
Home » Uncategorized (Page 171)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Phase 3-LY2439821 (ixekizumab) for psoriasis and psoriatic arthritis.
 |
|
|---|---|
http://www.ama-assn.org/resources/doc/usan/ixekizumab.pdf
USAN IXEKIZUMAB
PRONUNCIATION ix” e kiz’ ue mab
THERAPEUTIC CLAIM Treatment of autoimmune diseases
CHEMICAL NAMES
1. Immunoglobulin G4, anti-(human interleukin 17A) (human monoclonal LY2439821γ4-chain), disulfide with human monoclonal LY2439821 κ-chain, dimer
2. Immunoglobulin G4, anti-(human interleukin-17A (IL-17, cytotoxic
T-lymphocyte-associated antigen 8)); humanized mouse monoclonal LY2439821 des-Lys446-[Pro227]γ4 heavy chain {H10S>P,CH3107K>-} (133-219′)-disulfide with humanized mouse monoclonal LY2439821 κ light chain, dimer (225-225”:228-228”)-bisdisulfide
MOLECULAR FORMULA C6492H10012N1728O2028S46
MOLECULAR WEIGHT 146.2 kDa
SPONSOR Eli Lilly and Co.
CODE DESIGNATION LY2439821
CAS REGISTRY NUMBER 1143503-69-8
Ixekizumab is a humanized monoclonal antibody used in the treatment of autoimmune diseases.[1]
Ixekizumab was developed by Eli Lilly and Co.
Lilly’s Anti-IL-17 Monoclonal Antibody, Ixekizumab, Met Primary Endpoint in Phase II Study in Patients With Chronic Plaque Psoriasis – March 28, 2012
more info
Inflammation represents a key event of many diseases, such as psoriasis, inflammatory bowel diseases, rheumatoid arthritis, asthma, multiple sclerosis,
atherosclerosis, cystic fibrosis, and sepsis. Inflammatory cells, such as neutrophils, eosinophils, basophils, mast cells, macrophages, endothelial cells, and platelets, respond to inflammatory stimuli and foreign substances by producing bioactive mediators. These mediators act as autocrines and paracrines by interacting with many cell types to promote the inflammatory response. There are many mediators that can promote inflammation, such as cytokines and their receptors, adhesion molecules and their receptors, antigens involved in lymphocyte activation, and IgE and its receptors. [0004] Cytokines, for example, are soluble proteins that allow for communication between cells and the external environment. The term cytokines includes a wide range of proteins, such as lymphokines, monokines, interleukins, colony stimulating factors, interferons, tumor necrosis factors, and chemokines. Cytokines serve many functions, including controlling cell growth, migration, development, and differentiation, and mediating and regulating immunity, inflammation, and hematopoiesis. Even within a given function, cytokines can have diverse roles. For example, in the context of mediating and regulating inflammation, some cytokines inhibit the inflammatory response (anti-inflammatory cytokines), others promote the inflammatory response (pro-inflammatory cytokines). And certain cytokines fall into both categories, i.e., can inhibit or promote inflammation, depending on the situation. The targeting of proinflammatory cytokines to suppress their natural function, such as with antibodies, is a well-established strategy for treating various inflammatory diseases.
Many inflammatory diseases are treated by targeting proinflammatory cytokines with antibodies. Most (if not all) of the anti-proinflammatory cytokine antibodies currently on the market, and those currently in clinical trials, are of the IgG class. See, for example, Nature Reviews, vol. 10, pp. 301-316 (2010); Nature Medicine, vol. 18, pp. 736-749 (2012); Nature Biotechnology, vol. 30, pp. 475-477 (2012); Anti-Inflammatory & Anti- Allergy Agents in Medicinal Chemistry, vol. 8, pp. 51-71 (2009);
FlOOO.com/Reports/Biology/content/1/70, F 1000 Biology Reports, 1 :70 (2009); mAbs 4: 1, pp. 1-3 (2012); mAbs 3: 1, pp. 76-99 (2011); clinicaltrials.gov (generally), and
clinicaltrialsregister.eu/ (generally). These IgG antibodies are administered systemically and thus are often associated with unwanted side effects, which can include one or more of, for example, infusion reactions and immunogenicity, hypersensitivity reactions,
immunosuppression and infections, heart problems, liver problems, and others. Additionally the suppression of the target cytokines at non-diseased parts of the body can lead to unwanted effects.
In an attempt to reduce side effects associated with systemic treatment and to eliminate the inconvenience and expense of infusions, an article proposed an oral anti-TNF therapy that could be useful in treating Crohn’ s disease. Worledge et al. “Oral Administration of Avian Tumor Necrosis Factor Antibodies Effectively Treats Experimental Colitis in Rats.” Digestive Diseases and Sciences 45(12); 2298-2305 (December 2000). This article describes immunizing hens with recombinant human TNF and an adjuvant, fractionating polyclonal yolk antibody (IgY, which in chickens is the functional equivalent to IgG), and administering the unformulated polyclonal IgY (diluted in a carbonate buffer to minimize IgY acid hydrolysis in the stomach) to rats in an experimental rodent model of colitis. The rats were treated with 600mg/kg/day of the polyclonal IgY. The uses of animal antibodies and polyclonal antibodies, however, are undesirable.
In a similar attempt to avoid adverse events associated with systemic administration, another group, Avaxia Biologies Inc., describes a topical (e.g., oral or rectal) animal-dervied polyclonal anti-TNF composition that could be useful in treating
inflammation of the digestive tract, such as inflammatory bowel disease. WO2011047328. The application generally states that preferably the polyclonal antibody composition is prepared by immunizing an animal with a target antigen, and the preferably the polyclonal antibody composition is derived from milk or colostrum with bovine colostrums being preferred (e.g., p. 14). The application also generally states that the animal derived polyclonal antibodies could be specific for (among other targets) other inflammatory cytokines (e.g., pp. 6-7). This application describes working examples in which cows were immunized with murine TNF and the colostrum was collected post-parturition to generate bovine polyclonal anti-TNF antibodies (designated as AVX-470). The uses of animal-derived antibodies and polyclonal antibodies, however, are undesirable.
IgA molecular forms have been proposed as treatments for various diseases, most notably as treatments for pollen allergies, as treatments against pathogens, and as treatments for cancer.
For example, one article describes anti-AmbCtl (a ragweed pollen antigen) humanized monomelic IgA and dimeric IgA antibodies made in murine cells (NSO and Sp2/0 cells). The dimeric IgA contains a mouse J-chain. The article proposes that the antibodies may be applied to a mucosal surface or the lower airway to inhibit entry of allergenic molecules across the mucosal epithelium and therefore to prevent the development of allergic response. Sun et al. “Human IgA Monoclonal Antibodies Specific for a Major Ragweed Pollen Antigen.” Nature Biotechnology 13, 779-786 (1995).
Several other articles propose the use of IgA antibodies as a defense against pathogens.
Two articles proposed the use of an anti-streptococcal antigen I II secretory IgA-G hybrid antibody. Ma et al. “Generation and Assembly of Secretory Antibodies in Plants.” Science 268(5211), 716-719 (May 1995); Ma et al. “Characterization of a
Recombinant Plant Monoclonal Secretory Antibody and Preventive Immunotherapy in Humans.” Nature Medicine 4(5); 601-606 (May 1998). The hybrid antibody contains murine monoclonal kappa light chain, hybrid Ig A-G heavy chain, murine J- Chain, and rabbit secretory component. The antibody was made by successive sexual crossing between four transgenic N. tabacum plants and filial recombinants to form plant cells that expressed all four protein chains simultaneously. The parent antibody (the source of the antigen binding regions, is identified as the IgG antibody Guy’s 13. The group proposes that although slgA may provide an advantage over IgG in the mucosal environment, such is not always the case (1998 Ma at p. 604, right column).
A related article identifies the anti-streptococcal antigen I/II secretory IgA-G hybrid antibody, which was derived from Guy’s 13 IgA, as CaroRx. Wycoff. “Secretory IgA Antibodies from Plants.” Current Pharmaceutical Design 10(00); 1-9 (2004). Planet Biotechnology Inc. This related article states that the CaroRx antibody was designed to block adherence to teeth of the bacteria that causes cavities. Apparently, the CaroRx antibody was difficult to purify; the affinity of Protein A for the murine Ig domain was too low and protein G was necessary for sufficient affinity chromatography. Furthermore, the article states that several other chromatographic media had shown little potential as purification steps for the hybrid slgA-G from tobacco leaf extracts. The article also indicates that the authors were unable to control for human-like glycosylation in tobacco, but that such was not a problem because people are exposed to plant glycans every day in food without ill effect.
WO9949024, which lists Wycoff as an inventor, Planet Biotechnology Inc. as the applicant, describes the use of the variable regions of Guy’s 13 to make a secretory antibody from tobacco. The application contains only two examples – the first a working example and the second a prophetic example. Working Example 1 describes the transient production of an anti-S. mutans SA I/III (variable region from Guy’s 13) in tobacco. The tobacco plant was transformed using particle bombardment of tobacco leaf disks. Transgenic plants were then screened by Western blot “to identify individual transformants expressing assembled human slgA” (p. 25). Prophetic Example 2 states that in a transformation system for Lemna gibba (a monocot), bombardment of surface-sterilized leaf tissue with DNA- coated particles “is much the same as with” tobacco (a dicot). The prophetic example also stops at screening by immunoblot analysis for antibody chains and assembled slgA, and states that the inventors “expect to find fully assembled slgA.” [0014] Another article proposed the use of an anti-RSV glycoprotein F IgA antibodies (mlgA, dlgA, and slgA). Berdoz et al. “In vitro Comparison of the Antigen-Binding and Stability Properties of the Various Molecular Forms of IgA antibodies Assembled and Produced in CHO Cells.” Proc. Natl. Acad. Sci. USA 96; 3029-3034 (March 1999). The slgA antibody was made in CHO cells sequentially transfected with chimeric heavy and light chains, human J-Chain, and human secretory component, respectively. Single clones were generated to express the mlgA (clone 22), the dlgA (clone F), and the slgA (clone 6) (p. 3031).
Still other articles proposed, for example: (1) anti-HSV mlgA made in maize (Karnoup et al. Glycobiology 15(10); 965-981 (May 2005)) (which states that at that time there had been little success in the application of IgA class antibodies to therapeutic use because of the difficulty in producing the dimeric form in mammalian cells at economic levels); (2) anti-C. difficile toxin A chimeric mouse-human monomeric and dimeric IgA made in CHO cells (Stubbe et al. Journal of Immunology 164; 1952-1960 (2000)); (3) anti-N. meningitidis chimeric IgA antibodies were produced in BHK cells cotransfected with human J-Chain and/or human secretory component (Vidarsson et al., Journal of Immunology 166; 6250-6256 (2001)); (4) mti-Pseudomonas aeruginosa 06 lipopolysaccharide chimeric mouse/human mlgAl made in CHO cells (Preston et al. Infection and Immunity 66(9); 4137- 4142 (September 1998)); (5) anti-Plasmodium mlgA made in CHO cells (Pleass et al. Blood 102(13); 4424-4429 (December 2003)) (which states that unlike their parental mouse IgG antibodies, the mlgA antibodies failed to protect against parasitic challenge in vivo); and (5) ^^-Helicobacter pylori urease subunit A slgA and dlgA (Berdoz et al. Molecular
Immunology 41(10); 1013-1022 (August 2004)). [0016] For a review article discussing passive and active protection against pathogens at mucosal surfaces, see Corthesy. “Recombinant Immunoglobulin A: Powerful Tools for Fundamental and Applied Research.” Trends in Biotechnology 20(2); 65-71 (February 2002).
Still other articles propose the use of IgA antibodies as a treatment for cancer.
For example, one article describes a Phase la trial of a muring anti-transferrin receptor IgA antibody (Brooks et al. “Phase la Trial of Murine Immunoglobulin A
Antitransferrin Receptor Antibody 42/6.” Clinical Cancer Research 1(11); 1259-1265 (November 1995)). Another article describes a human anti-Ep-CAM mIgA made in BHK (baby hamster kidney) cells (Huls et al. “Antitumor Immune Effector Mechanisms Recruited by Phase Display-Derived Fully Human IgGl and IgAl Monoclonal Antibodies.” Cancer Research 59; 5778-5784 (November 1999)). Still another article describes an anti-HLA Class II chimeric mIgA antibody made in BHK cells (Dechant et al. “Chimeric IgA Antibodies Against HLA Class II Effectively Trigger Lymphoma Cell Killing.” Blood 100(13); 4574- 4580 (December 2002)). Yet other articles describe anti-EGFR mIgA or dlgA antibodies made in CHO, including Dechant et al. “Effector Mechanisms of Recombinant IgA
Antibodies Against Epidermal Growth Factor Receptor.” Journal of Immunology 179; 2936- 2943 (2007), Beyer et al. “Serum- Free Production and Purification of Chimeric IgA
Antibodies.” Journal of Immunology 346; 26-37 (2009) (stating that as of 2009, IgA antibodies have not been commercially explored for problems including lack of production and purification methods), and Lohse et al. “Recombinant Dimeric IgA Antibodies Against the Epidermal Growth Factor Receptor Mediate Effective Tumor Cell Killing.” Journal of Immunology 186; 3770-3778 (February 2011).
For a review article on anti-cancer IgA antibodies, see Dechant et al. “IgA antibodies for Cancer Therapy. ” Critical Reviews in Oncology/Hematology 39; 69-77 (2001); states that compared with infectious diseases, the role of IgA in cancer immunotherapy is even less investigated).
IL17 and IFN-garama inhibition for the treatment of autoimmune inflammation
The IL-17 family of cytokines has been associated with the pathogenesis of autoimmune diseases and is generally blamed for the pathogenic symptoms of autoimmune inflammation. Overexpression of IL-17 is a hallmark for autoimmune diseases like rheumatoid arthritis, systemic lupus erythematomatosus, inflammatory bowel disease, multiple sclerosis, and psoriasis (Yao Z et. al., J Immunol, 155(12), 1995, 5483-6. Chang S H, et.al, Cytokine, 46, 2009, 7-11; Hisakata Yamada et.al, Journal of Inflamm. Res., 3, 2010, 33-44)).
The IL-17 cytokine family comprises six members, out of which IL-17 A and IL-17F are the best characterized. IL-17A and IL-17F exist as homo- as well as as heterodimers (IL-17AA, IL-17AF, IL-17FF). IL-17A and IL-17F are clearly associated with inflammation (Gaffen S H, Cytokine, 43, 2008, 402-407; Torchinsky M B et al, Cell. Mol. Life Sci., 67, 2010, 1407- 1421).
The secretion of IL-17 is predominantly caused by a specific subtype of T helper cells termed TH-17 cells. IL-23, TGFp and IL-6 were shown to be important factors leading to conversion of nai‘ve CD4+ T-cells to THl 7 cells. It was also reported that TGF and IL-6 potently induce in synergy THl 7 differentiation. Important transcription factors for the secretion of IL-17 from TH17 cells are RORyt and STAT3 (IvanovJ et.al. Cell 126, 2006, 1121-1133). IL-17 induces pro-inflammatory cytokines (IL-6, TNF- and IL-lb) and Chemokines (CXCL1,GCP-2,CXCL8 or IL-8,CINC,MCP-1). It increases the production of nitric oxide prostaglandin E2 and matrix-metalloproteinases. As a consequence of these events neutrophil infiltration, tissue damage and chronic inflammation occurs (PECK A et.al, Clin Immunol., 132(3), 2009, 295-304).
Before the recognition of the importance of IL-17 in autoimmune inflammation, IFN-gamma derived from THl cells was believed to be an important cytokine that drives autoimmune disorders (Takayanagi H et. al. Nature, 408, 2000, 600-605. Huang W. et. al. Arthritis Res. Ther., 5, 2002, R49-R59) The secretion of IFN-gamma is a key feature of the THl effector cell lineage and the secretion is regulated by the transcription factors T-bet and STAT4 (Bluestone JA et. al. Nat Rev Immunol, 11, 2009, 811-6). Infiltration of activated T-cells and elevation of M-CSF, IL-10 and TNF support this notion (Yamanda H et.al Ann. Rheu. Dis., 67, 2008, 1299-1304; Kotake S et.al. Eur. J. Immunol, 35, 2005, 3353-3363).
Recently, a more complex situation was proposed, where hybrid TH17/TH1 cells induced by IL-23 and IL-6 in concert with IL-1 secrete IL-17 and IFN-gamma. These cells are under the control of the transcription factors RORyt and T-bet, confirming the notion, that these are true hybrids of THl and THl 7 cells. It was also demonstrated that these double producing cells are the pathogenic species in IBD and EAE (Buonocore S et.al. Nature, 464, 2010, 1371-5; Ghoreshi K. et. al. Nature, 467, 2010, 967-971).
Compounds which target and suppress both IL-17 and IFN-gamma are predisposed for the treatment of autoimmune disorders.
The effectiveness of blocking IL-17 signaling as therapeutic treatment in autoimmune diseases has already been proven in clinical trials with e.g. monoclonal antibodies against IL- 17A (AIN457, secukinumab; Ly2439821,ixekizumab; RG4934) and/or the IL-17 receptor IL- 17RA (AMG827, brodalumab).
Positive results have been reported for the treatment of rheumatoid arthritis, psoriasis and uveitis (Hueber W et al, Sci. Transl. Med., 2, 2010, 52ra72, DOI: 10.1126/scitranslmed.3001107; van den Berg W B e/ al, Nat. Rev. Rheumatol, 5, 2009, 549-553), ankylosing spondylitis and spondyloarthritides (Song I-H et al, Curr. Opin. Rheumatol., 23, 2011, 346-351).
Secukinumab is currently under investigation in clinical trials for psoriatic arthritis, Behcet disease, uveitits, inflammatory bowel disease, Crohn’s disease, multiple sclerosis (Kopf M et al., Nat. Rev. Drug Disc, 9, 2010, 703-718; Song I-H et al, Curr. Opin. Rheumatol., 23, 2011, 346-351).
Brodalumab, Ixekizumab and RG4934 are currently in clinical trials for the treatment of rheumatoid arthritis, psoriasis and/or psoriatic arthritis (Kopf M et al, Nat. Rev. Drug Disc, 9, 2010, 703-718; clinicaltrials.gov; Medicines in development for skin diseases, 201 1, published by PhRMA, www .phrma. com) .
With regard to blocking of IFN-gamma signaling as therapeutic treatment in autoimmune diseases, the IFN-gamma-specific monoclonal antibody AMG811 is currently under clinical investigations for the treatment of systemic lupus erythematosus (Kopf M et al., Nat. Rev. Drug Disc, 9, 2010, 703-718).
LASTACAFT, ALCAFTADINE.. Drug Patent Expiration, 21st Nov 2013

ALCAFTADINE
Alcaftadine is used to prevent eye irritation brought on by allergic conjunctivitis. It is a H1histamine receptor antagonist.
It was approved by the U.S. Food and Drug Administration in 2010 under the trade name Lastacaft.
LASTACAFT, ALLERGAN
Drug Patent Expiration and Exclusivity
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| ALCAFTADINE | SOLUTION/DROPS; OPHTHALMIC | 0.25% | RX | 022134 | 001 |
Patents
There are 1 patent(s) protecting ALLERGAN’s LASTACAFT.
The last patent expires on 2013-11-21.
| Patent | Expiration | |
|---|---|---|
| US5468743 | Imidazo[2,1-b]benzazepine derivatives, compositions and method of use
The present invention is concerned with novel imidazo[2, 1-b][3]benzazepines of formula ##STR1## the pharmaceutically acceptable addition salts and stereochemically isomeric forms thereof, wherein each of the dotted lines independently represents an optional bond; R.sup.1 represents hydrogen, halo, C.sub.1-4 alkyl or C.sub.1-4 alkyloxy; R.sup.2 represents hydrogen, halo, C.sub.1-4 alkyl or C.sub.1-4 alkyloxy; R.sup.3 represents hydrogen, C.sub.1-4 alkyl, ethenyl substituted with hydroxycarbonyl or C.sub.1-4 alkyloxycarbonyl, C.sub.1-4 alkyl substituted with hydroxycarbonyl or C.sub.1-4 alkyloxycarbonyl, hydroxyC.sub.1-4 alkyl, formyl or hydroxycarbonyl; R.sup.4 represents hydrogen, C.sub.1-4 alkyl, hydroxyC.sub.1-4 alkyl, phenyl or halo; R.sup.5 represents hydrogen, C.sub.1-4 alkyl or halo; L represents hydrogen; C.sub.1-6 alkyl; C.sub.1-6 alkyl substituted with one substituent selected from the group consisting of hydroxy, halo, C.sub.1-4 alkyloxy, hydroxycarbonyl, C.sub.1-4 alkyloxycarbonyl, C.sub.1-4 alkyloxycarbonyl-C.sub.1-4 alkyloxy, hydroxycarbonylC.sub.1-4 alkyloxy, C.sub.1-4 alkyloxycarbonylamino, C.sub.1-4 alkylaminocarbonyl, C.sub.1-4 alkylaminocarbonylamino, C.sub.1-4 alkylaminothiocarbonylamino, aryl, aryloxy and arylcarbonyl; C.sub.1-6 alkyl substituted with both hydroxy and aryloxy; C.sub.3-6 alkenyl; C.sub.3-6 alkenyl substituted with aryl; or, L represents a radical of formula –Alk–Y–Het.sup.1 (a-1),–Alk–NH–CO–Het.sup.2 (a-2)or –Alk–Het.sup.3 (a-3); provided that 6,11-dihydro-11-(4-piperidinylidene)-5H-imidazo[2,1-b][3]benzazepine is ecxluded, which are useful antiallergic compounds.Compositions comprising said compounds, methods of using and processes for preparing the same.
|
2013-11-21 |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the ALLERGAN.
Exclusivity ends on 2015-07-28.
| Date | Supplement No. | Action | Documents |
|---|---|---|---|
| 2010-07-28 | 000 | Approval |
Medicine Can be Sweet
Medicine Can be Sweet
 Glycosylated analogues of pramlintide were synthesized by a combination of solid-phase peptide synthesis and enzymatic glycosylation
Glycosylated analogues of pramlintide were synthesized by a combination of solid-phase peptide synthesis and enzymatic glycosylation
http://www.chemistryviews.org/details/ezine/5275441/Medicine_Can_be_Sweet.html
Medicine Can be Sweet



RABEPRAZOLE

Pariprazole sodium;Rabeprazole sodium;LY-307640;E-3810;Aciphex;Pariet
Rabeprazole /ˌræ.ˈbɛp.ræ.zɔːl/ is an antiulcer drug in the class of proton pump inhibitors. It was developed by Eisai Co. and is marketed by Janssen-Cilag as the sodium salt under the brand names AcipHex (/ˈæsɨfɛks/, referring to pH) in the US, Pariet in Europe, Brazil, Canada, Japan, Russia and Australia, Acigard, Cyra, Rabium, Esoon,Orporo, Parit, Rabemac, Rabiloz, Razo, Rabifast, Rablet and Rabsiv in India, and Zechin in Pakistan.
Rabeprazole, 2-[[[4-(3-Methoxypropoxy)-3-methyl-2-pyridinyl]methyl]sulfinyl]-1H-benzimidazole has the following structural formula

Rabeprazole belongs to a class of antisecretory compounds (substituted benzimidazole proton-pump inhibitors) that do not exhibit anticholinergic or histamine H2-receptor antagonist properties, but suppress gastric acid secretion by inhibiting the gastric H+, K+ATPase at the secretory surface of the gastric parietal cell. Because this enzyme is regarded as the acid (proton) pump within the parietal cell, rabeprazole has been characterized as a gastric proton-pump inhibitor. Rabeprazole blocks the final step of gastric acid secretion. So that it can effectively inhibit the secretion of an acid and is therefore effective in the therapy or prevention of human and animal peptic ulcer.
-
US 5045552 discloses the preparation of Rabeprazole sodium by known traditional procedures, such as dissolution of the product in a mixture of stoichiometric quantity of aqueous sodium hydroxide and ethanol, then removal of water azeotropically, thereafter drying the residue at low pressure and then crystallization of the residue with less polar solvent such as diethyl ether, tert-butyl methyl ether.
The U.S. Pat. No. 5,045,552 discloses the Rabeprazole and many other substituted benzimidazole-type compounds having anti-ulcer activity. This patent further discloses the process for preparation of Rabeprazole by oxidation of Rabeprazole sulfide using 85% m-chloroperbenzoic acid in a mixture of dichloromethane and diethyl ether followed by work up to get product as oil. The obtained oil is crystallized from a mixture of dichloromethane/ether. Optionally the oily crude is dissolved in aqueous solution of sodium hydroxide. The obtained solution is subjected to azeotropic distillation with ethanol to remove water and adding ether to get crystalline Rabeprazole base.

According to the prior art, Rabeprazole base is crystallized using dichloromethane/ether to obtain crystalline off white product. The HPLC purity is less than or equal to 99% and the isolation procedure involves azeotropic distillation of water, during which the product is exposed to high temperature and leads to certain impurities. Repeated crystallization is needed to remove impurities to get desired quality. Using large volumes of chlorinated solvents in the plant leads to environmental hazardous.
Japanese patent application JP2001039975 teaches that the product obtained by example 33 of U.S. Pat. No. 5,045,552 with a melting range of 140-141° C. corresponds to amorphous rabeprazole sodium
The U.S. Pat. No. 6,919,459 patent also discloses the process for the preparation of Rabeprazole by oxidation of Rabeprazole sulfide using m-Chloroperbenzoic acid (m-CPBA) in a suitable solvent. The reaction mass is subjected to repeated washings at different pH levels and isolate the product from aqueous layer.

Rabeprazole is not stable at acidic conditions and decomposes to form unknown impurities. To remove these impurities repeated crystallizations are required to get desire quality of the final product.
The WO2006/117802 PCT application discloses the process for the preparation of Rabeprazole sodium by oxidation of Rabeprazole sulfide with sodium hypo halite solution in water or a mixture of water and water miscible solvent medium using alkali metal hydroxide and catalyst. The reaction mass is saturated by inorganic saturating agents and the Rabeprazole sodium salt is extracted with water immiscible organic solvent. Organic solvent is distilled and the residue is dissolved in second organic solvent to get clear solution, which is precipitated by adding antisolvent.
The WO2006/120701 PCT application discloses process for manufacture of amorphous Rabeprazole sodium by the reaction of Rabeprazole base with aqueous sodium hydroxide. Ethanol is added to the obtained solution. Solvents are distilled from the solution to get thick mass. Organic solvent is added to the obtained residue to get clear solution, to which antisolvent is added to get amorphous Rabeprazole sodium.
The prior art methods cited above have many disadvantages, these methods involve more number of organic solvents and lack successive extractions and washings of the layers during work up procedure. It leads to many impurities that ultimately affect on purity and yield loss of final product.
The U.S. Pat. No. 6,180,652 and WO 2003101452 PCT application discloses the process for the preparation of amorphous rabeprazole sodium, which is obtained by lyophilization of an aqueous solution of rabeprazole sodium acetone complex and an aqueous NaOH solution of Rabeprazole respectively.

Lyophilization technique is not suitable for production at industrial scale and it needs more time cycle and involves the cost.
We observed that rabeprazole is rapidly degraded in chlorinated solvent like dichloromethane to form unknown impurities, due to impurities while distillation gummy material is formed. It leads to yellowish color in final product, finally it leads to yield loss in final product.
According to prior art methods,
-
- (a) Dichloromethane/ether is used for final crystallization gives off white product with HPLC purity less than or equal to 99% and
- (b) Rabeprazole sodium is isolated by using azeotropic distillation. It needs high temperature to remove water and the reaction mass is exposed to high temperature to form unknown impurities, to remove these impurities repeated crystallizations are required to get desire quality of the final product
US 6,313,303 discloses the preparation of sulfoxides by oxidizing thio ether with a peroxoborate salt in the presence of an acid anhydride or a metal catalyst; and the preparation of sulfoxides by oxidizing thio ether with an N- halosuccinimide, l,3-dihalo-5,5-dimethyl-hydantoin or dichloroisocyanuric acid salt in the presence of a base.
IN 192030 discloses the purification process of Rabeprazole, in which sulfone enriched Rabeprazole is treated with an amino alcohol e.g. ethanolamine in the presence of an organic solvent, further the reaction mixture washed with water to remove the sulfone impurities. US 7,439,367 (IN218648, 058/MUM/2003, 193/MUM/2003) discloses the preparation of Rabeprazole by oxidizing its corresponding sulfide compound, where aqueous hypohalite solution is used as an oxidizing agent. The said oxidation is carried out at a controlled temperature and pH. During said oxidation the pH of the reaction mixture is maintained in the range of 9 to 12. This process utilizes catalyst such as pyridine, di-isopropyl ethyl amine and N,N-dimethyl amino pyridine.
US 7,060,837 discloses the purification of lansoprazole using ammonia, ammonium hydroxide, diethylamine, triethylamine and methylamine in the presence of solvent. The said patent utilizes acid for the isolation of lanzoprazole in pure form.
US 2008/0161579 (IN190/MUM/2005) discloses a process for the preparation of Rabeprazole sodium comprising oxidation of Rabeprazole sulfide with sodium hypohalite in water or a mixture of water and water miscible solvent using alkali metal hydroxide and catalyst. It also discloses a process for the preparation of Rabeprazole sulfide.
WO 2008/045777 (1856/CHE/2006) discloses the preparation of
Rabeprazole by oxidizing the corresponding sulfide compound using about 0.8 to 1.25 equivalents of an oxidizing agent in the presence of less than or about 2.25 equivalents of a base where aqueous sodium hypohalite used as an oxidizing agent.
WO 2006/024890 discloses a process for the preparation of Rabeprazole in which the Rabeprazole obtained was treated with the triethylamine in hexane. The use of n-hexane in the final stage is not suitable for manufacturing point of view as it is difficult to remove residual hexane solvent. There are several disadvantages associated with such known processes; all the methods reported in these prior arts leads to the formation of many impurities which ultimately affects the purity of the final product.
US 5,045,552 patent discloses the preparation of Rabeprazole by oxidizing the Rabeprazole sulfide using m-chloroperbenzoic acid as shown in scheme-I. The crude Rabeprazole was dissolved in sodium hydroxide and the resulting solution was azeotropically distilled together with ethanol thrice to remove the water. Finally ether was added to get the crystals of Rabeprazole sodium
WO 03/101452 discloses a method for the preparation of Rabeprazole sodium comprising dissolving Rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization.

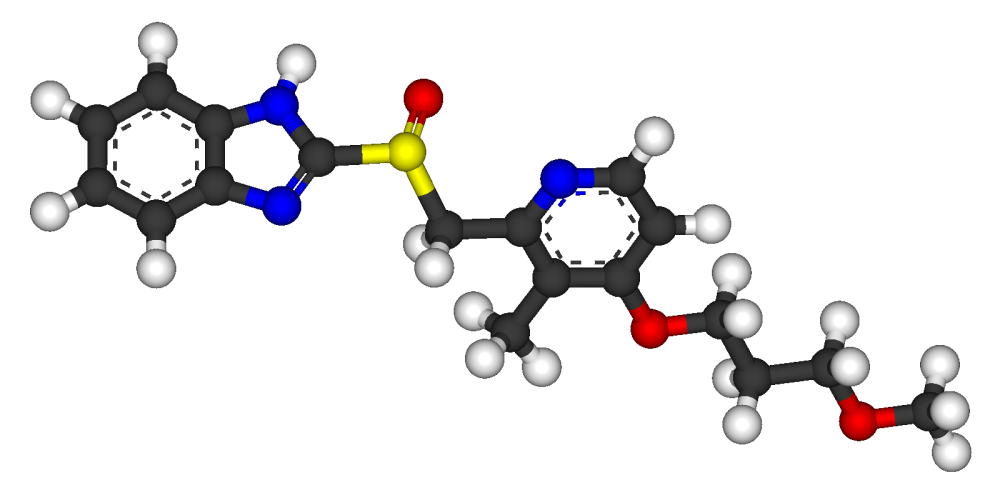


Souda, S.; Ueda, N.; Miyazawa, S.; Tagami, K.; Nomoto, S.; Okita, M.; Shimomura, N.; Kaneko, T.; Fujimoto, M.; Murakami, M.; Oketani, K.; Fujisaki, H.; Shibata, H.; Wakabayashi, T. (Eisai Co., Ltd.); Pyridine derivs., pharmaceutical compsns. comprising the same, the use of the same for the manufacture of medicaments having therapeutic or preventative value, and a process for preparing the same. AU 8781138; EP 0268956; EP 0475456; EP 0654471; EP 0786461; JP 1989006270; JP 1993247035; JP 1995291967; US 5045552; US 5998445 .
Castaner, J.; Prous, J.; E-3810. Drugs Fut 1991, 16, 1, 19.
Sohda, S.; Tagami, K.; Chiku, S.; Synthesis of 14C-labelled sodium pariprazole (E3810). J Label Compd Radiopharm 1993, 33, 9, 849.

Rabeprazole as “CYRA” (Systopic Labs Pvt Ltd), “Elpizole” (Orchid Chemicals & Pharmaceuticals), Elpizole-20 (Orchid Chemicals & Pharmaceuticals), Rablet (Lupin), Acigard (3D), AcipHex, Rabeloc, Pariet, Rabider (Duta Formulations) Rabsiv 20 (Saharsh Biologicals) is supplied in:
- Tablet, enteric-coated; 10 mg
- Tablet, enteric-coated; 20 mg
- Pali-Schöll I, Jensen-Jarolim E (April 2011). “Anti-acid medication as a risk factor for food allergy”. Allergy 66 (4): 469–77. doi:10.1111/j.1398-9995.2010.02511.x. PMID 21121928.
HPLC METHOD
Rabeprazole with more impurities, particularly at 2.12 RRT (393 mass), 3.51 RRT (491 mass), 4.47 RRT (457 mass), 4.85 RRT (684 mass) and 4.54 RRT (893 mass). The mass (molecular or formula weight) number of the impurities were identified using LCMS. Particularly, the obtained product contains unknown impurities of higher molecular weight in the range of 0.1-1.0 % at relative retention time (RRT) of 2.12, 3.51, 4.47, 4.85, and 4.54 RRT as measured by high performance liquid chromatography (HPLC) method provided below.
The purity of the product obtained is determined by high performance liquid chromatography method under the conditions mentioned below.
Column: Prontosil Kromabond 100-5-C18 (250 x 4.6 mm), 5μ,
Mobile phase A: 1.36g KH2PO4 to 1 litre water, 0.5ml OfEt3N, Mobile phase B: Methanol: ACN (95:5),
Diluent: Mobile phase A and ACN (70:30),
Flow Rate: 1.0 mL/min,
Detection: UV at 280 nm,
Injection Volume: 20 μL, Run Time: 60 min.
Column oven temperature: 3O0C. Surprisingly the applicant identified a method in which, crude Rabeprazole was treated with diethylamine and optionally addition of TBAB (tetrabutylammmonium bromide) as catalyst, where the impurity level reduced. Though the reported amines like triethyl amine, ethanolamine, and ammonia are effectively used to minimize sulfone impurity, those are failed or unsatisfactory to remove the impurities at 2.12 RRT, 3.51 RRT, 4.47 RRT, 4.85 RRT and 4.54 RRT.
SPECTRAL DATA
EP 1869015 B1 FOR RABEPRAZOLE SODIUM
IR Spectra (KBr, cm-1): 3382, 2927, 1583, 1462, 1384, 1298, 1269, 1190, 1157, 1093, 1018, 745.
H NMR Spectra [200 M Hz, CD3OD] δ (ppm): 8.23 – 8.25 (1H, d, ArH); 7.57 – 7.62 (2H, m, ArH); 7.0 – 7.09 (2H, m, ArH); 6.87 – 6.90 (1H, d, ArH); 4.57 – 4.63 (2H, d, O=S-CH2-Ar); 4.0 – 4.1 (2H, t, -O-CH2-CH2-); 3.49 – 3.55 (2H, t, -CH2-O-CH3); 3.31 (3H, s, -OCH3); 2.1 (3H, s, Ar-CH3); 1.96 – 2.0 (2H, t, -CH2-CH2-CH2-).
MP
As per the process described and exemplified in the U. S. Patent No.
5,045,552, rabeprazole sodium is prepared by oxidizing 2-[[4-(3- methoxyporpoxy)-3-methylpyridine-2-yl]rnethylthio]-1 H-benzimidazole with m- chloroperbenzoic acid to afford the rabeprazole base which is further converted to its sodium salt by using 0.1 N aqueous solution of sodium hydroxide, followed by addition of ethanol. The water is removed by azeotropic distillation and the product is precipitated by using ether as solvent such as diethyl ether, tert-butyl methyl ether. The melting point of the disclosed rabeprazole sodium salt is 140- 1410C. The isolation process described in the U. S. Patent No. 5,045,552 has numerous disadvantages such as large volume of solvents is required for azeotropic removal of water during which the product is exposed to high temperature and leads to certain impurities. Based on these drawbacks the isolation process finds to be unsuitable for preparation of amorphous rabeprazole sodium at commercial scale operations.
Japanese patent application JP 2001039975 indicates that the product obtained by example 33 of the U. S. Patent No. 5,045,552 with a melting point of
140-1410C corresponds to amorphous rabeprazole sodium. In this application, the X-ray powder diffraction pattern of the amorphous rabeprazole sodium is shown.
The PCT patent publication No. WO 03/101452 discloses a method for the preparation of rabeprazole sodium comprising dissolving rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization. U.S. Patent No. 6,180,652 B1 (the ‘652 patent) describes acetone complex of rabeprazole sodium, process for its production and characterizes it by powder X-ray diffraction, infra-red spectroscopy and 1H-NMR spectroscopy. The ‘652 patent further reports a process for preparation of amorphous rabeprazole sodium by lyophilizing (freeze-drying) an aqueous solution of rabeprazole sodium acetone complex.
However, lyophilization is a technique, which is not suitable for production at industrial scale because this process presents serious limitations on cost, time, equipment capability and environmental protection.
According to PCT patent publication No. WO 2004/085424A1 , amorphous rabeprazole sodium is obtained by heating the rabeprazole sodium acetone complex at elevated temperature, preferably between 100 and 1100C. It is well known that exposing rabeprazole-type compounds to high temperatures increases the risk of decomposition to form impurities and as such, heat treatment of rabeprazole sodium acetone complex into amorphous rabeprazole sodium is not adequate for the production of a rabeprazole which is suitable for pharmaceutical use.
PCT patent publication No. WO 2007/023393 A2 reports a process for preparation of amorphous rabeprazole sodium, the said process comprises: i) contacting rabeprazole sodium acetone complex with a first solvent system which includes a hydrocarbon solvent or an ether solvent or an alcohol solvent or mixtures thereof; ii) filtering the solid from the solvent system used in step i) or distilling the solvent system used in step i) under reduced or atmospheric pressure, to thereby obtain a residue; iii) contacting the wet solid or the residue of step ii) with a second solvent system which includes a hydrocarbon solvent or an ether solvent; and iv) filtering to obtain a wet solid from the solvent system used in step iii) to obtain a wet solid.
The methods for preparation of amorphous rabeprazole sodium as described in the patents U.S. Patent No. 6,180,652 B1 , PCT patent publication No. WO 2004/085424A1 and PCT patent publication No. WO 2007/023393 A2 involves lengthy process i.e., proceeds via rabeprazole sodium acetone complex intermediate and also the yields obtained in these processes are very low.
U.S. Patent Application No. US2004/0180935A1 teaches a process for production of amorphous rabeprazole sodium by dissolving rabeprazole acid in a mixture of sodium hydroxide and methanol at 25-350C, removing the solvent by evaporation and precipitating the product by adding petroleum ether.
PCT patent publication No. WO 2006/120701 A1 teaches a process for manufacture of amorphous rabeprazole sodium with mean particle diameter between 10 to 55 μm, the said process comprises, addition of rabeprazole to aqueous sodium hydroxide; addition of ethyl alcohol to the solution; distillation of solvents from the solution thus obtained till thick mass is obtained; addition of an organic solvent selected from ethyl acetate, dichloromethane, chloroform, butyl acetate, ethanol, isopropyl alcohol, methanol, tetrahydrofuran, to the residue to obtain a clear solution; addition of this clear solution to an anti-solvent includes diisopropyl ether, diethyl ether, methyl tert-butyl ether, under agitation and isolation of the product.
Since a solvent may play an important role in increasing the yield rate or in determination of physical properties of drug substance such as crystal form, purity, solubility, etc., even if such a solvent is known to be toxic, there may be many cases that the use thereof in the preparation of drug substance cannot be avoided in terms of risk benefits. In such cases, this guideline (ICH guidelines Q3C(R3)) decrees that a concentration of a residual solvent in drug substance should be not more than a specified value, which is toxicologically acceptable. The methods for preparation of amorphous rabeprazole sodium as described in the patents, U.S. Patent Application No. US2004/0180935A1 and PCT patent publication No. WO 2006/120701 A1 suffers with residual solvent problem and thereby commercially not viable. These methods utilize the solvents like diisopropyl ether and petroleum ether as precipitating solvents. These solvents are difficult to remove completely by practical manufacturing techniques. According to the ICH guidelines Q3C(R3), there is no adequate toxicological data for the solvents like diisopropyl ether and petroleum ether on which to base a PDE was found. However, a need still remains for an improved and commercially viable process of preparing pure amorphous rabeprazole sodium that would solve the aforesaid problems associated with processes described in the prior art, which will be suitable for largr-scale preparation, in terms of simplicity, chemical yield and purity of the product, and which would carry out with comparatively smaller volume of solvent
GSK and Genmab seek FDA approval for ofatumumab combination therapy for CLL first-line treatment
GlaxoSmithKline (GSK) and Genmab have submitted a supplemental Biologics License Application (sBLA) to the US Food and Drug Administration (FDA) seeking the use of Arzerra (ofatumumab) in combination with an alkylator-based therapy in patients with chronic lymphocytic leukaemia (CLL) who have not received prior treatment.
READ AT
GSK and Genmab seek FDA approval for ofatumumab combination therapy for CLL first-line treatment

{kind=link}