
Home » Phase3 drugs (Page 20)
Category Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
MEPOLIZUMAB….GSK to file severe asthma drug by year end
The first non-inhaled treatment for a difficult-to-treat form of severe asthma is getting closer to market after GlaxoSmithKline said it would initiate global filings for the drug at the end of this year, on the back of strong late-stage clinical data.
Mepolizumab – a monoclonal antibody that inhibits interleukin 5 – is being investigated as a treatment for severe eosinophilic asthma in patients who experience exacerbations despite high-dose oral or inhaled corticosteroids (ICS) and an additional controller such as long-acting beta-2 agonist.
Read more at: http://www.pharmatimes.com/Article/14-03-13/GSK_to_file_severe_asthma_drug_by_year_end.aspx#ixzz2vuANtYaK
Follow us: @PharmaTimes on Twitter
Mepolizumab (proposed trade name Bosatria) is a humanized monoclonal antibody that recognizes interleukin-5 (IL-5), and is used to treat certain kinds of asthma and white blood cell diseases.
 IL 5
IL 5
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | IL-5 |
Recent studies have concluded that mepolizumab may improve exacerbations in patients with severe eosinophilic asthma, an adult-onset asthma which represents less than 5% of all asthma.
IL-5 is a chemical messenger in the immune system that stimulates the growth of eosinophils. In eosinophilic asthma, eosinophils are present in the lungs. When mepolizumab was given to people with eosinophilic asthma, it eliminated eosinophils from the bloodstream,and reduced eosinophils in the lungs and bone marrow. Mepolizumab also reduced the number of asthma exacerbations, and reduced the need for corticosteroids.[1]Mepolizumab improved the quality of life, but the improvement was “not clinically meaningful,” according to a reviewer.[2] [3]
In a recent multi-centre, double-blinded, randomised, controlled trial study of Mepolizumab in severe eosinophilic asthma, Mepolizumab reduced the number of clinically significant exacerbations compared to a placebo. Additionally Mepolizumab reduced sputum and blood eosinophil counts and was shown to be safe for up to 12 months.[4]

Mepolizumab is also in development for the management of hypereosinophilic syndrome by GlaxoSmithKline (GSK) and has received orphan drug designation by the FDA.[5] Mepolizumab has been shown to reduce the need for corticosteroids and improve symptoms in FIP1L1/PDGFRA negative hypereosinophilic syndrome.[6]
UK pharma giant GlaxoSmithKline (LSE: GSK) says that a pivotal Phase III study of mepolizumab, an investigational IL-5 antagonist monoclonal antibody, met its primary endpoint of reduction in the frequency of exacerbations, in patients with severe eosinophilic asthma.
Mepolizumab could add £400 million ($668 million) to GSK’s revenue by 2021, according to estimates from Barclays reported by The Wall Street Journal. Analysts from Deutsche Bank forecast £300 million in mepolizumab sales by 2018 for the company, already a leader in the asthma treatment sector.

The study (MEA115588) evaluated the efficacy of two-dose regimens of mepolizumab in the treatment of patients with severe eosinophilic asthma. Patients remained on their current asthma maintenance therapy throughout the study and were randomized to receive either mepolizumab 75mg intravenous (IV), 100mg subcutaneous (SC), or placebo every four weeks.
For the primary end point, both mepolizumab treatment arms showed statistically significant reductions in the frequency of clinically significant exacerbations of asthma compared to placebo (75mg IV, 47%, p<0.001; 100mg SC, 53%, p<0.001).
Adverse events reported in the study were similar across all treatment groups. The most common reported adverse events across all treatment groups were nasopharyngitis, headache, upper respiratory tract infection and asthma. The frequency of adverse events was 83% in the placebo group, 84% in the mepolizumab 75mg IV and 78% in the mepolizumab 100mg SC group. The frequency of serious adverse events was 14% in the placebo group, 7% in the mepolizumab 75mg IV and 8% in the mepolizumab 100mg SC group.
Backs up earlier studies; regulatory filing mooted at year end
Dave Allen, head of GSK Respiratory Therapy Area Unit, R&D, said: “We are really pleased to have generated further positive data on mepolizumab, consistent with the findings from our earlier exacerbation study. We now have two studies showing a reduction in exacerbations in a specific group of patients with a severe form of asthma who continue to exacerbate despite treatment with high doses of their current maintenance therapies. This is very positive news for patients. For GSK it is exciting that this is the first non-inhaled treatment for severe asthma and we will be progressing towards global filings at the end of the year.”
In addition, a second Phase III study (MEA115575) designed to evaluate the use of mepolizumab 100mg SC, every four weeks in comparison to placebo in reducing daily oral corticosteroid use while maintaining asthma control also met its primary endpoint. The study showed that patients on mepolizumab 100mg SC were able to achieve greater reductions in their maintenance oral corticosteroid dose during weeks 20-24 compared to patients on placebo (p =0.008), while maintaining asthma control.
In this study adverse events were similar across treatment groups. The most common reported adverse events in the two treatment groups were headache, nasopharyngitis, bronchitis, sinusitis, fatigue and asthma. The frequency of adverse events was 92% in the placebo and 84% in the mepolizumab treatment group. Frequency of serious adverse events was 18% in the placebo group and 1% in the mepolizumab group.
Mepolizumab Useful in Refractory Eosinophilic Asthma, a Rare Subtype of Asthma
Eosinophil.
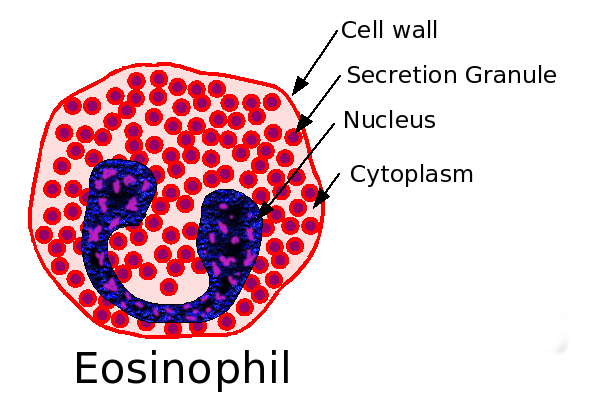
Eosinophil.Eeosinophilic form of asthma represents less than 5% of cases of adult-onset asthma and is difficult to treat.
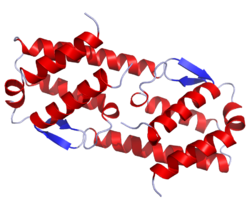
Crystal structure of human IL-5. .
Mepolizumab reduced the number of blood and sputum eosinophils and allowed prednisone sparing in patients who had asthma with sputum eosinophilia despite prednisone treatment.
Mepolizumab therapy reduced exacerbations by 43% and improved Asthma Quality of Life Questionnaire (AQLQ) scores in patients with refractory eosinophilic asthma.
Eosinophils may have a role as important effector cells in the pathogenesis of severe exacerbations of asthma in patients with eosinophilic asthma.
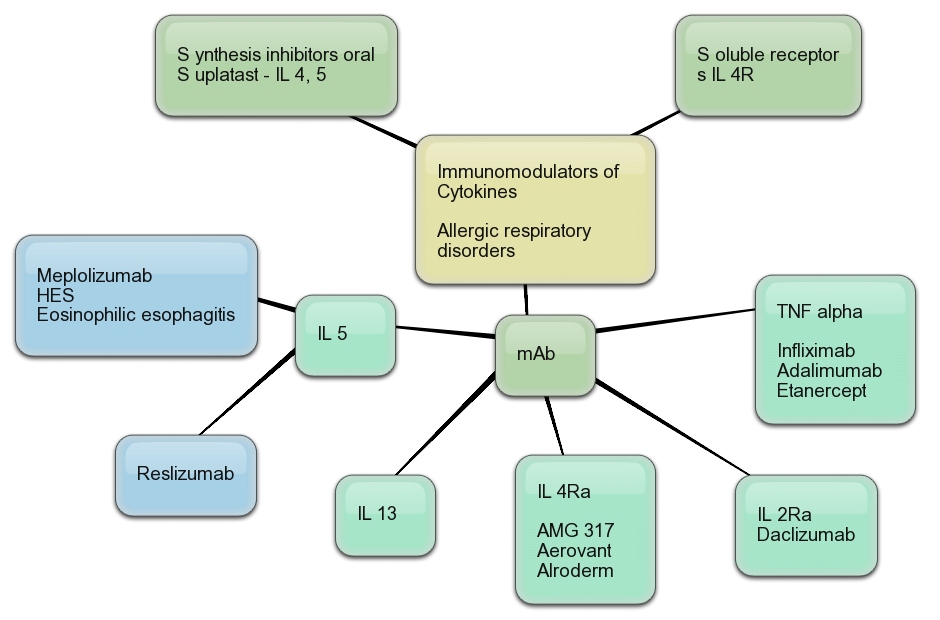
Cytokine targets for immunomodulators for allergic disorders.
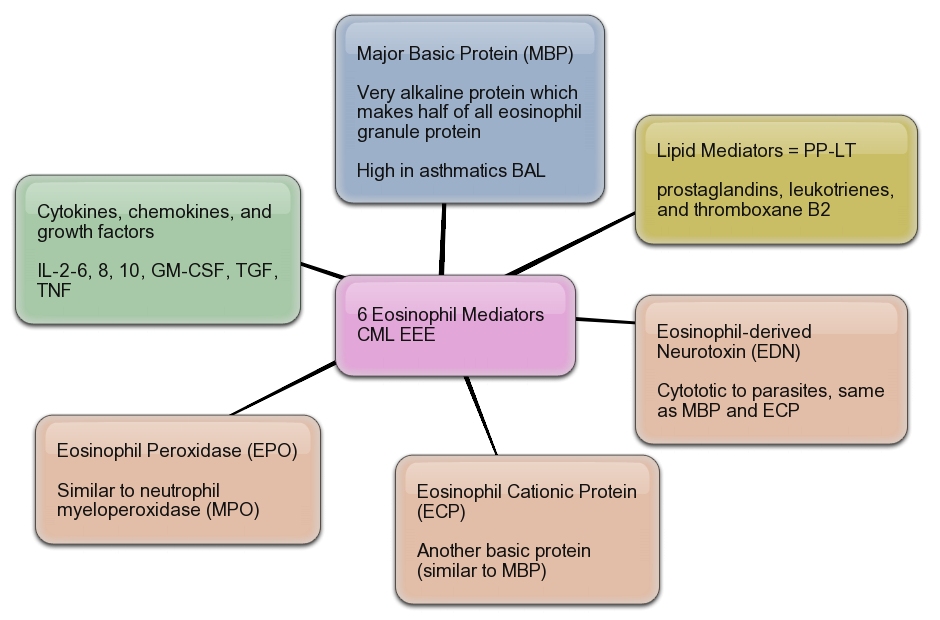
Mediators from Eosinophils
References
- Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009 Mar 5;360(10):973-84.
- Nair P, Pizzichini MM, Kjarsgaard M, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009 Mar 5;360(10):985-93.
- Eosinophils in asthma – closing the loop or opening the door? Sally E. Wenzel, N Engl J Med. 2009 Mar 5;360(10):1026-7.
- Pavord, Ian D; Korn, Stephanie; Howarth, Peter; Bleecker, Eugene R; Buhl, Roland; Keene, Oliver N; Ortega, Hector; Chanez, Pascal (August 2012). “Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial”. The Lancet 380 (9842): 651–659. doi:10.1016/S0140-6736(12)60988-X.
- Phase III study of Bosatria (mepolizumab) showed disease control with reduced corticosteroid use in hypereosinophilic syndrome
- http://content.nejm.org/cgi/content/abstract/358/12/1215 Rothenberg et al 2008

PIRODAVIR



A mixture of 10.4 parts of 3-chloro-6-methylpyridazine, 22.4 parts of ethyl 4-[2-(4-piperidinyl)ethoxy]benzoate butanedioate (1:1), 8.6 parts of sodium carbonate and 0.9 parts of N,N-dimethylformamide was stirred for 3 hours in an oil bath at .+-.150.degree. C. After cooling, water and dichloromethane were added and the layers were separated. The organic layer was dried, filtered and evaporated. The residue was purified by column chromatography over silica gel using a mixture of trichloromethane and ethanol (99:1 by volume) as eluent. The pure fractions were collected and the eluent was evaporated. The residue was crystallized from a mixture of 2,2′-oxybispropane and 2-propanone (75:25 by volume). The precipitated product was filtered off and dried, yielding 17 parts (56.8%) of ethyl 4-[2-[1-(6-methyl-3-pyridazinyl)-4-piperidinyl]-ethoxy]benzoate; mp. 130.1.degree. C. (comp. 1).

Scheme 1. Synthesis of Pirodavir (3) and Related Compounds
| US2985657 * | Oct 12, 1959 | May 23, 1961 | Paul A J Janssen | 1-(aroylalkyl)-4-heterocyclylpiperazines |
| US4068383 * | Sep 30, 1976 | Jan 17, 1978 | Hoechstmass Balzer Gmbh & Co. | Tape measure reel |
| US4451476 * | Oct 17, 1983 | May 29, 1984 | Sterling Drug Inc. | Isoxazoles as antiviral agents |
| US4604127 * | May 15, 1985 | Aug 5, 1986 | Eli Lilly And Company | Herbicidal pyridazinylimidazolidinone compounds |
| EP0137242A2 * | Aug 20, 1984 | Apr 17, 1985 | Sterling Winthrop Inc. | (Substituted) Phenyl-aliphatic-isoxazoles useful as antiviral agents and preparation thereof |
| EP0156433A2 * | Mar 15, 1985 | Oct 2, 1985 | Janssen Pharmaceutica N.V. | Anti-virally active pyridazinamines |
| EP0211457A2 * | Jul 9, 1986 | Feb 25, 1987 | Janssen Pharmaceutica N.V. | Novel (4-substituted-piperazinyl)pyridazines |
| JPS5877866A * | Title not available |
BARDOXOLONE METHYL

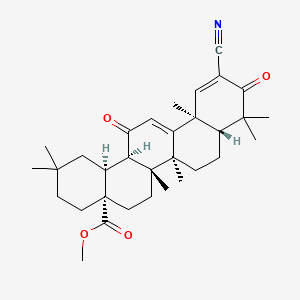
BARDOXOLONE METHYL
- Molecular FormulaC32H43NO4
- Average mass505.688 Da
Methyl 2-cyano-3,12-dioxooleana-1,9(11)dien-28-oate
methyl 2-cyano-3, 12-dioxooleana-1,9(11)-dien-28-oate
2-Cyano-3,12-dioxoolean-1,9(11)-dien-28-oic acid methyl ester
(6aR,6bS,8aR,12aS,14aR,14bS)-11-Cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,3,4,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-hexadecahydropicene-4a(2H)-carboxylic acid methyl ester
BARD
CDDO-Me
Methyl-CDDO
NSC-713200
RTA-402
TP-155C
218600-53-4 CAS
218600-44-3 (free acid)
Treatment of pulmonary arterial hypertension (PAH), diabetic nephropathies and hereditary nephritis, Phase 3

Compounds were synthesized as below:

Scheme 1

Scheme 2
a: HCO2Et/MeONa/THF,b: PhSeCl/AcOEt; 30%H202/THF,c: NH2OH-HCI EtOH/H2O, d: MeONa/MeOH/Et2O,e: KOH/MeOH,f: Jones,g:HCO2Et/MeONa/PhH,h: Lil/DMF Compound 10 was prepared by formylation of OA (Compound 9) (Simonsen and Ross, 1957) with ethyl formate in the presence of sodium methoxide in THF (Clinton et al., 1961). Compound 7 was obtained by introduction of a double bond at C-l of Compound 10 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30%) hydrogen peroxide (Sharpless et al, 1973). Compound 11 was synthesized from Compound 10 by addition of hydroxylamine in aqueous ethanol; cleavage of Compound 11 with sodium methoxide gave Compound 12 (Johnson and Shelberg, 1945). Compound 14 was prepared from Compound 13 (Picard et al, 1939) by alkali hydrolysis followed by Jones oxidation. Compound 15 was prepared by formylation of Compound 14 with ethyl formate in the presence of sodium methoxide in benzene. Compound 16 was synthesized from Compound 15 by addition of hydroxylamine. Cleavage of 16 with sodium methoxide gave Compound 17. Compound 6 (CDDO) was prepared by introduction of a double bond at C-l of Compound 17 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30% hydrogen peroxide, followed by halogenolysis with lithium iodide in DMF (Dean, P.D.G., 1965).
Bardoxolone methyl (also known as “RTA 402” and “CDDO-methyl ester”) is an orally-available first-in-class synthetic triterpenoid. It is an inducer of the Nrf2 pathway, which can suppress oxidative stress and inflammation, and is undergoing clinical development for the treatment of advanced chronic kidney disease (CKD) in type 2 diabetes mellitus patients.
Bardoxolone methyl was previously being investigated by Reata Pharmaceuticals, Inc. in partnership with Abbott Laboratories and Kyowa Hakko Kirin, as an experimental therapy for advanced chronic kidney disease (CKD) in type 2 diabetes mellitus patients. Reata, in consultation with the BEACON Steering Committee, has decided to terminate the Phase 3 BEACON trial of bardoxolone methyl in patients with stage 4 chronic kidney disease and type 2 diabetes. This decision was made based upon a recommendation of the Independent Data Monitoring Committee (IDMC) to stop the trial “for safety concerns due to excess serious adverse events and mortality in the bardoxolone methyl arm.” [1][2][3][4]
RTA-402 is a triterpenoid anti-inflammatory agent in phase II trials at Reata Pharmaceuticals for the treatment of pulmonary arterial hypertension.
This company and M.D. Anderson Cancer Center had been evaluating clinically the product for the treatment of lymphoma. Reata had been evaluating the compound in combination with gemcitabine in patients with unresectable pancreatic cancer and melanoma. Preclinical studies were also being conducted by Reata for the treatment of inflammatory bowel disease (IBD) and autoimmune disease. Reata Pharmaceuticals and Kyowa Hakko Kirin had been conducting phase II clinical studies for the treatment of diabetic nephropathy. Reata and Abbott also had been conducting phase III clinical trials for delaying progression to end-stage renal disease in patients with chronic kidney disease and type 2 diabetes; however, in 2012 these trials were discontinued due to serious adverse events and mortality. Phase II clinical trials for this indication were discontinued by Kyowa Hakko Kirin in Japan. The compound had been in early clinical studies for the treatment of multiple myeloma; however, no recent development has been reported for this indication. Phase I clinical trials for the treatment of solid tumors have been completed.
RTA-402 has demonstrated a wide variety of potentially therapeutic mechanisms, including inhibition of inducible nitric oxide synthase and cyclooxygenase expression, stimulation of expression of cytoprotective enzymes such as NAD(P)H quinine oxidoreductase and hemeoxygenase-1, and reduction in pSTAT3 levels. In cancer patients, the drug candidate exploits fundamental physiological differences between cancerous and non-cancerous cells by modulating oxidative stress response pathways. Due to this mechanism, RTA-402 is toxic to cancer cells, but induces protective antioxidant and anti-inflammatory responses in normal cells. In previous studies, the compound was shown to inhibit growth and cause regression of cancerous tumors as a single agent and, in combination with radiation and chemotherapy, to suppress radiation and chemotherapy-induced toxicities in normal tissues and cause minimal toxicity in non-human primates when dosed orally at very high doses for 28 consecutive days.
An analog of RTA-401, RTA-402 is a compound found in medicinal plants with a greater potency than the natural product.
RTA-401 was originally developed at Dartmouth College and M.D. Anderson Cancer Center. In November 2004, Reata completed a license agreement with these organizations, and was granted exclusive worldwide rights to this new class of anticancer compounds. In 2008, orphan drug designation was assigned by the FDA for the treatment of pancreatic cancer. In 2010, the compound was licensed to Kyowa Hakko Kirin by Reata Pharmaceuticals in China, Japan, Korea, Thailand and Southeast Asian countries for the treatment of chronic kidney disease. Abbott acquired rights to develop and commercialize the drug outside US, excluding certain Asian markets.
Phase 1
Bardoxolone methyl was first advanced into the clinic to assess its anticancer properties. In two Phase 1 trials that included 81 oncology patients, bardoxolone methyl reduced serum creatinine levels, with a corresponding improvement in estimated glomerular filtration rate (eGFR). Improvements were more pronounced in a subset of patients with established CKD and were maintained over time in patients who continued on bardoxolone methyl therapy for 5 months. Based on these observed effects and the well-described role of oxidative stress and inflammation in CKD, especially in type 2 diabetes, it was hypothesized that bardoxolone methyl could improve renal function in CKD patients with type 2 diabetes.[5]
Phase 2
A multi-center, double-blind, placebo-controlled Phase 2b clinical trial (BEAM) conducted in the US studied 227 patients with moderate to severe CKD (eGFR 20 – 45 ml/min/1.73m²) and type 2 diabetes. The primary endpoint was change in estimated GFR following 24 weeks of treatment. Following 24 weeks, patients treated with bardoxolone methyl experienced a mean increase in estimated GFR of over 10 ml/min/1.73m², compared with no change in the placebo group. Approximately three-quarters of bardoxolone methyl treated patients experienced an improvement in eGFR of 10 percent or more, including one-quarter who saw a significant improvement of 50% or more compared to less than 2% of patients on placebo. Adverse events were generally manageable and mild to moderate in severity. The most frequently reported adverse event in the bardoxolone methyl group was muscle spasm. Final data was published in The New England Journal of Medicine.
Concerns have been raised whether there is a true improvement in kidney function because of the significant weight loss of the patients in the active-treatment-group that ranged from 7.7-10.1 kg (7-10% of the initial body weight) and whether this weight loss in patients receiving bardoxolone included muscle wasting with a commensurate decrease in the serum creatinine level. In that case the decrease in creatinine would not necessarily be a true improvement in kidney function.[6][7][8][9][10]
Phase 3
A multinational, double-blind, placebo-controlled Phase 3 outcomes study (BEACON) was started in June 2011, testing bardoxolone methyl’s impact on progression to ESRD or cardiovascular death in 1600 patients with Stage 4 CKD (eGFR 15 – 30 ml/min/1.73m²) and type 2 diabetes. This phase 3 trail was halted in October 2012 because of adverse effects (namely a higher cardiovascular mortality in the treatment arm).[11]
Mechanism of action
Bardoxolone methyl is an inducer of the KEAP1–Nrf2 pathway.
PAPER
http://modernsteroid.blogspot.com/2012/04/synthetic-oleane-triterpenoids-as.html


Click to access ol400399x_si_001.pdf

1. To a stirred solution of oleanolic acid (22.8 grams, 0.05 mol, 1.0 equiv) in dimethyl formamide (200 mL) was
added powdered K2CO3 (20.7 grams, 0.15 mol, 3.0 equiv) slowly upon stirring, and the reaction mixture was allowed to
cool to 0 o
C. To the stirred suspension was added iodomethane (3.4 mL, 0.055 mol, 1.1 equiv) slowly, and after the
completion of addition, the reaction was allowed to warm to room temperature overnight. After the completion of the
reaction, dimethyl formamide was removed by distillation. The resulting solid mixture was dissolved in methylene
chloride (1 L) and washed with water (4 x 100 mL) and brine (1 x 100 mL). The organics was dried over Na2SO4 and the
solvent was removed to give the crude product 8 as a white solid, which was used directly for the next step without
further purifications.
2. To a stirred suspension of ester 8 (11.8 grams, 0.025 mol, 1.0 equiv) obtained above in anhydrous dimethyl
sulfoxide (250 mL) was added iodoxybenzoic acid (21.0 grams, 0.075 mol, 3.0 equiv) and fluorobenzene (5 mL). The
resulting suspension was heated to 85 o
C under nitrogen for 24 hours. After the completion of the reaction, it was
quenched with 20% aqueous sodium thiosulfate (200 mL). The resulting mixture was extracted with methylene chloride
(4 x 150 mL), the combined organic extracts were washed with saturated NaHCO3 (100 mL) and brine (100 mL), and
dried over Na2SO4. The solvent was removed to give the crude product 14 as yellowish solid, which was used directly for
the next step without further purifications.
3. To a stirred solution of 14 (9.32 grams, 0.02 mol, 1.0 equiv) in methylene chloride (100 mL) was slowly added mchloroperbenzoic
acid (6.4 grams, ~70% purity, 0.026 mol, 1.3 equiv) at 0 o
C. After the completion of addition, the
reaction was allowed to warm to room temperature and kept stirring for 24 hours. After the completion of the reaction,
the reaction mixture was diluted with methylene chloride (300 mL), and the resulting mixture was washed with 20%
aqueous sodium thiosulfate (3 x 100 mL), 10% potassium carbonate (2 x 100 mL), and brine (100 mL). The organics were
dried over Na2SO4 and the solvent was removed to give crude mixture of 15 and 16 as yellowish solid, which was used
directly for the next step without purifications.
4. To the resulting solution of 15 and 16 obtained above in acetic acid (50 mL) was added dropwise hydrobromic
acid (1.0 mL, 0.009 mol, 0.44 equiv) at room temperature. The reaction mixture was then heated to 35 o
C, and bromine
(5.8 mL, 0.05 mol, 2.4 equiv) was thus added dropwise. The resulting reaction mixture was kept stirring for another 24 h.
After completion of the reaction, the acid was removed under vacuum. And the residue was then quenched with 20%
aqueous sodium thiosulfate (100 mL), and extracted with methylene chloride (4 x 100 mL). The combined organic
extracts were washed with saturated sodium bicarbonate (2 x 50 mL), brine (1 x 50 mL), and dried over Na2SO4. The
solvent was removed to give crude bromo enone 17 as yellowish to yellow solid, which can be used directly for the next
step without further purification or subjected to flash column chromatography to give pure bromo enone 17 as a
yellowish solid.
5. To a stirred solution of bromo enone 17 (5.8 grams, 10.0 mmol, 1.0 equiv) in anhydrous dimethyl formamide (80
mL) was added copper (I) cyanide (1.0 grams, 11.0 mmol, 1.1 equiv) and potassium iodide (328 mg, 2.0 mmol, 0.20
equiv), and the resulting reaction mixture was heated to 120 o
C for 24 h. After the completion of reaction, it was cooled
to room temperature, quenched with water (200 mL), and diluted with ethyl acetate (500 mL). The organic phase was
washed with saturated NaHCO3 (2 x 80 mL), brine (80 mL), and dried over Na2SO4. Removal of solvent and flash column
chromatography over silica gel using hexanes:EtOAc (2:1) to give bardoxolone methyl (1) as a yellowish solid.
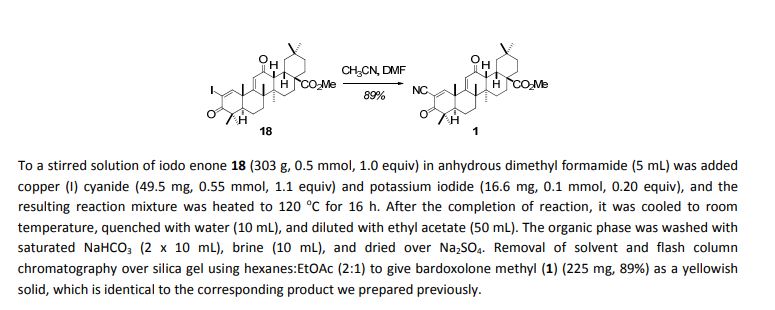
After the completion of the reaction, it was cooled to room temperature and
quenched with 20% aqueous sodium thiosulfate (20 mL). It was extracted with methylene chloride (3 x 20 mL), the
combined organic extracts were washed with saturated aqueous NaHCO3 (10 mL), brine (10 mL), and dried over Na2SO4.
Removal of solvent and flash column chromatography over silica gel using hexanes:EtOAc (4:1 & 2:1) to give iodo enone
18 (509 mg, 84%) as a yellowish solid. 1H NMR (500 MHz, CDCl3) δ 8.12 (s, 1H), 6.00 (s, 1H), 3.70 (s, 3H), 3.04 (dd, 1H, J1 =
10.0 Hz, J2 = 3.7 Hz), 2.92 (d, 1H, J = 4.6 Hz), 1.63-1.94 (m, 9H), 1.46-1.62 (m, 3H), 1.43 (s, 3H), 1.18-1.36 (m, 3H), 1.30 (s,
3H), 1.23 (s, 3H), 1.17 (s, 3H), 1.02 (s, 3H), 1.00 (s, 3H), 0.90 (s, 3H); 13C NMR (500 MHz, CDCl3) δ 199.6, 196.9, 178.4,
170.3, 163.5, 124.1, 102.3, 52.1, 49.9, 48.4, 47.4, 46.4, 45.9, 45.4, 42.3, 36.0, 34.7, 33.5, 33.0, 32.1, 31.7, 30.9, 28.3, 28.2,
27.3, 24.8, 23.3, 22.9, 22.4, 21.9, 18.8; FT-IR (solution, CDCl3, cm-1): 2952, 2869, 2253, 1717, 1659, 1469, 1386, 907, 732,
651, 623, 443; HRMS-ESI (calcd. for C31H44IO4 [M+H]+
) 607.2284, found 607.2280.
CLIP

PATENT
In a preferred embodiment, such compounds include derivatives of ursolic acid and oleanoic acid. In a particularly preferred embodiment, derivatives of OA, e.g., 2-cyano-3,12-dioxoolean-l,9-dien-28oic acid (CDDO):

have been found to be effective in suppression of human breast cancer cell growth, and highly potent in many vitro assay systems such as: suppression of nitric oxide and prostaglandin production in macrophages, inhibition of growth of human breast cancer cells, suppression of nitric oxide formation in rat prostate cells, and suppression of prostaglandin formation in human colon fibroblasts, as detailed in the Figures.
Compounds were synthesized as below:
Scheme 1
Scheme 2
a: HCO2Et/MeONa/THF,b: PhSeCl/AcOEt; 30%H202/THF,c: NH2OH-HCI EtOH/H2O, d: MeONa/MeOH/Et2O,e: KOH/MeOH,f: Jones,g:HCO2Et/MeONa/PhH,h: Lil/DMF Compound 10 was prepared by formylation of OA (Compound 9) (Simonsen and Ross, 1957) with ethyl formate in the presence of sodium methoxide in THF (Clinton et al., 1961). Compound 7 was obtained by introduction of a double bond at C-l of Compound 10 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30%) hydrogen peroxide (Sharpless et al, 1973). Compound 11 was synthesized from Compound 10 by addition of hydroxylamine in aqueous ethanol; cleavage of Compound 11 with sodium methoxide gave Compound 12 (Johnson and Shelberg, 1945). Compound 14 was prepared from Compound 13 (Picard et al, 1939) by alkali hydrolysis followed by Jones oxidation. Compound 15 was prepared by formylation of Compound 14 with ethyl formate in the presence of sodium methoxide in benzene. Compound 16 was synthesized from Compound 15 by addition of hydroxylamine. Cleavage of 16 with sodium methoxide gave Compound 17. Compound 6 (CDDO) was prepared by introduction of a double bond at C-l of Compound 17 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30% hydrogen peroxide, followed by halogenolysis with lithium iodide in DMF (Dean, P.D.G., 1965).
PATENT
WO2009/146216 A2,

Compounds 401, 402, 404, 402-04, 402-35 and 402-56 can be prepared according to the methods taught by Honda et al. (1998), Honda et al. (2000b), Honda et al. (2002), Yates et al. (2007), and U.S. Patent 6,974,801, which are all incorporated herein by reference. The synthesis of the other compounds are disclosed in the following applications, each of which is incorporated herein by reference: U.S. Application Nos. 61/046,332, 61/046,342, 61/046,363, 61/046,366, 61/111,333, 61/111,269, and 61/111,294. The synthesis of the other compounds are also disclosed in the following separate applications filed concurrently herewith, each of which is incorporated herein by reference in their entireties: U.S. Patent Application by Eric Anderson, Xin Jiang, Xiaofeng Liu; Melean Visnick, entitled “Antioxidant Inflammation Modulators: Oleanolic Acid Derivatives With Saturation in the C- Ring,” filed April 20, 2009; U.S. Patent Application by Eric Anderson, Xin Jiang and Melean Visnick, entitled “Antioxidant Inflammation Modulators: Oleanolic Acid Derivatives with Amino and Other Modifications At C-17,” filed April 20, 2009; U.S. Patent Application by Xin Jiang, Xioafeng Liu, Jack Greiner, Stephen S. Szucs, Melean Visnick entitled, “Antioxidant Inflammation Modulators: C-17 Homologated Oleanolic Acid Derivatives,” filed April 20, 2009.
PAPER
Chemical Communications, 2011 , vol. 47, 33 p. 9495 – 9497
http://pubs.rsc.org/en/Content/ArticleLanding/2011/CC/c1cc11633a#!divAbstract
http://www.rsc.org/suppdata/cc/c1/c1cc11633a/c1cc11633a.pdf NMR GIVEN
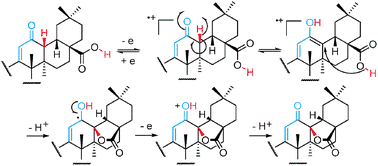
2-Cyano-3,12-dioxooleana-1,9(11)-dien-28-oate (CDDO)
A mixture of 1 (0.25 g, 0.51 mmol) and DDQ (0.12 g, 0.51 mmol) in anhydrous benzene (20 mL) was
refluxed for 15 min. After filtration, the filtrate was evaporated in vacuo to give a residue, which was
subjected to flash column chromatography (petroleum ether/EtOAc) to give CDDO as an amorphous
solid (0.23 g, 91%). The title compound was known as CAS 218600-44-3
m.p. 180-182 °C;
ESI-MS: 490 [M-H]-, 492 [M+H]+;
1H NMR (300M Hz, CDCl3, 25 °C, TMS): δ 8.05 (1H, s), 5.99 (1H, s), 3.03-2.98 (2H, m), 1.55,1.38,
1.34, 1.22, 1.00, 0.91, 0.85 (each 3H,s ,CH3) ppm.
PAPER
SYNTHESIS
Journal of Medicinal Chemistry, 2000 , vol. 43, 22 p. 4233 – 4246
http://pubs.acs.org/doi/full/10.1021/jm0002230

Bioorganic and Medicinal Chemistry Letters, 1998 , vol. 8, 19 p. 2711 – 2714
http://www.sciencedirect.com/science/article/pii/S0960894X9800479X

PAPER
Bioorganic and Medicinal Chemistry Letters, 2005 , vol. 15, # 9 p. 2215 – 2219
http://www.sciencedirect.com/science/article/pii/S0960894X05003306

PATENT
Method of synthesis of CDDO. CDDO may be synthesized by the scheme outlined below.

Methyl-CDDO. Methyl-CDDO (CDDO-Me), the C-28 methyl ester of CDDO, also exerts strong antiproliferative and apoptotic effects on leukemic cell lines and in primary AML samples in vitro as well as induces monocytic differentiation of leukemic cell lines and some primary AMLs. Thus, CDDO-Me provides chemotherapy for the treatment of leukemias. The present invention demonstrates that this effect is profoundly increased by combination of CDDO-Me with other chemotherapeutic agents. These include retinoids such as ATRA, 9-cis retinoic acid, , LG100268, LGD1069 (Targretin, bexarotene), fenretinide [N-(4- hydroxyphenyl)retinamide, 4-HPR], CD437 and other RXR and RAR-specific ligands. This combination also increases ara-C cytotoxicity, further reduces AML colony formation, inhibits ERK phosphorylation and promotes Bcl-2 dephosphorylation, and inhibits in vitro angiogenesis. The ability of CDDO-Me in combination with retinoids to induce differentiation in leukemic cells in vitro show that these compounds may have similar in vivo effects. The anti-angiogenic properties of CDDO-Me further increase its potent anti-leukemia activity in combination with retinoids. Furthermore, CDDO-Me was found to be more potent at lower concentrations than CDDO.
Method of synthesis of CDDO-Me.
CDDO-Me may be synthesized by the scheme outlined below.

The present invention provides combinations of CDDO-compounds and chemotherapeutic agents that are useful as treatments for cancers and hematological malignancies. In one embodiment, the chemotherapeutics are retinoids. As CDDO- compounds are PPARγ ligands and PPARγ is known to be altered in many types of cancers, the inventors contemplate, that ligation of PPARγ in combination with retinoids such as, RXR-specific ligands, provides a mechanistic basis for maximal increase in transcriptional activity of the target genes that control apoptosis and differentiation. The CDDO-compounds and retinoids in combination demonstrate an increased ability to induce differentiation, induce cytotoxicity, induce apoptosis, induce cell killing, reduce colony formation and inhibit the growth of several types of leukemic cells.
PAPER
Efficient and scalable synthesis of bardoxolone methyl (cddo-methyl ester).
Bardoxolone methyl (2-cyano-3,12-dioxooleane-1,9(11)-dien-28-oic acid methyl ester; CDDO-Me) (1), a synthetic oleanane triterpenoid with highly potent anti-inflammatory activity (levels below 1 nM), has completed a successful phase I clinical trial for the treatment of cancer and a successful phase II trial for the treatment of chronic kidney disease in type 2 diabetes patients. Our synthesis of bardoxolone methyl (1) proceeds in ∼50% overall yield in five steps from oleanolic acid (2), requires only one to two chromatographic purifications, and can provide gram quantities of 1.

References
- “Bardoxolone methyl – Oral, Once Daily AIM for Renal/Cardiovascular/Metabolic Diseases”. Reata Pharmaceuticals. Archived from the original on 15 July 2011. Retrieved June 2, 2011.
- “Abbott and Reata Pharmaceuticals Announce Agreement to Develop and Commercialize Bardoxolone Methyl for Chronic Kidney Disease Outside the U.S.” (Press release). Reata Pharmaceuticals. September 23, 2010. Retrieved June 2, 2011.
- “Reata Pharmaceuticals Licenses Chronic Kidney Disease Drug Bardoxolone Methyl to Kyowa Hakko Kirin”(Press release). Reata Pharmaceuticals. January 7, 2010. Retrieved June 2, 2011.
- “Company Statement: Termination of Beacon Trial”.Reata Pharmaceuticals. Retrieved October 18, 2012.
- Pergola, P. E.; Krauth, M.; Huff, J. W.; Ferguson, D. A.; Ruiz, S.; Meyer, C. J.; Warnock, D. G. (2011). “Effect of Bardoxolone Methyl on Kidney Function in Patients with T2D and Stage 3b–4 CKD”. American Journal of Nephrology 33 (5): 469–476. doi:10.1159/000327599. PMID 21508635.
- Pergola, P. E.; Raskin, P.; Toto, R. D.; Meyer, C. J.; Huff, J. W.; Grossman, E. B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; Wittes, J.; Warnock, D. G.; Beam Study, I. (2011). “Bardoxolone Methyl and Kidney Function in CKD with Type 2 Diabetes” (pdf). New England Journal of Medicine 365 (4): 327–336.doi:10.1056/NEJMoa1105351. PMID 21699484. edit
- van Laecke, S.; Vanholder, R. (2011). “Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1745, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047578.
- Rogacev, K. S.; Bittenbring, J. T.; Fliser, D. (2011).“Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1745–1746, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047579.
- Upadhyay, A.; Sarnak, M. J.; Levey, A. S. (2011).“Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1746, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047580.
- McMahon, G. M.; Forman, J. P. (2011). “Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1746, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047581.
- ClinicalTrials.gov NCT01351675 Bardoxolone Methyl Evaluation in Patients With Chronic Kidney Disease and Type 2 Diabetes (BEACON)
- Design and synthesis of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages
Bioorg Med Chem Lett 1998, 8(19): 2711 - Novel synthetic oleanate triterpenoids: A series of highly active inhibitors of nitric production in mouse macrophages
Bioorg Med Chem Lett 1999, 9(24): 3429 - WO 1999065478
- WO 2013169553
- CN 102875634
- US 2012330050
- US 2012071684
- WO 2011130302
- WO 2010093944
- WO 2009089545
- WO 2009023232
- WO 2008111497
- Anderson, Amy C.; Browning, R. Greg; Couch, Robin D.; Gribble, Gordon W.; Honda, Tadashi; Wright, Dennis L.; Sporn, Michael B.
Bioorganic and Medicinal Chemistry Letters, 2005 , vol. 15, 9 p. 2215 – 2219 - Journal of Medicinal Chemistry, 2004 , vol. 47, 20 p. 4923 – 4932
- Journal of Medicinal Chemistry, 2000 , vol. 43, 22 p. 4233 – 4246
- Bioorganic and Medicinal Chemistry Letters, 2002 , vol. 12, 7 p. 1027 – 1030
- Journal of Medicinal Chemistry, 2000 , vol. 43, 22 p. 4233 – 4246
- Chemical Communications, 2011 , vol. 47, 33 p. 9495 – 9497
- Reata Pharmaceuticals, Inc. – bardoxolone methyl
- Bardoxolone Methyl and Kidney Function in CKD with Type 2 Diabetes
- Effect of Bardoxolone Methyl on Kidney Function in Patients with T2D and Stage 3b – 4 CKD
- American Diabetes Association presentation
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8440854 * | Jan 23, 2012 | May 14, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with amino acid and other modifications at C-17 |
| US8513436 | Dec 19, 2011 | Aug 20, 2013 | Reata Pharmaceuticals, Inc. | Pyrazolyl and pyrimidinyl tricyclic enones as antioxidant inflammation modulators |
| WO2002047611A2 * | Nov 28, 2001 | Jun 20, 2002 | Univ Texas | Cddo-compounds and combination therapies thereof |
| WO2008064132A2 * | Nov 16, 2007 | May 29, 2008 | Dartmouth College | Synthetic triterpenoids and tricyclic-bis-enones for use in stimulating bone and cartilage growth |
| WO2009118441A1 * | Feb 12, 2009 | Oct 1, 2009 | Consejo Superior De Investigaciones Cientifícas | Use of pentacyclic triterpene for the preparation of a pharmaceutical compound intended for the treatment of multiple sclerosis |
| WO2013083659A1 | Dec 5, 2012 | Jun 13, 2013 | Cambridge Enterprise Limited | Combination treatment comprising ho – 1 inhibitor and immunotherapeutic agent |
| US7176237 | Jan 15, 2003 | Feb 13, 2007 | The Trustees Of Dartmouth College | Tricyclic-bis-enone derivatives and methods of use thereof |
| US7435755 | Nov 28, 2001 | Oct 14, 2008 | The Trustees Of Dartmouth College | CDDO-compounds and combination therapies thereof |
| US7678830 | Feb 7, 2007 | Mar 16, 2010 | Trustees Of Dartmouth College | Tricyclic-bis-enone derivatives and methods of use thereof |
| US7714012 | Nov 16, 2007 | May 11, 2010 | Trustees Of Dartmouth University | Synthesis and biological activities of new tricyclic-bis-enones (TBEs) |
| US7795305 | Oct 10, 2008 | Sep 14, 2010 | Board Of Regents, The University Of Texas System | CDDO-compounds and combination therapies thereof |
| US7863327 | May 3, 2005 | Jan 4, 2011 | Trustees Of Dartmouth College | Therapeutic compounds and methods of use |
| US7915402 | Apr 20, 2009 | Mar 29, 2011 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with saturation in the C-ring |
| US7943778 | Apr 20, 2009 | May 17, 2011 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: C-17 homologated oleanolic acid derivatives |
| US8034955 | Oct 29, 2007 | Oct 11, 2011 | Trustees Of Dartmouth College | Therapeutic compounds and methods of use |
| US8067394 | May 10, 2010 | Nov 29, 2011 | Trustees Of Dartmouth College | Synthesis and biological activities of new tricyclic-bis-enones (TBEs) |
| US8067465 | Mar 11, 2010 | Nov 29, 2011 | The Trustees Of Dartmouth College | Tricyclic-bis-enone derivatives and methods of use thereof |
| US8071632 | Apr 20, 2009 | Dec 6, 2011 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: novel derivatives of oleanolic acid |
| US8124656 | Feb 23, 2011 | Feb 28, 2012 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with saturation in the C-ring |
| US8124799 | Apr 20, 2009 | Feb 28, 2012 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with amino and other modifications at C-17 |
| US8129429 | Jan 12, 2009 | Mar 6, 2012 | Reata Pharmaceuticals, Inc. | Synthetic triterpenoids and methods of use in the treatment of disease |
| US8258329 | Apr 20, 2009 | Sep 4, 2012 | Reata Pharmaceuticals, Inc. | Dehydroandrosterone analogs including an anti-inflammatory pharmacore and methods of use |
| US8299046 | Nov 16, 2007 | Oct 30, 2012 | Trustees Of Dartmouth College | Synthetic triterpenoids and tricyclic-bis-enones for use in stimulating bone and cartilage growth |
| US8314137 | Jul 22, 2009 | Nov 20, 2012 | Trustess Of Dartmouth College | Monocyclic cyanoenones and methods of use thereof |
| US8338618 | Nov 11, 2011 | Dec 25, 2012 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: novel derivatives of oleanolic acid |
| US8394967 | Feb 23, 2011 | Mar 12, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: C-17 homologated oleanolic acid derivatives |
| US8440820 | Jan 11, 2012 | May 14, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with saturation in the C-ring |
| US8440854 | Jan 23, 2012 | May 14, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with amino acid and other modifications at C-17 |
| US8455544 | Jan 26, 2012 | Jun 4, 2013 | Reata Pharmaecuticals, Inc. | Synthetic triterpenoids and methods of use in the treatment of disease |
| US8513436 | Dec 19, 2011 | Aug 20, 2013 | Reata Pharmaceuticals, Inc. | Pyrazolyl and pyrimidinyl tricyclic enones as antioxidant inflammation modulators |
| US8586775 | Aug 24, 2011 | Nov 19, 2013 | Trustees Of Dartmouth College | Therapeutic compounds and methods of use |
 |
| Professor Honda received his B.S. degree in Chemistry in 1974, his M.S. degree in Organic Chemistry in 1976, and his Ph.D. in Organic Chemistry in 1979 from the University of Tokyo. In 1979, he joined the Department of Drug Discovery Chemistry at Suntory Institute for Biomedical Research in Japan and worked there as a drug synthetic chemist (finally senior researcher) for 13 years. In 1991, he joined the Central Pharmaceutical Research Institute at Japan Tobacco Inc. and worked as a chief senior researcher for 3 years. In 1995, he joined Dr. Gribble’s laboratory at Dartmouth College as a research associate. In 1998, he joined the research faculty of Dartmouth College. In 2005, he was promoted to Research Associate Professor. |
Dr. Honda and his collaborators have further explored new structures based on CDDO and different five-ringed triterpenoids.
During the course of these investigations, Dr. Honda has designed three-ringed compounds with similar enone functionalities in rings A and C to those of CDDO, but having a much simpler structure than five-ringed triterpenoids. He and his collaborators have found that they are also a novel class of potent anti-inflammatory, cytoprotective, growth suppressive, and pro-apoptotic compounds. Amongst such three-ringed compounds, TBE-31 with the C-8a ethynyl group is much more potent than CDDO in various bioassays in vitro and in vivo. Thus, further investigation (design, synthesis, biological evaluation, etc.) of new TBE-31 analogues is currently being performed in order to discover analogues having different and/or better features than TBE-31, for example, higher potency and lower toxicity, better bioavailability and different distributions in organs, high water-solubility and so on.

Mechanism studies suggest that CDDO regulates various molecules regarding inflammation, differentiation, apoptosis, and proliferation by reversible Michael addition between the cyano enone functionality of CDDO and the sulfhydryl groups of cysteine moieties on these molecules. Based on this fact and the structure of TBE-31, Dr. Honda has designed single-ringed compounds, which represent the ideal simple structure. The synthesis of these new compounds is currently in progress.

 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| ChEMBL | |
| ECHA InfoCard | 100.132.153 |
| Chemical and physical data | |
| Formula | C32H43NO4 |
| Molar mass | 505.69 g/mol |
| 3D model (JSmol) | |
///////////////Bardoxolone Methyl, CDDO-Me; CDDO methyl ester; 218600-53-4; Bardoxolone (methyl); RTA 402 CDDO-Me, CDDO methyl ester, 218600-53-4, Bardoxolone (methyl), RTA 402 , PHASE 3,NSC 713200
CC1(CCC2(CCC3(C(C2C1)C(=O)C=C4C3(CCC5C4(C=C(C(=O)C5(C)C)C#N)C)C)C)C(=O)OC)C
Actelion’s novel antibiotic Cadazolid receives US FDA Qualified Infectious Disease Product designation for the treatment of Clostridium difficile-associated diarrhea .

CADAZOLID, ACT-179811
1-Cyclopropyl-6-fluoro-7-[4-({2-fluoro-4-[(5R)-5-(hydroxymethyl)-2-oxo-1,3-oxazolidin-3-yl]phenoxy}methyl)-4-hydroxypiperidin-1-yl]-4-oxo-1,4-dihydroquinolin-3-carboxylic acid
l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-(R)-5-hydroxymethyl-2-oxo- oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro- quinoline-3-carboxylic acid
| Formula | C29H29F2N3O8 |
|---|---|
| Mol. mass | 585.55 g/mol |
Actelion Pharmaceuticals Ltd / Actelion’s novel antibiotic cadazolid receives US FDA Qualified Infectious Disease Product designation for the treatment of Clostridium difficile-associated diarrhea .
ALLSCHWIL/BASEL, SWITZERLAND – 27 February 2014 – Actelion Ltd (six:ATLN) today announced that the US Food and Drug Administration (FDA) has designated cadazolid as both a Qualified Infectious Disease Product (QIDP) and a Fast Track development program for the treatment of Clostridium difficile-associated diarrhea (CDAD).
The QIDP designation for cadazolid means that – among other incentives – cadazolid would receive a nine-month priority review upon successful completion of the ongoing global Phase III IMPACT program. The Fast Track designation is intended to promote communication and collaboration between the FDA and the Company on the development of the drug.
The designations are based on the 2012 US Generating Antibiotic Incentives Now (GAIN) Act. The GAIN act is a legislative effort to incentivize the development of new antibiotic agents that target serious life-threatening infections.
Guy Braunstein, M.D. and Head of Clinical Development commented: “Clostridium difficile-associated diarrhea is a very serious and potentially life-threatening infection. There is a great need for an antibiotic that allows effective treatment of CDAD with low recurrence rates, particularly in infections caused by hypervirulent strains. The GAIN act highlights the importance of research in this area and we are very happy to receive the advantages that this designation for cadazolid will afford us.”
ABOUT THE IMPACT PROGRAM
IMPACT is an International Multi-center Program Assessing Cadazolid Treatment in patients suffering from Clostridium difficile-associated diarrhea (CDAD). The program comprises two Phase III studies comparing the efficacy and safety of cadazolid (250 mg administered orally twice daily for 10 days) versus vancomycin (125 mg administered orally four times daily for 10 days).
The IMPACT studies are designed to determine whether the clinical response after administration of cadazolid is non-inferior to vancomycin in subjects with CDAD, and whether administration of cadazolid is superior to vancomycin in the sustained clinical response. The program is expected to enroll approximately 1’280 subjects worldwide, and commenced enrollment in the fourth quarter of 2013.
ABOUT CADAZOLID
The novel antibiotic cadazolid is a strong inhibitor of Clostridium difficile protein synthesis leading to strong suppression of toxin and spore formation. In preclinical studies cadazolid showed potent in vitro activity against Clostridium difficile clinical isolates and a low propensity for resistance development. In a human gut model of CDAD, cadazolid had a very limited impact on the normal gut microflora.
Cadazolid absorption is negligible resulting in high gut lumen concentrations and low systemic exposure, even in severe cases of CDAD where the gut wall can be severely damaged and permeability to drugs potentially increased.
Cadazolid is an experimental antibiotic of the oxazolidinone class made by Actelion Pharmaceuticals Ltd. which is effective against Clostridium difficile, a major cause of drug resistant diarrhea in the elderly.[1] Current drug treatments for this infection involve orally delivered antibiotics, principally fidaxomicin, metronidazole and vancomycin; the last two drugs are the principal therapeutic agents in use, but fail in approximately 20 to 45% of the cases. The drug is presently in Phase III trials.[1] The drug works by inhibiting synthesis of proteins in the bacteria, thus inhibiting the production of toxins and the formation of spores.[2]
Structure
The chemical structure of cadazolid combines the pharmacophores of oxazolidinone and fluoroquinolone.[2]
In a study published in the journal Anaerobe, cadazolid has been shown to be effective in vitro against 133 strains of Clostridium difficile all collected from Sweden.[3]
In phase I tests, sixty four male patients reacted favourably to cadazolid which primarily acted and remained in the colon while displaying little toxicity even in regimes involving large doses.[1]
ABOUT CADAZOLID IN THE PHASE II STUDY
Cadazolid was studied in a Phase II multi-center, double-blind, randomized, active reference, parallel group, therapeutic exploratory study. The study evaluated the efficacy, safety and tolerability of a 10-day, twice daily oral administration of 3 doses (250 mg, 500 mg or 1,000 mg b.i.d.) of cadazolid in subjects with Clostridium difficile-associated diarrhea (CDAD). As the current standard of care for CDAD, oral vancomycin (125 mg qid for 10 days) was used as the active reference. The study was completed in December of 2012, after having enrolled 84 subjects with CDAD.
The results of the Phase II study indicate that the effect of all doses of cadazolid were numerically similar to, or better than vancomycin on key endpoints including CDAD clinical cure rates as well as sustained cure rates. Clinical cure rate was defined as the resolution of diarrhea and no further need for CDAD therapy at test-of-cure 24 to 72 hours after the last dose of treatment, while sustained cure rate was defined as clinical cure with no recurrence of CDAD up to 4 weeks post-treatment. Recurrence rates were numerically lower for all doses of cadazolid as compared to vancomycin. Cadazolid was safe and well tolerated.
ABOUT THE GAIN ACT (INCLUDING FAST TRACK DESIGNATION)
The Food and Drug Administration Safety and Innovation Act (FDASIA) was signed into law in July 2012. The GAIN Act is Title VIII to FDASIA. The purpose of the GAIN Act is to encourage pharmaceutical research of certain antibiotics by designation of products as QIDPs. These products are intended to treat serious or life-threatening infections and include those to treat certain specifically identified pathogens, which are listed in the GAIN Act. C. difficile is one such specifically identified pathogen and drugs to treat CDAD would be eligible for designation as a QIDP.
The GAIN Act also provides that qualifying drugs (QIDPs) are eligible for inclusion in the FDA’s Fast Track program. This program is intended to facilitate development and expedite review of new drugs and includes close early communication between the FDA and a drug’s sponsor.
ABOUT FAST TRACK DRUG DEVELOPMENT PROGRAMS
For further information regarding Fast Track Drug Development Programs, please refer to the FDA document “Guidance for Industry on Fast Track Drug Development Programs: Designation, Development, and Application Review”. This document is available on the Internet at:
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM079736.pdf
ABOUT CLOSTRIDIUM DIFFICILE-ASSOCIATED DIARRHEA
Clostridium difficile is a Gram-positive, anaerobic, spore-forming bacterium that is the leading cause of nosocomial diarrhea. Clostridium difficile-associated diarrhea (CDAD or CDI for Clostridium difficile infection) can be a severe and life-threatening disease and results from the overgrowth in the colon of toxigenic strains of Clostridium difficile, generally during or after therapy with broad-spectrum antibiotics. CDAD is a major healthcare problem and a leading cause of morbidity in elderly hospitalized patients. The frequency and severity of CDAD in the western world has increased in recent years, and new hypervirulent and epidemic strains of Clostridium difficile have been discovered that are characterized by overproduction of toxins and other virulence factors, and by acquired resistance to fluoroquinolones such as moxifloxacin.
Current antibiotic therapy for CDAD includes vancomycin and metronidazole. While clinical cure rates are generally 85-90%, recurrences rates of 15-30 % with either drug are problematic as Clostridium difficile produces spores that are resistant to antibiotic treatment and routine disinfection. Spores surviving in the gut of patients and/or in the hospital environment may play a major role in re-infection and recurrence of CDAD after antibiotic treatment. Vancomycin and metronidazole are reported to promote spore formation in vitro at sub-inhibitory concentrations.
Actelion Ltd.
Actelion Ltd. is a leading biopharmaceutical company focused on the discovery, development and commercialization of innovative drugs for diseases with significant unmet medical needs.
Actelion is a leader in the field of pulmonary arterial hypertension (PAH). Our portfolio of PAH treatments covers the spectrum of disease, from WHO Functional Class (FC) II through to FC IV, with oral, inhaled and intravenous medications. Although not available in all countries, Actelion has treatments approved by health authorities for a number of specialist diseases including Type 1 Gaucher disease, Niemann-Pick type C disease, Digital Ulcers in patients suffering from systemic sclerosis, and mycosis fungoides in patients with cutaneous T-cell lymphoma.
Founded in late 1997, with now over 2,400 dedicated professionals covering all key markets around the world including the US, Japan, China, Russia and Mexico, Actelion has its corporate headquarters in Allschwil / Basel, Switzerland
…………………..
Preparation of the compound of formula II
The compound of formula II can be obtained by hydrogenation of the compound of formula VIII

VIII
over a noble metal catalyst such as palladium or platinum on charcoal in a solvent such as THF, MeOH or EA between 00C and 400C or by hydrolysis of in presence of a solution of HBr in water or AcOH between 00C and 800C in a solvent such as AcOH.
The compounds of formula III can be prepared as summarized in Scheme 1 hereafter.

IX VI IIIA: R1= H IIIS: ^ = SO2R5
Scheme 1
The compounds of formula V can be prepared as summarized in Scheme 2 hereafter.

II X XI

Scheme 2
The compounds of formula X can be prepared from the methylidene derivatives of formula XII as summarized in Scheme 3 hereafter.

Xc XII Xa: R1 = H

Scheme 3
Example 1:
l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-((/f)-5-hydroxymethyl-2-oxo- oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro- quinoline-3-carboxylic acid:
1 i. (R)-3-(3-fluoro-4-hydroxy-phenyl)-5-hydroxymethyl-oxazolidin-2-one:
A solution of (7?y)-3-(4-benzyloxy-3-fluoro-phenyl)-5-hydroxymethyl-oxazolidin-2-one (6.34 g, prepared according to WO 2004/096221) in THF/MeOH (1 :1; 200 ml) was hydrogenated over Pd/C 10% (1 g) overnight. The catalyst was filtered off, the filtrate evaporated under reduced pressure and the residue stirred in EA. The crystals were collected by filtration, affording 3.16 g (70% yield) of a colourless solid. 1H NMR (DMSOd6; δ ppm): 3.5 (m, IH), 3.64 (m, IH), 3.74 (dd, J = 8.8, 6.4, IH), 3.99 (t, J = 8.8, IH), 4.64 (m, IH), 5.16 (t, J = 5.6, IH), 6.93 (dd, J = 9.7, 8.8, IH), 7.08 (ddd, J = 8.8, 2.6, 1.2, IH), 7.45 (dd, J = 13.5, 2.6, IH), 9.66 (s, IH). MS (ESI): 228.1.
1. ii. 4-[2-fluoro-4- ((R)-5-hydroxymethyl-2-oxo-oxazolidin-3-yl)-phenoxymethyl]- 4-hydroxy-piperidine-l-carboxylic acid benzyl ester:
A solution of intermediate l.i (1.27 g) and l-oxa-6-aza-spiro[2.5]octane-6-carboxylic acid benzyl ester (1.60 g; prepared according to US 4244961) were dissolved in DMF (15 ml) and treated with Na2CO3 (1.16 g). The mixture was heated at 1000C overnight. The residue obtained after workup (DCM) was stirred in EA, and the solid was collected by filtration and sequentially washed with EA and Hex, affording 2.52 g (94.5% yield) of a beige solid.
1H NMR (DMSOd6; δ ppm): 1.57 (m, 4H), 3.14 (m, 2H), 3.54 (m, IH), 3.64 (m, IH), 3.79 (m, 5 H), 4.03 (t, J = 9.1, 1 H), 4.66 (m, 1 H), 4.78 (s, 1 H), 5.05 (s, 2 H), 5.16 (t,
J = 5.6, 1 H), 7.18 (m, 2 H), 7.32 (m, 5 H), 7.55 (d, J = 12, 1 H).
MS (ESI): 475.0.
1. iii. (R)-3-[3-fluoro-4-(4-hydroxy-piperidin-4-ylmethoxy)-phenyl]-5-hydroxymethyl- oxazolidin-2-one:
A suspension of intermediate l.ii (2.5 g) in EA/MeOH (1 :1; 100 ml) was hydrogenated over Pd/C for 48 h. The suspension was heated at 400C and the catalyst was filtered off.
The filtrate was evaporated under reduced pressure affording 1.61 g (89% yield) of a yellow powder.
1H NMR (DMSOd6; δ ppm): 1.4-1.63 (m, 4H), 2.67 (m, 2H), 2.83 (m, 2H), 3.53 (dd, J = 4.0, 12.0, IH); 3.66 (dd, J = 3.3, 12.0, IH), 3.71 (s, 2H); 3.80 (m, IH), 4.05 (t, J = 9.0,
IH), 4.48 (s, IH), 4.68 (m, IH), 5.20 (s, IH), 7.20 (m, 2H), 7.57 (d, IH).
MS (ESI): 341.5.
l.iv. l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-((R)-5-hydroxymethyl-2-oxo-oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid:
A solution of intermediate l.iii (200 mg), 7-chloro-l-cyclopropyl-6-fiuoro-l,4-dihydro- 4-0X0-3 -quinolinecarboxylic acid boron diacetate complex (241 mg; prepared according to WO 88/07998) and DIPEA (100 μl) in NMP (2 ml) was stirred at 85°C for 5 h. The reaction mixture was evaporated under reduced pressure and the residue was taken up in 5M HCl in MeOH (3 ml) and stirred. The resulting solid was collected by filtration and washed with MeOH to afford 230 mg (67% yield) of a yellow solid.
1H NMR (DMSOd6; δ ppm): 1.66-1.35 (m, 4H), 1.75 (d, J = 12.8, 2H), 1.95 (m, 2H), 3.33 (t broad, J = 11.0, 2H), 3.57 (m, 3H), 3.67 (dd, J = 12.3, 3.3, IH), 3.83 (m, 2H), 3.92 (s, 2H), 4.06 (t, J = 9.0, IH), 4.69 (m, IH), 7.24 (m, 2H), 7.60 (m, 2H), 7.90 (d, J = 13.3, IH), 8.66 (s, IH).
MS (ESI): 585.9.
References
- Boschert, Sherry (19 Sep 2012). “Promising C. difficile Antibiotic in Pipeline”. Internal Medicine News. International Medical News Group. Retrieved 22 May 2013.
- “Cadazolid”. .actelion.com. Retrieved 2013-05-22.
- “Anaerobe – In vitro activity of cadazolid against Clostridium difficile strains isolated from primary and recurrent infections in Stockholm, Sweden”. ScienceDirect.com. 2013-02-26. Retrieved 2013-05-22.
- WO 2008056335
- WO 2009136379
Sonidegib/Erismodegib..Novartis Cancer Drug LDE225 Meets Primary Endpoint in Phase 2


Sonidegib/Erismodegib
CODE DESIGNATION ..LDE225, NVP-LDE-225
Treatment of medulloblastoma PHASE3 2014 FDA FILING
Treatment of advanced basal cell carcinoma PHASE3 2014 FDA FILING
Treatment of SOLID TUMORS..PHASE1 2017 FDA FILING
READMalignant Solid Tumors of Childhood
THERAPEUTIC CLAIM Oncology, Antineoplastics & Adjunctive Therapies

CHEMICAL NAMES
1. [1,1′-Biphenyl]-3-carboxamide, N-[6-[(2R,6S)-2,6-dimethyl-4-morpholinyl]-3-pyridinyl]-2-
methyl-4′-(trifluoromethoxy)-, rel-
2. N-{6-[(2R,6S)-2,6-dimethylmorpholin-4-yl]pyridin-3-yl}-2-methyl-4′-
(trifluoromethoxy)biphenyl-3-carboxamide
N-[6-[(2S,6R)-2,6-dimethylmorpholin-4-yl]pyridin-3-yl]-2-methyl-3-[4-(trifluoromethoxy)phenyl]benzamide
N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-(trifluoromethoxy)biphenyl-3-carboxamide
MOLECULAR FORMULA C26H26F3N3O3
MOLECULAR WEIGHT 485.5
SPONSOR Novartis Pharma AG
CAS REGISTRY NUMBER 956697-53-3 free form
NOTE… DIPHOSPHATE SALT IS THE DRUG WITH CAS 1218778-77-8
sonidegib – European Medicines Agency READ THIS..
Summary EudraCT Number: 2012-004022-21 Sponsor’s Protocol … READ THIS
About the Study
The Phase II, randomized, double-blind BOLT (Basal cell carcinoma Outcomes in LDE225 Trial) study was designed to assess the safety and efficacy of two oral dose levels of LDE225 (200 mg and 800 mg) in patients with locally advanced or metastatic basal cell carcinoma[4], which are subtypes of advanced basal cell carcinoma.
The primary endpoint was the proportion of patients achieving an objective response rate, defined as a confirmed complete response and partial response as their best overall response per modified RECIST criteria, within six months of starting treatment with LDE225. Key secondary endpoints of the study included assessing the duration of tumor responseand the rate of complete response. Other secondary endpoints included progression-free survival, time to tumor response and overall surviva

Sonidegib (INN) or Erismodegib (USAN), also known as LDE225 is a Hedgehog signalling pathway inhibitor (via smoothened antagonism) being developed as an anticancer agent by Novartis.[1][2] It has been investigated as a potential treatment for:
- Pancreatic cancer[3][4][5][6]
- Breast cancer[7][8]
- Basal cell carcinoma of the skin[9][10][11]
- Small cell lung cancer[12]
- Medulloblastoma[13][14]
- Advanced solid tumours (including ovarian, breast, pancreatic, stomach, oesophageal cancers and glioblastoma multiforme)[15][16][17]
- Acute leukaemia[18]
- Chronic myeloid leukaemia[19]
- Myelofibrosis and Essential thrombocythaemia[20]
NVP-LDE-225, a product candidate developed by Novartis, is in phase III clinical trials for the treatment of medulloblastoma and basal cell carcinoma. Phase II trials are in progress for the treatment of adult patients with relapsed or refractory or untreated elderly patients with acute leukemia.
Early clinical trials are ongoing for the oral treatment of advanced solid tumors, for the treatment of myelofibrosis in combination with ruxolitinib and for the treatment of small cell lung cancer. A phase II clinical trial for the treatment of basal cell carcinomas in Gorlin’s syndrome patients with a cream formulation of NVP-LDE-225 was discontinued in 2011 since the formulation did not demonstrate tumor clearance rate sufficient to support further development.
Dana-Farber Cancer Institute and the Massachusetts General Hospital are conducting phase I clinical trials for the treatment of locally advanced or metastatic pancreatic cancer in combination with chemotherapy. In 2009, orphan drug designation was assigned in the E.U. for the treatment of Gorlin syndrome.
It has demonstrated significant efficacy against melanoma in vitro and in vivo.[21] It also demonstrated efficacy in a mouse model of pancreatic cancer.[22]
NVP-LDE225 Diphosphate salt (Erismodegib, Sonidegib)

- Synonym:Erismodegib, Sonidegib
- CAS Number:1218778-77-8
- Mol. Formula:C26H26F3N3O3 ∙ 2H3PO4
- MW:681.5
- nmr.http://www.chemietek.com/Files/Line2/Chemietek,%20NVP-LDE225%20[02],%20NMR.pdf
- hplc–http://www.chemietek.com/Files/Line3/Chemietek,%20NVP-LDE225%20[02],%20HPLC.pdf
Brief Description:
About LDE225
LDE225 (sonidegib) is an oral, investigational, selective smoothened inhibitor being studied in a variety of cancers. Smoothened (SMO) is a molecule that regulates the hedgehog (Hh) signaling pathway, which plays a critical role in stem cell maintenance and tissue repair. LDE225 is currently in clinical development for a variety of diseases including myelofibrosis, leukemia and solid tumors.
Given that LDE225 is an investigational compound, the safety and efficacy profile has not yet been fully established. Access to this investigational compound is available only through carefully controlled and monitored clinical trials. These trials are designed to better understand the potential benefits and risks of the compound. Given the uncertainty of clinical trials, there is no guarantee that LDE225 will ever be commercially available anywhere in the world.
Possibility (LDE225) is effective in medulloblastoma relapsed or refractory hedgehog pathway inhibitor sonidegib has been revealed. That the anti-tumor effect was observed in some patients and tolerability in 1/2 test phase.
4th Quadrennial Meeting of the World Federation of Neuro-Oncology in conjunction with the 18th Annual Meeting of the Society for Neuro-Oncology, which was held in San Francisco November 21 to 24 in (WFNO-SNO2013), rice Dana-Farber It was announced by Mark Kieran Mr. Children’s Hospital Cancer Center.
The research group, announced the final results of the Phase 1 trial that target advanced solid cancer in children of sonidegib. 1 dose increased multi-test phase, was initiated from 372mg/m2 once-daily dosing to target children under the age of 18 more than 12 months. (233mg/m2 group 11 people, 16 people 372mg/m2 group, 11 people group 425mg/m2, 680mg/m2 group 21 women) who participated 59 people, including medulloblastoma 38 patients. 12 median age was (2-17).
Creatine phosphokinase elevation of grade 4 only were seen at 372mg/m2 as dose-limiting toxicity only, and became two recommended dose phase and 680mg/m2. Nausea muscle pain creatine kinase rise malaise (22.0%) (15.3%) (15.3%), (13.6%), vomiting side effects were many, was (13.6%). Hypersensitivity vomiting creatine kinase increased (3.4%) (1.7%) (1.7%), rhabdomyolysis side effects of grade 3/4 was (1.7%). (One group 372mg/m2, 425mg/m2 group one) complete response was obtained in two people, a strong correlation was found between the activation of the hedgehog pathway and effect.
Phase III clinical trials that target medulloblastoma the activated hedgehog pathway currently are underway.
About Novartis
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, eye care, cost-saving generic pharmaceuticals, preventive vaccines and diagnostic tools, over-the-counter and animal health products. Novartis is the only global company with leading positions in these areas. In 2013, the Group achieved net sales of USD 57.9 billion, while R&D throughout the Group amounted to approximately USD 9.9 billion (USD 9.6 billion excluding impairment and amortization charges). Novartis Group companies employ approximately 136,000 full-time-equivalent associates and operate in more than 140 countries around the world.

The following Examples serve to illustrate the invention without limiting the scope thereof, it is understood that the invention is not limited to the embodiments set forth herein, but embraces ali such forms thereof as come within the scope of the disclosure,

Step 1:
To a solution of 2-chloro-5-nitro-pyridine 1 (5.58 g, 35.2 mmoL) and c/s-2,6- dimethylmorpholine (4.05 g, 35.2 mmoL) in anhydrous DMF (30 mi.) was added K2CO3 (9.71 g, 70.4 mnrtoL). The mixture was heated at 50ºC overnight. After concentration, the residue is partitioned between EtOAc and water. The EtOAc layer is dried over anhydrous Na2SO4 and concentrated to give crude product 3 as a yellow solid, after purification by silica gel chromatography, obtained pure product (7.80 g, 93.2%). LC-MS m/z: 238.2 (M+ 1).
Step 2:
The above material 3 (7.3Og. 30.8 mmoL) was hydrogenated in the presence of 10% Pd-C (1.0 g) in MeOH (120 ml) under hydrogen overnight. The suspension was filtered through celite and the filtrate was concentrated to give the crude product 4 (5.92 g) as a dark brown oil which was used directly in the next step without further purification. LC-MS m/z. 208.2 (M+1).
Step 3:
To a solution of 3-bromo-2-methyl benzoic acid (2.71 g, 12.6 mmoL), 6-((2S,6R)-2,6- dimethylmorpholino)pyridin-3-arnine 4 (2.61 g, 12.6 mmoL), and HATU (4.80 g, 12.6 mmoL) in anhydrous DMF (30 mL) was added diisopropylethylamine (6.58 mL, 37.8 mmoL) dropwise. The resulting mixture was stirred overnight at room temperature. The reaction mixture was diluted with water (50 mL), and then extracted with EtOAc (3×120 mL). The organic layer was dried and concentrated to give the crude product. This crude product was then purified by flash column chromatography using 30% EtOAc in hexane as eiuent to give 5 as a white solid (4.23 g, 83.0%). LC-MS m/z: 404.1 (M+1).
Step 4:
A mixture of 4-(trif!uoromethoxy)phenylboronic acid (254 mg, 1.24 mmol), 3-bromo- N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-ylJ-4-methyl-benzamide 5 (250 mg, 0.62mmol), Pd(PPh3)4 (36 mg, 0.03 mmol), Na2CO3 (2.0M aqueous solution, 1.23 mL, 2.4 mmol) and DME (4.5 mL) in a sealed tube was heated at 130ºC overnight. The reaction mixture was diluted with EtOAc and water. The aqueous layer was extracted with EtOAc. The combined organic layer was washed with brine and concentrated to give the crude product which was then purified by preparative mass triggered HPLC (C18 column, etuted with CH3CN-H2O containing 0.05% TFA) to give N-(6-((2S,6R)-2,6-dimethyfmorpholino)pyridin-3-yl)-2-rnethyl- 4′-(trifluoromethoxy)biphenyi-3-carboxamide (183.5 mg, 61.1% yield). LC-MS m/z: 486.2 (M+1).
The resultant crystalline product (Form A) was converted to the amorphous form by dissolving in 3% w/w aqueous ethanol, and the resultant solution spray dried at about 150ºC.
Form B was prepared by heating the amorphous form in an oven at 110ºC for 2 hours. In a further embodiment, the invention relates to a process step or steps, or an intermediate as described herein.

…………………………..
SYNTHESIS

| LC-MS m/z 486.2 (M + 1) |

Step 1: To a solution of 3-iodo-4-methyl-benzoic acid (10.0 g, 38.2 mmol) in methanol (70 ml) is added concentrated sulfuric acid (0.5 ml). The reaction mixture is heated at 70° C. for 48 hours, cooled to room ambient temperature and then concentrated. After that, ethyl acetate (100 ml) and aqueous NaHCO3 (saturated, 100 ml) solution are added to the residue. The organic layer is separated and washed again with aqueous NaHCO3 (saturated, 100 ml) solution. The organic layer is separated, dried over anhydrous Na2SO4 and concentrated to yield 3-iodo-4-methyl-benzoic acid methyl ester 1. It is used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ 8.31 (s, 1H), 7.87 (d, 1H, J=8.4 Hz), 7.48 (d, 1H, J=8.4 Hz), 3.85 (s, 3H), 3.35 (s, 3H); LC-MS m/z: 277.0 (M+1).
Step 2: To a round-bottom flask containing 3-iodo-4-methyl-benzoic acid methyl ester (1.38 g, 5.00 mmol), 4-cyanophenylboronic acid (1.10 g, 7.48 mmol), palladium acetate (168 mg, 0.748 mmol), 2-(dicyclohexylphosphino)biphenyl (0.526 g, 1.50 mmol) and potassium fluoride (0.870 g, 15.0 mmol) is added anhydrous 1,4-dioxane (15 ml). The flask is purged with argon and sealed. The mixture is stirred at 130° C. for 18 hours, cooled to ambient temperature and then water (20 ml) and ethyl acetate (20 ml) are added. Solid is removed under vacuum filtration. The filtrate is extracted with EtOAc (20 ml×2). The organic layers are combined, washed with aqueous HCl (5%, 20 ml) and saturated NaHCO3 (20 ml). It is dried over MgSO4, and concentrated. The residue is purified by silica gel column chromatography (EtOAc/Hexane, gradient) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid methyl ester 2; LC-MS m/z: 252.1 (M+1).
Step 3: To a solution of 4′-cyano-6-methyl-biphenyl-3-carboxylic acid methyl ester 2 (2.56 g, 10.3 mmol) in 1,4-dioxane-H2O (1:1 mixture, 20 ml) is added NaOH (1.22 g, 30.2 mmol)). The reaction is stirred at ambient temperature for 24 hours. To this mixture is added aqueous HCl (1 N, 36 ml) and it is then extracted with ethyl acetate (40 ml×3). The organic layers are combined, dried over anhydrous Na2SO4. The solver is removed. The solid obtained is washed with small amount of acetonitrile and air dried to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid 3: 1H NMR (DMSO-d6) δ 7.94 (d, 2H, J=8.0 Hz), 7.84 (dd, 1H, J1=8.4 Hz, J2=1.2 Hz), 7.75 (d, 1H, J=1.2 Hz), 7.61 (d, 2H, J=8.0 Hz), 7.48 (d, 1H, J=8.4 Hz), 2.29 (s, 3 H); LC-MS m/z 238.1 (M+1).
Step 4: To a suspension of 4′-cyano-6-methyl-biphenyl-3-carboxylic acid 3 (40 mg, 0.17 mmol) in anhydrous methylene chloride (5 ml) is added 2 drops of DMF. Then oxalyl chloride (32 mg, 22 μl, 0.25 mmol) is added. The mixture is stirred at ambient temperature until it turns clear. After that, it is concentrated, re-dissolved in anhydrous methylene chloride (3 ml), and added to a solution of 4-(morpholine-4-sulfonyl)-phenylamine (61 mg, 0.25 mmol) and triethylamine (34 mg, 47 μl, 0.33 mmol) in methylene chloride (2 ml). The mixture is stirred for 2 hours, concentrated and the residue is purified by preparative mass triggered HPLC (C18 column, eluted with CH3CN—H2O containing 0.05% TFA) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [4-(morpholine-4-sulfonyl)-phenyl]-amide: 1H NMR (DMSO-d6) δ 10.64 (s, 1H), 8.07 (d, 2H, J=8.8 Hz), 7.97 (d, 2H, J=8.4 Hz), 7.95 (d, 1H, J=8.8 Hz), 7.89 (s, 1H), 7.43 (d, 2H, J=8.4 Hz), 7.67 (d, 2H, J=8.8 Hz), 7.53 (d, 2H, J=8.8 Hz), 3.63 (m, 4H), 2.84 (m, 4H) 2.32 (s, 3H); LC-MS m/z: 462.1 (M+1).
Example 2 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-amide

Step 1: To a solution of 2-chloro-5-nitro-pyridine 4 (2.38 g, 15 mmol.) and cis-2,6-dimethylmorpholine (1.73 g, 15 mmol.) is added K2CO3 (4.14 g, 30 mmol.). The mixture was heated at 50° C. overnight. After concentration, the residue is partitioned between EtOAc and water. The EtOAc layer is dried over anhydrous Na2SO4 and concentrated to give crude product 6 as a yellow solid. The crude product is used directly in next step without further purification. LC-MS m/z: 238.1 (M+1).
Step 2: The above crude material 6 is hydrogenated in the presence of Pd—C (0.2 g) in MeOH (100 mL) under hydrogen over 10 h. The suspension is filtered through celite and the filtrate is concentrated to give the crude product 7 as a dark brown oil which is used directly in the next step without further purification. LC-MS m/z: 208.1 (M+1).
Step 3: To a solution of 3-bromo-4-methyl benzoic acid (108 mg, 0.5 mmol.), 6-(2,6-Dimethyl-morpholin-4-yl)-pyridin-3-ylamine 7 (104 mg, 0.5 mmol.), amd HATU (190 mg, 0.5 mmol.) in dry DMF (5 mL) is added triethylamine (139 uL, 1.0 mmol.) dropwise. The resulting mixture is stirred at room temperature for 2 h. After concentration, the residue is partitioned between EtOAc and water. The organic layer is dried and concentrated to give the crude product. The final compound is purified by flash column chromatography using 50% EtOAc in hexane as eluent to give 8 as a white solid. LC-MS m/z: 404.1 (M+1).
Step 4: A mixture of 4-cyanophenyl boronic acid (18 mg, 0.12 mmol), 3-bromo-N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamide 8 (40 mg, 0.1 mmol), Pd(PPh3)4 (11 mg, 0.01 mmol), and Na2CO3 (42 mg, 0.4 mmol) in a combined solvent system of toluene (0.2 mL) and water (0.2 mL) and ethanol (0.05 mL) is heated at 140° C. under microwave irradiation for 30 min. The reaction mixture is diluted with EtOAc and water. The aqueous layer is extracted with EtOAc. The combined organic layer is washed with brine and concentrated to give the crude product which is purified by preparative mass triggered HPLC (C18 column, eluted with CH3CN—H2O containing 0.05% TFA) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-amide. LC-MS m/z: 427.2 (M+1).

4-(Trifluoromethoxy)phenylboronic acid
- CAS Number 139301-27-2
- Linear Formula CF3OC6H4B(OH)2
- Molecular Weight 205.93
CONDENSE WITH …3-bromo-N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamideACS Medicinal Chemistry Letters, 2010 , vol. 1, 3 p. 130 – 134

A mixture of 4-(trifluoromethoxy)phenylboronic acid (254 mg, 1.24 mmol), 3-bromo-N-[6-(2,6-
dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamide E (250 mg, 0.62mmol), Pd(PPh3)4
(36 mg, 0.03 mmol), Na2CO3 (2.0M aqueous solution, 1.23 mL, 2.4 mmol) and DME (4.5 mL)
in a sealed tube was heated at 1300C overnight. The reaction mixture was diluted with EtOAc
and water. The aqueous layer was extracted with EtOAc. The combined organic layer was
washed with brine and concentrated to give the crude product which was then purified by
preparative mass triggered HPLC (C18 column, eluted with CH3CN-H2O containing 0.05% TFA)
to give N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-
(trifluoromethoxy)biphenyl-3-carboxamide (5m, 183.5 mg, 61.1% yield). LC-MS m/z: 486.2 (M+1).
HRMS (m/z): [M+H]+
calcd for C26H27N3O3F3 486.2005; found 486.1986,
1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.15 (s, 1H), 8.43 (d, 1H), 7.94 (dd, 1H), 7.52-7.43
(m, 5H), 7.38 (m, 1H), 7.33 (m, 1H), 6.86 (d, 1H), 4.06 (d, 2H), 3.62 (m, 2H), 2,34 (m, 2H), 2.22
(s, 3H), 1.16 (d, 6H).
http://pubs.acs.org/doi/suppl/10.1021/ml1000307/suppl_file/ml1000307_si_001.pdf
Reference
- “LDE225 – PubChem”. PubChem. National Institutes of Health. Retrieved 16 February 2014.
- Pan, S; Wu, X; Jiang, J; Gao, W; Wan, Y; Cheng, D; Han, D; Liu, J; Englund, NP; Wang, Y; Peukert, S; Miller-Moslin, K; Yuan, J; Guo, R; Matsumoto, M; Vattay, A; Jiang, Y; Tsao, J; Sun, F; Pferdekamper, AC; Dodd, S; Tuntland, T; Maniara, W; Kelleher, JF; Yao, Y; Warmuth, M; Williams, J; Dorsch, M (10 June 2010). “Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist”. ACS Medicinal Chemistry Letters 1 (3): 130–134. doi:10.1021/ml1000307.
- “A Biomarker Study to Identify Predictive Signatures of Response to LDE225 (Hedgehog Inhibitor) In Patients With Resectable Pancreatic Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Gemcitabine + Nab-paclitaxel With LDE-225 (Hedgehog Inhibitor) as Neoadjuvant Therapy for Pancreatic Adenocarcinoma”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Dose-escalation, and Safety Study of LDE225 and Gemcitabine in Locally Advanced or Metastatic Pancreatic Cancer Patients”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Pilot Study of a Hedgehog Pathway Inhibitor (LDE-225) in Surgically Resectable Pancreas Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Study With LDE225 in Combination With Docetaxel in Triple Negative (TN) Advanced Breast Cancer (ABC) Patients (EDALINE)”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014.
- “LDE225 in Treating Patients With Stage II-III Estrogen Receptor- and HER2-Negative Breast Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase II Study of Efficacy and Safety in Patients With Locally Advanced or Metastatic Basal Cell Carcinoma (BOLT)”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “To Evaluate the Safety, Local Tolerability, PK and PD of LDE225 on Sporadic Superficial and Nodular Skin Basal Cell Carcinomas(sBCC)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Trial to Evaluate the Safety, Local Tolerability, Pharmacokinetics and Pharmacodynamics of LDE225 on Skin Basal Cell Carcinomas in Gorlin Syndrome Patients”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Combination of the Hedgehog Inhibitor, LDE225, With Etoposide and Cisplatin in the First-Line Treatment of Patients With Extensive Stage Small Cell Lung Cancer (ES-SCLC)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase III Study of Oral LDE225 Versus (vs) Temozolomide (TMZ) in Patients With Hedge-Hog (Hh)-Pathway Activated Relapsed Medulloblastoma (MB)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase I Dose Finding and Safety Study of Oral LDE225 in Children and a Phase II Portion to Assess Preliminary Efficacy in Recurrent or Refractory MB”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Phase Ib, Dose Escalation Study of Oral LDE225 in Combination With BKM120 in Patients With Advanced Solid Tumors”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Dose Finding and Safety of Oral LDE225 in Patients With Advanced Solid Tumors”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “LDE225 and Paclitaxel in Solid Tumors”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Study of Efficacy and Safety of LDE225 in Adult Patients With Relapsed/Refractory Acute Leukemia”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Nilotinib and LDE225 in the Treatment of Chronic or Accelerated Phase Myeloid Leukemia in Patients Who Developed Resistance to Prior Therapy”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase Ib/II Dose-finding Study to Assess the Safety and Efficacy of LDE225 + INC424 in Patients With MF”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- Jalili, A; Mertz, KD; Romanov, J; Wagner, C; Kalthoff, F; Stuetz, A; Pathria, G; Gschaider, M; Stingl, G; Wagner, SN (30 July 2013). “NVP-LDE225, a potent and selective SMOOTHENED antagonist reduces melanoma growth in vitro and in vivo.” (PDF). PloS one 8 (7): e69064. doi:10.1371/journal.pone.0069064. PMC 3728309.PMID 23935925.
- Fendrich, V; Wiese, D; Waldmann, J; Lauth, M; Heverhagen, AE; Rehm, J; Bartsch, DK (November 2011). “Hedgehog inhibition with the orally bioavailable Smo antagonist LDE225 represses tumor growth and prolongs survival in a transgenic mouse model of islet cell neoplasms.”. Annals of Surgery 254 (5): 818–23.doi:10.1097/SLA.0b013e318236bc0f. PMID 22042473.
- ChemMedChem, 2013 , vol. 8, 8 p. 1261 – 1265
- ACS Med. Chem. Lett., 2010, 1 (3), pp 130–134.
- MORE REF
sonidegib
Skin Cancer Foundation. “Skin Cancer Facts.” Available at:http://www.skincancer.org/skin-cancer-information/skin-cancer-facts . Accessed on February 14, 2014.
Rubin AI, Chen EH, Ratner D (2005). Current Concepts: Basal-Cell Carcinoma. N Engl J Med; 353:2262-9.
ClinicalTrials.gov. “A Phase II Study of Efficacy and Safety in Patients With Locally Advanced or Metastatic Basal Cell Carcinoma (BOLT)” Available at:http://clinicaltrials.gov/ct2/show/NCT01327053?term=%22LDE225%22+and+%22BOLT%22&rank=1. Accessed on February 14, 2014.
National Cancer Institute Dictionary of Cancer Terms. “Complete Response.” Available at: http://www.cancer.gov/dictionary?CdrID=45652 . Accessed on February 14, 2014.
National Cancer Institute Dictionary of Cancer Terms. “Partial Response.” Available at: http://www.cancer.gov/dictionary?CdrID=45819 . Accessed on February 14, 2014.
Wong C S M, Strange R C, Lear J T (2003). Basal cell carcinoma. BMJ; 327:794-798.
Copcu E, Aktas A. Simultaneous two organ metastases of the giant basal cell carcinoma of the skin. Int Semin Surg Oncol. 2005;2:1-6. Available at:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC544837/ . Accessed on February 14, 2014.
Skin Cancer Foundation. “Basal Cell Carcinoma Treatment Options.” Available athttp://www.skincancer.org/skin-cancer-information/basal-cell-carcinoma/bcc-treatment-options . Accessed on February 14, 2014.
Stuetz A, et al. LDE225, a specific smoothened inhibitor, for the topical treatment of nevoid basal cell carcinoma syndrome (Gorlin’s syndrome). Melanoma Research. 2010; 20:e40. Available at:http://journals.lww.com/melanomaresearch/Fulltext/2010/06001/FC24_LDE225,_a_specific_smoothened_inhibitor,_for.87.aspx#FC24_LDE225%2C_a_specific_smoothened_inhibitor%2C_for.87.aspx?s=2&_suid=139234380607909969110518506816.
Novartis.com. “The Pipeline of Novartis Oncology: LDE225.” Available at:http://www.novartisoncology.com/research-innovation/pipeline.jsp #. Accessed on February 14, 2014.
Children’s Medical Research Center, Children’s Memorial Hospital/Northwestern University Feinberg School of Medicine. “The Sonic hedgehog/patched/gli signal transduction pathway.” Available at http://www.childrensmrc.org/iannaccone/gli/ . Accessed on February 14, 2014.
Gupta S, Takebe N, LoRusso P. Targeting the Hedgehog pathway in cancer. Ther Adv Med Oncol. 2010 July; 2(4): 237-250. Available at:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3126020/ . Accessed on February 14, 2014.
| WO2004078163A2 | Feb 26, 2004 | Sep 16, 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2007113120A1 | Mar 22, 2007 | Oct 11, 2007 | Frank Hoffmann | Stamping apparatus with feed device |
| WO2007131201A2 * | May 4, 2007 | Nov 15, 2007 | Irm Llc | Compounds and compositions as hedgehog pathway modulators |
| WO2008154259A1 | Jun 4, 2008 | Dec 18, 2008 | Irm Llc | Biphenylcarboxamide derivatives as hedgehog pathway modulators |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link
blogs are

MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ


‘Female Viagra’ Flibanserin now on track for Q3 filing in USA

Flibanserin, girosa
167933-07-5 cas no
147359-76-0 (monoHCl)
- Bimt 17
- BIMT 17 BS
- Bimt-17
- Flibanserin
- Girosa
- UNII-37JK4STR6Z
Women with low libido in the US will have to wait even longer for approval of the first ever treatment for the condition after regulators requested more data on the forerunner flibanserin, delaying its submission until later this year.
The US Food and Drug Administration has asked manufacturer Sprout Pharmaceuticals for data on how flibanserin interacts with other medicines and also how it affects driving ability, after around 10% of patients experienced sleepiness while on the drug
Read more at: http://www.pharmatimes.com/Article/14-02-11/Female_Viagra_now_on_track_for_Q3_filing_in_USA.aspx#ixzz2tAWxwzRD
December 11, 2013 – Sprout Pharmaceuticals today announced that it has received and appealed the Food and Drug Administration’s (FDA) Complete Response Letter (CRL) for flibanserin through the Formal Dispute Resolution process.
Flibanserin is an investigational, once-daily treatment for Hypoactive Sexual Desire Disorder, or HSDD, in premenopausal women. HSDD is the most commonly reported form of female sexual dysfunction
read all here
A new drug being developed by Boehringer Ingelheim could give a boost to the sex drive of women with low libido. The drug, known as flibanserin, has been shown in clinical trials to increase their sexual desire when taken once a day at bedtime.
The results from four pivotal Phase III clinical trials on women with hypoactive sexual desire disorder (HSDD) were presented this week at the European Society for Sexual Medicine’s congress in Lyon, France. The trials showed that participants taking flibanserin had a significant improvement in their sexual desire compared to those given a placebo. They also experienced less of the distress associated with sexual dysfunction.
The drug was initially being investigated as a treatment for depression, and acts on the serotonin receptors in the brain – it is both a 5-HT1A receptor agonist and a 5-HT2A receptor antagonist. It is also a partial agonist at the dopamine D4 receptor.

Neurotransmitters such as serotonin are believed to be involved in sexual function, and antidepressants are commonly associated with a loss of libido, so this was an obvious side-effect to look out for during clinical trials in depression. But far from suppressing the libido in women, it appeared to have the opposite effect, so trials in women with HSDD were initiated.
Hormone replacement can improve the libido of women who have had their ovaries removed, but there is no available drug to treat those who have not. There have been accusations that pharma companies invent new diseases like HSDD in order to sell more medicines, but according to Kathleen Segraves, an assistant professor at Case Western Reserve University in the US who has worked in the field of sexual functioning for many years, this is not the case here. HSDD is a very real disorder, she says, and the potential for a treatment for these women is very exciting.

Flibanserin (code name BIMT-17; proposed trade name Girosa) is a drug that was investigated by Boehringer Ingelheim as a novel, non-hormonal treatment for pre-menopausal women with Hypoactive Sexual Desire Disorder (HSDD).[1][2] Development was terminated in October 2010 following a negative report by the U.S. Food and Drug Administration.[3]
HSDD is the most commonly reported female sexual complaint and characterized by a decrease in sexual desire that causes marked personal distress and/or personal difficulties. According to prevalence studies about 1 in 10 women reported low sexual desire with associated distress, which may be HSDD.[4] The neurobiological pathway of female sexual desire involves interactions among multiple neurotransmitters, sex hormones and various psychosocial factors. Sexual desire is modulated in distinct brain areas by a balance between inhibitory and excitatory neurotransmitters, serotonin acting as an inhibitor while dopamine and norepinephrine act as a stimulator of sexual desire.[5][6]Flibanserin is a 5-HT1A receptor agonist and 5-HT2A receptor antagonist that had initially been investigated as an antidepressant. Preclinical evidence suggested that flibanserin targets these receptors preferentially in selective brain areas and helps to restore a balance between these inhibitory and excitatory effects.[6] HSDD has been recognized as a distinct sexual function disorder for more than 30 years.
The proposed mechanism of action refers back to the Kinsey dual control model. Several sex steroids, neurotransmitters, and hormones have important excitatory or inhibitory effects on the sexual response. Among the neurotransmitters, the excitatory activity is driven by dopamine and norepinephrine, while the inhibitory activity is driven by serotonin. The balance between these systems is relevant for a healthy sexual response. By modulating these neurotransmitters in selective brain areas, flibanserin, a 5-HT1A receptoragonist and 5-HT2A receptor antagonist, is likely to restore the balance between these neurotransmitter systems.[6]
Several large pivotal Phase III studies with Flibanserin were conducted in the USA, Canada and Europe. They involved more than 5,000 pre-menopausal women with generalized acquired Hypoactive Sexual Desire Disorder (HSDD). The results of the Phase III North American Trials demonstrated that
Although the two North American trials that used the flibanserin 100 mg qhs dose showed a statistically significant difference between flibanserin and placebo for the endpoint of [satisfying sexual events], they both failed to demonstrate a statistically significant improvement on the co-primary endpoint of sexual desire. Therefore, neither study met the agreed-upon criteria for success in establishing the efficacy of flibanserin for the treatment of [Hypoactive Sexual Desire Disorder].
These data were first presented on November 16, 2009 at the congress of the European Society for Sexual Medicine in Lyon, France. The women receiving Flibanserin reported that the average number of times they had “satisfying sexual events” rose from 2.8 to 4.5 times a month. However, women receiving placebo reported also an increase of “satisfying sexual events” from 2.7 to 3.7 times a month.
Evaluation of the overall improvement of their condition and whether the benefit was meaningful to the women, showed a significantly higher rate of a meaningful benefit in the flibanserin-treated patient group versus the placebo group.The onset of the Flibanserin effect was seen from the first timepoint measured after 4 weeks of treatment and maintained throughout the treatment period.
The overall incidence of adverse events among women taking flibanserin was low, the majority of adverse events being mild to moderate and resolved during the treatment. The most commonly reported adverse events included dizziness, nausea, fatigue, somnolence and insomnia.

On June 18, 2010, a federal advisory panel to the U.S. Food and Drug Administration (FDA) unanimously voted against recommending approval of Flibanserin.
Earlier in the week, a FDA staff report also recommended non-approval of the drug. While the FDA still might approve Flibanserin, in the past, negative panel votes tended to cause the FDA not to approve.
On October 8, 2010, Boehringer Ingelheim announced that it would discontinue its development of flibanserin in light of the FDA advisory panel’s recommendation.
On June 27, 2013, Sprout Pharmaceuticals confirmed they had resubmitted flibanserin for FDA approval.
Flibanserin, chemically 1 -[2-(4-(3-trifluoromethylphenyl)piperazin-1 – yl)ethyl]-2,3-dihydro-1 H-benzimidazole-2-one was disclosed in form of its hydrochloride in European Patent No. 526,434 (‘434) and has the following chemical structure:

Process for preparation of flibanserin were disclosed in European Patent No. ‘434, U.S. Application Publication No. 2007/0032655 and Drugs of the future 1998, 23(1): 9-16.
According to European Patent No. ‘434 flibanserin is prepared by condensing 1-(2-chloroethyl)-2,3-dihydro-1 H-benzimidazol-one with m- trifluoromethyl phenyl piperazine. According to U.S. Application Publication No. 2007/0032655 flibanserin is prepared by condensing 1-[(3-trifluoromethyl)phenyl]-4-(2- chloroethyl)piperazine with 1 -(2-propenyl)-1 ,3-dihydro-benzimidazol-2H-one.
According to Drugs of the future 1998, 23(1): 9-16 flibanserin is prepared by reacting 1-(2-chloroethyl)-2,3-dihydro-1 H-benzimidazol-one with m- trifluoromethylphenylpiperazine.
PATENT
1-[2-(4-(3-trifluoromethyl-phenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1H-benzimidazol-2-one
Compound 3
Hydrochloride salt (isopropanol) M.p. 230-231°C
Analysis

¹H NMR (DMSO-d₆/CDCL₃ 5:2) 11.09 (b, 1H), 11.04 (s, 1H), 7.5-6.9 (8H), 4.36 (t, 2H), 4.1-3.1 (10H)
CLIP

The compound 1-[2-(4-(3-trifluoromethyl-phenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H- benzimidazol-2-one (flibanserin) is disclosed in form of its hydrochlorid in European Patent Application EP-A-526434 and has the following chemical structure:

Flibanserin shows affinity for the 5-HTιA and 5-HT2-receptor. It is therefore a promising therapeutic agent for the treatment of a variety of diseases, for instance depression, schizophrenia, Parkinson, anxiety, sleep disturbances, sexual and mental disorders and age associated memory impairment.

EXAMPLE……… EP1518858A1
375 kg of 1-[(3-trifluoromethyl)phenyl]-4-(2-cloroethyl)piperazin are charged in a reactor with 2500 kg of water and 200 kg of aqueous Sodium Hydroxide 45%. Under stirring 169.2 kg of 1-(2-propenyl)-1,3-dihydro-benzimidazol-2H-one, 780 kg of isopropanol, 2000 kg of water and 220 kg of aqueous Sodium Hydroxide 45% are added. The reaction mixture is heated to 75-85° C. and 160 kg of concentrated hydrochloric acid and 200 kg of water are added.
The reaction mixture is stirred at constant temperature for about 45 minutes. After distillation of a mixture of water and Isopropanol (about 3000 kg) the remaining residue is cooled to about 65-75° C. and the pH is adjusted to 6.5-7.5 by addition of 125 kg of aqueous Sodium Hydroxide 45%. After cooling to a temperature of 45-50° C., the pH value is adjusted to 8-9 by addition of about 4 kg of aqueous Sodium Hydroxide 45%. Subsequently the mixture is cooled to 30-35° C. and centrifuged. The residue thus obtained is washed with 340 l of water and 126 l of isopropanol and then with water until chlorides elimination.
The wet product is dried under vacuum at a temperature of about 45-55° C. which leads to 358 kg of crude flibanserin polymorph A. The crude product thus obtained is loaded in a reactor with 1750 kg of Acetone and the resulting mixture is heated under stirring until reflux. The obtained solution is filtered and the filtrate is concentrated by distillation. The temperature is maintained for about 1 hour 0-5° C., then the precipitate solid is isolated by filtration and dried at 55° C. for at least 12 hours.
The final yield is 280 kg of pure flibanserin polymorph A.
CLIP
Flibanserin may be prepared by reacting 1-(phenylvinyl)-2,3-dihydro-1H-benzimidazol-2-one (I) with 1,2-dichloroethane (II) in the presence of NaH in warm dimethylformamide. The resulting 1-(2-chloroethyl)-2,3-dihydro-1H-benzimidazol-one (III) is in turn coupled with commercially available m-trifluoromethylphenylpiperazine hydrochloride (IV) in the presence of sodium carbonate and catalytic potassium iodide in refluxing ethanol. The crude flibanserin hydrochloride (V) is then dissolved in aqueous ethanol and the pure base is precipitated upon addition of sodium hydroxide.
PICK UP INTERMEDIATES FROM CHEM24H.COM
1-(1-phenylvinyl)-1,3-dihydro-2H-benzimidazol-2-one (I)
1,2-dichloroethane (II)
1-(2-chloroethyl)-1,3-dihydro-2H-benzimidazol-2-one (III)
1-[3-(trifluoromethyl)phenyl]piperazine; N-[3-(trifluoromethyl)phenyl]piperazine (IV)
1-(2-[4-[3-(trifluoromethyl)phenyl]piperazino]ethyl)-1,3-dihydro-2H-benzimidazol-2-one (V)
PATENT


wherein R2 is as defined in formula X; with a compound of formula Xl:

According to another aspect of the present invention there is provided a novel compound or a salt thereof selected from the compounds of formula I, IV and VII:


Wherein R is hydrogen or an amino protecting group.
Preferable the amino protecting groups are selected from butyl, 1 ,1- diphenylmethyl, methoxymethyl, benzyloxymethyl, trichloroethoxymethyl, pyrrolidinomethyl, cyanomethyl, pivaloyloxymethyl, allyl, 2-propenyl, t- butyldimethylsilyl, methoxy, thiomethyl, phenylvinyl, 4-methoxyphenyl, benzyl, A- methoxybenzyl, 2,4-dimethoxybenzyl, 2-nitrobenzyl, t-butoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, 4-chlorophenoxycarbonyl, A- nitrophenoxycarbonyl, methoxycarbonyl and ethoxycarbonyl. Still more preferable protecting groups are selected from t- butoxycarbonyl, ethoxycarbonyl, methoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, phenylvinyl and 2-propenyl.
R1 is independently selected from chlorine, bromine, iodine, methanesulphonate, trifluoromethanesulphonate, paratoluenesulphonate or benzenesulphonate. Preferable R1 is independently selected from chlorine, bromine or iodine and more preferable R1 is chlorine.
Wherein R2 is hydrogen or an amino protecting group.
The amino protecting group may be any of the groups commonly used to protect the amino function such as alkyl, substituted alkyl, hetero substituted alkyl, substituted or unsubstituted unsaturated alkyl, alkyl substituted hetero atoms, substituted or unsubstituted phenyl, substituted or unsubstituted benzyl, alkyoxy carbonyl groups and aryloxy carbonyl groups.
Preferable the amino protecting groups are selected from butyl, 1 ,1 – diphenylmethyl, methoxymethyl, benzyloxymethyl, trichloroethoxymethyl, pyrrolidinomethyl, cyanomethyl, pivaloyloxymethyl, allyl, 2-propenyl, t- butyldimethylsilyl, methoxy, thiomethyl, phenylvinyl, 4-methoxyphenyl, benzyl, A- methoxybenzyl, 2,4-dimethoxybenzyl, 2-nitrobenzyl, t-butoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, 4-chlorophenoxycarbonyl, A- nitrophenoxycarbonyl, methoxycarbonyl and ethoxycarbonyl. Still more preferable protecting groups are selected from t- butoxycarbonyl, ethoxycarbonyl, methoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, phenylvinyl and 2-propenyl. The following examples are given for the purpose of illustrating the present invention and should not be considered as limitations on the scope and spirit of the invention.
EXAMPLES Example 1
A mixture of sodium hydroxide (47 gm) and i-(α-methylvinyl) benzimidazol-2-one (100 gm) in dimethylformamide (400 ml) was .stirred for 1 hour at room temperature. Dibromoethane (217 gm) was slowly added to the mixture and stirred at 1 hour 30 minutes. The resulting solution after addition water (500 ml) was extracted with ethyl acetate. The combined ethyl acetate extract washed with water. After drying the solvent was removed under vacuum to yield 132 gm of 1 ,3-dihydro-1-(2-bromoethyl)-3-isopropenyl-2H-benzimidazol- 2-one as a yellow oily liquid.
Example 2 A mixture of 1 ,3-dihydro-1-(2-bromoethyl)-3-isopropenyl-2H- benzimidazol-2-one (100 gm), diethanolamine (175 ml), sodium carbonate (40 gm) and potassium iodide (10 gm) was heated to 90 to 95 deg C and stirred for 2 hours. The reaction mass was cooled to room temperature and added water (500 ml). The resulting mixture extracted into ethyl acetate and the organic layer washed with water. After drying the solvent was removed under vacuum to yield 105 gm of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3-isopropenyl- 2H-benzimidazol-2-one as a thick yellow oily liquid.
Example 3
To the mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3- isopropenyl-2H-benzimidazol-2-one (100 gm) obtained as in example 2 and chloroform (300 ml), thionyl chloride (95 ml) was slowly added. The mixture was heated to reflux and stirred for 2 hours. The excess thionyl chloride and chloroform was distilled off to yield 98 gm of 1 ,3-dihydro-1-[2-[N-[bis-(2- chloroethyl)amino]ethyl]-3-isopropenyl-2H-benzimidazol-2-one as a brown coloured sticky residue.
Example 4
1 ,3-dihydro-1-[2-[N-[bis-(2-chloroethyl)amino]ethyl]-3-isopropenyl-2H- benzimidazol-2-one (98 gm) obtained as in example 3 was added to water (500 ml) and concentrated hydrochloric acid (200 ml) mixture. The mixture was heated to 60 to 65 deg C and stirred for 1 hour. The contents of the flask cooled to room temperature and pH of the solution adjusted to 9 – 10 with 10% sodium hydroxide solution. The resulting solution extracted with ethyl acetate and washed the organic layer with water. Evaporate the solvent under reduced pressure to yield 82 gm of 1 ,3-dihydro-1-[2-[N-bis-(2-chloroethyl)amino]ethyl]- 2H-benzimidazol-2-one as a dark brown coloured oily liquid
Example 5
A mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-chloroethyl)amino]ethyl]-1,2-H- benzimidazol-2-one (82 gm) obtained as in example 4, xylene (300 ml) and m- trifluoromethyl aniline (58 gm) was refluxed for 64 hours. The reaction mass was cooled to room temperature and filtered to obtain 1-[2-(4-(3- thfluoromethylphenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H-benzimidazole-2-one hydrochloride (Flibanserin hydrochloride) as a light brown coloured solid.
The crude flibanserin hydrochloride was purified in isopropyl alcohol to give 85 gm of pure flibanserin hydrochloride as off white solid.
Example 6
Piperazine (12 gm), toluene(60 ml) and tetra butyl ammonium bromide (1 gm) mixture was heated to 60 deg C, added 1 ,3-dihydro-1-(2-bromoethyl)-3- isopropenyl-2H-benzimidazol-2-one (10 gm) and stirred for 4 hours at 90 to 95 deg C. The mixture was cooled to 60 deg C and added water (50 ml). The separated toluene layer distilled under vacuum to give 8.5 gm of 1 ,3-dihydro-1- (2-piperazinyl)ethyl-3-isopropenyl-2H-benzimidazol-2-one as a white solid.
Example 7
To the mixture of concentrated hydrochloric acid (20 ml) and water (100 ml) was added 1 ,3-dihydro-1-(2-piperazinylethyl)-3-isopropenyl-2H- benzimidazol-2-one (10 gm) obtained as in example 6 and heated to 60 to 65 deg C 1 hour. The mixture was cooled to room temperature and pH of the solution was adjusted to 9 – 10 with 10% sodium hydroxide solution, extracted with ethyl acetate and the organic layer was washed with water. After drying the solvent was removed under vacuum to yield 8.5 gm of 1 ,3-dihydro-1-(2- piperazinyl ethyl)-2H-benzimidazol-2-one as a white solid.
Example 8
3-trifluoromethylaniline (40 gm) and hydrobromic acid (85 ml; 48- 50%w/w) mixture was cooled to 0 to 5 deg C. To this mixture added sodium nitrite solution (18.5 gm in 25 ml of water) at 5 to 10 deg C and copper powder (1 gm). The temperature was slowly raised to 50 to 55 deg C and stirred for 30 minutes. Added water (200 ml) to reaction mass and applied steam distillation, collected m-trifluoromethylbromobenzene as oily liquid. The oily liquid washed with sulfuric acid for two times (2 X 10 ml) followed by washed with water (2 X 20 ml) and dried the liquid with sodium sulphate to give 22 gm of m- trifluoromethylbromobenzene.
Example 9
To a mixture of 1 ,3-dihydro-1-(2-piperazinyl ethyl)-2H-benzimidazol-2- one (10 gm) obtained as in example 7, m-trifluoromethylbromobenzene (9 gm) obtained as in example 8, sodium tert-butoxide (5.5 gm), palladium acetate (4.5 mg) and xylene (80 ml) was added tri-tert.-butylphosphine (0.2 ml). The mixture was heated to 120 deg C and stirred for 3 hours. The reaction mass was cooled, added water (100 ml) and extracted with ethyl acetate and the organic layer was washed with water. After drying the solvent was removed under vacuum to yield
10 gm of 1-[2-(4-(3-trifluoromethylphenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H- benzimidazole-2-one (Flibanserin).
Example 10
To a mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3- isopropenyl-2H-benzimidazol-2-one (100 gm) obtained as in example 3, cyclohexane (400 ml) and sodium carbonate (35 gm) was added benzene sulfonyl chloride (116 gm) at room temperature. The mixture was heated to 80 to
85 deg C and stirred for 8 hours . The contents were cooled to room temperature and added water (500 ml). Distilled the organic layer to give 182 gm of 1 ,3-dihydro-1-[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzimidazol-2-one.
Example 11
1 ,3-dihydro-1 -[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzitηidazol-2-one (100 gm) obtained as in example 10, dimethylformamide (500 ml) and sodium corbonate (18 gm) was mixed and heated to 70 deg C. To the mixture was added m-trifluoromethyl aniline (27 gm) and heated to 80 to 85 deg C, stirred for 5 hours. The reaction mass was cooled and added water (2000 ml), filtered the solid to yield 1 ,3-dihydro-1-[2-[4-(3- trifluoromethylphenyl)piperazinyl]ethyl]-3-isopropenyl-2H benzimidazol-2-one. Example 12
1 ,3-dihydro-1-[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzimidazol-2-one (100 gm) obtained as in example 11 added to the mixture of water (500 ml) and concentrated hydrochloric acid (200 ml), heated to 65 deg C and stirred for 1 hour. The reaction mass was cooled to room temperature and pH adjusted to 10 to 10-5 with 10% sodium hydroxide solution. The resulting mixture was extracted with ethyl acetate and the organic
■ layer was washed with water. After drying the solvent was removed under vacuum to yield 87 gm of 1-[2-(4-(3-trifluoromethylphenyl)piperazin-1-yl)ethyl]- 2,3-dihydro-1 H-benzimidazole -2-one (Flibanserin).
Paper
Journal of Pharmaceutical and Biomedical Analysis, v.57, 2012 Jan 5, p.104(5)
Isolation and structural elucidation of flibanserin as an adulterant in a health supplement used for female sexual performance enhancement
Low, Min-Yong et al
http://www.sciencedirect.com/science/article/pii/S0731708511004833

This proposed formula and structure was further confirmed by 1H and 13C NMR data which indicated the presence of 20 carbon atoms and 21 protons.
1H NMR
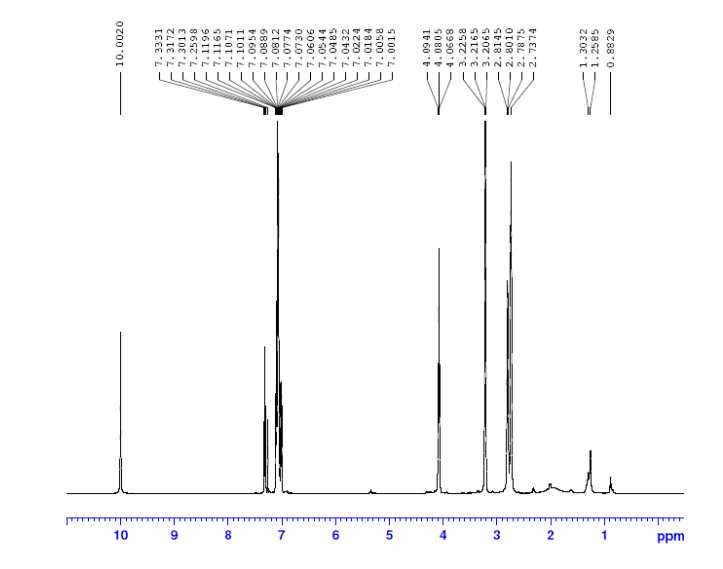
13C NMR
1D and 2DNMR data were used to assign the protons and carbon atoms.
In the1H NMR spectrum , a sharp singlet at 10.00 ppm integrating for one
proton is a typical proton attached to nitrogen. HMBC correlated this proton to C-2, C-4, and C-9 suggesting that it was H-3.
Complex signals were observedbetween 7.00 to 7.31 ppm, integrating for eight protons. A triplet at 7.31 ppm,integrating for a proton has a coupling constant of 8.0 Hz. HMBC correlated thisproton with C-16, C-19, and C-21 suggesting that it was H-20.
A double-doubletsplitting pattern at chemical shift 7.11 ppm, integrating for a proton, has couplingconstants of 6.3 Hz and 1.6 Hz.
HMBC correlated this proton to C-6, C-7, and C-9 showing that it was H-8. Overlapped signals were observed from 7.04 ppm to7.10 ppm, integrating for five protons. A double-doublet splitting pattern at 7.01ppm with coupling constant 8.0 Hz and 2.0 Hz, integrating for a proton was
observed.
HMBC correlated this proton to C-17 suggesting that it was either H-19or H-21. Four triplet signals were also observed from 2.73 ppm to 4.08 ppm,integrating for a total of twelve protons.
Two of these triplet signals at 2.74 ppmand 3.22 ppm integrated for four protons each, suggesting overlapping signals ofmethylene protons. This was further confirmed by 13C and DEPT NMR.
13C and DEPT NMR data showed the signals of four methylene, eight methineand six quaternary carbon atoms. The DEPT signals at 53.1 ppm and 48.6 ppmhave intensities which were double of those from the rest of the methylene carbonsignals, suggesting two methylene carbon atoms each contributing to the signal at 53.1 ppm and 48.6 ppm.
DEPT

HMQC results further indicated that these two methylene carbon signals at 53.1 ppm and 48.6 ppm were correlated to the protons signal at 2.73 ppm and 4.08 ppm respectively, which corresponded to four protons each. The finding confirmed overlapping methylene carbon signals (at 53.1 ppm and 48.6 ppm) and methylene proton signals (at 2.73 ppm and 4.08 ppm). Hence, the unknown compound has six methylene carbon atoms with a total of twelve methylene protons.
The chemical shifts of the twelve methylene protons suggested that they were attached to relatively electronegative atoms. It was speculated that the six methylene groups were attached to the nitrogen atoms and the electron withdrawing effect of these electronegative nitrogen atoms resulted in the deshielding of the protons. HMBC and COSY correlations were used to assign the rest of the protons
HMBC

HMQC
COSY
The 13C NMR data showed that there were two quaternary carbon at
155.6 ppm and 151.3 ppm. The carbon with chemical shift 155.6 ppm was C-2. Inthe structure of imidazolone, carbonyl carbon C-2 was attached to two nitrogenatoms which helped to withdraw electrons from oxygen to C-2. Hence, C-2 wasless deshielded as compared to a normal carbonyl carbon which has chemical shiftabove 170 ppm.
Eight methine carbons and two quaternary carbons with chemicalshifts above 108 ppm suggested the presence of two aromatic rings. Thequaternary carbon with chemical shift 125.4 ppm was C-22 which was attached tothree fluorine atoms. Due to the strong electron withdrawing effect of the fluorineatoms, C-22 was highly deshielded and had a high chemical shift.
The IR spectrum of the isolated compound showed absorption bands of amide (νC=O 1685 cm-1, νN-H (stretch) 3180 cm-1, νN-H (bending) 1610 cm-1), alkyl fluoride (νC-F1077 cm-1, 1112 cm-1, 1158 cm-1), aromatic ring (ν Ar-H 3028 cm-1, 3078 cm-1 andνC=C 1401 cm-1, 1446 cm-1, 1453 cm-1, 1468 cm-1, 1487 cm-1) and alkane (νC-H2891 cm-1, 2930 cm-1 2948 cm-).
FOR MASS, HMBC ETC SEE………http://orgspectroscopyint.blogspot.in/2015/06/flibanserin.html
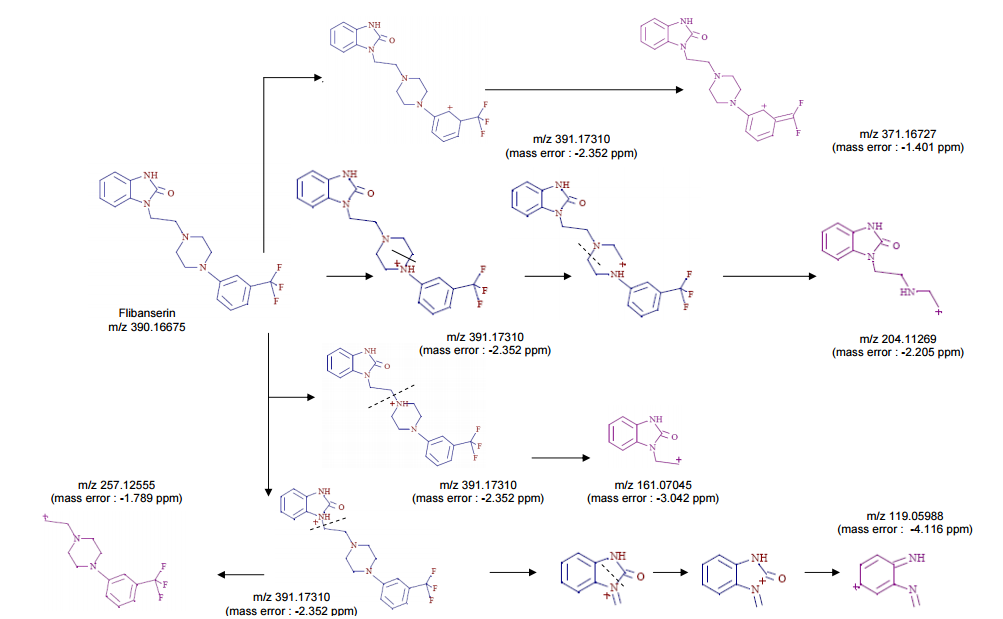
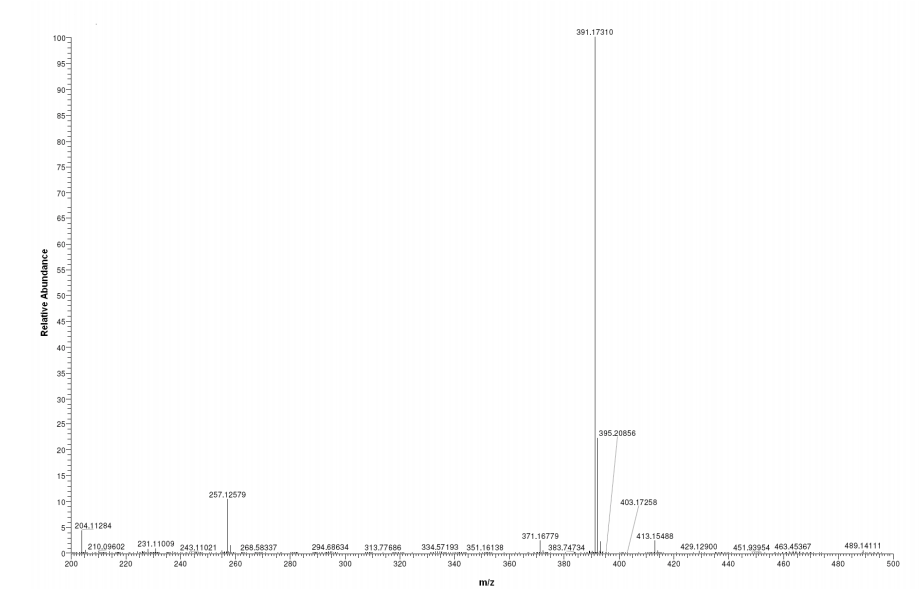
NMR PREDICT
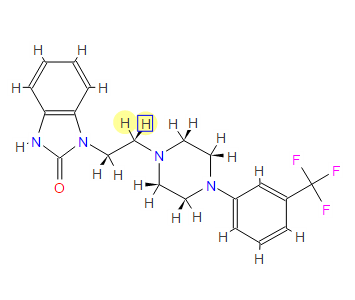
1H NMR PREDICT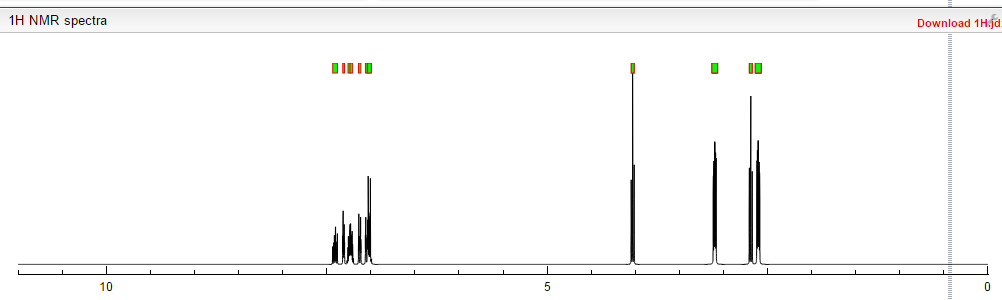
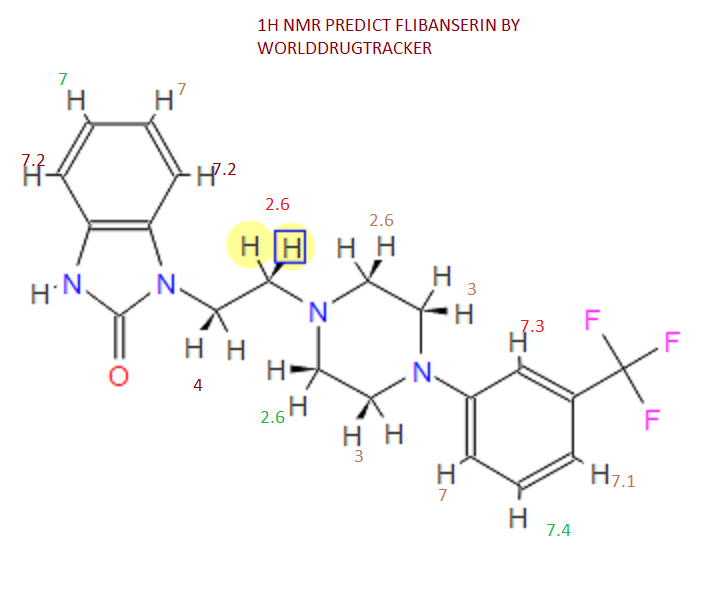
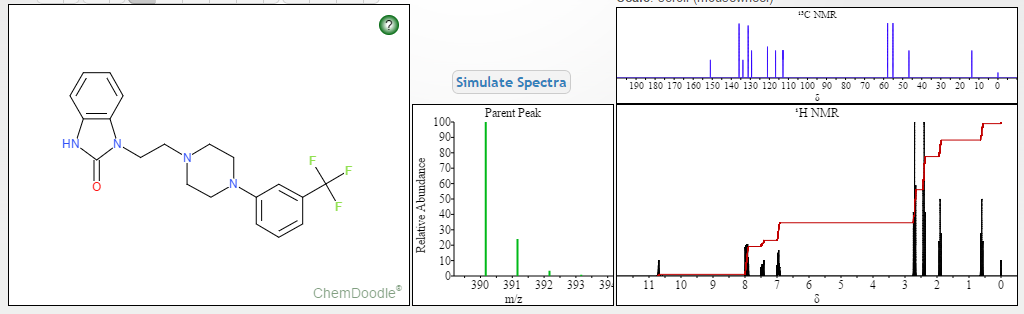
13C NMR PREDICT
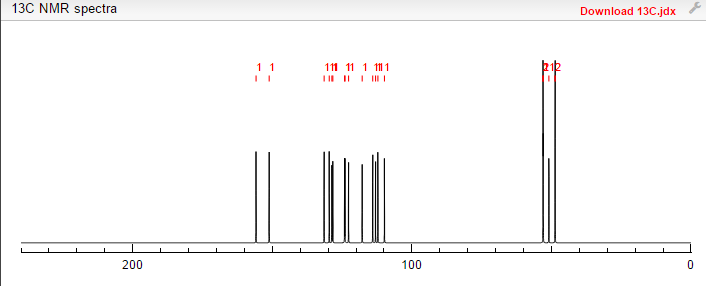
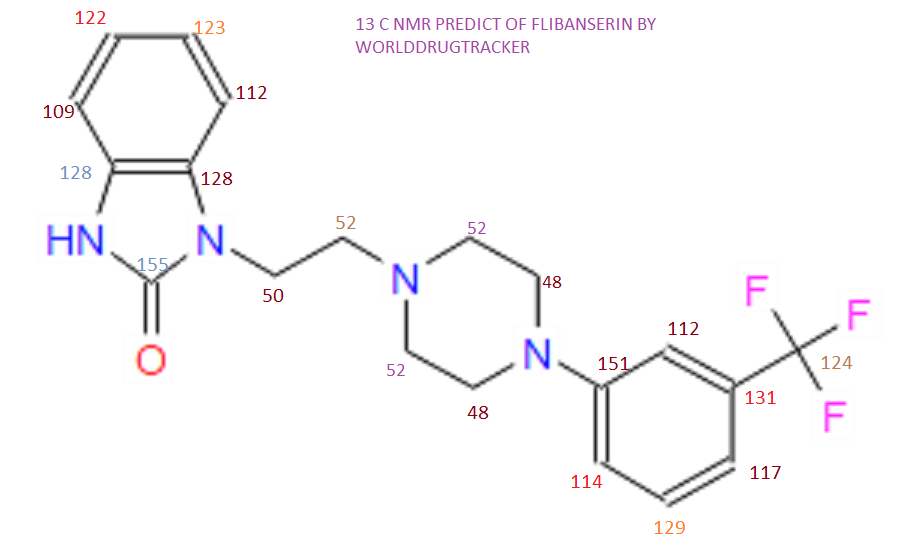
COSY PREDICT
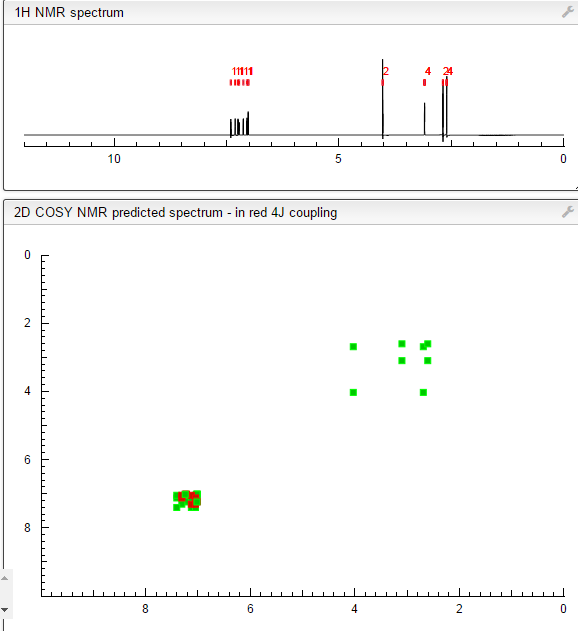
NMR PREDICT FROM MOLBASE
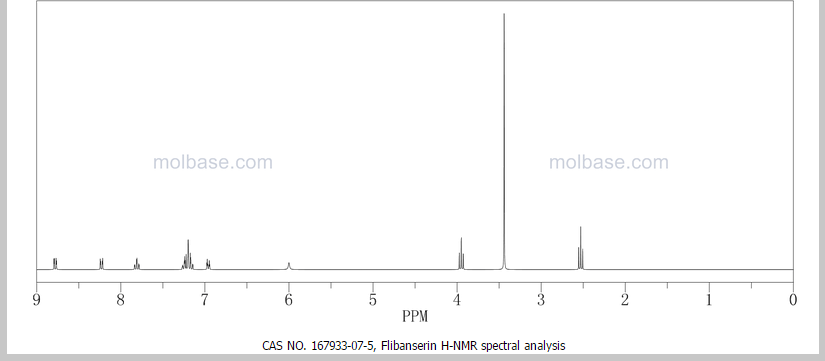
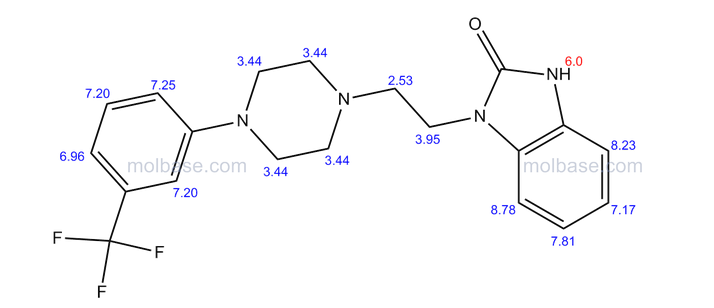
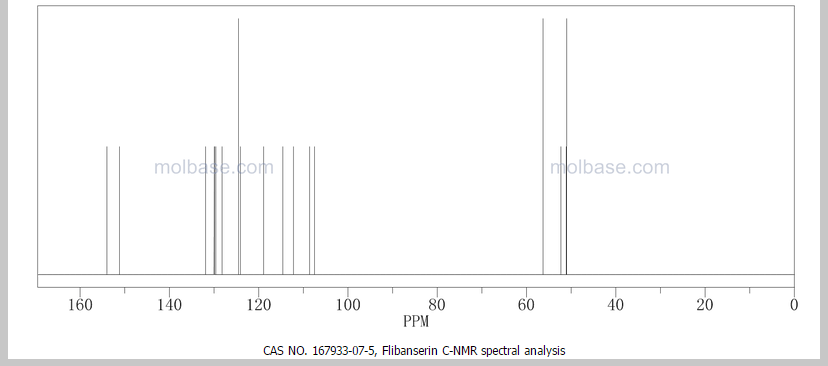

PATENT
US5576318, 1996
1 H NMR (DMSO-d6 /CDCL3 5:2) 11.09 (b, 1H), 11.04 (s, 1H), 7.5-6.9 (SH), 4.36 (t, 2H), 4.1-3.1 (10 H)
UPDATES………..
A Facile Route of Synthesis for Making Flibanserin

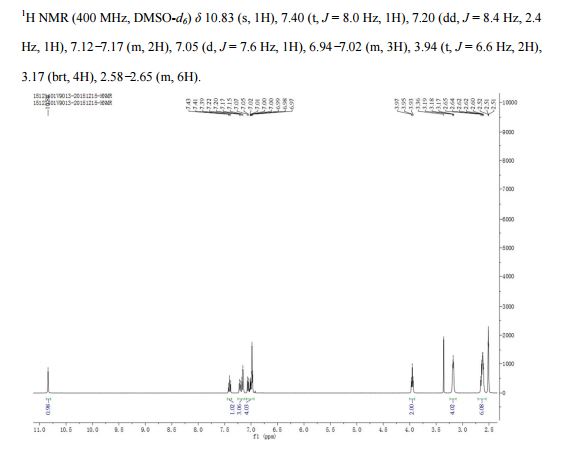
REFERENCES
- Borsini F, Evans K, Jason K, Rohde F, Alexander B, Pollentier S (summer 2002). “Pharmacology of flibanserin”. CNS Drug Rev. 8 (2): 117–142. doi:10.1111/j.1527-3458.2002.tb00219.x. PMID 12177684.
- Jolly E, Clayton A, Thorp J, Lewis-D’Agostino D, Wunderlich G, Lesko L (April 2008). “Design of Phase III pivotal trials of flibanserin in female Hypoactive Sexual Desire Disorder (HSDD)”. Sexologies 17 (Suppl 1): S133–4. doi:10.1016/S1158-1360(08)72886-X.
- Spiegel online: Pharmakonzern stoppt Lustpille für die Frau, 8 October 2010 (in German)
- Nygaard I (November 2008). “Sexual dysfunction prevalence rates: marketing or real?”. Obstet Gynecol 112 (5): 968–9.doi:10.1097/01.AOG.0000335775.68187.b2. PMID 18978094.
- Clayton AH (July 2010). “The pathophysiology of hypoactive sexual desire disorder in women”. Int J Gynaecol Obstet 110 (1): 7–11.doi:10.1016/j.ijgo.2010.02.014. PMID 20434725.
- Pfaus JG (June 2009). “Pathways of sexual desire”. J Sex Med 6 (6): 1506–33. doi:10.1111/j.1743-6109.2009.01309.x.PMID 19453889.
- Yves Aubert, Thesis, Leiden University. (Dec 11, 2012) Sex, aggression and pair-bond: a study on the serotonergic regulation of female sexual function in the marmoset monkey
- Viagra for women?
- Marazziti D, Palego L, Giromella A, et al. (June 2002). “Region-dependent effects of flibanserin and buspirone on adenylyl cyclase activity in the human brain”. Int. J. Neuropsychopharmacol. 5 (2): 131–40. doi:10.1017/S1461145702002869.PMID 12135537.
- Podhorna J, Brown RE (June 2000). “Flibanserin has anxiolytic effects without locomotor side effects in the infant rat ultrasonic vocalization model of anxiety”. Br J Pharmacol 130 (4): 739–746. doi:10.1038/sj.bjp.0703364. PMC 1572126.PMID 10864879.
- Brambilla A, Baschirotto A, Grippa N, Borsini F (December 1999). “Effect of flibanserin (BIMT 17), fluoxetine, 8-OH-DPAT and buspirone on serotonin synthesis in rat brain”. Eur Neuropsychopharmacol 10 (1): 63–7. doi:10.1016/S0924-977X(99)00056-5.PMID 10647099.
| EP0200322A1 * | Mar 18, 1986 | Nov 5, 1986 | H. Lundbeck A/S | Heterocyclic compounds |
| BE904945A1 * | Title not available | |||
| GB2023594A * | Title not available | |||
| US3472854 * | May 29, 1967 | Oct 14, 1969 | Sterling Drug Inc | 1-((benzimidazolyl)-lower-alkyl)-4-substituted-piperazines |
| US4954503 * | Sep 11, 1989 | Sep 4, 1990 | Hoechst-Roussel Pharmaceuticals, Inc. | 3-(1-substituted-4-piperazinyl)-1H-indazoles |
update………..
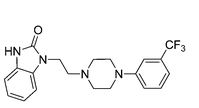
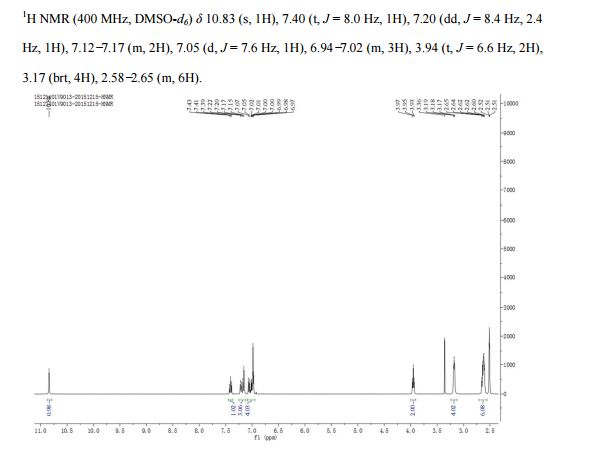
1-(2-(4-(3-(Trifluoromethyl)phenyl)piperazin-1-yl)ethyl)-1H-benzo[d]imidazol-2(3H)-one (1)

A novel and efficient route of synthesis for making flibanserin via 2-ethoxy-1H-benzo[d]imidazole (12) was described with excellent yield. This protocol provided a more facile approach toflibanserin.
A Facile Route of Synthesis for Making Flibanserin
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.6b00108

aReagents and conditions: (a) ethyl benzoylacetate, 200 °C; (b) dichloroethane, NaH, DMF; (c) conc HCl (aq); (d) 1-(3-(trifluoromethyl)phenyl)piperazine hydrochloride, Na2CO3, KI, EtOH; (e)
- 3.Bietti, G.; Borsini, F.; Turconi, M.; Giraldo, E.; Bignotti, M. For treatment of central nervous system disorders. U.S. Patent 5,576,318, 1996.
- 4.Mohan Rao, D.; Krishna Reddy, P.; Venkat Reddy, B. Preparing benzoimidazol-2-one compound, useful to prepare flibanserin, comprises reacting benzoimidazol-2-one compound with 2-(2-hydroxy-ethylamino)-ethanol to give (bis-(hydroxy-ethyl)-amino)-ethyl-benzoimidazol-2-one compound. PCT. Int.WO2,010,128,516, 2010.5.
- 5.Vernin, G.; Domlog, H.; Siv, C.; Metzger, J.; El-Shafei, A. K.Synthesis of 1-alkyl and 1, 3-dialkyl-2-benzimidazolones from 1-alkenyl-2-benzimidazolones using phase-transfer catalysis technique J. Heterocycl. Chem. 1981, 18, 85– 89, DOI: 10.1002/jhet.5570180118
-
A patent application for the new synthetic route has been filed in China (CN201610527244.4).

aReagents and conditions: (a) ethyl acetoacetate, KOH, EtOH, xylene, reflux, 56%; (b) 1,2-dibromoethane, K2CO3, DMF, 50 °C, 50%; (c) K2CO3, CH3CN, 70 °C, 80%; (d) conc. HCl (aq), isopropanol, 70 °C; (e) NaOH (aq), rt, 72% over two steps.

aReagents and conditions: (a) tetraethyl orthocarbonate, AcOH, 70 °C, 94%; (b) 1-bromo-2-chloroethane, K2CO3, acetone, reflux, 75%; (c) K2CO3, NaI, H2O, reflux, 92%; (d) conc. HCl (aq), isopropanol, 70 °C; (e) NaOH (aq), 68% over two steps.
//////////////
Route to Benzimidazol-2-ones via Decarbonylative Ring Contraction of Quinoxalinediones: Application to the Synthesis of Flibanserin, A Drug for Treating Hypoactive Sexual Desire Disorder in Women and Marine Natural Product Hunanamycin Analogue
![]()


Route to Benzimidazol-2-ones via Decarbonylative Ring Contraction of Quinoxalinediones: Application to the Synthesis of Flibanserin, A Drug for Treating Hypoactive Sexual Desire Disorder in Women and Marine Natural Product Hunanamycin Analogue

INTRODUCTION
Benzimidazol-2-ones 1 are an important class of heterocycles and a privileged scaffold in medicinal chemistry. They consist of cyclic urea fused with the aromatic backbone, which can potentially interact in a biological system by various noncovalent interactions such as hydrogen bonding and π stacking. Benzimidazolone derivatives exhibit a wide range of biological activities, and they are useful in treating various diseases including cancer, type II diabetes, central nervous system disorders, pain management, and infectious disease.1 Selected compounds embedded with a benzimidazol-2-one moiety along with their use are captured in Figure 1. It is worth mentioning that oxatomide drug with a benzimidazol-2-one core was approved for marketing a few years ago.2a Very recently, US Food and Drug Administration approved a new drug called flibanserin for the treatment of hypoactive sexual desire disorder (HSDD) in females, which contains benzimidazol-2- one motif.2b
CONCLUSIONS
We have developed a mild and new protocol for the synthesis of benzimidazol-2-ones from quinoxalinediones through decarbonylation. The present methodology can be an addition to the toolbox to prepare benzimidazolones, and it will be useful in medicinal chemistry, particularly, late-stage functionalization of natural products, drug scaffolds, or an intermediate containing quinoxaline-2,3-diones. As direct application of this method, we have successfully developed a new route for the synthesis of recently approved drug flibanserin and a urea analogue of antibiotic natural product hunanamycin A. Later application demonstrates the utility of the present method in late-stage functionalization
Synthesis of 1-(2-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)ethyl)-1,3-dihydro-2Hbenzo[d]imidazol-2-one (Flibanserin)
Flibanserin hydrochloride as white solid.
1H NMR (400MHz ,DMSO-d6) 11.06 (s, 1 H), 10.93 (br. s., 1 H), 7.54 – 7.41 (t, J = 7.9 Hz, 1 H), 7.36 – 7.22 (m, 3 H), 7.15 (d, J = 7.6 Hz, 1 H), 7.09 – 7.01 (m, 3 H), 4.30 (t, J = 6.7 Hz, 2 H), 4.01 (d, J = 11.6 Hz, 2 H), 3.75 (d, J = 10.4 Hz, 2 H), 3.54 – 3.43 (d, J = 4.2 Hz 2 H), 3.31 – 3.10 (m, 4 H);
HRMS (ESI): m/z calculated for C20H22ON4F3[M+H]+ 391.1740 found 391.1743;
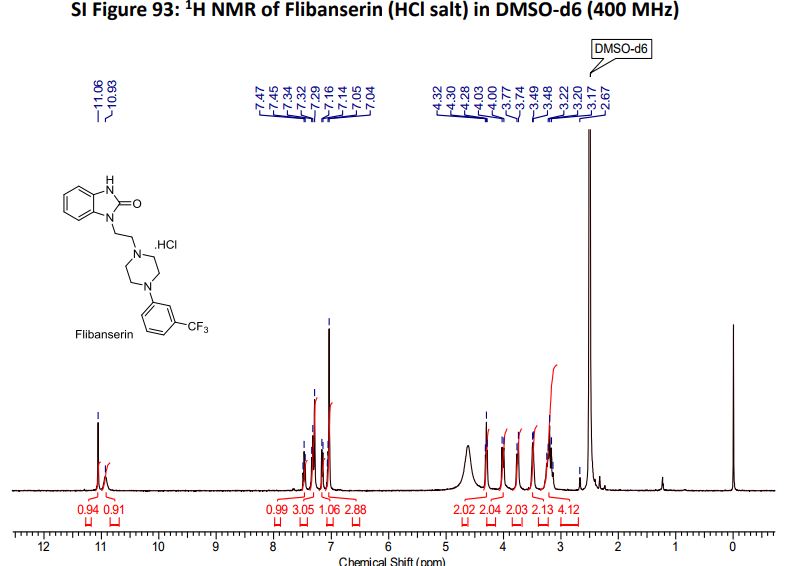
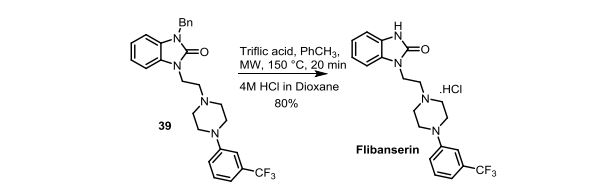

Scheme 4. Synthesis of Flibanserin through Ring Contraction
The same methodology was applied for the synthesis of flibanserin, also known as “female viagra”, which is the first approved medication for treating HSDD in women and is classified as a multifunctional serotonin agonist antagonist.(14, 15) Our synthesis of flibanserin commenced with 1-benzyl-1,4-dihydroquinoxaline-2,3-dione 36,(16) which was reacted with known chloride 37(17) under the basic condition in DMF to give the desired product 38 in good yield. Compound 38 was subjected for the decarbonylative cyclization under the optimized condition to afford the product 39 in 59% yield. Finally, the benzyl group was deprotected using trifluoromethanesulfonic acid in toluene under microwave irradiation,(8b, 18) which gave flibanserin in excellent yield (Scheme 4). The final product was isolated as HCl salt, and all of the spectral data are in agreement with the published data.(15c)

Rahul D. Shingare completed his M.Sc (Chemistry) from Fergusson College, Pune in 2008. He worked as a research associate in Ranbaxy and Lupin New drug discovery center, Gurgaon and Pune respectively until 2012 and currently pursuing his doctoral research in NCL – Pune from 2012.
Current Research Interests: Antibacterial Natural Product Hunanamycin A: Total Synthesis, SAR and Related Chemistry.
e-mail: rd.shingare@ncl.res.in

Akshay Kulkarni completed his M.Sc. from Ferguson College, Pune University in the year 2015 and joined our group as a Project Assistant in the month of October, 2015.
Current research interest: Synthesis of silicon incorporated biologically active antimalerial compounds.
e-mail : as.kulkarni@ncl.res.in
Dr.D. Srinivasa Reddy
Organic Chemistry Division
CSIR-National Chemical Laboratory
-
See, previous synthesis of Flibanserin:
(a) Bietti, G.; Borsini, F.; Turconi, M.; Giraldo, E.; Bignotti, M. For treatment of central nervous system disorders. U.S. Patent 5,576,318, 1996.
(b) Mohan, R. D.; Reddy, P. K.;Reddy, B. V. Process for the preparation of Flibanserin involving novel intermediates. WO2010128516 A2,2010.
(c) Yang, F.; Wu, C.; Li, Z.; Tian, G.; Wu, J.; Zhu, F.; Zhang, J.; He, Y.; Shen, J. A Facile route of synthesis for making Flibanserin Org. Process Res. Dev. 2016, 20, 1576 DOI: 10.1021/acs.oprd.6b00108
[ACS Full Text ], [CAS]
], [CAS] -
Xueong, X. Preparation method of Flibanserin. CN104926734 A, 2015.
//////////
ERTUGLIFLOZIN

ERTUGLIFLOZIN, PFIZER
THERAPEUTIC CLAIM Treatment of type 2 diabetes
CHEMICAL NAMES
1. β-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-(hydroxymethyl)-
2. (1S,2S,3S,4R,5S)-5-{4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl}-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
PF-04971729, MK 8835
M. Wt: 436.88
Formula: C22H25ClO7
CAS No:. 1210344-57-2
Diabetes looms as a threat to human health worldwide. As a result, considerable research efforts are devoted to identify new and efficacious anti-diabetic agents lacking the side effects associated with some of the current drugs (hypoglycemia, weight gain).Inhibition of sodium-dependent glucose cotransporter 2 (SGLT2), a transporter located in the kidney, is a mechanism that promotes glucosuria and therefore, reduction of plasma glucose concentration. Since the mechanism operates in a glucose-dependent and insulin-independent manner, and is associated with weight loss, it has emerged as a very promising approach to the pathophysiologic treatment of type 2 diabetes. Ertugliflozin (PF-04971729), an anti-diabetic agent currently in development (Phase 3 clinical trials) and belonging to a new class of SGLT2 inhibitors bearing a dioxa-bicyclo[3.2.1]octane bridged ketal motif.

SYNTHESIS
Scheme 1 outlines the general procedures one could use to provide compounds of the present invention.

Scheme 1 AIIyI 2,3,4-tιϊ-O-benzyl-D-glucopyranoside (La, where Pg1 is a benzyl group) can be prepared by procedures described by Shinya Hanashima, et al., in Bioorganic & Medicinal Chemistry, 9, 367 (2001 ); Patricia A. Gent et al. in Journal of the Chemical Society, Perkin 1, 1835 (1974); Hans Peter Wessel in the Journal of Carbohydrate Chemistry, 7, 263, (1988); or Yoko Yuasa, et al., in Organic Process Research & Development, 8, 405-407
(2004). In step 1 of Scheme 1 , the hydroxymethylene group can be introduced onto the glycoside by means of a Swern oxidation followed by treatment with formaldehyde in the presence of an alkali metal hydroxide (e.g., sodium hydroxide). This is referred to as an aldol-Cannizzaro reaction. The Swern oxidation is described by Kanji Omura and Daniel Swern in Tetrahedron, 34, 1651 (1978). Modifications of this process known to those of skill in the art may also be used. For example, other oxidants, like stabilized 2- iodoxybenzoic acid described by Ozanne, A. et al. in Organic Letters, 5, 2903 (2003), as well as other oxidants known by those skilled in the art can also be used. The aldol Cannizzaro sequence has been described by Robert Schaffer in the Journal of The American Chemical Society, 81 , 5452 (1959) and Amigues, E.J., et al., in Tetrahedron, 63,10042 (2007).
In step 2 of Scheme 1 , protecting groups (Pg2) can be added by treating intermediate (MD) with the appropriate reagents and procedures for the particular protecting group desired. For example, p-methoxybenzyl (PMB) groups may be introduced by treatment of intermediate (MD) with p-methoxybenzyl bromide or p-methoxybenzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide (DMF). Conditions involving para-methoxybenzyltrichloroacetimidate in presence of a catalytic amount of acid (e.g., trifluoromethanesulfonic acid, methanesulfonic acid, or camphorsulfonic acid) in a solvent such as dichloromethane, heptane or hexanes can also be used. Benzyl (Bn) groups may be introduced by treatment of intermediate (MD) with benzyl bromide or benzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide. Conditions involving benzylthchloroacetimidate in presence of a catalytic amount of acid (e.g., trifluoromethanesulfonic acid, methanesulfonic acid, or camphorsulfonic acid) in a solvent such as dichloromethane, heptane or hexanes can also be used. In step 3 of Scheme 1 , the allyl protection group is removed (e.g., by treatment with palladium chloride in methanol; cosolvent like dichloromethane may also be used; other conditions known by those skilled in the art could also be used, see T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991 ) to form the lactol (Ld).
In step 4 of Scheme 1 , oxidation of the unprotected hydroxyl group to an oxo group (e.g., Swern oxidation) then forms the lactone (l-e).
In step 5 of Scheme 1 , the lactone (Le) is reacted with Λ/,O-dimethyl hydroxylamine hydrochloride to form the corresponding Weinreb amide which may exist in equilibrium in a closed/opened form, (l-f/l-g). The “Weinreb amide” (LgJ can be made using procedures well known to those of skill in the art. See, Nahm, S., and S. M. Weinreb, Tetrahedron Letters. 22 (39), 3815-1818 (1981 ). For example, intermediate (l-f/l-α) can be prepared from the commercially available Λ/,O-dimethylhydroxylamine hydrochloride and an activating agent (e.g., trimethylaluminum). In step 6 of Scheme 1 , the arylbenzyl group (Ar) is introduced using the desired organometallic reagent (e.g., organo lithium compound (ArLi) or organomagnesium compound (ArMgX)) in tetrahydrofuran (THF) at a temperature ranging from about -780C to about 2O0C followed by hydrolysis (upon standing in protic conditions) to the corresponding lactol (N) which may be in equilibrium with the corresponding ketone (Ni). The bridged ketal motif found in (A) and (B) can be prepared by removing the protecting groups (Pg2) using the appropriate reagents for the protecting groups employed. For example, the PMB protecting groups may be removed by treatment with trifluoroacetic acid in the presence of anisole and dichloromethane (DCM) at about O0C to about 230C (room temperature). The remaining protecting groups (Pg1) may then be removed using the appropriate chemistry for the particular protecting groups. For example, benzyl protecting groups may be removed by treating with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature to produce the final products (A) and (B). When R1 is CN, the use of a Lewis acid like boron trichloride at a temperature ranging from about -780C to about room temperature in a solvent like dichloromethane or 1 ,2-dichloroethane may also be used to remove benzyl protective and/or para- methoxybenzyl protective groups. When R1 is CN and R2 is (Ci-C4)alkoxy in intermdediate (l-i) or in products (A) or (B), upon treatment with a Lewis acid such as boron trichloride or boron tribomide, partial to complete de-alkylation to the corresponding phenol may occur to lead to the corresponding compound (A) or (B) where R1 is CN and R2 is OH. If this occurs, the (d- C4)alkoxy group may be re-introduced via selective alkylation using a (CrC4) alkyl iodide under mildly basic conditions, for example, potassium carbonate in acetone at a temperature ranging from about room temperature to about 56 degrees Celsius.
When R1 and/or R2 is (CrC4)alkyl-SO2- it is understood by one skilled in the art that the organometallic addition step 6 (Scheme 1 ) will be carried out on the corresponding (d- C4)alkyl-S- containing organometallic reagent. The thio-alkyl is then oxidized at a later stage to the corresponding sulfone using conventional methods known by those skilled in the art.
The compounds of the present invention may be prepared as co-crystals using any suitable method. A representative scheme for preparing such co-crystals is described in Scheme 2.

Scheme 2
In Scheme 2, wherein Me is methyl and Et is ethyl, in step 1 , 1-(5-bromo-2- chlorobenzyl)-4-ethoxybenzene is dissolved in 3:1 , toluene: tetrahydrofuran followed by cooling the resulting solution to <-70°C. To this solution is added hexyllithium while maintaining the reaction at <-65°C followed by stirring for 1 hour. (3R,4S,5R,6R)-3,4,5- ths(thmethylsilyloxy)-6-((trimethylsilyloxy)methyl)-tetrahydropyran-2-one (ll-a) is dissolved in toluene and the resulting solution is cooled to -150C. This solution is then added to the – 7O0C aryllithium solution followed by stirring for 1 hour. A solution of methanesulfonic acid in methanol is then added followed by warming to room temperature and stirring for 16 to 24 hours. The reaction is deemed complete when the α-anomer level is < 3%. The reaction is then basified by the addition of 5 M aqueous sodium hydroxide solution. The resulting salts are filtered off followed by concentration of the crude product solution. 2- methyltetrahydrofuran is added as a co-solvent and the organic phase is extracted twice with water. The organic phase is then concentrated to 4 volumes in toluene. This concentrate is then added to a 5:1 , heptane: toluene solution causing precipitate to form. The solids are collected and dried under vacuum to afford a solid.
In step 2 of Scheme 2, to (ll-b) in methylene chloride is added imidazole followed by cooling to O0C and then addition of trimethylsilylchlohde to give the persilylated product.
The reaction is warmed to room temperature and quenched by the addition of water, and the organic phase is washed with water. This crude methylene chloride solution of (ll-c) is dried over sodium sulfate and then taken on crude into the next step.
In step 3 of Scheme 2, the crude solution of (ll-c) in methylene chloride is concentrated to low volume and then the solvent is exchanged to methanol. The methanol solution of (ll-c) is cooled to O0C, then 1 mol% of potassium carbonate is added as a solution in methanol followed by stirring for 5 hours. The reaction is then quenched by addition of 1 mol% acetic acid in methanol, followed by warming to room temperature, solvent exchange to ethyl acetate, and then filtration of the minor amount of inorganic solids. The crude ethyl acetate solution of (ll-d) is taken directly into the next step.
In step 4 of Scheme 2, the crude solution of (ll-d) is concentrated to low volume, then diluted with methylene chloride and dimethylsulfoxide. Triethylamine is added followed by cooling to 1O0C and then sulfur trioxide pyridine complex is added in 3 portions as a solid at 10 minute intervals. The reaction is stirred an additional 3 hours at 1O0C before quenching with water and warming to room temperature. The phases are separated followed by washing the methylene chloride layer with aqueous ammonium chloride. The crude methylene chloride solution of (ll-e) is taken directly into the next step.
In step 5 of Scheme 2, the crude solution of (ll-e) is concentrated to low volume and then the solvent is exchanged to ethanol. Thirty equivalents of aqueous formaldehyde is added followed by warming to 550C. An aqueous solution of 2 equivalents of potassium phosphate, tribasic is added followed by stirring for 24 hours at 550C. The reaction temperature is then raised to 7O0C for an additional 12 hours. The reaction is cooled to room temperature, diluted with te/t-butyl methyl ether and brine. The phases are separated followed by solvent exchange of the organic phase to ethyl acetate. The ethyl acetate phase is washed with brine and concentrated to low volume. The crude concentrate is then purified by silica gel flash chromatography eluting with 5% methanol, 95% toluene. Product containing fractions are combined and concentrated to low volume.
Methanol is added followed by stirring until precipitation occurs. The suspension is cooled and the solids are collected and rinsed with heptane followed by drying. Product (ll-f) is isolated as a solid.
In step 6 of Scheme 2, compound (ll-f) is dissolved in 5 volumes of methylene chloride followed by the addition of 1 mol% SiliaBonc/® tosic acid and stirring for 18 hours at room temperature. The acid catalyst is filtered off and the methylene chloride solution of (ll-g) is taken directly into the next step co-crystallization procedure.
In step 7 of Scheme 2, the methylene chloride solution of (ll-g) is concentrated and then the solvent is exchanged to 2-propanol. Water is added followed by warming to 550C. An aqueous solution of L-pyroglutamic acid is added followed by cooling the resulting solution to room temperature. The solution is then seeded and granulated for 18 hours. After cooling, the solids are collected and rinsed with heptane followed by drying. Product (ll-h) is isolated as a solid.
An alternative synthesis route for compounds (A) of the present invention is depicted in Scheme 3 and described below.

Scheme 3
The synthesis of (lll-a), where R3 is an alkyl or fluoro substituted alkyl (except for the carbon adjacent to the oxygen atom) can be prepared in a similar way as described in step 1 of Scheme 2. In step 1 of Scheme 3, the primary hydroxyl group is selectively protected by an appropriate protective group. For example, a trityl group (Pg3 = Tr) can be introduced by treatment of intermediate (lll-a) with chlorotriphenylmethane in presence of a base like pyridine in a solvent like toluene, tetrahydrofuran or dichloromethane at a temperature ranging from about 0 degrees Celsius to about room temperature. Additional examples of such protective groups and experimental conditions are known by those skilled in the art and can be found in T. W. Greene, Protective Groups in Organic Synthesis. John Wiley & Sons, New York, 1991.
In step 2 of Scheme 3, the secondary hydroxyl groups can be protected by the appropriate protecting groups. For example, benzyl groups (Pg4 is Bn) can be introduced by treatment of intermediate (lll-b) with benzyl bromide or benzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide at a temperature ranging from about 0 degrees Celsius to about 80 degrees Celsius. Acetyl or benzoyl groups (Pg4 = Ac or Bz) may be introduced by treatment of intermediate (lll-b) with acetyl chloride, acetyl bromide or acetic anhydride or benzoyl chloride or benzoic anhydride in the presence of a base like triethylamine, Λ/,Λ/-diisopropylethylamine or 4-
(dimethylamino)pyridine in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or dichloromethane at a temperature ranging from about 0 degrees Celsius to about 80 degrees Celsius.
In step 3 of Scheme 3, the primary hydroxyl group is deprotected to lead to intermediate (lll-d). When Pg3 is Tr, intermediate (lll-c) is treated in the presence of an acid like para-toluenesulfonic acid in a alcoholic solvent like methanol at a temperature ranging from about -20 degrees Celsius to about room temperature to provide intermediate (lll-d). Cosolvents like chloroform may be used.
In step 4 of Scheme 3, a hydroxymethylene group is introduced through a process similar to the one already described in Scheme 1 (step 1 ) and Scheme 2 (steps 4 and 5).
Other sources of formaldehyde, like paraformaldehyde in a solvent like ethanol at a temperature ranging from about room temperature to about 70 degrees Celsius in the presence of an alkali metal alkoxide can also be used in this step. When Pg4is Bn, this step provides intermediate (lll-e) and when Pg4 is Ac or Bz, this step provides intermediate (lll-f).
In step 5 of Scheme 3, intermediate (lll-e) is treated with an acid like trifluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce intermediate (lll-g).
In step 6 of Scheme 3, the remaining protecting groups (Pg4) may then be removed using the appropriate chemistry for the particular protecting groups. For example, benzyl protecting groups may be removed by treating with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature to produce the final product (A).
In step 7 of Scheme 3, intermediate (lll-f) is treated with an acid like trifluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce the final product (A). Another alternative scheme for synthesizing product (A) is depicted in Scheme 4 and described below.

Scheme 4 In step 1 of Scheme 4, intermediate (lll-a) is treated with the appropriate arylsulfonyl chloride R4SO2CI or arylsulfonic anhydride R4S(O)2OS(O)2R4 (wherein R4 is an optionally substituted aryl group, such as found in the arylsulfonyl chlorides 4-methyl-benzenesulfonyl chloride, 4-nitro-benzenesulfonyl chloride, 4-fluoro-benzenesulfonyl chloride, 2,6-dichloro- benzenesulfonyl chloride, 4-fluoro-2-methyl-benzenesulfonyl chloride, and 2,4,6-trichloro- benzenesulfonyl chloride, and in the arylsulfonic anhydride, p-toluenesulfonic anhydride) in presence of a base like pyridine, triethylamine, Λ/,Λ/-diisopropylethylamine in a solvent like tetrahydrofuran, 2-methyltetrahydrofuran at a temperature ranging from about -20 degrees Celsius to about room temperature. Some Lewis acids like zinc(ll) bromide may be used as additives. In step 2 of Scheme 4, intermediate (IV-a) is submitted to a Kornblum-type oxidation
(see, Kornblum, N., et al., Journal of The American Chemical Society, 81 , 4113 (1959)) to produce the corresponding aldehyde which may exist in equilibrium with the corresponding hydrate and/or hemiacetal form. For example intermediate (IV-a) is treated in the presence of a base like pyridine, 2,6-lutidine, 2,4,6-collidine, Λ/,Λ/-diisopropylethylamine, A- (dimethylamino)pyridine in a solvent like dimethyl sulfoxide at a temperature ranging from about room temperature to about 150 degrees Celsius. The aldehyde intermediate produced is then submitted to the aldol/Cannizzaro conditions described for step 1 (Scheme 1 ) and step 5 (Scheme 2) to produce intermediate (IV-b). In step 3 of Scheme 4, intermediate (IV-b) is treated with an acid like thfluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce the final product (A).
When R2 is (C2-C4)alkynyl the process may be performed using Scheme 5, wherein R6 is H or (CrC2)alkyl.

Scheme 5
In step 1 of Scheme 5, which provides intermediate (V-i), the organometallic addition step is carried out in a similar way to the one described in Schemel , step 6, using the organometallic reagent derived from (V-a), where Pg5 is a suitable protective group for the hydroxyl group. For instance Pgs can be a te/t-butyldimethylsilyl group (TBS) (see
US2007/0054867 for preparation of for instance {4-[(5-bromo-2-chloro-phenyl)-methyl]- phenoxy}-te/t-butyl-dimethyl-silane).
In step 2 of Scheme 5, when Pg2 = PMB, intermediate (V-i) is treated with an acid like trifluoroacetic acid, methanesulfonic acid or an acidic resin in presence of anisole in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce intermediate (V-j).
In step 3 of Scheme 5, protecting groups (Pg5) and (Pg1) can be removed to provide (V-k). Typically (Pg5) is TBS and Pg1 is Bn. In this circumstance, the protecting groups are removed by sequential treatment of (V-j) with 1 ) tetrabutylammonium fluoride in a solvent like tetrahydrofuran or 2-methyltetrahydrofuran at a temperature ranging from 0 degrees
Celsius to about 40 degrees Celsius and 2) treatment with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature. In this sequence, the order of the 2 reactions is interchangeable.
In step 4 of Scheme 5, intermediate (V-k) is treated with N,N-bis- (trifluoromethanesulfonyl)-aniline in presence of a base like triethylamine or 4- dimethyaminopyridine in a solvent like dichloromethane or 1 ,2-dichloroethane at a temperature ranging from 0 degrees Celsius to about 40 degrees Celsius to produce intermediate (V-I).
In step 5 of Scheme 5, intermediate (V-I) is subjected to a Sonogashira-type reaction (see, Sonogashira, K. Coupling Reactions Between sp2 and sp Carbon Centers. In
Comprehensive Organic Synthesis (eds. Trost, B. M., Fleming, I.), 3, 521-549, (Pergamon, Oxford, 1991 )).

IS ERTUGLIFLOZIN
Example 4
(1 S.2S.3S.4R.5S)-5-[4-chloro-3-(4-ethoxy-benzyl)-Dhen yll- 1 -h vdroxymeth yl-6.8-dioxa- bicvclo[3.2.1loctane-2,3Λ-triol (4A) and (1S,2S,3SΛS,5S)-5-[4-chloro-3-(4-ethoxy- benzvD-phen yll- 1 -h vdroxymeth yl-6, 8-dioxa-bicvclo[3.2.1 loctane-2, 3, 4-triol (4B):

To a solution of {(2S,3S)-2,3,4-tris-benzyloxy-5-[4-chloro-3-(4-ethoxy-benzyl)-phenyl]-6,8- dioxa-bicyclo[3.2.1]oct-1-yl}-methanol (l-4k: 335 mg) in ethanol/tetrahydrofuran (10 ml_, 4/1 volume) was added successively formic acid (420 microL, 22 equivalents) and palladium black (208 mg, 4 equivalents) and the resulting mixture was stirred at room temperature. After 1 hour, additional formic acid (420 microL, 22 equivalents) and palladium black (208 mg, 4 equivalents) were added and the mixture was allowed to stir for an additional hour at room temperature. The palladium was filtered and the crude mixture obtained after evaporation of solvent was purified by HPLC preparative.
HPLC preparative: reverse phase C18 Gemini column 5 micrometer 30 x 100 mm, 40 mL/minute, gradient of acetonitrile/0.1 % formic acid : water/0.1 % formic acid; 25 to 50% of acetonitrile/0.1 % formic acid over 18 minutes; UV detection: 220 nm. The HPLC indicated a ratio of diastereomers of 1.1 :1 (4A:4B).
4A: (60 mg, 29% yield); Rt = 12.4 minutes; the fractions containing the product were concentrated under reduced pressure. The crude material was precipitated from ethyl acetate and heptane. The resulting white solid was washed with heptane 2 times and dried under reduced pressure.
MS (LCMS) 437.3 (M+H+; positive mode); 481.3 (M+HCO2 ~; negative mode). 1H NMR (400 MHz, methanol-d4) delta 7.43 (d, 1 H, J = 1.9 Hz), 7.36 (dd, 1 H, J = 8.3 and 2Hz), 7.32 (d, 1 H, J = 8.3 Hz), 7.08-7.04 (m, 2H), 6.79-6.75 (m, 2H), 4.12 (d, 1 H, J = 7.5 Hz), 4.00 (s, 2H), 3.96 (q, 2H, J = 7.0 Hz), 3.81 (d, 1 H, J = 12.5 Hz), 3.75 (dd, 1 H, J = 8.3 and 1.3 Hz), 3.65 (d, 1 H, J = 12.5 Hz), 3.63 (t, 1 H, J = 8.2 Hz), 3.57 (dd, 1 H, J = 7.5 and 1.3 Hz), 3.52 (d, 1 H, J = 8.0 Hz), 1.33 (t, 3H, J = 6.9 Hz). HRMS calculated for C22H26O7CI (M+H+) 437.1361 , found 437.1360.
4B: (30 mg, 15% yield); Rt = 13.2 minutes; the fractions containing the product were concentrated under reduced pressure. The crude material was precipitated from ethyl acetate and heptane. The resulting white solid was washed with heptane 2 times and dried under reduced pressure.
MS (LCMS) 437.3 (M+H+; positive mode) 481.3 (M+HCO2 “, negative mode). 1H NMR (400 MHz, methanol-d4) delta 7.48 (d, 1 H, J = 1.9 Hz) 7.40 (dd, 1 H, J = 8.1 and 1.9 Hz), 7.32 (d, 1 H, J = 8.3 Hz), 7.08-7.03 (m, 2H), 6.80-6.74 (m, 2H), 4.04-3.99 (m, 3H), 3.95 (q, 2H, J = 7 Hz), 3.89-3.81 (m, 4H), 3.73 (d, 1 H, J = 12.5 Hz), 3.49 (d, 1 H, J = 7.3 Hz), 1.32 (t, 3H, J = 7 Hz). HRMS calculated for C22H26O7CI (M+H+) 437.1361 , found 437.1358.
Merck & Co., Inc. and Pfizer Enter Worldwide Collaboration Agreement to Develop and Commercialize Ertugliflozin, an Investigational Medicine for Type 2 Diabetes
 ERTUGLIFLOZIN
ERTUGLIFLOZIN
Merck & Co., Inc. (NYSE: MRK), known as MSD outside the United States and Canada (“Merck”), and Pfizer Inc. (NYSE:PFE) today announced that they have entered into a worldwide (except Japan) collaboration agreement for the development and commercialization of Pfizer’s ertugliflozin (PF-04971729), an investigational oral sodium glucose cotransporter (SGLT2) inhibitor being evaluated for the treatment of type 2 diabetes. Ertugliflozin is Phase III ready, with trials expected to begin later in 2013.
“We are pleased to join forces with Merck in the battle against type 2 diabetes and the burden that it poses on global health,” said John Young, president and general manager, Pfizer Primary Care. “Through this collaboration, we believe we can build on Merck’s leadership position in diabetes care with the introduction of ertugliflozin, an innovative SGLT2 inhibitor discovered by Pfizer scientists.”
Under the terms of the agreement, Merck, through a subsidiary, and Pfizer will collaborate on the clinical development and commercialization of ertugliflozin and ertugliflozin-containing fixed-dose combinations with metformin and JANUVIA® (sitagliptin) tablets. Merck will continue to retain the rights to its existing portfolio of sitagliptin-containing products. Pfizer has received an upfront payment and milestones of $60 million and will be eligible for additional payments associated with the achievement of pre-specified future clinical, regulatory and commercial milestones. Merck and Pfizer will share potential revenues and certain costs on a 60/40 percent basis.
“Merck continues to build upon our leadership position in the oral treatment of type 2 diabetes through our own research and business development,” said Nancy Thornberry, senior vice president and Diabetes and Endocrinology franchise head, Merck Research Laboratories. “We believe ertugliflozin has the potential to complement our strong portfolio of investigational and marketed products, and we look forward to collaborating with Pfizer on its development.”
……………….
Development of an Early-Phase Bulk Enabling Route to Sodium-Dependent Glucose Cotransporter 2 Inhibitor Ertugliflozin

The development and optimization of a scalable synthesis of sodium-dependent glucose cotransporter 2 inhibitor, ertugliflozin, for the treatment of type-2 diabetes is described. Highlights of the chemistry are a concise, four-step synthesis of a structurally complex API from known intermediate 4 via persilylation–selective monodesilylation, primary alcohol oxidation, aldol-crossed-Cannizzaro reaction, and solid-phase acid-catalyzed bicyclic ketal formation. The final API was isolated as the l-pyroglutamic acid cocrystal.
1= ertugliflozin
| PF-04971729, a potent and selective inhibitor of the sodium-dependent glucose cotransporter 2, is currently in phase 2 trials for the treatment of diabetes mellitus. Inhibitory effects against the organic cation transporter 2-mediated uptake of [14C] metformin by PF- 04971729 also were very weak (IC50 900μM). The disposition of PF-04971729, an orally active selective inhibitor of the sodium-dependent glucose cotransporter 2, was studied after a single 25-mg oral dose of [14C]-PF-04971729 to healthy human subjects. The absorption of PF-04971729 in humans was rapid with a Tmax at ~ 1.0 h. Of the total radioactivity excreted in feces and urine, unchanged PF-04971729 collectively accounted for ~ 35.3% of the dose, suggestive of moderate metabolic elimination in humans. |
| References on PF-04971729: [1]. 1. Amit S. Kalgutkar, Meera Tugnait, Tong Zhu, et al.Preclinical Species and Human Disposition of PF-04971729, a Selective Inhibitor of the Sodium-Dependent Glucose cotransporter 2 and Clinical Candidate for the Treatment of Type 2 . Diabetes Mellitus Drug Metabolism and Diposition, 2011, 39 (9):. 1609-1619 Abstract (1S, 2S, 3S, 4R, 5S) -5 – [4-Chloro-3-(4-ethoxybenzyl) phenyl] -1 -hydroxymethyl-6 ,8-dioxabicyclo [3.2.1] octane-2 ,3,4-triol (PF-04971729), a potent and selective inhibitor of the sodium-dependent glucose cotransporter 2, is currently in phase 2 trials for the treatment of diabetes mellitus. This article describes the preclinical species and in vitro human disposition characteristics of PF-04971729 that were used in experiments performed to support the first-in-human study. Plasma clearance was low in rats (4.04 ml · min? 1 · kg? 1) and dogs (1.64 ml · min? 1 · kg? 1), resulting in half-lives of 4.10 and 7.63 h, respectively. Moderate to good bioavailability in rats (69%) and dogs (94%) was . observed after oral dosing The in vitro biotransformation profile of PF-04971729 in liver microsomes and cryopreserved hepatocytes from rat, dog, and human was qualitatively similar;. prominent metabolic pathways included monohydroxylation, O-deethylation, and glucuronidation No human-specific metabolites of PF-04971729 were detected in in vitro studies. Reaction phenotyping studies using recombinant enzymes indicated a role of CYP3A4/3A5, CYP2D6, and UGT1A9/2B7 in the metabolism of PF-04971729. No competitive or time-dependent inhibition of the major human cytochrome P450 enzymes was discerned with PF-04971729. Inhibitory effects against the organic cation transporter 2-mediated uptake of [14C] metformin by PF-04971729 also were very weak (IC50 =? 900 μM). Single-species allometric scaling of rat pharmacokinetics of PF-04971729 was used to predict human clearance, distribution volume, and oral bioavailability. Human pharmacokinetic predictions were consistent with the potential for a low daily dose. First-in-human studies after oral administration indicated that the human pharmacokinetics / dose predictions for PF -04971729 were in the range that is likely to yield a favorable pharmacodynamic response.. [2] … Timothy Colin Hardman, Simon William Dubrey Development and potential role of type-2 sodium-glucose transporter Inhibitors for Management of type 2 Diabetes Diabetes Ther 2011 September; 2 (3):. 133-145 Abstract There is a recognized need for new treatment options for type 2 diabetes mellitus (T2DM). Recovery of glucose from the glomerular filtrate represents an important mechanism in maintaining glucose homeostasis and represents a novel target for the management of T2DM. Recovery of glucose from the glomerular filtrate is executed principally by the type 2 sodium-glucose cotransporter (SGLT2). Inhibition of SGLT2 promotes glucose excretion and normalizes glycemia in animal models. First reports of specifically designed SGLT2 inhibitors began to appear in the second half of the 1990s. Several candidate SGLT2 inhibitors are currently under development, with four in the later stages of clinical testing. The safety profile of SGLT2 inhibitors is expected to be good, as their target is a highly specific membrane transporter expressed almost exclusively within the renal tubules. One safety concern is that of glycosuria , which could predispose patients to increased urinary tract infections. So far the reported safety profile of SGLT2 inhibitors in clinical studies appears to confirm that the class is well tolerated. Where SGLT2 inhibitors will fit in the current cascade of treatments for T2DM has yet to be established. The expected favorable safety profile and insulin-independent mechanism of action appear to support their use in combination with other antidiabetic drugs. Promotion of glucose excretion introduces the opportunity to clear calories (80-90 g [300-400 calories] of glucose per day) in patients that are generally overweight, and is expected to work synergistically with weight reduction programs. Experience will most likely lead to better understanding of which patients are likely to respond best to SGLT2 inhibitors, and under what circumstances.[3]. Zhuang Miao, Gianluca Nucci, Neeta Amin. Pharmacokinetics, Metabolism and Excretion of the Anti-Diabetic Agent Ertugliflozin (PF-04971729) in Healthy Male the Subjects. Drug Metabolism and Diposition. Abstract The Disposition of ertugliflozin (PF-04971729) , an orally active selective inhibitor of the sodium-dependent glucose cotransporter 2, was studied after a single 25-mg oral dose of [14C]-PF-04971729 to healthy human subjects. Mass balance was achieved with approximately 91% of the administered dose recovered in urine and feces. The total administered radioactivity excreted in feces and urine was 40.9% and 50.2%, respectively. The absorption of PF-04971729 in humans was rapid with a Tmax at ~ 1.0 h. Of the total radioactivity excreted in feces and urine, unchanged PF-04971729 collectively accounted for ~ 35.3% of the dose, suggestive of moderate metabolic elimination in humans. The principal biotransformation pathway involved glucuronidation of the glycoside hydroxyl groups to yield three regioisomeric metabolites M4a, M4b and M4c (~ 39.3% of the dose in urine) of which M4c was the major regioisomer (~ 31.7% of the dose). The structure of M4a and M4c were confirmed to be PF-04971729-4-O-β-and-3-O-β-glucuronide , respectively, via comparison of the HPLC retention time and mass spectra with authentic standards. A minor metabolic fate involved oxidation by cytochrome P450 to yield monohydroxylated metabolites M1 and M3 and des-ethyl PF-04971729 (M2), which accounted for ~ 5.2% of the dose in excreta. In plasma, unchanged PF-04971729 and the corresponding 4-O-β-(M4a) and 3-O-β-(M4c) glucuronides were the principal components, which accounted for 49.9, 12.2 and 24.1% of the circulating radioactivity. Overall, these data suggest that PF-04971729 is well absorbed in humans, and eliminated largely via glucuronidation.. [4] .. Tristan S. Maurer, Avijit Ghosh, Nahor Haddish-Berhane pharmacodynamic Model of Sodium-Glucose Transporter 2 (SGLT2) Inhibition: Implications for Quantitative Translational Pharmacology AAPS J. 2011; 13 (4): 576-584 Abstract Sodium-glucose co-transporter-2 (SGLT2) inhibitors are an emerging class of agents for use in the treatment of type 2 diabetes mellitus (T2DM). Inhibition of SGLT2 leads to improved glycemic control through increased urinary glucose excretion (UGE). In this study, a biologically based pharmacokinetic / pharmacodynamic (PK / PD) model of SGLT2 inhibitor-mediated UGE was developed. The derived model was used to characterize the acute PK / PD relationship of the SGLT2 inhibitor, dapagliflozin, in rats. The quantitative translational pharmacology of dapagliflozin was examined through both prospective simulation and direct modeling of mean literature data obtained for dapagliflozin in healthy subjects. Prospective simulations provided time courses of UGE that were of consistent shape to clinical observations, but were modestly biased toward under prediction. Direct modeling provided an improved characterization of the data and precise parameter estimates which were reasonably consistent with those predicted from preclinical data. Overall, these results indicate that the acute clinical pharmacology of SGLT2 inhibitors in healthy subjects can be reasonably well predicted from preclinical data through rational accounting of species differences in pharmacokinetics, physiology, and SGLT2 pharmacology. Because these data can be generated at the earliest stages of drug discovery, the proposed model is useful in the design and development of novel SGLT2 inhibitors. In addition, this model is expected to serve as a useful foundation for future efforts to understand and predict the effects of SGLT2 inhibition under chronic administration and in other patient populations.[5]. Yoojin Kim, Ambika R Babu Clinical potential of sodium-glucose cotransporter 2 Inhibitors in the Management of type 2 Diabetes Diabetes Obes Metab Syndr 2012; 5:…. 313-327 Abstract Background The Kidney plays an Important role in glucose metabolism, and has been considered a target for therapeutic intervention. The sodium-glucose cotransporter type 2 (SGLT2) mediates most of the glucose reabsorption from the proximal renal tubule. Inhibition of SGLT2 leads to glucosuria and provides a unique mechanism to lower elevated blood glucose levels in diabetes. The purpose of this review is to explore the physiology of SGLT2 and discuss several SGLT2 inhibitors which have clinical data in patients with type 2 diabetes. Methods We performed a PubMed search using the terms “SGLT2″ and “SGLT2 inhibitor” through April 10, 2012. Published articles, press releases, and abstracts presented at national and international meetings were considered. Results SGLT2 inhibitors correct a novel pathophysiological defect, have an insulin-independent action, are efficacious with glycosylated hemoglobin reduction ranging from 0.5% to 1.5%, promote weight loss, have a low incidence of hypoglycemia, complement the action of other antidiabetic agents, and can be used at any stage of diabetes. They are generally well tolerated. However, due to side effects, such as repeated urinary tract and genital infections, increased hematocrit, and decreased blood pressure, appropriate patient selection for drug initiation and close monitoring after initiation will be important. Results of ongoing clinical studies of the effect of SGLT2 inhibitors on diabetic complications and cardiovascular safety are crucial to determine the risk -benefit ratio. A recent decision by the Committee for Medicinal Products for Human Use of the European Medicines Agency has recommended approval of dapagliflozin for the treatment of type 2 diabetes as an adjunct to diet and exercise, in combination with other glucose-lowering medicinal products , including insulin, and as a monotherapy for metformin-intolerant patients. Clinical research also remains to be carried out on the long-term effects of glucosuria and other potential effects of SGLT2 inhibitors, especially in view of the observed increase in the incidence of bladder and breast cancer SGLT2 inhibitors represent a promising approach for the treatment of diabetes, and could potentially be an addition to existing therapies Keywords:.. sodium-glucose cotransporter type 2, SGLT2, inhibitors, kidney, glucosuria, oral diabetes agent, weight loss.[6]. Clinical Trials with PF-04971729 |
Example 6 Manufacturing Process for Tablets US20130137646
 |
Idrabiotaparinux for anticoagulant therapy.
Idrabiotaparinux
(biotinylated idraparinux, SSR-126517, SSR-126517E)
405159-59-3 9 x Na salt
774531-07-6 (free acid)
Idrabiotaparinux has an attached biotin moiety at the non-reducing end unit, which allows its neutralisation with avidin, an egg-derived protein with low antigenicity. This compound is currently investigated in clinical trials for prevention of recurrent VTE in patients with acute pulmonary embolism. The future of idrabiotaparinux depends also on the safety and efficacy of avidin.
Symptomatic deep vein thrombosis (DVT) and/or pulmonary embolism (PE) – treatment and secondary prevention of recurrent venous thromboembolism (VTE).
SSR-126517, a biotinylated idraparinux, had been in phase III clinical trials at Sanofi (formerly known as sanofi-aventis) for the treatment of pulmonary embolism, deep venous thrombosis (DVT) and atrial fibrillation. However, in 2009, development of the compound was discontinued.

Idrabiotaparinux (biotinylated idraparinux, SSR-126517, SSR-126517E) is a long-acting selective pentasaccharide indirect factor Xa coagulation inhibitor, administered by once weekly subcutaneous (SC) injection at a dose of 3mg in patients without severe renal insufficiency and, after an initial dose of 3mg, at 1.8mg in those with renal insufficiency.
Warfarin, heparin and their derivatives have been the traditional anticoagulants used for prophylaxis and treatment of venous thromboembolism. While the modern clinician is familiar with the efficacy and pharmacokinetics of these agents, their adverse effects have provided the impetus for the development of newer anticoagulants with improved safety, ease of administration, more predictable pharmacodynamics and comparable efficacy. Research into haemostasis and the coagulation cascade has made the development of these newer anticoagulants possible.
These drugs include the factor Xa inhibitors and IIa (thrombin) inhibitors. Direct and indirect factor Xa inhibitors are being developed with a relative rapid onset of action and stable pharmacokinetic profiles negating the need for close monitoring; this potentially makes them a more attractive option than heparin or warfarin. Examples of direct factor Xa inhibitors include apixaban, rivaroxaban, otamixaban, betrixaban and edoxaban. Examples of indirect factor Xa inhibitors include fondaparinux, idraparinux and idrabiotaparinux.
Direct thrombin inhibitors (factor IIa inhibitors) were developed with the limitations of standard heparin and warfarin in mind. Examples include recombinant hirudin (lepirudin), bivalirudin, ximelagatran, argatroban, and dabigatran etexilate. This review will discuss emerging novel anticoagulants and their use for the prophylaxis and management of venous thromboembolism, for stroke prevention in nonvalvular atrial fibrillation and for coronary artery disease.
Idrabiotaparinux is intended as a substitute for current long-term oral anticoagulation (e.g. with warfarin) and has no known food or drug interactions, no need for overlapping with other anticoagulants or for laboratory blood monitoring.
Idrabiotaparinux has superseded the development and marketing of the non-biotinylated idraparinux. Idrabiotaparinux is also in phase III clinical trials for the prevention of stroke in patients with atrial fibrillation (AF).
Idrabiotaparinux will be the first once a week anticoagulant for the treatment of patients with VTE. It is intended to provide a predictable response with fixed dosing, no interactions with food, no requirement for overlapping with other therapy and no routine laboratory monitoring.
Developer Sanofi-aventis.
Standard treatment of venous thromboembolism,including deep vein thrombosis and pulmonary
embolism, is started with a rapidly acting parenteral anticoagulant such as heparin or low-molecular-weight
heparin for at least 5 days and is overlapped with a Vitamin K antagonist such as warfarin.
Warfarin is then continued for at least 3 months. Although eff ective, this drug has important limitations. Lifestyle changes are necessary because of interactions with food, alcohol,and other drugs, and the unpredictable anticoagulant eff ect of warfarin necessitates frequent coagulation monitoring and dose adjustments to optimise the balance between effi cacy and safety. Warfarin reduces the risk of recurrent venous thromboembolism by up to 90%, but there is a catch-up eff ect if warfarin is stopped in patients with unprovoked venous thromboembolism. This eff ect means that, by 2 years,the risk of recurrence in patients treated for 3 months is akin to that in patients treated for 12 months.

Consequently, some experts recommend life-long warfarin therapy for patients with unprovoked venousthromboembolism. The complexity of such treatment has prompted the development of new oral and parenteral anticoagulants that are more convenient to administer than is warfarin Idraparinux is a synthetic pentasaccharide that accel erates antithrombin-dependent inhibition of factor Xa and has a half-life of about 80 h. When compared with conventional anticoagulation therapy,
idraparinux given once-weekly by subcutaneous injection was non-inferior for treatment of deep vein thrombosis,but was inferior for treatment of pulmonary embolism. In patients with venous thromboembolism who received a 6 month course of anticoagulant treatment, idraparinux was better than was placebo for prevention of recurrent venous thromboembolism.6However, when compared with warfarin for stroke prevention in patients with atrial fi brillation, there was an excess of major bleeding with idraparinux (including intracranial haemorrhage).7Prompted by these safety concerns, idrabiotaparinux was developed as a replacement for idraparinux.

Idrabiotaparinux (International Non-proprietary Name), or SSR126517 (laboratory code), is developed by sanofi-aventis as the first long-acting anticoagulant administered once-weekly by subcutaneous route, with the unique property to be almost instantly and specifically neutralizable by intravenous administration of avidin. It is developed as an alternative to vitamin K antagonists (VKA). Idrabiotaparinux is the biotinylated pentasaccharide corresponding to the structure depicted below.

The pentasaccharide structure of idrabiotaparinux is the same as idraparinux, another antithrombotic agent developed by sanofi-aventis (see structure below). However in idrabiotaparinux, the presence of a biotin hook covalently linked to the first saccharidic unit enables the compound to be neutralized by avidin or streptavidin, as described in the international patent application WO 02/24754.

Idraparinux
In the EQUINOX trial, which enrolled 757 patients with DVT treated for 6 months with equimolar doses of either idrabiotaparinux or idraparinux, the administration of idrabiotaparinux was demonstrated to provide bioequipotent results to idraparinux in terms of pharmacokinetics and pharmacodynamics, in patients with symptomatic deep venous thrombosis (Journal of Thrombosis and Haemostasis, 2010, Vol. 9, p. 92-99). The results of this bioequipotency trial indicated that idrabiotaparinux could be a suitable treatment for patients with deep venous thrombosis. However, the apparent failure of idraparinux in patients with pulmonary embolism indicated the need for a formal evaluation of idrabiotaparinux in this patient group (N. Eng. J. Med., 2007, Vol. 357, p. 1094-104).
IDRABIOTAPARINUX
It has now been demonstrated, in a phase III study involving 3202 patients with pulmonary embolism, that idrabiotaparinux is a safe and effective drug in the treatment of pulmonary embolism in patients with or without deep venous thrombosis and in the secondary prevention of venous thromboembolic events in said patients. The invention therefore relates to idrabiotaparinux for use in the treatment of pulmonary embolism in patients with or without deep venous thrombosis and the secondary prevention of venous thromboembolic events in said patients, wherein the efficacy and safety of said uses are clinically proven by a phase III clinical trial. According to the instant invention, the terms below have the following meanings:
“idrabiotaparinux” designates the sodium salt of this compound, as defined above, or any other pharmaceutically acceptable salt thereof;
-a “phase III clinical trial” refers to an international, multicenter, randomized, double-blind, double-dummy, parallel group study involving a large patients group (3202 patients in the instant invention), aiming at being the definitive assessment of how effective and safe the drug is, in comparison with current standard treatment; – “deep venous thrombosis” refers to a blood clot in a deep vein of the lower limbs;
new polysaccharides of the invention, are comparable to the oligosaccharides of the prior art antithrombotic activity. But they also have the advantage of being quickly neutralized by a specific antidote in an emergency. This specific antidote avidin (The Merck Index, Twelfth Edition, 1996, MN 920, pages 151-152) or streptavidin, two tetrameric protein with respective masses equal to approximately 66 000 and 60 000 Da, which have a very high affinity for biotin. In general, the invention relates to synthetic polysaccharides antithrombotic activity has at least one covalent bond with biotin or a biotin derivative. As a derivative of biotin include the biotin derivatives listed in the catalog Pierce 1999-2000 pages 62-81, for example 6-biotinamido hexanoate,
you,

or 2-biotinamido éthanethiole

Patent application WO 02/24754 describes synthetic polysaccharides which have a covalent bond with biotin (hexahydro-2-oxo-1H-thieno[3,4-d]imidazole-4-pentanoic acid) or with a biotin derivative. Such polysaccharides have an antithrombotic activity which means that they can be used as anticoagulants, and also have the advantage of being able to be rapidly neutralized with a specific antidote, in an emergency situation. This specific antidote is avidin (The Merck Index, Twelfth edition, 1996, M.N. 920, pages 151-152) or streptavidin, two tetrameric proteins of respective weights equal to approximately 66 000 and 60 000 Da, which have a very strong affinity for biotin.
Patent application WO 02/24754 describes in particular the following compound, known as idrabiotaparinux:

In the mammalian body, idrabiotaparinux is partly metabolized at the level of the amide bond adjacent to the biotin group, thus producing a pentasaccharide compound bearing an amine chain —NH—CO—(CH2)5—NH2 on the first glucosamine unit, as described in patent application WO 2010/023374.
It may be desirable, in particular in the context of clinical developments of molecules of pharmaceutical interest, to limit or even prevent the metabolization of compounds of this type.
Novel polysaccharides with structures analogous to some of those described in patent application WO 02/24754 have now been identified, which polysaccharides have antithrombotic properties and a neutralization capacity, for example via avidin, which are comparable to those described in that patent application, but which also have improved metabolic stability.
Generally, the invention therefore relates to synthetic polysaccharides with antithrombotic activity having at least one covalent bond with biotin or a biotin derivative, characterized in that said covalent bond is resistant to metabolic cleavage and comprises a linkage X chosen from —O—, —N(R)—, —N(R)—CO— and —N(R′)—CO—N(R″)—, in which R is an alkyl group and R′ and R″, which may be identical or different, are, independently of one another, hydrogen atoms or alkyl groups.
For the purposes of the present invention, and unless otherwise mentioned in the text, the term “alkyl” is intended to mean a linear or branched, saturated aliphatic group comprising from 1 to 6 carbon atoms, and advantageously a methyl group.
Biotin, or hexahydro-2-oxo-1H-thieno[3,4-d]imidazole-4-pentanoic acid, is the compound having the following formula:

By way of biotin derivatives, mention may be made of those indicated in the Pierce catalog 1999-2000, pages 62 to 81, or in patent application WO 02/24754.
Idrabiotaparinux sodium;
Molecular Formula:C53H88N4O51S8.9NaCAS
Registry Number:405159-59-3
nonasodium methyl (2-deoxy-3 ,4-di-O-methyl-2-{6 – [5 – (2-oxohexahydro-1H-thieno [3,4-d] imidazol-4-yl) pentanamido] hexanamido} -6-O-sulfo-α-D-glucopyranosyl) – (1 → 4) – (2,3-di-O-methyl-β-D-glucopyranosyluronate) – (1 → 4) – (2,3,6 -tri-O-sulfo-α-D-glucopyranoside) – (1 → 4) – (2,3-di-O-methyl-α-L-idopyranosyluronate) – (1 → 4) -2,3,6 – tri-O-sulfo-α-D-glucopyranoside

………………..
SYNTHESIS
FIGURE 9
Synthesis of the pentasaccharide 39

39
PREPARATION 34
Methyl (6-O-acetyl-2-azido-2-deoxy-3) 4-di-0-methyl-O-glucopyranosyl) – (1 → 4) – (benzyl 2,3-di-O-methyl- β-D-glucopyranosyluronate) – (1 – → 4) – (3,6-di-O-acetyl-2-0-benzyl–D-glucopyranosyl) – (1 -> 4) – (benzyl 2,3 -di-O-methyl-a-idopyranosyl-uronate) – (1 → 4) -2,3,6-tri-0-benzyLa-D-glucopyranoside (39)
Compound 6-0-acetyl-2 was dissolved -azido-2-deoxy-3 ,4-di-0-methyl-, β-D-glucopyranose trichloroacetimidate 38 (265 mg, 0.631 mmol) (obtained by J. Basten, and Chem. Lett Bioorg. Med. al.. (1992), 2 (9), 901)
and
compound 32 (584 mg, 0.420 mmol) (obtained by P. Westerduin and Med. Bioorg Chem. al., 1994, 2, 1267) in a dichloromethane / diethyl ether 1/2 (v / v) (28.5 mL).
After addition of 4 Å molecular sieves powder, the mixture is cooled to -20 ° C. and a 0.1 M solution of trimethylsilyl trifluoromethanesulfonate in dichloromethane (94.6 uL). After 10 minutes, again added the imidate (53.8 mg) and a 0.1 M solution of trimethylsilyl trifluoromethanesulfonate in dichloromethane (19.2 uL). After 10 minutes, the mixture was neutralized by addition of solid sodium hydrogen carbonate. After filtration and concentration, the residue was purified by column chromatography on silica gel (toluene / ethyl acetate 3/1 (v / v)) to give 499 mg of compound 39. [Α] = +66 (c = 1, 07, dichloromethane).
FIGURE 10 -Summary of the pentasaccharide 44 (Method I)
COMPOUND 39

COMPD 40

COMPD 44
……………………
In scheme 1, the starting, intermediate and final compounds are the following:
-
- compound (I): N-succinimidyl N-biotinyl-6-aminocaproate,
- compound (II): N-biotinyl-6-aminocaproic acid,
- compound (II′): N-biotinyl-6-aminocaproate carboxylate,
- compound (III): biotin,
- compound (III′): cyanomethyl biotinate.

EXAMPLE 1 Preparation of the Compound (I)

The reactions are monitored by LC with the following conditions: Symmetry C18 150×4.6 mm 5μ column (Waters); eluent A: 0.01 M KH2PO4 buffer adjusted to pH=3; eluent B: acetonitrile; flow rate 1 ml/min; gradient: t=0 min A/B 85/15, t=9 min A/B 65/35, t=10 min A/B 85/15, t=15 min A/B 85/15. This method makes it possible to visualize the biotin (compound (III), tR=4.5 min), the intermediate activated ester (III′) (tR=8.4 min), the N-biotinyl-6-aminocaproic acid (compound (II), tR=5.5 to 5.6 min), the intermediate mixed anhydride (II′) (tR=11.2 min) and the N-succinimidyl N-biotinyl-6-aminocaproate (compound (I), tR=7.9 to 8.2 min).
1.1: Preparation of the Compound (II)


7.5 kg of biotin (III), triethylamine (15 l, 2 V, 3.5 eq), NMP (15 l, 2 V) and, finally, chloroacetonitrile (3.5 kg, 0.47 OU, 1.5 eq) are charged to a reactor. The medium is heated to 60° C. After this temperature has been maintained for 2 h, an LC analysis shows that all the biotin has been converted into compound (III′) (<2%). The medium is cooled to 50° C. and then transferred into another reactor, containing aminocaproic acid (9.05 kg, 1.206 OU, 2.2 eq). Rinsing is carried out with NMP (0.1 V). The medium is heated to 100° C. and maintained at this temperature for 2 h. An LC analysis shows that less than 2% of activated biotin (III′) remains. The medium is cooled to 60° C. Acetonitrile (60 l, 8 V) preheated to 55° C. is run in. The mixture is stirred for 30 minutes at 60° C., and then cooled to 20° C. Stirring is carried out for 1 h. The suspension is filtered, then rinsing is carried out with 3 times acetonitrile (5 V) and then with THF (5 V). Drying is carried out under vacuum at a maximum of 60° C. until there is no change in weight. 12.0 kg of the compound (II) are thus obtained, with a yield of 109% and an organic purity, measured by LC, of 98.6%.
10.0 kg of the compound (II) are recharged to a reactor. Hydrochloric acid (90 l, 9 V of water+10 l, 1 V of 36% HCl) is then added. The suspension is stirred at 20° C. for 30 min. The suspension is filtered and rinsing is carried out 3 times with water (4 V, 40 l), then twice with THF (3.5 V). Drying is carried out under vacuum at a maximum of 45° C. until there is no change in weight. 6.1 kg of the compound (II) are thus obtained, with a yield of 66%.
1.2: Preparation of the Compound (I)
In a reactor, 3 kg of the compound (II) are suspended in DMF (25 l, 8.3 V) and the temperature is brought to −5° C. Triethylamine (1.02 kg, 0.34 OU, 1.2 eq) is then added. After stirring for 15 minutes, ethyl chloroformate (1.1 kg, 0.365 OU, 1.2 eq) is added gently (over the course of at least 1 h). Rinsing is carried out with DMF (0.9 l, 0.3 V). The medium is stirred at −5° C. for at least 2 h. The suspension becomes finer and yellow. An LC analysis shows that all the compound (II) (<3%) has reacted.
N-Hydroxysuccinimide (1.04 kg, 0.386 OU, 1.2 eq) in solution in DMF (3 l, 1 V) is then introduced in 1 step (over the course of at least 20 min). Rinsing is carried out with DMF (1.5 l, 0.5 V). The medium is stirred for 1 h 30 at −5° C. An LC analysis shows that the presence of residual compound (II) is less than 3%. The temperature is brought to 22° C., the suspension is taken up in DCM (12 V, 36 l) and the resulting organic phase is washed with water (15 l, 5 V). The organic phase is drawn off and the aqueous phase is extracted twice with DCM (30 l, 3 V). The organic phases are mixed and are washed with water (1.5 l, 0.5 V). The organic phase is concentrated to 6 V, i.e. 181. Heating is carried out at 40° C. and MTBE (6.25 V, 19 l) is added over the course of a minimum of 1 h. The mixture is maintained at 40° C. for 1 h, and then MTBE (8.75 V, 26 l) is added over the course of a minimum of 2 h. The mixture is maintained at 40° C. for at least 30 minutes, and then cooled to 20° C. over the course of a minimum of 2 h, and maintained at this temperature for 30 minutes. The suspension is filtered by suction and the cake is washed with acetone (5 V, 15 l) and then twice more with acetone (2 V, 6 l). The resulting product is filtered by suction and dried in an oven under vacuum at a maximum of 40° C. until there is no change in weight.
3 kg of the compound (I) are thus obtained in the form of a cream powder, with a yield of 80% and an organic purity, measured by LC, of 96.0%. Except for the compound (II), the presence of which is not problematic for a subsequent coupling reaction with a polysaccharide since it will be inert during this coupling, the purity of the compound (I) is 98%.
The biotinylated polysaccharides, the preparation of which is described above, are for example such as those described in patent applications WO 02/24754 and WO 2006/030104. They may in particular be the biotinylated pentasaccharide known under the International Nonproprietary Name “idrabiotaparinux” and described in patent application WO 02/24754, or the biotinylated hexadecasaccharides described in examples 1 and 2 of patent application WO 2006/030104.
In order to prepare these biotinylated polysaccharides, the compound (I) is coupled, respectively, with the pentasaccharide 44 described in patent application WO 02/24754
pentasaccharide 44: methyl (2-amino-2-deoxy-3,4-di-O-methyl-6-O-sulfonato-α-D-glucopyranosyl)-(1→4)-(2,3-di-O-methyl-β-D-glucopyranosyluronic acid)-(1→4)-(2,3,6-tri-O-sulfonato-α-D-glucopyranosyl)-(1→4)-(2,3-di-O-methyl-α-L-idopyranosyluronic acid)-(1→4)-2,3,6-tri-O-sulfonato-α-D-glucopyranoside
EXAMPLE 2
Preparation of a biotinylated polysaccharide, idrabiotaparinux

A solution of 1.22 kg of the crude pentasaccharide 44 (containing salts), as described in patent application WO 02/24754, is prepared in 8.51 of water (7 V). 0.5 kg (1.6 eq) of the compound (I), 0.12 kg (2.0 eq) of NaHCO3 and 0.37 kg of NaCl are added thereto. The solution is in the form of a white suspension. 3.7 l of acetone are added thereto and the reaction medium is stirred at approximately 25° C. for at least 22 h. This suspension is then slowly run into a mixture of ethanol (120 l) and MTBE (60 l) cooled beforehand to approximately 4° C., which makes it possible to precipitate the biotinylated pentasaccharide. The resulting suspension is then filtered and rinsed successively with absolute ethanol and acetone. The precipitate is oven-dried under a vacuum until there is no change in weight. 1.60 kg of crude idrabiotaparinux (containing salts) are thus obtained in the form of a cream powder, with an organic purity of 99%, and with a yield of 109% with respect to the pentasaccharide 44 and a chemical yield of 70% over the last 3 stages.
………………………….
Compound of Preparation Example 1:
Methyl (2 – [N-(6-aminohexanoyl)]-2-deoxy-3 ,4-di-O-methyl-6-O-sulfonato-UD-glucopyranosyl) – (1 → 4) – (acid 2, 3 -di-O-methyl-β-D-glucopyranosyluronic) – (1-rf) – (2,3,6-tri-O-sulphonate-D-glucopyranosyl) – (1 → 4) – (2,3 – di-O-methyl-alpha-L-idopyranosyluronique) – (1 → 4) -2,3,6-tri-Osulfonato-D-glucopyranoside, sodium salt

Compound 1
1) Preparation of 6 – (benzyloxycarbonylamino) hexanoate succinimidyl
To a solution of 6 – (benzyloxycarbonyl amino) hexanoic acid (1.00 g, 3.77 mmol) in dimethylformamide (20 mL) was added triethylamine (0.63 mL, 4.52 mmol) and stirring the mixture at room temperature under argon for 30 minutes. The solution was cooled to 0 ° C and added dropwise ethyl chloroformate (0.43 mL, 4.52 mmol). After two hours at room temperature, N-hydroxysuccinimide (0.52 g, 4.52 mmol) and stirring the mixture overnight at room temperature. Evaporated to dryness before the residue in water to which is added with ethyl acetate. The phases were separated and the aqueous phase is extracted with ethyl acetate. The organic phases are combined, dried over sodium sulfate, filtered and evaporated to dryness before purification on a column of silica gel with pentane mixture of ethyl acetate / (75/25 v / v) as eluent. Once the fractions evaporated to give 1.13 g 6 – (benzyloxycarbonylamino) succinimidyl hexanoate as an oil. TLC: R f = 0.22 on silica gel plate with a mixture of n-heptane/ethyl acetate (30/70 v / v) as eluent.
2) Preparation of compound the
Grafting the amine is carried out on the chain 44 pentasaccharide, or methyl (2 – amino-2-deoxy-3 ,4-di-0-methyl-6-0-sulfonato-α-D-glucopyranosyl) – (1 → 4) – (2,3 – di-0-methyl-β-D-glucopyranosyluronic) – (1 -> 4) – (2,3,6-tri-0-sulphonato-α-D-gluco-pyranosyl) – (1 → 4) – (2,3-di-O-methyl-α-L-idopyranosyl-uronic acid) – (1 → 4) – 2,3,6-tri-O-sulfo-α-nato- D-glucopyranoside, sodium salt, the preparation of which is described in patent application WO 02/24754:

44
To a solution of 6 – (benzyloxycarbonylamino) succinimidyl hexanoate (783 mg, 2.16 mmol) in N, N-dimethylformamide (10 mL) was added the pentasaccharide 44 (505 mg, 0.29 mmol). After stirring for 24 hours in an inert atmosphere and at room temperature, the solvent was evaporated under reduced pressure and the residue (40 mL) before washing the solution with chloroform (2 x 30 mL) dissolved in water. The chloroform phase is washed with water (10 mL) and aqueous phases were combined and evaporated to dryness under reduced pressure. The solid residue was triturated with 2-propanol (10 mL) and the suspension centrifuged for 5 minutes at 2500 rpm. The alcoholic phase is removed and replaced with 2-propanol (10 mL) and centrifugation was repeated. After having extracted the solvent, the crude product was dried under vacuum.
-2-deoxy-3 ,4-di-0-methyl-6-0-sulfonato-α-D [N-(benzyloxycarbonyl-6-aminohexanoyl)] – thus obtained 399 mg of the compound “, or methyl (2 -glucopyranosyl) – (1 → 4) – (the acid 2 3-di-0-methyl-β-D-glucopyranosyluronic) – (1 → 4) – (2,3,6-tri-0-sulfonato-α- D-glucopyranosyl) – (1 – »4) – (2,3-di-0-methyl-α-L-idopyranosyluronique) – (1 – → 4) -2,3,6 – tri-O-sulphonate- α-D-glucopyranoside, wherein Pg is benzyloxycarbonyl:

January 1 compound
Proton NMR at 200 MHz in deuterated water: The structure of the expected product is confirmed that the spectrum is identical to that performed on a product synthesized according to Example 5 of WO 02/24754 without the signals due to the biotin portion atoms but with signals of 7.4 to 7.5 ppm due to the benzyloxy group.
3) Preparation of compound 1
The product ‘s obtained at the end of the previous step (399 mg) is dissolved in deuterated water (10 mL). The solution of palladium on charcoal are treated with 10% (25 mg) and the solution was allowed to stir 20 hours in the presence of hydrogen at atmospheric pressure. Mixed with water (15 mL) was diluted, the catalyst was filtered and the solution was washed with chloroform (2 x 15 mL) before evaporating to dryness under reduced pressure. An aliquot (98 mg to 320 mg) of this product was purified on a column of Sephadex G-25 (2.5 x 50 cm) with water as eluent to give 25 mg of compound 1.
HPLC: Tr = 15.4 min column X-Terra RP-18 15W x 4.6 mm, 5μ particles of Waters in SA. With detection at 211 nm UV lamp.Eluent 1: water containing 0.02 M ammonium acetate and 0.05 M di-n-butylamine, adjusted to pH 7 with acetic acid. 2 Eluant: acetonitrile / water (90/10 v / v) containing 0.05 M di-n-butylamine and 0.08M acetic acid. The proportions of eluents are programmed so that the eluent composition is 10% 2 0 min. , 20% at 25 min. , 50% at 40 min. , 50% at 43 min. and 5% to 50 minutes. Proton NMR at 600 MHz in deuterated water: The structure of the expected product is confirmed that the spectrum is identical to that performed on a product synthesized according to Example 5 of WO 02/24754 without the signals due to the biotin portion atoms.
…………………………
 IDRABIOTAPARINUX
IDRABIOTAPARINUX
REFERENCES
JOURNAL OF THROMBOSIS AND HAEMOSTASIS vol. 9, 2010, pages 92 – 99
BULLER HARRY R ET AL: “Idraparinux versus standard therapy for venous thromboembolic disease“, NEW ENGLAND JOURNAL OF MEDICINE, vol. 357, no. 11, September 2007 (2007-09) , pages 1094-1104,
EQUINOX INVESTIGATORS: “Efficacy and safety of once weekly subcutaneous idrabiotaparinux in the treatment of patients with symptomatic deep venous thrombosis.“, JOURNAL OF THROMBOSIS AND HAEMOSTASIS : JTH JAN 2011 LNKD- DOI:10.1111/J.1538-7836.2010.04100.X PUBMED:20946157, vol. 9, no. 1, January 2011 (2011-01), pages 92-99,
N. ENG. J. MED. vol. 357, 2007, pages 1094 – 104
PRANDONI P ET AL: “Idraparinux: review of its clinical efficacy and safety for prevention and treatment of thromboembolic disorders“, EXPERT OPINION ON INVESTIGATIONAL DRUGS, ASHLEY PUBLICATIONS LTD., LONDON, GB, vol. 17, no. 5, 1 May 2008 (2008-05-01), pages 773-777,
SAVI P ET AL: “Reversible biotinylated oligosaccharides: A new approach for a better management of anticoagulant therapy“, JOURNAL OF THROMBOSIS AND HAEMOSTASIS, BLACKWELL PUBLISHING, OXFORD, GB, vol. 6, no. 10, 1 January 2008 (2008-01-01), pages 1697-1706,
Emerging anticoagulants.Kennedy B, Gargoum FS, Kennedy L, Khan F, Curran DR, O’Connor TM.Curr Med Chem. 2012;19(20):3388-416. Review.
| 1 | * | ANONYMOUS: “Bioequipotency Study of SSR126517E and Idraparinux in Patients With Deep Venous Thrombosis of the Lower Limbs (EQUINOX)” INTERNET CITATION, [Online] 10 April 2008 (2008-04-10), pages 1-4, XP002503606 Retrieved from the Internet: URL:http://www.clinicaltrials.gov/ct2/show/NCT00311090?term=equinox&rank=1> [retrieved on 2008-11-11] |
| 2 | * | BULLER HARRY ROGER ET AL: “Idrabiotaparinux, a Biotinylated Long-Acting Anticoagulant, in the Treatment of Deep Venous Thrombosis (EQUINOX Study): Safety, Efficacy, and Reversibility by Avidin” BLOOD, vol. 112, no. 11, November 2008 (2008-11), page 18, XP009118800 & 50TH ANNUAL MEETING OF THE AMERICAN- SOCIETY-OF-HEMATOLOGY; SAN FRANCISCO, CA, USA; DECEMBER 06 -09, 2008 ISSN: 0006-4971 |
| 3 | * | HIRSH J ET AL: “Beyond unfractionated heparin and warfarin: Current and future advances” CIRCULATION, LIPPINCOTT WILLIAMS & WILKINS, US, vol. 116, no. 5, 1 July 2007 (2007-07-01), pages 552-560, XP002503605 ISSN: 0009-7322 |
| 4 | * | PRANDONI P ET AL: “Idraparinux: review of its clinical efficacy and safety for prevention and treatment of thromboembolic disorders” EXPERT OPINION ON INVESTIGATIONAL DRUGS, ASHLEY PUBLICATIONS LTD., LONDON, GB, vol. 17, no. 5, 1 May 2008 (2008-05-01), pages 773-777, XP008098574 ISSN: 1354-3784 |
| 5 | * | SAVI P ET AL: “Reversible biotinylated oligosaccharides: A new approach for a better management of anticoagulant therapy” JOURNAL OF THROMBOSIS AND HAEMOSTASIS, BLACKWELL PUBLISHING, OXFORD, GB, vol. 6, no. 10, 19 July 2008 (2008-07-19), pages 1697-1706, XP002503607 ISSN: 1538-7933 |
PATENTS
| WO2002024754A1 | Sep 20, 2001 | Mar 28, 2002 | Akzo Nobel Nv | Polysaccharides with antithrombotic activity comprising at least a covalent bond with biotin or a biotin derivative |
| EP2145624A1 * | Jul 18, 2008 | Jan 20, 2010 | Sanofi-Aventis | Use of idrabiotaparinux for decreasing the incidence of bleedings during an antithrombotic treatment |
| WO2008113918A1 * | Feb 12, 2008 | Sep 25, 2008 | Sanofi Aventis | Heparins including at least one covalent bond with biotin or a biotin derivative, method for preparing same and use thereof |
| WO2008113919A1 * | Feb 12, 2008 | Sep 25, 2008 | Sanofi Aventis | Low molecular weight heparins including at least one covalent bond with biotin or a biotin derivative, method for making same and use thereof |
| WO2010007530A1 * | Jul 17, 2009 | Jan 21, 2010 | Sanofi-Aventis | Use of idrabiotaparinux for decreasing the incidence of bleedings during an antithrombotic treatment |
| WO2010023375A1 * | Aug 24, 2009 | Mar 4, 2010 | Sanofi-Aventis | Hexadecasaccharides with antithrombotic activity, including a covalent bond and an amino chain |
| WO2011061449A1 | Nov 19, 2010 | May 26, 2011 | Sanofi-Aventis | Method for preparing n-succinimidyl n-biotinyl-6-aminocaproate |
| EP2145624A1 * | Jul 18, 2008 | Jan 20, 2010 | Sanofi-Aventis | Use of idrabiotaparinux for decreasing the incidence of bleedings during an antithrombotic treatment |
| EP2233143A1 * | Mar 24, 2009 | Sep 29, 2010 | Sanofi-Aventis | Use of idrabiotaparinux for decreasing the incidence of bleedings during an antithrombotic treatment |


IDRAPARINUX… Sanofi (PHASE III)

IDRAPARINUX
Nonasodium (2S,3S,4S,5R,6R)-6-[(2R,3R,4S,5R,6R)-6-[(2R,3S,4S,5R,6R)-2-carboxy-4,5-dimethoxy-6-[(2R,3R,4S,5R,6S)-6-methoxy-4,5-disulfooxy-2-(sulfooxymethyl)oxan-3-yl]oxyoxan-3-yl]oxy-4,5-disulfooxy-2-(sulfooxymethyl)oxan-3-yl]oxy-4,5-dimethoxy-3-[(2R,3R,4S,5R,6R)-3,4,5-trimethoxy-6-(sulfooxymethyl)oxan-2-yl]oxyoxane-2-carboxylic acid |
| CAS number | 149920-56-9 |
|---|
| Formula | C38H55Na9O49S7 |
|---|---|
| Mol. mass | 1727.17683 g/mol |
CAS 162610-17-5 (free acid)
SANORG34006, SR-34006, SanOrg 34006, SanOrg-34006, UNII-H84IXP29FN, AC1MJ0N4, Org-34006
Methyl O-2,3,4-tri-O-methyl-6-O-sulfo-alpha-D-glucopyranosyl-(1–4)-O-2,3-di-O-methyl-beta-D-glucopyranuronosyl-(1–4)-O-2,3,6-tri-O-sulfo-alpha-D-glucopyranosyl-(1–4)-O-2,3-di-O-methyl-alpha-L-idopyranuronosyl-(1–4)-2,3,6-tri-O-sulfo-alpha-D-glucopyran
Sanofi-Syn(Originator), Organon (Codevelopment), PHASE 3
Methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose
methyl O-2,3,4-tri-O-methyl-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-O-di-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt. [α]D²⁰ = +46.2° (c=1; water). Anomeric protons chemical shifts: 5.43; 5.37; 5.16; 5.09; and 5.09 ppm.
Idraparinux sodium, or methyl O-2,3,4-tri-O-methyl-6-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranose, is a pentasaccharide with antithrombotic activity.
The preparation of idraparinux by sulfatation of a deprotected pentasaccharide is described in Bioorganic & Medicinal Chemistry, 1994, Vol. 2, No. 11, pp. 1267-1280, and also in patent EP 0 529 715 B1.
Idraparinux sodium is an anticoagulant medication in development by Sanofi-Aventis.[1]
It has a similar chemical structure and the same method of action as fondaparinux, but with an elimination half-life about five to six times longer (an increase from fondaparinux’s 17 hours to approximately 80 hours), which means that the drug should only need to be injected once a week.
As of July 2007, it has completed the Phase III clinical trial AMADEUS.
Idraparinux selectively blocks coagulation factor Xa.[2]
See Heparin: Mechanism of anticoagulant action for a comparison of the mechanism of heparin, low-molecular-weight heparins, fondaparinux and idraparinux.
Idraparinux sodium is a synthetic pentasaccharide with indirect coagulation factor Xa inhibitor activity. The drug candidate had been in phase III clinical development at Sanofi (formerly known as sanofi-aventis) for the once-weekly long-term treatment and secondary prevention of venous thromboembolic events in patients with pulmonary embolism (PE) and deep vein thrombosis (DVT), as well as for the prevention of thromboembolic complications related to atrial fibrillation (AF).
However, no recent development has been reported for this research. The oligosaccharide is delivered by subcutaneous injection. Unlike other products, idraparinux is administered once weekly rather than daily, thereby increasing patient convenience.
Originally developed under a collaboration between sanofi-sventis and Akzo Nobel’s human healthcare business Organon, all rights to idraparinux were transferred to Sanofi in January 2004 in exchange for revenues based on future sales.
Several synthetic pentasaccharides have been developed, such as Idraparinux, where all hydroxyl groups are methylated or sulphated, as illustrated below:

Initially, the firm Organon developed a way of synthesis for the preparation of the “active pentasaccharide”. This synthesis, using the 3-0-benzyl-1 ,2-0-isopropylidene-a-D- glucofuranose as substrate (Van Boeckel et al., J. Carbohydr. Chem. 1985, 4, p.293-321 ), comprises more than 50 steps, and the inversion of configuration of the C5 carbon is carried out by the opening of an epoxide. After a step of protection followed by a bromination, the G unit is thus obtained. It is well known that the synthesis of said G unit is very tedious, due to the number of steps for obtaining such unit and the known tendency of L-idose derivatives to exist as furanoses. After being coupled to the H unit, successive steps of protection-deprotection then an oxidation reaction carried out on C6 carbon, lead to the GH disaccharide.
In the preparation of Idraparinux, the synthesis of the disaccharide GH is nearly similar to the above synthesis of early synthetic pentasaccharides. The major innovation lies in the obtaining of disaccharide EF by epimerization of disaccharide GH. The coupling of both disaccharides leads to the tetrasaccharide EFGH, which is further coupled to the D unit for obtaining said pentasaccharide. The preparation of the disaccharide EF from GH allows notably the decrease of the total number of the steps to approximatively 25 (Petitou, M.; Van Boeckel, C.A. Angew. Chem., Int.Ed. 2004, 43, p.31 18-3133).
Hence, all current syntheses of the “active pentasaccharide” comprise a large number of steps and more particularly involves the complex synthesis of key L-iduronic acid derivative (G unit). Indeed, the preparation of the G unit of the “active pentasaccharide” of heparin has always been a limiting step in the synthesis of antithrombotic heparin derivatives.
Thus, there is still a need for a new efficient process of preparation of L-iduronic acid derivative, which would not possess the drawbacks established above and would be compatible with industrial scales. Besides, there is a need for such process which would in addition lead to an improved process of preparation of the “active pentasaccharide” constituting the heparin derivatives.
-
Idrabiotaparinux, developed by sanofi-aventis, is the biotinylated pentasaccharide corresponding to the structure depicted below. The pentasaccharide structure of idrabiotaparinux is the same as idraparinux, another antithrombotic agent developed by sanofi-aventis (see structure below). However in idrabiotaparinux, the presence of a biotin hook covalently linked to the first saccharidic unit enables the compound to be neutralized by avidin or streptavidin, as described in the international patent application WO 02/24754 .


-
In the EQUINOX trial, which enrolled patients with DVT treated for 6 months with equimolar doses of either idrabiotaparinux or idraparinux, idrabiotaparinux, with the same anti-activated factor X pharmacological activity (hereafter “anti-Xa activity”) as idraparinux, was shown to have a similar efficacy, but, surprisingly, a better safety with less observed bleedings, in particular major bleedings.
-
Therefore, the subject-matter of the invention is the use of idrabiotaparinux for the manufacture of a medicament useful for the treatment and secondary prevention of thrombotic pathologies, wherein the use of idrabiotaparinux involves a decrease in the incidence of bleedings during said treatment.
-
In other words, the invention relates to the use of idrabiotaparinux as an antithrombotic treatment, wherein said use minimizes the risk of bleedings during the antithrombotic treatment. Indeed, idrabiotaparinux enables to increase the benefit-risk ratio during the antithrombotic treatment.

The L-ioduronic acid methyl ester derivative (XII) is then converted into its D-glucuronic acid methyl ester counterpart (XIII) by epimerization with NaOMe in refluxing MeOH, followed by esterification with MeI and KHCO3 in DMF.
Protection of the ester (XIII) with levulinic acid (IX) by means of DCC and DMAP in dioxane, followed by acetolysis of the anomeric center with sulfuric acid in acetic anhydride furnishes the disaccharide (XIV), which is then saponified with piperidine and subjected to reaction with trichloroacetonitrile and Cs2CO3 in THF to yield the imidate (XV).
Glycosylation of the disaccharide (XII) with the imidate (XV) by means of trimethylsilyl triflate in CH2Cl2, followed by removal of the levulinoyl group by means of hydrazine acetate, furnishes the tetrasaccharide (XVI), which is coupled with the glucosyl trichloroacetimidate (XVIII) by means of trimethylsilyl trifluoromethanesulfonate in CH2Cl2 providing the pentasaccharide (XVII).
Glucosyl imidate (XVIII) is prepared by methylation of 1,6-anhydroglucose (XIX) with MeI and NaH in DMF, followed by acetolysis with Ac2O/TFA to give compound (XX), which is treated with piperidine in THF and finally with trichloroacetonitrile in dichloromethane in the presence of Cs2CO3.
The pentasaccharide (XVII) is deprotected by saponification with LiOH in THF/H2O2, and then hydrogenated over Pd/C in tert-butanol/water to provide a fully deprotected pentamer, which is finally subjected to sulfation with triethylamine sulfur trioxide complex in DMF and converted into the corresponding sodium salt by elution in a Dowex 50 XW4-Na+ or a Mono-Q anion-exchange column.

……………..
Glycosylation of sugar (I) with the idopyranosyl fluoride (II) by means of BF3.Et2O and molecular sieves in dichloromethane gives the disaccharide fragment (III), which is then converted into acetonide (V) by saponification of the ester functions with t-BuOK, followed by reaction with 2,2-dimethoxypropane (IV) in DMF and acidification with p-toluensulfonic acid. Methylation of acetonide (V) with MeI and NaH in DMF/MeOH provides the disaccharide (VI), which is then treated with HOAc to yield the 4′,6′-diol (VII). Selective silylation of the diol (VII) with tert-butyldimethylsilyl chloride (TBDMSCl) in pyridine leads to the 6′-O-TBDMS derivative (VIII), which is condensed with levulinic acid (IX) by means of dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) in dioxane to give the ester (X). Compound (X) is then submitted to simultaneous Jones oxidation and TBDMS removal with CrO3 and H2SO4/H2O in acetone to provide the iduronic acid derivative (XI), which is converted into the key intermediate (XII), first by esterification with MeI and KHCO3 in DMF and then by removal of the 4′-O-levulinoyl protecting group with HOAc and hydrazine hydrate in pyridine.

………………………
Idraparinux sodium, or methyl O-2,3,4-tri-O-methyl-6-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronate sodium-(1→4)-O-2,3,6-tri-O-sodium sulfonato-α-D-glucopyranose, is a pentasaccharide with antithrombotic activity.
The preparation of idraparinux by sulfatation of a deprotected pentasaccharide is described in Bioorganic & Medicinal Chemistry, 1994, Vol. 2, No. 11, pp. 1267-1280, and also in patent EP 0 529 715 B1.
A crystalline form of the pentasaccharide methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose has now been isolated. This compound in its crystalline form has proven to be very useful for the preparation of idraparinux, since it makes it possible to obtain this product in a particularly interesting chemical yield and with a significant gain in quality, the purity being improved as regards the crude product obtained, as will be detailed hereinbelow. These gains in reaction yield and in purity for the production of idraparinux are considerable advantages from an industrial viewpoint, since improving the robustness of a process is a constant cause for concern, especially in the case of large-scale syntheses.
One subject of the invention is thus the compound methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic glucopyranose in crystalline form.
Methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose, referred to hereinbelow as the compound of formula (I), corresponds to the following formula:

The compound of formula (I) in crystalline form according to the invention has a powder X-ray diffractogram whose characteristic lines are approximately at 12.009; 7.703; 7.300; 7.129; 5.838; 4.665; 4.476 and 3.785 angströms (interplanar distances). It also has a melting point of about 203° C. (203° C.±1° C.).
EXAMPLE 1 Preparation of the Compound of Formula (I) in Crystalline Form (Scheme 1)

Methyl O-2,3,4-tri-O-methyl-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-β-D-glucopyranosyluronic acid-(1→4)-O-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranosyluronic acid-(1→4)-O-α-D-glucopyranose, referred to hereinbelow as the compound of formula (I)
1.1: Preparation of the Compound of Formula (I′)
The compound of formula (I″) is obtained, for example, according to the teaching of patent EP 0 529 715 B1 or of the articles “Bioorg. Med. Chem.” (1994, Vol. 2, No. 11, pp. 1267-1280), “Bioorg. Med. Chem. Letters” (1992, Vol. 2, No. 9, pp. 905-910) or “Magnetic Resonance in Chemistry” (2001, Vol. 39, pp. 288-293). The compound of formula (I″) (5 g, 3.06 mmol) is dissolved in acetonitrile (10 mL). Deionized water (12.2 mL) and aqueous 30% sodium hydroxide solution (4.1 g) are then added. The mixture is heated to 40° C. and maintained at this temperature for 5 hours. The reaction medium is then cooled to 20° C. and acidified to pH 6.25 with aqueous 1N hydrochloric acid solution (about 17.7 g) before extraction with MTBE of certain impurities, the saponified product remaining in the aqueous phase. The residual acetonitrile, contained in the aqueous phase, is then removed by concentration, followed by diluting with deionized water (125 mL). The saponified product is finally precipitated at pH 1.5 by adding aqueous 1N hydrochloric acid solution (about 17.6 g) at 20° C. The suspension is maintained for 4 hours at 20° C. before filtration. The wet solid is finally dried in a vacuum oven at 30° C. to give 2.93 g (93.6%) of compound of formula (I).
NMR (anomeric protons of the saccharide units D, E, F, G, H): 5.79, 5.14, 5.55, 5.92, 4.94 ppm.
1.2 Preparation of the Crude Compound of Formula (I)
The compound of formula (I′) obtained after the preceding step is dissolved in tetrahydrofuran (18 mL). Palladium-on-charcoal (0.3 g) is added. The reaction medium is hydrogenated at 0.3 bar of hydrogen (relative pressure) for 4 hours. After filtering and evaporating, 2.12 g (99%) of the crude compound of formula (I) are obtained.
1.3: Preparation of the Compound of Formula (I) in Crystalline Form Using an Isopropanol/MTBE Mixture
The crude hydrogenated product obtained after the preceding step is dissolved in isopropanol (13 mL) at 65° C., and then crystallized at room temperature. The suspension is then cooled to 40° C., followed by addition of MTBE (13 mL), and is then cooled slowly to 10° C. After maintenance at 10° C. for 2 hours, the crystalline hydrogenated product is filtered off, washed and dried. 1.66 g of the compound of formula (I) in crystalline form are thus obtained, in the form of a cream-white powder. The reaction yield for the production of the compound of formula (I) in crystalline form, from the compound of formula (I′), is 92.5%. When expressed relative to the starting compound (I″), the reaction yield for the production of the compound of formula (I) in crystalline form is 86.6%.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (I) in crystalline form: 5.77, 5.11, 5.51, 5.84, 5.01 ppm.
1.4: Preparation of the Compound of Formula (I) in Crystalline Form Using Isopropanol
The crude hydrogenated product obtained after step 1.2 is dissolved in isopropanol (5 volumes) at 75° C. The medium is then cooled slowly until crystals appear, according to the known standard techniques for crystallization. The process is performed, for example, by a first step of cooling at 65° C. for 1 hour, and than a second step of cooling to a final temperature of 25° C. over 4 hours or of 5° C. over 6 hours, and finally maintenance at this final temperature for 30 minutes. The suspension is then filtered and rinsed with isopropanol (2×0.1 V) and compound (I) is isolated in the form of white crystals, which appear under a microscope in the form of needles. The 1H NMR analysis of these crystals is identical to that described after step 1.3 above.
EXAMPLE 4 Preparation of Idraparinux from the Compound of Formula (I) in Crystalline Form (Scheme 2)
The preparation of idraparinux (II) from the compound of formula (I) is summarized in Scheme 2.

The compound of formula (I) in crystalline form, as obtained according to Example 1.3, is dissolved in N,N′-dimethylformamide (6.6 mL) and then heated to 30° C. Under an inert atmosphere, 3.8 g of pyridine-sulfur trioxide complex are added slowly, followed by maintenance at 30° C. for 4 hours. The reaction medium is then poured into aqueous 23.8% sodium hydrogen carbonate solution (16.3 g) maintained at a maximum of 25° C., to obtain the compound of formula (II). The reaction medium is kept stirring for hours. The solution of sulfated product is then poured onto an MTBE/isopropanol/ethanol mixture (171 mL/70 mL/70 mL). Precipitation of the product is observed, and, after filtering off, washing and drying the cake, 4.99 g (96.8%) of compound of formula (II) are obtained, and are then purified by anion-exchange chromatography according to the usual techniques.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (II): 5.48, 4.68, 5.44, 5.08, 5.18 ppm.
It thus appears that the process according to the invention makes it possible to obtain idraparinux (compound of formula (II)) in a chemical yield of about 84% (precisely 83.8% according to the protocols described above) starting from the compound of formula (I″), i.e. a gain in yield of about 30% relative to the process described in patent EP 0 529 715 B1.
………………..
methyl O-2,3,4-tri-O-methyl-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-di-O-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2,3-O-di-methyl-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt. [α]D²⁰ = +46.2° (c=1; water). Anomeric protons chemical shifts: 5.43; 5.37; 5.16; 5.09; and 5.09 ppm.
WAS PREPARED AS PER
- Example 3
methyl O-4-O-(4-sulfoaminophenyl)-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt.
NOTE THIS IS ANALOGOUS PROCEDURE AND NOT SIMILAR
-
Methyl O-4-O-(4-nitrophenyl)-6-O-acetyl-2,3-O-di-phenylmethyl-α-D-glucopyranosyl-(1→4)-O-(methyl 3-O-methyl-2-O-acetyl-α-L-idopyranosyluronate)-(1→4)-O-2,3,6-tri-O-acetyl-α-D-glucopyranoside (100 mg, 0.09 mmol), obtained by the known imidate coupling of the trichloroacetimidate of O-4-O-(4-nitrophenyl)-6-O-acetyl-2,3-O-di-phenylmethyl-α-D-glucopyranoside and methyl O-(methyl 3-O-methyl-2-O-acetyl-α-L-idopyranosyluronate)-(1→4)-O- 2,3,6-tri-O-acetyl-α-D-glucopyranoside, was dissolved in tetrahydrofuran (9 ml) and cooled to -5 °C. At this temperature a 30% aq. solution of hydrogen peroxide (4.5 ml) was added to the reaction mixture, and after 10 min a 1.25 M lithium hydroxide solution (4.7 ml) was added. The mixture was stirred for 1 h at -5 °C, after which time the temperature was raised to 0 °C and the mixture was stirred overnight. The reaction mixture was acidified with 6N hydrogen chloride at 0 °C to pH 1.5, after which the saponified compound was extracted with ethyl acetate. The organic layers were pooled, dried over magnesium sulfate, and evaporated to give 63 mg (84%) of methyl O-4-O-(4-nitrophenyl)-2,3-O-di-phenylmethy1-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-α-L-idopyranuronosyl-(1→4)-O-α-D-glucopyranoside, which was dissolved in methanol (8 ml). 10% Pd on charcoal (63 mg) was added and the mixture hydrogenolyzed overnight. After filtration and evaporation 27 mg (50%) of methyl O-4-O-(4-aminophenyl)-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-α-L-idopyranuronosyl-(1→4)-O-α-D-glucopyranoside were obtained.
13 mg of methyl O-4-O-(4-aminophenyl)-O-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-α-L-idopyranuronosyl-(1→4)-O-α-D-glucopyranoside were dissolved in 2 ml of dry N,N-dimethylformamide, and under an atmosphere of nitrogen 148 mg of triethylamine sulfurtrioxide complex were added. The mixture was stirred overnight at 50 °C, after which an aq. solution of sodium hydrogen carbonate was added under ice cooling. The mixture was stirred for 1 h at room temperature, concentrated to a small volume and desalted on a Sephadex G-10 column with water. The crude product obtained was purified by HPLC using a Mono-Q anion exchange column to give 11 mg (37%) of methyl O-4-O-(4-sulfoaminophenyl)-2,3,6-tri-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-methyl-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-O-2,3,6-tri-O-sulfo-α-D-glucopyranoside nonakis sodium salt. [α]D²⁰ = +52.2° (c=0.67; water). Anomeric protons chemical shifts: 5.5; 5.17; and 5.15 ppm.
………………………………..
BMCL Volume 19, Issue 14, 15 July 2009, Pages 3875–3879
http://www.sciencedirect.com/science/article/pii/S0960894X0900482X


Final elaboration of the pentasaccharide 1. Reagents and conditions: (a) TMSOTf, Et2O, 4 Å MS, rt, 66% (28α), 15% (28β); (b) CAN, CH3CN, toluene, H2O, rt, 72%; (c) CCl3CN, DBU, CH2Cl2, rt, 98%; (d) TMSOTf, 4 Å MS, CH2Cl2, rt, 51% (73% based on recovery of 4); (e) Pd/C (10%), H2, t-BuOH, H2O, rt; (f) SO3·Et3N, DMF, 50 °C, 93% (2 steps).
The final elaboration of the pentasaccharide 1 was illustrated in IN ABOVE SCHEME Coupling of the glucopyranosyl trichloroacetimidate 6 with disaccharide acceptor 5 in the presence of trimethylsilyl trifluoromethylsulfonate and powdered 4 Å molecular sieves at room temperature in diethyl ether afforded the desired α-coupled trisaccharide 28α in a yield of 66%, together with 15% of the separable β-coupled product 28β. The anomeric 4-methoxyphenyl group in trisaccharide 28α was removed with CAN, and the resulting lactol was readily converted into the trisaccharide trichloroacetimidate 3. Coupling of donor 3 with the disaccharide acceptor 4 in the presence of trimethylsilyl trifluoromethylsulfonate and powdered 4 Å molecular sieves at room temperature in dichloromethane afforded the fully protected pentasaccharide 2 in 51% yield (73% based on recovery of 4). Finally, pentasaccharide 2 was subject to hydrogenolysis of the benzyl protecting groups. The highly polar product without purification was O-sulfated directly with triethylamine-sulfur trioxide complex to afford the sulfated pentasaccharide 1 in an excellent yield of 93% (for two steps).
Summarizing, the potent anti-thromboembolic pentasaccharide Idraparinux (1) was synthesized in total 51 steps and in 4% overall yield from d-glucose and methyl α-d-glucopyranoside.18 The synthetic route is convergent with a linear sequence of 27 steps, and the transformations are scalable. The 4-methoxyphenol glycoside intermediates are easy to be purified by crystallization.
Compound 1: ![]() 54.2 (c 1.0, H2O);
54.2 (c 1.0, H2O);
1H NMR (400 MHz, D2O) δ 3.27 (t, J = 8.4 Hz, 1H), 3.30–3.38 (m, 2H), 3.47 (s, 3H), 3.53 (s, 3H), 3.56 (s, 6H), 3.58 (s, 3H), 3.62 (s, 3H), 3.63 (s, 3H), 3.64 (s, 6H), 3.75 (d, J = 10.0 Hz, 1H), 3.83–3.97 (m, 4H), 3.98 (t, J = 8.8 Hz, 1H), 4.06–4.18 (m, 3H), 4.19–4.45 (m, 8H), 4.56 (br t, J = 9.6 Hz, 1H), 4.65 (t, J = 9.2 Hz, 1H), 4.66 (d, J = 7.6 Hz, 1H), 5.00 (br s, 1H), 5.11 (br s, 1H), 5.17 (d, J = 3.6 Hz, 1H), 5.43 (d,J = 3.2 Hz, 1H), 5.47 (d, J = 4.0 Hz, 1H);
ESI-MS m/z 774.1 [M−8Na+6H]2−, 763.0; [M−9Na+7H]2−, 508.5 [M−9Na+6H]3−.
………………….
The process of preparation of Idraparinux having the following formula:

may comprise the following steps :
1 ) preparation a compound of formula (IXB)

(IXB) wherein Ra is methyl, Rb is methyl, Rc is methyl, T-i is benzyl, T2 is benzyl, T3 is benzyl and T is methyl, by the process according to the invention;
2) epimerisation of the disaccharide (IXB) so as to form disaccharide D of formula :

3) protection of the 4′-OH of D with a levulinoyl ester;
4) acetolysis of the disaccharide resulting from step 3), followed by preparation of the corresponding imidate;
5) coupling the disaccharide imidate resulting from step 4) with (IXB) obtainable by the process of the invention, wherein Ra is methyl, Rb is methyl, Rc is methyl, T-i is benzyl, T2 is benzyl, T3 is benzyl and T is methyl, to obtain a tetrasaccharide;
6) coupling the fully protected tetrasaccharide with a monosaccharide glycosyl imidate;
7) deprotection of the protecting groups by the successive saponification and hydrogenolysis;
8) sulfation of the hydroxyl groups.
In one embodiment, the present invention concerns a process of preparation of Idraparinux:

said process comprising the following steps:
preparation of a compound of formula (VI) such as defined above, from a compound of formula (V) such as defined above; preparation of a compound of formula (VII) such as defined above, from a compound of formula (VI) such as defined above;
preparation of a compound of formula (VIII) such as defined above, from a compound of formula (VII) such as defined above;
– preparation of a compound of formula (IX) such as defined above, from a compound of formula (VIII) such as defined above;
wherein in compounds of formulae (V), (VI), (VII), (VIII) and (IX), R-i , R2, R3 and X are as defined above, Ra is methyl, Rb is methyl, Rc is methyl, Rd is methyl and R’ is the monosaccharide of formula :

wherein T-i is benzyl, T2 is benzyl, T3 is benzyl and T is methyl. The inventors advantageously found that the process of preparation of Idraparinux comprising the decarboxylation/intramolecular cyclisation tandem reaction, which allows the inversion of configuration of C5 carbon of the compound of formula (VI), is more efficient than the processes previously described in the literature. Indeed, the process according to the invention allows advantageously a significant decrease of the number of steps and thus an improvement of the overall yield. Thus, the process of preparation of Idraparinux may be carried out in industrial scales. The inventors found an efficient process of preparation of Idraparinux.
According to another object, the present invention concerns the use of compounds of formulae (V), (VI), (VI I), (VIII) and (IX), as intermediates for the preparation of Idraparinux. In particular, the present invention concerns the use of a compounds of formulae (VB), (VI B), (VI IB), (VI I IB) and (IXB), as intermediates for the preparation of Idraparinux The invention is further illustrated but not restricted by the description in the following examples. Example 1 :
Preparation of Methyl-4,6-0-benzylidene-a-D-glucopyranoside (la)
CHC13)

Tf = 166-167°C (litt. 165-166°C) To a solution of benzaldehyde (400 mL, 3.94 mol, 5.9 eq.) was added zinc chloride (100.3 g, 0.74 mol, 1 .1 eq.) under vigorous stirring. After homogenization of the solution methyl- a-D-glucopyranoside (129.6 g, 0.67 mol, 1.0 eq.) was added portionwise. After 16 hours stirring at room temperature the reaction mixture was diluted with diethyl ether (100 mL). The mixture was then poured dropwise and under vigorous stirring in a solution containing ice water (1 .5 L) and hexane (350 mL). The precipitate was filtered, washed with diethyl ether (3 x 300 mL) and dried under vacuum over KOH. The product was then recrystallised from CH2CI2 (720 mL) and washed with a Et20/CH2CI2 solution (75:25, 2 x 200 mL). The filtrate was repeatedly recrystallised five times from CH2CI2 to afford compound la as white crystals (136.97 g, 0.49 mol, 72%).
1H NMR (CDCI3, 250 MHz): δ 2.35 (d, JCH-OH = 9.2 Hz, 1 H, OH), 2.83 (d, JCH-OH = 2.2 Hz, 1 H, OH), 3.46 (s, 3H, -OCH3), 3.43-3.46 (m, 1 H, H-4), 3.63 (td, JCH–OH = ^2,3 = 9.2 Hz, J1 2 = 3.9 Hz, 1 H, H-2), 3.70-3.81 (m, 2H, H-5, H-6), 3.93 (td, J = 9.2 Hz, JCH–OH = 2.2 Hz, 1 H, H- 3), 4.29 (m, 1 H, H-6 ), 4.79 (d, J1i2 = 3.9 Hz, 1 H, H-1 ), 5.54 (s, 1 H, Ph-CH), 7.35-7.38 (m, 3H, HAr), 7.47-7.51 (m, 2H, HAr).
13C NMR (CDCI3, 62.9 MHz): δ 55.6 (-OCH3), 62.5 (C-5), 69.0 (C-6), 71.1 (C-3), 72.9 (C- 2), 81 .0 (C-4), 99.9 (C-1 ), 102.0 (Ph-CH), 126.4, 128.5, 129.4, 137.1 (6xCAr). IR (film) v (cm“1): 3369 (O-H). Preparation of Methyl-2,3-di-0-methyl-4,6-0-benzylidene-a-D-glucopyranoside (Ma)
13)

°C)
To a solution of compound la (47.60 g, 0.17 mol, 1.0 eq.) in anhydrous THF (750 mL) under an argon atmosphere and cooled to 0°C, was added portionwise sodium hydride (60%, 16.93 g, 0.42 mol, 2.5 eq.). After 20 minutes methyl iodide was added dropwise (30 mL, 0.48 mol, 2.8 eq.) and the reaction mixture was allowed to reach room temperature. After 16 hours, methanol was added portionwise (75 mL) and the solution was stirred for another 15 minutes before being concentrated. The resulting residue was dissolved in EtOAc (400 mL) and washed with water (2 x 250 mL). The organic layer was dried (MgS04), filtered and concentrated. The resulting solid was dissolved in diethyl ether (1000 mL), hexane was added (400 mL) and the solvent was partially evaporated at low temperature. The crystals obtained were washed with hexane and the filtrate once again partially evaporated, filtered and the precipitate washed with hexane. The combined precipitates afforded compound Ma as white crystals (46.10 g, 0.15 mol, 88%).
1H NMR (CDCI3, 250 MHz): δ 3.30 (dd, J2-3 = 9.1 Hz, J1-2 = 3.7 Hz, 1 H, H-2), 3.44 (s, 3H, – OCH3), 3.49-3.87 (m, 4H, H-4, H-5, H-6, H-3), 3.55 (s, 3H, -OCH3), 3.64 (s, 3H, -OCH3), 4.28 (dd, J6-6. = 9.1 Hz, J5-6 = 3.7 Hz, 1 H, H-6 ), 4.85 (d, J1-2 = 3.7 Hz, 1 H, H-1 ), 5.54 (s, 1 H, Ph-CH), 7.36-7.41 (m, 3H, HAr), 7.48-7.52 (m, 2H, HAr).
13C NMR (CDCI3, 62.9 MHz): δ 55.4, 59.5, 61.1 (3x-OCH3), 62.3 (C-5), 69.1 (C-6), 79.9 (C-3), 81 .5 (C-4), 82.2 (C-2), 98.5 (C-1 ), 101.4 (Ph-CH), 126.2, 128.3, 129.0, 137.4 (6xCAr).
Preparation of Methyl-2,3-di-0-methyl-a-D-glucopyranoside (Ilia)
13)

To a suspension of compound Ma (10.33 g, 33.29 mmol, 1.0 eq.) in methanol (150 mL) was added para-toluenesulfonic acid monohydrate (322 mg, 1 .69 mmol, 0.05 eq.). After 4 hours stirring at room temperature, sodium carbonate (300 mg) was added and the reaction mixture was stirred an additional 15 minutes before filtration through a pad of Celite®. Then the filtrate was concentrated and the residue obtained was dissolved in a mixture of distilled water/diethyl ether (3:1 , 150 mL). The organic layer was extracted with water (2 x 50 mL) then the combined aqueous phases were concentrated and dried one night under vacuum over KOH. The resulting residue was recrystallised from toluene using petroleum ether as a co-solvent. The crystals obtained were washed with hexane and dried under vacuum over KOH to obtain compound Ilia as white crystals (6.63 g, 29.83 mmol, 90%).
1H NMR (DMSO, 400 MHz): δ 3.03 (dd, J2-3 = 9.3 Hz, J1-2 = 3.5 Hz, 1 H, H-2), 3.12-3.20 (m, 2H, H-3, H-4), 3.27 (s, 3H, -OCH3), 3.32 (s, 3H, -OCH3), 3.30-3.33 (m, 1 H, H-5), 3.44 (s, 3H, -OCH3), 3.40-3.46 (m, 1 H, H-6), 3.62 (ddd, J6-6‘ = 1 1.6 Hz, JCH-OH = 5.7 Hz, J5-6 = 1 ,9 Hz, 1 H, H-6′), 4.52 (t, J = 5.7 Hz, 1 H, OH), 4.78 (d, Ji-2 = 3.5 Hz, 1 H, H-1 ), 5.09 (d,
13C NMR (DMSO, 100.6 MHz): δ 54.1 , 57.4, 60.0 (3x-OCH3), 60.6 (C-6), 69.5 (C-3), 72.5 (C-5), 80.9 (C-2), 82.8 (C-4), 96.4 (C-1 ).
IR (film) v (cm“1): 3419 (O-H).
Preparation of Methyl methyl-2,3-di-0-methyl-a-D-glucopyranosiduronate (IVa)
C13)

To a solution of compound Ilia (500 mg, 2.25 mmol, 1.0 eq.) in distilled water (15 mL) were successively added NaBr (50 mg, 0.49 mmol, 0.2 eq.) and TEMPO (7 mg, 0.05 mmol, 0.02 eq.). The reaction mixture was cooled with the aid of an ice bath then a solution of NaOCI (13% v/v, 5.2 mL, 9.1 mmol, 4.0 eq.) was added. After 5 hours stirring at 0°C ethanol was added (96% v/v, 8 mL), then the pH was reduced to 2-3 by addition of HCI (10 % v/v). The solvent was evaporated and the residue obtained was suspended in methanol, filtered in order to remove the remaining salts and washed several times with dichloromethane and methanol. The filtrate was concentrated then dissolved, under an argon atmosphere, in dry methanol (40 mL). para-toluenesulfonic acid (85 mg, 0.45 mmol, 0.2 eq.) was added then the reaction mixture was heated under reflux overnight. The solvent was evaporated and the residue obtained was dissolved in EtOAc (60 mL). The organic layer was washed with a 5% aqueous NaHC03 solution (2 χ 20 mL) and with brine (1 χ 20 mL). The aqueous phase was extracted with dichloromethane (3 χ 20 mL). The combined organics were dried (MgS04), filtered and evaporated. Column chromatography (hexane/ethyl acetate 50:50) gave compound IVa as a colourless oil (503 mg, 2.00 mmol, 89%).
1H NMR (CDCIs, 400 MHz): δ 3.10 (d, JCH-OH = 3.0 Hz, 1 H, OH), 3.26 (dd, J2-3 = 9.3 Hz, J1 -2 = 3.4 Hz, 1 H, H-2), 3.47 (s, 3H, -OCH3), 3.49-3.52 (m, 1 H, H-3), 3.50 (s, 3H, -OCH3), 3.62 (s, 3H, -OCH3), 3.74 (td, J = 9.5 Hz, JCH-OH = 3.0 Hz, 1 H, H-4), 3.82 (s, 3H, -OCH3), 4.14 (d, J4.5 = 9.6 Hz, 1 H, H-5), 4.91 (d, Ji-2 = 3.4 Hz, 1 H, H-1 ).
13C NMR (CDCI3, 100.6 MHz): δ 52.9, 56.0, 59.1 , 61 .3 (4x-OCH3), 70.6 (C-5), 71.7 (C-4), 80.9 (C-2), 81.8 (C-3), 98.1 (C-1 ), 170.9 (C=0).
IR (film) v (cm“1): 3475 (O-H), 1750 (C=0).
Preparation of Methyl methyl^-O-il’-ethoxy^’-propyn-l’-ylJ^.S-di-O-methyl-a-D- glucopyranosiduronate (Va)

To a solution of compound IVa (4.56 g, 18.21 mmol, 1.0 eq.) in chloroform (stabilised with amylene, 200 mL) were added, under an argon atmosphere, P205 (5.31 g, 36.29 mmol, 2.0 eq.) and propargylaldehyde diethylacetal (5.2 mL, 36.27 mmol, 2.0 eq.), then the reaction mixture was heated at 60°C. After 4 hours stirring, the reaction mixture was filtered through a pad of Celite® then the solvent was removed under vacuum. The crude mixture was suspended in EtOAc (300 mL), washed with a 5% NaHC03 aqueous solution (1 x 30 mL) and brine (1 x 30 ml_). The organic layer was dried (MgS04), filtered, and evaporated. Column chromatography (gradient hexane/ethyl acetate 80:20 – 20:80) afforded compound Va as a colourless oil (4.07 g, 12.24 mmol, 67%) in a diastereomeric mixture (64:36) (the relative composition of the mixture was determined by 1H NMR from integrations of protons EtO-CH), along with some unreacted compound IVa (1.17 g, 4.68 mmol, 26%).
1H NMR (CDCI3, 400 MHz): δ 1.18-1.25 (m, 3H, -OCH2CH3) (diastereomeric mixture), 2.56 (m, 1 H, H-C≡C-) (mixture), 3.26-3.31 (m, 1 H, H-2) (mixture), 3.43 (s, 3H, -OCH3) (major), 3.44 (s, 3H, -OCH3) (minor), 3.50 (s, 3H, -OCH3) (mixture), 3.59 (s, 3H, -OCH3) (minor), 3.62 (s, 3H, -OCH3) (major), 3.47-3.62 (m, 2H, H-3, -OCHaHbCH3) (mixture), 3.65-3.73 (m, 1 H, -OCHaHbCH3) (mixture), 3.78 (s, 3H, -OCH3) (major), 3.80 (s, 3H, -OCH3) (minor), 3.78-3.86 (m, 1 H, H-4) (mixture), 4.15 (d, J4-5 = 10.0 Hz, 1 H, H-5) (major), 4.18 (d, J4-5 = 10.0 Hz, 1 H, H-5) (minor), 4.86-4.88 (m, 1 H, H-1 ) (mixture), 5.35 (d, J = 1.7 Hz, 1 H, EtO- CH) (minor), 5.58 (d, J = 1.7 Hz, 1 H, EtO-CH) (major).
13C NMR (CDCI3, 100.6 MHz): δ 15.0 (-OCH2CH3) (mixture), 52.6 (-OCH3) (major), 52.7 (- OCH3) (minor), 55.8 (-OCH3) (mixture), 59.2 (-OCH3) (major), 59.3 (-OCH3) (minor), 60.4 (-OCH2CH3) (major), 61 .3 (-OCH2CH3) (minor), 61.4 (-OCH3) (mixture), 70.1 (C-5) (minor), 70.2 (C-5) (major), 74.0 (H-C≡C-) (major), 74.2 (H-C≡C-) (minor), 76.4 (C-4) (minor), 76.7 (C-4) (major), 78.6 (H-C≡C-) (minor), 78.9 (H-C≡C-) (major), 81 .5 (C-2) (major), 81 .8 (C-2) (minor), 81.9 (C-3) (minor), 82.9 (C-3) (major), 92.6 (EtO-CH) (mixture), 97.9 (C-1 ) (minor), 98.0 (C-1 ) (major), 169.6 (C=0) (major), 169.9 (C=0) (minor). Elemental analysis: Calculated: C: 54.21 ; H: 7.28. Found: C: 54.17 ; H: 7.13.
ESI-MS (pos. mode): m/z = 355 [M+Na]+.
IR (film) v (cm“1): 1752 (C=0), 3266 (≡C-H). Preparation of 4-0-(1′-ethoxy-2′-propyn-1,-yl)-1 ,2,3-tri-0-methyl-a-D-gluco- pyranosiduronic acid (Via)

To a solution of compound Va (1.12 g, 3.37 mmol, 1.0 eq.) in EtOH/H20 (3:1 , 100 mL) was added sodium hydroxide (156 mg, 3.90 mmol, 1 .3 eq.). After 5 hours stirring at room temperature the solvent was evaporated. The residue obtained was dissolved in water (50 mL). The pH of the aqueous layer was reduced to 2-3 with a 5% citric acid aqueous solution, then the layer was saturated with sodium chloride before extraction with dichloromethane (10 x 20 mL). If necessary the pH was adjusted by addition of more citric acid aqueous solution. The combined organics were dried (MgS04), filtered and removed under vacuum. Compound Via was obtained without further purification as a colourless oil (1.020 g, 3.20 mmol, 95%), in a mixture of diastereomers (75:25) (the relative composition of the mixture was determined by 1H NMR from integrations of protons EtO-CH).
1H NMR (CDCIs, 400 MHz): δ 1.16-1.24 (m, 3H, -OCH2CH3) (diastereomeric mixture), 2.59 (d, J = 1 .6 Hz, 1 H, H-C≡C-) (major), 2.62 (s I, 1 H, H-C≡C-) (minor), 3.25-3.33 (m, 1 H, H- 2), 3.44 (s, 3H, -OCH3) (mixture), 3.51 (s, 3H, -OCH3) (mixture), 3.62 (s, 3H, -OCH3) (mixture), 3.54-3.62 (m, 2H, H-3, -OCHaHbCH3) (mixture), 3.68-3.77 (m, 1 H, -OCHaHbCH3) (mixture), 3.81-3.87 (m, 1 H, H-4) (mixture), 4.13-4.18 (m, 1 H, H-5) (mixture), 4.88-4.90 (m, 1 H, H-1 ), 5.45 (s I, 1 H, EtO-CH) (minor), 5.63 (d, J = 1.6 Hz, 1 H, EtO-CH) (major).
13C NMR (CDCI3, 100.6 MHz): δ 14.9 (-OCH2CH3) (mixture), 55.9 (-OCH3) (mixture), 59.2 (-OCH3) (minor), 59.3 (-OCH3) (major), 60.7 (-OCH2CH3) (mixture), 61 .2 (-OCH3) (minor), 61.3 (-OCH3) (major), 70.1 (C-5) (mixture), 74.3 (H-OC-) (major), 74.8 (H-OC-) (minor), 75.7 (C-4) (minor), 76.4 (C-4) (major), 78.5 (H-C≡C-) (minor), 78.8 (H-C≡C-) (major), 81.4 (C-2) (major), 81 .7 (C-2) (minor), 81 .8 (C-3) (minor), 82.9 (C-3) (major), 92.5 (EtO-CH) (mixture), 97.8 (C-1 ) (minor), 98.0 (C-1 ) (major), 173.8 (C=0) (major), 174.0 (C=0) (minor).
ESI-MS (pos. mode): m/z = 341 [M+Na]+. IR (film) v (cm“1): 1751 (C=0), 3268 (≡C-H).
Preparation of Methyl-4,7-anhydro-6-deoxy-6-methylene-7-ethoxy-2,3-di-0-methyl- a-L-/‘d -heptopyranoside (Vila)

To a solution of compound Via (1.89 g, 5.92 mmol, 1 .0 eq.) in anhydrous THF (40 mL) and cooled to 0°C, were added IBCF (0.84 mL, 6.48 mmol, 1.1 eq.) and N- methylmorpholine (0.72 mL, 6.55 mmol, 1.1 eq.). After 20 minutes stirring the flask was covered with aluminium foil, 2-mercaptopyridine /V-oxide sodium salt (1 .77 g, 1 1.80 mmol, 2.0 eq.) was added and the reaction mixture was stirred at ambient temperature. After 2 hours, anhydrous THF (80 mL) then ie f-butylthiol (0.28 mL, 2.61 mmol, 1.6 eq.) were added. The aluminium foil was removed and the reaction mixture was irradiated and heated 30 minutes with a UV lamp (300W). The thiol excess was neutralized with a NaOCI aqueous solution (13% v/v, 10 mL). The reaction mixture was concentrated then dissolved in EtOAc (100 mL), washed successively with a 5% NaHC03 aqueous solution (2 x 15 mL), a 5 % citric acid aqueous solution (1 x 15 mL) and brine (1 x 25 mL), then the aqueous layer was extracted with dichloromethane (2 x 20 mL). The combined organics were dried (MgS04), filtered and concentrated. Column chromatography (gradient dichloromethane/ethyl acetate 95:5 – 75:25) afforded compound Vila as a colourless oil (218 mg, 0.79 mmol, 48%), in a mixture of diastereomers (67:33) (the relative composition of the mixture was determined by 1H NMR from integrations of protons H-2).
1H NMR (CDCIs, 400 MHz): δ 1.23 (t, J = 7.1 Hz, 3H, -OCH2CH3) (diastereomeric mixture), 3.13 (dd, J2-3 = 9.6 Hz, J1-2 = 3.0 Hz, 1 H, H-2) (minor), 3.30 (dd, J2-3 = 5.0 Hz, J1-2 = 1.6 Hz, 1 H, H-2) (major), 3.41 (s, 3H, -OCH3) (minor), 3.47 (s, 3H, -OCH3) (major), 3.50 (s, 3H, -OCH3) (mixture), 3.53 (s, 3H, -OCH3) (major), 3.55-3.61 (m, 1 H, -OCHaHbCH3) (mixture), 3.72 (dd, J2-3 = 5.0 Hz, J3-4 = 2.8 Hz, 1 H, H-3) (major), 3.78-3.90 (m, 1 H, – OCHaHbCH3) (mixture), 3.93-4.07 (m, 2H, H-3, H-4) (mixture), 4.59 (d I, J4-5 = 4.0 Hz, 1 H, H-5) (major), 4.62 (d, J1-2 = 1.7 Hz, 1 H, H-1 ) (major), (td, J4-5 = 7.9 Hz, J = 2.6 Hz, 1 H, H- 5), (minor), 4.79 (d, J1-2 = 3.0 Hz, 1 H, H-1 ) (minor), 5.35-5.57 (m, 3H, H-7, -C=CH2) (mixture). 13C NMR (CDCI3, 100.6 MHz): δ 15.3 (-OCH2CH3) (minor), 15.4 (-OCH2CH3) (major), 56.5 (-OCH3) (minor), 56.8 (-OCH3) (major), 58.6 (-OCH3) (major), 59.1 (-OCH3) (minor), 59.9 (- OCH3) (major), 60.3 (-OCH3) (minor), 63.3 (-OCH2CH3) (minor), 63.8 (-OCH2CH3) (major), 74.2 (C-5) (minor), 74.7 (C-5) (major), 76.4 (C-3) (major), 77.0 (C-4) (major), 77.7 (C-2) (major), 79.0 (C-3) (minor), 79.7 (C-4) (minor), 80.1 (C-2) (minor), 99.3 (C-1 ) (major), 99.5 (C-1 ) (minor), 102.2 (C-7) (major), 103.0 (C-7) (minor), 1 1 1.6 (-C=CH2) (minor), 1 15.2 (- C=CH2) (major), 147.4 (C-6) (minor), 148.1 (C-6) (major).
ESI-MS (pos. mode): m/z = 297 [M+Na]+.
Preparation of Methyl-4,7-anhydro-7-ethoxy-2,3-di-0-methyl-a-L-/ o-hepto- pyranosid-6-ulose (Villa)

Through a solution of compound Vila (449 mg, 1 .64 mmol, 1.0 eq.) in anhydrous dichloromethane (10 ml_), under an argon atmosphere and cooled to -78°C, was bubbled ozone (0.2 L/min, 1 10 V). When the solution had turned dark blue, oxygen was bubbled through in order to remove the excess ozone. When the solution became colorless dimethylsulfide (5 drops) was added and the solution was brought to room temperature. After 1 h15 the reaction mixture was concentrated. Column chromatography (gradient dichloromethane/ethyl acetate 95:5 – 80:20) afforded compound Villa as a white solid (364 mg, 1.32 mmol, 80%), in a mixture of diastereomers (79:21 ) (the relative composition of the mixture was determined by 1H NMR from integrations of protons H-2).
1H NMR (CDCI3, 400 MHz): δ 1.24-1 .28 (m, 3H, -OCH2CH3) (diastereomeric mixture), 3.10 (dd, J2-3 = 10.2 Hz, J1-2 = 2.9 Hz, 1 H, H-2) (minor), 3.17 (dd, J2-3 = 9.4 Hz, J1-2 = 2.8 Hz, 1 H, H-2) (major), 3.38 (s, 3H, -OCH3) (major), 3.42 (s, 3H, -OCH3) (minor), 3.50 (s, 3H, – OCH3) (mixture), 3.63 (s, 3H, -OCH3) (major), 3.66 (s, 3H, -OCH3) (minor), 3.48-3.73 (m, 3H, H-3 major, -OCHaHbCH3 major, -OCHaHbCH3 minor), 3.77-3.95 (m, 2H, -OCHaHbCH3 minor, -OCHaHbCH3 major), 4.07 (dd, J2-3 = 10.2 Hz, J3-4 = 7.7 Hz, 1 H, H-3) (minor), 4.34 (d, J4-5 = 9.1 Hz, 1 H, H-5) (minor), 4.39-4.44 (m, 2H, H-4 minor, H-5 major), 4.50 (dd, J3-4 = 9.5 Hz, J4-5 = 6.2 Hz, 1 H, H-4) (major), 4.76 (d, Ji-2 = 2.8 Hz, 1 H, H-1 ) (major), 4.79 (d, J1-2 = 2.9 Hz, 1 H, H-1 ) (minor), 4.89 (d, J = 1 ,1 Hz, 1 H, H-7) (minor), 4.93 (s I, 1 H, H-7) (major).
13C NMR (CDCI3, 100.6 MHz): δ 15.1 (-OCH2CH3) (minor), 15.2 (-OCH2CH3) (major), 56.7 (-OCH3) (minor), 57.2 (-OCH3) (major), 59.3 (-OCH3) (mixture), 59.8 (-OCH3) (major), 60.6 (-OCH3) (minor), 65.0 (-OCH2CH3) (minor), 65.5 (-OCH2CH3) (major), 70.2 (C-5) (major), 72.4 (C-5) (minor), 75.9 (C-4) (major), 79.2 (C-4) (minor), 79.4 (C-3) (major), 79.8 (C-2 major, C-3 minor), 80.2 (C-2) (minor), 96.1 (C-7) (major), 97.2 (C-7) (minor), 98.7 (C-1 ) (major), 99.0 (C-1 ) (minor), 205.3 (C-6) (minor), 205.6 (C-6) (major).
IR (film) v (cm“1): 1783 (C=0).
ESI-MS (pos. mode): m/z = 299 [M+Na]+, 331 [M+Na+MeOH]+.
Preparation of Methyl methyl-2,3-di-0-methyl-a-L-idopyranosiduronate (IXa)
; CHCI3)

To a solution of compound Villa (50 mg, 0.18 mmol, 1 .0 eq.) in dichloromethane (3 mL), under an argon atmosphere and cooled to 0°C, were added m-CPBA (77%, 120 mg, 0.54 mmol, 3.0 eq.) and NaHC03 (20 mg, 0.23 mmol, 1 .3 eq.). After 3 hours stirring the solvent was removed under vacuum. The resulting residue was dissolved in EtOAc (30 mL), extracted with distilled water (2 x 10 mL), and the aqueous phase was concentrated. The crude mixture was dissoved in methanol (10 mL), para-toluenesulfonic acid monohydrate was added (4 mg, 0.02 mmol, 0.1 eq.) then the reaction mixture was heated to reflux and the reaction monitored by 1H NMR in deuterated methanol. After 8 hours the solvent was evaporated. The residue obtained was dissolved in DMF (5 mL) then triethylamine (28 μί, 0.20 mmol, 1 .1 eq.) and methyl iodide (56 μί, 0.90 mmol, 5 eq.) were added. After 3h30 the reaction mixture was concentrated, dissolved in EtOAc (30 mL) and the organic phase was washed with a 5% NaHC03 aqueous solution (2 x 10 mL), a 5% citric acid aqueous solution (2 x 10 mL) and brine (1 x 10 mL). The aqueous phase was extracted with dichloromethane (5 x 10 mL) and the combined organics were dried (MgS04), filtered and concentrated. Column chromatography (dichloromethane/ethyl acetate 85:15) afforded compound Xla as a colourless oil (25 mg, 0.10 mmol, 56%). 1H NMR (CDCI3, 400 MHz): δ 3.41 (d I, J2-3 = 3.5 Hz, 1 H, H-2), 3.47 (s, 3H, -OCH3), 3.56 (s, 3H, -OCH3), 3.57 (s, 3H, -OCH3), 3.69 (t, J2-3 = J3-4 = 3.5 Hz, 1 H, H-3), 3.75-3.78 (m, 1 H, OH), 3.80 (s, 3H, -OCH3), 3.97 (m, 1 H, H-4), 4.42 (d, J4-5 = 1 .6 Hz, 1 H, H-5), 4.61 (d,
13C NMR (CDCI3, 100.6 MHz): δ 52.4, 57.5, 58.4, 60.8 (4x-OCH3), 67.7 (C-4), 74.8 (C-5), 77.2 (C-2), 77.5 (C-3), 100.9 (C-1 ), 169.6 (C=0).
Elemental analysis: Calculated: C: 48.00 ; H: 7.25. Found: C: 47.62 ; H: 7.15.
ESI-MS (pos. mode): m/z = 272 [M+Na]+.
IR (film) v (cm“1): 3491 (O-H), 1765 (C=0). Example 2 :
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(2′,3′-di-0-methyl-p-D-glucopyranosyl- uronate)-a-D-glucopyranoside (IVb)

To a solution of the co

Me
(3.279 g, 5.00 mmol, 1.0 eq.) in a water/acetonitrile mixture (1 :1 , 300 mL) were added NaBr (105 mg, 1 .02 mmol, 0.2 eq.) and TEMPO (33 mg, 0.21 mmol, 0.04 eq.). The reaction mixture was cooled with the aid of an ice bath then a solution of NaOCI (13% v/v, 1 1.5 mL, 20.08 mmol, 4.0 eq.) was added. After 3 hours stirring at 0°C, NaOCI was added anew (13% v/v, 1 1 .5 mL, 20.08 mmol, 4.0 eq.). After two more hours ethanol was added (96% v/v, 20 mL), then the pH was reduced to 2-3 by addition of HCI (10% v/v). The solvent was evaporated and the residue obtained was suspended in DMF (40 mL) then triethylamine (2.8 mL, 2.032 g, 20.0 mmol, 4.0 eq.) and methyl iodide (6.2 mL, 14.136 g, 99.6 mmol, 20.0 eq.) were added. After 4 hours stirring at room temperature the solvent was evaporated and the residue obtained was dissolved in EtOAc (200 mL). The organic layer was washed with a 5% citric acid aqueous solution (1 χ 20 mL) and brine (1 χ 20 mL). The aqueous layer was extracted with dichloromethane (2 χ 20 mL). The combined organics were dried (MgS04), filtered and evaporated. The residue obtained was dissolved in DMF (20 mL), then triethylamine (1.4 mL, 1 .016 g, 10.0 mmol, 2.0 eq.) and methyl iodide (3.1 mL, 7.068 g, 49.8 mmol, 10.0 eq) were added. After 60 hours stirring at room temperature the solvent was evaporated and the residue obtained was dissolved in EtOAc (200 mL). The organic layer was washed with a 5% citric acid aqueous solution (2 x 20 mL) and brine (1 χ 20 mL). The organic layer was dried (MgS04), filtered and evaporated. Column chromatography (gradient hexane/ethyl acetate 80:20 – 50:50) gave compound IVb as a colourless oil which was dissolved in a diethyl ether/hexane mixture and evaporated at room temperature to afford a white solid (2.500 g, 3.66 mmol, 73%).
1H NMR (CDCI3, 400 MHz): δ 2.92 (d, 1 H), 2.93-2.98 (m, 2H), 3.41 (s, 3H), 3.48-3.76 (m, 5H), 3.51 (s, 3H), 3.60 (s, 3H), 3.63 (s, 3H), 3.87-3.98 (m, 3H), 4.36-4.41 (m, 1 H), 4.49- 5.08 (m, 6H), 4.61 (d, 1 H), 7.24-7.42 (m, 15H).
Elemental analysis: Calculated: C: 65.09 ; H: 6.79. Found: C: 65.29 ; H: 6.96.
ESI-MS (pos. mode): m/z = 705 [M+Na]+.
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′-0-(1 “-ethoxy-2”-propyn-1 “-yl)-2′,3′- di-0-methyl-p-D-glucopyranosyluronate)-a-D-glucopyranoside (Vb)

E F To a solution of compound IVb (385 mg, 0.56 mmol, 1 .0 eq.) in chloroform (stabilised with amylene, 30 mL) were added, under an argon atmosphere, P205 (410 mg, 2.80 mmol, 5.0 eq.) and propargylaldehyde diethylacetal (0.4 mL, 2.79 mmol, 5.0 eq.), then the reaction mixture was heated at reflux. After 5 hours stirring, the reaction mixture was filtered through a pad of Celite® then the solvent was removed under vacuum. The crude mixture was suspended in EtOAc (60 mL), washed with a 5% NaHC03 aqueous solution (1 x 15 mL) and brine (1 x 15 mL). The organic layer was dried (MgS04), filtered, and evaporated. Column chromatography (gradient hexane/ethyl acetate 90:10 – 70:30) afforded compound Vb as a colourless oil (275 mg, 0.36 mmol, 64%) in a diastereomeric mixture (64:36).
1H NMR (CDCI3, 250 MHz): δ 1.17-1 .27 (m, 3H), 2.55 (d, 0.36H), 2.57 (d, 0.64H), 2.92- 3.08 (m, 2H), 3.38 (s, 3H), 3.49 (s, 1.92H), 3.50 (s, 1.08H), 3.57 (s, 1.08H), 3.59 (s, 1.92H), 3.60 (s, 1.92H), 3.62 (s, 1.08H), 3.44-3.97 (m, 10H), 4.35 (t, 1 H), 4.46-4.76 (m, 6H), 5.03 (d, 1 H), 5.32 (d, 0.36H), 5.56 (d, 0.64H), 7.21-7.42 (m, 15H).
Elemental analysis: Calculated: C: 65.95 ; H: 6.85. Found: C: 65.92 ; H: 6.75.
ESI-MS (pos. mode): m/z = 787 [M+Na]+.
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′-0-(1 “-ethoxy-2″-propyn-1 ” 1 ‘,2′,3’-tri-0-methyl-a-D-glucopyranosiduronic acid)-a-D-glucopyranoside (Vlb)

E F
To a solution of compound Vb (1.02 g, 1.33 mmol, 1.0 eq.) in EtOH/H20 (1 :1 , 100 mL) was added sodium hydroxide (82 mg, 2.05 mmol, 1.5 eq.). After 3 hours stirring at room temperature sodium hydroxide was added anew (27 mg, 0.68 mmol, 0.5 eq.). After an additional hour stirring the solvent was evaporated. The residue obtained was dissolved in water (40 mL). The pH of the aqueous layer was reduced to 2-3 with a 10% HCI aqueous solution then the layer was saturated with sodium chloride before extraction with dichloromethane (3 x 20 mL). The combined organics were dried (MgS04), filtered and removed under vacuum. Compound VIb was obtained without further purification as a white solid (930 mg, 1.24 mmol, 93%), in a diastereomeric mixture (63:37).
1H NMR (CDCIs, 400 MHz): δ 1.17-1.28 (m, 3H), 2.65 (d, 0.63H), 2.68 (d, 0.37H), 2.90- 3.09 (m, 2H), 3.37 (s, 3H), 3.46 (s, 1.1 1 H), 3.57 (s, 1.89H), 3.58 (s, 1.1 1 H), 3.60 (s, 1.89H), 3.42-3.90 (m, 10H), 4.29 (d, 1 H), 4.46-4.97 (m, 7H), 5.44 (d, 0.37H), 5.61 (d, 0.63H), 7.28-7.44 (m, 15H).
ESI-MS (pos. mode): m/z = 773 [M+Na]+ , 795 [M-H+2Na]+ . ESI-MS (neg. mode): m/z = 749 [M-H]\
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′,7′-anhydro-6′-deoxy-6′-methylene-7′- ethoxy-2 3′-di-0-methyl-α-L-/‘ o-heptopyranosyl)-α-D-glucopyranoside (Vllb)

To a solution of compound VIb (647 mg, 0.86 mmol, 1.0 eq.) in anhydrous THF (20 mL) and cooled to 0°C, were added IBCF (0.1 1 mL, 0.85 mmol, 1.0 eq.) and N- methylmorpholine (0.10 mL, 0.91 mmol, 1.1 eq.). After 10 minutes stirring, the flask was covered with aluminium foil, 2-mercaptopyridine /V-oxide sodium salt (512 mg, 3.43 mmol, 4.0 eq.) was added and the reaction mixture was stirred at ambient temperature. After 20 minutes anhydrous THF (100 mL) then ie f-butylthiol (0.18 mL, 1 .68 mmol, 2.0 eq.) were added. The aluminium foil was removed and the reaction mixture was irradiated and heated 15 minutes with a UV lamp (300W). The thiol excess was neutralized with a NaOCI aqueous solution (13%, 10 mL). The reaction mixture was concentrated then dissolved in EtOAc (100 mL), washed successively with a 5% NaHC03 aqueous solution (1 x 15 mL), a 5 % citric acid aqueous solution (1 x 15 mL) and brine (1 x 15 mL), then the aqueous layer was extracted with dichloromethane (2 x 20 mL). The combined organics were dried (MgS04), filtered and concentrated. Column chromatography (gradient hexane/ethyl acetate 90 :10 – 70:30) afforded compound Vllb as a colourless oil (251 mg, 0.36 mmol, 42%) in a mixture of diastereomers (61 :39).
1H NMR (CDCI3, 250 MHz): δ 1.18-1.29 (m, 3H), 2.90-3.07 (m, 1 H), 3.37 (s, 1.17H), 3.38 (s, 1 .83H), 3.46 (s, 3H), 3.54 (s, 1.83H), 3.60 (s, 1 .17H), 3.29-4.00 (m, 10H), 4.1 1 -4.26 (m, 1 H), 4.50-4.96 (m, 8H), 5.09-5.48 (m, 3H), 7.23-7.39 (m, 15H).
ESI-MS (pos. mode): m/z = 720 [M+Na]+ .
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(4′,7′-anhydro-7′-ethoxy-2′,3′-di-0- methyl-a-L-/‘ o-heptopyranosid-6′-ulosyl)-a-D-glucopyranoside (Vlllb)

Through a solution of compound Vllb (145 mg, 0.21 mmol, 1 .0 eq.) in anhydrous dichloromethane (10 mL), under an argon atmosphere and cooled to -78°C, was bubbled ozone (0.2 L/min, 1 10 V). When the solution had turned dark blue, oxygen was bubbled through in order to remove the excess ozone. When the solution became colorless dimethylsulfide (4 drops) was added and the solution was brought to room temperature. After 30 min stirring the reaction mixture was concentrated. Column chromatography (gradient hexane/ethyl acetate 90:10 – 60:40) afforded compound Vlllb as a white solid (100 mg, <67%) in a mixture of diastereomers (67:33).
1H NMR (CDCI3, 400 MHz): δ 1.21 -1 .28 (m, 3H), 2.93-3.07 (m, 1 H), 3.31-4.26 (m, 20H), 4.52-5.02 (m, 9H), 7.20-7.45 (m, 15H).
ESI-MS (pos. mode): m/z = 731 [M+Na]+ .
Preparation of Methyl-2,3,6-tri-0-benzyl-4-0(methyl 2′,3′-di-0-methyl-a-L- idopyranosiduronate)-a-D-glucopyranoside (IXb)

To a solution of compound Vlllb (62 mg, 87 μηηοΙ, 1 .0 eq.) in dichloromethane (5 mL), under an argon atmosphere and cooled to 0°C, were added m-CPBA (77%, 58 mg, 259 μηηοΙ, 3.0 eq.) and NaHC03 (1 1 mg, 130 μηηοΙ, 1 .5 eq.). After 5 hours stirring the solvent was removed under vacuum. The reaction mixture was then dissolved in EtOAc (50 mL) and washed successively with a 5% NaHC03 aqueous solution (1 x 10 mL), a 5 % citric acid aqueous solution (1 x 10 mL) and brine (1 x 10 mL). The organic layer was dried (MgS04), filtered and concentrated. The crude mixture was dissolved in anhydrous methanol (10 mL) and sodium methoxide was added to reach pH = 10. After 30 minutes stirring at room temperature the reaction mixture was neutralized with Dowex®, filtered through a pad of Celite®, and concentrated. The residue obtained was dissolved in DMF (10 mL) then triethylamine (13 μί, 93 μηηοΙ, 1.1 eq.) and methyl iodide (27 μί, 434 μηηοΙ, 5.0 eq.) were added. After 2h30 stirring the reaction mixture was concentrated, dissolved in EtOAc (40 mL) and washed with a 5% citric acid aqueous solution (2 x 10 mL), a 5% NaHC03 aqueous solution (2 x 10 mL), and brine (1 x 10 mL). The organic layer was dried (MgS04), filtered and concentrated. Column chromatography (gradient hexane/ethyl acetate 60:40-50:50) afforded compound IXb as a colourless oil (12 mg, 18 μηηοΙ, 20% over two steps).
1H NMR (CDCIs, 400 MHz): δ 3.23 (s, 3H), 3.20-3.25 (m, 1 H), 3.36 (s, 3H), 3.43 (s, 3H), 3.46 (s, 3H), 3.33-3.58 (m, 3H), 3.60-3.69 (m, 2H), 3.72-3.95 (m, 4H), 4.53-4.60 (m, 4H), 4.68-4.97 (m, 4H), 5.14 (s, 1 H), 7.24-7.37 (15H).
ESI-MS (pos. mode): m/z = 705 [M+Na]+ .
……………
Volume 69, Issue 15, 15 April 2013, Pages 3149–3158
http://www.sciencedirect.com/science/article/pii/S0040402013003025
Abstract
Idraparinux, the fully O-sulfated, O-methylated, heparin-related pentasaccharide possessing selective factor Xa inhibitory activity, was prepared by two novel synthetic pathways. Each route was based on a 2+3 block synthesis utilizing the same l-iduronic acid-containing trisaccharide acceptor, which was glycosylated with either a glucuronide disaccharide donor or its non-oxidized precursor. The latter route, involving the oxidation of the glucose unit into d-glucuronic acid at a pentasaccharide level proved to be much more efficient, providing the target pentasaccharide in a reasonable overall yield.
Graphical abstract

- ……………………………
- SYNTHESIS
- US 20120041189 A1,
- http://www.patexia.com/us-publications/20120041189
- EXAMPLE 1Preparation of the Compound of Formula (I) in Crystalline Form (Scheme 1)
1.1: Preparation of the Compound of Formula (I′)
The compound of formula (I″) is obtained, for example, according to the teaching of patent EP 0 529 715 B1 or of the articles “Bioorg. Med. Chem.” (1994, Vol. 2, No. 11, pp. 1267-1280), “Bioorg. Med. Chem. Letters” (1992, Vol. 2, No. 9, pp. 905-910) or “Magnetic Resonance in Chemistry” (2001, Vol. 39, pp. 288-293). The compound of formula (I″) (5 g, 3.06 mmol) is dissolved in acetonitrile (10 mL). Deionized water (12.2 mL) and aqueous 30% sodium hydroxide solution (4.1 g) are then added. The mixture is heated to 40° C. and maintained at this temperature for 5 hours. The reaction medium is then cooled to 20° C. and acidified to pH 6.25 with aqueous 1N hydrochloric acid solution (about 17.7 g) before extraction with MTBE of certain impurities, the saponified product remaining in the aqueous phase. The residual acetonitrile, contained in the aqueous phase, is then removed by concentration, followed by diluting with deionized water (125 mL). The saponified product is finally precipitated at pH 1.5 by adding aqueous 1N hydrochloric acid solution (about 17.6 g) at 20° C. The suspension is maintained for 4 hours at 20° C. before filtration. The wet solid is finally dried in a vacuum oven at 30° C. to give 2.93 g (93.6%) of compound of formula (I).
NMR (anomeric protons of the saccharide units D, E, F, G, H): 5.79, 5.14, 5.55, 5.92, 4.94 ppm.
1.2 Preparation of the Crude Compound of Formula (I)
The compound of formula (I′) obtained after the preceding step is dissolved in tetrahydrofuran (18 mL). Palladium-on-charcoal (0.3 g) is added. The reaction medium is hydrogenated at 0.3 bar of hydrogen (relative pressure) for 4 hours. After filtering and evaporating, 2.12 g (99%) of the crude compound of formula (I) are obtained.
1.3: Preparation of the Compound of Formula (I) in Crystalline Form Using an Isopropanol/MTBE Mixture
The crude hydrogenated product obtained after the preceding step is dissolved in isopropanol (13 mL) at 65° C., and then crystallized at room temperature. The suspension is then cooled to 40° C., followed by addition of MTBE (13 mL), and is then cooled slowly to 10° C. After maintenance at 10° C. for 2 hours, the crystalline hydrogenated product is filtered off, washed and dried. 1.66 g of the compound of formula (I) in crystalline form are thus obtained, in the form of a cream-white powder. The reaction yield for the production of the compound of formula (I) in crystalline form, from the compound of formula (I′), is 92.5%. When expressed relative to the starting compound (I″), the reaction yield for the production of the compound of formula (I) in crystalline form is 86.6%.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (I) in crystalline form: 5.77, 5.11, 5.51, 5.84, 5.01 ppm.
1.4: Preparation of the Compound of Formula (I) in Crystalline Form Using Isopropanol
The crude hydrogenated product obtained after step 1.2 is dissolved in isopropanol (5 volumes) at 75° C. The medium is then cooled slowly until crystals appear, according to the known standard techniques for crystallization. The process is performed, for example, by a first step of cooling at 65° C. for 1 hour, and than a second step of cooling to a final temperature of 25° C. over 4 hours or of 5° C. over 6 hours, and finally maintenance at this final temperature for 30 minutes. The suspension is then filtered and rinsed with isopropanol (2×0.1 V) and compound (I) is isolated in the form of white crystals, which appear under a microscope in the form of needles. The 1H NMR analysis of these crystals is identical to that described after step 1.3 above.
EXAMPLE 4
Preparation of Idraparinux from the Compound of Formula (I) in Crystalline Form (Scheme 2)
The preparation of idraparinux (II) from the compound of formula (I) is summarized in Scheme 2.
The compound of formula (I) in crystalline form, as obtained according to Example 1.3, is dissolved in N,N’-dimethylformamide (6.6 mL) and then heated to 30.degree. C. Under an inert atmosphere, 3.8 g of pyridine-sulfur trioxide complex are added slowly, followed by maintenance at 30.degree. C. for 4 hours. The reaction medium is then poured into aqueous 23.8% sodium hydrogen carbonate solution (16.3 g) maintained at a maximum of 25.degree. C., to obtain the compound of formula (II). The reaction medium is kept stirring for hours. The solution of sulfated product is then poured onto an MTBE/isopropanol/ethanol mixture (171 mL/70 mL/70 mL). Precipitation of the product is observed, and, after filtering off, washing and drying the cake, 4.99 g (96.8%) of compound of formula (II) are obtained, and are then purified by anion-exchange chromatography according to the usual techniques.
NMR (anomeric protons of the saccharide units D, E, F, G, H) of the compound of formula (II): 5.48, 4.68, 5.44, 5.08, 5.18 ppm.It thus appears that the process according to the invention makes it possible to obtain idraparinux (compound of formula (II)) in a chemical yield of about 84% (precisely 83.8% according to the protocols described above) starting from the compound of formula (I”), i.e. a gain in yield of about 30% relative to the process described in patent EP 0 529 715 B1.

IDRAPARINUX
References
- Bousser MG, Bouthier J, Büller HR, et al. (January 2008). “Comparison of idraparinux with vitamin K antagonists for prevention of thromboembolism in patients with atrial fibrillation: a randomised, open-label, non-inferiority trial”. Lancet 371 (9609): 315–21. doi:10.1016/S0140-6736(08)60168-3.PMID 18294998.
- Buller HR, Cohen AT, Davidson B, et al. (September 2007). “Idraparinux versus standard therapy for venous thromboembolic disease”. N. Engl. J. Med. 357 (11): 1094–104. doi:10.1056/NEJMoa064247. PMID 17855670.
- Bioorg Med Chem1994,2,(11):1267
- Drugs Fut2002,27,(7):639
- EP 0454220, JP 1992225994, US 5378829, US 5382570, US 5529985, US 5773605
- EP 0529715
-
Bioorg Med Chem Lett. 2009 Jul 15;19(14):3875-9. doi: 10.1016/j.bmcl.2009.03.155. Epub 2009 Apr 5.
- Chemistry – A European Journal, 2012 , vol. 18, 34 p. 10643 – 10652
- Magnetic Resonance in Chemistry, 2001 , vol. 39, 5 p. 288 – 293
- Tetrahedron, 2013 , vol. 69, 15 p. 3149 – 3158…….. MP 210-15 DEG CENT
-
- I. Capila, R.J. Linhardt
Angew. Chem., Int. Ed., 41 (2002), p. 390
- I. Capila, R.J. Linhardt
-
- L. Roden
D.A. Lane, U. Lindahl (Eds.), Chemical and Biological Properties, Clinical Applications, CRC Press, Boca Raton, FL (1989), p. 1
- L. Roden
-
- (a) C.A.A. van Boeckel, M. Petitou
Angew. Chem., Int. Ed. Engl., 32 (1993), p. 1671
- (b) M. Petitou, C.A.A. van Boeckel
Angew. Chem., Int. Ed., 43 (2004), p. 3118
- (a) C.A.A. van Boeckel, M. Petitou
-
- M. Petitou, B. Casu, U. Lindahl
Biochimie, 85 (2003), p. 83Article |
- M. Petitou, B. Casu, U. Lindahl
-
- (a) M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, J.C. Jacquinet, P. Sinay, G. Torri
Carbohydr. Res., 167 (1987), p. 67Article |
- (b) P.-A. Driguez, I. Lederman, J.-M. Strassel, J.-M. Herbert, M. Petitou
J. Org. Chem., 64 (1999), p. 9512
- (c) J. Choay, M. Petitou, J.C. Lormeau, P. Sinay, B. Casu, G. Gatti
Biochem. Biophys. Res. Commun., 116 (1983), p. 492Article |
- (d) P. Sinay, J.C. Jacquinet, M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, G. Torri
Carbohydr. Res., 132 (1984), p. C5
- (a) M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, J.C. Jacquinet, P. Sinay, G. Torri
-
- (a) P. Westerduin, C.A.A. van Boeckel, J.E.M. Basten, M.A. Broekhoven, H. Lucas, A. Rood, H. van der Heijden, R.G.M. van Amsterdam, T.G. van Dinther, D.G. Meuleman, A. Visser, G.M.T. Vogel, J.B.L. Damm, G.T. Overklift
- Bioorg. Med. Chem., 2 (1994), p. 1267Article |
 PDF (1579 K)
PDF (1579 K)
- (b) J.M. Herbert, J.P. Herault, A. Bernat, R.G.M. van Amsterdam, J.C. Lormeau, M. Petitou, C. van Boeckel, P. Hoffmann, D.G. Meuleman
Blood, 91 (1998), p. 4197
-
- (a) P. Prandoni, D. Tormene, M. Perlati, B. Brandolin, L. Spiezia
Expert Opin. Investig. Drugs, 17 (2008), p. 773
- (b) A.S. Go, D.E. Singer
Lancet, 371 (2008), p. 278Article|
- (a) P. Prandoni, D. Tormene, M. Perlati, B. Brandolin, L. Spiezia
-
- I.M. Pinilla, M.B. Martinez, J.A. Galbis
Carbohydr. Res., 338 (2003), p. 549Article |
- I.M. Pinilla, M.B. Martinez, J.A. Galbis
-
- (a) K. Yoza, N. Amanokura, Y. Ono, T. Akao, H. Shinmori, M. Takeuchi, S. Shinkai, D.N. Reinhoudt
Chem. Eur. J., 5 (1999), p. 2722
- (b) J. Elhalabi, K.G. RiceCarbohydr. Res., 335 (2001), p. 159Article| PDF (177 K)
- (a) K. Yoza, N. Amanokura, Y. Ono, T. Akao, H. Shinmori, M. Takeuchi, S. Shinkai, D.N. Reinhoudt
-
- S.D. Debenham, E.J. Toone
Tetrahedron: Asymmetry, 11 (2000), p. 385Article |
- S.D. Debenham, E.J. Toone
-
- A. Meijer, U. Ellervik
J. Org. Chem., 69 (2004), p. 6249 and references therein
- A. Meijer, U. Ellervik
-
- P. Duchaussoy, G. Jaurand, P.-A. Driguez, I. Lederman, F. Gourvenec, J.-M. Strassel, P. Sizun, M. Petitou, J.-M. Herbert
Carbohydr. Res., 317 (1999), p. 63Article |
- P. Duchaussoy, G. Jaurand, P.-A. Driguez, I. Lederman, F. Gourvenec, J.-M. Strassel, P. Sizun, M. Petitou, J.-M. Herbert
-
- J. Lee, X.-A. Lu, S.S. Kulkarni, Y. Wen, S.-C. Hung
J. Am. Chem. Soc., 126 (2004), p. 476
- J. Lee, X.-A. Lu, S.S. Kulkarni, Y. Wen, S.-C. Hung
-
- L.A.G.M. van den Broek, D.J. Vermaas, B.M. Heskamp, C.A.A. van Boeckel
Recl. Trav. Chim. Pays-Bas., 112 (1993), p. 82
- R.R. Schmidt, J. Michel
Angew. Chem., Int. Ed. Engl., 19 (1980), p. 731
- L.A.G.M. van den Broek, D.J. Vermaas, B.M. Heskamp, C.A.A. van Boeckel
-
- (a) F. Lin, W. Peng, W. Xu, X. Han, B. Yu
Carbohydr. Res., 339 (2004), p. 1219
- (b) P.L. Anelli, C. Biffi, F. Montanari, S. Quici
J. Org. Chem., 52 (1987), p. 2559
- (c) P.L. Anelli, S. Banfi, F. Montanari, S. Quici
J. Org. Chem., 54 (1989), p. 2970
- (a) F. Lin, W. Peng, W. Xu, X. Han, B. Yu
-
- (a) P. Bourhis, F. Machetto, P. Duchaussoy, J.-P. Herault, J.-M. Mallet, J.-M. Herbert, M. Petitou, P. Sinay
- For other examples of 4,6-locked idose glycosyl donors, see: Bioorg. Med. Chem. Lett., 7 (1997), p. 2843
- (b) N. Barroca, J.-C. Jacquinet
Carbohydr. Res., 337 (2002), p. 673
- (c) J. Kuszmann, G. Medgyes, S. Boros
Carbohydr. Res., 339 (2004), p. 1569
- (d) J. Tatai, P. Fugedi
Org. Lett., 9 (2007), p. 4647
| WO2002024754A1 | Sep 20, 2001 | Mar 28, 2002 | Akzo Nobel Nv | Polysaccharides with antithrombotic activity comprising at least a covalent bond with biotin or a biotin derivative |
| WO2006030104A1 * | Sep 7, 2005 | Mar 23, 2006 | Sanofi Aventis | Biotinylated hexadecasaccharides, preparation and use thereof |
| WO2007042469A2 * | Oct 6, 2006 | Apr 19, 2007 | Organon Nv | Anticoagulant antithrombotic dual inhibitors comprising a biotin label |
| EP0230023A2 * | Dec 19, 1986 | Jul 29, 1987 | Marion Merrell Dow Inc. | Pharmaceutical compositions for the enhancement of wound healing |
| EP0300099A1 * | Jul 20, 1987 | Jan 25, 1989 | Akzo N.V. | New pentasaccharides |
| EP0301618A2 * | Jul 4, 1988 | Feb 1, 1989 | Akzo N.V. | New pentasaccharides |
| EP0454220A1 * | Apr 16, 1991 | Oct 30, 1991 | Akzo Nobel N.V. | Carbohydrate derivatives comprising a trisaccharide unit |
| GB1110939A * | Title not available | |||
| US3017407 * | Aug 18, 1958 | Jan 16, 1962 | Riker Laboratories Inc | Process for producing polysulfuric acid esters of polysaccharides |


Eluxadoline …Diarrhea-predominant irritable bowel syndrome
Eluxadoline
5 JAN 2014
Furiex Pharmaceuticals Inc. more than doubled in its best day of trading after its experimental drug alleviated diarrhea and abdominal pain caused by irritable bowel syndrome in two studies.
The drug eluxadoline met targets for improvements in stool consistency and abdominal pain that were developed in conjunction with U.S. and European regulators, the company said today. Furiex will apply for approval in June, Chairman Fred Eshelman said in an investor call today. He estimated annual sales of $750 million to $1 billion.
“By our math, it looks like a pretty doggone good market,” Eshelman said on the call, noting that there is only one currently approved drug available in the U.S. for the condition.
Diarrhea-predominant irritable bowel syndrome is a chronic disorder that affects about 28 million patients in the U.S. and Europe, Furiex said in the statement.Furiex said it would apply by mid-year for U.S. approval of the drug, eluxadoline, to treat diarrhea-predominant irritable bowel syndrome (IBS-d), a debilitating bowel disorder that affects about 28 million people in the United States and major European markets.
Furiex said it expected to seek European approval in early 2015.
“We believe that there are a lot of patients out there who need this drug. There is a huge unmet need,” Furiex Chief Medical Officer June Almenoff said in a telephone interview.
Currently approved drugs for IBS address constipation associated with the disorder, but there are few options for diarrhea predominant IBS.
Furiex founder and chairman Fred Eshelman said he believes the drug has the potential for blockbuster sales, which he defined as annual sales of between $750 million and $1 billion.
Eluxadoline was tested at two doses against a placebo over the course of 12 weeks to meet requirements by the U.S. Food and Drug Administration, and for 26 weeks for European health regulators, in Phase III studies involving 2,428 patients, Furiex said.
For the combined goal of improvement in abdominal pain and stool consistency for at least half the days in the study, eluxadoline achieved a statistically significant improvement at the 100 milligram and 75 mg doses through 12 weeks in both studies.
On the 26-week measure, the higher dose succeeded in both studies but the lower dose missed statistical significance in one of the two trials, according to initial results released by the company.
The success appeared to be driven by the percentage of patients reporting improvements in diarrhea, which ranged from 30 percent to 37 percent versus 22 percent and 20.9 percent for the placebo groups.
When the composite goal was broken into its two components, researchers found a numerical improvement in pain response rates that did not achieve statistical significance.
The drug appeared to be safe and well-tolerated in both studies, Furiex said. The most commonly reported side effects were constipation and nausea.
The company plans to present a far more detailed analysis of the late stage studies at an upcoming medical meeting.
“We’re very excited about the path ahead and about how this can transform patients’ lives,” Almenoff said.
Eluxadoline
5-({[(2S)-2-amino-3-(4-carbamoyl-2,6-dimethylphenyl)propanoyl][(1S)-1-(4-phenyl-1H-imidazol-2-yl)ethyl]amino}methyl)-2-methoxybenzoic acid
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
864821-90-9 CAS
JNJ-27018966
Molecular Formula: C32H35N5O5
Molecular Weight: 569.6508
Agents for Irritable Bowel Syndrome, mu-Opioid Agonists, delta-Opioid Antagonists
Mu Delta is a locally active mu opioid receptor agonist and delta opioid receptor antagonist in phase III clinical evaluation at Furiex Pharmaceuticals for the oral treatment of diarrheal predominant irritable bowel syndrome (d-IBS).
The product candidate holds an advantage over currently marketed products for this indication because it acts locally on the enteric nervous system, possibly decreasing adverse effects on the central nervous system. In 2011, fast track designation was assigned in the U.S. for the treatment of d-IBS. In 2011, Mu Delta was licensed to Furiex Pharmaceuticals by Janssen for the treatment of d-IBS, granting an option to Furiex to continue development and commercialization following phase II proof of concept studies.
The opioid receptors were identified in the mid-1970’s, and were quickly categorized into three sub-sets of receptors (mu, delta and kappa). More recently the original three types of receptors have been further divided into sub-types. Also known is that the family of opioid receptors are members of the G-protein coupled receptor (GPCR) super-family. More physiologically pertinent are the well established facts that opioid receptors are found throughout the central and peripheral nervous system of many mammalian species, including humans, and that modulation of the respective receptors can elicit numerous, albeit different, biological effects, both desirable and undesirable (D. S. Fries, “Analgesics”, inPrinciples of Medicinal Chemistry, 4th ed.; W. O. Foye, T. L. Lemke, and D. A. Williams, Eds.; Williams and Wilkins: Baltimore, Md., 1995; pp. 247-269; J. V. Aldrich, “Analgesics”, Burger’s Medicinal Chemistry and Drug Discovery, 5thEdition, Volume 3: Therapeutic Agents, John Wiley & Sons, Inc., 1996, pp. 321-441). In the most current literature, the likelihood of heterodimerization of the sub-classes of opioid receptors has been reported, with respective physiological responses yet undetermined (Pierre J. M. Riviere and Jean-Louis Junien, “Opioid receptors: Targets for new gastrointestinal drug development”, Drug Development 2000, pp. 203-238).
A couple biological effects identified for opioid modulators have led to many useful medicinal agents. Most significant are the many centrally acting mu opioid agonist modulators marketed as analgesic agents to attenuate pain (e.g., morphine), as well as peripherally acting mu agonists to regulate motility (e.g., loperamide). Currently, clinical studies are continuing to evaluate medicinal utility of selective delta, mu, and kappa modulators, as well as compounds possessing combined sub-type modulation. It is envisioned such explorations may lead to agents with new utilities, or agents with minimized adverse side effects relative to currently available agents (examples of side effects for morphine includes constipation, respiratory depression, and addiction potential). Some new GI areas where selective or mixed opioid modulators are currently being evaluated includes potential treatment for various diarrheic syndromes, motility disorders (post-operative ileus, constipation), and visceral pain (post operative pain, irritable bowel syndrome, and inflammatory bowel disorders) (Pierre J. M. Riviere and Jean-Louis Junien, “Opioid receptors: Targets for new gastrointestinal drug development” Drug Development, 2000, pp. 203-238).
Around the same time the opioid receptors were identified, the enkephalins were identified as a set of endogenous opioid ligands (D. S. Fries, “Analgesics”, inPrinciples of Medicinal Chemistry, 4th ed.; W. O. Foye; T. L. Lemke, and D. A. Williams, Eds.; Williams and Wilkins: Baltimore, Md., 1995; pp. 247-269). Schiller discovered that truncating the original pentapeptide enkephalins to simplified dipeptides yielded a series of compounds that maintained opioid activity (Schiller, P. WO 96/06855). However one potential drawback cited for such compounds is the likelihood of their inherent instability (P. W. Schiller et al., Int. J. Pept. Protein Res. 1993, 41 (3), pp. 313-316).
More recently, a series of opioid pseudopeptides containing heteroaromatic or heteroaliphatic nuclei were disclosed, however this series is reported showing a different functional profile than that described in the Schiller works. (L. H. Lazarus et al., Peptides 2000, 21, pp. 1663-1671).
Most recently, works around morphine related structures were reported by Wentland, et al, where carboxamido morphine derivatives and it’s analogs were prepared (M. P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, pp. 1717-1721; M. P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, pp. 623-626). Wentland found that substitution for the phenol moiety of the morphine related structures with a primary carboxamide led anywhere from equal activities up to 40 fold reduced activities, depending on the opioid receptor and the carboxamide. It was also revealed that any additional N-substitutions on the carboxamide significantly diminished the desired binding activity.
Compounds of the present invention have not been previously disclosed and are believed to provide advantages over related compounds by providing improved pharmacological profiles.
Opioid receptor modulators, agonists or antagonists are useful in the treatment and prevention of various mammalian disease states, for example pain and gastrointestinal disorders such as diarrheic syndromes, motility disorders including post-operative ileus and constipation, and visceral pain including post-operative pain, irritable bowel syndrome and inflammatory bowel disorders.
It is an object of the present invention to provide opioid receptor modulators. It is a further object of the invention to provide opioid receptor agonists and opioid receptor antagonists. It is an object of the present invention to provide opioid receptor ligands that are selective for each type of opioid receptor, mu, delta and kappa. It is a further object of the present invention to provide opioid receptor ligands that modulate two or three opioid receptor types, mu, delta and kappa, simultaneously.
It is an object of the invention to provide certain instant compounds that are also useful as intermediates in preparing new opioid receptor modulators. It is also an object of the invention to provide a method of treating or ameliorating a condition mediated by an opioid receptor. And, it is an object of the invention to provide a useful pharmaceutical composition comprising a compound of the present invention useful as an opioid receptor modulator.
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1 h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid is an opoid receptor modulator (mu receptor agonist and delta receptor antagonist) and may be useful for treating irritable bowel syndrome, pain or other opioid receptor disorders.
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid and methods of making this molecule are disclosed in
US application 2005/02033143. Example 9 of US application 2005/02033143 makes the hydrochloride salt of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
Applicants have discovered a process of making the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid and two novel crystals of this zwitterion. In Applicant’s hands, these novel crystals provide improved properties and can be purified at higher purity. Applicant’s new process results in improved and less costly process manufacturing conditions than the procedure disclosed in US application 2005/02033143.
………………..
FIG. 6 is the molecular structure of the zwitterion 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.

…………………..
SYNTHESIS OF 5-formyl-2- methoxy-benzoic acid methyl ester
Example 8: 2-Methoxy-5-formylbenzoic acid

Lithium hydroxide (1.04g, 0.043mol, 3eq) in water (lOmL) was added to a stirred solution of methyl 2-methoxy-5-formylbenzoate (2.8g, 0.014mol, leq) in a mixture of tetrahydrofuran (30mL) and methanol (20mL). The solution was stirred overnight, acidified to pH 1 with 10% HCl and the organic solvents removed in vacuo. The aqueous solution was extracted with ethyl acetate (lOOmL) and the organic solution washed with brine (lOOmL), then extracted with saturated aqueous sodium bicarbonate (3 x lOOmL). The basic solution was washed with ethyl acetate (lOOmL), then acidified to pH 1 with 10% HCl and back extracted with dichloromethane (3 x lOOmL). The organic solution was dried over sodium sulfate and evaporated in vacuo to give a cream coloured powder (2.01g, 77%). 1H NMR (CDC13) δ 9.99 (s, IH, O=C- H), 4.14 (s, 3H, CH3).
………………
ANALOGOUS METHOD TO PREPARE..2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester

USE 5-formyl-2- methoxy-benzoic acid methyl ester for 3,4- dimethoxybenzaldehyde, TO GET 2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester
Example 4
(3,4-Dimethoxy-benzyl)-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amine

A solution of 1-(4-phenyl-1 W-imidazol-2-yl)-ethylamine (0.061 g, 0.33 mmol) of Example 3, and 0.55 g (0.33 mmol) of 3,4-dimethoxybenzaldehyde in 5 ml_ of anhydrous methanol was stirred at room temperature for 1 h and then cooled to about 0-100C in an ice bath for 1 h. The reaction was treated carefully with 0.019 g (0.49 mmol) of sodium borohydride in one portion and maintained at about 0-100C for 21 h. Cold 2M aqueous HCI was added dropwise (30 drops), the mixture was stirred for 5 min, and then partially concentrated in vacuo unheated. The residual material was taken up in EtOAc to yield a suspension that was treated with 5 ml_ of cold 3M aqueous NaOH and stirred vigorously until clear. The phases were separated and the aqueous layer was extracted three times additional with EtOAc. The combined extracts were dried over MgSO4, filtered, and concentrated to yield (3,4-dimethoxy- benzyl)-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amine as a light yellow oil (HPLC: 87% @ 254nm and 66% @ 214 nm).
MS (ES+) (relative intensity): 338.1 (100) (M+1)
This sample was of sufficient quality to use in the next reaction without further purification.
…………………..
SYNTHESIS
In an embodiment, the present invention is directed to processes for the preparation of the compound of formula (IV)

also known as, 5-({[2-amino-3-(4-carbamoyl-2,5-dimethyl-phenyl)- propionyl]-[1 -(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy- benzoic acid
Example 1
(S)-2-ferf-Butoxycarbonylamino-3-(4-carbamoyl-2.6-dimethyl-phenyl)- propionic acid


STEP A: Trifluoromethanesulfonic acid 4-bromo-3,5-dimethyl-phenyl ester
To a cooled (0°C) solution of 4-bromo-3,5-dimethylphenol (3.05 g, 15.2 mmol) in pyridine (8 ml_) was added trifluoromethanesulfonic anhydride (5.0 g, 17.7 mmol) dropwise. After completion of addition, the resulting mixture was stirred at 0°C for 15 min, and then at room temperature overnight. The reaction was quenched by addition of water, and then extracted with EtOAc. The organic extracts were washed sequentially with water, 2N HCI (2x ), brine, and then dried over MgSO4. Filtration and evaporation to dryness yielded compound 1 b as a colorless oil.
1H NMR (300 MHz, CDCI3): δ 2.45 (6H, s), 7.00 (2H, s).
Step B: 4-Bromo-3,5-dimethylbenzoic acid
Into a solution of compound 1 b (6.57 g, 19.7 mmol) in DMF (65 ml_) were added K2CO3 (13.1 g, 94.7 mmol), Pd(OAc)2 (0.44 g, 1.97 mmol) and 1 ,1′-bis(diphenylphosphino)ferrocene (2.29 g, 4.14 mmol). The resulting mixture was bubbled in gaseous CO for 10 min and was heated to 60°C for 7.5h with a CO(9) balloon. The cooled mixture was partitioned between aqueous NaHCO3 and EtOAc, and filtered. The aqueous phase was separated, acidified with aqueous 6N HCI, extracted with EtOAc, and then dried over Na2SO4. Filtration and concentration of the filtrate yielded crude compound 1c as a brown residue, which was used in the next step without further purification. STEP C: Method A: 4-Bromo-3,5-dimethyl-benzamide
Into a suspension of compound 1c in DCM (40 ml_) was added SOCI2 (3.1 rnL, 42 mmol) and the mixture was heated at reflux for 2 h. Upon removal of the solvent by evaporation, the residue was dissolved in DCM (40 ml_) and then ammonium hydroxide (28% NH3 in water, 2.8 ml_) was added. The reaction mixture was heated at 5O0C for 2 h and concentrated. The residue was diluted with H2O, extracted with EtOAc, and the organic portion was dried over Na2SO4. After filtration and evaporation, the residue was purified by flash column chramotagraphy (eluent: EtOAc) to yield compound 1 d as an off-white solid.
1H NMR (300 MHz, CD3CN): δ 2.45 (6H, s), 5.94 (1 H, br s), 6.71 (1 H, br s), 7.57 (2H, s)
MS(ES+)(relative intensity): 228.0 (100%) (M+1).
Step C: Method B: 4-Bromo-3,5-dimethyl-benzamide
A mixture of compound 1 b (3.33 g, 10 mmol), PdCI2 (0.053 g, 0.3 mmol), hexamethyldisilazane (HMDS, 8.4 ml_, 40 mmol), and DPPP (0.12 g, 0.3 mmol) was bubbled with a gaseous CO for 5 min and then stirred in a CO balloon at 80°C for 4 h. To the reaction mixture was added MeOH (5 ml_). The reaction mixture was stirred for 10 min, diluted with 2N H2SO4 (200 ml_), and then extracted with EtOAc. The EtOAc extract was washed with saturated aqueous NaHCO3, brine, and then dried over Na2SO4. Filtration and evaporation of the resultant filtrate yielded a residue, which was purified by flash column chromatography (eluent: EtOAc) to yield compound 1d as a white solid.
Step D: 2-terf-Butoxycarbonylaminoacrylic acid methyl ester
To a suspension of /V-Boc-serine methyl ester (Compound 1e, 2.19 g, 10 mmol) and EDCI (2.01 g, 10.5 mmol) in DCM (70 ml_) was added CuCI (1.04 g, 10.5 mmol). The reaction mixture was stirred at room temperature for 72 h. Upon removal of the solvent, the residue was diluted with EtOAc, washed sequentially with water and brine and then dried over MgSO4. The crude product was purified by flash column chromatography (eluent: EtOAc:hexane ~1 :4) to yield compound 1f as a colorless oil.
1H NMR (300 MHz, CDCI3): δ 1.49 (9H, s), 3.83 (3H, s), 5.73 (1 H, d, J = 1.5 Hz), 6.16 (1 H1 S), 7.02 (1 H, s).
STEP E: (2)-2-fert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)acrylic acid methyl ester
A flask charged with compound 1d (0.46 g, 2.0 mmol), compound 1f (0.80 g, 4.0 mmol), tri-o-tolylphosphine (0.098 g, 0.32 mmol) and DMF (8 ml_) was purged with N2(g) 3 times. After the addition of tris(dibenzylideneacetone)dipalladium (0) (0.074 g, 0.08 mmol) and TEA (0.31 ml_, 2.2 mol), the reaction mixture was heated at 110°C for 24 h. At that time, the reaction was quenched by addition of water, and then extracted with EtOAc. The organic phase was washed with 1 N HCI, saturated aqueous NaHCO3, brine, and dried over MgSO4. The mixture was concentrated to a residue, which was purified by flash column chromatography (eluent: EtOAc:hexane~1 :1 to EtOAc only) to yield compound 1g as a white solid.
1H NMR (300 MHz, CD3OD): δ 1.36 (9H, s), 2.26 (6H, s), 3.83 (3H, s), 7.10 (1 H, s), 7.56 (2H, s); 13C NMR (75 MHz, DMSO-d6): δ 17.6, 25.7, 50.2, 78.7, 124.9, 126.4,
128.3, 131.2, 135.2, 135.5, 152.8, 164.3, 169.6;
MS (ES+) (relative intensity): 349.1 (38%)(M+1).
STEP F: (S)-2-ferf-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid methyl ester
Into a reactor charged with a solution of compound 1g (0.56 g, 1.6 mmol) in degassed MeOH (80 mL) was added [Rh(COd)(H1R-DIPAMP)J+BF4 “ under a stream of argon. The reactor was sealed and flushed with H2, stirred at 6O0C under 1000 psi of H2 for 14 days. The crude product was purified by flash column chromatography (eluent: EtOAc:hexane ~1 :1) to yield compound 1 h as a white solid. ee: >99%; 1H NMR (300 MHz, CDCI3): δ 1.36 (9H, s), 2.39 (6H, s), 3.11 (2H, J = 7.2 Hz), 3.65 (3H, s), 4.53-4.56 (1 H, m), 5.12 (1 H, d, J = 8.7 Hz), 5.65 (1 H, br s), 6.09 (1 H, br s), 7.46 (2H, s);
MS(ES+) (relative intensity): 250.9 (100) (M-BoC)+.
STEP G: (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid
Into an ice-cooled solution of compound “I h (0.22 g, 0.63 mmol) in THF (3.5 ml_) was added an aqueous LiOH solution (1 N, 3.5 ml_) and the reaction mixture stirred at 0°C. Upon completion of the reaction, the reaction mixture was concentrated and the aqueous phase was neutralized with cooled aqueous 1 N HCI at 0°C, and then extracted with EtOAc. The combined extracts were dried over Na2SO4 overnight. Filtration and evaporation of the filtrate to dryness yielded compound 1j as a white solid. 1H NMR (300 MHz, DMSO-cfe): δ 1.30 (9H, s), 2.32 (6H, s), 2.95(1 H, dd,
J= 8.8, 13.9 Hz), 3.10 (1 H, dd, J= 6.2, 14.0 Hz), 4.02-4.12 (1 H, m), 7.18-7.23 (2H, m), 7.48 (2H1 s), 7.80 (1 H, s);
MS(ES+) (relative intensity): 236.9 (6) (M-BoC)+.
Example 5
5-((r2-Amino-3-(4-carbamoyl-2.6-dimethyl-phenyl)-propionvn-n-(4-phenyl- 1 H-imidazol-2-yl)-ethvπ-aminol-methyl)-2-methoxy-benzoic acid


STEP A. 2-Methoxy-5-{[1-(4-phenyl-1 W-imidazol-2-yl)-ethylamino]-methyl}- benzoic acid methyl ester
Using the procedures described for Example 4, substituting 5-formyl-2- methoxy-benzoic acid methyl ester (WO 02/22612) for 3,4- dimethoxybenzaldehyde, 2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester was prepared.
STEP B. 5-({[2-ferf-ButoxycarbonylmethyI-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl]-[1 -(4-phenyl-1 H-imidazoI-2-yl)-ethyl]-amino}-methyl)-2- methoxy-benzoic acid methyl ester
Using the procedure of Example 3 for the conversion of Cpd 3d to Cpd 3e, substituting 2-methoxy-5-{[1-(4-phenyl-1 /-/-imidazol-2-yl)-ethylamino]- methylj-benzoic acid methyl ester for Cpd 3d and substituting 2-tert- Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionic acid for 2- tø/t-Butoxycarbonylamino-3-(4-hydroxy-2,6-dimethyl-phenyl)-propionic acid, Cpd 5a was prepared.
STEP C. 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl]-[1 -(4-phenyl-1 W-imidazol-2-yl)-ethyl]-amino}-methyl)-2- methoxy-benzoic acid
5-({[2-tørf-Butoxycarbonylmethyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)- propionyl]-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy- benzoic acid methyl ester was dissolved in an ice-chilled (0-10°C), mixed solvent system of THF (10 ml_) and MeOH (5 ml_). A LiOH H2O/water suspension (2.48 M; 3.77 ml_) was added dropwise, then the reaction was allowed to warm to room temperature and stirred overnight. The resulting mixture was cooled in an ice bath and the basic solution was neutralized with 2N citric acid until slightly acidic. The mixture was concentrated under reduced pressure to remove the volatile materials, after which time the remaining aqueous phase was extracted with EtOAc (3 x 26 ml_). These combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure to yield a pale yellowish white solid. This crude material was dissolved in a 10% MeOH/CH2CI2 solution and adsorbed onto 30 g of silica. The adsorbed material was divided and chromatographed on an ISCO normal phase column over two runs, using a 40 g Redi-Sep column for both runs. The solvent system was a gradient MeOHZCH2CI2 system as follows: Initial 100% CH2CI2, 98%-92% over 40 min; 90% over 12 min, and then 88% over 13 min. The desired product eluted cleanly between 44-61 min. The desired fractions were combined and concentrated under reduced pressure to yield 5-({[2-terf- butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4- phenyl-1 /-/-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid, Cpd 5b, as a white solid.
STEP D. 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1 – (4-phenyl-1 W-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A portion of Cpd 5b (0.27g, 0.41 mmol) was dissolved in EtOAc (39 ml_)/THF (5 ml_), filtered, and subsequently treated with gaseous HCI for 15 min. After completion of the HCI addition, the reaction was slowly warmed to room temperature and a solid precipitate formed. After 5 h the reaction appeared >97% complete by LC (@214nm; 2.56 min.). The stirring was continued over 3 d, then the solid was collected and rinsed with a small amount of EtOAc. The resulting solid was dried under high vacuum under refluxing toluene for 2.5 h to yield Cpd 5c as a white solid di-HCI salt.
Example 2
Racemic 2-terf-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethvl- phenvD-propionic acid

STEP A: Racemic 2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)propionic acid methyl ester
To a reactor charged with a solution of compound 1g (0.68 g, 1.95 mmol) in MeOH (80 mL) was added 10% Pd-C (0.5 g). The reactor was connected to a hydrogenator and shaken under 51 psi of H2 overnight. The mixture was filtered through a pad of Celite and the filtrate was concentrated to dryness to yield compound 2a as a white solid.
The 1H NMR spectrum was identical to that of (S)-2-tert- butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)propionic acid methyl ester, compound 1 h.
STEP B: Racemic 2-terf-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)propionic acid
Following the procedure described for Example 1 , STEP G (preparation of (S)-2-teAt-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid), compound 2b – racemic 2-te/?-butoxycarbonylamino-3- (4-carbamoyl-2,6-dimethyl-phenyl)propionic acid – was prepared.
…………….
POLYMORPHS
Example 1 Preparation of the zwitterion of 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A 1 L three-necked round-bottomed flask equipped with a mechanical stirrer, addition funnel and a thermocouple was charged without agitation. 34.2 g of 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid (see Example 9 of US 2005/0203143), 340 ml of acetone, and 17 ml of 204 mmolar concentrated HCl were combined in the flask. The stirring was started and the resulting slurry formed a clear solution. This solution was heated to 45° C. under vigorous stirring and aged at this temperature for a period of two hours. After the completion, the reaction mass was cooled to ambient temperature and the supernatant was removed by suction. The vessel along with the residue was rinsed with 20 ml of acetone and then removed as previously. 170 ml of water was added and the reaction mass and was aged under stirring until a homogeneus solution resulted. This solution was then added over a period of ˜½ hr to a solution of 90 ml of 1N NaOH and water. The pH was adjusted to 6.5-7.0 accordingly. The resulting slurry was aged for about 2 hrs at ambient temperature, cooled to 10-15° C., aged at that temperature for about 1 hr, and then filtered. The solid was washed with 10 ml water, air-dried for a period of 4 to 5 hrs, and then placed in a vacuum oven at 50-55° C. until the water content was less than 3%.
Example 2 Preparation of the Form α Crystal
The Form α crystal can be prepared by storing the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid at 0-25% relative humidity for 3 days. Representative PXRD, TGA, and DSC data are shown in FIGS. 1-3 respectively.
Example 3 Preparation of the Form β crystal
The Form β crystal can be prepared by storing the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid at greater than 60% relative humidity for 3 days. Representative PXRD, TGA, and DSC data are shown in FIGS. 1, 4, and 5 respectively.
…………….
SYNTHESIS
Example 9 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid

A. 2-Methoxy-5{[1-(4-phenyl-1 H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester.
Using the procedures described for Example 3, substituting 5-formyl-2-methoxy-benzoic acid methyl ester (WO 02/22612) for 3,4-dimethoxybenzaldehyde, 2-methoxy-5-{[1-(4-phenyl-1H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester was prepared.
B. 5-({[2-tert-Butoxycarbonyl methyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid methyl ester.
Using the procedure of Example 1 for the conversion of Cpd 1d to Cpd 1e, substituting 2-methoxy-5-{[1-(4-phenyl-1H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester for Cpd 1 d and substituting 2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl-propionic acid of Example 8 for 2-tert-Butoxycarbonylamino-3-(4-hydroxy-2,6-dimethyl-phenyl)-propionic acid, Cpd 9a was prepared.
C. 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[11-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
5-({[2-tert-Butoxycarbonyl methyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid methyl ester was dissolved in an ice-chilled (0-10° C.), mixed solvent system of THF (10 mL) and MeOH (5 mL). A LiOH.H2O/water suspension (2.48 M; 3.77 mL) was added dropwise, then the reaction was allowed to warm to room temperature and stirred overnight. The resulting mixture was cooled in an ice bath and the basic solution was neutralized with 2N citric acid until slightly acidic. The mixture was concentrated under reduced pressure to remove the volatile materials, after which time the remaining aqueous phase was extracted with EtOAc (3×26 mL). These combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure to give 2.26 g (146% of theory) of pale yellowish white solid. This crude material was dissolved in a 10% MeOH/CH2Cl2 solution and adsorbed onto 30 g of silica. The adsorbed material was divided and chromatographed on an ISCO normal phase column over two runs, using a 40 g Redi-Sep column for both runs. The solvent system was a gradient MeOH/CH2Cl2 system as follows: Initial 100% CH2Cl2, 98%-92% over 40 min; 90% over 12 min, and then 88% over 13 min. The desired product eluted cleanly between 44-61 min. The desired fractions were combined and concentrated under reduced pressure to yield 1.74 g (113% of theory) of 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid, Cpd 9b, as a white solid.
D. 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
A portion of Cpd 9b (0.27g, 0.41 mmol) was dissolved in EtOAc (39 mL)/THF (5 mL), filtered, and subsequently treated with gaseous HCl for 15 min. After completion of the HCl addition, the reaction was slowly warmed to room temperature and a solid precipitate formed. After 5 h the reaction appeared >97% complete by LC (@214 nm; 2.56 min.). The stirring was continued over 3 d, then the solid was collected and rinsed with a small amount of EtOAc. The resulting solid was dried under high vacuum under refluxing toluene for 2.5 h to yield 0.19 g (71%) of desired Cpd 9c as a white solid di-HCl salt.
Example 8 (S)-2-tert-Butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester

A. (S)-2-tert-Butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester. Into a cool solution of Boc-L-(2,6-diMe)Tyr-OMe (7.0 g, 21.6 mmol; Sources: Chiramer or RSP AminoAcidAnalogues) and N-phenyltrifluoromethanesulfonimide (7.9 g, 22.0 mmol) in dichloromethane (60 mL) was added triethylamine (3.25 mL, 23.3 mmol). The resulting solution was stirred at 0° C. for 1 h and slowly warmed to rt. Upon completion, the reaction was quenched by addition of water. The separated organic phase was washed with 1 N NaOH aqueous solution, water and dried over Na2SO4 overnight. After filtration and evaporation, the residue was purified by flash column chromatography (eluent: EtOAc-hexane: 3:7) to give the desired product (9.74 g, 99%) as a clear oil; 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.39 (6H, s), 3.06 (2H, d, J=7.7 Hz), 3.64 (3H, s), 4.51-4.59 (1H, m), 5.12 (1H, d, J=8.5 Hz), 6.92 (2H, s); MS (ES+) (relative intensity): 355.8 (100) (M−Boc)+.
B. (S)4-(2-tert-Butoxycarbonylamino-2-methoxycarbonylethyl)-3,5-dimethylbenzoic acid. To a suspension of (S)-2-tert-butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester (9.68 g, 21.3 mmol), K2CO3 (14.1 g, 0.102 mol), Pd(OAc)2 (0.48 g, 2.13 mmol) and 1,1′-bis(diphenylphosphino)ferrocene (2.56 g, 4.47 mmol) in DMF (48 mL) was bubbled in gaseous CO for 15 min. The mixture was heated to 60° C. for 8 h with a CO balloon. The cool mixture was partitioned between NaHCO3 and EtOAc, and filtered. The aqueous layer was separated, acidified with 10% citric acid aqueous solution, extracted with EtOAc, and finally dried over Na2SO4. Filtration and concentration of the filtrate resulted in a residue. The residue was recrystallized from EtOAc-hexanes to afford the desired product (7.05 g, 94%); 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.42 (6H, s), 3.14 (2H, J=7.4 Hz), 3.65 (3H, s), 4.57-4.59 (1H, m), 5.14 (1H, d, J=8.6 Hz), 7.75 (2H, s); MS(ES+) (relative intensity): 251.9 (100) (M−Boc)+.
C. (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid methyl ester. Into a stirring solution of (S)-4-(2-tert-butoxycarbonylamino-2-methoxycarbonylethyl)-3,5-dimethyl benzoic acid (3.00 g, 8.54 mmol), PyBOP (6.68 g, 12.8 mmol) and HOBt (1.74 g, 12.8 mmol) in DMF (36 mL) was added DIPEA (5.96 mL, 34.2 mmol) and NH4Cl (0.92 g, 17.1 mmol). The resulting mixture was stirred at rt for 40 min before being partitioned between aqueous NH4Cl solution and EtOAc. The separated organic phase was washed sequentially with 2N citric acid aqueous solution, saturated aqueous NaHCO3 solution, and brine, then dried over Na2SO4 overnight. After filtration and concentration, the residue was purified by flash column chromatography (eluent: EtOAc) to give the product. (3.00 g, 100%); 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.39 (6H, s), 3.11 (2H, J=7.2 Hz), 3.65 (3H, s), 4.53-4.56 (1H, m), 5.12 (1H, d, J=8.7 Hz), 5.65 (1H, brs), 6.09 (1H, br s), 7.46 (2H, s); MS(ES+) (relative intensity): 250.9 (100) (M−Boc)+.
D. (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid. Into an ice-cooled solution of methyl ester from Step C (2.99 g, 8.54 mmol) in THF (50 mL) was added an aqueous LiOH solution (1N, 50 mL) and stirred at 0° C. Upon consumption of the starting materials, the organic solvents were removed and the aqueous phase was neutralized with cooled 1N HCl at 0° C., and extracted with EtOAc, and dried over Na2SO4 overnight. Filtration and evaporation to dryness led to the title acid (S)-2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid (2.51 g, 87%); 1H NMR (300 MHz, DMSO-d6): δ 1.30 (9H, s), 2.32 (6H, s), 2.95 (1H, dd, J=8.8, 13.9 Hz), 3.10 (1H, dd, J=6.2, 14.0 Hz), 4.02-4.12 (1H, m), 7.18-7.23 (2H, m), 7.48 (2H, s), 7.80 (1H, s); MS(ES+) (relative intensity): 236.9 (6) (M−Boc)+.
…………………..
PATENTS
1.WO 2005090315
2..WO 2006099060
3.WO 2009009480
4. WO 2010062590
5.US 2011263868 *
|
12-24-2010
|
NOVEL COMPOUNDS AS OPIOID RECEPTOR MODULATORS
|
|
|
8-32-2010
|
Compounds as opioid receptor modulators
|
|
|
6-23-2010
|
Compounds as opioid receptor modulators
|
|
|
2-12-2010
|
PROCESS FOR THE PREPARATION OF OPIOD MODULATORS
|
|
|
12-9-2009
|
Process for the preparation of opioid modulators
|
| US7629488 * | Mar 6, 2006 | Dec 8, 2009 | Janssen Pharmaceutica N.V. | Process for the preparation of opioid modulators |
| US7741356 * | Mar 14, 2005 | Jun 22, 2010 | Janssen Pharmaceutica N.V. | Compounds as opioid receptor modulators |
| US7786158 * | Oct 24, 2007 | Aug 31, 2010 | Janssen Pharmaceutica N.V. | Compounds as opioid receptor modulators |
| US7994206 | Jul 7, 2008 | Aug 9, 2011 | Janssen Pharmaceutica, N.V. | Crystals and process of making 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid |
| CN1950342A | Mar 14, 2005 | Apr 18, 2007 | 詹森药业有限公司 | Novel compounds as opioid receptor modulators |
Update july 2015
Eluxadoline
Trade Name: Viberzi®
Research Code: JNJ-27018966, JNJ27018966, JNJ 27018966
Chemical Name: 5 – [[[(2S) -2-amino-3- [4- (aminocarbonyl) -2,6-dimethylphenyl ] -1- oxopropyl] [(1S) -1- (4-phenyl-1H-imidazol-2-yl) ethyl] amino] methyl] -2-methoxybenzoic acid
CAS No: 864821-90-9
MOA: mu opioid receptor agonist
Indication: Irritable bowel syndrome with diarrhea (IBS-D)
Approval Date: May 27, 2015 (US)
Originator: Furiex Pharmaceuticals Inc ( Furiex acquired Eluxadoline from Janssen in 2011 )
Developer: Forest Laboratories Inc. (acquired by Actavis PLC in 2014 )


{kind=link}