



如果您有关于yaopha.com的问题或建议请与我们联系,具体方式如下:
E-mail:contactus@yaopha.com
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » Phase3 drugs (Page 23)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

faldaprevir , 801283-95-4 cas no, BI-201335
(1R,2S)-1-{[(2S,4R)-4-[{8-bromo-7-methoxy-2-[2-(2-methylpropanamido)-1,3-thiazol-4-yl]quinolin-4-yl}oxy]-1-[(2S)-2-{[(cyclopentyloxy)carbonyl]amino}-3,3-dimethylbutanoyl]pyrrolidine-2-carboxamido]-2-ethenylcyclopropane-1-carboxylic acid
Molecular Formula: C40H49BrN6O9S
Molecular Weight: 869.82 g.mol-1
2 nd nov 2013
Boehringer Ingelheim today announced new data from its Phase III clinical trial programme, STARTVerso™, which evaluates faldaprevir* in combination with pegylated interferon and ribavirin (PegIFN/RBV). Patients with genotype-1 (GT-1) hepatitis C (HCV) who have not received previous treatment (treatment-naïve: STARTVerso™1&2),1 treatment-experienced patients (STARTVerso™3),2 and HIV co-infected patients (STARTVerso™4)3 participated in this study programme. The results from these and additional studies will be presented at the 64th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD), also known as The Liver Meeting®, taking place 1-5 November in Washington, D.C.
Faldaprevir (formerly BI 201335) is an experimental drug candidate for the treatment of hepatitis C. It is being developed byBoehringer-Ingelheim and is currently in Phase III trials.[1]
Faldaprevir is a hepatitis C virus protease inhibitor.
Faldaprevir is being tested in combination regimens with pegylated interferon and ribavirin, and in interferon-free regimens with other direct-acting antiviral agents including BI 207127.
Data from the SOUND-C2 study, presented at the 2012 AASLD Liver Meeting, showed that a triple combination of faldaprevir, BI 207127, and ribavirin performed well in HCV genotype 1b patients.[2] Efficacy fell below 50%, however, for dual regimens without ribavirin and for genotype 1a patients.
The following Compound 1):

(1)
wherein B is

; L° is MeO-; L1 is Br; and R2 is and having the chemical name: l-{ [4-[8-Bromo-2-(2-isopropylcarbamoyl-thiazol-4-yl)-7- methoxy-quinolin-4-yloxy]-l-(R)-(2-cyclopentyloxycarbonyl amino-3,3-(S)-dimethyl- butyryl)-pyrrolidine-(S)-2-carbonyl]-amino}-2-(S)-vinyl-cyclopropane-(R)-carboxylic acid, is known as a selective and potent inhibitor of the HCV NS3 serine protease and useful in the treatment of HCV infection. Compound (1) falls within the scope of the acyclic peptide series of HCV inhibitors disclosed in U.S. Patents RE 40,525, 7,514,557 and 7,585,845. Compound (1) is disclosed specifically as Compound # 1055 in U.S. Patent 7,585,845, and as Compound # 1008 in U.S. Patent 7,514,557. Compound (1), and pharmaceutical formulations thereof, can be prepared according to the general procedures found in the above-cited references, all of which are herein incorporated by reference in their entirety. Preferred forms of Compound (1) include the crystalline forms, in particular the crystalline sodium salt form, which can be prepared as described in U.S. Patent Application Publication No. 2010/0093792, also incorporated herein by reference. Data demonstrating the activity of Compound (1) as an inhibitor of the HCV NS3 serine protease and its corresponding demonstrated utility in the treatment of HCV infection in mono-infected patients, can be found in U.S. Patent 7,585,845, as well as in numerous publications presenting the preclinical characterization or clinical trial results with Compound (1). See, e.g., Sulkowski MS, et al, Hepatol (2009), Vol. 50, pg. 2A, Abtract LB3; Sulkowski MS, et al., J Hepatol (2010) Vol. 52, Supp. 1, pgs. S462-S463, Abstract 1190; Berg et al., Hepatol (2010), Vol. 52, Supp. SI, Abstract 804; and White PW, et al., Antimicrob Agents Chemother (2010) 54(11):4611-4618.
Combination therapy regimens directed to administering Compound (1) with an interferon- alpha and ribavirin for the treatment of HCV infection are described in U.S. Patent Application Publication Nos. 2010/0068182 and 2011/0268700.
HIV/HCV coinfected persons tend to have higher HCV viral loads and are less likely to clear the HCV spontaneously. The urgency for treatment of persons who are coinfected is greater than it is for those with HCV infection alone. The course of liver disease is more rapid in HIV/HCV-coinfected persons, including an approximately 2-fold increased risk of cirrhosis, more rapid progression to decompensated liver disease and increased risk for hepatocellular carcinoma (Graham CS, et al., Clin Infect Dis (2001 );33:562-569) .
Treatment of HCV might improve the tolerability of highly active antiretroviral therapy (HAART) because HCV infection increases the risk of mitochondrial toxicity and hepatotoxicity from HAART (Sulkowski MS, et al., JAMA (2000);283:74-80; Lafeuil!ade A, et al., Lancet (2001);357:280-281 ). Although there is much less published information on treatment outcomes in those who are HIV/HCV-coinfected than in HCV mono-infected patients, all accumulated data demonstrate that sustained virological response (SVR) and cure from HCV infection with pegylated interferon alpha and ribavirin is achieved in a substantially lower proportion of HIV/HCV coinfected patients when compared to HCV mono-infected patients. Factors associated with a poor treatment response (e.g., a high baseline HCV viral load, cirrhosis, and African American race) are present in a higher proportion of HIV/HCV coinfected populations, when compared to HCV monoinfected populations. It is not clear to what extent HIV infection itself diminishes the SVR rate, and to what extent advanced immunosuppression (e.g., CD4+ T lymphocyte count <200/mm3) further reduces response to HCV treatment (Toriani FJ, et al., N Engl J Med (2004);351(5): 438 -50; Nunez M, et al., ARHR (2007); 23(8):972-982).
Thus, there is a continuing high unmet need in the art for therapies that are effective against HCV in patients that are co-infected with HIV.

Pimavanserin, ACP 103
N-(4-fluorophenylmethyl)-N-(1-methylpiperidin-4-yl)-N’-(4-(2-methylpropyloxy)phenylmethyl)carbamide, 706779-91-1 cas
 706782-28-7 (tartrate) |
THURSDAY Oct. 31, 2013 — Many people living with Parkinson’s disease suffer from hallucinations and delusions, but an experimental drug might offer some relief without debilitating side effects.
READ ALL AT
http://www.drugs.com/news/new-shows-early-promise-treating-parkinson-s-psychosis-48630.html

The drug — pimavanserin — appears to significantly relieve these troubling symptoms, according to the results of a phase 3 trial to test its effectiveness.
Pimavanserin (ACP-103) is a drug developed by Acadia Pharmaceuticals which acts as an inverse agonist on the serotonin receptor subtype 5-HT2A, with 40x selectivity over 5-HT2C, and no significant affinity or activity at 5-HT2B or dopamine receptors.[1] As of September 3 2009, pimavanserin has not met expectations for Phase III clinical trials for the treatment of Parkinson’s disease psychosis,[2] and is in Phase II trials for adjunctive treatment of schizophrenia alongside an antipsychotic medication.[3] It is expected to improve the effectiveness and side effect profile of antipsychotics.[4][5][6]
3-D MODEL OF DRUG PIMAVANSERIN, THE DEVELOPMENT OF WHICH HAS BEEN EXPEDITED BY THE FDA

Psychiatrist Herb Meltzer sadly watched the agitated woman accuse her son of trying to poison her. Although not her physician, Dr. Meltzer certainly recognized the devastating effects of his mother-in-law’s Parkinson’s disease psychosis (PDP). Occurring in up to half of all patients with Parkinson’s, symptoms of the psychotic disorder may include hallucinations and delusions. The development of PDP often leads to institutionalization and increased mortality.
“I was on the sidelines,” explains Dr. Meltzer, professor of psychiatry and physiology and director of the Translational Neuropharmacology Program at Northwestern University Feinberg School of Medicine. “I told my brother-in-law it was the disease talking, not his mother.”
Ironically, Dr. Meltzer has been far from the sidelines and right on the PDP playing field for quite a while. In fact, he may soon see a drug he helped develop become the first approved treatment for the disorder. In early April, Dr. Meltzer celebrated, along with colleagues at ACADIA Pharmaceuticals in San Diego for which he has been a clinical advisor, the stunning announcement: the Food and Drug Administration (FDA) had expedited the company’s path to filing a new drug application (NDA) for pimavanserin, a selective serotonin 5-HT2Areceptor blocker. Typically, the FDA requires data from two successful pivotal Phase III clinical studies affirming a drug candidate’s safety and efficacy before the agency will even consider an NDA. Just as ACADIA was planning to launch another Phase III study this spring to fulfill this requirement, the FDA decided the company had amassed enough data to support an NDA filing.

HERBERT MELTZER, MD, DESIGNED ACADIA PHARMACEUTICAL’S INITIAL PROOF OF CONCEPT TRIAL OF THE DRUG PIMAVANSERIN TO TREAT PARKINSON’S DISEASE PSYCHOSIS.
“This action on the part of the FDA is extremely unusual,” says Dr. Meltzer, who designed ACADIA’s initial proof-of-concept trial of pimavanserin, a drug he had initially suggested ACADIA develop to treat schizophrenia, with PDP as a secondary indication. “The FDA staff decided that results from my small clinical study and the first successful Phase III study were sufficient to establish efficacy and safety.”
Bringing a safe and effective drug to market is a monumental achievement. Pimavanserin is not yet there but has significantly moved within striking distance with this recent nod from the regulatory agency.
The neuropharmacologist’s collaboration with ACADIA began in 2000. The company wanted to develop a drug targeting the serotonin 5-HT 2A receptor, a neurotransmitter ACADIA believed played a key role in schizophrenia based upon basic research from Meltzer and their own studies. A distinguished schizophrenia investigator, then at Case Western Reserve University, he welcomed ACADIA’s offer to translate his ideas about developing safer and more effective drug treatments for psychosis. Through his provocative and groundbreaking research, Dr. Meltzer originally championed the idea that blocking the 5-HT2A receptor would lead to better antipsychotic drugs with fewer side effects. Existing drugs often impaired motor function because they targeted the dopamine D2 receptor. Of the 14 different types of serotonin receptors in this complex area of study, Dr. Meltzer zeroed in on the 5-HT2A type—the same receptor that leads to hallucinogenic properties of LSD and mescaline. It was an ideal target to complement weak D2 receptor blockade in schizophrenia and as a standalone treatment for PD psychosis.
………………………………………….

DULAGLUTIDE
PRONUNCIATION doo” la gloo’ tide
THERAPEUTIC CLAIM Treatment of type II diabetes
CHEMICAL NAMES
1. 7-37-Glucagon-like peptide I [8-glycine,22-glutamic acid,36-glycine] (synthetic
human) fusion protein with peptide (synthetic 16-amino acid linker) fusion protein with immunoglobulin G4 (synthetic human Fc fragment), dimer
2. [Gly8,Glu22,Gly36]human glucagon-like peptide 1-(7-37)-peptidyltetraglycyl-Lseryltetraglycyl-L-seryltetraglycyl-L-seryl-L-alanyldes-Lys229-[Pro10,Ala16,Ala17]human immunoglobulin heavy constant γ4 chain H-CH2-CH3 fragment, (55-55′:58-58′)-bisdisulfide dimer
STRUCTURAL FORMULA
Monomer
HGEGTFTSDV SSYLEEQAAK EFIAWLVKGG GGGGGSGGGG SGGGGSAESK 50
YGPPCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSQEDP 100
EVQFNWYVDG VEVHNAKTKP REEQFNSTYR VVSVLTVLHQ DWLNGKEYKC 150
KVSNKGLPSS IEKTISKAKG QPREPQVYTL PPSQEEMTKN QVSLTCLVKG 200
FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSRLT VDKSRWQEGN 250
VFSCSVMHEA LHNHYTQKSL SLSLG 275
Disulfide bridges location
55-55′ 58-58′ 90-150 90′-150′ 196-254 196′-254′
MOLECULAR FORMULA C2646H4044N704O836S18
MOLECULAR WEIGHT 59.67 kDa
MANUFACTURER Eli Lilly and Company
CODE DESIGNATION LY2189265
CAS REGISTRY NUMBER 923950-08-7
http://www.ama-assn.org/resources/doc/usan/dulaglutide.pdf
LY2189265 (dulaglutide), a glucagon-like peptide-1 analog, is a biologic entity being studied as a once-weekly treatment for type 2 diabetes.
Dulaglatuide works by stimulating cells to release insulin only when blood sugar levels are high.
Gwen Krivi, Ph.D., vice president, product development, Lilly Diabetes, said of the drug, “We believe dulaglutide, if approved, can bring significant benefits to people with type 2 diabetes.”
In fact, it might help to control both diabetics’ blood sugar and their high blood pressure.
Eli Lilly CEO John Lechleiter believes the drug has the potential to be a blockbuster. Lilly could be ready to seek approval by 2013.
For more information on dulaglutide clinical studies, click here.
Data Preseted at 49th EASD Annual Meeting Show Treatment with Lilly’s Investigational Dulaglutide Resulted in Improved Patient-Reported Health Outcomes – September 26, 2013
Lilly Announces Positive Results of Phase III Trials of Dulaglutide in Type 2 Diabetes – April 16, 2013
Lilly Diabetes Announces Positive Results of Phase III Trials of Dulaglutide in Type 2 Diabetes – October 22, 2012
 |
|
|---|---|
http://www.ama-assn.org/resources/doc/usan/ixekizumab.pdf
USAN IXEKIZUMAB
PRONUNCIATION ix” e kiz’ ue mab
THERAPEUTIC CLAIM Treatment of autoimmune diseases
CHEMICAL NAMES
1. Immunoglobulin G4, anti-(human interleukin 17A) (human monoclonal LY2439821γ4-chain), disulfide with human monoclonal LY2439821 κ-chain, dimer
2. Immunoglobulin G4, anti-(human interleukin-17A (IL-17, cytotoxic
T-lymphocyte-associated antigen 8)); humanized mouse monoclonal LY2439821 des-Lys446-[Pro227]γ4 heavy chain {H10S>P,CH3107K>-} (133-219′)-disulfide with humanized mouse monoclonal LY2439821 κ light chain, dimer (225-225”:228-228”)-bisdisulfide
MOLECULAR FORMULA C6492H10012N1728O2028S46
MOLECULAR WEIGHT 146.2 kDa
SPONSOR Eli Lilly and Co.
CODE DESIGNATION LY2439821
CAS REGISTRY NUMBER 1143503-69-8
Ixekizumab is a humanized monoclonal antibody used in the treatment of autoimmune diseases.[1]
Ixekizumab was developed by Eli Lilly and Co.
more info
Inflammation represents a key event of many diseases, such as psoriasis, inflammatory bowel diseases, rheumatoid arthritis, asthma, multiple sclerosis,
atherosclerosis, cystic fibrosis, and sepsis. Inflammatory cells, such as neutrophils, eosinophils, basophils, mast cells, macrophages, endothelial cells, and platelets, respond to inflammatory stimuli and foreign substances by producing bioactive mediators. These mediators act as autocrines and paracrines by interacting with many cell types to promote the inflammatory response. There are many mediators that can promote inflammation, such as cytokines and their receptors, adhesion molecules and their receptors, antigens involved in lymphocyte activation, and IgE and its receptors. [0004] Cytokines, for example, are soluble proteins that allow for communication between cells and the external environment. The term cytokines includes a wide range of proteins, such as lymphokines, monokines, interleukins, colony stimulating factors, interferons, tumor necrosis factors, and chemokines. Cytokines serve many functions, including controlling cell growth, migration, development, and differentiation, and mediating and regulating immunity, inflammation, and hematopoiesis. Even within a given function, cytokines can have diverse roles. For example, in the context of mediating and regulating inflammation, some cytokines inhibit the inflammatory response (anti-inflammatory cytokines), others promote the inflammatory response (pro-inflammatory cytokines). And certain cytokines fall into both categories, i.e., can inhibit or promote inflammation, depending on the situation. The targeting of proinflammatory cytokines to suppress their natural function, such as with antibodies, is a well-established strategy for treating various inflammatory diseases.
Many inflammatory diseases are treated by targeting proinflammatory cytokines with antibodies. Most (if not all) of the anti-proinflammatory cytokine antibodies currently on the market, and those currently in clinical trials, are of the IgG class. See, for example, Nature Reviews, vol. 10, pp. 301-316 (2010); Nature Medicine, vol. 18, pp. 736-749 (2012); Nature Biotechnology, vol. 30, pp. 475-477 (2012); Anti-Inflammatory & Anti- Allergy Agents in Medicinal Chemistry, vol. 8, pp. 51-71 (2009);
FlOOO.com/Reports/Biology/content/1/70, F 1000 Biology Reports, 1 :70 (2009); mAbs 4: 1, pp. 1-3 (2012); mAbs 3: 1, pp. 76-99 (2011); clinicaltrials.gov (generally), and
clinicaltrialsregister.eu/ (generally). These IgG antibodies are administered systemically and thus are often associated with unwanted side effects, which can include one or more of, for example, infusion reactions and immunogenicity, hypersensitivity reactions,
immunosuppression and infections, heart problems, liver problems, and others. Additionally the suppression of the target cytokines at non-diseased parts of the body can lead to unwanted effects.
In an attempt to reduce side effects associated with systemic treatment and to eliminate the inconvenience and expense of infusions, an article proposed an oral anti-TNF therapy that could be useful in treating Crohn’ s disease. Worledge et al. “Oral Administration of Avian Tumor Necrosis Factor Antibodies Effectively Treats Experimental Colitis in Rats.” Digestive Diseases and Sciences 45(12); 2298-2305 (December 2000). This article describes immunizing hens with recombinant human TNF and an adjuvant, fractionating polyclonal yolk antibody (IgY, which in chickens is the functional equivalent to IgG), and administering the unformulated polyclonal IgY (diluted in a carbonate buffer to minimize IgY acid hydrolysis in the stomach) to rats in an experimental rodent model of colitis. The rats were treated with 600mg/kg/day of the polyclonal IgY. The uses of animal antibodies and polyclonal antibodies, however, are undesirable.
In a similar attempt to avoid adverse events associated with systemic administration, another group, Avaxia Biologies Inc., describes a topical (e.g., oral or rectal) animal-dervied polyclonal anti-TNF composition that could be useful in treating
inflammation of the digestive tract, such as inflammatory bowel disease. WO2011047328. The application generally states that preferably the polyclonal antibody composition is prepared by immunizing an animal with a target antigen, and the preferably the polyclonal antibody composition is derived from milk or colostrum with bovine colostrums being preferred (e.g., p. 14). The application also generally states that the animal derived polyclonal antibodies could be specific for (among other targets) other inflammatory cytokines (e.g., pp. 6-7). This application describes working examples in which cows were immunized with murine TNF and the colostrum was collected post-parturition to generate bovine polyclonal anti-TNF antibodies (designated as AVX-470). The uses of animal-derived antibodies and polyclonal antibodies, however, are undesirable.
IgA molecular forms have been proposed as treatments for various diseases, most notably as treatments for pollen allergies, as treatments against pathogens, and as treatments for cancer.
For example, one article describes anti-AmbCtl (a ragweed pollen antigen) humanized monomelic IgA and dimeric IgA antibodies made in murine cells (NSO and Sp2/0 cells). The dimeric IgA contains a mouse J-chain. The article proposes that the antibodies may be applied to a mucosal surface or the lower airway to inhibit entry of allergenic molecules across the mucosal epithelium and therefore to prevent the development of allergic response. Sun et al. “Human IgA Monoclonal Antibodies Specific for a Major Ragweed Pollen Antigen.” Nature Biotechnology 13, 779-786 (1995).
Several other articles propose the use of IgA antibodies as a defense against pathogens.
Two articles proposed the use of an anti-streptococcal antigen I II secretory IgA-G hybrid antibody. Ma et al. “Generation and Assembly of Secretory Antibodies in Plants.” Science 268(5211), 716-719 (May 1995); Ma et al. “Characterization of a
Recombinant Plant Monoclonal Secretory Antibody and Preventive Immunotherapy in Humans.” Nature Medicine 4(5); 601-606 (May 1998). The hybrid antibody contains murine monoclonal kappa light chain, hybrid Ig A-G heavy chain, murine J- Chain, and rabbit secretory component. The antibody was made by successive sexual crossing between four transgenic N. tabacum plants and filial recombinants to form plant cells that expressed all four protein chains simultaneously. The parent antibody (the source of the antigen binding regions, is identified as the IgG antibody Guy’s 13. The group proposes that although slgA may provide an advantage over IgG in the mucosal environment, such is not always the case (1998 Ma at p. 604, right column).
A related article identifies the anti-streptococcal antigen I/II secretory IgA-G hybrid antibody, which was derived from Guy’s 13 IgA, as CaroRx. Wycoff. “Secretory IgA Antibodies from Plants.” Current Pharmaceutical Design 10(00); 1-9 (2004). Planet Biotechnology Inc. This related article states that the CaroRx antibody was designed to block adherence to teeth of the bacteria that causes cavities. Apparently, the CaroRx antibody was difficult to purify; the affinity of Protein A for the murine Ig domain was too low and protein G was necessary for sufficient affinity chromatography. Furthermore, the article states that several other chromatographic media had shown little potential as purification steps for the hybrid slgA-G from tobacco leaf extracts. The article also indicates that the authors were unable to control for human-like glycosylation in tobacco, but that such was not a problem because people are exposed to plant glycans every day in food without ill effect.
WO9949024, which lists Wycoff as an inventor, Planet Biotechnology Inc. as the applicant, describes the use of the variable regions of Guy’s 13 to make a secretory antibody from tobacco. The application contains only two examples – the first a working example and the second a prophetic example. Working Example 1 describes the transient production of an anti-S. mutans SA I/III (variable region from Guy’s 13) in tobacco. The tobacco plant was transformed using particle bombardment of tobacco leaf disks. Transgenic plants were then screened by Western blot “to identify individual transformants expressing assembled human slgA” (p. 25). Prophetic Example 2 states that in a transformation system for Lemna gibba (a monocot), bombardment of surface-sterilized leaf tissue with DNA- coated particles “is much the same as with” tobacco (a dicot). The prophetic example also stops at screening by immunoblot analysis for antibody chains and assembled slgA, and states that the inventors “expect to find fully assembled slgA.” [0014] Another article proposed the use of an anti-RSV glycoprotein F IgA antibodies (mlgA, dlgA, and slgA). Berdoz et al. “In vitro Comparison of the Antigen-Binding and Stability Properties of the Various Molecular Forms of IgA antibodies Assembled and Produced in CHO Cells.” Proc. Natl. Acad. Sci. USA 96; 3029-3034 (March 1999). The slgA antibody was made in CHO cells sequentially transfected with chimeric heavy and light chains, human J-Chain, and human secretory component, respectively. Single clones were generated to express the mlgA (clone 22), the dlgA (clone F), and the slgA (clone 6) (p. 3031).
Still other articles proposed, for example: (1) anti-HSV mlgA made in maize (Karnoup et al. Glycobiology 15(10); 965-981 (May 2005)) (which states that at that time there had been little success in the application of IgA class antibodies to therapeutic use because of the difficulty in producing the dimeric form in mammalian cells at economic levels); (2) anti-C. difficile toxin A chimeric mouse-human monomeric and dimeric IgA made in CHO cells (Stubbe et al. Journal of Immunology 164; 1952-1960 (2000)); (3) anti-N. meningitidis chimeric IgA antibodies were produced in BHK cells cotransfected with human J-Chain and/or human secretory component (Vidarsson et al., Journal of Immunology 166; 6250-6256 (2001)); (4) mti-Pseudomonas aeruginosa 06 lipopolysaccharide chimeric mouse/human mlgAl made in CHO cells (Preston et al. Infection and Immunity 66(9); 4137- 4142 (September 1998)); (5) anti-Plasmodium mlgA made in CHO cells (Pleass et al. Blood 102(13); 4424-4429 (December 2003)) (which states that unlike their parental mouse IgG antibodies, the mlgA antibodies failed to protect against parasitic challenge in vivo); and (5) ^^-Helicobacter pylori urease subunit A slgA and dlgA (Berdoz et al. Molecular
Immunology 41(10); 1013-1022 (August 2004)). [0016] For a review article discussing passive and active protection against pathogens at mucosal surfaces, see Corthesy. “Recombinant Immunoglobulin A: Powerful Tools for Fundamental and Applied Research.” Trends in Biotechnology 20(2); 65-71 (February 2002).
Still other articles propose the use of IgA antibodies as a treatment for cancer.
For example, one article describes a Phase la trial of a muring anti-transferrin receptor IgA antibody (Brooks et al. “Phase la Trial of Murine Immunoglobulin A
Antitransferrin Receptor Antibody 42/6.” Clinical Cancer Research 1(11); 1259-1265 (November 1995)). Another article describes a human anti-Ep-CAM mIgA made in BHK (baby hamster kidney) cells (Huls et al. “Antitumor Immune Effector Mechanisms Recruited by Phase Display-Derived Fully Human IgGl and IgAl Monoclonal Antibodies.” Cancer Research 59; 5778-5784 (November 1999)). Still another article describes an anti-HLA Class II chimeric mIgA antibody made in BHK cells (Dechant et al. “Chimeric IgA Antibodies Against HLA Class II Effectively Trigger Lymphoma Cell Killing.” Blood 100(13); 4574- 4580 (December 2002)). Yet other articles describe anti-EGFR mIgA or dlgA antibodies made in CHO, including Dechant et al. “Effector Mechanisms of Recombinant IgA
Antibodies Against Epidermal Growth Factor Receptor.” Journal of Immunology 179; 2936- 2943 (2007), Beyer et al. “Serum- Free Production and Purification of Chimeric IgA
Antibodies.” Journal of Immunology 346; 26-37 (2009) (stating that as of 2009, IgA antibodies have not been commercially explored for problems including lack of production and purification methods), and Lohse et al. “Recombinant Dimeric IgA Antibodies Against the Epidermal Growth Factor Receptor Mediate Effective Tumor Cell Killing.” Journal of Immunology 186; 3770-3778 (February 2011).
For a review article on anti-cancer IgA antibodies, see Dechant et al. “IgA antibodies for Cancer Therapy. ” Critical Reviews in Oncology/Hematology 39; 69-77 (2001); states that compared with infectious diseases, the role of IgA in cancer immunotherapy is even less investigated).
IL17 and IFN-garama inhibition for the treatment of autoimmune inflammation
The IL-17 family of cytokines has been associated with the pathogenesis of autoimmune diseases and is generally blamed for the pathogenic symptoms of autoimmune inflammation. Overexpression of IL-17 is a hallmark for autoimmune diseases like rheumatoid arthritis, systemic lupus erythematomatosus, inflammatory bowel disease, multiple sclerosis, and psoriasis (Yao Z et. al., J Immunol, 155(12), 1995, 5483-6. Chang S H, et.al, Cytokine, 46, 2009, 7-11; Hisakata Yamada et.al, Journal of Inflamm. Res., 3, 2010, 33-44)).
The IL-17 cytokine family comprises six members, out of which IL-17 A and IL-17F are the best characterized. IL-17A and IL-17F exist as homo- as well as as heterodimers (IL-17AA, IL-17AF, IL-17FF). IL-17A and IL-17F are clearly associated with inflammation (Gaffen S H, Cytokine, 43, 2008, 402-407; Torchinsky M B et al, Cell. Mol. Life Sci., 67, 2010, 1407- 1421).
The secretion of IL-17 is predominantly caused by a specific subtype of T helper cells termed TH-17 cells. IL-23, TGFp and IL-6 were shown to be important factors leading to conversion of nai‘ve CD4+ T-cells to THl 7 cells. It was also reported that TGF and IL-6 potently induce in synergy THl 7 differentiation. Important transcription factors for the secretion of IL-17 from TH17 cells are RORyt and STAT3 (IvanovJ et.al. Cell 126, 2006, 1121-1133). IL-17 induces pro-inflammatory cytokines (IL-6, TNF- and IL-lb) and Chemokines (CXCL1,GCP-2,CXCL8 or IL-8,CINC,MCP-1). It increases the production of nitric oxide prostaglandin E2 and matrix-metalloproteinases. As a consequence of these events neutrophil infiltration, tissue damage and chronic inflammation occurs (PECK A et.al, Clin Immunol., 132(3), 2009, 295-304).
Before the recognition of the importance of IL-17 in autoimmune inflammation, IFN-gamma derived from THl cells was believed to be an important cytokine that drives autoimmune disorders (Takayanagi H et. al. Nature, 408, 2000, 600-605. Huang W. et. al. Arthritis Res. Ther., 5, 2002, R49-R59) The secretion of IFN-gamma is a key feature of the THl effector cell lineage and the secretion is regulated by the transcription factors T-bet and STAT4 (Bluestone JA et. al. Nat Rev Immunol, 11, 2009, 811-6). Infiltration of activated T-cells and elevation of M-CSF, IL-10 and TNF support this notion (Yamanda H et.al Ann. Rheu. Dis., 67, 2008, 1299-1304; Kotake S et.al. Eur. J. Immunol, 35, 2005, 3353-3363).
Recently, a more complex situation was proposed, where hybrid TH17/TH1 cells induced by IL-23 and IL-6 in concert with IL-1 secrete IL-17 and IFN-gamma. These cells are under the control of the transcription factors RORyt and T-bet, confirming the notion, that these are true hybrids of THl and THl 7 cells. It was also demonstrated that these double producing cells are the pathogenic species in IBD and EAE (Buonocore S et.al. Nature, 464, 2010, 1371-5; Ghoreshi K. et. al. Nature, 467, 2010, 967-971).
Compounds which target and suppress both IL-17 and IFN-gamma are predisposed for the treatment of autoimmune disorders.
The effectiveness of blocking IL-17 signaling as therapeutic treatment in autoimmune diseases has already been proven in clinical trials with e.g. monoclonal antibodies against IL- 17A (AIN457, secukinumab; Ly2439821,ixekizumab; RG4934) and/or the IL-17 receptor IL- 17RA (AMG827, brodalumab).
Positive results have been reported for the treatment of rheumatoid arthritis, psoriasis and uveitis (Hueber W et al, Sci. Transl. Med., 2, 2010, 52ra72, DOI: 10.1126/scitranslmed.3001107; van den Berg W B e/ al, Nat. Rev. Rheumatol, 5, 2009, 549-553), ankylosing spondylitis and spondyloarthritides (Song I-H et al, Curr. Opin. Rheumatol., 23, 2011, 346-351).
Secukinumab is currently under investigation in clinical trials for psoriatic arthritis, Behcet disease, uveitits, inflammatory bowel disease, Crohn’s disease, multiple sclerosis (Kopf M et al., Nat. Rev. Drug Disc, 9, 2010, 703-718; Song I-H et al, Curr. Opin. Rheumatol., 23, 2011, 346-351).
Brodalumab, Ixekizumab and RG4934 are currently in clinical trials for the treatment of rheumatoid arthritis, psoriasis and/or psoriatic arthritis (Kopf M et al, Nat. Rev. Drug Disc, 9, 2010, 703-718; clinicaltrials.gov; Medicines in development for skin diseases, 201 1, published by PhRMA, www .phrma. com) .
With regard to blocking of IFN-gamma signaling as therapeutic treatment in autoimmune diseases, the IFN-gamma-specific monoclonal antibody AMG811 is currently under clinical investigations for the treatment of systemic lupus erythematosus (Kopf M et al., Nat. Rev. Drug Disc, 9, 2010, 703-718).
http://www.ama-assn.org/resources/doc/usan/vercirnon.pdf
|
vercirnon
|
|
| Trade Name: | Traficet-EN |
| Synonym: | CCX282-B, GSK1605786, GSK 1605786 |
4-(2-(4-(tert-butyl)phenylsulfonamido)-5-chlorobenzoyl)pyridine 1-oxide
698394-73-9 [RN]
Anti-inflammatory intended to treat Crohn’s
disease and inflammatory bowel disease
GSK1605786A (formerly CCX282-B) targets chemokine receptor CCR9, which is expressed selectively on intestinal lymphocytes and dendritic cells. CCR9 mediates migration of immune cells to the intestine, and blockade of the receptor inhibits migration.
GSK1605786A is being studied in CD at a dose of 500 mg by mouth once daily or 500 mg by mouth twice daily versus placebo. Final data is anticipated to be collected mid-2012 for a study evaluating efficacy over a 12-week treatment period.
A study reviewing maintenance of remission is expected to be complete in July 2014 and a long-term safety study is scheduled for completion in July 2015.
GSK-1605786 (CCX-282; Traficet-EN), a selective antagonist of the CC chemokine receptor (CCR9), is being developed by GlaxoSmithKline plc under license from ChemoCentryx Inc for the potential treatment of inflammatory bowel disease, including Crohn’s disease and celiac disease. CCR9 is a tissue-specific lymphocyte trafficking molecule that selectively attracts both B- and T-cells to the small gut. Inhibition of CCR9 by GSK-1605786 may inhibit B- and T-cell entry to the small gut and ameliorate inflammation while leaving immune function at other anatomical sites unaffected. GSK-1605786 was assessed as a treatment for moderate-to-severe Crohn’s disease in the phase II/III PROTECT-1 trial and as a treatment for celiac disease in a phase II trial. Data suggest that GSK-1605786 is efficacious in patients with Crohn’s disease with the advantage of being orally bioavailable.

http://www.who.int/medicines/publications/druginformation/issues/Proposed-List_109.pdf str is available in this link
20,20,21,21,21-pentafluoro-17-hydroxy-11β-[4-
(methanesulfonyl)phenyl]-19-nor-17α-pregna-4,9-dien-3-one
progesterone receptor antagonist
BAY 1002670, vilaprisan
1262108-14-4
C27H29F5O4S, 544.574
http://www.who.int/medicines/publications/druginformation/issues/Proposed-List_109.pdf str is available in this link
Bayer has also made good progress in the development of new treatment options for patients with gynecological diseases: sPRM (BAY 1002670) is a novel oral progesterone receptor modulator that holds the promises of long-term treatment of patients with symptomatic uterine fibroids. Based on promising early clinical data the initiation of a Phase III study is planned for mid-2014.
A selective progesterone receptor modulator (SPRM) is an agent that acts on the progesterone receptor. A characteristic that distinguishes such substances from receptor full agonists (such as progesterone) and full antagonists (such as mifepristone) is that their action differs in different tissues (agonist in some while antagonist in others). This mixed agonist/antagonist profile of action leads to selective stimulation or inhibition progesterone-like action in different tissues and furthermore raises the possibility of dissociation of desirable therapeutic effects from undesirable side effects in synthetic progesterone receptor drug candidates


amcrasto@gmail.com
email me if u like my posts

Isavuconazole (BAL4815; trade name Cresemba) is a triazole antifungal drug. Its prodrug, isavuconazonium sulfate (BAL8557), was granted approval by the U.S. Food and Drug Administration (FDA) on March 6, 2015[1]
During its Phase III drug trials, Astellas partnered with Basilea Pharmaceutica, the developer of the drug, for rights to co-development and marketing of isavuconazole. [2]
On May 28, 2013, Basilea Pharmaceutica announced it had been granted orphan drug status by the FDA for treatment of aspergillosis.[3] Since then, it has also been granted orphan drug status for the treatment of invasive candidiasis.[4]

Isavuconazonium sulfate (BAL8557)—a prodrug of isavuconazole.
CLINICAL TRIALS…LINK
PATENTS
|
6-27-2012
|
Process for the manufacture of enantiomerically pure antifungal azoles as ravuconazole and isavuconazole
|
|
|
11-18-2011
|
Antifungal Composition
|
|
|
9-29-2010
|
PROCESS FOR PREPARATION OF WATER-SOLUBLE AZOLE PRODRUGS
|
|
|
12-3-2008
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
3-14-2007
|
N-phenyl substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
11-3-2004
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
10-10-2001
|
Azoles for treatment of fungal infections
|
Several azoles are currently used for systemic mycoses. However, none of them fulfills the needs of clinical requirement in full extent, particularly with regard 0 to broad antifungal spectrum including aspergillus fumigatus, less drug-drug interaction, and appropriate plasma half-life for once a day treatment. Other clinical requirements which are not fulfilled by the azoles currently used, are efficacy against major systemic mycoses including disseminated aspergillosis, safety, and oral or parenteral formulations. Particularly, demand of a 5 parenteral administration of the azoles is increasing for the treatment of serious systemic mycoses. Most of the azoles on the market as well as under development are highly lipophilic molecules that make the parenteral formulation difficult.

Isavuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol; formula I, R1 and R3 represent fluorine and R2 represents hydrogen] as well as Ravuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol; formula I, R1 and R2 represent fluorine and R3 represents hydrogen] are useful antifungal drugs as reported in U.S. Pat. No. 5,648,372 from Feb. 1, 1995 or in U.S. Pat. No. 5,792,781 from Sep. 18, 1996 or in U.S. Pat. No. 6,300,353 from Oct. 9, 2001 (WO99/45008).
Since compounds of general formula I contain two adjacent chiral centers, synthesis of enantiomerically pure compound is complex and until now, all patented syntheses are not efficient enough and do not allow cost effective manufacturing on a technical scale:
Thus, U.S. Pat. Nos. 5,648,372 or 5,792,781 describe enantioselective synthesis of compounds of formula I (specifically Ravuconazole) from chiral 3-hydroxy-2-methyl propionic acid in 12 steps with overall yield lower than 5%. In another approach including 13 steps and low overall yield, (R)-lactic acid was used as the starting material (Chem. Pharm. Bull. 46(4), 623 (1998) and ibid. 46(7), 1125 (1998)).
Because both starting materials contain only one chiral center, in a number of inefficient steps, the second, adjacent chiral center has to be created by a diastereoselective reaction (using either Corey or Sharpless epoxidation method) which is not sufficiently selective leading mostly to a mixture of two diastereomers which have to be separated.
The second approach, based on (R)-methyl lactate, was recently very thoroughly optimized by BMS on a multi kilogram scale but it still does not fulfill requirements for cost effective manufacturing process (Organic Process Research & Development 13, 716 (2009)). The overall yield of this optimized 11 steps process is still only 16% (Scheme 1).

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (U.S. Pat. No. 6,300,353 from Oct. 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to U.S. Pat. No. 6,300,353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec. 1, 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in U.S. Pat. No. 6,133,485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid.
This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (U.S. Pat. No. 6,300,353). It is obvious that this process using less expensive (S)-lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from Jun. 26, 2001) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(II)-catalyst and diethyl zinc (Scheme 2).

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 1611 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
All known processes for enantiomerically pure compounds of formula I have definitely too many operation steps and specifically very low overall yield. The chiral starting materials used, either 3-hydroxy-2-methyl propionic acid or (S)- or (R)-methyl lactate, contain only one chiral center and consequently, in number of steps, the second adjacent chiral center has to be ineffectively generated which makes the entire process long and expensive. The only known process, which generates both chiral centers simultaneously, requires again expensive chiral starting material (R)-2-butynol.
ISAVUCONAZOLE
…………………………………………….
synthetic scheme A, starting from 4-[(2R)-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propionyl]morpholine [which can be prepared by a same procedure as described in Chem. Pharm. Bull. 41, 1035, 1993.]. This synthesis route has been described for example in European Patent Application No. 99101360.8.


(a)
………………………………………………………………………
Example 1 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1R)-10-camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41% yield, 99.1% ee);
m.p. 164-166° C.; [α]=−30° (c=1, methanol, 25° C.);
NMR (CDCl3): 1.23 (3H, d, J=8 Hz), 4.09 (1H, q, J=8 Hz), 4.26 (1H, d, J=14 Hz), 4.92 (1H, d, J=14 Hz), 5.75 (1H, s), 6.75-6.85 (2H, m), 7.45-7.54 (2H, m), 7.62 (1H, s), 7.69 (1H, s), 7.75 (1H, d, J=8 Hz), 7.86 (1H, s), 8.03 (1H, d, J=8 Hz).
The analytical data were identical with published (U.S. Pat. No. 5,648,372 and Chem. Pharm. Bull. 1998, 46, 623-630).
Example 2 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
Racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (44 g) and (1R)-10-camphorsulfonic acid (20 g) were suspended in methanol (ca. 300 ml), the slurry was stirred intensively, warmed up to ca. 70° C. and a small addition of acetic acid was added to obtain a clear solution. After cooling of the solution to rt and then to 0° C., the mixture was seeded with enantiomerically pure salt and stirred for another 2 hrs. The crystalline solid was collected by filtration, washed with cooled methanol and dried under reduced pressure. The crystals were partitioned between methylenechloride (300 ml) and saturated aqueous sodium bicarbonate solution (200 ml). The organic layer was washed twice with water (50 ml), dried with magnesium sulphate, filtrated and evaporated under reduced pressure to give the title compound (16.9 g, 38% yield, 95% ee). The analytical data were identical with published (U.S. Pat. No. 5,648,372 or Chem. Pharm. Bull. 1998, 46, 623).
Example 3 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (10 g) in acetone (ca. 200 ml) a solution of (1R)-10-camphorsulfonic acid (3.9 g) in methanol (50 ml) was added and the mixture was heated shortly under reflux until a clear solution was obtained. The solution was then slowly cooled to rt, seeded with crystals of the desired enantiomeric salt and let overnight. The solid precipitate was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This salt was then taken up in methylenechloride and water and basified with aqueous sodium bicarbonate solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride. The organic phases were combined, dried with sodium sulphate, filtrated and the solvent removed under reduced pressure. The crude product was then dissolved in ethanol, the slurry heated for 20 min, small amount of water was added, the solution slowly cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with cold ethanol and dried under reduced pressure to provide the title enantiomerically pure compound (3.9 g, 39% yield, 96% ee). The analytical date were identical with published in U.S. Pat. No. 6,300,353 B1 and WO 99/45008.
Example 4 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (100 g) in acetone (1000 ml) a solution of (1R)-10-camphorsulfonic acid (47 g) in methanol (500 ml) was added at rt, then slurry was heated under stirring to almost reflux for ca. 30 min, then cooled slowly to rt, seeded with the pure enantiomeric salt and stirred over night. The solid was collected by filtration, washed with methanol/acetone mixture, dried under reduced pressure. The residue was taken up with a solvent mixture of methylenechloride/water and after addition of saturated aqueous sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated, the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 39 g (39% yield, 92% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
Example 5 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
A solution of the racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (4.4 g) and (1R)-10-camphorsulfonic acid (2 g) in toluene (40 ml) containing glacial acetic acid (0.6 ml) was warmed up to approximately 70° C., then allowed to cool slowly to 20° C., seeded with the pure enantiomeric salt whereupon the pure enantiomeric salt start to crystallize out. After ca. 2 hrs at this temperature the solid was collected, washed with cold toluene and dried. The crystals were taken with a solvent mixture of methylenechloride/water and after addition of aqueous saturated sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated and the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 2 g (45% yield, 99% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
…………………………………..
WO 1999045008
The following synthetic scheme 1 illustrates the manufacture of one of the compounds of formula I′:



……………………………….
Bioorganic and medicinal chemistry letters, 2003 , vol. 13, 2 p. 191 – 196
http://www.sciencedirect.com/science/article/pii/S0960894X02008922
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.


We synthesized a series of new triazolium derivatives of Figure 1, Figure 3 and Scheme 1. CompoundsScheme 1 and Scheme 2, 6, 9, 10 and 11 were first prepared as outlined in Scheme 2 in order to analyze their stability and ability to release Figure 1, Figure 3 and Scheme 1. Next, aromatic analogues 18, 19, 20,21 and Figure 1, Figure 3 and Scheme 3 were synthesized for optimization of 11 to increase its water solubility and conversion rate. Compounds in the second series had sarcosine esters6 to make them water soluble, and they were also designed to generate acetaldehyde7 instead of formaldehyde for a better safety profile. The synthetic procedures for the second series of the derivatives are outlined in Scheme 3.

Scheme 2.
(a) ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt (quant); (b) Figure 1, Figure 3 and Scheme 1, CH3CN, 80 °C (60%); (c) (1) ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (30%, two steps); (d) (1) NaI, CH3CN, 50 °C ; (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 50 °C (88%, two steps); Synthesis of Scheme 1 and Scheme 2: (1) N-3-hydroxypropyl-N-methylamine, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) AcCl, Et3N, CH2Cl2, rt (20%, two steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (82%); Synthesis of 10: (1) l-prolinol, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (<10%, 2 steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (92%); Synthesis of 11: (1) 2-hydroxymethyl-N-methylaniline, ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt; (2) Ac2O, diisopropylethylamine, rt (20%, two steps); (3)Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux (63%).
Figure options

Scheme 3.
(a) (1) oxalyl chloride, DMF, 0 °C; (2) KOtBu, THF, −5 °C (97%, two steps); (b) CH3NH2, MeOH, rt (90%); (c) LiAlH4, THF, 0 °C (80%); (d) (1) ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (2) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (84%, two steps); (e) (1) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C; (2) DOWEX-1 Cl− form, aqueous MeOH, rt (65%, two steps); (f) (1) HCl, EtOAc, rt; (2) lyophilization (69%, two steps); Synthesis of 18: (1) (i) (4,5-difluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (quant, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 80 °C; (50%,); (3) HCl, EtOAc, rt (90%); Synthesis of 19: (1) (i) 2-fluoro-6-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (74%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux; (3) HCl, EtOAc, rt (29%, two steps); Synthesis of 20: (1) (i) (5-fluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (91%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 70 °C (72%); (3) HCl, EtOAc, rt (88%); Synthesis of 21: (1) (i) (4-chloro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (71%, two steps); (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 65 °C; (3) HCl, EtOAc, rt (65%, two steps).
read more at
Boyd, B.; Castaner, J. BAL-4815/BAL-8557
Drugs Fut 2006, 31(3): 187
Antimicrobial Agents and Chemotherapy, 2008 , vol. 52, 4 p. 1396 – 1400
Ohwada, J.; Tsukazaki, M.; Hayase, T.; Oikawa, N.; Isshiki, Y.; Umeda, I.; Yamazaki, T.; Ichihara, S.; Shimma, N.Development of novel water antifungal, RO0098557
21st Med Chem Symp (November 28-30, Kyoto) 2001, Abst 1P-06
Ohwada, J.; Tsukazaki, M.; Hayase, T.; et al.
RO0098557, a novel water soluble azole prodrug for parenteral and oral administration (I). Design, synthesis, physicochemical properties and bioconversion42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-820
Tasaka et al., Chem. Pharm. Bull. 41(6) pp. 1035-1042 (1993).
There have been three phase III clinical trials of isavuconazole, ACTIVE, VITAL and SECURE. As of June 2015, SECURE and VITAL have been presented in abstract form and results from ACTIVE have not been released.[9]
The SECURE trial compared voriconazole and isavuconazole in invasive fungal infections due to aspergillus. Isuvaconazole was found to be non-inferior to voriconazole, anothertriazole antifungal, with all cause mortality at 18.6%, compared to 20.2% in the voriconazole group. It additionally demonstrated a similar side effect profile.[10]
Data from the VITAL study showed that isavuconazole could be used in treatment of invasive mucormycosis, but did not evaluate its clinical efficacy for this indication.[11]
The ACTIVE trial is a comparison of isuvaconazole and caspofungin for invasive candida infections and results are anticipated in the second half of 2015.[12][13]
| US4861879 | Feb 9, 1988 | Aug 29, 1989 | Janssen Pharmaceutica N.V. | [[4-[4-Phenyl-1-piperazinyl)phenoxymethyl]-1-3-dioxolan-2-yl]-methyl]-1H-imidazoles and 1H-1,2,4-triazoles |
| US5900486 | Sep 9, 1997 | May 4, 1999 | Hoffmann-La Roche Inc. | N-benzylazolium derivatives |
| AU4536497A | Title not available | |||
| EP0667346A2 | Feb 3, 1995 | Aug 16, 1995 | Eisai Co., Ltd. | Azole antifungal agents, process for the preparation there of and intermediates |
| WO1992017474A1 | Mar 26, 1992 | Oct 15, 1992 | Pfizer | Triazole antifungal agents |
| US5648372 | Feb 1, 1995 | Jul 15, 1997 | Eisai Co., Ltd. | Antifungal agents, and compositions |
| US5686646 * | May 23, 1995 | Nov 11, 1997 | Schering-Plough Corporation | Chiral hydrazine derivatives |
| US5746840 * | Mar 28, 1997 | May 5, 1998 | Janssen Pharmaceutica, N.V. | Process for preparing enantiomerically pure 6-{4-chlorophenyl) (1 H-1,2,4-triazol-1-YL) methyl}-1-methyl-1 H-benzotriazole |
| US5792781 | Sep 18, 1996 | Aug 11, 1998 | Eisai Co., Ltd. | Antifungal agents, processes for the preparation thereof, and intermediates |
| US6020497 | Oct 9, 1998 | Feb 1, 2000 | Merck & Co., Inc. | 3-substitutes isoxazolidines as chiral auxiliary agents |
| US6133485 | Apr 15, 1998 | Oct 17, 2000 | Synphar Laboratories, Inc. | Asymmetric synthesis of 2-(2,4-difluorophenyl)-1-heterocycl-1-yl butan-2,3-diols |
| US6300353 | Mar 5, 1999 | Oct 9, 2001 | Basilea Pharmaceutica Ag, A Swiss Company | Azoles for treatment of fungal infections |
| US6383233 | Mar 7, 1997 | May 7, 2002 | Reuter Chemicscher Apparatebau Kg | Separation process |
| US6812238 * | Oct 31, 2000 | Nov 2, 2004 | Basilea Pharmaceutica Ag | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7151182 * | Sep 3, 2004 | Dec 19, 2006 | Basilea Pharmaceutica Ag | Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7803949 * | Dec 20, 2006 | Sep 28, 2010 | Eisai R&D Management Co., Ltd. | Process for preparation of water-soluble azole prodrugs |
| US20030236419 | Dec 31, 2002 | Dec 25, 2003 | Sumika Fine Chemicals Co., Ltd. | Production methods of epoxytriazole derivative and intermediate therefor |
| US20040176432 | Jun 17, 2002 | Sep 9, 2004 | Milan Soukup | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
| WO2003002498A1 * | Jun 17, 2002 | Jan 9, 2003 | Basilea Pharmaceutica Ag | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{2-[(1R,2R)-(2,5-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-1,3-thiazol-4-yl}benzonitrile
|
|
| Clinical data | |
| Trade names | Cresemba (prodrug form) |
| AHFS/Drugs.com | entry |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, intravenous |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 6918485 |
| ChemSpider | 5293682 |
| UNII | 60UTO373KE |
| ChEBI | CHEBI:85979 |
| ChEMBL | CHEMBL409153 |
| NIAID ChemDB | 416566 |
| Chemical data | |
| Formula | C22H17F2N5OS |
| Molecular mass | 437.47 g/mol |


/////


FORIGERIMOD
CHEMICAL NAMES
1. L-Tyrosine, L-arginyl-L-isoleucyl-L-histidyl-L-methionyl-L-valyl-L-tyrosyl-L-seryl-L-lysyl-L-arginyl-O-phosphono-L-serylglycyl-L-lysyl-L-prolyl-L-arginylglycyl-L-tyrosyl-L-alanyl-L-phenylalanyl-L-isoleucyl-L-α-glutamyl-
2. O3,140-phosphono(human U1 small nuclear ribonucleoprotein 70 kDa (snRNP70))-(131-151)-peptide
MOLECULAR FORMULA C117H181N34O32PS
MOLECULAR WEIGHT 2639
TRADEMARK Lupuzor
SPONSOR Cephalon, Inc.
CODE DESIGNATION IPP 201101
CAS REGISTRY NUMBER 497156-60-2
STRUCTURAL FORMULA

stucture, http://www.ama-assn.org/ama1/pub/upload/mm/365/forigerimod.pdf
STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN. FORIGERIMOD.
…………………………………………………………………………………………………………..
FORIGERIMOD ACETATE
CAS REGISTRY NUMBER 1160237-55-7 of acetate
http://www.ama-assn.org/resources/doc/usan/forigerimod-acetate.pdf
STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN. FORIGERIMOD ACETATE

Forigerimod, also known as Lupuzor or CEP-33457, (SyB L-1001) is a CD4 T-cell modulator being investigated for the treatment of systemic lupus erythematosus (SLE). In the Phase II trials, Lupuzor was administered subcutaneously at a dose of 200 mcg once a month for 3 months. The Phase III study is anticipated to be complete in September 2012 and will measure the proportion of patients achieving a combined clinical response using the SLE responder index.

Positive final Lupuzor trial results. Marketwire. www.marketwire.com/press-release/Positive-Final-Lupuzor-Trial-Results-AIM-IMM-1176375.htm. Published November 19, 2009. Accessed June 18, 2011.
Rigerimod (IPP-201101, Lupuzor) is a polypeptide corresponding to the sequence 131-151 of the 70k snRNP protein with a serine phosphorylated in position 140.[1]
It gave encouraging results in a phase IIb trial for severe lupus.[1] Another phase IIb trial has started recruiting in the US.[2]
Lupuzor™ is a potential treatment for lupus, currently given the approval by the US FDA to start Phase III with a Special Protocol Assessment (SPA) and Fast Track designation. ImmuPharma holds all worldwide rights in this lead compound.
Background
Lupus (or Systemic Lupus Erythematosus) is a chronic, potentially life-threatening autoimmune disease. An estimated 1.4 million people are diagnosed in the 7 major world markets (the USA, Japan, Germany, France, Spain, the UK and Italy). Lupus is an inflammatory disease, which attacks multiple organs such as the skin, joints, kidneys, blood cells, heart and lungs. There is currently no cure.
The development of ImmuPharma’s Lupuzor™
ImmuPharma’s compound Lupuzor™ (previously known as IPP-201101 and also referred to as rigerimod or P140) has a novel mechanism of action aimed at modulating the body’s immune system so it does not attack healthy cells, without causing adverse side effects. It has the potential to halt the progression of the disease in a substantial proportion of patients.
Lupuzor™ has successfully completed Phase I, Phase IIa and Phase IIb studies and has now been given the approval by the US FDA to enter Phase III, the final testing phase.
The latest highlights of Lupuzor’s™ development as a treatment for lupus include:
How Lupuzor™ works in the treatment of lupus
Lupuzor™ is a drug that specifically modulates the immune system of lupus patients by modifying the behaviour of some of the key cells involved in the pathogenesis of the disease. The clinical profile of lupus patients is generally assessed by standardised scales such as SLEDAI (SLE Disease Activity Index): the lower the score, the better the condition of the patient. During this Phase II study, the SLEDAI scores were assessed on multiple occasions even though the study was not designed or powered to demonstrate clinical benefit as the primary endpoint due to the short treatment period.
| forigerimod | IPP-201101 | oligopeptide | therapeutic | nucleolin |
| forigerimod acetate | CEP-33457, P-140, IPP-201101 | oligopeptide (salt) | therapeutic | nucleolin |

MACITENTAN
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide,
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl] -N’-propylsulfamide
CAS NO 441798-33-0
ACT-064992, Opsumit,UNII-Z9K9Y9WMVL
Mechanism of Action: Endothelin receptor antagonist (ERA)
Date of Approval: October 18, 2013(US)
Indication: Pulmonary Hypertension (PAH)
Company: Actelion Pharmaceuticals Ltd
PCT patent application: WO2002053557
FDA N204410, MACITENTANTABLET; ORAL10MG, OPSUMIT, ACTELION PHARMS LTD
Macitentan is achiral
Macitentan is a crystalline powder that is insoluble in water. In the solid state macitentan is very stable, is not hygroscopic, and is not light sensitive.
Macitentan (Opsumit® )is a novel dual endothelin receptor antagonist that resulted from a tailored drug discovery process. Macitentan has a number of potentially key beneficial characteristics – i.e., increased in vivo preclinical efficacy vs. existing ERAs resulting from sustained receptor binding and tissue penetration properties. A clinical pharmacology program indicated a low propensity of macitentan for drug-drug interactions.
Macitentan (ACT-064992) is a tissue-targeting dual ET(A)/ET(B) endothelin (ET) receptor antagonist designed for tissue targeting. Macitentan inhibited ET-1-induced contractions in isolated endothelium-denuded rat aorta (ET(A) receptors) and sarafotoxin S6c-induced contractions in isolated rat trachea (ET(B) receptors). In diabetic rats, chronic administration of macitentan decreased blood pressure and proteinuria and prevented end-organ damage. Treatment with macitentan enhanced the cytotoxicity mediated by paclitaxel as measured by the degree of apoptosis in tumor cells and tumor-associated endothelial cells. A Phase III clinical trial of macitentan was successfully completed in 2012.


Macitentan is an investigational drug being studied for the treatment of pulmonary arterial hypertension. It acts as a dualendothelin receptor antagonist and is being developed by Actelion.[1] A Phase III clinical trial was successfully completed in 2012.[2]
on 22 October 2012 – Actelion (SIX: ATLN) announced that it has submitted a New Drug Application (NDA) to the US Food and Drug Administration (FDA) seeking approval for macitentan (Opsumit®) for the treatment of patients with pulmonary arterial hypertension
Actelion’s experimental lung drug macitentan prolonged overall survival by more than a third according to detailed study data, which the company hopes will convince investors it has a viable follow-up product to secure its commercial future.
Europe’s largest standalone biotech company wants the drug, which treats pulmonary arterial hypertension — a disease that causes high blood pressure in the arteries of the lungs — to replace blockbuster Tracleer.
Tracleer currently makes up 87 percent of sales but loses patent protection in 2015 and has also seen its market share eroded by Gilead’s Letairis.

Macitentan has an active metabolite, ACT-132577, which is an oxidative depropylation product. Both macitentan and ACT-132577 are mainly excreted in form of hydrolysis products via urine (about 2/3 of all metabolites) and faeces (1/3).[3]
Co-administration of ciclosporin has only a slight effect on the concentrations of macitentan and its active metabolite, whilerifampicin decreases the area under the curve (AUC) of the drug’s blood plasma concentration by 79%, and ketoconazoleapproximately doubles it. This corresponds to the finding that macitentan is mainly metabolised via the liver enzyme CYP3A4.[4]

SYNTHESIS
The synthesis begins with the reaction of chlorosulfonyl isocyanate (1) (dissolved in dichloromethane at 0 ° C) with one equivalent of tert-butanol. This produces a by BOC protected Aminosulfonylchlorid (2). With one equivalent of n-propylamine (dissolved in 3 eq. Of triethylamine, dichloromethane, at 0 ° C, RT 16 h) is produced by a hydrochloric acid elimination BOC-protected sulfamide (3). This is dissolved in 5 M HCl and dioxane (4-8 h), the BOC protecting group is cleaved. The sulfamide formed (4) is potassium tert-butoxide-(dissolved in MeOH, 3h) is converted to the potassium salt (5). Tert-butoxide potassium acts as a very strong base for deprotonation. This sulfamide potassium salt reacts with the nucleophilic substituents on the heteroaromatic Dichlorpyrimidinderivat (6) (dissolved in dimethyl sulfoxide, at room temperature, RT 42-72 h) under KCl-cleavage to a Monochlorpyrimidin intermediate (7). By treatment with ethylene glycol (dissolved in dimethyl ether, potassium-tert-butoxide,), the ethylene glycol side chain is generated (8). With 2-chloro-5-bromo-pyrimidine (dissolved in tetrahydrofuran, close, at 60-75 ° C) is formed under elimination of HCl in an S N 1 reaction Macitentan (9)…………Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 .

Synthesis of Macitentan
…………………………………………….
SYNTHESIS


YOU CAN READ AT YAOPHA.COM, lovely site to see for drugs


如果您有关于yaopha.com的问题或建议请与我们联系,具体方式如下:
E-mail:contactus@yaopha.com
………………………….
SYNTHESIS
(WO2006/051502A2, JMC2012, 7849). Chlorosulfonyl isocyanate ( 1 ) reaction with tert-butyl alcohol 2 , which is then reacted with n-propylamine 3 . 3 de-boc protected through the acid after reaction with potassium t-butoxide 4 . Another compound 5 with NaH after acidic protons off with dimethyl carbonate ( 6 ) to obtain 7 . 7 and formamidine hydrochloride ( 8 ) to ring chlorinated later POCl3 9 . 9 and 4 SNAr reaction occurs 10 . 10under basic conditions with ethylene glycol SNAr reaction occurs again in alkaline conditions with11 SNAr reaction occurs MACITENTAN.

………………………
http://www.google.com/patents/WO2014155304A1?cl=en
LC-MS (Agilent MS detector G1956B with Agilent 1200 Binary Pump and DAD).
Parameters of the LC-MS method:
Injection volume: 2 |jL
Column: Kinetex C18, 2.6 μιη, 2.1 x 50 mm
Column flow rate: 1 mL/min
Eluents: Eluent A: water + 0.08% TFA
Eluent B: MeCN + 0.012% TFA
Gradient: 2.0 min 95% B
2.8 min 95% B
3.0 min 5% B
Temperature: 40°C Detector wavelength 210 nm

Preparation B: N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]- 4-pyrimidinyl] -N’-propylsulfamide (macitentan):
N-(5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl)propane- 1-sulfamide (200 g; 0.46 mol; see Example 2 or 3) and 5-bromo-2-chloropyrimidine (117 g; 0.60 mol; 1.3 eq) were dissolved in toluene (3 L) and DMF (400 mL). The reaction mixture was warmed up to 50°C and toluene (approx. 400 mL) was distilled our under reduced pressure. The mixture was cooled to 0 °C and tBuOK (156 g, 3 eq, 1.38 mol) was added portionwise. It was stirred at 20 °C for 1 h. Water (1 L) was added and the pH of the solution was adjusted to 3-5 using 33% aq. HC1. The mixture was heated to 50°C and the layers were separated. The org. phase was treated with charcoal at 50°C and filtered over Celite. The filter cake was rinsed with toluene. At 50°C, water (1 L) was added to the org. layer. The layers were separated. The org. layer was concentrated under reduced pressure to a total volume of 1 L and cooled to 0°C. The solid obtained was filtered off. It was rinsed with toluene and MeOH. The crude material was suspended in EA (1 L) and heated to 50°C. 300 mL of EA were distilled out and MeOH (400 mL) was added. The suspension was cooled down to 0°C. The solid was filtered off, rinsed with MeOH and dried under reduced pressure to afford the title compound as a white solid (225 g; 83% yield).

……………………
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm3009103

Starting from the structure of bosentan (1), we embarked on a medicinal chemistry program aiming at the identification of novel potent dual endothelin receptor antagonists with high oral efficacy. This led to the discovery of a novel series of alkyl sulfamide substituted pyrimidines. Among these, compound 17 (macitentan, ACT-064992) emerged as particularly interesting as it is a potent inhibitor of ETA with significant affinity for the ETB receptor and shows excellent pharmacokinetic properties and high in vivo efficacy in hypertensive Dahl salt-sensitive rats. Compound 17 successfully completed a long-term phase III clinical trial for pulmonary arterial hypertension
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (17)
……………
WO 2015004265 click
Example 3 : N-(5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl)pr opane- 1- sulfamide (reaction in and work-up with MIBK):
EG (124 mL, 3.7 mol, 6.0 eq.) was added to a warm (40-50°C) suspension of the compound of Preparation A (150 g, 0.37 mol) in MIBK (600 mL). Solid KOtBu (114 g, 1.11 mol, 3.0 eq.) was added portionwise so that IT < 60°C. The mixture was stirred for
2- 3 h at 100-105°C. After completion of the reaction (LC-MS control), it was cooled to 50 °C. A 40%) aq. solution of citric acid monohydrate (300 mL) was added until pH 4 was reached. The layers were separated. The org. phase was washed with water (450 mL) and the layers were separated. Water (450 mL) was added and the mixture was warmed to 50°C. It was stirred at 50°C for 5 min. The layers were separated. The org. phase was concentrated under vacuum at 50°C until 200 mL of MIBK were removed. Hept (800 mL) was added dropwise at 70-75°C until turbidity was observed. The mixture was seeded with an analytically pure sample of N-(5-(4-bromophenyl)-6-(2 hydroxy ethoxy)pyrimidin-4-yl)propane-l-sulfamide and stirred at 60-65°C for 30 min. It was allowed to cool to 5°C within 5 h. It was filtered off, rinsed with a cold MIBK/Hept mixture (300 mL, 1 : 1) and dried under vacuum at 50°C to yield the title compound as a white solid (121 g; 76% yield).
The product had NMR data equivalent to those reported in Bolli et al, J. Med. Chem. (2012), 55, 7849-7861. [M+H]+ = 430 and 432. LC-MS: tR = 1.46 min; purity: 98.4% a/a. Residual ethylene glycol (GC-FID): 530 ppm.
…….
CN 104447572 click
(l) Martin H. Bolli et al. Reported the synthesis of Marcy cefotetan follows:

[0008] The method W 5- (4- desert phenyl) -4,6-dichloro-chewing clever as a starting material, N- propyl amine Lai ugly bell in DMS0 as a reaction solvent, an alcohol bell as t a base under substitution reaction conditions, the reaction temperature needs of 24-7 to give


The intermediate compound 15, compound 15 in hexylene glycol dimethyl off as the reaction solvent, a tertiary alcohol under conditions with a strong base clock as hexanediol substitution reaction, l〇 (TC Reaction of 18-2 to give compound 17, Compound 17 was then reacted with 5-chloro-chewing desert -2 clever substitution reaction at tetraammine Qiao Nan as a reaction solvent, ammoniated axis as the alkali conditions, the reaction to give the final product of Marcy cefotetan The route every step the higher the yield, the experimental use of N- propyl amine Lai ugly bell hygroscopic, unstable and a long time before the two-step reaction, the reaction at the second step requires l〇 (TC high temperature 18-2 technology is not suitable for industrial production.
[0009] International Patent W02002 / 053557 discloses some preparation methods and other Massey cefotetan column derivative method at each step of the preparation of the reaction times are longer, some reactions up to 4 days, and the resulting intermediate are purified by column chromatography method is not suitable for industrial production.



[00 pairs (3) N- [5- (4- desert) -6-mouth – [(5-desert -2- chew clever-yl) oxy] hexyl oxy] -4-chewing clever yl] -N ‘- Lai ugly propyl amine (Formula I) Synthesis
[0036] Weigh 20gN-5- (4- desert) -6- (2-2- light hexyl group -) 4- chew clever group -N ‘- Lai ugly propyl amine, 200ml dried DMS0 added to 1L H jar, add 20g of alcohol t-clock was added in portions, then add 17. 7g5- desert – dichloro chew clever, 30-4 (TC reduction reaction, the reaction and the reaction solution. a 10% sample skillfully acid to adjust PH value 3 to 4, the reaction mixture was added to 1000ml water, olive mix, suction. suction Massey cefotetan get wet crude product 42g, 450ml of methanol was added at room temperature and then beating 20min, filtration and dried 45C to give white solid was dried under vacuum to give 23.2 Marcy cefotetan yield;.. 85%
[0037] The compound (Formula I) relating to the physical and chemical properties, spectroscopic data are as follows:
[0038] branded point; 135-136 ° C; we NMR (300MHz, DMS0) 5 (egg m):… 9 8 (s, lH), 8 7 (s, 2H), 8 5 (s, l H,) 7. 5 (s, 2H), 7. 2 (s, IH), 7. 1 (s, 2H,) 4. 7 (s, 2H), 4. 6 (s, 2H,) 2. 8 (s, 2H,), 1. 5 (m, 2H,), 0. 81 (m, 3H), MS Qiaoqiao m / z 589 ([M + Tin +).
…………
see
WO 2002053557
http://www.google.com/patents/WO2002053557A1?cl=en
………..


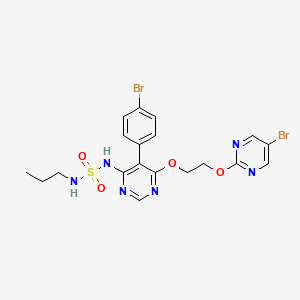
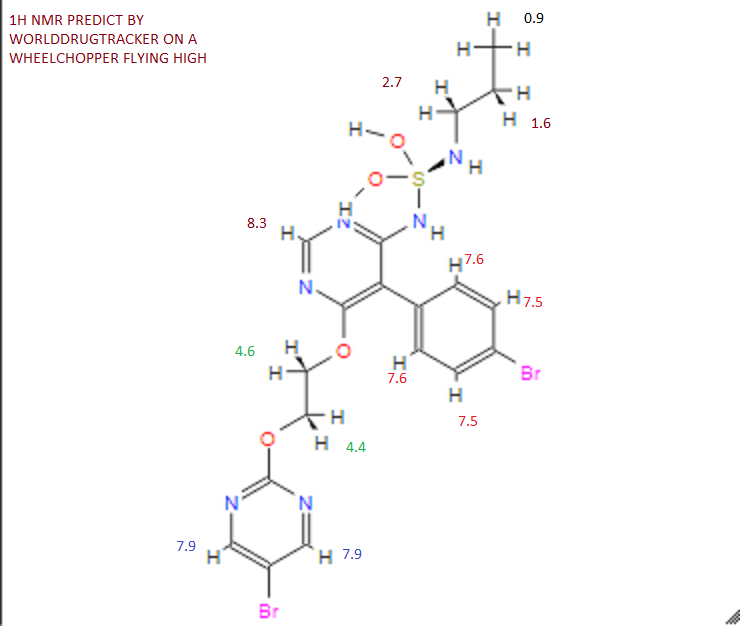
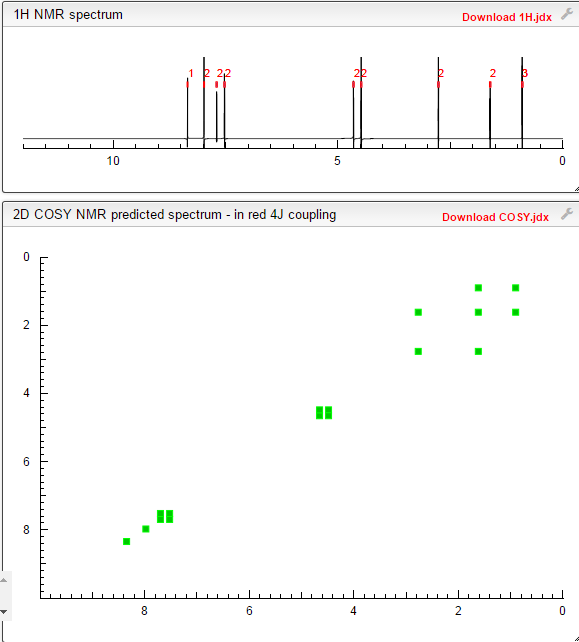
Assignment of the signals mentioned in the text of the H-NMR spectrum of the drug Macitentan
Solvent: CDCl 3
δ 8.51 (s, 2H, CH) 11 , 8.49 (s, 1 H, CH) 10 , 7.58 to 7.63 (m, 2H, CH) 9 , 7.16 to 7.21 ( m, 2H, CH) 8 , 6.88 (s, 1H, NH) 7 , 5.61 (t, J = 6.2 Hz, 1H, NH) 6 , 4.72 to 4.76 (m, 2H , CH 2 ) 5 , 4.62 to 4.66 (m, 2H, CH 2 ) 4 , 2.99 (q, J = 6.8 Hz, 2H, CH 2 ) 3 , 1.61 (h, J = 7.3 Hz, 2H, CH2 ) 2 , 0.97 (t, J = 7.4 Hz, 3H, CH 3 ) 1 . [Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 .]

Solvent: CDCl 3
δ 11.6, 22.7, 46.1, 65.3, 65.9, 104.8, 112.4, 123.7, 128.0, 131.7, 133.0, 155.7, 156 , 4, 159.7, 163.5, 166.3. [ Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 . ]
NMR PREDICT BY ME
1H NMR PREDICT

13C NMR PREDICT BY ME


COSY PREDICT BY ME, WORLDDRUGTRACKER ON A WHEELCHOPPER SCALING NEW HEIGHTS
REFERENCES
Actelion Ltd
Actelion Ltd is a biopharmaceutical company with its corporate headquarters in Allschwil/Basel, Switzerland. Actelion’s first drug Tracleer®, an orally available dual endothelin receptor antagonist, has been approved as a therapy for pulmonary arterial hypertension. Actelion markets Tracleer through its own subsidiaries in key markets worldwide, including the United States (based in South San Francisco), the European Union, Japan, Canada, Australia and Switzerland. Actelion, founded in late 1997, is a leading player in innovative science related to the endothelium – the single layer of cells separating every blood vessel from the blood stream. Actelion’s over 2,400 employees focus on the discovery, development and marketing of innovative drugs for significant unmet medical needs. Actelion shares are traded on the SIX Swiss Exchange (ticker symbol: ATLN) as part of the Swiss blue-chip index SMI (Swiss Market Index SMI®).
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide
|
|
| Clinical data | |
| Trade names | Opsumit |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Metabolism | Hydrolysis, oxidation (CYP3A4) |
| Excretion | 2/3 urine, 1/3 faeces |
| Identifiers | |
| CAS Registry Number | 441798-33-0 |
| ATC code | C02KX04 |
| PubChem | CID: 16004692 |
| ChemSpider | 13134960 |
| ChEBI | CHEBI:76607 |
| Synonyms | ACT-064992 |
| Chemical data | |
| Formula | C19H20Br2N6O4S |
| Molecular mass | 588.273 g/mol |
| Patent | Submitted | Granted |
|---|---|---|
| Sulfamides and their use as endothelin receptor antagonists [US7094781] | 2004-04-22 | 2006-08-22 |
| Sulfamides and their use as endothelin receptor antagonists [US7285549] | 2006-08-10 | 2007-10-23 |
| Stable Pharmaceutical Compositions Comprising a Pyrimidine – Sulfamide [US2008233188] | 2008-09-25 | |
| Combination Comprising Paclitaxel for Treating Ovarian Cancer [US2010311774] | 2010-12-09 | |
| Stable pharmaceutical compositions comprising a pyrimidine-sulfamide [US2010004274] | 2010-01-07 | |
| SULFONYLUREA MODULATORS OF ENDOTHELIN RECEPTOR [US2011082151] | 2011-04-07 | |
| ENDOTHELIN RECEPTOR ANTAGONISTS FOR EARLY STAGE IDIOPATHIC PULMONARY FIBROSIS [US2010022568] | 2007-04-12 | 2010-01-28 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2011136818] | 2011-06-09 | |
| Therapeutic Compositions Comprising a Specific Endothelin Receptor Antagonist and a PDE5 Inhibitor [US2009318459] | 2009-12-24 |
Patent and Exclusivity
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
|
|---|---|---|---|---|---|---|---|
| N204410 | 001 | US7094781 | Oct 12, 2022 | Y | Y | ||
| N204410 | 001 | US8268847 | Apr 18, 2029 | U – 1446 | |||
| N204410 | 001 | US8367685 | Oct 4, 2028 | Y | U – 1445 |
| Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N204410 | 001 | ODE | Oct 18, 2020 |
| N204410 | 001 | NCE | Oct 18, 2018 |
U1446 METHOD OF TREATING PULMONARY HYPERTENSION COMPRISING ADMINISTERING MACITENTAN IN COMBINATION WITH A COMPOUND HAVING PHOSPHODIESTERASE-5 INHIBITORY PROPERTIES
U1445 METHOD OF TREATING PULMONARY ARTERIAL HYPERTENSION BY ADMINISTERING A PHARMACEUTICAL COMPOSITION COMPRISING MACITENTAN AND A POLYSORBATE, WHERIN THE POLYSORBATE REPRESENTS 0.1 TO 1% OF THE WEIGHT OF SAID PHARMACEUTICAL COMPOSITION
OPSUMIT (macitentan) is an endothelin receptor antagonist. The chemical name of macitentan is N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide. It has a molecular formula of C19H20Br2N6O4S and a molecular weight of 588.27. Macitentan is achiral and has the following structural formula:
 |
Macitentan is a crystalline powder that is insoluble in water. In the solid state macitentan is very stable, is not hygroscopic, and is not light sensitive.
OPSUMIT is available as a 10 mg film-coated tablet for once daily oral administration. The tablets include the following inactive ingredients: lactose monohydrate, magnesium stearate, microcrystalline cellulose, polysorbate 80, povidone, and sodium starch glycolate Type A. The tablets are film-coated with a coating material containing polyvinyl alcohol, soya lecithin, talc, titanium dioxide, and xanthan gum.
//////

omecamtiv mecarbil
Cytokinetics, Inc. (CYTK): Cytokinetics: Clinical Investigators’ Opinions On …
Seeking Alpha
Pending the results of the ongoing COSMIC-HF trial in 1H, 2014, I believe that the ATOMIC-AHF results support moving omecamtiv mecarbil into Phase III. With success in the Phase III, omecamtiv mecarbil would likely be a several billion drug. I regard …http://seekingalpha.com/article/1672892-cytokinetics-clinical-investigators-opinions-on-atomic-ahf-trial-results?source=google_news
Omecamtiv mecarbil (INN), previously codenamed CK-1827452, is a cardiac specific myosin activator. It is clinically tested for its role in the treatment of left ventricular systolic heart failure.[1] Systolic heart failure is characterised as a decreased cardiac output (<40% ejection fraction), due to decreased stroke volume, resulting in the inability to meet the metabolic demands of the body.[2] The loss of contraction is caused by a reduced number of effective actin-myosin cross bridges in the left ventricular myocytes. One possible underlying mechanism is altered signal transduction that interferes with excitation-contraction coupling.[3] A decreased cardiac output causes peripheral hypotension and activation of the sympathetic nervous system.[2] This in turn stimulates the cardiac myocytes excessively, eventually leading to left ventricular hypertrophy, characteristic of chronic heart failure. Some symptoms of systolic heart failure are fatigue, peripheral oedema, dyspnoea, exercise intolerance and breathlessness.[2] Current inotropic drug therapies such as dobutamine, are palliative and not a cure. They also cause many adverse effects including arrhythmias related to increased myocardical oxygen consumption, desensitization of adrenergic receptors and altering intracellular calcium levels.[4] Thus systolic heart failure is considered malignant, however the novel mechanism of Omecamtiv Mecarbil is a hopeful long-term resolution.
Cardiac myocytes contract through a cross-bridge cycle between the myofilaments, actin and myosin. Chemical energy in the form of ATP is converted into mechanical energy which allows myosin to strongly bind to actin and produce a power stroke resulting in sarcomere shortening/contraction.[3] Omecamtiv Mecarbil specifically targets and activates myocardial ATPase and improves energy utilization. This enhances effective myosin cross-bridge formation and duration, while the velocity of contraction remains the same.[5] It also increases the rate of phosphate release from myosin, thereby accelerating the rate-determining step of the cross-bridge cycle, which is the transition of the actin-myosin complex from the weakly bound to the strongly bound state.[1] The overall result of Omecamtiv Mecarbil is an increase in left ventricular systolic ejection time, sarcomere shortening and stroke volume, while the systolic pressure remains the same.[5] This causes a decrease in heart rate while myocardial oxygen consumption is unaffected. The increased cardiac output is independent of intracellular calcium and cAMP levels.[4][6] Thus Omecamtiv Mecarbil improves systolic function by increasing the systolic ejection duration/stroke volume, without consuming more ATP energy, oxygen or altering intracellular calcium levels causing an overall improvement in cardiac efficiency.[5]
Experimental studies on rats and dogs, proved the efficacy and mechanism of action of Omecamtiv Mecarbil.[4] Current clinical studies on humans have shown there is a direct linear relationship between dose and systolic ejection time.[1][7][8] The dose-dependent effects persisted throughout the entire trial, suggesting that desensitization does not occur. The maximum tolerated dose was observed to be an infusion of 0.5 mg/kg/h. Adverse effects, such as ischemia, were only seen at doses beyond this level, due to extreme lengthening of systolic ejection time.[1] Thus due to the unique cardiac myosin activation mechanism, Omecamtiv Mecarbil could safely improve cardiac function within tolerated doses. Omecamtiv Mecarbil effectively relieves symptoms and enhances the quality of life of systolic heart failure patients. It drastically improves cardiac performance in the short term, however the hopeful long term effects of reduced mortality have yet to be studied.[2][1]

{kind=link}