
Home » Uncategorized (Page 29)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pabinafusp alfa
(Heavy chain)
EVQLVQSGAE VKKPGESLKI SCKGSGYSFT NYWLGWVRQM PGKGLEWMGD IYPGGDYPTY
SEKFKVQVTI SADKSISTAY LQWSSLKASD TAMYYCARSG NYDEVAYWGQ GTLVTVSSAS
TKGPSVFPLA PSSKSTSGGT AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL
YSLSSVVTVP SSSLGTQTYI CNVNHKPSNT KVDKKVEPKS CDKTHTCPPC PAPELLGGPS
VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVKFNWYV DGVEVHNAKT KPREEQYNST
YRVVSVLTVL HQDWLNGKEY KCKVSNKALP APIEKTISKA KGQPREPQVY TLPPSRDELT
KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPVLD SDGSFFLYSK LTVDKSRWQQ
GNVFSCSVMH EALHNHYTQK SLSLSPGKGS SETQANSTTD ALNVLLIIVD DLRPSLGCYG
DKLVRSPNID QLASHSLLFQ NAFAQQAVCA PSRVSFLTGR RPDTTRLYDF NSYWRVHAGN
FSTIPQYFKE NGYVTMSVGK VFHPGISSNH TDDSPYSWSF PPYHPSSEKY ENTKTCRGPD
GELHANLLCP VDVLDVPEGT LPDKQSTEQA IQLLEKMKTS ASPFFLAVGY HKPHIPFRYP
KEFQKLYPLE NITLAPDPEV PDGLPPVAYN PWMDIRQRED VQALNISVPY GPIPVDFQRK
IRQSYFASVS YLDTQVGRLL SALDDLQLAN STIIAFTSDH GWALGEHGEW AKYSNFDVAT
HVPLIFYVPG RTASLPEAGE KLFPYLDPFD SASQLMEPGR QSMDLVELVS LFPTLAGLAG
LQVPPRCPVP SFHVELCREG KNLLKHFRFR DLEEDPYLPG NPRELIAYSQ YPRPSDIPQW
NSDKPSLKDI KIMGYSIRTI DYRYTVWVGF NPDEFLANFS DIHAGELYFV DSDPLQDHNM
YNDSQGGDLF QLLMP
(Light chain)
DIVMTQTPLS LSVTPGQPAS ISCRSSQSLV HSNGNTYLHW YLQKPGQSPQ LLIYKVSNRF
SGVPDRFSGS GSGTDFTLKI SRVEAEDVGV YYCSQSTHVP WTFGQGTKVE IKRTVAAPSV
FIFPPSDEQL KSGTASVVCL LNNFYPREAK VQWKVDNALQ SGNSQESVTE QDSKDSTYSL
SSTLTLSKAD YEKHKVYACE VTHQGLSSPV TKSFNRGEC
(Disulfide bridge: H22-H96, H145-H201, H221-L219, H227-H’227, H230-H’230, H262-H322, H368-H426, H596-H609, H847-H857, H’22-H’96, H’145-H’201, H’221-L’219, H’262-H’322, H’368-H’426, H’596-H’609, H’847-H’857, L23-L93, L139-L199, L’23-L’93, L’139-L’199)
Pabinafusp alfa
CAS 2140211-48-7
PMDA 2021/3/23, JAPAN
Pabinafusp alfa (genetical recombination) (JAN)
2140211-48-7, UNII: TRF8S0U6ON
Immunoglobulin G1, anti-(human transferrin receptor) (human-mus musculus monoclonal JR-141 gamma1-chain) fusion protein with peptide (synthetic 2-amino acid linker) fusion protein with human iduronate-2-sulfatase, disulfide with human-mus musculus mono
Immunoglobulin G1-kappa, anti-(human transferrin receptor 1, tfr1) humanized monoclonal antibody, fused with human iduronate-2-sulfatase, glycoform alfa:
Pabinafusp alfa is under investigation in clinical trial NCT03568175 (A Study of JR-141 in Patients With Mucopolysaccharidosis II).
JR-141
NEW DRUG APPROVALS
ONE TIME
$10.00
JCR Pharmaceuticals Announces Approval of IZCARGO® (Pabinafusp Alfa) for Treatment of MPS II (Hunter Syndrome) in Japan
– First Approved Enzyme Replacement Therapy for MPS II to Penetrate Blood-Brain Barrier via Intravenous Administration, Validating JCR’s J-Brain Cargo® Technology –March 23, 2021 07:30 AM Eastern Daylight Time
HYOGO, Japan–(BUSINESS WIRE)–JCR Pharmaceuticals Co., Ltd. (TSE 4552; “JCR”) today announced that the Ministry of Health, Labour and Welfare (MHLW) in Japan has approved IZCARGO® (pabinafusp alfa 10 mL, intravenous drip infusion) for the treatment of mucopolysaccharidosis type II (MPS II, or Hunter syndrome). IZCARGO® (formerly known as JR-141) is a recombinant iduronate-2-sulfatase enzyme replacement therapy (ERT) that relies on J-Brain Cargo®, a proprietary technology developed by JCR, to deliver therapeutics across the blood-brain barrier (BBB). It is the first-ever approved ERT that penetrates the BBB via intravenous administration, a potentially life-changing benefit for individuals with lysosomal storage disorders (LSDs) such as MPS II.
“Subsequent to this approval in Japan, I look forward to further accumulation of clinical evidence for pabinafusp alfa in Brazil, the US and EU”Tweet this
Many patients with MPS II show complications not only in somatic symptoms but also in the central nervous system (CNS), which are often severe, with significant effects on patients’ neurocognitive development, independence, and quality of life. By delivering the enzyme to both the body and the brain, IZCARGO® treats the neurological complications of Hunter syndrome that other available therapies have been unable or inadequate to address so far.
“Approval of IZCARGO® in Japan under SAKIGAKE designation is a key milestone in JCR Pharmaceuticals’ global expansion. It comes on the heels of Fast Track designation from the US FDA, orphan designation from the European Medicines Agency, and the FDA’s acceptance of the JR-141 Investigational New Drug application, enabling JCR to begin our Phase 3 trial in the US,” said Shin Ashida, chairman and president of JCR Pharmaceuticals. “These critical regulatory milestones in Japan, where we have such a strong record of success, and those in the US and Europe, provide important validation of the value of our J-Brain Cargo® technology to deliver therapies across the blood-brain barrier, which we believe is essential to addressing the central nervous system complications of lysosomal storage disorders. We will continue our uncompromising effort to take on the challenge of providing new treatment options for patients with lysosomal storage disorders around the world as soon as possible.”
The MHLW’s approval of IZCARGO® is based on totality of evidence from non-clinical and clinical studies1-4. In a phase 2/3 clinical trial conducted in Japan, all 28 patients experienced significant reductions in heparan sulfate (HS) concentrations in the cerebrospinal fluid (CSF) – a biomarker for effectiveness against CNS symptoms of MPS II – after 52 weeks of treatment, thus meeting the trial’s primary endpoint. IZCARGO® maintained somatic disease control in patients who switched from standard ERT to IZCARGO®. The study also confirmed an improvement in somatic symptoms in participants who had not previously received standard ERT prior to the start of the trial. Additionally, a neurocognitive development assessment demonstrated maintenance or improvement of age-equivalent function in 21 of the 28 patients. There were no reports of serious treatment-related adverse events in the trial, suggestive of a favorable safety and tolerability profile for IZCARGO®.4
“Subsequent to this approval in Japan, I look forward to further accumulation of clinical evidence for pabinafusp alfa in Brazil, the US and EU,” said Dr. Paul Harmatz of University of California – San Francisco (UCSF) Benioff Children’s Hospital Oakland, Oakland, CA, United States. “The availability of an enzyme replacement therapy that crosses the blood-brain barrier is expected to treat both CNS and somatic symptoms associated with this devastating and life-threatening disorder, including developmental and cognitive delays, bone deformities, and abnormal behavior, which have, historically, been unaddressed.”
JCR recently filed an application with the Brazilian Health Surveillance Agency (Agência Nacional de Vigilância Sanitária [ANVISA]) for marketing approval of IZCARGO® for the treatment of patients with MPS II. JCR is also preparing to launch a Phase 3 trial of IZCARGO® in the US, Brazil, the UK, Germany, and France.
About pabinafusp alfa
Pabinafusp alfa (10 mL, intravenous drip infusion) is a recombinant fusion protein of an antibody against the human transferrin receptor and idursulfase, the enzyme that is missing or malfunctioning in subjects with Hunter syndrome. It incorporates J-Brain Cargo®, JCR’s proprietary BBB-penetrating technology, to cross the BBB through transferrin receptor-mediated transcytosis, and its uptake into cells is mediated through the mannose-6-phosphate receptor. This novel mechanism of action is expected to make pabinafusp alfa effective against the CNS symptoms of Hunter syndrome.
In pre-clinical trials, JCR has confirmed both high-affinity binding of pabinafusp alfa to transferrin receptors, and passage across the BBB into neuronal cells, as evidenced by electron microscopy. In addition, JCR has confirmed enzyme uptake in various brain tissues. The company has also confirmed a reduction of substrate accumulation in the CNS and peripheral organs in an animal model of Hunter syndrome.1
In several clinical trials of pabinafusp alfa, JCR obtained evidence of reduced HS concentrations in the CSF, a biomarker for assessing effectiveness against CNS symptoms. The results were consistent with those obtained in pre-clinical studies. Clinical studies have also demonstrated positive effects of pabinafusp alfa on CNS symptoms.2
About J-Brain Cargo® Technology
JCR’s first-in-class proprietary technology, J-Brain Cargo®, enables the development of therapies that cross the BBB and penetrate the CNS. The CNS complications of diseases are often severe, resulting in developmental delays, an impact on cognition and, above all, poor prognosis, which affect patients’ independence as well as the quality of life of patients and their caregivers. With J-Brain Cargo®, JCR seeks to address the unresolved clinical challenges of LSDs by delivering the enzyme to both the body and the brain.
About Mucopolysaccharidosis II (Hunter Syndrome)
Mucopolysaccharidosis II (Hunter syndrome) is an X-linked recessive LSD caused by a deficiency of iduronate-2-sulfatase, an enzyme that breaks down complex carbohydrates called glycosaminoglycans (GAGs, also known as mucopolysaccharides) in the body. Hunter syndrome, which affects an estimated 7,800 individuals worldwide (according to JCR research), gives rise to a wide range of somatic and neurological symptoms. The current standard of care for Hunter syndrome is ERT. CNS symptoms related MPS II have been unmet medical needs so far.
About JCR Pharmaceuticals Co., Ltd.
JCR Pharmaceuticals Co., Ltd. (TSE 4552) is a global specialty pharmaceuticals company that is redefining expectations and expanding possibilities for people with rare and genetic diseases worldwide. We continue to build upon our 45-year legacy in Japan while expanding our global footprint into the US, Europe, and Latin America. We improve patients’ lives by applying our scientific expertise and unique technologies to research, develop, and deliver next-generation therapies. Our approved products in Japan include therapies for the treatment of growth disorder, Fabry disease, acute graft-versus host disease, and renal anemia. Our investigational products in development worldwide are aimed at treating rare diseases including MPS I (Hurler syndrome, Hurler-Scheie, and Scheie syndrome), MPS II (Hunter syndrome), Pompe disease, and more. JCR strives to expand the possibilities for patients while accelerating medical advancement at a global level. Our core values – reliability, confidence, and persistence – benefit all our stakeholders, including employees, partners, and patients. Together we soar. For more information, please visit https://www.jcrpharm.co.jp/en/site/en/.
1 Sonoda H, Morimoto H, Yoden E, et al. A blood-brain-barrier-penetrating anti-human transferrin receptor antibody fusion protein for neuronopathic mucopolysaccharidosis II. Molecular Therapy. 2018;26(5):1366-1374.
2 Morimoto H, Kida K, Yoden E, et al. Clearance of heparan sulfate in the brain prevents neurodegeneration and neurocognitive impairment in MPS II mice. Molecular Therapy. 2021;S1525-0016(21)00027-7.
3 Okuyama T, Eto Y, Sakai N, et al. Iduronate-2-sulfatase with anti-human transferrin receptor antibody for neuropathic mucopolysaccharidosis II: a phase 1/2 trial. Molecular Therapy. 2019;27(2):456-464.
4 Okuyama T, Eto Y, Sakai N, et al. A phase 2/3 trial of pabinafusp alfa, IDS fused with anti-human transferrin receptor antibody, targeting neurodegeneration in MPS-II. Molecular Therapy. 2021;29(2):671-679.
//////////Pabinafusp alfa, JR-141, JR 141,APPROVALS 21, JAPAN 2021
#Pabinafusp alfa, #JR-141, #JR 141, #APPROVALS 21, #JAPAN 2021
DASATINIB

DASATINIB
ダサチニブ水和物
BMS 354825
863127-77-9 HYDRATE, USAN, BAN INN, JAN
UNII: RBZ1571X5H
302962-49-8 FREE FORM Dasatinib anhydrous USAN, INN
Molecular Formula, C22-H26-Cl-N7-O2-S.H2-O, Molecular Weight, 506.0282T6N DNTJ A2Q D- DT6N CNJ B1 FM- BT5N CSJ DVMR BG F1[WLN]X78UG0A0RNдазатиниб [Russian] [INN]دازاتينيب [Arabic] [INN]达沙替尼 [Chinese] [INN]1132093-70-9[RN]302962-49-8[RN]5-Thiazolecarboxamide, N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-87129966762[Beilstein]
A pyrimidine and thiazole derived ANTINEOPLASTIC AGENT and PROTEIN KINASE INHIBITOR of BCR-ABL KINASE. It is used in the treatment of patients with CHRONIC MYELOID LEUKEMIA who are resistant or intolerant to IMATINIB.
An orally bioavailable synthetic small molecule-inhibitor of SRC-family protein-tyrosine kinases. Dasatinib binds to and inhibits the growth-promoting activities of these kinases. Apparently because of its less stringent binding affinity for the BCR-ABL kinase, dasatinib has been shown to overcome the resistance to imatinib of chronic myeloid leukemia (CML) cells harboring BCR-ABL kinase domain point mutations. SRC-family protein-tyrosine kinases interact with a variety of cell-surface receptors and participate in intracellular signal transduction pathways; tumorigenic forms can occur through altered regulation or expression of the endogenous protein and by way of virally-encoded kinase genes. (NCI Thesaurus)
5-Thiazolecarboxamide, N-(2-chloro-6-methylphenyl)-2-((6-(4-(2-hydroxyethyl)-1-piperazinyl)-2-methyl-4-pyrimidinyl)amino)-, monohydrate
Synthesis ReferenceUS6596746
DASATINIB ANHYDROUS
- KIN 001-5
- NSC 759877
- Sprycel
- 302962-49-8 Dasatinib anhydrous
- 5-THIAZOLECARBOXAMIDE, N-(2-CHLORO-6-METHYLPHENYL)-2-((6-(4-(2-HYDROXYETHYL)-1-PIPERAZINYL)-2-METHYL-4-PYRIMIDINYL)AMINO)-
- BMS-354825
- DASATINIB [INN]
- DASATINIB [MI]
- DASATINIB [WHO-DD]
- DASATINIB ANHYDROUS
| No. | NDA No. | Major Technical Classification | Patent No. | Estimated Expiry Date | Drug Substance Claim | Drug Product Claim | Patent Use Code (All list) |
| 1 | N021986 | Formula | 6596746 | 2020-06-28 | Y | Y | U – 748 |
| 2 | N021986 | Formula | 6596746 | 2020-06-28 | Y | Y | U – 780 |
| 3 | N021986 | Uses(Indication) | 7125875 | 2020-04-13 | U – 779 | ||
| 4 | N021986 | Uses(Indication) | 7125875 | 2020-04-13 | U – 780 | ||
| 5 | N021986 | Uses(Indication) | 7153856 | 2020-04-28 | U – 780 | ||
| 6 | N021986 | Crystal | 7491725 | 2026-03-28 | Y | Y | |
| 7 | N021986 | Formulation | 8680103 | 2025-02-04 | Y |



SPRYCEL (dasatinib) is an inhibitor of multiple tyrosine kinases.
The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2- methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate. The molecular formula is C22H26ClN7O2S • H2O, which corresponds to a formula weight of 506.02 (monohydrate).
The anhydrous free base has a molecular weight of 488.01. Dasatinib has the following chemical structure: Dasatinib is a white to off-white powder and has a melting point of 280°–286° C.
The drug substance is insoluble in water and slightly soluble in ethanol and methanol. SPRYCEL tablets are white to off-white, biconvex, film-coated tablets containing dasatinib, with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol
| DASATINIBDASATINIB (DASATINIB) | ANDA #202103 | TABLET;ORAL | Discontinued | APOTEX INC |
| SPRYCELSPRYCEL (DASATINIB) | NDA #021986 | TABLET;ORAL | Prescription | BRISTOL MYERS SQUIBBSPRYCEL (DASATINIB) | NDA #022072 | TABLET; ORAL | Prescription | BRISTOL MYERS SQUIBB |
Clip
https://www.pharmainbrief.com/files/2017/09/A-106-17-20170918-Reasons.pdfhttps://www.accessdata.fda.gov/drugsatfda_docs/appletter/2016/202103Orig1s000ltr.pdfU.S. Patent Number Expiration Date 6,596,746 (the ‘746 patent) June 28, 20207,125,875 (the ‘875 patent) April 13, 20207,153,856 (the ‘856 patent) April 28, 20207,491,725 (the ‘725 patent) March 28, 20268,680,103 (the ‘103 patent) February 4, 2025
Drug Name:Dasatinib HydrateResearch Code:BMS-354825Trade Name:Sprycel®MOA:Kinase inhibitorIndication:Acute lymphoblastic leukaemia (ALL); Chronic myeloid leukemia (CML )Status:ApprovedCompany:Bristol-Myers Squibb (Originator)Sales:$1,620 Million (Y2015);
$1,493 Million (Y2014);
$1,280 Million (Y2013);
$1,019 Million (Y2012);
$803 Million (Y2011);ATC Code:L01XE06Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2006-06-28 | Marketing approval | Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet, Film coated | Eq. 20 mg/50 mg/70 mg/80 mg/100 mg/140 mg Dasatinib | Bristol-Myers Squibb | Priority; Orphan |
| 2006-06-28 | Additional approval | Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet, Film coated | 70 mg | Bristol-Myers Squibb | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2006-11-20 | Marketing approval | Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet, Film coated | 20 mg/50 mg/70 mg/80 mg/100 mg/140 mg | Bristol-Myers Squibb | Orphan |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-06-16 | Modified indication | Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet, Film coated | 20 mg/50 mg | Bristol-Myers Squibb, Otsuka | |
| 2009-01-21 | Marketing approval | Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet, Film coated | 20 mg/50 mg | Bristol-Myers Squibb, Otsuka |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2013-09-17 | Marketing approval | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 20 mg | 南京正大天晴制药 | ||
| 2013-09-17 | Marketing approval | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 50 mg | 南京正大天晴制药 | ||
| 2013-09-17 | Marketing approval | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 70 mg | 南京正大天晴制药 | ||
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 50 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 50 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 50 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 20 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 20 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 20 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 70 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 70 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 70 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 100 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 100 mg | Bristol-Myers Squibb | |
| 2011-09-07 | Marketing approval | 施达赛/Sprycel | Acute lymphoblastic leukaemia (ALL), Chronic myeloid leukemia (CML ) | Tablet | 100 mg | Bristol-Myers Squibb |
SPRYCEL (dasatinib) is a kinase inhibitor. The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate. The molecular formula is C22H26ClN7O2S • H2O, which corresponds to a formula weight of 506.02 (monohydrate). The anhydrous free base has a molecular weight of 488.01. Dasatinib has the following chemical structure:
 |
Dasatinib is a white to off-white powder. The drug substance is insoluble in water and slightly soluble in ethanol and methanol.
SPRYCEL tablets are white to off-white, biconvex, film-coated tablets containing dasatinib, with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol.
Dasatinib hydrate was first approved by the U.S. Food and Drug Administration (FDA) on June 28, 2006, then approved by European Medicine Agency (EMA) on Nov 20, 2006, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jan 21, 2009. It was developed and marketed as Sprycel® by Bristol Myers Squibb in the US.
Dasatinibhydrate is a kinase inhibitor.It is indicated for the treatment ofchronic myeloid leukemia and acutelymphoblastic leukemia.
Sprycel® is available as film-coatedtabletfor oral use, containing 20, 50, 70, 80, 100 or 140 mg offreeDasatinib. The recommended dose is 100 mg once daily forchronic myeloid leukemia. Another dose is 140 mg once daily for accelerated phase chronic myeloid leukemia, myeloid or lymphoid blast phase chronic myeloid leukemia, or Ph+ acutelymphoblastic leukemia.
Dasatinib, also known as BMS-354825, is an orally bioavailable synthetic small molecule-inhibitor of SRC-family protein-tyrosine kinases. Dasatinib binds to and inhibits the growth-promoting activities of these kinases. Apparently because of its less stringent binding affinity for the BCR-ABL kinase, dasatinib has been shown to overcome the resistance to imatinib of chronic myeloid leukemia (CML) cells harboring BCR-ABL kinase domain point mutations.
Dasatinib, sold under the brand name Sprycel among others, is a targeted therapy medication used to treat certain cases of chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL).[3] Specifically it is used to treat cases that are Philadelphia chromosome-positive (Ph+).[3] It is taken by mouth.[3]
Common adverse effects include low white blood cells, low blood platelets, anemia, swelling, rash, and diarrhea.[3] Severe adverse effects may include bleeding, pulmonary edema, heart failure, and prolonged QT syndrome.[3] Use during pregnancy may result in harm to the baby.[3] It is a tyrosine-kinase inhibitor and works by blocking a number of tyrosine kinases such as Bcr-Abl and the Src kinase family.[3]
Dasatinib was approved for medical use in the United States and in the European Union in 2006.[3][2] It is on the World Health Organization’s List of Essential Medicines.
Medical uses
Dasatinib is used to treat people with chronic myeloid leukemia and people with acute lymphoblastic leukemia who are positive for the Philadelphia chromosome.[5]
In the EU dasatinib is indicated for children with
- newly diagnosed Philadelphia chromosome-positive chronic myelogenous leukaemia in chronic phase (Ph+ CML CP) or Ph+ CML CP resistant or intolerant to prior therapy including imatinib.[2]
- newly diagnosed Ph+ acute lymphoblastic leukaemia (ALL) in combination with chemotherapy.[2]
- newly diagnosed Ph+ CML in chronic phase (Ph+ CML-CP) or Ph+ CML-CP resistant or intolerant to prior therapy including imatinib.[2]
and adults with
- newly diagnosed Philadelphia-chromosome-positive (Ph+) chronic myelogenous leukaemia (CML) in the chronic phase;[2]
- chronic, accelerated or blast phase CML with resistance or intolerance to prior therapy including imatinib mesilate;[2]
- Ph+ acute lymphoblastic leukaemia (ALL) and lymphoid blast CML with resistance or intolerance to prior therapy.[2]
Adverse effects
The most common side effects are infection, suppression of the bone marrow (decreasing numbers of leukocytes, erythrocytes, and thrombocytes),[6] headache, hemorrhage (bleeding), pleural effusion (fluid around the lungs), dyspnea (difficulty breathing), diarrhea, vomiting, nausea (feeling sick), abdominal pain (belly ache), skin rash, musculoskeletal pain, tiredness, swelling in the legs and arms and in the face, fever.[2] Neutropenia and myelosuppression were common toxic effects. Fifteen people (of 84, i.e. 18%) in the above-mentioned study developed pleural effusions, which was a suspected side effect of dasatinib. Some of these people required thoracentesis or pleurodesis to treat the effusions. Other adverse events included mild to moderate diarrhea, peripheral edema, and headache. A small number of people developed abnormal liver function tests which returned to normal without dose adjustments. Mild hypocalcemia was also noted, but did not appear to cause any significant problems. Several cases of pulmonary arterial hypertension (PAH) were found in people treated with dasatinib,[7] possibly due to pulmonary endothelial cell damage.[8]
On October 11, 2011, the U.S. Food and Drug Administration (FDA) announced that dasatinib may increase the risk of a rare but serious condition in which there is abnormally high blood pressure in the arteries of the lungs (pulmonary hypertension, PAH).[9] Symptoms of PAH may include shortness of breath, fatigue, and swelling of the body (such as the ankles and legs).[9] In reported cases, people developed PAH after starting dasatinib, including after more than one year of treatment.[9] Information about the risk was added to the Warnings and Precautions section of the Sprycel drug label.[9]
Pharmacology

Crystal structure[10] (PDB 2GQG) of Abl kinase domain (blue) in complex with dasatinib (red).
Dasatinib is an ATP-competitive protein tyrosine kinase inhibitor. The main targets of dasatinib are BCR/Abl (the “Philadelphia chromosome”), Src, c-Kit, ephrin receptors, and several other tyrosine kinases.[11] Strong inhibition of the activated BCR-ABL kinase distinguishes dasatinib from other CML treatments, such as imatinib and nilotinib.[11][12] Although dasatinib only has a plasma half-life of three to five hours, the strong binding to BCR-ABL1 results in a longer duration of action.[12]
History
See also: Discovery and development of Bcr-Abl tyrosine kinase inhibitors
Dasatinib was developed by collaboration of Bristol-Myers Squibb and Otsuka Pharmaceutical Co., Ltd,[13][14][15] and named for Bristol-Myers Squibb research fellow Jagabandhu Das, whose program leader says that the drug would not have come into existence had he not challenged some of the medicinal chemists‘ underlying assumptions at a time when progress in the development of the molecule had stalled.[16]
Society and culture
Legal status
Dasatinib was approved for used in the United States in June 2006 and in the European Union in November 2006[17][2]
In October 2010, dasatinib was approved in the United States for the treatment of newly diagnosed adults with Philadelphia chromosome positive chronic myeloid leukemia in chronic phase (CP-CML).[18]
In November 2017, dasatinib was approved in the United States for the treatment of children with Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in the chronic phase.[19]
Approval was based on data from 97 pediatric participants with chronic phase CML evaluated in two trials—a Phase I, open-label, non-randomized, dose-ranging trial and a Phase II, open-label, non-randomized trial.[19] Fifty-one participants exclusively from the Phase II trial were newly diagnosed with chronic phase CML and 46 participants (17 from the Phase I trial and 29 from the Phase II trial) were resistant or intolerant to previous treatment with imatinib.[19] The majority of participants were treated with dasatinib tablets 60 mg/m2 body surface area once daily.[19] Participants were treated until disease progression or unacceptable toxicity.[19]
Economics
The Union for Affordable Cancer Treatment objected to the price of dasatinib, in a letter to the U.S. trade representative. The average wholesale price in the U.S. is $367 per day, twice the price in other high income countries. The price in India, where the average annual per capita income is $1,570, and where most people pay out of pocket, is Rs6627 ($108) a day. Indian manufacturers offered to supply generic versions for $4 a day, but, under pressure from the U.S., the Indian Department of Industrial Policy and Promotion refused to issue a compulsory license.[20]
Bristol-Myers Squibb justified the high prices of cancer drugs with the high R&D costs, but the Union of Affordable Cancer Treatment said that most of the R&D costs came from the U.S. government, including National Institutes of Health funded research and clinical trials, and a 50% tax credit. In England and Wales, the National Institute for Health and Care Excellence recommended against dasatinib because of the high cost-benefit ratio.[20]
The Union for Affordable Cancer Treatment said that “the dasatinib dispute illustrates the shortcomings of US trade policy and its impact on cancer patients”[20]
Brand names
In Bangladesh dasatinib is available under the trade name Dasanix by Beacon Pharmaceuticals.In India, It is marketed by brand name NEXTKI by EMCURE PHARMACEUTICALS[medical citation needed]
Research
Dasatinib has been shown to eliminate senescent cells in cultured adipocyte progenitor cells.[21] Dasatinib has been shown to induce apoptosis in senescent cells by inhibiting Src kinase, whereas quercetin inhibits the anti-apoptotic protein Bcl-xL.[21] Administration of dasatinib along with quercetin to mice improved cardiovascular function and eliminated senescent cells.[22] Aged mice given dasatinib with quercetin showed improved health and survival.[22]
Giving dasatinib and quercetin to mice eliminated senescent cells and caused a long-term resolution of frailty.[23] A study of fourteen human patients suffering from idiopathic pulmonary fibrosis (a disease characterized by increased numbers of senescent cells) given dasatinib and quercetin showed improved physical function and evidence of reduced senescent cells.[21]Route 1
Reference:1. WO2005077945A2 / US2012302750A1.Route 2
Reference:1. WO0062778A1 / US6596746B1.Route 3
Reference:1. J. Med. Chem. 2004, 47, 6658-6661.
2. J. Med. Chem. 2006, 49, 6819-6832.Route 4
Reference:1. CN104292223A.Route 5
Reference:1. CN103420999A.
Syn 1

Reference
Balaji, N.; Sultana, Sayeeda. Trace level determination and quantification of potential genotoxic impurities in dasatinib drug substance by UHPLC/infinity LC. International Journal of Pharmacy and Pharmaceutical Sciences. Department of Chemistry. St. Peter’s University. Tamil Nadu, India 600054. Volume 8. Issue 10. Pages 209-216. 2016
SYN 2

Reference
Zhang, Shaoning; Wei, Hongtao; Ji, Min. Synthesis of dasatinib. Zhongguo Yiyao Gongye Zazhi. Dept. of Pharmaceutical Engineering, School of Chemistry & Chemical Engineering. Southeast University. Nanjing, Jiangsu Province, Peop. Rep. China 210096. Volume 41. Issue 3. Pages 161-163. 2010
SYN 3

Reference
Suresh, Garbapu; Nadh, Ratnakaram Venkata; Srinivasu, Navuluri; Yennity, Durgaprasad. A convenient new and efficient commercial synthetic route for dasatinib (Sprycel). Synthetic Communications. Division of Chemistry, Department of Science and Humanities. Vignan’s Foundation for Science Technology and Research University. Guntur, India. Volume 47. Issue 17. Pages 1610-1621. 2017
SYN 4

Reference
Chen, Bang-Chi; Zhao, Rulin; Wang, Bei; Droghini, Roberto; Lajeunesse, Jean; Sirard, Pierre; Endo, Masaki; Balasubramanian, Balu; Barrish, Joel C. A new and efficient preparation of 2-aminothiazole-5-carbamides: applications to the synthesis of the anticancer drug dasatinib. ARKIVOC (Gainesville, FL, United States). Discovery Chemistry. Bristol-Myers Squibb Research and Development. Princeton, USA 08543. Issue 6.Pages 32-38. 2010
SYN 5

Reference
An, Kang; Guan, Jianning; Yang, Hao; Hou, Wen; Wan, Rong. Improvement on the synthesis of Dasatinib. Jingxi Huagong Zhongjianti. College of Science. Nanjing University of Technology. Nanjing, Jiangsu Province, Peop. Rep. China 211816. Volume 41. Issue 2. Pages 42-44. 2011
PATENT
https://patents.google.com/patent/US7491725B2/en
EXAMPLESExample 1Preparation of Intermediate:
(S)-1-sec-Butylthiourea

To a solution of S— sec-butyl-amine (7.31 g, 0.1 mol) in chloroform (80 mL) at 0° C. was slowly added benzoyl isothiocyanate (13.44 mL, 0.1 mol). The mixture was allowed to warm to 10° C. and stirred for 10 min. The solvent was then removed under reduced pressure, and the residue was dissolved in MeOH (80 mL). An aqueous solution (10 mL) of NaOH (4 g, 0.1 mol) was added to this solution, and the mixture was stirred at 60° C. for another 2 h. The MeOH was then removed under reduced pressure, and the residue was stirred in water (50 mL). The precipitate was collected by vacuum filtration and dried to provide S-1-sec-butyl-thiourea (12.2 g, 92% yield). mp 133-134° C.; 1H NMR (500 MHz, DMSO-D6) δ 7.40 (s, 1H), 7.20 (br s, 1H), 6.76 (s, 1H), 4.04 (s, 1H), 1.41 (m, 2H), 1.03 (d, J=6.1 Hz, 3H), 0.81 (d, J=7.7 Hz, 3H); 13C NMR (125 MHz, DMSO-D6) δ 182.5, 50.8, 28.8, 19.9, 10.3; LRMS m/z 133.2 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.49; H, 8.88; N, 21.32; S, 24.27.
Example 2Preparation of Intermediate:
(R)-1-sec-Butylthiourea

(R)-1-sec-Butylthiourea was prepared in 92% yield according to the general method outlined for Example 1. mp 133-134° C.; 1H NMR(500 MHz, DMSO) δ 0.80(m, 3H, J=7.7), 1.02(d, 3H, J=6.1), 1.41(m, 2H), (3.40, 4.04)(s, 1H), 6.76(s, 1H), 7.20(s, br, 1H), 7.39(d, 1H, J=7.2); 13C NMR (500 MHz, DMSO) δ: 10.00, 19.56, 28.50, 50.20, 182.00; m/z 133.23 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.32; H, 9.15; N, 21.14; S, 24.38.
Example 3Preparation of:

To a solution of 3-amino-N-methyl-4-methylbenzamide hydrochloride (1.0 g, 5 mmol) in acetone (10 mL) at 0° C. was added pyridine (1.2 mL, 15 mmol) dropwise via syringe. 3-Methoxyacryloyl chloride (0.72 mL. 6.5 mmol) was added and the reaction stirred at room temperature for 1 h. The solution was cooled again to 0° C. and 1N HCl (1.5 mL) was added dropwise via pipet. The reaction mixture was stirred for 5 min, then water (8.5 mL) was added via an addition funnel. The acetone was removed in vacuo and the resulting solution stirred for 4h. Crystallization began within 15 min. After stirring for 4 h, the vessel was cooled in an ice bath for 30 min, filtered, and rinsed with ice cold water (2×3 mL) to give compound 3A (0.99 g, 78% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.12 (br s, 1H), 7.76 (s, 1H), 7.29 (m, 2H), 7.05 (d, J=7.9 Hz, 1H), 5.47 (d, J=12.3 Hz, 1H), 3.48 (s, 3H), 2.54 (d, J=4.7 Hz, 3H), 2.03 (s, 3H); HPLC rt 2.28 min (Condition A).
3B. Example 3To a 50 mL RBF containing the above compound 3A (0.5 g, 2.0 mmol) was added THF (2.5 mL) and water (2 mL), followed by NBS (0.40 g, 2.22 mmol), and the solution was stirred for 90 min. R-sec-butylthiourea (Ex. 2) (267 mg), was added, and the solution was heated to 75° C. for 8 h. Conc. NH4OH was added to adjust the pH to 10 followed by the addition of EtOH (15 mL). Water (15 mL) was added and the slurry stirred for 16 h, filtered, and washed with water to give Example 3 as a light brown solid (0.48 g, 69% yield, 98% purity). MS 347.1; HPLC 2.59.
Example 4Preparation of:

Example 4 is prepared following the methods of Example 3 but using the appropriate acryl benzamide and Example 1.
Example 5Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (The Compound of Formula (IV))

5A. 1-(6-Chloro-2-methylpyrimidin-4-yl)thiourea

To a stirring slurry of 4-amino-5-chloro-2-methylpyrimidine (6.13 g, 42.7 mmol) in THF (24 mL) was added ethyl isothiocyanatoformate (7.5 mL, 63.6 mmol), and the mixture heated to reflux. After 5h, another portion of ethyl isothiocyanato formate (1.0 mL, 8.5 mmol) was added and after 10h, a final portion (1.5 mL, 12.7 mmol) was added and the mixture stirred 6h more. The slurry was evaporated under vacuum to remove most of the solvent and heptane (6 mL) added to the residue. The solid was collected by vacuum filtration and washed with heptane (2×5 mL) giving 8.01 g (68% yield) of the intermediate ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate.A solution of ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate (275 mg, 1.0 mmol) and 1N sodium hydroxide (3.5 eq) was heated and stirred at 50° C. for 2h. The resulting slurry was cooled to 20-22° C. The solid was collected by vacuum filtration, washed with water, and dried to give 185 mg of 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea (91% yield). 1H NMR (400 MHz, DMSO-d6): δ2.51 (S, 3H), 7.05 (s, 1H), 9.35 (s,1H), 10.07 (s, 1H), 10.91 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 25.25, 104.56, 159.19, 159.33, 167.36, 180.91.
5B. (E)-N-(2-Chloro-6-methylphenyl)-3-ethoxyacrylamide

To a cold stirring solution of 2-chloro-6-methylaniline (59.5 g 0.42 mol) and pyridine (68 ml, 0.63 mol) in THF (600 mL) was added 3-ethoxyacryloyl chloride (84.7 g, 0.63 mol) slowly keeping the temp at 0-5° C. The mixture was then warmed and stirred for 2 h. at 20° C. Hydrochloric acid (1N, 115 mL) was added at 0-10° C. The mixture was diluted with water (310 mL) and the resulting solution was concentrated under vacuum to a thick slurry. The slurry was diluted with toluene (275 mL) and stirred for 15 min. at 20-22° C. then 1 h. at 0° C. The solid was collected by vacuum filtration, washed with water (2×75 mL) and dried to give 74.1 g (73.6% yield) of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide). 1H NMR (400 Hz, DMSO-d6) δ 1.26 (t, 3H, J=7 Hz), 2.15 (s, 3H), 3.94 (q, 2H, J=7 Hz), 5.58 (d, 1H, J=12.4 Hz), 7.10-7.27 (m, 2H, J=7.5 Hz), 7.27-7.37 (d, 1H, J=7.5 Hz), 7.45(d, 1H, J=12.4 Hz), 9.28 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 14.57, 18.96, 67.17, 97.99, 126.80, 127.44, 129.07, 131.32, 132.89, 138.25, 161.09, 165.36.
5C. 2-Amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide

To a mixture of compound 5B (5.00 g, 20.86 mmol) in 1,4-dioxane (27 mL) and water (27 mL) was added NBS (4.08 g, 22.9 mmol) at −10 to 0° C. The slurry was warmed and stirred at 20-22° C. for 3h. Thiourea (1.60 g, 21 mmol) was added and the mixture heated to 80° C. After 2h, the resulting solution was cooled to 20-22° and conc. ammonium hydroxide (4.2 mL) was added dropwise. The resulting slurry was concentrated under vacuum to about half volume and cooled to 0-5° C. The solid was collected by vacuum filtration, washed with cold water (10 mL), and dried to give 5.3 g (94.9% yield) of 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6) δ δ 2.19 (s, 3H), 7.09-7.29 (m, 2H, J=7.5), 7.29-7.43 (d, 1H, J=7.5), 7.61 (s, 2H), 7.85 (s, 1H), 9.63 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 18.18, 120.63, 126.84, 127.90, 128.86, 132.41, 133.63, 138.76, 142.88, 159.45, 172.02.
5D. 2-(6-Chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide

To a stirring solution of compound 5C (5.00 g, 18.67 mmol) and 4,6-dichloro-2-methylpyrimidine (3.65 g 22.4/mmol) in THF (65 mL) was added a 30% wt. solution of sodium t-butoxide in THF (21.1 g, 65.36 mmol) slowly with cooling to keep the temperature at 10-20° C. The mixture was stirred at room temperature for 1.5 h and cooled to 0-5° C. Hydrochloric acid, 2N (21.5 mL) was added slowly and the mixture stirred 1.75 h at 0-5° C. The solid was collected by vacuum filtration, washed with water (15 mL) and dried to give 6.63 g (86.4% yield) of compound 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).
5E. Example 5To a mixture of compound 5D (4.00 g, 10.14 mmol) and hydroxyethylpiperazine (6.60 g, 50.69 mmol) in n-butanol (40 mL) was added DIPEA (3.53 mL, 20.26 mmol). The slurry was heated at 118° C. for 4.5 h, then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with n-butanol (5 mL), and dried. The product (5.11 g) was dissolved in hot 80% EtOH—H2O (80 mL), and the solution was clarified by filtration. The hot solution was slowly diluted with water (15 mL) and cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with 50% ethanol-water (5 mL) and dried affording 4.27 g (83.2% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide as monohydrate. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.40 (s, 3H), 2.42 (t, 2H, J=6), 2.48 (t, 4H, J=6.3), 3.50 (m, 4H), 3.53 (q, 2H, J=6), 4.45 (t, 1H, J=5.3), 6.04 (s, 1H), 7.25 (t, 1H, J=7.6), 7.27 (dd, 1H, J=7.6, 1.7), 7.40 (dd, 1H, J=7.6, 1.7), 8.21 (s, 1H), 9.87 (s, 1H), 11.47.
Example 6Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide

To a slurry of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide 5B (120 mg, 0.50 mmol) in THF (0.75 ml) and water (0.5 mL) was added NBS (98 mg, 0.55 mmol) at 0° C. The mixture was warmed and stirred at 20-22° C. for 3h. To this was added 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea 5A (100 mg, 0.49 mmol), and the slurry heated and stirred at reflux for 2h. The slurry was cooled to 20-22° C. and the solid collected by vacuum filtration giving 140 mg (71% yield) of 2-(6-chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).Compound 5D was elaborated to N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide, following Step 5E.
Example 7Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide7A. 2-[4-(6-Chloro-2-methyl-pyrimidin-4-yl)-piperazin-1-yl]-ethanol
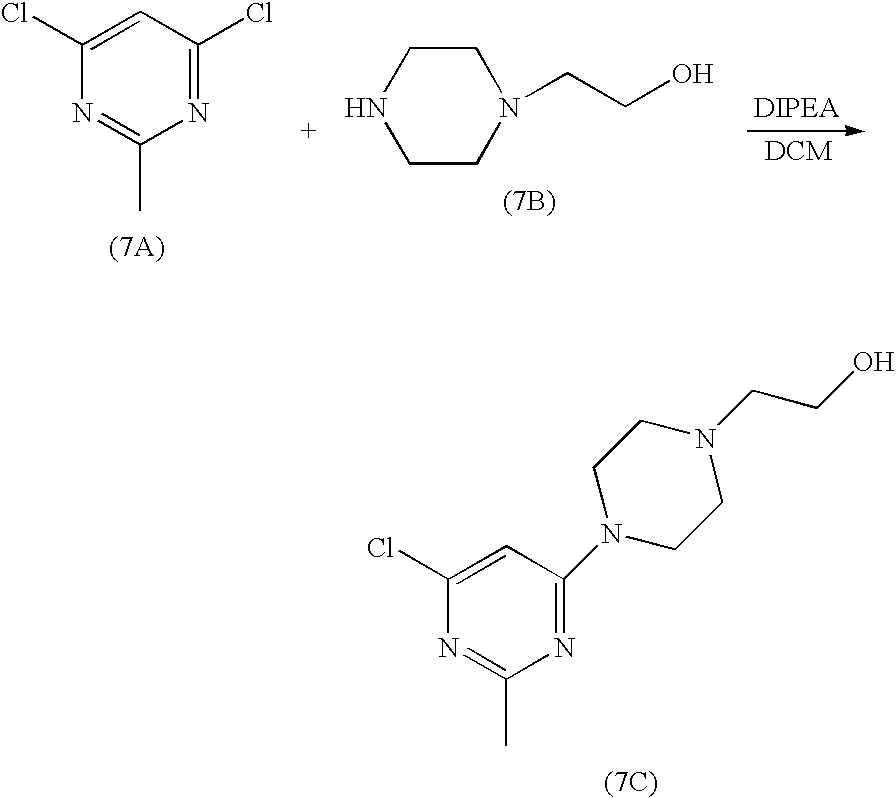
2-piperazin-1-yl-ethanol (8.2 g, 63.1 mmol) was added to a solution of 4,6-dichloro-2-methylpyrimidine (5.2 g, 31.9 mmol) in dichloromethane (80 ml) at rt. The mixture was stirred for two hours and triethylamine (0.9 ml) was added. The mixture was stirred at rt for 20h. The resultant solid was filtered. The cake was washed with dichloromethane (20 ml). The filtrate was concentrated to give an oil. This oil was dried under high vacuum for 20h to give a solid. This solid was stirred with heptane (50 ml) at rt for 5h. Filtration gave 7C (8.13 g) as a white solid
7B. Example 7

To a 250 ml of round bottom flask were charged compound 5C (1.9 g, 7.1 mmol), compound 7C (1.5 g, 5.9 mmol), K2CO3 (16 g, 115.7 mmol), Pd (OAc)2 (52 mg, 0.23 mmol) and BINAP (291 mg, 0.46 mmol). The flask was placed under vacuum and flushed with nitrogen. Toluene was added (60 ml). The suspension was heated to 100-110° C. and stirred at this temperature for 20h. After cooling to room temperature, the mixture was applied to a silica gel column. The column was first eluted with EtOAC, and then with 10% of MeOH in EtOAC. Finally, the column was washed with 10% 2M ammonia solution in MeOH/90% EtOAC. The fractions which contained the desired product were collected and concentrated to give compound IV as a yellow solid (2.3 g).
Analytical MethodsSolid State Nuclear Magnetic Resonance (SSNMR)All solid-state C-13 NMR measurements were made with a Bruker DSX-400, 400 MHz NMR spectrometer. High resolution spectra were obtained using high-power proton decoupling and the TPPM pulse sequence and ramp amplitude cross-polarization (RAMP-CP) with magic-angle spinning (MAS) at approximately 12 kHz (A. E. Bennett et al, J. Chem. Phys., 1995, 103, 6951), (G. Metz, X. Wu and S. O. Smith, J. Magn. Reson. A., 1994, 110, 219-227). Approximately 70 mg of sample, packed into a canister-design zirconia rotor was used for each experiment. Chemical shifts (δ) were referenced to external adamantane with the high frequency resonance being set to 38.56 ppm (W. L. Earl and D. L. VanderHart, J. Magn. Reson., 1982, 48, 35-54).X-Ray Powder DiffractionOne of ordinary skill in the art will appreciate that an X-ray diffraction pattern may be obtained with a measurement error that is dependent upon the measurement conditions employed. In particular, it is generally known that intensities in a X-ray diffraction pattern may fluctuate depending upon measurement conditions employed. It should be further understood that relative intensities may also vary depending upon experimental conditions and, accordingly, the exact order of intensity should not be taken into account. Additionally, a measurement error of diffraction angle for a conventional X-ray diffraction pattern is typically about 5% or less, and such degree of measurement error should be taken into account as pertaining to the aforementioned diffraction angles. Consequently, it is to be understood that the crystal forms of the instant invention are not limited to the crystal forms that provide X-ray diffraction patterns completely identical to the X-ray diffraction patterns depicted in the accompanying Figures disclosed herein. Any crystal forms that provide X-ray diffraction patterns substantially identical to those disclosed in the accompanying Figures fall within the scope of the present invention. The ability to ascertain substantial identities of X-ray diffraction patterns is within the purview of one of ordinary skill in the art.X-Ray powder diffraction data for the crystalline forms of Compound (IV) were obtained using a Bruker GADDS (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) (General Area Detector Diffraction System) manual chi platform goniometer. Powder samples were placed in thin walled glass capillaries of 1 mm or less in diameter; the capillary was rotated during data collection. The sample-detector distance was 17 cm. The radiation was Cu Kα (45 kV 111 mA, λ=1.5418 Å). Data were collected for 3<2θ<35° with a sample exposure time of at least 300 seconds.Single Crystal X-RayAll single crystal data were collected on a Bruker-Nonius (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) Kappa CCD 2000 system using Cu Kα radiation (λ=1.5418 Å) and were corrected only for the Lorentz-polarization factors. Indexing and processing of the measured intensity data were carried out with the HKL2000 software package (Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W. C. Jr & Sweet, R. M. (Academic, NY), Vol. 276, pp. 307-326) in the Collect program suite (Data collection and processing user interface: Collect: Data collection software, R. Hooft, Nonius B. V., 1998).The structures were solved by direct methods and refined on the basis of observed reflections using either the SDP (SDP, Structure Determination Package, Enraf-Nonius, Bohemia NY 11716 Scattering factors, including f′ and f″, in the SDP software were taken from the “International Tables for Crystallography”, Kynoch Press, Birmingham, England, 1974; Vol IV, Tables 2.2A and 2.3.1) software package with minor local modifications or the crystallographic package, MAXUS (maXus solution and refinement software suite: S. Mackay, C. J. Gilmore, C. Edwards, M. Tremayne, N. Stewart, K. Shankland. maXus: a computer program for the solution and refinement of crystal structures from diffraction data).The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were varied.The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were variedDifferential Scanning CalorimetryThe DSC instrument used to test the crystalline forms was a TA Instruments® model Q1000. The DSC cell/sample chamber was purged with 100 ml/min of ultra-high purity nitrogen gas. The instrument was calibrated with high purity indium. The accuracy of the measured sample temperature with this method is within about +/−1° C., and the heat of fusion can be measured within a relative error of about +/−5%. The sample was placed into an open aluminum DSC pan and measured against an empty reference pan. At least 2 mg of sample powder was placed into the bottom of the pan and lightly tapped down to ensure good contact with the pan. The weight of the sample was measured accurately and recorded to a hundredth of a milligram. The instrument was programmed to heat at 10° C. per minute in the temperature range between 25 and 350° C.The heat flow, which was normalized by a sample weight, was plotted versus the measured sample temperature. The data were reported in units of watts/gram (“W/g”). The plot was made with the endothermic peaks pointing down. The endothermic melt peak was evaluated for extrapolated onset temperature, peak temperature, and heat of fusion in this analysis.Thermogravimetric Analysis (TGA)The TGA instrument used to test the crystalline forms was a TAInstruments® model Q500. Samples of at least 10 milligrams were analyzed at a heating rate of 10° C. per minute in the temperature range between 25° C. and about 350° C.
Example 8Preparation of:
crystalline monohydrate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)An example of the crystallization procedure to obtain the crystalline monohydrate form is shown here:
- Charge 48 g of the compound of formula (IV).
- Charge approximately 1056 mL (22 mL/g) of ethyl alcohol, or other suitable alcohol.
- Charge approximately 144 mL of water.
- Dissolve the suspension by heating to approximately 75° C.
- Optional: Polish filter by transfer the compound of formula (IV) solution at 75° C. through the preheated filter and into the receiver.
- Rinse the dissolution reactor and transfer lines with a mixture of 43 mL of ethanol and 5 mL of water.
Heat the contents in the receiver to 75-80° C. and maintain 75-80° C. to achieve complete dissolution.Charge approximately 384 mL of water at a rate such that the batch temperature is maintained between 75-80° C.Cool to 75° C., and, optionally, charge monohydrate seed crystals. Seed crystals are not essential to obtaining monohydrate, but provide better control of the crystallization.
- Cool to 70° C. and maintain 70° C. for ca. 1 h.
- Cool from 70 to 5 C over 2 h, and maintain the temperature between 0 at 5° C. for at least 2 h.
- Filter the crystal slurry.
- Wash the filter cake with a mixture of 96 mL of ethanol and 96 mL of water.
- Dry the material at ≦50° C. under reduced pressure until the water content is 3.4 to 4.1% by KF to afford 41 g (85 M %).
Alternately, the monohydrate can be obtained by:- 1) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate and heated at 80° C. to give bulk monohydrate.
- 2) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate. On standing several days at room temperature, bulk monohydrate had formed.
- 3) An aqueous suspension of compound IV was seeded with monohydrate and heated at 70° C. for 4 hours to give bulk monohydrate. In the absence of seeding, an aqueous slurry of compound IV was unchanged after 82 days at room temperature.
- 4) A solution of compound IV in a solvent such as NMP or DMA was treated with water until the solution became cloudy and was held at 75-85° C. for several hours. Monohydrate was isolated after cooling and filtering.
- 5) A solution of compound IV in ethanol, butanol, and water was heated. Seeds of monohydrate were added to the hot solution and then cooled. Monohydrate was isolated upon cooling and filtration.
One of ordinary skill in the art will appreciate that the monohydrate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 1 or by a representative sampling of peaks as shown in Table 1.Representative peaks taken from the XRPD of the monohydrate of the compound of formula (IV) are shown in Table 1.TABLE 1 2-Theta d(Å) Height 17.994 4.9257 915 18.440 4.8075 338 19.153 4.6301 644 19.599 4.5258 361 21.252 4.1774 148 24.462 3.6359 250 25.901 3.4371 133 28.052 3.1782 153The XRPD is also characterized by the following list comprising 2θ values selected from the group consisting of: 4.6±0.2, 11.2±0.2, 13.8±0.2, 15.2±0.2, 17.9±0.2, 19.1±0.2, 19.6±0.2, 23.2±0.2, 23.6±0.2. The XRPD is also characterized by the list of 2θ values selected from the group consisting of: 18.0±0.2, 18.4±0.2, 19.2±0.2, 19.6±0.2, 21.2±0.2, 24.5±0.2, 25.9±0.2, and 28.0±0.2.Single crystal x-ray data was obtained at room temperature (+25° C.). The molecular structure was confirmed as a monohydrate form of the compound of Formula (IV).The following unit cell parameters were obtained for the monohydrate of the compound of formula (IV) from the x-ray analysis at 25° C.:a(Å)=13.8632(7); b(Å)=9.3307(3); c(Å)=38.390(2);V(Å3) 4965.9(4); Z′=1; Vm=621Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.354Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).Single crystal x-ray data was also obtained at −50° C. The monohydrate form of the compound of Formula (IV) is characterized by unit cell parameters approximately equal to the following:Cell dimensions:
- a(Å)=13.862(1);
- b(Å)=9.286(1);
- c(Å)=38.143(2);
Volume=4910(1) Å3Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.369wherein the compound is at a temperature of about −50° C.The simulated XRPD was calculated from the refined atomic parameters at room temperature.The monohydrate of the compound of formula (IV) is represented by the DSC as shown in FIG. 2. The DSC is characterized by a broad peak between approximately 95° C. and 130° C. This peak is broad and variable and corresponds to the loss of one water of hydration as seen in the TGA graph. The DSC also has a characteristic peak at approximately 287° C. which corresponds to the melt of the dehydrated form of the compound of formula (IV).The TGA for the monohydrate of the compound of Formula (IV) is shown in FIG. 2 along with the DSC. The TGA shows a 3.48% weight loss from 50° C. to 175° C. The weight loss corresponds to a loss of one water of hydration from the compound of Formula (IV).The monohydrate may also be prepared by crystallizing from alcoholic solvents, such as methanol, ethanol, propanol, i-propanol, butanol, pentanol, and water.
Example 9Preparation of:
crystalline n-butanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)The crystalline butanol solvate of the compound of formula (IV) is prepared by dissolving compound (IV) in 1-butanol at reflux (116-118° C.) at a concentration of approximately 1 g/25 mL of solvent. Upon cooling, the butanol solvate crystallizes out of solution. Filter, wash with butanol, and dry.The following unit cell parameters were obtained from the x-ray analysis for the crystalline butanol solvate, obtained at room temperature:a(Å)=22.8102(6); b(Å)=8.4691(3); c(Å)=15.1436(5); β=95.794(2);V(Å3) 2910.5(2); Z′=1; Vm=728Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.283Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the butanol solvate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 3 or by a representative sampling of peaks. Representative peaks for the crystalline butanol solvate are 2θ values of: 5.9±0.2, 12.0±0.2, 13.0±0.2, 17.7±0.2, 24.1±0.2, and 24.6±0.2.
Example 10Preparation of:
crystalline ethanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)

To a 100-mL round bottom flask was charged 4.00 g (10.1 mmol) of 5D (contained 2.3 Area % 5C) 6.60 g (50.7 mmol) of 7B, 80 mL of n-butanol and 2.61 g (20.2 mmol) of DIPEA. The resulting slurry was heated to 120° C. and maintained at 120° C. for 4.5 h whereby HPLC analysis showed 0.19 relative Area % of residual 5D to compound IV. The homogeneous mixture was cooled to 20° C. and left stirring overnight. The resulting crystals were filtered. The wet cake was washed twice with 10-mL portions of n-butanol to afford a white crystalline product. HPLC analysis showed this material to contain 99.7 Area % compound IV and 0.3 Area % 5C.The resulting wet cake was returned to the 100-mL reactor, and charged with 56 mL (12 mL/g) of 200 proof ethanol. At 80° C. an additional 25 mL of ethanol was added. To this mixture was added 10 mL of water resulting in rapid dissolution. Heat was removed and crystallization was observed at 75-77° C. The crystal slurry was further cooled to 20° C. and filtered. The wet cake was washed once with 10 mL of 1:1 ethanol: water and once with 10 mL of n-heptane. The wet cake contained 1.0% water by KF and 8.10% volatiles by LOD. The material was dried at 60° C./30 in Hg for 17 h to afford 3.55 g (70 M %) of material containing only 0.19% water by KF, 99.87 Area % by HPLC. The 1H NMR spectrum, however revealed that the ethanol solvate had been formed.The following unit cell parameters were obtained from the x-ray analysis for the crystalline ethanol solvate (di-ethanolate, E2-1), obtained at −40° C.:a(Å)=22.076(1); b(Å)=8.9612(2); c(Å)=16.8764(3); β=114.783(1);V(Å3) 3031.1(1); Z′=1; Vm=758Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.271Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 4 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 5.8±0.2, 11.3±0.2, 15.8±0.2, 17.2±0.2, 19.5±0.2, 24.1±0.2, 25.3±0.2, and 26.2±0.2.In addition, during the process to form the ethanolate (diethanolate) the formation of another ethanol solvate (½ ethanolate, T1E2-1) has been observed. To date this additional ethaonol solvate is known strictly as a partial desolvation product of the original diethanolate form E2-1, and has only been observed on occasion during crystallization of E2-1The following unit cell parameters were obtained from the x-ray analysis for the crystalline ½ ethanol solvate T1E2-1, obtained at −10° C.:a(Å)=22.03(2); b(Å)=9.20(1); c(Å)=12.31(1);β=93.49(6)V(Å3) 2491(4)); Z′=1; Vm=623;Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.363Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (T1E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 7 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 7.20±0.2, 12.01±0.2, 12.81±0.2, 18.06±0.2, 19.30±0.2, and 25.24±0.2.
Example 11Preparation of:
crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (Neat form N-6)To a mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL) was added DIPEA (155 mL, 0.89 mol). The suspension was heated at 110° C. (solution obtained) for 25 min., then cooled to about 90° C. The resulting hot solution was added dropwise into hot (80° C.) water (8010) mL, keeping the temperature at about 80° C. The resulting suspension was stirred 15 min at 80° C. then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with water (2×1600 mL) and dried in vacuo at 55-60° C. affording 192.45 g (88.7% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H), 2.41 (s, 3H), 2.43 (t, 2H, J=6), 2.49 (t, 4H, J=6.3), 3.51 (m, 4H), 3.54 (q, 2H, J=6), 4.46 (t, 1H, J=5.3), 6.05 (s, 1H), 7.26 (t, 1H, J=7.6), 7.28 (dd, 1H, J=7.6, 1.7), 7.41 (dd, 1H, J=7.6, 1.7), 8.23 (s, 1H), 9.89 (s, 1H), 11.48. KF0.84; DSC: 285.25° C. (onset), 286.28° C. (max).The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline compound IV, obtained at 23° C.:a(Å)=22.957(1); b(Å)=8.5830(5); c(Å)=13.803(3); β=112.039(6);V(Å3)=2521.0(5); Z′=1; Vm=630Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.286Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the crystalline form of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 5 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (N-6) are 2θ values of: 6.8±0.2, 11.1±0.2, 12.3±0.2, 13.2±0.2, 13.7±0.2, 16.7±0.2, 21.0±0.2, 24.3±0.2, and 24.8±0.2.
Example 12Preparation of:
crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (neatform T1H1-7)The title neat form may be prepared by heating the monohydrate form of the compound of formula (IV) above the dehydration temperature.The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline (T1H1-7) compound IV, obtained at 25° C.:a(Å)=13.4916; b(Å)=9.3992(2); c(Å)=38.817(1);V(Å3)=4922.4(3); Z′=1; Vm=615Space group PbcaDensity (calculated) (g/cm3) 1.317Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the neat crystalline form (T1H1-7) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 6 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (T1H1-7)) are 2θ values of: 8.0±0.2, 9.7±0.2, 11.2±0.2, 13.3±0.2, 17.5±0.2, 18.9±0.2, 21.0±0.2, 22.0±0.2.Obviously, numerous modifications and variations of the present invention are possible in light of the above teachings. It is therefore to be understood that within the scope of the appended claims, the invention may be practiced otherwise than as specifically described herein.PATENThttps://patents.google.com/patent/US8680103B2/enAminothiazole-aromatic amides of formula I

wherein Ar is aryl or heteroaryl, L is an optional alkylene linker, and R2, R3, R4, and R5, are as defined in the specification herein, are useful as kinase inhibitors, in particular, inhibitors of protein tyrosine kinase and p38 kinase. They are expected to be useful in the treatment of protein tyrosine kinase-associated disorders such as immunologic and oncological disorders [see, U.S. Pat. No. 6,596,746 (the ‘746 patent), assigned to the present assignee and incorporated herein by reference], and p38 kinase-associated conditions such as inflammatory and immune conditions, as described in U.S. patent application Ser. No. 10/773,790, filed Feb. 6, 2004, claiming priority to U.S. Provisional application Ser. No. 60/445,410, filed Feb. 6, 2003 (hereinafter the ‘410 application), both of which are also assigned to the present assignee and incorporated herein by reference.The compound of formula (IV), ′N-(2-Chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, is an inhibitor of SRC/ABL and is useful in the treatment of oncological diseases.

Other approaches to preparing 2-aminothiazole-5-carboxamides are described in the ‘746 patent and in the ‘410 application. The ‘746 patent describes a process involving treatment of chlorothiazole with n-BuLi followed by reaction with phenyl isocyanates to give chlorothiazole-benzamides, which are further elaborated to aminothiazole-benzamide final products after protection, chloro-to-amino substitution, and deprotection, e.g.,

The ‘410 application describes a multi-step process involving first, converting N-unsubstituted aminothiazole carboxylic acid methyl or ethyl esters to bromothiazole carboxylic acid esters via diazotization with tert-butyl nitrite and subsequent CuBr2 treatment, e.g.,

then, hydrolyzing the resulting bromothiazole esters to the corresponding carboxylic acids and converting the acids to the corresponding acyl chlorides, e.g.,

then finally, coupling the acyl chlorides with anilines to afford bromothiazole-benzamide intermediates which were further elaborated to aminothiazole-benzamide final products, e.g.,

Other approaches for making 2-aminothiazole-5-carboxamides include coupling of 2-aminothiazole-5-carboxylic acids with amines using various coupling conditions such as DCC [Roberts et al, J. Med. Chem. (1972), 15, at p. 1310], and DPPA [Marsham et al., J. Med. Chem. (1991), 34, at p. 1594)].The above methods present drawbacks with respect to the production of side products, the use of expensive coupling reagents, less than desirable yields, and the need for multiple reaction steps to achieve the 2-aminothiazole-5-carboxamide compounds.Reaction of N,N-dimethyl-N′-(aminothiocarbonyl)-formamidines with α-haloketones and esters to give 5-carbonyl-2-aminothiazoles has been reported. See Lin, Y. et al, J. Heterocycl. Chem. (1979), 16, at 1377; Hartmann, H. et al, J. Chem. Soc. Perkin Trans. (2000), 1, at 4316; Noack, A. et al; Tetrahedron (2002), 58, at 2137; Noack, A.; et al. Angew. Chem. (2001), 113, at 3097; and Kantlehner, W. et al., J. Prakt. Chem./Chem.-Ztg. (1996), 338, at 403. Reaction of β-ethoxy acrylates and thioureas to prepare 2-aminothiazole-5-carboxylates also has been reported. See Zhao, R., et al., Tetrahedron Lett. (2001), 42, at 2101. However, electrophilic bromination of acrylanilide and crotonanilide has been known to undergo both aromatic bromination and addition to the α,β-unsaturated carbon-carbon double bonds. See Autenrieth, Chem. Ber. (1905), 38, at 2550; Eremeev et al., Chem. Heterocycl. Compd. Engl. Transl. (1984), 20, at 1102.New and efficient processes for preparing 2-aminothiazole-5-carboxamides are desired.
SUMMARY OF THE INVENTION
This invention is related to processes for the preparation of 2-aminothiazole-5-aromatic amides having the formula (I),

wherein L, Ar, R2, R3, R4, R5, and m are as defined below, comprising reacting a compound having the formula (II),

wherein Q is the group —O—P*, wherein P* is selected so that, when considered together with the oxygen atom to which P* is attached, Q is a leaving group, and Ar, L, R2, R3, and m are as defined below,
with a halogenating reagent in the presence of water followed by a thiourea compound having the formula (III),

wherein, R4 and R5 are as defined below,
to provide the compound of formula (I),

wherein,Ar is the same in formulae (I) and (II) and is aryl or heteroaryl;L is the same in formulae (I) and (II) and is optionally-substituted alkylene;R2 is the same in formulae (I) and (II), and is selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo;R3 is the same in formulae (I) and (II), and is selected from hydrogen, halogen, cyano, haloalkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, aryl, heteroaryl, cycloalkyl, and heterocyclo;R4 is (i) the same in each of formulae (I) and (III), and (ii) is independently selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo, or alternatively, R4 is taken together with R5, to form heteroaryl or heterocyclo;R5 is (i) the same in each of formulae (I) and (III), and (ii) is independently selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo, or alternatively, R5 is taken together with R4, to form heteroaryl or heterocyclo; andm is 0 or 1.Applicants have surprisingly discovered said process for converting β-(P*)oxy acryl aromatic amides and thioureas to 2-aminothiazole derivatives, wherein the aromatic amides are not subject to further halogenation producing other side products. Aminothiazole-aromatic amides, particularly, 2-aminothiazole-5-benzamides, can thus be efficiently prepared with this process in high yield.In another aspect, the present invention is directed to crystalline forms of the compound of formula (IV).
EXAMPLESExample 1Preparation of Intermediate:
(S)-1-sec-Butylthiourea

To a solution of S-sec-butyl-amine (7.31 g, 0.1 mol) in chloroform (80 mL) at 0° C. was slowly added benzoyl isothiocyanate (13.44 mL, 0.1 mol). The mixture was allowed to warm to 10° C. and stirred for 10 min. The solvent was then removed under reduced pressure, and the residue was dissolved in MeOH (80 mL). An aqueous solution (10 mL) of NaOH (4 g, 0.1 mol) was added to this solution, and the mixture was stirred at 60° C. for another 2 h. The MeOH was then removed under reduced pressure, and the residue was stirred in water (50 mL). The precipitate was collected by vacuum filtration and dried to provide S-1-sec-butyl-thiourea (12.2 g, 92% yield). mp 133-134° C.; 1H NMR (500 MHz, DMSO-D6) δ 7.40 (s, 1H), 7.20 (br s, 1H), 6.76 (s, 1H), 4.04 (s, 1H), 1.41 (m, 2H), 1.03 (d, J=6.1 Hz, 3H), 0.81 (d, J=7.7 Hz, 3H); 13C NMR (125 MHz, DMSO-D6) δ 182.5, 50.8, 28.8, 19.9, 10.3; LRMS m/z 133.2 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.49; H, 8.88; N, 21.32; S, 24.27.
Example 2Preparation of Intermediate:
(R)-1-sec-Butylthiourea

(R)-1-sec-Butylthiourea was prepared in 92% yield according to the general method outlined for Example 1. mp 133-134° C.; 1H NMR (500 MHz, DMSO) δ 0.80 (m, 3H, J=7.7), 1.02 (d, 3H, J=6.1), 1.41 (m, 2H), (3.40, 4.04) (s, 1H), 6.76 (s, 1H), 7.20 (s, br, 1H), 7.39 (d, 1H, J=7.2); 13C NMR (500 MHz, DMSO) δ: 10.00, 19.56, 28.50, 50.20, 182.00; m/z 133.23 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.32; H, 9.15; N, 21.14; S, 24.38.
Example 3Preparation of:

To a solution of 3-amino-N-methyl-4-methylbenzamide hydrochloride (1.0 g, 5 mmol) in acetone (10 mL) at 0° C. was added pyridine (1.2 mL, 15 mmol) dropwise via syringe. 3-Methoxyacryloyl chloride (0.72 mL 6.5 mmol) was added and the reaction stirred at room temperature for 1 h. The solution was cooled again to 0° C. and 1N HCl (1.5 mL) was added dropwise via pipette. The reaction mixture was stirred for 5 min, then water (8.5 mL) was added via an addition funnel. The acetone was removed in vacuo and the resulting solution stirred for 4 h. Crystallization began within 15 min. After stirring for 4 h, the vessel was cooled in an ice bath for 30 min, filtered, and rinsed with ice cold water (2×3 mL) to give compound 3A (0.99 g, 78% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.12 (br s, 1H), 7.76 (s, 1H), 7.29 (m, 2H), 7.05 (d, J=7.9 Hz, 1H), 5.47 (d, J=12.3 Hz, 1H), 3.48 (s, 3H), 2.54 (d, J=4.7 Hz, 3H), 2.03 (s, 3H); HPLC rt 2.28 min (Condition A).
3B. Example 3To a 50 mL RBF containing the above compound 3A (0.5 g, 2.0 mmol) was added THF (2.5 mL) and water (2 mL), followed by NBS (0.40 g, 2.22 mmol), and the solution was stirred for 90 min. R-sec-butylthiourea (Ex. 2) (267 mg), was added, and the solution was heated to 75° C. for 8 h. Conc. NH4OH was added to adjust the pH to 10 followed by the addition of EtOH (15 mL). Water (15 mL) was added and the slurry stirred for 16 h, filtered, and washed with water to give Example 3 as a light brown solid (0.48 g, 69% yield, 98% purity). MS 347.1; HPLC 2.59.
Example 4Preparation of:

Example 4 is prepared following the methods of Example 3 but using the appropriate acryl benzamide and Example 1.
Example 5Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (The compound of Formula (IV))

5A. 1-(6-Chloro-2-methylpyrimidin-4-yl)thiourea

To a stirring slurry of 4-amino-5-chloro-2-methylpyrimidine (6.13 g, 42.7 mmol) in THF (24 mL) was added ethyl isothiocyanatoformate (7.5 mL, 63.6 mmol), and the mixture heated to reflux. After 5 h, another portion of ethyl isothiocyanato formate (1.0 mL, 8.5 mmol) was added and after 10 h, a final portion (1.5 mL, 12.7 mmol) was added and the mixture stirred 6 h more. The slurry was evaporated under vacuum to remove most of the solvent and heptane (6 mL) added to the residue. The solid was collected by vacuum filtration and washed with heptane (2×5 mL) giving 8.01 g (68% yield) of the intermediate ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate.A solution of ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate (275 mg, 1.0 mmol) and 1N sodium hydroxide (3.5 eq) was heated and stirred at 50° C. for 2 h. The resulting slurry was cooled to 20-22° C. The solid was collected by vacuum filtration, washed with water, and dried to give 185 mg of 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea (91% yield). 1H NMR (400 MHz, DMSO-d6): δ2.51 (S, 3H), 7.05 (s, 1H), 9.35 (s, 1H), 10.07 (s, 1H), 10.91 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 25.25, 104.56, 159.19, 159.33, 167.36, 180.91.
5B. (E)-N-(2-Chloro-6-methylphenyl)-3-ethoxyacrylamide

To a cold stirring solution of 2-chloro-6-methylaniline (59.5 g 0.42 mol) and pyridine (68 ml, 0.63 mol) in THF (600 mL) was added 3-ethoxyacryloyl chloride (84.7 g, 0.63 mol) slowly keeping the temp at 0-5° C. The mixture was then warmed and stirred for 2 h. at 20° C. Hydrochloric acid (1N, 115 mL) was added at 0-10° C. The mixture was diluted with water (310 mL) and the resulting solution was concentrated under vacuum to a thick slurry. The slurry was diluted with toluene (275 mL) and stirred for 15 min. at 20-22° C. then 1 h. at 0° C. The solid was collected by vacuum filtration, washed with water (2×75 mL) and dried to give 74.1 g (73.6% yield) of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide). 1H NMR (400 Hz, DMSO-d6) δ 1.26 (t, 3H, J=7 Hz), 2.15 (s, 3H), 3.94 (q, 2H, J=7 Hz), 5.58 (d, 1H, J=12.4 Hz), 7.10-7.27 (m, 2H, J=7.5 Hz), 7.27-7.37 (d, 1H, J=7.5 Hz), 7.45 (d, 1H, J=12.4 Hz), 9.28 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 14.57, 18.96, 67.17, 97.99, 126.80, 127.44, 129.07, 131.32, 132.89, 138.25, 161.09, 165.36.
5C. 2-Amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide

To a mixture of compound 5B (5.00 g, 20.86 mmol) in 1,4-dioxane (27 mL) and water (27 mL) was added NBS (4.08 g, 22.9 mmol) at −10 to 0° C. The slurry was warmed and stirred at 20-22° C. for 3 h. Thiourea (1.60 g, 21 mmol) was added and the mixture heated to 80° C. After 2 h, the resulting solution was cooled to 20-22° and conc. ammonium hydroxide (4.2 mL) was added dropwise. The resulting slurry was concentrated under vacuum to about half volume and cooled to 0-5° C. The solid was collected by vacuum filtration, washed with cold water (10 mL), and dried to give 5.3 g (94.9% yield) of 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6) δ δ 2.19 (s, 3H), 7.09-7.29 (m, 2H, J=7.5), 7.29-7.43 (d, 1H, J=7.5), 7.61 (s, 2H), 7.85 (s, 1H), 9.63 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 18.18, 120.63, 126.84, 127.90, 128.86, 132.41, 133.63, 138.76, 142.88, 159.45, 172.02.
5D. 2-(6-Chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide

To a stirring solution of compound 5C (5.00 g, 18.67 mmol) and 4,6-dichloro-2-methylpyrimidine (3.65 g 22.4/mmol) in THF (65 mL) was added a 30% wt. solution of sodium t-butoxide in THF (21.1 g, 65.36 mmol) slowly with cooling to keep the temperature at 10-20° C. The mixture was stirred at room temperature for 1.5 h and cooled to 0-5° C. Hydrochloric acid, 2N (21.5 mL) was added slowly and the mixture stirred 1.75 h at 0-5° C. The solid was collected by vacuum filtration, washed with water (15 mL) and dried to give 6.63 g (86.4% yield) of compound 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).
5E. Example 5To a mixture of compound 5D (4.00 g, 10.14 mmol) and hydroxyethylpiperazine (6.60 g, 50.69 mmol) in n-butanol (40 mL) was added DIPEA (3.53 mL, 20.26 mmol). The slurry was heated at 118° C. for 4.5 h, then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with n-butanol (5 mL), and dried. The product (5.11 g) was dissolved in hot 80% EtOH—H2O (80 mL), and the solution was clarified by filtration. The hot solution was slowly diluted with water (15 mL) and cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with 50% ethanol-water (5 mL) and dried affording 4.27 g (83.2% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide as monohydrate. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.40 (s, 3H), 2.42 (t, 2H, J=6), 2.48 (t, 4H, J=6.3), 3.50 (m, 4H), 3.53 (q, 2H, J=6), 4.45 (t, 1H, J=5.3), 6.04 (s, 1H), 7.25 (t, 1H, J=7.6), 7.27 (dd, 1H, J=7.6, 1.7), 7.40 (dd, 1H, J=7.6, 1.7), 8.21 (s, 1H), 9.87 (s, 1H), 11.47.
Example 6Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide

To a slurry of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide 5B (120 mg, 0.50 mmol) in THF (0.75 ml) and water (0.5 mL) was added NBS (98 mg, 0.55 mmol) at 0° C. The mixture was warmed and stirred at 20-22° C. for 3 h. To this was added 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea 5A (100 mg, 0.49 mmol), and the slurry heated and stirred at reflux for 2 h. The slurry was cooled to 20-22° C. and the solid collected by vacuum filtration giving 140 mg (71% yield) of 2-(6-chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).Compound 5D was elaborated to N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide, following Step 5E.
Example 7Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide7A. 2-[4-(6-Chloro-2-methyl-pyrimidin-4-yl)-piperazin-1-yl]-ethanol

2-Piperazin-1-yl-ethanol (8.2 g, 63.1 mmol) was added to a solution of 4,6-dichloro-2-methylpyrimidine (5.2 g, 31.9 mmol) in dichloromethane (80 ml) at rt. The mixture was stirred for two hours and triethylamine (0.9 ml) was added. The mixture was stirred at rt for 20 h. The resultant solid was filtered. The cake was washed with dichloromethane (20 ml). The filtrate was concentrated to give an oil. This oil was dried under high vacuum for 20 h to give a solid. This solid was stirred with heptane (50 ml) at rt for 5 h. Filtration gave 7C (8.13 g) as a white solid
7B. Example 7

To a 250 ml of round bottom flask were charged compound 5C (1.9 g, 7.1 mmol), compound 7C (1.5 g, 5.9 mmol), K2CO3 (16 g, 115.7 mmol), Pd (OAc)2 (52 mg, 0.23 mmol) and BINAP (291 mg, 0.46 mmol). The flask was placed under vacuum and flushed with nitrogen. Toluene was added (60 ml). The suspension was heated to 100-110° C. and stirred at this temperature for 20 h. After cooling to room temperature, the mixture was applied to a silica gel column. The column was first eluted with EtOAC, and then with 10% of MeOH in EtOAC. Finally, the column was washed with 10% 2M ammonia solution in MeOH/90% EtOAC. The fractions which contained the desired product were collected and concentrated to give compound IV as a yellow solid (2.3 g).
Analytical MethodsSolid State Nuclear Magnetic Resonance (SSNMR)All solid-state C-13 NMR measurements were made with a Bruker DSX-400, 400 MHz NMR spectrometer. High resolution spectra were obtained using high-power proton decoupling and the TPPM pulse sequence and ramp amplitude cross-polarization (RAMP-CP) with magic-angle spinning (MAS) at approximately 12 kHz (A. E. Bennett et al, J. Chem. Phys., 1995, 103, 6951), (G. Metz, X. Wu and S. O, Smith, J. Magn. Reson. A, 1994, 110, 219-227). Approximately 70 mg of sample, packed into a canister-design zirconia rotor was used for each experiment. Chemical shifts (6) were referenced to external adamantane with the high frequency resonance being set to 38.56 ppm (W. L. Earl and D. L. VanderHart, J. Magn. Reson., 1982, 48, 35-54).X-Ray Powder DiffractionOne of ordinary skill in the art will appreciate that an X-ray diffraction pattern may be obtained with a measurement error that is dependent upon the measurement conditions employed. In particular, it is generally known that intensities in a X-ray diffraction pattern may fluctuate depending upon measurement conditions employed. It should be further understood that relative intensities may also vary depending upon experimental conditions and, accordingly, the exact order of intensity should not be taken into account. Additionally, a measurement error of diffraction angle for a conventional X-ray diffraction pattern is typically about 5% or less, and such degree of measurement error should be taken into account as pertaining to the aforementioned diffraction angles. Consequently, it is to be understood that the crystal forms of the instant invention are not limited to the crystal forms that provide X-ray diffraction patterns completely identical to the X-ray diffraction patterns depicted in the accompanying Figures disclosed herein. Any crystal forms that provide X-ray diffraction patterns substantially identical to those disclosed in the accompanying Figures fall within the scope of the present invention. The ability to ascertain substantial identities of X-ray diffraction patterns is within the purview of one of ordinary skill in the art.X-Ray powder diffraction data for the crystalline forms of Compound (IV) were obtained using a Bruker GADDS (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) (General Area Detector Diffraction System) manual chi platform goniometer. Powder samples were placed in thin walled glass capillaries of 1 mm or less in diameter; the capillary was rotated during data collection. The sample-detector distance was 17 cm. The radiation was Cu Kα (45 kV 111 mA, λ=1.5418 Å). Data were collected for 3<2θ<35° with a sample exposure time of at least 300 seconds.Single Crystal X-RayAll single crystal data were collected on a Bruker-Nonius (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) Kappa CCD 2000 system using Cu Kα radiation (λ=1.5418 Å) and were corrected only for the Lorentz-polarization factors. Indexing and processing of the measured intensity data were carried out with the HKL2000 software package (Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W. C. Jr. & Sweet, R. M. (Academic, NY), Vol. 276, pp. 307-326) in the Collect program suite (Data collection and processing user interface: Collect: Data collection software, R. Hooft, Nonius B. V., 1998).The structures were solved by direct methods and refined on the basis of observed reflections using either the SDP (SDP, Structure Determination Package, Enraf-Nonius, Bohemia N.Y. 11716 Scattering factors, including f′ and f″, in the SDP software were taken from the “International Tables for Crystallography”, Kynoch Press, Birmingham, England, 1974; Vol IV, Tables 2.2A and 2.3.1) software package with minor local modifications or the crystallographic package, MAXUS (maXus solution and refinement software suite: S. Mackay, C. J. Gilmore, C. Edwards, M. Tremayne, N. Stewart, K. Shankland. maXus: a computer program for the solution and refinement of crystal structures from diffraction data).The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were varied.The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were variedDifferential Scanning CalorimetryThe DSC instrument used to test the crystalline forms was a TA INSTRUMENTS° model Q1000. The DSC cell/sample chamber was purged with 100 ml/min of ultra-high purity nitrogen gas. The instrument was calibrated with high purity indium. The accuracy of the measured sample temperature with this method is within about +/−1° C., and the heat of fusion can be measured within a relative error of about +/−5%. The sample was placed into an open aluminum DSC pan and measured against an empty reference pan. At least 2 mg of sample powder was placed into the bottom of the pan and lightly tapped down to ensure good contact with the pan. The weight of the sample was measured accurately and recorded to a hundredth of a milligram. The instrument was programmed to heat at 10° C. per minute in the temperature range between 25 and 350° C.The heat flow, which was normalized by a sample weight, was plotted versus the measured sample temperature. The data were reported in units of watts/gram (“W/g”). The plot was made with the endothermic peaks pointing down. The endothermic melt peak was evaluated for extrapolated onset temperature, peak temperature, and heat of fusion in this analysis.Thermogravimetric Analysis (TGA)The TGA instrument used to test the crystalline forms was a TA INSTRUMENTS® model Q500. Samples of at least 10 milligrams were analyzed at a heating rate of 10° C. per minute in the temperature range between 25° C. and about 350° C.
Example 8Preparation of:
Crystalline monohydrate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)An example of the crystallization procedure to obtain the crystalline monohydrate form is shown here:Charge 48 g of the compound of formula (IV).Charge approximately 1056 mL (22 mL/g) of ethyl alcohol, or other suitable alcohol.Charge approximately 144 mL of water.Dissolve the suspension by heating to approximately 75° C.Optional: Polish filter by transfer the compound of formula (IV) solution at 75° C. through the preheated filter and into the receiver.Rinse the dissolution reactor and transfer lines with a mixture of 43 mL of ethanol and 5 mL of water.Heat the contents in the receiver to 75-80° C. and maintain 75-80° C. to achieve complete dissolution.Charge approximately 384 mL of water at a rate such that the batch temperature is maintained between 75-80° C.Cool to 75° C., and, optionally, charge monohydrate seed crystals. Seed crystals are not essential to obtaining monohydrate, but provide better control of the crystallization.Cool to 70° C. and maintain 70° C. for ca. 1 h.Cool from 70 to 5 C over 2 h, and maintain the temperature between 0 at 5° C. for at least 2 h.Filter the crystal slurry.Wash the filter cake with a mixture of 96 mL of ethanol and 96 mL of water.Dry the material at ≦50° C. under reduced pressure until the water content is 3.4 to 4.1% by KF to afford 41 g (85 M %).Alternately, the monohydrate can be obtained by:1) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate and heated at 80° C. to give bulk monohydrate.2) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate. On standing several days at room temperature, bulk monohydrate had formed.3) An aqueous suspension of compound IV was seeded with monohydrate and heated at 70° C. for 4 hours to give bulk monohydrate. In the absence of seeding, an aqueous slurry of compound IV was unchanged after 82 days at room temperature.4) A solution of compound IV in a solvent such as NMP or DMA was treated with water until the solution became cloudy and was held at 75-85° C. for several hours. Monohydrate was isolated after cooling and filtering.5) A solution of compound IV in ethanol, butanol, and water was heated. Seeds of monohydrate were added to the hot solution and then cooled. Monohydrate was isolated upon cooling and filtration.One of ordinary skill in the art will appreciate that the monohydrate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 1 or by a representative sampling of peaks as shown in Table 1.Representative peaks taken from the XRPD of the monohydrate of the compound of formula (IV) are shown in Table 1.TABLE 1 2-Theta d(Å) Height 17.994 4.9257 915 18.440 4.8075 338 19.153 4.6301 644 19.599 4.5258 361 21.252 4.1774 148 24.462 3.6359 250 25.901 3.4371 133 28.052 3.1782 153The XRPD is also characterized by the following list comprising 2θ values selected from the group consisting of: 4.6±0.2, 11.2±0.2, 13.8±0.2, 15.2±0.2, 17.9±0.2, 19.1±0.2, 19.6±0.2, 23.2±0.2, 23.6±0.2. The XRPD is also characterized by the list of 2θ values selected from the group consisting of: 18.0±0.2, 18.4±0.2, 19.2±0.2, 19.6±0.2, 21.2±0.2, 24.5±0.2, 25.9±0.2, and 28.0±0.2.Single crystal x-ray data was obtained at room temperature (+25° C.). The molecular structure was confirmed as a monohydrate form of the compound of Formula (IV).The following unit cell parameters were obtained for the monohydrate of the compound of formula (IV) from the x-ray analysis at 25° C.:a(Å)=13.8632(7); b(Å)=9.3307(3); c(Å)=38.390(2);V(Å3) 4965.9(4); Z′=1; Vm=621Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.354wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).Single crystal x-ray data was also obtained at −50° C. The monohydrate form of the compound of Formula (IV) is characterized by unit cell parameters approximately equal to the following:Cell dimensions: a(Å)=13.862(1);
- b(Å)=9.286(1);
- c(Å)=38.143(2);
Volume=4910(1) Å3Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.369wherein the compound is at a temperature of about −50° C.The simulated XRPD was calculated from the refined atomic parameters at room temperature.The monohydrate of the compound of formula (IV) is represented by the DSC as shown in FIG. 2. The DSC is characterized by a broad peak between approximately 95° C. and 130° C. This peak is broad and variable and corresponds to the loss of one water of hydration as seen in the TGA graph. The DSC also has a characteristic peak at approximately 287° C. which corresponds to the melt of the dehydrated form of the compound of formula (IV).The TGA for the monohydrate of the compound of Formula (IV) is shown in FIG. 2 along with the DSC. The TGA shows a 3.48% weight loss from 50° C. to 175° C. The weight loss corresponds to a loss of one water of hydration from the compound of Formula (IV).The monohydrate may also be prepared by crystallizing from alcoholic solvents, such as methanol, ethanol, propanol, i-propanol, butanol, pentanol, and water.
Example 9Preparation of:
Crystalline n-butanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)The crystalline butanol solvate of the compound of formula (IV) is prepared by dissolving compound (IV) in 1-butanol at reflux (116-118° C.) at a concentration of approximately 1 g/25 mL of solvent. Upon cooling, the butanol solvate crystallizes out of solution. Filter, wash with butanol, and dry.The following unit cell parameters were obtained from the x-ray analysis for the crystalline butanol solvate, obtained at room temperature:a(Å)=22.8102(6); b(Å)=8.4691(3); c(Å)=15.1436(5); β=95.794(2);V(Å3) 2910.5(2); Z′=1; Vm=728Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.283wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the butanol solvate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 3 or by a representative sampling of peaks. Representative peaks for the crystalline butanol solvate are 2θ values of: 5.9±0.2, 12.0±0.2, 13.0±0.2, 17.7±0.2, 24.1±0.2, and 24.6±0.2.
Example 10Preparation of:
Crystalline ethanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)

To a 100-mL round bottom flask was charged 4.00 g (10.1 mmol) of 5D (contained 2.3 Area % 5C) 6.60 g (50.7 mmol) of 7B, 80 mL of n-butanol and 2.61 g (20.2 mmol) of DIPEA. The resulting slurry was heated to 120° C. and maintained at 120° C. for 4.5 h whereby HPLC analysis showed 0.19 relative Area % of residual 5D to compound IV. The homogeneous mixture was cooled to 20° C. and left stirring overnight. The resulting crystals were filtered. The wet cake was washed twice with 10-mL portions of n-butanol to afford a white crystalline product. HPLC analysis showed this material to contain 99.7 Area % compound IV and 0.3 Area % 5C.The resulting wet cake was returned to the 100-mL reactor, and charged with 56 mL (12 mL/g) of 200 proof ethanol. At 80° C. an additional 25 mL of ethanol was added. To this mixture was added 10 mL of water resulting in rapid dissolution. Heat was removed and crystallization was observed at 75-77° C. The crystal slurry was further cooled to 20° C. and filtered. The wet cake was washed once with 10 mL of 1:1 ethanol:water and once with 10 mL of n-heptane. The wet cake contained 1.0% water by KF and 8.10% volatiles by LOD. The material was dried at 60° C./30 in Hg for 17 h to afford 3.55 g (70 M %) of material containing only 0.19% water by KF, 99.87 Area % by HPLC. The 1H NMR spectrum, however revealed that the ethanol solvate had been formed.The following unit cell parameters were obtained from the x-ray analysis for the crystalline ethanol solvate (di-ethanolate, E2-1), obtained at −40° C.:a(Å)=22.076(1); b(Å)=8.9612(2); c(Å)=16.8764(3); β=114.783(1);V(Å3) 3031.1(1); Z′=1; Vm=758Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.271wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 4 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 5.8±0.2, 11.3±0.2, 15.8±0.2, 17.2±0.2, 19.5±0.2, 24.1±0.2, 25.3±0.2, and 26.2±0.2.In addition, during the process to form the ethanolate (diethanolate) the formation of another ethanol solvate (½ ethanolate, T1E2-1) has been observed. To date this additional ethanol solvate is known strictly as a partial desolvation product of the original diethanolate form E2-1, and has only been observed on occasion during crystallization of E2-1The following unit cell parameters were obtained from the x-ray analysis for the crystalline ½ ethanol solvate T1E2-1, obtained at −10° C.:a(Å)=22.03(2); b(Å)=9.20(1); c(Å)=12.31(1);β=93.49(6)V(Å3) 2491(4)); Z′=1; Vm=623;Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.363wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (T1E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 7 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 7.20±0.2, 12.01±0.2, 12.81±0.2, 18.06±0.2, 19.30±0.2, and 25.24±0.2.
Example 11Preparation of:
Crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (Neat form N-6)To a mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL) was added DIPEA (155 mL, 0.89 mol). The suspension was heated at 110° C. (solution obtained) for 25 min., then cooled to about 90° C. The resulting hot solution was added dropwise into hot (80° C.) water (8010) mL, keeping the temperature at about 80° C. The resulting suspension was stirred 15 min at 80° C. then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with water (2×1600 mL) and dried in vacuo at 55-60° C. affording 192.45 g (88.7% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H), 2.41 (s, 3H), 2.43 (t, 2H, J=6), 2.49 (t, 4H, J=6.3), 3.51 (m, 4H), 3.54 (q, 2H, J=6), 4.46 (t, 1H, J=5.3), 6.05 (s, 1H), 7.26 (t, 1H, J=7.6), 7.28 (dd, 1H, J=7.6, 1.7), 7.41 (dd, 1H, J=7.6, 1.7), 8.23 (s, 1H), 9.89 (s, 1H), 11.48. KF0.84; DSC: 285.25° C. (onset), 286.28° C. (max).The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline compound IV, obtained at 23° C.:a(Å)=22.957(1); b(Å)=8.5830(5); c(Å)=13.803(3); β=112.039(6);V(Å3)=2521.0(5); Z′=1; Vm=630Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.286wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the crystalline form of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 5 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (N-6) are 2θ values of: 6.8±0.2, 11.1±0.2, 12.3±0.2, 13.2±0.2, 13.7±0.2, 16.7±0.2, 21.0±0.2, 24.3±0.2, and 24.8±0.2.
Example 12Preparation of:
Crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (neat form T1H1-7)The title neat form may be prepared by heating the monohydrate form of the compound of formula (IV) above the dehydration temperature.The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline (T1H1-7) compound IV, obtained at 25° C.:a(Å)=13.4916; b(Å)=9.3992(2); c(Å)=38.817(1);V(Å3)=4922.4(3); Z′=1; Vm=615Space group PbcaDensity (calculated) (g/cm3) 1.317wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the neat crystalline form (T1H1-7) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 6 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (T1H1-7)) are 2θ values of: 8.0±0.2, 9.7±0.2, 11.2±0.2, 13.3±0.2, 17.5±0.2, 18.9±0.2, 21.0±0.2, 22.0±0.2.Obviously, numerous modifications and variations of the present invention are possible in light of the above teachings. It is therefore to be understood that within the scope of the appended claims, the invention may be practiced otherwise than as specifically described herein.
PAPERhttps://pubs.acs.org/doi/abs/10.1021/jm060727j
2-Aminothiazole (1) was discovered as a novel Src family kinase inhibitor template through screening of our internal compound collection. Optimization through successive structure−activity relationship iterations identified analogs 2 (Dasatinib, BMS-354825) and 12m as pan-Src inhibitors with nanomolar to subnanomolar potencies in biochemical and cellular assays. Molecular modeling was used to construct a putative binding model for Lck inhibition by this class of compounds. The framework of key hydrogen-bond interactions proposed by this model was in agreement with the subsequent, published crystal structure of 2 bound to structurally similar Abl kinase. The oral efficacy of this class of inhibitors was demonstrated with 12m in inhibiting the proinflammatory cytokine IL-2 ex vivo in mice (ED50 ∼ 5 mg/kg) and in reducing TNF levels in an acute murine model of inflammation (90% inhibition in LPS-induced TNFα production when dosed orally at 60 mg/kg, 2 h prior to LPS administration). The oral efficacy of 12m was further demonstrated in a chronic model of adjuvant arthritis in rats with established disease when administered orally at 0.3 and 3 mg/kg twice daily. Dasatinib (2) is currently in clinical trials for the treatment of chronic myelogenous leukemia.

PATENT
https://patents.google.com/patent/WO2019209908A1/enDasatinib (DAS), having the chemical designation N-(2-chloro-6-methylphenyl)-2- [[6-[4-(2-hydroxyethyl)-l-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5- thiazolecarboxamide, monohydrate, is an orally bioavailable inhibitor of the receptor tyrosine kinase (RTK) epidermal growth factor receptor (ErbB; EGFR) family, with antineoplastic activity. Dasatinib has the following structure:

Dasatinib is commercially marketed under the name SPRY CEL® and is indicated for the treatment of patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase, for the treatment of patients chronic, accelerated, or myeloid or lymphoid blast phase Philadelphia chromosome-positive chronic myeloid leukemia with resistance or intolerance to prior therapy and for the treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia with resistance or intolerance to prior therapy.Solid forms of dasatinib are described in U.S. Patent Nos. 7491725 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 8680103 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 7973045 (anhydrous), 8067423 (isopropyl alcohol solvate), 8242270 (butanol solvate, monohydrate, diethanolate, hemi- ethanolate, anhydrous), 8884013 (monohydrates), 9249134 (amorphous), 9456992 (solid dispersion nanoparticles), 9556164 (saccharin salt crystal) and 9884857 (saccharinate, glutarate, nicotinate); in U.S. Publication Nos. 20160250153 (solid dispersion nanoparticles), 20160264565 (Form-SDI), 20160361313 (solid dispersion nanoparticles), 20170183334 (salts) and 20140031352 (anti-oxidative acid); in International Publication Nos.W02010067374 (solvated forms and Form I), W02010139980, W02010139981,W02013065063 (anhydrous), W02017103057, W02017108605 (solid dispersion),WO2017134617 (amorphous), WO2014086326 (NMP, isoamyl-OH, 1, 3-propanediol process), WO2015107545, WO2015181573, WO2017134615 (PG solvate), W02010062715 (isosorbide dimethyl ether, N,N’-dimethylethylene urea, N,N’-dimethyl-N,N’-propylene urea), WO2010139979 (DCM, DMSP, monohydrate), WO2011095588 (anhydrate, hydrochloride, hemi-ethanol), W02012014149 (N-methylformamide) and W02017002131 (propandiol, monohydrate); and in Chinese Patent Nos. CN102643275, CN103059013, CN103819469, CN104341410. None of the references describe an ethyl formate solvate of dasatinib.Dasatinib co-crystals are described in U.S. Patent No. 9,340,536 (co-crystals selected from methyl-4-hydroxybenzoate, nicotinamide, ethyl gallate, methyl gallate, propyl gallate, ethyl maltol, vanillin, menthol, and (lR,2S,5R)-(-)-menthol) and International Publication No. W02016001025 (co-crystal selected from menthol or vanillin). None of the references describe dasatinib co-crystal comprising dasatinib and a second compound, as a co-crystal former, wherein the second compound is selected from butyl paraben, propyl paraben and ethyl vanillin.Dasatinib (DAS), having the chemical designation N-(2-chloro-6-methylphenyl)-2- [[6-[4-(2-hydroxyethyl)-l-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5- thiazolecarboxamide, monohydrate, is an orally bioavailable inhibitor of the receptor tyrosine kinase (RTK) epidermal growth factor receptor (ErbB; EGFR) family, with antineoplastic activity. Dasatinib has the following structure:
Dasatinib is commercially marketed under the name SPRY CEL® and is indicated for the treatment of patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase, for the treatment of patients chronic, accelerated, or myeloid or lymphoid blast phase Philadelphia chromosome-positive chronic myeloid leukemia with resistance or intolerance to prior therapy and for the treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia with resistance or intolerance to prior therapy.Solid forms of dasatinib are described in U.S. Patent Nos. 7491725 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 8680103 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 7973045 (anhydrous), 8067423 (isopropyl alcohol solvate), 8242270 (butanol solvate, monohydrate, diethanolate, hemi- ethanolate, anhydrous), 8884013 (monohydrates), 9249134 (amorphous), 9456992 (solid dispersion nanoparticles), 9556164 (saccharin salt crystal) and 9884857 (saccharinate, glutarate, nicotinate); in U.S. Publication Nos. 20160250153 (solid dispersion nanoparticles), 20160264565 (Form-SDI), 20160361313 (solid dispersion nanoparticles), 20170183334 (salts) and 20140031352 (anti-oxidative acid); in International Publication Nos.W02010067374 (solvated forms and Form I), W02010139980, W02010139981,W02013065063 (anhydrous), W02017103057, W02017108605 (solid dispersion),WO2017134617 (amorphous), WO2014086326 (NMP, isoamyl-OH, 1, 3-propanediol process), WO2015107545, WO2015181573, WO2017134615 (PG solvate), W02010062715 (isosorbide dimethyl ether, N,N’-dimethylethylene urea, N,N’-dimethyl-N,N’-propylene urea), WO2010139979 (DCM, DMSP, monohydrate), WO2011095588 (anhydrate, hydrochloride, hemi-ethanol), W02012014149 (N-methylformamide) and W02017002131 (propandiol, monohydrate); and in Chinese Patent Nos. CN102643275, CN103059013, CN103819469, CN104341410. None of the references describe an ethyl formate solvate of dasatinib.Dasatinib co-crystals are described in U.S. Patent No. 9,340,536 (co-crystals selected from methyl-4-hydroxybenzoate, nicotinamide, ethyl gallate, methyl gallate, propyl gallate, ethyl maltol, vanillin, menthol, and (lR,2S,5R)-(-)-menthol) and International Publication No. W02016001025 (co-crystal selected from menthol or vanillin). None of the references describe dasatinib co-crystal comprising dasatinib and a second compound, as a co-crystal former, wherein the second compound is selected from butyl paraben, propyl paraben and ethyl vanillin. hereafter. ClaimsHide Dependent What is claimed is:1. A dasatinib co-crystal comprising dasatinib and a second compound, wherein the second compound is selected from butyl paraben, propyl paraben and ethyl vanillin.2. The dasatinib co-crystal according to claim 1, wherein a molar ratio of the dasatinib to the second compound is about 1: 1.3. The dasatinib co-crystal according to claim 1, wherein the second compound is butyl paraben.4. The dasatinib co-crystal according to claim 3, wherein a molar ratio of the dasatinib to the butyl paraben is about 1 : 1.5. The dasatinib co-crystal according to claim 1, which is Form I co-crystal of dasatinib and butyl paraben.6. The dasatinib co-crystal according to claim 5, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 4.9, 9.8, 11.3, 14.9, 17.5, 20.8, 21.6, 22.6 and 25.4° 2Q degrees.7. The dasatinib co-crystal according to claim 5, characterized by a thermal event at about 287.3 °C, as measured by differential scanning calorimetry.8. The dasatinib co-crystal according to claim 5, characterized by a weight loss of 8.1% from about 70 °C through about 165 °C, as measured by thermal gravimetric analysis.9. The dasatinib co-crystal of claim 5 monoclinic, P2i/C.10. The dasatinib co-crystal d of claim 5 which has single crystal parametersa = 18.630 (2) Ab = 8.725 (1) Ac = 22.331 (2) Aa = g = 90°, b = 104.575 (8)°.11. The dasatinib co-crystal of claim 5 which has a cell volume is about 3512.9 A3.12. The dasatinib co-crystal according to claim 1, wherein the second compound is ethyl vanillin.13. The dasatinib co-crystal according to claim 9, wherein a molar ratio of the dasatinib to the ethyl vanillin is about 1 : 1.14. The dasatinib co-crystal according to claim 1, which is Form II co-crystal of dasatinib and ethyl vanillin.15. The dasatinib co-crystal according to claim 14, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 5.7, 10.9, 13.5, 17.1, 18.4, 19.4, 23.7 and 26.3° 2Q degrees.16. The dasatinib co-crystal according to claim 14, characterized by one or more thermal events selected from about 140 °C, about 181 °C, and about 293 °C, as measured by differential scanning calorimetry.17. The dasatinib co-crystal according to claim 14, characterized by a weight loss of 24.3% from about 120 through 250 °C, as measured by thermal gravimetric analysis.18. The dasatinib co-crystal of claim 14 monoclinic, P2i/n.19. The dasatinib co-crystal d of claim 14 which has single crystal parametersa = 18.452 (1) Ab = 9.441 (6) Ac = 19.377 (1) Aa = g = 90°, b = 108.78 (1)°.20. The dasatinib co-crystal of claim 5 which has a cell volume is about 3195.71 A3.21. The dasatinib co-crystal according to claim 1, wherein the second compound is propyl paraben.22. The dasatinib co-crystal according to claim 21, wherein a molar ratio of the dasatinib to the propyl paraben is about 1 : 1.23. The dasatinib co-crystal according to claim 1, which is Form III co-crystal ofdasatinib and propyl paraben.24. The dasatinib co-crystal according to claim 23, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 4.8, 9.6, 11.9, 14.8, 18.4, 22.2, 23.9 and 26.1° 2Q degrees.25. The dasatinib co-crystal of claim 23 monoclinic, P2i/n.26. The dasatinib co-crystal of claim 23 which has single crystal parametersa = 18.859 (9) Ab = 8.131 (6) Ac = 22.473 (1) Aa = g = 90°, b = 103.87(1)°.27. The dasatinib co-crystal of claim 23 which has a cell volume is about 3345.51 A3.28. An ethyl formate solvate of dasatinib.29. The ethyl formate solvate of dasatinib according to claim 28, wherein a molar ratio of the dasatinib to the ethyl formate is about 1 : 1.30. The ethyl formate solvate of dasatinib according to claim 1, which is Form I of ethyl formate solvate of dasatinib.31. The ethyl formate solvate of dasatinib according to claim 30, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 6.0, 12.1, 15.1, 18.0, 23.8 and 24.8° 2Q degrees.32. The ethyl formate solvate of dasatinib according to claim 30, characterized by athermal event at about 287.3 °C, as measured by differential scanning calorimetry.33. The ethyl formate solvate of dasatinib according to claim 30, characterized by aweight loss of 8.1% from about 70 °C through about 165 °C, as measured by thermal gravimetric analysis.34. The ethyl formate solvate of dasatinib of claim 23 orthorhombic, P2i/c.35. The ethyl formate solvate of dasatinib of claim 23 which has single crystal parameters a = 14.8928 (5) Ab = 8.3299 (3) Ac = 22.18990 (6) Aa = g =b = 90°.36. The ethyl formate solvate of dasatinib of claim 23 which has a cell volume is about 2731.9 A3.37. A pharmaceutical composition comprising a pharmaceutically effective amount of the dasatinib co-crystal according to claim 1 and pharmaceutically acceptable excipient.38. A method of treating disease in a patient comprising administering a pharmaceutical formulation according to claim 37 to the patient in need thereof.39. A method of treating disease according to claim 38, wherein the disease ismyelogenous leukemia.40. A method of treating disease according to claim 38, wherein the disease isPhiladelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase.41. A method of treating disease according to claim 38, wherein the disease Ph+ acute lymphoblastic leukemia (Ph+ ALL).42. A method of making the dasatinib co-crystal according to claim 1, comprisingdissolving dasatinib and a second compound, wherein the second compound is selected from the group consisting of butyl paraben, propyl paraben and ethyl vanillin, in heated methanol (-10: 1 – wt(mg)DAs:v(mL)MeOH and molD,\s:mohnci compound is 1 : 1.1) to form a clear solution, heating the solution under vacuum for about l8-20h to yield the dasatinib co-crystal.43. A process for the preparation Form II co-crystal of dasatinib and ethyl vanillin,according to claim 14, comprising: (g) dissolving Form I of ethyl formate solvate of dasatinib and ethyl vanillin in N-methyl-2-pyrrolidone to form a solution;(h) adding water to the solution;(i) stirring the solution for about 12-24 hours to form a slurry;(j) filtering the slurry to yield a precipitate;(k) washing the precipitate with water; and(l) drying the precipitate under vacuum with warming to yield Form II co crystal of dasatinib and ethyl vanillin.44. A process for the preparation of Form I of ethyl formate solvate of dasatinib,according to claim 30, comprising:(d) dissolving dasatinib in ethyl formate to form a solution;(e) stirring the solution for about 12-24 hours form a slurry;(f) filtering the slurry to yield Form I of ethyl formate solvate of dasatinib.45. A process for the preparation of Form I of ethyl formate solvate of dasatinib,according to claim 30, comprising:(g) dissolving dasatinib in N-Methyl-2-pyrrolidone to form a solution;(h) adding ethyl formate to the solution to form a slurry;(i) adding additional ethyl formate to the slurry;(j) stirring the slurry for about 2 hours;(k) filtering the slurry to yield a precipitate; and(l) washing the precipitate with ethyl formate to yield Form I of ethyl formate solvate of dasatinib.
ATENThttps://patents.google.com/patent/WO2013065063A1/en
Dasatinib, N-(2-chloro-6-methylphenyl)-2- [(6-[4-(2-hydroxyl)- 1 -piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5- thiazolecarboxamide compound having the following chemical structure of Formula (I)

Formula IAlso known as BMS-354825, it is a drug produced by Bristol Myers Squibb and sold under the trade name Sprycel. Dasatinib is an oral dual BCR/ABL and SRC family tyrosine kinase inhibitor approved for use in patients with chronic myelogenous leukemia (CML) after Imatinib treatment has failed and Philadelphia chromosome- positive acute lymphoblastic leukemia (Ph + ALL). It is also being assessed for use in metastatic melanoma.A preparation of Dasatinib is described in US patent No. 6596746 (B l ), where the process is done by reacting compound of the following formula III with N-(2- hydroxyethyl) piperazine at 80° C.

Formula IIIThe compound of Formula (I) and its preparation is described in US Patent No. 6596746, US patent application No. 2005/0176965 Al , and US patent application No. 2006/0004067 Al .l Polymorphism is defined as “the ability of a substance to exist as two or more crystalline phases that have different arrangement and /or conformations of the molecules in the crystal Lattice. Thus, in the strict sense, polymorphs are different crystalline forms of the same pure substance in which the molecules have different arrangements and / or different configurations of the molecules”. Different polymorphs may differ in their physical properties such as melting point, solubility, X-ray diffraction patterns, 1R etc. Polymorphic forms of a compound can be distinguished in the laboratory by analytical methods such as X-ray diffraction (XRD), Differential Scanning Calorimetry (DSC) and Infrared spectrometry (IR). Solvent medium and mode of crystallization play very important role in obtaining a crystalline form.The discovery of new polymorphic forms is a continuing goal of formulators. The new polymorphs may be advantageous for dosage form development and enhancing bioavailability owing to the altered physiochemical properties. Some form may turn out to be more efficacious. Discovering novel processes to prepare known polymorphic forms is also a primary goal of the pharmaceutical development scientists. New processes can provide novel intermediates or synthetic pathways that result in product with increased chemical and polymorphic purity in addition to providing cost and other advantages. There is thus a need to provide novel synthetic routes and intermediates that can realize these goals.Several crystalline forms of Dasatinib are described in the literature; these are designated as HI -7, BU-2, E2-1 , N-6, T1 H1 -7 and TIE2-1. Crystalline Dasatinib monohydrate (H I -7) and butanol solvate (BU-2) along with the processes for their preparation are described in WO 2005077945. In addition US 2006/0004067, which is continuation of US 2005215795 also describe two ethanol solvates (E2-1 ; TIE2-1) and two anhydrous forms (N-6 and T1 H1 -7).WO 2009053854 discloses various Dasatinib solvates including their crystalline form, amorphous form and anhydrous form.US patent No. 7973045 discloses the anhydrous form of Dasatinib and process for preparation thereof. The anhydrous form disclosed therein have typical characteristic XRD peaks at about 7.2, 1 1.9, 14.4, 16.5, 17.3, 19.1 , 20.8, 22.4, 23.8, 25.3 and 29.1 on the 2- theta value. WO 2010062715 discloses isosorbide dimethyl ether solvate, Ν,Ν’- dimethylethylene urea solvate and N,N’-dimethyl-N,N’-propylene urea solvate of Dasatinib.WO 2010067374 discloses novel crystalline form I, solvates of DMF, DMSO, toluene, isopropyl acetate and processes for their preparation.WO 2010139979 discloses MDC solvate and process of preparation, for use in the manufacture of pure Dasatinib.WO 2010139980 discloses a process for the preparation of crystalline Dasatinib monohydrate.The present invention is a step forward in this direction and provides a novel anhydrous form and process for its preparation, which can be used for the preparation of pure Dasatinib, in particularly Dasatinib monohydrate.The process for preparing Dasatinib monohydrate is described in US 2006/0004067. Further studies by the inventors have shown that the preparation of Dasatinib by using the method, which is disclosed in US 2006/0004067 yields the monohydrate with ~ 90% purity. Therefore the present invention provides a novel anhydrous form which can be used to get Dasatinib monohydrate with high yield and purity.Preparing API with increased purity is always an aim of the pharmaceutical development team. The inventors of the present invention have found that preparingDasatinib monohydrate using the novel anhydrous form of the present invention resulted in a highly pure product with a good yield.Scheme 1 shows a general process for the preparation of Dasatinib as disclosed in US 2006/0004067. Intermediate 3 and N-(2-hydroxyethyl) piperazine are heated together in a solvent system comprising n-butanol as a solvent and diisopropyl ethylamine (DIPEA) as a base. On cooling of the reaction mixture, Dasatinib precipitates out which is isolated by filtration.



DasatinibScheme 1Example – 1In a reaction vessel, N-(2-chloro-6-methylphenyl)-2-[(6-chloro-2-methyl-4- pyrimidinyl) amino] -5-thiazolecarboxamide (1 gm, 2.54 mmol) and N-(2- hydroxyethyl) piperazine (5.3 gm, 40.70 mmol) was added under stirring. The reaction mixture was heated at 80 °C for 2H. Acetonitrile was added into reaction mixture at 80 °C and stirred for 30 min. Cooled the suspension to room temperature and stirred for 30 min. Filtered, washed with acetonitrile and dried at 60 °C under vacuum to get 950 mg anhydrous N-(2-chloro-6-methylphenyl)-2-[(6-[4-(2-hydroxy 1)- 1 -piperaziny l]-2- methyl-4-pyrimidinyl]amino]-5-thiazole carboxamide (76.73 % Yield).HPLC Purity 99.90 %M/C by KF 0.12 %DSC 278.17 °CTGA 2.05 %XRD as provided in Fig. 2
Patent
Publication numberPriority datePublication dateAssigneeTitleUS7491725B22004-02-062009-02-17Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-aromatic carboxamides as kinase inhibitorsWO2009147238A12008-06-062009-12-10Boehringer Ingelheim International GmbhSolid pharmaceutical formulations comprising bibw 2992WO2010062715A22008-11-032010-06-03Teva Pharmaceutical Industries Ltd.Polymorphs of dasatinib and process for preparation thereofWO2010067374A22008-12-082010-06-17Hetero Research FoundationPolymorphs of dasatinibWO2010139980A12009-06-032010-12-09Generics [Uk] LimitedProcess for preparing crystalline dasatinib monohydrateWO2010139979A22009-06-032010-12-09Generics [Uk] LimitedProcesses for preparing crystalline formsWO2010139981A22009-06-032010-12-09Generics [Uk] LimitedProcesses for preparing crystalline formsWO2011003853A22009-07-062011-01-13Boehringer Ingelheim International GmbhProcess for drying of bibw2992, of its salts and of solid pharmaceutical formulations comprising this active ingredientUS7973045B22007-10-232011-07-05Teva Pharmaceutical Industries Ltd.Anhydrous form of dasatinib and process for preparation thereofWO2011095588A12010-02-042011-08-11Ratiopharm GmbhPharmaceutical composition comprising n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamidWO2012014149A12010-07-302012-02-02Ranbaxy Laboratories LimitedN-methylformamide solvate of dasatinibCN102643275A2011-02-212012-08-22江苏先声药物研究有限公司A new preparation method for Dasatinib N-6 crystal formCN103059013A2011-10-182013-04-24北京本草天源药物研究院New crystal of Dasatinib monohydrate and preparation method thereofWO2013065063A12011-11-032013-05-10Cadila Healthcare LimitedAnhydrous form of dasatinib, process for its preparation and its useUS20140031352A12012-07-242014-01-30Laurus Labs Private LimitedSolid forms of tyrosine kinase inhibitors, process for the preparation and their pharmaceutical composition thereofCN103819469A2012-11-162014-05-28重庆医药工业研究院有限责任公司Crystal form of dasatinib and preparation method for crystal form of dasatinibWO2014086326A12012-12-062014-06-12Zentiva, K.S.A method for the preparation and purification of new and known polymorphs and solvates of dasatinibUS8884013B22010-02-082014-11-11Nan Jing Cavendish Bio-Engineering Technology Co., Ltd.Polymorphs of Dasatinib, preparation methods and pharmaceutical compositions thereofCN104341410A2013-08-092015-02-11上海科胜药物研发有限公司New Dasatinib crystal form and preparation method thereofWO2015107545A12013-12-182015-07-23Dharmesh Mahendrabhai ShahWater soluble salts of dasatinib hydrateWO2015181573A12014-05-262015-12-03Egis Gyógyszergyár Zrt.Dasatinib saltsWO2016001025A12014-06-302016-01-07Basf SeMulticomponent crystals of dasatinib with menthol or vanillinUS9249134B22013-03-262016-02-02Cadila Healthcare LimitedProcess for preparation of amorphous form of dasatinibUS9340536B22012-06-152016-05-17Basf SeMulticomponent crystals comprising dasatinib and selected co-crystal formersUS20160250153A12012-01-132016-09-01Xspray Microparticles AbNovel methodsUS20160264565A12013-11-082016-09-15Shilpa Medicare LimitedCrystalline dasatinib processWO2017002131A12015-06-292017-01-05Msn Laboratories Private LimitedCrystalline forms of n-(2-chloro-6-methy]phenvn-2-[f6-[4-(2-hvdroxvethvl)-l- piperazinvil-2-methvl-4-pvrimidinvllaminol-5-thiazolecarboxamide and their process thereofUS9556164B22013-07-252017-01-31Basf SeSalts of Dasatinib in crystalline formWO2017103057A12015-12-162017-06-22Synthon B.V.Pharmaceutical composition comprising anhydrous dasatinibWO2017108605A12015-12-222017-06-29Synthon B.V.Pharmaceutical composition comprising amorphous dasatinibWO2017134615A12016-02-032017-08-10Dr. Reddy’s Laboratories LimitedSolid state forms of dasatinib and processes for their preparationWO2017134617A12016-02-032017-08-10Dr. Reddy’s Laboratories LimitedProcess for the preparation of amorphous dasatinibUS9884857B22013-07-252018-02-06Basf SeSalts of dasatinib in amorphous form
PATENTSPublication numberPriority datePublication dateAssigneeTitleUS20060079563A1 *1999-04-152006-04-13Jagabandhu DasCyclic protein tyrosine kinase inhibitorsUS20070219370A1 *2006-03-152007-09-20Bristol-Myers Squibb CompanyProcess for preparing n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino] -5-thiazolecarboxamide and related metabolites thereofUS20080275009A1 *2005-09-212008-11-06Bristol-Myers Squibb CompanyOral administration of n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-1,3-thiazole-5-carboxamide and salts thereofUS20090030203A1 *2005-08-052009-01-29Bristol-Myers Squibb CompanyPreparation of 2-amino-thiazole-5-carboxylic-acid derivativesUS20090118297A1 *2007-10-232009-05-07Ondrej SimoPolymorphs of dasatinib and process for preparation thereofWO2012014149A12010-07-302012-02-02Ranbaxy Laboratories LimitedN-methylformamide solvate of dasatinibUS20120309968A1 *2010-02-082012-12-06Nan Jing Cavendish Bio-Engineering Technology Co., Ltd.Polymorphs of dasatinib, preparation methods and pharmaceutical compositions thereofUS8530492B22009-04-172013-09-10Nektar TherapeuticsOligomer-protein tyrosine kinase inhibitor conjugatesUS8680103B22004-02-062014-03-25Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-aromatic carboxamides as kinase inhibitorsWO2014102759A22012-12-312014-07-03Ranbaxy Laboratories LimitedProcess for the preparation of dasatinib and its intermediatesUS8816077B22009-04-172014-08-26Nektar TherapeuticsOligomer-protein tyrosine kinase inhibitor conjugatesUS20150057446A1 *2012-04-202015-02-26Shilpa Medicare LimitedProcess for preparing dasatinib monohydrateWO2016001025A12014-06-302016-01-07Basf SeMulticomponent crystals of dasatinib with menthol or vanillinUS9340536B22012-06-152016-05-17Basf SeMulticomponent crystals comprising dasatinib and selected co-crystal formersUS9556164B22013-07-252017-01-31Basf SeSalts of Dasatinib in crystalline formUS9884857B22013-07-252018-02-06Basf SeSalts of dasatinib in amorphous formWO2018078392A12016-10-292018-05-03Cipla LimitedPolymorphs of dasatinibWO2018100585A12016-12-012018-06-07Natco Pharma LimitedAn improved process for the preparation of dasatinib polymorphWO2018134189A12017-01-202018-07-26Cerbios-Pharma SaCo-crystal of an antitumoral compoundWO2018134190A12017-01-202018-07-26Cerbios-Pharma SaCo-crystals of an antitumoral compoundUS10174018B22016-12-132019-01-08Princeton Drug Discovery IncProtein kinase inhibitorsWO2019209908A12018-04-252019-10-31Johnson Matthey Public Limited CompanyCrystalline forms of dasatinibUS10722484B22016-03-092020-07-28K-Gen, Inc.Methods of cancer treatmentUS10799459B12019-05-172020-10-13Xspray Microparticles AbRapidly disintegrating solid oral dosage forms containing dasatinibFamily To Family CitationsUS7396935B22003-05-012008-07-08Bristol-Myers Squibb CompanyAryl-substituted pyrazole-amide compounds useful as kinase inhibitorsUS7652146B2 *2004-02-062010-01-26Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-carboxamides useful as kinase inhibitorsTW200600513A *2004-06-302006-01-01Squibb Bristol Myers CoA method for preparing pyrrolotriazine compoundsPE20061394A1 *2005-03-152006-12-15Squibb Bristol Myers CoMetabolites of n- (2-chloro-6-methylphenyl) -2 – [[6- [4- (2-hydroxyethyl) -1-piperazinyl] -2-methyl-4-pyrimidinyl] amino] -5-thiazolecarboxamidesUS20060235006A1 *2005-04-132006-10-19Lee Francis YCombinations, methods and compositions for treating cancerPL1885339T32005-05-052015-12-31Bristol Myers Squibb Holdings IrelandFormulations of a src/abl inhibitorWO2008076883A22006-12-152008-06-26Abraxis Bioscience, Inc.Triazine derivatives and their therapeutical applicationsWO2010139979A22009-06-032010-12-09Generics [Uk] LimitedProcesses for preparing crystalline formsWO2010139980A1 *2009-06-032010-12-09Generics [Uk] LimitedProcess for preparing crystalline dasatinib monohydrateEP2359813A12010-02-042011-08-24Ratiopharm GmbHPharmaceutical composition comprising N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamidCN102250084A *2010-02-082011-11-23南京卡文迪许生物工程技术有限公司Dasatinib polymorphic substance as well as preparation method and pharmaceutical composition thereofCN102643275B *2011-02-212016-04-20江苏先声药物研究有限公司The preparation method that a kind of Dasatinib N-6 crystal formation is newWO2013065063A12011-11-032013-05-10Cadila Healthcare LimitedAnhydrous form of dasatinib, process for its preparation and its useUS20150087687A12012-03-232015-03-26Dennis BrownCompositions and methods to improve the therapeutic benefit of indirubin and analogs thereof, including meisoindigoSG10201610869TA2012-06-262017-02-27Del Mar PharmaceuticalsMethods for treating tyrosine-kinase-inhibitor-resistant malignancies in patients with genetic polymorphisms or ahi1 dysregulations or mutations employing dianhydrogalactitol, diacetyldianhydrogalactiCN103664929B *2012-08-302016-08-03石药集团中奇制药技术(石家庄)有限公司Dasatinib polycrystalline form medicament and preparation methodCN102838595B *2012-09-132014-09-24江苏奥赛康药业股份有限公司Preparation method of high-purity dasatinib and by-product of dasatinibCN103819469A *2012-11-162014-05-28重庆医药工业研究院有限责任公司Crystal form of dasatinib and preparation method for crystal form of dasatinibCZ306598B62012-12-062017-03-22Zentiva, K.S.A method of preparation and purification of new and known polymorphs and dasatinib solvatesCN105764502A2013-07-262016-07-13现代化制药公司Combinatorial methods to improve the therapeutic benefit of bisantrene and analogs and derivatives thereofCN103408542B *2013-08-132016-06-29南京优科生物医药研究有限公司A kind of preparation method of highly purified Dasatinib anhydrideWO2015049645A2 *2013-10-042015-04-09Alembic Pharmaceuticals LimitedAn improved process for the preparation of dasatinibCZ306732B62013-12-192017-05-31Zentiva, K.S.A method of preparation of the anhydrous polymorphic form of N-6 DasatinibCN104788445B *2015-04-102017-06-23山东新时代药业有限公司A kind of synthetic method of Dasatinib intermediateCN106668022B *2015-11-052020-09-15武汉应内药业有限公司Application of aminothiazole MyD88 specific inhibitor TJM2010-5* Cited by examiner, † Cited by third party, ‡ Family to family citation
References[edit]
- ^ “Sprycel (Dasatinib)” (PDF). Therapeutic Goods Administration(TGA). Retrieved 18 July 2020.
- ^ Jump up to:a b c d e f g h i j “Sprycel EPAR”. European Medicines Agency(EMA). Retrieved 28 April 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g h “Dasatinib”. The American Society of Health-System Pharmacists. Retrieved 8 December 2017.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Keating GM (January 2017). “Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia”. Drugs. 77 (1): 85–96. doi:10.1007/s40265-016-0677-x. PMID 28032244. S2CID 207489056.
- ^ Olivieri, A.; Manzione, L. (2007). “Dasatinib: a new step in molecular target therapy”. Annals of Oncology. 18 Suppl 6: vi42–vi46. doi:10.1093/annonc/mdm223. PMID 17591830.
- ^ “NHS – Healthcare News”. nelm.nhs.uk. Archived from the original on 5 May 2013. Retrieved 27 September 2011.
- ^ Yurttaş NO, Eşkazan AE (2018). “Dasatinib-induced pulmonary arterial hypertension”. British Journal of Clinical Pharmacology. 84 (5): 835–845. doi:10.1111/bcp.13508. PMC 5903230. PMID 29334406.
- ^ Jump up to:a b c d “Sprycel (dasatinib) and risk of pulmonary arterial hypertension”. U.S. Food and Drug Administration (FDA). 23 September 2011. Retrieved 28 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Tokarski JS, Newitt JA, Chang CY, Cheng JD, Wittekind M, Kiefer SE, et al. (June 2006). “The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants”. Cancer Research. 66 (11): 5790–7. doi:10.1158/0008-5472.CAN-05-4187. PMID 16740718.
- ^ Jump up to:a b Piscitani L, Sirolli V, Morroni M, Bonomini M (2020). “Nephrotoxicity Associated with Novel Anticancer Agents (Aflibercept, Dasatinib, Nivolumab): Case Series and Nephrological Considerations”. International Journal of Molecular Sciences. 21(14): e4878. doi:10.3390/ijms21144878. PMC 7402330. PMID 32664269.
- ^ Jump up to:a b Braun TP, Eide CA, Druker BJ (2020). “Response and Resistance to BCR-ABL1-Targeted Therapies”. Cancer Cell. 37(4): 530–542. doi:10.1016/j.ccell.2020.03.006. PMC 7722523. PMID 32289275.
- ^ “Otsuka and Bristol-Myers Squibb Announce a Change in Contract Regarding Collaboration in Japan in the Oncology Therapy Area”.
- ^ “FDA Approves U.S. Product Labeling Update for Sprycel (dasatinib) to Include Three-Year First-Line and Five-Year Second-Line Efficacy and Safety Data in Chronic Myeloid Leukemia in Chronic Phase”. Bristol-Myers Squibb (Press release).
- ^ “Bristol-Myers Squibb Announces Extension of U.S. Agreement for ABILIFY and Establishment of an Oncology Collaboration with Otsuka”. Bristol-Myers Squibb (Press release).
- ^ Drahl C (16 January 2012). “How Jagabandhu Das made dasatinib possible”. The Safety Zone blog. Chemical & Engineering News. Retrieved 29 August 2016.
- ^ “Drug Approval Package: Sprycel (Dasatinib) NDA #021986 & 022072”. U.S. Food and Drug Administration (FDA). 6 September 2006. Retrieved 28 April 2020.
- ^ “2010 Notifications”. U.S. Food and Drug Administration (FDA). 18 November 2010. Retrieved 28 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e “FDA approves dasatinib for pediatric patients with CML”. U.S. Food and Drug Administration (FDA). 9 November 2017. Retrieved 28 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c Cohen D (November 2014). “US trade rep is pressing Indian government to forbid production of generic cancer drug, consortium says”. BMJ. 349: g6593. doi:10.1136/bmj.g6593. PMID 25370846. S2CID 206903723.
- ^ Jump up to:a b c Kirkland JL, Tchkonia T (2020). “Senolytic drugs: from discovery to translation”. Journal of Internal Medicine. 288 (5): 518–536. doi:10.1111/joim.13141. PMC 7405395. PMID 32686219.
- ^ Jump up to:a b Paez-Ribes M, González-Gualda E, Doherty GJ, Muñoz-Espín D (2019). “Targeting senescent cells in translational medicine”. EMBO Molecular Medicine. 11 (12): e10234. doi:10.15252/emmm.201810234. PMC 6895604. PMID 31746100.
- ^ Wyld L, Bellantuono I, Tchkonia T, Danson S, Kirkland JL (2020). “Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies”. Cancers. 12 (8): e2134. doi:10.3390/cancers12082134. PMC 7464619. PMID 32752135.
Further reading[edit]
- Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. (December 2004). “Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays”. Journal of Medicinal Chemistry. 47 (27): 6658–61. doi:10.1021/jm049486a. PMID 15615512.
External links[edit]
- “Dasatinib”. Drug Information Portal. U.S. National Library of Medicine.
/////////////DASATINIB, BMS 35482503, KIN 001-5, NSC 759877, Sprycel, BMS, APOTEX, ダサチニブ水和物 , X78UG0A0RN, дазатиниб , دازاتينيب , 达沙替尼 ,
#DASATINIB, #BMS 35482503, #KIN 001-5, #NSC 759877, #Sprycel, #BMS, #APOTEX, #ダサチニブ水和物 , #X78UG0A0RN, #дазатиниб , #دازاتينيب , #达沙替尼 ,
O.Cc1nc(Nc2ncc(s2)C(=O)Nc3c(C)cccc3Cl)cc(n1)N4CCN(CCO)CC4
PATENT
https://patents.google.com/patent/US8884013B2/enDasatinib, with the trade name SPRYCEL™, is a oral tyrosine kinase inhibitor and developed by BMS Company. It is used to cure adult chronic myelogenous leukemia (CML), acute lymphatic leukemia (ALL) with positive Philadelphia chromosome, etc. Its chemical name is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidyl]amino]-5-thiazolformamide and its chemical structure is as following:

Five polymorphs of Dasatinib and the preparation methods thereof were described by Bristol-Myers Squibb in the Chinese Patent Application No. CN200580011916.6 (publication date is 13 Jun. 2007). The preparation methods instructed in this document are:Monohydrate: Dasatinib (48 g) was added into ethanol (1056 mL 22 ml/g) and water (144 mL), and dissolved by heating to 75° C.; the mixture was purified, filtrated and transferred to the receiver. The solution reactor and transferring pipes were washed with the mixture of ethanol (43 mL) and water (5 mL). The solution was heated to 75˜80° C. to be soluble completely and water (384 mL) was heated and the temperature of the solution was kept between 75° C. and 80° C. The seed crystal of monohydrate (preferable) was added when cooling to 75° C., and keep the temperature at 70° C. for 1 h; cooling to 5° C. within 2 h and keeping the temperature at 0˜5° C. for 2 h. The slurry was filtrated and the filter cake was washed by the mixture of ethanol (96 mL) and water (96 mL); after being dried under vacuum≦50° C. 41 g of solid was obtained.Butanol solvate: under refluxing (116° C.˜118° C.), Dasatinib was dissolved in 1-butanol (about 1 g/25 mL) to yield crystalline butanol solvate of Dasatinib. When cooling, this butanol solvate was recrystallized from solution. The mixture was filtrated and the filter cake was dried after being washed with butanol.Ethanol solvate: 5D (4 g, 10.1 mmol), 7B (6.6 g, 50.7 mmol), n-bubanol (80 mL) and DIPEA (2.61 g, 20.2 mmol)) were added into a 100 ml round flask. The obtained slurry was heated to 120° C. and kept the temperature for 4.5 h, and then cooled to 20° C. and stirred over night. The mixture was filtrate, and the wet filter cake was washed with n-butanol (2×10 mL) to yield white crystal product. The obtained wet filter cake was put back to the 100 ml reactor and 56 mL (12 mL/g) of 200 proof ethanol was added. Then additional ethanol (25 mL) was added at 80° C., and water (10 mL) was added into the mixture to make it dissolved rapidly. Heat was removed and crystallization was observed at 75° C.˜77° C. The crystal slurry was further cooled to 20° C. and filtrated. The wet filter cake was washed with ethanol:water (1:1, 10 mL) once and then washed with n-heptane (10 mL) once. After that it was dried under the condition of 60° C./30 in Hg for 17 h to yield 3.55 g of substance only containing 0.19% water.Neat form of N-6: DIPEA (155 mL, 0.89 mmol) was added into the mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL). The suspension was heated at 110° C. for 25 min to be solution, which was then cooled down to about 90° C. The obtained solution was added dropwise into hot water (80° C., 8010 mL), and the mixture was stirred at 80° C. with heat preservation for 15 min and cooled to room temperature slowly. The solid was filtrated under vacuum and collected, washed by water (2×1600 mL) and dried under vacuum at 55° C.˜60° C. to give 192.45 of compound.Neat form of T1H1-7 (neat form and pharmaceutically acceptable carrier): monohydrate of Dasatinib was heated over dehydrate temperature to yield.Because Dasatinib is practically insoluble in water or organic solvent (e.g. methanol, ethanol, propanol, isopropanol, butanol, pentanol, etc.), even in the condition of heating, a large amount (over 100 times) of solvent is needed, which is disadvantageous in industrial production; in addition, with the method described in the Patent document of CN200580011916.6, the related substances in products can not be lowed effectively during the process of crystal preparation to improve the products quality.In terms of polymorphs of drug, each polymorph has different chemical and physical characteristics, including melting point, chemical stability, apparent solubility, rate of dissolution, optical and mechanical properties, vapor pressure as well as density. Such characteristics can directly influence the work-up or manufacture of bulk drug and formulation, and also affect the stability, solubility and bioavailability of formulation. Consequently, polymorph of drug is of great importance to quality, safety and efficacy of pharmaceutical preparation. When it comes to Dasatinib, there are still needs in the art for new polymorphs suitable for industrial production and with excellent physical and chemical properties as well.Example 1Preparation of the Polymorph IA. Dasatinib (10 g) and DMSO (40 ml) were added into a flask and heated up to 60˜70° C. by stirring, after dissolving, the mixture (120 mL) of water and acetone (1:1) was added under heat preservation. When crystal was precipitated, cooled it down to 0° C. to grow the grains for 10 minutes. Filtrate it and the cake was washed by water and then by the mixture of water and acetone (1:1). After that it was dried under −0.095 MPa at about 50° C. using phosphorus pentoxide as drying aid to give 7.7 g of white solid. Yield was 77%.Contrasts Index of raw material Items before transformation Index of Polymorph I Appearance off-white powder White crystal powder Related substance 0.85% 0.07% KF moisture 0.67% 3.59% 70~150 0.72% 3.63% TGA weight loss
The following items of products prepared by Method A were detected: microscope-crystal form (See. FIG. 1); XRPD Test (See. FIG. 2), IR Test (See. FIG. 3), DSC-TGA Test (See. FIG. 4-1, 4–2), 13C Solid-state NMR Test (See. FIG. 5).B. Dasatinib (10 g) and DMSO (40 ml) were added into a flask and heated slowly up to 60˜70° C. by stirring, after dissolving, the mixture (160 mL) of ethanol and water (1:1) was added under heat preservation. When crystal was precipitated, cooled it down to 0° C. to grow the grains for 10 minutes. Filtrate it and the cake was washed by the mixture of ethanol and water (1:1) and dried under −0.095 MPa at about 50° C. using phosphorus pentoxide as drying aid to give 7.7 g of white solid. Yield was 87%.Contrasts Index of raw material Items before transformation Index of Polymorph I Appearance off-white powder White crystal powder Related substance 0.85% 0.08% KF moisture 0.67% 3.58% 70~150 0.72% 3.67% TGA weight lossHPLC.Related Substances DeterminationHPLC conditions and system applicability: octadecylsilane bonded silica as the filler; 0.05 mol/L of potassium dihydrogen phosphate (adjusted to pH 2.5 by phosphoric acid, 0.2% triethylamine)-methanol (45:55) as the mobile phase; detection wavelength was 230 nm; the number of theoretical plates should be not less than 2000, calculated according to the peak of Dasatinib. The resolution of the peak of Dasatinib from the peaks of adjacent impurities should meet requirements.Determination method: sample was dissolved in mobile phase to be the solution containing 0.5 mg per milliliter. 20 μL of such solution was injected into liquid chromatograph, and chromatogram was recorded until the sixfold retention time of major component peak. If there were impurities peaks in the chromatogram of sample solution, total impurities and any single impurity were calculated by normalization method on the basis of peak area.Stability of Polymorph in the FormulationsThe XRPD patterns of capsules and tablets respectively prepared in the Example 3 and Example 4 have been tested, and compared with XRPD characteristic peaks of Polymorph I of Dasatinib prepared by the Method A in the Example 1 in the present invention, as listed in the following table:Bulk Drug Capsules 1 Capsules 2 Tablets 2 (Polymorph (Polymorph (Polymorph Tablets 1 (Polymorph I) I) I) (Polymorph I) I) 2θ 2θ 2θ 2θ 2θ 9.060 9.080 9.070 9.060 9.070 11.100 11.120 11.110 11.100 11.110 13.640 13.670 13.650 13.640 13.650 15.100 15.120 15.110 15.100 15.110 17.820 17.840 17.830 17.820 17.820 19.380 19.400 19.390 19.380 19.390 22.940 22.970 22.950 22.950 22.950The results in the above-mentioned comparative table have shown that the crystal form had substantially no change after Polymorph I of Dasatinib in the invention were prepared into capsules or tablets by the formulation process.In addition, The relative substances of capsules and tablets respectively prepared in the Example 3 and Example 4 have been tested, and compared with those of Polymorph I of Dasatinib prepared by the Method A in the Example 1 in the present invention, as listed in the following table:Bulk Drug (Polymorph I) Capsules 1 Capsules 2 Tablets 1 Tablets 2 0.07% 0.08% 0.08% 0.07% 0.08%The results in the above-mentioned comparative table have shown that the Polymorph I of Dasatinib was stable, and there were no significantly changes in respect to the relative substances, after Polymorph I of Dasatinib in the invention were prepared into capsules or tablets by the formulation process.INDUSTRIAL APPLICATIONThe present invention provides novel polymorphs of Dasatinib, preparing methods, and pharmaceutical composition comprising them. These polymorphs have better physicochemical properties, are more stable and are more suitable for industrial scale production, furthermore, are suitable for long-term storage, and are advantageous to meet the requirements of formulation process and long-term storage of formulations. The preparation technique of this invention was simple, quite easy for operation and convenient for industrial production, and the quality of the products was controllable with paralleled yields. In addition, by the methods of polymorph preparation in this invention, the amount of organic solvent used in crystal transformation could be reduced greatly, which led to reduced cost of products; organic solvents in Class III with low toxicity could be used selectively to prepare the polymorphs of this invention, reducing the toxic effects of the organic solvents potentially on human body to some extent.PATENThttps://patents.google.com/patent/WO2010067374A2/enDasatinib are antineoplastic agents, which were disclosed in WO Patent Publication No. 00/62778 and U.S. Patent No. 6,596,746. Dasatinib, chemically N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4- pyrimidinyl]amino]-5-thiazolecarboxamide, is represented by the following structure:

Polymorphism is defined as “the ability of a substance to exist as two or more crystalline phases that have different arrangement and /or conformations of the molecules in the crystal Lattice. Thus, in the strict sense, polymorphs are different crystalline forms of the same pure substance in which the molecules have different arrangements and / or different configurations of the molecules”. Different polymorphs may differ in their physical properties such as melting point, solubility, X-ray diffraction patterns, etc. Although those differences disappear once the compound is dissolved, they can appreciably influence pharmaceutically relevant properties of the solid form, such as handling properties, dissolution rate and stability. Such properties can significantly influence the processing, shelf life, and commercial acceptance of a polymorph. It is therefore important to investigate all solid forms of a drug, including all polymorphic forms, and to determine the stability, dissolution and flow properties of each polymorphic form. Polymorphic forms of a compound can be distinguished in the laboratory by analytical methods such as X-ray diffraction (XRD), Differential Scanning Calorimetry (DSC) and Infrared spectrometry (IR).Solvent medium and mode of crystallization play very important role in obtaining a crystalline form over the other. Dasatinib can exist in different polymorphic forms, which differ from each other in terms of stability, physical properties, spectral data and methods of preparation.U.S. Patent Application No. 2005/0215795 A1 (herein after referred to as the 795 patent application) described five crystalline forms of dasatinib (monohydrate, butanol solvate, ethanol solvate, neat form (N-6) and neat form (T1H1-7)), characterized by powder X-ray diffraction (P-XRD) pattern.According to the ‘795 patent application, dasatinib monohydrate is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 18.0, 18.4, 19.2, 19.6, 21.2, 24.5, 25.9 and 28.0 ± 0.2 degrees. As per the process exemplified in the ‘795 patent application, dasatinb monohydrate can be obtained in dasatinib, by heating and dissolving the dasatinib in an ethanol and water mixture. Crystallizing the monohydrate from the ethanol and water mixture and cooled to get dasatinib monohydrate.According to the ‘795 patent application, dasatinib crystalline butanol solvate is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 5.9, 12.0, 13.0, 17.7, 24.1 and 24.6 ± 0.2 degrees.According to the 795 patent application, dasatinib crystalline ethanol solvate is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 5.8, 11.3, 15.8, 17.2, 19.5, 24.1, 25.3 and 26.2 ± 0.2 degrees.According to the 795 patent application, dasatinib crystalline neat form (N-6) is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 6.8, 11.1, 12.3, 13.2, 13.7, 16.7, 21.0, 24.3 and 24.8 ± 0.2 degrees.According to the 795 patent application, dasatinib crystalline neat form (T1H1-7) is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 8.0, 9.7, 11.2, 13.3, 17.5, 18.9, 21.0 and 22.0 ± 0.2 degrees.U.S. Patent application No. 2006/0094728 disclosed ethanolate form (T1E2-1) of dasatinib, characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 7.2, 12.0, 12.8, 18.0, 19.3 and 25.2 ± 0.2 degrees. We have discovered novel crystalline form of dasatinib, dasatinib dimethylformamide solvate, dasatinib dimethyl sulfoxide solvate, dasatinib toluene solvate and dasatinib isopropyl acetate solvate.Another object of the present invention is to provide process for preparing the novel crystalline form of dasatinib, dasatinib dimethylformamide solvate, dasatinib dimethyl sulfoxide solvate, dasatinib toluene solvate, dasatinib isopropyl acetate solvate and known crystalline dasatinib monohydrate.Still another object of the present invention is to provide pharmaceutical compositions containing the novel crystalline form of dasatinib.Reference Example2-(6-Cholro-2-methylpyrimidin-4-yl-amino)-N-(2-chloro-6-methylphenyl) thiazole-5-carboxamide (15 gm) was added to 1-(2-hydroxyethyl)piperazine at 250C and heated to 850C, stirred for 2 hours 30 minutes at 850C. To the solution was added water (500 ml) at 800C and slowly cooled to 250C, stirred for 1 hour at 250C. The solid was collected by filtration and the solid was washed with water (50 ml), and then dried the solid at 550C under vacuum to obtain 15 gm of dasatinib.Example 1Dasatinib (5 gm) obtained according to reference example was dissolved in ethyl acetate (300 ml) at 250C and heated to reflux temperature. To the solution was added methanol (100 ml) and stirred for 30 minutes at reflux temperature to form clear solution. The solution was slowly cooled to room temperature and then cooled to O0C, stirred for 1 hour at O0C. The solid was collected by filtration and the solid was washed with mixture of ethyl acetate and methanol (20 ml, 3:1), and then dried the solid at 500C under vacuum to obtain 3.5 gm of crystalline dasatinib form I.Example 2Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in acetone (100 ml) and methanol (250 ml) and heated to reflux temperature, stirred for 30 minutes at reflux temperature to form clear solution. The solution was cooled to room temperature and then cooled to 200C, stirred for 1 hour at 200C. The solid was collected by filtration and the solid was washed with mixture of acetone (10 ml) and methanol (25 ml), and then dried the solid at 500C under vacuum to obtain 4 gm of crystalline dasatinib form I (HPLC purity: 99.85%).Example 3Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in dimethylformamide (25 ml) at 250C and heated to 650C to form clear solution. To the solution was slowly added acetone (50 ml) at 650C and stirred for 1 hour at 650C. The solution was slowly cooled to 250C and stirred for 1 hour at 250C. The contents are filtered and the solid obtained was washed with mixture of dimethylformamide and acetone (15 ml, 1:2), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethylformamide solvate (HPLC purity: 99.94%).Example 4Dasatinib (5 gm) was dissolved in dimethylformamide (25 ml) at 250C and heated to 650C to form clear solution. Ethyl acetate (50 ml) was added slowly to the solution at 650C and stirred for 1 hour at 650C. The solution was slowly cooled to 250C, stirred for 1 hour at 250C and filtered. The solid obtained was washed with mixture of dimethylformamide and ethyl acetate (30 ml, 1:2), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethylformamide solvate.Example 5Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in dimethylformamide (25 ml) and heated to 650C to form a clear solution. The solution was cooled to 250C and then cooled to 50C, stirred for 4 hour at 50C. The solid was collected by filtration and the solid was washed with chilled dimethylformamide (10 ml), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethylformamide solvate (HPLC purity: 99.9%).Example 6Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in dimethylformamide (25 ml) and heated to 650C to form a clear solution. Water (50 ml) was added slowly to the solution at 650C and stirred for 1 hour at 650C. The solution was cooled to 250C and stirred for 30 minutes at 250C. The solid was collected by filtration and the solid was washed with mixture of dimethylformamide and water (15 ml, 1 :2), and then dried the solid at 500C under vacuum to obtain 4.7 gm of dasatinib dimethylformamide solvate (HPLC purity: 99.93%).Example 7Dasatinib dimethylformamide solvate (4.7 gm) obtained as in example 6 was dissolved in water (50 ml) and heated to 750C, stirred for 4 hours at 750C. The solution was cooled to 250C, stirred for 30 minutes at 250C and filtered. The solid obtained was washed with water (15 ml), and then dried at 500C under vacuum to obtain 4.7 gm of dasatinib monohydrate.Example 8Dasatinib (20 gm) was dissolved in dimethyl sulfoxide (100 ml) at 250C and heated to 650C to form clear solution. To the solution was slowly added water (200 ml) at 650C and stirred for 1 hour at 650C. The solution was slowly cooled to 250C and stirred for 30 minutes at 250C. The solid was collected by filtration and the solid was washed with mixture of dimethyl sulfoxide and water (30 ml, 1 :2), and then dried the solid at 500C under vacuum to obtain 19.5 gm of dasatinib monohydrate.Example 9Dasatinib (5 gm) was dissolved in isopropyl acetate (65 ml) and heated to 800C, stirred for 1 hour at 800C to form a clear solution. The solution was cooled to 250C, stirred for 1 hour at 250C and filtered. The solid obtained was washed with isopropyl acetate (15 ml) to obtain 5 gm of dasatinib isopropyl acetate solvate.Example 10Dasatinib (6 gm) was dissolved in toluene (100 ml) and heated to reflux temperature, stirred for 2 hours at reflux temperature to form a clear solution. The solution was slowly cooled to 250C. The contents are filtered and the solid obtained was washed with toluene (20 ml) to obtain 5.5 gm of dasatinib toluene solvate.Example 11Dasatinib (5 gm) was dissolved in dimethyl sulfoxide (20 ml) at 250C and heated to 650C. To the solution was slowly added ethyl acetate (200 ml) at 650C and the solution was slowly cooled to O0C, stirred for 2 hours at O0C. The solid was collected by filtration and the solid was washed with mixture of dimethyl sulfoxide and ethyl acetate (55 ml, 1 :10), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethyl sulfoxide solvate.
PATENThttps://patents.google.com/patent/WO2014086326A1/enDasatinib, N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)- 1 -piperazinyl]-2- methyl-4-pyrimidmyl]amino]-5-thiazole carboxamide of formula I, also known as BMS- 354825, is a cancer treatment drug developed by Bristol-Myers Squibb and sold under the trade name Sprycel®. Dasatinib is a multi- BCR/ABL and Src family tyrosine kinase inhibitor and it is used for treatment of chronic myelogenous leukaemia (CML) as a secondary drug after primary treatment with imatinib (Gleevec®). It is also used for treatment of acute lymphoblastic leukaemia caused by mutation/translocation of chromosomes and development of the so-called Philadelphia chromosome (Ph+ ALL). However, its potential is so wide that the possibility of using it for treatment of other types of cancer, including advanced stages of prostate cancer, is still being investigated.

(I)In accordance with the basic patent WO2000062778A1, dasatinib is prepared by reaction of the key intermediate of formula II with l-(2-hydroxyethyl)piperazine in the presence of a base and a suitable solvent (Scheme 1). A similar preparation method was later used in a number of other process patents, only varying the corresponding base or solvent. Through the selection of a suitable solvent or procedure a great number of solvates or polymorphs can be prepared. Polymorphs have been one of the most frequently studied physical characteristics of active pharmaceutical substances (API) recently. Thus, different polymorphs of one API may have entirely different physical-chemical properties such as solubility, melting point, mechanical resistance of crystals but they may also influence the chemical and physical stability. Then, these properties may have an impact on further processes such as handling of the particular API, grinding or formulation method. These various physical-chemical characteristics of polymorphs influence the resulting bioavailability of the solid dosage form. Therefore, looking for new polymorphs and solvates is becoming an important tool for obtaining a polymorph form with the desired physical-chemical characteristics.

The process patent WO2005077945A2 describes preparation of the following solvates of dasatinib: monohydrate, butanol solvate, as well as two anhydrous forms (N-6 and T1H1- 7). A related patent also mentions two ethanol solvates, the hemi-ethanol and diethanol solvates (US 8 242 270 B2). Salts, various combinations of salts and their solvates have been described in detail in the patent application WO2007035874A1.Another process patent, WO2009053854A2, dealt with the preparation of a number of solvates or mixed solvates out of which especially the isopropanol and mixed isopropanol/dimethyl sulfoxide solvates, as well as a new solid form B, another anhydrous polymorph of dasatinib, are worth mentioning. Other patent applications have also dealt with the preparation of other solvates/mixed solvates (WO2010067374A2), or processes for the preparation and purification of the monohydrate/anhydrous form (WO2010139981A2) and its polymorphs (WO2011095059 Al).API solvates or salts are used in drug formulations in many cases. In the case of solvates the limits for individual solvents, their contents or maximum daily doses have to be strictly observed. Then, these limits can dramatically restrict their effective use. Thus, the clearly most convenient option is the use of sufficiently stable polymorphs of API that do not contain any solvents bound in the crystalline structure.Some of the above mentioned patent documents describe preparation of a stable anhydrous form of dasatinib (N-6). In accordance with individual patent documents the main disadvantages of the preparation of N-6 is the necessity of desolvation of the solvated form of the API at high temperatures (WO2009053854A2), or application of an increased temperature (50°C and more) and vacuum for a relatively long time (8-12h; WO2010139981A2 and WO2005077945A2). These procedures are very demanding from the point of view of general technology, energy and time, to say nothing of the necessity to work under an inert atmosphere to prevent possible oxidation-degradation reactions of the API. This is because dasatinib may be oxidized by atmospheric oxygen to the corresponding N-oxide (oxidation occurs in the piperazine ring), which may undergo the Cope elimination at increased temperatures. This secondary reaction may subsequently impair the purity of the prepared API.With a view to the above mentioned facts it is obvious that completely new methods and processes have to be developed even for polymorphs or solvates that are already well- known. Generally, the development of technologically and economically more efficient procedures is the main decisive parameter in their industrial utilization for the preparation of the API.Dasatinib of formula I is prepared by a reaction of the intermediate of formula II with l-(2- hydroxyethyl)piperazine in the presence of diisopropylethylamine (DIPEA) in an organic solvent from the group of dipolar aprotic solvents, higher alcohols or diols.If a dipolar aprotic solvent from the group of N-methyl-2-pyrrolidone (NMP), N^iV-dimethyl formamide (DMF), AyV-dimethyl acetamide (DMA), dimethyl sulfoxide (DMSO), formamide (FA), N,N -dimethyl propylene urea (DMPU) and l,3-dimethyl-2-imidazolidinone (DMI) is used, the reaction is carried out at 50-110°C under an inert atmosphere for 1/2-6 hours. In a preferable embodiment, NMP, DMSO, DMPU or DMI is used and the reaction is carried out at 90°C for 1-3 hours. The result of the reaction is crude dasatinib in the form of a solution in the corresponding solvent.If an alcohol from the group of isoamyl alcohol or 1,3-propanediol is used as a solvent for preparation of the crude dasatinib, the reaction mixture is heated at 120-160°C for 2-12 hours, in a preferable embodiment at 135°C for 3-6 hours.If dipolar aprotic solvents (NMP, DMF, DMA, DMSO, FA, DMPU and DMI) are used, in step a) a precipitant is added to the hot solution (90°C) under continuous stirring in an inert atmosphere in a 2- 15 fold, most preferably 4-10fold (by volume) amount with respect to the dipolar aprotic solvent. Suitable precipitants comprise especially acetonitrile, propionitrile, most preferably acetonitrile.After addition of the precipitant the obtained solution is withdrawn from the heating bath and is slowly left to cool down to 22°C under continuous stirring in an inert atmosphere. Crystallization occurs within 1-120 minutes (depending on the volume, until complete cooling). After having cooled down to 22°C (laboratory temperature), the suspension is stirred for another hour. The corresponding solvate of dasatinib is aspirated by well-known techniques in an inert atmosphere at 10-35 °C, most preferably at 22°C, and washed with the respective co-solvent.The solvate of dasatinib obtained this way can be directly used in the next step – recrystallization, without the necessity of drying. If necessary, the product may be dried at 10- 35°C, most preferably at 25°C, and at the pressure of 10-200 kPa, most preferably 50 kPa, for 6-24 hours, most preferably 12 hours.If NMP is used as the solvent in step a), the corresponding NMP solvate is isolated. The obtained dried crystalline NMP solvate (NM) of dasatinib has a characteristic XRPD pattern, which is presented in Figure no. 1. The NMP solvate (NM) has the following characteristic peaks: 5.88; 6.73; 10.73; 11.92; 13.39; 14.97; 16.72; 18.95; 20.17; 21.46; 22.81; 24.65; 25.18; 26.02 and 28.06 ± 0.2° 2-theta.If isoamyl alcohol or 1,3-propanediol are used as the solvents in step a), the reaction mixture is left to cool down to 22°C after expiration of the reaction time (3-6 h). Crystallization generally begins when the inner temperature of the reaction mixture drops to 100°C. After cooling down to 22°C (laboratory temperature), the suspension is further stirred for another 1 hour. Crystalline dasatinib is aspirated by well-known techniques in an inert atmosphere at 10-35°C, most preferably at 22°C, and washed with the corresponding solvent.The obtained product is dried at 10-35°C, most preferably at 25°C, and at the pressure of 10-200 kPa, most preferably 50 kPa, for 6-24 hours, most preferably 12 hours.The obtained crystalline isoamyl alcohol solvate (SI) of dasatinib has a characteristic XRPD pattern, which is shown in Figure no. 2. The solvate (SI) has the following characteristic peaks: 5.72; 10.35; 11.42; 12.61; 13.14; 14.27; 15.33; 17.18; 17.44; 17.97; 19.12; 19.95; 20.38; 22.05; 22.42; 23.01; 23.46; 23.68; 25.26; 26.20; 26.45; 26.62 and 27.78 ± 0.2° 2-theta.The obtained crystalline 1,3-propanediol solvate (SP) of dasatinib has a characteristic XRPD pattern, which is shown in Figure no. 3. The solvate (SP) has the following characteristic peaks: 6.04; 12.01; 15.10; 17.95; 18.35; 18.77; 21.25; 21.51; 22.96; 24.08; 24.62; 25.80; 26.16; 28.16 and 33.6578 ± 0.2° 2-theta.These solvates (or polymorph forms) are then easily converted to the desired anhydrous polymorph N-6 or another solvate in steps b) and c). All the forms prepared this way are sufficiently stable and can easily be isolated in the chemical purities of 99% and higher (in accordance with HPLC).The anhydrous polymorph form N-6 is prepared in the following way: any solvate or another polymorph is dissolved under an inert atmosphere at 90°C (reflux) in a 10-30 times, most preferably 20 times, the (weight) amount of the crystallization solvent. Suitable crystallization solvents include especially methanol, ethanol, isopropanol, most preferably methanol.A co-solvent is added in 0.1-10 times, most preferably ½-l times, the volume of the crystallization solvent used in an inert atmosphere at 90°C. The co-solvent can be, e.g., acetonitrile, propionitrile and their mixtures, most preferably acetonitrile. After addition of the co-solvent the obtained solution is withdrawn from the heating bath and is slowly left to cool down to 22°C under continuous stirring in an inert atmosphere. Crystallization occurs during 1-120 minutes (depending on the volume, until complete cooling). After having cooled down to 22°C (laboratory temperature), the suspension is stirred for another hour. Crystalline dasatinib is aspirated by well-known techniques in an inert atmosphere at 10-35°C, most preferably at 22°C, and washed with the corresponding co-solvent. The chemical purity of the obtained product is 99% (in accordance with HPLC); it is the polymorph form N-6 and its XRPD pattern is shown in Figure no. 4. The polymorph form N-6 has the following characteristic peaks: 6.77; 12.31; 13.16; 13.75; 16.70; 17.20; 18.54; 19.34; 20.25; 20.95; 21.94; 24.28; 24.82; and 27.80 ± 0.2° 2-theta.Brief Description of Drawings:Figure 1: shows an X-ray powder diffraction pattern of the crystalline solvate NM. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation.Figure 2: shows an X-ray powder diffraction pattern of the isoamyl alcohol crystalline solvate SI. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation. Figure 3: shows an X-ray powder diffraction pattern of the 1,3 propanediol crystalline solvate SP. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation.Figure 4: shows an X-ray powder diffraction pattern of the crystalline anhydrous form N-6. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation.Examples: The following working examples illustrate methods for the preparation of dasatinib of formula I, its polymorph form N-6 and its solvates NM, SI, SP.The polymorph forms and solvates of dasatinib were characterized with X-ray powder diffraction using the following methods:The diffraction patterns were measured using an X’PERT PRO MPD PANalytical diffractometer with a graphite monochromator, radiation used CuKa (λ=1.542 A), excitation voltage: 45 kV, anode current: 40 mA, measured range: 2 – 40° 2Θ, increment: 0.01° 2Θ. The measurement was carried out using a flat powder sample that was placed on a Si plate. For the primary optic setting programmable divergence diaphragms with the irradiated sample area of 10 mm, Soller diaphragms 0.02 rad and an anti-dispersion diaphragm ¼ were used. For the secondary optic setting an X’Celerator detector with the maximum opening of the detection slot, Soller diaphragms 0.02 rad and an anti-dispersion diaphragm 5.0 mm were used. HPLC method:Stock solution of samples: dissolve 5.0 mg of the sample in 10.0 ml of 50% acetonitrile R with water.Dimensions of the chromatographic HPLC column: / = 0.10 m, d= 3 mm- stationary phase: Zorbax Eclipse Plus Phenyl-Hexyl RRHD 1.8 μιη; temperature: 35 °C. Mobile phase: A: phosphate buffer (0.01 M sodium dihydrogen phosphate, pH treated by addition of sodium hydroxide to 7.00 ± 0.05); B: acetonitrile R.Gradient (A/B; flow 0.6 ml/min): 0 min 80/20; 10 min 50/50; 11 min 50/50; 12 min 80/20. Detection at the wavelength of 220 nm.Feed: 2 μΐ of the sample stock solution Example 1.Preparation of the NMP solvate (NM) of dasatinib:The intermediate of formula II (1.00 g; 2.54 mmol) and l-(2-hydroxyethyl)piperazine (1.66 g; 12.77 mmol) were dissolved in N-methylpyrrolidone (5 ml) under an inert atmosphere and diisopropylethylamine (0.9 ml, 5.18 mmol) was added to the reaction mixture. The reaction mixture was stirred and heated up to 90°C for 70 minutes and then acetonitrile (30 ml) was added to the reaction. The mixture was withdrawn from the heating bath and stirred intensively. Crystallization started after 5 minutes, the suspension was left to cool down under continuous stirring. After achieving the laboratory temperature it was stirred for another 2 hours. The crystalline substance was aspirated on frit S3, washed with acetonitrile (5 ml) and dried by suctioning under an inert nitrogen atmosphere for 15 minutes. The XRPD pattern of the sample obtained this way corresponds to the NMP solvate (NM) and can be used in the subsequent steps without the necessity of drying. Drying after 6 hours in an exsiccator at the laboratory temperature in vacuo (50 kPa) provided 1.2 g of crystalline dasatinib; 80% of the theoretical yield. HPLC purity 99.12%. The 1H NMR and 13C NMR spectra correspond to the data known from the literature. The XRPD pattern of the dried product corresponds to the NMP solvate (NM). The NM solvate is characterized by the reflections presented in Table 1 :Table 1 – NM forminterplanarpos. distance[°2Th.] [nm] rel. int. [%]5.88 1.5024 81.86.73 1.3131 100.010.73 0.8236 10.611.92 0.7420 59.213.39 0.6606 19.614.97 0.5915 38.416.72 0.5298 45.018.95 0.4679 10.920.17 0.4399 13.921.46 0.4138 13.422.81 0.3895 21.024.65 0.3608 13.325.18 0.3534 14.426.02 0.3422 11.928.06 0.3177 5.8
Norepinephrine bitartrate

Norepinephrine bitartrate
Arterenol bitartrate
RN: 3414-63-9
FREE FORM 138-65-8
UNIIIFY5PE3ZRW
R FORM CAS Number108341-18-0,
- 1,2-Benzenediol, 4-(2-amino-1-hydroxyethyl)-, (R)-, [R-(R*,R*)]-2,3-dihydroxybutanedioate (1:1) (salt), monohydrate
- 1,2-Benzenediol, 4-[(1R)-2-amino-1-hydroxyethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (1:1) (salt), monohydrate (9CI)
- Arterenol, tartrate, monohydrate (6CI)
- L-Noradrenaline bitartrate monohydrate
- Levarterenol bitartrate monohydrate
WeightAverage: 337.281
Chemical FormulaC12H19NO10
(+-)-Arterenol bitartrate
(+-)-Noradrenaline bitartrate
(+-)-Norepinephrine bitartrate
(2R,3R)-2,3-dihydroxybutanedioic acid 4-[(1R)-2-amino-1-hydroxyethyl]benzene-1,2-diol hydrate
ORD +41.3 °, water, 4% ; Wavlen: 589.3 nm; Temp: 25 °C, AND MP 163-165 °C, GB 747768 1956 NorepinephrineCAS Registry Number: 51-41-2CAS Name: 4-[(1R)-2-Amino-1-hydroxyethyl]-1,2-benzenediolAdditional Names: (-)-a-(aminomethyl)-3,4-dihydroxybenzyl alcohol; l-3,4-dihydroxyphenylethanolamine; noradrenaline; levarterenolTrademarks: Adrenor; Levophed (Winthrop)Molecular Formula: C8H11NO3Molecular Weight: 169.18Percent Composition: C 56.79%, H 6.55%, N 8.28%, O 28.37%Literature References: Demethylated precursor of epinephrine, q.v. Occurs in animals and man, and is a sympathomimetic hormone of both adrenal origin and adrenergic orthosympathetic postganglionic origin in man. Physiologic review: Malmejac, Physiol. Rev.44, 186 (1964). It has also been found in plants, e.g., Portulaca olerocea L., Portulacaceae: Fing et al.,Nature191, 1108 (1961). Synthesis of dl-form: Payne, Ind. Chem.37, 523 (1961). Historic review of synthesis: Loewe, Arzneim.-Forsch.4, 583 (1954). Resolution of dl-form: Tullar, J. Am. Chem. Soc.70, 2067 (1948); idem,US2774789 (1956 to Sterling Drug). Configuration: Pratesi et al.,J. Chem. Soc.1959, 4062. Comprehensive description: C. F. Schwender, Anal. Profiles Drug Subs.1, 149-173 (1972); T. D. Wilson, ibid.11, 555-586 (1982).Properties: Microcrystals, dec 216.5-218°. [a]D25 -37.3° (c = 5 in water with 1 equiv HCl).Optical Rotation: [a]D25 -37.3° (c = 5 in water with 1 equiv HCl)
Derivative Type: HydrochlorideCAS Registry Number: 329-56-6Trademarks: Arterenol (HMR)Molecular Formula: C8H11NO3.HClMolecular Weight: 205.64Percent Composition: C 46.73%, H 5.88%, N 6.81%, O 23.34%, Cl 17.24%Properties: Crystals, mp 145.2-146.4°. [a]D25 -40° (c = 6). Freely sol in water. Solns slowly oxidize under the influence of light and oxygen in a manner comparable to epinephrine hydrochloride.Melting point: mp 145.2-146.4°Optical Rotation: [a]D25 -40° (c = 6)
Derivative Type:d-BitartrateCAS Registry Number: 69815-49-2Additional Names: Levarterenol bitartrateTrademarks: Aktamin; BinodrenalMolecular Formula: C8H11NO3.C4H6O6Molecular Weight: 319.26Percent Composition: C 45.14%, H 5.37%, N 4.39%, O 45.10%Properties: Obtained as the monohydrate, crystals, mp 102-104°. [a]D25 -10.7° (c = 1.6 in H2O). When anhydr, mp 158-159° (some decompn). Freely sol in water.Melting point: mp 102-104°; mp 158-159° (some decompn)Optical Rotation: [a]D25 -10.7° (c = 1.6 in H2O)
Derivative Type:dl-FormProperties: Crystals, dec 191°. Sparingly sol in water; very slightly sol in alc, ether; readily sol in dilute acids, caustic.
Therap-Cat: Adrenergic (vasopressor); antihypotensive.Therap-Cat-Vet: Sympathomimetic; vasopressor in shock.Keywords: a-Adrenergic Agonist; Antihypotensive.
Precursor of epinephrine that is secreted by the adrenal medulla and is a widespread central and autonomic neurotransmitter. Norepinephrine is the principal transmitter of most postganglionic sympathetic fibers and of the diffuse projection system in the brain arising from the locus ceruleus. It is also found in plants and is used pharmacologically as a sympathomimetic.
Norepinephrine (sometimes referred to as l-arterenol/Levarterenol or l-norepinephrine) is a sympathomimetic amine which differs from epinephrine by the absence of a methyl group on the nitrogen atom.
Norepinephrine Bitartrate is (-)-α-(aminomethyl)-3,4-dihydroxybenzyl alcohol tartrate (1:1) (salt) monohydrate and has the following structural formula:
 |
LEVOPHED is supplied in sterile aqueous solution in the form of the bitartrate salt to be administered by intravenous infusion following dilution. Norepinephrine is sparingly soluble in water, very slightly soluble in alcohol and ether, and readily soluble in acids. Each mL contains the equivalent of 1 mg base of norepinephrine, sodium chloride for isotonicity, and not more than 2 mg of sodium metabisulfite as an antioxidant. It has a pH of 3 to 4.5. The air in the ampuls has been displaced by nitrogen gas.
Norepinephrine, also known as noradrenaline, is a medication used to treat people with very low blood pressure.[2] It is the typical medication used in sepsis if low blood pressure does not improve following intravenous fluids.[3] It is the same molecule as the hormone and neurotransmitter norepinephrine.[2] It is given by slow injection into a vein.[2]
Common side effects include headache, slow heart rate, and anxiety.[2] Other side effects include an irregular heartbeat.[2] If it leaks out of the vein at the site it is being given, norepinephrine can result in limb ischemia.[2] If leakage occurs the use of phentolamine in the area affected may improve outcomes.[2] Norepinephrine works by binding and activating alpha adrenergic receptors.[2]
Norepinephrine was discovered in 1946 and was approved for medical use in the United States in 1950.[2][4] It is available as a generic medication.[2]
Medical uses
Norepinephrine is used mainly as a sympathomimetic drug to treat people in vasodilatory shock states such as septic shock and neurogenic shock, while showing fewer adverse side-effects compared to dopamine treatment.[5][6]
Mechanism of action
It stimulates α1 and α2 adrenergic receptors to cause blood vessel contraction, thus increases peripheral vascular resistance and resulted in increased blood pressure. This effect also reduces the blood supply to gastrointestinal tract and kidneys. Norepinephrine acts on beta-1 adrenergic receptors, causing increase in heart rate and cardiac output.[7] However, the elevation in heart rate is only transient, as baroreceptor response to the rise in blood pressure as well as enhanced vagal tone ultimately result in a sustained decrease in heart rate.[8] Norepinephrine acts more on alpha receptors than the beta receptors.[9]
Names
Norepinephrine is the INN while noradrenaline is the BAN.
SYN
Chemical Synthesis
Norepinephrine, L-1-(3,4-dihydroxyphenyl)-2-aminoethanol (11.1.4), is synthesized by two methods starting from 3,4-dihydroxybenzaldehyde. According to the first method, the indicated aldehyde is transformed into the cyanohydrin (11.1.3) by reaction with hydrogen cyanide, which is then reduced into norepinephrine (11.1.5).
The second method consists of the condensation of diacetate of the same aldehyde with nitromethane, which forms (3,4-diacetoxyphenyl)-2-nitroethanol (11.1.5). Then the nitro group is reduced and the product (11.1.6) is hydrolyzed into the desired norepinephrine (11.1.4) [4,9,13,14].
Purification Methods
Recrystallise adrenor from EtOH and store it in the dark under N2. [pKa, Lewis Brit J Pharmacol Chemother 9 488 1954, UV: Bergstr.m et al. Acta Physiol Scand 20 101 1950, Fluorescence: Bowman et al. Science NY 122 32 1955, Tullar J Am Chem Soc 70 2067 1948.] The L-tartrate salt monohydrate has m 102-104.5o, [] D -11o (c 1.6, H2O), after recrystallisation from H2O or EtOH. [Beilstein 13 III 2382.]
PATENT
https://patents.google.com/patent/WO2013008247A1/en4-[(lR)-2-amino-l-hydroxyethyl]benzene-l,2-diol, commonly known as (R)-(-)- norepinephrine or noradrenaline is a catecholamine with multiple roles including as a hormone and a neurotransmitter. As a stress hormone, norepinephrine affects parts of the brain where attention and responding actions are controlled. Along with epinephrine, norepinephrine also underlies the fight-or-flight response, directly increasing heart rate, triggering the release of glucose from energy stores, and increasing blood flow to skeletal muscle. Norepinephrine also has a neurotransmitter role when released diffusely in the brain as an antiinflammatory agent.When norepinephrine acts as a drug it increases blood pressure by increasing vascular tone through a-adrenergic receptor activation. The resulting increase in vascular resistance triggers a compensatory reflex that overcomes its direct stimulatory effects on the heart, called the baroreceptor reflex, which results in a drop in heart rate called reflex bradycardia.(R)-(-)-Norepinephrine has a following structure:

(R)-(-)-Norepinephrine was first time disclosed in the US patent US2774789, where it was obtained by resolution of dl-norepinephrine, with optically active acids such as d- tartaric acid, 1-malic acid or N-benzoyl-l-threonine. The patent does not disclose the preparation of dl-norepinephrine. The patent GB747768 describes reduction of amino ketones where 3,4-dihydroxy-a- aminoacetophenone hydrochloride was converted into its d-tartrate salt; followed by reduction of the d-tartrate salt. This process leads to formation of excessive amount of d- adrenaline d-tartrate (which is a bi-product) as it crystallized first; whereas the desired 1- adrenaline d-tartrate crystallizes after 2 days and in smaller yield. Also the patent does not disclose the source of 3,4-dihydroxy-a-aminoacetophenone hydrochloride.It has been unsuccessfully tried to treat dihydroxy-a-chloroacetophenone with hexamethylenetetramine (commonly known as hexamine) and to treat the reaction product with an acid to obtain arterenone (see Mannich, Hahn B., Berichte der deutschen chemischen Gesellschaft, volume 44, issue 2, Pages 1542 – 1552 (1911)). Mannich found that the treatment of this and similar halogen ketones with hexamine did not produce an addition compound but resulted in splitting of halogen acid which made the process impossible. Mannich also found that an addition compound of the halogen ketone and hexamine is formed only when the two phenolic hydroxyl groups are closed i.e. protected by acylation or etherification. Hence according to Mannich, the reaction is not at all possible for the compounds containing two unprotected phenolic hydroxyl groups. The US patent US 1680055 discloses the preparation of monohydroxy-a-substituted- aminoacetophenones either by reacting monohydroxy-a-bromoacetophenones with a substituted amine or by reacting protected monohydroxy-a-bromoacetophenones with a substituted amine followed by deprotection. The patent does not disclose the preparation of dihydroxy-a-aminoacetophenones (where amino group is unsubstituted).It is disclosed in the US patent US2786871 that when chloroaceto pyrocatechol is treated with ammonia, arterenone is obtained in 50% yield. However when the reaction is carried out in basic medium, darkening of the reaction mass takes place which results in coloured product. The patent also discloses preparation of amino-methyl-(monohydroxyphenyl)- ketones by reacting halogen ketone with hexamine. It is also disclosed in the patent that the process is applicable only to the halogenomethyl-monohydroxyphenyl-ketones.Following are some of the methods for preparation of 3,4-dihydroxy-a- aminoacetophenone, reported in the literature. J. Am. Pharm. Association (1946) 35, 306 – 309 discloses preparation of 3,4-dihydroxy- a-aminoacetophenone by reacting 3,4-dihydroxy-a-chloroacetophenone with dibenzyl amine followed by hydrogenation of resulting dibenzylamino ketone. The main disadvantage of this reaction is formation of derivatives of dibenzyl amines, which remain in the final product in the form of impurities.Acta Chimica Academiae Scientiarum Hungaricae (1951), 1, 395-402, discloses preparation of 3,4-dihydroxy-a-aminoacetophenone from 3,4-dihydroxyphenyloxo acetaldehyde and benzyl amine followed by reduction of benzylamino ketone intermediate. The main disadvantage of this method is that the starting acetaldehyde derivative is very expensive and not easily available.It is disclosed in Recueil des Travaux Chimiques des Pays-Bas et de la Belgique (1952), 71, 933-44, that 3,4-dihydroxy-a-aminoacetophenone hydrochloride is formed by demethylation of 3,4-dimethoxy-a-aminoacetophenone hydrochloride using 48% HBr. The reaction results in less than 10% yield of the aminoacetophenone.Monatshefte fuer Chemie (1953), 84 1021-32, discloses preparation of 3,4-dihydroxy-a- aminoacetophenone by reacting 3,4-dihydroxy-a-chloroacetophenone with sodium azide followed by hydrogenation of azide intermediate using 4% palladium on carbon as a catalyst. In the hydrogenation step, 1.6 gm of azide intermediate requires 1.4 gm of catalyst, which is not economical and industrially feasible.
Preparation of 3,4-dihydroxy-a-aminoacetophenones hydrochloride is disclosed in J. Am. Chem. Soc, 1955, volume 77, issue 10, pages 2896 – 2897. The following scheme is disclosed in the article:


It is clear from the above scheme that the process requires additional steps of protection and deprotection of hydroxyl and amino groups, and use of potassium phthalimide requires anhydrous reaction conditions. Therefore the process is time consuming and not economical.Chinese patent CN101798271A describes reduction of 3,4-dihydroxy-a- aminoacetophenone hydrochloride in water as solvent followed by neutralization with aqueous ammonia. Since dl-norepinephrine has partial solubility in aqueous basic medium result in to loss of product. Also it is necessary to maintain low volume of solvent throughout the process for better yields making the process stringent.European patent EP1930313 discloses preparation of a-amino ketones. The preparation is carried out by reacting an organic sulfide in a polar solvent with a compound containing a leaving group attached to a primary or secondary carbon atom to form a sulfonium salt, which is reacted with a ketone in presence of a base and a polar solvent. Oxiranes obtained are further converted into the corresponding aminoketone, by aminolysis followed by selective oxidation. The following scheme is disclosed in the patent.

It is clear from the above scheme that the process requires many steps and hence is time consuming. The patent does not exemplify the synthesis of dihydroxy-a- aminoacetophenones.Thus, the search for a suitable manufacturing process for (R)-norepinephrine intermediates remains undoubtedly of interest. We were surprised to find that hardly any literature discloses the process for preparation of dihydroxy-a-aminoacetophenones acid addition salts. We have found that the reaction of dihydroxy-a-haloacetophenone with hexamine is feasible and results in high yield of product although both the hydroxyl groups on the phenyl ring of acetophenone are unprotected. Object of the invention:It is therefore an object of the invention is to overcome or ameliorate at least one disadvantage of the prior art or to provide a useful alternative.Another object of the invention is to provide a novel, safe, efficient, concise, ecological, high yielding, industrially feasible and simpler process for preparation of (R)-(-)- norepinephrine intermediates.Another object of the invention is to provide a process for synthesis of 3,4-dihydroxy-a- aminoacetophenone salt, which is feasible without protecting both the hydroxyl group on the phenyl ring of acetophenone.Yet another object of the invention is to provide an improved process for hydrogenation of 3,4-dihydroxy-a-aminoacetophenone salt to prepare (dl)-norepinephrine salt.Summary of the invention:In accordance with the above objectives, the present invention provides a process for preparation of (dl)-norepinephrine intermediate of formula (III) comprising reacting 3,4- dihydroxy-a-haloacetophenone of formula (I) with hexamine to provide a quaternary ammonium salt of formula (II); followed by hydrolyzing the quaternary ammonium salt of formula (II) with an acid.In a second aspect, the present invention provides a novel quaternary ammonium salt of formula (II) and its preparation.In a third aspect, the present invention provides a novel process for hydrogenation of 3,4- dihydroxy-a-aminoacetophenone acid salt to provide (dl)-norepinephrine acid addition salt.Example 1Preparation of quaternary ammonium saltA 5000 ml four neck round bottom flask with water condenser and calcium chloride tube was charged with Hexamine (210.28 gm), chloroform (1200 ml), 3,4-dihydroxy-a- chloroacetophenone (250 gm) and isopropanol (1000 ml) at room temperature. The reaction mass was gently heated at 63°C for 4 hours. The reaction was monitored by TLC. The reaction mass was cooled to room temperature and filtered to get solid. The solid was washed with acetone and dried at 50°C for 4 hours to obtain quaternary ammonium salt which was used in the next step without purification.Yield – 410 gm (93.65%)Nature – off white solidm.p. – 180 to l82°CNMR (DMSO-d6): – δ =4.51 – 4.75 (m, 8H), 5.39 (s, 6H), 6.92 (d, 1H, J= 7.5 Hz), 7.37 – 7.42 (m, 2H), 9.67 (s, br, 1H), 10.44 (s, br, 1H)Example 2Preparation of 3,4-dihydroxy-a-aminoacetophenone hydrochlorideA 2000 ml four neck round bottom flask with water condenser and calcium chloride tube was charged with the quaternary ammonium salt obtained in the example 1 (120 gm), methanol (862.5 ml) and cone, hydrochloric acid (194.4 ml). The reaction mixture was heated to 60 to 65°C and aged at same temperature for 3 to 4 hours. The reaction was monitored by TLC. The reaction mass was cooled and neutralized using base to give 3,4- dihydroxy-a-aminoacetophenone. The solid was filtered, washed with water and dried at 50°C. This base was further converted in to its hydrochloride salt with IPA-HC1 mixture. Yield – 72 gm (96.3%)Nature – off white solidHPLC – 99.7%1H NMR(CD30D) – 5 = 3.62(s, 1H), 6.80 (d, J = 8 Hz, 1H), 7.38 (d, J = 1.3 Hz, 1H), 7.63 (d, J = 8 Hz, 1H).Example 3Preparation of (dl)-norepinephrine hydrochlorideA 500 ml hydrogenation flask was charged with 3,4-dihydroxy-a-aminoacetophenone hydrochloride obtained in the example 2 (55 gm), 10% palladium on carbon (5 gm) and methanol (300 ml). The reaction mixture was heated to 45°C with hydrogen gas pressure of 4 to 5 kg m2. The reaction mixture was stirred at 45°C for 5 hours. The catalyst was removed by filtration. The filtrate was cooled to 5 to 10 °C and ammonia gas was passed through the solvent for 2 h till the pH of the solution was around 9. The solid obtained was filtered, washed with methanol and dried in air to obtain (dl)-norepinephrine. Yield – 43.5 gm (96.7%)Nature white crystalline solidHPLC 99.6%Example 4Preparation of (dl)-norepinephrine hydrochlorideA 500 ml hydrogenation flask was charged with 3,4-dihydroxy-a-aminoacetophenone hydrochloride obtained from process similar to example 2 (55 gm), 10% palladium on carbon (5 gm) and methanol (300 ml). The reaction mixture was aged at 25 °C with hydrogen gas pressure of 4 to 3 kg/m2. The reaction mixture was stirred at 25°C for 15 hours. The reaction was monitored by TLC. The catalyst was removed by filtration. The filtrate was cooled to 5 to 10 °C and ammonia solution was added to the reaction mixture till the pH of the solution around 9. The solid obtained was filtered, washed with methanol and dried in air to obtain (dl)-norepinephrine.Yield – 41.5 gm (92.2%)Nature – white crystalline solidHPLC – 99.5%
PATENTUS-10865180https://patentscope.wipo.int/search/en/detail.jsf?docId=US283323778&_cid=P11-KMEC1N-93277-1
| Norepinephrine Bitartrate (Arterenol Bitartrate) is chemically known as (−)-α-(aminomethyl)-3, 4-dihydroxybenzyl alcohol tartrate (1:1) (salt) monohydrate is a catecholamine family that functions in the brain and body as a hormone and neurotransmitter. As a stress hormone, Norepinephrine affects parts of the brain where attention and responding actions are controlled. Along with epinephrine, Norepinephrine also underlies the fight-or-flight response, directly increasing heart rate, triggering the release of glucose from energy stores, and increasing blood flow to skeletal muscle. Norepinephrine also has a neurotransmitter role when released diffusely in the brain as an anti-inflammatory agent. |
| LEVOPHED® (l-Norepinephrine) is supplied in sterile aqueous solution in the form of the bitartrate salt to be administered by intravenous infusion following dilution. Norepinephrine is sparingly soluble in water, very slightly soluble in alcohol and ether, and readily soluble in acids. Each ml contains the equivalent of 1 mg base of Norepinephrine, sodium chloride for isotonicity, and not more than 2 mg of sodium metabisulfite as an antioxidant. |
| Norepinephrine Bitartrate is (−)-α-(amino methyl)-3,4-dihydroxybenzyl alcohol tartrate (1:1) (salt) monohydrate and has the following structural formula: |
| (l)-Norepinephrine was first disclosed in 1947 by Sterling Drugs. U.S. Pat. No. 2,774,789 discloses the resolution of dl-Norepinephrine with optically active acids such as d-tartaric acid, 1-malic acid or N-benzoyl-l-threonine. The patent does not disclose the basic synthesis of dl-Norepinephrine. |
| Journal of the American Chemical Society, Volume 70 (6), 1948 describes the resolution of dl-Norepinephrine in to d-arterenol-d-bitartrate and l-arterenol-d-bitartrate in water and aqueous methanol. Further it also describes isolation of d-arterenol and l-arterenol form above tartrate salts. |
| U.S. Pat. No. 2,786,871 discloses the process for the preparation of arterenol wherein chloroacetopyrocatechol is treated with ammonia and arterenol is obtained in 50% yield. |
| J. Am. Pharm. Association (1946) 35, 306-309 discloses preparation of 3,4-dihydroxyaminoacetophenone by reacting 3,4-dihydroxy-α-chloroacetophenone with dibenzyl amine, followed by hydrogenation of the resulting dibenzylamino ketone. The main disadvantage of this reaction is the formation of derivatives of dibenzyl amines, which carried over to final product in the form of impurities. |
| Acta Chimica Academiae Scientiarum Hungaricae (1951), 1, 395-402 discloses preparation of 3, 4-dihydroxy-α-aminoacetophenone from 3,4-dihydroxyphenyloxo acetaldehyde and benzyl amine followed by reduction of the benzylamino ketone intermediate. The main disadvantage of this method is that the starting acetaldehyde derivative is very expensive and not easily available. |
| CN101798271A describes reduction of 3,4-dihydroxy-α-aminoacetophenone hydrochloride in water as solvent followed by neutralization with aqueous ammonia. Since dl-Norepinephrine has partial solubility in aqueous basic medium, this process results in a loss of product. Also, it is necessary to maintain low volume of solvent throughout the process for better yields making the process stringent. |
| WO2009004593 describes the process for the preparation of Epinephrine wherein (−) epinephrine is obtained by chiral separation of dl-epinephrine using the chiral acid such as L-tartaric acid with an optical purity of 95.24%. |
| WO2013008247 discloses a process for preparation of (dl)-norepinephrine hydrochloride salt by reacting 3,4-dihydroxy-a-haloacetophenone with hexamethylenetetramine to provide hexamine salt; followed by hydrolysis and hydrogenation. However, this process fails to teach the resolution of (dl)-norepinephrine hydrochloride and preparation of l-Norepinephrine Bitartrate monohydrate. |
| WO2016038422 discloses a process for the preparation of optically enriched adrenaline or adrenaline tartrate comprising the steps of: (a) reacting a mixture of (−)-adrenaline and (+)-adrenaline with L(+)-tartaric acid to form adrenaline tartrate; (b) contacting the adrenaline tartrate with less than 1 equivalent of ammonium hydroxide. However, the product achieved is with purity of only 98%. |
| CN107298646 describes the process for the preparation of Norepinephrine wherein L-Norepinephrine tartrate is obtained by chiral separation of dl-Norepinephrine using the chiral acid such as L-tartaric acid. The chiral separation step using L-tartaric acid is repeated once to obtain pure Norepinephrine. However, there is no information on bitartrate salt and its optical purity. |
| In light of the above, there remains a need in the art for highly pure l-Norepinephrine Bitartrate having high enantiomeric purity i.e. greater than 99.0% so as to provide enhanced therapeutic efficacy and safety when administered. Surprisingly the present inventors have found out a process for the preparation of (l)-Norepinephrine Bitartrate having enantiomeric purity greater than 99.5%, for which protection is sought. |
Reference Example-1(U.S. Pat. No. 2,774,789, Example-A)
Preparation of l-Norepinephrine Bitartrate
| To a four necked 100 ml flask charged racemic Norepinephrine base (20 gm), d-(−) tartaric acid (18.34 gm), and water (35 ml) at room temperature. The reaction mass was stirred to obtain clear solution, cooled to 0-5° C. After 5 hours slight turbidity was observed. Turbidity increases slowly to get thick white slurry after 6 hours, reaction mass becomes very thick which was difficult to filter, washed solid wet cake by 4.0 ml water followed by two 12 ml portions of 95% ethanol. Suck dried the solid completely, dried at 45° C. to get l-Norepinephrine Bitartrate (28 gm) which is in crude form. |
| Crude l-Norepinephrine Bitartrate (20 gm) dissolved in 14 ml of water at 50° C. Clear solution was obtained. Activated charcoal was added to this solution and stirred the reaction mass for more for 30 min. Filtered through Hyflo and cooled to 0-5° C. After 2 hours, clear solution obtained gets converted to thick solid mass. Filtered and washed the solid with 1.5 ml of chilled water followed 14 ml of 95% ethanol. |
| This dry solid 8 gm (after 1 st purification) was then dissolved in 8 ml of water at 50° C. to get clear solution. This reaction mass was then cooled to 0-5° C. After 1 hour, a clear solution gets converted to a thick solid mass. Maintained the reaction mass for more than 2 hours at the same conditions. Filtered the thick solid and washed with 95% ethanol. Dried the solid at 45° C. to obtain l-Norepinephrine Bitartrate. |
| Chiral Purity by HPLC: l-Norepinephrine Bitartrate=68.45%, and d-isomer=31.55% |
| Specific Optical Rotation: −6.33° |
Reference Example-2 (JAGS, 1948, Page-2067-68, Example-a)
| To a four necked flask charged racemic Norepinephrine base (20 gm), d-(−) tartaric acid (18.34 gm), and water (35.20 ml) at room temperature. After 5 minutes reaction mass becomes clear liquid. Cooled the reaction mass to 2-3° C. After 30 minutes, reaction mass was observed to be turbid and further the reaction mass becomes very thick. This mass was, stirred for 2 hours at 0-5° C. Then filtered reaction mass at same temperature and washed solid wet cake with 3.5 ml water followed by two 11.8 ml portions of 95% ethanol. Dried the solid at air oven at 45° C. to get crude tartrate salt (15 gm). |
| Crude tartrate salt (15 gm) was dissolved in 5 ml of water at 50° C. to get clear solution. Cooled to 2-3° C. After 30 minutes, a clear solution gets converted to a thick solid mass. Filtered the solid and washed with 1.5 ml of chilled water and then 15 ml of 95% ethanol. Dried the solid at 45° C. to obtain semi pure l-Norepinephrine Bitartrate (8 gm). |
| This semi pure l-Norepinephrine Bitartrate (8 gm) was dissolved in 8 ml of water at 50° C. to get clear solution. Cooled the mass to 2-3° C. After 30 minutes clear solution gets converted to thick solid mass. Filtered the solid and washed with 8 ml of 95% ethanol. Dried the solid at 45° C. to obtain pure l-Norepinephrine Bitartrate (3 gm). |
| Chiral Purity: l-Norepinephrine Bitartrate=77.14%, d-isomer=22.86% |
| Specific Optical Rotation: −10.4° |
Example-1: Preparation of 2-Chloro-1-(3, 4-Dihydroxyacetophenone)
| In round bottom flask, charged Methylene Chloride (1000 ml), Aluminium chloride (300 gm) and cooled to 0-5° C. Pyrocatechol (100 gm) was added lot wise. Chloroacetyl chloride (108 gm) was added drop wise at 0-5° C. Then stirred the reaction mass at 25-30° C. for 20-24 hours. After completion of the reaction, reaction mass was quenched in aq. HCl, filtered the reaction mass and wet cake was charged in water containing acetic acid. Filtered the reaction mass and cooled to 15-20° C., filtered solid and washed with water. |
| Yield: 110 gm. |
| HPLC Purity: 99.5% |
Example-2: Preparation of Hexamine Salt
| In a round bottom flask charged 2-chloro-1-(3, 4-dihydroxyacetophenone) (100 gm), Hexamine (87 gm), IPA (500 ml), Chloroform (400 ml). Stirred the reaction mass at reflux temperature for 6 hours. After completion of the reaction, cooled to 25-30° C., filtered and washed the wet cake with IPA and Methanol. |
| Yield: 160 gm. |
| HPLC Purity: 99.3% |
Example-3: Preparation of 2-Amino-1-(3,4-Dihydroxyphenyl)Ethanone Hydrochloride
| In a round bottom flask charged Hexamine salt (100 gm), Methanol (600 ml), aqueous HCl and heated the reaction mass to 55-60° C. After completion of the reaction, the mass was dissolved in water, by adjusting pH with liquor ammonia. Filtered the solid and washed with water, dried the material at 45-50° C. |
| This free base was charged in 900 ml methanol and pH was adjusted to 1-1.5 by IPA.HCl and distilled off methanol completely to get white solid which was isolated by filtration. |
| Yield: 37 gm |
| HPLC Purity: 99.5% |
Example-4: Preparation of [4-(2-Amino-1-Hydroxyethyl) Benzene-1, 2-Diol] (Racemic Norepinephrine Base)
| Charged 2-amino-1-(3, 4-dihydroxyphenyl) ethanone hydrochloride (100 gm), 10% Pd/C(10 gm), methanol (700 ml) and water (300 ml) mixture in autoclave. Stirred the reaction mass at 40-45° C. After completion of reaction, Pd/C was removed by filtration. Collected filtrate and distilled off methanol. pH was adjusted by liquor ammonia. Isolated the solid by filtration and washed with water followed by methanol. Dried the solid at 40-45° C. |
| Yield: 67 gm |
| Purity: 99.2% |
Example-5: Preparation of l-Norepinephrine Base
| Charged racemic Norepinephrine base (100 gm), D-(−)-Tartaric acid (142 gm), water (100 ml) in a round bottom flask. The reaction mass was stirred to get clear solution. After some time, solid started to crystallize. Reaction mass was diluted with methanol (900 ml). Maintained the reaction mass under stirring for 24 hours at 25-30° C. Filtered and washed the wet cake with methanol to obtain Crude l-Norepinephrine tartrate salt. |
| Yield: 85 gm |
| The crude l-Norepinephrine tartrate salt was converted into its free base by dissolving this crude tartrate salt in water (500 ml) and adjusted pH to 8-8.5 by liquor ammonia and isolated the solid by filtration. Dried the material at 40-45° C. to obtain pure l-Norepinephrine free base (43 gm). |
| Yield: 43 gm (l-Norepinephrine pure base). |
| HPLC Purity: 99.7% |
| Chiral Purity: 98.0% |
Example-6: Preparation of Pure l-Norepinephrine Base
| Charged l-Norepinephrine base (100 gm) obtained from Example-5, D-(−)-Tartaric acid (142 gm), water (100 ml) in a round bottom flask. The reaction mass was stirred to get clear solution. After some time, a solid started to crystallize. Reaction mass was diluted with methanol (900 ml). Maintained the reaction mass under stirring for 24 hours at 25-30° C. Filtered and washed the wet cake with methanol to obtain l-Norepinephrine tartrate salt. |
| Yield: 88 gm |
| The l-Norepinephrine tartrate salt was converted into its free base by dissolving this crude tartrate salt in water (500 ml) and adjusted the pH to 8-8.5 by liquor ammonia and isolated the solid by filtration. Dried the material at 40-45° C. to obtain pure l-Norepinephrine free base (44 gm). |
| Yield: 44 gm (l-Norepinephrine pure base). |
| HPLC Purity: 99.7% |
| Chiral Purity: 99.1% |
Example-7: Preparation of Highly Pure Norepinephrine Bitartrate Monohydrate
| Charged Norepinephrine pure base (100 gm), L-(+) tartaric acid (100 gm), water (100 ml) and methanol (900 ml), Stirred the reaction mass to get clear solution. After some time, a solid started to crystallize then the reaction mass was diluted with methanol (900 ml). Maintained the reaction mass under stirring at 25-30° C. for 24 hours. Filtered and washed the wet cake with methanol to obtain Norepinephrine Bitartrate Monohydrate (90 gm). |
| HPLC Purity: 99.8% |
| Chiral Purity: 99.4% |
Example-8: Purification of l-Norepinephrine Bitartrate Monohydrate
| Charged 100 gm tartrate salt obtained from example-6, purified water (100 ml) and heated the reaction mass to 40-45° C. to obtain clear solution, cooled to 0-5° C. Charged IPA (100 ml) slowly and the mass was stirred for one hour. The solid was isolated by filtration and washed with IPA. Dried the material at 40-45° C. to obtain l-Norepinephrine Bitartrate Monohydrate (82 gm) having high enantiomeric purity. |
| HPLC Purity: 99.85% |
| Chiral Purity: 99.87% |
| Specific Optical rotation: −11.0° |
Example-9
| The following table sets forth the high purity of the l-Norepinephrine Bitartrate monohydrate of the invention as compared with prior art references. |
| [TABLE-US-00001] Referencel-Norepinephrine Example-2Bitartrate U.S. Pat. No.(JACS, 1948,monohydrate 2,774,789Page-2067-68,of the presentPurity CriteriaExample-AExample-a)invention Optical purity of l-68.45%77.14%99.87%NorepinephrineBitartratemonohydrateSpecific Optical−6.33°−10.4°−11.0°rotation(Limit: −10°to −12°) |
| It is evident from the above table that the compound of the present invention has substantially improved optical purity. |
PATENTCN-102525895
Publication numberPriority datePublication dateAssigneeTitleCN101053557A *2006-04-132007-10-17邵长青Noradrenaline bitartrate medicine composition frozen dried powder injectionCN102335123A *2010-07-162012-02-01上海禾丰制药有限公司Noradrenaline bitartrate injection and preparation technology thereofPublication numberPriority datePublication dateAssigneeTitleEP3110399B12014-02-272018-01-10Sintetica S.A.Process for producing a stable low concentration, injectable solution of noradrenalineFamily To Family CitationsCN109394683A *2018-12-072019-03-01远大医药(中国)有限公司A kind of preparation method of noradrenaline bitartrate injection
References
- ^ Andersen, A. M. (1975). “Structural Studies of Metabolic Products of Dopamine. IV. Crystal and Molecular Structure of (−)-Noradrenaline”. Acta Chem. Scand. 29b: 871–876. doi:10.3891/acta.chem.scand.29b-0871.
- ^ Jump up to:a b c d e f g h i j “Norepinephrine Bitartrate”. The American Society of Health-System Pharmacists. Archived from the original on 26 March 2017. Retrieved 26 March 2017.
- ^ Latifi, Rifat (2016). Surgical Decision Making: Beyond the Evidence Based Surgery. Springer. p. 67. ISBN 9783319298245. Archived from the original on 2017-03-27.
- ^ Encyclopedia of the Neurological Sciences. Academic Press. 2014. p. 224. ISBN 9780123851581. Archived from the original on 2017-03-27.
- ^ Rhodes, Andrew; Evans, Laura E (March 2017). “Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock 2016” (PDF). Critical Care Medicine. 45 (3): 486–552. doi:10.1097/CCM.0000000000002255. hdl:10281/267577. PMID 28098591. S2CID 52827184.
We recommend norepinephrine as the first-choice vasopressor (strong recommendation, moderate quality of evidence).
- ^ De Backer D, Biston P, Devriendt J, Madl C, Chochrad D, Aldecoa C, Brasseur A, Defrance P, Gottignies P, Vincent JL (March 2010). “Comparison of dopamine and norepinephrine in the treatment of shock”. The New England Journal of Medicine. 362 (9): 779–89. doi:10.1056/nejmoa0907118. PMID 20200382.
- ^ I Moore, Joanne (6 December 2012). Pharmacology (3 ed.). Springer Science and Business Media. p. 39. ISBN 9781468405248. Retrieved 19 November 2017.
- ^ “CV Physiology | Circulating Catecholamines”. cvphysiology.com. Retrieved 2019-02-27.
- ^ Sacha, Pollard; Stephenie, B Edwin; Cesar, Alaniz (July 2015). “Vasopressor and Inotropic Management Of Patients With Septic Shock”. Physical Therapy. 40 (7): 449–450. PMC 4495871. PMID 26185405.
External links
- “Norepinephrine”. Drug Information Portal. U.S. National Library of Medicine.
- “Norepinephrine bitartrate”. Drug Information Portal. U.S. National Library of Medicine.
////////Norepinephrine bitartrate, ARTERELOL, a-Adrenergic Agonist, Antihypotensive, levarterenol, Adrenor, Levophed,
#Norepinephrine bitartrate, #ARTERELOL, #a-Adrenergic Agonist, #Antihypotensive, #levarterenol, #Adrenor, #Levophed,
O.O[C@H]([C@@H](O)C(O)=O)C(O)=O.NC[C@H](O)C1=CC(O)=C(O)C=C1
Pyridostigmine
Pyridostigmine
- Molecular FormulaC9H13N2O2
- Average mass181.211 Da
155-97-5[RN]3-[(Dimethylcarbamoyl)oxy]-1-methylpyridinium
3-Dimethylcarbamoyloxy-1-methyl-pyridinium5-21-02-00078 (Beilstein Handbook Reference)[Beilstein]
Pyridostigmine BromideCAS Registry Number: 101-26-8CAS Name: 3-[[(Dimethylamino)carbonyl]oxy]-1-methylpyridinium bromideAdditional Names: 3-hydroxy-1-methylpyridinium bromide dimethylcarbamate; 1-methyl-3-hydroxypyridinium bromide dimethylcarbamate; 3-(dimethylcarbamyloxy)-1-methylpyridinium bromideManufacturers’ Codes: Ro-1-5130Trademarks: Kalymin (Temmler); Mestinon (Roche); Regonol (Organon)Molecular Formula: C9H13BrN2O2Molecular Weight: 261.12Percent Composition: C 41.40%, H 5.02%, Br 30.60%, N 10.73%, O 12.25%Literature References: Reversible inhibitor of acetylcholinesterase.
Prepn: Urban, US2572579 (1951 to Hoffmann-La Roche). Mechanism of protective effect in soman poisoning: X. Deyi et al.,Fundam. Appl. Toxicol.1, 217 (1981). Evaluation of effect on neuromuscular function: M. Glikson et al.,ibid.16, 288 (1991). Evaluation of side effects profile under desert conditions: J. E. Cook et al.,Mil. Med.157, 250 (1992). Review of prophylactic effect in nerve agent poisoning: R. M. Dawson, J. Appl. Toxicol.14, 317 (1994).Properties: Shiny, hygroscopic crystals from abs ethanol, mp 152-154°. Freely sol in water, alcohol. Practically insol in ether, acetone, benzene. Aq solns may be sterilized by autoclaving with steam.Melting point: mp 152-154°Therap-Cat: Cholinergic; in treatment of myasthenia gravis. Pre-exposure antidote to chemical warfare agents.Keywords: Cholinergic.
Pyridostigmine is a medication used to treat myasthenia gravis.[1] It is also used together with atropine to end the effects of neuromuscular blocking medication of the non-depolarizing type.[2] It is typically given by mouth but can also be used by injection.[2] The effects generally begin within 45 minutes and last up to 6 hours.[2]
Common side effects include nausea, diarrhea, frequent urination, and abdominal pain.[2] More severe side effects include low blood pressure, weakness, and allergic reactions.[2] It is unclear if use in pregnancy is safe for the fetus.[2] Pyridostigmine is an acetylcholinesterase inhibitor in the cholinergic family of medications.[2] It works by blocking the action of acetylcholinesterase and therefore increases the levels of acetylcholine.[2]
Pyridostigmine was patented in 1945 and came into medical use in 1955.[3] It is on the World Health Organization’s List of Essential Medicines.[4] Pyridostigmine is available as a generic medication.[2]
Medical uses
Pyridostigmine is used to treat muscle weakness in people with myasthenia gravis or forms of congenital myasthenic syndrome and to combat the effects of curariform drug toxicity. Pyridostigmine bromide has been FDA approved for military use during combat situations as an agent to be given prior to exposure to the nerve agent Soman in order to increase survival. Used in particular during the first Gulf War, pyridostigmine bromide has been implicated as a causal factor in Gulf War syndrome.[5]
Pyridostigmine sometimes is used to treat orthostatic hypotension.[6] It may also be of benefit in chronic axonal polyneuropathy.[7]
It is also being prescribed ‘off-label’ for the postural tachycardia syndrome as well as complications resulting from Ehlers–Danlos syndrome.[7][8]
Contraindications
Pyridostigmine bromide is contraindicated in cases of mechanical intestinal or urinary obstruction and should be used with caution in patients with bronchial asthma.[9][10]
Side effects
Common side effects include:[9]
- Sweating
- Diarrhea
- Nausea
- Vomiting
- Abdominal cramps
- Increased salivation
- Tearing
- Increased bronchial secretions
- Constricted pupils
- Facial flushing due to vasodilation
- Erectile dysfunction
Additional side effects include:[9]
- Muscle twitching
- Muscle cramps and weakness
Mechanism of action
Pyridostigmine inhibits acetylcholinesterase in the synaptic cleft, thus slowing down the hydrolysis of acetylcholine. It is a quaternary carbamate inhibitor of cholinesterase that does not cross the blood–brain barrier which carbamylates about 30% of peripheral cholinesterase enzyme. The carbamylated enzyme eventually regenerates by natural hydrolysis and excess ACh levels revert to normal.
The ACh diffuses across the synaptic cleft and binds to receptors on the post synaptic membrane, causing an influx of Na+, resulting in depolarization. If large enough, this depolarization results in an action potential. To prevent constant stimulation once the ACh is released, an enzyme called acetylcholinesterase is present in the endplate membrane close to the receptors on the post synaptic membrane, and quickly hydrolyses ACh.
Names
Pyridostigmine bromide is available under the trade names Mestinon (Valeant Pharmaceuticals), Regonol and Gravitor (SUN Pharma).
Chemistry
Pyridostigmine, 3-[(dimethylaminocarbonyl)oxy]-1-methyl pyridinium bromide, is synthesized from 3-hydroxypyridine, which is reacted with dimethylaminocarbamoyl chloride, which gives 3-(dimethylaminocarbamoyl)pyridine. The last is reacted with methylbromide, giving pyridostigmine.
- R. Urban, U.S. Patent 2,572,579 (1951).
Syn
youtube
SYN
Method of synthesis
i. 3-hydroxypiridine is reacted with dimethylaminocarbamoyl chloride to give 3-(dimethylaminocarbamoyl)pyridine.
ii. The above formed compound is reacted with methylbromide to produce pyridostigmine. [2]


CLIP

Paper
Journal of Biological Chemistry (1961), 236, 1498-500.
Zeitschrift fuer Klinische Medizin (1985) (1986), 41(7), 495-8
Zhonghua Yaoxue Zazhi (1993), 45(6), 601-14.
Trends in Organic Chemistry (2011), 15, 25-31.
PATENT
WO 9822458
PATENT
WO 2008074816
https://patents.google.com/patent/WO2008074816A1/en
References
- ^ World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. p. 429. hdl:10665/44053. ISBN 9789241547659.
- ^ Jump up to:a b c d e f g h i “Neostigmine Bromide”. The American Society of Health-System Pharmacists. Archived from the original on 21 December 2016. Retrieved 8 December 2016.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 540. ISBN 9783527607495. Archived from the original on 2016-12-20.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Golomb BA (March 2008). “Acetylcholinesterase inhibitors and Gulf War illnesses”. Proceedings of the National Academy of Sciences of the United States of America. 105 (11): 4295–300. Bibcode:2008PNAS..105.4295G. doi:10.1073/pnas.0711986105. JSTOR 25461411. PMC 2393741. PMID 18332428. Lay summary – Reuters (March 10, 2008).
- ^ Gales BJ, Gales MA (2007). “Pyridostigmine in the treatment of orthostatic intolerance”. Annals of Pharmacotherapy. 41 (2): 314–8. doi:10.1345/aph.1H458. PMID 17284509. S2CID 22855759.
- ^ Jump up to:a b Gales BJ, Gales MA (February 2007). “Pyridostigmine in the treatment of orthostatic intolerance”. The Annals of Pharmacotherapy. 41 (2): 314–8. doi:10.1345/aph.1H458. PMID 17284509. S2CID 22855759.
- ^ Kanjwal K, Karabin B, Sheikh M, et al. (June 2011). “Pyridostigmine in the treatment of postural orthostatic tachycardia: a single-center experience”. Pacing and Clinical Electrophysiology. 34 (6): 750–5. doi:10.1111/j.1540-8159.2011.03047.x. PMID 21410722. S2CID 20405336.
- ^ Jump up to:a b c Mestinon | Home Archived 2008-05-13 at the Wayback Machine
- ^ Mestinon Official FDA information, side effects and uses Archived 2008-05-24 at the Wayback Machine
External links[
- “Pyridostigmine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Mestinon, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682229 |
| Pregnancy category | AU: C |
| Routes of administration | by mouth, intravenous |
| ATC code | N07AA02 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | 7.6 +/- 2.4% |
| Elimination half-life | 1.78 +/- 0.24hrs |
| Excretion | kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 155-97-5 |
| PubChem CID | 4991 |
| DrugBank | DB00545 |
| ChemSpider | 4817 |
| UNII | 19QM69HH21 |
| KEGG | D00487 |
| ChEMBL | ChEMBL1115 |
| CompTox Dashboard (EPA) | DTXSID20165786 |
| Chemical and physical data | |
| Formula | C9H13N2O2 |
| Molar mass | 181.215 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESO=C(Oc1ccc[n+](c1)C)N(C)C | |
| hideInChIInChI=1S/C9H13N2O2/c1-10(2)9(12)13-8-5-4-6-11(3)7-8/h4-7H,1-3H3/q+1 Key:RVOLLAQWKVFTGE-UHFFFAOYSA-N |
/////////////Pyridostigmine,
Buspirone
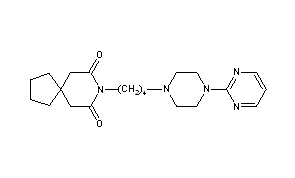
Buspirone
- Molecular FormulaC21H31N5O2
- Average mass385.503 Da
- буспиронبوسبيرون丁螺酮
251-489-4[EINECS]253-072-2[EINECS]36505-84-7[RN]8-[4-(4-Pyrimidin-2-yl-piperazin-1-yl)-butyl]-8-aza-spiro[4.5]decane-7,9-dione8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dione
- 8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dione
- Buspin
- Buspirone
- Spitomin
BuspironeCAS Registry Number: 36505-84-7CAS Name: 8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dioneMolecular Formula: C21H31N5O2Molecular Weight: 385.50Percent Composition: C 65.43%, H 8.11%, N 18.17%, O 8.30%Literature References: Non-benzodiazepine anxiolytic; 5-hydroxytryptamine (5-HT1) receptor agonist. Prepn: Y. H. Wu et al.,J. Med. Chem.15, 477 (1972); Y. H. Wu, J. W. Rayburn, DE2057845 (1971 to Bristol-Myers); eidem,US3717634 (1973 to Mead-Johnson). Pharmacology: L. E. Allen et al.,Arzneim.-Forsch.24, 917 (1974). Comparison with diazepam in treatment of anxiety: H. L. Goldberg, R. J. Finnerty, Am. J. Psychiatry136, 1184 (1979); A. F. Jacobson et al.,Pharmacotherapy5, 290 (1985). Nonsynergistic effect with alcohol: T. Seppala et al.,Clin. Pharmacol. Ther.32, 201 (1982). Disposition and metabolism: S. Caccia et al.,Xenobiotica13, 147 (1983). Series of articles on chemistry, pharmacology, addictive potential, and clinical trials: J. Clin. Psychiatry43, pp 1-116 (1982); on pharmacology, safety and clinical comparison with clorazepate: Am. J. Med.80, Suppl. 3B, 1-51 (1986). Review of pharmacology and therapeutic efficacy: K. L. Goa, A. Ward, Drugs32, 114-129 (1986). Review: M. W. Jann, Pharmacotherapy8, 100-116 (1988); D. P. Taylor, FASEB J.2, 2445-2452 (1988).
Derivative Type: HydrochlorideCAS Registry Number: 33386-08-2Trademarks: Ansial (Vita); Ansiced (Abello); Axoren (Glaxo Wellcome); Bespar (BMS); Buspar (BMS); Buspimen (Menarini); Buspinol (Zdravlje); Buspisal (Lesvi); Narol (Almirall)Molecular Formula: C21H31N5O2.HClMolecular Weight: 421.96Percent Composition: C 59.77%, H 7.64%, N 16.60%, O 7.58%, Cl 8.40%Properties: Crystals from abs ethanol, mp 201.5-202.5°. LD50 i.p. in rats: 136 mg/kg (Allen).Melting point: mp 201.5-202.5°Toxicity data: LD50 i.p. in rats: 136 mg/kg (Allen)
Therap-Cat: Anxiolytic.Keywords: Anxiolytic; Arylpiperazines; Serotonin Receptor Agonist.
Buspirone, sold under the brand name Buspar, among others, is a medication primarily used to treat anxiety disorders, particularly generalized anxiety disorder.[9][10] Benefits support its short term use.[11] It has not been found to be effective in treating psychosis.[9] It is taken by mouth, and it may take up to four weeks to have an effect.[9][10]
Common side effects of buspirone include nausea, headaches, dizziness, and difficulty concentrating.[9][11] Serious side effects may include hallucinations, serotonin syndrome, and seizures.[11] Its use in pregnancy appears to be safe but has not been well studied, while use during breastfeeding is not recommended.[11][12] It is a serotonin 5-HT1A receptor agonist.[2]
Buspirone was first made in 1968 and approved for medical use in the United States in 1986.[9][10] It is available as a generic medication.[11] In 2018, it was the 92nd most-commonly prescribed medication in the United States, with more than 8 million prescriptions.[13][14]
Medical uses
Anxiety
Buspirone is used for the short-term treatment of anxiety disorders or symptoms of anxiety.[15][16][17][18][19] It is generally less preferred than selective serotonin reuptake inhibitors (SSRIs).[10]
Buspirone has no immediate anxiolytic effects, and hence has a delayed onset of action; its full clinical effectiveness may require 2–4 weeks to manifest itself.[20] The drug has been shown to be similarly effective in the treatment of generalized anxiety disorder (GAD) to benzodiazepines including diazepam, alprazolam, lorazepam, and clorazepate.[2] Buspirone is not known to be effective in the treatment of other anxiety disorders besides GAD,[21] although there is some limited evidence that it may be useful in the treatment of social phobia as an adjunct to selective serotonin reuptake inhibitors (SSRIs).[2][22]
Other uses
Sexual dysfunction
There is some evidence that buspirone on its own may be useful in the treatment of hypoactive sexual desire disorder (HSDD) in women.[23]
Miscellaneous
Buspirone is not effective as a treatment for benzodiazepine withdrawal, barbiturate withdrawal, or alcohol withdrawal/delirium tremens.[24]
SSRI and SNRI antidepressants such as paroxetine and venlafaxine may cause jaw pain/jaw spasm reversible syndrome (although it is not common), and buspirone appears to be successful in treating bruxism on SSRI/SNRI-induced jaw clenching.[25][26]
Contraindications
Buspirone has these contraindications:[27][28]
- Hypersensitivity to buspirone
- Metabolic acidosis, as in diabetes
- Should not be used with MAO inhibitors
- Severely compromised liver and/or kidney function
Side effects
Main article: List of side effects of buspirone
Known side effects associated with buspirone include dizziness, headaches, nausea, nervousness, and paresthesia.[2] Buspirone is relatively well tolerated, and is not associated with sedation, cognitive and psychomotor impairment, muscle relaxation, physical dependence, or anticonvulsant effects.[2] In addition, buspirone does not produce euphoria[20] and is not a drug of abuse.[16]
It is unclear if there is a risk of tardive dyskinesia or other movement disorders with buspirone.[9]
Overdose
Buspirone appears to be relatively benign in cases of single-drug overdose, although no definitive data on this subject appear to be available.[29] In one clinical trial, buspirone was administered to healthy male volunteers at a dosage of 375 mg/day, and produced side effects including nausea, vomiting, dizziness, drowsiness, miosis, and gastric distress.[15][16][18] In early clinical trials, buspirone was given at dosages even as high as 2,400 mg/day, with akathisia, tremor, and muscle rigidity observed.[30] Deliberate overdoses with 250 mg and up to 300 mg buspirone have resulted in drowsiness in about 50% of individuals.[30] One death has been reported in association with 450 mg buspirone together with alprazolam, diltiazem, alcohol, cocaine.[30]
Interactions
Buspirone has been shown in vitro to be metabolized by the enzyme CYP3A4.[8] This finding is consistent with the in vivo interactions observed between buspirone and these inhibitors or inducers of cytochrome P450 3A4 (CYP3A4), among others:[27]
- Itraconazole: Increased plasma level of buspirone
- Rifampicin: Decreased plasma levels of buspirone
- Nefazodone: Increased plasma levels of buspirone
- Haloperidol: Increased plasma levels of haloperidol
- Carbamazepine: Decreased plasma levels of buspirone
- Grapefruit: Significantly increases the plasma levels of buspirone.[31] See grapefruit–drug interactions.
- Fluvoxamine: Moderately increase plasma levels of buspirone.[32]
Elevated blood pressure has been reported when buspirone has been administered to patients taking monoamine oxidase inhibitors (MAOIs).[27]
Pharmacology
Pharmacodynamics
| Site | Ki (nM) | Species | Ref |
|---|---|---|---|
| 5-HT1A | 3.98–214 21 (median) | Human | [33][34] |
| 5-HT1B | >100,000 | Rat | [35] |
| 5-HT1D | 22,000–42,700 | Human | [36][37] |
| 5-HT2A | 138 759–1,300 | Human Rat | [38] [35][38] |
| 5-HT2B | 214 | Human | [38] |
| 5-HT2C | 490 1,100–6,026 | Human Rat/pig | [38] [35][38] |
| 5-HT3 | >10,000 | Rat | [39][40] |
| 5-HT4 | >10,000 | Rat | [40] |
| 5-HT6 | 398 | Mouse | [41] |
| 5-HT7 | 375–381 | Rat | [42][43] |
| α1 | 1,000 | Rat | [35] |
| α2 | 6,000 | Rat | [44] |
| α2A | 7.3 (1-PP) | Human | [35] |
| β | 8,800 | Rat | [35] |
| D1 | 33,000 | Rat | [35] |
| D2 | 484 240 | Human Rat | [45] [35] |
| D3 | 98 | Human | [45] |
| D4 | 29 | Human | [45] |
| mACh | 38,000 | Rat | [35] |
| GABAA (BDZ) | >100,000 | Rat | [35] |
| Values are Ki (nM). The smaller the value, the more strongly the drug binds to the site. |
Buspirone acts as an agonist of the serotonin 5-HT1A receptor with high affinity.[2][35] It is a partial agonist of both presynaptic 5-HT1A receptors, which are inhibitory autoreceptors, and postsynaptic 5-HT1A receptors.[2] It is thought that the main effects of buspirone are mediated via its interaction with the presynaptic 5-HT1A receptor, thus reducing the firing of serotonin-producing neurons.[2] Buspirone also has lower affinities for the serotonin 5-HT2A, 5-HT2B, 5-HT2C, 5-HT6, and 5-HT7 receptors.[33]
In addition to binding to serotonin receptors, buspirone is an antagonist of the dopamine D2 receptor with weak affinity.[2][35] It preferentially blocks inhibitory presynaptic D2 autoreceptors, and antagonizes postsynaptic D2 receptors only at higher doses.[2] In accordance, buspirone has been found to increase dopaminergic neurotransmission in the nigrostriatal pathway at low doses, whereas at higher doses, postsynaptic D2 receptors are blocked and antidopaminergic effects such as hypoactivity and reduced stereotypy, though notably not catalepsy, are observed in animals.[2] Buspirone has also been found to bind with much higher affinity to the dopamine D3 and D4 receptors, where it is similarly an antagonist.[45]
A major metabolite of buspirone, 1-(2-pyrimidinyl)piperazine (1-PP), occurs at higher circulating levels than buspirone itself and is known to act as a potent α2-adrenergic receptor antagonist.[44][46][47] This metabolite may be responsible for the increased noradrenergic and dopaminergic activity observed with buspirone in animals.[46][48] In addition, 1-PP may play an important role in the antidepressant effects of buspirone.[48] Buspirone also has very weak and probably clinically unimportant affinity for the α1-adrenergic receptor.[35][49] However, buspirone has been reported to have shown “significant and selective intrinsic efficacy” at the α1-adrenergic receptor expressed in a “tissue- and species-dependent manner”.[49]
Unlike benzodiazepines, buspirone does not interact with the GABAA receptor complex.[2][50]
Pharmacokinetics
Buspirone has a low oral bioavailability of 3.9% relative to intravenous injection due to extensive first-pass metabolism.[2] The time to peak plasma levels following ingestion is 0.9 to 1.5 hours.[2] It is reported to have an elimination half-life of 2.8 hours,[2] although a review of 14 studies found that the mean terminal half-life ranged between 2 and 11 hours, and one study even reported a terminal half-life of 33 hours.[4] Buspirone is metabolized primarily by CYP3A4, and prominent drug interactions with inhibitors and inducers of this enzyme have been observed.[7][8] Major metabolites of buspirone include 5-hydroxybuspirone, 6-hydroxybuspirone, 8-hydroxybuspirone, and 1-PP.[4][5][6] 6-Hydroxybuspirone has been identified as the predominant hepatic metabolite of buspirone, with plasma levels that are 40-fold greater than those of buspirone after oral administration of buspirone to humans.[5] The metabolite is a high-affinity partial agonist of the 5-HT1A receptor (Ki = 25 nM) similarly to buspirone, and has demonstrated occupancy of the 5-HT1A receptor in vivo.[5] As such, it is likely to play an important role in the therapeutic effects of buspirone.[5] 1-PP has also been found to circulate at higher levels than those of buspirone itself and may similarly play a significant role in the clinical effects of buspirone.[46][48]

Phase I Metabolism of buspirone in humans[51][52][8]
History
Buspirone was first synthesized, by a team at Mead Johnson, in 1968,[21] but was not patented until 1975.[54][55] It was initially developed as an antipsychotic drug acting on the D2 receptor, but was found to be ineffective in the treatment of psychosis; it was then used as an anxiolytic instead.[2] In 1986, Bristol-Myers Squibb gained FDA approval for buspirone in the treatment of GAD.[21][56] The patent placed on buspirone expired in 2001 and it is now available as a generic drug.
Society and culture

Buspar (buspirone) 10-mg tablets
Generic names
Buspirone is the INN, BAN, DCF, and DCIT of buspirone, while buspirone hydrochloride is its USAN, BANM, and JAN.[1][57][58][59]
Brand name
Buspirone was primarily sold under the brand name Buspar.[57][59] Buspar is currently listed as discontinued by the US Federal Drug Administration.[60] In 2010, in response to a citizen petition, the US FDA determined that Buspar was not withdrawn for sale because of reasons of safety or effectiveness.[61]
2019 shortage
Due to interrupted production at a Mylan Pharmaceuticals plant in Morgantown, West Virginia, the United States experienced a shortage of buspirone in 2019.[62]
Research
Some tentative research supports other uses such as the treatment of depression and behavioral problems following brain damage.[2]
Chemistry
Buspirone is a member of the azapirone chemical class, and consists of azaspirodecanedione and pyrimidinylpiperazine components linked together by a butyl chain.
Analogues
Structural analogues of buspirone include other azapirones like gepirone, ipsapirone, perospirone, and tandospirone.[53]
Synthesis

Buspirone synthesis:[54] DE 2057845 U.S. Patent 3,717,634 U.S. Patent 3,907,801 U.S. Patent 3,976,776
Alkylation of 1-(2-pyrimidyl)piperazine (1) with 3-chloro-1-cyanopropane (2, 4-chlorobutyronitrile) gives 3, which is reduced either by hydrogenation over Raney nickel catalyst, or with LAH. The resulting 1° amine (4) from the previous step is then reacted with 3,3-tetramethyleneglutaric anhydride (5, 8-Oxaspiro[4.5]decane-7,9-dione) in order to yield buspirone (6).
PAPERS
- Koziol, Anna E.; Acta Crystallographica, Section E: Structure Reports Online 2006, V62(12), Po5616-o5618
- Mou, Jie; Organic Preparations and Procedures International 2008, V40(4), P391-394
- Kairisalo, Pekka Juhani; FI 72975 B 1987
- Journal of medicinal chemistry (1983), 26(2), 194-203
- Journal of medicinal chemistry (1986), 29(8), 1476-82.
- Medicinal research reviews (1990), 10(3), 283-326.
- Heterocycles (1993), 36(7), 1463-9
- Journal of medicinal chemistry (1996), 39(5), 1125-9.
- Journal of medicinal chemistry (1996), 39(16), 3195-202.
- Nature Catalysis, 3(10), 843-850; 2020
PAPER
https://pubs.rsc.org/en/content/articlelanding/2019/GC/C8GC03328E#!divAbstract
- Green Chemistry, 21(1), 59-63; 2019
Abstract
A continuous flow method for the direct conversion of alcohols to amines via a hydrogen borrowing approach is reported. The method utilises a low loading (0.5%) of a commercial catalyst system ([Ru(p-cymene)Cl2]2 and DPEPhos), reagent grade solvent and is selective for primary alcohols. Successful methylation of amines using methanol and the direct dimethylamination of alcohols using commercial dimethylamine solution are reported. The synthesis of two pharmaceutical agents Piribedil (5) and Buspirone (25) were accomplished in good yields employing these new methods.

http://www.rsc.org/suppdata/c8/gc/c8gc03328e/c8gc03328e2.pdf
8-(4-hydroxybutyl)-8-azaspiro[4.5]decane-7,9-dione (23): A solution of 3,3-tetramethyleneglutaric anhydride (0.25 mol/L in THF) was combined in a tee piece with a solution of 4-amino-1-butanol (0.25 mol/L in THF) and reacted in a 20 mL reactor coil (stainless steel, 20 min residence time) heated at 250 °C. The output was concentrated in vacuo and the residue purified by column chromatography on silica gel to afford the product in 84% yield (Rf = 0.31, 63% DCM/AcOEt). 1H NMR (400 MHz, CDCl3) δ = 3.78 (t, J = 7.2 Hz, 2H), 3.65 (t, J = 6.0 Hz, 2H), 2.58 (s, 4H), 1.77 – 1.64 (m, 4H), 1.64 – 1.53 (m, 4H), 1.53 – 1.43 (m, 4H). 13C NMR (100 MHz, CDCl3) δ = 172.33, 62.28, 44.87, 39.47, 39.14, 37.54, 29.81, 24.35, 24.17. HRMS for [C13H22NO3] + calculated 240.1594 found 240.1605.

8-(4-(4-(pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione (Buspirone, 25): The flow system was flushed with THF, the back-pressure regulator was set to 50 bar, and the coil reactor heated to 250 °C. Then a solution (10 mL overall volume) containing 1-(2-pyrimidyl)piperazine (2 mmol), 8-(4-hydroxybutyl)- 8-azaspiro[4.5]decane-7,9-dione (23) (2 mmol), dichloro(p-cymene)ruthenium(II) dimer (0.08 mmol) and bis[(2- diphenylphosphino)phenyl] ether (DPEPhos, 0.17 mmol) was pumped at 0.8 ml/min through a heated coil (8 mL, Phoenix reactor). The output solution obtained in steady state (monitored using the FlowUV) was concentrated in vacuo and purified by column chromatography on silica gel to afford the desired product in 76% yield (Rf = 0.29, 5% MeOH/DCM). 1H NMR (400 MHz, CDCl3) δ = 8.31 (d, J = 4.7 Hz, 2H), 6.48 (t, J = 4.7 Hz, 1H), 3.84 (t, J = 5.1 Hz, 4H), 3.79 (t, J = 6.8 Hz, 2H), 2.60 (s, 4H), 2.50 (t, J = 5.1 Hz, 4H), 2.40 (t, J = 6.8 Hz, 2H), 1.79 – 1.65 (m, 4H), 1.65 – 1.42 (m, 8H). 13C NMR (100 MHz, CDCl3) δ = 172.19, 161.63, 157.68, 109.77, 58.31, 53.06, 44.92, 43.60, 39.48, 39.35, 37.56, 26.04, 24.19, 24.19. HRMS for [C21H32N5O2] + calculated 386.2551 found 386.2570.


PAPER
Organic Preparations and Procedures International, 40(4), 391-394; 2008
https://www.tandfonline.com/doi/abs/10.1080/00304940809458099



PATENTS
US 3907801
ES 526304
EP 395192
EP 565274
EP 634411
EP 680961
US 5521313
Indian Pat. Appl., 2011MU01860,
PATENTS
WO 2014152737
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014152737
Syn
J Med Chem 1972,15(5),477-479
DE 2057845; FR 2073406; GB 1332194; US 3717634
The condensation of 1-(2-pyrimidinyl)piperazine (I) with 3-chloro-1-cyanopropane (II) by means of Na2CO3 in n-butanol gives 4-(2-pyrimidinyl)-1-(3-cyanopropyl)piperazine (III). This product is reduced with LiAlH4 or with H2 and Raney-Ni yielding 4-(2-pyrimidinyl)-1-(4-aminobutyl)piperazine (IV), which is finally condensed with 8-oxaspiro[4.5]decane-7,9-dione-(3,3-tetramethylene-glutaric anhydride) (V) in pyridine.
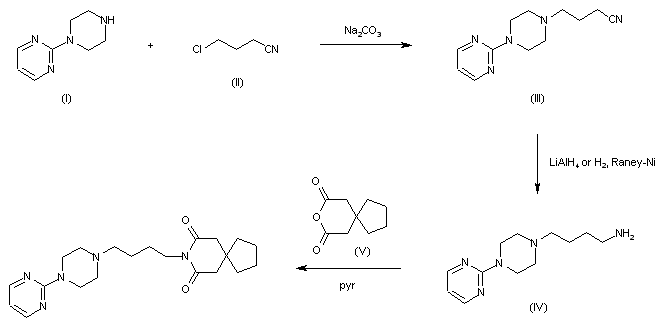
CLIP
Anxiolytics (Tranquilizers)
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Buspirone
Buspirone, 8-[4-[4-(2-pyrimidyl)-1-piperazinyl]butyl]-8-azaspiro [4,5] decan-7,9-dione (5.2.6), is synthesized by the reaction of 1-(2-pyrimidyl)-4-(4-aminobutyl)piperazine (5.2.4) with 8-oxaspiro[4,5]decan-7,9-dione (5.2.5). In turn, 1-(2-pyrimidyl)-4-(4-aminobutyl)piperazine (5.2.4) is synthesized by the reaction of 1-(2-pyrimidyl)piperazine with 4-chlorobutyronitrile, giving 4-(2-pyrimidyl)-1-(3-cyanopropyl)piperazine (5.2.3), which is hydrogenated with Raney nickel into buspirone (5.2.4) [51–55].

Buspirone is an extremely specific drug that could possibly represent a new chemical class of anxiolytics—azaspirones. As an anxiolytic, its activity is equal to that of benzodiazepines; however, it is devoid of anticonvulsant and muscle relaxant properties, which are characteristic of benzodiazepines. It does not cause dependence or addiction. The mechanism of its action is not conclusively known. It does not act on the GABA receptors, which occurs in benzodiazepine use; however, it has a high affinity for seratonin (5-HT) receptors and a moderate affinity for dopamine (D2) receptors. Buspirone is effective as an anxiolytic. A few side effects of buspirone include dizziness, drowsiness, headaches, nervousness, fatigue, and weakness. This drug is intended for treatment of conditions of anxiety in which stress, muscle pain, rapid heart rate, dizziness, fear, etc. are observed; in other words, conditions of anxiety not associated with somewhat common, usual, and everyday stress. Synonyms for buspirone are anizal, axoren, buspar, buspimen, buspinol, narol, travin, and others.
CLIP
Applications of Biocatalysis for Pharmaceuticals and Chemicals
Ramesh N. Patel, in Organic Synthesis Using Biocatalysis, 2016
5.2 Enzymatic Preparation of 6-Hydroxybuspirone
Buspirone (Buspar®, 59, Figure 11.17) is a drug used for the treatment of anxiety and depression, thought to produce its effects by binding to the serotonin 5HT1A receptor [114–116]. Mainly as a result of hydroxylation reactions, it is extensively converted to various metabolites and blood concentrations return to low levels a few hours after dosing [117]. A major metabolite, 6-hydroxybuspirone, produced by the action of liver cytochrome P450 CYP3A4, was present at much higher concentrations in human blood than buspirone itself. For development of 6-hydroxybuspirone as a potential antianxiety drug, preparation and testing of the two enantiomers as well as the racemate was of interest. An enantioselective microbial reduction process was developed for the reduction of 6-oxobuspirone 60 to (R)-6-hydroxybuspirone 61a or (S)-6-hydroxybuspitone 61b. About 150 microbial cultures were screened for the enantioselective reduction of 60. Rhizopus stolonifer SC 13898, Neurospora crassa SC 13816, Mucor racemosus SC 16198, and Pseudomonas putida SC 13817 gave >50% reaction yields and >95% ee of (S)-6-hydroxybuspirone 61a. The yeast strains Hansenula polymorpha SC 13845 and Candida maltosa SC 16112 gave (R)-6-hydroxybuspirone in >60% reaction yield and >97% ee [118]. The NADPH-dependent (R)-reductase (RHBR) from H. polymorpha SC 13845 was purified to homogeneity, its N-terminal and internal amino acid sequences were determined and the corresponding gene was cloned and expressed in E. coli. To regenerate the NADPH required for reduction, glucose-6-phosphate dehydrogenase gene from Saccharomyces cerevisiae was cloned and coexpressed in the same E. coli strain. Recombinant cultures coexpressing (R)-reductase (RHBR) and glucose 6-phosphate dehydrogenase catalyzed the reduction of 6-ketobuspirone to (R)-6-hydroxybuspirone 61a in 99% yield and 99.9% ee at 50 g/L substrate input [119].

The NADH-dependent (S)-reductase (SHBR) from P. putida SC 16269 was also purified to homogeneity, its N-terminal and internal amino acid sequences were determined and the corresponding gene was cloned and expressed in E. coli. To regenerate the NADH required for reduction, the NAD+ dependent formate dehydrogenase gene from Pichia pastoris was also cloned and co-expressed in the same E. coli strain. Recombinant E. coli coexpressing (S)-reductase and formate dehydrogenase was used to catalyze the reduction of 6-ketobuspirone to (S)-6-hydroxybuspirone 61b, in >98% yield and >99.8% ee at 50 g/L substrate input [119].
PATENT
https://patents.google.com/patent/US6686361
The present invention relates to methods of treating anxiety and depression using R-6-hydroxy-buspirone and pharmaceutical compositions containing R-6-hydroxy-buspirone.
Buspirone, chemically: 8-[4-[4-(2-pyrimidinyl)1-piperazinyl]butyl-8-azaspiro(4,5)-decane-7,9-dione, is approved for the treatment of anxiety disorders and depression by the United States Food and Drug Administration. It is available under the trade name BUSPAR® from Bristol-Myers Squibb Company.
Studies have shown that buspirone is extensively metabolized in the body. (See, for example, Mayol, et al., Clin. Pharmacol. Ther., 37, p. 210, 1985). One of the metabolites is 6-hydroxy-8-[4-[4-(2-pyrimidinyl)1-piperazinyl]butyl-8-azaspiro(4,5)-decane-7,9-dione having Formula I. This metabolite is also known as BMS 28674, BMS 442608, or

as 6-hydroxy-buspirone. This compound is believed to be the active metabolite of buspirone and its use in treating anxiety disorders and depression is disclosed in U.S. Pat. No. 6,150,365. The specific stereochemistry of 6-hydroxy-buspirone has not been described previously. Neither racemic 6-hydroxy-buspirone nor its enantiomers are commercially available at the present time.
Preclinical studies demonstrate that 6-hydroxy-buspirone, like buspirone, demonstrates a strong affinity for the human 5-HT1A receptor. In functional testing, 6-hydroxy-buspirone produced a dose-dependent anxiolytic response in the rat pup ultrasonic vocalization test, a sensitive method for assessment of anxiolytic and anxiogenic effects (Winslow and Insel, 1991, Psychopharmacology, 105:513-520).
Clinical studies in volunteers orally dosed with buspirone demonstrate that 6-hydroxy-buspirone blood plasma levels were not only 30 to 40 times higher but were sustained compared to buspirone blood plasma levels. The time course of 6-hydroxy-buspirone blood plasma levels, unlike buspirone blood plasma levels, correlate more closely with the sustained anxiolytic effect seen following once or twice a day oral dosing with buspirone.
Although buspirone is an effective treatment for anxiety disorders and depression symptomatology in a significant number of patients treated, about a third of patients get little to no relief from their anxiety and responders often require a week or more of buspirone treatment before experiencing relief from their anxiety symptomatology. Further, certain adverse effects are reported across the patient population. The most commonly observed adverse effects associated with the use of buspirone include dizziness, nausea, headache, nervousness, lightheadedness, and excitement. Also, since buspirone can bind to central dopamine receptors, concern has been raised about its potential to cause unwanted changes in dopamine-mediated neurological functions and a syndrome of restlessness, appearing shortly after initiation of oral buspirone treatment, has been reported in small numbers of patients. While buspirone lacks the prominent sedative effects seen in more typical anxiolytics such as the benzodiazepines, patients are nonetheless advised against operating potentially dangerous machinery until they experience how they are affected by buspirone.
It can be seen that it is desirable to find a medicament with buspirone’s advantages but which demonstrates more robust anxiolytic potency with a lack of the above described adverse effects.
Formation of 6-hydroxy-buspirone occurs in the liver by action of enzymes of the P450 system, specifically CYP3A4. Many substances such as grapefruit juice and certain other drugs; e.g. erythromycin, ketoconazole, cimetidine, etc., are inhibitors of the CYP3A4 isozyme and may interfere with the formation of this active metabolite from buspirone. For this reason it would be desirable to find a compound with the advantages of buspirone but without the drug—drug interactions when coadministered with agents affecting the activity level of the CYP3A4 isozyme.
EXAMPLE 3One-Step Synthesis of 6-Hydroxy-buspirone (I)
Buspirone (19.3 g, 50 mmole) was dissolved in dry THF (400 mL) and the resulting solution was cooled to −78° C. A solution of KN(SiMe3)2 in toluene (100 mL, 1 M) was added slowly. After the reaction mixture was stirred at −78° C. for 1 h, a solution of 2-(phenylsulfonyl)-3-phenyloxaziridine (Davis reagent, prepared according to literature method: F. A. Davis, et al., Org. Synth., 1988, 66, 203) (17.0 g, 65 mmole) in dry THF (150 mL, precooled to −78° C.) was added quickly via a cannular. After stirred for 30 mins at −78° C., the reaction was quenched with 1 N HCl solution (500 mL). It was extracted with EtOAc (3×500 mL). The aqueous layer was separated, neutralized with saturated sodium bicarbonate solution, and extracted with EtOAc (3×500 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a white solid residue which was subjected to column chromatography using CH2Cl2/MeOH/NH4OH (200:10:1) as the eluent to give pure 6-hydroxy-buspirone (I, 7.2 g) and a mixture of buspirone and 6-hydroxy-buspirone (I). The mixture was purified by above column chromatography to afford another 3.3 g of pure 6-hydroxy-buspirone (I).
1H NMR (CDCl3) δ8.30 (d, J=4.7 Hz, 2H), 6.48 (t, J=4.7 Hz, 1H), 4.20 (s, 1H), 3.83-3.72 (m, 5H), 3.55 (s, 1H), 2.80 (d, J=17.5 Hz, 1H), 2.55-2.40 (m, 7H), 2.09-2.03 (m, 1H), 1.76-1.54 (m, 10 H), 1.41-1.36 (m, 1H), 1.23-1.20 (m, 1H).
References
- ^ Jump up to:a b Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 192–. ISBN 978-1-4757-2085-3.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r Loane C, Politis M (June 2012). “Buspirone: what is it all about?”. Brain Research. 1461: 111–8. doi:10.1016/j.brainres.2012.04.032. PMID 22608068. S2CID 11734819.
- ^ Jump up to:a b c “buspirone (Rx) – BuSpar, Buspirex, more.” Medscape Reference. WebMD. Retrieved 14 November 2013.
- ^ Jump up to:a b c Gammans RE, Mayol RF, LaBudde JA (March 1986). “Metabolism and disposition of buspirone”. The American Journal of Medicine. 80 (3B): 41–51. doi:10.1016/0002-9343(86)90331-1. PMID 3515929.
- ^ Jump up to:a b c d e Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing Textbook of Psychopharmacology. American Psychiatric Pub. pp. 490–. ISBN 978-1-58562-309-9.
- ^ Jump up to:a b Wong H, Dockens RC, Pajor L, Yeola S, Grace JE, Stark AD, et al. (August 2007). “6-Hydroxybuspirone is a major active metabolite of buspirone: assessment of pharmacokinetics and 5-hydroxytryptamine1A receptor occupancy in rats”. Drug Metabolism and Disposition. 35 (8): 1387–92. doi:10.1124/dmd.107.015768. PMID 17494642. S2CID 25558546.
- ^ Jump up to:a b c Mahmood I, Sahajwalla C (April 1999). “Clinical pharmacokinetics and pharmacodynamics of buspirone, an anxiolytic drug”. Clinical Pharmacokinetics. 36 (4): 277–87. doi:10.2165/00003088-199936040-00003. PMID 10320950. S2CID 1102318.
- ^ Jump up to:a b c d Zhu M, Zhao W, Jimenez H, Zhang D, Yeola S, Dai R, et al. (April 2005). “Cytochrome P450 3A-mediated metabolism of buspirone in human liver microsomes”. Drug Metabolism and Disposition. 33 (4): 500–7. doi:10.1124/dmd.104.000836. PMID 15640381. S2CID 10142905.
- ^ Jump up to:a b c d e f “Buspirone Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 3 March 2019.
- ^ Jump up to:a b c d Wilson, T. K.; Tripp, J. (January 2018). “Buspirone”. StatPearls. PMID 30285372.
- ^ Jump up to:a b c d e British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. p. 338. ISBN 9780857113382.
- ^ “Buspirone Pregnancy and Breastfeeding Warnings”. Drugs.com. Retrieved 3 March 2019.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Buspirone Hydrochloride – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Jump up to:a b “BUSPIRONE HCL (buspirone hydrochloride) tablet [Watson Laboratories, Inc.]”. DailyMed. Watson Laboratories, Inc. July 2013. Retrieved 14 November 2013.
- ^ Jump up to:a b c “BUSPAR® (buspirone hydrochloride) Tablets 5 mg & 10 mg PRODUCT INFORMATION” (PDF). TGA eBusiness Services. Aspen Pharma Pty Ltd. January 2010. Retrieved 14 November2013.
- ^ Rossi S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- ^ Jump up to:a b “Buspirone 10mg Tablets”. electronic Medicines Compendium. Actavis UK Ltd. 10 September 2012. Retrieved 14 November 2013.
- ^ Joint Formulary Committee. British National Formulary (BNF). Pharmaceutical Press. p. 224.
- ^ Jump up to:a b Sadock BJ, Sadock VA, Ruiz P (22 September 2014). Kaplan and Sadock’s Synopsis of Psychiatry: Behavioral Sciences/Clinical Psychiatry. Wolters Kluwer Health. pp. 3211–. ISBN 978-1-4698-8375-5.
- ^ Jump up to:a b c Howland RH (November 2015). “Buspirone: Back to the Future”. Journal of Psychosocial Nursing and Mental Health Services. 53 (11): 21–4. doi:10.3928/02793695-20151022-01. PMID 26535760.
- ^ Masdrakis VG, Turic D, Baldwin DS (2013). “Pharmacological treatment of social anxiety disorder”. Anxiety Disorders. Modern Trends in Pharmacopsychiatry. 29. pp. 144–53. doi:10.1159/000351960. ISBN 978-3-318-02463-0. PMID 25225024.
- ^ Goldstein I, Kim NN, Clayton AH, DeRogatis LR, Giraldi A, Parish SJ, et al. (January 2017). “Hypoactive Sexual Desire Disorder: International Society for the Study of Women’s Sexual Health (ISSWSH) Expert Consensus Panel Review”. Mayo Clinic Proceedings. 92 (1): 114–128. doi:10.1016/j.mayocp.2016.09.018. PMID 27916394.
- ^ Sontheimer DL, Ables AZ (March 2001). “Is imipramine or buspirone treatment effective in patients wishing to discontinue long-term benzodiazepine use?”. The Journal of Family Practice. 50(3): 203. PMID 11252203.
- ^ Garrett AR, Hawley JS (April 2018). “SSRI-associated bruxism: A systematic review of published case reports”. Neurology. Clinical Practice. 8 (2): 135–141. doi:10.1212/CPJ.0000000000000433. PMC 5914744. PMID 29708207.
- ^ Prisco V, Iannaccone T, Di Grezia G (2017-04-01). “Use of buspirone in selective serotonin reuptake inhibitor-induced sleep bruxism”. European Psychiatry. Abstract of the 25th European Congress of Psychiatry. 41: S855. doi:10.1016/j.eurpsy.2017.01.1701.
- ^ Jump up to:a b c “Buspirone monograph”. Drugs.com. Retrieved 2011-08-27.
- ^ Geddes J, Gelder MG, Mayou R (2005). Psychiatry. Oxford [Oxfordshire]: Oxford University Press. p. 237. ISBN 978-0-19-852863-0.
- ^ Fulton B, Brogden RN (1997). “Buspirone”. CNS Drugs. 7 (1): 68–88. doi:10.2165/00023210-199707010-00007. ISSN 1172-7047.
- ^ Jump up to:a b c Dart RC (2004). Medical Toxicology. Lippincott Williams & Wilkins. pp. 886–. ISBN 978-0-7817-2845-4.
- ^ Lilja JJ, Kivistö KT, Backman JT, Lamberg TS, Neuvonen PJ (December 1998). “Grapefruit juice substantially increases plasma concentrations of buspirone”. Clinical Pharmacology and Therapeutics. 64 (6): 655–60. doi:10.1016/S0009-9236(98)90056-X. PMID 9871430. S2CID 22009095.
- ^ Lamberg TS, Kivistö KT, Laitila J, Mårtensson K, Neuvonen PJ (1998). “The effect of fluvoxamine on the pharmacokinetics and pharmacodynamics of buspirone”. European Journal of Clinical Pharmacology. 54 (9–10): 761–6. doi:10.1007/s002280050548. PMID 9923581. S2CID 21939719.
- ^ Jump up to:a b c Roth BL, Driscol J. “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 14 August 2017.
- ^ Boess FG, Martin IL (1994). “Molecular biology of 5-HT receptors”. Neuropharmacology. 33 (3–4): 275–317. doi:10.1016/0028-3908(94)90059-0. PMID 7984267. S2CID 35553281.
- ^ Jump up to:a b c d e f g h i j k l m Hamik A, Oksenberg D, Fischette C, Peroutka SJ (July 1990). “Analysis of tandospirone (SM-3997) interactions with neurotransmitter receptor binding sites”. Biological Psychiatry. 28 (2): 99–109. doi:10.1016/0006-3223(90)90627-e. PMID 1974152. S2CID 25608914.
- ^ Peroutka SJ, Switzer JA, Hamik A (1989). “Identification of 5-hydroxytryptamine1D binding sites in human brain membranes”. Synapse. 3 (1): 61–6. doi:10.1002/syn.890030109. PMID 2521959.
- ^ Waeber C, Schoeffter P, Palacios JM, Hoyer D (June 1988). “Molecular pharmacology of 5-HT1D recognition sites: radioligand binding studies in human, pig and calf brain membranes”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 337 (6): 595–601. doi:10.1007/bf00175783. PMID 2975354. S2CID 21344978.
- ^ Jump up to:a b c d e Bonhaus DW, Weinhardt KK, Taylor M, DeSouza A, McNeeley PM, Szczepanski K, et al. (1997). “RS-102221: a novel high affinity and selective, 5-HT2C receptor antagonist”. Neuropharmacology. 36 (4–5): 621–9. doi:10.1016/s0028-3908(97)00049-x. PMID 9225287. S2CID 24930608.
- ^ Nelson DR, Thomas DR (May 1989). “[3H]-BRL 43694 (Granisetron), a specific ligand for 5-HT3 binding sites in rat brain cortical membranes”. Biochemical Pharmacology. 38 (10): 1693–5. doi:10.1016/0006-2952(89)90319-5. PMID 2543418.
- ^ Jump up to:a b Borsini F, Giraldo E, Monferini E, Antonini G, Parenti M, Bietti G, Donetti A (September 1995). “BIMT 17, a 5-HT2A receptor antagonist and 5-HT1A receptor full agonist in rat cerebral cortex”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 352 (3): 276–82. doi:10.1007/bf00168557. PMID 8584042. S2CID 19340842.
- ^ Plassat JL, Amlaiky N, Hen R (August 1993). “Molecular cloning of a mammalian serotonin receptor that activates adenylate cyclase”. Molecular Pharmacology. 44 (2): 229–36. PMID 8394987.
- ^ Lovenberg TW, Baron BM, de Lecea L, Miller JD, Prosser RA, Rea MA, et al. (September 1993). “A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms”. Neuron. 11 (3): 449–58. doi:10.1016/0896-6273(93)90149-l. PMID 8398139. S2CID 28729004.
- ^ Ruat M, Traiffort E, Leurs R, Tardivel-Lacombe J, Diaz J, Arrang JM, Schwartz JC (September 1993). “Molecular cloning, characterization, and localization of a high-affinity serotonin receptor (5-HT7) activating cAMP formation”. Proceedings of the National Academy of Sciences of the United States of America. 90 (18): 8547–51. Bibcode:1993PNAS…90.8547R. doi:10.1073/pnas.90.18.8547. PMC 47394. PMID 8397408.
- ^ Jump up to:a b Blier P, Curet O, Chaput Y, de Montigny C (July 1991). “Tandospirone and its metabolite, 1-(2-pyrimidinyl)-piperazine–II. Effects of acute administration of 1-PP and long-term administration of tandospirone on noradrenergic neurotransmission”. Neuropharmacology. 30 (7): 691–701. doi:10.1016/0028-3908(91)90176-c. PMID 1681447. S2CID 44297577.
- ^ Jump up to:a b c d Bergman J, Roof RA, Furman CA, Conroy JL, Mello NK, Sibley DR, Skolnick P (March 2013). “Modification of cocaine self-administration by buspirone (buspar®): potential involvement of D3 and D4 dopamine receptors”. The International Journal of Neuropsychopharmacology. 16 (2): 445–58. doi:10.1017/S1461145712000661. PMC 5100812. PMID 22827916.
- ^ Jump up to:a b c Tunnicliff G (September 1991). “Molecular basis of buspirone’s anxiolytic action”. Pharmacology & Toxicology. 69 (3): 149–56. doi:10.1111/j.1600-0773.1991.tb01289.x. PMID 1796057.
- ^ Zuideveld KP, Rusiç-Pavletiç J, Maas HJ, Peletier LA, Van der Graaf PH, Danhof M (December 2002). “Pharmacokinetic-pharmacodynamic modeling of buspirone and its metabolite 1-(2-pyrimidinyl)-piperazine in rats”. The Journal of Pharmacology and Experimental Therapeutics. 303 (3): 1130–7. doi:10.1124/jpet.102.036798. PMID 12438536. S2CID 14139919.
- ^ Jump up to:a b c Fava M (2007). “The combination of buspirone and bupropion in the treatment of depression”. Psychotherapy and Psychosomatics. 76 (5): 311–2. doi:10.1159/000104708. PMID 17700052. S2CID 46284917.
- ^ Jump up to:a b Stern TA, Fava M, Wilens TE, Rosenbaum JF (27 April 2015). Massachusetts General Hospital Psychopharmacology and Neurotherapeutics E-Book. Elsevier Health Sciences. pp. 29–. ISBN 978-0-323-41323-7.
- ^ Nutt DJ, Ballenger JC (15 April 2008). Anxiety Disorders. John Wiley & Sons. pp. 395–. ISBN 978-0-470-98683-7.
- ^ Dockens RC, Salazar DE, Fulmor IE, Wehling M, Arnold ME, Croop R (November 2006). “Pharmacokinetics of a newly identified active metabolite of buspirone after administration of buspirone over its therapeutic dose range”. Journal of Clinical Pharmacology. 46(11): 1308–12. doi:10.1177/0091270006292250. PMID 17050795.
- ^ Jajoo HK, Mayol RF, LaBudde JA, Blair IA (1989). “Metabolism of the antianxiety drug buspirone in human subjects”. Drug Metabolism and Disposition. 17 (6): 634–40. PMID 2575499.
- ^ Taylor DP, Moon SL (July 1991). “Buspirone and related compounds as alternative anxiolytics”. Neuropeptides. 19 Suppl: 15–9. doi:10.1016/0143-4179(91)90078-w. PMID 1679210. S2CID 13730683.
- ^ Jump up to:a b Allen LE, Ferguson HC, Kissel JW (May 1972). “Psychosedative agents. 2. 8-(4-Substituted 1-piperazinylalkyl)-8-azaspiro(4.5)decane-7,9-diones”. Journal of Medicinal Chemistry. 15 (5): 477–9. doi:10.1021/jm00275a009. PMID 5035267.
- ^ US Patent 3907801 N-(8 (4-pyridyl-piperazino)-alkyl(9 -azaspiroalkanediones
- ^ United States Federal Drug Administration (September 9, 1986). Approval Type-1 New Molecular Entry.https://www.accessdata.fda.gov/drugsatfda_docs/nda/pre96/018731Orig1s000rev.pdf
- ^ Jump up to:a b Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 149–. ISBN 978-3-88763-075-1.
- ^ Morton IK, Hall JM (6 December 2012). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 57–. ISBN 978-94-011-4439-1.
- ^ Jump up to:a b “Buspirone”.
- ^ “Drugs@FDA: FDA Approved Drug Products”. http://www.accessdata.fda.gov. Retrieved 2019-09-20.
- ^ “Determination That BUSPAR (Buspirone Hydrochloride) Tablets, 10 Milligrams, 15 Milligrams, and 30 Milligrams, Were Not Withdrawn From Sale for Reasons of Safety or Effectiveness”. Federal Register. 2010-10-19. Retrieved 2019-09-20.
- ^ Rabin RC (2019-02-01). “Shortage of Anxiety Drug Leaves Patients Scrambling”. The New York Times. ISSN 0362-4331. Retrieved 2019-09-20.
External links
- Media related to Buspirone at Wikimedia Commons
- “Buspirone”. Drug Information Portal. U.S. National Library of Medicine.
////////////Buspirone, буспирон , بوسبيرون , 丁螺酮 , Anxiolytic,Arylpiperazines, Serotonin Receptor Agonist, Ansial, Vita, Ansiced, Abello, Axoren, Glaxo Wellcome, Bespar, BMS, Buspar, Buspimen, Menarini, Buspinol, Zdravlje, Buspisal, Lesvi, Narol, Almirall,
#Buspirone, #буспирон , #بوسبيرون , #丁螺酮 , #Anxiolytic, #Arylpiperazines, #Serotonin Receptor Agonist, #Ansial, #Vita, #Ansiced, #Abello, #Axoren, #Glaxo Wellcome, #Bespar, #BMS, #Buspar, #Buspimen, Menarini, Buspinol, Zdravlje, Buspisal, Lesvi, Narol, Almirall,
EVEROLIMUS
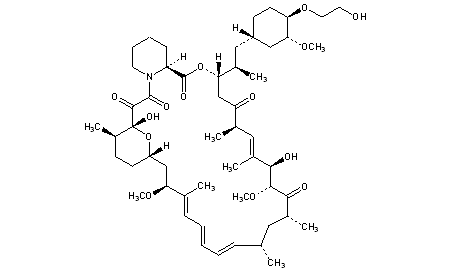
Everolimus
159351-69-6[RN]
23,27-Epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydr oxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,26R,27R,34aS)-
23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-
42-O-(2-Hydroxyethyl)rapamycin
- Synonyms:RAD-001, SDZ-RAD, Afinitor
- ATC:L04AA18
Use:immunosuppressantChemical name:42-O-(2-hydroxyethyl)rapamycinFormula:C53H83NO14
- MW:958.24 g/mol
- CAS-RN:159351-69-6
EverolimusCAS Registry Number: 159351-69-6CAS Name: 42-O-(2-Hydroxyethyl)rapamycinAdditional Names: 40-O-(2-hydroxyethyl)rapamycinManufacturers’ Codes: RAD-001; SDZ RADTrademarks: Certican (Novartis)Molecular Formula: C53H83NO14Molecular Weight: 958.22Percent Composition: C 66.43%, H 8.73%, N 1.46%, O 23.38%Literature References: Macrolide immunosuppressant; derivative of rapamycin, q.v. Inhibits cytokine-mediated lymphocyte proliferation. Prepn: S. Cottens, R. Sedrani, WO9409010; eidem, US5665772 (1994, 1997 both to Sandoz). Pharmacology: W. Schuler et al., Transplantation64, 36 (1997). Whole blood determn by LC/MS: N. Brignol et al., Rapid Commun. Mass Spectrom.15, 898 (2001); by HPLC: S. Baldelli et al., J. Chromatogr. B816, 99 (2005). Clinical pharmacokinetics in combination with cyclosporine: J. M. Kovarik et al., Clin. Pharmacol. Ther.69, 48 (2001). Clinical study in prevention of cardiac-allograft vasculopathy: H. J. Eisen et al.,N. Engl. J. Med.349, 847 (2003). Review: F. J. Dumont et al., Curr. Opin. Invest. Drugs2, 1220-1234 (2001); B. Nashan, Ther. Drug Monit.24, 53-58 (2002).Therap-Cat: Immunosuppressant.Keywords: Immunosuppressant.эверолимус[Russian][INN]إيفيروليموس[Arabic][INN]依维莫司[Chinese][INN]Trade Name:Certican® / Zortress® / Afinitor®MOA:mTOR inhibitorIndication:Rejection of organ transplantation; Renal cell carcinoma; Advanced renal cell carcinoma (RCC); Advanced breast cancer; Pancreatic cancer; Renal angiomyolipoma; Tuberous sclerosis complex (TSC); Rejection in heart transplantation; Rejection of suppression renal transplantation; Subependymal giant cell astrocytoma; neuroendocrine tumors (NET); Advanced gastrointestinal tumorsStatus:ApprovedCompany:Novartis (Originator)Sales:$1,942 Million (Y2015);
$1,902 Million (Y2014);
$1,558 Million (Y2013);
$1,007 Million (Y2012);
$630 Million (Y2011);ATC Code:L04AA18Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-08-29 | New dosage form | Afinitor Disperz | Renal cell carcinoma , Advanced breast cancer, Pancreatic cancer, Renal angiomyolipoma, Tuberous sclerosis complex (TSC) | Tablet, For suspension | 2 mg/3 mg/5 mg | Novartis | Priority |
| 2010-04-20 | New strength | Zortress | Advanced renal cell carcinoma (RCC) | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2009-03-30 | Marketing approval | Afinitor | Advanced renal cell carcinoma (RCC) | Tablet | 2.5 mg/5 mg/7.5 mg/10 mg | Novartis | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2016-06-02 | New indication | Afinitor | neuroendocrine tumors (NET), Advanced gastrointestinal tumors | Tablet | Novartis | ||
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis | Orphan; Conditional Approval |
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet, Orally disintegrating | 2 mg/3 mg/5 mg | Novartis | Orphan; Conditional Approval |
| 2009-08-03 | Marketing approval | Afinitor | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-12-22 | New indication | Certican | Rejection of suppression renal transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2007-01-26 | Marketing approval | Certican | Rejection in heart transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-02-13 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 2.5 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 10 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 5 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2003-07-18 | Marketing approval | Certican | Rejection of organ transplantation, Renal cell carcinoma | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
clip
Active Substance The active substance Everolimus is a hydroxyethyl derivative of rapamycin, which is a macrolide, isolated from the micro-organism Streptomyces hygroscopicus. The guideline, impurities in new active substances ICHQ 3A (R), does not apply to active substance of fermented origin. Everolimus (INN) or 42-O-(2-hydroxyethyl)-rapamycin (chemical name) or C5 3H8 3N O1 4 has been fully described. The molecule is amorphous and is stabilised with an antioxidant. Its physico-chemical properties including parameters such as solubility, pH, specific rotation, potential polymorphism and potential isomerism have been fully characterised. Everolimus is a white to faintly yellow amorphous powder. It is almost insoluble in water, is unstable at temperatures above 25 °C and is sensitive to light. In addition, possible isomerism has been investigated. Everolimus contains 15 asymmetric carbon atoms and 4 substituted double bonds. The configuration of the asymmetric carbon atoms and the double bonds is guaranteed by the microbial origin of Rapamycin. The configuration is not affected by the chemical synthesis. Polymorphism has been comprehensively discussed and it was demonstrated that the molecule domain remains amorphous.

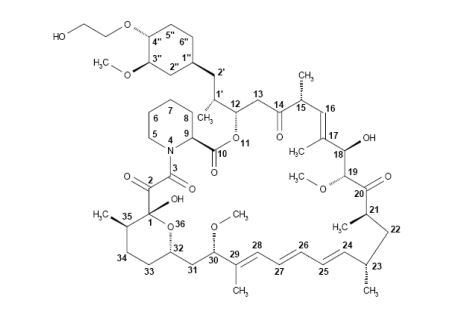
Synthesis of Everolimus The manufacturing process consists of four main steps, (1) fermentation, (2) extraction of rapamycin from the fermentation broth, (3) chemical modification of rapamycin starting material, (4) purification of crude everolimus and stabilisation with BHT. The choice of the stabilizer has been sufficiently explained and justified by experimental results. Interactions products of Everolimus and the antioxidant were not detected, or were below detection limit. Rapamycin, obtained by a fermentation process, was used as the starting material. Reaction conditions and the necessary in-process controls are described in detail. Adequate specifications for starting materials and isolated intermediates and descriptions of the test procedures have been submitted. Control of the quality of solvents, reagents and auxiliary materials used in the synthesis has been adequately documented. It is stated by the manufacturer of rapamycin solution that no starting material of animal or human origin is used in the fermentation. Elucidation of structure and other characteristics The structure of Everolimus has been fully elucidated using several spectroscopic techniques such as ultraviolet absorption spectroscopy (UV), Infra-red spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance spectroscopy (1 H and 13C NMR), mass spectroscopy, diffractometry (X-ray) and elemental analysis. Related substances An extensive discussion was presented on the related substances. The complex structure of Everolimus allows several possible degradation pathways to occur at various positions of the molecule. Everolimus alone is extremely sensitive to oxidation. By the addition of an antioxidant, the sensitivity to oxidation is significantly reduced (the antioxidant is known to react as a scavenger of peroxide radicals). It is assumed that oxidation of Everolimus proceeds via a radical mechanism. All the requirements set in the current testing instruction valid for Everolimus are justified on the basis of the results obtained during development and manufactured at the production scale.
fda
Everolimus was first approved by Swiss Agency for therapeutic products,Swissmedic on July 18, 2003, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on April 23, 2004, and approved by the U.S. Food and Drug Administration (FDA) on Mar 30, 2009, approved by European Medicine Agency (EMA) on Aug 3, 2009. It was developed and marketed as Certican® by Novartis in SE.
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR). It is indicated for the treatment of renal cell cancer and other tumours and currently used as an immunosuppressant to prevent rejection of organ transplants.
Certican® is available as tablet for oral use, containing 0.25, 0.5 or 0.75 mg of free Everolimus. The recommended dose is 10 mg once daily with or without food for advanced HR+ breast cancer, advanced progressive neuroendocrine tumors, advanced renal cell carcinoma or renal angiomyolipoma with tuberous sclerosis complex.
Everolimus, also known as RAD001, is a derivative of the natural macrocyclic lactone sirolimus with immunosuppressant and anti-angiogenic properties. In cells, everolimus binds to the immunophilin FK Binding Protein-12 (FKBP-12) to generate an immunosuppressive complex that binds to and inhibits the activation of the mammalian Target of Rapamycin (mTOR), a key regulatory kinase. Inhibition of mTOR activation results in the inhibition of T lymphocyte activation and proliferation associated with antigen and cytokine (IL-2, IL-4, and IL-15) stimulation and the inhibition of antibody production.
Everolimus is a medication used as an immunosuppressant to prevent rejection of organ transplants and in the treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors as targeted therapy for use in a number of cancers.[medical citation needed]
It is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is marketed by Novartis under the trade names Zortress (USA) and Certican (European Union and other countries) in transplantation medicine, and as Afinitor (general tumours) and Votubia (tumours as a result of TSC) in oncology. Everolimus is also available from Biocon, with the brand name Evertor.
Medical uses
Everolimus is approved for various conditions:
- Advanced kidney cancer (US FDA approved in March 2009)[3]
- Prevention of organ rejection after renal transplant(US FDA April 2010)[4]
- Subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis (TS) in patients who are not suitable for surgical intervention (US FDA October 2010)[5]
- Progressive or metastatic pancreatic neuroendocrine tumors not surgically removable (May 2011)[6]
- Breast cancer in post-menopausal women with advanced hormone-receptor positive, HER2-negative type cancer, in conjunction with exemestane (US FDA July 2012)[7]
- Prevention of organ rejection after liver transplant(Feb 2013)
- Progressive, well-differentiated non-functional, neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease (US FDA February 2016).[8]
- Tuberous sclerosis complex-associated partial-onset seizures for adult and pediatric patients aged 2 years and older. (US FDA April 2018).[9]
UK National Health Service
NHS England has been criticised for delays in deciding on a policy for the prescription of everolimus in the treatment of Tuberous Sclerosis. 20 doctors addressed a letter to the board in support of the charity Tuberous Scelerosis Association saying ” around 32 patients with critical need, whose doctors believe everolimus treatment is their best or only option, have no hope of access to funding. Most have been waiting many months. Approximately half of these patients are at imminent risk of a catastrophic event (renal bleed or kidney failure) with a high risk of preventable death.”[10] In May 2015 it was reported that Luke Henry and Stephanie Rudwick, the parents of a child suffering from Tuberous Sclerosis were trying to sell their home in Brighton to raise £30,000 to pay for treatment for their daughter Bethany who has tumours on her brain, kidneys and liver and suffers from up to 50 epileptic fits a day.[11]
Clinical trials
As of October 2010, Phase III trials are under way in gastric cancer, hepatocellular carcinoma, and lymphoma.[12] The experimental use of everolimus in refractory chronic graft-versus-host disease was reported in 2012.[13]
Interim phase III trial results in 2011 showed that adding Afinitor (everolimus) to exemestane therapy against advanced breast cancer can significantly improve progression-free survival compared with exemestane therapy alone.[14]
A study published in 2012, shows that everolimus sensitivity varies between patients depending on their tumor genomes.[15] A group of patients with advanced metastasic bladder carcinoma (NCT00805129) [16] treated with everolimus revealed a single patient who had a complete response to everolimus treatment for 26 months. The researchers sequenced the genome of this patient and compared it to different reference genomes and to other patients’ genomes. They found that mutations in TSC1 led to a lengthened duration of response to everolimus and to an increase in the time to cancer recurrence. The mutated TSC1 apparently had made these tumors vulnerable to treatment with everolimus.[medical citation needed]
A phase 2a randomized, placebo-controlled everolimus clinical trial published in 2014 showed that everolimus improved the response to an influenza vaccine by 20% in healthy elderly volunteers.[17] A phase 2a randomized, placebo-controlled clinical trial published in 2018 showed that everolimus in combination with dactolisib decreased the rate of reported infections in an elderly population.[17]
Mechanism
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex.[18] This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2 positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types.[medical citation needed] Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
mTORC1 inhibition by everolimus has been shown to normalize tumor blood vessels, to increase tumor-infiltrating lymphocytes, and to improve adoptive cell transfer therapy.[19]
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.[18]
TSC1 and TSC2, the genes involved in tuberous sclerosis, act as tumor suppressor genes by regulating mTORC1 activity. Thus, either the loss or inactivation of one of these genes lead to the activation of mTORC1.[20]
Everolimus binds to its protein receptor FKBP12, which directly interacts with mTORC1, inhibiting its downstream signaling. As a consequence, mRNAs that code for proteins implicated in the cell cycle and in the glycolysis process are impaired or altered, and tumor growth is inhibited.[20]
Adverse reactions
A trial using 10 mg/day in patients with NETs of GI or lung origin reported “Everolimus was discontinued for adverse reactions in 29% of patients and dose reduction or delay was required in 70% of everolimus-treated patients. Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). The most common adverse reactions (incidence greater than or equal to 30%) were stomatitis, infections, diarrhea, peripheral edema, fatigue and rash. The most common blood abnormalities found (incidence greater than or equal to 50%) were anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase (AST) and fasting hyperglycemia.”.[8]
Role in heart transplantation
Everolimus may have a role in heart transplantation, as it has been shown to reduce chronic allograft vasculopathy in such transplants. It also may have a similar role to sirolimus in kidney and other transplants.[21]
Role in liver transplantation
Although, sirolimus had generated fears over use of m-TOR inhibitors in liver transplantation recipients, due to possible early hepatic artery thrombosis and graft loss, use of everolimus in the setting of liver transplantation is promising. Jeng et al.,[22] in their study of 43 patients, concluded the safety of everolimus in the early phase after living donor liver transplantation. In their study, no hepatic artery thrombosis or wound infection was noted. Also, a possible role of everolimus in reducing the recurrence of hepatocellular carcinoma after liver transplantation was correlated. A target trough level of 3 ng/mL at 3 months was shown to be beneficial in recipients with pre-transplant renal dysfunction. In their study, 6 of 9 renal failure patients showed significant recovery of renal function, whereas 3 showed further deterioration, one of whom required hemodialysis.[23] Recently published report by Thorat et al. showed a positive impact on hepatocellular carcinoma (HCC) when everolimus was used as primary immunosuppression starting as early as first week after living donor liver transplantation (LDLT) surgery.[24] In their retrospective and prospective analysis at China Medical University Hospital in Taiwan, the study cohort (n=66) was divided in two groups depending upon the postoperative immunosuppression. Group A: HCC patients that received Everolimus + Tacrolimus based immunosuppressive regimen (n=37). Group B: HCC patients that received standard Tacrolimus based immunosuppressive regimen without everolimus (n=29). The target trough level for EVR was 3 to 5 ng/ml while for TAC it was 8–10 ng/ml. The 1-year, 3-year and 4-year overall survival achieved for Group A patients (Everolimus group) was 94.95%, 86.48% and 86.48%, respectively while for Group B patients it was 82.75%, 68.96%, and 62.06%, respectively (p=0.0217). The first 12-month report of ongoing Everolimus multicenter prospective trial in LDLT (H2307 trial), Jeng LB et al. have shown a 0% recurrence of HCC in everolimus group at 12 months.[25] Jeng LB concluded that an early introduction of everolimus + reduced tacrolimus was non-inferior to standard tacrolimus in terms of efficacy and renal function at 12 months, with HCC recurrence only in tacrolimus control patients.
Use in vascular stents
Everolimus is used in drug-eluting coronary stents as an immunosuppressant to prevent restenosis. Abbott Vascular produce an everolimus-eluting stent (EES) called Xience Alpine. It utilizes the Multi-Link Vision cobalt chromium stent platform and Novartis’ everolimus. The product is widely available globally including the US, the European Union, and Asia-Pacific (APAC) countries. Boston Scientific also market EESes, recent offerings being Promus Elite and Synergy.[citation needed]
Use in aging
Inhibition of mTOR, the molecular target of everolimus, extends the lifespan of model organisms including mice,[26] and mTOR inhibition has been suggested as an anti-aging therapy. Everolimus was used in a clinical trial by Novartis, and short-term treatment was shown to enhance the response to the influenza vaccine in the elderly, possible by reversing immunosenescence.[27] Everolimus treatment of mice results in reduced metabolic side effects compared to sirolimus.[18]Route 1
Reference:1. US5665772A.
2. Drug. Future 1999, 24, 22-29.Route 2
Reference:1. WO2014203185A1.Route 3
Reference:1. WO2012103959A1.Route 4
Reference:1. CN102731527A.
SYN

Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 2

Synthetic Reference
Polymer compositions containing a macrocyclic triene compound; Shulze, John E.; Betts, Ronald E.; Savage, Douglas R.; Assignee Sun Bow Co., Ltd., Bermuda; Sun Biomedical Ltd. 2003; Patent Information; Nov 06, 2003; WO 2003090684 A2
SYN 3

Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 4

Synthetic Reference
Zabudkin, Oleksandr; Schickaneder, Christian; Matviienko, Iaroslav; Sypchenko, Volodymyr. Method for the synthesis of rapamycin derivatives. Assignee Synbias Pharma AG, Switz. EP 3109250. (2016).
SYN 5

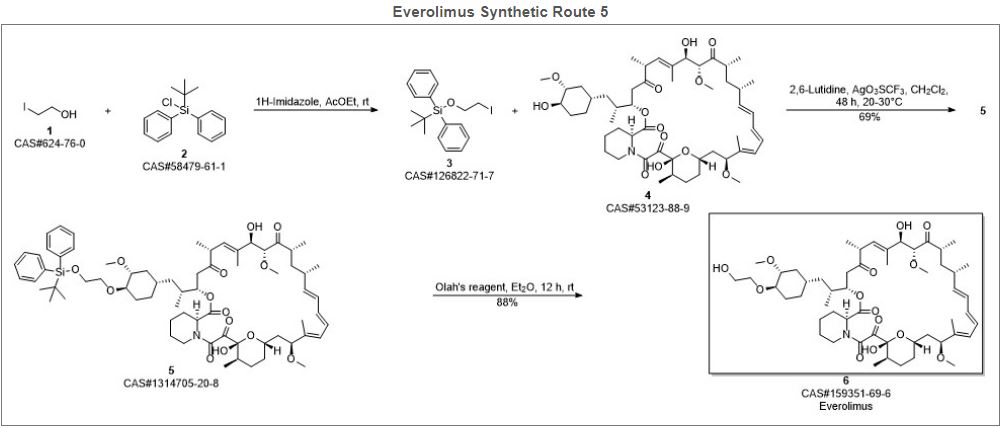
Synthetic Reference
Lu, Shiyong; Zhang, Xiaotian; Chen, Haohan; Ye, Weidong. Preparation of sirolimus 40-ether derivative. Assignee Zhejiang Medicine Co., Ltd. Xinchang Pharmaceutical Factory, Peop. Rep. China. CN 105237549. (2016).
SYN 6

Synthetic Reference
Seo, Jeong U.; Ham, Yun Beom; Kang, Heung Mo; Lee, Gwang Mu; Kim, In Gyu; Kim, Jeong Jin; Park, Ji Su. Preparation of everolimus and synthetic intermediate thereof. Assignee CKD Bio Corp., S. Korea. KR 1529963 (2015).
SYN
EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol.
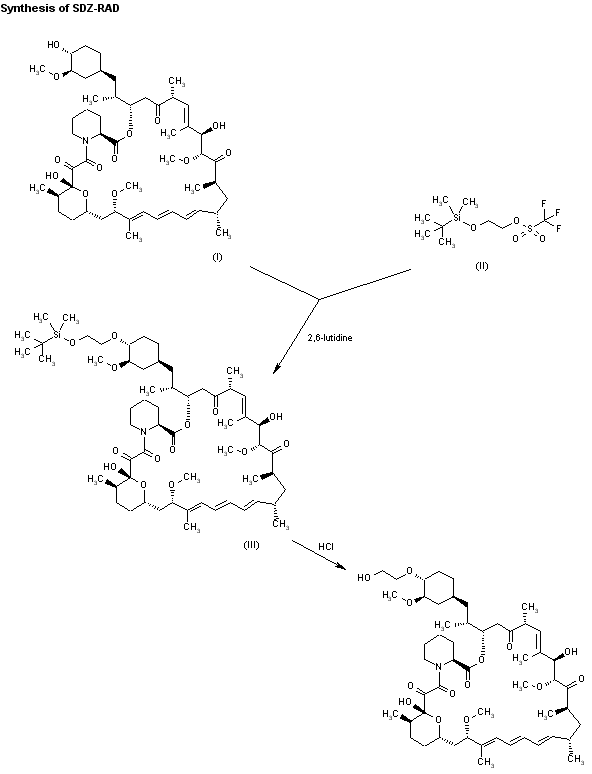
SYN
J Label Compd Radiopharm 1999,42(1),29
The compound has been obtained biosynthetically by an optimized fermentation process using Streptomyces hygroscopicus mutant RSH 1701 with a complex culture medium were [14C]-labeled (1R,3R,4R)-2,3-dichydroxycyclo-hexanecarboxylic acid (I) and [14C]-labeled (S)-pipecolic acid (II) have been added. This fermentation process yielded [14C]-labeled rapamycin (III), which was finally selectively O-alkylated at the C-40 position with monosilylated ethylene glycol triflate in DMSO/dimethoxyethane.
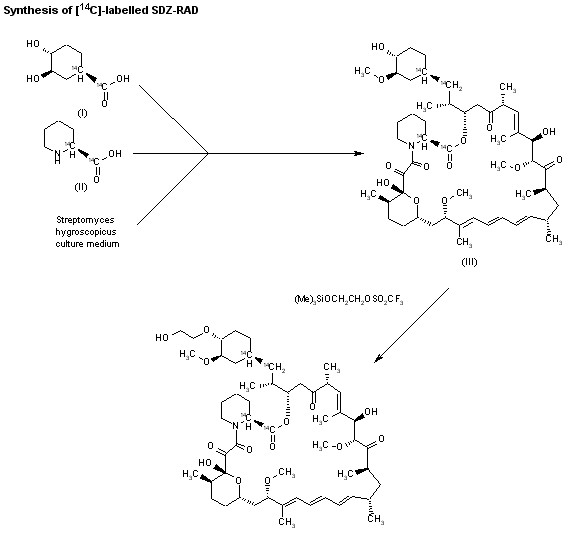
SYN
The reaction of the labeled acylated (+)-bornane-10,2-sultam (IV) with triethyl phosphite gives the phosphonate (V), which is treated with paraformaldehyde, galvinoxyl and K2CO3 yielding the acrylate derivative (VI). The cyclization of (VI) with butadiene (VII) by means of diethylaluminum chloride and galvinoxyl (as radical scavenger) affords the cyclohexene-carboxamide derivative (VIII), which is hydrolyzed with LiOH in THF/water giving the (1R)-3-cyclohexenecarboxylic acid (IX). The oxidation of (IX) with m-chloroperbenzoic acid and triethylamine in CCl4 yielded regioselectively the hydroxylactone (X), which is finally hydrolyzed with HCl to the labeled intermediate (I).
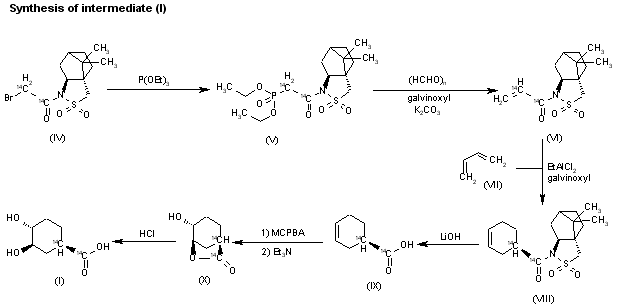
SYN
The reaction of the labeled acylated (-)-bornane-10,2-sultam (XI) with benzophenone imine (XII) gives the glycylsultam derivative (XIII), which is alkylated with 4-iodobutyl chloride (XIV) by means of butyllithium and DMPU in THF yielding intermediate (XV). The selective hydrolysis of (XV) with HCl affords the omega-chloro-L-norleucine derivative (XVI), which is cyclized by means of tetrabutylammonium fluoride and DIEA in hot acetonitrile giving the (2S)-piperidyl derivative (XVII). Finally, this compound is hydrolyzed with LiOH in THF/water to the labeled intermediate (II).
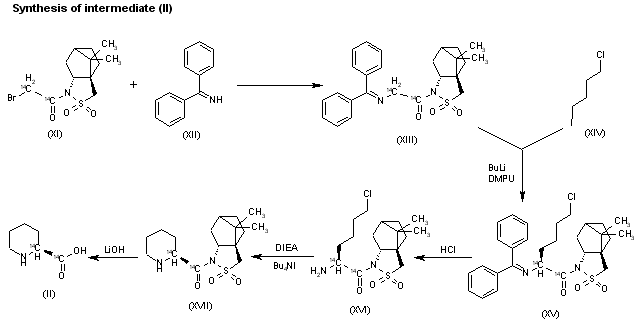
clipRapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:

See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.

(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis

Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
…………..
SYNTHESIS
https://www.google.com/patents/WO2012103960A1
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
RAP-OH + L-CH2-CH2-0-P — –> RAP-O-CH2-CH2-O-P — – > RAP-O-CH2-CH2-OH
(2) (4) (1)
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):

According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.

(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).

Only 8% yield of the corresponding compound (4B)

and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
xample 6: 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
……………………….
SYNTHESIS
https://www.google.co.in/patents/WO1994009010A1
Example 8: 40-O-(2-Hydroxy)ethyl-rapamycin
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
PATENT
https://patents.google.com/patent/WO2016020664A1/en
Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),

It is a derivative of sirolimus of formula III),

and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress™ (USA) and Certican™ (Europe and other countries) in transplantation medicine, and Afinitor™ in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV),

wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),

was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Sche

Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2- Hydroxy)ethyl rapamycin] of formula (I). However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),

as the alkylation agent. The overall yield reported was 25%. International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),

wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus. European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step. The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity
Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I) Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC. Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS- ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to – 25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol- triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.
Patent
Publication numberPriority datePublication dateAssigneeTitleUS5665772A *1992-10-091997-09-09Sandoz Ltd.O-alkylated rapamycin derivatives and their use, particularly as immunosuppressantsEP1518517A2 *2002-04-242005-03-30Sun Biomedical, Ltd.Drug-delivery endovascular stent and method for treating restenosisWO2012103960A12011-02-042012-08-09Synthon BvProcess for making trisubstituted silyloxyethyl triflatesCN102786534A2012-05-252012-11-21上海现代制药股份有限公司Preparation method of everolimusCN103788114A *2012-10-312014-05-14江苏汉邦科技有限公司Preparation method for everolimusEP3166950A12014-08-042017-05-17Cipla LimitedProcess for the synthesis of everolimus and intermediates thereof
CN107417718A *2017-08-182017-12-01常州兰陵制药有限公司The preparation method of everolimus intermediateUS9938297B22014-08-042018-04-10Cipia LimitedProcess for the synthesis of everolimus and intermediates thereofCN108676014A *2018-06-152018-10-19国药集团川抗制药有限公司The method for purifying the method for everolimus intermediate and preparing everolimus
Clip


References
- a WO 9 409 010 (Sandoz-Erfindungen; 28.4.1994; GB-prior. 9.10.1992).
- b US 6 277 983 (American Home Products; 21.8.2001; USA-prior. 27.9.2000).
- US 6 384 046 (Novartis; 7.5.2002; GB-prior. 27.3.1996).
- US 20 040 115 (Univ. of Pennsylvania; 15.1.2004; USA-prior. 9.7.2002).
- fermentation of rapamycin (sirolimus):
- Chen, Y. et al.: Process Biochemistry (Oxford, U. K.) (PBCHE5) 34, 4, 383 (1999).
- The Merck Index, 14th Ed., 666 (3907) (Rahway 2006).
- US 3 929 992 (Ayerst McKenna & Harrison Ltd.; 30.12.1975; USA-prior. 29.9.1972).
- WO 9 418 207 (Sandoz-Erfindungen; 18.8.1994; GB-prior. 2.2.1993).
- EP 638 125 (Pfizer; 17.4.1996; J-prior. 27.4.1992).
- US 6 313 264 (American Home Products; 6.11.2001; USA-prior. 8.3.1994).
clip
https://doi.org/10.1039/C7MD00474EIssue 1, 2018
Ascomycins and rapamycins The ascomycin tacrolimus (44, FK-506) and the two rapamycins sirolimus (45, rapamycin) and everolimus (46) are macrolides that contain 21- and 29-membered macrocyclic rings, respectively (Figure 7).[3] Their MWs range from just over 800 Da for tacrolimus (44) to >900 Da for sirolimus (45) and everolimus (46) and they have >10 HBAs. Like other natural product derived drugs in bRo5 space, they are above average complexity (SMCM 119–134) due to their 14–15 chiral centres. All three are immunosuppressants that are mainly used to prevent rejection of transplanted organs. They bind to overlapping, but slightly different parts of a shallow pocket at the surface of the immunophilin FK506 binding protein (FKBP12, Figure 8 A). Whereas tacrolimus (44) only binds in the pocket on FKBP12 (Figure 8 B),[67] sirolimus (45) and everolimus (46) promote binding of mammalian target of rapamycin (mTOR) so that they bind in a groove formed by FKBP12 and mTOR (Figure 8 C).[68] The complex between tacrolimus (44) and FKBP12 inhibits calcineurin, which results in reduced production of interleukin-2 and inactivation of T cells. Formation of the ternary complexes between FKBP12, sirolimus (45) [or everolimus (46)] and mTOR inhibits mTOR, which arrests growth of T lymphocytes by reducing their sensitivity to interleukin 2. Both tacrolimus (44) and sirolimus (45) have low (15–20 %) and variable bioavailabilities, whereas the bioavailability of everolimus (46) has been increased somewhat as compared to sirolimus (45).[3] Tacrolimus (44) was isolated from Streptomyces tsukubaensis in 1987,[69, 70] while sirolimus (45) was first identified from a Streptomycete strain found in a soil sample from Easter Island.[71] Later it was also isolated from fermentation of another Streptomycete strain.[72, 73] Both drugs are now produced through fermentation.[74, 75] Sirolimus suffers from low bioavailability as well as toxicity, and semi-synthetic derivatives were therefore prepared to minimise these issues. This led to the discovery of everolimus (46), synthesised by selective alkylation of one of the two secondary hydroxyl groups of sirolimus (45) with 2-(tert-butyldimethylsilyl)oxyethyltriflate followed by silyl ether deprotection with HCl (Scheme 8).[76, 77]

Figure 7. Structures of the ascomycin tacrolimus (44) and the rapamycins sirolimus (45) and everolimus (46) that are used mainly to prevent rejection of organ transplants.
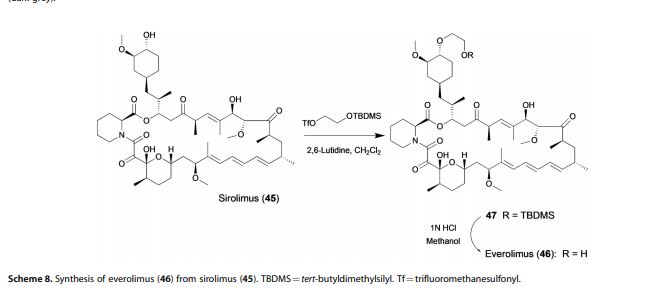
[67] G. D. Van Duyne, R. F. Standaert, P. A. Karplus, S. L. Schreiber, J. Clardy, Science 1991, 252, 839 – 842. [68] A. M. Marz, A.-K. Fabian, C. Kozany, A. Bracher, F. Hausch, Mol. Cell. Biol. 2013, 33, 1357 – 1367.
[69] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249 – 1255. [70] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T. Taga, J. Am. Chem. Soc. 1987, 109, 5031 – 5033. [71] C. Vzina, A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721 – 726. [72] S. N. Sehgal, H. Baker, C. Vzina, J. Antibiot. 1975, 28, 727 – 732. [73] S. N. Sehgal, T. M. Blazekovic, C. Vzina, 1975, US3929992A. [74] C. Barreiro, M. Mart nez-Castro, Appl. Microbiol. Biotechnol. 2014, 98, 497 – 507. [75] S. R. Park, Y. J. Yoo, Y.-H. Ban, Y. J. Yoon, J. Antibiot. 2010, 63, 434 – 441. [76] F. Navarro, S. Petit, G. Stone, 2007, US20020032213A1. [77] S. Cottens, R. Sedrani, 1997, US5665772A.
clip
Ferreting out why some cancer drugs struggle to shrink tumors
Study shows how stopping one enzyme could help drugs treat an important class of cancers more effectively
by Stu Borman
JUNE 27, 2018 | APPEARED IN VOLUME 96, ISSUE 27
In several types of cancer, including most cases of breast cancer, a cell-signaling network called the PI3K pathway is overactive. Drug designers have tried to quiet this pathway to kill cancer, but they haven’t had much success and, more frustratingly, haven’t understood why the problem is so hard to solve.

“There have been more than 200 clinical trials with experimental drugs that target the PI3K pathway, and probably more than $1 billion invested,” says Sourav Bandyopadhyay of the University of California, San Francisco. Just a handful of drugs have been approved by the U.S. FDA and one, Novartis’s Afinitor (everolimus), deters cancer growth but doesn’t shrink tumors, and it prolongs patient survival only a few months.
Bandyopadhyay, his UCSF colleague John D. Gordan, and coworkers used a proteomics approach to ferret out why previous attempts to target the PI3K pathway have had limited success and, using that information, devised and tested a possible fix (Nat. Chem. Biol. 2018, DOI: 10.1038/s41589-018-0081-9).
The stubborn pathway involves a series of kinases—enzymes that modify other proteins by adding phosphate groups—starting with one called PI3K. Overactivation of the pathway produces the transcription factor MYC, which turns on protein synthesis and can spark cancer growth.
The UCSF team used kinase-affinity beads and tandem mass spectrometry to survey all kinases active in breast cancer cells before and after treatment with a variety of cancer drugs. The team studied this so-called kinome to look for kinases associated with the cells’ tendency to resist drug treatments.
The researchers found that a kinase called AURKA undermines everolimus and other pathway-targeted drugs by reversing their effects. While the drugs try to turn off the PI3K pathway, AURKA, activated separately by other pathways, keeps the PI3K pathway turned on. To add insult to injury, MYC boosts AURKA production, maintaining a plentiful supply of the drug spoiler.

When the researchers coadministered everolimus with the AURKA inhibitor MLN8237, also called alisertib, everolimus could inhibit the PI3K pathway as it was designed to do, without interference. The combination treatment killed most types of cancer cells in culture and shrank tumors in mice with breast cancer, whereas everolimus alone permitted slow tumor growth to continue.
References

- ^ Jump up to:a b Use During Pregnancy and Breastfeeding
- ^ Formica RN, Lorber KM, Friedman AL, Bia MJ, Lakkis F, Smith JD, Lorber MI (March 2004). “The evolving experience using everolimus in clinical transplantation”. Transplantation Proceedings. 36 (2 Suppl): 495S–499S. doi:10.1016/j.transproceed.2004.01.015. PMID 15041395.
- ^ “Afinitor approved in US as first treatment for patients with advanced kidney cancer after failure of either sunitinib or sorafenib” (Press release). Novartis. 30 March 2009. Retrieved 6 April 2009.
- ^ “Novartis receives US FDA approval for Zortress (everolimus) to prevent organ rejection in adult kidney transplant recipients” (Press release). Novartis. 22 April 2010. Archived from the original on 25 April 2010. Retrieved 26 April 2010.
- ^ “Novartis’ Afinitor Cleared by FDA for Treating SEGA Tumors in Tuberous Sclerosis”. 1 November 2010.
- ^ https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm254350.htm
- ^ “US FDA approves Novartis drug Afinitor for breast cancer”. Reuters. 20 July 2012.
- ^ Jump up to:a b Everolimus (Afinitor). Feb 2016
- ^ Everolimus (Afinitor). April 2018
- ^ Lintern, Shaun (14 April 2015). “Policy delays risk ‘preventable deaths’, doctors warn NHS England”. Health Service Journal. Retrieved 20 April 2015.
- ^ “Couple forced to sell home after NHS refuse to fund daughter’s treatment for rare illness”. Daily Express. 11 May 2015. Retrieved 12 May 2015.
- ^ http://www.genengnews.com/gen-news-highlights/novartis-afinitor-cleared-by-fda-for-treating-sega-tumors-in-tuberous-sclerosis/81244159/
- ^ Lutz M, Kapp M, Grigoleit GU, Stuhler G, Einsele H, Mielke S (April 2012). “Salvage therapy with everolimus improves quality of life in patients with refractory chronic graft-versus-host disease” (PDF). Bone Marrow Transplant. 47 (S1): S410–S411.
- ^ “Positive Trial Data Leads Novartis to Plan Breast Cancer Filing for Afinitor by Year End”. 2011.
- ^ Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, Viale A, Heguy A, Peng L, Chan TA, Bochner B, Bajorin DF, Berger MF, Taylor BS, Solit DB (October 2012). “Genome sequencing identifies a basis for everolimus sensitivity”. Science. 338 (6104): 221. Bibcode:2012Sci…338..221I. doi:10.1126/science.1226344. PMC 3633467. PMID 22923433.
- ^ [1]
- ^ Jump up to:a b Zhavoronkov A (2020). “Geroprotective and senoremediative strategies to reduce the comorbidity, infection rates, severity, and lethality in gerophilic and gerolavic infections”. Aging. 12 (8): 6492–6510. doi:10.18632/aging.102988. PMC 7202545. PMID 32229705.
- ^ Jump up to:a b c Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, Pumper CP, Kimple ME, Lamming DW (February 2016). “Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system”. Aging Cell. 15 (1): 28–38. doi:10.1111/acel.12405. PMC 4717280. PMID 26463117.
- ^ Wang S, Raybuck A, Shiuan E, Jin J (2020). “Selective inhibition of mTORC1 in tumor vessels increases antitumor immunity”. JCI Insight. 5 (15): e139237. doi:10.1172/jci.insight.139237. PMC 7455083. PMID 32759497.
- ^ Jump up to:a b “Archived copy”. Archived from the original on 8 March 2014. Retrieved 26 February 2014.
- ^ Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sørensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P (August 2003). “Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients”. The New England Journal of Medicine. 349 (9): 847–58. doi:10.1056/NEJMoa022171. PMID 12944570.
- ^ Jeng LB, Thorat A, Hsieh YW, Yang HR, Yeh CC, Chen TH, Hsu SC, Hsu CH (April 2014). “Experience of using everolimus in the early stage of living donor liver transplantation”. Transplantation Proceedings. 46 (3): 744–8. doi:10.1016/j.transproceed.2013.11.068. PMID 24767339.
- ^ Jeng L, Thorat A, Yang H, Yeh C-C, Chen T-H, Hsu S-C. Impact of Everolimus On the Hepatocellular Carcinoma Recurrence After Living Donor Liver Transplantation When Used in Early Stage: A Single Center Prospective Study [abstract]. Am J Transplant. 2015; 15 (suppl 3). http://www.atcmeetingabstracts.com/abstract/impact-of-everolimus-on-the-hepatocellular-carcinoma-recurrence-after-living-donor-liver-transplantation-when-used-in-early-stage-a-single-center-prospective-study/. Accessed 1 September 2015.
- ^ Thorat A, Jeng LB, Yang HR, Yeh CC, Hsu SC, Chen TH, Poon KS (November 2017). “Assessing the role of everolimus in reducing hepatocellular carcinoma recurrence after living donor liver transplantation for patients within the UCSF criteria: re-inventing the role of mammalian target of rapamycin inhibitors”. Annals of Hepato-Biliary-Pancreatic Surgery. 21 (4): 205–211. doi:10.14701/ahbps.2017.21.4.205. PMC 5736740. PMID 29264583.
- ^ Jeng LB, Lee SG, Soin AS, Lee WC, Suh KS, Joo DJ, Uemoto S, Joh J, Yoshizumi T, Yang HR, Song GW, Lopez P, Kochuparampil J, Sips C, Kaneko S, Levy G (December 2017). “Efficacy and safety of everolimus with reduced tacrolimus in living-donor liver transplant recipients: 12-month results of a randomized multicenter study”. American Journal of Transplantation. 18 (6): 1435–1446. doi:10.1111/ajt.14623. PMID 29237235.
- ^ Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (July 2009). “Rapamycin fed late in life extends lifespan in genetically heterogeneous mice”. Nature. 460 (7253): 392–5. Bibcode:2009Natur.460..392H. doi:10.1038/nature08221. PMC 2786175. PMID 19587680.
- ^ Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB (December 2014). “mTOR inhibition improves immune function in the elderly”. Science Translational Medicine. 6 (268): 268ra179. doi:10.1126/scitranslmed.3009892. PMID 25540326. S2CID 206685475.
Further reading
- Sedrani R, Cottens S, Kallen J, Schuler W (August 1998). “Chemical modification of rapamycin: the discovery of SDZ RAD”. Transplantation Proceedings. 30 (5): 2192–4. doi:10.1016/S0041-1345(98)00587-9. PMID 9723437.
External links
- “Everolimus”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | Everolimus /ˌɛvəˈroʊləməs/ |
| Trade names | Afinitor, Zortress |
| Other names | 42-O-(2-hydroxyethyl)rapamycin, RAD001 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609032 |
| License data | EU EMA: by INNUS DailyMed: EverolimusUS FDA: Everolimus |
| Pregnancy category | AU: C[1] |
| Routes of administration | By mouth |
| ATC code | L01EG02 (WHO) L04AA18 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyEU: Rx-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Elimination half-life | ~30 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 159351-69-6 |
| PubChem CID | 6442177 |
| DrugBank | DB01590 |
| ChemSpider | 21106307 |
| UNII | 9HW64Q8G6G |
| KEGG | D02714 |
| ChEMBL | ChEMBL1908360 |
| CompTox Dashboard (EPA) | DTXSID0040599 |
| ECHA InfoCard | 100.149.896 |
| Chemical and physical data | |
| Formula | C53H83NO14 |
| Molar mass | 958.240 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESOCCO[C@@H]1CC[C@H](C[C@H]1OC)C[C@@H](C)[C@@H]4CC(=O)[C@H](C)/C=C(\C)[C@@H](O)[C@@H](OC)C(=O)[C@H](C)C[C@H](C)\C=C\C=C\C=C(/C)[C@@H](OC)C[C@@H]2CC[C@@H](C)[C@@](O)(O2)C(=O)C(=O)N3CCCC[C@H]3C(=O)O4 | |
| hideInChIInChI=1S/C53H83NO14/c1-32-16-12-11-13-17-33(2)44(63-8)30-40-21-19-38(7)53(62,68-40)50(59)51(60)54-23-15-14-18-41(54)52(61)67-45(35(4)28-39-20-22-43(66-25-24-55)46(29-39)64-9)31-42(56)34(3)27-37(6)48(58)49(65-10)47(57)36(5)26-32/h11-13,16-17,27,32,34-36,38-41,43-46,48-49,55,58,62H,14-15,18-26,28-31H2,1-10H3/b13-11+,16-12+,33-17+,37-27+/t32-,34-,35-,36-,38-,39+,40+,41+,43-,44+,45+,46-,48-,49+,53-/m1/s1 Key:HKVAMNSJSFKALM-GKUWKFKPSA-N |
//////////////// RAD-001, SDZ RAD, Certican, Novartis, Immunosuppressant, Everolimus, Afinitor, эверолимус , إيفيروليموس , 依维莫司 ,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Everolimus
159351-69-6[RN]
23,27-Epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydr oxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,26R,27R,34aS)-
23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-
42-O-(2-Hydroxyethyl)rapamycin
- Synonyms:RAD-001, SDZ-RAD, Afinitor
- ATC:L04AA18
Use:immunosuppressantChemical name:42-O-(2-hydroxyethyl)rapamycinFormula:C53H83NO14
- MW:958.24 g/mol
- CAS-RN:159351-69-6
EverolimusCAS Registry Number: 159351-69-6CAS Name: 42-O-(2-Hydroxyethyl)rapamycinAdditional Names: 40-O-(2-hydroxyethyl)rapamycinManufacturers’ Codes: RAD-001; SDZ RADTrademarks: Certican (Novartis)Molecular Formula: C53H83NO14Molecular Weight: 958.22Percent Composition: C 66.43%, H 8.73%, N 1.46%, O 23.38%Literature References: Macrolide immunosuppressant; derivative of rapamycin, q.v. Inhibits cytokine-mediated lymphocyte proliferation. Prepn: S. Cottens, R. Sedrani, WO9409010; eidem, US5665772 (1994, 1997 both to Sandoz). Pharmacology: W. Schuler et al., Transplantation64, 36 (1997). Whole blood determn by LC/MS: N. Brignol et al., Rapid Commun. Mass Spectrom.15, 898 (2001); by HPLC: S. Baldelli et al., J. Chromatogr. B816, 99 (2005). Clinical pharmacokinetics in combination with cyclosporine: J. M. Kovarik et al., Clin. Pharmacol. Ther.69, 48 (2001). Clinical study in prevention of cardiac-allograft vasculopathy: H. J. Eisen et al.,N. Engl. J. Med.349, 847 (2003). Review: F. J. Dumont et al., Curr. Opin. Invest. Drugs2, 1220-1234 (2001); B. Nashan, Ther. Drug Monit.24, 53-58 (2002).Therap-Cat: Immunosuppressant.Keywords: Immunosuppressant.эверолимус[Russian][INN]إيفيروليموس[Arabic][INN]依维莫司[Chinese][INN]Trade Name:Certican® / Zortress® / Afinitor®MOA:mTOR inhibitorIndication:Rejection of organ transplantation; Renal cell carcinoma; Advanced renal cell carcinoma (RCC); Advanced breast cancer; Pancreatic cancer; Renal angiomyolipoma; Tuberous sclerosis complex (TSC); Rejection in heart transplantation; Rejection of suppression renal transplantation; Subependymal giant cell astrocytoma; neuroendocrine tumors (NET); Advanced gastrointestinal tumorsStatus:ApprovedCompany:Novartis (Originator)Sales:$1,942 Million (Y2015);
$1,902 Million (Y2014);
$1,558 Million (Y2013);
$1,007 Million (Y2012);
$630 Million (Y2011);ATC Code:L04AA18Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-08-29 | New dosage form | Afinitor Disperz | Renal cell carcinoma , Advanced breast cancer, Pancreatic cancer, Renal angiomyolipoma, Tuberous sclerosis complex (TSC) | Tablet, For suspension | 2 mg/3 mg/5 mg | Novartis | Priority |
| 2010-04-20 | New strength | Zortress | Advanced renal cell carcinoma (RCC) | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2009-03-30 | Marketing approval | Afinitor | Advanced renal cell carcinoma (RCC) | Tablet | 2.5 mg/5 mg/7.5 mg/10 mg | Novartis | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2016-06-02 | New indication | Afinitor | neuroendocrine tumors (NET), Advanced gastrointestinal tumors | Tablet | Novartis | ||
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis | Orphan; Conditional Approval |
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet, Orally disintegrating | 2 mg/3 mg/5 mg | Novartis | Orphan; Conditional Approval |
| 2009-08-03 | Marketing approval | Afinitor | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-12-22 | New indication | Certican | Rejection of suppression renal transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2007-01-26 | Marketing approval | Certican | Rejection in heart transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-02-13 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 2.5 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 10 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 5 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2003-07-18 | Marketing approval | Certican | Rejection of organ transplantation, Renal cell carcinoma | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
clip
Active Substance The active substance Everolimus is a hydroxyethyl derivative of rapamycin, which is a macrolide, isolated from the micro-organism Streptomyces hygroscopicus. The guideline, impurities in new active substances ICHQ 3A (R), does not apply to active substance of fermented origin. Everolimus (INN) or 42-O-(2-hydroxyethyl)-rapamycin (chemical name) or C5 3H8 3N O1 4 has been fully described. The molecule is amorphous and is stabilised with an antioxidant. Its physico-chemical properties including parameters such as solubility, pH, specific rotation, potential polymorphism and potential isomerism have been fully characterised. Everolimus is a white to faintly yellow amorphous powder. It is almost insoluble in water, is unstable at temperatures above 25 °C and is sensitive to light. In addition, possible isomerism has been investigated. Everolimus contains 15 asymmetric carbon atoms and 4 substituted double bonds. The configuration of the asymmetric carbon atoms and the double bonds is guaranteed by the microbial origin of Rapamycin. The configuration is not affected by the chemical synthesis. Polymorphism has been comprehensively discussed and it was demonstrated that the molecule domain remains amorphous.
Synthesis of Everolimus The manufacturing process consists of four main steps, (1) fermentation, (2) extraction of rapamycin from the fermentation broth, (3) chemical modification of rapamycin starting material, (4) purification of crude everolimus and stabilisation with BHT. The choice of the stabilizer has been sufficiently explained and justified by experimental results. Interactions products of Everolimus and the antioxidant were not detected, or were below detection limit. Rapamycin, obtained by a fermentation process, was used as the starting material. Reaction conditions and the necessary in-process controls are described in detail. Adequate specifications for starting materials and isolated intermediates and descriptions of the test procedures have been submitted. Control of the quality of solvents, reagents and auxiliary materials used in the synthesis has been adequately documented. It is stated by the manufacturer of rapamycin solution that no starting material of animal or human origin is used in the fermentation. Elucidation of structure and other characteristics The structure of Everolimus has been fully elucidated using several spectroscopic techniques such as ultraviolet absorption spectroscopy (UV), Infra-red spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance spectroscopy (1 H and 13C NMR), mass spectroscopy, diffractometry (X-ray) and elemental analysis. Related substances An extensive discussion was presented on the related substances. The complex structure of Everolimus allows several possible degradation pathways to occur at various positions of the molecule. Everolimus alone is extremely sensitive to oxidation. By the addition of an antioxidant, the sensitivity to oxidation is significantly reduced (the antioxidant is known to react as a scavenger of peroxide radicals). It is assumed that oxidation of Everolimus proceeds via a radical mechanism. All the requirements set in the current testing instruction valid for Everolimus are justified on the basis of the results obtained during development and manufactured at the production scale.
fda
Everolimus was first approved by Swiss Agency for therapeutic products,Swissmedic on July 18, 2003, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on April 23, 2004, and approved by the U.S. Food and Drug Administration (FDA) on Mar 30, 2009, approved by European Medicine Agency (EMA) on Aug 3, 2009. It was developed and marketed as Certican® by Novartis in SE.
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR). It is indicated for the treatment of renal cell cancer and other tumours and currently used as an immunosuppressant to prevent rejection of organ transplants.
Certican® is available as tablet for oral use, containing 0.25, 0.5 or 0.75 mg of free Everolimus. The recommended dose is 10 mg once daily with or without food for advanced HR+ breast cancer, advanced progressive neuroendocrine tumors, advanced renal cell carcinoma or renal angiomyolipoma with tuberous sclerosis complex.
Everolimus, also known as RAD001, is a derivative of the natural macrocyclic lactone sirolimus with immunosuppressant and anti-angiogenic properties. In cells, everolimus binds to the immunophilin FK Binding Protein-12 (FKBP-12) to generate an immunosuppressive complex that binds to and inhibits the activation of the mammalian Target of Rapamycin (mTOR), a key regulatory kinase. Inhibition of mTOR activation results in the inhibition of T lymphocyte activation and proliferation associated with antigen and cytokine (IL-2, IL-4, and IL-15) stimulation and the inhibition of antibody production.
Everolimus is a medication used as an immunosuppressant to prevent rejection of organ transplants and in the treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors as targeted therapy for use in a number of cancers.[medical citation needed]
It is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is marketed by Novartis under the trade names Zortress (USA) and Certican (European Union and other countries) in transplantation medicine, and as Afinitor (general tumours) and Votubia (tumours as a result of TSC) in oncology. Everolimus is also available from Biocon, with the brand name Evertor.
Medical uses
Everolimus is approved for various conditions:
- Advanced kidney cancer (US FDA approved in March 2009)[3]
- Prevention of organ rejection after renal transplant(US FDA April 2010)[4]
- Subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis (TS) in patients who are not suitable for surgical intervention (US FDA October 2010)[5]
- Progressive or metastatic pancreatic neuroendocrine tumors not surgically removable (May 2011)[6]
- Breast cancer in post-menopausal women with advanced hormone-receptor positive, HER2-negative type cancer, in conjunction with exemestane (US FDA July 2012)[7]
- Prevention of organ rejection after liver transplant(Feb 2013)
- Progressive, well-differentiated non-functional, neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease (US FDA February 2016).[8]
- Tuberous sclerosis complex-associated partial-onset seizures for adult and pediatric patients aged 2 years and older. (US FDA April 2018).[9]
UK National Health Service
NHS England has been criticised for delays in deciding on a policy for the prescription of everolimus in the treatment of Tuberous Sclerosis. 20 doctors addressed a letter to the board in support of the charity Tuberous Scelerosis Association saying ” around 32 patients with critical need, whose doctors believe everolimus treatment is their best or only option, have no hope of access to funding. Most have been waiting many months. Approximately half of these patients are at imminent risk of a catastrophic event (renal bleed or kidney failure) with a high risk of preventable death.”[10] In May 2015 it was reported that Luke Henry and Stephanie Rudwick, the parents of a child suffering from Tuberous Sclerosis were trying to sell their home in Brighton to raise £30,000 to pay for treatment for their daughter Bethany who has tumours on her brain, kidneys and liver and suffers from up to 50 epileptic fits a day.[11]
Clinical trials
As of October 2010, Phase III trials are under way in gastric cancer, hepatocellular carcinoma, and lymphoma.[12] The experimental use of everolimus in refractory chronic graft-versus-host disease was reported in 2012.[13]
Interim phase III trial results in 2011 showed that adding Afinitor (everolimus) to exemestane therapy against advanced breast cancer can significantly improve progression-free survival compared with exemestane therapy alone.[14]
A study published in 2012, shows that everolimus sensitivity varies between patients depending on their tumor genomes.[15] A group of patients with advanced metastasic bladder carcinoma (NCT00805129) [16] treated with everolimus revealed a single patient who had a complete response to everolimus treatment for 26 months. The researchers sequenced the genome of this patient and compared it to different reference genomes and to other patients’ genomes. They found that mutations in TSC1 led to a lengthened duration of response to everolimus and to an increase in the time to cancer recurrence. The mutated TSC1 apparently had made these tumors vulnerable to treatment with everolimus.[medical citation needed]
A phase 2a randomized, placebo-controlled everolimus clinical trial published in 2014 showed that everolimus improved the response to an influenza vaccine by 20% in healthy elderly volunteers.[17] A phase 2a randomized, placebo-controlled clinical trial published in 2018 showed that everolimus in combination with dactolisib decreased the rate of reported infections in an elderly population.[17]
Mechanism
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex.[18] This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2 positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types.[medical citation needed] Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
mTORC1 inhibition by everolimus has been shown to normalize tumor blood vessels, to increase tumor-infiltrating lymphocytes, and to improve adoptive cell transfer therapy.[19]
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.[18]
TSC1 and TSC2, the genes involved in tuberous sclerosis, act as tumor suppressor genes by regulating mTORC1 activity. Thus, either the loss or inactivation of one of these genes lead to the activation of mTORC1.[20]
Everolimus binds to its protein receptor FKBP12, which directly interacts with mTORC1, inhibiting its downstream signaling. As a consequence, mRNAs that code for proteins implicated in the cell cycle and in the glycolysis process are impaired or altered, and tumor growth is inhibited.[20]
Adverse reactions
A trial using 10 mg/day in patients with NETs of GI or lung origin reported “Everolimus was discontinued for adverse reactions in 29% of patients and dose reduction or delay was required in 70% of everolimus-treated patients. Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). The most common adverse reactions (incidence greater than or equal to 30%) were stomatitis, infections, diarrhea, peripheral edema, fatigue and rash. The most common blood abnormalities found (incidence greater than or equal to 50%) were anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase (AST) and fasting hyperglycemia.”.[8]
Role in heart transplantation
Everolimus may have a role in heart transplantation, as it has been shown to reduce chronic allograft vasculopathy in such transplants. It also may have a similar role to sirolimus in kidney and other transplants.[21]
Role in liver transplantation
Although, sirolimus had generated fears over use of m-TOR inhibitors in liver transplantation recipients, due to possible early hepatic artery thrombosis and graft loss, use of everolimus in the setting of liver transplantation is promising. Jeng et al.,[22] in their study of 43 patients, concluded the safety of everolimus in the early phase after living donor liver transplantation. In their study, no hepatic artery thrombosis or wound infection was noted. Also, a possible role of everolimus in reducing the recurrence of hepatocellular carcinoma after liver transplantation was correlated. A target trough level of 3 ng/mL at 3 months was shown to be beneficial in recipients with pre-transplant renal dysfunction. In their study, 6 of 9 renal failure patients showed significant recovery of renal function, whereas 3 showed further deterioration, one of whom required hemodialysis.[23] Recently published report by Thorat et al. showed a positive impact on hepatocellular carcinoma (HCC) when everolimus was used as primary immunosuppression starting as early as first week after living donor liver transplantation (LDLT) surgery.[24] In their retrospective and prospective analysis at China Medical University Hospital in Taiwan, the study cohort (n=66) was divided in two groups depending upon the postoperative immunosuppression. Group A: HCC patients that received Everolimus + Tacrolimus based immunosuppressive regimen (n=37). Group B: HCC patients that received standard Tacrolimus based immunosuppressive regimen without everolimus (n=29). The target trough level for EVR was 3 to 5 ng/ml while for TAC it was 8–10 ng/ml. The 1-year, 3-year and 4-year overall survival achieved for Group A patients (Everolimus group) was 94.95%, 86.48% and 86.48%, respectively while for Group B patients it was 82.75%, 68.96%, and 62.06%, respectively (p=0.0217). The first 12-month report of ongoing Everolimus multicenter prospective trial in LDLT (H2307 trial), Jeng LB et al. have shown a 0% recurrence of HCC in everolimus group at 12 months.[25] Jeng LB concluded that an early introduction of everolimus + reduced tacrolimus was non-inferior to standard tacrolimus in terms of efficacy and renal function at 12 months, with HCC recurrence only in tacrolimus control patients.
Use in vascular stents
Everolimus is used in drug-eluting coronary stents as an immunosuppressant to prevent restenosis. Abbott Vascular produce an everolimus-eluting stent (EES) called Xience Alpine. It utilizes the Multi-Link Vision cobalt chromium stent platform and Novartis’ everolimus. The product is widely available globally including the US, the European Union, and Asia-Pacific (APAC) countries. Boston Scientific also market EESes, recent offerings being Promus Elite and Synergy.[citation needed]
Use in aging
Inhibition of mTOR, the molecular target of everolimus, extends the lifespan of model organisms including mice,[26] and mTOR inhibition has been suggested as an anti-aging therapy. Everolimus was used in a clinical trial by Novartis, and short-term treatment was shown to enhance the response to the influenza vaccine in the elderly, possible by reversing immunosenescence.[27] Everolimus treatment of mice results in reduced metabolic side effects compared to sirolimus.[18]Route 1
Reference:1. US5665772A.
2. Drug. Future 1999, 24, 22-29.Route 2
Reference:1. WO2014203185A1.Route 3
Reference:1. WO2012103959A1.Route 4
Reference:1. CN102731527A.
SYN
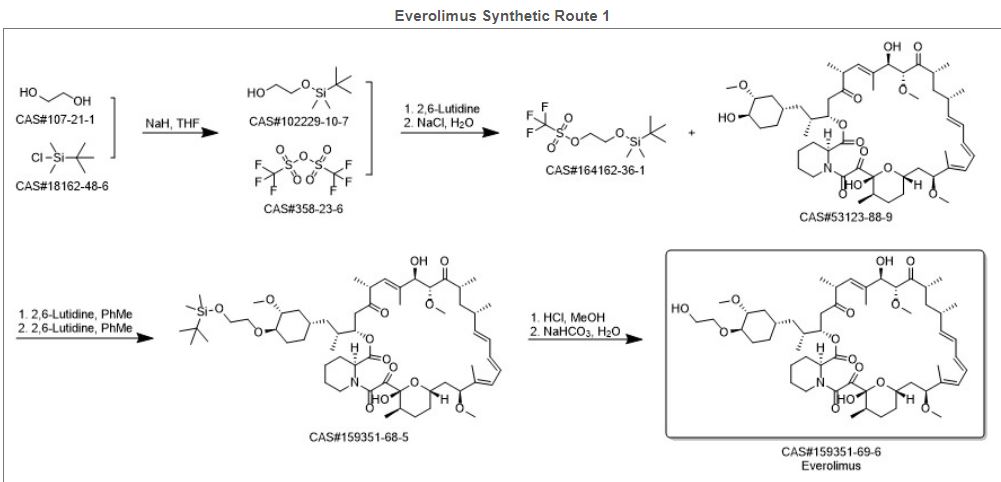{kind=link}
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 2
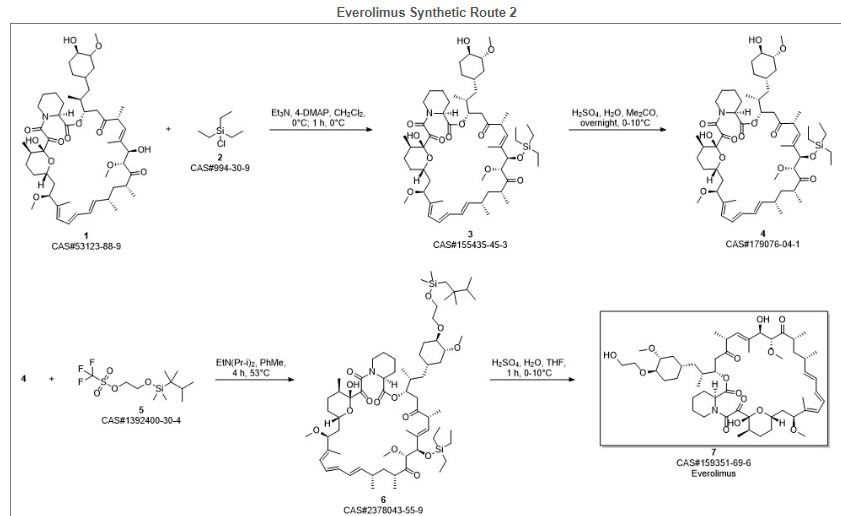
Synthetic Reference
Polymer compositions containing a macrocyclic triene compound; Shulze, John E.; Betts, Ronald E.; Savage, Douglas R.; Assignee Sun Bow Co., Ltd., Bermuda; Sun Biomedical Ltd. 2003; Patent Information; Nov 06, 2003; WO 2003090684 A2
SYN 3
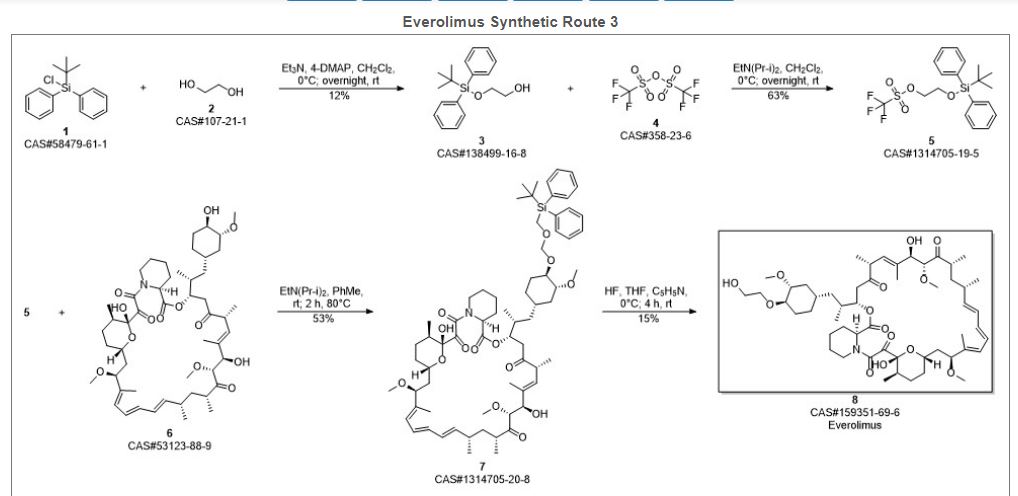
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 4
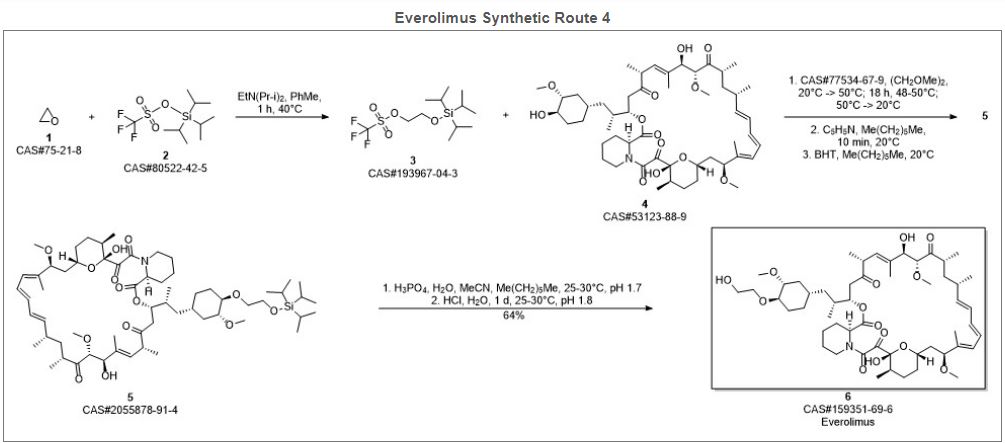
Synthetic Reference
Zabudkin, Oleksandr; Schickaneder, Christian; Matviienko, Iaroslav; Sypchenko, Volodymyr. Method for the synthesis of rapamycin derivatives. Assignee Synbias Pharma AG, Switz. EP 3109250. (2016).
SYN 5
Synthetic Reference
Lu, Shiyong; Zhang, Xiaotian; Chen, Haohan; Ye, Weidong. Preparation of sirolimus 40-ether derivative. Assignee Zhejiang Medicine Co., Ltd. Xinchang Pharmaceutical Factory, Peop. Rep. China. CN 105237549. (2016).
SYN 6
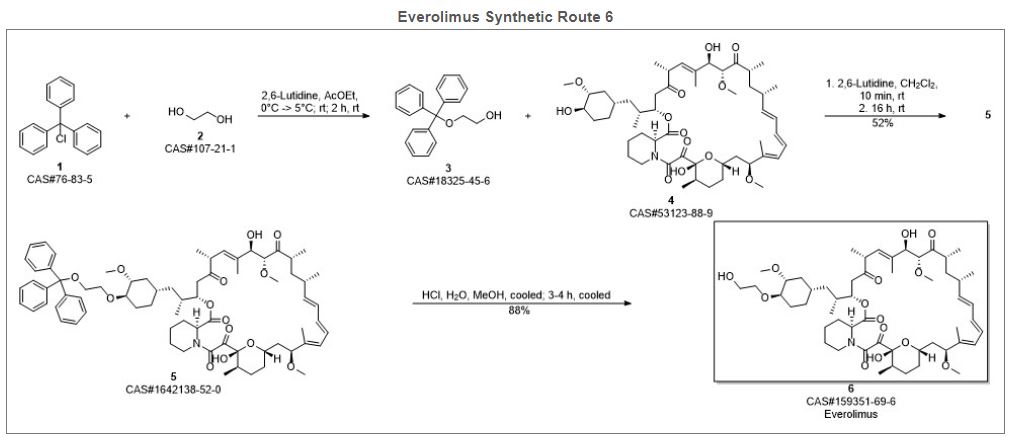
Synthetic Reference
Seo, Jeong U.; Ham, Yun Beom; Kang, Heung Mo; Lee, Gwang Mu; Kim, In Gyu; Kim, Jeong Jin; Park, Ji Su. Preparation of everolimus and synthetic intermediate thereof. Assignee CKD Bio Corp., S. Korea. KR 1529963 (2015).
SYN
EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol.
SYN
J Label Compd Radiopharm 1999,42(1),29
The compound has been obtained biosynthetically by an optimized fermentation process using Streptomyces hygroscopicus mutant RSH 1701 with a complex culture medium were [14C]-labeled (1R,3R,4R)-2,3-dichydroxycyclo-hexanecarboxylic acid (I) and [14C]-labeled (S)-pipecolic acid (II) have been added. This fermentation process yielded [14C]-labeled rapamycin (III), which was finally selectively O-alkylated at the C-40 position with monosilylated ethylene glycol triflate in DMSO/dimethoxyethane.
SYN
The reaction of the labeled acylated (+)-bornane-10,2-sultam (IV) with triethyl phosphite gives the phosphonate (V), which is treated with paraformaldehyde, galvinoxyl and K2CO3 yielding the acrylate derivative (VI). The cyclization of (VI) with butadiene (VII) by means of diethylaluminum chloride and galvinoxyl (as radical scavenger) affords the cyclohexene-carboxamide derivative (VIII), which is hydrolyzed with LiOH in THF/water giving the (1R)-3-cyclohexenecarboxylic acid (IX). The oxidation of (IX) with m-chloroperbenzoic acid and triethylamine in CCl4 yielded regioselectively the hydroxylactone (X), which is finally hydrolyzed with HCl to the labeled intermediate (I).
SYN
The reaction of the labeled acylated (-)-bornane-10,2-sultam (XI) with benzophenone imine (XII) gives the glycylsultam derivative (XIII), which is alkylated with 4-iodobutyl chloride (XIV) by means of butyllithium and DMPU in THF yielding intermediate (XV). The selective hydrolysis of (XV) with HCl affords the omega-chloro-L-norleucine derivative (XVI), which is cyclized by means of tetrabutylammonium fluoride and DIEA in hot acetonitrile giving the (2S)-piperidyl derivative (XVII). Finally, this compound is hydrolyzed with LiOH in THF/water to the labeled intermediate (II).
clipRapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:
See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.
(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
…………..
SYNTHESIS
https://www.google.com/patents/WO2012103960A1
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
RAP-OH + L-CH2-CH2-0-P — –> RAP-O-CH2-CH2-O-P — – > RAP-O-CH2-CH2-OH
(2) (4) (1)
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):
According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.
(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).
Only 8% yield of the corresponding compound (4B)
and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
xample 6: 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
……………………….
SYNTHESIS
https://www.google.co.in/patents/WO1994009010A1
Example 8: 40-O-(2-Hydroxy)ethyl-rapamycin
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
PATENT
https://patents.google.com/patent/WO2016020664A1/en
Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),
It is a derivative of sirolimus of formula III),
and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress™ (USA) and Certican™ (Europe and other countries) in transplantation medicine, and Afinitor™ in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV),
wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),
was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Sche
Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2- Hydroxy)ethyl rapamycin] of formula (I). However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),
as the alkylation agent. The overall yield reported was 25%. International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),
wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus. European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step. The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity
Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I) Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC. Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS- ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to – 25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol- triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.
Patent
Publication numberPriority datePublication dateAssigneeTitleUS5665772A *1992-10-091997-09-09Sandoz Ltd.O-alkylated rapamycin derivatives and their use, particularly as immunosuppressantsEP1518517A2 *2002-04-242005-03-30Sun Biomedical, Ltd.Drug-delivery endovascular stent and method for treating restenosisWO2012103960A12011-02-042012-08-09Synthon BvProcess for making trisubstituted silyloxyethyl triflatesCN102786534A2012-05-252012-11-21上海现代制药股份有限公司Preparation method of everolimusCN103788114A *2012-10-312014-05-14江苏汉邦科技有限公司Preparation method for everolimusEP3166950A12014-08-042017-05-17Cipla LimitedProcess for the synthesis of everolimus and intermediates thereof
CN107417718A *2017-08-182017-12-01常州兰陵制药有限公司The preparation method of everolimus intermediateUS9938297B22014-08-042018-04-10Cipia LimitedProcess for the synthesis of everolimus and intermediates thereofCN108676014A *2018-06-152018-10-19国药集团川抗制药有限公司The method for purifying the method for everolimus intermediate and preparing everolimus
Enzymes
Synthesis Path
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | Certican | Novartis ,2004 | |
| F | Certican | Novartis | |
| I | Certican | Novartis | |
| J | Certican | Novartis |
Formulations
- tabl. 0.25 mg, 0.5 mg, 0.75 mg
References
- a WO 9 409 010 (Sandoz-Erfindungen; 28.4.1994; GB-prior. 9.10.1992).
- b US 6 277 983 (American Home Products; 21.8.2001; USA-prior. 27.9.2000).
- US 6 384 046 (Novartis; 7.5.2002; GB-prior. 27.3.1996).
- US 20 040 115 (Univ. of Pennsylvania; 15.1.2004; USA-prior. 9.7.2002).
- fermentation of rapamycin (sirolimus):
- Chen, Y. et al.: Process Biochemistry (Oxford, U. K.) (PBCHE5) 34, 4, 383 (1999).
- The Merck Index, 14th Ed., 666 (3907) (Rahway 2006).
- US 3 929 992 (Ayerst McKenna & Harrison Ltd.; 30.12.1975; USA-prior. 29.9.1972).
- WO 9 418 207 (Sandoz-Erfindungen; 18.8.1994; GB-prior. 2.2.1993).
- EP 638 125 (Pfizer; 17.4.1996; J-prior. 27.4.1992).
- US 6 313 264 (American Home Products; 6.11.2001; USA-prior. 8.3.1994).
clip
https://doi.org/10.1039/C7MD00474EIssue 1, 2018
Ascomycins and rapamycins The ascomycin tacrolimus (44, FK-506) and the two rapamycins sirolimus (45, rapamycin) and everolimus (46) are macrolides that contain 21- and 29-membered macrocyclic rings, respectively (Figure 7).[3] Their MWs range from just over 800 Da for tacrolimus (44) to >900 Da for sirolimus (45) and everolimus (46) and they have >10 HBAs. Like other natural product derived drugs in bRo5 space, they are above average complexity (SMCM 119–134) due to their 14–15 chiral centres. All three are immunosuppressants that are mainly used to prevent rejection of transplanted organs. They bind to overlapping, but slightly different parts of a shallow pocket at the surface of the immunophilin FK506 binding protein (FKBP12, Figure 8 A). Whereas tacrolimus (44) only binds in the pocket on FKBP12 (Figure 8 B),[67] sirolimus (45) and everolimus (46) promote binding of mammalian target of rapamycin (mTOR) so that they bind in a groove formed by FKBP12 and mTOR (Figure 8 C).[68] The complex between tacrolimus (44) and FKBP12 inhibits calcineurin, which results in reduced production of interleukin-2 and inactivation of T cells. Formation of the ternary complexes between FKBP12, sirolimus (45) [or everolimus (46)] and mTOR inhibits mTOR, which arrests growth of T lymphocytes by reducing their sensitivity to interleukin 2. Both tacrolimus (44) and sirolimus (45) have low (15–20 %) and variable bioavailabilities, whereas the bioavailability of everolimus (46) has been increased somewhat as compared to sirolimus (45).[3] Tacrolimus (44) was isolated from Streptomyces tsukubaensis in 1987,[69, 70] while sirolimus (45) was first identified from a Streptomycete strain found in a soil sample from Easter Island.[71] Later it was also isolated from fermentation of another Streptomycete strain.[72, 73] Both drugs are now produced through fermentation.[74, 75] Sirolimus suffers from low bioavailability as well as toxicity, and semi-synthetic derivatives were therefore prepared to minimise these issues. This led to the discovery of everolimus (46), synthesised by selective alkylation of one of the two secondary hydroxyl groups of sirolimus (45) with 2-(tert-butyldimethylsilyl)oxyethyltriflate followed by silyl ether deprotection with HCl (Scheme 8).[76, 77]
Figure 7. Structures of the ascomycin tacrolimus (44) and the rapamycins sirolimus (45) and everolimus (46) that are used mainly to prevent rejection of organ transplants.

[67] G. D. Van Duyne, R. F. Standaert, P. A. Karplus, S. L. Schreiber, J. Clardy, Science 1991, 252, 839 – 842. [68] A. M. Marz, A.-K. Fabian, C. Kozany, A. Bracher, F. Hausch, Mol. Cell. Biol. 2013, 33, 1357 – 1367.
[69] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249 – 1255. [70] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T. Taga, J. Am. Chem. Soc. 1987, 109, 5031 – 5033. [71] C. Vzina, A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721 – 726. [72] S. N. Sehgal, H. Baker, C. Vzina, J. Antibiot. 1975, 28, 727 – 732. [73] S. N. Sehgal, T. M. Blazekovic, C. Vzina, 1975, US3929992A. [74] C. Barreiro, M. Mart nez-Castro, Appl. Microbiol. Biotechnol. 2014, 98, 497 – 507. [75] S. R. Park, Y. J. Yoo, Y.-H. Ban, Y. J. Yoon, J. Antibiot. 2010, 63, 434 – 441. [76] F. Navarro, S. Petit, G. Stone, 2007, US20020032213A1. [77] S. Cottens, R. Sedrani, 1997, US5665772A.
clip
Ferreting out why some cancer drugs struggle to shrink tumors
Study shows how stopping one enzyme could help drugs treat an important class of cancers more effectively
by Stu Borman
JUNE 27, 2018 | APPEARED IN VOLUME 96, ISSUE 27
In several types of cancer, including most cases of breast cancer, a cell-signaling network called the PI3K pathway is overactive. Drug designers have tried to quiet this pathway to kill cancer, but they haven’t had much success and, more frustratingly, haven’t understood why the problem is so hard to solve.
“There have been more than 200 clinical trials with experimental drugs that target the PI3K pathway, and probably more than $1 billion invested,” says Sourav Bandyopadhyay of the University of California, San Francisco. Just a handful of drugs have been approved by the U.S. FDA and one, Novartis’s Afinitor (everolimus), deters cancer growth but doesn’t shrink tumors, and it prolongs patient survival only a few months.
Bandyopadhyay, his UCSF colleague John D. Gordan, and coworkers used a proteomics approach to ferret out why previous attempts to target the PI3K pathway have had limited success and, using that information, devised and tested a possible fix (Nat. Chem. Biol. 2018, DOI: 10.1038/s41589-018-0081-9).
The stubborn pathway involves a series of kinases—enzymes that modify other proteins by adding phosphate groups—starting with one called PI3K. Overactivation of the pathway produces the transcription factor MYC, which turns on protein synthesis and can spark cancer growth.
The UCSF team used kinase-affinity beads and tandem mass spectrometry to survey all kinases active in breast cancer cells before and after treatment with a variety of cancer drugs. The team studied this so-called kinome to look for kinases associated with the cells’ tendency to resist drug treatments.
The researchers found that a kinase called AURKA undermines everolimus and other pathway-targeted drugs by reversing their effects. While the drugs try to turn off the PI3K pathway, AURKA, activated separately by other pathways, keeps the PI3K pathway turned on. To add insult to injury, MYC boosts AURKA production, maintaining a plentiful supply of the drug spoiler.
When the researchers coadministered everolimus with the AURKA inhibitor MLN8237, also called alisertib, everolimus could inhibit the PI3K pathway as it was designed to do, without interference. The combination treatment killed most types of cancer cells in culture and shrank tumors in mice with breast cancer, whereas everolimus alone permitted slow tumor growth to continue.
References
- ^ Jump up to:a b Use During Pregnancy and Breastfeeding
- ^ Formica RN, Lorber KM, Friedman AL, Bia MJ, Lakkis F, Smith JD, Lorber MI (March 2004). “The evolving experience using everolimus in clinical transplantation”. Transplantation Proceedings. 36 (2 Suppl): 495S–499S. doi:10.1016/j.transproceed.2004.01.015. PMID 15041395.
- ^ “Afinitor approved in US as first treatment for patients with advanced kidney cancer after failure of either sunitinib or sorafenib” (Press release). Novartis. 30 March 2009. Retrieved 6 April 2009.
- ^ “Novartis receives US FDA approval for Zortress (everolimus) to prevent organ rejection in adult kidney transplant recipients” (Press release). Novartis. 22 April 2010. Archived from the original on 25 April 2010. Retrieved 26 April 2010.
- ^ “Novartis’ Afinitor Cleared by FDA for Treating SEGA Tumors in Tuberous Sclerosis”. 1 November 2010.
- ^ https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm254350.htm
- ^ “US FDA approves Novartis drug Afinitor for breast cancer”. Reuters. 20 July 2012.
- ^ Jump up to:a b Everolimus (Afinitor). Feb 2016
- ^ Everolimus (Afinitor). April 2018
- ^ Lintern, Shaun (14 April 2015). “Policy delays risk ‘preventable deaths’, doctors warn NHS England”. Health Service Journal. Retrieved 20 April 2015.
- ^ “Couple forced to sell home after NHS refuse to fund daughter’s treatment for rare illness”. Daily Express. 11 May 2015. Retrieved 12 May 2015.
- ^ http://www.genengnews.com/gen-news-highlights/novartis-afinitor-cleared-by-fda-for-treating-sega-tumors-in-tuberous-sclerosis/81244159/
- ^ Lutz M, Kapp M, Grigoleit GU, Stuhler G, Einsele H, Mielke S (April 2012). “Salvage therapy with everolimus improves quality of life in patients with refractory chronic graft-versus-host disease” (PDF). Bone Marrow Transplant. 47 (S1): S410–S411.
- ^ “Positive Trial Data Leads Novartis to Plan Breast Cancer Filing for Afinitor by Year End”. 2011.
- ^ Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, Viale A, Heguy A, Peng L, Chan TA, Bochner B, Bajorin DF, Berger MF, Taylor BS, Solit DB (October 2012). “Genome sequencing identifies a basis for everolimus sensitivity”. Science. 338 (6104): 221. Bibcode:2012Sci…338..221I. doi:10.1126/science.1226344. PMC 3633467. PMID 22923433.
- ^ [1]
- ^ Jump up to:a b Zhavoronkov A (2020). “Geroprotective and senoremediative strategies to reduce the comorbidity, infection rates, severity, and lethality in gerophilic and gerolavic infections”. Aging. 12 (8): 6492–6510. doi:10.18632/aging.102988. PMC 7202545. PMID 32229705.
- ^ Jump up to:a b c Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, Pumper CP, Kimple ME, Lamming DW (February 2016). “Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system”. Aging Cell. 15 (1): 28–38. doi:10.1111/acel.12405. PMC 4717280. PMID 26463117.
- ^ Wang S, Raybuck A, Shiuan E, Jin J (2020). “Selective inhibition of mTORC1 in tumor vessels increases antitumor immunity”. JCI Insight. 5 (15): e139237. doi:10.1172/jci.insight.139237. PMC 7455083. PMID 32759497.
- ^ Jump up to:a b “Archived copy”. Archived from the original on 8 March 2014. Retrieved 26 February 2014.
- ^ Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sørensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P (August 2003). “Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients”. The New England Journal of Medicine. 349 (9): 847–58. doi:10.1056/NEJMoa022171. PMID 12944570.
- ^ Jeng LB, Thorat A, Hsieh YW, Yang HR, Yeh CC, Chen TH, Hsu SC, Hsu CH (April 2014). “Experience of using everolimus in the early stage of living donor liver transplantation”. Transplantation Proceedings. 46 (3): 744–8. doi:10.1016/j.transproceed.2013.11.068. PMID 24767339.
- ^ Jeng L, Thorat A, Yang H, Yeh C-C, Chen T-H, Hsu S-C. Impact of Everolimus On the Hepatocellular Carcinoma Recurrence After Living Donor Liver Transplantation When Used in Early Stage: A Single Center Prospective Study [abstract]. Am J Transplant. 2015; 15 (suppl 3). http://www.atcmeetingabstracts.com/abstract/impact-of-everolimus-on-the-hepatocellular-carcinoma-recurrence-after-living-donor-liver-transplantation-when-used-in-early-stage-a-single-center-prospective-study/. Accessed 1 September 2015.
- ^ Thorat A, Jeng LB, Yang HR, Yeh CC, Hsu SC, Chen TH, Poon KS (November 2017). “Assessing the role of everolimus in reducing hepatocellular carcinoma recurrence after living donor liver transplantation for patients within the UCSF criteria: re-inventing the role of mammalian target of rapamycin inhibitors”. Annals of Hepato-Biliary-Pancreatic Surgery. 21 (4): 205–211. doi:10.14701/ahbps.2017.21.4.205. PMC 5736740. PMID 29264583.
- ^ Jeng LB, Lee SG, Soin AS, Lee WC, Suh KS, Joo DJ, Uemoto S, Joh J, Yoshizumi T, Yang HR, Song GW, Lopez P, Kochuparampil J, Sips C, Kaneko S, Levy G (December 2017). “Efficacy and safety of everolimus with reduced tacrolimus in living-donor liver transplant recipients: 12-month results of a randomized multicenter study”. American Journal of Transplantation. 18 (6): 1435–1446. doi:10.1111/ajt.14623. PMID 29237235.
- ^ Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (July 2009). “Rapamycin fed late in life extends lifespan in genetically heterogeneous mice”. Nature. 460 (7253): 392–5. Bibcode:2009Natur.460..392H. doi:10.1038/nature08221. PMC 2786175. PMID 19587680.
- ^ Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB (December 2014). “mTOR inhibition improves immune function in the elderly”. Science Translational Medicine. 6 (268): 268ra179. doi:10.1126/scitranslmed.3009892. PMID 25540326. S2CID 206685475.
Further reading
- Sedrani R, Cottens S, Kallen J, Schuler W (August 1998). “Chemical modification of rapamycin: the discovery of SDZ RAD”. Transplantation Proceedings. 30 (5): 2192–4. doi:10.1016/S0041-1345(98)00587-9. PMID 9723437.
External links
- “Everolimus”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | Everolimus /ˌɛvəˈroʊləməs/ |
| Trade names | Afinitor, Zortress |
| Other names | 42-O-(2-hydroxyethyl)rapamycin, RAD001 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609032 |
| License data | EU EMA: by INNUS DailyMed: EverolimusUS FDA: Everolimus |
| Pregnancy category | AU: C[1] |
| Routes of administration | By mouth |
| ATC code | L01EG02 (WHO) L04AA18 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyEU: Rx-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Elimination half-life | ~30 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 159351-69-6 |
| PubChem CID | 6442177 |
| DrugBank | DB01590 |
| ChemSpider | 21106307 |
| UNII | 9HW64Q8G6G |
| KEGG | D02714 |
| ChEMBL | ChEMBL1908360 |
| CompTox Dashboard (EPA) | DTXSID0040599 |
| ECHA InfoCard | 100.149.896 |
| Chemical and physical data | |
| Formula | C53H83NO14 |
| Molar mass | 958.240 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESOCCO[C@@H]1CC[C@H](C[C@H]1OC)C[C@@H](C)[C@@H]4CC(=O)[C@H](C)/C=C(\C)[C@@H](O)[C@@H](OC)C(=O)[C@H](C)C[C@H](C)\C=C\C=C\C=C(/C)[C@@H](OC)C[C@@H]2CC[C@@H](C)[C@@](O)(O2)C(=O)C(=O)N3CCCC[C@H]3C(=O)O4 | |
| hideInChIInChI=1S/C53H83NO14/c1-32-16-12-11-13-17-33(2)44(63-8)30-40-21-19-38(7)53(62,68-40)50(59)51(60)54-23-15-14-18-41(54)52(61)67-45(35(4)28-39-20-22-43(66-25-24-55)46(29-39)64-9)31-42(56)34(3)27-37(6)48(58)49(65-10)47(57)36(5)26-32/h11-13,16-17,27,32,34-36,38-41,43-46,48-49,55,58,62H,14-15,18-26,28-31H2,1-10H3/b13-11+,16-12+,33-17+,37-27+/t32-,34-,35-,36-,38-,39+,40+,41+,43-,44+,45+,46-,48-,49+,53-/m1/s1 Key:HKVAMNSJSFKALM-GKUWKFKPSA-N |
//////////////// RAD-001, SDZ RAD, Certican, Novartis, Immunosuppressant, Everolimus, Afinitor, эверолимус , إيفيروليموس , 依维莫司 ,
#RAD-001, #SDZ RAD, #Certican, #Novartis, #Immunosuppressant, #Everolimus, #Afinitor, #эверолимус , #إيفيروليموس , #依维莫司 ,
Fluvoxamine

Fluvoxamine
- Molecular FormulaC15H21F3N2O2
- Average mass318.335 Da
- 54739-18-3
(E)-5-Methoxy-1-[4-(trifluoromethyl)phenyl]-1-pentanone O-(2-Aminoethyl)oxime1-Pentanone, 5-methoxy-1-[4-(trifluoromethyl)phenyl]-, O-(2-aminoethyl)oxime, (1E)-2-[({(1E)-5-Methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene}amino)oxy]ethanamine
2-{[(E)-{5-Methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene}amino]oxy}ethanamine1-Pentanone, 5-methoxy-1-(4-(trifluoromethyl)phenyl)-, O-(2-aminoethyl)oxime, (E)-
387954739-18-3[RN]5583954[Beilstein]5-Methoxy-4′-(trifluoromethyl)valerophenone (E)-O-(2-aminoethyl)oximeA selective serotonin reuptake inhibitor that is used in the treatment of DEPRESSION and a variety of ANXIETY DISORDERS.
Fluvoxamine, sold under the brand name Luvox among others, is an antidepressant of the selective serotonin reuptake inhibitor (SSRI) class[5] which is used primarily for the treatment of obsessive–compulsive disorder (OCD).[6] It is also used to treat depression and anxiety disorders, such as panic disorder, social anxiety disorder, and post-traumatic stress disorder.[7][8]
FLUVOXAMINE MALEATE
C19H25F3N2O6, 434.4 g/mol
1-Pentanone, 5-methoxy-1-(4-(trifluoromethyl)phenyl)-, O-(2-aminoethyl)oxime, (E)-, (Z)-2-butenedioate (1:1)
(Z)-but-2-enedioic acid;2-[(E)-[5-methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene]amino]oxyethanamine
Luvox
61718-82-9
CAS 54739-20-7
Fevarin, Luvox CR
Synonyms
- 5-Methoxy-4′-(trifluoromethyl)valerophenone (E)-O-(2-aminoethyl)oxime, maleate (1:1)
- 5-Methoxy-4′-trifluoromethylvalerophenone (E)-O-2-aminoethyloxime monomaleate
- DU23000
- Fevarin
- Fluvoxamine maleate
- Luvox
- Luvox CR
- SME 3110
- UNII-5LGN83G74V
| Originator Company | Solvay SA |
| Active Companies | AbbVie Inc; Abbott Laboratories; Meiji Seika Pharma Co Ltd; Solvay SA |
| Launched (Obsessive compulsive disorder – EU – Dec-1983) |
In the EU, the product is indicated for the treatment of obsessive compulsive disorder (OCD) and for the treatment of major depressive disorder (MDD)
In Japan, Luvox is indicated for the treatment of adult or pediatric OCD, social anxiety disorder (SAD) and MDD
USFDA The drug was approved for the treatment of OCD and SAD in April 2008
CHINA
In 2000, the drug was launched in China for the treatment of OCD and MDD
Patents and Generics
FDA exclusivity expired in the US in June 2000. Generic versions have been on the market since that time. Generic fluvoxamine was still available in the US by May 2007, despite the fact the Solvay/Jazz product had not been relaunched . By October 2004, the drug was also off patent in most European countries .
Medical uses
Fluvoxamine is approved in the United States for OCD,[9][6] and social anxiety disorder.[10] In other countries (e.g., Australia,[11][12] the UK,[13] and Russia[14]) it also has indications for major depressive disorder. In Japan it is currently[when?] approved to treat OCD, SAD and MDD.[15][16] Fluvoxamine is indicated for children and adolescents with OCD.[17] The drug works long-term, and retains its therapeutic efficacy for at least one year.[18] It has also been found to possess some analgesic properties in line with other SSRIs and tricyclic antidepressants.[19][20][21]
There is tentative evidence that fluvoxamine is effective for social phobia in adults.[22] Fluvoxamine is also effective for GAD, SAD, panic disorder and separation anxiety disorder in children and adolescents.[23] There is tentative evidence that fluvoxamine may help some people with negative symptoms of chronic schizophrenia.[24][25]
A double-blind controlled study found that fluvoxamine may prevent clinical deterioration in outpatients with symptomatic COVID-19. The study had important limitations: it was run fully remotely; it had a small sample size (150) and short follow-up duration (15 days).[26] The accompanying editorial noted that, although this study is important enough to choose out of more than 10,000 other COVID-19 related submissions, it “presents only preliminary information” and “the findings should be interpreted as only hypothesis generating; they should not be used as the basis for current treatment decisions.”[27] Similarly, the study authors themselves cautioned that “the trial’s results should not be treated as a measure of fluvoxamine’s effectiveness against COVID-19 but as an encouraging indicator that the drug warrants further testing.”[28] A prospective open-labelled cohort study showed similar results.[29]
Adverse effects
Gastrointestinal side effects are more common in those receiving fluvoxamine than with other SSRIs.[30] Otherwise, fluvoxamine’s side-effect profile is very similar to other SSRIs.[2][9][11][13][31][32]Common (1–10% incidence) adverse effects
- Nausea
- Vomiting
- Weight loss
- Yawning
- Loss of appetite
- Agitation
- Nervousness
- Anxiety
- Insomnia
- Somnolence (drowsiness)
- Tremor
- Restlessness
- Headache
- Dizziness
- Palpitations
- Tachycardia (high heart rate)
- Abdominal pain
- Dyspepsia (indigestion)
- Diarrhea
- Constipation
- Hyperhidrosis (excess sweating)
- Asthenia (weakness)
- Malaise
- Sexual dysfunction (including delayed ejaculation, erectile dysfunction, decreased libido, etc.)
- Xerostomia (dry mouth)
Uncommon (0.1–1% incidence) adverse effects
- Arthralgia
- Hallucination
- Confusional state
- Extrapyramidal side effects (e.g. dystonia, parkinsonism, tremor, etc.)
- Orthostatic hypotension
- Cutaneous hypersensitivity reactions (e.g. oedema [buildup of fluid in the tissues], rash, pruritus)
Rare (0.01–0.1% incidence) adverse effects
- Mania
- Seizures
- Abnormal hepatic (liver) function
- Photosensitivity (being abnormally sensitive to light)
- Galactorrhoea (expulsion of breast milk unrelated to pregnancy or breastfeeding)
Unknown frequency adverse effects
- Hyperprolactinaemia (elevated plasma prolactin levels leading to galactorrhoea, amenorrhoea [cessation of menstrual cycles], etc.)
- Bone fractures
- Glaucoma
- Mydriasis
- Urinary incontinence
- Urinary retention
- Bed-wetting
- Serotonin syndrome — a potentially fatal condition characterised by abrupt onset muscle rigidity, hyperthermia (elevated body temperature), rhabdomyolysis, mental status changes (e.g. coma, hallucinations, agitation), etc.
- Neuroleptic malignant syndrome — practically identical presentation to serotonin syndrome except with a more prolonged onset
- Akathisia — a sense of inner restlessness that presents itself with the inability to stay still
- Paraesthesia
- Dysgeusia
- Haemorrhage
- Withdrawal symptoms
- Weight changes
- Suicidal ideation and behaviour
- Violence towards others[33]
- Hyponatraemia
- Syndrome of inappropriate antidiuretic hormone secretion
- Ecchymoses
Interactions[edit]

Luvox (fluvoxamine) 100 mg film-coated scored tablets
Fluvoxamine inhibits the following cytochrome P450 enzymes:[34][35][36][37][38][39][40][41][42]
- CYP1A2 (strongly) which metabolizes agomelatine, amitriptyline, caffeine, clomipramine, clozapine, duloxetine, haloperidol, imipramine, phenacetin, tacrine, tamoxifen, theophylline, olanzapine, etc.
- CYP3A4 (moderately) which metabolizes alprazolam, aripiprazole, clozapine, haloperidol, quetiapine, pimozide, ziprasidone, etc.[43]
- CYP2D6 (weakly) which metabolizes aripiprazole, chlorpromazine, clozapine, codeine, fluoxetine, haloperidol, olanzapine, oxycodone, paroxetine, perphenazine, pethidine, risperidone, sertraline, thioridazine, zuclopenthixol, etc.[44]
- CYP2C9 (moderately) which metabolizes nonsteroidal anti-inflammatory drugs, phenytoin, sulfonylureas, etc.
- CYP2C19 (strongly) which metabolizes clonazepam, diazepam, phenytoin, etc.
- CYP2B6 (weakly) which metabolizes bupropion, cyclophosphamide, sertraline, tamoxifen, valproate, etc.
By so doing, fluvoxamine can increase serum concentration of the substrates of these enzymes.[34]
The plasma levels of oxidatively metabolized benzodiazepines (e.g., triazolam, midazolam, alprazolam and diazepam) are likely to be increased when co-administered with fluvoxamine. However the clearance of benzodiazepines metabolized by glucuronidation (e.g., lorazepam, oxazepam, temazepam)[45][46] is unlikely to be affected by fluvoxamine.[47] It appears that benzodiazepines metabolized by nitro-reduction (clonazepam, nitrazepam) are unlikely to be affected by fluvoxamine.[48] Using fluvoxamine and alprazolam together can increase alprazolam plasma concentrations.[49] If alprazolam is coadministered with fluvoxamine, the initial alprazolam dose should be reduced to the lowest effective dose.[50][51]
Fluvoxamine and ramelteon coadministration is not indicated.[52][53]
Fluvoxamine has been observed to increase serum concentrations of mirtazapine, which is mainly metabolized by CYP1A2, CYP2D6, and CYP3A4, by 3- to 4-fold in humans.[54] Caution and adjustment of dosage as necessary are warranted when combining fluvoxamine and mirtazapine.[54]
Fluvoxamine seriously affects the pharmacokinetics of tizanidine and increases the intensity and duration of its effects. Because of the potentially hazardous consequences, the concomitant use of tizanidine with fluvoxamine, or other potent inhibitors of CYP1A2, should be avoided.[55]
Fluvoxamine’s interaction with St John’s wort can lead to increased serotonin levels and potentially lead to serotonin syndrome.[citation needed]
Pharmacology
| Site | Ki (nM) |
|---|---|
| SERT | 2.5 |
| NET | 1,427 |
| 5-HT2C | 5,786 |
| α1-adrenergic | 1,288 |
| σ1 | 36 |
Fluvoxamine is a potent selective serotonin reuptake inhibitor with around 100-fold affinity for the serotonin transporter over the norepinephrine transporter.[35] It has negligible affinity for the dopamine transporter or any other site, with the sole exception of the σ1 receptor.[59][60] It behaves as a potent agonist at this receptor and has the highest affinity (36 nM) of any SSRI for doing so.[59] This may contribute to its antidepressant and anxiolytic effects and may also afford it some efficacy in treating the cognitive symptoms of depression.[61] Unlike fluoxetine, fluvoxamine’s metabolites are inactive, without a significant effect on serotonin or norepinephrine uptake.[62]
History
Fluvoxamine was developed by Kali-Duphar,[63] part of Solvay Pharmaceuticals, Belgium, now Abbott Laboratories, and introduced as Floxyfral in Switzerland in 1983.[63] It was approved by the U.S. Food and Drug Administration (FDA) in 1994, and introduced as Luvox in the US.[64] In India, it is available, among several other brands, as Uvox by Abbott.[65] It was one of the first SSRI antidepressants to be launched, and is prescribed in many countries to patients with major depression.[66] It was the first SSRI, a non-TCA drug, approved by the U.S. FDA specifically for the treatment of OCD.[67] At the end of 1995, more than ten million patients worldwide had been treated with fluvoxamine.[68][failed verification] Fluvoxamine was the first SSRI to be registered for the treatment of obsessive compulsive disorder in children by the FDA in 1997.[69] In Japan, fluvoxamine was the first SSRI to be approved for the treatment of depression in 1999[70][71] and was later in 2005 the first drug to be approved for the treatment of social anxiety disorder.[72] Fluvoxamine was the first SSRI approved for clinical use in the United Kingdom.[73]
Society and culture
Manufacturers include BayPharma, Synthon, and Teva, among others.[74]
SYN


SYN
J. Zhejiang Univ. (Medical Sci.) (2003), 32 (5), 441-442

PATENT
WO 2014178064
The present invention relates to an improved and industrially applicable process for the preparation of fluvoxamine maleate of formula I,
Fluvoxamine or (E)-5-methoxy-1 -[4-(trifluoromethyl)phenyl]pentan- 1 -one-O-2-aminoethyl oxime is an antidepressant which functions as a selective serotonin reuptake inhibitor (SSRI). Fluvoxamine is used for the treatment of major depressive disorder (MDD), obsessive compulsive disorder (OCD), and anxiety disorders such as panic disorder and post-traumatic stress disorder (PTSD). Fluvoxamine CR (controlled release) is approved to treat social anxiety disorder.
Fluvoxamine maleate and compounds were first disclosed in US patent 4,085,225. According to said patent, Fluvoxamine maleate prepared by alkylation reaction of 5-methoxy-4′-trifluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in dimethylformamide in the presence of a base such as potassium hydroxide powder for two days at 25°C.
Subsequently the solvent is removed under vacuum then the residue is acidified and extracted with ether to remove the unreacted oxime followed by basification. The obtained fluvoxamine base in ether extract is washed with sodium bicarbonate solution. The fluvoxamine base is then treated with maleic acid in absolute ethanbl and the residue obtained by concentration under vacuum is recrystallized from acetonitrile to obtain fluvoxamine maleate. The process is very much tedious, time consuming as it requires two days for the reaction completion. Operations like removal of dimethylformamide, ether, ethanol makes process cumbersome at plant level. Requirement of
various solvents lead the process to be non-eco-friendly. Moreover the patent is silent about yield and purity of the product.
In an alternate route described in US patent 4,085,225, the oxime of formula III is converted to formula I in a five step process i.e. alkylation of formula III with ethylene oxide. The reaction solvent is ethanol in which lithium is already dissolved. The reaction further involves addition of acetic acid to give the hydroxyethyl compound of formula A as oil. The compound of formula A is purified chromatographically over the silica gel, which is converted to a mesylate compound of formula B by treating with methanesulfonyl chloride and triethylamine at -5 to 0°C, then aminated with ammonia in methanol at 100°C using autoclave for 16 hours followed by removal of methanol and extraction in ether to give fluvoxamine base.
The base is then converted to the maleate salt formula I, which is finally purified by recrystallization from acetonitrile.
There are lots of disadvantages involve like more unit operations, use of various solvents and handling of ethylene oxide which is also known for its carcinogen effect. More unit operations lead to long occupancy of reactors in the plant as well as man power, high energy consumption and require bigger plant. These all parameters make the process commercially unviable as wel l as environmentally non-feasible. Further, purification of the compound of formula A requires cumbersome technique i.e chromatography over silica gel as well as lengthy work-up procedure in U.S. Pat. No. 4,085,225 requires complete removal of organic solvents at various stages.
US patent 6,433,225 discloses the process for preparing fluvoxamine maleate, prepared by alkylating 5-methoxy-4′-trtfluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in toluene and PEG-400 (polyethyleneglycol-400) as facilitator in the presence of a base potassium hydroxide powder at 30-35°C to obtain fluvoxamine base in
toluene layer is then treated with maleic acid in water. The precipitated fluvoxamine maleate is filtered and washed with toluene and dried. The obtained dried cake recrystallized with water to get fluvoxamine maleate. The process disclosed in the patent is silent about actual purity of the product. As per our scientist’s observation alkylation reaction at the temperature of 30-35°C may lead to non completion of reaction and results lower yield. Additional step of purification may further lead to loss of yield.
Thus, present invention fulfills the need of the art and provides an improved and industrially applicable process for preparation of fluvoxamine maleate, which provides fluvoxamine maleate in high purity and overall good yield.
EXAMPLES:
Stage – 1 : Preparation of (1E)-N-hydroxy-5-methoxy-1-(4-trifluoromethyI pheny 1) pentan-1-imine formula III
To a stirred solution of 5-methoxy- 1 -(4-trifluoromethylphenyl) pentan-1 -one ( 150 gm) in methanol (750 ml), sodium carbonate (granule) (72 gm) and hydroxylamine hydrochloride (59.64 gm) were added at temperature 25-30°C. The reaction mass was heated 45-50°C for 10- 15 minutes followed by maintaining the reaction mass at temperature 45-50°C for 8-9 hours under stirring. The reaction mass was cooled to 25-30°C and filtered under vacuum to remove unreacted inorganic matter, then distilled out the methanol completely from the collected filtrate under vacuum at temperature below 50°C. The obtained slurry was cooled to 25-30°C and water (300 ml) was added into the residue followed by the addition of hexane (300×2 ml) and stirred for 30 minutes. The layers were separated. The collected organic layer was stirred for 5- 10 minutes at temperature 25-30°C followed by cooling the mass at temperature -5°C to – 10°C, stirred for 30-40 minutes and filtered at the same temperature. The product was suck dried at -5 to -10°C and further in vacuum at 25-30°C for 2-3 hours to give 138 – 142 gm of title compound. HPLC purity: >98.5%
Stage – 2: Preparation of crude fluvoxamine maleate formula I
To a prepared solution of dimethyl sulphoxide (575 ml), potassium hydroxide flakes ( 1 14.64 gm) and water (69 ml), stage-1 (1 15 gm) was added at temperature 40-45°C. The reaction mixture was stirred to get clear solution followed by adding 2-chloroethylamine hydrochloride (86.36 gm) drop wise into the reaction mixture at temperature 40-45°C and maintained for 1 -2 hour. Water (1 150 ml) was added in to the reaction mixture at temperature 25-30°C and stirred for 20-25 minutes. Then toluene (575 ml x 2) was added and stirred for 30 minutes and preceded for separation of layers followed by washing the toluene layer with water ( 1 1 50 x 5 ml). The solution of maleic acid (48.47 gm) dissolved in water (98 ml) was added into above obtained toluene layer and stirred at temperature 25-30°C for 2-3 hours. The reaction mixture was cooled to 0-5°C and maintained for 30-40 minutes at the same temperature. The obtained material was washed with toluene, filtered and suck dried. The wet cake was then added hexane (600 ml) and stirred for 30 minutes at temperature 25-30°C, filtered, washed with hexane and dried to get 161 gm of title compound. HPLC purity: >98.5%
Stage – 3: Preparation of pure fluvoxamine maleate formula I
In to the reaction assembly, water (600 ml) was added and heated to 40-45°C. Stage -2 ( 1 50 gm) was added into the hot water under stirring. The reaction mixture was stirred for 5- 10 minutes, filtered and cooled to 25°C. Toluene (68 ml) was added into the reaction mixture at temperature 25°C and stirred for 30 minutes. Filtered the solid, washed with 10-15°C chilled water and dried to get the pure 127.5 gm fluvoxamine maleate. HPLC purity: >99.8%
Process for isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one formula II
To a solution of cone. HCl (600 ml) and water ( 160 ml), organic residue (250 gm) of ( 1 £)+( 1 Z) of 1 -N-hydroxy-5-methoxy- 1 -[4-(trifluoromethyl) phenyl]pentan-1 -imine and traces of 5-methoxy- 1 -[4-(trifluoromethyl)phenyl]pentan- 1-one (obtained after hexane recovery from stage-1 filtrate) was added at temperature 25-30°C under stirring. The reaction mixture was heated to 67-75°C and maintained for 13-14 hours followed by cool ing the reaction mixture at temperature 25-30°C. Then after hexane (500 x 2 ml) was added into the reaction mixture and stirred for 15 minutes at 25-30°C. The organic layers were separated and sodium bicarbonate solution (25 gm sodium bicarbonate dissolved in 250 ml water) was added into the hexane layer and stirred for 15 minutes. The layers were separated and water (250ml) was added into hexane layer and stirred for 15 minutes at temperature 25-30°C. Further the layers were separated and hexane layer was added activated charcoal ( 12.5 gm) and stirred for 20-30 minutes at temperature 30-35°C. The reaction mixture was filtered and stirred for 5-10 minutes at 25-30°C followed by cooling at 0 to -5°C and stirred for 30-40 minutes at 0 to -5°C. The reaction mixture was filtered and dried to get 150 to l 75 gm of title compound. HPLC purity: >99%.
PATENT
US 20140243544
IN 2013MU01290/WO 2014178064
WO 2014035107
PATENT
https://patents.google.com/patent/US9783492B2/en
Fluvoxamine or (E)-5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one-O-2-aminoethyl oxime is an antidepressant which functions as a selective serotonin reuptake inhibitor (SSRI). Fluvoxamine is used for the treatment of major depressive disorder (MDD), obsessive compulsive disorder (OCD), and anxiety disorders such as panic disorder and post-traumatic stress disorder (PTSD). Fluvoxamine CR (controlled release) is approved to treat social anxiety disorder.
Fluvoxamine maleate and compounds were first disclosed in U.S. Pat. No. 4,085,225. According to said patent, Fluvoxamine maleate prepared by alkylation reaction of 5-methoxy-4′-trifluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in dimethylformamide in the presence of a base such as potassium hydroxide powder for two days at 25° C.

Subsequently the solvent is removed under vacuum then the residue is acidified and extracted with ether to remove the unreacted oxime followed by basification. The obtained fluvoxamine base in ether extract is washed with sodium bicarbonate solution. The fluvoxamine base is then treated with maleic acid in absolute ethanol and the residue obtained by concentration under vacuum is recrystallized from acetonitrile to obtain fluvoxamine maleate. The process is very much tedious, time consuming as it requires two days for the reaction completion. Operations like removal of dimethylformamide, ether, ethanol makes process cumbersome at plant level. Requirement of various solvents lead the process to be non-eco-friendly. Moreover the patent is silent about yield and purity of the product.
In an alternate route described in U.S. Pat. No. 4,085,225, the mine of formula III is converted to formula I in a five step process i.e. alkylation of formula III with ethylene oxide. The reaction solvent is ethanol in which lithium is already dissolved. The reaction further involves addition of acetic acid to give the hydroxyethyl compound of formula A as oil. The compound of formula A is purified chromatographically over the silica gel, which is converted to a mesylate compound of formula B by treating with methanesulfonyl chloride and triethylamine at −5 to 0° C., then aminated with ammonia in methanol at 100° C. using autoclave for 16 hours followed by removal of methanol and extraction in ether to give fluvoxamine base.

The base is then converted to the maleate salt formula I, which is finally purified by recrystallization from acetonitrile.
There are lots of disadvantages in like more unit operations, use of various solvents and handling of ethylene oxide which is also known for its carcinogen effect. More unit operations lead to long occupancy of reactors in the plant as well as man power, high energy consumption and require bigger plant. These all parameters make the process commercially unviable as well as environmentally non-feasible. Further, purification of the compound of formula A requires cumbersome technique i.e chromatography over silica gel as well as lengthy work-up procedure in U.S. Pat. No. 4,085,225 requires complete removal of organic solvents at various stages.
U.S. Pat. No. 6,433,225 discloses the process for preparing fluvoxamine maleate, prepared by alkylating 5-methoxy-4′-trifluoromethylvalerophenone oxime compound of formula III with 2-chloroethylamine hydrochloride in toluene and PEG-400 (polyethyleneglycol-400) as facilitator in the presence of a base potassium hydroxide powder at 30-35°C. to obtain fluvoxamine base in toluene layer is then treated with maleic acid in water. The precipitated fluvoxamine maleate is filtered and washed with toluene and dried. The obtained dried cake recrystallized with water to get fluvoxamine maleate. The process disclosed in the patent is silent about actual purity of the product. As per our scientist’s observation alkylation reaction at the temperature of 30-35° C. may lead to non completion of reaction and results lower yield. Additional step of purification may further lead to loss of yield.
EXAMPLES
Stage-1: Preparation of (1 E)-N-hydroxy-5-methoxy-1-(4-trifluoromethyl phenyl)pentan-1-imine Formula III
To a stirred solution of 5-methoxy-1-(4-trifluoromethylphenyl)pentan-1one (150 gm) in methanol (750 ml), sodium carbonate (granule) (72 gm) and hydroxylamine hydrochloride (59.64 gm) were added at temperature 25-30° C. The reaction mass was heated 45-50° C. for 10-15 minutes followed by maintaining the reaction mass at temperature 45-50° C. for 8-9 hours under stirring. The reaction mass was cooled to 25-30° C. and filtered under vacuum to remove unreacted inorganic matter, then distilled out the methanol completely from the collected filtrate under vacuum at temperature below 50° C. The obtained slurry was cooled to 25-30° C. and water (300 ml) was added into the residue followed by the addition of hexane (300×2 ml) and stirred for 30 minutes. The layers were separated. The collected organic layer was stirred for 5-10 minutes at temperature 25-30° C. followed by cooling the mass at temperature −5° C. to −10° C., stirred for 30-40 minutes and filtered at the same temperature. The product was suck dried at −5 to −10° C. and further in vacuum at 25-30° C. for 2-3 hours to give 138-142 gm of title compound. HPLC purity: >98.5%
Stage-2: Preparation of Crude Fluvoxamine Maleate Formula I
To a prepared solution of dimethyl sulphoxide (575 ml), potassium hydroxide flakes (114.64 gm) and water (69 ml), stage-1 (115 gm) was added at temperature 40-45° C. The reaction mixture was stirred to get clear solution followed by adding 2-chloroethylamine hydrochloride (8636 gm) drop wise into the reaction mixture at temperature 40-45° C. and maintained for 1-2 hour. Water (1150 ml) was added in to the reaction mixture at temperature 25-30° C. and stirred for 20-25 minutes. Then toluene (575 ml×2) was added and stirred for 30 minutes and preceded for separation of layers followed by washing the toluene layer with water (1150×5 ml). The solution of maleic acid (48.47 gm) dissolved in water (98 ml) was added into above obtained toluene layer and stirred at temperature 25-30° C. for 2-3 hours. The reaction mixture was cooled to 0-5° C. and maintained for 30-40 minutes at the same temperature. The obtained material was washed with toluene, filtered and such dried. The wet cake was then added hexane (600 ml) and stirred for 30 minutes at temperature 25-30° C., filtered, washed with hexane and dried to get 161 gm of title compound. HPLC purity: >98.5%
Stage-3: Preparation of Pure Fluvoxamine Maleate Formula I
In to the reaction assembly, water (600 ml) was added and heated to 40-45° C. Stage-2 (150 gm) was added into the hot water under stirring. The reaction mixture was stirred for 5-10 minutes, filtered and cooled to 25° C. Toluene (68 ml) was added into the reaction mixture at temperature 25° C. and stirred for 30 minutes. Filtered the solid, washed with 10-15° C. chilled water and dried to get the pure 127.5 gm fluvoxamine maleate. HPLC purity: >99.8%
Process for isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one Formula II
To a solution of conc. HCl (600 ml) and water (160 organic residue (250 gm) of (1 E)+(1 Z) of 1-N-hydroxy-5-methoxy-1-[4trifluoromethyl)phenyl]pentan-1-imine and traces of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one (obtained after hexane recovery from stage-1 filtrate) was added at temperature 25-30° C. under stirring. The reaction mixture was heated to 67-75° C. and maintained for 13-14 hours followed by cooling the reaction mixture at temperature 25-30° C. Then after hexane (500×2 ml) was added into the reaction mixture and stirred for 15 minutes at 25-30° C. The organic layers were separated and sodium bicarbonate solution (25 gm sodium bicarbonate dissolved in 250 ml water) was added into the hexane layer and stirred for 15 minutes. The layers were separated and water (250 ml) was added into hexane layer and stirred for 15 minutes at temperature 25-30° C. Further the layers were separated and hexane layer was added activated charcoal (12.5 gm) and stirred for 20-30 minutes at temperature 30-35° C. The reaction mixture was filtered and stirred for 5-10 minutes at 25-30° C. followed by cooling at 0 to −5° C. and stirred for 30-40 minutes at 0 to −5° C. The reaction mixture was filtered and dried to get 150 to 175 gm of title compound. HPLC purity: >99%.
Claims (5)Hide Dependent
We claim:1. An improved process for the preparation of fluvoxamine maleate of formula I,

wherein the improvements comprises the steps of:a). condensing the compound of formula II,

with hydroxylamine hydrochloride in the presence of sodium carbonate granules at temperature 45-50° C. in suitable solvent to form a compound of formula III, wherein the compound of formula III comprises a mixture of (1E)+(1Z) isomers of 1-N-hydroxy-5-methoxy-1-[4(trifluoromethyl)phenyl]pentan-1-imine, and wherein the mixture of (1E)+(1Z) isomers of 1-N-hydroxy-5-methoxy-1-[4(trifluoromethyl)phenyl]pentan-1-imine comprises 98% of E-isomer and 2% of Z-isomer;

b). isolating compound of formula III;c). treating compound of formula III with 2-chloroethylamine hydrochloride in the presence of base in suitable solvent at 40-45° C. to form compound of formula IV;

d). extracting compound of formula IV with suitable solvent to form an organic layer;e). treating organic layer of step d) with maleic acid;f). isolating crude fluvoxamine maleate of formula I; andg). optionally purifying fluvoxamine maleate of formula I.
2. The process according to claim 1, wherein in step a), said suitable solvent is selected from the group consisting of alcohol, ketone, nitrile, and hydrocarbons in any suitable proportion or mixtures thereof;in step c), said base is selected from the group consisting of sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, lithium carbonate, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate, triethylamine and diisopropylethyamine;in step c), said solvent is selected from the group consisting of dimethylformamide (DMF), dimethylsulphoxide (DMSO) and hexamethylphosphoramide (HMPA) in any suitable proportion or mixtures thereof; andin step d) said suitable solvent is selected from the group consisting of toluene and xylene.3. A process for the isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one of formula II from mixture of (1E)+(1Z) of 1-N -hydroxy-5-methoxy-1-[4-(trifluoromethyl) phenyl]pentan-1-imine of formula III by treating compound of formula III with aqueous hydrochloric acid, wherein the mixture of (1E)+(1Z) of 1-N-hydroxy-5-methoxy-1-[4-(trifluoromethyl) phenyl]pentan-1-imine of formula III comprises 98% of E-isomer and 2% of Z-isomer.4. The process according to claim 3, wherein the reaction is performed at temperature 65-75°C.5. The process according to claim 1, wherein in step a), said suitable solvent is methanol.
Publication numberPriority datePublication dateAssigneeTitleUS4081551A *1975-03-201978-03-28U.S. Philips CorporationOxime ethers having anti-depressive activityUS4085225A1975-03-201978-04-18U.S. Philips CorporationOxime ethers having anti-depressive activityCN1079733A *1993-04-081993-12-22中国科学院成都有机化学研究所The synthetic method of a-benzoin oximeUS6433225B11999-11-122002-08-13Sun Pharamaceutical Industries, Ltd.Process for the preparation of fluvoxazmine maleateCN101654419A *2009-09-122010-02-24西北师范大学Preparation method of fluvoxamine maleate
Syn
US 6433225 SUN
https://patents.google.com/patent/US6433225B1/en
EXAMPLE 1
To a stirred mixture of toluene (1.20 lit.), PEG-400 (0.4 lit) and powdered potassium hydroxide (86.0 g on 100% basis, 1.53 mol.) at ambient temperature is added 5-methoxy-4′-trifluoromethylvalerophenone oxime (100 g, 0.363 mol.), followed by 2-chloroethyl amine hydrochloride (50.56 g, 0.435 mol.). The mixture is stirred at 30-35° C. for 2 hours. Water (1.2 lit.) is then added, stirred for 30 mins. and the aqueous layer is separated out. The organic layer is washed with water (˜3×500 ml) until the washings are neutral. To the washed organic layer is added a solution of maleic acid (14.14 g, 0.363 mol.) in water (65 ml) and the mixture is stirred at 25-30° C. temperature for 2 hours, then cooled to 5-10° C. when the maleate salt crystallizes out. The crystallized fluvoxamine maleate is filtered, washed with toluene (200 ml) and sucked to dryness. The crude fluvoxamine maleate thus obtained is dissolved in water (300 ml) at 50-55° C. to get a clear solution, then gradually cooled to 5-8° C. and then further stirred at this temperature for 2 hours. The recrystallised fluvoxamine maleate is filtered, washed with chilled water (5° C., 100 ml) and sucked dry. The product is finally dried at 50-55° C. to constant weight. The fluvoxamine maleate obtained complies with the specifications of British Pharmacopoeia, 1999.EXAMPLE 2
This process when scaled up in pilot plant on 4.0 kg scale input of 5-methoxy-4′-trifluoromethylvalerophenone oxime gave 4.5 kg (71.2%) of fluvoxamine maleate, complying to the specifications of British Pharmacopoeia, 1999.

SYN
US 4085225
https://patents.google.com/patent/US4085225A/en
EXAMPLE 15-Methoxy-4′-trifluoromethylvalerophenone O-(2-aminoethyl) oxime maleate (1:1).
20.4 Mmol (5.3 g) of 5-methoxy-4′-trifluoromethylvalerophenone (melting point 43°-44° C), 20.5 mmol (3.1 g) of 2-aminooxyethylaminedihydrochloride and 10 ml of pyridine were refluxed for 15 hours in 20 ml of absolute ethanol. After evaporating the pyridine and the ethanol in vacuo, the residue was dissolved in water. This solution was washed with petroleum ether and 10 ml of 50% sodium hydroxide solution were then added. Then three extractions with 40 ml of ether were carried out. The ether extract was washed successively with 20 ml of 5% sodium bicarbonate solution and 20 ml of water. After drying on sodium sulphate, the ether layer was evaporated in vacuo. Toluene was then evaporated another three times (to remove the pyridine) and the oil thus obtained was dissolved in 15 ml of absolute ethanol. An equimolar quantity of maleic acid was added to said solution and the solution was then heated until a clear solution was obtained. The ethanol was then removed in vacuo and the residue was crystallized from 10 ml of acetonitrile at +5° C. After sucking off and washing with cold acetonitrile, it was dried in air. The melting point of the resulting title compound was 120°-121.5° C.

SYN
GB 1535226

References
- ^ Jump up to:a b Use During Pregnancy and Breastfeeding
- ^ Jump up to:a b c d e f “Product Information Luvox”. TGA eBusiness Services. Abbott Australasia Pty Ltd. 15 January 2013. Retrieved 21 October 2013.
- ^ van Harten J (March 1993). “Clinical pharmacokinetics of selective serotonin reuptake inhibitors”. Clinical Pharmacokinetics. 24 (3): 203–20. doi:10.2165/00003088-199324030-00003. PMID 8384945. S2CID 84636672.
- ^ “Luvox”. ChemSpider. Royal Society of Chemistry. Archived from the original on 15 November 2013. Retrieved 21 October 2013.
- ^ “Fluvoxamine Maleate Information”. U.S. Food and Drug Administration(FDA). 15 July 2015. Archived from the original on 29 November 2019. Retrieved 28 November 2019.
- ^ Jump up to:a b McCain JA (July 2009). “Antidepressants and suicide in adolescents and adults: a public health experiment with unintended consequences?”. P T. 34(7): 355–78. PMC 2799109. PMID 20140100.
- ^ Figgitt DP, McClellan KJ (October 2000). “Fluvoxamine. An updated review of its use in the management of adults with anxiety disorders”. Drugs. 60 (4): 925–54. doi:10.2165/00003495-200060040-00006. PMID 11085201.
- ^ Irons J (December 2005). “Fluvoxamine in the treatment of anxiety disorders”. Neuropsychiatric Disease and Treatment. 1 (4): 289–99. PMC 2424117. PMID 18568110.
- ^ Jump up to:a b “Fluvoxamine Maleate tablet, coated prescribing information”. DailyMed. 14 December 2018. Retrieved 28 November 2019.
- ^ “Luvox CR approved for OCD and SAD”. MPR. 29 February 2008. Retrieved 2 March 2019.
- ^ Jump up to:a b Rossi S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- ^ “Luvox Tablets”. NPS MedicineWise. Retrieved 22 October 2018.
- ^ Jump up to:a b Joint Formulary Committee (2013). British National Formulary (BNF)(65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- ^ “Summary of Full Prescribing Information: Fluvoxamine”. Drug Registry of Russia (RLS) Drug Compendium (in Russian). Retrieved 21 March 2015.
- ^ “2005 News Releases”. Astellas Pharma. Retrieved 16 September 2018.
- ^ “International Approvals: Ebixa, Depromel/Luvox, M-Vax”. http://www.medscape.com. Retrieved 16 September 2018.
- ^ “US-FDA Fluvoxamine Product Insert”. March 2005.
- ^ Wilde MI, Plosker GL, Benfield P (November 1993). “Fluvoxamine. An updated review of its pharmacology, and therapeutic use in depressive illness”. Drugs. 46(5): 895–924. doi:10.2165/00003495-199346050-00008. PMID 7507038.
- ^ Kwasucki J, Stepień A, Maksymiuk G, Olbrych-Karpińska B (2002). “[Evaluation of analgesic action of fluvoxamine compared with efficacy of imipramine and tramadol for treatment of sciatica–open trial]”. Wiadomosci Lekarskie. 55 (1–2): 42–50. PMID 12043315.
- ^ Schreiber S, Pick CG (August 2006). “From selective to highly selective SSRIs: a comparison of the antinociceptive properties of fluoxetine, fluvoxamine, citalopram and escitalopram”. European Neuropsychopharmacology. 16 (6): 464–8. doi:10.1016/j.euroneuro.2005.11.013. PMID 16413173. S2CID 39278756.
- ^ Coquoz D, Porchet HC, Dayer P (September 1993). “Central analgesic effects of desipramine, fluvoxamine, and moclobemide after single oral dosing: a study in healthy volunteers”. Clinical Pharmacology and Therapeutics. 54 (3): 339–44. doi:10.1038/clpt.1993.156. PMID 8375130. S2CID 8229797.
- ^ Williams T, Hattingh CJ, Kariuki CM, Tromp SA, van Balkom AJ, Ipser JC, Stein DJ (October 2017). “Pharmacotherapy for social anxiety disorder (SAnD)”. The Cochrane Database of Systematic Reviews. 10 (10): CD001206. doi:10.1002/14651858.CD001206.pub3. PMC 6360927. PMID 29048739.
- ^ Cheer SM, Figgitt DP (2002). “Spotlight on fluvoxamine in anxiety disorders in children and adolescents”. CNS Drugs. 16 (2): 139–44. doi:10.2165/00023210-200216020-00006. PMID 11825104. S2CID 26774895.
- ^ Silver H (2001). “Fluvoxamine as an adjunctive agent in schizophrenia”. CNS Drug Reviews. 7 (3): 283–304. doi:10.1111/j.1527-3458.2001.tb00200.x. PMC 6741705. PMID 11607044.
- ^ Polcwiartek C, Nielsen J (March 2016). “The clinical potentials of adjunctive fluvoxamine to clozapine treatment: a systematic review”. Psychopharmacology. 233 (5): 741–50. doi:10.1007/s00213-015-4161-1. PMID 26626327. S2CID 12168939.
- ^ Lenze EJ, Mattar C, Zorumski CF, Stevens A, Schweiger J, Nicol GE, Miller JP, Yang L, Yingling M, Avidan MS, Reiersen AM (December 2020). “Fluvoxamine vs Placebo and Clinical Deterioration in Outpatients With Symptomatic COVID-19: A Randomized Clinical Trial”. JAMA. 324 (22): 2292–2300. doi:10.1001/jama.2020.22760. PMID 33180097.
- ^ Seymour CW, Bauchner H, Golub RM (December 2020). “COVID-19 Infection-Preventing Clinical Deterioration”. JAMA. 324 (22): 2300. doi:10.1001/jama.2020.21720. PMID 33180115.
- ^ [+https://scitechdaily.com/antidepressant-fluvoxamine-may-prevent-covid-19-infections-from-worsening/ “Antidepressant Fluvoxamine May Prevent COVID-19 Infections From Worsening”] Check
|url=value (help). - ^ https://academic.oup.com/ofid/advance-article/doi/10.1093/ofid/ofab050/6124100
- ^ Brayfield A, ed. (13 August 2013). Fluoxetine Hydrochloride. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 24 November 2013.
- ^ Taylor D, Paton C, Shitij K (2012). The Maudsley prescribing guidelines in psychiatry. West Sussex: Wiley-Blackwell. ISBN 978-0-470-97948-8.
- ^ “Faverin 100 mg film-coated tablets – Summary of Product Characteristics (SPC)”. electronic Medicines Compendium. Abbott Healthcare Products Limited. 14 May 2013. Retrieved 21 October 2013.
- ^ “Top Ten Legal Drugs Linked to Violence”. Time. 7 January 2011. Retrieved 10 September 2014.
- ^ Jump up to:a b Ciraulo DA, Shader RI (2011). Ciraulo DA, Shader RI (eds.). Pharmacotherapy of Depression (2nd ed.). Springer. p. 49. doi:10.1007/978-1-60327-435-7. ISBN 978-1-60327-435-7.
- ^ Jump up to:a b Brunton L, Chabner B, Knollman B (2010). Goodman and Gilman’s The Pharmacological Basis of Therapeutics (12th ed.). New York: McGraw-Hill Professional. ISBN 978-0-07-162442-8.
- ^ Baumann P (December 1996). “Pharmacokinetic-pharmacodynamic relationship of the selective serotonin reuptake inhibitors”. Clinical Pharmacokinetics. 31 (6): 444–69. doi:10.2165/00003088-199631060-00004. PMID 8968657. S2CID 31923953.
- ^ DeVane CL, Gill HS (1997). “Clinical pharmacokinetics of fluvoxamine: applications to dosage regimen design”. The Journal of Clinical Psychiatry. 58 Suppl 5 (Suppl 5): 7–14. PMID 9184622.
- ^ DeVane CL (1998). “Translational pharmacokinetics: current issues with newer antidepressants”. Depression and Anxiety. 8 Suppl 1 (Suppl 1): 64–70. doi:10.1002/(SICI)1520-6394(1998)8:1+<64::AID-DA10>3.0.CO;2-S. PMID 9809216.
- ^ Bondy B, Spellmann I (March 2007). “Pharmacogenetics of antipsychotics: useful for the clinician?”. Current Opinion in Psychiatry. 20 (2): 126–30. doi:10.1097/YCO.0b013e328017f69f. PMID 17278909. S2CID 23859992.
- ^ Kroon LA (September 2007). “Drug interactions with smoking”. American Journal of Health-System Pharmacy. 64 (18): 1917–21. doi:10.2146/ajhp060414. PMID 17823102.
- ^ Waknine Y (13 April 2007). “Prescribers Warned of Tizanidine Drug Interactions”. Medscape News. Medscape. Retrieved 1 February 2008.
- ^ “Fluvoxamine (Oral Route) Precautions”. Mayo Clinic. Retrieved 2 November2018.
- ^ Hemeryck A, Belpaire FM (February 2002). “Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update”. Current Drug Metabolism. 3 (1): 13–37. doi:10.2174/1389200023338017. PMID 11876575.
- ^ “Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers”.
- ^ Raouf M (2016). Fudin J (ed.). “Benzodiazepine Metabolism and Pharmacokinetics” (PDF).
- ^ Peppers MP (1996). “Benzodiazepines for alcohol withdrawal in the elderly and in patients with liver disease”. Pharmacotherapy. 16 (1): 49–57. doi:10.1002/j.1875-9114.1996.tb02915.x. PMID 8700792. S2CID 1389910.
- ^ “fluvoxamine maleate: PRODUCT MONOGRAPH” (PDF). 2016.
- ^ “Luvox Data Sheet” (PDF). Medsafe, New Zealand. 2017.
- ^ Suzuki Y, Shioiri T, Muratake T, Kawashima Y, Sato S, Hagiwara M, Inoue Y, Shimoda K, Someya T (April 2003). “Effects of concomitant fluvoxamine on the metabolism of alprazolam in Japanese psychiatric patients: interaction with CYP2C19 mutated alleles”. European Journal of Clinical Pharmacology. 58 (12): 829–33. doi:10.1007/s00228-003-0563-9. PMID 12698310. S2CID 32559753.
- ^ Gerlach M, Warnke A, Greenhill L (2014). Psychiatric Drugs in Children and Adolescents: Basic Pharmacology and Practical Applications. Springer-Verlag Wien. p. 131. ISBN 978-3-7091-1500-8.
- ^ Fleishaker JC, Hulst LK (1994). “A pharmacokinetic and pharmacodynamic evaluation of the combined administration of alprazolam and fluvoxamine”. European Journal of Clinical Pharmacology. 46 (1): 35–9. doi:10.1007/bf00195913. PMID 8005185. S2CID 2161450.
- ^ Obach RS, Ryder TF (August 2010). “Metabolism of ramelteon in human liver microsomes and correlation with the effect of fluvoxamine on ramelteon pharmacokinetics”. Drug Metabolism and Disposition. 38 (8): 1381–91. doi:10.1124/dmd.110.034009. PMID 20478852. S2CID 8421997.
- ^ Pandi-Perumal SR, Spence DW, Verster JC, Srinivasan V, Brown GM, Cardinali DP, Hardeland R (12 April 2011). “Pharmacotherapy of insomnia with ramelteon: safety, efficacy and clinical applications”. Journal of Central Nervous System Disease. 3: 51–65. doi:10.4137/JCNSD.S1611. PMC 3663615. PMID 23861638.
- ^ Jump up to:a b Anttila AK, Rasanen L, Leinonen EV (October 2001). “Fluvoxamine augmentation increases serum mirtazapine concentrations three- to fourfold”. The Annals of Pharmacotherapy. 35 (10): 1221–3. doi:10.1345/aph.1A014. PMID 11675851. S2CID 44807359.
- ^ Granfors MT, Backman JT, Neuvonen M, Ahonen J, Neuvonen PJ (April 2004). “Fluvoxamine drastically increases concentrations and effects of tizanidine: a potentially hazardous interaction”. Clinical Pharmacology and Therapeutics. 75(4): 331–41. doi:10.1016/j.clpt.2003.12.005. PMID 15060511. S2CID 25781307.
- ^ Ishikawa M, Ishiwata K, Ishii K, Kimura Y, Sakata M, Naganawa M, et al. (October 2007). “High occupancy of sigma-1 receptors in the human brain after single oral administration of fluvoxamine: a positron emission tomography study using [11C]SA4503”. Biological Psychiatry. 62 (8): 878–83. doi:10.1016/j.biopsych.2007.04.001. PMID 17662961. S2CID 728565.
- ^ Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing textbook of psychopharmacology (4th ed.). Arlington, VA: American Psychiatric Pub. p. 354. ISBN 978-1-585-62386-0. OCLC 320111564.
- ^ Yahata M, Chiba K, Watanabe T, Sugiyama Y (September 2017). “Possibility of Predicting Serotonin Transporter Occupancy From the In Vitro Inhibition Constant for Serotonin Transporter, the Clinically Relevant Plasma Concentration of Unbound Drugs, and Their Profiles for Substrates of Transporters”. Journal of Pharmaceutical Sciences. 106 (9): 2345–2356. doi:10.1016/j.xphs.2017.05.007. PMID 28501470.
- ^ Jump up to:a b Hashimoto K (September 2009). “Sigma-1 receptors and selective serotonin reuptake inhibitors: clinical implications of their relationship”. Central Nervous System Agents in Medicinal Chemistry. 9 (3): 197–204. doi:10.2174/1871524910909030197. PMID 20021354.
- ^ Westenberg HG, Sandner C (April 2006). “Tolerability and safety of fluvoxamine and other antidepressants”. International Journal of Clinical Practice. 60 (4): 482–91. doi:10.1111/j.1368-5031.2006.00865.x. PMC 1448696. PMID 16620364.
- ^ Hindmarch I, Hashimoto K (April 2010). “Cognition and depression: the effects of fluvoxamine, a sigma-1 receptor agonist, reconsidered”. Human Psychopharmacology. 25 (3): 193–200. doi:10.1002/hup.1106. PMID 20373470. S2CID 26491662.
- ^ Hrdina PD (July 1991). “Pharmacology of serotonin uptake inhibitors: focus on fluvoxamine”. Journal of Psychiatry & Neuroscience. 16 (2 Suppl 1): 10–8. PMC 1188307. PMID 1931931.
- ^ Jump up to:a b Sittig’s Pharmaceutical Manufacturing Encyclopedia (PDF) (3rd ed.). William Andrew. 2008. p. 1699. ISBN 978-0-8155-1526-5. Retrieved 17 October2013.
- ^ Leslie LK, Newman TB, Chesney PJ, Perrin JM (July 2005). “The Food and Drug Administration’s deliberations on antidepressant use in pediatric patients”. Pediatrics. 116 (1): 195–204. doi:10.1542/peds.2005-0074. PMC 1550709. PMID 15995053.
- ^ “Brand Index―Fluvoxamine India”. Archived from the original on 19 October 2013. Retrieved 18 October 2013.
- ^ Omori IM, Watanabe N, Nakagawa A, Cipriani A, Barbui C, McGuire H, Churchill R, Furukawa TA (March 2010). “Fluvoxamine versus other anti-depressive agents for depression”. The Cochrane Database of Systematic Reviews (3): CD006114. doi:10.1002/14651858.CD006114.pub2. PMC 4171125. PMID 20238342.
- ^ “OCD Medication”. Archived from the original on 14 October 2013. Retrieved 17 October 2013.
- ^ “Fluvoxamine Product Monograph” (PDF). 1999.
- ^ “Luvox Approved For Obsessive Compulsive Disorder in Children and Teens”. Archived from the original on 16 January 2009. Retrieved 8 February 2014.
- ^ Higuchi T, Briley M (February 2007). “Japanese experience with milnacipran, the first serotonin and norepinephrine reuptake inhibitor in Japan”. Neuropsychiatric Disease and Treatment. 3 (1): 41–58. doi:10.2147/nedt.2007.3.1.41. PMC 2654524. PMID 19300537.
- ^ “Human Metabolome Database: Showing metabocard for Fluvoxamine (HMDB0014322)”. http://www.hmdb.ca. Retrieved 15 September 2018.
- ^ “Solvay’s Fluvoxamine maleate is first drug approved for the treatment of social anxiety disorder in Japan”.
- ^ Walker R, Whittlesea C, eds. (2007) [1994]. Clinical Pharmacy and Therapeutics (4th ed.). Edinburgh: Churchill Livingstone Elsevier. ISBN 978-0-7020-4293-5.
- ^ “Fluvoxamine”. http://www.drugbank.ca. Retrieved 22 October 2019.
External links
- “Fluvoxamine”. Drug Information Portal. U.S. National Library of Medicine.
{kind=link}
{kind=link}
/////////DU23000, Fevarin, Fluvoxamine maleate, Luvox, Luvox CR, SME 3110, UNII-5LGN83G74V, Fluvoxamine, sme 3110, DU 23000
#DU23000, #Fevarin, #Fluvoxamine maleate, #Luvox, #Luvox CR, #SME 3110, #UNII-5LGN83G74V, #Fluvoxamine, #sme 3110, #DU 23000
OI 338
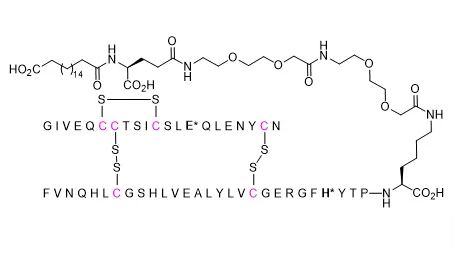
OI 338
OI338GT (NN1953)
NNC0123-0000-0338
Insulin oral (NN 1953); Insulin-338-GIPET-I; LAI 338; NN 1438; NN-1953; NNC-0123-0000-0338; NNC0123-0338; OI-338GT; Oral insulin 338 C10
- OriginatorNovo Nordisk
- ClassAntihyperglycaemics; Insulins; Pancreatic hormones
- Mechanism of ActionOrnithine decarboxylase stimulants; Phosphokinase stimulants; Protein tyrosine kinase stimulants
- Phase IIType 1 diabetes mellitus; Type 2 diabetes mellitus
- 28 Jul 2018No recent reports of development identified for phase-I development in Type-1 diabetes mellitus in Germany (SC, Injection)
- 28 Jul 2018No recent reports of development identified for phase-I development in Type-2-diabetes-mellitus in Denmark (SC, Injection)
- 11 Sep 2017Efficacy and adverse events data from a phase II trial in Type-2 diabetes mellitus presented at the 53rd Annual Meeting of the European Association for the Study of Diabetes (EASD-2017)
OI-338GT is a long-acting oral basal insulin analogue which had been in phase II clinical trials at Novo Nordisk for the treatment of patients with type 2 and type 1 diabetes. In 2016, the company discontinued the development of the product as the emergent product profile and required overall investments were not commercially viable in the increasingly challenging payer environment.
PAPERJ. Med. Chem. 2021, 64, 1, 616–628
Publication Date:December 28, 2020
https://doi.org/10.1021/acs.jmedchem.0c01576https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01576

Recently, the first basal oral insulin (OI338) was shown to provide similar treatment outcomes to insulin glargine in a phase 2a clinical trial. Here, we report the engineering of a novel class of basal oral insulin analogues of which OI338, 10, in this publication, was successfully tested in the phase 2a clinical trial. We found that the introduction of two insulin substitutions, A14E and B25H, was needed to provide increased stability toward proteolysis. Ultralong pharmacokinetic profiles were obtained by attaching an albumin-binding side chain derived from octadecanedioic (C18) or icosanedioic acid (C20) to the lysine in position B29. Crucial for obtaining the ultralong PK profile was also a significant reduction of insulin receptor affinity. Oral bioavailability in dogs indicated that C18-based analogues were superior to C20-based analogues. These studies led to the identification of the two clinical candidates OI338 and OI320 (10 and 24, respectively).
Oral insulin 338 (I338) is a long-acting, basal insulin analogue formulated in a tablet with the absorption-enhancer sodium caprate. We investigated the efficacy and safety of I338 versus subcutaneous insulin glargine (IGlar) in patients with type 2 diabetes. METHODS: This was a phase 2, 8-week, randomised, double-blind, double-dummy, active-controlled, parallel trial completed at two research institutes in Germany. Insulin-naive adult patients with type 2 diabetes, inadequately controlled on metformin monotherapy or combined with other oral antidiabetic drugs (HbA1c 7·0-10·0%; BMI 25·0-40·0 kg/m(2)), were randomly assigned (1:1) to receive once-daily I338 plus subcutaneous placebo (I338 group) or once-daily IGlar plus oral placebo (IGlar group). Randomisation occurred by interactive web response system stratified by baseline treatment with oral antidiabetic drugs. Patients and investigators were masked to treatment assignment. Weekly insulin dose titration aimed to achieve a self-measured fasting plasma glucose (FPG) concentration of 4·4-7·0 mmol/L. The recommended daily starting doses were 2700 nmol I338 or 10 U IGlar, and maximum allowed doses throughout the trial were 16 200 nmol I338 or 60 U IGlar. The primary endpoint was treatment difference in FPG concentration at 8 weeks for all randomly assigned patients receiving at least one dose of trial product (ie, the full analysis set). The trial has been completed and is registered at ClinicalTrials.gov, number NCT02470039. FINDINGS: Between June 1, 2015, and Oct 19, 2015, 82 patients were screened for eligibility and 50 patients were randomly assigned to the I338 group (n=25) or the IGlar group (n=25). Mean FPG concentration at baseline was 9·7 (SD 2·8) in the I338 group and 9·1 (1·7) in the IGlar group. Least square mean FPG concentration at 8 weeks was 7·1 mmol/L (95% CI 6·4-7·8) in the I338 group and 6·8 mmol/L (6·5-7·1) in the IGlar group, with no significant treatment difference (0·3 mmol/L [-0·5 to 1·1]; p=0·46). I338 and IGlar were well tolerated by patients. Adverse events were reported in 15 (60%) patients in the I338 group and 17 (68%) patients in the IGlar group. The most common adverse events were diarrhoea (three [12%] patients in each group) and nasopharyngitis (five [20%] in the I338 group and two [8%] in the IGlar group). Most adverse events were graded mild (47 of 68 events), and no severe adverse events were reported. One patient in the IGlar group had a treatment-emergent serious adverse event (urogenital haemorrhage of moderate intensity, assessed by the investigator as unlikely to be related to treatment; the patient recovered). Incidence of hypoglycaemia was low in both groups (n=7 events in the I338 group; n=11 in the IGlar group), with no severe episodes. INTERPRETATION: I338 can safely improve glycaemic control in insulin-naive patients with type 2 diabetes with no evidence of a difference compared with insulin glargine, a widely used subcutaneously administered basal insulin. Further development of this particular oral insulin project was discontinued because I338 doses were high and, therefore, production of the required quantities of I338 for wide public use was deemed not commercially viable. Improvement of technologies involved in the product’s development is the focus of ongoing research. FUNDING: Novo Nordisk…..Halberg, I. B.; Lyby, K.; Wassermann, K.; Heise, T.; Zijlstra, E.; Plum-Mörschel, L. Efficacy and safety of oral basal insulin versus subcutaneous insulin glargine in type 2 diabetes: a randomised, double-blind, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 179– 188, DOI: 10.1016/s2213-8587(18)30372-3
ral insulin 338 is a novel tablet formulation of a long-acting basal insulin. This randomised, open-label, four-period crossover trial investigated the effect of timing of food intake on the single-dose pharmacokinetic properties of oral insulin 338. Methods: After an overnight fast, 44 healthy males received single fixed doses of oral insulin 338 administered 0, 30, 60 or 360 min before consuming a standardised meal (500 kcal, 57 energy percent [E%] carbohydrate, 13 E% fat, 30 E% protein). Blood samples for pharmacokinetic assessment were taken up to 288 h post-dose. Results: Total exposure (area under the concn.-time curve from time zero to infinity [AUCIns338,0-∞]) and max. concn. (Cmax,Ins338) of insulin 338 were both significantly lower for 0 vs. 360 min post-dose fasting (ratio [95% confidence interval (CI)]: 0.36 [0.26-0.49], p < 0.001, and 0.35 [0.25-0.49], p < 0.001, resp.). There were no significant differences in AUCIns338,0-∞ and Cmax,Ins338 for 30 or 60 vs. 360 min post-dose fasting (ratio [95% CI] 30 vs. 360 min: 0.85 [0.61-1.21], p = 0.36, and 0.86 [0.59-1.26], p = 0.42; ratio [95% CI] 60 vs. 360 min: 0.96 [0.72-1.28], p = 0.77, and 0.99 [0.75-1.31], p = 0.95). The mean half-life was ∼ 55 h independent of the post-dose fasting period. Oral insulin 338 was well-tolerated with no safety issues identified during the trial. Conclusions: Oral insulin 338 pharmacokinetics are not affected by food intake from 30 min after dosing, implying that patients with diabetes mellitus do not need to wait more than 30 min after a morning dose of oral insulin 338 before having their breakfast. This is considered important for convenience and treatment compliance. ClinicalTrials.gov identifier: NCT02304627./……Halberg, I. B.; Lyby, K.; Wassermann, K.; Heise, T.; Plum-Mörschel, L.; Zijlstra, E. The effect of food intake on the pharmacokinetics of oral basal insulin: A randomised crossover trial in healthy male subjects. Clin. Pharmacokinet. 2019, 58, 1497– 1504, DOI: 10.1007/s40262-019-00772-2
///////////////OI 338, OI338GT, NN1953, NNC0123-0000-0338, Insulin oral (NN 1953), Insulin-338-GIPET-I, LAI 338, NN 1438, NN-1953, NNC-0123-0000-0338, NNC0123-0338, OI-338GT, Oral insulin 338 C10
Esketamine

Esketamine
- Molecular FormulaC13H16ClNO
- Average mass237.725 Da
(+)-Ketamine(2S)-2-(2-Chlorophenyl)-2-(methylamino)cyclohexanone
(S)-Ketamine33643-46-8[RN]7884Cyclohexanone, 2-(2-chlorophenyl)-2-(methylamino)-, (2S)-Cyclohexanone, 2-(2-chlorophenyl)-2-(methylamino)-, (S)-
KetamineCAS Registry Number: 6740-88-1CAS Name: 2-(2-Chlorophenyl)-2-(methylamino)cyclohexanoneMolecular Formula: C13H16ClNOMolecular Weight: 237.73Percent Composition: C 65.68%, H 6.78%, Cl 14.91%, N 5.89%, O 6.73%Literature References: Prepn: C. L. Stevens, BE634208; idem,US3254124 (1963, 1966 both to Parke, Davis). Isoln of optical isomers: T. W. Hudyma et al.,DE2062620 (1971 to Bristol-Myers), C.A.75, 118119x (1971). Clinical pharmacology of racemate and enantiomers: P. F. White et al.,Anesthesiology52, 231 (1980). Toxicity: E. J. Goldenthal, Toxicol. Appl. Pharmacol.18, 185 (1971). Enantioselective HPLC determn in plasma: G. Geisslinger et al.,J. Chromatogr.568, 165 (1991). Comprehensive description: W. C. Sass, S. A. Fusari, Anal. Profiles Drug Subs.6, 297-322 (1977). Review of pharmacology and use in veterinary medicine: M. Wright, J. Am. Vet. Med. Assoc.180, 1462-1471 (1982). Review of pharmacology and clinical experience: D. L. Reich, G. Silvay, Can. J. Anaesth.36, 186-197 (1989); in pediatric procedures: S. M. Green, N. E. Johnson, Ann. Emerg. Med.19, 1033-1046 (1990).Properties: Crystals from pentane-ether, mp 92-93°. uv max (0.01N NaOH in 95% methanol): 301, 276, 268, 261 nm (A1%1cm 5.0, 7.0, 9.8, 10.5). pKa 7.5. pH of 10% aq soln 3.5.Melting point: mp 92-93°pKa: pKa 7.5Absorption maximum: uv max (0.01N NaOH in 95% methanol): 301, 276, 268, 261 nm (A1%1cm 5.0, 7.0, 9.8, 10.5)
Derivative Type: HydrochlorideCAS Registry Number: 1867-66-9Manufacturers’ Codes: CI-581Trademarks: Ketalar (Pfizer); Ketanest (Pfizer); Ketaset (Fort Dodge); Ketavet (Gellini); Vetalar (Bioniche)Molecular Formula: C13H16ClNO.HClMolecular Weight: 274.19Percent Composition: C 56.95%, H 6.25%, Cl 25.86%, N 5.11%, O 5.84%Properties: White crystals, mp 262-263°. Soly in water: 20 g/100 ml. LD50 in adult mice, rats (mg/kg): 224 ±4, 229 ±5 i.p. (Goldenthal).Melting point: mp 262-263°Toxicity data: LD50 in adult mice, rats (mg/kg): 224 ±4, 229 ±5 i.p. (Goldenthal)
NOTE: This is a controlled substance (depressant): 21 CFR, 1308.13.Therap-Cat: Anesthetic (intravenous).Therap-Cat-Vet: Anesthetic (intravenous).Keywords: Anesthetic (Intravenous).Esketamine hydrochloride, S enantiomer of ketamine, is in phase III clinical trials by Johnson & Johnson for the treatment of depression.Drug Name:Esketamine HydrochlorideResearchCode:JNJ-54135419MOA:Dopamine reuptake inhibitor; NMDA receptor antagonistIndication:DepressionStatus:Phase III (Active)Company:Johnson & Johnson (Originator)
| Molecular Weight | 274.19 |
| Formula | C13H16ClNO•HCl |
| CAS No. | 33643-46-8 (Esketamine); 33643-47-9 (Esketamine Hydrochloride); |
{kind=link}
Reference:1. US6040479.
https://patents.google.com/patent/US6040479A/en
EXAMPLE 1
50 g (0.21 mol) R,S-ketamine are dissolved in 613 ml of acetone at the boiling point and subsequently mixed with 31.5 g (0.21 mol) L-(+)-tartaric acid. In order to obtain a clear solution, 40 ml of water are added thereto at the boiling point and subsequently the clear solution is filtered off while still hot. After the addition of seed crystals obtained in a small preliminary experiment, the whole is allowed to cool to ambient temperature while stirring. After standing overnight, the crystals formed are filtered off with suction and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.).
Yield (tartrate): 64.8 g
m.p.: 161° C.
[α]D : +26.1° (c=2/H2 O)
Thereafter, the crystallisate is recrystallised in a mixture of 1226 ml acetone and 90 ml water. After cooling to ambient temperature and subsequently stirring for 4 hours, the crystals are filtered off with suction and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C). There are obtained 38.8 g of tartrate (95.29% of theory).
m.p.: 175.3° C.
[α]D : +68.9° (c=2/H2 O)
The base is liberated by taking up 38.8 g of tartrate in 420 ml of aqueous sodium hydroxide solution and stirring with 540 ml of diethyl ether. The ethereal phase is first washed with water and subsequently with a saturated solution of sodium chloride. The organic phase is dried over anhydrous sodium sulphate. After filtering, the solution is evaporated to dryness on a rotary evaporator, a crystalline, colourless product remaining behind.
Yield (crude base): 21.5 g=86.0% of theory
m.p.: 118.9° C. (literature: 120-122° C.)
[α]D : -55.8° (c=2/EtOH) (literature: [α]D : -56.35° ).
In order possibly to achieve a further purification, the base can be recrystallised from cyclohexane. For this purpose, 10.75 g of the crude base are dissolved in 43 ml cyclohexane at the boiling point. While stirring, the clear solution is slowly cooled to about 10° C. and then stirred at this temperature for about 1 hour. The crystallisate which precipitates out is filtered off with suction and dried to constant weight.
Yield (base): 10.3 g=82.4% of theory
m.p.: 120° C. (literature: 120-122° C.)
[α]D : -56.8° (c=2/EtOH) (literature: [α]D : -56.35° )
EXAMPLE 2
125 ml of water are taken and subsequently 31.5 g (0.21 mol) L-(+)-tartaric acid and 50 g (0.21 mol) R,S-ketamine added thereto. While stirring, this mixture is warmed to 50-60° C. until a clear solution results. After cooling to ambient temperature while stirring and subsequently stirring overnight, the crystals formed are filtered off with suction. Subsequently, the crystallisate is first washed with water (1-6° C.) and subsequently washed twice with, in each case, 20 ml of acetone. Drying in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.) gives 31.79 g of tartrate (78.23%) of theory).
EXAMPLE 3
150 ml of water are taken and subsequently mixed with 39.8 g (0.27 mol) L-(+)-tartaric acid and 50 g (0.21 mol) R,S-ketamine. While stirring, this mixture is warmed to 50-60° C. until a clear solution results.
After cooling to ambient temperature while stirring and subsequently stirring overnight, the crystals formed are filtered off with suction. Subsequently, the crystallisate is successively washed with 8 ml of water (1-6° C.) and thereafter twice with, in each case, 20 ml acetone.
Drying in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.) gives 32.58 g of tartrate (80.02% of theory).
EXAMPLE 4
150 ml of water and 50 ml isopropanol are taken. After the addition of 39.8 g (0.21 mol) L-(+)-tartaric acid and 50 g (0.21 mol) R,S-ketamine, the mixture is heated to reflux temperature while stirring until a solution results (possibly add water until all is dissolved).
Subsequently, while stirring, the solution is allowed to cool to ambient temperature and stirred overnight. The crystals are filtered off with suction and subsequently washed with a 1:2 mixture of 20 ml of water/isopropanol and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.). There are obtained 24.45 g of tartrate (62.63% of theory).
EXAMPLE 5
50 g (0.21 mol) R,S-ketamine are dissolved at the boiling point in 300 ml acetone and subsequently mixed with 31.5 g (0.21 mol) L-(+)-tartaric acid and 100 ml of water. The whole is allowed to cool while stirring and possibly seeded.
After standing overnight, the crystals formed are filtered off with suction, then washed twice with, in each case, 20 ml acetone and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.). There are obtained 30.30 g of tartrate (74.57% of theory).
EXAMPLE 6
75 ml of water and 50 ml isopropanol are taken and subsequently 39.8 g (0.27 mol) L-(+)-tartaric acid added thereto. While stirring, the mixture is heated to reflux temperature until a clear solution results. After cooling to ambient temperature while stirring and subsequently stirring overnight, the crystals formed are filtered off with suction. Subsequently, the crystallisate is washed with a 1:2 mixture of 20 ml water/isopropanol. After drying in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.), there are obtained 34.84 g of tartrate (85.74% of theory).
EXAMPLE 7
20 g of the S-(+)-tartrate obtained in Example 4 are dissolved in 100 ml of water at 30-40° C. With about 7 ml of 50% sodium hydroxide solution, an S-(-)-ketamine base is precipitated out up to about pH 13. It is filtered off with suction and washed neutral with water to pH 7-8. Subsequently, it is dried for about 24 hours at 50° C. in a circulating air drying cabinet. There are obtained 11.93 g S-(-)-ketamine (97.79% of theory).
EXAMPLE 8
5 g of the S-(-)-ketamine obtained in Example 7 are dissolved in 50 ml isopropanol at about 50° C. and possibly filtered off with suction over kieselguhr. Subsequently, gaseous hydrogen chloride is passed in at 50-60° C. until a pH value of 0-1 is reached. The reaction mixture is allowed to cool to ambient temperature, filtered off with suction and washed with about 5 ml isopropanol. The moist product is dried overnight at about 50° C. in a circulating air drying cabinet. There are obtained 5.09 g S-(+)-ketamine hydrochloride (88.06% of theory).
Route 2
{kind=link}
Reference:1. J. Am. Chem. Soc. 2015, 137, 3205-3208.
https://pubs.acs.org/doi/10.1021/jacs.5b00229
Here we report the direct asymmetric amination of α-substituted cyclic ketones catalyzed by a chiral phosphoric acid, yielding products with a N-containing quaternary stereocenter in high yields and excellent enantioselectivities. Kinetic resolution of the starting ketone was also found to occur on some of the substrates under milder conditions, providing enantioenriched α-branched ketones, another important building block in organic synthesis. The utility of this methodology was demonstrated in the short synthesis of (S)-ketamine, the more active enantiomer of this versatile pharmaceutical.



CLIP

Initial reagent: cyclopentyl Grignard Step 0: Producing cyclopentyl Grignard Reacting cyclopentyl bromide with magnesium in solvent (ether or THF) Best results: distill solvent from Grignard under vacuum and replace with hydrocarbon solvent (e.g. benzene) Step 1: processing to (o-chlorophenyl)-cyclopentyl ketone Adding o-chlorobenzonitrile to cyclopentyl Grignard in solvent, stirring for long period of time (typically three days) Hydrolyzing reaction with mixture containing crushed ice, ammonium chloride and some ammonium hydroxide Extraction with organic solvent gives (o-chlorophenyl)-cyclopentyl ketone
Step 2: processing to alpha-bromo (o-chlorophenyl)-cyclopentyl ketone ketone processed with bromine in carbon tetrachloride at low temperature (typical T = 0°C), addition of bromine dropwise forming orange suspension Suspension washed in dilute aquerous solution of sodium bisufide and evaporated giving 1-bromocyclopentyl-(o-chlorophenyl)-ketone Note: bromoketone is unstable, immeadiate usage. Bromination carried out with NBromosuccinimide result higher yield (roughly 77%) Step 3: processing to 1-hydroxycyclopentyl-(o-chlorophenyl)-ketone-N-methylimine Dissolving bromoketone in liquid methylamine freebase (or benzene as possible solvent) After time lapse (1h): excess methylamine evaporated, residue dissolved in pentane and filtered evaporation of solvent yields 1-hydroxy-cyclopentyl-(o-chlorophenyl)-ketone N-methylimine Note: longer time span (4-5d) for evaporation of methylaminemay increase yield Step 4: processing to 2-Methylamino-2-(o-chlorophenyl)-cyclohexanone (Ketamine) Method: Thermal rearragement (qualitative yield after 30min in 180°C) N-methylimine dissolved in 15ml decalin, refluxed for 2.5h Evaporation of solvent under reduced temperature followed by extraction of residue with dilute hydrochloric acid Treatment with decolorizing charcoal (solution: acidic => basic) Recrystallization from pentane-ether Note – alternative to use of decalin: pressure bomb
racemic compound, in pharmaceutical preparation racemic more active enantiomere esketamine (S-Ketamine) available as Ketanest S, but Arketamine (R-Ketamine) never marketed for clinical use, Optical rotation: varies between salt and free base form free base form: (S)-Ketamine dextrorotation (S)-(+)-ketamine hydrochloridesalt: levorotation(S)-(-)-ketamine Reason found in molecular level: different orientation of substituents: freebase: o-chlorophenyl equatorial, methylamino axia
Sources: http://creationwiki.org/Ketamine#Synthesis http://www.lycaeum.org/rhodium/chemistry/pcp/ketamine.html https://pubchem.ncbi.nlm.nih.gov/compound/ketamine https://pubchem.ncbi.nlm.nih.gov/compound/ketamine#section=Drug-Warning http://www.rsc.org/chemistryworld/2014/02/ketamine-special-k-drugs-podcast http://drugabuse.com/library/the-effects-of-ketamine-use/ http://www.drugfreeworld.org/drugfacts/prescription/ketamine.html http://onlinelibrary.wiley.com/doi/10.1002/1615-9314(20021101)25:15/17%3C1155::AID-JSSC1155%3E3.0.CO;2-M/pdf
CLIP
Process Research and Impurity Control Strategy of Esketamine Organic Process Research & Development ( IF 3.023
) Pub Date: 2020-03-18 , DOI: 10.1021/acs.oprd.9b00553
Shenghua Gao; Xuezhi Gao; Zhezhou Yang; Fuli Zhang
An improved synthesis of ( S )-ketamine (esketamine) has been developed, which was cost-effective, and the undesired isomer could be recovered by racemization. Critical process parameters of each step were identified as well as the process-related impurities. The formation mechanisms and control strategies of most impurities were first discussed. Moreover, the ( S )-ketamine tartrate is a dihydrate, which was disclosed for the first time. The practicable racemization catalyzed by aluminum chloride was carried out in quantitative yield with 99% purity . The ICH-grade quality ( S)-ketamine hydrochloride was obtained in 51.1% overall yield (14.0% without racemization) by chiral resolution with three times recycling of the mother liquors. The robust process of esketamine could be industrially scalable.
Process Research and ketamine impurity control strategy
has been developed an improved ( S ) – ketamine (esketamine) synthesis, the high cost-effective way, the undesired isomer may be recycled by racemization. Determine the key process parameters and process-related impurities for each step. First, the formation mechanism and control strategy of most impurities are discussed. In addition, ( S )-ketamine tartrate is a dihydrate, which is the first time it has been published. The feasible racemization catalyzed by aluminum chloride proceeds in a quantitative yield with a purity of 99%. ICH grade quality ( S) 5-ketamine hydrochloride can be obtained through chiral resolution and three times the mother liquor recovery rate. The total yield is 51.1% (14.0% without racemization). The robust process of ketamine can be used in Industrial promotion.

CLIP

CLIP
https://link.springer.com/article/10.1007/s13738-018-1404-1#citeas
Taghizadeh, M.J., Gohari, S.J.A., Javidan, A. et al. A novel strategy for the asymmetric synthesis of (S)-ketamine using (S)-tert-butanesulfinamide and 1,2-cyclohexanedione. J IRAN CHEM SOC 15, 2175–2181 (2018). https://doi.org/10.1007/s13738-018-1404-1
Abstract
We present a novel asymmetric synthesis route for synthesis of (S)-ketamine using a chiral reagent according to the strategy (Scheme 1), with good enantioselectivity (85% ee) and yield. In this procedure, the (S)-tert-butanesulfinamide (TBSA) acts as a chiral auxiliary reagent to generate (S)-ketamine. A series of new intermediates were synthesized and identified for the first time in this work (2–4). The monoketal intermediate (1) easily obtained after partial conversion of one ketone functional group of 1,2-cyclohexanedione into a ketal using ethylene glycol. The sulfinylimine (2) was obtained by condensation of (S)-tert-butanesulfinamide (TBSA) with (1), 4-dioxaspiro[4.5]decan-6-one in 90% yield. The (S)-N–tert-butanesulfinyl ketamine (3) was prepared on further reaction of sulfinylimine (2) with appropriate Grignard reagent (ArMgBr) in which generated chiral center in 85% yield and with 85% diastereoselectivity. Methylation of amine afforded the product (4). Finally, the sulfinyl- and ketal-protecting groups were removed from the compound (4) by brief treatment with stoichiometric quantities of HCl in a protic solvent gave the (S)-ketamine in near quantitative yield.

Esketamine, sold under the brand name Spravato[4] among others,[6][7] is a medication used as a general anesthetic and for treatment-resistant depression.[4][1] Esketamine is used as a nasal spray or by injection into a vein.[4][1]
Esketamine acts primarily as a non-competitive N-methyl-D-aspartate (NMDA) receptor antagonist.[1][8] It also acts to some extent as a dopamine reuptake inhibitor but, unlike ketamine, does not interact with the sigma receptors.[1] The compound is the S(+) enantiomer of ketamine, which is an anesthetic and dissociative similarly.[1] It is unknown whether its antidepressant action is superior, inferior or equal to racemic ketamine and its opposite enantiomer, arketamine, which are both being investigated for the treatment of depression.
Esketamine was introduced for medical use in 1997.[1] In 2019, it was approved for use with other antidepressants, for the treatment of depression in adults in the United States.[9]
In August 2020, it was approved by the U.S. Food and Drug Administration (FDA) with the added indication for the short-term treatment of suicidal thoughts.[10]
Medical uses
Anesthesia
Esketamine is a general anesthetic and is used for similar indications as ketamine.[1] Such uses include induction of anesthesia in high-risk patients such as those with hemorrhagic shock, anaphylactic shock, septic shock, severe bronchospasm, severe hepatic insufficiency, cardiac tamponade, and constrictive pericarditis; anesthesia in caesarian section; use of multiple anesthetics in burns; and as a supplement to regional anesthesia with incomplete nerve blocks.[1]
Depression
See also: List of investigational antidepressants
Similarly to ketamine, esketamine appears to be a rapid-acting antidepressant.[8][11] It received a breakthrough designation from the FDA for treatment-resistant depression (TRD) in 2013 and major depressive disorder (MDD) with accompanying suicidal ideation in 2016.[12][11] The medication was studied for use in combination with an antidepressant in people with TRD who had been unresponsive to treatment;[12][8][11] six phase III clinical trials for this indication were conducted in 2017.[12][8][11] It is available as a nasal spray.[12][8][11]
In February 2019, an outside panel of experts recommended that the FDA approve the nasal spray version of esketamine,[13] provided that it be given in a clinical setting, with people remaining on site for at least two hours after. The reasoning for this requirement is that trial participants temporarily experienced sedation, visual disturbances, trouble speaking, confusion, numbness, and feelings of dizziness during immediately after.[14]
In January 2020, esketamine was rejected by the National Health Service of Great Britain. NHS questioned the benefits and claimed that it was too expensive. People who have been already using the medication were allowed to complete treatment if their doctors consider this necessary.[15]
Side effects
Most common side effects when used in those with treatment resistant depression include dissociation, dizziness, nausea, sleepiness, anxiety, and increased blood pressure.[16]
Pharmacology
Esketamine is approximately twice as potent as an anesthetic as racemic ketamine.[17] It is eliminated from the human body more quickly than arketamine (R(–)-ketamine) or racemic ketamine, although arketamine slows its elimination.[18]
A number of studies have suggested that esketamine has a more medically useful pharmacological action than arketamine or racemic ketamine[citation needed] but, in mice, that the rapid antidepressant effect of arketamine was greater and lasted longer than that of esketamine.[19] The usefulness of arketamine over eskatamine has been supported by other researchers.[20][21][22]
Esketamine inhibits dopamine transporters eight times more than arketamine.[23] This increases dopamine activity in the brain. At doses causing the same intensity of effects, esketamine is generally considered to be more pleasant by patients.[24][25] Patients also generally recover mental function more quickly after being treated with pure esketamine, which may be a result of the fact that it is cleared from their system more quickly.[17][26] This is however in contradiction with R-ketamine being devoid of psychotomimetic side effects.[27]
Unlike arketamine, esketamine does not bind significantly to sigma receptors. Esketamine increases glucose metabolism in frontal cortex, while arketamine decreases glucose metabolism in the brain. This difference may be responsible for the fact that esketamine generally has a more dissociative or hallucinogenic effect while arketamine is reportedly more relaxing.[26] However, another study found no difference between racemic and (S)-ketamine on the patient’s level of vigilance.[24] Interpretation of this finding is complicated by the fact that racemic ketamine is 50% (S)-ketamine.
History
Esketamine was introduced for medical use as an anesthetic in Germany in 1997, and was subsequently marketed in other countries.[1][28] In addition to its anesthetic effects, the medication showed properties of being a rapid-acting antidepressant, and was subsequently investigated for use as such.[8][12] In November 2017, it completed phase III clinical trials for treatment-resistant depression in the United States.[8][12] Johnson & Johnson filed a Food and Drug Administration (FDA) New Drug Application (NDA) for approval on September 4, 2018;[29] the application was endorsed by an FDA advisory panel on February 12, 2019, and on March 5, 2019, the FDA approved esketamine, in conjunction with an oral antidepressant, for the treatment of depression in adults.[9]
In the 1980s and ’90s, closely associated ketamine was used as a club drug known as “Special K” for its trip-inducing side effects.[30][31]
Society and culture
Names
Esketamine is the generic name of the drug and its INN and BAN, while esketamine hydrochloride is its BANM.[28] It is also known as S(+)-ketamine, (S)-ketamine, or (–)-ketamine, as well as by its developmental code name JNJ-54135419.[28][12]
Esketamine is marketed under the brand name Spravato for use as an antidepressant and the brand names Ketanest, Ketanest S, Ketanest-S, Keta-S for use as an anesthetic (veterinary), among others.[28]
Availability
Esketamine is marketed as an antidepressant in the United States;[9] and as an anesthetic in the European Union.[28]
Legal status
Esketamine is a Schedule III controlled substance in the United States.[4]
References
- ^ Jump up to:a b c d e f g h i j Himmelseher S, Pfenninger E (December 1998). “[The clinical use of S-(+)-ketamine–a determination of its place]”. Anasthesiologie, Intensivmedizin, Notfallmedizin, Schmerztherapie. 33 (12): 764–70. doi:10.1055/s-2007-994851. PMID 9893910.
- ^ “Spravato 28 mg nasal spray, solution – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 24 November 2020.
- ^ “Vesierra 25 mg/ml solution for injection/infusion – Summary of Product Characteristics (SmPC)”. (emc). 21 February 2020. Retrieved 24 November2020.
- ^ Jump up to:a b c d e “Spravato- esketamine hydrochloride solution”. DailyMed. 6 August 2020. Retrieved 26 September 2020.
- ^ “Spravato EPAR”. European Medicines Agency (EMA). 16 October 2019. Retrieved 24 November 2020.
- ^ “Text search results for esketamine: Martindale: The Complete Drug Reference”. MedicinesComplete. London, UK: Pharmaceutical Press. Retrieved 20 August 2017.[dead link]
- ^ Brayfield A, ed. (9 January 2017). “Ketamine Hydrochloride”. MedicinesComplete. London, UK: Pharmaceutical Press. Retrieved 20 August2017.[dead link]
- ^ Jump up to:a b c d e f g Rakesh G, Pae CU, Masand PS (August 2017). “Beyond serotonin: newer antidepressants in the future”. Expert Review of Neurotherapeutics. 17 (8): 777–790. doi:10.1080/14737175.2017.1341310. PMID 28598698. S2CID 205823807.
- ^ Jump up to:a b c “FDA approves new nasal spray medication for treatment-resistant depression; available only at a certified doctor’s office or clinic”. U.S. Food and Drug Administration (FDA) (Press release). Retrieved 2019-03-06.
- ^ “FDA Approves A Nasal Spray To Treat Patients Who Are Suicidal”. NPR. 4 August 2020. Retrieved 27 September 2020.
- ^ Jump up to:a b c d e Lener MS, Kadriu B, Zarate CA (March 2017). “Ketamine and Beyond: Investigations into the Potential of Glutamatergic Agents to Treat Depression”. Drugs. 77 (4): 381–401. doi:10.1007/s40265-017-0702-8. PMC 5342919. PMID 28194724.
- ^ Jump up to:a b c d e f g “Esketamine – Johnson & Johnson – AdisInsight”. Retrieved 7 November 2017.
- ^ Koons C, Edney A (February 12, 2019). “First Big Depression Advance Since Prozac Nears FDA Approval”. Bloomberg News. Retrieved February 12, 2019.
- ^ Psychopharmacologic Drugs Advisory Committee (PDAC) and Drug Safety and Risk Management (DSaRM) Advisory Committee (February 12, 2019). “FDA Briefing Document” (PDF). Food and Drug Administration. Retrieved February 12, 2019.
Meeting, February 12, 2019. Agenda Topic: The committees will discuss the efficacy, safety, and risk-benefit profile of New Drug Application (NDA) 211243, esketamine 28 mg single-use nasal spray device, submitted by Janssen Pharmaceutica, for the treatment of treatment-resistant depression.
- ^ “Anti-depressant spray not recommended on NHS”. BBC News. 28 January 2020.
- ^ “Esketamine nasal spray” (PDF). U.S. Food and Drug Administration (FDA). Retrieved 21 October 2019.
- ^ Jump up to:a b Himmelseher S, Pfenninger E (December 1998). “[The clinical use of S-(+)-ketamine–a determination of its place]”. Anasthesiologie, Intensivmedizin, Notfallmedizin, Schmerztherapie (in German). 33 (12): 764–70. doi:10.1055/s-2007-994851. PMID 9893910.
- ^ Ihmsen H, Geisslinger G, Schüttler J (November 2001). “Stereoselective pharmacokinetics of ketamine: R(–)-ketamine inhibits the elimination of S(+)-ketamine”. Clinical Pharmacology and Therapeutics. 70 (5): 431–8. doi:10.1067/mcp.2001.119722. PMID 11719729.
- ^ Zhang JC, Li SX, Hashimoto K (January 2014). “R (-)-ketamine shows greater potency and longer lasting antidepressant effects than S (+)-ketamine”. Pharmacology, Biochemistry, and Behavior. 116: 137–41. doi:10.1016/j.pbb.2013.11.033. PMID 24316345. S2CID 140205448.
- ^ Muller J, Pentyala S, Dilger J, Pentyala S (June 2016). “Ketamine enantiomers in the rapid and sustained antidepressant effects”. Therapeutic Advances in Psychopharmacology. 6 (3): 185–92. doi:10.1177/2045125316631267. PMC 4910398. PMID 27354907.
- ^ Hashimoto K (November 2016). “Ketamine’s antidepressant action: beyond NMDA receptor inhibition”. Expert Opinion on Therapeutic Targets. 20 (11): 1389–1392. doi:10.1080/14728222.2016.1238899. PMID 27646666. S2CID 1244143.
- ^ Yang B, Zhang JC, Han M, Yao W, Yang C, Ren Q, Ma M, Chen QX, Hashimoto K (October 2016). “Comparison of R-ketamine and rapastinel antidepressant effects in the social defeat stress model of depression”. Psychopharmacology. 233 (19–20): 3647–57. doi:10.1007/s00213-016-4399-2. PMC 5021744. PMID 27488193.
- ^ Nishimura M, Sato K (October 1999). “Ketamine stereoselectively inhibits rat dopamine transporter”. Neuroscience Letters. 274 (2): 131–4. doi:10.1016/s0304-3940(99)00688-6. PMID 10553955. S2CID 10307361.
- ^ Jump up to:a b Doenicke A, Kugler J, Mayer M, Angster R, Hoffmann P (October 1992). “[Ketamine racemate or S-(+)-ketamine and midazolam. The effect on vigilance, efficacy and subjective findings]”. Der Anaesthesist (in German). 41 (10): 610–8. PMID 1443509.
- ^ Pfenninger E, Baier C, Claus S, Hege G (November 1994). “[Psychometric changes as well as analgesic action and cardiovascular adverse effects of ketamine racemate versus s-(+)-ketamine in subanesthetic doses]”. Der Anaesthesist (in German). 43 Suppl 2: S68-75. PMID 7840417.
- ^ Jump up to:a b Vollenweider FX, Leenders KL, Oye I, Hell D, Angst J (February 1997). “Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET)”. European Neuropsychopharmacology. 7 (1): 25–38. doi:10.1016/s0924-977x(96)00042-9. PMID 9088882. S2CID 26861697.
- ^ Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, Ma M, Dong C, Hashimoto K (September 2015). “R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects”. Translational Psychiatry. 5 (9): e632. doi:10.1038/tp.2015.136. PMC 5068814. PMID 26327690.
- ^ Jump up to:a b c d e “Esketamine”. Drugs.com.
- ^ “Janssen Submits Esketamine Nasal Spray New Drug Application to U.S. FDA for Treatment-Resistant Depression”. Janssen Pharmaceuticals, Inc.
- ^ Marsa, Linda (January 2020). “A Paradigm Shift for Depression Treatment”. Discover. Kalmbach Media.
- ^ Hoffer, Lee (7 March 2019). “The FDA Approved a Ketamine-Like Nasal Spray for Hard-to-Treat Depression”. Vice. Retrieved 11 February 2020.
External links
- “Esketamine”. Drug Information Portal. U.S. National Library of Medicine.
- “Esketamine hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
{kind=link}
{kind=link}
/////////////Esketamine, JNJ 54135419, phase 3
Inclisiran
Inclisiran
CAS 1639324-58-5
- ALN-60212
- ALN-PCSsc
US FDA APPROVED
12/22/2021 |
To treat heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease as an add-on therapy, Leqvio
Inclisiran was first developed by Alnylam Pharmaceuticals, Inc. (Cambridge, Massachusetts, US). Development has now been assumed by The Medicines Company (Parsippany, New Jersey, US). One phase I and two phase II trials have been completed. Topline results of two phase III trials were also recently presented while other phase III trials are still ongoing as part of the ORION clinical development program. …..https://www.ncbi.nlm.nih.gov/books/NBK555477/
Inclisiran is a long-acting, synthetic small interfering RNA (siRNA) directed against proprotein convertase subtilisin-kexin type 9 (PCSK9), which is a serine protease that regulates plasma low-density lipoprotein cholesterol (LDL-C) levels. By binding to PCSK9 messenger RNA, inclisiran prevents protein translation of PCSK9, leading to decreased concentrations of PCSK9 and plasma concentrations of LDL cholesterol.1,2 Lowering circulating plasma LDL-C levels offers an additional benefit of reducing the risk of cardiovascular disease (CVD) and improving cardiovascular outcomes, as hypercholesterolemia is a major known risk factor for CVD.1,2
On December 11, 2020, the European Commission (EC) granted authorization for marketing inclisiran as the first and only approved siRNA for the treatment of adults with primary hypercholesterolemia (heterozygous familial and non-familial) or mixed dyslipidemia, alone or in combination with other lipid-lowering therapies. It is marketed under the trade name Leqvio 8 and is also currently under review by the FDA.
Inclisiran, sold under the brand name Leqvio, is a medication for the treatment of people with atherosclerotic cardiovascular disease (ASCVD), ASCVD risk equivalents and heterozygous familial hypercholesterolemia (HeFH). It is a small interfering RNA that inhibits translation of the protein PCSK9.[2][3][4] It is being developed by The Medicines Company which licensed the rights to inclisiran from Alnylam Pharmaceuticals.[5]
On 15 October 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Leqvio, intended for the treatment for primary hypercholesterolaemia or mixed dyslipidaemia.[6] Inclisiran was approved for use in the European Union in December 2020.[1]
History
In 2019 The Medicines Company announced positive results from pivotal phase III study (all primary and secondary endpoints were met with efficacy consistent with Phase I and II studies). The company anticipates regulatory submissions in the U.S. in the fourth quarter of 2019, and in Europe in the first quarter of 2020.[7] The Medicines Company is being acquired by Novartis.[8]

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b “Leqvio EPAR”. European Medicines Agency. 13 October 2020. Retrieved 6 January 2021.
- ^ Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. (January 2017). “A Highly Durable RNAi Therapeutic Inhibitor of PCSK9”. The New England Journal of Medicine. 376 (1): 41–51. doi:10.1056/NEJMoa1609243. PMC 5778873. PMID 27959715.
- ^ Spreitzer H (11 September 2017). “Neue Wirkstoffe: Inclisiran”. Österreichische Apotheker-Zeitung (in German) (19/2017).
- ^ “Proposed INN: List 114” (PDF). WHO Drug Information. WHO. 29 (4): 531f. 2015.
- ^ Taylor NP (26 August 2019). “Medicines Company’s PCSK9 drug hits phase 3 efficacy goals”. FierceBiotech.
- ^ “Leqvio: Pending EC decision”. European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “The Medicines Company Announces Positive Topline Results from First Pivotal Phase 3 Trial of Inclisiran”. The Medicines Company. Retrieved 29 August 2019.
- ^ “Novartis acquires medicines company”. Novartis. Retrieved 15 January 2020.
Further reading
- Ray KK, Landmesser U, Leiter LA, et al. (April 2017). “Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol” (PDF). N. Engl. J. Med. 376 (15): 1430–1440. doi:10.1056/NEJMoa1615758. hdl:10044/1/45416. PMID 28306389. S2CID 205101529.
- Ray KK, Wright RS, Kallend D, et al. (March 2020). “Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol”. N. Engl. J. Med. 382 (16): 1507–1519. doi:10.1056/NEJMoa1912387. PMID 32187462.
External links
- “Inclisiran”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03399370 for “Inclisiran for Participants With Atherosclerotic Cardiovascular Disease and Elevated Low-density Lipoprotein Cholesterol (ORION-10)” at ClinicalTrials.gov
- Clinical trial number NCT03400800 for “Inclisiran for Subjects With ACSVD or ACSVD-Risk Equivalents and Elevated Low-density Lipoprotein Cholesterol (ORION-11)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Leqvio |
| Other names | ALN-PCSsc, ALN-60212 |
| Routes of administration | Subcutaneous injection |
| ATC code | C10AX16 (WHO) |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 1639324-58-5 |
| DrugBank | DB14901 |
| UNII | UOW2C71PG5 |
| KEGG | D11931 |
| Chemical and physical data | |
| Formula | C520H679F21N175O309P43S6 |
| Molar mass | 16248.27 g·mol−1 |
General References
- Kosmas CE, Munoz Estrella A, Sourlas A, Silverio D, Hilario E, Montan PD, Guzman E: Inclisiran: A New Promising Agent in the Management of Hypercholesterolemia. Diseases. 2018 Jul 13;6(3). pii: diseases6030063. doi: 10.3390/diseases6030063. [PubMed:30011788]
- German CA, Shapiro MD: Small Interfering RNA Therapeutic Inclisiran: A New Approach to Targeting PCSK9. BioDrugs. 2020 Feb;34(1):1-9. doi: 10.1007/s40259-019-00399-6. [PubMed:31782112]
- Doggrell SA: Inclisiran, the billion-dollar drug, to lower LDL cholesterol – is it worth it? Expert Opin Pharmacother. 2020 Nov;21(16):1971-1974. doi: 10.1080/14656566.2020.1799978. Epub 2020 Aug 4. [PubMed:32749892]
- Goldstein JL, Brown MS: Regulation of low-density lipoprotein receptors: implications for pathogenesis and therapy of hypercholesterolemia and atherosclerosis. Circulation. 1987 Sep;76(3):504-7. doi: 10.1161/01.cir.76.3.504. [PubMed:3621516]
- Pratt AJ, MacRae IJ: The RNA-induced silencing complex: a versatile gene-silencing machine. J Biol Chem. 2009 Jul 3;284(27):17897-901. doi: 10.1074/jbc.R900012200. Epub 2009 Apr 1. [PubMed:19342379]
- Leiter LA, Teoh H, Kallend D, Wright RS, Landmesser U, Wijngaard PLJ, Kastelein JJP, Ray KK: Inclisiran Lowers LDL-C and PCSK9 Irrespective of Diabetes Status: The ORION-1 Randomized Clinical Trial. Diabetes Care. 2019 Jan;42(1):173-176. doi: 10.2337/dc18-1491. Epub 2018 Nov 28. [PubMed:30487231]
- Cupido AJ, Kastelein JJP: Inclisiran for the treatment of hypercholesterolaemia: implications and unanswered questions from the ORION trials. Cardiovasc Res. 2020 Sep 1;116(11):e136-e139. doi: 10.1093/cvr/cvaa212. [PubMed:32766688]
- Novartis: Novartis receives EU approval for Leqvio (inclisiran), a first-in-class siRNA to lower cholesterol with two doses a year [Link]
- Summary of Product Characteristics: Leqvio (inclisiran), solution for subcutaneous injection [Link]
Summary
- Atherosclerotic cardiovascular disease (ASCVD) remains one of the leading causes of death in Canada. Cholesterol, specifically low-density lipoprotein cholesterol (LDL-C), is a major risk factor for cardiovascular disease (CVD) and is thereby targeted to reduce the likelihood of a cardiovascular event, such as a myocardial infarction (MI) and stroke.
- Inclisiran, first developed by Alnylam Pharmaceuticals, Inc. (Cambridge, Massachusetts, US) then by The Medicines Company (Parsippany, New Jersey, US), is a small interfering ribonucleic acid (siRNA) molecule being investigated for the treatment of hypercholesterolemia.
- ORION-1 was a phase II, double-blind, placebo-controlled, multi-centre, randomized controlled trial of 501 patients. Patients were included in the trial if they had a history of ASCVD or were at high risk of ASCVD. The treatment arms were administered 200 mg, 300 mg, or 500 mg of inclisiran on day 1, or 100 mg, 200 mg, or 300 mg of inclisiran on days 1 and 90. The comparator was either placebo on day 1 or placebo on days 1 and 90. The primary end point was percentage change in LDL-C at day 180 from baseline.
- The ORION-1 study demonstrated that inclisiran, administered at various doses and intervals, compared with placebo, resulted in a statistically significant reduction in LDL-C levels (P < 0.001 for all comparisons versus placebo). The greatest reduction in LDL-C levels was obtained with the 300 mg dose of inclisiran given at days 1 and 90 with a 52.6% (95% confidence interval [CI]: −57.1 to −48.1) reduction at day 180 compared with baseline, and a mean absolute reduction in LDL-C levels of 1.66 (standard deviation 0.54) mmol/L. Results from the ORION-1 trial provided the necessary data to make a decision regarding the dosing regimen to be used in subsequent phase III trials, in particular the ORION-11 phase III trial.
- The ORION-11 study was a phase III international, multi-centre, and double-blind trial which randomized 1,617 participants (87% with established ASCVD) to inclisiran 300 mg (n = 810) or placebo (n = 807). An initial inclisiran dose of 300 mg given subcutaneously was administered at day 1, day 90, and then every six months for two doses, that is at days 270 and 450. The mean baseline LDL-C level was 2.8 mmol/L (inclisiran) and 2.7 mmol/L (placebo); 96% of participants were on high-dose statin therapy. There was a 50% time-averaged reduction in LDL-C levels from day 90 to day 540 (P < 0.00001). Pre-specified exploratory cardiovascular composite end point (cardiac death, cardiac arrest, MI, or stroke) occurred in 7.8% of inclisiran treated patients versus 10.3% of patients on placebo; this lower rate was mainly driven by a reduction in MI and stroke. With respect to adverse effects, 4.69% of patients on inclisiran reported an injection site reaction, compared with 0.5% of patients on placebo. All reactions were transient. There was no evidence of liver, kidney, muscle, or platelet toxicity.
- Inclisiran may be an option in the future as a cholesterol-lowering medication, where it would likely be used in patients who are unable to achieve their LDL-C targets despite maximally tolerated statin therapy or who are intolerant to statin therapy. However, results from the inclisiran cardiovascular outcome trial (ORION-4), are needed to confirm its efficacy in reducing CVD and its long-term safety.
- Inclisiran is not yet approved by any regulatory authority, but its ORION clinical development program identifies the year 2021 as the goal to reach worldwide markets.
///////////Inclisiran, LEQVIO, ALN 60212, ALN PCSsc , NOVARTIS, Leqvio, APPROVALS 2021, FDA 2021

NEW DRUG APPROVALS
ONE TIME
$10.00