acid. It occurs as a white crystalline substance insoluble in water but soluble in alcohol and acetic acid, with melting point at 165–167 °C. Salts of this carboxylic acid are called chenodeoxycholates. Chenodeoxycholic acid is one of the main bile acids produced by the liver.[1]
It was first isolated from the bile of the domestic goose, which gives it the “cheno” portion of its name (Greek: χήν = goose).[2]
Chenodeoxycholic acid and cholic acid are the two primary bile acids in humans. Some other mammals have muricholic acid or deoxycholic acid rather than chenodeoxycholic acid.[1]
Chenodeoxycholic acid is synthesized in the liver from cholesterol by a process which involves several enzymatic steps.[1] Like other bile acids, it can be conjugated in the liver with taurine or glycine, forming taurochenodeoxycholate or glycochenodeoxycholate. Conjugation results in a lower pKa. This means the conjugated bile acids are ionized at the usual pH in the intestine and will stay in the gastrointestinal tract until reaching the ileum where most will be reabsorbed. Bile acids form micelles which facilitate lipid digestion. After absorption, they are taken up by the liver and resecreted, so undergoing an enterohepatic circulation. Unabsorbed chenodeoxycholic acid can be metabolised by bacteria in the colon to form the secondary bile acid known as lithocholic acid.
Chenodeoxycholic acid is the most potent natural bile acid at stimulating the nuclear bile acid receptor, farnesoid X receptor (FXR).[3]The transcription of many genes is activated by FXR.
Indication
Chenodiol is indicated for patients with radiolucent stones in well-opacifying gallbladders, in whom selective surgery would be undertaken except for the presence of increased surgical risk due to systemic disease or age. Chenodiol will not dissolve calcified (radiopaque) or radiolucent bile pigment stones.
Associated Conditions
Pharmacodynamics
It acts by reducing levels of cholesterol in the bile, helping gallstones that are made predominantly of cholesterol to dissolve. Chenodeoxycholic acid is ineffective with stones of a high calcium or bile acid content.
Mechanism of action
Chenodiol suppresses hepatic synthesis of both cholesterol and cholic acid, gradually replacing the latter and its metabolite, deoxycholic acid in an expanded bile acid pool. These actions contribute to biliary cholesterol desaturation and gradual dissolution of radiolucent cholesterol gallstones in the presence of a gall-bladder visualized by oral cholecystography. Bile acids may also bind the the bile acid receptor (FXR) which regulates the synthesis and transport of bile acids.
EMA
On 16 December 2014, orphan designation (EU/3/14/1406) was granted by the European Commission to Sigma-Tau Pharma Ltd, United Kingdom, for chenodeoxycholic acid for the treatment of inborn errors in primary bile acid synthesis.
The sponsorship was transferred to sigma-tau Arzneimittel GmbH, Germany, in May 2015.
Chenodeoxycholic acid has been authorised in the EU as Chenodeoxycholic acid sigma-tau since 10 April 2017.
The name of the product changed to Chenodeoxycholic acid Leadiant in May 2017.
The sponsorship was transferred to Leadiant GmbH, Germany, in June 2017.
On 16 February 2017, the Committee for Orphan Medicinal Products (COMP) concluded its review of the designation EU/3/14/1406 for Chenodeoxycholic acid sigma-tau (chenodeoxycholic acid) as an orphan medicinal product for the treatment of inborn errors in primary bile acid synthesis. The COMP assessed whether, at the time of marketing authorisation, the medicinal product still met the criteria for orphan designation. The Committee looked at the seriousness and prevalence of the condition, and the existence of other methods of treatment. As other methods of treatment are authorised in the European Union (EU), the COMP also considered whether the medicine is of significant benefit to patients with inborn errors in primary bile acid synthesis. The COMP recommended that the orphan designation of the medicine be maintained1.
1 The maintenance of the orphan designation at time of marketing authorisation would, except in specific situations, give an orphan medicinal product 10 years of market exclusivity in the EU. This means that in the 10 years after its authorisation similar products with the same therapeutic indication cannot be placed on the market.
http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2015/02/WC500183233.pdf
Therapeutic applications
Chenodeoxycholic acid has been used as medical therapy to dissolve gallstones.[4]
Chenodeoxycholic acid can be used in the treatment of cerebrotendineous xanthomatosis.[5]
The Australian biotechnology company Giaconda has tested a treatment for Hepatitis C infection that combines chenodeoxycholic acid with bezafibrate.[6]
As diarrhea is a complication of chenodeoxycholic acid therapy, it has also been used to treat constipation.[7][8]
In supramolecular chemistry, molecular tweezers based on a chenodeoxycholic acid scaffold is a urea receptor that can contain anionsin its binding pocket in order of affinity: H2PO4− (dihydrogen phosphate) > Cl− > Br− > I− reflecting their basicities (tetrabutylammonium counter ion).[9]

- PAPER
- 1H and 13C NMR characterization and stereochemical assignments of bile acids in aqueous media
Lipids (2005), 40, (10), 1031-1041. - https://onlinelibrary.wiley.com/doi/abs/10.1007/s11745-005-1466-1

PAPER
Improved Chemical Synthesis, X-Ray Crystallographic Analysis, and NMR Characterization of (22R)-/(22S)-Hydroxy Epimers of Bile Acids
Lipids (2014), 49, (11), 1169-1180.
Improved Chemical Synthesis, X‐Ray Crystallographic Analysis, and NMR Characterization of (22R)‐/(22S)‐Hydroxy Epimers of Bile Acids
A Practical and Eco-friendly Synthesis of Oxo-bile Acids
By Han, Young Taek and Yun, HwayoungFrom Organic Preparations and Procedures International, 48(1), 55-61; 2016
DOI:10.1080/00304948.2016.1127101
General Procedure
An aqueous solution of 0.2 M NaBrO3 (1.5 equiv. per hydroxy group) was added dropwise to a slurry of bile acid (1 equiv.) and ceric ammonium nitrate (0.05 equiv.) in 20% aqueous acetonitrile (0.2 M) at 80°C over 20 min. The bile acid slowly dissolved in a few minutes, and then the color of the reaction mixture changed to orange. The reaction mixture was stirred at the same temperature and the progress of the reaction was monitored by TLC on silica gel (1:20 MeOH-CH2Cl2) until disappearance of the starting material and partially oxidized intermediates. It was then cooled in an ice bath and quenched with aqueous Na2S2O3 solution. Water was added slowly to the resulting white suspension until no more oxo-bile acid precipitated. The white solid was collected, washed with water until the filtrate was colorless, and then dried in vacuo at 50°C. Methyl 3,7α-Diacetoxy-12-oxo-5β-cholanoate(3),21 was obtained in 92% yield (275 mg) as a white solid from 300 mg (0.590 mmol) of 2 via the general procedure. mp. 176-178°C, lit.22 mp. 178-179°C, IR (thin film, neat): 2947 (m), 2873 (s), 1736 (w), 1706 (w), 1436 (s), 1365 (m) cm-1; 1H-NMR (400 MHz, CDCl3): δ 4.96 (m, 1H, 7-CH), 4.55 (m, 1H, 3-CH), 3.64 (s, 3H), 2.49 (t, 1H, J = 12.6 Hz), 2.41-0.80 (m, 23H), 2.01 (s, 3H), 2.00 (s, 3H), 1.01 (s, 3H, 18-CH3), 1.00 (s, 3H, 19-CH3), 0.83 (d, 3H, J = 6.6 Hz, 21-CH3); 13C-NMR (CDCl3, 100 MHz): δ 214.0 (12-C), 174.6 (24-C), 170.7 (C = O), 170.2 (C = O), 73.5 (3-C), 70.5 (7-C), 57.1 (13-C), 53.1 (14-C), 51.5 (CH3O), 46.3 (17-C), 40.5 (5-C), 37.9 (11-C), 37.8 (4-C), 37.6 (8-C), 35.54 (9-C), 35.52 (20-C), 34.9 (1-C), 34.5 (10-C), 31.3 (6-C), 31.2 (22-C), 30.4 (23-C), 27.4 (16-C), 26.5 (2-C), 23.8 (15-C), 22.1 (19-C), 21.51 (CH3CO2), 21.46 (CH3CO2), 18.6 (21-C), 11.5 (18-C); LR-MS (FABC) m/z 505 (M+H +). HR-MS (FABC): Calcd for C29H45O7 (M+H +): 505.3165. Found 505.3161.
next step
R:KOH, R:N2H4

NOTE STARTING IS BILE ACID AS BELOW
Cholan-24-oic acid, 3,7-bis(acetyloxy)-12-oxo-, methyl ester, (3α,5β,7α)-
- 5β-Cholan-24-oic acid, 3α,7α-dihydroxy-12-oxo-, methyl ester, diacetate (8CI)
- 5β-Cholanic acid, 3α,7α-dihydroxy-12-oxo-, methyl ester, diacetate (6CI,7CI)
- 3α,7α-Diacetoxy-12-oxo-5β-cholan-24-oic acid methyl ester
- Methyl 3α,7α-diacetoxy-12-oxo-5β-cholan-24-oate
- Methyl 3α,7α-diacetoxy-12-oxo-5β-cholanate

-
- The structures of the principal human bile acids







PAPER
https://pubs.acs.org/doi/pdf/10.1021/jo01091a623
Journal of Organic Chemistry
Volume24
Pages1367-8
Journal
1959
DOI:10.1021/jo01091a623
Chenodeoxycholic acid (V). Five hundred mg. of the above ester IV was hydrolyzed with 80 ml. of ethanolic 5% potassium hydroxide for 4 hr. After partial concentration of the volume and addition of water, the reaction product was acidified with hydrochloric acid. The resulting precipitate was collected, dried, and crystallized from ethyl acetate. A quantitative crop (400 mg.) of prisms melting at 143- 145° were obtained. Recrystallization from the same solvent yielded a product of m.p. 145-146°, [ ]2 +10.7° (dioxane). Anal. Caled, for C24H40O4: C, 73.43; H, 10.27. Found: C, 73.49; H, 10.31.

NOTE I IS BILE ACID
Cholan-24-oic acid, 3,7-bis(acetyloxy)-12-oxo-, methyl ester, (3α,5β,7α)-
- 5β-Cholan-24-oic acid, 3α,7α-dihydroxy-12-oxo-, methyl ester, diacetate (8CI)
- 5β-Cholanic acid, 3α,7α-dihydroxy-12-oxo-, methyl ester, diacetate (6CI,7CI)
- 3α,7α-Diacetoxy-12-oxo-5β-cholan-24-oic acid methyl ester
- Methyl 3α,7α-diacetoxy-12-oxo-5β-cholan-24-oate
- Methyl 3α,7α-diacetoxy-12-oxo-5β-cholanate
-
- The structures of the principal human bile acids
PATENT
https://patents.google.com/patent/CN102060902A/en
chenodeoxycholic acid (3 α, 7 α – dihydroxy _5 β – cholestane-24-oic acid) Chenodeoxycholic Ac id (referred to as CDCA), clinically used to correct dissolving cholesterol calculi and bile saturation drugs, the main function is to reduce the cholesterol in the bile saturation, large doses can inhibit the synthesis of cholesterol CDCA and increasing bile gallstone patients cholesterol level in a non-saturated, thereby preventing the formation of cholesterol gallstones of cholesterol and promote stone dissolve and fall off. It also has significant anti-asthmatic, anti-inflammatory, antitussive and expectorant effects.
[0003] Synthesis of chenodeoxycholic acid or ursodeoxycholic acid (3 α, 7β_ -5β_ dihydroxy-cholestane-24-oic acid, ursodeoxycholic Acid, referred UDCA), a key intermediate. Ursodeoxycholic acid is the main active ingredient of precious Chinese medicine bear bile, used in a variety of clinical hepatobiliary disease and dyspepsia. Currently we bear bile resources are scarce, mainly used synthetic chemical ursodeoxycholic acid as a clinical treatment. Therefore, the preparation of chenodeoxycholic acid is also important for the preparation of ursodeoxycholic acid.
[0004] CDCA mainly come from poultry or livestock bile extraction. Traditional extraction process complicated operation, low yield, (pharmaceutical industry, 1987,18 (9), 416; Chinese Journal of Biochemical Pharmaceutics, 1996,17 (1), 17; Applied Technology, 1998, (4), 9; CN1850846A ) can not meet the needs of modern industry. Chemical synthesis of chenodeoxycholic acid have also been reported (Japanese Journal of Chemistry 1955,76 (3), 297 -J Org Chem 1982,47 (2): 2331; Journal of Biochemical Pharmaceutics 1987,1,6 -, Tap Chi Duoc ^ oc2004 , 44 (1), 11; CN1869043A), but lower yield widespread pollution major problem, especially in the oxidation reaction is often used to expensive, and polluting agents.Therefore, to reduce pollution, reduce environmental hazards, streamline operations, improve yield, reduce costs, important for the synthesis of chenodeoxycholic acid.
n particular by the following steps:
(1) Preparation of cholate: bile acid in alcohol, concentrated hydrochloric acid as catalyst, at reflux, cooling and crystallization, filtration, and washed with methanol.
[0008] (2) Preparation of 3α, 7α- diacetyl hydroxy -12α- cholate: bile acid ester was dissolved in dichloromethane and triethylamine was added with stirring acetic anhydride and the catalyst N, N- dimethyl pyridine, methylene chloride was distilled off, poured into water, filtered to give 3α, 7α- diacetyl -12 α – hydroxy cholate.
[0009] (3) 3α, 7α- diacetyl -12– Preparation oxo chenodeoxycholic acid ester: Take 3 α, 7 α – diacetyl -12 α – hydroxy cholate dissolved in ethyl acetate and methanol, bromide and tetrabutylammonium bromide as catalyst, and acetic acid was added dropwise under stirring hypochlorite, the organic solvent was distilled off and filtered, to give 12-oxo-3,7-diacetyl Chenodeoxy cholate.
[0010] (4) i2 – Preparation oxo chenodeoxycholic acid: 3,7-diacetyl-12-oxo-chenodeoxycholic acid ester added ethanol – sodium hydroxide solution, at reflux.PH adjusted with hydrochloric acid value of the reaction system acidic, ethanol was distilled off, and filtered to give 12- oxo crude chenodeoxycholic acid, fine recrystallization.
[0011] Preparation of chenodeoxycholic acid (5): 12- oxo take chenodeoxycholic acid, ethylene glycol and solid sodium hydroxide, hydrated corpus, refluxed for 2 hours, gradually warming evaporated partially hydrated corpus, continue to heat up to 150 ° C, continued to reflux, cooled to room temperature, poured into water, adjusting the PH with hydrochloric acid, the white precipitate was filtered, washed with water to give crude chenodeoxycholic acid, recrystallization
Azusa mouth
M ο not mesh
[0012] Step (1): cholic acid to alcohol weight to volume ratio of 1: 2 ~ 5, the volume ratio of concentrated hydrochloric acid to alcohol is 10 wide: 100, 5-5 hours reflux time was 0.5.
[0013] Step (2): cholate: acetic anhydride molar ratio = 1: 2 ~ 5, the reaction temperature, time; Tl2O hours; cholate was added per mole of N, N- dimethylpyridine wide 5g.
[0014] Step (; 3): The hypochlorite is sodium hypochlorite or calcium hypochlorite; bromide is sodium bromide, potassium bromide and the like.
[0015] Step (4): recrystallization from a solvent with an alcohol such: as methanol or ethanol.
[0016] Step (5): recrystallization solvent is a water-miscible organic solvents, such as: methanol, ethanol, acetonitrile, acetone and the like.
[0017] Step (cholate was used ¾ of methyl cholate, ethyl cholate, cholic acid or cholic acid propyl ester; Step (3) used as 3 [alpha], 7 α – diacetyl -12 α – hydroxy cholate as 3 α, 7 α – diacetyl -12 α – hydroxy methyl cholate, 3 α, 7α- diacetyl -12 α – hydroxy bile acid ethyl ester, 3 α, 7α- diacetyl yl -12 α – hydroxy acid or ester 3α, 7α- diacetyl -12 α – hydroxy acid ester.
[0018] The invention has the advantages: in cholic acid as raw materials, and the choice of bromide tetrabutylammonium bromide as catalyst, in a non-polluting oxidizing agent is hypochlorite, Intermediate 3 α, 7 α – Diacetyl _12_ oxo chenodeoxycholic acid ester yield of 90% or more, thereby improving the yield of the final product of chenodeoxycholic acid, 99% yield, low cost and no pollution, very convenient for industrial production. detailed description
[0019] The present invention will be better described, for example is as follows:
(1) Preparation of methyl cholate: bile acid 5. lg, 15ml of anhydrous methanol, heating the whole solution. Refluxed for 3 hours, was added 0. 4ml concentrated hydrochloric acid, the reaction was stopped after 30min, after slow cooling, and filtered to give methyl cholate 5. 05g, 95% yield. 1HNMR (CDCl3):. Δ 0. 70 (s, 3H, 18- CH3), 0.90 (s, 3H, 19- CH3), 0.98 (d, 3H, 21-CH3), 3 50 (m, 1H, 3 β -H), 3. 67 (s, 3H, OCH3), 3. 87 (s, 1H, 7 β -H), 3. 99 (s, 1H, 12 β -H).
[0020] (2) Preparation of 3α, 7α- methyl cholate diacetyl-hydroxy -12α-: bile acid methyl ester 4. 71g (Ilmmol) IOOml was placed in a flask, was added methylene chloride 30ml, triethylamine 3 . Chiu 1, stirred at room temperature, was added dropwise acetic anhydride 2. 7ml (28. 6mmo 1), followed by addition of 20mg N, N- dimethylpyridine catalyst, the reaction time of 7 hours, methylene chloride was distilled off, into the water, filtered to give a white solid. The crude product was recrystallized from methanol to give white crystals 4. 05g, yield 67.2%. 1H NMR (CDCl3) δ: 4.90 (m, 1H, 7 β -H), 4. 59 (s, 1H, 3 β -H), 4 01 (s, 1H, 12 β -H), 3 67.. (s, 3Η, OCH3), 2. 08 (s, 3Η, CH3CO), 2. 02 (s, 3Η, CH3CO), 0. 98 (s, 3Η, 21-CH3), 0. 93 (s, 3Η , 19-CH3), 0.69 (s, 3Η, 18_CH3).
[0021] (3) 3α, 7α – 12-oxo-diacetyl chenodeoxycholic acid methyl ester prepared: Take 3 α, 7 α – diacetyl -12 α- hydroxy methyl cholate 1.917 g ( 3. 79mmol) was placed in a 50ml round bottom flask, 12ml of ethyl acetate was added, 5ml methanol, stirring at room temperature, was added 0. 25g 0. Ig of potassium bromide and tetrabutylammonium bromide. Was added dropwise a solution of acetic acid and 6g of sodium hypochlorite (7%) (5.62mmol), for 10 hours. Methanol was distilled off under reduced pressure and ethyl acetate, filtered, washed with water, and dried to give crude 1.915g, 1.75g as a white solid after recrystallization from methanol, yield 91.2%. 1H bandit R (CDCl3) δ:.. 4. 99 (d, 1H, 7 β-H), 4 60 (m, 1H, 3 β-H), 3 67 (s, 3H, OCH3), 2. 07 (s, 6H, CH3CO), 1. 03 (s, 6H, I8-CH3 and 19-CH3), 0. 82 (d, 3H, 21-CH3) ο
[0022] (4) 12- oxo chenodeoxycholic acid Preparation: Take 3 α, 7 α – diacetyl _12_ oxo chenodeoxycholic acid methyl ester 1. 56g, was dissolved in 30ml 95% ethanol was added 3. 2g of sodium hydroxide, heated at reflux for 5 hours. PH adjusted with hydrochloric acid value of the reaction system, most of the ethanol was distilled off, filtered, washed with water, and dried to give a white solid 12- oxo-1 crude chenodeoxycholic acid, recrystallized from methanol ^ g 1. 25g, yield rate of 96%. Tun bandit R (CDCl3) δ:. 3.96 (d, 1H, 7 β-H), 3 47 (m, 1H, 3 β-H), 1.03 (s, 3H, 19_CH3), 0.89 (s, 3H, 18_CH3 ), 0 · 70 (d, 3 H, 21_CH3).
[0023] Preparation of chenodeoxycholic acid (5): 12- oxo take chenodeoxycholic acid 0. 9g, 15ml ethylene glycol was added solid sodium hydroxide and 1. 5g, 15ml hydrated corpus (80%) , 120 ° C reflux for 2 hours, change return device is a distillation apparatus, was gradually warmed evaporated amount hydrated corpus, continue to heat up to 150 ° C, continuing reflux for 4h, cooled to room temperature, poured into water, adjusted with HCl of PH3, white precipitated, was filtered cake was washed with water, and dried to give crude chenodeoxycholic acid 0. 92g, recrystallized from methanol to give 0. 86g, 99 (s, 1H, C00H).
Paper
https://pubs.acs.org/doi/abs/10.1021/ja01168a045
Reactions of 2-Arylcyclohexanones. IV. Michael Addition of Malonic Ester to 2-Phenyl-Δ2-cyclohexenone
The Preparation of Chenodeoxycholic Acid and Its Glycine and Taurine Conjugates.Hofmann, Alan F.


Preparation of Chenodeoxycholic Acid
Further studies on the synthesis of thienamycin: a facile and stereoselective synthesis of a bicyclic .beta.-keto ester by 1,3-dipolar cycloaddition




Chenodeoxycholic acid (3α, 7α- -5β- dihydroxy-cholestane acid) Chenodeoxycholic Acid (referred to as CDCA), a medicine for treating gallstones. 1848 first discovered in goose bile, 1924, known as the CDCA. By reducing cholesterol absorption, synthesis, the bile cholesterol decreased, thereby suppressing cholesterol gallstone formation and promote dissolution, and can reduce cholesterol saturation.
Chenodeoxycholic acid addition pharmaceutically itself, but also as the preparation of ursodeoxycholic acid (3α, 7β- -5β- dihydroxy bile acid, abbreviated UDCA) starting material. Ursodeoxycholic acid is the main active ingredient contained bile valuable medicine, in clinical treatment of various gastrointestinal diseases and bladder diseases. But the limited sources of bear bile medicine, and contrary to the principles of animal protection. So, dwindling source of natural bear bile, can not meet the medical requirements. Therefore, the preparation of chenodeoxycholic acid is also of great significance for further preparation of ursodeoxycholic acid.
CDCA bile extracted mainly from poultry or animal bile extraction methods in the past as it involves toxic chemicals (animal biological pharmacy, 1981, People’s Medical Publishing House, P259; pharmaceutical industry, 1987,18 (2): 75-76; ) or unsafe to use a large amount of organic solvent (Chinese Journal of biochemical Pharmaceutics, 1996,17 (1): 17; application technology, 1998,4: 9-10; US Patent, 3,965,131; US Patent, 4,331,607; USPatent, 4,163,017), can not be meet the requirements of modern industry, CDCA and low purity prepared costly.
PATENT
https://patents.google.com/patent/WO2007069814A1/en
Chenodeoxycholic acid is generally contained in bile of cow, swine, bear, or poultry such as chicken or goose, as well as in bile of human. Chenodeoxycholic acid is used as starting material for the preparation of ursodeoxycholic acid which is effective to alleviate biliary system diseases, hyperlipidemia, cholelithiasis, and chronic liver diseases, and a typical process for preparing ursodeoxycholic acid known in the art is as follows.
A typical process for preparing chenodeoxycholic acid comprises the steps of: esterifying cholic acid (3α,7α,12θ!-trihydroxy cholic acid) with methyl; protecting the hydroxyl group of 3α and Ia position by acetylating them with anhydrous acetic acid; oxidizing the hydroxyl group of 12α position to carbonyl group by using chromic acid, and then removing the carbonyl group by Wolff-kichner reduction reaction; hydrolyzing and deprotecting the obtained product to yield chenodeoxycholic acid. The above process requires the reaction to be maintained at a high temperature of more than 200 °C , and the supply of raw material may be interrupted by bovine spongiform encephalopathy, etc. Bile ,of poultry contains chenodeoxycholic acid, lithocholic acid, and a small amount of cholic acid. Thus, the process for separating chenodeoxycholic acid from poultry is well known in the art, but is not economically reasonable due to the supply decrease of raw material and low yield [see, Windhaus et al, I Physiol. Chem., 140, 177-185 (1924)].
US Patent No. 4,186,143 disclosed a process for purely separating and purifying chenodeoxycholic acid from chenodeoxycholic acid mixture derived from natural swine bile. This process comprises the major steps of: pre-treatment to remove 3ohydroxy-6- oxo-5/3-cholic acid by saponification of bile; esterification of bile acid; acetylation of bile acid ester; removal of intermediate product by using non-polar organic solvent; crystallization of acetylated ester of formula I; deprotection; and production of the compound of formula I by using crystallization in organic solvent. However, this patent does not describe HPLC content for acetylated ester of formula I, and the purity of the final product is very low since the specific rotatory power is [ofo25 +13.8° (c=l, CHCl3), and the melting point is 119-121 °C [STD: [α]D 25 +15.2°(c=l, CHCl3), melting point 127- 129 “C]. Also, the crystallization for purifying the final product requires a very long time (i.e., 16-48 hours), and the entire process is complex as eight (8) steps. Thus, when purifying the compound of formula I by using the above process, the yield of the final product becomes low, and the reaction time is as long as 12 days. Therefore, the process is not economically reasonable.
Step 6: Deprotection and crystallization of chenodeoxycholic acid
To 220ml of water were added 24.5g of chenodeoxycholic acid-diacetate-ester and 29.5g of sodium hydroxide, and then the solution was stirred with reflux for 4 hours. To the solution was added 370ml of water. The solution’s pH is adjusted to 2.0-3.0 by using 59ml of hydrochloric acid. Then, the solution was stirred at 35-45 °C for 1 hour, and then filtered. The filtered material was washed with 24.5ml of water and dried in vacuum at 70 °C to obtain 19.5g of pure chenodeoxycholic acid, m.p.: 160-161 °C, [α]o25 +13.0°(c=l, CHCl3).
Step 8: Production of the compound of formula I
The reaction solution was extracted by using ethyl acetate, and aqueous layer was discarded therefrom. Ethyl acetate layer in the solution was washed with 6% saline, and the solution was distilled to about 90ml. This solution was cooled, kept cool for one day after adding 90ml of hexane, and filtered. Thus filtered material was washed with 20ml of hexane, and dried in vacuum at 60 °C to produce 12.7g of chenodeoxycholic acid. m.p. 142-1450C; [α]D 25 +13.0°(c=l, CHCl3). INDUSTRIAL APPLICABILITY The present invention can purify chenodeoxycholic acid of formula I from swine bile solid in high yield and purity. Also, the present invention is suitable for industrial purification by reducing the purification time.
PATENT
https://patents.google.com/patent/CN102060902A/en
chenodeoxycholic acid (3 α, 7 α – dihydroxy _5 β – cholestane-24-oic acid) Chenodeoxycholic Ac id (referred to as CDCA), clinically used to correct dissolving cholesterol calculi and bile saturation drugs, the main function is to reduce the cholesterol in the bile saturation, large doses can inhibit the synthesis of cholesterol CDCA and increasing bile gallstone patients cholesterol level in a non-saturated, thereby preventing the formation of cholesterol gallstones of cholesterol and promote stone dissolve and fall off. It also has significant anti-asthmatic, anti-inflammatory, antitussive and expectorant effects.
[0003] Synthesis of chenodeoxycholic acid or ursodeoxycholic acid (3 α, 7β_ -5β_ dihydroxy-cholestane-24-oic acid, ursodeoxycholic Acid, referred UDCA), a key intermediate. Ursodeoxycholic acid is the main active ingredient of precious Chinese medicine bear bile, used in a variety of clinical hepatobiliary disease and dyspepsia. Currently we bear bile resources are scarce, mainly used synthetic chemical ursodeoxycholic acid as a clinical treatment. Therefore, the preparation of chenodeoxycholic acid is also important for the preparation of ursodeoxycholic acid.
[0004] CDCA mainly come from poultry or livestock bile extraction. Traditional extraction process complicated operation, low yield, (pharmaceutical industry, 1987,18 (9), 416; Chinese Journal of Biochemical Pharmaceutics, 1996,17 (1), 17; Applied Technology, 1998, (4), 9; CN1850846A ) can not meet the needs of modern industry. Chemical synthesis of chenodeoxycholic acid have also been reported (Japanese Journal of Chemistry 1955,76 (3), 297 -J Org Chem 1982,47 (2): 2331; Journal of Biochemical Pharmaceutics 1987,1,6 -, Tap Chi Duoc ^ oc2004 , 44 (1), 11; CN1869043A), but lower yield widespread pollution major problem, especially in the oxidation reaction is often used to expensive, and polluting agents.Therefore, to reduce pollution, reduce environmental hazards, streamline operations, improve yield, reduce costs, important for the synthesis of chenodeoxycholic acid.
Preparation of chenodeoxycholic acid.
[0007]

Cholic acid esters prepared by (1) Weigh 50 g of cholic acid, dissolved in 150 ml of anhydrous methanol was added 5 ml of concentrated hydrochloric acid was refluxed for 30 minutes, cooled slowly into the freezer, the available capacity methyl cholate It was 95%.
(2) hydroxy -12α- diacetyl – Preparation of methyl cholate methyl cholate weighed 50 g, was dissolved in 100 ml of pyridine was purified, dissolved completely, 100 ml of acetic anhydride was stirred at room temperature for 3 to 4 hours, poured into 500 ml of water, a white precipitate in the refrigerator, filtered the next day, diacetyl -12α- available hydroxy – methyl cholate, yield 40%.
(3) 3α, 7α–diacetoxy-12-oxo – Preparation of methyl cholanic acid prepared above was weighed 25 g of crude product, dissolved in 250 ml of acetone, filtered to remove insolubles, the stirring conditions , the Jones reagent was slowly added, at room temperature for 30 minutes, filtered, water was added to the filtrate precipitated white precipitate was filtered available 3α, 7α–diacetoxy-12-oxo – methyl-cholanic acid. The yield was 100%.
(4) 12- oxo – Preparation of chenodeoxycholic acid in ethanol 10% – sodium hydroxide solution and saponified for 1 hour at room temperature, the solution was acidified, poured into water to give 12- oxo – chenodeoxycholic acid , 100% yield.Recrystallized in absolute ethanol.
Preparation of chenodeoxycholic acid (5) was weighed 12- oxo – chenodeoxycholic acid, 20 grams, was added 300 ml of ethylene glycol and 30 g of solid sodium hydroxide and 300 ml of hydrazine hydrate (85%), 100 ℃ refluxed for 2 hours, warming gradually raised to 130. ℃, generated by hydrazine hydrate was distilled off, continue to heat up to 185 ~ 190 ℃, continued reflux for 4 hours, cooled to a lower temperature, poured into water and heat, PH adjusted with hydrochloric acid (20%) 3, a white precipitate was filtered cake was washed with water to give chenodeoxycholic acid.
(6) Purification of chenodeoxycholic acid obtained weighed amount of chenodeoxycholic acid, dissolved with a small amount of ethanol, was impregnated on a silica gel column petroleum ether, liquid flow linear velocity by column chromatography 1 ~ 5cm / control points, with petroleum ether: acetone = 2, begins to elute, detected by TLC chromatography therebetween, Junichi appearance of spots to be chenodeoxycholic acid appears to start collecting the eluate until no Chenodeoxy acid spots, distillation under reduced pressure and dried to give pure higher chenodeoxycholic acid.
PATENTS
References
- ^ Jump up to:a b c Russell DW (2003). “The enzymes, regulation, and genetics of bile acid synthesis”. Annu. Rev. Biochem. 72: 137–74. doi:10.1146/annurev.biochem.72.121801.161712. PMID 12543708.
- Jump up^ Carey MC (December 1975). “Editorial: Cheno and urso: what the goose and the bear have in common”. N. Engl. J. Med. 293 (24): 1255–7. doi:10.1056/NEJM197512112932412. PMID 1186807.
- Jump up^ Parks DJ, Blanchard SG, Bledsoe RK, et al. (May 1999). “Bile acids: natural ligands for an orphan nuclear receptor”. Science. 284 (5418): 1365–8. doi:10.1126/science.284.5418.1365. PMID 10334993.
- Jump up^ Thistle JL, Hofmann AF (September 1973). “Efficacy and specificity of chenodeoxycholic acid therapy for dissolving gallstones”. N. Engl. J. Med. 289 (13): 655–9. doi:10.1056/NEJM197309272891303. PMID 4580472.
- Jump up^ Berginer VM, Salen G, Shefer S (December 1984). “Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid”. N. Engl. J. Med. 311 (26): 1649–52. doi:10.1056/NEJM198412273112601. PMID 6504105.
- Jump up^ Giaconda. “Press release”. Retrieved 5 April 2014.
- Jump up^ Bazzoli F, Malavolti M, Petronelli A, Barbara L, Roda E (1983). “Treatment of constipation with chenodeoxycholic acid”. J. Int. Med. Res. 11 (2): 120–3. PMID 6852359.
- Jump up^ Rao AS, Wong BS, Camilleri M, et al. (November 2010). “Chenodeoxycholate in females with irritable bowel syndrome-constipation: a pharmacodynamic and pharmacogenetic analysis”. Gastroenterology. 139 (5): 1549–58, 1558.e1. doi:10.1053/j.gastro.2010.07.052. PMC 3189402
 . PMID 20691689.
. PMID 20691689. - Jump up^ Ki Soo Kim, Hong-Seok Kim Molecular Tweezer Based on Chenodeoxycholic Acid:Synthesis, Anion Binding Properties. Bulletin of the Korean Society 1411-1413 2004 Article ArchivedSeptember 27, 2007, at the Wayback Machine.
 |
|
 |
|
| Names | |
|---|---|
| IUPAC names
chenodiol
OR 3α,7α-dihydroxy-5β-cholanic acid OR 5β-cholanic acid-3α,7α-diol OR (R)-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic acid |
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| DrugBank | |
| ECHA InfoCard | 100.006.803 |
| EC Number | 207-481-8 |
| KEGG | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C24H40O4 | |
| Molar mass | 392.57 g/mol |
| Melting point | 165 to 167 °C (329 to 333 °F; 438 to 440 K) |
| Pharmacology | |
| A05AA01 (WHO) | |
| License data | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
////////////////////Chenodeoxycholic acid, ケノデオキシコール酸 , orphan designation
[H][C@@]1(CC[C@@]2([H])[C@]3([H])[C@H](O)C[C@]4([H])C[C@H](O)CC[C@]4(C)[C@@]3([H])CC[C@]12C)[C@H](C)CCC(O)=O
FDA and USDA announce key step to advance collaborative efforts to streamline produce safety requirements for farmers
DRUG REGULATORY AFFAIRS INTERNATIONAL

june 5, 2018

Release
As part of the U.S. Food and Drug Administration and the U.S. Department of Agriculture’s ongoing effort…
View original post 899 more words
Penciclovir

Penciclovir
- Molecular FormulaC10H15N5O3
- Average mass253.258 Da
Cas 39809-25-1
97845-62-0 (Na salt)
Launched – 1996 PERRIGO, Herpes labialis
BRL-39123; penciclovir; BRL 39123A; penciclovir sodium; Denavir; Vectavir; Euraxvir; Fenivir
Penciclovir [USAN:INN:BAN]
- BRL 39123
- BRL-39123
- CCRIS 9213
- Denavir
- HSDB 8123
- Penciclovir
- Penciclovirum
- Penciclovirum [INN-Latin]
- UNII-359HUE8FJC
Penciclovir was approved for medical use in 1996.[2]
Developed and launched by SmithKline Beecham (SB; now GlaxoSmithKline) and now marketed in the US by Prestium Pharma and ex-US by Novartis, penciclovir (Vectavir; Fenivir; Denavir; Euraxvir) is a 1% topical cream indicated for the treatment of recurrent herpes labialis (cold sores) in adults and children 12 years of age and older
APPROVALS
THE US
In September 1996, the compound was approved by the US FDA for cold sore treatment , and was launched in the US in 1997.
EUROPE
In December 1995, SB filed for European approvals of the drug . In 1997, the drug was approved in Belgium Iceland Denmark Norway Ireland . In January 2003, the drug was launched in Sweden . In May 2007, the drug was launched in Portugal .
JAPAN
In December 1995, SB filed for Japanese approval of the drug .
CHINA
In September 1999, the compound was approved in China
FDA
Click to access 020629s016lbl.pdf
Chemically, penciclovir is known as 9-[4-hydroxy-3-(hydroxymethyl)butyl] guanine. Its molecular formula is C10H15N5O3; its molecular weight is 253.26. It is a synthetic acyclic guanine derivative
Penciclovir is a white to pale yellow solid. At 20°C it has a solubility of 0.2 mg/mL in methanol, 1.3 mg/mL in propylene glycol, and 1.7 mg/mL in water. In aqueous buffer (pH 2) the solubility is 10.0 mg/mL. Penciclovir is not hygroscopic. Its partition coefficient in n-octanol/water at pH 7.5 is 0.024 (logP = -1.62).
Medical use
In herpes labialis, the duration of healing, pain and detectable virus is reduced by up to one day,[3] compared with the total duration of 2–3 weeks of disease presentation.
Mechanism of action
Penciclovir is inactive in its initial form. Within a virally infected cell a viral thymidine kinase adds a phosphate group to the penciclovir molecule; this is the rate-limiting step in the activation of penciclovir. Cellular (human) kinases then add two more phosphate groups, producing the active penciclovir triphosphate. This activated form inhibits viral DNA polymerase, thus impairing the ability of the virus to replicate within the cell.
The selectivity of penciclovir may be attributed to two factors. First, cellular thymidine kinases phosphorylate the parent form significantly less rapidly than does the viral thymidine kinase, so the active triphosphate is present at much higher concentrations in virally infected cells than in uninfected cells. Second, the activated drug binds to viral DNA polymerase with a much higher affinity than to human DNA polymerases. As a result, penciclovir exhibits negligible cytotoxicity to healthy cells.
The structure and mode of action of penciclovir are very similar to that of other nucleoside analogues, such as the more widely used aciclovir. A difference between aciclovir and penciclovir is that the active triphosphate form of penciclovir persists within the cell for a much longer time than the activated form of aciclovir, so the concentration within the cell of penciclovir will be higher given equivalent cellular doses.
SYN
Choudary, B.M.; Geen, G.R.; Grinter, T.J.; MacBeath, F.S.; Parratt, M.J.
Influence of remote structure upon regioselectivity in the N-alkylation of 2-amino-6-chloropurine: Application to the synthesis of penciclovir
Nucleosides Nucleotides 1994, 13(4): 979
PATENT
US 6573378
PATENT
CN 102070636
PAPER
https://www.tandfonline.com/doi/abs/10.1081/SCC-120026312?journalCode=lsyc20Selective and Practical Synthesis of Penciclovir


9-[4-Hydroxy-3-(hydroxymethyl)butyl]guanine
(Penciclovir)[3a,b] (1)
………………………as a colorless crystalline solid: m.p. 268.4–269.2C [Lit.[3a,b]
275–277C]. UV (H2O) max 252 and 273 (sh) nm [Lit[3a,b] 253 and 270
(sh) nm].
1HNMR (DMSO-d6) 1.42 (pseudo septet, 1H, J¼7 Hz, H-30),
1.69 (pseudo q, 2H, J¼7 Hz, H-20), 3.37 (ddd, 2H, J¼11, 7 and 7 Hz)
and 3.41 (ddd, 2H, J¼11, 7 and 7 Hz) (CH2OH), 3.98 (t, 2H, J¼7 Hz,
H-10), 4.42 (t, 2H, J¼7 Hz, OH), 6.42 (br s, 2H, NH2), 7.67 (s, 1H, H-8),
10.50 (br s, 1H, H-1).
The 1HNMR spectrum is in good agreement with
the literature data.[3a,b]
3 (a) Harnden, M.R.; Jarvest, R.L.Tetrahedron Lett. 1985, 26, 4265–4268;
(b) Harnden, M.R.;Jarvest, R.L.; Bacon, T.H.; Boyd, M.R. J. Med. Chem. 1987, 30,1636–1642;
PAPER
Improved industrial syntheses of penciclovir and famciclovir using N2-acetyl-7-benzylguanine and a cyclic side chain precursor.




| Journal of Zhejiang University SCIENCE A 2013 Vol.14 No.10 P.760-766 |
Accelerated effect on Mitsunobu reaction via bis-N-tert-butoxycarbonylation protection of 2-amino-6-chloropurine and its application in a novel synthesis of penciclovir
| Author(s): Li-yan Dai, Qiu-long Shi, Jing Zhang, Xiao-zhong Wang, Ying-qi Chen | ||
| Affiliation(s): . Department of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China | ||
| Corresponding email(s): dailiyan@zju.edu.cn | ||
| Key Words: 2-amino-6-chloropurine, Mitsunobu reaction, bis-Boc protection, Penciclovir (PCV) | ||
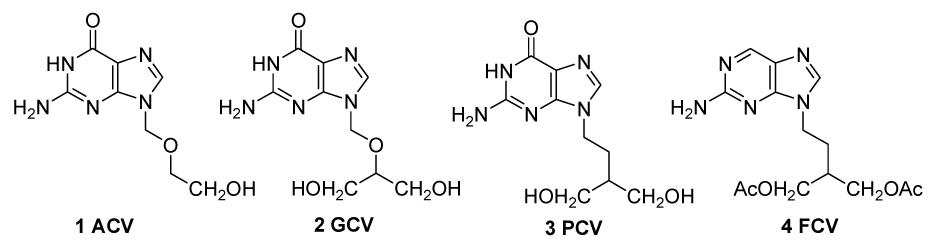
Purine derivatives
To synthesize 3 and 4, 2-amino-6-chloropurine (ACP) is commonly used as a starting material, coupling with alkyl halide side chains (Geen et al., 1990; Geen et al., 1992; Kim et al., 1998; Brand et al., 1999; Toyokuni et al., 2003). However, considering its isomerization at N7 and N9 positions under acidic or alkaline conditions, the most challengeable issue is the selectivity of a N-alkylation at the N7 or N9 position of ACP. Normally, alkylation takes place at the N9 position as well as at the N7 position of the purine moiety, and the N9/N7 ratio is usually less than 6:1 (Kim et al., 1998). Accordingly, to improve this ratio, several approaches have been reported, mainly involving changing the structure of the side chains (Geen et al., 1992) and modification of the ACP (Brand et al., 1999). For example, as reported by Zheng et al. (2004) (Fig. 2), a side chain 6 was synthesized and separated readily at 0 °C. After coupling 6 with 2-amino-6-chloropurine 7, the ratio of the product 9-isomer purine (8a) and the 7-isomer purine (8b) could reach about 10:1. However, the reaction temperature must be strictly controlled as 6 decomposes easily even at room temperature and then an extra careful column chromatography separation procedure would be required to obtain pure 8a. Thus, finding a more practical and efficient method, which could avoid the formation of N7-alkylated compound and shorten the synthetic steps to obtain ACP, becomes attractive.
Synthesis of penciclovir (PCV) with conventional method
The Mitsunobu reaction might be an alternative (potential) approach (Mitsunobu, 1981; Swamy et al., 2009). This reaction has become a very popular chemical transformation due to its mildness, occurring under essentially neutral conditions, and its stereospecificity, proceeding with complete Walden inversion of stereochemistry (Mitsunobu, 1981). Moreover, it permits C-O, C-S, C-N, or C-C bonds formed by the condensation of an acidic component with a primary or a secondary alcohol. Actually, some literature has already reported successful Mitsunobu coupling of ACP and adenine with allylic and benzylic alcohol, showing a good N9 selectivity (Yang et al., 2005; Kitade et al., 2006; Yin et al., 2006). However, a poor to modest yield (20%–50%) and a limited substrate scope were observed. In order to improve these yields, Lu et al. (2007) developed a modified Mitsunobu method to couple purine with alcohols in a higher temperature (70 °C), along with two rounds of the Mitsunobu reaction; yet its long reaction procedure and poor atom economy weaken its potential. The poor solubility of ACP or its derivatives in THF, the preferred solvent for Mitsunobu reactions, is likely the primary reason for these defects being observed.
A possible process to improve the solubility of ACP is to make use of the tert-butoxycarbonyl group (Boc), which can serve as the protection of the exocylic amino groups functionality and increase the lipophilicity of the base portion of the purine. Another advantage of the Boc protection group is that its acidolytic removal is less sensitive to steric factors and can also be removed under neutral conditions (Hwu et al., 1996; Siro et al., 1998). In contrast, a few studies have recently been reported that apply the Boc group in the protection of nucleobase (Sikchi and Hultin, 2006; Porcheddu et al., 2008). As described by Porcheddu et al. (2008), solubility of nucleobases, including guanine, was increased in some organic solvents after protected by Boc groups. In addition, some results in our previous study (Yang et al., 2011) demonstrated a very good improvement in coupling purine derivatives under Mitsunobu conditions. Thus, it could be safer to presume that protecting amino groups of ACP with Boc would be an ideal way for its application in the synthesis of PCV 3 and offer similar results as shown under Mitsunobu conditions.
In this study, we firstly synthesized a bis-Boc protected ACP, namely, bis-Boc-2-amino-6-chloropurine 9 (Fig. 3) and investigated its solubility in several different Mitsunobu solvents, then coupling bis-Boc-2-amino-6-chloropurine 9 with a large scope of alcohols confirmed its good reactivity for a Mitsunobu reaction and successfully developed a new and efficient method for the preparation of PCV using Mitsunobu coupling reaction as the key step.

Synthesis of bis-Boc-6-chloropurine 9
a: 2-amino-6-chloropurine, 4,4-dimethylaminopyridine (DMAP), THF and Boc2O, 25 °C, N2; b: MeOH, NaHCO3, 55 °C
To a 250 ml N2-flushed flask with dry THF (100 ml), equipped with a magnetic stir bar, 2-amino-6-chloropurine (2.0 g, 11.8 mmol) and DMAP (0.14 g, 1.18 mmol) were added. Boc2O (10.3 g, 47.2 mmol) was added to the stirred suspension under an N2atmosphere, then the reaction mixture was stirred for 6 h at room temperature (TLC analysis indicated the disappearance of 2-mino-6-chloropurine). The excess amount of THF was removed, and the crude product was dissolved in AcOEt (400 ml), washed with HCl aqueous (2 mol/L, 1×30 ml) and brine (2×50 ml), dried with Na2SO4 and concentrated in vacuo to give a white solid (5.2 g, 94.5%). mp 51–52 °C; 1H NMR (500 MHz, CDCl3): δ=1.47 (s, 18H, C(CH3)3), 1.69 (s, 9H, C(CH3)3), 8.58 (s, 1H, CH); 13C NMR (125 MHz, CDCl3) δ=153.8, 152.0, 151.8, 150.6, 145.5, 144.7, 130.8, 88.0, 83.9, 28.0.
2. Bis-Boc-2-amino-6-chloropurine (9)
A solution of the white solid obtained above (14 g, 30 mmol) in MeOH (400 ml) was added to saturated NaHCO3 aqueous (200 ml), then the turbid solution was stirred at 55 °C for 2 h, at which point clean conversion to bis-Boc protected adenine was observed by TLC. After evaporation of MeOH, the residue mixture was cooled, added 5 mol/L hydrochloric acid to get pH=7 (approximate). A large amount of white solid formed, the reaction mixture was filtrated and then dried under a vacuum to give a white solid 9 (10.5 g, 95.5%). mp 101.3–103.3 °C; 1H NMR (500 MHz, CDCl3): δ=1.50 (s, 18H, C(CH3)3), 8.41 (s, 1H, CH); 13C NMR (125 MHz, CDCl3) δ=153.5, 151.9, 151.6, 151.3, 145.6, 128.5, 82.7, 28.5.
2.3. 5-(2-hydroxyethyl)-2,2-dimethyl-1,3-dioxane (5)
2-hydroxymethyl-1,4-butanediol 11 (8.10 g, 67.4 mmol) and 2,2-dimethoxypropane (13 ml, 105.7 mmol) were dissolved in dry THF (20 ml). The mixture was stirred and p-toluenesulfonic acid monohydrate (0.64 g, 3.4 mmol) was added, the clear solution was stirred at room temperature for 12 h, triethylamine (10 ml) was added to quench the reaction, and the solution was stirred for 30 min. Then solvents were removed to leave a colorless liquid, the residue was subject to column chromatography on silica gel eluted with 2:1 EtOAc/hexane to give a colorless liquid 5 (6.2 g, 61.5%), R f=0.46 (2:1 EtOAc/hexane). 1H NMR (500 MHz, CDCl3): δ=3.99 (dd, 2H, Heq. J 1=11.80 Hz, J 2=4.45 Hz, CH2); 3.80 (t, 2H, J=6.71 Hz, CH2), 3.34 (dd, 2H, Hax. J 1=11.80 Hz, J 2=8.11 Hz, CH2), 1.90–1.98 (m, 2H, CH and OH), 1.62 (q, 2H, J=6.85 Hz, CH2 ); 13C NMR (125 MHz, CDCl3): δ=100.5, 69.8, 60.4, 31.9, 30.3, 21.2.
2.4. Bis-Boc-2-amino-6-chloro-9-[2-(2,2-dimethyl-1,3-dioxan-5-yl)ethyl] purine (12)
Bis-Boc-2-amino-6-chloropurine 9 (1.0 equivalent) was added to a solution of the side chain 5 (1.1 equivalent) and phosphine reagent (1.1 equivalent) in anhydrous THF under N2 atmosphere at 0 °C, the resulting solution was treated with di-p-nitrobenzyl azocarboxylate (DNAD) (1.1 equivalent) dropwise and the reaction mixture was continued at room temperature for 8 h, then the solvent was evaporated and the residue dissolved in cyclohexane. The triphenylphosphane oxide precipitated and was filtered off and then the filtrate evaporated under reduced pressure. The product was purified by a column chromatography on silica gel to obtain the pure products as a white solid. mp>280 °C (dec); 1H NMR (500 MHz, CDCl3): δ=8.36 (s, 1H, CH), 4.02 (t, 2H, J=7.23 Hz, CH2), 3.79 (dd, 2H, Heq. J 1=11.57 Hz, J 2=4.46 Hz, CH2), 3.56 (dd, 2H, Hax. J 1=11.57 Hz, J 2=8.77 Hz, CH2), 1.67 (q, 2H, J=7.22 Hz, CH2), 1.53–1.61 (m, 1H, CH), 1.47 (s, 18H, C(CH3)3), 1.39 (s, 3H, CH3), 1.36 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ=154.3, 151.7, 151.5, 151.1, 128.0, 104.8, 81.7, 71.5, 50.8, 33.7, 28.6, 26.2, 25.7.
2.5. 2-amino-6-chloro-9-[2-(2,2-dimethyl-1,3-dioxan-5-yl) ethyl]purine (8a)
A mixture of compound 12 (2.56 g, 5.0 mmol), 2,6-dimethyl pyridine (1.18 ml, 10 mmol) and dry DCM (20 ml) was stirred at 0 °C, then TBTMS-OTf was added dropwise; after the addition, the reaction mixture was stirred at room temperature until TLC showed that compound 12 had completely disappeared. Then 30 ml saturated ammonium chloride solution was added, separated the organic layer, extracted with DCM (2×20 ml), combined and washed by saturated NaCl (2×40 ml), dried with anhydrous sodium sulfate and evaporated to give a white solid (1.21 g, 78%). mp 125–126 °C; 1H NMR (500 MHz, CDCl3): δ=8.07 (s, 1H, CH) , 6.99 (s, 2H, NH2), 4.12 (t, 2H, J=7.31 Hz, CH2), 3.82 (dd, 2H, 4′-Heq, J 1=11.79 Hz, J 2=4.50 Hz, CH2), 3.53 (dd, 2H, 4′-Hax, J 1=11.79 Hz, J 2=8.80 Hz, CH2), 1.74 (q, 2H, J=7.30 Hz, CH2), 1.53–1.65 (m, 1H, CH), 1.36 (s, 3H, CH3), 1.31 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ=159.94, 150.31, 150.26, 141.84, 132.11, 100.52, 68.14, 52.90, 31.32, 26.84, 26.05.
2.6. 9-[4-hydroxy-3-(hydroxymethyl)butyl] guanine (PCV 3)
Compound 12 (5.12 g, 10 mmol) was dissolved in THF (20 ml) hydrochloric acid (2 mol/L, 20 ml). The mixture was stirred for 2 h at 70 °C, and then slowly warmed to reflux for 2 h. After evaporation of the THF under reduced vacuum, 10% aqueous NaOH solution was added to neutralize the residual liquid, and a large amount of off-white solid formed, filtered, washed with acetone and then water, and dried under vacuum to give an off-white solid 3 (2.07 g, 82%). mp 274.6–276.9 °C.
Table 1
Mole fraction solubility x of bis-Boc-2-amino-6-chloropurine 9 in different Mitsunobu solvents
| T (K) (±0.05 K) | Solubility x a (%)
|
|||
| THFb | DCMb | Methylbenzeneb | Acetonitrileb | |
| 273.15 | 0.1141 | 0.0493 | 0.0213 | 0.0150 |
| 278.15 | 0.1191 | 0.0552 | 0.0253 | 0.0178 |
| 283.15 | 0.1251 | 0.0613 | 0.0303 | 0.0210 |
| 288.15 | 0.1299 | 0.0664 | 0.0349 | 0.0244 |
| 293.15 | 0.1352 | 0.0734 | 0.0405 | 0.0288 |
| 298.15 | 0.1399 | 0.0809 | 0.0470 | 0.0347 |
| 303.15 | 0.1463 | 0.0894 | 0.0544 | 0.0417 |
| 308.15 | 0.1523 | 0.0983 | 0.0634 | 0.0501 |
| 313.15 | 0.1581 | 0.1081 | 0.0734 | 0.0617 |
- a: the solubility of bis-Boc-2-amino-6-chloropurine 9 was measured by our previous method with temperature ranging from 273.15 K to 313.15 K (Wang et al., 2008) at atmospheric pressure. The laser monitoring observation technique was used to determine the disappearance of the solid phase in a solid and liquid mixture
b: all the solvents were further purified by distillation in dry agent (Na/benzophenone) and the sample bis-boc-2-amino-6-chloropurine 9 was dried in vacuum for over 2 d
As shown in Table 1, THF, which is the most common solvent in Mitsunobu reaction, has great solubility for bis-Boc-2-amino-6-chloropurine 9. Afterwards, the best solvent THF was taken for coupling 9 with a number of alcohols under normal Mitsunobu conditions to investigate its reactivity. The results were illustrated in Table 2. We clearly learned that bis-Boc-2-amino-6-chloropurine 9, as an excellent nucleophilic precursor, was able to react with a large number of alcohols, including primary alcohol, secondary alcohol, allyl alcohol, benzyl alcohol, etc., with high N9 selectivity and yields. Moreover, tert-Butyl alcohol still could not react with a protected purine as in the previous study (Yang et al., 2011), owing to its steric hindrance in tertiary carbon.
Table 2
Investigation of the reactivity of bis-Boc-2-amino-6-chloropurine 9 with different alcohols
| Entry | Alcohol | Product | Isolated yield (%) |
| 1 | 10a | 90.2 | |
| 2 | 10b | 86.6 | |
| 3 | 10c | 83.3 | |
| 4 | 10d | 84.8 | |
| 5 | 10e | 86.4 | |
| 6 | 10f | 81.2 | |
| 7 | 10g | 81.5 | |
| 8 | 10h | 80.7 | |
| 9 | 10i | 0 |
- a): a mixture of 9 (1.0 equivalent), alcohol (1.1 equivalent) and phosphine reagent (1.1 equivalent) in anhydrous THF stirring under N2 atmosphere at 0 °C, then treated with azo-reagent DNAD (1.1 equivalent) warmed to room temperature; b): the mixture of the products from procedure a, THF (20 ml) and aqueous hydrochloric acid (2 mol/L, 20 ml) was refluxed for 2 h at 70 °C
- According to the research results above, it is more reasonable and assuring to prepare PCV via a Mitsunobu reaction. This novel method for the preparation of PCV is indicated in Fig. 4. First, the side chain of 5-(2-hydroxyethyl)-2,2-dimethyl-1,3 -dioxane 5 was achieved through the commercially available starting material 2-hydroxymethyl-1,4-butanediol 11 reacting with 2,2-dimethoxypropane catalyzed by p-toluenesulfonic acid. The free –OH group of compound 5 is not necessary to be converted to the other leaving group such as chlorine, tosylate or methanesulphonate, which is always taken as a necessary step in the previous method or many other previous studies for the preparation of PVC till now (Harnden and Jarvest, 1985; Harnden et al., 1987; Zheng et al., 2004), making the synthesis of the side chain part of our method much more convenient and practical.

Synthesis of penciclovir (PCV) with new method
a: 2,2-dimethoxypropane, p-toluenesulfonic acid, THF; b: 1.1 equivalent of the side chain 5, 1.1 equivalent of PPh3, and 1.1 equivalent of azodicarboxylate reagent at rt. in THF; c: TBDMS-OTf, DCM; d: aqueous hydrochloric acid (2 mol/L), THF; e: aqueous hydrochloric acid (2 mol/L)
Our next objective was the synthesis of PCV. As was expected, bis-Boc-2-amino-6-chloropurine 9 combined with the side chain 5(1.1 equivalent) under normal Mitsunobu conditions successfully obtained the desired N9-alkylated compound 12 in 92% yield without the undesired N7 alkylation by-product being formed. Importantly, the reaction conditions were significantly milder than those reported in recent studies (Geen et al., 1990; 1992; Kim et al., 1998; Brand et al., 1999; Toyokuni et al., 2003), requiring only 1.1 equivalent of each of the alcohol, PPh3 and DNAD, and proceeding to completion within 60 min at room temperature. This is mainly due to the enhanced solubility of the compound 9 as mentioned above. By process c in Fig. 4, compound 8a was obtained under neutral conditions. It is 1H and 13C NMR spectra further indicated that no 7-isomer purine (8b) was formed. Subsequently, we could obtain PCV 3 in an acid condition as procedure e; or directly starting from 12, where hydrolytic dechlorination and deprotection step(s) were accomplished in one pot under mild acid conditions (2mol/L, hydrochloric acid in THF at room temperature) to afford the target PCV 3 in 80%–85% yield (process d). The overall yield of PCV from 11 was 44.5% higher than that in previous study (16%) (Zheng et al., 2004).
References
 [1] Ashton, W.T., Karkas, J.D., Field, A.K., Tolman, R.L., 1982. Activation by thymidine kinase and potent antiherpetic activity of 2’-nor-2’-deoxyguanosine (2’NDG). Biochemical and Biophysical Research Communications, 108(4):1716-1721.[2] Brand, B., Reese, C.B., Song, Q., Visintin, C., 1999. Convenient syntheses of 9-[4-hydroxy-3-(hydroxymethyl)butyl] guanine (penciclovir) and 9-[4-acetoxy-3-(acetoxymethyl) butyl]-2-amino-9H-purine (famciclovir). Tetrahedron, 55(16):5239-5252.[3] De Clercq, E., 1991. Broad-spectrum anti-DNA virus and anti-retrovirus activity of phosphonylmethoxyalkylpurines and pyrimidines. Biochemical Pharmacology, 42(5):963-972.[4] Dey, S., Garner, P., 2000. Synthesis of tert-butoxycarbonyl (Boc)-protected purines. The Journal of Organic Chemistry, 65(22):7697-7699.[5] Geen, G.R., Grinter, T.J., Kincey, P.M., Jarvest, R.L., 1990. The effect of the C-6 substituent on the regioselectivity of N-alkylation of 2-aminopurines. Tetrahedron, 46(19):6903-6914.[6] Geen, G.R., Kincey, P.M., Choudary, B.M., 1992. Regiospecific Michael additions with 2-aminopurines. Tetrahedron Letters, 33(32):4609-4612.[7] Harnden, M.R., Jarvest, R.L., 1985. An improved synthesis of the antiviral acyclonucleoside 9-(4-hydroxy-3-hydroxymethylbut-1-yl) guanine. Tetrahedron Letters, 26(35):4265-4268.[8] Harnden, M.R., Jarvest, R.L., Bacon, T.H., Boyd, M.R., 1987. Synthesis and antiviral activity of 9-[4-hydroxy-3-(hydroxymethyl) but-1-yl] purines. Journal of Medicinal Chemistry, 30(9):1636-1642.[9] Hwu, J.R., Jain, M.L., Tsay, S.C., Hakimelahi, G.H., 1996. Ceric ammonium nitrate in the deprotection of tert-butoxycarbonyl group. Tetrahedron Letters, 37(12):2035-2038.[10] Kim, D.K., Lee, N., Kim, Y.W., Chang, K.Y., Kim, J.S., Im, G.J., Choi, W.S., Jung, I.H., Kim, T.S., Hwang, Y.Y., 1998. Synthesis and evaluation of 2-amino-9-(3-hydroxymethyl-4-alkoxycarbonylo-xybut-1-yl) purines as potential prodrugs of penciclovir. Journal of Medicinal Chemistry, 41(18):3435-3441.[11] Kitade, Y., Ando, T., Yamaguchi, T., Hori, A., Nakanishi, M., Ueno, Y., 2006. 4’-fluorinated carbocyclic nucleosides: synthesis and inhibitory activity against S-adenosyl-l-homocysteine hydrolase. Bioorganic & Medicinal Chemistry, 14(16):5578-5583.[12] Korba, B.E., Boyd, M.R., 1996. Penciclovir is a selective inhibitor of hepatitis B virus replication in cultured human hepatoblastoma cells. Antimicrobial Agents and Chemotherapy, 40(13):1282-1284.[13] Lu, W., Sengupta, S., Petersen, J.L., Akhmedov, N.G., Shi, X., 2007. Mitsunobu coupling of nucleobases and alcohols: an efficient, practical synthesis for novel nonsugar carbon nucleosides. Journal of Organic Chemistry, 72(13):5012-5015.[14] Martin, J.C., Dvorak, C.A., Smee, D.F., Matthews, T.R., Verheyden, J.P.H., 1983. 9-(1,3-dihydroxy-2-propoxymethyl) guanine: a new potent and selective antiherpes agent. Journal of Medicinal Chemistry, 26(5):759-761.[15] Mitsunobu, O., 1981. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis, 1981(1):1-28.[16] Ogilvie, K.K., Cheriyan, U.O., Radatus, B.K., Smith, K.O., Galloway, K.S., Kennell, W.L., 1982. Biologically active acyclonucleoside analogues. II. The synthesis of 9-[[2-hydroxy-1-(hydroxymethyl)ethoxy]methyl] guanine (BIOLF-62). Canadian Journal of Chemistry, 60(24):3005-3010.[17] Porcheddu, A., Giacomelli, G., Piredda, I., Carta, M., Nieddu, G., 2008. A Practical and efficient approach to PNA monomers compatible with Fmoc-mediated solid-phase synthesis protocols. European Journal of Organic Chemistry, 2008(34):5786-5797.[18] Schaeffer, H.J., Beauchamp, L., Miranda, P.D., Elion, G.B., Bauer, D.J., Collins, P., 1978. 9-(2-hydroxyethoxymethyl) guanine activity against viruses of the herpes group. Nature, 272(5654):583-585.[19] Shaw, T., Amor, P., Civitico, G., Boyd, M., Locarnini, S., 1994. In vitro antiviral activity of penciclovir, a novel purine nucleoside, against duck hepatitis B virus. Antimicrobial Agents and Chemotherapy, 38(4):719-723.[20] Smith, K.O., Galloway, K.S., Kennell, W.L., Ogilvie, K.K., Radatus, B.K., 1982. A new nucleoside analog, 9-[[2-hydroxy-1-(hydroxymethyl)ethoxyl]methyl] guanine, highly active in vitro against herpes simplex virus types 1 and 2. Antimicrobial Agents and Chemotherapy, 22(1):55-61.[21] Tippie, M.A., Martin, J.C., Smee, D.F., Matthews, T.R., Verheyden, J.P.M., 1984. Antiherpes simplex virus activity of 9-[4-hydroxy-3-(hydroxymethyl)-1-butyl] guanine. Nucleosides and Nucleotides, 3(5):525-535.[22] Toyokuni, T., Walsh, J.C., Namavari, M., Shinde, S.S., Moore, J.R., Barrio, J.R., Satyamurthy, N., 2003. Selective and practical synthesis of penciclovir. Synthetic Communications, 33(22):3897-3905.[23] Sikchi, S.A., Hultin, P.G., 2006. Solventless protocol for efficient Bis-N-Boc protection of adenosine, cytidine, and guanosine derivatives. Journal of Organic Chemistry, 71(16):5888-5891.[24] Siro, J.G., Martin, J., Garcia-Navio, J.L., Remuinan, M.J., Vaquero, J.J., 1998. Easy microwave assisted deprotection of N-Boc derivatives. Synlett, 1998(2):147-148.[25] Swamy, K.C.K., Kumar, N.N.B., Balaraman, E., Kumar, K.V.P.P., 2009. Mitsunobu and related reactions: advances and applications. Chemical Reviews, 109(6):2551-2651.[26] Wang, L., Dai, L.Y., Lei, M., Chen, Y., 2008. Solubility of hexamethylenetetramine in a pure water, methanol, acetic acid, and ethanol+water mixture from (299.38 to 340.35) K. Journal of Chemical & Engineering Data, 53(12):2907-2909.[27] Yang, J., Dai, L., Wang, X., Chen, Y., 2011. Di-p-nitrobenzyl azodicarboxylate (DNAD): an alternative azo-reagent for the Mitsunobu reaction. Tetrahedron, 67(7):1456-1462.[28] Yang, M.M., Schneller, S.W., Korba, B., 2005. 5’-homoneplanocin a inhibits hepatitis B and hepatitis C. Journal of Medicinal Chemistry, 48(15):5043-5046.[29] Yin, X.Q., Li, W.K., Schneller, S.W., 2006. An efficient Mitsunobu coupling to adenine-derived carbocyclic nucleosides. Tetrahedron Letters, 47(52):9187-9189.[30] Zheng, Q.H., Wang, J.Q., Liu, X., Fei, X.S., Mock, B.H., Glick-Wilson, B.E., Sullivan, M.L., Raikwar, S.P., Gardner, T.A., Kao, C.H., 2004. An improved total synthesis of PET HSV-tk gene reporter probe 9-(4-[18F] fluoro-3-hydroxymethylbutyl) guanine ([18F]FHBG). Synthetic Communications, 34(4):689-704.
[1] Ashton, W.T., Karkas, J.D., Field, A.K., Tolman, R.L., 1982. Activation by thymidine kinase and potent antiherpetic activity of 2’-nor-2’-deoxyguanosine (2’NDG). Biochemical and Biophysical Research Communications, 108(4):1716-1721.[2] Brand, B., Reese, C.B., Song, Q., Visintin, C., 1999. Convenient syntheses of 9-[4-hydroxy-3-(hydroxymethyl)butyl] guanine (penciclovir) and 9-[4-acetoxy-3-(acetoxymethyl) butyl]-2-amino-9H-purine (famciclovir). Tetrahedron, 55(16):5239-5252.[3] De Clercq, E., 1991. Broad-spectrum anti-DNA virus and anti-retrovirus activity of phosphonylmethoxyalkylpurines and pyrimidines. Biochemical Pharmacology, 42(5):963-972.[4] Dey, S., Garner, P., 2000. Synthesis of tert-butoxycarbonyl (Boc)-protected purines. The Journal of Organic Chemistry, 65(22):7697-7699.[5] Geen, G.R., Grinter, T.J., Kincey, P.M., Jarvest, R.L., 1990. The effect of the C-6 substituent on the regioselectivity of N-alkylation of 2-aminopurines. Tetrahedron, 46(19):6903-6914.[6] Geen, G.R., Kincey, P.M., Choudary, B.M., 1992. Regiospecific Michael additions with 2-aminopurines. Tetrahedron Letters, 33(32):4609-4612.[7] Harnden, M.R., Jarvest, R.L., 1985. An improved synthesis of the antiviral acyclonucleoside 9-(4-hydroxy-3-hydroxymethylbut-1-yl) guanine. Tetrahedron Letters, 26(35):4265-4268.[8] Harnden, M.R., Jarvest, R.L., Bacon, T.H., Boyd, M.R., 1987. Synthesis and antiviral activity of 9-[4-hydroxy-3-(hydroxymethyl) but-1-yl] purines. Journal of Medicinal Chemistry, 30(9):1636-1642.[9] Hwu, J.R., Jain, M.L., Tsay, S.C., Hakimelahi, G.H., 1996. Ceric ammonium nitrate in the deprotection of tert-butoxycarbonyl group. Tetrahedron Letters, 37(12):2035-2038.[10] Kim, D.K., Lee, N., Kim, Y.W., Chang, K.Y., Kim, J.S., Im, G.J., Choi, W.S., Jung, I.H., Kim, T.S., Hwang, Y.Y., 1998. Synthesis and evaluation of 2-amino-9-(3-hydroxymethyl-4-alkoxycarbonylo-xybut-1-yl) purines as potential prodrugs of penciclovir. Journal of Medicinal Chemistry, 41(18):3435-3441.[11] Kitade, Y., Ando, T., Yamaguchi, T., Hori, A., Nakanishi, M., Ueno, Y., 2006. 4’-fluorinated carbocyclic nucleosides: synthesis and inhibitory activity against S-adenosyl-l-homocysteine hydrolase. Bioorganic & Medicinal Chemistry, 14(16):5578-5583.[12] Korba, B.E., Boyd, M.R., 1996. Penciclovir is a selective inhibitor of hepatitis B virus replication in cultured human hepatoblastoma cells. Antimicrobial Agents and Chemotherapy, 40(13):1282-1284.[13] Lu, W., Sengupta, S., Petersen, J.L., Akhmedov, N.G., Shi, X., 2007. Mitsunobu coupling of nucleobases and alcohols: an efficient, practical synthesis for novel nonsugar carbon nucleosides. Journal of Organic Chemistry, 72(13):5012-5015.[14] Martin, J.C., Dvorak, C.A., Smee, D.F., Matthews, T.R., Verheyden, J.P.H., 1983. 9-(1,3-dihydroxy-2-propoxymethyl) guanine: a new potent and selective antiherpes agent. Journal of Medicinal Chemistry, 26(5):759-761.[15] Mitsunobu, O., 1981. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis, 1981(1):1-28.[16] Ogilvie, K.K., Cheriyan, U.O., Radatus, B.K., Smith, K.O., Galloway, K.S., Kennell, W.L., 1982. Biologically active acyclonucleoside analogues. II. The synthesis of 9-[[2-hydroxy-1-(hydroxymethyl)ethoxy]methyl] guanine (BIOLF-62). Canadian Journal of Chemistry, 60(24):3005-3010.[17] Porcheddu, A., Giacomelli, G., Piredda, I., Carta, M., Nieddu, G., 2008. A Practical and efficient approach to PNA monomers compatible with Fmoc-mediated solid-phase synthesis protocols. European Journal of Organic Chemistry, 2008(34):5786-5797.[18] Schaeffer, H.J., Beauchamp, L., Miranda, P.D., Elion, G.B., Bauer, D.J., Collins, P., 1978. 9-(2-hydroxyethoxymethyl) guanine activity against viruses of the herpes group. Nature, 272(5654):583-585.[19] Shaw, T., Amor, P., Civitico, G., Boyd, M., Locarnini, S., 1994. In vitro antiviral activity of penciclovir, a novel purine nucleoside, against duck hepatitis B virus. Antimicrobial Agents and Chemotherapy, 38(4):719-723.[20] Smith, K.O., Galloway, K.S., Kennell, W.L., Ogilvie, K.K., Radatus, B.K., 1982. A new nucleoside analog, 9-[[2-hydroxy-1-(hydroxymethyl)ethoxyl]methyl] guanine, highly active in vitro against herpes simplex virus types 1 and 2. Antimicrobial Agents and Chemotherapy, 22(1):55-61.[21] Tippie, M.A., Martin, J.C., Smee, D.F., Matthews, T.R., Verheyden, J.P.M., 1984. Antiherpes simplex virus activity of 9-[4-hydroxy-3-(hydroxymethyl)-1-butyl] guanine. Nucleosides and Nucleotides, 3(5):525-535.[22] Toyokuni, T., Walsh, J.C., Namavari, M., Shinde, S.S., Moore, J.R., Barrio, J.R., Satyamurthy, N., 2003. Selective and practical synthesis of penciclovir. Synthetic Communications, 33(22):3897-3905.[23] Sikchi, S.A., Hultin, P.G., 2006. Solventless protocol for efficient Bis-N-Boc protection of adenosine, cytidine, and guanosine derivatives. Journal of Organic Chemistry, 71(16):5888-5891.[24] Siro, J.G., Martin, J., Garcia-Navio, J.L., Remuinan, M.J., Vaquero, J.J., 1998. Easy microwave assisted deprotection of N-Boc derivatives. Synlett, 1998(2):147-148.[25] Swamy, K.C.K., Kumar, N.N.B., Balaraman, E., Kumar, K.V.P.P., 2009. Mitsunobu and related reactions: advances and applications. Chemical Reviews, 109(6):2551-2651.[26] Wang, L., Dai, L.Y., Lei, M., Chen, Y., 2008. Solubility of hexamethylenetetramine in a pure water, methanol, acetic acid, and ethanol+water mixture from (299.38 to 340.35) K. Journal of Chemical & Engineering Data, 53(12):2907-2909.[27] Yang, J., Dai, L., Wang, X., Chen, Y., 2011. Di-p-nitrobenzyl azodicarboxylate (DNAD): an alternative azo-reagent for the Mitsunobu reaction. Tetrahedron, 67(7):1456-1462.[28] Yang, M.M., Schneller, S.W., Korba, B., 2005. 5’-homoneplanocin a inhibits hepatitis B and hepatitis C. Journal of Medicinal Chemistry, 48(15):5043-5046.[29] Yin, X.Q., Li, W.K., Schneller, S.W., 2006. An efficient Mitsunobu coupling to adenine-derived carbocyclic nucleosides. Tetrahedron Letters, 47(52):9187-9189.[30] Zheng, Q.H., Wang, J.Q., Liu, X., Fei, X.S., Mock, B.H., Glick-Wilson, B.E., Sullivan, M.L., Raikwar, S.P., Gardner, T.A., Kao, C.H., 2004. An improved total synthesis of PET HSV-tk gene reporter probe 9-(4-[18F] fluoro-3-hydroxymethylbutyl) guanine ([18F]FHBG). Synthetic Communications, 34(4):689-704.
SYN
EP 0141927; ES 8602791; ES 8603887; ES 8603888; JP 1994293764; US 5075445
This compound has been obtained by two similar ways: 1) The reaction of 6-chloropurine-2-amine (I) with 6,6-dimethyl-5,7-dioxaspiro[2.5]octane-4,8-dione (II) by means of K2CO3 in DMF gives the expected condensation product (III), which is methanolized with HCl/methanol yielding 2-[2-(2-amino-6-methoxypurin-9-yl)ethyl]malonic acid dimethyl ester (IV). The reduction of (IV) with NaBH4 in tert-butanol/methanol affords the corresponding diol (V), which is finally converted into pecnciclovir by hydrolysis with 2N NaOH. 2) The reaction of purine (I) with 3-bromopropane-1,1,1-tricarboxylic acid triethyl ester (VI) by means ofK2CO3 in DMF gives the expected condensation product (VII), which is partially decarboxylated with sodium methoxide in methanol yielding 2-[2-(2-amino-6-chloropurin-9-yl)ethyl]malonic acid diethyl ester (VIII). The reduction of (VIII) with NaBH4 in tert-butanol/methanol followed by acetylation with acetic anhydride affords the corresponding diol diacetate (IX), which is finally converted into penciclovir by hydrlysis with 2N HCl.
References
- Jump up^ “Penciclovir”. Merriam-Webster Dictionary. Retrieved 2016-01-22.
- Jump up^ Long, Sarah S.; Pickering, Larry K.; Prober, Charles G. (2012). Principles and Practice of Pediatric Infectious Disease. Elsevier Health Sciences. p. 1502. ISBN 1437727026.
- Jump up^ Farmaceutiska Specialiteter i Sverige – the Swedish official drug catalog. [http://www.fass.se Fass.se –> Vectavir. Retrieved on August 12, 2009. Translated from “Tiden för läkning, smärta och påvisbart virus förkortas med upp till ett dygn.”
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ˌpɛnˈsaɪkloʊˌvɪər/[1] |
| Trade names | Denavir |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a697027 |
| Pregnancy category |
|
| Routes of administration |
Topical |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 1.5% (oral), negligible (topical) |
| Protein binding | <20% |
| Metabolism | Viral thymidine kinase |
| Elimination half-life | 2.2–2.3 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.189.687 |
| Chemical and physical data | |
| Formula | C10H15N5O3 |
| Molar mass | 253.258 g/mol |
| 3D model (JSmol) | |
/////////////Penciclovir, BRL-39123, BRL 39123A, penciclovir sodium, Denavir, Vectavir, Euraxvir, Fenivir,
C1=NC2=C(N1CCC(CO)CO)NC(=NC2=O)N
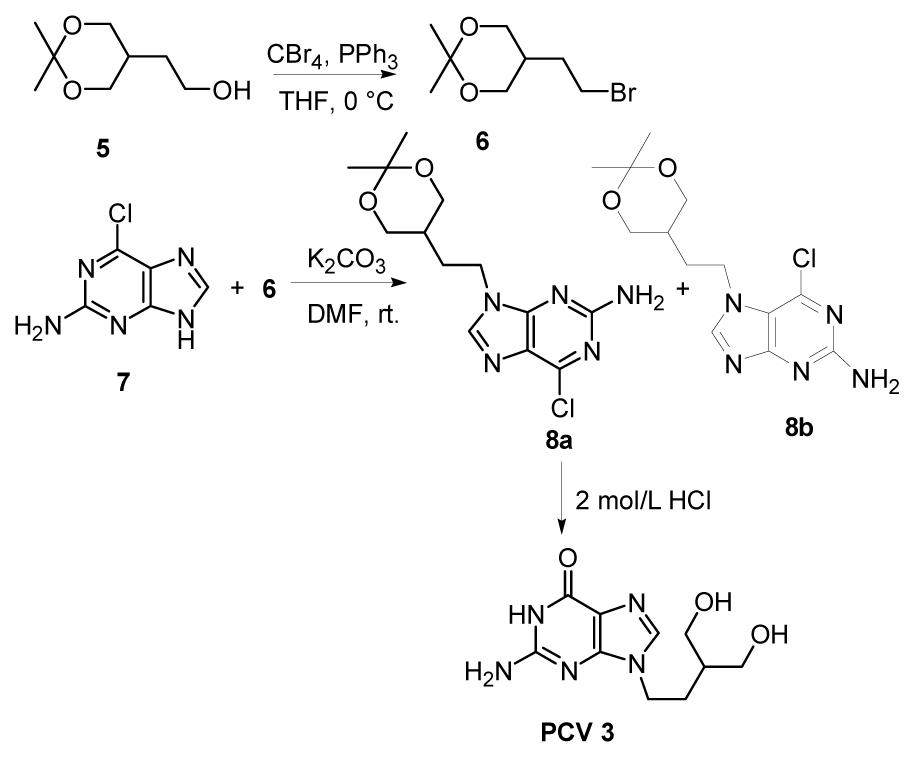
Doxepin, ドキセピン

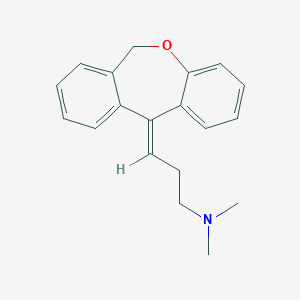
Doxepin
1668-19-5
1229-29-4 (hydrochloride), 4698-39-9 ((E)-isomer); 25127-31-5 ((Z)-isomer)
Launched – 1964
| Doxepin Hydrochloride | 3U9A0FE9N5 | 1229-29-4 |
NSC-108160
P-3693A
SO-101
Aponal
Quitaxon
Silenor
Sinequan
Sinquan
Xepin
Zonalon
USP
USP32/pub/data/v32270/usp32nf27s0_m28110
N,N-Dimethyldibenz[b,e]oxepin-D11(6H),![]() -propylamine hydrochloride
-propylamine hydrochloride ![]()
![]()
![]() [1229-29-4; 4698-39-9 ((E)-isomer); 25127-31-5 ((Z)-isomer)].
[1229-29-4; 4698-39-9 ((E)-isomer); 25127-31-5 ((Z)-isomer)].
DESCRIPTION
SINEQUAN® (doxepin hydrochloride) is one of a class of psychotherapeutic agents known as dibenzoxepin tricyclic compounds. The molecular formula of the compound is C19H21NO•HCl having a molecular weight of 316. It is a white crystalline solid readily soluble in water, lower alcohols and chloroform.
Inert ingredients for the capsule formulations are: hard gelatin capsules (which may contain Blue 1, Red 3, Red 40, Yellow 10, and other inert ingredients); magnesium stearate; sodium lauryl sulfate; starch.
Inert ingredients for the oral concentrate formulation are: glycerin; methylparaben; peppermint oil; propylparaben; water.
Chemistry
SINEQUAN (doxepin HCl) is a dibenzoxepin derivative and is the first of a family of tricyclic psychotherapeutic agents. Specifically, it is an isomeric mixture of: 1-Propanamine, 3-dibenz[b,e]oxepin-11(6H)ylidene-N,N-dimethyl-, hydrochloride.
 |
For Consumers
WHAT ARE THE POSSIBLE SIDE EFFECTS OF DOXEPIN (SINEQUAN) (SINEQUAN)?
Get emergency medical help if you have any of these signs of an allergic reaction: hives; difficulty breathing; swelling of your face, lips, tongue, or throat.
Report any new or worsening symptoms to your doctor, such as: mood or behavior changes, anxiety, panic attacks, trouble sleeping, or if you feel impulsive, irritable, agitated, hostile, aggressive, restless, hyperactive (mentally or physically), more depressed, or have thoughts about suicide or hurting yourself.
Synthesis Reference
Luigi Schioppi, Brian Talmadge Dorsey, Michael Skinner, John Carter, Robert Mansbach, Philip Jochelson, Roberta L. Rogowski, Cara Casseday, Meredith Perry, Bryan Knox, “LOW-DOSE DOXEPIN FORMULATIONS AND METHODS OF MAKING AND USING THE SAME.” U.S. Patent US20090074862, issued March 19, 2009.

DOI: 10.1007/BF00904459
DOI: 10.1007/BF00901313 US 3420851
DE 1232161
SYN 2
Synth Commun 1989, 19(19): 3349, US 3438981

Condensation of dibenzo-oxepinone (I) with 3-(dimethylamino)propylmagnesium chloride (II), followed by a dehydration of the resultant tertiary alcohol with hot HCl gives the target 3-(dimethylamino)propylidene derivative.
SYN 3

Chlorination of 2-(phenoxymethyl)benzoic acid (I) with SOCl2 at 50 °C gives 2-(phenoxymethyl)benzoyl chloride (II), which undergoes cyclization in the presence of FeCl3 in toluene to furnish dibenzo[b,e]oxepin-11-one (III)
Grignard reaction of intermediate (III) with tert-butyl 3-chloropropyl ether (IV) using Mg in refluxing THF or Et2O provides 11-(3-tert-butoxypropyl)-6,11-dihydrodibenzo[b,e]oxepin-11-ol (V), which upon elimination by means of HCl in refluxing EtOH affords alkene (VI).
Treatment of tert-butyl ether (VI) with SOCl2 in refluxing toluene gives 11-(3-chloropropylidene)-6,11-dihydrodibenzo[b,e]oxepine (VII), which is then coupled with dimethylamine (VIII) in the presence of Ni(OAc)2, PPh3 and K2CO3 in DMF or in EtOH at 100 °C to furnish doxepin (VII) .
Finally, treatment of tertiary amine (VII) with HCl at 140 °C yields the target doxepin hydrochloride .
US 2014309437, CN 102924424
Doxepin is a tricyclic antidepressant (TCA) used as a pill to treat major depressive disorder, anxiety disorders, chronic hives, and for short-term help with trouble remaining asleep after going to bed (a form of insomnia).[8][7][9] As a cream it is used for short term treatment of itchiness due to atopic dermatitis or lichen simplex chronicus.[10]
At doses used to treat depression, doxepin appears to inhibit the reuptake of serotonin and norepinephrine and to have antihistamine, adrenergic and serotonin receptor antagonistic, and anticholinergic activities; at low doses used to treat insomnia it appears to be selective for the histamine H1 receptor.[11]
It was introduced under the brand names Quitaxon and Aponal by Boehringer, which discovered it, and as Sinequan by Pfizer,[12] and has subsequently been marketed under many other names worldwide.[2]
Medical uses
Doxepin is used as a pill to treat major depressive disorder, anxiety disorders, chronic hives, and for short-term help with trouble remaining asleep after going to bed (a form of insomnia).[8][7][9] As a cream it is used for short term treatment of itchiness to due atopic dermatitis or lichen simplex chronicus.[10]
In 2016 the American College of Physicians advised that insomnia be treated first by treating comorbid conditions, then with cognitive behavioral therapy and behavioral changes, and then with drugs; doxepin was among those recommended for short term help maintaining sleep, on the basis of weak evidence.[13][14] The 2017 American Academy of Sleep Medicine recommendations focused on treatment with drugs were similar.[13] A 2015 AHRQ review of treatments for insomnia had similar findings.[15]
A 2010 review found that topical doxepin is useful to treat itchiness.[16]
A 2010 review of treatments for chronic hives found that doxepin had been superseded by better drugs but was still sometimes useful as a second line treatment.[17]
Chemistry
Doxepin is a tricyclic compound, specifically a dibenzoxepin, and possesses three rings fused together with a side chain attached in its chemical structure.[38] It is the only TCA with a dibenzoxepin ring system to have been marketed.[64] Doxepin is a tertiary amine TCA, with its side chain–demethylated metabolite nordoxepin being a secondary amine.[40][41] Other tertiary amine TCAs include amitriptyline, imipramine, clomipramine, dosulepin (dothiepin), and trimipramine.[65][66] Doxepin is a mixture of (E) and (Z) stereoisomers (the latter being known as cidoxepin or cis-doxepin) and is used commercially in a ratio of approximately 85:15.[3][67] The chemical name of doxepin is (E/Z)-3-(dibenzo[b,e]oxepin-11(6H)-ylidene)-N,N-dimethylpropan-1-amine[38][68] and its free base form has a chemical formula of C19H21NO with a molecular weight of 279.376 g/mol.[68] The drug is used commercially almost exclusively as the hydrochloride salt; the free base has been used rarely.[3][69] The CAS Registry Number of the free base is 1668-19-5 and of the hydrochloride is 1229-29-4.[3][69]

clip
https://www.sciencedirect.com/science/article/pii/S0040402007016079

History
Doxepin was discovered in Germany in 1963 and was introduced in the United States as an antidepressant in 1969.[38] It was subsequently approved at very low doses in the United States for the treatment of insomnia in 2010.[44][69]
Society and culture
Generic names
Doxepin is the generic name of the drug in English and German and its INN and BAN, while doxepin hydrochloride is its USAN, USP, BANM, and JAN.[3][69][70][2] Its generic name in Spanish and Italian and its DCIT are doxepina, in French and its DCF are doxépine, and in Latin is doxepinum.[2]
The cis or (Z) stereoisomer of doxepin is known as cidoxepin, and this is its INN while cidoxepin hydrochloride is its USAN.[3]
Brand names
It was introduced under the brand names Quitaxon and Aponal by Boehringer and as Sinequan by Pfizer.[12]
As of October 2017, doxepin is marketed under many brand names worldwide: Adnor, Anten, Antidoxe, Colian, Dofu, Doneurin, Dospin, Doxal, Doxepini, Doxesom, Doxiderm, Flake, Gilex, Ichderm, Li Ke Ning, Mareen, Noctaderm, Oxpin, Patoderm, Prudoxin, Qualiquan, Quitaxon, Sagalon, Silenor, Sinepin, Sinequan, Sinequan, Sinquan, and Zonalon.[2] It is also marketed as a combination drug with levomenthol under the brand name Doxure.[2]
Approvals
The oral formulations of doxepin are FDA-approved for the treatment of depression and sleep-maintenance insomnia and its topical formulations are FDA-approved the short-term management for some itchy skin conditions.[71] Whereas in Australia and the United Kingdom, the only licensed indication(s) is/are in the treatment of major depression and pruritus in eczema, respectively.[20][72]
Research
Antihistamine
As of 2017 there was no good evidence that topical doxepin was useful to treat localized neuropathic pain.[73] Cidoxepin is under development by Elorac, Inc. for the treatment of chronic urticaria (hives).[74] As of 2017, it is in phase II clinical trials for this indication.[74] The drug was also under investigation for the treatment of allergic rhinitis, atopic dermatitis, and contact dermatitis, but development for these indications was discontinued.[74]
Headache
Doxepin was under development by Winston Pharmaceuticals in an intranasal formulation for the treatment of headache.[75] As of August 2015, it was in phase II clinical trials for this indication.[75]
PATENT
https://patents.google.com/patent/US9486437B2/en
Doxepin:
Doxepin HCl is a tricyclic compound currently approved and available for treatment of depression and anxiety. Doxepin has the following structure:

For all compounds disclosed herein, unless otherwise indicated, where a carbon-carbon double bond is depicted, both the cis and trans stereoisomers, as well as mixtures thereof are encompassed.
Doxepin belongs to a class of psychotherapeutic agents known as dibenzoxepin tricyclic compounds, and is currently approved and prescribed for use as an antidepressant to treat depression and anxiety. Doxepin has a well-established safety profile, having been prescribed for over 35 years.
Doxepin, unlike most FDA approved products for the treatment of insomnia, is not a Schedule IV controlled substance. U.S. Pat. Nos. 5,502,047 and 6,211,229, the entire contents of which are incorporated herein by reference, describe the use of doxepin for the treatment chronic and non-chronic (e.g., transient/short term) insomnias at dosages far below those used to treat depression.
It is contemplated that doxepin for use in the methods described herein can be obtained from any suitable source or made by any suitable method. As mentioned, doxepin is approved and available in higher doses (75-300 milligrams) for the treatment of depression and anxiety. Doxepin HCl is available commercially and may be obtained in capsule form from a number of sources. Doxepin is marketed under the commercial name SINEQUAN® and in generic form, and can be obtained in the United States generally from pharmacies in capsule form in amounts of 10, 25, 50, 75, 100 and 150 mg dosage, and in liquid concentrate form at 10 mg/mL. Doxepin HCl can be obtained from Plantex Ltd. Chemical Industries (Hakadar Street, Industrial Zone, P.O. Box 160, Netanya 42101, Israel), Sifavitor S.p.A. (Via Livelli 1—Frazione, Mairano, Italy), or from Dipharma S.p.A. (20021 Baranzate di Bollate, Milano, Italy). Also, doxepin is commercially available from PharmacyRx (NZ) (2820 1st Avenue, Castlegar, B.C., Canada) in capsule form in amounts of 10, 25, 50, 75, 100 and 150 mg. Furthermore, Doxepin HCl is available in capsule form in amounts of 10, 25, 50, 75, 100 and 150 mg and in a 10 mg/ml liquid concentrate from CVS Online Pharmacy Store (CVS.com).
Also, doxepin can be prepared according to the method described in U.S. Pat. No. 3,438,981, which is incorporated herein by reference in its entirety. It should be noted and understood that although many of the embodiments described herein specifically refer to “doxepin,” other doxepin-related compounds can also be used, including, for example, pharmaceutically acceptable salts, prodrugs, metabolites, in-situ salts of doxepin formed after administration, and solid state forms, including polymorphs and hydrates.
Metabolites:
In addition, doxepin metabolites can be prepared and used. By way of illustration, some examples of metabolites of doxepin can include, but are not limited to, desmethyldoxepin, hydroxydoxepin, hydroxyl-N-desmethyldoxepin, doxepin N-oxide, N-acetyl-N-desmethyldoxepin, N-desmethyl-N-formyldoxepin, quaternary ammonium-linked glucuronide, 2-O-glucuronyldoxepin, didesmethyldoxepin, 3-O-glucuronyldoxepin, or N-acetyldidesmethyldoxepin. The metabolites of doxepin can be obtained or made by any suitable method, including the methods described above for doxepin.
Desmethyldoxepin has the following structure:

Desmethyldoxepin is commercially available as a forensic standard. For example, it can be obtained from Cambridge Isotope Laboratories, Inc. (50 Frontage Road, Andover, Mass.). Desmethyldoxepin for use in the methods discussed herein can be prepared by any suitable procedure. For example, desmethyldoxepin can be prepared from 3-methylaminopropyl triphenylphosphonium bromide hydrobromide and 6,11-dihydrodibenz(b,e)oxepin-11-one according to the method taught in U.S. Pat. No. 3,509,175, which is incorporated herein by reference in its entirety.
Hydroxydoxepin has the following structure:
2-Hydroxydoxepin can be prepared by any suitable method, including as taught by Shu et al. (Drug Metabolism and Disposition (1990) 18:735-741), which is incorporated herein by reference in its entirety.
Hydroxyl-N-desmethyldoxepin has the following structure:

2-Hydroxy-N-desmethyldoxepin can be prepared any suitable method.
Doxepin N-oxide has the following structure:

Doxepin N-oxide can be prepared by any suitable method. For example, doxepin N-oxide can be prepared as taught by Hobbs (Biochem Pharmacol (1969) 18:1941-1954), which is hereby incorporated by reference in its entirety.
N-acetyl-N-desmethyldoxepin has the following structure:

N-acetyl-N-desmethyldoxepin can be prepared by any suitable means. For example, (E)-N-acetyl-N-desmethyldoxepin has been produced in filamentous fungus incubated with doxepin as taught by Moody et al. (Drug Metabolism and Disposition (1999) 27:1157-1164), hereby incorporated by reference in its entirety.
N-desmethyl-N-formyldoxepin has the following structure:

N-desmethyl-N-formyldoxepin can be prepared by any suitable means. For example, (E)-N-desmethyl-N-formyldoxepin has been produced in filamentous fungus incubated with doxepin as taught by Moody et al. (Drug Metabolism and Disposition (1999) 27:1157-1164), hereby incorporated by reference in its entirety.
N-acetyldidesmethyldoxepin has the following structure:

N-acetyldidesmethyldoxepin can be prepared by any suitable means. For example, (E)-N-acetyldidesmethyldoxepin has been produced in filamentous fungus incubated with doxepin as taught by Moody et al. (Drug Metabolism and Disposition (1999) 27:1157-1164), hereby incorporated by reference in its entirety.
Didesmethyldoxepin has the following structure:

Didesmethyldoxepin can be prepared by any suitable means. For example, (Z)- and (E)-didesmethyldoxepin have been isolated from plasma and cerebrospinal fluid of depressed patients taking doxepin, as taught by Deuschle et al. (Psychopharmacology (1997) 131:19-22), hereby incorporated by reference in its entirety.
3-O-glucuronyldoxepin has the following structure:

3-O-glucuronyldoxepin can be prepared by any suitable means. For example, (E)-3-O-glucuronyldoxepin has been isolated from the bile of rats given doxepin, as described by Shu et al. (Drug Metabolism and Disposition (1990) 18:1096-1099), hereby incorporated by reference in its entirety.
2-O-glucuronyldoxepin has the following structure:

2-O-glucuronyldoxepin can be prepared by any suitable means. For example, (E)-2-O-glucuronyldoxepin has been isolated from the bile of rats given doxepin, and also in the urine of humans given doxepin, as described by Shu et al. (Drug Metabolism and Disposition (1990) 18:1096-1099), hereby incorporated by reference in its entirety.
Quaternary ammonium-linked glucuronide of doxepin (doxepin N+-glucuronide) has the following structure:

N+-glucuronide can be obtained by any suitable means. For example, doxepin N+-glucuronide can be prepared as taught by Luo et al. (Drug Metabolism and Disposition, (1991) 19:722-724), hereby incorporated by reference in its entirety.
PATENT
https://patents.google.com/patent/CN105330638A/en
doxepin hydrochloride, the chemical name is N, N- dimethyl-3-dibenzo (b, e) _ oxepin -11 (6H) -1-propanamine salt subunit cistron iso the mixture body configuration. CAS Number 1229-29-4 thereof, of the formula
[0003]

[0004] Doxepin hydrochloride is a drug for the treatment of depression and anxiety neurosis that act to inhibit the central nervous system serotonin and norepinephrine reuptake, such that these two synaptic cleft neurotransmitter concentration increased and antidepressant effect, but also has anti-anxiety and sedative effects. Doxepin hydrochloride oral absorption, bioavailability of 13-45%, half-life (Shu 1/2) is 8-12 hours, to apparent volume of distribution (1) ^ 9-33171.Primarily metabolized in the liver to active metabolites thereof demethylation.Metabolite excretion from the kidney, elderly patients decline of metabolism and excretion ability of this product
[0005] Chinese Patent CN102924486A discloses a method for preparing a hydrochloride of doxepin. The method comprises the coupling reaction CN, i.e., the use of Ni (0Α〇) 2 / ΡΡ1 ^ φ to the amine-based compound. Although Ni catalyst the reaction step (OAc) 2 is more readily available and inexpensive, but the low yield of this step, and low product purity.
SUMMARY
[0006] Accordingly, the present invention provides a method of o-toluic acid synthesized multi doxepin hydrochloride, the higher the yield and purity of the obtained product was purified by this method.
[0007] – o-methylbenzoate method for the synthesis of doxepin hydrochloride, comprising the steps of:
[0008] (1) o-methylbenzoic acid with N- halosuccinimide benzylation halogenation reaction occurs in an acetonitrile solvent in the light conditions, to give o-halo-methylbenzoic acid (Compound J), the following reaction formula,
[0009]

[0010] (2) Compound J celite load cesium fluoride intramolecular substitution reaction, to give phthalide (Compound H) in an acetonitrile solvent and as a catalyst, the following reaction formula,
[0011]

[0012] (3) The phenol compound J with sodium methoxide in an alcohol solvent substitution reaction, to give a compound I, the following reaction formula,
[0013]

[0014] (4) The cyclization reaction of Compound I in a solvent in the catalytic DMS0 anhydrous aluminum chloride to give 6, 11-dihydro-dibenzo [b, e] oxepin -11- one (compound A), the following reaction formula,
[0015]

[0016] (5) 6, 11-dihydro-dibenzo [b, e] oxepin-11-one (Compound A) and 3-chloropropyl alkyl tert-butyl ether (compound B) is added magnesium powder and with THF and / or a nucleophilic addition of anhydrous diethyl ether under the conditions of the reaction solvent to give the hydroxy compound (compound C), the following reaction formula,
[0017]
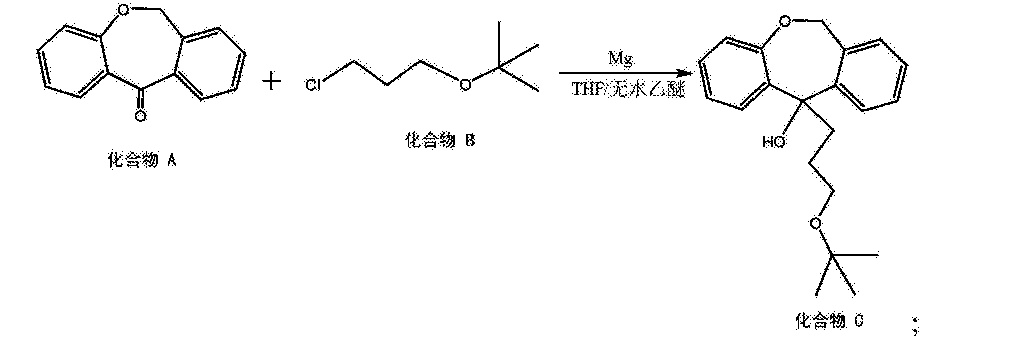
[0018] (6) heating elimination reaction to give an olefin compound (Compound D) in a strong base in an alcoholic solvent to the hydroxy compound, the following reaction formula,
[0019]

[0020] (7) to the olefinic compound in the nucleophilic substitution reaction of a hydrogen halide acid, to give halide (Compound E), the following reaction formula,

[0022] wherein the compound E X is a C1, Br, or a a I;
[0023] (8) the halide with dimethylamine in a solvent under an organic lithium compound is added in ether to nucleophilic substitution reaction to yield doxepin (Compound F.), The following reaction formula,
[0024]

[0025] (9) the doxepin neutralization reaction with hydrochloric acid to give sulfasalazine (Compound G), the following reaction formula,

Example 1
[0043] placed in a 20L reaction vessel acetonitrile, o-methylbenzoic acid, N- bromosuccinimide, using a water bath temperature controlled at 10 ° C, under stirring for 4h. A known separation method, separation of o-bromomethyl-benzoic acid. This compound is named J.
[0044] placed in a 20L reaction container, Compound J, diatomaceous earth in an amount of 0.05 to load cesium fluoride (compound J as a mass basis), acetonitrile in an amount of 2.5 (in Compound J 1 is a mass basis), and the temperature was adjusted to 30 ° C, with stirring under reflux for 20h adjustment. Then, a known means for separating the reaction phthalide.
After [0045] placed in a 20L reaction vessel phthalide, 3 an amount of sodium methoxide in ethanol solvent (total mass of phenol phthalide and 1 meter), the reaction solution temperature adjusted to 50 ° C, was added dropwise start phenol was 1.05 mass (in mass was 1 meter phthalide), dropwise over lh. After the dropwise addition, the reaction temperature after 5h using known separation methods, to give o-methyl benzyl phenyl ether, this compound is named I.
[0046] The above compound I, in an amount of 10% anhydrous aluminum chloride (mass of Compound I was 100% basis), the amount of DMS0 3 (mass basis Compound I 1) into a reaction vessel , the temperature was adjusted to 95 ° C. The reaction time is to be 12h. Using known separation means for separating the 6, 11-dihydro-dibenzo [b, e] oxepin-11-one.
[0047] placement 6, 11-dihydro-dibenzo in a reaction vessel and 20L [b, e] oxepin-11-one, 1.1-dihydro-fold of the mole of diphenyl at 6, 11 and [ b, e] oxepin-11-one 3-chloropropyl alkyl tert-butyl ether, 2 times the mass 6, 11-dihydro-dibenzo [b, e] oxepin-11-one magnesium in , taking all of fifths THF (5 to 6 times by mass, 11-dihydro-dibenzo [b, e] THF oxepin-11-one) and heated to 35 ° C and allowed to react. After the reaction started, the remaining 3/5 of THF was added dropwise.Was added dropwise to the system to be completed into hydrogen, reflux. After a total reaction 5h, the reaction was stopped. After the system was cooled and then poured into saturated ammonium chloride solution, extracted twice with ethyl acetate was added, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to give hydroxy compound.
[0048] placed in a 20L reaction vessel above hydroxy compound, an ethanol solution of 1.5 times the mass of hydroxy compound class of sodium hydroxide (concentration l〇wt mass%), was heated to 65 ° C, 2h elimination reaction after the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the olefinic compounds.
[0049] placed in a 20L reaction vessel of the olefin compound, in an aqueous solution plus 1 times the mass of the olefinic compound hydrochloride (concentration of 5wt%), and heated to 50 ° C, so that a nucleophilic substitution reaction . The reaction time is to be after 4h, the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the halides.
[0050] placed in a 20L reaction vessel above halide, 0.1 times the mass of methyl lithium halides to 2 times the mass of the halide in diethyl ether, heated to 40 ° C, so that the nucleophilic substitution reaction. The reaction time is to be after 5h, the reaction was stopped, reaction was complete and extracted with ethylacetate three times, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to obtain doxepin.
[0051] 20L is placed in a pressure reactor above doxepin, 1.05 times the mass of material in the doxepin hydrochloride (concentration of 30wt%), the control pressure to 3 ~ 4MPa, and heated to 130 ° C , and among the responses. Time after to be reacted for 20 h, cooled to room temperature and should be finished by filtration, and dried to give doxepin hydrochloride. In this embodiment overall yield 37.9%, measured by HPLC obtaining 99.2% purity.
[0052] Example 2
[0053] placed in a 20L reaction vessel acetonitrile, o-methylbenzoic acid, N- bromosuccinimide, using a water bath temperature controlled at 20 ° C, under stirring for 2h. A known separation method, separation of o-toluic acid halide.
[0054] placed in a 20L reaction container, Compound J, an amount of load of cesium fluoride Celite ~ 0.05 0.15 (in mass Compound J is 1 meter), in an amount of 2.5 to 8 acetonitrile (compound J as a mass basis), and the temperature was adjusted to 30 ~ 50 ° C, 12 ~ 20h at reflux with stirring under regulation. Then, a known means for separating the reaction phthalide.
After [0055] phthalide placed in 20L reaction vessel, an amount of sodium methoxide in 10 ethanol solvent (total mass of phenol phthalide and 1 meter), adjusting the temperature of the reaction solution was 60 ° C, was added dropwise start phenol was 1.15 mass (in mass was 1 meter phthalide), dropwise over lh.After the dropwise addition, the reaction temperature after 5h using known separation methods, to give o-methyl benzyl phenyl ether, this compound is named I.
[0056] The above compound I, in an amount of 40% anhydrous aluminum chloride (mass of Compound I was 100% basis), in an amount of DMS0 8 (in compound I is a mass basis) into a reaction vessel , the temperature was adjusted to 105 ° C. The reaction time is to be for 6h. Using known separation means for separating the 6, 11-dihydro-dibenzo [b, e] oxepin-11-one.
[0057] placement 6, 11-dihydro-dibenzo in a reaction vessel and 20L [b, e] oxepin-11-one, 1.5-dihydro-fold of the mole of diphenyl at 6, 11 and [ b, e] oxepin-11-one 3-chloropropyl alkyl tert-butyl ether, 2.4 times the mass in 6, 11-dihydro-dibenzo [b, e] oxepin-11-one of magnesium, taking all fifths THF (5 to 7 times the mass in 6, 11-dihydro-dibenzo [b, e] THF oxepin-11-one) is to make, and heated to 40 ° C reaction.After the reaction started, the remaining 3/5 of THF was added dropwise. Was added dropwise to the system to be completed into hydrogen, reflux. When the total reaction 2h, the reaction was stopped. After the system was cooled and then poured into saturated ammonium chloride solution, extracted twice with ethyl acetate was added, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to give hydroxy compound.
[0058] placed in a 20L reaction vessel above hydroxy compound, an ethanol solution of 5 times the mass of hydroxy compound class of sodium hydroxide (concentration of 70wt%), was heated to 80 ° C, the reaction was stopped after the elimination reaction LH, cooling, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the olefinic compounds.
[0059] placed in a 20L reaction vessel of the olefin compound, in an aqueous solution of 2 times the mass of the olefinic compound added hydrobromic acid (concentration of 30wt%), and heated to 60 ° C, so that nucleophilic Substitution reaction. The reaction time is to be after the 1. 5h, the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the halides.
[0060] placed in a 20L reaction vessel above halide, 0.8 times the mass of phenyl lithium halide to 8 times the mass of the halide in diethyl ether, heated to 50 ° C, so that the nucleophilic substitution reaction. The reaction time is to be after 2h, the reaction was stopped, reaction was complete and extracted with ethylacetate three times, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to obtain doxepin.
[0061] 20L is placed in a pressure reactor above doxepin, 1.2 times the mass of material in the doxepin hydrochloride (concentration of 38wt%), the control pressure to 3 ~ 4MPa, and heated to 150 ° C , and among the responses. Time after to be reacted for 16 h, cooled to room temperature and should be finished by filtration, and dried to give doxepin hydrochloride. In this embodiment overall yield 39.7%, measured by HPLC obtaining 99.4% purity.
[0062] Example 3
[0063] placed in a 20L reaction vessel acetonitrile, o-methylbenzoic acid, N- bromosuccinimide, using a water bath temperature controlled at 15 ° C, under stirring for 3h. A known separation method, separation of o-bromomethyl-benzoic acid.
[0064] placed in a 20L reaction container, Compound J, an amount of load of cesium fluoride Celite ~ 0.05 0.15 (in mass Compound J is 1 meter), in an amount of 2.5 to 8 acetonitrile (compound J as a mass basis), and the temperature was adjusted to 30 ~ 50 ° C, 12 ~ 20h at reflux with stirring under regulation. Then, a known means for separating the reaction phthalide.
After [0065] phthalide placed in 20L reaction vessel, an amount of sodium methoxide in ethanol solvent 6 (total mass of phenol phthalide and 1 meter), adjusting the temperature of the reaction solution was 55 ° C, was added dropwise start phenol was 1.10 mass (in mass was 1 meter phthalide), dropwise over lh.After the dropwise addition, the reaction temperature after 3. 5h using known separation methods, to give o-methyl benzyl phenyl ether, this compound is named I.
[0066] Anhydrous aluminum above compound I, in an amount of 25% of the chloride (compound I mass is 100% basis), in an amount of DMS0 6. 5 (in compound I is a mass basis) into the reaction vessel temperature is adjusted to 100 ° C. The reaction time is to be 9h. Using known separation means for separating the 6, 11-dihydro-dibenzo [b, e] oxepin-11-one.
[0067] placement 6, 11-dihydro-dibenzo in a reaction vessel and 20L [b, e] oxepin-11-one, 1.3-dihydro-fold of the mole of diphenyl at 6, 11 and [ b, e] oxepin-11-one 3-chloropropyl alkyl tert-butyl ether, 2.2 times the mass in 6, 11-dihydro-dibenzo [b, e] oxepin-11-one of magnesium, taking all fifths THF (5 to 7 times the mass in 6, 11-dihydro-dibenzo [b, e] THF oxepin-11-one) is to make, and heated to 38 ° C reaction.After the reaction started, the remaining 3/5 of THF was added dropwise. Was added dropwise to the system to be completed into hydrogen, refluxed for 2h. After a total reaction 3. 5h, the reaction was stopped. After the system was cooled and then poured into saturated ammonium chloride solution, extracted twice with ethyl acetate was added, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to give hydroxy compound.
[0068] placed in a 20L reaction vessel above hydroxy compound, an ethanol solution of 3-hydroxysteroid times the mass of the compound of sodium hydroxide (concentration of 40wt%), and heated to 75 ° C, 1. 5h the reaction stopped after elimination the reaction was cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the olefinic compounds.
[0069] placed in a 20L reaction vessel of the olefin compound, an aqueous solution of 1.5-fold increase in the mass of hydroiodic olefinic compounds (concentration of 18wt%), was heated to 55 ° C, so nucleophilic substitution reaction. The reaction time is to be after 2h, the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the halides.
[0070] placed in a 20L reaction vessel above halide, 0.4 times the mass of the halide in n-butyllithium, in diethyl ether five times the mass of halide and heated to 45 ° C, so that a nucleophilic substitution reaction . The reaction time is to be 3. After 5h, the reaction was stopped, reaction was complete and extracted with ethylacetate three times, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to obtain doxepin.
[0071] 20L is placed in a pressure reactor above doxepin, 1.12 times the mass of material in the doxepin hydrochloride (concentration of 34wt%), the control pressure to 3 ~ 4MPa, and heated to 140 ° C , and among the responses. Time after to be reacted for 18 h, cooled to room temperature and should be finished by filtration, and dried to give doxepin hydrochloride. In this embodiment overall yield 40.2%, measured by HPLC obtaining 99.5% purity.
[0072] Example 4
[0073] placed in a 20L reaction vessel acetonitrile, o-methylbenzoic acid, N- bromosuccinimide, using a water bath temperature controlled at 15 ° C, under stirring for 4h. A known separation method, separation of o-toluic acid halide.
[0074] placed in a 20L reaction container, Compound J, an amount of load of cesium fluoride Celite ~ 0.05 0.15 (in mass Compound J is 1 meter), in an amount of 2.5 to 8 acetonitrile (compound J as a mass basis), and the temperature was adjusted to 30 ~ 50 ° C, 12 ~ 20h at reflux with stirring under regulation. Then, a known means for separating the reaction phthalide.
After [0075] phthalide placed in 20L reaction vessel, 5 an amount of sodium methoxide in ethanol solvent (total mass of phenol phthalide and 1 meter), adjusting the temperature of the reaction solution was 55 ° C, was added dropwise start phenol was 1.15 mass (in mass was 1 meter phthalide), dropwise over lh.After the dropwise addition, the reaction temperature after 5h using known separation methods, to give o-methyl benzyl phenyl ether, this compound is named I.
[0076] The above compound I, in an amount of 25% anhydrous aluminum chloride (mass of Compound I was 100% basis), in an amount of DMS0 8 (in compound I is a mass basis) into a reaction vessel , the temperature was adjusted to 100 ° C. The reaction time is to be 12h. Using known separation means for separating the 6, 11-dihydro-dibenzo [b, e] oxepin-11-one.
[0077] placement 6, 11-dihydro-dibenzo in a reaction vessel and 20L [b, e] oxepin-11-one, 1.3-dihydro-fold of the mole of diphenyl at 6, 11 and [ b, e] oxepin-11-one 3-chloropropyl alkyl tert-butyl ether, 2.4 times the mass in 6, 11-dihydro-dibenzo [b, e] oxepin-11-one of magnesium, taking all fifths THF (5 to 7 times the mass in 6, 11-dihydro-dibenzo [b, e] THF oxepin-11-one) is to make, and heated to 40 ° C reaction.After the reaction started, the remaining 3/5 of THF was added dropwise. Was added dropwise to the system to be completed into hydrogen, reflux. When the total reaction 2h, the reaction was stopped. After the system was cooled and then poured into saturated ammonium chloride solution, extracted twice with ethyl acetate was added, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to give hydroxy compound.
[0078] placed in a 20L reaction vessel above hydroxy compound, an ethanol solution of 5 times the mass of hydroxy compound class of sodium hydroxide (concentration of 70wt%), was heated to 80 ° C, the reaction was stopped after the elimination reaction LH, cooling, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the olefinic compounds.
[0079] placed in a 20L reaction vessel of the olefin compound, an aqueous solution of 1.5-fold increase in the mass of hydroiodic olefinic compounds (concentration of 30wt%), and heated to 60 ° C, so nucleophilic substitution reaction. The reaction time is to be after the 1. 5h, the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the halides.
[0080] placed in a 20L reaction vessel above halide, 0.8 times in mass n-butyl lithium halide, eight times the mass of the halide in diethyl ether, heated to 50 ° C, so that a nucleophilic substitution reaction . The reaction time is to be after 2h, the reaction was stopped, reaction was complete and extracted with ethylacetate three times, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to obtain doxepin.
[0081] 20L is placed in a pressure reactor above doxepin, 1.2 times the mass of material in the doxepin hydrochloride (concentration of 38wt%), the control pressure to 3 ~ 4MPa, and heated to 150 ° C , and among the responses. Time after to be reacted for 16 h, cooled to room temperature and should be finished by filtration, and dried to give doxepin hydrochloride. In this embodiment overall yield 41.6%, measured by HPLC obtaining 99.7% purity.
[0082] Example 5
[0083] placed in a 20L reaction vessel acetonitrile, o-methylbenzoic acid, N- bromosuccinimide, using a water bath temperature controlled at 15 ° C, the reaction 2. 5h under stirring. A known separation method, separation of o-bromomethyl-benzoic acid.
[0084] placed in a 20L reaction vessel o-bromomethyl benzoic acid, diatomaceous earth in an amount of load of cesium fluoride 0.05 ~ 0.15 (in mass Compound J is 1 meter), in an amount of 2. 5-8 acetonitrile (compound J as a mass basis), and the temperature was adjusted to 30 ~ 50 ° C, 12 ~ 20h at reflux with stirring under regulation. Then, a known means for separating the reaction phthalide.
After [0085] phthalide placed in 20L reaction vessel, 5 an amount of sodium methoxide in ethanol solvent (total mass of phenol phthalide and 1 meter), adjusting the temperature of the reaction solution was 55 ° C, was added dropwise start was 1.08 mass of phenol (mass was phthalide 1 meter), dropwise over lh.After the dropwise addition, the reaction temperature after 3h using known separation methods, to give o-methyl benzyl phenyl ether, this compound is named I.
[0086] Anhydrous aluminum above compound I, in an amount of 25% of the chloride (compound I mass is 100% basis), in an amount of DMS0 5 (in compound I is a mass basis) into a reaction vessel , the temperature was adjusted to 100 ° C.The reaction time is to be 8h. Using known separation means for separating the 6, 11-dihydro-dibenzo [b, e] oxepin-11-one.
[0087] placement 6, 11-dihydro-dibenzo in a reaction vessel and 20L [b, e] oxepin-11-one, 1.2-dihydro-fold of the mole of diphenyl at 6, 11 and [ b, e] oxepin-11-one 3-chloropropyl alkyl tert-butyl ether, 2.2 times the mass in 6, 11-dihydro-dibenzo [b, e] oxepin-11-one of magnesium, taking all fifths THF (5 to 7 times the mass in 6, 11-dihydro-dibenzo [b, e] THF oxepin-11-one) is to make, and heated to 38 ° C reaction.After the reaction started, the remaining 3/5 of THF was added dropwise. Was added dropwise to the system to be completed into hydrogen, reflux. When the total reaction 2h, the reaction was stopped. After the system was cooled and then poured into saturated ammonium chloride solution, extracted twice with ethyl acetate was added, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to give hydroxy compound.
[0088] placed in a 20L reaction vessel above hydroxy compound, an ethanol solution of 2 times the mass of hydroxy compound class of sodium hydroxide (concentration of 40wt%), was heated to 70 ° C, the reaction was stopped after the elimination reaction 2h, cooling, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the olefinic compounds.
[0089] placed in a 20L reaction vessel of the olefin compound, an aqueous solution of 1.5-fold increase in the mass of hydroiodic olefinic compounds (concentration of 15wt%), and heated to 50 ° C, so nucleophilic substitution reaction. The reaction time is to be after 4h, the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the halides.
[0090] placed in a 20L reaction vessel above halide, 0.4 times the mass of the halide in n-butyl lithium, 2 to 8 times the mass of the halide in diethyl ether, heated to 45 ° C, so that nucleophilic Substitution reaction. The reaction time is to be after 3h, the reaction was stopped, reaction was complete and extracted with ethylacetate three times, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to obtain doxepin.
[0091] 20L is placed in a pressure reactor above doxepin, 1.12 times the mass of material in the doxepin hydrochloride (mass concentration 37. 6wt%), the control pressure to 3 ~ 4MPa, heated to 140 ° C, allowing the reaction among. Time after to be reacted for 20 h, cooled to room temperature and should be finished by filtration, and dried to give doxepin hydrochloride. In this embodiment overall yield 43.9%, measured by HPLC obtaining 99.9% purity.
PATENTS
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Sinequan, many others[2] |
| Synonyms | NSC-108160[3] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682390 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth, topical, intravenous, intramuscular injection[1] |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 13–45% (mean 29%)[5][6] |
| Protein binding | 76%[7] |
| Metabolism | Hepatic (CYP2D6, CYP2C19)[4][5] |
| Metabolites | Nordoxepin, glucuronide conjugates[4] |
| Elimination half-life | Doxepin: 8–24 hours (mean 17 hours)[7] Nordoxepin: 31 hours[7] |
| Excretion | Urine: ~50%[4][5] Feces: minor[5] |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C19H21NO |
| Molar mass | 279.376 g/mol |
| 3D model (JSmol) | |
- Virtanen R, Iisalo E, Irjala K: Protein binding of doxepin and desmethyldoxepin. Acta Pharmacol Toxicol (Copenh). 1982 Aug;51(2):159-64. [PubMed:7113722]
- Virtanen R, Scheinin M, Iisalo E: Single dose pharmacokinetics of doxepin in healthy volunteers. Acta Pharmacol Toxicol (Copenh). 1980 Nov;47(5):371-6. [PubMed:7293791]
- Negro-Alvarez JM, Carreno-Rojo A, Funes-Vera E, Garcia-Canovas A, Abellan-Aleman AF, Rubio del Barrio R: Pharmacologic therapy for urticaria. Allergol Immunopathol (Madr). 1997 Jan-Feb;25(1):36-51. [PubMed:9111875]
- Sansone RA, Sansone LA: Pain, pain, go away: antidepressants and pain management. Psychiatry (Edgmont). 2008 Dec;5(12):16-9. [PubMed:19724772]
- Kirchheiner J, Meineke I, Muller G, Roots I, Brockmoller J: Contributions of CYP2D6, CYP2C9 and CYP2C19 to the biotransformation of E- and Z-doxepin in healthy volunteers. Pharmacogenetics. 2002 Oct;12(7):571-80. [PubMed:12360109]
- ZONALON® (doxepin hydrochloride) CREAM, 5% [Link]
- FDA Label: SilenorTM (doxepin) tablets for oral administration [Link]
//////////////Doxepin, ドキセピン , NSC-108160 , P-3693A , SO-101
[H]C(CCN(C)C)=C1C2=CC=CC=C2COC2=CC=CC=C12
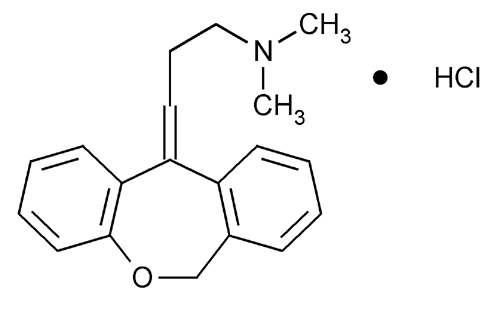
C19H21NO·HCl ![]()
![]()
![]()
![]()
![]() 315.84
315.84
N,N-Dimethyldibenz[b,e]oxepin-D11(6H),
USP Doxepin Hydrochloride RS.
USP Doxepin Related Compound A RS
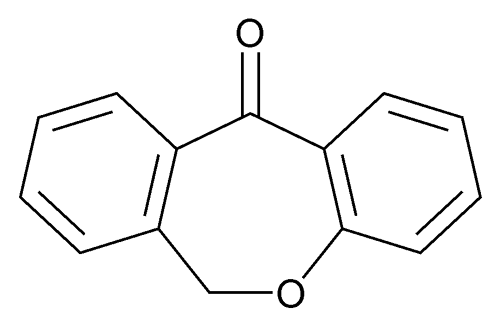 .
. USP Doxepin Related Compound B RS
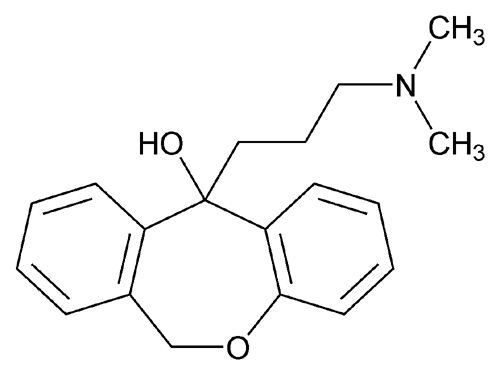 .
. USP Doxepin Related Compound C RS.
Identification—
Related compounds—
Chromatographic system (see Chromatography ![]() 621
621![]() )— The liquid chromatograph is equipped with a 215-nm detector and a 4.6-mm × 25-cm column that contains 5-µm packing L1. The flow rate is about 1 mL per minute. The column temperature is maintained at 30
)— The liquid chromatograph is equipped with a 215-nm detector and a 4.6-mm × 25-cm column that contains 5-µm packing L1. The flow rate is about 1 mL per minute. The column temperature is maintained at 30![]() . Chromatograph about 20 µL of the Standard solution, and record the peak areas as directed for Procedure: the resolution, R, between doxepin related compound A and doxepin related compound C is not less than 1.5; the resolution between doxepin related compound C and doxepin related compound B is not less than 1.5; and the signal-to-noise ratio for all the peaks is not less than 10. [NOTE—Use the approximate relative retention times given in Table 1 for the purpose of peak identification. The doxepin related compound C peak will be the largest peak in the Standard solution chromatogram.]
. Chromatograph about 20 µL of the Standard solution, and record the peak areas as directed for Procedure: the resolution, R, between doxepin related compound A and doxepin related compound C is not less than 1.5; the resolution between doxepin related compound C and doxepin related compound B is not less than 1.5; and the signal-to-noise ratio for all the peaks is not less than 10. [NOTE—Use the approximate relative retention times given in Table 1 for the purpose of peak identification. The doxepin related compound C peak will be the largest peak in the Standard solution chromatogram.]
| Name | Relative Retention Time (RRT) |
Limit (%) |
| Doxepin related compound A | 0.48 | 0.10 |
| Doxepin related compound C | 0.55 | 0.20 |
| Doxepin related compound B | 0.63 | 0.10 |
| Doxepin hydrochloride | 1.0 | — |
| Unknown impurity | — | 0.10 each |
Procedure— Inject a volume (about 20 µL) of the Test solution into the chromatograph, record the chromatogram for up to 2.2 times the retention time of doxepin, and measure the peak responses. Calculate the percentage of each individual doxepin related compound in the portion of Doxepin Hydrochloride taken by the formula:
in which rU is the individual peak response for each doxepin related compound obtained from the Test solution; rS is the response of the corresponding peak in theStandard solution; CS is the concentration, in mg per mL, of each doxepin related compound in the Standard solution; and CT is the concentration, in mg per mL, of Doxepin Hydrochloride in the Test solution. The related substance limits are listed in Table 1. [NOTE—Discard any peak with a relative retention time less than 0.25. This method is not intended to resolve the E- and Z-isomers of doxepin hydrochloride. Minor variations in the mobile phase composition could result in a shoulder in the trailing edge of doxepin. In cases where there may be separation, both the E- and Z-isomers should be used in the appropriate calculations.] Use the response of the doxepin peak obtained from the Standard solution and the concentration of doxepin hydrochloride in the Standard solution to calculate the percentage of unknown individual impurities.
Assay—
Procedure— Separately inject equal volumes (about 20 µL) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the responses for the major peaks. Calculate the quantity, in mg, of C19H21NO·HCl in the portion of Doxepin Hydrochloride taken by the formula:
in which C is the concentration, in µg per mL, of USP Doxepin Hydrochloride RS in the Standard preparation, and rU(Z) and rU(E) are the respective peak responses of the (Z)- and (E)-isomers obtained from the Assay preparation, and rS(Z) and rS(E) are the respective peak responses of the (Z)- and (E)-isomers obtained from the Standard preparation. Calculate the percentage of the (Z)-isomer in the Assay preparation taken by the formula:
in which WS is the weight, in mg, of USP Doxepin Hydrochloride RS in the Standard preparation, WT is the weight, in mg, in the portion of Doxepin Hydrochloride taken, and PZ is the labeled percentage of (Z)-isomer in USP Doxepin Hydrochloride RS. Similarly calculate the percentage of (E)-isomer in the Assay preparationtaken by the formula:
in which PE is the labeled percentage of (E)-isomer in USP Doxepin Hydrochloride RS.
Auxiliary Information— Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
| Monograph | Ravi Ravichandran, Ph.D. Senior Scientist 1-301-816-8330 |
(MDPP05) Monograph Development-Psychiatrics and Psychoactives |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
Pharmacopeial Forum: Volume No. 32(2) Page 330
Chromatographic Column—
ONC201 disrupts mitochondrial function and kills breast cancer cells, reveals study — Med-Chemist

TRAIL, a member of the TNF family of ligands, causes caspase-dependent apoptosis through activation of its receptors, death receptor 4 and DR5.ONC201 was originally identified as a small molecule that inhibits both Akt and ERK, resulting in dephosphorylation of Foxo3a and thereby induces TRAIL transcription.Recently, two independent groups, Wafik El Deiry at Fox Chase and…
via ONC201 disrupts mitochondrial function and kills breast cancer cells, reveals study — Med-Chemist
FDA approves new treatment Xeljanz (tofacitinib) for moderately to severely active ulcerative colitis
| The U.S. Food and Drug Administration today expanded the approval of Xeljanz (tofacitinib) to include adults with moderately to severely active ulcerative colitis. Xeljanz is the first oral medication approved for chronic use in this indication. Other FDA-approved treatments for the chronic treatment of moderately to severely active ulcerative colitis must be administered through an intravenous infusion or subcutaneous injection. |
May 30, 2018
Release
The U.S. Food and Drug Administration today expanded the approval of Xeljanz (tofacitinib) to include adults with moderately to severely active ulcerative colitis. Xeljanz is the first oral medication approved for chronic use in this indication. Other FDA-approved treatments for the chronic treatment of moderately to severely active ulcerative colitis must be administered through an intravenous infusion or subcutaneous injection.
“New treatments are needed for patients with moderately to severely active ulcerative colitis,” said Julie Beitz, M.D., director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research. “Today’s approval provides an alternative therapy for a debilitating disease with limited treatment options.”
Ulcerative colitis is a chronic, inflammatory bowel disease affecting the colon. Patients experience recurrent flares of abdominal pain and bloody diarrhea. Other symptoms include fatigue, weight loss and fever. More than 900,000 patients are affected in the U.S., many of them experiencing moderately to severely active ulcerative colitis, and there is currently no cure.
The efficacy of Xeljanz for the treatment of moderately to severely active ulcerative colitis was demonstrated in three controlled clinical trials. This included two 8-week placebo-controlled trials that demonstrated that 10 mg of Xeljanz given twice daily induces remission in 17 to 18 percent of patients by week eight. In a placebo-controlled trial among patients who achieved a clinical response by week eight, Xeljanz, at a 5 mg or 10 mg dose given twice daily, was effective in inducing remission by week 52 in 34 percent and 41 percent of patients, respectively. Among patients who achieved remission after 8 weeks of treatment, 35 percent and 47 percent achieved sustained corticosteroid-free remission when treated with 5 mg and 10 mg, respectively.
The safety of chronic use of Xeljanz for ulcerative colitis was studied in the 52-week placebo- controlled trial. Additional supportive safety information was collected from patients who received treatment in an open-label long-term study.
The most common adverse events associated with Xeljanz treatment for ulcerative colitis were diarrhea, elevated cholesterol levels, headache, herpes zoster (shingles), increased blood creatine phosphokinase, nasopharyngitis (common cold), rash and upper respiratory tract infection.
Less common serious adverse events included malignancy and serious infections such as opportunistic infections. Xeljanz has a boxed warning for serious infections and malignancy. Patients treated with Xeljanz are at increased risk for developing serious infections that may lead to hospitalization or death. Lymphoma and other malignancies have been observed in patients treated with Xeljanz.
Use of Xeljanz in combination with biological therapies for ulcerative colitis or with potent immunosuppressants, such as azathioprine and cyclosporine, is not recommended.
Xeljanz, made by Pfizer Labs, was previously approved in 2012 for rheumatoid arthritis and in 2017 for psoriatic arthritis.
/////////////Xeljanz, tofacitinib, pfizer, fda 2017, psoriatic arthritis, ulcerative colitis
Specific Stereoisomeric Conformations Determine the Drug Potency of Cladosporin Scaffold against Malarial Parasite
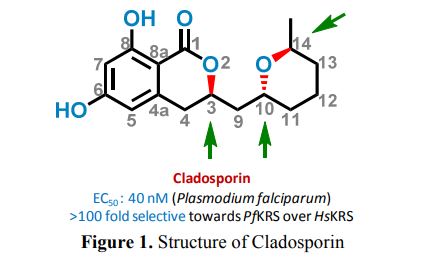
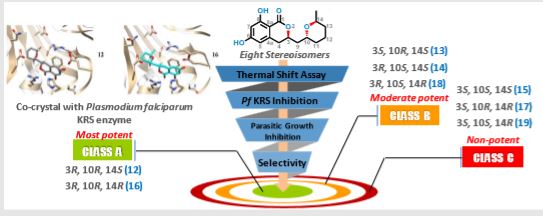

Specific Stereoisomeric Conformations Determine the Drug Potency of Cladosporin Scaffold against Malarial Parasite
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.8b00565

Dr. D. Srinivasa Reddy has been appointed as an editor of Bioorganic & Medicinl Chemistry Letters, Elsevier Publications. Congratulation Sir !
Click here for details. https://www.journals.elsevier.com/bioorganic-and-medicinal-chemistry-letters
The research interests of his group lie in issues related to application of oriented organic synthesis, in particular total synthesis of biologically active natural products, medicinal chemistry and crop protection. This team has been credited with having accomplished total synthesis of more than 25 natural products with impressive biological activities. “Some of our recent achievements include identification of potential leads, like antibiotic compound based on hunanamycin natural product for treating food infections, anti-diabetic molecule in collaboration with an industry partner and anti-TB compound using a strategy called ‘re-purposing of a drug scaffold’,” said Reddy.
A total of two awardees out of four were from CSIR institutes. In addition to Reddy, Rajan Shankarnarayanan, CSIR – CCMB, Hyderabad (basic sciences), also was conferred with the award. Vikram Mathews, CMC, Vellore (medical research) and Prof Ashish Suri, AIIMS, New Delhi (clinical research), were the others to receive the awards.
With more than 80 scientific publications and 35 patents, Reddy is one of the most prominent scientists in the city and has already been honoured with the Shanti Swarup Bhatnagar prize in chemical sciences. Reddy is also a nominated member of the scientific body of Indian Pharmacopoeia, government of India and was elected as a fellow of the Telangana and Maharashtra Academies of Sciences in addition to the National Academy of Sciences, India (NASI).
FDA Approves Tavalisse (fostamatinib disodium hexahydrate) for Chronic Immune Thrombocytopenia — Med-Chemist

Rigel Pharmaceuticals, Inc. announced that the U.S. Food and Drug Administration (FDA) approved Tavalisse (fostamatinib disodium hexahydrate) for the treatment of thrombocytopenia in adult patients with chronic immune thrombocytopenia (ITP) who have had an insufficient response to a previous treatment. Tavalisse is an oral spleen tyrosine kinase (SYK) inhibitor that targets the underlying autoimmune cause of the…
Mibefradil, a new class of compound to study TRPM7 channel function — Sussex Drug Discovery Centre

Transient receptor potential (TRPM) is a family of non-selective cation channels that are widely expressed in mammalian cells. TRP channels are composed of six transmembrane domains and the family consists of eight different channels, TRPM1–TRPM8. TRPM7 is compromised of an ion channel moiety essential for the ion channel function, which serves to increase intracellular calcium […]
via Mibefradil, a new class of compound to study TRPM7 channel function — Sussex Drug Discovery Centre
