
Home » Phase3 drugs (Page 22)
Category Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TAVABOROLE, AN 2690, 他伐硼罗 Таваборол تافابورول
TAVABOROLE
- AN 2690
- AN-2690
- AN2690
- UNII-K124A4EUQ3
5-Fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole
5-Fluoro-2,1-benzoxaborol-1(3H)-ol;
1,3-Dihydro-5-fluoro-1-hydroxy-2,1-benzoxaborole
MOLECULAR FORMULA C7H6BFO2
MOLECULAR WEIGHT 151.9
SPONSOR Anacor Pharmaceuticals, Inc.
CAS REGISTRY NUMBER 174671-46-6
Mp 118-120° C…..US20070265226
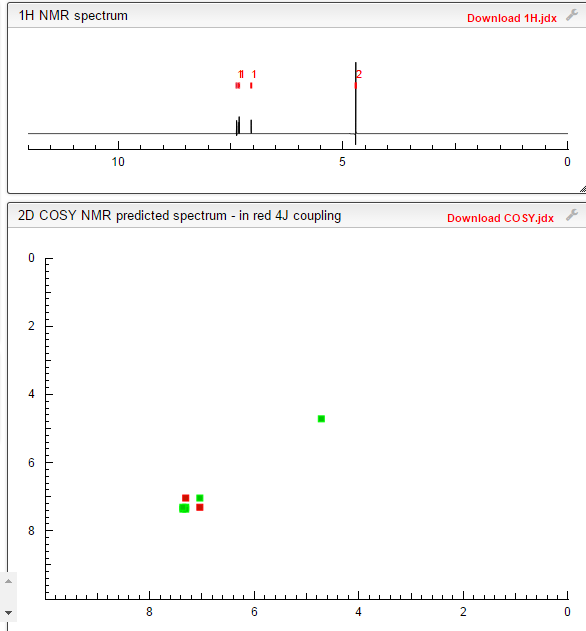
1H NMR (300 MHz, DMSO-d6) δ (ppm) 4.95 (s, 2H), 7.15 (m, 1H), 7.24 (dd, J=9.7, 1.8 Hz, 1H), 7.74 (dd, J=8.2, 6.2 Hz, 1H), 9.22 (s, 1H)
FDA APPROVED JULY 2 2014………..“FDA Approves Anacor Pharmaceuticals’ KERYDIN™ (Tavaborole) Topical Solution, 5% for the Treatment of Onychomycosis of the Toenails”. Market Watch. July 8, 2014.
Has antifungal activity.
The US Food and Drug Administration (FDA) 2014 JULY 8 ratified the Anacor’s Kerydin (5% Tavaborole solution) for the topical treatment of nail fungal infections. Tavaboroleindications of toenail fungus Trichophyton rubrum or Trichophyton rubrum infections.Instructions recommended once a day for toenail infections, treatment for 48 weeks, on the recommendation of Anacor, and do not need to nail debridement.
I tis an oxaborole antifungal used topically, as a 5% w/w solution, for the treatment of onychomycosis of the toenails due to Trichophyton rubrumor T. mentagrophytes. It is applied to the affected toenail once daily for 48 weeks.
Ingrowing toenails and application site reactions including exfoliation, erythema, and dermatitis have been reported during use.
1H NMR FROM NET

CLICK ON IMAGE FOR CLEAR VIEW
COSY NMR PREDICT
Tavaborole (AN2690, trade name Kerydin) is a topical antifungal medication for the treatment of onychomycosis, a fungal infectionof the nail and nail bed. Tavaborole began its Phase 3 trials in December 2010[1] and was approved in July 2014.[2] Tavaborole inhibits an essential fungal enzyme, Leucyl-tRNA synthetase, or LeuRS, required for protein synthesis. The inhibition of protein synthesis leads to termination of cell growth and cell death, eliminating the fungal infection. No treatment-related systemic side effects were observed in any of its clinical trials.
Tavaborole is the first oxygen boron used to treat toenail infections dioxolane (oxaborole) antifungal agents, located in Palo Alto, Anacor focuses on boron-based drug development and production, according to the latest news, Tavaborole future also be used to infect fingernails. Wedbush Securities analyst predicts that next year the drug sales in the United States for $ 16 million, by 2021 will reach peak sales of $ 347 million.
Gram-negative bacteria cause approximately 70% of the infections in intensive care units. A growing number of bacterial isolates responsible for these infections are resistant to currently available antibiotics and to many in development. Most agents under development are modifications of existing drug classes, which only partially overcome existing resistance mechanisms. Therefore, new classes of Gram-negative antibacterials with truly novel modes of action are needed to circumvent these existing resistance mechanisms. We have previously identified a new a way to inhibit an aminoacyl-tRNA synthetase, leucyl-tRNA synthetase (LeuRS), in fungi via the oxaborole tRNA trapping (OBORT) mechanism.
Herein, we show how we have modified the OBORT mechanism using a structure-guided approach to develop a new boron-based antibiotic class, the benzoxaboroles, which inhibit bacterial leucyl-tRNA synthetase and have activity against Gram-negative bacteria by largely evading the main efflux mechanisms in Escherichia coli and Pseudomonas aeruginosa. The lead analogue, is active against Gram-negative bacteria, including Enterobacteriaceaebearing NDM-1 and KPC carbapenemases, as well as P. aeruginosa. This novel boron-based antibacterial, has good mouse pharmacokinetics and was efficacious against E. coli and P. aeruginosa in murine thigh infection models, which suggest that this novel class of antibacterials has the potential to address this unmet medical need.
Anacor continued development on that drug, tavaborole, and filed for FDA approval in July. The FDA will review the phase 3 trial data and issue a decision on July 29, 2014.
If approved, Anacor hopes tavaborole’s ability to clear onychomycosis in 10% of treated patients will be enough to win market share away from generic Lamisil and generic topical Pentac. While Lamisil cleared the fungus in 38% of patients, it’s been associated with rare cases of liver failure. And Pentac requires frequent debridement of the nail and only clears the fungus in 5.5% to 8.5% of patients.
Tavaborole is a novel, topical antifungal medication being developed for the topical treatment of onychomycosis, a nail fungus infection, which affects seven to ten percent of the U.S. population. Early studies show AN-2690 penetrates the nail effectively and has robust activity against dermatophytes, which cause onychomycosis.

1H NMR PREDICT

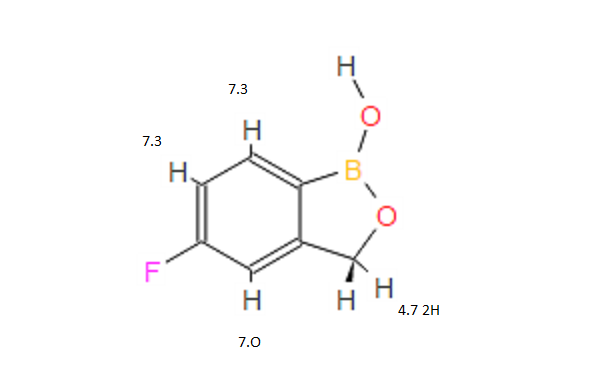
……………………………………………………………………………
13 C NMR PREDICT

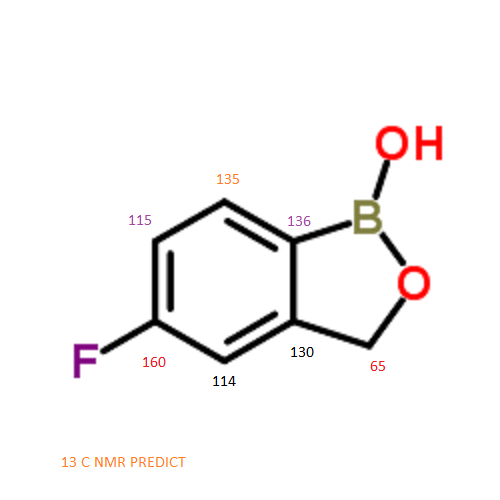
ARTICLE
July 18, 2013
Anacor Pharmaceuticals to Present Pivotal Phase 3 Data of Tavaborole for the Topical Treatment of Toenail Onychomycosis
Abstract Accepted for Oral Presentation at the 2013 American Podiatric Medical Association Annual Scientific Meeting
PALO ALTO, Calif.–(BUSINESS WIRE)– Anacor Pharmaceuticals (NASDAQ:ANAC) announced today that its abstract “Pivotal Phase 3 Safety and Efficacy Results of Tavaborole (Formerly AN2690), a Novel Boron-Based Molecule for the Topical Treatment of Toenail Onychomycosis” was accepted for oral presentation at the 2013 APMA Annual Scientific Meeting (The National) to be held in Las Vegas, Nevada. Max Weisfeld, DPM, will present the data from tavaborole’s Phase 3 studies on Monday, July 22, 2013 during the Evidence-Based Medicine and Oral Abstracts session.
As announced earlier this year, tavaborole achieved statistically significant and clinically meaningful results on all primary and secondary endpoints in two Phase 3 pivotal studies without concomitant debridement. Anacor is seeking approval for tavaborole from the Food and Drug Administration (FDA) and will file a New Drug Application imminently. Currently, there is only one FDA-approved topical treatment for onychomycosis, a fungal infection of the nail and nail bed, which affects approximately 35 million people in the United States.
“I’m impressed with tavaborole’s safety and efficacy data. There is no FDA-approved topical treatment for onychomycosis with tavaborole’s range of efficacy and ability to penetrate the nail to reach the site of the infection,” said Dr. Weisfeld. “Tavaborole’s Phase 3 results demonstrate its ability to clear the nail and eliminate the infection which is important to both patients and the physicians who treat them. In addition, tavaborole is easy to apply and dries quickly which makes it convenient for patients to use.”
“We are pleased to present these positive data at the APMA’s Annual Scientific Meeting, the leading annual meeting of podiatrists. As we seek FDAapproval for tavaborole, we look forward to developing relationships with podiatrists to potentially offer them a new treatment option for the large number of patients who seek treatment for onychomycosis,” said David Perry, Chief Executive Officer of Anacor Pharmaceuticals.
About the Studies
Anacor conducted two separate Phase 3 studies of tavaborole on patients with distal subungual onychomycosis affecting 20 to 60 percent of the target great toenail. Approximately 600 patients aged 18 years and older with no upper age limit (the oldest subject was 88 years old) were enrolled in each study and randomized two-to-one to receive either tavaborole or the vehicle control. Patients were instructed to apply tavaborole solution or the vehicle to the toenail once daily for 48 weeks.
A copy of the presentation will be available on Anacor’s website following the oral session.
About Anacor Pharmaceuticals
Anacor is a biopharmaceutical company focused on discovering, developing and commercializing novel small-molecule therapeutics derived from its boron chemistry platform. Anacor has discovered eight compounds that are currently in development. Its two lead product candidates are topically administered dermatologic compounds — tavaborole, a topical antifungal for the treatment of onychomycosis, and AN2728, a topical anti-inflammatory PDE-4 inhibitor for the treatment of atopic dermatitis and psoriasis. In addition to its two lead programs, Anacor has discovered three other wholly-owned clinical product candidates — AN2718 and AN2898, which are backup compounds to tavaborole and AN2728, respectively, and AN3365 an antibiotic for the treatment of infections caused by Gram-negative bacteria. We have discovered three other compounds that we have out-licensed for further development — two compounds for the treatment of animal health indications that are licensed to Eli Lilly and Company and AN5568, also referred to as SCYX-7158, for human African trypanosomiasis (HAT, or sleeping sickness), which is licensed to Drugs for Neglected Diseases initiative, or DNDi. We also have a pipeline of other internally discovered topical and systemic boron-based compounds in development. For more information, visit http://www.anacor.com.
Patents
WO 1995033754
WO 2004009578….
WO 2006089067
WO 2008025543
…………………………..
SYNTHESIS

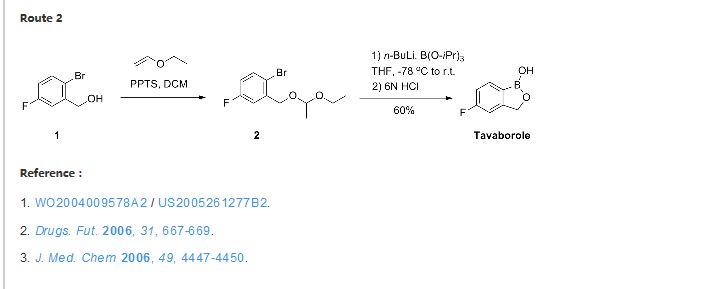
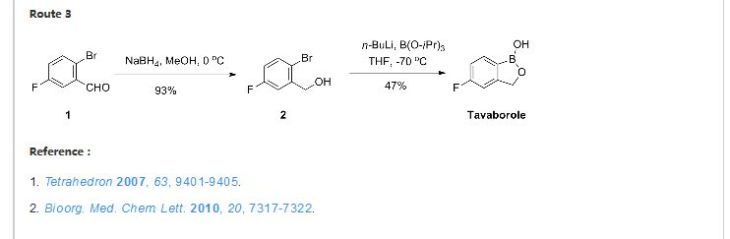
Reference:
ELI LILLY AND COMPANY Patent: WO2004/9578 A2, 2004 ; Location in patent: Page 36-37 ; WO 2004/009578 A2
PATENT
Anacor Pharmaceuticals Patent: US2007/265226 A1, 2007 ; Location in patent: Page/Page column 59 ;
http://www.google.com/patents/US20070265226

1,3-Dihydro-5-fluoro-1-hydroxy-2,1-benzoxaborole (19b)
To a solution of 5b (73.2 g, 293 mmol) in dry THF (400 mL) was added n-butyllithium (1.6 M in hexanes; 200 mL) over 45 min at −78° C. under nitrogen atmosphere. Anion precipitated. After 5 min, (i-PrO)3B (76.0 mL, 330 mmol) was added over 10 min, and the mixture was allowed to warm to room temperature over 1.5 h. Water and 6 N HCl (55 mL) were added, and the solvent was removed under reduced pressure to about a half volume. The mixture was poured into ethyl acetate and water. The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure. To a solution of the residue in tetrahydrofuran (360 mL) was added 6 N HCl (90 mL), and the mixture was stirred at 30° C. overnight. The solvent was removed under reduced pressure to about a half volume. The mixture was poured into ethyl acetate and water. The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was treated with i-Pr2O/hexane to give 19b (26.9 g, 60%) as a white powder:
mp 118-120° C.;
1H NMR (300 MHz, DMSO-d6) δ (ppm) 4.95 (s, 2H), 7.15 (m, 1H), 7.24 (dd, J=9.7, 1.8 Hz, 1H), 7.74 (dd, J=8.2, 6.2 Hz, 1H), 9.22 (s, 1H);
ESI-MS m/z 151 (M−H)−;
HPLC purity 97.8%; Anal (C7H6BFO2) C, H.
…………………
Gunasekera, Dinara S.; Gerold, Dennis J.; Aalderks, Nathan S.; Chandra, J. Subash; Maanu, Christiana A.; Kiprof, Paul; Zhdankin, Viktor V.; Reddy, M. Venkat Ram Tetrahedron, 2007 , vol. 63, # 38 p. 9401 – 9405
…………………….
Baker, Stephen J.; Zhang, Yong-Kang; Akama, Tsutomu; Lau, Agnes; Zhou, Huchen; Hernandez, Vincent; Mao, Weimin; Alley; Sanders, Virginia; Plattner, Jacob J. Journal of Medicinal Chemistry, 2006 , vol. 49, # 15 p. 4447 – 4450
………………..
Ding, Charles Z.; Zhang, Yong-Kang; Li, Xianfeng; Liu, Yang; Zhang, Suoming; Zhou, Yasheen; Plattner, Jacob J.; Baker, Stephen J.; Liu, Liang; Duan, Maosheng; Jarvest, Richard L.; Ji, Jingjing; Kazmierski, Wieslaw M.; Tallant, Matthew D.; Wright, Lois L.; Smith, Gary K.; Crosby, Renae M.; Wang, Amy A.; Ni, Zhi-Jie; Zou, Wuxin; Wright, Jon Bioorganic and Medicinal Chemistry Letters, 2010 , vol. 20, # 24 p. 7317 – 7322
…………..
PATENT
PREPARATION 13 5-Fluoro-3H-benzo[c][1,2)oxaborol-1-ol

Dissolve 1-bromo-2-(1-ethoxy-ethoxymethyl)-4-fluoro-benzene(5.4 g, 19.5 mmol) in dry THF (100 mL) and cool to −78° C. under nitrogen. Add butyl lithium (2.5M in Hexanes, 10.2 mL, 25.4 mmol) dropwise at −78° C. Upon complete addition, stir the reaction at −78° C. for 10 minutes and then add trimethyl borate (4.4 mL, 39 mmol) and warm the reaction to room temperature. Pour the reaction into 1N HCl (100 mL) and stir for 1 hour. Extract the biphasic mixture with ether three times. Dry the combined organic layers with sodium sulfate, filter and concentrate in vacuo. Triturate the oily residue with cold hexanes to yield 2.1 g (70%) of the title compoud as a white solid.
1H NMR (d6-DMSO)
9.18 (s, 1H),
7.70 (dd, J=8.2, 5.8 Hz, 1H),
7.20 (dd, J=9.5, 2.7 Hz, 1H),
7.11 (m, 1H), 4.92 (s, 1H).
…………………
SEE
http://jpet.aspetjournals.org/content/early/2012/11/28/jpet.112.200030.full.pdf
………………………………..
SEE
Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, Mao W, Alley MR, Sanders V, Plattner JJ.
J Med Chem. 2006 Jul 27;49(15):4447-50.
Boron-containing inhibitors of synthetases.
Baker SJ, Tomsho JW, Benkovic SJ.
Chem Soc Rev. 2011 Aug;40(8):4279-85. doi: 10.1039/c0cs00131g. Epub 2011 Feb 7. Review.
- Benzoxaborole antimalarial agents. Part 2: Discovery of fluoro-substituted 7-(2-carboxyethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaboroles.
Zhang YK, Plattner JJ, Freund YR, Easom EE, Zhou Y, Ye L, Zhou H, Waterson D, Gamo FJ, Sanz LM, Ge M, Li Z, Li L, Wang H, Cui H.
Bioorg Med Chem Lett. 2012 Feb 1;22(3):1299-307. doi: 10.1016/j.bmcl.2011.12.096. Epub 2011 Dec 28.
Tavaborole Market Opportunity
Anacor is developing tavaborole specifically to address the current limitations of existing treatment options for onychomycosis. This includes designed leaps forward in both the potential safety and efficacy profile aimed to make the drug a best-in-class therapy. Additionally, management has used the company’s expertise in medicinal chemistry to improve delivery of the compound through the nail plate to the nail bed, the site of onychomycosis infection. For example, preclinical studies indicate that tavaborole is able to penetrate the nail plate 250 times more effectively than ciclopirox.
Tavaborole novel mechanism of action inhibits an essential fungal enzyme, leucyl transfer RNA synthetase, or LeuRS required for protein synthesis. The inhibition of protein synthesis leads to termination of cell growth and cell death, eliminating the fungal infection.
Likewise, the topical dosing was designed to eliminate systemic absorption. Previous preclinical and clinical data shows topical treatment with tavaborole resulted in little or no detectable levels of drug in the blood or urine. No treatment related systemic side effects have been observed in any clinical trials to date. Safety data from the company’s studies to date was recently presented at the 100th National APMA meeting in Washington, DC.
Anacor’s topical solution currently in two phase III trials for onychomycosis. Phase II data with tavaborole suggests efficacy superior to ciclopirox with little to no systemic exposure.
Data from an open-label phase 2 program with tavaborole showed 50% patients using a 7.5% solution saw 2 mm clear nail growth and negative fungal cultures after six months. Roughly 25% of the patients saw 5 mm clear nail growth and negative fungal cultures after six months.
Anacor and partner Merck (NYSE:MRK) met with the U.S. FDA in 2009 to discuss the phase II data. Merck has since returned the rights to tavaborole to Anacor. The original deal was with Schering-Plough in 2007. Merck most likely felt as though tavaborole clashed with existing products or did not have peak sales potential large enough to continue the partnership with Anacor. We see tavaborole as a specialty promoted product, into podiatrists and dermatologists. For a company like Anacor, it’s an attractive first product.
Anacor’s first phase III trial completed enrollment in November 2011. The second phase III trial completed enrollment in December 2011. Data from these trials are expected around the middle of January 2013. Data from the second study is expected six weeks later. Given the positive phase II data noted above, we think odds favor a positive outcome. A benchmark for the trial is the efficacy of Lamisil, which is a complete cure rate of around 35% to 40%, and a mycological cure of around 70% after a typical course of treatment.
I note that on Anacor’s third quarter conference call management noted that they are pleased with the conduct of the trial to date. Specifically, the compliance rate appears to better than management had expected. The trial was designed with a 20% drop-out rate. It looks as though the drop-out rate is only around 13%, at a minimum suggestive of good safety and tolerability, but potentially also a sign that the drug is working.
I see onychomycosis as a significant market opportunity for Anacor. An estimated 35 million Americans have nail fungus, with about 95% of the infections in the toenail. With efficacy similar to Lamisil, we think Anacor can capture 20% of the market. With a price per course of treatment at around $1,200, I think peak sales of tavaborole are $500 million.
Conclusion
I’ll note two more important pieces of information for investors. Firstly, besides optimism for tavaborole, Anacor has apipeline of anti-infectant drugs. For this article I discussed only tavaborole. A second article can be dedicated entirely to AN2728 for the treatment of psoriasis and atopic dermatitis. Anacor also has an animal health collaboration with Eli Lilly (NYSE:LLY).
The second important thing to note is Anacor’s cash position. The company reported financial results on November 7, 2012. The company held $36.6 million in cash on the balance sheet as of September 30, 2012. However, in October 2012, the company completed an underwritten public offering of 4.0 million shares of common stock at $6.00 per share to raise net proceeds of $22.7 million. I view the current cash position as sufficient to report data from both phase 3 trials and, if positive, file the new drug application (NDA) around the middle of 2013.
With phase 3 data expected in less than two months, good prior evidence of both safety and efficacy, and a solid cash position, I think Anacor could be an attractive investment at today’s price. The stock is down meaningfully over the past month and investors can buy sizably below the October offering.


SYNTHESIS

References
- Clinical trial number NCT01270971 at ClinicalTrials.gov
- “FDA Approves Anacor Pharmaceuticals’ KERYDIN™ (Tavaborole) Topical Solution, 5% for the Treatment of Onychomycosis of the Toenails”. Market Watch. July 8, 2014.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204427s000lbl.pdf
- http://www.molbase.com/en/hnmr_174671-46-6-moldata-1568017.html#tabs
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-Fluoro-2,1-benzoxaborol-1(3H)-ol
|
|
| Clinical data | |
| Trade names | Kerydin |
| Legal status |
|
| Routes of administration |
Topical use only |
| Identifiers | |
| CAS Registry Number | 174671-46-6 |
| ATC code | None |
| PubChem | CID: 11499245 |
| ChemSpider | 9674047 |
| Synonyms | AN2690 |
| Chemical data | |
| Formula | C7H6BFO2 |
| Molecular mass | 151.93 g/mol |
NMR PREDICT
H-NMR spectral analysis
 CAS NO. 174671-46-6, 5-fluoro-1-hydroxy-3H-2,1-benzoxaborole H-NMR spectral analysis |

C-NMR spectral analysis
 CAS NO. 174671-46-6, 5-fluoro-1-hydroxy-3H-2,1-benzoxaborole C-NMR spectral analysis |

more

 Anacor
Anacor
BIG TEAM Hernandez, front row, fifth from left, poses during a research meeting at Naeja’s headquarters.
Anacor Pharmaceuticals is out to change that. The Palo Alto, Calif.-based biotechnology company is developing a family of boron-containing small-molecule drugs. And with the assistance of Naeja Pharmaceutical, a Canadian contract research organization, Anacor has licensed one of those molecules to GlaxoSmithKline and taken another one into Phase III clinical trials.
Anacor was founded in 2002 to develop technology created by Lucy Shapiro, a Stanford University bacterial geneticist, and Stephen J. Benkovic, a Pennsylvania State University organic chemist. Through a long-standing scientific collaboration, the two researchers had discovered boron-containing compounds that inhibited specific bacterial targets………..https://pubs.acs.org/cen/coverstory/89/8912cover3.html
UPDATED………….
mp 118-120….http://www.syninnova.com/catalog/product/SL-264
antifugal AN2690 by Anacor
Tavaborole inhibits an essential fungal enzyme, Leucyl-tRNA synthetase, or LeuRS, required for protein synthesis.
Minimum Inhibitory Concentration: 1, 1, 0.5, 0.25, and 0.25 μg/mL for T.rubrum, T.mentagrophytes, C.albicans, C.neoformans, A.fumigatus, respectivley.
AN2690 is a new boron-containing antifungal agent for the potential treatment of onychomycosis. Onychomycosis is caused mainly by dermatophytes, a class of fungus that dwells on skin, hair, and nails and is the cause of other cutaneous fungal infections such as athlete’s foot.
In vitro: AN2690 showed the most active against fungi and especially against the dermatophytes T. rubrum and T. mentagrophytes, the primary fungal pathogens causing onychomycosis. In addition, AN2690 was identified as having a unique profile of in vitro antidermatophyte activity, maintenance of this activity in the presence of keratin, and exceedingly good penetration of human nails [1].
Ex vivo: AN2690 was found to have superior penetration compared to ciclopirox, and achieves levels within and under the nail plate that suggest it has the potential to be an effective topical treatment for onychomycosis [2].
Clinical trial: The efficacy of tavaborole as a topical treatment for onychomycosis has been evaluated in two identical randomised, double-blind phase III studies, NCT01270971 (301) and NCT01302119 (302), enrolling 593 and 601 patients, respectively. Completely or almost clear nail and negative mycology was achieved in 15.3 and 17.9 % of tavaborole recipients compared with 1.5 and 3.9 % of vehicle recipients [3]
References:
[1] Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, Mao W, Alley MR, Sanders V, Plattner JJ. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole (AN2690), for the potential treatment of onychomycosis. J Med Chem. 2006;49(15):4447-50.
[2] Hui X, Baker SJ, Wester RC, Barbadillo S, Cashmore AK, Sanders V, Hold KM, Akama T, Zhang YK, Plattner JJ, Maibach HI. In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci. 2007;96(10):2622-31.
[3] Markham A. Tavaborole: first global approval. Drugs. 2014;74(13):1555-8.
UPDATE
http://www.google.im/patents/EP1976536A2?cl=en
EXAMPLE 23
Alternative Preparation of 4 from 3
A 22.0 L 3-neck flask was equipped with a stir motor, N2 inlet, addition funnel, heating mantle, and condenser. The flask was charged with 3500 g (17.1 moi) of 2-bromo-5-fluorobenzyl alcohol followed by the addition of 3556 g of tetrahydrofuran and 16.4 g (0.17 mol) of methanesulfonic acid. Next, 400 g (4.7 mol) of 3,4-dihydro-2H-pyran was added at 100C. This step is exothermic so no additional charges should be made until exotherm subsides. The temperature was increased to 27°C, stirred for 15 min and then charged with 400 g (4.7 mol) of 3,4-dihydro-2H- pyran at 240C. Again the temperature increased (24°C to 380C). The mixture was stirred for 15 min. Once the exotherm subsided, the flask was again charged with 40Og (4.7 mol) of 3,4-dihydro-2H-pyran at 350C. The temperature again increased to 470C over a 20 min period. Once the exotherm subsided, the mixture was stirred for 15 min. Finally the remaining 400 g (4.7 mol) of 3,4-dihydro-2H-pyran was added at 440C. The temperature increased to 510C. After stirring for one hour, a sample was removed to check for removal of starting material. Upon reaction completion, contents were cooled to 20 ± 5 0C.
EXAMPLE 24

Alternative Preparation of 5 from 4
To a 22.0 L 3-neck flask equipped with a stir motor, N2 inlet, addition funnel, cooling bath, and condenser was charged 436 g (17.96 mol) of magnesium turnings. 5334 g of tetrahydrofuran was then added followed by 291 g (0.51 mol) of diisobutylaluminum hydride (DIBAL) (25%wt) in toluene. The mixture was stirred for 60 min at 20 ± 5 0C. Some gas evolution was seen. Next, 260-430 g -3-5% (by weight if solution of 4 was dropped to drums) of 4 in THF was added. The mixture was stirred for 15-30 min at which time a slight exotherm should be seen (ΔT = 10- 150C). Once the exotherm was observed, the reaction mixture was cooled to 5 ± 5 0C. To this mixture, the remaining 8.22-8.39 kg of 4 in THF was added at a rate such that the temperature was kept below 300C (t = 3h). The reaction was stirred at 20-25 0C for 30 min, at which time an aliquot was removed, quench with 3 N HCl (10 mL), and analyzed.
Upon completion, the contents were cooled to -25 ± 5°C. A solution of trimethylborate in THF was prepared by mixing 2665 g (25.7 mol) of trimethyl borate and 6666 g of tetrahydrofuran. This solution can be prepared in a drum with stirring. [0618] Next, the 9331 g of trimethyl borate in THF was added at a rate such that the temperature was kept between -35 and -20 °C (t = 2.5h). The mixture became very thick so THF was added. After stirring at -25 ± 5°C for 10 min, 50 mL aliquot was removed, quenched with 25 mL of 3N HCl, and submitted for CoR. Stirring continued at -25 ± 50C for Ih, and then the mixture was allowed to warm to ambient temperature, where it was stirred for at least 12h. Pull two samples (one at 6h and the other at 12h).
Results:
1H-NMR (300 MHz, DMSO-d6) δ (ppm) 1.45-1.75 (m, 6H), 3.53 (s, 6H), 3.45 (m, IH), 3.75 (m, IH), 4.69 (t, J=3 Hz, IH), 4.97 (d, J=14.1 Hz, IH), 5.14 (d, J=14.1 Hz, IH), 7.03 ((td, J=8.4, 2.7 Hz, IH), 7.24 (dd, J=10.8, 2.1 Hz, IH), 7.89 (t, J=7.8 Hz, IH), 8.76 (s, IH).
EXAMPLE 25
Alternative Preparation of I from 5

To the reaction mixture above was added 5.3 kg of USP water. After stirring for 30 min, the mixture was charged 5.3 kg of acetic acid. Gas evolution was seen. After stirring for 30 min, an aliquot was removed for analysis. Mixture was then heated to reflux for 36-48 hours. During the reflux period, 12-13 L of THF were removed.
When the reaction was complete, the contents were cooled by the reactor to <40°C by setting jacket and by charging 10.5 kg of USP water. THF was removed until distillate did not remain. Contents of the reactor were transferred to Rosenmund filter dryer and allowed to cool to 20 ± 5°C. Reactor was rinsed with water, filtered, and then washed again with 10.5 kg of USP water. The flask was charged with 10.5 kg of 10% ACN in water (v/v) and agitated for Ih. After filtering, the cake was washed with 10.5 kg of 10% ACN in water (v/v), and then charged with 10.5 kg 10% ACN in water (v/v). The contents were agitated for Ih. The contents were subsequently washed with 10.5 kg of USP water, charged with 7.0 L of 5% Methyl t- Butyl Ether (MTBE)/Heptane (v/v), agitated for Ih, filtered, charged with 7.0 L of 5% MTBE/Heptanes (v/v) and again agitated for Ih. After filtering, the contents were charged again with 7.0 L of heptane and filtered. Solids were dried at <45°C to constant weight. Solids were recrystallized from toluene :heptane 75:25.
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
///////
Zosuquidar

LY335979, RS-33295-198 (Zosuquidar)
Roche Palo Alto (Originator)
LY335979 (Zosuquidar) is a selective Pgp (P-glycoprotein) inhibitor with a Ki of 59 nM. LY335979 significantly enhanced the survival of mice implanted with Pgp-expressing murine leukemia (P388/ADR) when administered in combination with either daunorubicin, doxorubicin or etoposide.
LY335979 (Zosuquidar)
M.Wt: 636.99
Formula: C32H31F2N3O2.3HCl
Name: Zosuquidar trihydrochloride
Elemental Analysis: C, 60.34; H, 5.38; Cl, 16.70; F, 5.97; N, 6.60; O, 5.02
CAS : 167465-36-3
167354-41-8 (free base)
Roche Bioscience (Originator), Eli Lilly and Company (Licensee).
US5654304, WO1994024107A1, WO2000075121, US6570016
Drug Des Discov 1992, 9(1): 69, Bioorg Med Chem Lett 1995, 5(21): 2473, Drugs Fut 2003, 28(2): 125
Zosuquidar is currently under development. It is now in “Phase 3” of clinical tests in the United States. Its action mechanism consists of the inhibition of P-glycoproteins; other drugs with this mechanism include tariquidar and laniquidar. P-glycoproteins are proteins which convert the energy derived from the hydrolysis of ATP to structural changes in protein molecules, in order to perform coupling, thus discharging medicine from cells. If P-glycoprotein coded with the MDR1 gene manifests itself in cancer cells, it discharges much of the antineoplastic drugs from the cells, making cancer cells medicine tolerant, and rendering antineoplastic drugs ineffective. This protein also manifests itself in normal organs not affected by the cancer (such as the liver, small intestine, and skin cells in blood vessels of the brain), and participates in the transportation of medicine. The compound Zosuquidar inhibits this P-glycoprotein, causing the cancer cells to lose their medicine tolerance, and making antineoplastic drugs effective
Clinicial trials: Clinical report published in 2010 showed that zosuquidar did not improve outcome in older acute myeloid leukemia, in part, because of the presence P-gp independent mechanisms of resistance. (Blood. 2010 Nov 18;116(20):4077-85.)
Zosuquidar is a potent P-glycoprotein inhibitor, which binds with high affinity to P-glycoprotein and inhibits P-glycoprotein-mediated multidrug resistance (MDR). P-glycoprotein, encoded by the MDR-1 gene, is a member of the ATP-binding cassette superfamily of transmembrane transporters and prevents the intracellular accumulation of many natural product-derived cytotoxic agents
Zosuquidar
U.S. Patent No. 5,112,817 to Fukazawa et al. discloses certain quinoline derivatives useful as anticancer drug potentiators for the treatment of multidrug resistance. One of the initially promising active agents there-disclosed is MS-073, which has the following structure:

MS-073
U.S. Pat. Nos. 5,643,909 and 5,654,304 disclose a series of 10,11- methanobenzosuberane derivatives useful in enhancing the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. One such derivative having good activity, oral bioavailability, and stability, is zosuquidar, a compound of formula (2R)-anti-5-
3 – [4-( 10, 11 -difluoromethanodibenzosuber-5-yl)piperazin- 1 -yl]-2-hydroxypropoxy) quinoline.

Zosuquidar
Given the limitations of previous generations of MDR modulators, three preclinical critical success factors were identified and met for zosuquidar: 1) it is a potent inhibitor of P-glycoprotein; 2) it is selective for P-glycoprotein; and 3) no pharmacokinetic interaction with co-administered chemotherapy is observed.
Zosuquidar is extremely potent in vitro (Kj = 59 nM) and is among the most active modulators of P-gp-associated resistance described to date. Zosuquidar has also demonstrated good in vivo activity in preclinical animal studies. In addition, the compound does not appear to be a substrate for P-gp efflux, resulting in a relatively long duration of reversal activity in resistant cells even after the modulator has been withdrawn.
Another significant attribute of zosuquidar as an MDR modulator is the minimal pharmacokinetic (PK) interactions with several oncolytics tested in preclinical models. Such minimal PK interaction permits normal doses of oncolytics to be administered and also a more straightforward interpretation of the clinical results.
Zosuquidar is generally administered in the form of the trihydrochloride salt. Conventional zosuquidar trihydrochloride formulations include those containing zosuquidar (50 mg as free base), glycine (15 mg), and mannitol (200 mg) dissolved in enough water for injection, to yield a free base concentration of 5 mg/mL. The formulation is filled into vials and lyophilized to give a vial containing 50 mg of free base. For such formulations, a 30 mL vial size is necessary to contain 50 mg of thezosuquidar formulation. For a typical >200 mg dose of zosuquidar, multiple 50 mg vials are needed to contain the formulation, greatly increasing manufacturing costs and reducing convenience for the end user {e.g., a pharmacist). Modified Cyclodextrins
Cyclodextrins are cyclic oligomers of glucose; these compounds form inclusion complexes with any drug whose molecule can fit into the lipophile-seeking cavities of the cyclodextrin molecule. See U.S. Pat. No. 4,727,064 for a description of various cyclodextrin derivatives. Cyclodextrins of preferred embodiments can include α-, β-, and χ-cyclodextrins. The α-cyclodextrins include six glucopyranose units, the β- cyclodextrins include seven glucopyranose units, and the χ-cyclodextrins include eight glucopyranose units. The β -cyclodextrins are generally preferred as having a suitable cavity size for zosuquidar. Cyclodextrin can be in any suitable form, including amorphous and crystalline forms, with the amorphous form generally preferred. Cyclodextrins suitable for use in the formulations of preferred embodiments include the hydroxypropyl, hydroxyethyl, glucosyl, maltosyl, and maltotrosyl derivatives of β- cyclodextrin, carboxyamidomethyl-β-cyclodextrin, carboxymethyl-β-cyclodextrin, and diethylamino-β-cyclodextrin.
Pharmaceutical complexes including various cyclodextrins and cyclodextrin derivatives are disclosed in the following United States patents: U.S. Pat. No. 4,024,223; U.S. Pat. No. 4,228,160; U.S. Pat. No. 4,232,009; U.S. Pat. No. 4,351,846; U.S. Pat. No. 4,352,793; U.S. Pat. No. 4,383,992; U.S. Pat. No. 4,407,795; U.S. Pat. No. 4,424,209; U.S. Pat. No. 4,425,336; U.S. Pat. No. 4,438,106; U.S. Pat. No. 4,474,881; U.S. Pat. No. 4,478,995; U.S. Pat. No. 4,479,944; U.S. Pat. No. 4,479,966; U.S. Pat. No. 4,497,803; U.S. Pat. No. 4,499,085; U.S. Pat. No. 4,524,068; U.S. Pat. No. 4,555,504; U.S. Pat. No. 4,565,807; U.S. Pat. No. 4,575,548; U.S. Pat. No. 4,598,070; U.S. Pat. No. 4,603,123; U.S. Pat. No. 4,608,366; U.S. Pat. No. 4,659,696; U.S. Pat. No. 4,623,641; U.S. Pat No. 4,663,316; U.S. Pat. No. 4,675,395; U.S. Pat. No. 4,728,509; U.S. Pat. No. 4,728,510; and U.S. Pat. No. 4,751,095.
Chemically modified and substituted α-, β-, and χ-cyclodextrins are generally preferred over unmodified α-, β-, and χ-cyclodextrins due to improved toxicity and solubility properties. The degree of substitution of the hydroxy 1 groups of the glucopyranose units of the cyclodextrin ring can affect solubility. In general, a higher average degree of substitution of substituent groups in the cyclodextrin molecule yields a cyclodextrin of higher solubility.
Examples for Pgp inhibitors are cyclosporine A, valpodar, elacridar, tariquidar, zosuquidar, laniquidar, biricodar, S-9788, MS-209, BIBW-22 (BIBW-22-BS) , toremifene, verapamil, dexverapamil , quinine, quinidine, trans- flupentixol, chinchonine and others (J. Roberts, C. Jarry (2003) : J. Med. Chem. 46, 4805 – 4817) . The list of inhibitors of P-glycoprotein is increasing (e.g. Wang et al . (2002) : Bioorg. Med. Chem. Lett. 12, 571 – 574) .
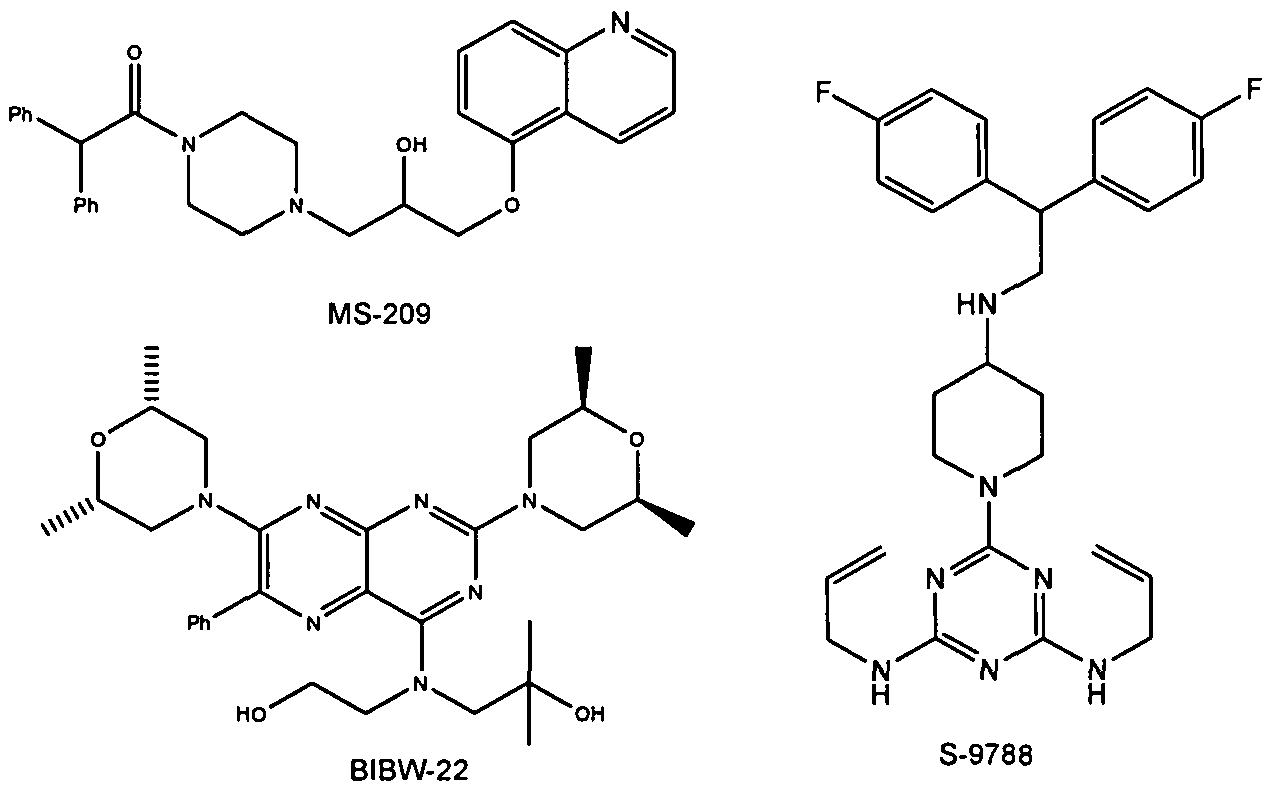
Figure 2: Structures of BIBW-22, MS-209 and S-9788
|
7-12-2000
|
10,11-methanodibenzosuberane derivatives
|
|
|
10-17-2007
|
Salt and crystalline forms of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline
|
|
|
9-2-2009
|
Salt and crystalline forms of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-YL)piperazin-1-YL]-2-hydroxypropoxy}quinoline
|
……………………

U.S. Pat. Nos. 5,643,909 and 5,654,304, incorporated herein by reference, disclose a series of 10,11-methanobenzosuberane derivatives useful in enhancing the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride disclosed therein, is currently under development as a pharmaceutical agent.
U.S. pat. No. 5,654,304 (‘304), incorporated by reference herein, discloses a series of 10,11-(optionally substituted)methanodibenzosuberane derivatives useful in enhancing, the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. (2R)-anti-5-{3-[4-(10,11-Difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinolone trihydrochloride is disclosed in ‘304 and is currently under development as a pharmaceutical agent. WO00/75121 discloses Form I, a crystalline form of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinolone trihydrochloride.
The art disclosed in U.S. Pat. No. 5,776,939, and U.S. Pat. No. 5,643,909 both incorporated herein by reference, and PCT Patent Applications (Publication numbers WO 94/24107 and 98/22112) teach the use of 1-formylpiperazine to introduce the piperazine group of the compound of formula II

Compound II is a mixture of syn isomer (III)

and anti isomer (IV)

The process as disclosed in U.S. Pat. Nos. 5,643,909 and 5,654,304 (represented by scheme A, below) involves (a) chromatographic separation(s) of the formyl piperazine compound; and (b) deformylation of the formyl piperazine compound to provide compound IV.https://www.google.co.in/patents/US6570016?cl=en
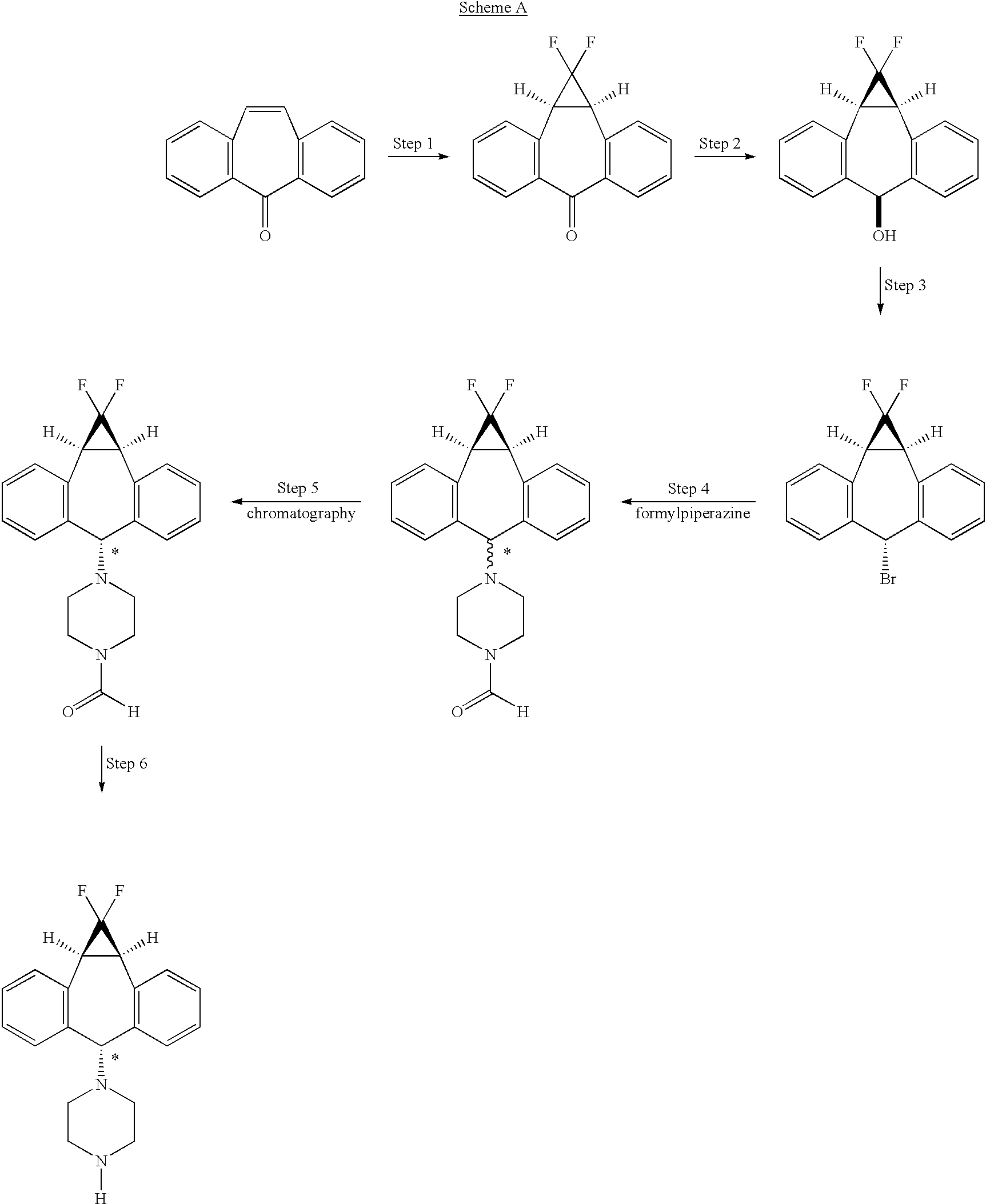
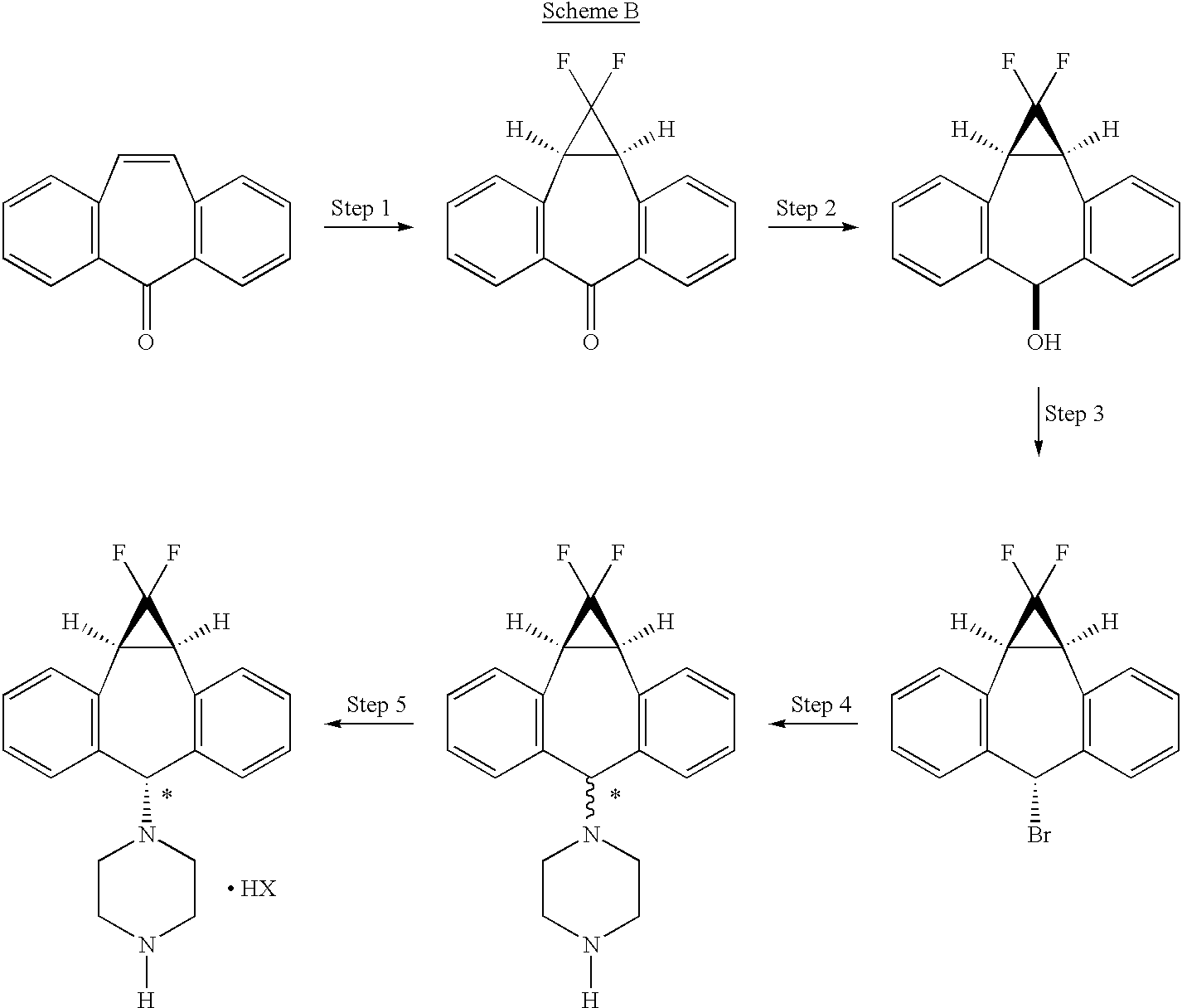
The process of the present invention uses piperazine to react with the (1aα,6α,10bα)-6-halo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cycloheptene compound or derivative, instead of formylpiperazine.
The process of the present invention is advantageous because piperazine is readily available in commercial quantities whereas 1-formylpiperazine, which was utilized in the process disclosed in U.S. Pat. No. 5,643,909 is often not readily available in commercial quantities. Additionally piperazine enjoys a significant cost advantage over 1-formylpiperazine.
The use of piperazine instead of 1-formylpiperazine is a significant advancement over the prior art because it obviates the need to deformylate or hydrolyze off the formyl group (step 6, scheme A), thereby providing fewer operational steps. U.S. Pat. No. 5,643,909 teaches the separation of the 1-formylpiperazine compounds by chromatography or repeated crystallization. The present invention obviates the need for chromatographic separations of the formylpiperazine diastereomeric addition compounds (see step 4, scheme A)
EXAMPLES
The following examples and preparations are illustrative only and are not intended to limit the scope of the invention in any way.
Preparation 1 R-1-(5-Quinolinyloxy)-2,3-epoxypropane
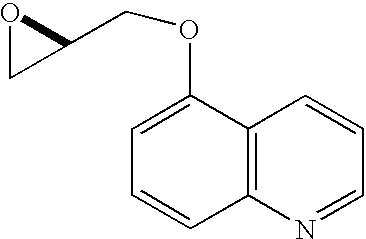
A mixture of 5-hydroxyquinoline (5.60 g, 38.6 mmol), R-glycidyl nosylate (10.0 g, 38.6 mmol), powdered potassium carbonate (11.7 g, 84.9 mmol), and N,N-dimethylformamide (100 mL) was stirred at ambient temperature until HPLC analysis (40% acetonitrile/60% of a 0.5% aqueous ammonium acetate solution, 1 mL/min, wavelength=230 nm, Zorbax RX-C8 25 cm×4.6 mm column) indicated complete disappearance of glycidyl nosylate (approximately 6 hours). The reaction mixture was filtered through paper and the filter cake was washed with 200 mL of a 3:1 mixture of MTBE and methylene chloride. The filtrate was washed with 200 mL of water and the aqueous layer was extracted four times with 100 mL of 3:1 MTBE/methylene chloride. The combined organic layers were dried over 30 grams of magnesium sulfate and the dried solution was then stirred with 50 grams of basic alumina for 30 minutes. The alumina was removed by filtration and the filter cake was washed with 200 mL of 3:1 MTBE/methylene chloride. The filtrate was concentrated to a volume of 100 mL, 300 mL of MTBE were added, and the solution was again concentrated to 80 mL. After heating to 50° C., the solution was treated with 160 mL of heptane dropwise over 15 minutes, allowed to cool to 40° C., and seeded, causing the formation of a crystalline precipitate. The mixture was stirred for two hours at ambient temperature and then at 0-5° C. for an additional 2 hours. The crystals were filtered, washed with cold heptane, and dried to provide 5.68 g (73.2%) of (2R)-1-(5-quinolinyloxy)-2,3-epoxypropane as white needles.
mp 79-81° C.;
[α]25 D−36.4° (c 2.1, EtOH);
1H NMR (500 MHz, CDCl3)δ 2.83 (dd, J=4.8, 2.7 Hz, 1H), 2.97 (m, 1H), 3.48 (m, 1H), 4.10 (dd, J=11.0, 6.0 Hz, 1H), 4.43 (dd, J=11.0, 2.7 Hz, 1H), 6.85 (d, J=7.8 Hz, 1H), 7.38 (dd, J=8.5 Hz, 4.1 Hz, 1H), 7.59 (m, 1H), 7.71 (d, J=8.5 Hz, 1H), 8.61 (m, 1H), 8.90 (m, 1H).
Example 1 (2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-qunolin-5-yloxy)-propan-2-ol Trihydrochloride

Preparation of the above compound is exemplified in the following preparative steps.
Step 1 1,1-Difluoro-1a,10b-dihydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6 (1H)-one

A solution of sodium chlorodifluoroacetate (350 g) in diglyme (1400 mL) was added dropwise over 4 to 8 hours, preferably over 6 hours, to a solution of 5H-dibenzo[a,d]cyclo-hepten-5-one (25 g) in diglyme (500 mL), with stirring, and under nitrogen, maintaining the reaction temperature at 160°-165° C. The cooled reaction mixture was poured into water (1.8 L) and extracted with ether (1.8 L). The organic phase was washed with water, dried over sodium sulfate (Na2SO4), and evaporated. The residue was recrystallized from ethanol, then from acetone/hexane to give 14 g of 1,1-difluoro-1a,10b-dihydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6(1H)-one.
mp 149.6° C.
Flash chromatography of the combined mother liquors on silica gel, eluting with 20% acetone/hexane, gave an additional 6.5 g of the target compound.
Step 2 (1aα,6β,10bα)-1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol

A solution of 1,1-difluoro-1a,10b-dihydro-dibenzo[a,e]cyclopropa[c]cyclohepten-6(1H)-one (20.4 g) in tetrahydrofuran/methanol (1:2, 900 mL) was cooled in an ice bath. Sodium borohydride (12 g) was added in portions. The cooling bath was removed and the reaction mixture was stirred at ambient temperature for 2 hours, then poured into water. The product was filtered off, washed with water, and dried to give 20 g of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol (ii).
mp 230.1°-230.6° C.
Step 2A Combined Steps 1 and 2 Procedure (1aα,6β,10bα)-1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol

To a solution of 103.1 g (0.500 mol) of 5H-dibenzo[a,d]cyclohepten-5-one (2) in 515 mL of triethylene glycol dimethyl ether heated to between 180° C. and 210° C. was added over 7 hours, 293.3 g (2.15 mol) of chlorodifluoroacetic acid lithium salt (as a 53% by weight solution in ethylene glycol dimethyl ether). The ethylene glycol dimethyl ether was allowed to distill from the reaction as the salt addition proceeded. The GC analysis of an aliquot indicated that all of the 5H-dibenzo[a,d]cyclohepten-5-one had been consumed. The reaction was cooled to ambient temperature and then combined with 400 mL of ethyl acetate and 75 g of diatomaceous earth. The solids were removed by filtration and washed with 300 mL of ethyl acetate. The washes and filtrate were combined and the ethyl acetate was removed by concentration under vacuum leaving 635 g of dark liquid. The dark liquid was cooled to 18° C. and to this was added, over 15 minutes, 6.62 g (0.175 mol) of sodium borohydride (as a 12% by wt solution in 14 M NaOH). After stirring for 2 h the reaction was quenched by careful addition of 900 mL of a 1:3.5:4.5 solution of conc. HCl-methanol-water. The suspension was stirred for 30 min and the crude product was collected by filtration, washed with 600 mL of 1:1 methanol-water and dried to 126.4 g of dark brown solid. The crude product was slurried in 600 mL of methylene chloride, filtered, washed twice with 150 mL portions of methylene chloride, and dried to 91.6 g (71%) of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol. Gas Chromatography (GC) Conditions; Column: JW Scientific DB-1, Initial Temperature 150° C. for 5 min, 10° C./min ramp, Final temp 250° C. for 5 min. tR: intermediate, 11.5 min; reaction product (alcohol), 11.9 min; starting material, 12.3 minutes.
Step 3 Preparation of (1aα,6α,10b)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa-[c]cycloheptene

A slurry of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol (3.0 g, 11.6 mmol, 1.0 equiv) in heptane (24 mL) was treated with 48% HBr (1.58 mL, 14.0 mmol, 1.2 equiv) and the reaction was heated at reflux with vigorous stirring for 2.5 hr. Solvent was then removed by atmospheric distillation (bp 95-98° C.) until approximately 9 mL of distillate was collected. The reaction was cooled and treated with EtOAc (15 mL), Na2SO4 and activated charcoal. The mixture was stirred at RT for 15 min and filtered through hyflo. The filter cake was washed with 50:50 EtOAc:heptane and the filtrate was concentrated in vacuo to provide the title product as a crystalline solid.
mp 119° C. (3.46 g corr., 93%);
1H NMR (500 MHz CDCl3) δ 7.20-7.41 (8H, m), 5.81 (1H, s), 3.41 (2H, d, J 12.5 Hz);
13CNMR (126 MHz CDCl3) δ 141.3, 141.2, 133.5, 130.1, 129.8, 128.3, 128.2, 112.9, 110.6, 110.5, 108.3, 53.6, 30.2, 30.1, 30.0.
Anal. Calcd. For C16H11BrF2: C, 59.84; H, 3.45. Found: C, 60.13; H, 3.50.
Step 3A Preparation of (1aα,6α,10bα)-6-Bromo-1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene

To a stirred suspension of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol, (18.4 g, 71.2 mmol) in 151 mL of methylene chloride which had been cooled to 10-17° C. was added phosphorous tribromide (9.6 g, 35.6 mmol) dropwise over 15 minutes. The cooling bath was removed and the reaction was stirred for 2 hours at ambient temperature. Analysis by gas chromatography indicated complete consumption of starting material. Cold water (92 mL) and activated carbon (1.84 g) were added and the resulting mixture was stirred for 30 minutes. The activated carbon was removed by filtration through Hyflo brand filter aid and the two phases were separated. The organic phase was washed with water (184 mL×2), brine (184 ml), dried over magnesium sulfate and concentrated to dryness under vacuum, affording 21.7 g (94.8%) of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene.
1H NMR (CDCl3, 300 MHz) δ 3.36 (s, 1H), 3.40 (s, 1H), 5.77 (s, 1H), 7.16-7.38 (m, 8H).
Steps 4 and 5 (1aα,6α,10bα)-1-(1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-yl)-piperazine, Hydrobromide Salt

To a solution of 237.5 g (0.739 mol) of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]-cyclopropa[c]cycloheptene in 3.56 L of acetonitrile was added 207.7 g (2.41 mol) of piperazine and the mixture was heated to reflux for 2 hours, at which time analysis by gas chromatography showed complete consumption of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene (iii) and formation of a mixture of syn and anti piperazine compounds (III and IV) in an anti-syn ratio of 55:45. The reaction was cooled to about 7° C. and stirred for 30 minutes at that temperature. The reaction mixture was filtered to remove the precipitated syn-isomer (III) and the filter cake was washed with 250 mL of acetonitrile. The combined filtrate and wash were concentrated under vacuum to 262.4 grams of a foam which was dissolved in 450 mL of acetonitrile with heating. The solution was cooled to about 12° C. in an ice bath and stirred for 1 hour at that temperature. The precipitated syn-piperazine compound of formula (III) was filtered and washed with 125 ml of acetonitrile. The combined filtrate and wash were concentrated under vacuum to 194.1 g and dissolved in 1.19 L of ethyl acetate. The organic solution was washed sequentially with 500 mL portions of 1N sodium hydroxide, water, and saturated sodium chloride. The ethyl acetate solution was dried over sodium sulfate and concentrated to give 137.0 grams of residue which was dissolved in 1.37 L of methylene chloride and seeded with (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine, hydrobromide salt, followed by the addition of 70.8 grams of 48% aqueous hydrobromic acid. The mixture was stirred for about 45 minutes, causing the anti-isomer to crystallize as its hydrobromide salt. The crystals were filtered, washed with methylene chloride, and dried to provide purified hydrobromide salt of compound (IVa), shown by HPLC to have an anti-syn ratio of 99.3:0.7. Treatment of the isolated hydrobromide salt of compound (IVa) with aqueous sodium hydroxide, extraction into methylene chloride, separation of the aqueous layer and concentration to dryness gave 80.1 grams (33.2% yield based on starting material) of (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine as the free base. Acidification of a solution of the free base in 800 mL of methylene chloride by addition of 41.2 g of 48% hydrobromic acid as described above afforded 96.4 g of pure hydrobromide salt (title compound) with an anti-syn ratio of 99.8:0.2 (HPLC), mp 282-284° C. 1H NMR (DMSO-d6) δ 2.41 (m, 4H), 3.11 (m, 4H), 3.48 (d, J=12.4 Hz, 2H), 4.13 (s, 1H), 7.2 (m, 8H), 8.65 (bs, 2H). 13C NMR (DMSO-d6) δ 28.0, 42.9, 48.0, 75.1, 108.5, 112.9, 117.3, 127.5, 128.0, 128.6, 129.6, 132.4, 141.3. IR: (KBr) 3019, 2481, 1587, 1497, 1298 cm−1. Anal. Calcd for C20H21BrF2N2: C, 58.98; H, 5.20; N, 6.88. Found: C, 58.75; H, 5.29; N, 7.05.
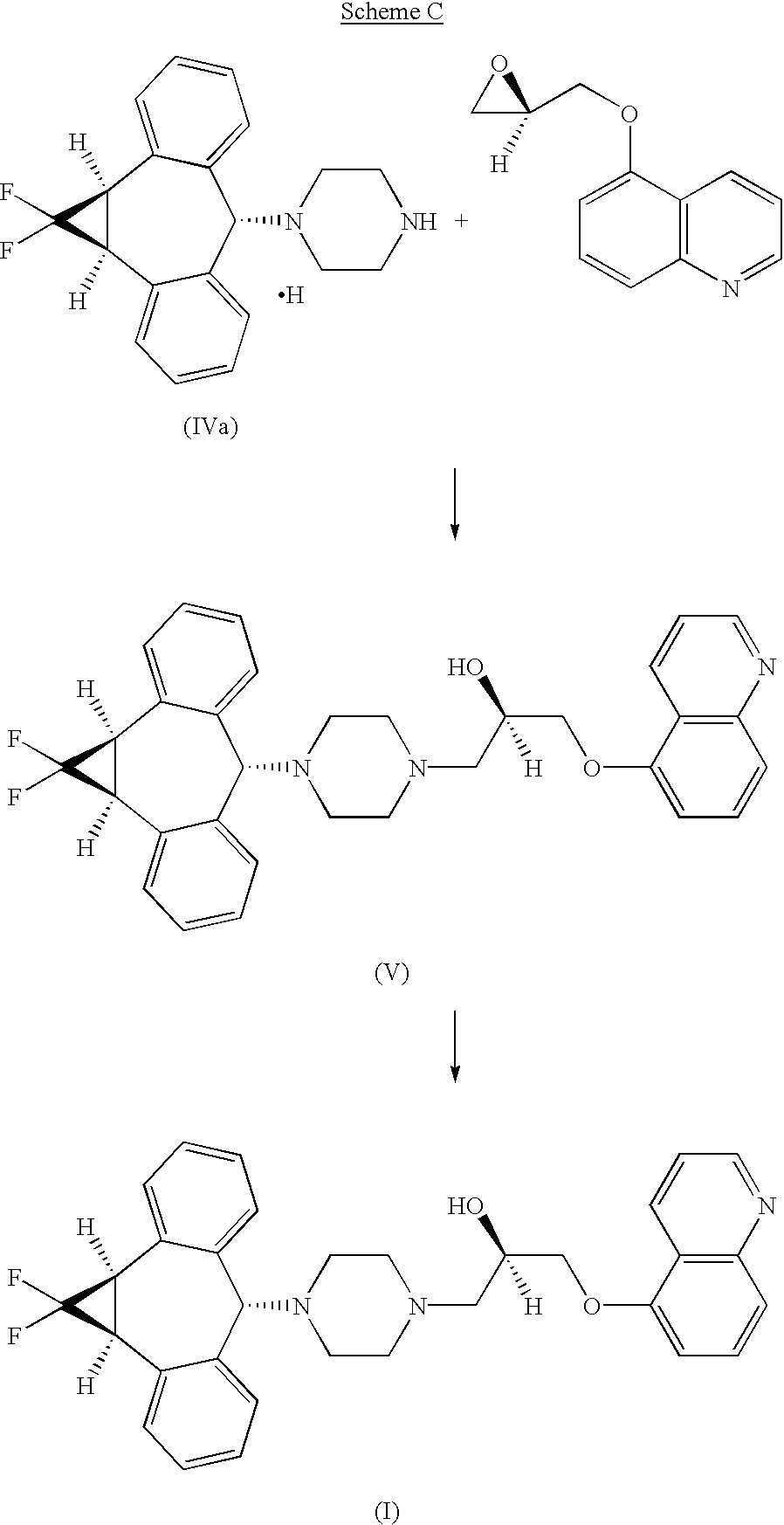
Step 6 Preparation of (2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-quinolin-5-yloxy)propan-2-ol Trihydrochloride
A suspension of (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine, hydrochloride compound of formula IVa (5.41 g, 14.9 mmol) and powdered sodium carbonate (3.16 g, 29.8 mmol) in 54 mL of 3A ethanol was stirred at ambient temperature for 1 hour. R-1-(5-quinolinyloxy)-2,3-epoxypropane (3.00 g, 14.9 mmol) was added in one portion and the reaction mixture was heated to 65° C. for 19 hours. HPLC analysis (Gradient system with solvent A (acetonitrile) and solvent B (0.02M sodium monophosphate buffer containing 0.1% triethylamine adjusted to pH 3.5 with phosphoric acid) as follows: 0-12 min, 30% solvent A/70% solvent B; 12-30 min, linear gradient from 30% to 55% solvent A/70% to 45% solvent B; 30-35 min, 55% solvent A/45% solvent B, 1 mL/min, 1=240 nm, Synchropak SCD-100 25 cm×4.6 mm column) indicated the total consumption of the piperazinyl compound of formula (IV). The mixture was allowed to cool to room temperature, filtered through a plug of silica gel, and eluted with an additional 90 mL of ethanol. The eluent was concentrated to a volume of approximately 60 mL and heated to 65° C. with stirring. A solution of HCl in ethanol (16.1 g at 0.135 g/g of solution, 59.6 mmol) was added dropwise over 10 minutes and the resultant product solution was seeded, causing the trihydrochloride salt to precipitate. The mixture was allowed to cool to ambient temperature and stirred slowly (less than 100 RPM) for 2 hours. The precipitate was filtered, washed with ethanol, and dried in vacuo at 50° C. to give the crude trihydrochloride salt which was further purified by recrystallization from methanol/ethyl acetate to provide 7.45 g (78.4%) of (2R)-anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-quinolin-5-yloxy)-propan-2-ol trihydrochloride.
Step 6a
The syn isomer compound of formula (III) isolated as described supra (combined steps 4 and 5), can be utilized to produce the corresponding syn-5-{3-[4-(10,11-difluoromethano-dibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride (XII) essentially as shown below for the free base of the anti isomer (IVa)in step 6.
https://www.google.co.in/patents/US6570016?cl=en
………………………………………
http://www.google.it/patents/WO1994024107A1?cl=en
REACTION SCHEME 1

FormuIa 1
Formula 1

Formula 2 Formula 2

Formula 3
Formula 3

Formula 4

Formula I
……………………………………….
http://www.google.com/patents/WO2000075121A3

1HNMR (500 MHz DMSO-d6) δ9.41 (2H, br. s), 7.17-7.31 (8H, m), 4.17 (1H, s), 3.52 (2H, d, J=12.4 Hz), 3.11 (4H, br. s), 2.48-2.51 (4H, m)
13CNMR (126 MHz DMSO-d6) δ142.3, 133.4, 130.5, 129.6, 129.0, 128.4, 115.9, 113.6, 111.3, 76.2, 49.0, 43.6, 29.2, 29.1, 29.0; FD MS: m/e 326 (M+).
Anal. Calcd. For C20H21ClF2N2: C, 66.20; H, 5.83; N, 7.72.
Found: C, 66.08; H, 5.90; N, 7.72.
…………………………………………..
http://www.google.com/patents/US6570016?cl=fr


(2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-qunolin-5-yloxy)-propan-2-ol Trihydrochloride
……………….
Chemical Shift Data and Peak Assignments for the Crystal Forms.

Form II has a solid-state 13C NMR spectrum comprised of isotropic peaks at the following chemical shifts: 29.9, 50.1, 55.3, 62.0, 66.5, 72.0, 75.8, 104.8, 107.5, 108.2, 109.1, 110.2, 112.0, 118.4, 119.5, 120.1, 123.1, 128.7, 131.1, 133.0, 134.8, 136.4, 136.9, 139.9, 140.0, 142.3, 144.5, 146.6, 149.0, 144.2, 153.0 and 153.6 ppm.
Form III has a solid-state 13C NMR spectrum comprised of isotropic peaks at the following chemical shifts: 30.3, 50.4, 59.1, 63.2, 72.8, 77.2, 109.1, 110.2, 112.2, 112.8, 118.7, 119.5, 119.9, 121.0, 122.2, 123.0, 128.9, 130.6, 132.7, 134.0, 136.4, 140.0, 141.0, 141.8, 142.5, 143.3, 146.1, 153.1, 153.8 and 154.7 ppm.
Empagliflozin

Empagliflozin
BI-10773
(2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-[(3S)-oxolan-3-yl]oxyphenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
M.Wt: 450.91
: C23H27ClO7
Sponsor/Developer: Eli Lilly and Boehringer Ingelheim
Mechanism of action: SGLT 2 inhibitor
Indication (Phase): Oral treatment of adults with type 2 diabetes (Phase III, expected to conclude by year’s end); Oral treatment of adults with type 2 diabetes plus high blood pressure (Phase IIb; trial results released Oct. 2)
NDA, MAA filings planned for 2013
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. Empagliflozin is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in the proximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra. The Empagliflozin phase III clinical trial program will include about 14,500 patients. The program consists of twelve ongoing international phase III clinical trials, including a large cardiovascular outcomes trial.
Empagliflozin is a novel SGLT2 inhibitor that is described for the treatment or improvement in glycemic control in patients with type 2 diabetes mellitus, for example in WO 05/092877, WO 06/117359, WO 06/120208, WO 2010/092126, WO 2010/092123, WO 2011/039107, WO 2011/039108. The use of a SGLT2 inhibitor in a method for treating obesity is described in WO 08/116,195
WO2005/092877
WO2006/117359 MP 149 DEG CENT
Empagliflozin is drug which is being investigated in clinical trials for the oral treatment oftype 2 diabetes by Boehringer Ingelheim and Eli Lilly and Company.[1][2] It is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in theproximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra.[3][4]
SGLT-2 inhibitors such as empagliflozin reduce blood glucose by blocking glucose reabsorption in the kidney and thereby excreting glucose (i.e., blood sugar) via the urine.[5]
As of December 2013, empagliflozin is in phase III clinical trials.[2]

When taken in dosages of 10 or 25 mg once a day, the incidence of adverse events was similar to placebo. However, there was a higher frequency of genital infections at both the 10 mg and the 25 mg dosages.
1-chloro-4-(β-D-glucopyranos-1-yl)-2-[4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-benzene of the formula

as described for example in WO 2005/092877. Methods of synthesis are described in the literature, for example WO 06/120208 and WO 2011/039108. According to this invention, it is to be understood that the definition of empagliflozin also comprises its hydrates, solvates and polymorphic forms thereof, and prodrugs thereof. An advantageous crystalline form of empagliflozin is described in WO 2006/117359 and WO 2011/039107 which hereby are incorporated herein in their entirety. This crystalline form possesses good solubility properties which enables a good bioavailability of the SGLT2 inhibitor. Furthermore, the crystalline form is physico-chemically stable and thus provides a good shelf-life stability of the pharmaceutical composition. Preferred pharmaceutical compositions, such as solid formulations for oral administration, for example tablets, are described in WO 2010/092126,

http://www.google.com/patents/WO2011039108A2
Example 1 : Synthesis of the fluoride VIII.1
Oxalylchloride (176kg; 1386mol; 1 ,14eq) is added to a mixture of 2-chloro-5-iodo benzoic acid (343kg; 1214mol) (compound IX.1 ), fluorobenzene (858kg) and N,N-dimethylformamide (2kg) within 3 hours at a temperature in the range from about 25 to 30°C (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30°C. The solvent (291 kg) is distilled off at a temperature between 40 and 45°C (p=200mbar). Then the reaction solution (91 1 kg) is added to aluminiumchloride AICI3 (181 kg) and fluorobenzene (192kg) at a temperature between about 25 and 30°C within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30°C and stirred for an additional hour. After phase separation the organic phase (1200kg) is separated into two halves (600kg each). From the first half of the organic phase solvent (172kg) is distilled off at a temperature of about 40 to 50°C (p=200mbar). Then 2-propanole (640kg) is added. The solution is heated to about 50°C and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a
fluorobenzene/2-propanole mixture (1 :4; 40kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50°C and p=200mbar. Then 2-propanole (240kg) is added at a temperature in the range between about 40 to 50°C. If the content of fluorobenzene is greater than 1 % as determined via GC, another 140kg of solvent are distilled off and 2- propanole (140kg) is added. Then the solution is cooled from about 50°C to 40°C within one hour and seeding crystals (50g) are added. The solution is further cooled from about 40°C to 20°C within 2 hours. Water (450kg) is added at about 20°C within 1 hour and the suspension is stirred at about 20°C for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1 :1 ; 800kg). The product is dried until a water level of <0.06%w/w is obtained. The second half of the organic phase is processed identically. A total of 410kg (94%yield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
Example 2: Synthesis of the ketone VII.1
To a solution of the fluoride VIII.1 (208kg), tetrahydrofuran (407kg) and (S)-3- hydroxytetrahydrofuran (56kg) is added potassium-ie f-butanolate solution (20%) in tetrahydrofuran (388kg) within 3 hrs at 16 to 25°C temperature. After completion of the addition, the mixture is stirred for 60min at 20°C temperature. Then the conversion is determined via HPLC analysis. Water (355kg) is added within 20 min at a temperature of 21 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20°C). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20°C). The phases are separated and solvent is distilled off from the organic phase at 19 to 45°C temperature under reduced pressure. 2-Propanol (703kg) is added to the residue at 40 to 46°C temperature and solvent is distilled off at 41 to 50°C temperature under reduced pressure. 2-Propanol (162kg) is added to the residue at 47°C temperature and solvent is distilled off at 40 to 47°C temperature under reduced pressure. Then the mixture is cooled to 0°C within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2- propanol (158kg) and subsequently with ie f.-butylmethylether (88kg) and dried at 19 to 43°C under reduced pressure. 227kg (91 ,8%) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
Example 3: Synthesis of the iodide V.1
To a solution of ketone VII.1 (217,4kg) and aluminium chloride (AICI3; 81 ,5kg) in toluene (366,8kg) is added 1 ,1 ,3,3-tetramethyldisiloxane (TMDS, 82,5kg) within 1 hr 30 min
(temperature: 18-26°C). After completion of the addition, the mixture is stirred for additional 1 hr at a temperature of 24°C. Then the conversion is determined via HPLC analysis.
Subsequently the reaction mixture is treated with acetone (15,0kg), stirred for 1 hr 5 min at 27°C temperature and the residual TMDS content is analyzed via GC. Then a mixture of water (573kg) and concentrated HCI (34kg) is added to the reaction mixture at a temperature of 20 to 51 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature:
51 °C). The stirrer is switched off and the mixture is left stand for 20 min (temperature: 52°C). The phases are separated and solvent is distilled off from the organic phase at 53-73°C temperature under reduced pressure. Toluene (52,8kg) and ethanol (435,7kg) are added to the residue at 61 to 70°C temperature. The reaction mixture is cooled to 36°C temperature and seeding crystals (0,25kg) are added. Stirring is continued at this temperature for 35 min. Then the mixture is cooled to 0 to 5°C and stirred for additional 30 min. The product is collected on a centrifuge, washed with ethanol (157kg) and dried at 15 to 37°C under reduced pressure. 181 kg (82,6%) of product are obtained as colourless solid. The identity of the product is determined via the HPLC retention time.
Example 4: Synthesis of the lactone IV.1
A suspension of the D-(+)-gluconic acid-delta-lactone IVa.1 (42,0kg), tetrahydrofuran (277,2kg), 4-methylmorpholine (NMM; 152,4kg) and 4-dimethylaminopyridine (DMAP;
1 ,44kg) is treated with chlorotrimethylsilane (TMSCI; 130,8kg) within 50 min at 13 to 19°C. After completion of the addition stirring is continued for 1 hr 30 min at 20 to 22°C and the conversion is determined via HPLC analysis. Then n-heptane (216,4kg) is added and the mixture is cooled to 5°C. Water (143kg) is added at 3 to 5°C within 15 min. After completion of the addition the mixture is heated to 15°C and stirred for 15 min. The stirrer is switched off and the mixture is left stand for 15 min. Then the phases are separated and the organic layer is washed in succession two times with water (143kg each). Then solvent is distilled off at 38°C under reduced pressure and n-heptane (130kg) is added to the residue. The resulting solution is filtered and the filter is rinsed with n-heptane (63kg) (filter solution and product solution are combined). Then solvent is distilled off at 39 to 40°C under reduced pressure. The water content of the residue is determined via Karl-Fischer analysis (result: 0,0%).
1 12,4kg of the product is obtained as an oil (containing residual n-heptane, which explains the yield of >100%). The identity of the product is determined via infrared spectrometry.
Example 5a: Synthesis of the glucoside 11.1
To a solution of the iodide V.1 (267kg) in tetrahydrofuran (429kg) is added Turbogrignard solution (isopropylmagnesium chloride/lithium chloride solution, 14 weight-% iPrMgCI in THF, molar ratio LiCI : iPrMgCI = 0,9 – 1 .1 mol/mol) (472kg) at -21 to -15°C temperature within 1 hr 50 min. On completion of the addition the conversion is determined via HPLC analysis. The reaction is regarded as completed when the area of the peak corresponding to the iodide V.1 is smaller than 5,0% of the total area of both peaks, iodide V.1 and the corresponding desiodo compound of iodide V.1 . If the reaction is not completed, additional Turbogrignard solution is added until the criterion is met. In this particular case the result is 3,45%. Then the lactone IV.1 (320kg) is added at -25 to -18°C temperature within 1 hr 25 min. The resulting mixture is stirred for further 1 hr 30 min at -13 to -18°C. On completion the conversion is determined via HPLC analysis (for information). On completion, a solution of citric acid in water (938L; concentration: 10 %-weight) is added to the reaction mixture of a volume of about 2500L at -13 to 19°C within 1 hr 25 min.
The solvent is partially distilled off from the reaction mixture (residual volume: 1816-1905L) at 20 to 30°C under reduced pressure and 2-methyltetrahydrofuran (532kg) is added. Then the stirrer is switched off and the phases are separated at 29°C. After phase separation the pH value of the organic phase is measured with a pH electrode (Mettler Toledo MT HA 405 DPA SC) or alternatively with pH indicator paper (such as pH-Fix 0-14, Macherey and Nagel). The measured pH value is 2 to 3. Then solvent is distilled off from the organic phase at 30 to 33°C under reduced pressure and methanol (1202kg) is added followed by the addition of a solution of 1 ,25N HCI in methanol (75kg) at 20°C (pH = 0). Full conversion to the acetale 111.1 is achieved by subsequent distillation at 20 to 32°C under reduced pressure and addition of methanol (409kg).
Completion of the reaction is obtained when two criteria are fulfilled:
1 ) The ratio of the sum of the HPLC-area of the alpha-form + beta-form of intermediate 111.1 relative to the area of intermediate llla.1 is greater or equal to 96,0% : 4,0%. 2) The ratio of the HPLC-area of the alpha-form of intermediate 111.1 to the beta-form of 111.1 is greater or equal to 97,0% to 3,0%.
In this particular case both criteria are met. Triethylamin (14kg) is added (pH = 7,4) and solvent is distilled off under reduced pressure, acetonitrile (835kg) is added and further distilled under reduced pressure. This procedure is repeated (addition of acetonitrile: 694kg) and methylene chloride (640kg) is added to the resulting mixture to yield a mixture of the acetale 111.1 in acetonitrile and methylene chloride. The water content of the mixture is determined via Karl Fischer titration (result: 0,27%).
The reaction mixture is then added within 1 hr 40 min at 10 to 19°C to a preformed mixture of AICI3 (176kg), methylene chloride (474kg), acetonitrile (340kg), and triethylsilane (205kg). The resulting mixture is stirred at 18 to 20°C for 70 min. After completion of the reaction, water (1263L) is added at 20 to 30°C within 1 hr 30 min and the mixture is partially distilled at 30 to 53°C under atmospheric pressure and the phases are separated. Toluene (698kg) is added to the organic phase and solvent is distilled off under reduced pressure at 22 to 33°C. The product is then crystallized by addition of seeding crystals (0,5kg) at 31 °C and water (267kg) added after cooling to 20°C. The reaction mixture is cooled to 5°C within 55 min and stirred at 3 to 5°C for 12 hrs. Finally the product is collected on a centrifuge as colourless, crystalline solid, washed with toluene (348kg) and dried at 22 to 58°C. 21 1 kg (73%) of product are obtained. The identity of the product is determined via the HPLC retention time.
Example 5b: Synthesis of the glucoside 11.1
To a solution of the iodide V.1 (30g) in tetrahydrofuran (55ml_) is added Turbogrignard solution (isopropylmagnesium chloride/lithium chloride solution, 14 weight-% iPrMgCI in THF, molar ratio LiCI : iPrMgCI = 0,9 – 1 .1 mol/mol) (53g) at -14 to -13°C temperature within 35 min. On completion of the addition the conversion is determined via HPLC analysis. The reaction is regarded as completed when the area of the peak corresponding to the iodide V.1 is smaller than 5,0% of the total area of both peaks, iodide V.1 and the corresponding desiodo compound of iodide V.1 . If the reaction is not completed, additional Turbogrignard solution is added until the criterion is met. In this particular case the result is 0,35%. Then the lactone IV.1 (36g) is added at -15 to -6°C temperature within 15 min. The resulting mixture is stirred for further 1 hr at -6 to -7°C. On completion, the conversion is determined via HPLC analysis (for information). On completion, a solution of citric acid in water (105mL;
concentration: 10 %-weight) is added to the reaction mixture at -15 to 10°C within 30 min. The solvent is partially distilled off from the reaction mixture (residual volume: 200mL) at 20 to 35°C under reduced pressure and 2-methyltetrahydrofuran (71 mL) is added. Then the mixture is stirred for 25min at 30°C. Then the stirrer is switched off and the phases are separated at 30°C. After phase separation the pH value of the organic phase is measured with a pH electrode (Mettler Toledo MT HA 405 DPA SC) or alternatively with pH indicator paper (such as pH-Fix 0-14, Macherey and Nagel). The measured pH value is 3. Then solvent is distilled off from the organic phase at 35°C under reduced pressure and methanol (126ml_) is added followed by the addition of a solution of 1 ,25N HCI in methanol (10,1 ml_) at 25°C (pH = 1 -2). Full conversion to the acetale 111.1 is achieved by subsequent distillation at 35°C under reduced pressure and addition of methanol (47ml_).
Completion of the reaction is obtained when two criteria are fulfilled:
1 ) The ratio of the sum of the HPLC-area of the alpha-form + beta-form of intermediate 111.1 relative to the area of intermediate llla.1 is greater or equal to 96,0% : 4,0%. In this particular case the ratio is 99,6% : 0,43%.
2) The ratio of the HPLC-area of the alpha-form of intermediate 111.1 to the beta-form of III.1 is greater or equal to 97,0% to 3,0%. In this particular case the ratio is 98,7% : 1 ,3%.
Triethylamin (2,1 mL) is added (pH = 9) and solvent is distilled off at 35°C under reduced pressure, acetonitrile (120ml_) is added and further distilled under reduced pressure at 30 to 35°C. This procedure is repeated (addition of acetonitrile: 102ml_) and methylene chloride (55ml_) is added to the resulting mixture to yield a mixture of the acetale 111.1 in acetonitrile and methylene chloride. The water content of the mixture is determined via Karl Fischer titration (result: 0,04%).
The reaction mixture is then added within 1 hr 5 min at 20°C to a preformed mixture of AICI3 (19,8g), methylene chloride (49ml_), acetonitrile (51 mL), and triethylsilane (23g). The resulting mixture is stirred at 20 to 30°C for 60 min. After completion of the reaction, water (156mL) is added at 20°C within 25 min and the mixture is partially distilled at 55°C under atmospheric pressure and the phases are separated at 33°C. The mixture is heated to 43°C and toluene (90mL) is added and solvent is distilled off under reduced pressure at 41 to 43°C. Then acetonitrile (1 OmL) is added at 41 °C and the percentage of acetonitrile is determined via GC measurement. In this particular case, the acetonitrile percentage is 27%- weight. The product is then crystallized by addition of seeding crystals (0,1 g) at 44°C and the mixture is further stirred at 44°C for 15min. The mixture is then cooled to 20°C within 60min and water (142mL) is added at 20°C within 30min. The reaction mixture is cooled to 0 to 5°C within 60 min and stirred at 3°C for 16 hrs. Finally the product is collected on a filter as colourless, crystalline solid, washed with toluene (80mL) and dried at 20 to 70°C. 20, 4g (62,6%) of product are obtained. The identity of the product is determined via the HPLC retention time.
- Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P (January 2012). “Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors”. Diabetes Obes Metab 14 (1): 83–90. doi:10.1111/j.1463-1326.2011.01517.x. PMID 21985634.
- “Empagliflozin”. clinicaltrials.gov. U.S. National Institutes of Health. Retrieved 22 September 2012.
- Nair S, Wilding JP (January 2010). “Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus”. J. Clin. Endocrinol. Metab. 95 (1): 34–42. doi:10.1210/jc.2009-0473. PMID 19892839.
- Bays H (March 2009). “From victim to ally: the kidney as an emerging target for the treatment of diabetes mellitus”. Curr Med Res Opin25 (3): 671–81. doi:10.1185/03007990802710422. PMID 19232040.
- Abdul-Ghani MA, DeFronzo RA (September 2008). “Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus”. Endocr Pract 14 (6): 782–90. PMID 18996802.
[1]. Grempler R, Thomas L, Eckhardt M et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012 Jan;14(1):83-90.
Abstract
AIMS: Empagliflozin is a selective sodium glucose cotransporter-2 (SGLT-2) inhibitor in clinical development for the treatment of type 2 diabetes mellitus. This study assessed pharmacological properties of empagliflozin in vitro and pharmacokinetic properties in vivo and compared its potency and selectivity with other SGLT-2 inhibitors. METHODS: [(14)C]-alpha-methyl glucopyranoside (AMG) uptake experiments were performed with stable cell lines over-expressing human (h) SGLT-1, 2 and 4. Two new cell lines over-expressing hSGLT-5 and hSGLT-6 were established and [(14)C]-mannose and [(14)C]-myo-inositol uptake assays developed. Binding kinetics were analysed using a radioligand binding assay with [(3)H]-labelled empagliflozin and HEK293-hSGLT-2 cell membranes. Acute in vivo assessment of pharmacokinetics was performed with normoglycaemic beagle dogs and Zucker diabetic fatty (ZDF) rats. RESULTS: Empagliflozin has an IC(50) of 3.1 nM for hSGLT-2. Its binding to SGLT-2 is competitive with glucose (half-life approximately 1 h). Compared with other SGLT-2 inhibitors, empagliflozin has a high degree of selectivity over SGLT-1, 4, 5 and 6. Species differences in SGLT-1 selectivity were identified. Empagliflozin pharmacokinetics in ZDF rats were characterised by moderate total plasma clearance (CL) and bioavailability (BA), while in beagle dogs CL was low and BA was high. CONCLUSIONS: Empagliflozin is a potent and competitive SGLT-2 inhibitor with an excellent selectivity profile and the highest selectivity window of the tested SGLT-2 inhibitors over hSGLT-1. Empagliflozin represents an innovative therapeutic approach to treat diabetes.
[2]. Thomas L, Grempler R, Eckhardt M et al. Long-term treatment with empagliflozin, a novel, potent and selective SGLT-2 inhibitor, improves glycaemic control and features of metabolic syndrome in diabetic rats. Diabetes Obes Metab. 2012 Jan;14(1):94-6.
Abstract
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. This series of studies was conducted to assess the in vivo pharmacological effects of single or multiple doses of empagliflozin in Zucker diabetic fatty rats. Single doses of empagliflozin resulted in dose-dependent increases in urinary glucose excretion and reductions in blood glucose levels. After multiple doses (5 weeks), fasting blood glucose levels were reduced by 26 and 39% with 1 and 3 mg/kg empagliflozin, respectively, relative to vehicle. After 5 weeks, HbA1c levels were reduced (from a baseline of 7.9%) by 0.3 and 1.1% with 1 and 3 mg/kg empagliflozin, respectively, versus an increase of 1.1% with vehicle. Hyperinsulinaemic-euglycaemic clamp indicated improved insulin sensitivity with empagliflozin after multiple doses versus vehicle. These findings support the development of empagliflozin for the treatment of type 2 diabetes.
[3]. Luippold G, Klein T, Mark M, Grempler R. Empagliflozin, a novel potent and selective SGLT-2 inhibitor, improves glycaemic control alone and in combination with insulin in streptozotocin-induced diabetic rats, a model of type 1 diabetes mellitus. Diabetes Obes Metab. 2012 Jul;14(7):601-7.
Abstract
AIM: Sodium glucose cotransporter-2 (SGLT-2) is key to reabsorption of glucose in the kidney. SGLT-2 inhibitors are in clinical development for treatment of type 2 diabetes mellitus (T2DM). The mechanism may be of value also in the treatment of type 1 diabetes mellitus (T1DM). This study investigated effects of the SGLT-2 inhibitor, empagliflozin, alone and in combination with insulin, on glucose homeostasis in an animal model of T1DM. METHODS: Sprague-Dawley rats were administered a single intraperitoneal injection of streptozotocin (STZ; 60 mg/kg). Acutely, STZ rats received two doses of insulin glargine with or without empagliflozin, and blood glucose was measured. In a subchronic study, STZ rats received empagliflozin alone, one or two insulin-releasing implants or a combination of one implant and empagliflozin over 28 days; blood glucose and HbA(1c) were measured. RESULTS: In the acute setting, empagliflozin in combination with 1.5 IU insulin induced a similar glucose-lowering effect as 6 IU insulin. Both interventions were more efficacious than monotherapy with 1.5 IU insulin. In the subchronic study, 12-h blood glucose profile on day 28 in the combination group was lower than with one implant, and similar to two implants. Plasma HbA(1c) was improved in the combination group and in animals with two implants. CONCLUSIONS: Empagliflozin reduced blood glucose levels in a T1DM animal model. Empagliflozin combined with low-dose insulin showed comparable glucose-lowering efficacy to treatment with high-dose insulin. Our data suggest that empagliflozin is an efficacious adjunctive-to-insulin therapy with the clinical potential for the treatment of T1DM.
[4]. Macha S, Rose P, Mattheus M et al. Lack of drug-drug interaction between empagliflozin, a sodium glucose cotransporter-2 inhibitor, and warfarin in healthy volunteers. Diabetes Obes Metab. 2012 Oct 24. doi: 10.1111/dom.12028. [Epub ahead of print]
Abstract
AIM: To investigate potential drug-drug interactions between empagliflozin and warfarin. MATERIALS AND METHODS: Healthy subjects (n=18) received empagliflozin 25 mg qd for 5 days (treatment A), followed by empagliflozin 25 mg qd for 7 days (days 6-12) with a single 25 mg dose of warfarin on day 6 (B), and a single 25 mg dose of warfarin alone (C), in an open-label, crossover study. Subjects received treatments in sequence AB_C or C_AB with a washout period of ≥14 days between AB and C or C and AB. RESULTS: Warfarin had no effect on empagliflozin area under concentration-time curve or maximum plasma concentration at steady-state (AUC(τ) (,ss) or C(max,ss) ): geometric mean ratios (GMRs) (90% confidence intervals [CI]) were 100.89% (96.86, 105.10) and 100.64% (89.79, 112.80), respectively. Empagliflozin had no effect on AUC from 0 hours to infinity (AUC(0) (-∞) ) or C(max) for R-warfarin or S-warfarin (GMRs [90% CI] for AUC(0) (-∞) : 98.49% [95.29, 101.80] and 95.88% [93.40, 98.43], respectively; C(max) : 97.89% [91.12, 105.15] and 98.88% [91.84, 106.47], respectively). Empagliflozin had no clinically relevant effects on warfarin’s anticoagulant activity (international normalised ratio [INR]) (GMR [95% CI] for peak INR: 0.87 [0.73, 1.04]; area under the effect-time curve from 0 to 168 hours: 0.88 [0.79, 0.98]. No drug-related adverse events were reported for empagliflozin after monotherapy or combined administration. The combination of empagliflozin and warfarin was well tolerated. CONCLUSIONS: No drug-drug interactions were observed between empagliflozin and warfarin, indicating that empagliflozin and warfarin can be co-administered without dosage adjustments of either drug.
[5]. Sarashina A, Koiwai K, Seman LJ et al. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single Doses of Empagliflozin, a Sodium Glucose Cotransporter-2 (SGLT-2) Inhibitor, in Healthy Japanese Subjects. Drug Metab Pharmacokinet. 2012 Nov 13. [Epub ahead of print]
Abstract
This randomized, placebo-controlled within dose groups, double-blind, single rising dose study investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of 1 mg to 100 mg doses of empagliflozin in 48 healthy Japanese male subjects. Empagliflozin was rapidly absorbed, reaching peak levels in 1.25 to 2.50 hours; thereafter, plasma concentrations declined in a biphasic fashion, with mean terminal elimination half-life ranging from 7.76 to 11.7 hours. Increase in empagliflozin exposure was proportional to dose. Oral clearance was dose independent and ranged from 140 to 172 mL/min. In the 24 hours following 100 mg empagliflozin administration, the mean (%CV) amount of glucose excreted in urine was 74.3 (17.1) g. The amount and the maximum rate of glucose excreted via urine increased with dose of empagliflozin. Nine adverse events, all of mild intensity, were reported by 8 subjects (7 with empagliflozin and 1 with placebo). No hypoglycemia was reported. In conclusion, 1 mg to 100 mg doses of empagliflozin had a good safety and tolerability profile in healthy Japanese male subjects. Exposure to empagliflozin was dose-proportional. The amount and rate of urinary glucose excretion were higher with empagliflozin than with placebo, and increased with empagliflozin dose.
…………………………….
formula A

Example I

(5-bromo-2-chloro-phenyl)-(4-methoxy-phenyl)-methanone
38.3 ml oxalyl chloride and 0.8 ml of dimethylformamide are added to a mixture of
100 g of 5-bromo-2-chloro-benzoic acid in 500 ml dichloromethane. The reaction mixture is stirred for 14 h, then filtered and separated from all volatile constituents in the rotary evaporator. The residue is dissolved in 150 ml dichloromethane, the solution is cooled to -5 0C, and 46.5 g of anisole are added. Then 51.5 g of aluminum trichloride are added batchwise so that the temperature does not exceed 5 0C. The solution is stirred for another 1 h at 1 to 5 0C and then poured onto crushed ice. The organic phase is separated, and the aqueous phase is extracted another three times with dichloromethane. The combined organic phases are washed with aqueous 1 M hydrochloric acid, twice with aqueous 1 M sodium hydroxide solution and with brine. Then the organic phase is dried, the solvent is removed and the residue is recrystallised in ethanol. Yield: 86.3 g (64% of theory)
Mass spectrum (ESI+): m/z = 325/327/329 (Br+CI) [M+H]+
Example Il
4-bromo-1-chloro-2-(4-methoxy-benzyl)-benzene
A solution of 86.2 g (5-bromo-2-chloro-phenyl)-(4-methoxy-phenyl)-methanone and 101.5 ml triethylsilane in 75 ml dichloromethane and 150 ml acetonitrile is cooled to 1O0C. Then with stirring 50.8 ml of boron trifluoride etherate are added so that the temperature does not exceed 2O0C. The solution is stirred for 14 h at ambient temperature, before another 9 ml triethylsilane and 4.4 ml boron trifluoride etherate are added. The solution is stirred for a further 3 h at 45 to 5O0C and then cooled to ambient temperature. A solution of 28 g potassium hydroxide in 70 ml of water is added, and the resulting mixture is stirred for 2 h. Then the organic phase is separated off and the aqueous phase is extracted another three times with diisopropylether. The combined organic phases are washed twice with aqueous 2 M potassium hydroxide solution and once with brine and then dried over sodium sulfate. After the solvent has been removed the residue is washed in ethanol, separated again and dried at 6O0C. Yield: 50.0 g (61 % of theory)
Mass spectrum (ESI+): m/z = 310/312/314 (Br+CI) [M+H]+
Example III

4-(5-bromo-2-chloro-benzyl)-phenol
A solution of 14.8 g 4-bromo-1-chloro-2-(4-methoxy-benzyl)-benzene in 150 ml dichloromethane is cooled in an ice bath. Then 50 ml of a 1 M solution of boron tribromide in dichloromethane are added, and the solution is stirred for 2 h at ambient temperature. The solution is then cooled in an ice bath again, and saturated aqueous potassium carbonate solution is added dropwise. At ambient temperature the mixture is adjusted with aqueous 1 M hydrochloric acid to a pH of 1 , the organic phase is separated, and the aqueous phase is extracted another three times with ethyl acetate. The combined organic phases are dried over sodium sulphate, and the solvent is removed completely. Yield: 13.9 g (98% of theory) Mass spectrum (ESI ): m/z = 295/297/299 (Br+CI) [M-HV
Example IV

r4-(5-bromo-2-chloro-benzyl)-phenoxyl-tert-butyl-dimethyl-silane
A solution of 13.9 g 4-(5-bromo-2-chloro-benzyl)-phenol in 140 ml dichloromethane is cooled in an ice bath. Then 7.54 g tert-butyldimethylsilylchlorid in 20 ml dichloromethane are added followed by 9.8 ml triethylamine and 0.5 g 4- dimethylaminopyridine. The solution is stirred for 16 h at ambient temperature and then diluted with 100 ml dichloromethane. The organic phase is washed twice with aqueous 1 M hydrochloric acid and once with aqueous sodium hydrogen carbonate solution and then dried over sodium sulfate. After the solvent has been removed the residue is filtered through silica gel (cyclohexane/ethyl acetate 100:1 ). Yield: 16.8 g (87% of theory) Mass spectrum (El): m/z = 410/412/414 (Br+CI) [M]+
Example V

2.3.4.6-tetrakis-O-(trimethylsilyl)-D-glucopyranone
A solution of 20 g D-glucono-1 ,5-lactone and 98.5 ml Λ/-methylmorpholine in 200 ml of tetrahydrofuran is cooled to -5 0C. Then 85 ml trimethylsilylchloride are added dropwise so that the temperature does not exceed 5 0C. The solution is then stirred for 1 h at ambient temperature, 5 h at 35 0C and again for 14 h at ambient temperature. After the addition of 300 ml of toluene the solution is cooled in an ice bath, and 500 ml of water are added so that the temperature does not exceed 100C. The organic phase is then separated and washed in each case once with aqueous sodium dihydrogen phosphate solution, water and brine. The solvent is removed, the residue is taken up in 250 ml of toluene, and the solvent is again removed completely. Yield: 52.5 g (approx. 90% pure)
Mass spectrum (ESI+): m/z = 467 [M+H]+
Example Vl

1-chloro-4-(β-D-qlucopyranos-1-yl)-2-(4-hvdroxybenzyl)-benzene
A solution of 4.0 g [4-(5-bromo-2-chloro-benzyl)-phenoxy]-te/Tf-butyl-dimethyl-silane in 42 ml dry diethyl ether is cooled to -800C under argon. 11.6 ml of a 1.7 M solution of te/if-butyllithium in pentane are slowly added dropwise to the cooled solution, and then the solution is stirred for 30 min at -80 0C. This solution is then added dropwise through a transfer needle, which is cooled with dry ice, to a solution of 4.78 g
2,3,4,6-tetrakis-O-(trimethylsilyl)-D-glucopyranone in 38 ml diethyl ether chilled to – 80 0C. The resulting solution is stirred for 3 h at -78 0C. Then a solution of 1.1 ml methanesulphonic acid in 35 ml of methanol is added and the solution is stirred for 16 h at ambient temperature. The solution is then neutralised with solid sodium hydrogen carbonate, ethyl acetate is added and the methanol is removed together with the ether. Aqueous sodium hydrogen carbonate solution is added to the remaining solution, and the resulting mixture is extracted four times with ethyl acetate. The organic phases are dried over sodium sulphate and evaporated down. The residue is dissolved in 30 ml acetonitrile and 30 ml dichloromethane and the solution is cooled to -10 0C. After the addition of 4.4 ml triethylsilane 2.6 ml boron trifluoride etherate are added dropwise so that the temperature does not exceed -5 0C. After the addition is complete the solution is stirred for another 5 h at -5 to -10 0C and then quenched by the addition of aqueous sodium hydrogen carbonate solution. The organic phase is separated, and the aqueous phase is extracted four times with ethyl acetate. The combined organic phases are dried over sodium sulfate, the solvent is removed, and the residue is purified by chromatography on silica gel (dichoromethane/methanol 1 :0->3:1 ). The product then obtained is an approx. 6:1 mixture of β/α which can be converted into the pure β-anomer by global acetylation of the hydroxy groups with acetic anhydride and pyridine in dichloromethane and recrystallization of the product from ethanol. The product thus obtained is converted into the title compound by deacetylation in methanol with aqueous 4 M potassium hydroxide solution. Yield: 1.6 g (46% of theory)
Mass spectrum (ESI+): m/z = 398/400 (Cl) [M+H]+
Preparation of the compound A:

1-chloro-4-(β-D-qlucopyranos-1-yl)-2-r4-(<fS)-tetrahvdrofuran-3-yloxy)-benzyll- benzene
0.19 g (f?)-3-(4-methylphenylsulfonyloxy)-tetrahydrofuran are added to a mixture of 0.20 g 1-chloro-4-(β-D-glucopyranos-1-yl)-2-(4-hydroxybenzyl)-benzene and 0.29 g cesium carbonate in 2.5 ml dimethylformamide. The mixture is stirred at 75 0C for 4 h, before another 0.29 g caesium carbonate and 0.19 g (f?)-3-(4-methylphenyl- sulfonyloxy)-tetrahydrofuran are added. After an additional 14 h stirring at 75 0C the mixture is cooled to ambient temperature and brine is added. The resulting mixture is extracted with ethyl acetate, the combined organic extracts are dried over sodium sulfate, and the solvent is removed. The residue is purified by chromatography on silica gel (dichloromethane/methanol 1 :0 -> 5:1 ). Yield: 0.12 g (49% of theory) Mass spectrum (ESI+): m/z = 451/453 (Cl) [M+H] +
The US Food and Drug Administration (FDA) has granted a six-month Priority Review designation to Genzyme’s New Drug Application (NDA) for Cerdelga (eliglustat)
13 dec 2013
Genzyme’s Cerdelga NDA receives US FDA priority review designationpharmabiz.comThe US Food and Drug Administration (FDA) has granted a six-month Priority Review designation to Genzyme’s New Drug Application (NDA) for Cerdelga (eliglustat), an investigational oral therapy for adult patients with Gaucher disease
ELIGLUSTAT TARTRATE
THERAPEUTIC CLAIM Treatment of lysosomal storage disorders
CHEMICAL NAMES
1. Octanamide, N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-
pyrrolidinylmethyl)ethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)
2. bis{N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(pyrrolidin-1-
ylmethyl)ethyl]octanamide} (2R,3R)-2,3-dihydroxybutanedioate
MOLECULAR FORMULA C23H36N2O4 . ½ C4H6O6
MOLECULAR WEIGHT 479.6
MANUFACTURER Genzyme Corp.
CODE DESIGNATION Genz-112638
CAS REGISTRY NUMBER 928659-70-5
old article cut paste
Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
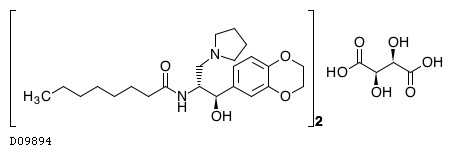
Eliglustat tartrate (USAN)
CAS:928659-70-5
February 15, 2013
Genzyme , a Sanofi company (EURONEXT: SAN and NYSE: SNY), today announced positive new data from the Phase 3 ENGAGE and ENCORE studies of eliglustat tartrate, its investigational oral therapy for Gaucher disease type 1. The results from the ENGAGE study were presented today at the 9th Annual Lysosomal Disease Network WORLD Symposium in Orlando, Fla. In conjunction with this meeting, Genzyme also released topline data from its second Phase 3 study, ENCORE. Both studies met their primary efficacy endpoints and together will form the basis of Genzyme’s registration package for eliglustat tartrateThe data presented at this year’s WORLD symposium reinforce our confidence that eliglustat tartrate may become an important oral option for patients with Gaucher disease”The company is developing eliglustat tartrate, a capsule taken orally, to provide a convenient treatment alternative for patients with Gaucher disease type 1 and to provide a broader range of treatment options for patients and physicians. Genzyme’s clinical development program for eliglustat tartrate represents the largest clinical program ever focused on Gaucher disease type 1 with approximately 400 patients treated in 30 countries.“The data presented at this year’s WORLD symposium reinforce our confidence that eliglustat tartrate may become an important oral option for patients with Gaucher disease,” said Genzyme’s Head of Rare Diseases, Rogerio Vivaldi MD. “We are excited about this therapy’s potential and are making excellent progress in our robust development plan for bringing eliglustat tartrate to the market.”ENGAGE Study Results:In ENGAGE, a Phase 3 trial to evaluate the safety and efficacy of eliglustat tartrate in 40 treatment-naïve patients with Gaucher disease type 1, improvements were observed across all primary and secondary efficacy endpoints over the 9-month study period. Results were reported today at the WORLD Symposium by Pramod Mistry, MD, PhD, FRCP, Professor of Pediatrics & Internal Medicine at Yale University School of Medicine, and an investigator in the trial.
The randomized, double-blind, placebo-controlled study had a primary efficacy endpoint of improvement in spleen size in patients treated with eliglustat tartrate. Patients were stratified at baseline by spleen volume. In the study, a statistically significant improvement in spleen size was observed at nine months in patients treated with eliglustat tartrate compared with placebo. Spleen volume in patients treated with eliglustat tartrate decreased from baseline by a mean of 28 percent compared with a mean increase of two percent in placebo patients, for an absolute difference of 30 percent (p<0.0001).
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.


Late-stage success for Sanofi/Regeneron RA drug sarilumab

SARILUMAB
PRONUNCIATION sar il’ ue mab
THERAPEUTIC CLAIM Treatment of rheumatoid arthritis and
ankylosing spondylitis
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human interleukin 6 receptor α) (human REGN88 heavy
chain), disulfide with human REGN88 light chain, dimer
2. Immunoglobulin G1, anti-(human interleukin-6 receptor subunit alpha (IL-6RA,
membrane glycoprotein 80, CD126)); human monoclonal RGN88 γ1 heavy chain (219-
214′)-disulfide with human monoclonal RGN88 κ light chain dimer (225-225”:228-
228”)-bisdisulfide
MOLECULAR FORMULA C6388H9918N1718O1998S44
MOLECULAR WEIGHT 144.13 kDa
SPONSOR Regeneron Pharmaceuticals, Inc.
CODE DESIGNATION REGN88, SAR153191
CAS REGISTRY NUMBER 1189541-98-7
sarilumab
Sarilumab (REGN88/SAR153191) is a fully-human monoclonal antibody directed against the IL-6 receptor (IL-6R). Sarilumab is a subcutaneously delivered inhibitor of IL-6 signaling, which binds with high affinity to the IL-6 receptor. It blocks the binding of IL-6 to its receptor and interrupts the resultant cytokine-mediated inflammatory signaling.
Sanofi and Regeneron’s investigational rheumatoid arthritis drug sarilumab has succeeded in a late-stage trial.
The year-long Phase III study enrolled 1,200 patients with active, moderate-to-severe RA who were inadequate responders to methotrexate. Patients were randomised to one of three subcutaneous treatment groups, all in combination with MTX and dosed every other week – sarilumab 200mg, 150mg or placebo.http://www.pharmatimes.com/Article/13-11-22/Late-stage_success_for_Sanofi_Regeneron_RA_drug.aspx
Sarilumab is a human monoclonal antibody against the interleukin-6 receptor.
Regeneron and Sanofi are currently co-developing the drug for the treatment of rheumatoid arthritis, for which it is in phase III trials. Development inankylosing spondylitis has been suspended after the drug failed to show clinical benefit over methotrexate in a phase II trial.[1][2]
On May 15th, 2013, both companies announced that 2 new trials were starting (COMPARE and ASCERTAIN) and the first patients had already been enrolled.[3]
On November 22nd, 2013, both companies On May 15th, 2013, both companies announced positive phase 3 results for the RA-MOBILITY trial
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Sarilumab”. American Medical Association.
- http://investor.regeneron.com/releasedetail.cfm?releaseid=590869
- http://en.sanofi.com/Images/33027_20130515_sari_en.pdf
fully human monoclonal antibody directed against the interleukin-6 receptor (IL-6R) in combination with methotrexate (MTX) therapy improved disease signs and symptoms as well as physical functionw while inhibiting progression of joint damage in adults with RA who saw little improvement through MTX therapy alone.
Sarilumab met all three primary endpoints of the 52-week SARIL-RA-MOBILITY Phase III trial by demonstrating clinically relevant and statistically significant improvements compared to the placebo group in the two groups treated with the drug candidate. The trial enrolled about 1,200 patients with active, moderate-to-severe rheumatoid arthritis who were inadequate responders to MTX therapy.
Of patients treated with the 200 mg dose of sarilumab plus MTX, 66% saw improvement in signs and symptoms of RA at 24 weeks, as measured by the American College of Rheumatology score of at-least 20% improvement. The percentage dipped to 58% of sarilumab 150 mg dose patients, and 33% of placebo patients.
Sarilumab 200 mg patients showed the least progression of structural damage after 52 weeks, registering a 0.25 change in the modified Van der Heijde total Sharp score. That contrasts with scores of 0.90 in patients taking sarilumab 150 mg, and 2.78 in the placebo group.
In addition, sarilumab 200 mg patients showed improvement in physical function, as measured by change from baseline in the Health Assessment Question-Disability at week 16. However, the companies did not quantify those results in their announcement. Sanofi and Regeneron said additional analyses of efficacy and safety data from SARIL-RA-MOBILITY will be presented “at a future medical conference.”
“We are encouraged by these Phase III results and the impact sarilumab demonstrated on inhibition of progression of structural damage assessed radiographically in this study,” Tanya M. Momtahen, Sanofi’s sarilumab global project head, said in a statement.
Sarilumab—known as SAR153191 and REGN88—blocks the binding of IL-6 to its receptor and interrupts the resultant cytokine-mediated inflammatory signaling characteristic of RA. Sarilumab was developed using Regeneron’s VelocImmune® antibody technology.
The positive results continue what has been mostly strong success in clinical trials for the partners, whose development collaborations include alirocumab (REGN727), dupilumab (REGN668), and enoticumab (REGN421). Alirocumab is a PCSK9 antibody being evaluated for its ability to manage LDL cholesterol, including in people who do not get to their target LDL levels using statin medicines alone. Dupilumab is an antibody to the receptors for interleukin-4 and interleukin-13 under evaluation in atopic dermatitis and eosinophilic asthma. Enoticumab is a fully human monoclonal antibody to delta-like ligand-4 (Dll4) now in Phase I study for advanced malignancies.
On its own, however, Sanofi’s R&D efforts have shown more mixed results, with the pharma giant earlier this month ending development of cancer drug candidate fedratinib (SAR302503) after it was placed on clinical hold by the FDA following reports that some patients in clinical trials developed symptoms consistent with Wernicke’s encephalopathy. Another cancer compound, iniparib, had its development halted earlier this year after a disappointing Phase III trial.
Daiichi Sankyo anticoagulant edoxaban succeeds in Phase III

Edoxaban, DU-176b
Daiichi Sankyo, APPROVED IN JAPAN as tosylate monohydrate salt in 2011 for the prevention of venous embolism in patients undergoing total hip replacement surgery
for synthesis see….http://www.sciencedirect.com/science/article/pii/S0968089613002642 Bioorganic & Medicinal Chemistry 21 (2013) 2795–2825, see s[pecific page 2808 for description ie 14/31 of pdf
WO 2010071121, http://www.google.com/patents/WO2010071121A1
WO 2007032498
N’-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide
NOV20, 2013
Daiichi Sankyo will file edoxaban on both sides of the Atlantic shortly after the bloodthinner proved as effective and safer than warfarin in a Phase III trial of patients with atrial fibrillation.
The company has presented data on edoxaban, a once-daily oral factor Xa inhibitor, at the American Heart Association meeting in Dallas, from a study involving 21,105 patients across 46 countries. The drug, evaluated in 60mg and 30mg doses, met its primary endpoint of non-inferiority compared to warfarin for the prevention of stroke or systemic embolic events in patients with non-valvular AF.http://www.pharmatimes.com/Article/13-11-20/Daiichi_Sankyo_anticoagulant_edoxaban_succeeds_in_Phase_III.aspx
Edoxaban (INN, codenamed DU-176b, trade name Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It is being developed by Daiichi Sankyo. It was approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1]
In animal studies, edoxaban is potent, selective for factor Xa and has good oral bioavailability.[2]
Daichi Sankyo’s edoxaban tosilate is an orally administered
coagulation factor Xa inhibitor that was approved and launched
in Japan for the preventive treatment of venous thromboembolic
events (VTE) in patients undergoing total knee arthroplasty, total
hip arthroplasty, or hip fracture surgery. Edoxaban has been
shown to have a rapid onset of anticoagulant effect due to short
Tmax (1–2 h) after dosing and sustained for up to 24 h post-dose.
Marketed under the brand name Lixiana, it is currently in phase
III studies in the US for the prevention of stroke and systemic embolic
events in patients with atrial fibrillation (AF) and venous
thromboembolism (VTE).
Several Phase II clinical trials have been conducted, for example for thromboprophylaxis after total hip replacement[3] (phase III early results compare well to enoxaparin[4]), and for stroke prevention in patients with atrial fibrillation[5][6].Those papers follow similar recent major trials showing similar results for the other new factor Xa inhibitors, rivaroxaban and apixaban.
A large phase III trial showed that edoxaban was non inferior to warfarin in preventing recurrent venous thromboembolic events with fewer episodes of major bleeding.[7]
- “First market approval in Japan for LIXIANA (Edoxaban)”. Press Release. Daiichi Sankyo Europe GmbH. 2011-04-22.
- Furugohri T, Isobe K, Honda Y, Kamisato-Matsumoto C, Sugiyama N, Nagahara T, Morishima Y, Shibano T (September 2008). “DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles”. J. Thromb. Haemost. 6 (9): 1542–9. doi:10.1111/j.1538-7836.2008.03064.x. PMID 18624979.
- Raskob, G.; Cohen, A. T.; Eriksson, B. I.; Puskas, D.; Shi, M.; Bocanegra, T.; Weitz, J. I. (2010). “Oral direct factor Xa inhibition with edoxaban for thromboprophylaxis after elective total hip replacement”. Thrombosis and Haemostasis 104 (3): 642–649. doi:10.1160/TH10-02-0142.PMID 20589317. edit
- “Phase III Trial Finds Edoxaban Outclasses Enoxaparin in Preventing Venous Thromboembolic Events”. 8 Dec 2010.
- Weitz JI, Connolly SJ, Patel I, Salazar D, Rohatagi S, Mendell J, Kastrissios H, Jin J, Kunitada S (September 2010). “Randomised, parallel-group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation”. Thromb. Haemost. 104 (3): 633–41. doi:10.1160/TH10-01-0066.
- Edoxaban versus Warfarin in Patients with Atrial Fibrillation Robert P. Giugliano, M.D., Christian T. Ruff, M.D., M.P.H., Eugene Braunwald, M.D., Sabina A. Murphy, M.P.H., Stephen D. Wiviott, M.D., Jonathan L. Halperin, M.D., Albert L. Waldo, M.D., Michael D. Ezekowitz, M.D., D.Phil., Jeffrey I. Weitz, M.D., Jindřich Špinar, M.D., Witold Ruzyllo, M.D., Mikhail Ruda, M.D., Yukihiro Koretsune, M.D., Joshua Betcher, Ph.D., Minggao Shi, Ph.D., Laura T. Grip, A.B., Shirali P. Patel, B.S., Indravadan Patel, M.D., James J. Hanyok, Pharm.D., Michele Mercuri, M.D., and Elliott M. Antman, M.D. for the ENGAGE AF-TIMI 48 InvestigatorsDOI: 10.1056/NEJMoa1310907
- “Edoxaban versus Warfarin for the Treatment of Symptomatic Venous Thromboembolism”. N. Engl. J. Med. August 2013. doi:10.1056/NEJMoa1306638. PMID 23991658.
- WO 03/000657 pamphlet WO 03/000680 pamphlet WO 03/016302 pamphlet WO 04/058715 pamphlet WO 05/047296 pamphlet WO 07/032498 pamphlet WO 08/129846 pamphlet WO 08/156159 pamphlet
- J Am Chem Soc 1978, 100(16): 5199
Drug formulation , lixiana, edoxaban tosylate monohydrate, CAS 912273-65-5, C24 H30 Cl N7 O4 S . C7 H8 O3 S . H2 O, 738.274
-
N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide p-toluenesulfonic acid monohydrate represented by the following formula (A) (hereinafter, also referred to as compound A) :
-


-
is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (FXa), and is useful as a preventive and/or therapeutic drug for thrombotic diseases (Patent Literature 1 to 8).
-
For example, a method comprising mixing the free form of compound A represented by the following formula (B) (hereinafter, also referred to as compound B):
-

-
with p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate, followed by crystallization from aqueous ethanol, is known as a method for obtaining compound A (Patent Literature 1 to 8). These literature documents do not make any mention about adding p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate in a stepwise manner in the step of obtaining compound A from compound B.
Citation ListPatent Literature
-
- Patent Literature 1: International Publication No. WO 03/000657
- Patent Literature 2: International Publication No. WO 03/000680
- Patent Literature 3: International Publication No. WO 03/016302
- Patent Literature 4: International Publication No. WO 04/058715
- Patent Literature 5: International Publication No. WO 05/047296
- Patent Literature 6: International Publication No. WO 07/032498
- Patent Literature 7: International Publication No. WO 08/129846
- Patent Literature 8: International Publication No. WO 08/156159
SIMILAR

OTHER SALTS

Edoxaban hydrochloride
CAS Number: 480448-29-1
Molecular Formula: C24H30ClN7O4S · HCl
Molecular Weight: 584.52 g.mol-1
Edoxaban is reported to be a member of the so-called “Xaban-group” and as such to be a low molecular inhibitor of the enzyme factor Xa, participating in the blood coagulation system. Therefore, edoxaban is classified as an antithrombotic drug and its possible medical indications are reported to be treatment of thrombosis and thrombosis prophylaxis after orthopaedic operations, such as total hip replacement, as well as for stroke prevention in patients with atrial fibrillation, the prophylaxis of the acute coronary syndrome and the prophylaxis after thrombosis and pulmonary embolism.
The IUPAC name for edoxaban is N’-(5-chloropyridin-2-yl)-N-[(15,2^,4S)-4- (dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[l ,3]thiazolo[5,4-c]pyridine-2- carbonyl)amino]cyclohexyl]oxamide. The chemical structure of edoxaban is shown in the formula (1) below:

formula ( 1 ) While Edoxaban is reported to be soluble in strongly acidic aqueous solutions, its solubility is considered to be very low in neutral or alkaline aqueous media. EP 2 140 867 A 1 claims an edoxaban-containing pharmaceutical composition comprising a water-swelling additive and/or a sugar alcohol. Further, it is alleged that compositions comprising lactose or cornstarch do not have good dissolution properties. The claimed pharmaceutical compositions in EP 2 140 867 Al are considered to show good dissolution properties in a neutral aqueous medium as well. Tablets comprising said composition were produced by wet granulation. However, it turned out that prior art pharmaceutical formulations comprising edoxaban being suitable for oral administration are still improvable with regards to dissolution rate and bioavailability. Further, stability and content uniformity of the known formulations could be improved. Further, due to the intolerance of many people to sugar alcohol(s), such as sorbitol, the use of sugar alcohol(s) should be avoided.
Amgen: Phase III Melanoma treatment, Talimogene Laherparepvec Improves Survival
 |
|
|---|---|
| Transmission electron micrograph of an unmodified herpes simplex virus |
Talimogene Laherparepvec
Amgen Presents Interim Overall Survival Data From Phase 3 Study Of Talimogene Laherparepvec In Patients With Metastatic Melanoma
T-VEC was engineered from herpes simplex 1 (HSV-1), a relatively innocuous virus that normally causes cold sores. A number of genetic modifications were made to the virus in order to:
- Attenuate the virus (so it can no longer cause herpes)
- Increase selectivity for cancer cells (so it destroys cancer cells while leaving healthy cells unharmed)
- Secrete the cytokine GM-CSF (a protein naturally secreted in the body to initiate an immune response)
T-VEC has a dual mechanism of action, destroying cancer both by directly attacking cancer cells and also by helping the immune system to recognize and destroy cancer cells. T-VEC is injected directly into a number of a patient’s tumors. The virus invades both cancerous and healthy cells, but it is unable to replicate in healthy cells and thus they remain unharmed. Inside a cancer cell, the virus is able to replicate, secreting GM-CSF in the process. Eventually overwhelmed, the cancer cell lyses (ruptures), destroying the cell and releasing new viruses, GM-CSF, and an array of tumor-specific antigens (pieces of the cancer cell that are small enough to be recognized by the immune system).
The GM-CSF attracts dendritic cells to the site. Dendritic cells are immune cells that process and present antigens to the immune system so that the immune system can then identify and destroy whatever produced the antigen. The dendritic cells pick up the tumor antigens, process them, and then present them on their surface to cytotoxic (killer) T cells. Now the T cells are essentially “programmed” to recognize the cancer as a threat. These T cells lead an immune response that seeks and destroys cancer cells throughout the body (eg, tumors and cancer cells that were not directly injected with T-VEC).

In this way, T-VEC has both a direct effect on injected tumors and a systemic effect throughout the entire body. Because the adaptive immune system “remembers” a target once it has been identified, there is high likelihood that the effect of an oncolytic virus like T-VEC will be durable (eg, prevent relapse). And it is for this reason that T-VEC does not need to be injected into every tumor, just a few in order to start the immune process.
Clinical efficacy in unresectable melanoma has been demonstrated in Phase II and Phase III clinical trials.
The Phase II clinical trial was published in the Journal of Clinical Oncology in 2009. 50 patients with advanced melanoma (most of whom had failed previous treatment) were treated with T-VEC. The overall response rate (patients with a complete or partial response per RECIST criteria) was 26% (16% complete responses, 10% partial responses). Another 4% of patients had a surgical complete response, and another 20% had stable disease for at least 3 months. On an extension protocol, 3 more patients achieved complete responses, and overall survival was 54% at 1 year and 52% at 2 years—demonstrating that responses to T-VEC are quite durable.
Consistent with other immunotherapies, some patients exhibited initial disease progression before responding to therapy because of the time it takes to generate the full immune response. Responses were seen in both injected and uninjected tumors (including those in visceral organs), demonstrating the systemic immunotherapeutic effect of T-VEC. Treatment was extremely well tolerated, with only Grade 1 or 2 drug-related side effects, the most common being mild flu-like symptoms.

|

|
Amgen announced the initial results of the Phase III OPTiM trial on Mar. 19, 2013. This global, randomized, open-label trial compared T-VEC with subcutaneously administered GM-CSF (2:1 randomization) in 430 patients with unresectable stage IIIB, IIIC or IV melanoma. The primary endpoint was durable response rate (DRR), defined as a complete or partial tumor response lasting at least 6 months and starting within 12 months of treatment.

T-VEC was proven to offer superior benefits in metastatic melanoma. DRR was achieved in 16% of patients receiving T-VEC compared with only 2% in the GM-CSF control group (P<.0001). The greatest benefit was seen in patient with stage IIIB or IIIC melanoma, with a 33% DRR vs 0% with GM-CSF. The objective response rate (any response) with T-VEC was 26%, with an impressive 11% of patients experiencing a complete response (complete disappearance of melanoma throughout the body). This demonstrated once again that T-VEC has a systemic immune effect that destroys distant, uninjected tumors. According to Financial Times one of the investigators involved questioned the ethics of the trial design, as the control arm received subcutaneous GM-CSF instead of standard care

A trend toward improved survival with T-VEC was observed in a pre-specified interim analysis of this endpoint, with the final survival data (event-driven) expected in late 2013. At the interim analysis, T-VEC was associated with a 21% reduced risk of death. The most common side effects with T-VEC were fatigue, chills, and fever. No serious side effect occurred in more than 3% of patients in either arm of the study.
The investigators concluded that “T-VEC represents a novel potential [treatment] option for melanoma with regional or distant metastases.” The success of T-VEC in the OPTiM trial represents the first Phase III proof of efficacy for a virus-based oncolytic immunotherapy.
Chugai Pharma to commercialize Helsinn’s Anamorelin for anorexia-cachexia syndrome in Germany, France, Benelux, UK and Ireland

LUGANO, Switzerland, November 12, 2013 /PRNewswire/ —
Swiss Pharma Group Helsinn seals an alliance with Japanese company Chugai by granting exclusive distribution & licensing rights to commercialize its innovative phase III ghrelin receptor agonist in Germany, France, Benelux, UK andIreland
Swiss-based Helsinn group has granted Chugai Pharma Marketing Ltd., a wholly-owned subsidiary of Chugai Pharmaceutical Co., Ltd., exclusive commercialization rights to their innovative ghrelin receptor agonist, anamorelin, for the three major European pharma markets.
Anamorelin is a new first-in-class, oral, once daily drug, currently in phase III for the treatment of anorexia-cachexia in NSCLC, a detrimental multifactorial disorder that affects over 50% of people with cancer and in which systemic inflammation, reduced food intake and altered metabolism contribute to loss of muscle mass and reduction of body weight leading to reduced quality of life, functional impairment and decreased survival.
- Anamorelin is a synthetic orally active ghrelin receptor agonist which is under development for the management of non-small lung cancer associated cachexia/anorexia.
- Development status: Phase 3
- Appetite loss; Aging; Bone injury; Cachexia

Anamorelin HCl
| M. Wt | 583.16 |
|---|---|
| Formula | C31H43ClN6O3 |
| CAS No. | 861998-00-7 |
| Synonyms | RC 1291; RC1291; RC 1291-HCl; ONO-7643; ONO7643; ONO 7643. |
| Chemical Name | 2-amino-N-((R)-1-((R)-3-benzyl-3-(1,2,2-trimethylhydrazinecarbonyl)piperidin-1-yl)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-methylpropanamide hydrochloride |
Necitumumab

Necitumumab
Necitumumab is a fully human IgG1 monoclonal antibody designed to block the ligand binding site of the human epidermal growth factor receptor (EGFR), which is a target in several anti-cancer treatments because it sparks cancer progression, both by promoting angiogenesis, or the formation of new blood vessels for tumors, and by inhibiting apoptosis, or cell death. Recently approved therapies for non-squamous NSCLC, including afatinib and erlotinib, target specific EGFR mutations, but those drugs are used to treat patients with nonsquamous histology.Lilly did not provide specific data regarding the results of the trial, but the company announced that it plans to present that data at a scientific meeting next year, and to request a review of the drug by regulatory authorities before the end of 2014.
Necitumumabis one of three monoclonal antibodies in Phase III
development that targets EGFR, the target of the approved antibodies
cetuximab and panitumumab. However, necitumumab is a fully human
IgG1 antibody, distinguishing it from both the approved agents.
Necitumumab is directed against the ligand binding site of EGFR and is
being co-developed by Eli Lilly and Bristol-Myers Squibb in the United
States, Canada, and Japan, while Eli Lilly alone is developing it for other
markets. Necitumumabfirst entered clinical development in 2004 and
is now in Phase III development for the treatment of non–small-cell
lung cancer and Phase II for the treatment of colorectal cancer. The
primary indication chosen further distinguishes necitumumabfrom both
cetuximab and panitumumab, but it is an indication for which EGFR
kinase inhibitors such as erlotinib are approved.
In December 2009, Eli Lilly stressed the long half-life of necitumumab
(7–10 days, which permits dosing at 2–3 week intervals) and its potential
both for reduced hypersensitivity reactions (i.e., better tolerability) and
for induced host-mediated anticancer activity. In addition, it highlighted
that necitumumabdisplays similar or superior activity to cetuximab
in anticancer models. Preliminary data were presented from the Phase
II study in colorectal cancer showing antitumor activity in 73% of 44
patients treated with necitumumabplus FOLFOX.
Both Phase III studies in non–small-cell lung cancer are in stage IV
disease and in groups of 947 patients treated with necitumumabplus
cisplatin and a second agent. The INSPIRE study in non-squamous
disease began in November 2009 and uses pemetrexed as the second
drug, while the SQUIRE study commenced in January 2010 in
squamous disease and uses gemcitabine. Both studies have primary
completion dates in late 2011 and study completion dates of mid-2012,
which points to BLA submission in 2013.

A Phase I study in patients with solid tumors suggested that skin
toxicity was the dose-limiting toxicity and suggested that 800 mg of
necitumumab (at weekly or fortnightly intervals) be the maximum dose
(Kuenen et al. 2010).16 This dose was employed in the initial colorectal
cancer study, at 14-day intervals, which revealed a 60% partial response
(Taberno et al. 2008).17
The development strategy for necitumumab appears to have been
designed to establish it initially in a major indication where it will not
be competing with established antibody products, while seeking
to exploit the reported advantages over cetuximab appears to be
a secondary priority. While the reported Phase II data are very
encouraging, it will be some time before a better assessment of the
commercial prospects of necitumumab can be made. However, it does
appear to have significant potential.
Necitumumab (proposed INN) is a monoclonal antibody and an antineoplastic. It binds to the epidermal growth factor receptor(EGFR).[1] As of October 2009, two Phase III clinical trials are planned to investigate its effects on non-small cell lung carcinoma.[2][3]
- International Nonproprietary Names for Pharmaceutical Substances (INN, prepublication copy), World Health Organization.
- ClinicalTrials.gov NCT00981058 Squamous Non-Small Cell Lung Cancer (NSCLC) Treatment With the Inhibitor of Epidermal Growth Factor Receptor (EGFR) (SQUIRE)
- ClinicalTrials.gov NCT00982111 NonSquamous Non-Small Cell Lung Cancer Treatment With the Inhibitor of Epidermal Growth Factor Receptor (INSPIRE)
Possible Efficacy Of Lilly’s Necitumumab (IMC-11F8) In Lung Cancer Subset
18.4 2013
Eli Lilly announced yesterday their very preliminary and non-quantitative conclusions on the SQUIRE study, a 1093-patient Phase III trial of their anti-epidermal growth factor receptor (EGFR) antibody, necitumumab (IMC-11F8), against Stage IV squamous, non-small cell lung carcinoma (NSCLC).http://www.forbes.com/sites/davidkroll/2013/08/14/possible-efficacy-of-necitumumab-imc-11f8-in-squamous-nsclc-lung-cancer-subset/

Reslizumab
Reslizumab(CINQUIL) is a humanized monoclonal antibody
targeted against IL-5 that is being developed by Cephalon for the
treatment of eosinophilic asthma. In September 2010, Cephalon
indicated that it hopes to file a BLA in 2013, focusing on this subset of
severe asthmatics. Such patients are ca. 30% of the asthmatic population,
with the 750,000 patients in the United States suggested to offer the potential for peak market sales of $1 billion. However, previous
attempts to develop recombinant IL-5 antagonists for the treatment of asthma saw very disappointing clinical results with both mepolizumab
(GlaxoSmithKline) and reslizumab (Schering-Plough and Celltech).
Schering-Plough (now Merck) had been developing reslizumab in
partnership with Celltech (now UCB), utilizing the latter’s antibody
technology, but terminated development in 2002 after disappointing
clinical results. The rights were acquired by Ception Therapeutics in
2007, with development reinitiated for both pediatric eosinophilic
esophagitis and eosinophilic asthma. Cephalon acquired an option to
acquire Ception in January 2009 and exercised this option in April 2010
despite unpromising results in the Phase II/III study of reslizumab in
pediatric eosinophilic esophagitis patients.
In its November 2009 R&D presentation, Cephalon presented data
(from Schering-Plough) showing that reslizumab treatment of asthmatics
results in a sustained suppression of eosinophil levels; the protocols
employed in the Phase II/III study in pediatric eosinophilic esophagitis
and a Phase II study in eosinophilic asthma were described. The Phase
III study in asthmatics has yet to commence, but a 190-patient openlabel Phase III extension study in eosinophilic esophagitis is ongoing.
The Phase II/III study showed no discernable symptom improvement
despite suppression of eosinophil levels at all three doses tested (see
Walsh 2010).43 The outcome of the 106-patient Phase II study, in
February 2009, in asthmatics prompted Cephalon to acquire Ception.
Reslizumab treatment produced significant improvement in lung
function and reduced airway inflammation.
Reslizumab is currently the most advanced of three anti–IL-5monoclonal antibodies in development, but the 2013 submission date for a BLA seems optimistic given that Phase III studies have yet to start.
Mepolizumab is now in Phase II studies for the treatment of severe
asthma and nasal polyposis (having previously been filed for approval
in Europe for the treatment of hypereosinophilic syndrome), but the
filing was withdrawn and development for that indication discontinued
in late 2009. MedImmune and Kyowa Hakko Kirin’s benralizumab has
successfully completed a Phase IIa study in asthma with data presented
in September 2010, and a 108-patient study in asthma was completed
in October 2010. A similar-size Phase II study in COPD commenced in
November 2010.
Reslizumab is a humanized monoclonal antibody intended for the treatment of eosinophil-meditated inflammations of the airways, skin and gastrointestinal tract.[1] As of September 2009, the drug is undergoing Phase II/III clinical trials.[2]
- Walsh, GM (2009). “Reslizumab, a humanized anti-IL-5 mAb for the treatment of eosinophil-mediated inflammatory conditions”. Current opinion in molecular therapeutics11 (3): 329–36. PMID 19479666.
- ClinicalTrials.gov
