VALSARTAN
CAS 137862-53-4
Molecular FormulaC24H29N5O3, Average mass435.519 Da
|
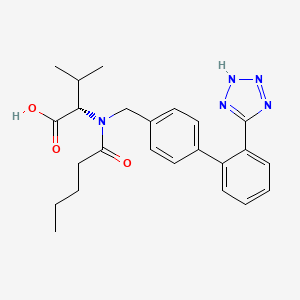
(2S)-3-methyl-2-[N-({4-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]phenyl}methyl)pentanamido]butanoic acid
|
PAPER
Greening the Valsartan Synthesis: Scale-up of Key Suzuki–Miyaura Coupling over SiliaCat DPP-Pd
† SiliCycle Inc., 2500 Parc-Technologique Blvd, Quebec City, Quebec, Canada G1P 4S6
‡ Istituto per lo Studio dei Materiali Nanostrutturati, CNR, via U. La Malfa 153, 90146 Palermo, Italy
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/op400118f
Publication Date (Web): June 17, 2013
The study of the scale-up of the heterogeneous Suzuki-Miyaura coupling reaction in batch conditions between 2-chlorobenzonitrile and 4-tolylboronic acid, a key step in valsartansynthesis, to produce 4′-methyl-2-biphenylcarbonitrile over the SiliaCat DPP-Pd catalyst in ethanol under reflux allows to identify the optimal reaction conditions.
The catalyst, regardless of limited Pd leaching, is not reusable, and the method can be effectively applied to the high yield synthesis of several coupling products, opening the route to efficient continuous coupling syntheses.
http://pubs.acs.org/doi/full/10.1021/op400118f
ABOUT VALSARTAN
Valsartan (Angiotan or Diovan) is an angiotensin II receptor antagonist (more commonly called an “ARB”, or angiotensin receptor blocker), with particularly high affinity for the type I (AT1) angiotensin receptor. By blocking the action of angiotensin, valsartan dilates blood vessels and reduces blood pressure.[1] In the U.S., valsartan is indicated for treatment ofhigh blood pressure, congestive heart failure (CHF), or post-myocardial infarction (MI).[2] In 2005, Valsartan was prescribed more than 12 million times in the United States[citation needed] and global sales were approximately $6.1 billion in 2010.[3] The patents for valsartan and valsartan/hydrochlorothiazide expired in September 2012.[4][5]
A study released in 2010, based on 819,491 cases in U.S. Department of Veterans Affairs database from 2002 to 2006, demonstrated a significant reduction in the incidence and progression of Alzheimer’s disease and dementia.[6] An earlier study released by theJournal of Clinical Investigation in 2007 found some efficacy in the use of valsartan in the treatment and prevention of Alzheimer’s disease (in a mouse model).[7]
Valsartan, also known as (S)—N-(1-Carboxy-2-methyl-prop-1-yl)-N-pentanoyl-N-[2′-(1H-tetrazol-5-yl)bi phenyl-4-ylmethyl]-amine, has the following structure:
and is marketed as the free acid under the name DIOVAN. DIOVAN is prescribed as oral tablets in dosages of 40 mg, 80 mg, 160 mg and 320 mg of valsartan.
Valsartan and/or its intermediates are disclosed in various references, including: U.S. Pat. Nos. 5,399,578, 5,965,592, 5,260,325, 6,271,375, WO 02/006253, WO 01/082858, WO 99/67231, WO 97/30036, Peter Bühlmayer, et. al., Bioorgan. & Med. Chem. Let., 4(1) 29–34 (1994), Th. Moenius, et. al., J. Labelled Cpd. Radiopharm., 43(13) 1245–1252 (2000), and Qingzhong Jia, et. al., Zhongguo Yiyao Gongye Zazhi, 32(9) 385–387 (2001).
Valsartan is an orally active specific angiotensin II antagonist acting on the AT1 receptor subtype. Valsartan is prescribed for the treatment of hypertension. U.S. Pat. No. 6,395,728 is directed to use of valsartan for treatment of diabetes related hypertension. U.S. Pat. Nos. 6,465,502 and 6,485,745 are directed to treatment of lung cancer with valsartan. U.S. Pat. No. 6,294,197 is directed to solid oral dosage forms of valsartan.
The synthesis of valsartan is discussed, inter alia, in U.S. Pat. No. 5,399,578. In the synthesis disclosed therein, the final synthetic step (exclusive of work-up and purification) involves the reaction of a cyano group on the biphenyl ring with an azide, for example, tributyl tin azide. The reaction scheme of the ‘578 patent is as follows:
Peter Bühlmayer, et. al., Bioorgan. & Med. Chem. Let., 4(1) 29–34 (1994)
In Moenius, et. al., J. Labelled Cpd. Radiopharm., 43(13) 1245–1252 (2000), various schemes for synthesis of valsartan are provided, with one being:
Another paper, Qingzhong Jia, et. al., Zhongguo Yiyao Gongye Zazhi, 32(9) 385–387 (2001), discloses a synthesis scheme for valsartan as follows:
There is a need in the art for an improved synthetic process for the preparation of valsartan and precursors of valsartan.
DOSE
Oral tablets, containing 40 mg (scored), 80 mg, 160 mg, or 320 mg of valsartan. Usual dosage ranges from 40–320 mg daily.
In some markets available as a hard gelatin capsule, containing 40 mg, 80 mg, or 160 mg of valsartan.
Diovan HCT contains a combination of valsartan and hydrochlorothiazide but, unlike Diovan, is only indicated for hypertension, not for CHF or post-MI. Diovan HCT is available in oral tablets, containing (valsartan/HCTZ mg) 80/12.5, 160/12.5, 160/25, 320/12.5, and 320/25.
Whether angiotensin receptor blockers may or may not increase the risk of myocardial infarction (heart attack) was announced in BMJ[8] and was debated in 2006 in the medical journal of the American Heart Association.[9][10] To date[when?], there is no consensus on whether ARBs have a tendency to increase MI, but there is also no substantive evidence to indicate that ARBs are able to reduce MI.
In the VALUE trial, the angiotensin II receptor blocker valsartan produced a statistically significant 19% (p=0.02) relative increase in the prespecified secondary end point of myocardial infarction (fatal and non-fatal) compared with amlodipine.[11]
The CHARM-alternative trial showed a significant +52% (p=0.025) increase in myocardial infarction with candesartan (versus placebo) despite a reduction in blood pressure.[12]
Indeed, as a consequence of AT1 blockade, ARBs increase Angiotensin II levels several-fold above baseline by uncoupling a negative-feedback loop. Increased levels of circulating Angiotensin II result in unopposed stimulation of the AT2 receptors, which are, in addition upregulated. Unfortunately, recent data suggest that AT2 receptor stimulation may be less beneficial than previously proposed and may even be harmful under certain circumstances through mediation of growth promotion, fibrosis, and hypertrophy, as well as proatherogenic and proinflammatory effects.[13][14][15]
In patients with impaired glucose tolerance, valsartan may decrease the incidence of developing diabetes mellitus type 2.[16] However, the absolute risk reduction is small (less than 1 percent per year) and diet, exercise or other drugs, may be more protective. In the same study, no reduction in the rate of cardiovascular events (including death) was shown.
There is a case report of a stillbirth in which valsartan is implicated.[18]In the US, UK and Australia, valsartan is marketed by Novartis under the trade name Diovan. In Pakistan, it is marketed by Efroze under the trade name Angiotan. In India, it is marketed by Cipla under the trade name Valtan and by Torrent Pharmaceuticals under the trade name Valzaar. In Egypt and in France, it is marketed by Novartis under the name of Tareg. In Ukraine, it is marketed by Фарма Старт under the trade name Диокор, Диокор Соло
- Marks JW (2007-02-15). “Valsartan, Diovan”. MedicineNet. Retrieved 2010-03-04.
- “Diovan prescribing information”. Novartis.
- J “Novartis Annual Report”. Novartis. 2010. Retrieved June 15, 2011.
- Philip Moeller (April 29, 2011). “Blockbuster Drugs That Will Go Generic Soon”. U.S.News & World Report.
- Eva Von Schaper (August 5, 2011). “Novartis’s Jimenez Has Blockbuster Plans For Diovan After Patent Expires”. Bloomberg.
- Li NC, Lee A, Whitmer RA, et al. (January 2010). “Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis”. BMJ 340: b5465. doi:10.1136/bmj.b5465.PMC 2806632. PMID 20068258.
- Wang J, Ho L, Chen L, et al. (November 2007). “Valsartan lowers brain β-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease” (PDF). J. Clin. Invest. 117 (11): 3393–402. doi:10.1172/JCI31547.PMC 2040315. PMID 17965777. Retrieved 2009-11-11.
- Verma S, Strauss M (November 2004). “Angiotensin receptor blockers and myocardial infarction: These drugs may increase myocardial infarction—and patients may need to be told”. BMJ329 (7477): 1248–9. doi:10.1136/bmj.329.7477.1248.PMC 534428. PMID 15564232.
- Strauss MH, Hall AS (August 2006). “Angiotensin receptor blockers may increase risk of myocardial infarction: unraveling the ARB-MI paradox”. Circulation 114 (8): 838–54.doi:10.1161/CIRCULATIONAHA.105.594986.PMID 16923768.
- Tsuyuki RT, McDonald MA (August 2006). “Angiotensin receptor blockers do not increase risk of myocardial infarction”. Circulation 114 (8): 855–60.doi:10.1161/CIRCULATIONAHA.105.594978.PMID 16923769.
- Julius S, Kjeldsen SE, Weber M, et al. (June 2004). “Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial”. The Lancet 363 (9426): 2022–31.doi:10.1016/S0140-6736(04)16451-9. PMID 15207952.
- Granger CB, McMurray JJ, Yusuf S, et al. (September 2003). “Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial”. The Lancet 362 (9386): 772–6.doi:10.1016/S0140-6736(03)14284-5. PMID 13678870.
- Levy BI (September 2005). “How to explain the differences between renin angiotensin system modulators”. Am. J. Hypertens. 18 (9 Pt 2): 134S–141S.doi:10.1016/j.amjhyper.2005.05.005. PMID 16125050.
- Levy BI (January 2004). “Can angiotensin II type 2 receptors have deleterious effects in cardiovascular disease? Implications for therapeutic blockade of the renin-angiotensin system”. Circulation 109 (1): 8–13.doi:10.1161/01.CIR.0000096609.73772.C5.PMID 14707017.
- Reudelhuber TL (December 2005). “The continuing saga of the AT2 receptor: a case of the good, the bad, and the innocuous”. Hypertension 46 (6): 1261–2.doi:10.1161/01.HYP.0000193498.07087.83.PMID 16286568.
- McMurray JJ, Holman RR, Haffner SM, et al. (April 2010).“Effect of valsartan on the incidence of diabetes and cardiovascular events” (PDF). The New England Journal of Medicine 362 (16): 1477–90. doi:10.1056/NEJMoa1001121.PMID 20228403.
- Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag.ISBN 3-85200-196-X.
- Briggs GG, Nageotte MP (2001). “Fatal fetal outcome with the combined use of valsartan and atenolol”. The Annals of Pharmacotherapy 35 (7–8): 859–61. doi:10.1345/aph.1A013.PMID 11485133.
UPDATE……

VALSARTAN
mp 114–118 °C;
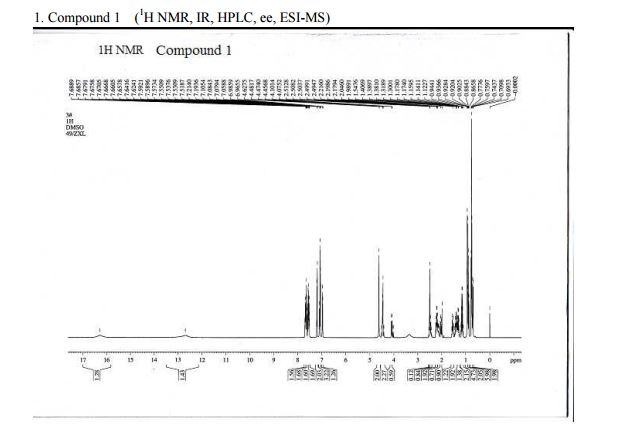
1H NMR (400 MHz, DMSO-d6): δ 12.6 (brs, 1H), 7.72 (m, 4H), 7.24 (m, 1H), 7.15 (m, 2H), 6.94 (m, 1H), 4.58 (m, 1H), 4.40 (m, 1H), 3.33 (m, 1H), 2.25 (m, 1H), 1.52 (m, 6H), 0.9 (m, 3H), 0.84 (m, 3H), 0.74 (m, 3H);
13C NMR (100 MHz, DMSO-d6): δ 174.0, 172.4, 171.8, 141.7, 138.2, 131.54, 131.1, 131.0, 129.3,128.8, 128.2, 127.4, 126.7, 70.3, 63.4, 49.9, 32.9, 28.05, 27.3, 22.2, 20.6, 14.2;
ESIMS: m/z calcd [M]+: 435; found: 436 [M+H]+; HRMS (ESI): m/z calcd [M]+: 435.5187; found: 435.5125 [M]+
US 7439261 B2
1H-NMR (CDCl3) (0.80-1.15 (m, 9H); 1.20-1.50 (m, 2H); 1.60-1.80 (m, 2H); 2.60 (t, 2H); 2.65-2.80 (m, 2H), 3.70 (d, 1H), 4.10 (d, 0.3 H), 4.30 (d, 0.7 H), 4.90 (d, 0.7H), 5.2 (d, 0.3H); 7.00 (d, 0.3H); 7.10-7.20 (m, 4H), 7.40-7.60 (m, 3H), 7.85 (d, 0.7 H).
SHORT DESCRIPTION
Valsartan, N-(1-oxopentyl)-N-[[2′-(1H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]-L-valine, is a known anti-hypertensive agent having the following formula (I):
Valsartan and its preparation are disclosed in U.S. Pat. No. 5,399,578, in particular in Example 16. One of the synthetic routes according to U.S. Pat. No. 5,399,578 can be schematically represented as follows:
The synthetic pathway comprises various steps, among which:
-
- coupling of compound (3) with 2-chlorobenzonitrile to obtain compound (4),
- radicalic bromination of compound (4) to give compound (5),
- transformation of the brominated derivative (5) into the respective aldehyde derivative (6),
- reductive alkylation of compound (6) to obtain intermediate (8),
- acylation of compound (8) to obtain intermediate (9),
- conversion of the cyano group to the tetrazole group to afford intermediate (10),
- deprotection of the carboxylic group by hydrogenolysis to obtain valsartan.
-
It is marketed as the free acid under the name DIOVAN. DIOVAN is prescribed as oral tablets in dosages of 40 mg, 80 mg, 160 mg and 320 mg ofvalsartan.
-
Valsartan and/or its intermediates are disclosed in various references, including: U.S. Pat. Nos. 5,399,578 ,5,965,592 , 5,260,325 , 6,271,375 , WO 02/006253 , WO 01/082858 , WO 99/67231 , WO 97/30036 , Peter Bühlmayer, et. al., Bioorgan. & Med. Chem. Let., 4(1) 29-34 (1994), Th. Moenius, et. al., J. Labelled Cpd. Radiopharm., 43(13) 1245 – 1252 (2000), and Qingzhong Jia, et. al., Zhongguo Yiyao Gongye Zazhi, 32(9) 385-387 (2001), all of which are incorporated herein by reference.
-
Valsartan is an orally active specific angiotensin II antagonist acting on the AT1 receptor subtype. Valsartan is prescribed for the treatment of hypertension. U.S. Pat. No. 6,395,728 is directed to use of valsartan for treatment of diabetes related hypertension. U.S. Pat. Nos. 6,465,502 and 6,485,745 are directed to treatment of lung cancer with valsartan. U.S. Pat. No. 6,294,197 is directed to solid oral dosage forms of valsartan
GOOD ARTICLES
http://users.uoa.gr/~tmavrom/2009/valsartan2009.pdf
http://www.acgpubs.org/JCM/2009/Volume%203/Issue%201/JCM-0908-14.pdf
https://www.beilstein-journals.org/bjoc/single/printArticle.htm?publicId=1860-5397-6-27 REPORTS
mp 114–118 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.6 (brs, 1H), 7.72 (m, 4H), 7.24 (m, 1H), 7.15 (m, 2H), 6.94 (m, 1H), 4.58 (m, 1H), 4.40 (m, 1H), 3.33 (m, 1H), 2.25 (m, 1H), 1.52 (m, 6H), 0.9 (m, 3H), 0.84 (m, 3H), 0.74 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 174.0, 172.4, 171.8, 141.7, 138.2, 131.54, 131.1, 131.0, 129.3,128.8, 128.2, 127.4, 126.7, 70.3, 63.4, 49.9, 32.9, 28.05, 27.3, 22.2, 20.6, 14.2; ESIMS: m/z calcd [M]+: 435; found: 436 [M+H]+; HRMS (ESI): m/z calcd [M]+: 435.5187; found: 435.5125 [M]+
Valsartan
Structural formula
UV – Spectrum
 |
| Conditions : Concentration – 1 mg / 100 ml |
| The solvent designation schedule |
methanol
 |
water
 |
0.1М HCl
 |
0.1M NaOH
 |
| maximum absorption |
249 nm |
250 nm |
248 nm |
251 nm |
 |
309 |
302 |
289 |
311 |
| e |
13400 |
13100 |
12600 |
13500 |
IR – spectrum
| Wavelength (μm) |
 |
| Wave number (cm -1 ) |
References
-
UV and IR Spectra. H.-W. Dibbern, R.M. Muller, E. Wirbitzki, 2002 ECV
-
NIST/EPA/NIH Mass Spectral Library 2008
-
Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman. Academic Press, 2000.
-
Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.

CLIP

(a) Et3N, CH2Cl2, 0 °C, 95%; (b) NaH, THF, 70%; (c) n-BuLi, 25 °C, THF, anhyd ZnCl2, −20 °C, Q-phos, Pd(OAc)2, 75 °C, 2 h, 80%; (d) 3 N NaOH, MeOH, reflux, 90%.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-6-27
valsartan 8; mp 114–118 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.6 (brs, 1H), 7.72 (m, 4H), 7.24 (m, 1H), 7.15 (m, 2H), 6.94 (m, 1H), 4.58 (m, 1H), 4.40 (m, 1H), 3.33 (m, 1H), 2.25 (m, 1H), 1.52 (m, 6H), 0.9 (m, 3H), 0.84 (m, 3H), 0.74 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 174.0, 172.4, 171.8, 141.7, 138.2, 131.54, 131.1, 131.0, 129.3,128.8, 128.2, 127.4, 126.7, 70.3, 63.4, 49.9, 32.9, 28.05, 27.3, 22.2, 20.6, 14.2; ESIMS: m/z calcd [M]+: 435; found: 436 [M+H]+; HRMS (ESI): m/z calcd [M]+: 435.5187; found: 435.5125 [M]+
PAPER
An Improved Synthesis of Valsartan
Department of Chemical Engineering, Anyang Institute of Technology, Anyang 455000, China
Org. Process Res. Dev., 2011, 15 (5), pp 986–988
DOI: 10.1021/op200032b
Publication Date (Web): July 5, 2011
Copyright © 2011 American Chemical Society
Abstract
Biphenyltetrazole group, an important component of sartans, is usually formed in excellent yield by the reaction of 4′-alkylbiphenyl-2-carbonitrile with excessive organotin azide. However, it is restricted in industrial scale because of the difficult post-treatment. In this article, an improved synthetic method for valsartan and the quantitative recovery of tri-n-butyltin chloride are reported. During this process, the tetrazole–Sn complex and excessive organotin azide were decomposed by HCl to furnish tri–n-butyltin chloride, and then reacted with NaF to lead to filterable polymer tributyltin fluoride which was converted again to tributyltin chloride by HCl in ethyl acetate. This approach is facile for the efficient manufacture of sartans using organotin azide to form the tetrazole group and is valuable for industry readers.
http://pubs.acs.org/doi/suppl/10.1021/op200032b
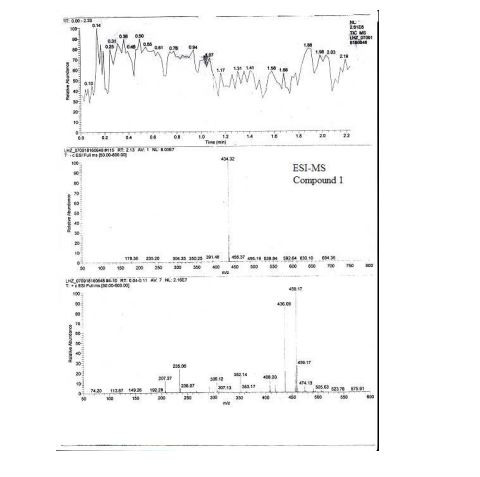
valsartan (1) (6.5 g, HPLC, 99.7%) as a white crystalline powder with a yield of 72.5% calculated on valstartan benzyl ester (2), mp 113117 C (lit.:14 mp 105115 C, from ethyl acetate). ESI-MS (-p): 434.32. HPLC purity 99.62%, ee =100% (OD-H, mobile phase: n-hexane and isopropyl alcohol in the ratio of 850:150). [R] 20 D = () 67.2 (1% w/v in methanol).
1 H NMR (DMSO-d6) δ: 0.690.94 (m, 9H), 1.101.20 (m, 1H), 1.281.58 (m, 3H), 1.982.10 (m, 1H), 2.172.50 (m, 2H), 4.074.63 (m, 3H), 6.967.21(m, 4H), 7.517.71 (m, 4H), 12.69 (br, 1H), 16.29 (br, 1H).
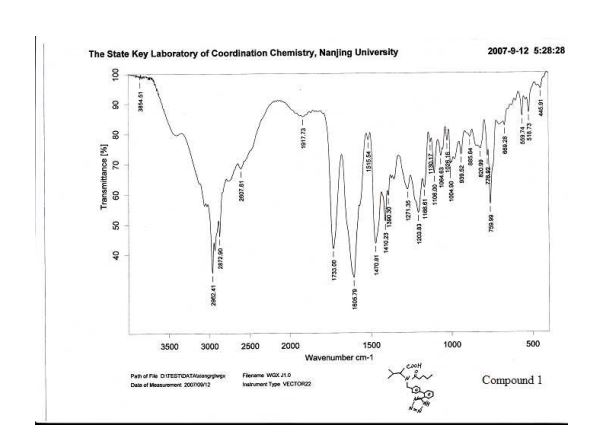
IR (KBr) νmax/cm1 : 3446(br, w), 3060(w), 2963(s), 2932(m), 2873(m), 2744(w), 2612(w), 1732(s), 1604(s), 1471(s), 1410(m), 1390(w), 1354(w), 1273(w), 1204(m), 1166(m), 1129(w), 1105(w), 1065(w), 1052(w), 1025(w), 996(w), 939(w), 901(w), 852(w), 822(w), 777(w), 760(m), 682(w), 670(w), 624(w), 559(w).
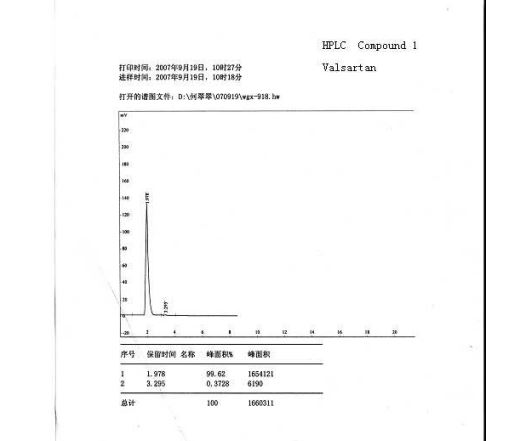
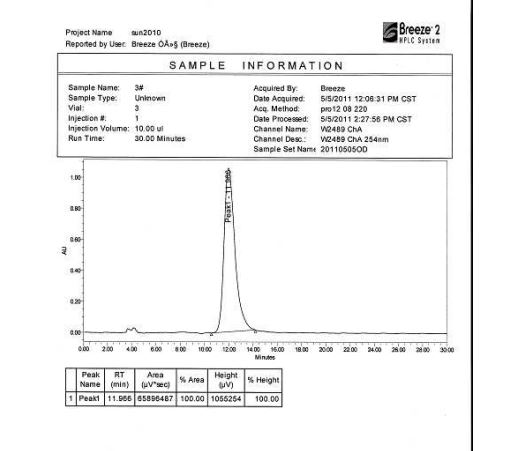
HPLC Conditions for Enantiomer Purity of Valsartan are listed below. Instrument: Water, Breeze 2 Column: Chiralcel OD-H Detection: UV, 220 nm Flow: 0.8 mL/min Injection volume: 10 µL Run time: 30 min Mobile phase: the ratio of n-hexane and isopropyl alcohol is 850:150 Retention time of valsartan: ∼12 min The enantiomeric purity of the crystallized Valsartan prepared in our experiments is nearly 100%. The peak occurred in 4 min can be attributed to the solvent peak in dead time.
Diovan (valsartan) is a nonpeptide, orally active, and specific angiotensin II receptor blocker acting on the AT1 receptor subtype.
Valsartan is chemically described as N-(1-oxopentyl)-N-[[2′-(1H-tetrazol-5-yl) [1,1′-biphenyl]-4- yl]methyl]-L-valine. Its empirical formula is C24H29N5O3, its molecular weight is 435.5, and its structural formula is:
Valsartan is a white to practically white fine powder. It is soluble in ethanol and methanol and slightly soluble in water.
Diovan is available as tablets for oral administration, containing 40 mg, 80 mg, 160 mg or 320 mg of valsartan. The inactive ingredients of the tablets are colloidal silicon dioxide, crospovidone, hydroxypropyl methylcellulose, iron oxides (yellow, black and/or red), magnesium stearate, microcrystalline cellulose, polyethylene glycol 8000, and titanium dioxide.
Valsartan
CAS Registry Number: 137862-53-4
CAS Name: N-(1-Oxopentyl)-N-[[2¢-(1H-tetrazol-5-yl)[1,1¢-biphenyl]-4-yl]methyl]-L-valine
Additional Names: N-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]-N-valeryl-L-valine; (S)-N-(1-carboxy-2-methylprop-1-yl)-N-pentanoyl-N-[2¢-(1H-tetrazol-5-yl)-biphenyl-4-ylmethyl]amine
Manufacturers’ Codes: CGP-48933
Trademarks: Diovan (Novartis); Tareg (Novartis)
Molecular Formula: C24H29N5O3
Molecular Weight: 435.52
Percent Composition: C 66.19%, H 6.71%, N 16.08%, O 11.02%
Literature References: Nonpeptide angiotensin II AT1-receptor antagonist. Prepn: P. Bühlmayer et al., EP 443983; eidem, US5399578 (1991, 1995 both to Ciba Geigy); idem et al., Bioorg. Med. Chem. Lett. 4, 29 (1994). Pharmacological profile: L. Criscione et al., Br. J. Pharmacol. 110, 761 (1993). HPLC determn in human plasma: A. Sioufi et al., J. Liq. Chromatogr. 17, 2179 (1994). Clinical pharmacology: P. Müller et al., Eur. J. Clin. Pharmacol. 47, 231 (1994). Clinical comparison with captopril, q.v., in high risk patients following myocardial infarction: M. A. Pfeffer et al., N. Engl. J. Med. 349, 1893 (2003). Review of pharmacology and clinical experience in heart failure: R. Latini et al., Expert Opin. Pharmacother. 5, 181-193 (2004).
Properties: Crystals from diisopropyl ether, mp 116-117°. Partition coefficient (n-octanol/aq phosphate buffer): 0.033. Sol in water at 25°.
Melting point: mp 116-117°
Log P: Partition coefficient (n-octanol/aq phosphate buffer): 0.033
Therap-Cat: Antihypertensive.
Keywords: Angiotensin II Receptor Antagonist; Antihypertensive; Biphenyltetrazole Derivatives.
////////
CCCCC(=O)N(CC1=CC=C(C=C1)C1=CC=CC=C1C1=NNN=N1)[C@@H](C(C)C)C(O)=O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....