
Home » Uncategorized (Page 170)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Atomoxetine

Atomoxetine
Atomoxetine hydrochloride (CAS NO.: 82248-59-7)
(R)-(-)-N-Methyl-gamma-(2-methylphenoxy)benzenepropanamine hydrochloride
| Patent No | PatentExpiry Date | |
|---|---|---|
| 5658590 | Nov 26, 2016 | |
| 5658590*PED | May 26, 2017 |
nda 021411 app 2002-11-26
TREATMENT OF ATTENTION-DEFICIT HYPERACTIVITY DISORDER
label
|
Country
|
Patent Number
|
Approved
|
Expires (estimated)
|
|---|---|---|---|
| United States | 5658590 | 1997-05-26 | 2017-05-26 |
The HCl salt of atomoxetine , with the (R)-configuration], which is marketed under the trade name Strattera, is used for treating attention-deficit hyperactivity disorder (ADHD
Atomoxetine is a drug approved for the treatment of attention-deficit hyperactivity disorder(ADHD).[1] It is a selective norepinephrine reuptake inhibitor (NRI),[1] not to be confused with serotonin norepinephrine reuptake inhibitors (SNRIs) or selective serotonin reuptake inhibitors (SSRIs), both of which are currently the most prescribed form of antidepressants.
his compound is manufactured, marketed and sold in theUnited States under the brand name Strattera by Eli Lilly and Company as a hydrochloride salt (atomoxetine HCl), the original patent filing company, and current U.S. patent owner. Generics of atomoxetine are sold in all other countries; they are manufactured by Torrent Pharmaceuticals using the label Tomoxetin, Ranbaxy Laboratories (through its Division: Solus) using the label Attentin, Sun Pharmaceuticals(through its Division: Milmet Pharmaceuticals), and Intas Biopharmaceuticals There is currently no generic manufactured directly in the United States since it is under patent until 2017.[2]
On August 12, 2010, Lilly lost a lawsuit that challenged Lilly’s patent on Strattera, increasing the likelihood of an earlier entry of a generic into the US market.[3] On September 1, 2010, Sun Pharmaceuticals announced it would begin manufacturing a generic in the United States.[4] In a July 29, 2011 conference call, however, Sun Pharmaceutical’s Chairman stated “Lilly won that litigation on appeal so I think [generic Strattera]’s deferred.”[5]
Atomoxetine is designated chemically as (−)-N-methyl-3-phenyl-3-(o-tolyloxy)-propylamine hydrochloride, and has a molecular mass of 291.82.[1] It has a solubility of 27.8 mg/mL in water.[1] Atomoxetine is a white solid that exists as a granular powder inside the capsule, along with pre-gelatinized starch and dimethicone.[1] The capsule shells contain gelatin, sodium lauryl sulfate, FD&C Blue No. 2, synthetic yellow iron oxide, titanium dioxide, red iron oxide, edible black ink, and trace amounts of other inactive ingredients.[1]
The compound (-)-N-methyl-3-(2-methylphenoxy)-3-phenylpropylamine, or (-)-Λ/-methyl-3-phenyl-3-(o-tolyloxy)-propylamine hydrochloride, is usually known by its adopted name “atomoxetine hydrochloride.” It is represented as shown in Formula 1 and is a selective norepinephrine reuptake inhibitor. A commercialatomoxetine hydrochloride product is sold as STRATTERA™ in the form of capsules containing 10, 18, 25, 40, 60, 80, or 100 mg of atomoxetine, for treating attention-deficit/hyperactivity disorder.
- “STRATTERA® (atomoxetine hydrochloride) CAPSULES for Oral Use. Full Prescribing Information.” Eli Lilly and Company, 2002, 2013. Revised August 5, 2013. [1]
- “Patent and Exclusivity Search Results”. Electronic Orange Book. US Food and Drug Administration. Retrieved 26 April 2009.
- “Drugmaker Eli Lilly loses patent case over ADHD drug, lowers revenue outlook”. Chicago Tribune.
- “Sun Pharma receives USFDA approval for generic Strattera capsules”. International Business Times.
- “Sun Pharma Q1 2011-12 Earnings Call Transcript 10.00 am, July 29, 2011”.
- Strattera by Eli Lilly and Company
- RxList.com – Strattera
- Detailed Strattera Consumer Information: Uses, Precautions, Side Effects
- All disclosed Lilly trials
- MSDS for Atomoxetine HCl
- Strattera Related Published Studies
Synthesis
Also known as: Atomoxetine hydrochloride, Strattera, Atomoxetine HCL, (R)-Tomoxetine hydrochloride, TOMOXETINE HYDROCHLORIDE, Tomoxetine, 82248-59-7
First step appears to be a Mannich reaction between acetophenone, paraformaldehyde and dimethylamine, although not formally written in the scheme.

Foster, B. J.; Lavagnino, E. R.; European Patent, 1982, EP 0052492.

Eli Lilly’s Strattera capsules.


Atomoxetine, designated chemically as (-)-N-methyl-3-phenyl-3-(0-tolyloxy)- propylamine hydrochloride, is structurally represented by the compound of Formula-I and is indicated for the potential treatment of attention-deficit hyperactivity disorder (ADHD). This compound is manufactured, marketed and sold in the United States under the brand name Strattera.

Formula-I Atomoxetine was first disclosed in US Patent No 4314081. The said patent disclosed Atomoxetine, its pharmaceutically acceptable salts and composition containing them.
4-hydroxy Atomoxetine, chemically known as R-(-)-N-methyl-3-(2-methyl-4- hydroxyphenyl)oxy)-3 -phenyl- 1-aminopropane, structurally represented by Formula-II, is a metabolite of Atomoxetine.

Fomnula-ll
4-hydroxy Atomoxetine hydrochloride was first disclosed in US Patent No 7384983, wherein 4-hydroxy Atomoxetine free base was dissolved in ethylacetate, treated the solution with 0.1N HC1; followed by lyophilization yielded a yellow solid which was dissolved in methanol and passed through a short column of activated carbon; the solvent was removed and finally the hydrochloride salt was recrystallized from water to afford 4-hydroxy Atomoxetine hydrochloride. However, this patent does not mention about the nature of the polymorph obtained through this process.
The asymmetric epoxidation of (E)-3-phenyl-2-propen-1-ol (I) by means of titanium tetraisopropoxide, (+)-diethyl tartrate (+)-(DET) and tBu-OOH in dichloromethane gives the chiral epoxide (II), which is opened by means of bis(2-methoxyethoxy)aluminum hydride (Red-Al) in DME to yield the chiral diol (III). The regioselective reaction of (III) with Ms-Cl and TEA in ethyl ether affords the primary mesylate (IV), which is condensed with 2-methylphenol (V) by means of PPh3 and DEAD in ethyl ether to provide the adduct (VI). Finally this compound is treated with methylamine in hot aq. THF to give rise to the target (R)-tomoxetine.

The reduction of omega-chloropropiophenone (I) with NaBH4 in ethanol gives 3-chloro-1-phenyl-1-propanol (II), which is treated with butyric anhydride and pyridine in dichloromethane to yield the corresponding racemic ester (III). The optical resolution of (III) with immobilized lipase B from Candida antarctica (CALB) affords a mixture of unreacted (S)-ester and (R)-alcohol (IV) that are separated by column chromatography. Condensation of th (R)-alcohol (IV) with 2-methylphenol (V) by means of PPh3 and diethyl azodicarboxylate (DEAD) in THF gives the corresponding ether (VI), which is finally treated with methylamine in refluxing ethanol.
more info
U.S. Patent No. 4,314,081 describes 3-Aryloxy-3-phenyl polyamines, which possess central nervous system activity. Atomoxetine is a member of the above class of compounds, and is a useful drug for the treatment of depression.Atomoxetine was claimed in U. S. Patent No. 4,314,081 and the patent describes a process for the preparation of atomoxetine and related compounds in two different ways as depicted below as Scheme A and Scheme B, respectively.
Scheme A
Atomoxetinc
Scheme B

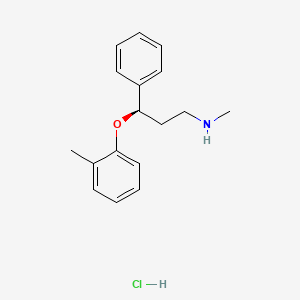
The process illustrated in Scheme A involves the preparation of the atomoxetineusing 3-phenyl chloropropyl amine (Formula 5) as a starting material. The process involves bromination of said starting compound (Formula 5) by using N-bromosuccinimide. Further the bromo derivative is condensed with o-cresol to result in a compound of Formula 7, which is then subjected to amination using methylamine. Though the process looks very simple, it involves the following disadvantages: i) N-bromosuccinimide being a corrosive and sensitive chemical, its usage demands special care; ii) the workup of the compound formula 7 involves high vacuum (0.03 torr) distillation at 135-1450C, which is a tedious and cumbersome process to carry out at the plant level; and iii) the reaction conditions involved in some of the steps are harsh, for example the amination reaction is conducted at 14O0C. at pressures of 10 kg/cm2 for 12 hours in an autoclave.
All the above points make the process not viable for practicing on a commercial scale. Further, as described in U.S. Patent 4,314,081 , the free base compounds exist as high boiling oils, but form white crystalline salts.
On the other hand, Scheme B describes the preparation of atomoxetine using β-dimethylaminopropiophenone produced by a Mannich reaction; which is reduced to the hydroxy derivative having Formula 9 using diborane; further the hydroxy compound (Formula 9) is converted to the corresponding chloro derivative of Formula 10 using dry HCI gas and thionyl chloride and is followed by condensation with o-cresol.
The said reaction is carried out in methanol at reflux for a duration of five days to achieve the compound of formula 11 and is followed by demethylation using cyanogen bromide to end up with atomoxetine. As can be clearly understood the process is associated with the following problems: i) the use of costly reagents such as diborane makes the process uneconomical; ii) the passage of dry HCI gas followed by thionyl chloride addition is ^ very cumbersome and is not advisable in the plant; iii) this is a time-consuming process, involving a reaction which requires five days for its completion; and iv) use of cyanogen bromide, which is highly toxic, is not desirable.
All of the above-quoted drawbacks make the process unfriendly to practice in a production plant as well as to the environment.
Further, M. Srebnik et al., Journal of Organic Chemistry, Vol. 53, pages2916-2920 (1988); E. Corey et al., Tetrahedron Letters, Vol. 30, pages 5207-5210 (1989);
U.S. Patent No. 4,868,344; Y. Gao et al., Journal of Organic Chemistry, Vol. 53, pages 4081-4084 (1988); J. Deeter et al.,
Tetrahedron Letters, Vol. 31, pages 7101-7104 (1990);
and U.S. Patent No. 4,950,791 disclose stereospecific methods for the preparation of 3-aryloxy-3-phenylpropylamines; the enantiomers of 3-hydroxy-3-phenylpropylamines are prepared by the stereospecific reduction of the corresponding ketones. The thus obtained (S)-3-hydroxy-3-phenyl propylamines are subjected to condensation with aryl alcohols using the Mitsunobo reaction. As can be seen in Scheme C, the reaction involves two critical steps.
Scheme C
Dusopinocampheny) chloroborane


OH
,CH,
DEAD/ tn phenyl phosphine

The first critical step is an asymmetric reduction of the ketone to its corresponding alcohol. The second critical step involves the condensation of the obtained enantiomeric alcohol with the corresponding aryl alcohol. The process suffers from the following disadvantages:
1) the reagent used for the asymmetric reduction of the ketone is highly expensive;
2) the reagent diethyl azodicarboxylate (“DEAD”) is expensive;
3) the DEAD reagent is known to be highly carcinogenic, thus creating problems in handling; and
4) the reaction involves the use of triphenylphosphine and DEAD and the resulting byproducts formed in the reaction, phoshineoxide and a hydrazine derivative, are very difficult to remove.
Therefore, commercial applicability of the said process is limited owing to the above noted disadvantages.
International Patent Publication No. WO 00/58262 relates to a stereo- specific process for the preparation of atomoxetineusing nucleophilic aromatic displacement of an aromatic ring having a functional group, which can be converted to a methyl group. As can be seen, the process is very lengthy and involves many steps and is thus not commercially desirable.
U.S. Patent No. 5,847,214 describes the nucleophilic aromatic displacement reaction of 3-hydroxy-3-arylpropylamines with activated aryl halides, for example the reaction of N-methyl-3-phenyl-3-hydroxypropylamine with 4- triflouromethyl-1-cholro benzene has been reported; the success of this reaction is mainly due to electron withdrawing group on benzene ring of the aryl halides.
U.S. Patent No. 6,541 ,668 describes a process for the preparation of atomoxetine and its pharmaceutically acceptable addition salts which comprises reacting an alkoxide of N-methyl-3-phenyl-3-hydroxy propyl amine or an N protected derivative thereof, with 2-flouro toluene in the presence of 1 ,3-Dimethyl – 2-imidazolidinone (“DMI”) or N-Methyl-3-pyrrolidinone (“NMP”) as the solvent. The process disclosed in the said patent can be shown as Scheme D. Further, the process disclosed in the said patent restricts itself to the solvents DMI and NMP.
Scheme D

Nevertheless, a new crystalline form of N-methyl-3-phenyl-3-(o- tolyloxy)propylamine oxalate and an isolation technique of (±)-atmoxetine free base in a solid form, an intermediate useful in the synthesis of atomoxetine hydrochloride, is desirable.

http://www.sciencedirect.com/science/article/pii/S0040403906025068
There have been several methods reported for preparing (R)-(−)-N-methyl-3-(2-methylphenoxy)-3-phenylpropylamine (Atomoxetine®). For example, U.S. Pat. No. 4,868,344 discloses a process as shown in the following scheme:

In this example, 3-chloropropiophenone is used as the starting material to be asymmetrically reduced with (−)-diisopinocamphenylchloroborane ((−)-IPc2BC1) to give the corresponding chiral alcohol. The resulting chiral alcohol is then reacted with o-cresol via Mitsunobu reaction to form the chiral ether compound. Subsequently, amination of the chiral ether compound with methylamine provided atomoxetine. In this process, the materials such as chiral-borane ((−)-IPc2BC1) and diethyl azodicarboxylate (DEAD) are expensive, and result in high manufacturing cost.
Further, WO 2006/009884 discloses another method for preparing atomoxetine, including the step of reacting N-methyl-3-phenyl-3-hydroxypropylamine with 2-fluorotoluene which is followed by resolution of the resulting product to provide optically pure atomoxetine as shown in the following scheme:

This process involving a chiral resolution step is inefficient due to low product yield, complicated and long time process that renders this process economically less competitive.
………………………………………………………………………………………………

see below
B.-F. Chen and co-inventors describe a synthesis of 5 that avoids costly reagents. It includes the preparation of the chiral amino alcohol 3 as a key intermediate. The route for preparing 5 starts with a Mannich reaction between benzophenone, N,O-dimethylhydroxylamine, and paraformaldehyde to give compound 1, isolated in 87.6% yield.
Ketone 1 is asymmetrically reduced to form alcohol 2 by using the chiral ruthenium catalyst RuCl2-[(S)-DMSEGPHOS)][(S)-DAIPEN]. The hydrogen pressure is described as “predetermined”, but no value is given. Product 2 is recovered as an oily product in 98.7% yield with 98.8% purity and 99% ee. It appears that the catalyst is not removed before the next step in which the oil is hydrogenated over a Raney nickel catalyst to form amino alcohol 3.
Intermediate 3 is also isolated as an oil in 96.4% yield, 96.5% % purity, and 99% ee. After recrystallization from toluene–heptane, the solid product is recovered with 100% ee. In the last step, 3 is treated with fluorotoluene 4 in the presence of t-BuOK to form atomoxetine, isolated as an oil in 91% yield with 97% ee. The purification of 5 and its conversion to the HCl salt are not described.
The inventors provide basic 1H-NMR data for all compounds except 5. The example describing the preparation of 1 lists one of the reactants as 2-acetylthiophene, which is clearly incorrect; and another reagent is called “32% hydrochloride”. These errors should have been spotted by anyone with a fundamental knowledge of chemistry who was involved in writing the patent—perhaps none were. (Sci Pharmtech [Taiwan]. US Patent 8,299,305, Oct. 30, 2012; Keith Turner)
View the full-text patent here.
| Patent Number: | US 8299305 |
| Title: | Process for preparation of atomoxetine |
| Inventor(s): | Chen, Bo-Fong; Li, Yan-Wei; Yeh, Jinun-Ban; Wong, Wei-Chyun |
| Patent Assignee(s): | SCI Pharmtech, Inc., Taiwan |
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab

NIVOLUMAB
Anti-PD-1;BMS-936558; ONO-4538
PRONUNCIATION nye vol’ ue mab
THERAPEUTIC CLAIM Treatment of cancer
CHEMICAL DESCRIPTION
A fully human IgG4 antibody blocking the programmed cell death-1 receptor (Medarex/Ono Pharmaceuticals/Bristol-Myers Squibb)
MOLECULAR FORMULA C6362H9862N1712O1995S42
MOLECULAR WEIGHT 143.6 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION MDX-1106, BMS-936558
CAS REGISTRY NUMBER 946414-94-4
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab
Nivolumab (nye vol’ ue mab) is a fully human IgG4 monoclonal antibody designed for the treatment of cancer. Nivolumab was developed by Bristol-Myers Squibb and is also known as BMS-936558 and MDX1106.[1] Nivolumab acts as an immunomodulator by blocking ligand activation of the Programmed cell death 1 receptor.
A Phase 1 clinical trial [2] tested nivolumab at doses ranging from 0.1 to 10.0 mg per kilogram of body weight, every 2 weeks. Response was assessed after each 8-week treatment cycle, and were evaluable for 236 of 296 patients. Study authors concluded that:”Anti-PD-1 antibody produced objective responses in approximately one in four to one in five patients with non–small-cell lung cancer, melanoma, or renal-cell cancer; the adverse-event profile does not appear to preclude its use.”[3]
Phase III clinical trials of nivolumab are recruiting in the US and EU.[4]
- Statement On A Nonproprietary Name Adopted By The USAN Council – Nivolumab, American Medical Association.
- A Phase 1b Study of MDX-1106 in Subjects With Advanced or Recurrent Malignancies (MDX1106-03), NIH.
- Topalian SL, et al. (June 2012). “Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer”. New England Journal of Medicine 366. doi:10.1056/NEJMoa1200690. Lay summary – New York Times.
- Nivolumab at ClinicalTrials.gov, A service of the U.S. National Institutes of Health.
The PD-1 blocking antibody nivolumab continues to demonstrate sustained clinical activity in previously treated patients with advanced non-small cell lung cancer (NSCLC), according to updated long-term survival data from a phase I trial.
Survival rates at one year with nivolumab were 42% and reached 24% at two years, according to the median 20.3-month follow up. Additionally, the objective response rate (ORR) with nivolumab, defined as complete or partial responses by standard RECIST criteria, was 17% for patients with NSCLC. Results from the updated analysis will be presented during the 2013 World Conference on Lung Cancer on October 29.
“Lung cancer is very difficult to treat and there continues to be a high unmet medical need for these patients, especially those who have received multiple treatments,” David R. Spigel, MD, the program director of Lung Cancer Research at the Sarah Cannon Research Institute and one of the authors of the updated analysis, said in a statement.
“With nivolumab, we are investigating an approach to treating lung cancer that is designed to work with the body’s own immune system, and these are encouraging phase I results that support further investigation in larger scale trials.”
In the phase I trial, 306 patients received intravenous nivolumab at 0.1–10 mg/kg every-other-week for ≤12 cycles (4 doses/8 week cycle). In all, the trial enrolled patients with NSCLC, melanoma, renal cell carcinoma, colorectal cancer, and prostate cancer.
The long-term follow up focused specifically on the 129 patients with NSCLC. In this subgroup, patients treated with nivolumab showed encouraging clinical activity. The participants had a median age of 65 years and good performance status scores, and more than half had received three or more prior therapies. Across all doses of nivolumab, the median overall survival was 9.9 months, based on Kaplan-Meier estimates.
In a previous update of the full trial results presented at the 2013 ASCO Annual Meeting, drug-related adverse events of all grades occurred in 72% of patients and grade 3/4 events occurred in 15%. Grade 3/4 pneumonitis related to treatment with nivolumab emerged early in the trial, resulting in 3 deaths. As a result, a treatment algorithm for early detection and management was developed to prevent this serious side effect.
Nivolumab is a fully human monoclonal antibody that blocks the PD-1 receptor from binding to both of its known ligands, PD-L1 and PD-L2. This mechanism, along with early data, suggested an associated between PD-L1 expression and response to treatment.
In separate analysis presented at the 2013 World Conference on Lung Cancer, the association of tumor PD-L1 expression and clinical activity in patients with NSCLC treated with nivolumab was further explored. Of the 129 patients with NSCLC treated with nivolumab in the phase I trial, 63 with NSCLC were tested for PD-L1 expression by immunohistochemistry (29 squamous; 34 non-squamous).
Bristol-Myers Squibb announced promising results from phase 2b study of its rheumatoid arthritis drug clazakizumab
NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL
CLAZAKIZUMAB
PRONUNCIATION klaz” a kiz’ ue mab
THERAPEUTIC CLAIM Autoimmune diseases, rheumatoid arthritis
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human interleukin 6) (human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 heavy chain), disulfide with human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 κ-chain, dimer
2. Immunoglobulin G1, anti-(human interleukin-6 (B-cell stimulatory factor 2, CTL differentiation factor, hybridoma growth factor, interferon beta-2)); humanized rabbit monoclonal BMS-945429/ALD518 [300-alanine(CH2-N67>A67)]1 heavy chain (223-217′)-disulfide with humanized rabbit monoclonal BMS-945429/ALD518 light chain dimer (229-229”:232-232”)-bisdisulfide, O-glycosylated
MOLECULAR FORMULA C6426H9972N1724O2032S42
MOLECULAR WEIGHT 145.2 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION BMS-945429, ALD518
CAS REGISTRY NUMBER 1236278-28-6
Monoclonal antibody
Type Whole antibody
Source Humanized
Target IL6
CAS number 1236278-28-6
Clazakizumab is a humanized monoclonal antibody designed for the treatment of rheumatoid arthritis.[1]
Clazakizumab was developed by Alder Biopharmaceuticals and Bristol-Myers Squibb.
gamma1 heavy chain (1-450) [humanized VH (Homo sapiens IGHV3-66*01 (83.50%) -(IGHD)-IGHJ3*02 M123>L (115)) [8.8.14] (1-120) -Homo sapiens IGHG1*03 CH

FDA approves GE’s imaging drug Vizamyl for Alzheimer’s
October 28, 2013 | By Anabela Farrica

Last Friday, FDA approved Vizamyl (flutemetamol F 18), a radioactive agent to be used with PET to help evaluate the brain of patients for Alzheimer’s disease or dementia. Vyzamil works by binding to beta-amyloid plaques, which can be found in the brain of people with Alzheimer’s disease or other dementias, as well as in the brain of elderly people who do not have neurological problems.
View original post 206 more words
Orphan drugs in Portugal
October 28 ,2013 | By Márcio Barra
What follows is a list of Orphan Drugs available in Portugal. This data has been compiled from two different databases, the INFOMED database, managed by the Portuguese National Competent Authrority on Medicines, INFARMED, and the OrphaNet database, from which sales numbers from Portugal were obtained, when available (note, sales numbers were, according to the Orphanet Website, last updated in September 28, 2013). The Portuguese Marketing approval date was also provided,
View original post 1,584 more words
Alexion obtains FDA breakthrough therapy status for cPMP to treat MoCD type A disorder

cyclic pyranopterin monophosphate (cPMP, ALXN1101)
Alexion Pharma International Sàrl has received a breakthrough therapy designation from the US Food and Drug Administration (FDA) for its cyclic pyranopterin monophosphate (cPMP, ALXN1101), an enzyme co-factor replacement therapy to treat patients with molybdenum cofactor deficiency (MoCD) type A.
Alexion obtains FDA breakthrough therapy status for cPMP to treat MoCD type A disorder
read all at
Cyclic pyranopterin monophosphate (cPMP) is an experimental treatment formolybdenum cofactor deficiency type A, which was developed by José Santamaría-Araujo and Schwarz at the German universities TU Braunschweig and the University of Cologne.[1][2]
cPMP is a precursor to molybdenum cofactor, which is required for the enyzme activity ofsulfite oxidase, xanthine dehydrogenase/oxidase and aldehyde oxidase.[3]
- Guenter Schwarz Laboratory, Institute for Biochemistry – University of Cologne (English, German)
- Günter Schwarz, José Angel Santamaria-Araujo, Stefan Wolf, Heon-Jin Lee, Ibrahim M. Adham, Hermann-Josef Gröne, Herbert Schwegler, Jörn Oliver Sass, Tanja Otte, Petra Hänzelmann, Ralf R. Mendel, Wolfgang Engel and Jochen Reiss (2004). “Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli“. Human Molecular Genetics 13 (12): 1249–1255. doi:10.1093/hmg/ddh136.PMID 15115759.
- Doctors risk untried drug to stop baby’s brain dissolving, TimesOnline, November 5, 2009
- José Angel Santamaria-Araujo, Berthold Fischer, Tanja Otte, Manfred Nimtz, Ralf R. Mendel, Victor Wray and Günter Schwarz (2004). “The Tetrahydropyranopterin Structure of the Sulfur-free and Metal-free Molybdenum Cofactor Precursor”. The Journal of Biological Chemistry 279 (16): 15994–15999.doi:10.1074/jbc.M311815200. PMID 14761975.
Molybdenum cofactor (Moco) deficiency is a pleiotropic genetic disorder. Moco consists of molybdenum covalently bound to one or two dithiolates attached to a unique tricyclic pterin moiety commonly referred to as molybdopterin (MPT). Moco is synthesized by a biosynthetic pathway that can be divided into four steps, according to the biosynthetic intermediates precursor Z (cyclicpyranopterin monophosphate; cPMP), MPT, and adenylated MPT. Mutations in the Moco biosynthetase genes result in the loss of production of the molybdenum dependent enzymes sulfite-oxidase, xanthine oxidoreductase, and aldehyde oxidase. Whereas the activities of all three of these cofactor-containing enzymes are impaired by cofactor deficiency, the devastating consequences of the disease can be traced to the loss of sulfite oxidase activity. Human Moco deficiency is a rare but severe disorder accompanied by serious neurological symptoms including attenuated growth of the brain, unbeatable seizures, dislocated ocular lenses, and mental retardation. Until recently, no effective therapy was available and afflicted patients suffering from Moco deficiency died in early infancy.
It has been found that administration of the molybdopterin derivative precursor Z, a relatively stable intermediate in the Moco biosynthetic pathway, is an effective means of therapy for human Moco deficiency and associated diseases related to altered Moco synthesis {see U.S. Patent No. 7,504,095). As with most replacement therapies for illnesses, however, the treatment is limited by the availability of the therapeutic active agent.
WO 2012112922 A1
In this synthesis, the deprotection may involve, for example, either sequential or one-pot deprotection of certain amino and hydroxyl protecting groups on a compound of formula (VII) to furnish the compound of formula (I). Suitable reagents and conditions for the deprotection of a compound of formula (VII) can be readily determined by those of ordinary skill in the art. For example, compound (I) may be formed upon treatment of a compound of formula (VII) under conditions so that hydroxyl protecting groups, such as acetate, isopropylidine, and benzylidine protecting groups, are removed from the formula (VII) structure. The acetate group can be cleaved, for example, under Zemplen conditions using catalytic NaOMe as a base in methanol. The benzylidene and isopropylidene groups can be cleaved by hydrogenation or using acidic hydrolysis as reported by R.M. Harm et ah, J. Am. Chem. Soc, 72, 561 (1950). In yet another example, the deprotection can be performed so that amino protecting groups, such as 9- fluorenylmethyl carbamate (Fmoc), t-butyl carbamate (Boc), and carboxybenzyl carbamate (cbz) protecting groups are cleaved from the compound of formula (VII). 9-fluorenylmethyl carbamate (Fmoc) can be removed under mild conditions with an amine base (e.g. , piperidine) to afford the free amine and dibenzofulvene, as described by E. Atherton et al, “The
Fluorenylmethoxycarbonyl Amino Protecting Group,” in The Peptides, S. Udenfriend and J. Meienhofer, Academic Press, New York, 1987, p. 1. t-butyl carbamate (Boc) can be removed, as reported by G.L. Stahl et al., J. Org. Chem., 43, 2285 (1978), under acidic conditions (e.g., 3 M HC1 in EtOAc). Hydrogenation can be used to cleave the carboxybenzyl carbamate (cbz) protecting group as described by J. Meienhofer et al., Tetrahedron Lett., 29, 2983 (1988).
To prevent oxidation of formula (I) during the reaction, the deprotection may be performed under anaerobic conditions. The deprotection may also be performed at ambient temperature or at temperatures of from about 20 – 60 °C (e.g. , 25, 30, 35, 40, 45, 50, or 55 °C).
The compound of formula (I) may be isolated in the form of a pharmaceutically acceptable salt. For example, the compound of formula (I) may be crystallized in the presence of HC1 to form the HC1 salt form of the compound. In some embodiments, the compound of formula (I) may be crystallized as the HBr salt form of the compound. The compound of formula (I) may also be isolated, e.g., by precipitation as a sodium salt by treating with NaOH. The compound of formula (I) is labile under certain reaction and storage conditions. In some embodiments, the final solution comprising the compound of formula (I) may be acidified by methods known in the art. For example, the compound of formula (I), if stored in solution, can be stored in an acidic solution.
In some embodiments, the compound of formula (I) may be prepared, for example, by: reacting a compound of formula (II- A):

with a compound of formula (III- A):

in the presence of a hydrazine to produce a compound of formula (IV- A):

selectively protecting the compound of formula (IV-A) to prepare a compound of formula (V-A):

wherein:
Rj is a protecting group, as defined above;
phosphorylating the compound of formula (V-A) to prepare a compound of formula (VI- A):

oxidizing the compound of formula (VI-A) to prepare a compound of formula (VII- A):

; and deprotecting the compound of formula (VII-A) to prepare the compound of formula (I). For example, a compound of formula (I) can be prepared as shown in Scheme 3.
Scheme 3.

5 R = Fraoc

In another embodiment, the compound of formula (I) is prepared by:
reacting a compound of formula (II- A):

with a compound of formula (III- A):

in the presence of a hydrazine to produce a compound of formula (IV-A):

selectively protecting the compound of formula (IV-A) to prepare a compound of formula (V-B):

wherein:
each Ri is independently a protecting group, as defined above;
phosphorylating the compound of formula (V-B) to prepare a compound of formula (VI-B):

oxidizing the compound of formula (VI-B) to prepare a compound of formula (VII-B):

; and deprotecting the compound of formula (VII-B) to prepare the compound of formula (I), example, a compound of formula (I) can be prepared as shown in Scheme 4.
Scheme 4.

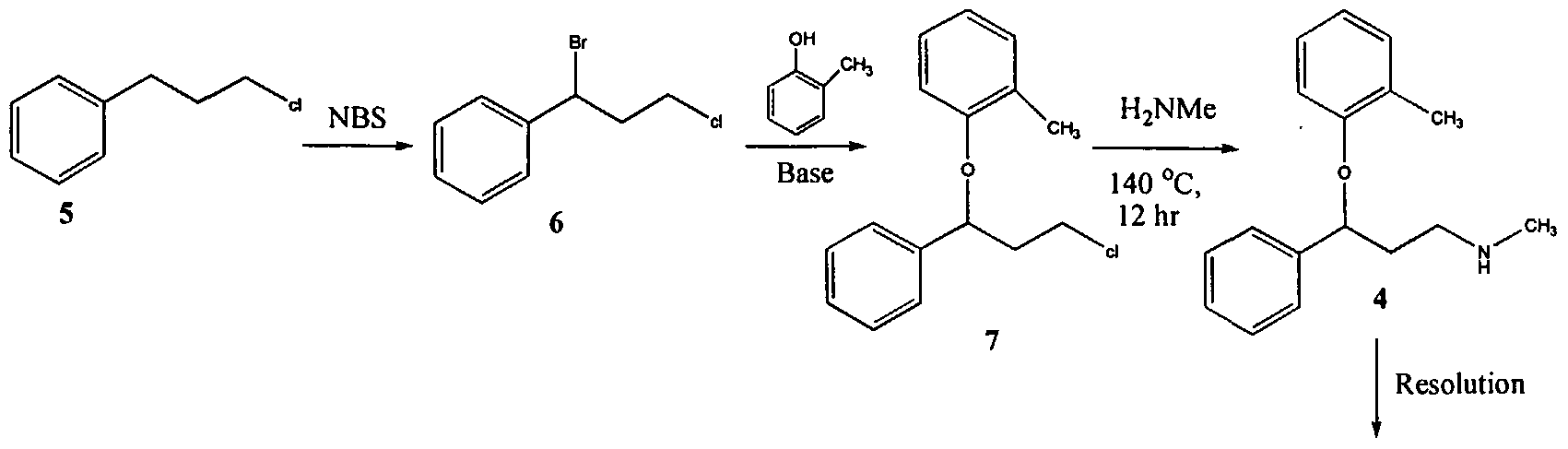
Alternatively, a compound of formula (I) can be formed as shown in Scheme 5. A diaminopyrimidinone compound of formula (II) can be coupled with a phosphorylated hexose sugar of formula (VIII), to give a compound of formula (IX). The piperizine ring nitrogen atoms can be protected to give a compound of formula (X) which can be oxidized to give a diol of formula (XI). The diol of formula (XI) can then be deprotected using appropriate conditions and converted to the compound of formula (I).
Scheme 5

In this embodiment, the phosphate may be introduced at the beginning of the synthesis to avoid undesirable equilibrium between the pyrano and furano isomers during subsequent steps of the synthesis. For example, a compound of formula (I) can be prepared as shown in Scheme 6.
Scheme 6.
ridine

A compound of formula (I) can also be formed as shown in Scheme 7. A diaminopyrimidinone compound of formula (II) can be coupled to a compound of formula (III) to afford the piperizine derivative of formula (IV). The piperizine ring nitrogen atoms of the compound of formula (IV) can be protected under standard conditions to give a derivative of formula (V). The formula (V) structure can be oxidized to afford compounds of formula (XII). Phosphorylation of a compound of formula (XII) gives a compound of formula (VII). Global deprotection of the compound of formula (VII) can afford the compound of formula (I).
Scheme 7
Piperizine ring protection
sphorylation
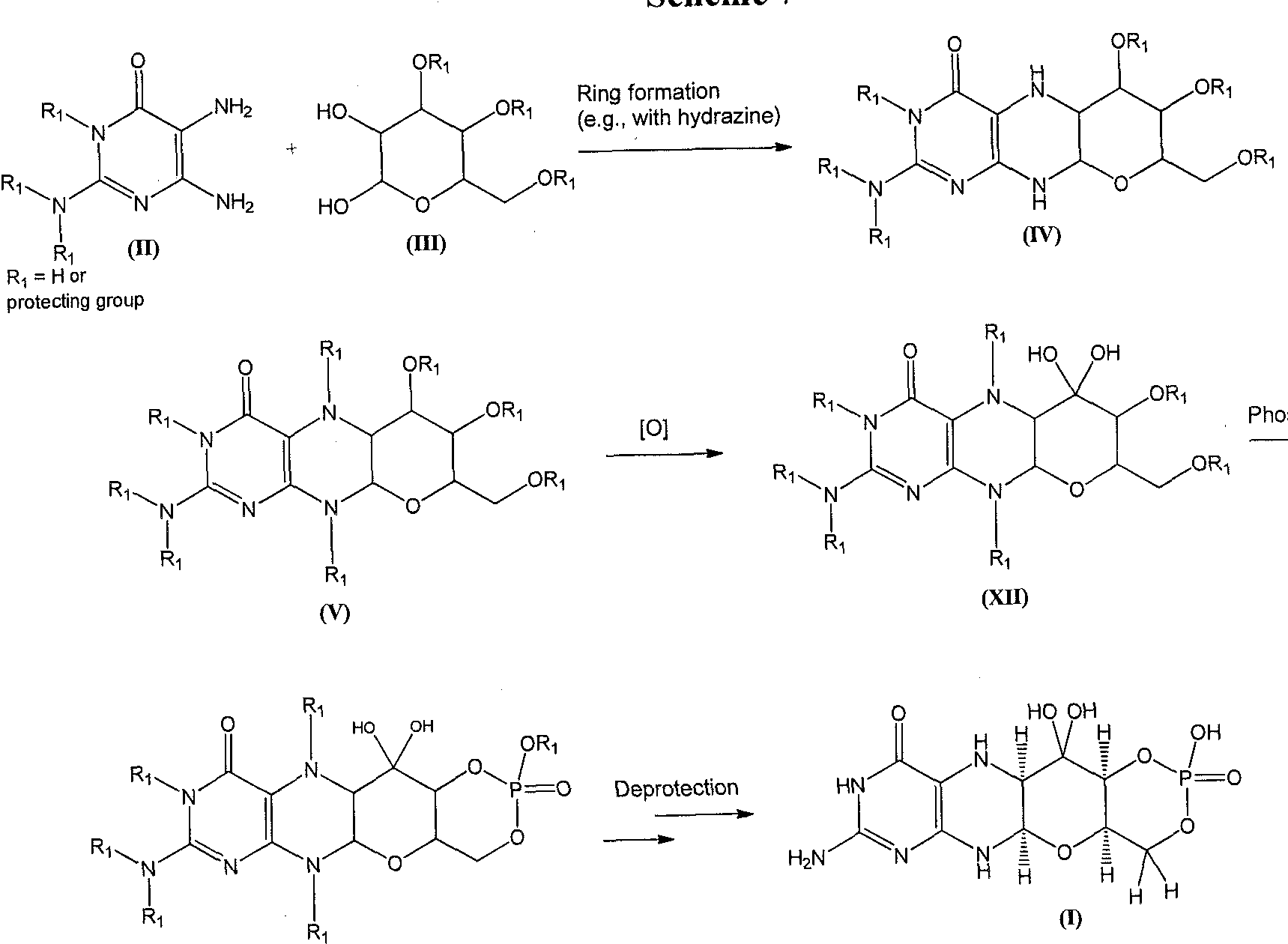
(VII)
For example, a compound of formula (I) can be prepared as shown in Scheme 8.
Scheme 8.

ATACAND, CANDESARTAN CILEXETIL, ASTRAZENECA.Drug Patent Expiration on 9 th Jan 2014

Candesartan cilexetil Candesartan cilexetil, Candesartan hexetil, H212/91, TCV-116, Kenzen, Blopress 16 mg Plus, Parapres, Ratacand, Blopress, Amias, Atacand
ATACAND
ATACAND (candesartan cilexetil), a prodrug, is hydrolyzed to candesartan during absorption from the gastrointestinal tract. Candesartan is a selective AT1 subtype angiotensin II receptor antagonist. Candesartan cilexetil, a nonpeptide, is chemically described as (±)-1-Hydroxyethyl 2-ethoxy-1-[p-(o-1H-tetrazol-5ylphenyl)benzyl]-7-benzimidazolecarboxylate, cyclohexyl carbonate (ester). Its empirical formula is C33H34N6O6, and its structural formula is:
 |
Candesartan cilexetil is a white to off-white powder with a molecular weight of 610.67. It is practically insoluble in water and sparingly soluble in methanol. Candesartan cilexetil is a racemic mixture containing one chiral center at the cyclohexyloxycarbonyloxy ethyl ester group. Following oral administration, candesartan cilexetil undergoes hydrolysis at the ester link to form the active drug, candesartan, which is achiral. ATACAND is available for oral use as tablets containing either 4 mg, 8 mg, 16 mg, or 32 mg of candesartan cilexetil and the following inactive ingredients: hydroxypropyl cellulose, polyethylene glycol, lactose, corn starch, carboxymethylcellulose calcium, and magnesium stearate. Ferric oxide (reddish brown) is added to the 8-mg, 16-mg, and 32-mg tablets as a colorant.
Drug Patent Expiration and Exclusivity
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| CANDESARTAN CILEXETIL | TABLET; ORAL | 4MG | RX | 020838 | 001 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 8MG | RX | 020838 | 002 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 16MG | RX | 020838 | 003 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 32MG | RX | 020838 | 004 |
Patents
There are 6 patent(s) protecting ASTRAZENECA’s ATACAND. The last patent 5534534*PED expires on 2014-01-09.View patent at USPTO
| Patent US | US | Expiration |
|---|---|---|
| 5534534*PED | 2014-1-9 | |
| 5534534 | Pharmaceutical compositions for oral use and method of preparing them
A pharmaceutical composition for oral use comprising an effective amount of a compound of the formula (I) having antagonistic action to angiotensin II ##STR1## (wherein the ring W is an optionally substituted N-containing heterocyclic residue; R.sup.3 is a group capable of forming an anion or a group convertible thereinto; X is a direct bond or a spacer having an atomic length of two or less between the phenylene group and the phenyl group; and n is an integer of 1 or 2) and an oily substance having a lower melting point, and a method for preparing a pharmaceutical composition for oral use comprising an effective amount of a compound of the formula (I) and an oily substance having a lower melting point, which comprises admixing the compound of the formula (I) with an oily substance having a lower melting point and then subjecting the mixture to molding.
|
2013-7-9(expired) |
| 5196444*PED | 2012-12-4(expired) | |
| 5196444 | 1-(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-c arboxylate and compositions and methods of pharmaceutical use thereof
1-(Cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-car boxylate or a pharmaceutically acceptable salt thereof has potent angiotensin II antihypertensive activity, thus being useful as therapeutic agents for treating circulatory system diseases such as hypertensive diseases, heart diseases (e.g. hypercardia, heart failure, cardiac infarction, etc.), strokes, cerebral apoplexy, nephritis, etc.
|
2012-6-4(expired) |
| 7538133*PED | 2011-10-18(expired) | |
| 5705517*PED | 2011-10-18(expired) |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the ASTRAZENECA.
| Date | Supplement No. | Action | Documents |
|---|---|---|---|
| 2013-04-26 | 038 | Labeling Revision | |
| 2012-04-27 | 035 | Labeling Revision | |
| 2012-04-13 | 032 | Labeling Revision | |
| 1998-06-04 | 000 | Approval | |
| 2011-06-24 | 033 | Labeling Revision | |
| 2009-10-22 | 031 | Patient Population Altered | |
| 2006-08-17 | 026 | Labeling Revision | |
| 2005-05-18 | 022 | New or Modified Indication | |
| 2005-02-22 | 024 | New or Modified Indication | |
| 2004-12-16 | 023 | Labeling Revision | |
| 2000-06-14 | 008 | Labeling Revision | |
| 2002-09-13 | 015 | Comparative Efficacy Claim | |
| 2003-01-22 | 017 | Labeling Revision | |
| 2003-04-23 | 019 | Labeling Revision | |
| 2013-02-21 | 037 | Manufacturing Change or Addition | |
| 1999-08-11 | 005 | Package Change | |
| 2000-12-27 | 009 | Manufacturing Change or Addition | |
| 2001-05-24 | 012 | Manufacturing Change or Addition | |
| 2001-11-28 | 016 | Labeling Revision | |
| 1999-07-28 | 004 | Control Supplement | |
| 2001-04-02 | 011 | Manufacturing Change or Addition | |
| 2001-10-04 | 014 | Control Supplement | |
| 1998-11-16 | 002 | Manufacturing Change or Addition | |
| 1999-12-08 | 006 | Package Change | |
| 2001-06-07 | 010 | Manufacturing Change or Addition | |
| 2001-03-29 | 013 | Package Change | |
| 1998-12-07 | 001 | Manufacturing Change or Addition |
 Candesartan is marketed as the cyclohexyl 1-hydroxyethyl carbonate (cilexetil) ester, known ascandesartan cilexetil. Candesartan cilexetil is metabolised completely by esterases in theintestinal wall during absorption to the active candesartan moieity. The use of a prodrug form increases the bioavailability of candesartan. Despite this, absolute bioavailability is relatively poor at 15% (candesartan cilexetil tablets) to 40% (candesartan cilexetil solution). Its IC50 is 15 µg/kg. U.S. Patent Nos. 5,196,444 and 5,578,733 describe the removal of a trityl protecting group of the N-protected tetrazolyl compounds using methanol in the presence of a mineral acid, such as hydrochloric acid, which requires complex extractions or chromatographic purification to produce pure candesartan cilexetil. U.S. Patent No. 7,345,072 describes the deprotection of tetrazolyl compounds, including candesartan cilexetil, in the presence of an anhydrous mineral acid or aqueous mineral acid at a concentration higher than 20% w/w. The strong acidic conditions produce more decomposition products and thereby reduces the overall purity of the final product. WO 05/021535 discloses the preparation of candesartan cilexetil by the deprotection of trityl moiety at a reflux temperature in the presence of anhydrous Ci to C5 alcohol under neutral or slightly basic conditions involving longer reaction time (for e.g. stirring for several hours, such as 18-24 hours); this is followed by removal of the triphenylmethylether moiety precipitated as a solid, and thereby increases the number of reaction steps. WO 05/037821 describes the deprotection of the trityl candesartan cilexetil by the use of methane sulphonic acid, p-toluene sulphonic acid, formic and trifluoroacetic acid in solvent mixture or by refluxing candesartan cilexetil in mixture of toluene, water, and methanol. The initial product obtained by these procedures is mostly a viscous oil or a semi solid, which is difficult to handle. WO 07/074399 and WO 07/042161 disclose the preparation of candesartancilexetil from trityl candesartan cilexetil involving Lewis acids such as boron trifluoride, zinc chloride, aluminium trihalide, or titanium tetrachloride which are costly and thus are not commercially viable.
Candesartan is marketed as the cyclohexyl 1-hydroxyethyl carbonate (cilexetil) ester, known ascandesartan cilexetil. Candesartan cilexetil is metabolised completely by esterases in theintestinal wall during absorption to the active candesartan moieity. The use of a prodrug form increases the bioavailability of candesartan. Despite this, absolute bioavailability is relatively poor at 15% (candesartan cilexetil tablets) to 40% (candesartan cilexetil solution). Its IC50 is 15 µg/kg. U.S. Patent Nos. 5,196,444 and 5,578,733 describe the removal of a trityl protecting group of the N-protected tetrazolyl compounds using methanol in the presence of a mineral acid, such as hydrochloric acid, which requires complex extractions or chromatographic purification to produce pure candesartan cilexetil. U.S. Patent No. 7,345,072 describes the deprotection of tetrazolyl compounds, including candesartan cilexetil, in the presence of an anhydrous mineral acid or aqueous mineral acid at a concentration higher than 20% w/w. The strong acidic conditions produce more decomposition products and thereby reduces the overall purity of the final product. WO 05/021535 discloses the preparation of candesartan cilexetil by the deprotection of trityl moiety at a reflux temperature in the presence of anhydrous Ci to C5 alcohol under neutral or slightly basic conditions involving longer reaction time (for e.g. stirring for several hours, such as 18-24 hours); this is followed by removal of the triphenylmethylether moiety precipitated as a solid, and thereby increases the number of reaction steps. WO 05/037821 describes the deprotection of the trityl candesartan cilexetil by the use of methane sulphonic acid, p-toluene sulphonic acid, formic and trifluoroacetic acid in solvent mixture or by refluxing candesartan cilexetil in mixture of toluene, water, and methanol. The initial product obtained by these procedures is mostly a viscous oil or a semi solid, which is difficult to handle. WO 07/074399 and WO 07/042161 disclose the preparation of candesartancilexetil from trityl candesartan cilexetil involving Lewis acids such as boron trifluoride, zinc chloride, aluminium trihalide, or titanium tetrachloride which are costly and thus are not commercially viable.
Synthesis
Candesartan is synthesised as follows:  kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
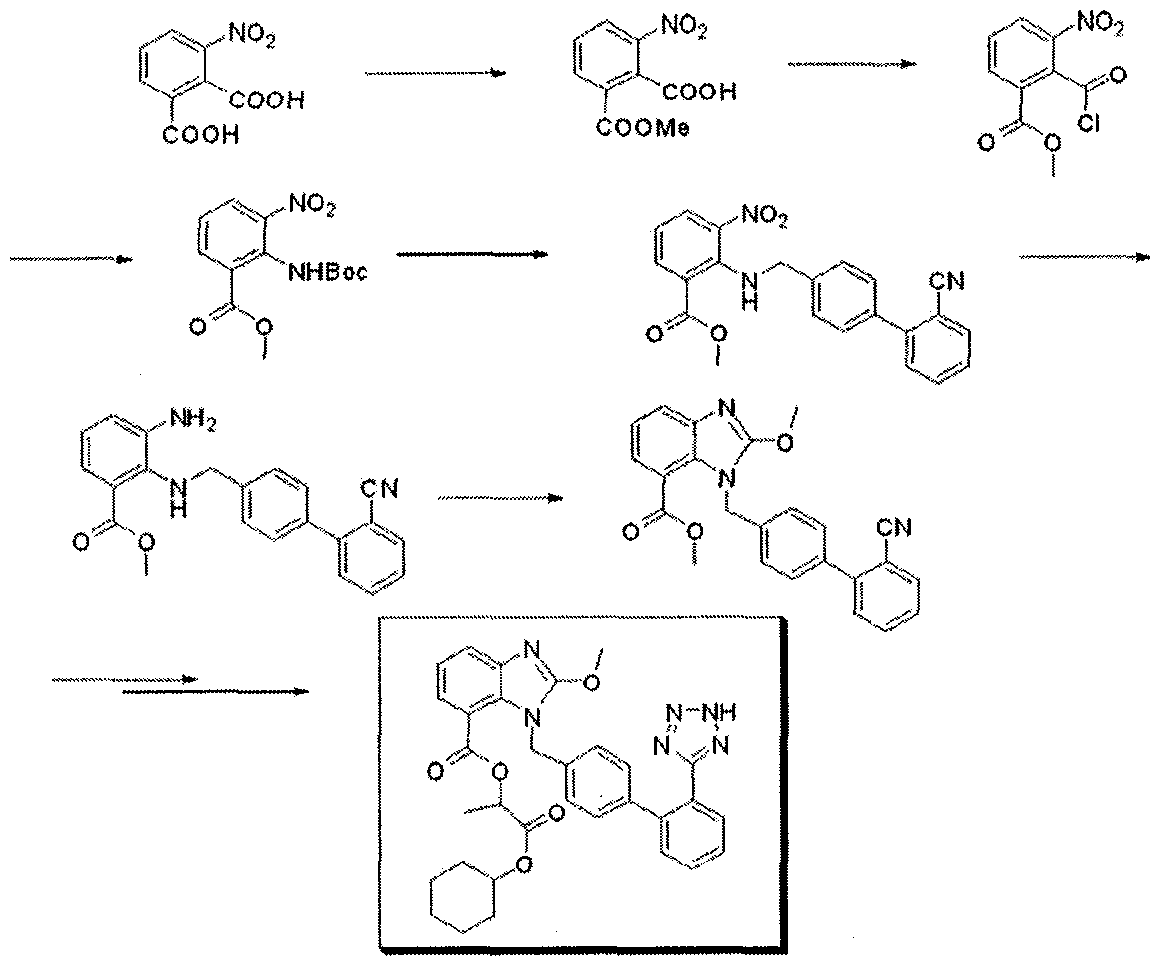
The method has technical problems as follows: a) the starting material is obtained by a minor reaction, b) its yield is relatively low and its industrial applicability is poor (due to N2 gas formation) because the Curtius rearrangement reaction is involved, and c) materials industrially hard to handle such as SOCI2 or NaH are used. In addition, methods for preparing an intermediate of candesartan cilexetil are disclosed in Organic Process Research & Development 11:490-493(2007), as represented by the following Reaction Scheme 2: Reaction Scheme 2

3:1 s

However, the preparation process has also shortcomings of a) undesired byproducts formed by nitrogenation at ortho- or para-position, b) safety problems from strong acids (sulfuric acid and nitric acid) used twice when introducing and rearranging nitrogen groups, and c) utilization of high-flammable Raney Ni.
cut paste
Novel and Practical Synthesis of Candesartan Cilexetil
Yongjun Mao, Ruisheng Xiong, Zheng Liu, Haihong Li, Jingkang Shen, and Jingshan Shen* *Chinese Academy of Sciences, Shanghai Institute of Materia Medica, 555 Zuchongzhi Rd., Zhangjiang Hi-Tech Park, Shanghai, 201203, China
Abstract
A novel and convergent synthetic route of candesartan cilexetil (API of Atacand), an effective angiotensin II receptor blocker, is described. Cleavage of the N-Boc and N-trityl protective group are implemented simultaneously and formation of the benzimidazole ring is conducted at the last step of this route, which gives candesartan cilexetil in 55% yield over six steps with 99.1% purity (HPLC). Full Text HTMLPDF (567KB)PDF with Links (932KB)
 This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.
This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.
| Benzimidazole derivs., their production and use | |
| Naka, T.; Nishikawa, K.; Kato, T. (Takeda Chemical Industries, Ltd.) | |
| EP 0459136; EP 0720982; JP 1992364171; JP 1996099960; US 5196444; US 5328919; US 5401764; US 5703110; US 5705517; US 5962491; US 6004989 |
more info Candesartan cilexetil of Formula I, disclosed in U.S. Patent No. 5,196,444 as crystalline form, i.e., Form-I (C-type crystals), is chemically described as 1- cyclohexyloxycarbonyloxyethyl 2-ethoxy-3-[[4-[2-(2H-tetrazol-5- yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate.

H Formula I It is useful in the treatment of cardiovascular complaints such as hypertension and heart failure. Candesartan cilexetil is poorly soluble in water, which is attributed to its hydrophobic nature. Solubility plays an important role in achieving the desired concentration of a drug in systemic circulation for accomplishing the pharmacological response. Various techniques are known in literature to increase the solubility of poorly-soluble drugs, including decreasing the particle size, complexation, changing the surface characteristics of the particles, and incorporation of drug particles into colloidal systems like nanoparticles and liposomes. Among these, the most commonly used technique to increase the solubility is particle size reduction. Sometimes the rate of dissolution of a poorly-soluble drug is the rate limiting factor in its rate of absorption by the body. These drugs may be more readily bioavailable if administered in a finely divided state. Particle size reduction increases the surface area causing an increase in the dissolution rate of the compound, and hence, its bioavailability. There are certain techniques reported in literature to reduce the particle size of such poorly-soluble drugs. PCT Publication No. WO 2006/122254 discloses stable candesartan cilexetil of fine particle size, wherein the stable micronized candesartan cilexetil is prepared by slurrying a sample of candesartan cilexetil of fine particle size in a suitable solvent for a suitable amount of time. In this application, candesartan cilexetil of fine particle size is obtained directly from the synthesis of candesartan cilexetil or by comminuting candesartan cilexetil using milling. PCT Publication No. WO 2005/123720 describes fine particles of candesartan cilexetil having improved pharmacokinetic profile and a process for their production, wherein fine particle size is obtained by a) dissolving candesartan cilexetil in an organic solvent; b) cooling the solution obtained in step a) under stirring to crystallize candesartan cilexetil from the solution; and c) isolating candesartan cilexetil having a particle size of with d90 not more than about 25 μ. U.S. Patent Application No. 2006/0165806 describes compositions comprising a candesartan, such as candesartan cilexitil. The candesartan particles of the composition have an effective average particle size of less than about 2000 nm. U.S. Patent Application No. 2008/0038359 describes a nanoparticle pharmaceutical formulation comprising a poorly soluble drug substance having an average particle size of less than about 1000 nm, a solid or semisolid dispersion vehicle, and optionally a non-surface modifying excipient. U.S. Patent No. 7,828,996 discloses the methods for forming nanoparticles of a material of narrow polydispersity with ultrasonic waves using a partially submersed sonicator that does not touch any part of the apparatus and the point of addition of organic solvent is in the wave funnel produced by sonication and within the selected distance from the wave-source depending on the desired particle size. U.S. Patent No. 7,780,989 discloses the preparation of a dispersion of nanocrystalline particles in an aqueous medium using ultrasound. U.S. Patent No. 5,314,506 describes a crystallization process in which a jet of a solution containing a substance is impinged with a second jet containing an anti-solvent for the substance. The rapid mixing produced by the impinging jets results in a reduction of the crystals so formed compared to conventional slow crystallization processes. The smallest crystals disclosed are about 3 μ and the majority are in the range of from about 3 μ to about 20 μ. PCT Publication No. WO 00/44468 describes a modification to the apparatus described in U.S. Patent No. 5,314,506, wherein ultrasound energy is applied at the point of impingement of the two jets to further enhance localized mixing and is stated to give direct formation of small crystals with a diameter of less than 1 μ. Generally, the crystalline particles described have an average size of 0.5 μ. Conventional particle size reduction methods such as high energy milling may result in loss of yield, noise and dusting, as well as unwanted exposure to highly potent pharmaceutical compounds. Also, in the case of crystalline compounds, stress generated on crystal surfaces during milling can adversely affect labile compounds. Therefore, there is a need for a process for particle size reduction of candesartan cilexetil, which is industrially advantageous, easy to handle and is cost effective.
-
Candesartan is a potent, selective AT1 subtype angiotensin II receptor antagonist and used for treatment of hypertension. Due to poor absorption of Candesartan in body, the prodrug candesartan cilexetilwas developed. The candesartan cilexetil is rapidly and completely hydrolyzed to candesartan in gastrointestinal tract.
-
[0004]U.S. Pat. No. 5,196,444 discloses Candesartan cilexetil and a process for its preparation by the reaction of 2-ethoxy-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid with trityl chloride in presence of triethyl amine in methylene chloride and purification by column chromatography gives 2-ethoxy-1-[[2′-(N-triphenylmethyltetrazol-5-yl)-biphenyl-4-yl]methyl] benzimidazole -7-carboxylic acid, which upon condensation with cyclohexyl 1-iodoethyl carbonate in presence of potassium carbonate in DMF followed by purification with column chromatography gives a colorless powder which is recrystallized in ethanol yields ‘C’ type crystals of Candesartancilexitil.
-
[0005]U.S. Pat. Application No. 2005/131027 discloses a process for preparation of candesartan cilexetil by reaction of trityl candesartanwith cilexetil halide and at least one base in a low boiling solvent in presence of phase transfer catalyst to give Trityl candesartan cilexetil, which upon deprotection with at least one organic acid in at least one organic solvent. U.S. Pat. Application 2005/131027 further discloses the deprotection of Trityl candesartan cilexetil in methanol without an acid.
-
[0006]The PCT publication WO 2005/021535 discloses the deprotection of Trityl candesartan cilexetil with neutral or slightly basic medium in alcohol.
-
[0007]Chem.Pharm.Bull. 47(2), 182-186 (1999) discloses two novel crystalline forms of Candesartan cilexetil, form-I and form-II.
-
[0008]PCT publication WO 04/085426 discloses Candesartan cilexetil 1,4-Dioxane solvate and two more crystalline forms, designated as form-III and form-IV. The disclosed process for preparation of form-III involves crystallization of Candesartan cilexetil in toluene and for form-IV involves crystallization in a mixture of methyl tert-butyl ether and methanol.
-
[0009]PCT publication WO 2005/077941 discloses several crystalline forms, solvates of Candesartan cilexetil along with a process for preparation of form-I (type-C).
-
[0010]The prior art disclosed methods for preparation of Candesartan cilexetilinvolves purification of Trityl candesartan and Candesartan cilexetil by column chromatography or involves the use of strong acids like IN HCl or the use of organic acids or without an acid in methanol for detrytilation of Trityl candesartan cilexetil.
-
[0011]There is a requirement of a process for preparation of Candesartancilexetil which yields a pure Candesartan cilexetil without involving the purification by column chromatography and the usage of strong acids for deprotection.
Candesartan cilexetil of formula (I) shown beiow is chemicaily described as (+/-)-1- [[(cyclohexyloxy)carbonyl]oxy]ethyl-2-ethoxy-1 -[[2′-(1 H-tetrazol-5-yl)-1 , 1 ‘-biphenyl- 4-yl]methyl]-1 H-benzimidazoie-7-carboxylate. An alternative designation is (+-)-1- hydroxyethyf 2~Ethoxy-1 -{p-(o-1 H-tetrazo!-5-yIphenyi)benzyJ)-7-benzϊmidazoie~ carboxyiic acid cyclohexyl carbonate (ester), with candesartan being the underlying carboxylic acid, i.e. 2-Ethoxy-1 -(p-(o-1 H-tetrazol-5-ylphenyl)benzy!)-7-benz- imidazolecarboxylic acid.

Because of its ability to inhibit the angiotensin-converting enzyme it is widely used for the treatment of hypertension and related diseases and conditions. As an angiotensin Ii receptor antagonist, candesartan ciiexetil avoids the side-effects of calcium antagonists, and shows high stability and obvious curative effects. Currently candesartan ciiexetil is soid as racemic mixture, it is produced according to published patents, e.g. EP 0 720 982 B1 and EP 0 459 136. in Chem. Pharm. Bull. 47(2), 182-186 (1999) two crystalline forms (Form I and II), together with an amorphous form, are disclosed and characterized by their DSC thermograms, X-ray diffraction patterns and IR spectra. US 5,196,444 disclosed the C-type crystal (Form I) of candesartan cilexetif, and processes for producing it under acidic conditions. WO 04/085426 discloses the dioxane solvate of candesartan ciiexetil, together with two additional crystalline forms. WO 2005/077941 discloses hydrates and solvates of candesartan ciiexetil, together with processes for their preparation. WO 2006/048237 also describes the preparation of new polymorphic forms ofcandesartan ciiexetil, together with processes for their preparation, including the preparation of amorphous candesartan ciiexetil by precipitating it with a liquid cyclic hydrocarbon from a solution of candesartan ciiexetil in a chlorinated solvent. in WO 2005/123721 processes for the preparation of amorphous candesartanciiexetil are provided, comprised of spray-drying and precipitation. HPLC CUT PASTE , READER TO PICK ONLY REQUIRED INFO Candesartan cilexetil (60 g) is dissolved in isopropanol (900 m!_) at 60-65 0C. Solution is hot filtered into reactor and quickly cooled to 35 0C. At this temperature nucleation is provoked with 300 mg of candesartan cilexetil form I and stirring is enforced. Suspension is cooled to 3O0C in 1 hour and rigorous stirring is continued at this temperature for additional 5 hours. Then stirring power is reduced and the suspension is cooled to 2O0C in 8 hours. The product is filtered, washed with isopropanoi and dried for 2 hours at 38°C. Yield: 48.7 g of candesartan cilexetil form I. Area % HPLC: candesartan cilexetil: 99.73%, alky ester of candesartan cilexetil 0.08%, candesartan cilexetii pyran below 0.05%, tritylcandesartan cϋexetil 0.09% Average particle size: 19 /vm, no agglomerates present (see Figure 2) B) Detection of impurities in candesartan cilexetil Example 6 Detection of candesartan cϊlexetil pyran in candesartan cilexetii by HPLC HPLC (external standard method) was performed using the following specifications : Column: Zorbax Eclipse XDB-C18, 50 mm x 4.6 mm i.d.τ 1.8 μm particles Eluent A: 0.01 M NaH2PO4, pH 2.5 Eluent B: acetonitriie Gradient of Eluent:

Flow rate: about 1.2 ml/min Diluent: acetonitriie : water = 70 : 30 (V/V). Detection: UV, wavelength 225 nm injection volume: 5 μl Column temperature : 500C Autosampler temperature: 7°C Example 7 Detection of cilexetil pyran in 1 -chloroethyl cyclohexylcarbonate by GC GC/FID (area percent method) was performed using the following specifications: Column: capillary (fused-silica) AT-WAX or adequate Length: 30 m ID: 0.32 mm Film thickness: 0.25 μm Carrier gas: helium Carrier gas flow rate: 2.0 ml/mi n Split ratio: 10 : 1 Air flow rate: 400 ml/min Hydrogen flow rate: 40 ml/min Make up gas flow ISb rate: 25 ml/min Column temperature 100°C (0 min) → 10°C/min → 2000C (10 min or prolonged if necessary) Injector temperature: 21 O0C Detector temperature: 250OC Injection volume : 1 μl Diluent: Acetonithle: chromatography grade. Chromatographic system suitability Signal/noise of 1 -chloroethyl cyclohexyl carbonate: not less than 10
………………
Seki M * Mitsubishi Tanabe Pharma Corporation, Osaka, Japan An Efficient C–H Arylation of a 5-Phenyl-1H-tetrazole Derivative: A Practical Synthesis of an Angiotensin II Receptor Blocker. Synthesis 2012; 44: 3231-3237 
Significance
Candesartan cilexetil (Atacand®) is an angiotensin II receptor antagonist that is prescribed for the treatment of hypertension. It is a prodrug that is hydrolyzed to candesartan in the gut. The synthesis depicted, features an efficient protocol for ruthenium-catalyzed C–H arylation of the tetrazole A. Comment A significant challenge in this small-scale synthesis was the final removal of the benzyl protecting group from the tetrazole unit using transfer hydrogenation. Best results were obtained using a ‘thickshell’ Pd/C catalyst from Evonik
Abbot’s device for leaky heart valves gains FDA approval
October 26, 2013 | By Anabela Farrica
The U.S. Food and Drug Administration has approved Abbot’s MitraClip, a device intended for patients with mitral valve regurgitation and for whom open-heart surgery for valve repair is deemed inadequate. Patients who find themselves in a too fragile state to endure such complex surgeries are generally treated with the available medicines and experience high rates of heart failure and rehospitalization.
View original post 235 more words
EMEDASTINE DIFUMARATE, EMADINE, 8 TH DEC 2013 PATENT EXPIRY

EMEDASTINE DIFUMARATE
Emedastine difumarate (Emadine) is a second generation antihistamine used in eye drops to treat allergic conjunctivitis. Its mechanism of action is a H1 receptor antagonist.

EMADINE
Drug Patent Expiration and Exclusivity
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| EMEDASTINE DIFUMARATE | SOLUTION/DROPS; OPHTHALMIC | 0.05% | RX | 020706 | 001 |
Patents
There are 1 patent(s) protecting ALCON’s EMADINE.
The last patent expires on 2013-12-08.
| Patent | Expiration | |
|---|---|---|
| US5441958 | Ophthalmic compositions comprising emedastine and methods for their use
Topical ophthalmic compositions comprising 1-(2-ethoxyethyl)-2-(4-methyl-1-homopiperazinyl)-benzimidazole and its ophthalmically acceptable acid addition salts have been found to be useful in treating allergic conjunctivitis and related ailments.
|
2013-12-8 |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the ALCON.
EMADINE ® (emedastine difumarate ophthalmic solution) 0.05% is a sterile ophthalmic solution containing emedastine, a relatively selective, H1-receptorantagonist for topical administration to the eyes. Emedastine difumarate is a white, crystalline, water-soluble fine powder with a molecular weight of 534.57. The chemical structure is presented below:
Structural Formula:
 |
Chemical Name:
lH-Benzimidazole, 1-(2-ethoxyethyl)-2-(hexahydro-4-methyl-1H-1,4-diazepin-1-yl), (E)-2-butenedioate (1:2)
Each mL of EMADINE contains: Active: 0.884 mg emedastine difumarate equivalent to 0.5 mg emedastine. Preservative: benzalkonium chloride0.01%. Inactives: tromethamine; sodium chloride; hydroxypropyl methylcellulose; hydrochloric acid/sodium hydroxide (adjust pH); and purified water. It has a pH of approximately 7.4 and an osmolality of approximately 300 mOsm/kg.
l-(2- ethoxyethyl)-2-(4-methyl-l-homopiperazinyl)-benzimidazole, otherwise known asemedastine, and its ophthalmically acceptable acid addition salts and methods for their use.
Allergic conjunctivitis is frequently characterized by ocular pruritus
(itching), erythema (inflammatory redness), edema and tearing. This condition is one of the most frequently treated by ophthalmologists, optometrists and allergists. To date, treatment has been primarily through the use of topically applied histamine t antagonists in combination with α-agonists. See, for example, the following articles:
1. Miller, J. and E.H. Wolf, “Antazoline phosphate and naphazoline hydrochloride, singly and in combination for the treatment of allergic conjunctivitis – a controlled, double-blind clinical trial.” Ann. Allergy, 35:81-86 (1975). 2. Vandewalker, M.L. et al., “Efficacy of Vasocon-A and its components with conjunctival provocation testing (CPT).” j± Allergy Clin. Immunol., 83:302 (1989). 3. Abelson, M.B. et al., “Effects of topically applied ocular decongestant and antihistamine.” Am. I. Ophthalmol., 90:254- 257 (1980).
Recent studies indicate that the antihistamine levocabastine exhibits clinical activity in patients with allergic conjunctivitis without the addition of a vasoconstrictor. See, Dechant, K.L. and K.L. Goa, “Levocabastine. A review of its pharmacological properties and therapeutic potential as a topical antihistamine in allergic rhinitis and conjunctivitis/’ Drugs, 41:202-224 (1991). In addition, it has recently been demonstrated that Hα antagonists are effective in relieving conjunctival injection (hyperemia) and erythema, as well as pruritus. See, Berdy, G.J. et al., “Allergic conjunctivitis: A survey of new antihistamines.” T. Ocular Pharmacol.. 7:313-324 (1991).
Although there are many different antihistamines available for systemic treatment of allergies and related ailments, many such antihistamines are not suitable for topical ophthalmic use because of limited ocular bioavailability. For example, terfenadine (Seldane®, made by Marion Merrell Dow), astemizole (Hismanal®, made by Janssen Pharmaceutica) and loratadine (Claritin®, made by Schering) all have good systemic activity; however, terfenadine has little or no local ocular activity, and astemizole and loratadine each have greatly reduced local ocular activity (as compared to its systemic activity).
Data on Bristol-Myers Squibb’s Anti IL-6 Antibody, Clazakizumab, Developed by Alder Biopharmaceuticals, to Be Presented at the American College of Rheumatology (ACR) 2013 Annual Meeting
This week, Bristol-Myers Squibb Company announced new data on the investigational anti-IL-6 antibody clazakizumab in adult patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate will be highlighted at the 2013 annual meeting of the American College of Rheumatology (ACR), taking place from October 25-30 in San Diego, Calif.
This asset was developed by Alder Biopharmaceuticals, and partnered with Bristol-Myers Squibb in 2009 in a deal worth $85 million up front and an additional $764 million in potential milestone payments. The therapeutic was developed using Alder’s yeast-based production technology, Mab Xpress, which enables the production of high quantities of antibodies.
Alder’s technology allows this class of therapeutics to enter disease areas that have previously been inaccessible for antibodies, such as migraine and cardiovascular disease. Alder is advancing an antibody therapeutic developed using this technology, ALD403, which targets the calcitonin gene-related peptide (CGRP) and holds promise for treating…
View original post 41 more words
