
Home » Phase3 drugs (Page 21)
Category Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Trelagliptin succinate (SYR-472) for the treatment of type 2 diabetes.

Trelagliptin succinate (SYR-472)
2-[[6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2, 4-dioxopyrimidin-1-yl]methyl]-4-fluorobenzonitrile; butanedioic acid
2-[6-[3(R)-Aminopiperidin-1-yl]-3-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-ylmethyl]-4-fluorobenzonitrile
2- [ [6- [ (3R) -3-amino-l-piperidinyl] -3, 4-dihydro-3- methyl-2, 4-dioxo-l (2H) -pyrimidinyl]methyl] -4-fluorobenzonitrile
succinic acid salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Sponsor/Developer: Takeda Pharmaceuticals and Furiex Pharmaceuticals
Mechanism of action: DPP-4 inhibitor
865759-25-7 cas FREE BASE
1029877-94-8 succinate
- SYR 111472 succinate
- SYR 472
- Syr-472
- Syr111472 succinate
- Trelagliptin succinate
- UNII-4118932Z90
- clinical trials….http://clinicaltrials.gov/search/intervention=SYR+472
Trelagliptin-succinate M. Wt: 475.47
Trelagliptin-succinate Formula: C22H26FN5O6
SYR-472 is an oral dipeptidyl peptidase IV inhibitor originated by Takeda. It is in phase III clinical trials for the treatment of type 2 diabetes.
- Diabetes affects 25.8 million people of all ages, or roughly 8.3 percent of the U.S. population.
- The World Health Organization predicts that there will be 366 million people worldwide affected by diabetes by the year 2030.
- The advent of trelagliptin succinate, a unique once weekly medication for patients with type 2 Diabetes is now the focus of clinical trials and exciting research and development.
- Phase III clinical trials of trelagliptin succinate commenced in September 2011, and are estimated to be complete by the second half of 2013.

Indication (Phase): Japan—Once-weekly oral treatment for type 2 diabetes (Phase III; study expected to be completed in second half of 2013)

trelagliptin succinate
Compound I, A, TRELAGLIPTIN which has the formula:

is a DPP-IV inhibitor that is described in U.S. patent application Ser. No. 11/080,992 filed Mar. 15, 2005 (see Compound 34). Its dosing, administration and biological activities are described in U.S. patent application Ser. No. 11/531,671 filed Sep. 13, 2006. U.S. patent application Ser. No. 11/080,992 and Ser. No. 11/531,671 are incorporated herein by reference in their entirety.
Dipeptidyl peptidase IV (IUBMB Enzyme Nomenclature EC.3.4.14.5) (referred herein as “DPP-IV”) is a type II membrane protein and a non-classical serine aminodipeptidase that removes Xaa-Pro dipeptides from the amino terminus (N-terminus) of polypeptides and proteins. DPP-IV is constitutively expressed on epithelial and endothelial cells of a variety of different tissues (e.g., intestine, liver, lung, kidney and placenta), and is also found in body fluids. DPP-IV is also expressed on circulating T-lymphocytes and has been shown to be synonymous with the cell-surface antigen, CD-26. DPP-IV has been implicated in a number of human disease states, including, but are not limit to, diabetes, particularly type II diabetes mellitus, diabetic dislipidemia, conditions of impaired glucose tolerance (IGT), conditions of impaired fasting plasma glucose (IFG), metabolic acidosis, ketosis, appetite regulation and obesity; autoimmune diseases such as inflammatory bowel disease, multiple sclerosis and rheumatoid arthritis; AIDS; and cancers.
DPP-IV inhibitors are believed to be useful agents for the prevention, delay of progression, and/or treatment of conditions mediated by DPP-IV.
Compound (A) or a salt thereof has been reported as an inhibitor of dipeptidyl peptidase (DPP-IV) , which is an enzyme that decomposes glucagon-like peptide-1 (GLP-1) , a hormone increasing insulin secretion (patent document 1) .
In addition, a method including administering 1 – 250 mg of compound (A) or a salt thereof to a patient once per week (patent documents 2, 3), crystal polymorphs of compound (A) (patent documents 4, 5) , and a preparation of compound (A)
(patent documents 6, 7) have also been reported. Compound (A) and a salt thereof are recommended for oral administration in view of the easiness of self-administration, and a tablet, particularly a tablet in the dosage form for administration once per week, is desired. [0006]
The dosage form of once per week is expected to improve drug compliance of patients, whereas it requires supply of compound (A) or a salt thereof to patients in a high dose as compared to, for example, the dosage form of once per day. Since a solid preparation containing compound (A) or a salt thereof in a high dose increases its size, it may conversely degrade the drug compliance for patients, particularly infants and elderly patients having difficulty in swallowing
……………………..
SYNTHESIS
Compound 34 IS TRELAGLIPTIN

4-Fluoro-2-methylbenzonitrile (31).
A mixture of 2-bromo-5-fluorotoluene (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was refluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 31 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
2-Bromomethyl-4-fluorobenzonitrile (32).
A mixture of 4-fluoro-2-methylbenzonitrile (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
Alternatively, 32 was made as follows.
4-Fluoro-2-methylbenzonitrile (1 kg) in DCE (2 L) was treated with AIBN (122 g) and heated to 75° C. A suspension of DBH (353 g) in DCE (500 mL) was added at 75° C. portionwise over 20 minutes. This operation was repeated 5 more times over 2.5 hours. The mixture was then stirred for one additional hour and optionally monitored for completion by, for example, measuring the amount of residual benzonitrile using HPLC. Additional AIBN (e.g., 12.5 g) was optionally added to move the reaction toward completion. Heating was stopped and the mixture was allowed to cool overnight. N,N-diisopropylethylamine (1.3 L) was added (at <10° C. over 1.5 hours) and then diethyl phosphite (1.9 L) was added (at <20° C. over 30 min). The mixture was then stirred for 30 minutes or until completion. The mixture was then washed with 1% sodium metabisulfite solution (5 L) and purified with water (5 L). The organic phase was concentrated under vacuum to afford 32 as a dark brown oil (3328 g), which was used without further purification (purity was 97% (AUC)).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (33).
A mixture of crude 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
Alternatively, 33 was made as follows.
To a solution of 6-chloro-3-methyluracil (750 g) and N,N-diisopropylethylamine (998 mL) in NMP (3 L) was added (at <30° C. over 25 min) a solution of 32 (2963 g crude material containing 1300 g of 32 in 3 L of toluene). The mixture was then heated at 60° C. for 2 hours or until completion (as determined, for example, by HPLC). Heating was then stopped and the mixture was allowed to cool overnight. Purified water (3.8 L) was added, and the resultant slurry was stirred at ambient temperature for 1 hour and at <5° C. for one hour. The mixture was then filtered under vacuum and the wet cake was washed with IPA (2×2.25 L). The material was then dried in a vacuum oven at 40±5° C. for 16 or more hours to afford 33 as a tan solid (>85% yield; purity was >99% (AUC)).
TFAsalt OF TRELAGLIPTIN
2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (34).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
FREE BASE NOF TRELAGLIPTIN
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 60° C. until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 60° C. and the mixture was allowed to cool to <25° C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2×3.6 L). The filtrate was concentrated at 45° C. under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34.
HCL salt OF TRELAGLIPTIN
The HCl salt of 34 was prepared from the TFA salt as follows. The TFA salt (34) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0° C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the HCl salt was prepared from the free base as follows. To a solution of free base in CH2Cl2 (12 L) was added (at <35° C. over 18 minutes) 2 M hydrochloric acid (3.1 L). The slurry was stirred for 1 hour and then filtered. The wet cake was washed with CH2Cl2 (3.6 L) and then THF (4.8 L). The wet cake was then slurried in THF (4.8 L) for one hour and then filtered. The filter cake was again washed with THF (4.8 L). The material was then dried in a vacuum oven at 50° C. (with a nitrogen bleed) until a constant weight (e.g., >26 hours) to afford 34 as the HCl salt as a white solid (1423 g, >85% yield).
Succinate salt OF TRELAGLIPTIN

The succinate salt of 34 was prepared from the HCl salt as follows. To a mixture of the HCl salt of 34 (1414 g), CH2Cl2 (7 L) and purified water (14 L) was added 50% NaOH solution (212 mL) until the pH of the mixture was >12. The biphasic mixture was stirred for 30 min and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (5.7 L) and the combined organic layers were washed with purified water (6 L). The organic layer was then passed through an in-line filter and concentrated under vacuum at 30° C. over three hours to afford the free base as an off-white solid. The free base was slurried in prefiltered THF (15 L) and prefiltered IPA (5.5 L). The mixture was then heated at 60° C. until complete dissolution of the free base was observed. A prefiltered solution of succinic acid (446 g) in THF (7 L) was added (over 23 min) while maintaining the mixture temperature at >57° C. After stirring at 60° C. for 15 min, the heat was turned off, the material was allowed to cool, and the slurry was stirred for 12 hours at 25±5° C. The material was filtered under vacuum and the wet cake was washed with prefiltered IPA (2×4.2 L). The material was then dried in a vacuum oven at 70±5° C. (with a nitrogen bleed) for >80 hours to afford the succinate salt of 34 as a white solid (1546 g, >90% yield).
The product was also converted to a variety of corresponding acid addition salts. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of 34 were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(−)-Mandelic and benzenesulfonic. The succinate was found to be crystalline as determined by x-ray powder diffraction analysis.
Methanesulfonate salt
In addition, the methanesulfonate salt was prepared as follows. A 10.5 g aliquot of the benzonitrile product was mixed with 400 mL of isopropylacetate. The slurry was heated to 75° C. and filtered through #3 Whatman filter paper. The solution was heated back to 75° C. and a 1M solution of methanesulfonic acid (30.84 mL) was added slowly over 10 minutes while stirring. The suspension was cooled to room temperature at a rate of about 20° C./hr. After 1 hr at room temperature, the solid was filtered and dried in an oven overnight to obtain the methanesulfonate salt.
…………………………
FORMULATION
COMPD A IS TRELAGLIPTIN
Examples (Comparative Example IA)
Succinate of compound (A) (26.6 mg) was weighed in a glass bottle and used as Comparative Example IA. (Comparative Example 2A)
The succinate of compound (A) and microcrystalline cellulose were uniformly mixed in a mortar at a ratio of 1:10, and the mixture (226.6 mg) was weighed in a glass bottle and used as Comparative Example 2A. (Comparative Example 3A)
The succinate of compound (A) and corn starch were uniformly mixed in a mortar at a ratio of 1:5, and the mixture (126.6 mg) was weighed in a glass bottle and used as Comparative Example 3A. (Example IA) Succinate of compound (A) , mannitol and corn starch according to the formulation of Table IA were uniformly mixed in a fluid bed granulator (LAB-I, POWREX CORPORATION) , and the mixture was granulated by spraying an aqueous solution of dissolved hypromellose 2910, and dried therein. The obtained granules were passed through a sieve -(16M) to give milled granules. To the milled granules were added croscarmellose sodium, microcrystalline cellulose and magnesium stearate, and they were mixed in a bag to give granules for tableting. The granules were punched by a rotary tableting machine (Correct 19K, Kikusui Seisakusho, Ltd.) with a 6.5 mmφ punch to give a plain tablet weighting 121 mg. On the other hand, titanium oxide, yellow ferric oxide and talc were dispersed in a hypromellose 2910 aqueous solution to prepare a film coating liquid. The aforementioned coating liquid was sprayed onto the above-mentioned plain tablet in a film coating machine (Hicoater HCP-75, Freund Corporation), to give 2500 film- coated tablets containing 3.125 mg of compound (A) (free form) per tablet. Table IA

………………………..
POLYMORPHS AND SYNTHESIS
FORM A
Form A may be prepared by crystallization from the various solvents and under the various crystallization conditions used during the polymorph screen (e.g., fast and slow evaporation, cooling of saturated solutions, slurries, and solvent/antisolvent additions). Tables B and C of Example 3 summarize the procedures by which Form A was prepared. For example, Form A was obtained by room temperature slurry of an excess amount of Compound I in acetone, acetonitrile, dichloromethane, 1,4-dioxane, diethyl ether, hexane, methanol, isopropanol, water, ethylacetate, tetrahydrofuran, toluene, or other like solvents on a rotating wheel for approximately 5 or 7 days. The solids were collected by vacuum filtration, and air dried in the hood. Also, Form A was precipitated from a methanol solution of Compound I by slow evaporation (SE).
[0091] Form A was characterized by XRPD, TGA, hot stage microscopy, IR, Raman spectroscopy, solution 1H-NMR, and solid state 13C-NMR.
[0092] Figure 1 shows a characteristic XRPD spectrum (CuKa, λ=1.5418A) of Form A. The XRPD pattern confirmed that Form A was crystalline. Major X-Ray diffraction lines expressed in °2Θ and their relative intensities are summarized in Table 1.
Table 1. Characteristic XRPD Peaks (CuKa) of Form A


Characterization Data of Form A of Compound I

8. Amorphous Form
[0137] The Amorphous Form of Compound I was prepared by lyophilization of an aqueous solution of Compound I (Example 10). The residue material was characterized by XRPD and the resulting XRPD spectrum displayed in Figure 26. The XRPD spectrum shows a broad halo with no specific peaks present, which confirms that the material is amorphous. The material was further characterized by TGA, DSC, hot stage microscopy, and moisture sorption analysis.
Table A. Approximate Solubilities of Compound I



Crystallization Experiments of Compound I from Solvents




a) FE = fast evaporation; SE = slow evaporation; RT = room temperature; SC = slow cool;CC = crash cool, MB = moisture sorption/desorption analysis b) qty = quantity; PO = preferred orientation
…………………………
SYNTHESIS
EXAMPLES
1. Preparation of 2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro- 2H-pyrimidin-l-ylmethyl]-4-fluoro-benzonitrile and pharmaceutically acceptable salts


4-Fluoro-2-methylbenzonitrile (3)
[0166] A mixture of 2-bromo-5fluorotoluene ( 2) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was re fluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 3 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, IH), 6.93-7.06 (m, 2H), 2.55 (s, 3H). 2-Bromomethyl-4-fluorobenzonitrile (4)
[0167] A mixture of 4-fluoro-2-methylbenzonitrile (3) (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification.1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J= 5.2, 8.4 Hz, IH), 7.28 (dd, J= 2.4, 8.8 Hz, IH), 7.12 (m, IH), 4.6 (s, 2H).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro- benzonitrile (6)
[0168] A mixture of crude 3-methyl-6-chlorouracil (5) (0.6 g, 3.8 mmol), 2- Bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO
(10 mL) was stirred at 60 C for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, 1=12, 8.4Hz, IH), 7.26 (d, J- 4.0Hz, IH), 7.11-7.17 (m, IH), 6.94 (dd, J=2.0, 9.0 Hz, IH), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for Ci3H9ClFN3O2, 293.68; found 293.68.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, TFA salt (1) (TFA salt of Compound I)

[0169] 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4- fluoro-benzonitrile (5) (300 mg, 1.0 mmol), (i?)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100 0C for 2 hrs. The final compound was obtained as a TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.16-7.27 (m, 2H), 5.46 (s, IH), 5.17-5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, IH), 2.67-2.92 (m, 2H), 2.07-2.17 (m, IH), 1.82-1.92 (m, IH), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, HCl salt

[0170] The TFA salt of Compound I was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0 C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.12-7.26 (m, 2H), 5.47 (s, IH), 5.21-5.32 (ABq, 2H, J = 32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, IH), 2.69-2.93 (m, 2H), 2.07-2.17 (m, IH), 1.83-1.93 (m, IH), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
General procedure for the preparation of salts of Compound I.
[0171] The benzonitrile product may be isolated as the free base if desired, but preferably, the product may be further converted to a corresponding acid addition salt. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of Compound I were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(-)-Mandelic and benzenesulfonic. [0172] The isolation and/or purification steps of the intermediate compounds in the above described process may optionally be avoided if the intermediates from the reaction mixture are obtained as relatively pure compounds and the by-products or impurities of the reaction mixture do not interfere with the subsequent reaction steps. Where feasible, one or more isolation steps may be eliminated to provide shorter processing times, and the elimination of further processing may also afford higher overall reaction yields.
…………………..
TABLET
2. Exemplary formulations comprising succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Provided are examples of tablet formulations that may be used to administer succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (Succinate salt of Compound I) according to the present invention. It is noted that the formulations provided herein may be varied as is known in the art.
The exemplary tablet formulations are as follows:
| 12.5 mg of Compound I (weight of free base form) per tablet | ||||
| Core Tablet Formulation | ||||
| (1) | 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4- | 17.0 | mg | |
| dioxo-3,4-dihydro-2H-pyrimidin-1- | ||||
| ylmethyl]-4-fluoro-benzonitrile (succinate salt) | ||||
| (2) | Lactose Monohydrate, NF, Ph, Eur | 224.6 | mg | |
| (FOREMOST 316 FAST FLO) | ||||
| (3) | Microcrystalline Cellulose, NF, Ph, Eur | 120.1 | mg | |
| (AVICEL PH 102) | ||||
| (4) | Croscarmellose Sodium, NF, Ph, Eur | 32.0 | mg | |
| (AC-DO-SOL) | ||||
| (5) | Colloidal Silicon Dioxide, NF, Ph, Eur | 3.2 | mg | |
| (CAB-O-SIL M-5P) | ||||
| (6) | Magnesium Stearate, NF, Ph, Eur | 3.2 | mg | |
| (MALLINCKRODT, Non-bovine Hyqual) | ||||
| TOTAL | 400.0 | mg | ||
| (per tablet) | ||||
…………..
US20080227798 AND US20120197018
POLYMORPHS AND SYNTHESIS
EXAMPLES Example 1 Preparation of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile succinate (Compound I)

Compound I may be prepared by the follow synthetic route (Scheme 1)

A. Preparation of 4-fluoro-2-methylbenzonitrile (Compound B)

Compound B was prepared by refluxing a mixture of 2-bromo-5-fluoro-toluene (Compound A) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product B (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
B. Preparation of 2-bromomethyl-4-fluorobenzonitrile (Compound C)

Compound C was prepared by refluxing a mixture of 4-fluoro-2-methylbenzonitrile (Compound B) (2 g, 14.8 mmol), N-bromosuccinimide (NBS) (2.64 g, 15 mmol) and azo-bis-isobutyronitrile (AIBN) (100 mg) in CCl4 under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give the crude product the form of an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
C. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound D)

Compound E was prepared by stirring a mixture of crude 3-methyl-6-chlorouracil D (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
D. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound F)

Compound F was prepared by mixing and stirring 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound E) (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as trifluoroacetate (TFA) salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J=35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
E. Preparation of Compound I: the succinic acid salt of 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile

The TFA salt prepared in the above step (Example 1, Step D) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The benzonitrile product (approximately 10 mg) was dissolved in MeOH (1 mL) and to which succinic acid in THF (1.05 equivalents) was added. The solutions were allowed to stand for three days open to the air. If a precipitate formed, the solid was collected by filtration. If no solid formed, the mixture was concentrated in vacuo, and the succinate salt was obtained after removing the solvent.
SUCCINATE SALT OF TRELAGLIPTIN
1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Compound I such prepared was found to be crystalline as determined by x-ray powder diffraction analysis (FIG. 1). The crystal material was designated Form A.
……………
patents
1. US 2013172377
2. WO 2011013639
3. WO 2009099172
4.WO 2009099171
5. WO 2008114807
6.WO 2008114800
7. WO 2008033851
8. WO 2007074884
9WO 2007035629
patent document 1: US2005/0261271
patent document 2: US2007/0060530
patent document 3: US2008/0287476
patent document 4: US2008/0227798
patent document 5: US2008/0280931
patent document 6: WO2008/114800
patent document 7: WO2011/013639
| US7906523 * | Oct 30, 2007 | Mar 15, 2011 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8084605 * | Nov 29, 2007 | Dec 27, 2011 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8188275 * | Oct 30, 2007 | May 29, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8222411 * | Sep 15, 2006 | Jul 17, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US20090275750 * | Sep 15, 2006 | Nov 5, 2009 | Jun Feng | Dipeptidyl peptidase inhibitors |
| WO2013183784A1 | Jun 4, 2013 | Dec 12, 2013 | Takeda Pharmaceutical Company Limited | Solid preparation |
| US20080227798 * | Nov 29, 2007 | Sep 18, 2008 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US20120197018 * | Feb 15, 2012 | Aug 2, 2012 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| WO2007033265A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033266A2 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033350A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetes |
| EP1586571A1 * | Dec 21, 2004 | Oct 19, 2005 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
13 NMR TRELAGLIPTIN SUCCINATE

1H NMR TRELAGLIPTIN SUCCINATE

Lodenafil Carbonate … an Erectile Dysfunction Drug in Phase III

Lodenafil carbonate
UNII-29X84F932D, CRIS-031
bis-(2-{4-[4-ethoxy-3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-benzenesulfonyl]piperazin-1-yl}-ethyl)carbonate
5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one. IS THE NAME OF MONOMER
398507-55-6 CAS
Cristalia (Originator)
| C47 H62 N12 O11 S2= MF | |
| Molecular Weight | 1035.199 |
Lodenafil is a drug belonging to a class of drugs called PDE5 inhibitor, which many other erectile dysfunction drugs such as sildenafil, tadalafil, and vardenafil also belong to. Like udenafil and avanafil it belongs to a new generation of PDE5 inhibitors.
- Sildenafil, tadalafil, vardenafil, and the newer udenafil and avanafil selectively inhibit PDE5, which is cGMP-specific and responsible for the degradation of cGMP in the corpus cavernosum. These phosphodiesterase inhibitors are used primarily as remedies for erectile dysfunction, as well as having some other medical applications such as treatment of pulmonary hypertension.
Lodenafil is formulated as a dimer, lodenafil carbonate, which breaks down in the body to form two molecules of the active drug lodenafil. This formulation has higher oral bioavailability than the parent drug.[1]
It is manufactured by Cristália Produtos Químicos e Farmacêuticos in Brazil and sold there under the brand-name Helleva.[2]

Helleva (Lodenafil Carbonate) is an oral PDE5 inhibitor prescribed to treat men suffering from erectile dysfunction. It operates by relaxing muscles and dilating blood vessels in the penis to increase circulation making it easier to attain and maintain an erection.
It has undergone Phase III clinical trials,[3][4][5] but is not yet approved for use in the United States by the U.S. Food and Drug Administration.

………..
SYNTHESIS
WO 2002012241 OR US7148350
MONOMER synthesis
 PIPERAZINE
PIPERAZINE
AND
 ETHYL CHLORO ACETATE
ETHYL CHLORO ACETATE
WILL GIVE
Ethyl 1-piperazinylacetate
SEE RXN 1 BELOW
Reaction 1:
Synthesis of Piperazine Ethyl Acetate
To a reaction blend containing 100 g (3 Eq, 0.515 mol, MW=194) of piperazine, 26.3 mL (1.1 Eq, 0.189 mol, MW=101, d=0.726) of triethylamine in 200 mL of isopropanol, add to a solution previously prepared of 18.4 mL (1 Eq., 0.172 mol, MW=122.55, d=1.15) of chloroacetate of ethyl in 140 mL of isopropanol under stirring, at room temperature. Keep the reaction medium under stirring, monitoring the reaction termination by means of a chromatography of the thin layer (about 2–3 hours). Add a solution of 40.6 g (0.344 mol) of succinic acid in 140 mL of isopropanol. Keep the system under stirring for about 30 minutes to assure total precipitation of the succinate salt of piperazine formed. Filter this salt and concentrate the filtrate containing the mono and dialkyled derivatives. We obtain a slightly yellowish oil, which is used in later phases without purification.
Mass obtained=33 g
GC/MS: Monoalkylated derivative 72%, and dialkylated 22%.
NEXT
Piperazine Ethyl Acetate
AND
![5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one Structure](https://i0.wp.com/www.chemicalbook.com/CAS/GIF/139756-22-2.gif)
5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one
WILL REACT TO GIVE… 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one AS IN RXN 4 BELOW
Reaction 4:
Synthesis of 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one.
Suspend 24.6 g (60 mmol, MW=410.9) of 5-(5-chlorosulfonyl-2-etoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one in 900 mL of ethanol absolute. Under stirring and at room temperature, add at only one time, a solution containing 31.0 g (3 Eq., 180 mmol MW=172) of N-piperazine ethyl acetate (Reaction 1) dissolved in 150 mL of ethanol absolute. In an interval of 2–10 minutes, all solid is consumed, forming a clean and homogeneous solution, and after that starts the precipitation of the expected product. At the end of the reaction, which lasts 2–3 hours (monitored by chromatography of thin layer), the product is vacuum filtered and the solid is washed with two portions of 50 mL of iced absolute ethanol. 29 g are obtained (yielding=89%) from the product as a white solid of MP=165.5–166.5° C.
Reaction 7:
Intermediate 1
5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one. IS MONOMER
please note during LAH redn …………. the PIP CH2-C=O-O CH2 CH3 BECOMES PIP-CH2CH2-OH
To a suspension of lithium aluminum hydride (0.74 g 2.2 Eq. MW=37.9) in 25 mL of THF, slowly add, under stirring and at room temperature, a suspension of 5.0 g (9.1 mmol, MW=546.6) of 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one in 50 mL of THF. The system is maintained under stirring, monitoring the consumption of the product by chromatography of thin layer, until the complete consumption of the starting reagent (about 5–6 hours). Slowly add water to the reaction medium and, when there is no longer release of H2, add HCl 1M regulating pH for 7. Extract the product with 3 200 mL-portions of chloroform, dry with anhydrous sodium sulfate and vacuum concentrate the product. It is obtained 3.8 g of the product as a cream solid MP=183–187° C. yielding 83%. The same was crystallized from methanol and DMF yielding a slightly yellowish solid with melting point at 189–192° C.

note …………. the PIP CH2-C=O-O CH2 CH3 BECOMES PIP-CH2CH2-OH
HOMODIMER CARBONATE
EXAMPLE 1B
Homodimer Carbonate of Intermediate 1—Alternative Method
A phosgene solution (3.5 g, 35 mmol) dissolved in 20 mL of toluene was added dropwise to a solution of 2.02 g (4 mmol) of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, suspended in 44 mL of toluene. The reaction mixture resulting is stirred and followed by chromatography analysis of thin layer every hour until the reagent conversion in its chloroformate was completed. When the analysis indicates the complete consumption of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, the volatile compounds of the reaction are vacuum removed (solvents and phosgene), yielding the esther chloroformate raw derivative of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one.
The raw chloroformate obtained above (4.0 mmol, 2.27 g) is dissolved in about 30 mL of dichloromethane, to which is added 2.07 g (4.1 mmol) of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, followed by the addition of 4 mL of dichloromethane containing 450 mg of triethylamine. The reaction mixture is maintained under stirring, being followed by chromatography of thin layer every hour until this indicates the end of the reaction (disappearing of chloroformate derivative). The reaction mixture is then diluted with 60 mL of dichloromethane, washed with NaCl saturated solution, after with sodium bicarbonate saturated solution and again with NaCl saturated solution. Organic phase is separated and dry with anhydrous sodium sulfate. The solvent is then evaporated to dry, yielding the dimer carbonate as a slightly yellowish solid.
This compound is re-crystallized from ethanol:DMF, yielding a pale white solid. Yielding m=3.2 g (76%)
Microanalysis: Theoretical C, (54.53%); H, (6.04%); N, (16.24%);
Obtained C, (54.45%); H, (6.02%); N, (16.17%).
INFO ABOUT INTERMEDIATE
5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one
| CAS No. | 139756-22-2 |
| Chemical Name: | 5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one |
| Synonyms: | Sildenafil Chlorosulfone IMpurity;Sildenafil Chlorosulfonyl IMpurity;5-(5-CHLOROSULFONYL-2-ETHOXY PHENYL)-1-METHYL-3-N-PROPYL-1;3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1 H-pyrazolo-(4-3-d)-pyrimidine-5;5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one;3-(4,7-Dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxy-benzenesulfonyl Chloride;4-Ethoxy-3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyriMidin-5-yl)benzene-1-sulfonyl chloride |
| CBNumber: | CB11175931 |
| Molecular Formula: | C17H19ClN4O4S |
http://www.chemicalbook.com/ChemicalProductProperty_EN_CB11175931.htm
…………..
SYNTHESIS OF

http://www.google.co.in/patents/US6362178

22.27 g (250 mmol) of D,L-alanine and 55.66 g (550 mmol) of triethylamine are dissolved in 250 ml of dichloromethane, and the solution is cooled to 0° C. 59.75 g (550 mmol) of trimethylsilyl chloride are added dropwise, and the solution is stirred for 1 hour at room temperature and for 1 hour at 40° C. After cooling to −10° C., 26.64 g (250 mmol) of butyryl chloride are added dropwise, and the resulting mixture is stirred for 2 hours at −10° C. and for one hour at room temperature.
With ice-cooling, 125 ml of water are added dropwise and the reaction mixture is stirred at room temperature for 15 minutes. The aqueous phase is evaporated to dryness, the residue is titrated with acetone and the mother liquor is filtered off with suction. The solvent is removed and the residue is chromatographed. The resulting product is dissolved in 3N aqueous sodium hydroxide solution and the resulting solution is evaporated to dryness. The residue is taken up in conc. HCl and once more evaporated to dryness. The residue is stirred with acetone, precipitated solid is filtered off with suction and the solvent is removed under reduced pressure. This gives 28.2 g (71%) of a viscous oil which crystallizes after some time.
200 MHz 1H-NMR (DMSO-d6): 0.84, t, 3H; 1.22, d, 3H; 1.50, hex, 2H; 2.07, t, 2H; 4.20, quin., 1H; 8.09, d, 1H.

25 g (210 mmol) of 2-hydroxybenzonitrile are refluxed with 87 g of potassium carbonate and 34.3 g (314.8 mmol) of ethyl bromide in 500 ml of acetone overnight. The solid is filtered off, the solvent is removed under reduced pressure and the residue is distilled under reduced pressure. This gives 30.0 g (97%) of a colourless liquid.
200 MHz 1H-NMR (DMSO-d6): 1.48, t, 3H; 4.15, quart., 2H; 6.99, dt, 2H; 7.51, dt, 2H.

21.4 g (400 mmol) of ammonium chloride are suspended in 375 ml of toluene, and the suspension is cooled to 0° C. 200 ml of a 2M solution of trimethylaluminium in hexane are added dropwise, and the mixture is stirred at room temperature until the evolution of gas has ceased. After addition of 29.44 g (200 mmol) of 2-ethoxybenzonitrile, the reaction mixture is stirred at 80° C. (bath) overnight.
With ice-cooling, the cooled reaction mixture is added to a suspension of 100 g of silica gel and 950 ml of chloroform, and the mixture is stirred at room temperature for 30 minutes. The mixture is filtered off with suction, and the filter residue is washed with the same amount of methanol. The mother liquor is concentrated, the resulting residue is stirred with a mixture of dichloromethane and methanol (9:1), the solid is filtered off with suction and the mother liquor is concentrated. This gives 30.4 g (76%) of a colourless solid.
200 MHz 1H-NMR (DMSO-d6): 1.36, t, 3H; 4.12, quart., 2H; 7.10, t, 1H; 7.21, d, 1H; 7.52, m, 2H; 9.30, s, broad, 4H.
EXAMPLE 10A 2-(2-Ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

7.16 g (45 mmol) of 2-butyrylamino-propionic acid and 10.67 g of pyridine are dissolved in 45 ml of THF and, after addition of a spatula tip of DMAP, heated to reflux. 12.29 g (90 mmol) of ethyl oxalyl chloride are slowly added dropwise, and the reaction mixture is refluxed for 3 hours. The mixture is poured into ice-water and extracted three times with ethyl acetate and the organic phase is dried over sodium sulphate and concentrated using a rotary evaporator. The residue is taken up in 15 ml of ethanol and refluxed with 2.15 g of sodium bicarbonate for 2.5 hours. The cooled solution is filtered.
With ice-cooling, 2.25 g (45 mmol) of hydrazine hydrate are added dropwise to a solution of 9.03 g (45 mmol) of 2-ethoxybenzamidine hydrochloride in 45 ml of ethanol, and the resulting suspension is stirred at room temperature for another 10 minutes. The ethanolic solution described above is added to this reaction mixture, and the mixture is stirred at a bath temperature of 70° C. for 4 hours. After filtration, the mixture is concentrated, the residue is partitioned between dichloromethane and water, the organic phase is dried over sodium sulphate and the solvent is removed under reduced pressure.
This residue is dissolved in 60 ml of 1,2-dichloroethane and, after addition of 7.5 ml of phosphorus oxychloride, refluxed for 2 hours. The mixture is diluted with dichloromethane and neutralized by addition of sodium bicarbonate solution and solid sodium bicarbonate. The organic phase is dried and the solvent is removed under reduced pressure. Chromatography using ethyl acetate and crystallization afford 4.00 g (28%) of a colourless solid, Rf=0.42 (dichloromethane/methanol=95:5)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.56, t, 3H; 1.89, hex, 2H; 2.67, s, 3H; 3.00, t, 2H; 4.26, quart., 2H; 7.05, m, 2H; 7.50, dt, 1H; 8.17, dd, 1H; 10.00, s, 1H.
EXAMPLE 15A 4-Ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride

At 0° C., 2.00 g (6.4 mmol) of 2-(2-ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are slowly added to 3.83 ml of chlorosulphonic acid. At room temperature, the reaction mixture is stirred ovemight, and then poured into ice-water and extracted with dichloromethane. This gives 2.40 g (91%) of a colourless foam.
200 MHz 1H-NMR (CDCl3): 1.03, t, 3H; 1.61, t, 2H; 1.92, hex, 2H; 2.67, s, 3H; 3.10, t, 2H; 4.42, quart., 2H; 7.27, t, 1H; 8.20, dd, 1H; 8.67, d, 1H; 10.18, s, 1H.
Example 22 2-[2-Ethoxy-5-(4-hydroxyethyl-1-amino-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one
By the same method, starting with 0.04 g (0.097 mmol) of 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride and 0.04 g (0.29 mmol) of 1-amino-4-hydroxyethylpiperazine, 46 mg (91%) of 2-[2-ethoxy-5-(4-hydroxyethyl-1-amino-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are obtained.
Rf=0.08 (dichloromethane/methanol=19:1)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.59, t, 3H; 1.90, sex., 2H; 2.49, m, 6H; 2.62, s, 3H; 2.71, m, 4H; 3.00, t, 2H; 3.55, t, 2H; 4.31, quart., 2H; 7.14, d, 1H; 8.05, dd, 1H; 8.60, d, 1H.
…………..
Methods of analysis
The development of lodenafil carbonate was reported by Toque et al. (2008). They observed the effects of lodenafil carbonate on rabbit and human corpus cavernosum relaxation, activity of PDE5 in human platelets, stability and metabolic studies in comparison with sildenafil and lodenafil, as well as the pharmacological evaluation of lodenafil carbonate after intravenous and oral administration in male beagles.
The determination of PDE activity, stability of lodenafil carbonate in human, dog and rat plasma and the pharmacokinetic parameters after a single intravenous or oral dose was carried out by LC-MS/MS analysis
Codevilla et al. (2011a) developed a stability-indicating reversed-phase liquid chromatography method using ultraviolet (UV) detection for the quantitative determination of lodenafil carbonate in tablets. The method can be useful for routine quality control assay and stability studies.
Another study for the determination of lodenafil carbonate in tablets was developed by Codevilla et al. (2011b). As an alternative to the LC method the authors suggested a UV-spectrophotometric method for the analysis of lodenafil carbonate in pharmaceutical form. The UV method offers advantages over other analytical methods due to its rapidity, simplicity, and lower cost. Recently, Codevilla et al. (2012) developed and validated a capillary zone electrophoresis (CZE) method for determination of lodenafil carbonate in drug products. There are some advantages to use the CZE method, such as rapid analysis, small sample and reagent consumption, high separation efficiency (Furlanetto et al., 2001; Yang et al., 2010). The results obtained from the UV-spectrophotometric method and CZE method were compared statistically with the LC method (Codevilla et al., 2011a) and the results showed no significant difference between these methods.
References
- Toque HA, Teixeira CE, Lorenzetti R, Okuyama CE, Antunes E, De Nucci G (September 2008). “Pharmacological characterization of a novel phosphodiesterase type 5 (PDE5) inhibitor lodenafil carbonate on human and rabbit corpus cavernosum”. European Journal of Pharmacology 591 (1–3): 189–95. doi:10.1016/j.ejphar.2008.06.055. PMID 18593576.
- Cristália Product page. Retrieved on September 16, 2009.
- ukmedix Lodenafil article. Retrieved on September 16, 2009.
- Glina S, Toscano I, Gomatzky C, de Góes PM, Júnior AN, Claro JF, Pagani E (February 2009). “Efficacy and tolerability of lodenafil carbonate for oral therapy in erectile dysfunction: a phase II clinical trial”. The Journal of Sexual Medicine 6 (2): 553–7. doi:10.1111/j.1743-6109.2008.01079.x.PMID 19040623.
- Glina S, Fonseca GN, Bertero EB, Damião R, Rocha LC, Jardim CR, Cairoli CE, Teloken C, Torres LO, Faria GE, da Silva MB, Pagani E (February 2010). “Efficacy and Tolerability of Lodenafil Carbonate for Oral Therapy of Erectile Dysfunction: A Phase III Clinical Trial”. The Journal of Sexual Medicine 7 (5): 1928–1936. doi:10.1111/j.1743-6109.2010.01711.x. PMID 20214718.
- Toque H A et al., (2008) European Journal of Pharmacology, 591(1-3):189-95.
- Exploring the role of PDE5 inhibition in the treatment of muscular dystrophy
Drugs Fut 2011, 36(4): 321
PANOBINOSTAT

Panobinostat
HDAC inhibitors, orphan drug
cas 404950-80-7
2E)-N-hydroxy-3-[4-({[2-(2-methyl-1H-indol-3-yl)ethyl]amino}methyl)phenyl]acrylamide
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (alternatively, N-hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide)
Molecular Formula: C21H23N3O2 Molecular Weight: 349.42622
- Faridak
- LBH 589
- LBH589
- Panobinostat
- UNII-9647FM7Y3Z
A hydroxamic acid analog histone deacetylase inhibitor from Novartis.
NOVARTIS, innovator
Histone deacetylase inhibitors
Is currently being examined in cutaneous T-cell lymphoma, CML and breast cancer.
clinical trials click here phase 3
DRUG SUBSTANCE–LACTATE AS IN http://www.google.com/patents/US7989639 SEE EG 31

Panobinostat (LBH-589) is an experimental drug developed by Novartis for the treatment of various cancers. It is a hydroxamic acid[1] and acts as a non-selective histone deacetylase inhibitor (HDAC inhibitor).[2]
panobinostat
Panobinostat is a cinnamic hydroxamic acid analogue with potential antineoplastic activity. Panobinostat selectively inhibits histone deacetylase (HDAC), inducing hyperacetylation of core histone proteins, which may result in modulation of cell cycle protein expression, cell cycle arrest in the G2/M phase and apoptosis. In addition, this agent appears to modulate the expression of angiogenesis-related genes, such as hypoxia-inducible factor-1alpha (HIF-1a) and vascular endothelial growth factor (VEGF), thus impairing endothelial cell chemotaxis and invasion. HDAC is an enzyme that deacetylates chromatin histone proteins. Check for
As of August 2012, it is being tested against Hodgkin’s Lymphoma, cutaneous T cell lymphoma (CTCL)[3] and other types of malignant disease in Phase III clinical trials, against myelodysplastic syndromes, breast cancer and prostate cancer in Phase II trials, and against chronic myelomonocytic leukemia (CMML) in a Phase I trial.[4][5]
Panobinostat is a histone deacetylase (HDAC) inhibitor which was filed for approval in the U.S. in 2010 for the oral treatment of relapsed/refractory classical Hodgkin’s lymphoma in adult patients. The company is conducting phase II/III clinical trials for the oral treatment of multiple myeloma, chronic myeloid leukemia and myelodysplasia. Phase II trials are also in progress for the treatment of primary myelofibrosis, post-polycythemia Vera, post-essential thrombocytopenia, Waldenstrom’s macroglobulinemia, recurrent glioblastoma (GBM) and for the treatment of pancreatic cancer progressing on gemcitabine therapy. Additional trials are under way for the treatment of hematological neoplasms, prostate cancer, colorectal cancer, renal cell carcinoma, non-small cell lung cancer (NSCLC), malignant mesothelioma, acute lymphoblastic leukemia, acute myeloid leukemia, head and neck cancer and gastrointestinal neuroendocrine tumors. Early clinical studies are also ongoing for the treatment of HER2 positive metastatic breast cancer. Additionally, phase II clinical trials are ongoing at Novartis as well as Neurological Surgery for the treatment of recurrent malignant gliomas as are phase I/II initiated for the treatment of acute graft versus host disease. The National Cancer Institute had been conducting early clinical trials for the treatment of metastatic hepatocellular carcinoma; however, these trials were terminated due to observed dose-limiting toxicity. In 2009, Novartis terminated its program to develop panobinostat for the treatment of cutaneous T-cell lymphoma. A program for the treatment of small cell lung cancer was terminated in 2012. Phase I clinical trials are ongoing for the treatment of metastatic and/or malignant melanoma and for the treatment of sickle cell anemia. The University of Virginia is conducting phase I clinical trials for the treatment of newly diagnosed and recurrent chordoma in combination with imatinib. Novartis is evaluating panobinostat for its potential to re-activate HIV transcription in latently infected CD4+ T-cells among HIV-infected patients on stable antiretroviral therapy.
Mechanistic evaluations revealed that panobinostat-mediated tumor suppression involved blocking cell-cycle progression and gene transcription induced by the interleukin IL-2 promoter, accompanied by an upregulation of p21, p53 and p57, and subsequent cell death resulted from the stimulation of caspase-dependent and -independent apoptotic pathways and an increase in the mitochondrial outer membrane permeability. In 2007, the compound received orphan drug designation in the U.S. for the treatment of cutaneous T-cell lymphoma and in 2009 and 2010, orphan drug designation was received in the U.S. and the E.U., respectively, for the treatment of Hodgkin’s lymphoma. This designation was also assigned in 2012 in the U.S. and the E.U. for the treatment of multiple myeloma.
Cardiovascular disease is the leading cause of morbidity and mortality in the western world and during the last decades it has also become a rapidly increasing problem in developing countries. An estimated 80 million American adults (one in three) have one or more expressions of cardiovascular disease (CVD) such as hypertension, coronary heart disease, heart failure, or stroke. Mortality data show that CVD was the underlying cause of death in 35% of all deaths in 2005 in the United States, with the majority related to myocardial infarction, stroke, or complications thereof. The vast majority of patients suffering acute cardiovascular events have prior exposure to at least one major risk factor such as cigarette smoking, abnormal blood lipid levels, hypertension, diabetes, abdominal obesity, and low-grade inflammation.
Pathophysiologically, the major events of myocardial infarction and ischemic stroke are caused by a sudden arrest of nutritive blood supply due to a blood clot formation within the lumen of the arterial blood vessel. In most cases, formation of the thrombus is precipitated by rupture of a vulnerable atherosclerotic plaque, which exposes chemical agents that activate platelets and the plasma coagulation system. The activated platelets form a platelet plug that is armed by coagulation-generated fibrin to form a biood clot that expands within the vessel lumen until it obstructs or blocks blood flow, which results in hypoxic tissue damage (so-called infarction). Thus, thrombotic cardiovascular events occur as a result of two distinct processes, i.e. a slowly progressing long-term vascular atherosclerosis of the vessel wall, on the one hand, and a sudden acute clot formation that rapidly causes flow arrest, on the other. This invention solely relates to the latter process.
Recently, inflammation has been recognized as an important risk factor for thrombotic events. Vascular inflammation is a characteristic feature of the atherosclerotic vessel wall, and inflammatory activity is a strong determinant of the susceptibility of the atherosclerotic plaque to rupture and initiate intravascular clotting. Also, autoimmune conditions with systemic inflammation, such as rheumatoid arthritis, systemic lupus erythematosus and different forms of vasculitides, markedly increase the risk of myocardial infarction and stroke.
Traditional approaches to prevent and treat cardiovascular events are either targeted 1) to slow down the progression of the underlying atherosclerotic process, 2) to prevent clot formation in case of a plaque rupture, or 3) to direct removal of an acute thrombotic flow obstruction. In brief, antiatherosclerotic treatment aims at modulating the impact of general risk factors and includes dietary recommendations, weight loss, physical exercise, smoking cessation, cholesterol- and blood pressure treatment etc. Prevention of clot formation mainly relies on the use of antiplatelet drugs that inhibit platelet activation and/or aggregation, but also in some cases includes thromboembolic prevention with oral anticoagulants such as warfarin. Post-hoc treatment of acute atherothrombotic events requires either direct pharmacological lysis of the clot by thrombolytic agents such as recombinant tissue-type plasminogen activator or percutaneous mechanical dilation of the obstructed vessel.
Despite the fact that multiple-target antiatherosclerotic therapy and clot prevention by antiplatelet agents have lowered the incidence of myocardial infarction and ischemic stroke, such events still remain a major population health problem. This shows that in patients with cardiovascular risk factors these prophylactic measures are insufficient to completely prevent the occurrence of atherothrombotic events.
Likewise, thrombotic conditions on the venous side of the circulation, as well as embolic complications thereof such as pulmonary embolism, still cause substantial morbidity and mortality. Venous thrombosis has a different clinical presentation and the relative importance of platelet activation versus plasma coagulation are somewhat different with an preponderance for the latter in venous thrombosis, However, despite these differences, the major underlying mechanisms that cause thrombotic vessel occlusions are similar to those operating on the arterial circulation. Although unrelated to atherosclerosis as such, the risk of venous thrombosis is related to general cardiovascular risk factors such as inflammation and metabolic aberrations.

Panobinostat can be synthesized as follows: Reduction of 2-methylindole-3-glyoxylamide (I) with LiAlH4 affords 2-methyltryptamine (II). 4-Formylcinnamic acid (III) is esterified with methanolic HCl, and the resulting aldehyde ester (IV) is reductively aminated with 2-methyltryptamine (II) in the presence of NaBH3CN (1) or NaBH4 (2) to give (V). The title hydroxamic acid is then obtained by treatment of ester (V) with aqueous hydroxylamine under basic conditions.
Panobinostat is currently being used in a Phase I/II clinical trial that aims at curing AIDS in patients on highly active antiretroviral therapy (HAART). In this technique panobinostat is used to drive the HI virus’s DNA out of the patient’s DNA, in the expectation that the patient’s immune system in combination with HAART will destroy it.[6][7]
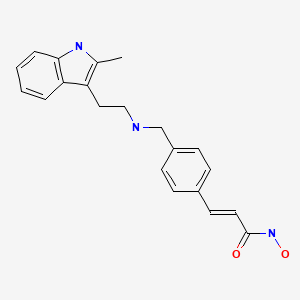 panobinostat
panobinostat
Panobinostat has been found to synergistically act with sirolimus to kill pancreatic cancer cells in the laboratory in a Mayo Clinic study. In the study, investigators found that this combination destroyed up to 65 percent of cultured pancreatic tumor cells. The finding is significant because the three cell lines studied were all resistant to the effects of chemotherapy – as are many pancreatic tumors.[8]
Panobinostat has also been found to significantly increase in vitro the survival of motor neuron (SMN) protein levels in cells of patients suffering fromspinal muscular atrophy.[9]
Panobinostat was able to selectively target triple negative breast cancer (TNBC) cells by inducing hyperacetylation and cell cycle arrest at the G2-M DNA damage checkpoint; partially reversing the morphological changes characteristic of breast cancer cells.[10]
Panobinostat, along with other HDAC inhibitors, is also being studied for potential to induce virus HIV-1 expression in latently infected cells and disrupt latency. These resting cells are not recognized by the immune system as harboring the virus and do not respond to antiretroviral drugs.[11]
Panobinostat inhibits multiple histone deacetylase enzymes, a mechanism leading to apoptosis of malignant cells via multiple pathways.[1]
The compound N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (alternatively, N-hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide) has the formula

as described in WO 02/22577. Valuable pharmacological properties are attributed to this compound; thus, it can be used, for example, as a histone deacetylase inhibitor useful in therapy for diseases which respond to inhibition of histone deacetylase activity. WO 02/22577 does not disclose any specific salts or salt hydrates or solvates of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide.
The compounds described above are often used in the form of a pharmaceutically acceptable salt. Pharmaceutically acceptable salts include, when appropriate, pharmaceutically acceptable base addition salts and acid addition salts, for example, metal salts, such as alkali and alkaline earth metal salts, ammonium salts, organic amine addition salts, and amino acid addition salts, and sulfonate salts. Acid addition salts include inorganic acid addition salts such as hydrochloride, sulfate and phosphate, and organic acid addition salts such as alkyl sulfonate, arylsulfonate, acetate, maleate, fumarate, tartrate, citrate and lactate. Examples of metal salts are alkali metal salts, such as lithium salt, sodium salt and potassium salt, alkaline earth metal salts such as magnesium salt and calcium salt, aluminum salt, and zinc salt. Examples of ammonium salts are ammonium salt and tetramethylammonium salt. Examples of organic amine addition salts are salts with morpholine and piperidine. Examples of amino acid addition salts are salts with glycine, phenylalanine, glutamic acid and lysine. Sulfonate salts include mesylate, tosylate and benzene sulfonic acid salts.
……………………………..
GENERAL METHOD OF SYNTHESIS
ADD YOUR METHYL AT RIGHT PLACE
As is evident to those skilled in the art, the many of the deacetylase inhibitor compounds of the present invention contain asymmetric carbon atoms. It should be understood, therefore, that the individual stereoisomers are contemplated as being included within the scope of this invention.
The hydroxamate compounds of the present invention can be produced by known organic synthesis methods. For example, the hydroxamate compounds can be produced by reacting methyl 4-formyl cinnamate with tryptamine and then converting the reactant to the hydroxamate compounds. As an example, methyl 4-formyl cinnamate 2, is prepared by acid catalyzed esterification of 4-formylcinnamic acid 3 (Bull. Chem. Soc. Jpn. 1995; 68:2355-2362). An alternate preparation of methyl 4-formyl cinnamate 2 is by a Pd- catalyzed coupling of methyl acrylate 4 with 4-bromobenzaldehyde 5.
CHO

Additional starting materials can be prepared from 4-carboxybenzaldehyde 6, and an exemplary method is illustrated for the preparation of aldehyde 9, shown below. The carboxylic acid in 4-carboxybenzaldehyde 6 can be protected as a silyl ester (e.g., the t- butyldimethylsilyl ester) by treatment with a silyl chloride (e.g., f-butyldimethylsilyl chloride) and a base (e.g. triethylamine) in an appropriate solvent (e.g., dichloromethane). The resulting silyl ester 7 can undergo an olefination reaction (e.g., a Horner-Emmons olefination) with a phosphonate ester (e.g., triethyl 2-phosphonopropionate) in the presence of a base (e.g., sodium hydride) in an appropriate solvent (e.g., tetrahydrofuran (THF)). Treatment of the resulting diester with acid (e.g., aqueous hydrochloric acid) results in the hydrolysis of the silyl ester providing acid 8. Selective reduction of the carboxylic acid of 8 using, for example, borane-dimethylsuflide complex in a solvent (e.g., THF) provides an intermediate alcohol. This intermediate alcohol could be oxidized to aldehyde 9 by a number of known methods, including, but not limited to, Swern oxidation, Dess-Martin periodinane oxidation, Moffatt oxidation and the like.

The aldehyde starting materials 2 or 9 can be reductively aminated to provide secondary or tertiary amines. This is illustrated by the reaction of methyl 4-formyl cinnamate 2 with tryptamine 10 using sodium triacetoxyborohydride (NaBH(OAc)3) as the reducing agent in dichloroethane (DCE) as solvent to provide amine 11. Other reducing agents can be used, e.g., sodium borohydride (NaBH ) and sodium cyanoborohydride (NaBH3CN), in other solvents or solvent mixtures in the presence or absence of acid catalysts (e.g., acetic acid and trifluoroacetic acid). Amine 11 can be converted directly to hydroxamic acid 12 by treatment with 50% aqueous hydroxylamine in a suitable solvent (e.g., THF in the presence of a base, e.g., NaOH). Other methods of hydroxamate formation are known and include reaction of an ester with hydroxylamine hydrochloride and a base (e.g., sodium hydroxide or sodium methoxide) in a suitable solvent or solvent mixture (e.g., methanol, ethanol or methanol/THF).

NOTE ….METHYL SUBSTITUENT ON 10 WILL GIVE YOU PANOBINOSTAT

……………………………….
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide
lactate
(34, panobinostat, LBH589)
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
for str see above link
α-methyl-β-(β-bromoethyl)indole (29) was made according to method reported by Grandberg et al.(2. Grandberg, I. I.; Kost, A. N.; Terent’ev, A. P. Reactions of hydrazine derivatives. XVII. New synthesis of α-methyltryptophol. Zhurnal Obshchei Khimii 1957, 27, 3342–3345. )
The bromide 29 was converted to amine 30 by using similar method used by Sletzinger et al.(3. Sletzinger, M.; Ruyle, W. V.; Waiter, A. G. (Merck & Co., Inc.). Preparation of tryptamine
derivatives. U.S. Patent US 2,995,566, Aug 8, 1961.)
To a 500 mL flask, crude 2-methyltryptamine 30 (HPLC purity 75%, 1.74 g, 7.29 mmol) and 3-(4-
formyl-phenyl)-acrylic acid methyl ester 31 (HPLC purity 84%, 1.65 g, 7.28 mmol) were added,
followed by DCM (100 mL) and MeOH (30 mL). The clear solution was stirred at room temp for 30
min, then NaBH3CN (0.439 g, 6.99 mmol) was added in small portions. The reaction mixture was
stirred at room temp overnight. After removal of the solvents, the residue was diluted with DCM and
added saturated NaHCO3 aqueous solution, extracted with DCM twice. The DCM layer was dried
and concentrated, and the resulting residue was purified by flash chromatography (silica, 0–10%
MeOH in DCM) to afford 33 as orange solid (1.52 g, 60%). LC–MS m/z 349.2 ([M + H]+). 33 was
converted to hydroxamic acid 34 according to procedure D (Experimental Section), and the freebase
34 was treated with 1 equiv of lactic acid in MeOH–water (7:3) to form lactic acid salt which was
further recrystallized in MeOH–EtOAc to afford the lactic acid salt of 34as pale yellow solid. LC–MS m/z 350.2 ([M + H − lactate]+).
= DELTA
1H NMR (DMSO-d6) 10.72 (s, 1H, NH), 7.54 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 16 Hz, 1H), 7.43 (d, J = 7.8 Hz, 2H), 7.38 (d, J = 7.6 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 6.97 (td, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d, J = 7.8Hz, 2H), 7.01 (t, J = 7.4, 0.9 Hz, 1H), 6.91 (td, J = 7.4, 0.9 Hz, 1H), 6.47 (d, J = 15.2 Hz, 1H), 3.94(q, J = 6.8 Hz, 1H, lactate CH), 3.92 (s, 2H), 2.88 and 2.81 (m, each, 4H, AB system, CH2CH2),2.31 (s, 3H), 1.21 (d, J = 6.8 Hz, 3H).;
13C NMR (DMSO-d6) 176.7 (lactate C=O), 162.7, 139.0,
137.9, 135.2, 134.0, 132.1, 129.1, 128.1, 127.4, 119.9, 119.0, 118.1, 117.2, 110.4, 107.0, 66.0, 51.3,
48.5, 22.9, 20.7, 11.2.
…………………………………………..
PANOBINOSTAT DRUG SUBSTANCE SYNTHESIS AND DATA
http://www.google.com/patents/US7989639

A flow diagram for the synthesis of LBH589 lactate is provided in FIG. A. A nomenclature reference index of the intermediates is provided below in the Nomenclature Reference Index:
| Nomenclature reference index | |
| Compound | Chemical name |
| 1 | 4-Bromo-benzaldehyde |
| 2 | Methyl acrylate |
| 3 | (2E)-3-(formylphenyl)-2-propenoic acid, methyl ester |
| 4 | 3-[4-[[[2-(2-Methyl-1H-indol-3- |
| yl)ethyl]amino]methyl]phenyl]-2- | |
| propenoic acid, methyl ester, monohydrochloride | |
| 5 | (2E)-N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3- |
| yl)ethyl]amino]methyl]phenyl]-2-propenamide | |
| 6 | 2-hydroxypropanoic acid, compd. with 2(E)-N- |
| hydroxy-3-[4-[[[2-(2-methyl-1H- | |
| indol-3-yl)ethyl]amino]methyl]phenyl]-2-propenamide | |
| Z3a | 2-Methyl-1H-indole-3-ethanamine |
| Z3b | 5-Chloro-2-pentanone |
| Z3c | Phenylhydrazine |
The manufacture of LBH589 lactate (6) drug substance is via a convergent synthesis; the point of convergence is the condensation of indole-amine Z3a with aldehyde 3.
The synthesis of indole-amine Z3a involves reaction of 5-chloro-2 pentanone (Z3b) with phenylhydrazine (Z3c) in ethanol at reflux (variation of Fischer indole synthesis).
Product isolation is by an extractive work-up followed by crystallization. Preparation of aldehyde 3 is by palladium catalyzed vinylation (Heck-type reaction; Pd(OAc)2/P(o-Tol)3/Bu3N in refluxing CH3CN) of 4-bromo-benzyladehyde (1) with methyl acrylate (2) with product isolation via precipitation from dilute HCl solution. Intermediates Z3a and 3 are then condensed to an imine intermediate, which is reduced using sodium borohydride in methanol below 0° C. (reductive amination). The product indole-ester 4, isolated by precipitation from dilute HCl, is recrystallized from methanol/water, if necessary. The indole ester 4 is converted to crude LBH589 free base 5 via reaction with hydroxylamine and sodium hydroxide in water/methanol below 0° C. The crude LBH589 free base 5 is then purified by recrystallization from hot ethanol/water, if necessary. LBH589 free base 5 is treated with 85% aqueous racemic lactic acid and water at ambient temperature. After seeding, the mixture is heated to approximately 65° C., stirred at this temperature and slowly cooled to 45-50° C. The resulting slurry is filtered and washed with water and dried to afford LBH589 lactate (6).
If necessary the LBH589 lactate 6 may be recrystallised once again from water in the presence of 30 mol % racemic lactic acid. Finally the LBH589 lactate is delumped to give the drug substance. If a rework of the LBH589 lactate drug substance 6 is required, the LBH589 lactate salt is treated with sodium hydroxide in ethanol/water to liberate the LBH589 free base 5 followed by lactate salt formation and delumping as described above.
All starting materials, reagents and solvents used in the synthesis of LBH589 lactate are tested according to internal specifications or are purchased from established suppliers against a certificate of analysis.
EXAMPLE 7 Formation of Monohydrate Lactate Salt
About 40 to 50 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base was suspended in 1 ml of a solvent as listed in Table 7. A stoichiometric amount of lactic acid was subsequently added to the suspension. The mixture was stirred at ambient temperature and when a clear solution formed, stirring continued at 4° C. Solids were collected by filtration and analyzed by XRPD, TGA and 1H-NMR.
| TABLE 7 | |||||
| LOD, % | |||||
| Physical | Crystallinity | (Tdesolvation) | |||
| Solvent | T, ° C. | Appear. | and Form | Tdecomposit. | 1H-NMR |
| IPA | 4 | FFP | excellent | 4.3 (79.3) | — |
| HA | 156.3 | ||||
| Acetone | 4 | FFP | excellent | 4.5 (77.8) | 4.18 (Hbz) |
| HA | 149.5 | ||||
The salt forming reaction in isopropyl alcohol and acetone at 4° C. produced a stoichiometric (1:1) lactate salt, a monohydrate. The salt is crystalline, begins to dehydrate above 77° C., and decomposes above 150° C.
EXAMPLE 18 Formation of Anhydrous Lactate Salt
DL-lactic acid (4.0 g, 85% solution in water, corresponding to 3.4 g pure DL-lactic acid) is diluted with water (27.2 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution is allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base (10.0 g) is placed in a 4-necked reaction flask with mechanical stirrer. Demineralized water (110.5 g) is added, and the suspension is heated to 65° C. (inner temperature) within 30 minutes. The DL-lactic acid solution is added to this suspension during 30 min at 65° C. During the addition of the lactate salt solution, the suspension converted into a solution. The addition funnel is rinsed with demineralized water (9.1 g), and the solution is stirred at 65° C. for an additional 30 minutes. The solution is cooled down to 45° C. (inner temperature) and seed crystals (10 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate monohydrate) are added at this temperature. The suspension is cooled down to 33° C. and is stirred for additional 20 hours at this temperature. The suspension is re-heated to 65° C., stirred for 1 hour at this temperature and is cooled to 33° C. within 1 hour. After additional stirring for 3 hours at 33° C., the product is isolated by filtration, and the filter cake is washed with demineralized water (2×20 g). The wet filter-cake is dried in vacuo at 50° C. to obtain the anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt as a crystalline product. The product is identical to the monohydrate salt (form HA) in HPLC and in 1H-NMR, with the exception of the integrals of water signals in the 1H-NMR spectra.
In additional salt formation experiments carried out according to the procedure described above, the product solution was filtered at 65° C. before cooling to 45° C., seeding and crystallization. In all cases, form A (anhydrate form) was obtained as product.
EXAMPLE 19 Formation of Anhydrous Lactate Salt
DL-lactic acid (2.0 g, 85% solution in water, corresponding to 1.7 g pure DL-lactic acid) is diluted with water (13.6 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution was allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base (5.0 g) is placed in a 4-necked reaction flask with mechanical stirrer. Demineralized water (54.85 g) is added, and the suspension is heated to 48° C. (inner temperature) within 30 minutes. The DL-lactic acid solution is added to this suspension during 30 minutes at 48° C. A solution is formed. Seed crystals are added (as a suspension of 5 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form A, in 0.25 g of water) and stirring is continued for 2 additional hours at 48° C. The temperature is raised to 65° C. (inner temperature) within 30 minutes, and the suspension is stirred for additional 2.5 hours at this temperature. Then the temperature is cooled down to 48° C. within 2 hours, and stirring is continued at this temperature for additional 22 hours. The product is isolated by filtration and the filter cake is washed with demineralized water (2×10 g). The wet filter-cake is dried in vacuo at 50° C. to obtain anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A) as a crystalline product.
EXAMPLE 20 Conversion of Monohydrate Lactate Salt to Anhydrous Lactate Salt
DL-lactic acid (0.59 g, 85% solution in water, corresponding to 0.5 g pure DL-lactic acid) is diluted with water (4.1 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution is allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
10 g of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt monohydrate is placed in a 4-necked reaction flask. Water (110.9 g) is added, followed by the addition of the lactic acid solution. The addition funnel of the lactic acid is rinsed with water (15.65 g). The suspension is heated to 82° C. (inner temperature) to obtain a solution. The solution is stirred for 15 minutes at 82° C. and is hot filtered into another reaction flask to obtain a clear solution. The temperature is cooled down to 50° C., and seed crystals are added (as a suspension of 10 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form, in 0.5 g of water). The temperature is cooled down to 33° C. and stirring is continued for additional 19 hours at this temperature. The formed suspension is heated again to 65° C. (inner temperature) within 45 minutes, stirred at 65° C. for 1 hour and cooled down to 33° C. within 1 hour. After stirring at 33° C. for additional 3 hours, the product is isolated by filtration and the wet filter cake is washed with water (50 g). The product is dried in vacuo at 50° C. to obtain crystalline anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A).
EXAMPLE 21 Formation of Anhydrous Lactate Salt
DL-lactic acid (8.0 g, 85% solution in water, corresponding to 6.8 g pure DL-lactic acid) was diluted with water (54.4 g), and the solution was heated to 90° C. (inner temperature) for 15 hours. The solution was allowed to cool down to room temperature and was used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (20 g) is placed in a 1 L glass reactor, and ethanol/water (209.4 g of a 1:1 w/w mixture) is added. The light yellow suspension is heated to 60° C. (inner temperature) within 30 minutes, and the lactic acid solution is added during 30 minutes at this temperature. The addition funnel is rinsed with water (10 g). The solution is cooled to 38° C. within 2 hours, and seed crystals (20 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form) are added at 38° C. After stirring at 38° C. for additional 2 hours, the mixture is cooled down to 25° C. within 6 hours. Cooling is continued from 25° C. to 10° C. within 5 hours, from 10° C. to 5° C. within 4 hours and from 5° C. to 2° C. within 1 hour. The suspension is stirred for additional 2 hours at 2° C., and the product is isolated by filtration. The wet filter cake is washed with water (2×30 g), and the product is dried in vacuo at 45° C. to obtain crystalline anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A).
EXAMPLE 28 Formation of Lactate Monohydrate Salt
3.67 g (10 mmol) of the free base monohydrate (N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide) and 75 ml of acetone were charged in a 250 ml 3-neck flask equipped with a magnetic stirrer and an addition funnel. To the stirred suspension were added dropwise 10 ml of 1 M lactic acid in water (10 mmol) dissolved in 20 ml acetone, affording a clear solution. Stirring continued at ambient and a white solid precipitated out after approximately 1 hour. The mixture was cooled in an ice bath and stirred for an additional hour. The white solid was recovered by filtration and washed once with cold acetone (15 ml). It was subsequently dried under vacuum to yield 3.94 g of the lactate monohydrate salt of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (86.2%).

References
- Revill, P; Mealy, N; Serradell, N; Bolos, J; Rosa, E (2007). “Panobinostat”. Drugs of the Future 32 (4): 315. doi:10.1358/dof.2007.032.04.1094476. ISSN 0377-8282.
- Table 3: Select epigenetic inhibitors in various stages of development from Mack, G. S. (2010). “To selectivity and beyond”. Nature Biotechnology 28 (12): 1259–1266.doi:10.1038/nbt.1724. PMID 21139608. edit
- ClinicalTrials.gov NCT00425555 Study of Oral LBH589 in Adult Patients With Refractory Cutaneous T-Cell Lymphoma
- ClinicalTrials.gov: LBH-589
- Prince, HM; M Bishton (2009). “Panobinostat (LBH589): a novel pan-deacetylase inhibitor with activity in T cell lymphoma”. Hematology Meeting Reports (Parkville, Australia: Peter MacCallum Cancer Centre and University of Melbourne) 3 (1): 33–38.
- Simons, J (27 April 2013). “Scientists on brink of HIV cure”. The Telegraph.
- ClinicalTrials.gov NCT01680094 Safety and Effect of The HDAC Inhibitor Panobinostat on HIV-1 Expression in Patients on Suppressive HAART (CLEAR)
- Mayo Clinic Researchers Formulate Treatment Combination Lethal To Pancreatic Cancer Cells
- Garbes, L; Riessland, M; Hölker, I; Heller, R; Hauke, J; Tränkle, Ch; Coras, R; Blümcke, I; Hahnen, E; Wirth, B (2009). “LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate”. Human Molecular Genetics 18 (19): 3645–3658. doi:10.1093/hmg/ddp313.PMID 19584083.
- Tate, CR; Rhodes, LV; Segar, HC; Driver, JL; Pounder, FN; Burow, ME; and Collins-Burow, BM (2012). “Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat”. Breast Cancer Research 14 (3).
- TA Rasmussen, et al. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Human Vaccines & Immunotherapeutics 9:5, 1-9, May 2013.
- Drugs of the Future 32(4): 315-322 (2007)
- WO 2002022577…
- WO 2007146718
- WO 2013110280
- WO 2010009285
- WO 2010009280
- WO 2005013958
- WO 2004103358
- WO 2003048774…
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
-
23009203 11-26-2012 Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. Journal of medicinal chemistry -
21634430 7-14-2011 Discovery of (2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an orally active histone deacetylase inhibitor with a superior preclinical profile. Journal of medicinal chemistry -
21417419 4-28-2011 Discovery, synthesis, and pharmacological evaluation of spiropiperidine hydroxamic acid based derivatives as structurally novel histone deacetylase (HDAC) inhibitors. Journal of medicinal chemistry -
19317450 4-23-2009 Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. Journal of medicinal chemistry -
15650931 1-1-2005 The American Society of Hematology–46th Annual Meeting and Exposition. HDAC, Flt and farnesyl transferase inhibitors. IDrugs : the investigational drugs journal -
US7989639 8-3-2011 PROCESS FOR MAKING SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2010286409 11-12-2010 SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2010179208 7-16-2010 Use of HDAC Inhibitors for the Treatment of Bone Destruction US2010160257 6-25-2010 USE OF HDAC INHIBITORS FOR THE TREATMENT OF MYELOMA US2010137398 6-4-2010 USE OF HDAC INHIBITORS FOR THE TREATMENT OF GASTROINTESTINAL CANCERS US2009306405 12-11-2009 PROCESS FOR MAKING N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE AND STARTING MATERIALS THEREFOR US2009281159 11-13-2009 USE OF HDAC INHIBITORS FOR THE TREATMENT OF LYMPHOMAS US2009264439 10-23-2009 Combination of a) N–4-(3-pyridyl)-2-pyrimidine-amine and b) a histone deacetylase inhibitor for the treatment of leukemia US2009197936 8-7-2009 SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2009012066 1-9-2009 Method of Use of Deacetylase Inhibitors
| US2008319045 | 12-26-2008 | Combination of Histone Deacetylase Inhibitors and Radiation |
| US2008221126 | 9-12-2008 | Use of Hdac Inhibitors for the Treatment of Myeloma |
| US2008176849 | 7-25-2008 | DEACETYLASE INHIBITORS |
| US2006189674 | 8-25-2006 | Deacetylase inhibitors |
| US7067551 | 6-28-2006 | Deacetylase inhibitors |
| US2006100140 | 5-12-2006 | Combination of a) n-{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]2-methylphenyl}-4- (3-pyridyl)-2-pyrimidine-amine and b) a histone deacetylase inhibitor for the treatment of leukemia |
| US6833384 | 12-22-2004 | Deacetylase inhibitors |
| US6552065 | 4-23-2003 | Deacetylase inhibitors |
| GB776693A | Title not available | |||
| GB891413A | Title not available | |||
| GB2185020A | Title not available | |||
| WO2002022577A2 | Aug 30, 2001 | Mar 21, 2002 | Kenneth Walter Bair | Hydroxamate derivatives useful as deacetylase inhibitors |
| WO2003016307A1 | Aug 6, 2002 | Aug 19, 1993 | Jolie Anne Bastian | β3 ADRENERGIC AGONISTS |
| WO2003039599A1 | Nov 5, 2002 | May 15, 2003 | Ying-Nan Pan Chen | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| WO2005105740A2 | Apr 26, 2005 | Nov 10, 2005 | Serguei Fine | Preparation of tegaserod and tegaserod maleate |
| WO2006021397A1 | Aug 22, 2005 | Mar 2, 2006 | Recordati Ireland Ltd | Lercanidipine salts |
…………………………………..
extras
5. Mocetinostat (MGCD0103), including pharmaceutically acceptable salts thereof. Balasubramanian et al., Cancer Letters 280: 211-221 (2009).
Mocetinostat, has the following chemical structure and name:

Vorinostat, including pharmaceutically acceptable salts thereof. Marks et al., Nature Biotechnology 25, 84 to 90 (2007); Stenger, Community Oncology 4, 384-386 (2007).
Vorinostat has the following chemical structure and name:

Belinostat (PXD-101 , PX-105684)
(2E)-3-[3-(anilinosulfonyl)phenyl]-N-hydroxyacrylamide

……………………………………………….
Dacinostat (LAQ-824, NVP-LAQ824,)
((E)-N-hydroxy-3-[4-[[2-hydroxyethyl-[2-(1 H-indol-3-yl)ethyl]amino]methyl]phenyl]prop-2-enamide

Entinostat (MS-275, SNDX-275, MS-27-275)
4-(2-aminophenylcarbamoyl)benzylcarbamate

(a) The HDAC inhibitor Vorinostat™ or a salt, hydrate, or solvate thereof.

Vorinostat………………..
(b) The HDAC inhibitor Givinostat or a salt, hydrate, or solvate thereof.

Givinostat or a salt, hydrate, or solvate thereof.
CLAZOSENTAN

Clazosentan
READ ALL AT
http://www.allfordrugs.com/2014/01/22/clazosentan/

READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
Tezosentan Disodium for pulmonary hypertension

TEZOSENTAN
180384-57-0 CAS OF FREE ACID
N-[6-(2-Hydroxyethoxy)-5-(2-methoxyphenoxy)-2-[2-(2H-tetrazol-5-yl)pyridin-4-yl]pyrimidin-4-yl]-5-propan-2-ylpyridine-2-sulfonamide
5-isopropyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5- (2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide
| Formula | C27H27N9O6S |
|---|---|
| Mol. mass | 605.624 |
…………………………………………………………………………….

Tezosentan disodium, Ro-61-0612, Veletri
5-isopropyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide sodium salt (1:2)
180384-58-1 of disodium salt, 180384-57-0 (free acid)

Tezosentan is a non-selective ETA and ETB receptor antagonist.[1] It acts as a vasodilator and was designed as a therapy for patients with acuteheart failure. Recent studies have shown however, that tezosentan does not improve dyspnea or reduce the risk of fatal or nonfatal cardiovascular events.[2]
Pulmonary disease (COPD), which may possibly be associated with pulmonary hypertension, as well as allergic and non-allergic rhinitis, provided that treatment with endothelin from a therapeutic standpoint is not contraindicated.
Tezosentan disodium is an endothelin ETB receptor antagonist in phase II clinical development for the treatment of stable, chronic pulmonary arterial hypertension. The drug was previously being evaluated for heart failure, but trials in that indication have been discontinued. The compound is being developed by Actelion.
………………………………..
SYNTHESIS

………………………………..
SYNTHESIS

Reaction of 4-cyano-pyridine (I) with Na in methanol followed by treatment with ammonium chloride provides 4-amidino-pyridine hydrochloride (II), which is then converted into 5-(2-methoxyphenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (IV) by condensation with diethyl malonate derivative (III) by means of Na in MeOH. By heating compound (IV) with phosphorus oxychloride (POCl3), 4,6-dichloro-5-(2-methoxyphenoxy)-2-pyridin-4-yl)pyrimidine (V) is obtained, which in turn is oxidized with peracetic acid in refluxing acetonitrile to afford N-oxide derivative (VI). Condensation of (VI) with 5-isopropylpyridine-2-sulfonamide potassium (VII) furnishes 5-isopropylpyridine-2-sulfonic acid 6-chloro-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl amide (VIII), which is then dissolved in dimethoxyethane and subjected to reaction with Na in hot ethylene glycol (IX) to provide N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl]-5-isopropylpyridine-2-sulfonamide (X). Refluxing of (X) with trimethylsilylcyanide and Et3N in acetonitrile yields cyano derivative (XI), which is then converted into the tetrazole derivative (XII) by reaction with sodium azide and NH4Cl in DMF at 70 C. Finally, the disodium salt of tezosentan is obtained by treatment of (XII) with Na/MeOH in THF. refEP 0799209; JP 1998509182; WO 9619459
…………………………………..
SYNTHESIS PROCEDURE as in EP0979822A1
Examples
- 1360 ml of formamide were added to 136 g (437 mmol) of 5-(2-methoxy-phenoxy)-2-pyridine-4-yl-pyrimidine-4,6-diole. Then, at a temperature of 0°C, 11.7 ml (219 mmol) of concentrated sulfuric acid and thereafter 36.5 g (130 mmol) of iron(II)sulfate heptahydrate were added to the suspension. After that, 89 ml (874 mmol) of 30% hydrogen peroxide were added dropwise within 1 hr at a temperature of 0°C to 5°C. The viscous yellow-brownish suspension was stirred at 0°C for 1.5 hr. Subsequently, a solution of 83 g (437 mmol) of sodium pyrosulfite in 680 ml of de-ionized water was added dropwise to the reaction mixture within 30 min. at 0°C to 5°C and the reaction mixture was stirred at 0°C to 5°C for 30 min. The suspension was then filtered under reduced pressure. The filtrate was first washed with 1750 ml of de-ionized water and thereafter with 700 ml of ethanol. Then the solid was dried at 80°C, 2000 Pa for 16 hr. There were obtained 132.4 g (91% of theory) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide with a HPLC purity of 91.4% (w/w).
-
Preparation of starting material:
- a) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hr 29.5 g of NH4Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydrochloride (decomposition point 245-247°C).
- b) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 min. to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in a) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridine-4-yl)-pyrimidine-4,6-diole (or tautomer), melting point above 250°C.
- Example 1
Example 2
- Within 20 min. 61 ml (633 mmol) of POCl3 were added dropwise to 34 ml (200 mmol) of diisopropyl ethylamine at 5°C to 10°C followed by stirring at 5°C to 10°C for 15 min. Then 23.5 g (66 mmol) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide were added in four portions under cooling followed by stirring at 90°C for 25 hr. The reaction mixture was cooled down to 20°C and transferred to a new flask together with 50 ml of dichloromethane. Volatile components (i.e. excess of POCl3) was removed by evaporation from 20°C to 70°C followed by re-distillation with 100 ml of toluene. After adding 250 ml of dichloromethane to the residue (88 g of a black oil) the solution was heated to 35°C to 40°C and 80 ml of de-ionized water were added dropwise within 30 min. whereby the pH was kept constant by the subsequent addition of 28% NaOH solution (60 ml) within 5 to 6 hr. The mixture was stirred at 35°C to 40°C for 30 min. followed by removal of dichloromethane by distillation. The resulting suspension was allowed to cool down to 20°C and was stirred for additional 2 hr. The solid was filtered off under suction, washed with 500 ml of water and dried at 70°C, 2000 Pa for 16 hr. There were obtained 21.3 g (86% of theory) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile with a HPLC purity of 94.3% (w/w).
- 8.95 g (24 mmol) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile were suspended in 100 ml of acetone. At a temperature of 20°C, 5.04 g (25 mmol) of 5-isopropyl-pyridine-2-sulfonamide, 1 ml of de-ionized water, 10.6 g (77 mmol) of potassium carbonate and 135 mg (1.2 mmol) 1,4-diazobicyclo[2.2.2]octane were added. The mixture was stirred at 40°C for 20 hr. Thereafter, another 240 mg (1.2 mmol) of 5-isopropyl-pyridine-2-sulfonamide and 80 mg (0.7 mmol) of 1,4-diazobicyclo[2.2.2]octane were added. The reaction mixture was stirred for 24 hr at 40°C followed by cooling to 20°C. Then 50 ml of de-ionized water and 45 ml of 3 N aqueous hydrochloric acid were added slowly until pH = 1. The acetone was removed by distillation and the resulting suspension was stirred at 20°C for 1.5 hr. The solid was filtered off under suction, washed first with 100 ml of de-ionized water and thereafter with 50 ml of t-butylmethylether. Then the solid was dried at 70°C, 2000 Pa for 20 hr. There were obtained 13.2 g (102% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 87.8% (w/w).
- Example 4
- 122 g (233 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide was suspended in 450 ml of N,N-dimethyl formamide and the mixture was cooled down to 15°C. At this temperature, 35 ml of hydrazine hydrate were added dropwise within 1 hr. The resulting solution was stirred at 15°C to 20°C for 16 hr and thereafter diluted with 600 ml of de-ionized water. Then 50 ml of glacial acetic acid were added dropwise at 0°C to 5°C until pH = 5.5. 600 g of ice were added and the suspension was stirred for 1 hr. The solid was filtered off under suction, washed with 3000 ml of water and dried at 40°C, 2000 Pa for 24 hr. There were obtained 126 g (97% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 91.8% (w/w).
- Example 6
- 20 g (35 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide were added to 160 ml of N,N-dimethyl formamide. The solution was kept at 15°C to 20°C and 23 ml of 6 N aqueous hydrochloric acid were added, followed by addition of a solution containing 4.8 g (7 mmol) of sodium nitrite in 20 ml de-ionized water within 10 min. The mixture was stirred at 20°C for 1 hr, then 140 ml of de-ionized water were added and the suspension was stirred at 0°C for 1 hr. The solid was filtered, firstly washed with 80 ml of de-ionized water and thereafter with 80 ml of t-butylmethylether. Then the solid was dried at 70°C and 2000 Pa for 16 hr. The crude product (23.4 g) was taken up with 117 ml of tetrahydrofuran for 1 hr. After filtration at 0°C the crystallized product was washed with 25 ml of t-butylmethylether and was then dried at 70°C, 2000 Pa for 16 hr. There were obtained 17.3 g (84% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 91.1% (w/w).
- Example 8
- Example 10
- 6.2 g of sodium hydroxide were added to 15 g (26 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amid and 75 ml of ethylene glycol. The mixture was heated to 85°C for 5 hr. Then 55 ml of de-ionized water were added and thereafter 55 ml of 3 N hydrochloric acid were added dropwise. The mixture was allowed to cool down to 20°C and was stirred for 1 hr. The solid was filtered off and dried at 70°C, 2000 Pa for 18 hr. There were obtained 16.2 g (103%) of 5-isopropyl-pyridine-2-sulfonic acid 16-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 92% (w/w). 80 ml of dioxane and 80 ml of ethanol were added to this solid. At a temperature of 60°C, gaseous ammonia was introduced into the liquid until pH = 9 to 10. The resulting suspension was allowed to cool down to 20°C and was stirred at 20°C for 20 hr and thereafter at 0°C for 2.5 hr. Then the solid was filtered off and dried at 70°C, 2000 Pa for 18 hr. There were obtained 14.2 g of mono ammonium salt with a HPLC purity of 96.2% (w/w). The solid was heated (reflux) in 70 ml of methanol, cooled down slowly to 20°C and stirred at 20°C for 19 hr and thereafter at 0°C for 2 hr. Then the solid was filtered off and dried at 70°C, 2000 Pa for 19 hr. There were obtained 11.5 g (66% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide sodium salt (1:2) with a HPLC purity of 98.6% (w/w).

Reaction of 2-chloro-5-ispropylpyridine (VII) with thiourea (A) in aqueous HCl gives 5-isopropyl- pyridine-2-thiol (VIII), which is chlorinated with chlorine in acetic acid to yield 5-isopropylpyridine-2-sulfochloride (IX). This compound is converted into 5-isopropylpyridine-2-sulfonamide potassium salt (X).
…………………………
synthesis
. Example 1
a) 200 ml of dimethoxyethane and 1 10.9 g of 4-[4-(4-tert- butyl-phenyl-sulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide are added all at once to a solution of 23.80 g of sodium in 660 ml of ethylene glycol. The solution is heated at 90°C for 20 hours while stirring, thereafter cooled, poured into 2500 ml of H2O and thereafter treated with CH3COOH to pH 5. The mixture is extracted three times with EtOAc, the organic phase is washed with H2O, dried with Na2Sθ4 and evaporated under reduced pressure. The residue is recrystall- ized from CH3CN and thereafter twice from a mixture of acetone and CH3CN. There is thus obtained 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide.
Preparation of the starting material:
b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After
6 hours 29.5 g of NH4CI are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydro- chloride (decomposition point 245-247°C).
c) 1 12.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydro- chloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)- pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
d) A suspension of 1 54.6 g of 5-(2-methoxy-phenoxy)-2- (pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCI3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2CI2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2CI2. The combined CH2CI2 extracts are dried with MgSθ4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2CI2 remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)- pyrimidine, melting point 1 78-1 80°C.
e) A solution of 1 7.4 g of 4,6-dichloro-5-(2-methoxy- phenoxy)-2-pyridin-4-yl)-pyrimidine in 100 ml of CH3CN is boiled at reflux for 3 hours with 1 5 ml of a 32% peracetic acid solution, thereafter cooled and stored in a refrigerator overnight. The crystals are filtered off under suction and dried at 50°C under reduced pressure. There is thus obtained 4-[4,6-dichloro- 5-(2-methoxy-phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide, melting point 189-1 90°C.
f) A solution of 36.4 g of 4-[4,6-dichloro-5-(2-methoxy- phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide and 52.8 g of p-tert- butylphenyl-sulphonamide potassium in 1 50 ml of abs. DMF is stirred at room temperature for 24 hours. Thereafter, it is poured into a mixture of 1 500 ml of H2O and 1000 ml of ether while stirring mechanically, whereby a precipitate forms. The suspension is adjusted to pH 5 with CH3COOH, suction filtered, the crystals are washed with cold water and thereafter with ether and dried at 50°C. There is thus obtained 4-[4-(4-tert- butyl-phenylsulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide as a colourless material of melting point 247-249°C.
Example 2
A solution of 78.45 g of 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide, 122.5 g of trimethylsilyl cyanide, 127.8 g of triethylamine and 1200 ml of CH3CN is boiled at reflux for 20 hours and thereafter evaporated under reduced pressure. The oily residue is taken up in 1000 ml of EtOAc and the solution is washed with CH3COOH:H2θ 9:1 and then with H2O. The EtOAc extracts are dried with Na2SO4. After evaporation of the solvent the residue is taken up in a mixture of CH3CN and CF3COOH (20:1 ), whereby a crystalline precipitate separates. There is thus obtained 4-tert-butyl-N-[2-(2-cyano-pyridin-4- yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4- yl]-benzenesulphonamide of melting point 176-1 79°C.
Example 3 for analogy only compd is different
A suspension of 50.0 g of 4-tert-butyl-N-[2-(2-cyano- pyridin-4-yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-4-yl]-benzenesulphonamide, 46.33 g of NH4CI and 56.47 g of NaN3 in 1600 ml of DMF is heated to 70°C for 24 hours while stirring vigorously. The majority of the solvent is distilled off under reduced pressure, the residue is dissolved in H2O, the solution is extracted four times at pH 6.5 with ether, thereafter treated with CH3COOH to pH = 4.5 and extracted with EtOAc. After working up there is obtained a residue which is treated with ether and filtered off under suction therefrom. There is thus obtained 4-tert-butyl-N-[6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-yl]-benzenesulphonamide, melting point 225-227°C.
Example 30 final product
In analogy to Example 3, from 5-isopropyl-pyridine-2- sulphonic acid 2-(2-cyano-pyridin-4-yl)-6-(2-hydroxy-ethoxy)- 5-(2-methoxy-phenoxy)-pyrimidin-4-ylamide there is obtained 5-isopropyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5- (2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide (tezosantan free base) as a white substance of melting point 1 98- 200°C from acetonitrile.
The corresponding disodium salt (tezosantan di sodium salt) is obtained as a white powder from this product using sodium methylate in analogy to Example 5
Example 5 for analogy only, compd is different
A solution of 47.8 g of 2-[6-(4-tert-butyl-phenylsulphonyl- amino)-5-(2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin- 4-yl)-pyrimidin-4-yloxy]-ethyl pyridin-2-ylcarbamate in 500 ml of abs. THF is treated dropwise with a cold solution of 2.8 g of sodium in 50 ml of methanol, whereby there forms gradually a solid precipitate which, after stirring at room temperature for 1 hour, is filtered off under suction, dried under greatly reduced pressure at 35°C for 3 days and thereafter at 50°C for 2 days. There is thus obtained the bis-sodium salt, decomposition point above 250°C.
References
- Urbanowicz, W; Sogni, P, Moreau, R, Tazi, K A, Barriere, E, Poirel, O, Martin, A, Guimont, M C, Cazals-Hatem, D, Lebrec, D (2004). “Tezosentan, an endothelin receptor antagonist, limits liver injury in endotoxin challenged cirrhotic rats”. Gut (BMJ Publishing Group Ltd & British Society of Gastroenterology) 53 (12): 1844–1849. doi:10.1136/gut.2003.036517. PMC 1774327. PMID 15542526.
- “Tezosentan does not appear to improve symptoms for patients with acute heart failure”. Medical Studies/Trials. news-medical.net. 7 Nov 2007. Retrieved 2007-11-24.
4 US2003/100507 A1
5 Drugs Fut 2003,28(8),754
6 WO 1996019459……
7 EP 0897914
8 WO 2011163085
9 WO 2004082637
| 10 WO 2002074034 |
11…
| 15055997 | 4-8-2004 | Discovery, modeling, and human pharmacokinetics of N-(2-acetyl-4,6-dimethylphenyl)-3-(3,4-dimethylisoxazol-5-ylsulfamoyl)thiophene-2-carboxamide (TBC3711), a second generation, ETA selective, and orally bioavailable endothelin antagonist. | Journal of medicinal chemistry |
12 ..
| 10610277 | 7-1-1999 | RO 610612 . | Drugs in R&D |
13….
| 3-27-2003 | Aqueous pharmaceutical composition comprising Tezosentan | |
| US6103902 | 8-16-2000 | Carbamoylation process |
| WO0036918 | 6-30-2000 | METHODS AND COMPOSITIONS FOR TREATMENT OF CELL PROLIFERATIVE DISORDERS METHODS AND COMPOSITIONS FOR TREATMENT OF CELL PROLIFERATIVE DISORDERS |
| US6063911 | 5-17-2000 | Methods and compositions for treatment of cell proliferative disorders |
READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
PSC 833 ( Valspodar )

Valspodar, SDZ-PSC-833, PSC-833, Amdray
P-Glycoprotein (MDR-1; ABCB1) Inhibitors , Multidrug Resistance Modulators
Valspodar is a cyclosporine derivative and a P-glycoprotein inhibitor currently in phase III clinical trials at the National Cancer Institute (NCI) in combination with chemotherapy for the treatment of leukemia. The drug was also being developed in combination with chemotherapy for the treatment of various other types of cancers, however, no recent developments on these trials have been reported.
P-glycoprotein is an ABC-transporter protein that has been implicated in conferring multidrug resistance to tumor cells. In previous trials, valspodar was associated with greater disease-free and overall survival in younger patients (45 years or below), and was shown to significantly increase the cellular uptake of daunorubicin in leukemic blast cells in vivo. However, in a phase III trial examining the drug candidate’s effects on AML in patients at least 60 years of age, valspodar was associated with excessive mortality and complete remission rates were higher in groups not treated with the compound.
Nonimmunosuppressive cyclosporin analog which is a potent multidrug resistance modifier; 7-10 fold more potent than cyclosporin A; a potent P glycoprotein inhibitor; MW 1215.
M.Wt: 1214.62
Formula: C63H111N11O12
CAS : 121584-18-7
IUPAC/Chemical name:
(3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-6,9,18,24-tetraisobutyl-3,21,30-triisopropyl-1,4,7,10,12,15,19,25,28-nonamethyl-33-((R,E)-2-methylhex-4-enoyl)-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontan-2,5,8,11,14,17,20,23,26,29,32-undecaone
6 – [(2S, 4R, 6E)-4-Methyl-2-(methylamino)-3-oxo-6-octenoic acid]-7-L-valine-cyclosporin A; Cyclo [[(2S, 4R, 6E) -4-methyl-2-(methylamino)-3-oxo-6-octenoyl]-L-valyl-N-methylglycyl-N-methyl-L-leucyl-L-valyl-N-methyl-L-leucyl-L- alanyl-D-alanyl-N-methyl-L-leucyl-Nm
[3′-oxo-4-butenyl-4-methyl-Thr1]-[Val2]-cyclosporine
Clinical trials
http://clinicaltrials.gov/search/intervention=psc+833
Synonyms
- 3′-Keto-bmt(1)-val(2)-cyclosporin A
- Amdray
- Psc 833
- PSC-833
- PSC833
- SDZ PSC 833
- Sdz-psc-833
- UNII-Q7ZP55KF3X
- Valspodar
Valspodar or PSC833 is an experimental cancer treatment and chemosensitizer drug.[1] It is a derivative of ciclosporin D.
Its primary use is that of a p-glycoprotein inhibitor. Previous studies in animal models have found it to be effective at preventing cancer cell resistance to chemotherapeutics, but these findings did not translate to clinical success.[2]
Valspodar, also known as PSC-833 is an analogue of cyclosporin-A. Valspodar inhibits p-glycoprotein, the multidrug resistance efflux pump, thereby restoring the retention and activity of some drugs in some drug-resistant tumor cells. This agent also induces caspase-mediated apoptosis.
PSC-833 is a non-immunosuppressive cyclosporin derivative that potently and specifically inhibits P-gp. In vitro experiments indicate that PSC-833interacts directly with P-gp with high affinity and probably interferes with the ATPase activity of P-gp. Studies in multidrug resistant tumor models confirm P-gp as the in vivo target of PSC-833 and demonstrate the ability of PSC-833 to reverse MDR leukemias and solid tumors in mice. Presently,PSC-833 is being evaluated in the clinic.
Valspodar can cause nerve damage.[1]
Valspodar

Synthesis By oxidation of cyclosporin D (I) with N-chlorosuccinimide and dimethylsulfide in toluene (1) Scheme 1 Description alpha (20, D) -..?. 255.1 (c 0.5, CHCl3) Manufacturer Sandoz Pharmaceuticals Corp (US).. . References 1 Bollinger, P., B flounder sterli, JJ, Borel, J.-F., Krieger, M., Payne, TG, Traber, RP, Wenger, R. (Sandoz AG; Sandoz Patent GmbH; Sandoz Erfindungen VmbH ). Cyclosporins and their use as pharmaceuticals.
AU 8817679, EP 296122, JP 89045396. AU 8817679; EP 0296122; JP 1989045396; JP 1996048696; US 5525590
……………………………..
- The cyclosporins comprise a class of structurally distinctive, cyclic, poly-N-methylated undecapeptides, generally possessing pharmacological, in particular immunosuppressive, anti-inflammatory and/or anti-parasitic activity, each to a greater or lesser degree. The first of the cyclosproins to be isolated was the naturally occurring fungal metabolite Ciclosporin or Cyclosporine, also known as cyclosporin A and now commercially available under the Registered Trade Mark SANDIMMUN®. Ciclosporin is the cyclosporin of formula A

wherein -MeBmt- represents the N-methyl-(4R)-4-but-2E-en-1-yl-4-methyl-(L)threonyl residue of formula B

in which -x-y- is trans -CH=CH- and the positive 2′, 3′ and 4′ have the configuration S, R and R respectively.
-
Since the original discovery of Ciclosporin, a wide variety of naturally occurring cyclosporins have been isolated and identified and many further non-natural cyclosporins have been prepared by total- or semi-synthetic means or by the application of modified culture techniques. The class comprised by the cyclosporins is thus now substantial and includes, for example, the naturally occurring cyclosporins A through Z [c.f. Traber et al. 1, Helv. Chim. Acta, 60, 1247-1255 (1977); Traber et al. 2, Helv. Chim. Acta, 65, 1655-1667 (1982); Kobel et al., Europ. J. Applied Microbiology and Biotechnology 14, 273-240 (1982); and von Wartburg et al. Progress in Allergy, 38, 28-45 (1986)], as well as various non-natural cyclosporin derivatives and artificial or synthetic cyclosporins including the dihydro- and iso-cyclosporins [in which the moiety -x-y- of the -MeBmt- residue (Formula B above) is saturated to give -x-y- = -CH₂-CH₂- / the linkage of the residue -MeBmt- to the residue at the 11-position of the cyclosporin molecule (Formula A above) is via the 3′-O-atom rather than the α-N-atom]; derivatised cyclosporins (e.g. in which the 3′-O-atom of the -MeBmt- residue is acylated or a further substituent is introduced at the α-carbon atom of the sarcosyl residue at the 3-position); cyclosporins in which the -MeBmt- residue is present in isomeric form (e.g. in which the configuration across positions 6′ and 7′ of the -MeBmt- residue is cis rather than trans); and cyclosporins wherein variant amino acids are incorporated at specific positions within the peptide sequence employing e.g. the total synthetic method for the production of cyclosporins developed by R. Wenger – see e.g. Traber et al. 1, Traber et al. 2 and Kobel et al. loc. cit.; U.S. Patents Nos 4 108 985, 4 210 581, 4 220 641, 4 288 431, 4 554 351 and 4 396 542; European Patent Publications Nos. 0 034 567 and 0 056 782; International Patent Publication No. WO 86/02080; Wenger 1, Transpl. Proc. 15, Suppl. 1:2230 (1983); Wenger 2, Angew. Chem. Int. Ed., 24, 77 (1985); and Wenger 3, Progress in the Chemistry of Organic Natural Products 50, 123 (1986).
-
The class comprised by the cyclosporins is thus now very large indeed and includes, for example [Thr]²-, [Val]²-, [Nva]²- and [Nva]²-[Nva]⁵-Ciclosporin (also known as cyclosporins C, D, G and M respectively), [3-O-acetyl-MeBmt]¹-Ciclosporin (also known as cyclosporin A acetate), [Dihydro-MeBmt]¹-[Val]²-Ciclosporin (also known as dihydro-cyclosporin D), [Iso-MeBmt]¹-[Nva]²-Ciclosporin (also known as isocyclosporin G), [(D)Ser]⁸-Ciclosporin, [MeIle]¹¹-Ciclosporin, [(D)MeVal]¹¹-Ciclosporin (also known as cyclosporin H), [MeAla]⁶-Ciclosporin, [(D)Pro]³-Ciclosporin and so on.
-
[In accordance with conventional nomenclature for cyclosporins, these are defined throughout the present specification and claims by reference to the structure of Ciclosporin (i.e. Cyclosporin A). This is done by first indicating the amino acid residues present which differ from those present in Ciclosporin (e.g. “[(D)Pro]³” to indicate that the cyclosporin in question has a -(D)Pro- rather than -Sar- residue at the 3-position) and then applying the term “Ciclosporin” to characterise remaining residues which are identical to those present in Ciclosporin.
-
The residue -MeBmt- at position 1 in Ciclosporin was unknown before the discovery of the cyclosporins. This residue and variants or modifications of it, e.g. as described below, are thus generally characteristic of the cyclosporins. In general, variants or alternatives to [MeBmt]¹ are defined by reference to the -MeBmt- structure. Thus for dihydrocyclosporins in which the moiety -x-y- (see formula B above) is reduced to -CH₂-CH₂-, the residue at the 1-position is defined as “-dihydro-MeBmt-“. Where the configuration across the moiety -x-y- is cis rather than trans, the resulting residue is defined as “-cis-MeBmt-“.
-
Where portions of the -MeBmt- residue are deleted, this is indicated by defining the position of the deletion, employing the qualifier “des” to indicate deletion, and then defining the group or atom omitted, prior to the determinant “-MeBmt-“, “-dihydro-MeBmt-“, “-cis-MeBmt-” etc.. Thus “-N-desmethyl-MeBmt-“, “-3′-desoxy-MeBmt-“, and “-3′-desoxy-4′-desmethyl-MeBmt-” are the residues of Formula B¹, B² and B³ respectively:

B¹ – X = CH₃, Y = OH, Z = H.
B² – X = CH₃, Y = H, Z = CH₃.
B³ – X = H, Y = H, Z = CH₃. -
Where positions or groups, e.g. in -MeBmt-, are substituted this is represented in conventional manner by defining the position and nature of the substitution. Thus -3′-O-acetyl-MeBmt- is the residue of formula B in which the 3′-OH group is acetylated (3′-O-COCH₃). Where substituents of groups, in e.g. -MeBmt-, are replaced, this is done by i) indicating the position of the replaced group by “des-terminology” as described above and ii) defining the replacing group. Thus -7′-desmethyl-7′-phenyl-MeBmt- is the residue of formula B above in which the terminal (8′) methyl group is replaced by phenyl. 3′-Desoxy-3′-oxo-MeBmt- is the residue of formula B above in which the 3′-OH group is replaced by =O.
-
In addition, amino acid residues referred to by abbreviation, e.g. -Ala-, -MeVal-, -αAbu- etc… are, in accordance with conventional practice, to be understood as having the (L)-configuration unless otherwise indicated, e.g. as in the case of “-(D)Ala-“. Residue abbreviations preceded by “Me” as in the case of “-MeLeu-“, represent α-N-methylated residues. Individual residues of the cyclosporin molecule are numbered, as in the art, clockwise and starting with the residue -MeBmt-, -dihydro-MeBmt- etc. … in position 1. The same numerical sequence is employed throughout the present specification and claims.]
-
[0010]Because of their unique pharmaceutical potential, the cyclosporins have attracted very considerable attention, not only in medical and academic circles, but also in the lay press. Cyclosporin itself is now commonly employed in the prevention of rejection following allogenic organ, e.g. heart, heart-lung, kidney and bone-marrow transplant, as well as, more recently, in the treatment of various auto-immune and related diseases and conditions. Extensive work has also been performed to investigate potential utility in the treatment of various parasitic diseases and infections, for example coccidiomycosis, malaria and schistosomiasis. Reports of investigative work into the potential utility of the very many other known cyclosporins in these or related indications now abound in the literature.



………………………………
References
- Wilkes, Gail; Ades, Terri B. (2004). Consumers Guide to Cancer Drugs. Jones & Bartlett Learning. p. 226. ISBN 9780763722548. Retrieved 29 May 2013.
- Tao, Jian’guo; Sotomayor, Eduardo. (2012). Hematologic Cancers: From Molecular Pathobiology to Targeted Therapeutics. Springer. p. 335. ISBN 9789400750289.
- PSC-833Drugs Fut 1995, 20(10): 1010
- US 5525590
- Synthesis of [S-[1-14C]Val(7)]VALSPODAR application of (+)/(-)-[13,14Cn]BABS and (+)/(-)-[13,14Cn]DPMGBS, part 4J Label Compd Radiopharm 2000, 43(3): 205
- WO 2006013094
- WO 2005013947
- WO 2002098418
- WO 1999017757
- Pharmaceutical Research, 2001 , vol. 18, 2 pg. 183 – 190
- US2003/158097 A1
- Valspodar; EP-B1 0 296 122:
- WO 94/07858
MIDOSTAURIN …with potential antiangiogenic and antineoplastic activities …

MIDOSTAURIN
READ …COMPLETE SYNTHESIS AT
AVOSENTAN

AVOSENTAN
N-[6-Methoxy-5-(2-methoxyphenoxy)-2-(4-pyridyl)pyrimidin-4-yl]-5-methylpyridine-2-sulfonamide
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidin-4-yl]-amide,
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide,
Endothelin ETA Receptor Antagonists
M.Wt: 479.51
Formula: C23H21N5O5S
Roche (Originator)
CAS No.: 290815-26-8
- RO 67-0565
- SPP 301
- UNII-L94KSX715K
PHASE 3
CLINICAL TRIALS
http://clinicaltrials.gov/search/intervention=spp301+OR+Avosentan
SPP-301 is an oral, once-daily, second-generation endothelin ETA receptor antagonist which had been in phase III clinical development at Speedel for the treatment of diabetic nephropathy. In December 2006, the company reported that the phase III trial had been stopped based on the recommendation from the trial’s Data Safety Monitoring Board (DSMB) to stop the trial following incidence of a significant imbalance in fluid retention in patients in the study arms. Speedel reported that the compound will be evaluated for potential new clinical development for the treatment of diabetic kidney disease and other indications.
Originally developed by Roche and specifically optimized for improved liver safety, SPP-301 was licensed to Speedel in October 2000. In 2003, Speedel exercised its option to license from Roche all rights to SPP-301, including exclusive worldwide rights for the full development and commercialization of the ETA antagonist. SPP-301 has fast track designation and has undergone a special protocol assessment (SPA) by the FDA. Speedel had been studying the drug for the treatment of hypertension.
AVOSENTAN
290815-26-8 CAS
PATENTS
2. WO 2004078104
3. WO 2005113543
4. WO 2007031501
5. WO 2008077916
Dutzler R, Ernstb B, Hediger MA, Keppler D, Mohr P, Neidhart W, Märki HP.Chimia (Aarau). 2010;64(9):662-6.
………………………
INTRODUCTION
-
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide corresponding to the formula

is an inhibitor of endothelin receptors. WO00/52007 describes the preparation of said compound which is crystallized from Me2Cl2.
-
Own investigations have shown that there exist two distinct crystalline forms, hereinafter referred to as form A and form B, as well as a number of further solvates, in particular the methanol, ethanol, isopropanol, dichloromethane, acetone, methyl ethyl ketone and tetrahydrofuran solvates.
-
It was further surprisingly found that the thermodynamically stable crystalline form – form B – can be prepared under controlled conditions and that said form B can be prepared with a reliable method in an industrial scale, which is easy to handle and to process in the manufacture and preparation of formulations.

………………..

4,6-Dichloro-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine (described in EP 0 799 209) can be transformed to the intermediate of formula (III)—according to scheme 1—on reaction with an appropriate sulfonamide of formula (II), wherein R1 is as defined in claim 1, in a suited solvent such as DMSO or DMF at room temperature or at elevated temperature and in the presence of a suited base such as potassium carbonate.


EXAMPLE 1
[0064] a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg2SO4, concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120° C. to give the desired 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as white crystals. Melting point 225-226° C. ISN mass spectrum, m/e 478.2 (M-1 calculated for C23H21N5O5S1: 478).
[0065] C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
[0066] Preparation of the starting material:
[0067] b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 19.66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40° C. until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60° C. for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide ( CHLORO STARTING MATERIAL) as yellow crystals. Melting point 177-179° C. ISN mass spectrum, m/e 482.2 (M-1 calculated for C22H18ClN5O5S1: 482).
……………………………….
http://www.google.com/patents/US6417360
EXAMPLE 1
a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg2SO4, concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120° C. to give the desired 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as white crystals. Melting point 225-226° C. ISN mass spectrum, m/e 478.2 (M-1 calculated for C23H21N5O5S1: 478).
C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
Preparation of the starting material:
b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 19.66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40° C. until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60° C. for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as yellow crystals. Melting point 177-179° C. ISN mass spectrum, m/e 482.2 (M-1 calculated for C22H18ClN5O5S1: 482).
…………………….
http://www.google.com/patents/EP0799209B1
SYNTHESIS OF
4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine
A BASIC STARTING MATERIAL FOR AVOSENTAN
- Preparation of the starting material
-
- b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hours 29.5 g of NH4Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydrochloride (decomposition point 245-247°C).
- c) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
- d) A suspension of 154.6 g of 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCl3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2Cl2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2Cl2. The combined CH2Cl2 extracts are dried with MgSO4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2Cl2remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine, melting point 178-180°C.
…………………………
http://www.google.com/patents/WO2000052007A1
Preparation of the starting material:
5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2- methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide IE THE 6 CHLORO COMPD
b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 1 .66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40°C until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60 °C for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2- methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide as yellow crystals. Melting point 177-179 °C. ISN mass spectrum, m/e 482.2 (M-l calculated for C22Hi8ClN5O5Sι: 482).

………………………………………………………………………………………….
NEXT

Example 1AVOSENTAN
a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of
5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl- pyrimidin-4-yl] -amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg SO , concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120 °C to give the desired 5-methyl-pyridine-2-sulfonic acid [6- methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide as white crystals. Melting point 225-226 °C. ISN mass spectrum, m/e 478.2 (M-l calculated for
C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
…………………………………………….

IS DESCRIBED IN
http://www.google.com/patents/EP2331513A1?cl=en
ALSO

-
Diabetic nephropathy is the principle cause of end stage renal disease in the western world. It is a major cause of morbidity and mortality in Type-I Diabetes, but is an increasing problem in Type-II Diabetes and because the incidence of this is five times that of Type-I Diabetes, it contributes at least 50% of diabetics with end stage renal disease.
-
The initial stage of subtle morphologic changes in the renal glomeruli is followed by microalbuminuria. This is associated with a modestly rising blood pressure and an increased incidence of cardiovascular disease. There follows a continued increase in urinary protein excretion and declining glomerular filtration rate. Diabetic nephropathy has many possible underlying pathophysiological causes including metabolic, glycosylation of proteins, haemodynamics, altered flow/pressure in glomeruli, the development of hypertension and cytokine production; all of these are associated with the development of extracellular matrix and increased vascular permeability leading to glomerular damage and proteinuria.
| WO2005113543A1 * | May 12, 2005 | Dec 1, 2005 | Alexander Bilz | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| WO2007031501A2 * | Sep 11, 2006 | Mar 22, 2007 | Speedel Pharma Ag | Pyridylsulfonamidyl-pyrimidines for the prevention of blood vessel graft failure |
| WO2008077916A1 * | Dec 21, 2007 | Jul 3, 2008 | Ovidiu Baltatu | Pharmaceutical composition using aliskiren and avosentan |
| EP1454625A1 * | Mar 6, 2003 | Sep 8, 2004 | Speedel Development AG | Pyridylsulfonamidyl-pyrimidines for the treatment of diabetic nephropathies |
| EP1595880A1 * | May 13, 2004 | Nov 16, 2005 | Speedel Pharma AG | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| EP1938812A1 * | Dec 22, 2006 | Jul 2, 2008 | Speedel Pharma AG | Pharmaceutical composition using aliskiren and avosentan |
| US6951856 | Jul 10, 2001 | Oct 4, 2005 | Actelion Pharmaceuticals Ltd. | Arylethene-sulfonamides |
| US7402587 | May 12, 2005 | Jul 22, 2008 | Speedel Pharma Ag | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| WO1996019459A1 * | Dec 8, 1995 | Jun 27, 1996 | Volker Breu | Novel sulfonamides |
| EP0713875A1 * | Nov 13, 1995 | May 29, 1996 | F. Hoffmann-La Roche AG | Sulfonamides |
| EP0897914A1 * | Aug 10, 1998 | Feb 24, 1999 | F. Hoffmann-La Roche Ag | Process for the preparation of 2,5-disubstitued pyridines |
READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html

APREMILAST, … ORALLY ACTIVE PDE4 INHIBITOR

APREMILAST
PDE4 inhibitor
N-{2-[(1S)-1-(3-Ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
(+)-2-[l-(3-ethoxy-4-methoxyphenyl)-2- methanesulfonylethyl]-4-acetylaminoisoindolin-l,3-dione,
(S)—N-{2-[1-(3-ethoxy-4-methoxy-phenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
(S)-N-{2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
Molecular Formula: C22H24N2O7S Molecular Weight: 460.50016
608141-41-9 CAS NO
Celgene (Originator)
CC-10004 (apremilast) is an oral compound that is being studied in multiple Phase III clinical trials for the treatment of psoriasis, psoriatic arthritis and other chronic inflammatory diseases. We successfully completed our early stage studies, demonstrating clinical activity and tolerability and meeting safety endpoints in a placebo controlled proof-of mechanism trial in moderate-to-severe psoriasis and psoriatic arthritis. With the initiation of six multi-center international clinical trials, we are advancing the clinical development of CC-10004.
CC-10004, , Apremilast (USAN), SureCN302992, Apremilast (CC-10004), QCR-202,
- Apremilast
- CC 10004
- CC-10004
- CC10004
- UNII-UP7QBP99PN
- CLINICAL TRIALS….http://clinicaltrials.gov/search/intervention=Apremilast+OR+CC-10004
Apremilast is an orally available small molecule inhibitor of PDE4 being developed byCelgene for ankylosing spondylitis, psoriasis, and psoriatic arthritis.[1][2] The drug is currently in phase III trials for the three indications. Apremilast, an anti-inflammatory drug, specifically inhibits phosphodiesterase 4. In general the drug works on an intra-cellular basis to moderate proinflammatory and anti-inflammatory mediator production.
 APREMILAST
APREMILAST
Apremilast is being tested for its efficacy in treating “psoriasis, psoriatic arthritis and other chronic inflammatory diseases such as ankylosing spondylitis, Behcet’s disease, and rheutmatoid arthritis.
“Apremilast is Celgene’s lead oral phosphodiesterase IV inhibitor and anti-TNF alpha agent in phase III clinical studies at Celgene for the oral treatment of moderate to severe plaque-type psoriasis and for the oral treatment of psoriatic arthritis.
Early clinical development is also ongoing for the treatment of acne, Behcet’s disease, cutaneous sarcoidosis, prurigo nodularis, ankylosing spondylitis, atopic or contact dermatitis and rheumatoid arthritis. No recent development has been reported for research for the treatment of skin inflammation associated with cutaneous lupus erythematosus.
In 2011, Celgene discontinued development of the compound for the management of vision-threatening uveitis refractory to other modes of systemic immunosuppression due to lack of efficacy.
Celgene had been evaluating the potential of the drug for the treatment of asthma; however, no recent development has been reported for this research. The drug candidate is also in phase II clinical development at the William Beaumont Hospital Research Institute for the treatment of chronic prostatitis or chronic pelvic pain syndrome and for the treatment of vulvodynia (vulvar pain).
In 2013, orphan drug designations were assigned to the product in the U.S. and the E.U. for the treatment of Behcet’s disease.
Celgene Corp has been boosted by more impressive late-stage data on apremilast, an oral drug for psoriatic arthritis, this time in previously-untreated patients.
The company is presenting data from the 52-week PALACE 4 Phase III study of apremilast tested in PsA patients who have not taken systemic or biologic disease modifying antirheumatic drugs (DMARDs) at the American College of Rheumatology meeting in San Diego. The results from the 527-patient trial show that at week 16, patients on 20mg of the first-in-class oral inhibitor of phosphodiesterase 4 (PDE4) achieved an ACR20 (ie a 20% improvement in the condition) response of 29.2% and 32.3% for 30mg aapremilast, compared with 16.9% for those on placebo.
After 52 weeks, 53.4% on the lower dose and 58.7% on 30mg achieved an ACR20 response. ACR50 and 70 was reached by 31.9% and 18.1% of patients, respectively, for apremilast 30mg. The compound was generally well-tolerated and discontinuation rates for diarrhoea and nausea were less than 2% over 52 weeks.
Commenting on the data, Alvin Wells, of the Rheumatology and Immunotherapy Center in Franklin, Wisconsin, noted that apremilast demonstrated long-term safety and tolerability and significant clinical benefit in treatment-naive patients. He added that “these encouraging results suggest that apremilast may have the potential to be used alone and as a first-line therapy”. Celgene is also presenting various pooled data from the first three trials in the PALACE programme which, among other things, shows that apremilast significantly improves swollen and tender joints.
Treatment for PSA, which affects about 30% of the 125 million people worldwide who have psoriasis, currently involves injectable tumour necrosis factor (TNF) inhibitors, notably AbbVie’s Humira (adalimumab) and Pfizer/Amgen’s Enbrel (etanercept), once patients have not responded to DMARDs (at least in the UK). While the biologics are effective, the side effect profile can be a concern, due to the risk of infection and tuberculosis and many observers believe that apremilast will prove popular with patients and doctors due to the fact that it is oral, not injectable.
Apremilast was filed for PsA with the US Food and Drug Administration in the first quarter and will be submitted on both sides of the Atlantic for psoriasis before year-end. The European filing will also be for PsA.
Apremilast impresses for Behcet’s disease
Celgene has also presented promising Phase II data on apremilast as a treatment for the rare inflammatory disorder Behcet’s disease. 71% of patients achieved complete response at week 12 in clearing oral ulcers
 APREMILAST
APREMILAST
- “Apremilast Palace Program Demonstrates Robust and Consistent Statistically Significant Clinical Benefit Across Three Pivotal Phase III Studies (PALACE-1, 2 & 3) in Psoriatic Arthritis” (Press release). Celgene Corporation. 6 September 2012. Retrieved 2012-09-10.
- “US HOT STOCKS: OCZ, VeriFone, Men’s Wearhouse, AK Steel, Celgene”. The Wall Street Journal. 6 September 2012. Retrieved 2012-09-06.
- Discovery of (S)-N-[2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4 and tumor necrosis factor-alpha inhibitor.
Man HW, Schafer P, Wong LM, Patterson RT, Corral LG, Raymon H, Blease K, Leisten J, Shirley MA, Tang Y, Babusis DM, Chen R, Stirling D, Muller GW.
J Med Chem. 2009 Mar 26;52(6):1522-4. doi: 10.1021/jm900210d.
- Therapeutics: Silencing psoriasis.Crow JM.Nature. 2012 Dec 20;492(7429):S58-9. doi: 10.1038/492S58a. No abstract available.
- NMR…http://file.selleckchem.com/downloads/nmr/S803401-Apremilast-HNMR-Selleck.pdf
- WO 2003080049
- WO 2013126495
- WO 2013126360
- WO 2003080049
- WO 2006065814
- US2003/187052 A1 …..MP 144 DEG CENT
- US2007/155791
-
J. Med. Chem., 2008, 51 (18), pp 5471–5489DOI: 10.1021/jm800582j
-
J. Med. Chem., 2011, 54 (9), pp 3331–3347DOI: 10.1021/jm200070e

…………………………………………
INTRODUCTION
2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione is a PDE4 inhibitor that is currently under investigation as an anti-inflammatory for the treatment of a variety of conditions, including asthma, chronic obstructive pulmonary disease, psoriasis and other allergic, autoimmune and rheumatologic conditions. S-enantiomer form of 2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione can be prepared by reacting (5)-aminosulfone 1 with intermediate 2.

Existing methods for synthesizing (S)-aminosulfone 1 involve resolution of the corresponding racemic aminosulfone by techniques known in the art. Examples include the formation and crystallization of chiral salts, and the use of chiral high performance liquid chromatography. See, e.g., Jacques, J., et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen, S. H., et al, Tetrahedron 33:2725 (1977); Eliel, E. L., Stereochemistry of Carbon Compounds (McGraw Hill, NY, 1962); and Wilen, S. H., Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972). In one example, as depicted in Scheme 1 below, (5)-aminosulfone 1 is prepared by resolution of racemic aminosulfone 3 with N-Ac-L-Leu. Racemic aminosulfone 3 is prepared by converting 3-ethoxy-4-methoxybenzonitrile 4 to enamine intermediate 5 followed by enamine reduction and borate hydrolysis. This process has been reported in U.S. Patent
Application Publication No. 2010/0168475.

CH2CI2, NaOH

Scheme 1
The procedure for preparing an enantiomerically enriched or enantiomerically pure aminosulfone, such as compound 1, may be inefficient because it involves the resolution of racemic aminosulfone 3. Thus, a need exists as to asymmetric synthetic processes for the preparation of an enantiomerically enriched or enantiomerically pure aminosulfone, particularly for manufacturing scale production. Direct catalytic asymmetric hydrogenation of a suitable enamine or ketone intermediate is of particular interest because it eliminates the need for either classic resolution or the use of stoichiometric amount of chiral auxiliary, and thus, may be synthetically efficient and economical.
……………………………………….
SYNTHESIS OF KEY INTERMEDIATE
Example 1
Synthesis of 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine

[00232] A slurry of dimethylsulfone (85 g, 903 mmol) in THF (480 ml) was treated with a
1.6M solution of n-butyllithium in hexane (505 ml, 808 mmol) at 0 – 5 °C. The resulting mixture was agitated for 1 hour then a solution of 3-ethoxy-4-methoxybenzonitrile (80 g, 451 mmol) in THF (240 ml) was added at 0 – 5 °C. The mixture was agitated at 0 – 5 °C for 0.5 hour, warmed to 25 – 30 °C over 0.5 hour and then agitated for 1 hour. Water (1.4 L) was added at 25 – 30 °C and the reaction mass was agitated overnight at room temperature (20 – 30 °C). The solid was filtered and subsequently washed with a 2: 1 mixture of water :THF (200 ml), water (200 ml) and heptane (2 x 200 ml). The solid was dried under reduced pressure at 40 – 45 °C to provide the product as a white solid (102 g, 83% yield); 1H NMR (DMSO-d6) δ 1.34 (t, J=7.0 Hz, 3H), 2.99 (s, 3H), 3.80 (s, 3H), 4.08 (q, J=7.0 Hz, 2H), 5.03 (s, 1H), 6.82 (s, 2H), 7.01 (d, J=8.5 Hz, 1H), 7.09 – 7.22 (m, 2H).
Example 2
Synthesis of (R)- 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine

[00233] A solution of bis(l,5-cyclooctadiene)rhodium(I) trifluoromethanesulfonate (36 mg, 0.074 mmol) and (i?)-l-[(5)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (40 mg, 0.074 mmol) in 25 mL of 2,2,2-trifluoroethanol was prepared under nitrogen. To this solution was then charged l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine (2.0 g, 7.4 mmol). The resulting mixture was heated to 50 °C and hydrogenated under 90 psig hydrogen pressure. After 18 h, the mixture was cooled to ambient temperature and removed from the hydrogenator. The mixture was evaporated and the residue was purified by chromatography on a CI 8 reverse phase column using a water-acetonitrile gradient. The appropriate fractions were pooled and evaporated to -150 mL. To this solution was added brine (20 mL), and the resulting solution was extracted with EtOAc (3 x 50 mL). The combined organic layers were dried (MgS04) and evaporated to provide the product as a white crystalline solid (1.4 g, 70% yield); achiral HPLC (Hypersil BDS C8, 5.0 μπι, 250 x 4.6 mm, 1.5 mL/min, 278nm, 90/10 gradient to 80/20 0.1% aqueous TFA/MeOH over 10 min then gradient to 10/90 0.1% aqueous TFA/MeOH over the next 15 min): 9.11 (99.6%); chiral HPLC (Chiralpak AD-H 5.0 μιη Daicel, 250 x 4.6 mm, 1.0 mL/min, 280 nm, 70:30:0.1 heptane-z-PrOH-diethylamine): 7.32 (97.5%), 8.26 (2.47%); 1H NMR (DMSO-de) δ 1.32 (t, J= 7.0 Hz, 3H), 2.08 (s, 2H), 2.96 (s, 3H), 3.23 (dd, J= 3.6, 14.4 Hz, 1H), 3.41 (dd, J= 9.4, 14.4 Hz, 1H), 3.73 (s, 3H), 4.02 (q, J= 7.0 Hz, 2H), 4.26 (dd, J= 3.7, 9.3 Hz, 1H), 6.89 (s, 2H), 7.02 (s, 1H); 13C NMR (DMSO-d6) δ 14.77, 41.98, 50.89, 55.54, 62.03, 63.68, 111.48, 111.77, 118.36, 137.30, 147.93, 148.09. Example 3
Synthesis of (6 -l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine N-Ac-L-Leu salt

[00234] A solution of bis(l,5-cyclooctadiene)rhodium(I) trifluoromethanesulfonate (17 mg, 0.037 mmol) and (5)-l-[(i?)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (20 mg, 0.037 mmol) in 10 mL of 2,2,2-trifluoroethanol was prepared under nitrogen. To this solution was then charged l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine (2.0 g, 7.4 mmol). The resulting mixture was heated to 50 °C and hydrogenated under 90 psig hydrogen pressure. After 18 h, the mixture was cooled to ambient temperature and removed from the hydrogenator. Ecosorb C-941 (200 mg) was added and the mixture was stirred at ambient temperature for 3 h. The mixture was filtered through Celite, and the filter was washed with additional trifluoroethanol (2 mL). Then, the mixture was heated to 55 °C, and a solution of N- acetyl-L-leucine (1.3 g, 7.5 mmol) was added dropwise over the course of 1 h. Stirring proceeded at the same temperature for 1 h following completion of the addition, and then the mixture was cooled to 22 °C over 2 h and stirred at this temperature for 16 h. The crystalline product was filtered, rinsed with methanol (2 x 5 mL), and dried under vacuum at 45 °C to provide the product as a white solid (2.6 g, 80% yield); achiral HPLC (Hypersil BDS Cg, 5.0 μιη, 250 x 4.6 mm, 1.5 mL/min, 278nm, 90/10 gradient to 80/20 0.1% aqueous TFA/MeOH over 10 min then gradient to 10/90 0.1% aqueous TFA/MeOH over the next 15 min): 8.57 (99.8%); chiral HPLC (Chiralpak AD-H 5.0 μιη Daicel, 250 x 4.6 mm, 1.0 mL/min, 280 nm, 70:30:0.1 heptane-z-PrOH-diethylamine): 8.35 (99.6%); 1H NMR (DMSO-<¾) δ 0.84 (d, 3H), 0.89 (d, J= 6.6 Hz, 3H), 1.33 (t, J= 7.0 Hz, 3H), 1.41 – 1.52 (m, 2H), 1.62 (dt, J= 6.7, 13.5 Hz, 1H), 1.83 (s, 3H), 2.94 (s, 3H), 3.28 (dd, J= 4.0, 14.4 Hz, 1H), 3.44 (dd, J= 9.1, 14.4 Hz, 1H), 3.73 (s, 3H), 4.02 (q, J= 6.9 Hz, 2H), 4.18 (q, J= 7.7 Hz, 1H), 4.29 (dd, J= 4.0, 9.1 Hz, 1H), 5.46 (br, 3H), 6.90 (s, 2H), 7.04 (s, 1H), 8.04 (d, J= 7.9 Hz, 1H); Anal. (C20H34N2O7S) C, H, N. Calcd C, 53.79; H, 7.67; N 6.27. Found C, 53.78; H, 7.57; N 6.18.
SUBSEQUENT CONVERSION
S-enantiomer form of 2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione can be prepared by reacting (5)-aminosulfone 1 with intermediate 2.
……………………………………
 APREMILAST
APREMILAST
GENERAL SYNTHESIS AND SYNTHESIS OF APREMILAST




(apremilast)
[0145] Preparation of 3-Ethoxy-4-methoxybenzonitrile (Compound 2). 3-Ethoxy-
4-methoxybenzaldehyde (Compound 1, 10.0 gm, 54.9 mmol, Aldrich) and hydroxylamine hydrochloride (4.67 gm, 65.9 mmol, Aldrich) were charged to a 250 mL three-necked flask at room temperature, followed by the addition of anhydrous acetonitrile (50 mL). The reaction mixture was stirred at room temperature for thirty minutes and then heated to reflux (oil bath at 85 °C). After two hours of reflux, the reaction mixture was cooled to room temperature, and added 50 mL of deionized water. The mixture was concentrated under reduced pressure to remove acetonitrile and then transferred to a separatory funnel with an additional 80 mL of deionized water and 80 mL dichloromethane. The aqueous layer was extracted with dichloromethane (3 x 50 mL). The combined organic layers were washed successively with water (80 mL) and saturated sodium chloride (80 mL). The organic layer was dried over anhydrous sodium sulfate (approximately 20 gm). The organic layer was filtered and concentrated under reduced pressure to give a yellow oil. Purification by silica gel chromatography (0 to 1 % MeOH/DCM ) afforded 3-Ethoxy-4-methoxybenzonitrile
(Compound 2) as a white solid (7.69 gm, 79 % yield). MS (ESI positive ion) m/z 178.1 (M + 1). HPLC indicated >99% purity by peak area. 1H-NMR (500 MHz, DMSO-c¾: δ ppm 1.32 (t, 3H), 3.83 (s, 3H), 4.05 (q, 2H), 7.10 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 7.40 (dd, J = 2.0 Hz, 1H).
[0146] Preparation of l-(3-Ethoxy-4-methoxyphenyi)-2-
(niethylsulfonyl)ethanamine (Compound 3). Dimethyl sulfone (2.60 gm, 27.1 mmol, Aldrich) and tetrahydrofuran (10 mL, Aldrich) were charged to a 250 mL three-necked flask at room temperature. The mixture was cooled to 0 – 5 °C, and the solution gradually turned white. n-Butyllithium (10.8 mL, 27.1 mmol, 2.5 M solution in hexanes, Aldrich) was added to the flask at a rate such that the reaction mixture was maintained at 5 – 10 °C. The mixture was stirred at 0 – 5 °C for one hour, turning light-yellow. 3-Ethoxy-4-methoxybenzonitrile (Compound 2, 4.01 gm, 22.5 mmol) in tetrahydrofuran (8 mL) was then charged to the flask at a rate such that the reaction mixture was maintained at 0 – 5 °C. The mixture was stirred at 0 – 5 °C for another 15 minutes. After warming to room temperature, the reaction mixture was stirred for another 1.5 hours and then transferred to a second 250 mL three-necked flask containing a suspension of sodium borohydride (1.13 gm, 29.3 mmol, Aldrich) in
tetrahydrofuran (1 1 mL), maintained at – 5 – 0 °C for 30 minutes. Trifluoroacetic acid (“TFA,” 5.26 mL, 68.3 mmol, Aldrich) was charged to the flask at a rate such that the reaction mixture was maintained at 0 – 5 °C. The mixture was stirred at 0 – 5 °C for 40 minutes and an additional 17 hours at room temperature. The reaction mixture was then charged with 2.7 mL of deionized water over five minutes at room temperature. The mxiture was stirred at room temperature for 15 hours. Aqueous NaOH (10 N, 4.9 mL) was charged to the flask over 15 minutes at 45 °C. The mixture was stirred at 45 °C for two hours, at 60 °C for 1.5 hours, and at room temperature overnight. After approximately 17 hours at room temperature the mixture was cooled to 0 °C for thirty minutes and then concentrated under reduced pressure. The residual material was charged with deionized water (3 mL) and absolute ethanol (3 mL) and stirred at 0 – 5 °C for 2 hours. The mixture was filtered under vacuum, and the filtered solid was washed with cold absolute ethanol (3 x 5 mL), followed by deionized water until the pH of the wash was about 8. The solid was air dried overnight, and then in a vacuum oven at 60 °C for 17 hours to afford Compound 3 as a white solid (4.75 gm, 77 %). MS (ESI positive ion) m/z 274.1 (M + 1). Ή-NMR (500 MHz, DMSO-c¾): δ ppm 1.32 (t, J = 7.0 Hz, 3H), 2.08 (bs, 2H), 2.95 (s, 3H), 3.23 (dd, J = 4.0 Hz, 1H), 3.40 (dd, J = 9.5 Hz, 1H), 3.72 (s, 3H), 4.01 (q, J = 7.0 Hz, 2H), 4.25 (dd, J = 3.5 Hz, 1H), 6.88 (s, 2H), 7.02 (s, 1H).
[0147] Preparation of 4-Nitroisobenzofuran-l,3-dione (Compound 5). Into a 250 mL round bottom flask, fitted with a reflux condenser, was placed 3-nitrophthalic acid (21.0 gm, 99 mmol, Aldrich) and acetic anhydride (18.8 mL, 199 mmol, Aldrich). The solid mixture was heated to 85 °C, under nitrogen, with gradual melting of the solids. The yellow mixture was heated at 85 °C for 15 minutes, and there was noticeable thickening of the mixture. After 15 minutes at 85 °C, the hot mixture was poured into a weighing dish, and allowed to cool. The yellow solid was grinded to a powder and then placed on a cintered funnel, under vacuum. The solid was washed with diethyl ether (3 x 15 mL), under vacuum and allowed to air dry overnight, to afford 4-nitroisobenzofuran-l ,3-dione, Compound 5, as a light-yellow solid (15.8 gm, 82 %). MS (ESI positive ion) m/z 194.0 (M + 1). TLC: Rf = 0.37 (10% MeOH/DCM with 2 drops Acetic acid) Ή-NMR (500 MHz, DMSO-i¾: δ ppm 8.21 (dd, J = 7.5 Hz, 1H), 8.39 (dd, J = 7.5 Hz, 1H), 8.50 (dd, J = 7.5 Hz, 1 H).
[0148] Preparation of 2-(l-(3-Ethoxy-4-methoxyphenyI)-2-
(methylsulfonyl)ethyl)-4-nitroisoindoline-l,3-dione (Compound 6). Into a 2 – 5 mL microwave vial was added 4-nitroisobenzofuran-l ,3-dione (Compound 5, 0.35 gm, 1.82 mmol), the amino-sulfone intermediate (Compound 3, 0.50 gm, 1.82 mmol) and 4.0 mL of glacial acetic acid. The mixture was placed in a microwave at 125 °C for 30 minutes. After 30 minutes the acetic acid was removed under reduced pressure. The yellow oil was taken up in ethyl acetate and applied to a 10 gm snap Biotage samplet. Purification by silica gel chromatography (0 to 20 % Ethyl Acetate/Hexanes) afforded Compound 6 as a light-yellow solid (0.67 gm, 82 %). MS (ESI positive ion) m/z 449.0 (M + 1). TLC: Rf = 0.19
(EtOAc:Hexanes, 1 : 1). HPLC indicated 99% purity by peak area. Ή-NMR (500 MHz, DMSO-c¾: δ ppm 1.32 (t, 3H), 2.99 (s, 3H), 3.73 (s, 3H), 4.02 (m, 2H), 4.21 (dd, J = 5.0 Hz, 1H), 4.29 (dd, J = 10.0 Hz, 1H), 5.81 (dd, J = 5.0 Hz, 1H), 6.93 (d, J – 8.5 Hz, 1H), 7.00 (dd, J = 2.0 Hz, 1H), 7.10 (d, J = 2.5 Hz, 1H), 8.07 (t, J = 15.5 Hz, 1H), 8.19 (dd, J = 8.5 Hz, 1H), 8.30 (dd, J = 9.0 Hz, 1H).
[0149] Preparation of 4-Amino-2-(l-(3-ethoxy-4-methoxyphenyl)-2-
(methylsulfonyl)ethyl)isoindoline-l,3-dione (Compound 7). Compound 6 (0.54 gm, 1.20 mmol) was taken up in ethyl acetate / acetone (1 : 1 , 24 mL) and flowed through the H-cube™ hydrogen reactor using a 10 % Pd/C CatCart™ catalyst cartridge system (ThalesNano, Budapest Hungary). After eluting, the yellow solvent was concentrated under reduced pressure to give Compound 7 as a yellow foam solid (0.48 gm, 95 %). MS (ESI positive ion) m/z 419.1 (M + 1). 1H-NMR (500 MHz, DMSO-<¾): δ ppm 1.31 (t, J = 7.0 Hz, 3H), 2.99 (s, 3H), 3.72 (s, 3H), 4.04 (q, J = 7.0 Hz, 2H), 4.09 (m, 1H), 4.34 (m, 1H), 5.71 (dd, J = 5.5 Hz, 1H), 6.52 (bs, 2H), 6.92-6.98 (m, 3H), 7.06 (bs, 1 H), 7.42 (dd, J = 7.0 Hz, 1H).
[0150] Preparation of N-(2-(l-(3-ethoxy-4-methoxyphenyl)-2-
(methylsuIfonyl)ethyl)-l,3-dioxoisoindolin-4-yl)acetamide (Apremilast, Compound 8).
Into a 2-5 mL microwave vial was placed Compound 7 (0.18 gm, 0.43 mmol), acetic anhydride (0.052 mL, 0.53 mmol) and acetic acid (4 mL). The microwave vial was placed into a Biotage microwave and heated to 125 °C for 30 minutes. The solvents were removed under reduced pressure and the residue was purified by silica gel chromatography (0 to 5% MeOH/DCM) to afford apremilast (Compound 8) as a yellow oil (0.14 gm, 71%). HPLC indicated 94.6% purity by peak area.
1H-NMR (500 MHz, DMSO-c 6): δ ppm 1.31 (t, 3H), 2.18 (s, 3H), 3.01 (s, 3H), 3.73 (s, 3H), 4.01 (t, J = 7.0 Hz, 2H), 4,14 (dd, J = 4.0 Hz, 1H), 4.33 (m, 1H), 5.76 (dd, J = 3.0 Hz, 1H), 6.95 (m, 2H), 7.06 (d, J = 1.5 Hz, 1H), 7.56 (d, J = 7.0 Hz, 1H), 7.79 (t, J = 7.7 Hz, 1H), 8.43 (d, J = 8.5 Hz, 1H), 9.72 (bs, 1H).
……………………..
SYNTHESIS
5. EXAMPLES
Certain embodiments provided herein are illustrated by the following non-limiting examples.
5.1 PREPARATION OF (+)-2-[l-(3-ETHOXY-4-METHOXYPHENYL)-2- METHANESULFONYLETHYLJ-4- ACETYL AMINOISOINDOLIN-1,3- DIONE (APREMILAST)

5.1.1 Preparation of 3-aminopthalic acid
10% Pd/C (2.5 g), 3-nitrophthalic acid (75.0 g, 355 mmol) and ethanol (1.5 L) were charged to a 2.5 L Parr hydrogenator under a nitrogen atmosphere. Hydrogen was charged to the reaction vessel for up to 55 psi. The mixture was shaken for 13 hours, maintaining hydrogen pressure between 50 and 55 psi. Hydrogen was released and the mixture was purged with nitrogen 3 times. The suspension was filtered through a celite bed and rinsed with methanol. The filtrate was concentrated in vacuo. The resulting solid was reslurried in ether and isolated by vacuum filtration. The solid was dried in vacua to a constant weight, affording 54 g (84%> yield) of 3-aminopthalic acid as a yellow product. 1H-NMR (DMSO-d6) δ: 3.17 (s, 2H), 6.67 (d, 1H), 6.82 (d, 1H), 7.17 (t, 1H), 8-10 (brs, 2H). 13C-NMR(DMSO-d6) δ: 112.00, 115.32, 118.20, 131.28, 135.86, 148.82, 169.15, 170.09.
5.1.2 Preparation of 3-acetamidopthalic anhydride
A I L 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser and charged with 3-aminophthalic acid (108 g, 596 mmol) and acetic anhydride (550 mL). The reaction mixture was heated to reflux for 3 hours and cooled to ambient temperature and further to 0-5. degree. C. for another 1 hour. The crystalline solid was collected by vacuum filtration and washed with ether. The solid product was dried in vacua at ambient temperature to a constant weight, giving 75 g (61% yield) of 3-acetamidopthalic anhydride as a white product. 1H-NMR (CDCI3) δ: 2.21 (s, 3H), 7.76 (d, 1H), 7.94 (t, 1H), 8.42 (d, 1H), 9.84 (s, 1H).
5.1.3 Resolution of 2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)- ethyl-2-amine
A 3 L 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser and charged with 2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)-eth-2-ylamine (137.0 g, 500 mmol), N-acetyl-L-leucine (52 g, 300 mmol), and methanol (1.0 L). The stirred slurry was heated to reflux for 1 hour. The stirred mixture was allowed to cool to ambient temperature and stirring was continued for another 3 hours at ambient temperature. The slurry was filtered and washed with methanol (250 mL). The solid was air-dried and then dried in vacuo at ambient temperature to a constant weight, giving 109.5 g (98% yield) of the crude product (85.8% ee). The crude solid (55.0 g) and methanol (440 mL) were brought to reflux for 1 hour, cooled to room temperature and stirred for an additional 3 hours at ambient temperature. The slurry was filtered and the filter cake was washed with methanol (200 mL). The solid was air-dried and then dried in vacuo at 30°C. to a constant weight, yielding 49.6 g (90%> recovery) of (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-ylamine-N-acety 1-L-leucine salt (98.4% ee). Chiral HPLC (1/99 EtOH/20 mM KH2P04 @pH 7.0, Ultron Chiral ES-OVS from Agilent Technologies, 150 mm.times.4.6 mm, 0.5 mL/min., @240 nm): 18.4 min (S-isomer, 99.2%), 25.5 min (R-isomer, 0.8%)
5.1.4 Preparation of (+)-2-[l-(3-ethoxy-4-methoxyphenyl)-2- methanesulfonylethyl] -4-acetylaminoisoindolin- 1 ,3-dione
A 500 mL 3 -necked round bottom flask was equipped with a mechanical stirrer,
thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98% ee), 3-acetamidophthalic anhydride (12.1 g, 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to <50°C. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL x
2), saturated aqeous NaHC03 (250 mL.times.2), brine (250 mL.times.2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL.times.2). The product was dried in vacuo at 60°C. to a constant weight, affording 19.4 g (75% yield) of Compound 3 APREMILAST with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2P04 @pH 3.5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm x 4.6 mm, 0.4 mL/min., @240 nm): 25.4 min (S-isomer, 98.7%), 29.5 min (R-isomer, 1.2%).
1H-NMR (CDC13) δ: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H).
13C-NMR(DMSO-d6) δ: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48.
…………………………………..
NMR
1H-NMR (CDCl3) δ: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H). 13C-NMR (DMSO-d6) δ: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48.
…………….

aReagents and conditions: (a) LiN(SiMe3)2, then Me2SO2/n-BuLi/BF3Et2O, −78 °C; (b) N-Ac-l-leucine, MeOH; (c) HOAc, reflux.
……………………
SARCOIDOSIS
Sarcoidosis is a disease of unknown cause. Sarcoidosis is characterized by the presence of granulomas in one or more organ systems. The most common sites of involvement are the lungs and the lymph nodes in the mediastinum and hilar regions. However, sarcoidosis is a systemic disease and a variety of organ systems or tissues may be the source of primary or concomitant clinical manifestations and morbidity. The clinical course of sarcoidosis is extremely variable, and ranges from a mild or even asymptomatic disease with spontaneous resolution to a chronic progressive disease leading to organ system failure and, in 1-5% of cases, death. See Cecil
Textbook of Medicine, 21st ed. (Goldman, L., Bennett, J. C. eds), W. B. Saunders Company, Philadelphia, 2000, p. 433-436.
While the cause of sarcoidosis is unknown, a substantial body of information suggests that immune mechanisms are important in disease pathogenesis. For example, sarcoidosis is
characterized by enhanced lymphocyte and macrophage activity. See Thomas, P.D. and
Hunninghake, G.W., Am. Rev. Respir. Dis., 1987, 135: 747-760. As sarcoidosis progresses, skin rashes, erythema nodosum and granulomas may form. Granulomas or fibrosis caused by sarcoidosis can occur throughout the body, and may affect the function of vital organs such as the lungs, heart, nervous system, liver or kidneys. In these cases, the sarcoidosis can be fatal. See
http://www.nlm.nih.gov/medlineplus/sarcoidosis.html (accessed November 12, 2009).
Moreover, a variety of exogenous agents, both infectious and non-infectious, have been hypothesized as a possible cause of sarcoidosis. See Vokurka et ah, Am. J. Respir. Crit. Care Med., 1997, 156: 1000-1003; Popper et al, Hum. Pathol, 1997, 28: 796-800; Almenoff et al, Thorax, 1996, 51 : 530-533; Baughman et al., Lancet, 2003, 361 : 1111-1118. These agents include mycobaceria, fungi, spirochetes, and the agent associated with Whipple’s disease. Id.
Sarcoidosis may be acute or chronic. Specific types of sarcoidosis include, but are not limited to, cardiac sarcoidosis, cutaneous sarcoidosis, hepatic sarcoidosis, oral sarcoidosis, pulmonary sarcoidosis, neurosarcoidosis, sinonasal sarcoidosis, Lofgren’s syndrome, lupus pernio, uveitis or chronic cutaneous sarcoidosis.
As the lung is constantly confronted with airborne substances, including pathogens, many researchers have directed their attention to identification of potential causative transmissible agents and their contribution to the mechanism of pulmonary granuloma formation associated with sarcoidosis. See Conron, M. and Du Bois, R.M., Clin. Exp. Allergy, 2001, 31 : 543-554; Agostini et al, Curr. Opin. Pulm. Med. , 2002, 8: 435-440.
Corticosteroid drugs are the primary treatment for the inflammation and granuloma formation associated with sarcoidosis. Rizatto et al. , Respiratory Medicine, 1997, 91 : 449-460. Prednisone is most often prescribed drug for the treatment of sarcoidosis. Additional drugs used to treat sarcoidosis include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, minocycline, doxycycline and chloroquin. TNF-a blockers such as thalidomide and infliximab have been reported to be effective in treating patients with sarcoidosis. Baughman et al, Chest, 2002, 122: 227-232; Doty et al, Chest, 2005, 127: 1064-1071. Antibiotics have also been studied for the treatment of sarcoidosis, such as penicillin antibiotics, cephalosporin antibiotics, macrolide antibiotics, lincomycin antibiotics, and tetracycline antibiotics. Specific examples include minocycline hydrochloride, clindamycin, ampicillin, or clarithromycin. See, e.g., U.S. Patent Publication No. 2007/0111956.
There currently lacks a Food and Drug Administration-approved therapeutic agent for the treatment of sarcoidosis, and many patients are unable to tolerate the side effects of the standard corticosteroid therapy. See Doty et al, Chest, 2005, 127: 1064-1071. Furthermore, many cases of sarcoidosis are refractory to standard therapy. Id. Therefore, a demand exists for new methods and compositions that can be used to treat patients with sarcoidosis.
……………..
PATENTS
|
8-15-2012
|
PROCESSES FOR THE PREPARATION OF AMINOSULFONE COMPOUNDS
|
|
|
11-4-2011
|
HETEROCYCLIC COMPOUNDS AS PHOSPHODIESTERASE INHIBITORS
|
|
|
5-27-2011
|
Nanosuspension of a Poorly Soluble Drug via Microfluidization Process
|
|
|
5-28-2010
|
METHODS AND COMPOSITIONS USING PDE4 INHIBITORS FOR THE TREATMENT AND MANAGEMENT OF CANCERS
|

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
NETUPITANT

NETUPITANT
- Ro 67-3189/000
- UNII-7732P08TIR
- Ro-67-3189
- Netupitant, an NK-1 antagonist is under development for the treatment of overactive bladder. HELSINN GROUP
CAS: 290297-26-6
290296-54-7 (di HCl)
U.S. Pat. Nos. 6,303,790, 6,531,597, 6,297,375 and 6,479,483, 6,719,996 and 6,593,472 to Hoffmann La Roche(originator).
IUPAC/Chemical name:
2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide
Chemical Formula: C30H32F6N4O
Exact Mass: 578.24803
Molecular Weight: 578.59
Elemental Analysis: C, 62.28; H, 5.57; F, 19.70; N, 9.68; O, 2.77
Netupitant is another selective NKi receptor antagonist under development by Helsinn Healthcare, having the formula 2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethyl-N-[4-(2- methylphenyl)-6-(4-methylpiperazin- l-yl)pyridin-3-yl]propanamide, or Benzeneacetamide, N,a,a-trimethyl-N-[4-(2-methylphenyl)-6-(4-methyl-l-piperazinyl)-3-pyridinyl]-3,5- bis(trifluoromethyl)-, and the below chemical structure:

Netupitant is a tachykinin NK-1 antagonist which had been in phase III clinical trials at Helsinn for the prophylaxis of chemotherapy-induced nausea and vomiting and in phase II clinical studies for the treatment of overactive bladder. However, no recent development has been reported for this research.
NK-1 receptor antagonists work by blocking the action of neurokinin-1 (Substance P), a naturally-occurring neurotransmitter in the brain that causes emesis. Netupitant was originally developed at Roche. In June 2005, Helsinn and Roche signed a licensing agreement granting Helsinn worldwide rights to the drug candidate.
Methods of synthesizing and formulating netupitant and its prodrugs are described in U.S. Patent Nos. 6,297,375, 6,719,996 and 6,593,472 to Hoffmann La Roche.
Netupitant is a highly selective NK1 receptor antagonist, which is thought to work by blocking the action of substance P, an endogenous neurotransmitter contained in high concentrations in the vomiting center of the brainstem that can stimulate the vomiting reflex. Netupitant is currently under phase III trials.
Chemotherapy is one of the treatment options utilized by oncologists in treating different types of cancers. Nausea and vomiting are the most common side-effects experienced by cancer patients when administered with chemotherapy. Netupitant-palonosetron, which is currently in Phase III trials helps in preventing CINV. The blockage of P/NK1 receptors by Netupitant in the central nervous system inhibits the binding of endogenous tachykinin neuropeptide substance and this result in preventing the chemotherapy-induced nausea and vomiting. Moreover, Palonosetron helps in the blockage of serotonin at 5-hydroxytryptamine type 3 (5-HT3) receptors and it also helps in the chemotherapy-induced nausea and vomiting.

Netupitant-Palonosetron FDC is estimated to answer significant unmet needs of the CINV market post its launch that is expected to be commercialized in 2014, as it would overcome the problems associated with current treatment with 5-HT3 receptor antagonists. Similar to Emend, Netupitant-Palonosetron FDC would gain considerable patient pool after its estimated launch in 2014, and subsequently match the patient share of Aloxi by 2018. Netupitant-Palonosetron FDC sales are expected to reach an estimated USD 515.0 million USD by 2018. FDC combination of 5-HT3 receptor antagonist and neurokinin-1 (NK1) receptor antagonist have shown better efficacy results in Phase II clinical trials for CINV patients and would thus lead to high uptake due to shifting physician and patient preference pattern towards better treatment for CINV.

Neurokinin 1 receptor antagonists are being developed for the treatment of a number of physiological disorders associated with an excess or imbalance of tachykinin, in particular substance P. Examples of conditions in which substance P has been implicated include disorders of the central nervous system such as anxiety, depression and psychosis (WO 95/16679, WO 95/18124 and WO 95/23798).
The neurokinin-1 receptor antagonists are further useful for the treatment of motion sickness and for treatment induced vomiting. The New England Journal of Medicine, Vol. 340, No. 3 190-195, 1999 has been described the reduction of cisplatin-induced emesis by a selective neurokinin-l-receptor antagonist. US5,972,938 describes a method for treating a psychoimmunologic or a psychosomatic disorder by administration of a tachykinin receptor, such as NK-1 receptor antagonist.
With the development of the 5-HT3 antagonist in the early 1990s, there emerged new strategies in the medical community to better control nausea and vomiting caused by various medical procedures, including chemotherapy (CINV), surgery (PONV), and radiation therapy (RINV). When added to steroids such as dexamethasone, several 5-HT3 antagonists have been demonstrated to significantly improve the standard of life for patients undergoing emetogenic medical procedures. Examples of 5-HT3 antagonists include ondansetron, marketed by
GlaxoSmithKline, and palonosetron, developed by Helsinn Healthcare.
Netupitant is another selective NKi receptor antagonist under development by Helsinn Healthcare, having the formula 2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethyl-N-[4-(2- methylphenyl)-6-(4-methylpiperazin- l-yl)pyridin-3-yl]propanamide, or Benzeneacetamide, N,a,a-trimethyl-N-[4-(2-methylphenyl)-6-(4-methyl-l-piperazinyl)-3-pyridinyl]-3,5- bis(trifluoromethyl)-, and the below chemical structure:
Methods of synthesizing and formulating netupitant and its prodrugs are described in U.S. Patent Nos. 6,297,375, 6,719,996 and 6,593,472 to Hoffmann La Roche.
Other representative NKi antagonists include ZD4974 (developed by AstraZeneca), CGP49823 (developed by Ciba-Geigy), Lanepitant and LY686017 (developed by Eli Lilly), FK888 (developed by Fujisawa), Vofopitant, Vestipitant and Orvepitant (developed by
GlaxoSmithKline), Befetupitant (developed by Hoffmann-La Roche), Rl 16031 (developed by Janssen), L-733060 and L-736281 (developed by Merck), TKA731, NKP608 and DNK333 (developed by Novartis), CP-96345, CP-99994, CP- 122721, CJ-17493, CJ-11974 and CJ-11972 (developed by Pfizer), RP67580 and Dapitant (developed by Rhone-Poulenc Rorer),
Nolpitantium and SSR240600 (developed by Sanofi-Aventis), SCH388714 and Rolapitant (developed by Schering-Plough), TAK637 (developed by Takeda), HSP117 (developed by Hisamitsu), KRP103 (developed by Kyorin Pharm) and SLV317 (developed by Solvay).
Chemical structures of the above-mentioned NKi antagonists are shown below and discussion of those compounds as well as other NKi antagonists is present in Expert Opin. Ther. Patents (2010) 20(8), pp 1019- 1045 by Huang et al.
………………………………………………
WO 2013057554
WO 2011061622
WO 2010119347
WO 2003006016
WO 2006002860///
WO 2002085458
US 2002091265…….
…………………………………………………..
http://pubs.acs.org/doi/full/10.1021/jo0523666
…………………………………………..
https://www.google.co.in/patents/US6297375
(2-(3,5-bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide) which has the formula Ib

and to pharmaceutically acceptable acid addition salts thereof.
The compound of formula Ib and its salts is also characterized by valuable therapeutic properties as a highly selective antagonist of the Neurokinin 1 (NK-1, substance P) The present compound of formula lb and its pharmaceutically acceptable salts can be prepared by methods known in the art, for example, by processes described below, which process comprises
a) reacting the compound of formula

with the compound of formula

to the compound of formula



EXAMPLE 14
2-(3,5-Bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide hydrochloride (1:2)
a) 1-Methyl-4-(5-nitro-pyridin-2-yl)-piperazine
To a solution of 20 g (126 mmol) of 2-chloro-5-nitropyridine in 200 ml tetrahydrofuran were added dropwise 35 ml (315 mmol) 1-methylpiperazine within 10 min. The reaction mixture was refluxed for additional 1.5 h. After cooling to room temperature, the solvent was removed in vacuo and the residue was re-dissolved in 200 ml ethyl acetate. The organic phase was washed with 200 ml 1 N sodium bicarbonate solution, dried (magnesium sulfate) and evaporated to give 27.9 g (quantitative) of the title compound as a yellow solid.
MS m/e (%):223 (M+H+, 100).
b)2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-propionamide
To a solution of 27.9 g (125.5 mmol) of 1-methyl-4-(5-nitro-pyridin-2-yl)-piperazine in 400 ml methanol were added 2.6 g of 10% of palladium on activated charcoal. The reaction mixture was hydrogenated (room temperature to ca. 45° C., 1 bar) until the theoretical amount of hydrogen was taken up (about 2 h). The catalyst was filtered off and was washed twice with 100 ml portions of methanol. The filtrate was evaporated in vacuo to give 28 g of a purple oil which consisted to ca. 90% of the desired aniline derivative according to analysis by thin layer chromatography.
This crude product was dissolved in a mixture of 400 ml tetrahydrofuran and 100 ml diethyl ether. After cooling to 0° C., 30 ml (215 mmol) of triethylamine were added in one portion. Stirring was continued while 26 g (215 mmol) of pivaloyl chloride were added dropwise within a period of 10 min. The ice bath was removed and the reaction mixture was stirred for 1 h at room temperature. Then, the solvent was removed in vacuo and the residue was suspended in 200 ml 1 N sodium bicarbonate solution. The product was extracted three times with 200 ml portions of dichloromethane, dried (sodium sulfate) and purified by flash chromatography to give 30 g (86%) of the title compound as pink crystals.
MS m/e (%):277 (M+H+, 100).
c) N-[4-Iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide
A solution of 30 g (108 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-propionamide and 58 ml (380 mmol) N,N,N′,N′-tetramethylethylenediamine under argon in 650 ml tetrahydrofuran was cooled in a dry ice bath to −78° C. Within lh, 239 ml (380 mmol) of a 1.6 N n-butyllithium solution in hexane were added dropwise. The reaction mixture was allowed to warm up to −30° C. overnight. After cooling again to −78° C., 43.6 g (170 mmol) iodine dissolved in 60 ml tetrahydrofuran were added dropwise during 15 min. The dry ice bath was replaced by an ice bath and a solution of 90 g (363 mmol) sodium thiosulfate pentahydrate in 250 ml water were added within 10 min when the temperature of the reaction mixture had reached 0° C. Then, 1000 ml diethyl ether were added and the organic layer was separated. The aqueous layer was extracted twice with 500 ml dichloromethane and the combined organic layers were dried (magnesium sulfate) and evaporated. Flash chromatography gave 18.5 g (42%) of the title compound as a light brown oil which crystallized upon standing at room temperature.
MS m/e (%): 403 (M+H+, 100).
d) 2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide
A mixture of 54 g (134 mmol) N-[4-iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide, 420 ml toluene, 150 ml 2 N sodium carbonate solution, 4.63 g (3.9 mmol) tetrakis(triphenylphosphine)palladium(0) and 20.16 g (147 mmol) o-tolylboronic acid was heated under argon at 80° C. for 12 h. After cooling to room temperature, the aqueous phase was separated and washed twice with toluene. The combined organic layers were washed with 50 ml brine, dried (sodium sulfate), evaporated and dried in vacuo to yield 49 g (quantitative) of the title compound as a brown oil.
MS m/e (%): 367 (M+H+, 100).
e) 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine
A suspension of 56 g (152 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide in 1300 ml 3 N hydrochloric acid solution was heated to 90-95° C. overnight. The reaction mixture was cooled to room temperature, washed with three 500 ml portions diethyl ether and filtered over celite. The filtrate was diluted with 500 ml water and was adjusted to pH 7-8 by addition of 28% sodium hydroxide solution under ice cooling. The product was extracted with four 1000 ml portions of dichloromethane. The combined organic layers were washed with 500 ml brine, dried (magnesium sulfate) and evaporated to give 35 g (82%) of the title compound as a light brown oil.
MS m/e (%):283 (M+H+, 100).
f) Methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine
A solution of 35 g (124 mmol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine in 270 ml trimethyl orthoformate and 8 drops trifluoroacetic acid was heated for 3 h at 130° C. The reaction mixture was evaporated and dried in vacuo for 30 min. The residual oil was dissolved in 100 ml tetrahydrofuran and was added dropwise under ice cooling to 9.4 g (248 mmol) lithium aluminum hydride in 300 ml tetrahydrofuran. The reaction mixture was stirred for lh at room temperature, cooled to 0° C. again and acidified (pH 1-2) by addition of 28% hydrochloric acid solution. After stirring for 5 min, 28% sodium hydroxide solution was added to reach pH 10. The solution was filtered over celite, evaporated and purified by flash chromatography to give 23.6 g (64%) of the title compound as a light brown oil.
MS m/e (%):297 (M+H+, 100).
g) 2-(3,5-Bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide
A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane. The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of the title compound as white crystals. M.p. 155-157° C.
MS m/e (%): 579 (M+H+, 100).
h)2-(3,5-Bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide hydrochloride (1:2)
To a solution of 31.6 g (54.6 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide in 250 ml diethyl ether were added under ice cooling 60 ml 3 N hydrochloric acid solution in diethyl ether. After stirring for 15 min at 0° C., the suspension was evaporated to dryness, re-suspended in 100 ml diethyl ether, filtered and dried in vacuo to give 34.8 g (98%) of the title compound as white crystals. M.p. 235-238° C.
MS m/e (%): 579 (M+H+, 100).
……………………………….
2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (Netupitant)

Other general procedures of preparing similar compounds to intermediate 1 of Scheme 1 are also disclosed in U.S. Pat. Nos. 6,303,790, 6,531,597, 6,297,375 and 6,479,483, the entirety of which are incorporated herein by reference.
Synthesis of methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine

Step 1:
13.0 g (82.5 mMol) 6-Chloro-nicotinic acid in 65 ml THF were cooled to 0° C. and 206.3 ml (206.3 mMol) o-tolylmagnesium chloride solution (1M in THF) were added over 45 minutes. The solution obtained was further stirred 3 hours at 0° C. and overnight at room temperature. It was cooled to −60° C. and 103.8 ml (1.8 Mol) acetic acid were added, followed by 35 ml THF and 44.24 g (165 mMol) manganese(III) acetate dihydrate. After 30 minutes at −60° C. and one hour at room temperature, the reaction mixture was filtered and THF removed under reduced pressure. The residue was partitioned between water and dichloromethane and extracted. The crude product was filtered on silica gel (eluent: ethyl acetate/toluene/formic acid 20:75:5) then partitioned between 200 ml aqueous half-saturated sodium carbonate solution and 100 ml dichloromethane. The organic phase was washed with 50 ml aqueous half-saturated sodium carbonate solution, The combined aqueous phases were acidified with 25 ml aqueous HCl 25% and extracted with dichloromethane. The organic extracts were dried (Na2SO4) and concentrated under reduced pressure to yield 10.4 g (51%) of 6-chloro-4-o-tolyl-nicotinic acid as a yellow foam. MS (ISN): 246 (M−H, 100), 202 (M-CO2H, 85), 166 (36).
Step 2:
To a solution of 8.0 g (32.3 mMol) 6-chloro-4-o-tolyl-nicotinic acid in 48.0 ml THF were added 3.1 ml (42.0 mMol) thionylchloride and 143 .mu.l (1.8 mMol) DMF. After 2 hours at 50° C., the reaction mixture was cooled to room temperature and added to a solution of 72.5 ml aqueous ammonium hydroxide 25% and 96 ml water cooled to 0″C. After 30 minutes at 0° C., THF was removed under reduced pressure and the aqueous layer was extracted with ethyl acetate. Removal of the solvent yielded 7.8 g (98%) 6-chloro-4-o-tolyl-nicotinamide as a beige crystalline foam. MS (ISP): 247 (M+H30 , 100).
Step 3:
1.0 g (4.05 mMol) 6-Chloro-4-o-tolyl-nicotinamidein 9.0 ml 1-methyl-piperazine was heated to 100° C. for 2 hours. The excess N-methyl-piperazine was removed under high vacuum and the residue was filtered on silica gel (eluent: dichloromethane) to yield 1.2 g (95%) 6-(4-methyl-piperazin-1yl)-4-o-tolyl-nicotinamide as a light yellow crystalline foam. MS (ISP): 311 (M+H+, 100), 254 (62).
Step 4:
A solution of 0.2 g (0.6 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 1.0 ml methanol was added to a solution of 103 mg (2.6 mMol) sodium hydroxide in 1.47 ml (3.2 mMol) NaOCl (13%) and heated for 2 hours at 70° C. After removal of methanol, the aqueous layer was extracted with ethyl acetate. The combined. organic extracts were dried (Na2SO4), concentrated under reduced pressure and the residue filtered on silica gel (eluent: dichloromethane/methanol 4:1) to yield 100 mg (70%) 6-(4-methyl-piperazine-1-yl)-4o-tolyl-pyridin-3-ylamine as a brown resin. MS (ISP): 283 (M+H+, 100), 226 (42).
Step 5:
2.15 ml (11.6 mMol) Sodium methoxide in methanol were added over 30 minutes to a suspension of 0.85 g (4.6 mMol) N-bromosuccinimide in 5.0 ml dichloromethane cooled to −5° C. The reaction mixture was stirred 16 hours at −5° C. Still at this temperature, a solution of 1.0 g (3.1 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 5.0 ml methanol was added over 20 minutes and stirred for 5 hours. 7.1 ml (7.1 mMol) Aqueous HCl 1N and 20 ml dichloromethane were added. The phases were separated and the organic phase was washed with deionized water. The aqueous phases were extracted with dichloromethane, brought to pH=8 with aqueous NaOH 1N and further extracted with dichloromethane. The latter organic, extracts were combined, dried (Na2SO4) and concentrated to yield 1.08 g (quant.) [6-(4-methyl-piperazin-1yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester as a grey foam. MS (ISP): 341 (M+H+, 100), 284 (35).
Step 6:
A solution of 0.5 g (1.4 mMol) [6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester in 3.0 ml dichloromethane was added over 10 minutes to a solution of 1.98 ml (6.9 mMol) Red-Al.RTM. (70% in toluene) and 2.5 ml toluene (exothermic, cool with a water bath to avoid temperature to go >50° C.). The reaction mixture was stirred 2 hours at 50° C. in CH2Cl2, extracted with ethyl acetate and cooled to 0° C. 4 ml Aqueous NaOH 1N were carefully (exothermic) added over 15 minutes, followed by 20 ml ethyl acetate. The phases were separated and the aqueous phase was extracted with ethyl acetate. The combined organic extracts were washed with deionized water and brine, dried (Na2SO4) and concentrated under reduced pressure to yield 0.37 g (89%) methyl-[6-4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine as an orange resin. MS (ISP): 297 (M+H+, 100).
Synthesis of 2-(3,5-bis-Trifluoromethyl-phenyl)-2-methyl-propionyl Chloride

15.0 g (50 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionic acid were dissolved in 127.5 ml dichloromethane in the presence of 0.75 ml DMF. 8.76 ml (2 eq.) Oxalyl chloride were added and after 4.5 hours, the solution was rotary evaporated to dryness. 9 ml Toluene were added and the resulting solution was again rotary evaporated, then dried under high vacuum yielding 16.25 g (quant.) of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride as a yellow oil of 86% purity according to HPLC analysis. NMR (250 MHz, CDCl3): 7.86 (br s, 1H); 7.77, (br s, 2H, 3 Harom); 1.77 (s, 6H, 2 CH3).
Synthesis of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (Netupitant)

A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane, The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1yl)-4-(o-tolyl)pyridin-3yl)propanamide as white crystals. M.P. 155-157° C.; MS m/e (%): 579 (M+H+, 100).
…………………………………..
http://www.google.com/patents/US20130231315
N OXIDE SYNTHESIS
Synthesis of 5-(2-(3,5-bis(trifluoromethyl)phenyl-N,2-dimethylpropanamido)2-(4-methylpiperazin-1yl)-4-(o-tolyl)pyridine 1-oxide

Step 1:
The solution of 6-chloropyridin-3-amine (115 g, 0.898 mol) and (Boc)2O (215.4 g, 0.988 mol) in 900 mL of dioxane was refluxed overnight. The resulting solution was poured into 1500 mL of water. The resulting solid was collected, washed with water and re-crystallized from EtOAc to afford 160 g tert-butyl (6-chloropyridin-3yl)carbamate as a white solid (Yield: 78.2%).
Step 2:
To the solution of tert-butyl (6-chloropyridin-3-yl)carbamate (160 g, 0.7 mol) in 1 L of anhydrous THF was added n-BuLi (600 mL, L5 ml) at −78° C. under N2 atmosphere. After the addition was finished, the solution was stirred at −78° C. for 30 min, and the solution of I2 (177.68 g, 0.7 mol) in 800 mL of anhydrous THF was added. Then the solution was stirred at −78° C. for 4 hrs, TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and purified by flash chromatography to afford 80 g of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate as a yellow solid (32.3%).
Step 3:
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate (61 g, 0.172 mol) in 300 of anhydrous THF was added 60% NaH (7.6 g, 0.189 mol) at 0° C. under N2 atmosphere. After the addition was finished, the solution was stirred for 30 min, and then the solution of MeI (26.92 g, 0.189 mol) in 100 mL of dry THF was added. Then the solution was stirred at 0° C. for 3 hrs. TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and concentrated to afford 63 g of crude tert-butyl (6-chloro-4-iodopyridin-3-yl)methyl)carbamate used into the following de-protection without the further purification.
Step 4:
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)(methyl)carbamate (62.5 g, 0.172 mol) in 500 mL of anhydrous DCM was added 180 mL of TFA. Then the solution was stirred at room temperature for 4 hrs. Concentrated to remove the solvent, and purified by flash chromatography to afford 45.1 g 6-chloro-4-iodo-N-methylpyridin-3-amine as a yellow solid (Yield: 97.3%).
Step 5:
To the solution of 6-chloro-4-iodo-N-methylpyridin-3-amine (40.3 g, 0.15 mol) and 2-methylbenzene boric acid (24.5 g, 0.18 mol) in 600 mL of anhydrous toluene was added 400 mL of 2 N aq. Na2CO3 solution, Pd(OAc)2 (3.36 g, 15 mmol) and PPh3(7.87 g, 0.03 mmol), The solution was stirred at 100° C. for 2 hrs. Cooled to room temperature, and diluted with water. EtOAc was added to extract twice. The combined organic phases were washed with water and brine consecutively, dried over Na2SO4, concentrated and purified by flash chromatography to afford 19 g 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine as a white solid (Yield: 54.6%).
Step 6:
To the solution of 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine (18.87 g, 81.3 mmol) and DMAP (29.8 g, 243.9 mmol) in 200 mL of anhydrous toluene was added the solution of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride (28.5 g, 89.4 mmol) in toluene under N2 atmosphere. The solution was heated at 120° C. for 23 hrs. Cooled to room temperature, poured into 1 L of 5% aq. NaHCO3 solution, and extracted with EtOAc twice. The combined organic phases were washed by water and brine consecutively, dried. over Na2SO4, filtered and purified by flash chromatography to afford 35 g 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(4-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide as a white solid (Yield: 83.9%).
Step 7:
To the solution of 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(o-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide (5.14 g, 10 mmol) in 60 mL of DCM was added m-CPBA (6.92 g, 40 mmol) at 0° C. under N2 atmosphere. Then the solution was stirred overnight at room temperature. 1 N aq. NaOH solution was added to wash twice for removing the excess m-CPBA. and a side product. The organic phase was washed by brine, dried over Na2SO4, filtered and concentrated to afford 5.11 g of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl-N,2-dimethylpropanamido)-2-chloro-4(o-tolyl)pyridine 1-oxide as a white solid (Yield: 96.4%).
Step 8:
To the solution of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-chloro-4-(o-tolyl)pyridine 1-oxide (5.1 g, 9.62 mmol) in 80 mL of n-BuOH was added N-methylpiperazine (7.41 g, 74.1 mmol) under N2 atmosphere. Then the solution was stirred at 80° C. overnight. Concentrated and purified by flash chromatography to afford 4.98 g 5-(2-(3,5-bis(trifluoromethyl)phenyl-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide as a white solid (Yield: 87.2%), 1HNMR (CDCl3, 400 MHz) δ 8.15 (s, 1H), 7.93 (s, 1H), 7.78 (s, 2H), 7.38 (m, 2H), 7.28 (m, 1H), 7.17 (m, 1H), 7.07 (s, 1H), 5.50 (s, 3H), 2.72 (d, J=4.4 Hz, 4H), 2.57 (m, 3H), 2.40 (s, 3H), 2.23 (s, 3H), 1.45-1.20 (m, 6H).
………………………………….
https://www.google.co.in/patents/US6479483


EXAMPLE 14 2-(3,5-bis-Trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperan-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide Hydrochloride (1:2)
a) 1-Methyl-4-(5-nitro-pyridin-2-yl)-piperazine
To a solution of 20 g (126 mmol) of 2-chloro-5-nitropyridine in 200 ml tetrahydrofuran were added dropwise 35 ml (315 mmol) 1-methylpiperazine within 10 min. The reaction mixture was refluxed for additional 1.5 h. After cooling to room temperature, the solvent was removed in vacuo and the residue was re-dissolved in 200 ml ethyl acetate. The organic phase was washed with 200 ml 1 N sodium bicarbonate solution, dried (magnesium sulfate) and evaporated to give 27.9 g (quantitative) of the title compound as a yellow solid.
MS m/e (%): 223 (M+H+, 100).
b) 2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl-propionamide
To a solution of 27.9 g (125.5 mmol) of 1-methyl-4-(5-nitro-pyridin-2-yl)-piperazine in 400 ml methanol were added 2.6 g of 10% of palladium on activated charcoal. The reaction mixture was hydrogenated (room temperature to ca. 45° C., 1 bar) until the theoretical amount of hydrogen was taken up (about 2 h). The catalyst was filtered off and was washed twice with 100 ml portions of methanol. The filtrate was evaporated in vacuo to give 28 g of a purple oil which consisted to ca. 90% of the desired aniline derivative according to analysis by thin layer chromatography.
This crude product was dissolved in a mixture of 400 ml tetrahydrofuran and 100 ml diethyl ether. After cooling to 0° C., 30 ml (215 mmol) of triethylamine were added in one portion. Stirring was continued while 26 g (215 mmol) of pivaloyl chloride were added dropwise within a period of 10 min. The ice bath was removed and the reaction mixture was stirred for 1 h at room temperature. Then, the solvent was removed in vacuo and the residue was suspended in 200 ml 1 N sodium bicarbonate solution. The product was extracted three times with 200 ml portions of dichloromethane, dried (sodium sulfate) and purified by flash chromatography to give 30 g (86%) of the title compound as pink crystals.
MS m/e (%): 277 (M+H+, 100).
c) N-[4-Iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide
A solution of 30 g (108 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-propionamide and 58 ml (380 mmol) N,N,N′,N′-tetramethylethylenediamine under argon in 650 ml tetrahydrofuran was cooled in a dry ice bath to −78° C. Within 1 h, 239 ml (380 mmol) of a 1.6 N n-butyllithium solution in hexane were added dropwise. The reaction mixture was allowed to warm up to −30° C. overnight. After cooling again to −78° C., 43.6 g (170 mmol) iodine dissolved in 60 ml tetrahydrofuran were added dropwise during 15 min. The dry ice bath was replaced by an ice bath and a solution of 90 g (363 mmol) sodium thiosulfate pentahydrate in 250 ml water were added within 10 min when the temperature of the reaction mixture had reached 0° C. Then, 1000 ml diethyl ether were added and the organic layer was separated. The aqueous layer was extracted twice with 500 ml dichloromethane and the combined organic layers were dried (magnesium sulfate) and evaporated. Flash chromatography gave 18.5 g (42%) of the tide compound as a light brown oil which crystallized upon standing at room temperature.
MS m/e (%): 403 (M+H+, 100).
d) 2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide
A mixture of 54 g (134 mmol) N-[4-iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide, 420 ml toluene, 150 ml 2 N sodium carbonate solution, 4.63 g (3.9 mmol) tetrakis(triphenylphosphine)palladium(0) and 20.16 g (147 mmol) o-tolylboronic acid was heated under argon at 80° C. for 12 h. After cooling to room temperature, the aqueous phase was separated and washed twice with toluene. The combined organic layers were washed with 50 ml brine, dried (sodium sulfate), evaporated and dried in vacuo to yield 49 g (quantitative) of the title compound as a brown oil.
MS m/e (%): 367 (M+H+, 100).
e) 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine
A suspension of 56 g (152 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide in 1300 ml 3 N hydrochloric acid solution was heated to 90-95° C. overnight. The reaction mixture was cooled to room temperature, washed with three 500 ml portions diethyl ether and filtered over celite. The filtrate was diluted with 500 ml water and was adjusted to pH 7-8 by addition of 28% sodium hydroxide solution under ice cooling. The product was extracted with four 1000 ml portions of dichloromethane. The combined organic layers were washed with 500 ml brine, dried (magnesium sulfate) and evaporated to give 35 g (82%) of the title compound as a light brown oil.
MS m/e (%):283 (M+H+, 100).
f) Methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine
A solution of 35 g (124 mmol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine in 270 ml trimethyl orthoformate and 8 drops trifluoroacetic acid was heated for 3 h at 130° C. The reaction mixture was evaporated and dried in vacuo for 30 min. The residual oil was dissolved in 100 ml tetrahydrofuran and was added dropwise under ice cooling to 9.4 g (248 mmol) lithium aluminum hydride in 300 ml tetrahydrofuran. The reaction mixture was stirred for 1 h at room temperature, cooled to 0° C. again and acidified (pH 1-2) by addition of 28% hydrochloric acid solution. After stirring for 5 min, 28% sodium hydroxide solution was added to reach pH 10. The solution was filtered over celite, evaporated and purified by flash chromatography to give 23.6 g (64%) of the title compound as a light brown oil.
MS m/e (%): 297 (M+H+, 100).
g) 2-(3,5-bis-Trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide
A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane. The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of the title compound as white crystals. M.p. 155-157° C.
MS m/e (%): 579 (M+H+, 100).
h) 2-(3,5-bis-Trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide Hydrochloride (1:2)
To a solution of 31.6 g (54.6 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide in 250 ml diethyl ether were added under ice cooling 60 ml 3 N hydrochloric acid solution in diethyl ether. After stirring for 15 min at 0° C., the suspension was evaporated to dryness, re-suspended in 100 ml diethyl ether, filtered and dried in vacuo to give 34.8 g (98%) of the title compound as white crystals. M.p. 235-238° C.
MS m/e (%): 579 (M+H+, 100).
…………………………….
Research and development of an efficient process for the construction of the 2,4,5-substituted pyridines of NK-1 receptor antagonists
Org Process Res Dev 2006, 10(6): 1157
Navari RM.
Drugs. 2013 Mar;73(3):249-62. doi: 10.1007/s40265-013-0019-1. Review.
-
Hoffmann-Emery F, Hilpert H, Scalone M, Waldmeier P.
J Org Chem. 2006 Mar 3;71(5):2000-8.
Hoffmann T, Bös M, Stadler H, Schnider P, Hunkeler W, Godel T, Galley G, Ballard TM, Higgins GA, Poli SM, Sleight AJ.
Bioorg Med Chem Lett. 2006 Mar 1;16(5):1362-5. Epub 2005 Dec 5.
http://www.sciencedirect.com/science/article/pii/S0960894X05014824
…………………………………….……………………………………………………….
| US6897226 * | 9 Jul 2003 | 24 May 2005 | Hoffmann-La Roche Inc. | NK-1 receptor active amine oxide prodrugs |
| US7211579 * | 15 Mar 2006 | 1 May 2007 | Hoffmann-La Roche Inc. | NK-1 receptor antagonists |
| US8426450 | 23 May 2012 | 23 Apr 2013 | Helsinn Healthcare Sa | Substituted 4-phenyl pyridines having anti-emetic effect |
| WO2011061622A1 | 18 Nov 2010 | 26 May 2011 | Helsinn Healthcare S.A. | Compositions for treating centrally mediated nausea and vomiting |
| WO2013057554A2 | 10 Oct 2012 | 25 Apr 2013 | Helsinn Healthcare Sa | Therapeutic combinations of netupitant and palonosetron |
| US8426450 | 23 May 2012 | 23 Apr 2013 | Helsinn Healthcare Sa | Substituted 4-phenyl pyridines having anti-emetic effect |
| WO2011061622A1 | 18 Nov 2010 | 26 May 2011 | Helsinn Healthcare S.A. | Compositions for treating centrally mediated nausea and vomiting |
| WO2013057554A2 | 10 Oct 2012 | 25 Apr 2013 | Helsinn Healthcare Sa | Therapeutic combinations of netupitant and palonosetron |
……………………………………………………………………………………….

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007
