
Home » Phase2 drugs (Page 24)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BELINOSTAT, FAST TRACK, ORPHAN DRUG, A hydroxamate-type inhibitor of histone deacetylase.

Belinostat (PXD101)
PHASE 2, FAST TRACK FDA , ORPHAN STATUS
- PDX101
- PX 105684
- PXD-101
- PXD101
- UNII-F4H96P17NZ
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
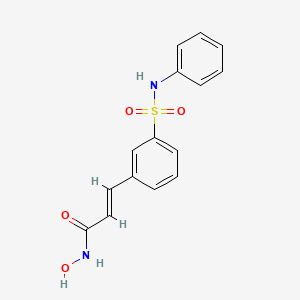
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
A hydroxamate-type inhibitor of histone deacetylase.
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
………………………………………………………………………..
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


…………………………………..
GENERAL SYNTHESIS
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A

Scheme 3B

Scheme 4


Scheme 8

Scheme 9

……………………………………………………………………..
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)

3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)

To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
……………………………………………………………………….
SYNTHESIS
US20100286279

…………………………………………………….
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,
J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1,
140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
………………………………………………..
SYNTHESIS
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
……………….
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.
………………………..

- Plumb, Jane A.; Finn, Paul W.; Williams, Robert J.; Bandara, Morwenna J.; Romero, M. Rosario; Watkins, Claire J.; La Thangue, Nicholas B.; Brown, Robert (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101”. Molecular Cancer Therapeutics 2 (8): 721–728. PMID 12939461.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
- Helvetica Chimica Acta, 2005 , vol. 88, 7 PG. 1630 – 1657, MP 172
- WO2009/40517 A2, ….
- WO2006/120456 A1, …..
- Synthetic Communications, 2010 , vol. 40, 17 PG. 2520 – 2524, MP 172
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 PG. 4694 – 4720, NMR IN SUP INFO
| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
………………………..
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
……………………………..


CLAZOSENTAN

Clazosentan
READ ALL AT
http://www.allfordrugs.com/2014/01/22/clazosentan/

READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
TEGOBUVIR ..IN PHASE II FOR HEPATITIS C

TEGOBUVIR
A non-structural protein 5B polymerase inhibitor
for Treatment of chronic hepatitis C
5-[6-[2,4-Bis(trifluoromethyl)phenyl]pyridazin-3-ylmethyl]-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridine
CHEMICAL NAMES
1. 5H-Imidazo[4,5-c]pyridine, 5-[[6-[2,4-bis(trifluoromethyl)phenyl]-3-pyridazinyl]methyl]-
2-(2-fluorophenyl)-
2. 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-
imidazo[4,5-c]pyridine
MOLECULAR FORMULA C25H14F7N5
MOLECULAR WEIGHT 517.4
MANUFACTURER Gilead Sciences, Inc.
CODE DESIGNATION
- GS 333126
- GS 9190
- GS-333126
- GS-9190
- Tegobuvir
- UNII-5NOK5X389M
CAS REGISTRY NUMBER 1000787-75-6
GS-9190, an RNA-directed RNA polymerase (NS5B) inhibitor, is in phase II clinical evaluation at Gilead for the treatment of hepatitis C virus (HCV) infection. A clinical trial with GS-9190 in combination with peginterferon alfa-2a and ribavirin and with GS-9451 or with GS-9256 in treatment-naive subjects with chronic genotype 1 HCV infection was discontinued due to serious adverse events.
Gilead (Originator)
Katholieke Universiteit Leuven (Originator)
……………………………………….
tegobuvir
PATENTS
WO 2005063744
WO 2008005519
WO 2009009001
WO 2010151488
WO 2010151487
WO 2010151472
WO 2011072370
WO 2011156757
WO 2012087596
WO 2013101550
Hebner CM, Han B, Brendza KM, Nash M, Sulfab M, Tian Y, Hung M, Fung W, Vivian RW, Trenkle J, Taylor J, Bjornson K, Bondy S, Liu X, Link J, Neyts J, Sakowicz R, Zhong W, Tang H, Schmitz U.
PLoS One. 2012;7(6):e39163. doi: 10.1371/journal.pone.0039163. Epub 2012 Jun 13.
Wong KA, Xu S, Martin R, Miller MD, Mo H.
Virology. 2012 Jul 20;429(1):57-62. doi: 10.1016/j.virol.2012.03.025. Epub 2012 Apr 28.
Zeuzem S, Buggisch P, Agarwal K, Marcellin P, Sereni D, Klinker H, Moreno C, Zarski JP, Horsmans Y, Mo H, Arterburn S, Knox S, Oldach D, McHutchison JG, Manns MP, Foster GR.
Hepatology. 2012 Mar;55(3):749-58. doi: 10.1002/hep.24744.
Shih IH, Vliegen I, Peng B, Yang H, Hebner C, Paeshuyse J, Pürstinger G, Fenaux M, Tian Y, Mabery E, Qi X, Bahador G, Paulson M, Lehman LS, Bondy S, Tse W, Reiser H, Lee WA, Schmitz U, Neyts J, Zhong W.
Antimicrob Agents Chemother. 2011 Sep;55(9):4196-203. doi: 10.1128/AAC.00307-11. Epub 2011 Jul 11.
- ……………………..
- http://www.google.com/patents/WO2013040492A2
- ompound 1 can be prepared using synthetic methods and intermediates like those described in US 7,754,720. Compound 1 can also be prepared as described in the following Example.
- Compound 1 is:

Compound 1 may also be referred to as 5-((6-(2,4-bis(trifluoromethyl)phenyl)pyridazin-3-yl)methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridine, 5-[[6-[2,4-bis (trifluoromethyl)phenyl]pyridazin=3-yl]methyl]-2-(2-fluorophenyl).
- Example 1 : 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H- imidazo[4,5-c]pyridi


Compound 103 was dissolved in dimethoxyethane (DME). To this solution was added 2,4-bis(trifluromethyl)phenylboronic acid 105 and a 2N aq. Na2C03 solution. To the resulting biphasic mixture was added Pd(PPh3)4 and the reaction was then heated at 80°C for 72 hrs. The reaction was cooled to room temperature and filtered through Celite and the Celite washed with EtOAc. The filtrate was concentrated in vacuo. The residue was purified on 6g Si02 using MeOH/CH2CI2 to elute compound. The compound thus obtained was contaminated with PPh3(0). The product was repurified on a 1 mm Chromatotron plate with 0 to 5%
MeOH/CH2CI2 in 1 % steps. The pure fractions were combined and concentrated in vacuo, then dried on high vacuum for 12 hrs. 11.8 mg of the free base of compound 1 was obtained with no PPh3 contamination. 1H NMR (300MHz,CD3OD) δ 6.20 (s, 2), 7.32 (m, 3), 7.52 (m, 1 ), 7.78 (d, 1), 7.89 (d, 1), 7.95 (s, 2), 8.15 (m, 3), 8.35 (d, 1), 9.12 (s, 1); LC/MS M+H = 518.
The intermediate compound 104 was prepared as follows, a. Preparation of Compound 10

101 102

To a solution of the commercially available starting material 101 in CHCI3, trichloroisocyanuric acid (TCCA) was added at 60°C. Then the solution was stirred for 1.5 hrs, cooled, and filtered with HiFlo-Celite. The filtrate was concentrated and dried with vacuum. The yield was 5.037 g of compound 102. b. Preparation of Compound 104.

102 104

To a solution of compound 103 in DMF (dimethylformamide), NaOH was added.
Compound 102 was dissolved in DMF (20 mL) and added to the solution slowly. The reaction was stirred for 3 hrs, was diluted with water and extracted with EtOAc. The organic layer was dried with Na2S0 . The solvent was removed and the product recrystallized with
dichloromethane. The yield was 5.7 g of compound 103.
- ……………………………
- US7754720
- Example 1a Synthesis of 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridineIn this method, dimethoxyethane or its related solvents, all having the general formula R1OR2O(R4O)aR3 wherein each of R1, R2, R3 and R4 are independently selected from C1-C6 alkyl and a is 0 or 1, have been found to be particularly advantageous over the conventional solvent DMF. Typically, each of R1, R2, R3 and R4 are independently C1-C2 alkyl and usually a is 0. C1-C6 alkyl includes fully saturated primary, secondary or tertiary hydrocarbon groups with 1 to 6 carbon atoms and thereby includes, but is not limited to methyl, ethyl, propyl, butyl, etc.Step 1



Compound MW Amount mmoles Equivalents SM 128.56 5 g 38.9 1 TCCA 232.41 3.62 g 15.6 0.4 CHCl3 130 ml To a solution of the commercially available starting material (SM) in CHCl3, trichloroisocyanuric acid (TCCA) was added at 60° C. Then the solution was stirred for 1.5 hrs., cooled down and filtered with HiFlo-Celite. The filtrate was concentrated and dried with vacuum. The yield was 5.037 g.
Step 2



Compound MW Amount mmoles Equivalents S.M. 163 5.073 g 31.12 1 Core 213.2 6.635 g 31.12 1 NaOH (10%) 40 1.245 g 31.12 1 DMF 320 ml To a solution of core (obtained as described in literature in DMF (dimethylformamide), NaOH was added. Then SM for this step (obtained from step 1) was dissolved in DMF (20 ml) and added to the solution slowly. The reaction was stirred for 3 hrs, was diluted with water and extracted with EtOAc. The organic layer was dried with Na2SO4. The solvent was removed and the product recrystallized with DCM (dichloromethane). The yield was 5.7 g.
Step 3



Compound MW Amount Moles Equivalents A 453.79 95 mg 0.209 1 DME 500 ul 2 N aq. Na2CO3 313ul 0.626 3 2,4-bisCF3– 257.93 80.9 mg 0.313 1.5 phenylboronic acid Pd(PPh3)4 1155 12 mg 0.0104 0.05 Compound A was dissolved in dimethoxyethane (DME). To this solution was added 2,4-bis(trifluromethyl)phenylboronic acid and a 2N aq. Na2CO3 solution. To the resulting biphasic mixture was added Pd(PPh3)4 and the reaction was then heated at 80° C. for 72 hrs. The reaction was cooled to room temperature and filtered through Celite and the Celite washed with EtOAc. The filtrate was concentrated in vacuo. The residue was purified on 6 g SiO2 using MeOH/CH2Cl2 to elute compound. The compound thus obtained was contaminated with PPh3(O). The product was repurified on a 1 mm Chromatotron plate with 0 to 5% MeOH/CH2Cl2 in 1% steps. The pure fractions were combined and concentrated in vacuo, then dried on high vacuum for 12 hrs. 11.8 mg of the free base of compound (1) was obtained with no PPh3 contamination.
1H NMR (300 MHz, CD3OD)
6.20 (s, 2)
7.32 (m, 3)
7.52 (m, 1)
7.78 (d, 1)
7.89 (d, 1)
7.95 (s, 2)
8.15 (m, 3)
8.35 (d, 1)
9.12 (s, 1)
LC/MS M+H=518
Example 1b Synthesis of 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridineThis example is directed to an additional method for making compound (1), employing the following schemes.

Methanesulfonic acid was added to 2-fluorobenzoic acid in a reactor with active cooling keeping T≦50° C. 3,4-Diaminopyridine was then added portionwise to this cooled slurry, keeping T≦35° C. The contents of the reactor were then heated to 50° C. Phosphorus pentoxide was added in a single charge. The reaction was then heated at 90-110° C. for at least 3 hours. The reaction was sampled for completion by HPLC analysis. The reaction was cooled to ambient temperature and water was added portionwise slowly to quench the reaction. The reaction was then diluted with water. In solubles were removed by filtration. The pH of the filtrate was adjusted to 5.5-5.8 with ammonium hydroxide. The reaction was allowed to self-seed and granulate for ˜4 hours at ambient temperature. The pH was then adjusted to 8.0-9.3 with ammonium hydroxide. The slurry was held at ambient temperature for at least 2 hours. The solids were isolated by filtration and washed with water, followed by IPE. The wet cake was dried in vacuo at not more than 60° C. until ≦1% water remains. The dry product is core (2).
Summary of Materials M.W. Wt. Ratio Mole ratio 3,4-Diaminopyridine 109.13 1.0 1.0 2-Fluorobenzoic acid 140.11 1.4 1.1 Methanesulfonic acid 96.1 7.0 8.0 Phosphorus pentoxide 141.94 1.3 1.0 Water 18.02 40 — Isopropyl ether 102.17 5.0 — Ammonium hydroxide 35.09 ~10 — 
A solution of compound (2a) in 1,2-dichloroethane was heated to 40-45° C. Trichloroisocyanuric acid was added and the mixture was heated at 60-70° C. for at least 2 hours. The reaction was sampled for completion by HPLC analysis. The reaction was cooled to ambient temperature. Celite was added to absorb insolubles, then solids were removed by filtration. The filtrate was washed with 0.5 N sodium hydroxide solution. The organic layer was concentrated to lowest stirrable volume and displaced with DMF. Core (2) and 10% aqueous sodium hydroxide solution were added. The reaction was stirred at ambient temperature for at least 8 hours. The reaction was sampled for completion by HPLC analysis. An additional 10% charge of 10% sodium hydroxide solution was added to the reaction. The reaction was then charged into water to isolate the crude product. After granulating for at least 1 hour, the solids were isolated and washed with water and isopropyl ether. Ethyl acetate was added and refluxed (internal T=70-77° C.) for 1-5 hours to dissolve product, then cooled to 18-23° C. slowly over 4-8 hours. The reactor contents were agitated at 18-23° C. for 8-20 hours and solids collected by filtration and rinsed with ethyl acetate. Low melt (i.e., DSC about 220 degrees C.) amorphous compound (1) was discharged. Amorphous compound (1) was dissolved in ethyl acetate by heating at reflux (internal T=70-77° C.) for 1-5 hours. Water content is controlled to about 0.2% by azeotropically removing water (with ethyl acetate the upper limit on water content is about 0.6% by weight; at about 0.9% by weight water the amorphous material will reprecipitate and crystals will not be obtained). The reactor contents are cooled slowly to 18-23° C. over 4-8 hours, then agitated at 18-23° C. for 8-20 hours and solids collected by filtration. The solids were rinsed with ethyl acetate and dried in vacuo at not more than 60° C. to obtain the dry crystalline compound (1).
Summary of Materials M.W. Wt. Ratio Mole ratio 3-chloro-6-methylpyridazine 128.56 1.0 1.0 2,4bis(trifluromethyl)phenylboronic 257.93 4.0 2.0 acid X-Phos 476.72 0.18 0.05 Palladium acetate 224.49 0.04 0.025 1,2-Dimethoxyethane 90.12 16.7 — Potassium carbonate 138.21 2.15 2.0 Water 18.02 7.8 — Copper iodide 190.45 0.037 0.025 Celite — 0.25 — Heptane 100.2 22.4 — Nuclear Magnetic Resonance (1H-, 13C-, and 19F-NMR) SpectraNuclear magnetic resonance (NMR) spectra of compound (1) is consistent with the proposed structure. The 13C, 19F, and 1H-NMR spectra of compound (1) in DMSO-d6 were measured using a Varian UnityInova-400 FT-NMR spectrometer. Spectra are shown in the table below. The NMR chemical shift assignments were established using 2D correlation experiments (COSY, HSQC, HMBC and HSQCTOCSY).
1H- and 13C-NMR Chemical Shift Assignments for Compound (1) Reference Standard
Atom δC/ppm (DMSO-d6) δF/ppm (DMSO-d6) δH/ppm (DMSO-d6) 1A 140.16 2A 128.32 (qa, JCF = 32 Hz) 3A 123.61, m 8.24 (m, 1 H) 4A 130.27 (q, JCF = 34 Hz) 5A 129.54 (q, JCF = 3 Hz) 8.22 (m, 1 H) 6A 133.36 7.88 (m, 1 H) 7A 123.20 (q, JCF = 273 Hz) −56.4b 8A 123.02 (q, JCF = 275 Hz) −62.0b 1B 158.76 2B 128.16 8.01 (d, 1 H, J = 8.4 Hz) 3B 126.20 7.95 (d, 1 H, J = 8.8 Hz) 4B 157.70 5B 60.49 6.17 (s, 2 H) 2C 131.86 8.31 (m, 1 H) 3C 112.63 7.86 (m, 1 H) 4C 155.44 6C 168.11 (d, JCF = 6 Hz) 8C 145.08 9C 133.06 9.25 (s, 1 H) 1D 123.11 (d, JCF = 10 Hz) 2D 160.46 (d, JCF = 254 Hz) −111.7 3D 116.59 (d, JCF = 22 Hz) 7.29 (m, 1 H) 4D 130.84 (d, JCF = 8 Hz) 7.46 (m, 1 H) 5D 124.13 (d, JCF = 4 Hz) 7.31 (m, 1 H) 6D 131.72 (d, JCF = 2 Hz) 8.35 (m, 1 H) amultiplicity, s: singlet, d: doublet, q: quartet, m: multiplet binterchangeable signals 
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html







DANOPREVIR (ITMN-191) …..a peptidomimetic inhibitor of the NS3/4A protease of hepatitis C virus (HCV)

Danoprevir
Danoprevir(ITMN-191) is a peptidomimetic inhibitor of the NS3/4A protease of hepatitis C virus (HCV) with IC50 of 0.2-3.5 nM, inhibition effect for HCV genotypes 1A/1B/4/5/6 is ~10-fold higher than 2B/3A. Phase 2.
Array BioPharma (Originator)
| RG7227 |
| ITMN-191 |
| RO5190591 |
2H-Isoindole-2-carboxylic acid, 4-fluoro-1,3-dihydro-, (2R,6S,12Z,13aS,14aR,16aS)-
14a-[[(cyclopropylsulfonyl)amino]carbonyl]-6-[[(1,1-dimethylethoxy)carbonyl]amino]-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydro-5,16-
dioxocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl ester
2. (2R,6S,12Z,13aS,14aR,16aS)-14a-[(cyclopropylsulfonyl)carbamoyl]-6-{[(1,1-
dimethylethoxy)carbonyl]amino}-5,16-dioxo-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-
a][1,4]diazacyclopentadecin-2-yl 4-fluoro-1,3-dihydro-2H-isoindole-2-carboxylate
Treatment of hepatitis C
MOLECULAR FORMULA C35H46FN5O9S
MOLECULAR WEIGHT 731.8
MANUFACTURER Genentech
CODE DESIGNATION R05190591
CAS REGISTRY NUMBER 850876-88-9, 916881-67-9
Danoprevir(ITMN-191) is a peptidomimetic
ITMN-191 (R-7227), a macrocyclic protease inhibitor, is in phase II clinical evaluation for the treatment of chronic hepatitis C virus (HCV) infection as monotherapy and in combination with Pegasys(R) (pegylated interferon alpha-2a) and Copegus(R) (ribavirin). The product candidate is also being evaluated in combination with R-7128 in treatment-naive patients infected with HCV genotype 1.
Danoprevir (ITMN-191; RG-7227), under development by InterMune Inc and Roche Holding AG, is a promising, potent NS3/4A protease inhibitor for the oral treatment of HCV infection. Preclinical data demonstrated that danoprevir binds with high affinity and dissociates slowly from the HCV NS3 protease, allowing high liver drug exposure with only modest plasma drug exposure.
In 2006, originator InterMune and licensee Roche entered into an exclusive worldwide collaboration agreement to develop and commercialize products from InterMune’s hepatitis C (HCV) protease inhibitor program, including ITMN-191. In 2010, the licensing agreement was terminated. Also in 2010, Roche acquired worldwide development and commercialization rights to R-7227 from InterMune. Preclinical pharmacokinetic results support the exploration of twice-daily oral dosing in HCV.
A phase Ib, ‘IFN-free’ clinical trial demonstrated that danoprevir, combined with the HCV polymerase inhibitor RG-7128 (Pharmasset Inc/Roche Holding AG), was effective in reducing HCV-RNA levels in a large proportion of treatment-naïve patients with HCV infection and in approximately half of previously non-responsive patients with HCV-1 infection, without resistance or safety concerns. In a phase IIb trial in treatment-naïve patients with HCV-1 infection, danoprevir plus pegylated IFNalpha2a and ribavirin resulted in undetectable levels of HCV-RNA in the majority of patients, without any evidence of viral resistance; however, the high-dose danoprevir arm was prematurely terminated because of grade 4 ALT elevations. Phase I trials have also demonstrated that ritonavir boosting improved the pharmacokinetic profile of danoprevir; therefore, at the time of publication, a phase IIb trial to evaluate ritonavir-boosted, low-dose danoprevir in combination with RG-7128 was planned. (source:
inhibitor of the NS3/4A protease of hepatitis C virus (HCV) with IC50 of 0.2-3.5 nM, inhibition effect for HCV genotypes 1A/1B/4/5/6 is ~10-fold higher than 2B/3A. Phase 2.
SODIUM SALT
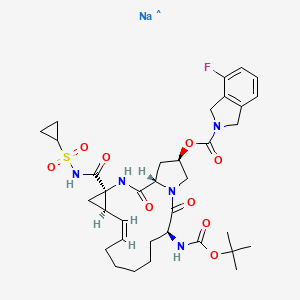
HERAPEUTIC CLAIM Treatment of hepatitis C
CHEMICAL NAMES
1. 2H-Isoindole-2-carboxylic acid, 4-fluoro-1,3-dihydro-, (2R,6S,12Z,13aS,14aR,16aS)-
14a-[[(cyclopropylsulfonyl)amino]carbonyl]-6-[[(1,1-dimethylethoxy)carbonyl]amino]-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydro-5,16-dioxocyclopropa
[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl ester, sodium salt (1:1)
2. sodium (cyclopropylsulfonyl){[(2R,6S,12Z,13aS,14aR,16aS)-6-{[(1,1-dimethylethoxy)
carbonyl]amino}-2-{[(4-fluoro-1,3-dihydro-2H-isoindol-2-yl)carbonyl]oxy}-5,16-dioxo-
1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydrocyclopropa[e]pyrrolo[1,2-
a][1,4]diazacyclopentadecine-14a(5H)-yl]formyl}azanide
MOLECULAR FORMULA C35H45FN5NaO9S
MOLECULAR WEIGHT 753.8
SPONSOR Genentech
CODE DESIGNATION
- Danoprevir sodium
- ITMN-191
- R 7227 sodium
- R7227
- RO 5190591-001
- RO5190591-001
- UNII-217RJI972K
CAS REGISTRY NUMBER 916826-48-7
DANOPREVIR SODIUM
The HCV protease mediates the cleavage of the HCV polyprotein to release the functional proteins that are essential for viral propagation. The inhibition of the HCV protease activity is expected to block HCV replication in infected host cells. Numberous HCV protease inhibitors have been identified. Non- limiting examples of HCV protease inhibitors are described in U.S. Patent Application Pub. Nos. 20040106559, 20040180815, 20040266668, 2004038872, 20050090432, 20050267018, 20070054842, 20070281885, 2007299078, 20080032936, 20080125444, 20080279821, 20090111757, 20090148407, 20090202480, 20090269305, 20090285773, 20090285774, 20100081700, 20100144608, 2010018355, 20100183551, 20100221217, 20100260710, 20100286185 and 20110135604, and U.S. Patent Nos. 6608027, 6767991, 7091184, 7119072, 7544798, 7642235 and 7829665, as well as WO2007014919, WO2007014926, WO2008046860, WO2008095058,
………………………………
danoprevir
patents and journal ref
1. WO 2005037214..
2. WO 2005095403
3. WO 2007015824..
4. WO 2008128921
5. WO 2009080542
6. WO 2009142842
7. WO 2010015545
8. WO 2013079424
9. WO 2012062685
10.WO 2013106631
11. Concise asymmetric synthesis of a (1R,2S)-1-amino-2-vinylcyclopropanecarboxylic acid-derived sulfonamide and ethyl ester
Org Biomol Chem 2013, 11(39): 6796http://pubs.rsc.org/en/content/articlelanding/2013/ob/c3ob41394b/unauth#!divAbstract
12.J. Med. Chem., Article ASAP,DOI: 10.1021/jm400164c
Kazmierski WM, Hamatake R, Duan M, Wright LL, Smith GK, Jarvest RL, Ji JJ, Cooper JP, Tallant MD, Crosby RM, Creech K, Wang A, Li X, Zhang S, Zhang YK, Liu Y, Ding CZ, Zhou Y, Plattner JJ, Baker SJ, Bu W, Liu L.
J Med Chem. 2012 Apr 12;55(7):3021-6. doi: 10.1021/jm201278q. Epub 2012 Apr 3.
14 . Discovery of novel P3-oxo inhibitor of hepatitis C virus NS3/4A serine protease.
Duan M, Kazmierski W, Crosby R, Gartland M, Ji J, Tallant M, Wang A, Hamatake R, Wright L, Wu M, Zhang YK, Ding CZ, Li X, Liu Y, Zhang S, Zhou Y, Plattner JJ, Baker SJ.
Bioorg Med Chem Lett. 2012 Apr 15;22(8):2993-6. doi: 10.1016/j.bmcl.2012.02.039. Epub 2012 Feb 22.
……………….

(2R,6S,13aS,14aR,16aS,Z)-6-(tert-Butoxycarbonylamino)-14a-(cyclopropylsulfonylcarbamoyl)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl 4-fluoroisoindoline-2-carboxylate (49)



For certain NS3 inhibitors shown in this section, additional chemical transformations are utilized to obtain the final products. The preparations of two such examples are described for compounds 153 and 154 below:

(2R,6S,13aS,14aR,16aS,Z)-6-(tert-botoxycarbonylamino)-2-(4-fluoroisoindoline-2-carbonyloxy)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-α][1,4]dizacyclopentadecine-14a-carboxylic acid (0.10 g, 0.16 mmol) and TEA (0.024 mL, 0.18 mmol) in THF (5 mL) was added ethyl carbonochlridate (0.016 mL, 0.17 mmol) at 0° C. The reaction was stirred at 0° C. for 2 hrs. Sodium boronhydride (0.012 g, 0.32 mmol) was added and the reaction was stirred at rt for 3 days. Water (5 mL) and ethyl acetate (10 mL) were added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was purified by column chromatography (ethyl acetate) to give the product (0.060 g, 61.4%) as white solid. 1H NMR (400 MHz, d6-DMSO) δ 8.47 (b, 1H), 7.35 (m, 1H), 7.10-7.20 (m, 2H), 7.03 (m, 1H), 5.47 (m, 1H), 5.28 (b, 1H), 4.98 (m, 1H), 4.67 (b, 4H), 4.56 (m, 1H), 4.46 (m, 1H), 4.26 (m, 1H), 3.92 (m, 1H), 3.66 (m, 2H), 3.16 (m, 1H), 2.67 (m, 1H), 2.21 (m, 2H), 1.80 (m, 1H), 1.68 (m, 1H), 1.30 (m, 8H), 1.11-1.20 (m, 9H), 0.85 (m, 1H), 0.77 (m, 1H).

A solution of oxalyl chloride 90.045 mL, 0.089 mmol) in DCM (5 mL) at −78° C. was added a solution of DMSO (0.015 g, 0.020 mmol) in DCM (2 mL) dropwise over 2 ninytes. The reaction was stirred at −78° C. for 10 minutes and the a solution of (2R,6S,13aS,14aR,16aS,Z)-6-(tert-botoxycarbonylamino)-14a-(hydroxymethyl)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-α][1,4]dizacyclopentadecin-2-yl-4-fluoroisoindoline-2-carboxylate (0.050 g, 0.081 mmol) in DCM (2 mL) was added. After stirred at −78° C. for 40 min, TEA (0.051 mL, 0.37 mmol) was added. The reaction was warmed to rt, water (5 mL) was added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was dissolved in MeOH (5 mL) and ammonium hydroxide (0.085 g, 2.45 mmol) and acetic acid (0.014 mL, 0.25 mmol) were added. The reaction stirred at rt for 3 minutes. NaCNBH3 90.015 g, 0.245 mmol) was added and stirred at rt for 30 minutes. The MeOH was removed. DCM (20 mL) and saturated sodium bicarbonate (5 mL) was added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was dissolved in DCM (5 mL). TEA (0.017 mL, 0.122 mmol) was added and followed by the cyclopropanesulfonyl chloride (0.015 g, 0.098 mmol). The reaction was stirred at rt for 5 hrs. The solvent was removed. The residue was purified by column chromatography (ethyl acetate) to give the product (0.017 g, 28.2%) as white solid. 1H NMR (400 MHz, d6-DMSO) δ 8.52 (m, 1H), 7.35 (m, 1H), 7.02-7.20 (m, 4H), 5.56 (m, 1H), 4.99 (m, 1H), 4.97 (m, 1H), 4.67 (m, 2H), 4.66 (s, 2H), 4.46 (m, 1H), 4.24 (m, 1H), 3.92 (m, 1H), 3.67 (m, 1H), 3.46 (m, 1H), 2.74 (m, 1 h), 2.67 (m, 1H), 2.22 (m, 2H), 1.84 (m, 1H), 1.68 (m, 1H), 1.08-1.36 (m, 20H), 0.89 (m, 2H), 0.81 (m, 2H).

Hoffmann-La Roche and Genentech’s danoprevir/r (RG7227) is a twice-daily, ritonavir-boosted HCV protease inhibitor with activity against HCV genotypes 1, 4 and 6. DAUPHINE, an ongoing phase II trial in 421 treatment-naive people with HCV genotypes 1 and 4, is comparing doses (200, 100, and 50 mg danoprevir, boosted with 100 mg ritonavir, twice-daily) and response-guided therapy with danoprevir/r plus PEG-IFN/RBV. At 12 weeks after treatment completion, HCV RNA was undetectable in 86% of the highest-dosing arm, 77% of the 100 mg arm, and 65% of the 50 mg arm.
Response to treatment in the 200 mg dosing arm did not differ according to HCV subtype or IL28B genotype; at 12 weeks after treatment completion, 88% of people with HCV subtype 1a and an IL28B non-CC genotype had undetectable HCV RNA. Across all dosing arms, HCV RNA remained undetectable 12 weeks after treatment completion in 100% of people with HCV genotype 4.
In the response-guided therapy arm, 76% of early responders (who were treated for 12 weeks) and 67% of late responders (treated for 24 weeks) maintained undetectable HCV RNA 12 weeks after treatment completion, bringing the overall total to 72%.
One death occurred during the trial—from sudden heart attack, in a participant with preexisting diabetes and hypertension—it was considered unrelated to study drugs. Adverse events were reported in virtually all study participants. Side effects from ritonavir, which is used to boost danoprevir levels, increased the likelihood of more than one serious adverse event among people in the danoprevir/r arms (range 4–9% vs. 1% for placebo). The rate of danoprevir/r-related treatment discontinuations was similar to the rate of PEG-IFN/RBV-associated discontinuations (3–7%, and 3–8%, respectively).
Common side effects (experienced by more than 15% of study participants) included fatigue, fever, chills, weakness, nausea, diarrhea, itching, rash, hair loss, headache, aching muscles and joints, insomnia, cough, and appetite loss. Diarrhea was the only side effect associated with danoprevir/r. Adding danoprevir/r did not increase rates of rash or anemia (known side effects of other HCV protease inhibitors). Most grade 3 and grade 4 lab abnormalities were neutropenia, reported in 22% to 38% of study participants.
Interferon-free DAA Combinations
Danoprevir/r and Mericitabine, plus Ribavirin (HCV Genotypes 1 and 4)
Roche’s phase IIb study, INFORM-SVR, is combining response-guided therapy with danoprevir/r, a twice-daily ritonavir-boosted HCV protease inhibitor, and mericitabine, a twice-daily nucleoside polymerase inhibitor, with or without ribavirin for 12 to 24 weeks in non-cirrhotic people with HCV genotype 1. The original study design was modified after high relapse rates were observed in the 12-week treatment and ribavirin-free arms. Treatment was extended to 24 weeks, and ribavirin was given to all participants.
The majority of INFORM-SVR participants were male, had HCV genotype 1a, and non-CC genotypes. Of the 64 people treated for 24 weeks with all three drugs, 41% experienced SVR-12. People with HCV genotype 1b were more likely to achieve SVR-12 (71% versus 26% in HCV genotype 1a). In contrast, SVR-12 was more likely among people with non-CC genotypes (32% for CC versus 44% for non-CC), although only 4 people had HCV genotype 1b and CC genotype. Breakthrough rates were higher in people who did not receive ribavirin, and in HCV genotype 1a versus 1b. Resistance to danoprevir/r was observed in all patients who experienced viral breakthrough; mericitabine resistance was found in one person.
Almost all participants had more than one adverse event; a total of 567 mild-to-moderate events were reported among 83 people. The most common side effects, occurring in >10% of people were headache, fatigue, nausea, diarrhea, colds, insomnia, itching, weakness, dizziness, irritability, shortness of breath, cough, upset stomach, painful joints, and vomiting. As for laboratory abnormalities, one person experienced grade 3 anemia, four people had grade 3 lipid elevations, and one case each of grade 3 elevations in phosphate and lipase were observed.
A single serious adverse event, multiple myeloma, occurred 53 days after treatment completion and one person discontinued due to pain in the back of the throat (it was not specified whether or not this was a treatment-related adverse event).
- Everson G, Cooper C, Shiffman ML, et al. Rapid and sustained achievement of undetectable HCV RNA during treatment with ritonavir-boosted danoprevir/PEG-IFNa-2A/RBV in HCV genotype 1 or 4 patients: Dauphine week 36 interim analysis (Abstract 1177). Paper presented at: 47th Annual Meeting of the European Association for the Study of the Liver; 2012 April 18–22; Barcelona, Spain. Available from: http://mobile.ilcapp.eu/EASL_161/poster_24544/program.aspx. (Accessed 2012 June 25)
- Gane EJ, Pockros P, Zeuzem S, et al. Interferon-free treatment with combination of mericitabine and danoprevir/r with or without ribavirin in treatment-naïve HCV genotype-1 infected patients (Abstract 1412). 47th Annual Meeting of the European Association for the Study of the Liver; 2012 April 18–22; Barcelona, Spain. Available from:http://mobile.ilcapp.eu/EASL_161/poster_24848/program.aspx. (Accessed 2012 June 25)
Non- limiting examples of suitable HCV protease inhibitors include ACH-1095
(Achillion), ACH-1625 (Achillion), ACH-2684 (Achillion), AVL-181 (Avila), AVL-192 (Avila), BI-201335 (Boehringer Ingelheim), BMS-650032 (BMS), boceprevir, danoprevir, GS- 9132 (Gilead), GS-9256 (Gilead), GS-9451 (Gilead), IDX-136 (Idenix), IDX-316 (Idenix), IDX- 320 (Idenix), MK-5172 (Merck), narlaprevir, PHX-1766 (Phenomix), telaprevir, TMC-435 (Tibotec), vaniprevir, VBY708 (Virobay), VX-500 (Vertex), VX-813 (Vertex), VX-985 (Vertex), or a combination thereof. Non-limiting examples of suitable HCV polymerase inhibitors include ANA-598 (Anadys), BI-207127 (Boehringer Ingelheim), BILB-1941 (Boehringer Ingelheim), BMS-791325 (BMS), filibuvir, GL59728 (Glaxo), GL60667 (Glaxo), GS-9669 (Gilead), IDX-375 (Idenix), MK-3281 (Merck), tegobuvir, TMC-647055 (Tibotec), VCH-759 (Vertex & ViraChem), VCH-916 (ViraChem), VX-222 (VCH-222) (Vertex & ViraChem), VX-759 (Vertex), GS-6620 (Gilead), IDX-102 (Idenix), IDX-184 (Idenix), INX-189 (Inhibitex), MK-0608 (Merck), PSI-938 (Pharmasset), RG7128 (Roche), TMC64912 (Medivir), GSK625433 (Glaxo SmithKline), BCX-4678 (BioCryst), ALS-2200 (Alios BioPharma/Vertex), ALS-2158 (Alios BioPharma/Vertex), or a combination thereof. A polymerase inhibitor may be a nucleotide polymerase inhibitor, such as GS-6620 (Gilead), IDX-102 (Idenix), IDX-184 (Idenix), INX-189 (Inhibitex), MK-0608 (Merck), PSI-938 (Pharmasset), RG7128 (Roche), TMC64912 (Medivir), ALS-2200 (Alios BioPharma/Vertex), ALS-2158 (Alios BioPharma/Vertex), or a combination therefore. A polymerase inhibitor may also be a non- nucleoside polymerase inhibitor, such as ANA-598 (Anadys), BI-207127 (Boehringer Ingelheim), BILB-1941 (Boehringer Ingelheim), BMS-791325 (BMS), filibuvir, GL59728 (Glaxo), GL60667 (Glaxo), GS-9669 (Gilead), IDX-375 (Idenix), MK-3281 (Merck), tegobuvir, TMC-647055 (Tibotec), VCH-759 (Vertex & ViraChem), VCH-916 (ViraChem), VX-222 (VCH-222) (Vertex & ViraChem), VX-759 (Vertex), or a combination thereof. Non-limiting examples of suitable NS5A inhibitors include GSK62336805 (Glaxo SmithKline), ACH-2928 (Achillion), AZD2836 (Astra-Zeneca), AZD7295 (Astra-Zeneca), BMS-790052 (BMS), BMS- 824393 (BMS), GS-5885 (Gilead), PPI-1301 (Presidio), PPI-461 (Presidio), or a combination thereof. Non-limiting examples of suitable cyclophilin inhibitors include alisporovir (Novartis & Debiopharm), NM-811 (Novartis), SCY-635 (Scynexis), or a combination thereof. Non-limiting examples of suitable HCV entry inhibitors include ITX-4520 (iTherx), ITX-5061 (iTherx), or a combination thereof.
WO 2007015824WO 2003053349WO 2005095403WO 2005037214WO 2005095403WO 2005037214WO 2003053349WO 2007015824WO 2008128921
| US8048862 | 14 Apr 2009 | 1 Nov 2011 | Intermune, Inc. | Macrocyclic inhibitors of hepatitis C virus replication |
| US8119592 | 10 Oct 2006 | 21 Feb 2012 | Intermune, Inc. | Compounds and methods for inhibiting hepatitis C viral replication |
| US8232246 | 30 Jun 2009 | 31 Jul 2012 | Abbott Laboratories | Anti-viral compounds |
| US8299021 | 19 Apr 2012 | 30 Oct 2012 | Intermune, Inc. | Macrocyclic inhibitors of hepatitis C virus replication |
| US8420596 | 10 Sep 2009 | 16 Apr 2013 | Abbott Laboratories | Macrocyclic hepatitis C serine protease inhibitors |
| WO2013106631A1 | 11 Jan 2013 | 18 Jul 2013 | Abbvie Inc. | Processes for making hcv protease inhibitors |
Danoprevir Clinical Trial Information( data from http://clinicaltrials.gov)
| NCT Number | Recruitment | Conditions | Sponsor /Collaborators |
Start Date | Phases |
|---|---|---|---|---|---|
| NCT01331850 | Completed | Hepatitis C, Chronic | Hoffmann-La Roche | 2011-05 | Phase 2 |
| NCT01531647 | Completed | Healthy Volunteer | Hoffmann-La Roche | 2012-01 | Phase 1 |
| NCT01588002 | Completed | Healthy Volunteer | Hoffmann-La Roche | 2012-04 | Phase 1 |
| NCT01592318 | Active, not recruiting | Healthy Volunteer | Hoffmann-La Roche | 2012-05 | Phase 1 |
| NCT01749150 | Recruiting | Hepatitis C, Chronic | Hoffmann-La Roche | 2013-04 | Phase 2 |

WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
Romosozumab (AMG 785) shines in phase II for osteoporosis
Romosozumab (AMG 785) is a humanized monoclonal antibody that targets sclerostin for the treatment of osteoporosis.[1]
Romosozumab was originally discovered by Celltech (now owned by UCB).[2] Celltech entered in a partnership with Amgen in 2002 for the product’s development.[3] As of January 2014, Phase 3 clinical trials are recruiting patients.[4]
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Romosozumab”. American Medical Association.
- Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003 Dec 1;22(23):6267-76.
- Celltech group Annual Report and Accounts 2002
- ClinicalTrials.gov: Romosozumab
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | Sclerostin |

Sclerostin / Source: Wikimedia Commons and JMROL
Biloine Young • Thu, November 1st, 2012
Good news for postmenopausal women came from a report given by Michael R. McClung, M.D., at the annual meeting of the American Society for Bone and Mineral Research. McClung and colleagues have found an antibody that targets the Wnt signaling pathway and its osteocyte-regulating molecule sclerostin, which increases bone formation while decreasing bone resorption, according to Nancy Walsh, staff writer for MedPage Today.
Walsh reports that one year of treatment with the antibody romosozumab (formerly AMG 785) led to an 11.3% absolute increase in bone mineral density (BMD) in postmenopausal women with low BMD (body mass index). That compared with BMD increases of only 7% with teriparatide (Forteo), 4% with alendronate (Fosamax), and no change with a placebo. “The discovery of sclerostin as an osteocyte-mediated stimulator of osteoblast function and bone formation opened the door for considering the inhibition of this protein and regulator as a target for osteoporosis treatment,” McClung said.
To further explore the therapeutic potential of this antibody, the researchers conducted a Phase II study that enrolled 419 women whose lumbar spine, total hip, or femoral neck T-scores were between −2 and −3.5. The mean age of the participating women was 67. Researchers randomized participants to receive romosozumab in dosages of 70 mg, 140 mg, or 210 mg each month, 140 or 210 mg every three months, or a placebo.
The total hip increase in BMD with romosozumab was 4.1% at 12 months, which was approximately double that seen with alendronate and teriparatide. The researchers also saw changes in biomarkers of bone metabolism, McClung noted. The pattern seen with romosozumab, McClung said, was increases for serum P1NP, favoring bone formation, and decreases in serum CTX, suggesting a slowing of bone resorption after one week of treatment.
Adverse events were similar in the treatment groups. The most common was back or extremity pain. Serious adverse events occurred in 9.8% of the romosozumab groups and in 14% of the placebo group. The only treatment-related adverse events that occurred in the romosozumab groups were injection site reactions, but these were mild and did not lead to a discontinuation of treatment, said McClung.
Amgen/UCB osteoporosis drug shines in Phase II
Amgen and UCB have been boosted by promising mid-stage data for their investigational osteoporosis drug romosozumab.
A Phase II trial, the results from which have been published in the New England Journal of Medicine,showed that romosozumab demonstrated a significant increase in bone mineral density. Specifically, the trial demonstrated that, compared with placebo, treatment for 12 months with the anti-sclerostin biologic significantly increased BMD at the lumbar spine, total hip and femoral neck.
Amgen and UCB noted that significant increases were also observed in the first BMD assessment at three months and moreover, in exploratory analyses, increases observed at the lumbar spine and hip “were significantly greater than those observed with current treatments”, namely Merck & Co’s Fosamax (alendronate) and Eli Lilly’s Forteo (teriparatide).
Iris Loew-Friedrich, chief medical officer at UCB, noted that romosozumab is designed to stimulate bone formation, “which makes it different from most available treatments that reduce bone resorption”. She added that “we are encouraged by the emerging efficacy and safety profile, and look forward to further investigating its potential in the ongoing global Phase III clinical programme”. Final data from the latter, which will enroll up to 10,000 patients, are expected by the end of 2015.
Sean Harper, Amgen R&D chief, noted that broken bones due to osteoporosis are common “yet the seriousness of this health event remains underappreciated, with only two in ten women receiving follow-up testing or treatment after they have broken a bone”. He added that “with its bone-forming ability, romosozumab may result in new treatment strategies”.
If all goes well in Phase III, many observers believe romosozumab could be a blockbuster.
Links


DR ANTHONY MELVIN CRASTO Ph.D
TOSEDOSTAT ….An aminopeptidase inhibitor with antineoplastic activity.

TOSEDOSTAT
An aminopeptidase inhibitor with antineoplastic activity.
- CHR 2797
- CHR-2797
- Tosedostat
- UNII-KZK563J2UW
- BB-76163Vernalis (Originator)
| CAS No. | 238750-77-1 |
| Chemical Name: | Tosedostat |
| Synonyms: | BB-76163;Chr-2797;tosedostat;CHR2797 (Tosedostat);Tosedostat (CHR2797);α-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]-benzeneaceticacidcyclopentlyester;alpha-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]benzeneacetic acid cyclopentyl ester;Benzeneacetic acid, alpha-(((2R)-2-((1S)-1-hydroxy-2-(hydroxyamino)-2-oxoethyl)-4-methyl-1-oxopentyl)amino)-, cyclopentyl ester, (alphas)- |
| Molecular Formula: | C21H30N2O6 |
| Formula Weight: | 406.47 |
CHR-2797 is an oral, once-daily experimental cancer therapy in phase II clinical development at Chroma Therapeutics for the oral treatment of refractory acute myeloid leukemia in elderly patients. It is also in early clinical development for the treatment of refractory solid tumors alone or in combination with chemotherapy.
No recent development has been reported for phase I/II studies evaluating CHR-2797 as monotherapy in hematologic/blood cancer. A phase I/II clinical trial of the compound in combination with erlotinib for non-small cell lung cancer was terminated in 2010 due to very poor recruitment of patients to the study.
Cell Therapeutics is also conducting phase II clinical trials of the compound for the treatment of myelodysplasia and acute myeloid leukemia.
CHR- 2797 is an inhibitor of aminopeptidases and has demonstrated strong preclinical efficacy as monotherapy in addition to demonstrating strong synergy with a number of leading cancer therapies in a range of cancer cells. It was originally licensed from Vernalis, where it was being evaluated for its potential in treating multiple sclerosis; however development in this indication has been discontinued.
In 2008, orphan drug designation was assigned to CHR-2797 in the U.S. for the treatment of acute myeloid leukemia. In 2011, the compound was licensed to Cell Therapeutics by Chroma Therapeutics in Central America, North America and South America for exclusive marketing and codevelopment for the oral treatment of blood-related cancers and other cancers.
In corporate news, biopharmaceutical company Cell Therapeutics, Inc. (CTIC) was up more than 6% and near 52 week highs after saying Thursday that the U.S. FDA has removed the partial clinical hold on tosedostat and all studies underway have been allowed to continue. Tosedostat is under development for the treatment of blood-related cancers. It is currently being studied in Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia and high-risk myelodysplastic syndromes.

Tosedostat is a proprietary orally bioavailable inhibitor of the M1 family of aminopeptidases with potential antineoplastic activity.
Tosedostat is converted intracellularly into a poorly membrane-permeable active metabolite (CHR-79888) which inhibits the M1 family of aminopeptidases, particularly puromycin-sensitive aminopeptidase (PuSA), and leukotriene A4 (LTA4) hydrolase; inhibition of these aminopeptidases in tumor cells may result in amino acid deprivation, inhibition of protein synthesis due to a decrease in the intracellular free amino acid pool, an increase in the level of the proapoptotic protein Noxa, and cell death.
Noxa is a member of the BH3 (Bcl-2 homology 3)-only subgroup of the proapoptotic Bcl-2 (B-cell CLL/lymphoma 2) protein family
Cell Therapeutics announced that it has received notification from the U.S. Food and Drug Administration (FDA) that the partial clinical hold on tosedostat (IND 075503) has been removed and all studies underway may continue. Tosedostat is a first-in-class selective inhibitor of aminopeptidases, which are required by tumor cells to provide amino acids necessary for growth and tumor cell survival, and is under development for the treatment of blood-related cancers.
Tosedostat is currently being studied in the United States and European Union in investigator-sponsored and cooperative group-sponsored Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia (AML) and high-risk myelodysplastic syndromes (MDS).
“We are pleased that the FDA has responded favorably to the tosedostat clinical trial data provided and removed the partial clinical hold to allow further development of tosedostat in ongoing and future studies,” said John Pagel, MD, PhD, Associate Member, Clinical Research Division, Fred Hutchinson Cancer Research Center; Associate Professor, Medical Oncology Division, University of Washington School of Medicine; and Principal Investigator in the tosedostat first-line AML/MDS trial.
Recently, WO 93/20047 disclosed a class of hydroxamic acid based MMP inhibitors which also are active in inhibiting TNF production.
As mentioned above, MMP inhibitors have been proposed with hydroxamic acid or carboxylic acid zinc binding groups. The following patent publications disclose hydroxamic acid-based MMP inhibitors:
US 4599361 (Searle) EP-A-0236872 (Roche) EP-A-0274453 (Bellon) WO 90/05716 (British Bio-technology) WO 90/05719 (British Bio-technology) WO 91/02716 (British Bio-technology) EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) EP-A-0497192 (Roche) WO 92/13831 (British Bio-technology) WO 92/17460 (SmithKline Beecham) WO 92/22523 – (Research Corporation Technologies) WO 93/09090 (Yamanouchi) WO 93/09097 (Sankyo) WO 93/20047 (British Bio-technology) WO 93/24449 (Celltech) WO 93/24475 (Celltech) EP-A-0574758 (Roche) The following patent publications disclose carboxylic acid-based MMP inhibitors:
EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) WO 93/24449 (Celltech) WO 93/24475 (Celltech)
|
|
|
TOSEDOSTAT
| WO1996033166A1 * | 17 Apr 1996 | 24 Oct 1996 | Du Pont Merck Pharma | Hydroxamic and carboxylic acids as metalloprotease inhibitors |
| WO1998011063A1 * | 8 Sep 1997 | 19 Mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| GB2268934A * | Title not available |
| US5652262 * | 14 mar 1994 | 29 lug 1997 | British Biotech Pharmaceutical, Ltd. | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| US5821262 * | 4 ott 1994 | 13 ott 1998 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as inhibitors of cytokine production |
| US5861436 * | 29 apr 1997 | 19 gen 1999 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| EP0423943A2 | 19 set 1990 | 24 apr 1991 | Beecham Group p.l.c. | Use of collagenase inhibitors in the treatment of demyelinating diseases, in particular multiple sclerosis |
| JPH03157372A | Titolo non disponibile | |||
| WO1997049674A1 | 20 giu 1997 | 31 dic 1997 | Francesca Abrate | Matrix metalloproteinase inhibitors |
| WO1998011063A1 | 8 set 1997 | 19 mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| WO1999040910A1 | 27 gen 1999 | 19 ago 1999 | Andrew Paul Ayscough | Anti-inflammatory agents |
| WO1999044602A1 | 5 mar 1999 | 10 set 1999 | British Biotech Pharm | Inflammatory cell inhibitors |
| WO1999046241A1 | 12 mar 1998 | 16 set 1999 | British Biotech Pharm | Cytostatic agents |
| WO2000044373A1 * | Jan 27, 2000 | Aug 3, 2000 | Raymond Paul Beckett | Antibacterial hydroxamic acid derivatives |
| US6545051 | Jan 27, 2000 | Apr 8, 2003 | British Biotech Pharmaceuticals, Ltd. | Antibacterial hydroxamic acid derivatives |
Drugs Fut 2009, 34(2): 115
PLoS One (2013), 8(2), e57641.
WO 1999046241
WO 1995019956
WO 1998011063
US 6462023
US 20100260674
WO 2000044373
WO 9940910
NMR
http://file.selleckchem.com/downloads/nmr/S152202-CHR-2797-NMR-Selleck.pdf
Anti-Metastatic and Anti-Invasive Agents Compounds which have the property of inhibiting the action of the metalioproteinase enzymes involved in connective tissue breakdown and remodelling, such as fibroblast collagenase (Type 1 ), PMN-collagenase, 72 kDa-gelatinase, 92 kDa- gelatinase, stromelysin, stromelysin-2 and PUMP-1 (known as “matrix metalloproteinases”, and herein referred to as MMPs) have been proposed and are being tested in the clinic for the treatment of solid tumours. Cancer cells are particularly adept at utilising the MMPs to achieve rapid remodelling of the extracellular matrix, thereby providing space for tumour expansion and permitting metastasis. MMP inhibitors should minimise these processes and thus slow or prevent cancer progression.
In view of the rapid emergence of multidrug-resistant bacteria, the development of antibacterial agents with novel modes of action that are effective against the growing number of resistant bacteria, particularly the vancomycin resistant enterococci and β-lactam antibiotic-resistant bacteria, such as methicillin-resistant Staphylocccus aureus, is of utmost importance.
The natural antibiotic actinonin (see for example J. C. S Perkin I, 1975, 819) is a hydroxamic acid derivative of Structure (A):

In ddition to actinonin, various structural analogues of actinonin have also been shown to have antibacterial activity (see for example Broughton et al. (Devlin et al. Journal of the Chemical Society. Perkin Transactions 1 (9):830-841, 1975; Broughton et al. Journal of the Chemical Society. Perkin Transactions 1 (9):857-860, 1975).
The matlystatin group of compounds, share a number of structural similarities with actinonin. Both are peptidic molecules with functional hydroxamic acid metal binding groups (Ogita et al., J. Antibiotics. 45(11):1723-1732; Tanzawa et al., J. Antibiotics. 45(11):1733-1737; Haruyama et al., J. Antibiotics. 47(12):1473-1480; Tamaki et al., J. Antibiotics. 47(12):1481-1492).
………………………………………………………….
EXAMPLE 44 2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl-pentanoylamine]-2-phenyl-ethanoic acid cyclopentyl ester

The above compound was prepared using procedures similar to those described in example 8 using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1H, s), 5.2-5.14 (1H, m), 4.02 (1H, d, J=6.9 Hz), 2.94-2.85 (1H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1H, m) and 0.86 (6H, dd, J=6.5, 11 5 Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1, 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; 8 (MeOD), 7.33-7.19 (5H, m), 5.3 (1H, s), 5.11-5.06 (1H, m), 3.81 (1H, d, J=7.3 Hz), 2.83-2.74 (lH, m), 1.83-1.45 (10H, bm), 1.12-1.03 (lH, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3 Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2
Example 1
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

HO Ξ CONHOH
Prepared using procedures similar to those described in Preparative Example A using phenylglycine cyclopentyl ester.
Diastereoisomer A
Η-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d,
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, 14 dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2,
58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
Η-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d, J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.

tosedostat
http://www.google.it/patents/US6545051

42
| WO98/11063 | WO99/46241 ex 1b | WO 98/11063 analogy ex 8 |

43
| WO98/11063 | WO99/46241 ex 1a | WO 98/11063 analogy ex 8 |
……………………………………………………………………
entry 65 in http://www.google.com/patents/WO2000044373A1
……………………………………………………………………………………………………….
http://www.google.com/patents/WO1999044602A1
Example 43
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester
TC

HO Ξ CONHOH
Prepared using procedures similar to those described in example 8 of WO 98/11063, using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d, 34
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d,
J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and
0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3,
129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.
……………………………..
3R-isobutyl-4S-methoxy-dihydrofuran-2,5-dione (WO 97/02239)
…………………………………………………………………………..
2(S)-Amino(phenyl)ethanoic acid cyclopentyl ester

…………………………………………………………………..
2(R)-[2,2-Dimethyl-5-oxo-1,3-dioxolan-4(S)-yl]-4-methylpentanoic acid pentafluorophenyl ester

…………………………………………………………..
intermediates
238750-91-9
α-amino-, cyclopentyl ester Benzeneacetic acid,
……………….
cas 240489-34-3
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

…………………..
will be updated very soon… keep watching
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family

Vedroprevir
{kind=link}
GS 9451, GS-9451, 1098189-15-1 USAN ZZ-81
VEDROPREVIR THERAPEUTIC CLAIM Treatment of hepatitis C
CHEMICAL NAMES 1. Cyclopropanecarboxylic acid, N-[[(1α,3β,5α)-bicyclo[3.1.0]hex-3- yloxy]carbonyl]-3-methyl-L-valyl-(4R)-4-[[8-chloro-2-[2-[(1-methylethyl)amino]- 4-thiazolyl]-7-[2-(4-morpholinyl)ethoxy]-4-quinolinyl]oxy]-L-prolyl-1-amino-2- ethyl-, (1R,2R)-
2. N-{[(1R,3r,5S)-bicyclo[3.1.0]hex-3-yloxy]carbonyl}-3-methyl-L-valyl-(4R)-4-[(8- chloro-2-{2-[(1-methylethyl)amino]thiazol-4-yl}-7-[2-(morpholin-4- yl)ethoxy]quinolin-4-yl)oxy]-L-prolyl-(1R,2R)-1-amino-2- ethylcyclopropanecarboxylic acid
MOLECULAR FORMULA C45H60ClN7O9S
MOLECULAR WEIGHT 910.5 daltons
SPONSOR Gilead Sciences, Inc.
CODE DESIGNATION GS-9451
CAS REGISTRY NUMBER1098189-15-1
WHO NUMBER9745
GS-9451 is a NS3 protease inhibitor in phase II clinical trials at Gilead for the oral treatment of hepatitis C.
…………………………………………………………………………
Discovery of GS-9451: An acid inhibitor of the hepatitis C virus NS3/4A protease Bioorg Med Chem Lett 2012, 22(7): 2629 ……………………………………………………………
PATENTS WO 2012087596 WO 2009005676 WO 2013106631 WO2013101550 ……………………….
WO2012087596A1 Compound 3 can be prepared using synthetic methods and intermediates like those described in USSN 12/215,605 (US 20090257978 A1). Compound 3 can also be prepared described in the following Example. Example 3: Preparation of Compound 3

Compound 315 (12 g, 13 mmol) was dissolved in THF (200 ml), LiOH (11g, 260 mmol) in H20 (200 ml) was added, followed by MeOH (200 ml). The mixture was kept stirring at room temperature for 20 hours. Upon completion of the reaction, 4 N HCI in H20 was added to adjust pH to 7 at 0 °C. The mixture was extracted with EtOAc (2 x 400 ml). The combined organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo to give compound 3 as a yellow solid (11 g, 93%). LC/MS = 911.52(M++1 ). 1H NMR (300MHz, CD3OD)57.95 (d, 1H), 7.90 (s, 1H), 7.48 (s, 1H), 7.31 (d, 1H), 5.42 (s, 1H), 4.37 (dd, 1H), 4.20 (m, 2H), 3.83-3.56 (m, 7H), 3.50 (m, 2H), 3.39 (m, 2H), 2.45 (m, 1H), 2.27(m, 1H), 1.62 (m, 2H), 1.50 (m, 1H), 1.33 (m, 2H), 1.18 (m, 1H), 1.05 (m, 8H), 0.90 (m, 3H), 0.76 (m, 11H), 0.14-0.04 (m, 2H) The intermediate compound 315 was prepared as follows.

301 302 a. Preparation of compound 301. To a dry, argon purged three-neck round bottom flask (1000 mL) were added anhydrous dichloromethane (100 mL) and Et2Zn (28 mL, 273 mmol) at 0 °C. (CAUTION: Source of argon can not be from needle. Use appropriate glass adapter only. A second bubbler can also be attached to the flask to prevent excessive pressure build up.) Cyclopenten-3-ol (10.0 mL, 119 mmol) was then added dropwise (large quantity of ethane gas was produced) to the flask and the reaction mixture was allowed to stir until the evolution of gas had ceased. Diiodomethane (22 mL, 242 mmol) was then added dropwise over a period of 30 minutes. The reaction was allowed to warm to room temperature and continued to stir overnight under a positive flow of argon, at which point TLC analysis had indicated complete disappearance of the starting alcohol. The reaction was then diluted with CH2CI2 and quenched with 2M HCI (white precipitate should be completely dissolved). The biphasic mixture was poured into a separatory funnel and the organic layer was collected. The solvent was removed under reduced pressure until 100 mL of material containing compound 301 remained. b. Preparation of compound 302. Anhydrous dichloromethane (525 mL) was added to the flask followed by the dropwise addition of triethylamine (34 mL, 245 mmol). The reaction continued to stir at room temperature under a positive flow of nitrogen at which point, disuccinimidylcarbonate (40.7 g, 159 mmol) was added to the flask portion wise. The reaction was allowed to stir until TLC analysis indicated complete disappearance of the starting material (2-3 days). Upon completion, the reaction mixture was quenched with 1 M HCI (200 mL x 2) and washed with H20 (200 mL x 2). The desired material was extracted using CH2CI2and the combined organic layers were dried using anhydrous MgS0 and passed through a silica plug. The solvent was removed under reduced pressure and the crude material was purified using flash chromatography (Rf = 0.33, 1 :1 Hex/EtOAc) to provide compound 302 (22 g, 75%): 1H NMR (300 MHz, CDCI3): δ 5. 24 (t, 1 H), 3.82 (s, 4H), 2.24 (m, 2H), 2.03 (d, 2H), 1.38 (m, 2H), 0.48 (m, 1 H), 0.40 (m, 1 H).


c. Preparation of compound 304. N-i-Boc-cis-4-Hydroxy-L-Proline methyl ester 303 (100.0 g, 407.7 mmol) and DABCO (1.5eq, 68.6g, 61 1.6 mmol) were dissolved in anhydrous toluene (200 mL) in a 2 L three necked round bottom flask with a mechanical stirrer and an addition funnel. After cooling the solution to 0 °C under N2, A solution of 4-Bromo-benzenesulfonyl chloride (1.3eq, 135.6g, 530.0 mmol) in 300 mL of toluene was added through addition funnel over 60 minutes. The reaction mixture was stirred and warmed to room temperature overnight (16 hours). The mixture was slowly poured into 2L 1 M Na2C03 (aq.), and the product was extracted with EtOAc (2L). After the organic phase was washed by 0.5 N HCI (2L), H20 (1 L), and brine (1 L), it was dried (MgS04), concentrated to give 195.45 g of a yellow oily brosylate product. To a solution of the above brosylate (407.7 mmol) in dichloromethane (300 mL) was slowly added 4.0 M HCI in dioxane (500 mL, 5eq) and the resulting solution was allowed to stir at room temperature for 2 hours. After ether (500mL) was added to the reaction mixture, the mixture was stirred for 15 minutes and the white precipitate was collected by filtration. The solid was washed with ether and hexane and then dried under vacuum overnight to obtain 153.0 g of the HCI amine salt of compound 304, 381.8 mmol, in 94% yield for two steps. d. Preparation of compound 305. To a solution of Boc-fert-butyl-glycine (97.0g, 420.0 mmol) in DMF (200mL) and DCM (200mL) were added HATU (217.76g, 572.7 mmol) and Hunig’s base (126 mL, 1 145.4 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of the previous HCI salt (153.0 g, 381.8 mmol) and Hunig’s base (126 mL, 1 145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3h, with monitoring by LCMS. The reaction mixture was concentrated to remove dichloromethane under reduced pressure and the white solid that formed was filtered off. The remaining DMF solution was diluted with ethyl acetate (1 L), washed successively with 3% LiCI (aq) (3x650mL), sat’d NH4CI (2x500mL), 0.5N HCI (aq) (2x600ml_), brine (500ml_), sat’d NaHC03 (3x500mL), and brine (500mL). The resulting organic fraction was dried (MgS04) and concentrated to afford compound 305 (111g). e. Preparation of compound 306. To a solution of the methyl ester 305 (120 g, 207.8 mmol) in THF (300 ml_), MeOH (75 mL) was added a solution of LiOH (26.18 g, 623.4 mmol) in H20 (150 ml_). The solution was allowed to stir at room temperature for 4 hours. The mixture was cooled in an ice-bath while acidifying with 3N HCI to pH about 5.5, stirred for 10minut.es, and the resulting white solids were collected by filtration. The solids were washed with more water, ether and hexane. The solids were dried under vacuum at 40°C overnight to give 95.78g (82%) of the acid 306. f. Preparation of compound 307. To a solution of the carboxylic acid 306 (81.4 g, 144.27 mmol) in DMF (200ml_) and dichloromethane (200mL) was added HATU (82.3g, 216.4 mmol) and Hunig’s base (47.5 mL, 432.8 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of amine (158.7 mmol) and Hunig’s base (47.5 mL, 1145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3 hours and monitored by LCMS. After the mixture was concentrated under reduced pressure to remove dichloromethane, the white solids that formed were filtered off. The remaining DMF solution was diluted with ethyl acetate (600mL) and successively washed with 3% LiCI (aq) (2x550mL), sat’d NH4CI (500mL), 1 N HCI (aq) (500mL), sat’d NaHC03(500mL), and brine (300mL). The resulting organic fraction was dried (Na2S04) and concentrated to afford compound 307 (111 g). g. Preparation of compound 308. Compound 307 was dissolved in 4N HCI in dioxane (300 mL) at room temperature and stirred for 2 hours. It was then concentrated under vacuum, and co-evaporated with dichloromethane (2 x 200mL) to dryness. The residue was dissolved in EtOAc (600mL) and sat’d aq. NaHC03 (1 L). It was stirred vigorously. After 10 minutes, carbonic acid bicyclo[3.1.0]hex-3-yl ester 2,5-dioxo-pyrrolidin-1-yl ester 302 (41.4 g, 173.1 mmol) was added in one portion. After the resulting mixture was stirred for another 30 minutes, the organic layer was collected and washed with brine (500mL), dried (Na2S04), and concentrated. The crude product was purified by flash chromatography on silica gel with ethyl acetate/hexane to afford 94.44 g (92%) of compound 308.



h. Preparation of compound 310.1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 309 (70.7 g, 354 mmol) was stirred in 48% aq. HBr (500 mL) at 110 °C for 72 hours. After the mixture was cooled to 0 °C with stirring, the solids were filtered and washed with water. The resulting solids were triturated with a saturated NaHC03 solution (-350 mL), filtered, washed with water, and dried under vacuum to give – 40 g (61%) of crude 310 as a dark brown solid. LC/MS = 186 (M++1). i. Preparation of compound 311. 1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 310 (40 g, 215 mmol) was dissolved in DMF (360 ml). Cesium carbonate (140 g, 430 mmol) was added, followed by bromoacetaldehyde dimethyl acetal (54.5 g, 323 mmol). The mixture was then vigorously stirred at 65 °C for 24 hours. Upon cooling to room temperature, EtOAc (1 L) and H20 (1 L) were added to the mixture. The organic layer was extracted with EtOAc (1 x 400 ml). The combined organic layer was washed with aqueous 3% LiCI solution (2 x 1 L), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give compound 311 as a white solid (39 g, 67%). j. Preparation of compound 312. To a mixture of 1-[2-Amino-3-chloro-4-(2,2-dimethoxy-ethoxy)-phenyl]-ethanone 311 ( 13 g, 47.5 mmol) and isopropylaminothiazole-4-carboxylic acid hydrobromide (12.64 g, 47.5 mmol) in pyridine (150 ml) was slowly added phosphorus oxychloride (9.47 g, 61.8 mmol) at -40 °C. The mixture was then stirred at 0 °C for 4 hours. Upon completion of the reaction, H20 (30 ml) was added dropwise to the mixture. The mixture was then stirred at 0 °C for another 15 minutes. The mixture was concentrated in vacuo. The residue was diluted with EtOAc, washed with a sat. NaHC03 aqueous solution. The organic layer was dried (Na2S04) and concentrated in vacuo. The residue was dissolved in CH2CI2, hexanes were added slowly to the solution, and a yellow solid started to crash out. More hexanes were added until not much product was left in the mother liquid to provide compound 312 (18 g, 85%). k. Preparation of compound 313. 2-lsopropylamino-thiazole-4-carboxylic acid [6-acetyl-2-chloro-3-(2,2-dimethoxy-ethoxy)- phenyl]-amide 312 (18 g, 40.7 mmol) was suspended in toluene (400 ml). NaH (2.4 g, 61 mmol) was added to the vigorously stirred mixture while monitoring H2evolution. The mixture became a clear solution during heating to reflux. The reaction was complete after refluxing for 3 hours. The mixture was cooled to room temperature. A solution of AcOH (69.2 mmol) in H20 (3 vol) was added to the mixture. After vigorous agitation for 1 hour at 0 °C, the solids were collected by filtration, rinsed forward with H20. The wet cake was dried under high vacuum to a constant weight to provide compound 313 ( 5 g, 86%). I. Preparation of compound 314. To a mixture of brosylate intermediate 303 (15 g, 35 mmol) and compound 313 (27.5 g, 38.5 mmol) in NMP (200 ml) was added cesium carbonate (25.1 g, 77 mmol). The mixture was stirred at 65 °C for 5 hours. The reaction was cooled to room temperature and EtOAc (600 ml) and an aqueous solution of 3% LiCI (600 ml) were added to the mixture. The organic layer was washed with aqueous 3% LiCI (1 x 600 ml), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired methyl ester as a yellow solid (23.6 g, 75%). LC/MS = 900. 1 3(M++ 1 ) . m. Preparation of compound 315. Methyl ester 314 (23.6 g, 26 mmol) was dissolved in glacial acetic acid (200 ml), 1.4 N HCI in H20 (75 ml) was added to the solution. The mixture was stirred at 60 °C for 1 hour. Upon completion of the reaction, the mixture was concentrated to remove the solvents, coevaporated with toluene (x 2) to remove residual acetic acid. The residue was then dissolved in EtOAc (500 ml) and sat. NaHC03 aqueous solution (enough to neutralize the mixture) while monitoring C02 evolution. The organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was further dried under high vacuum for 1 h and used as is for the next step. The crude was dissolved in CH2CI2 (360 ml), morpholine (3.4 g, 39 mmol) and sodium triacetoxyborohydride (7.2 g, 34 mmol) were added to the mixture at 0 °C. Then glacial acetic acid (0.47 g, 7.8 mmol) was added dropwise to the mixture. The reaction was complete in 10 minutes at 0 °C. Sat. NaHC03 aqueous solution was added to quench the reaction. After stirring for another 20 minutes, the organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired amine product 315 as a yellow solid (12 g, 50%). LC/MS = 924.63(M++ 1 )
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
GRAZOPREVIR, MK 5172
- Grazoprevir hydrate
- UNII-4O2AB118LA
- MK 5172
MW804.99
quinoxalinyl)pentyl]cyclopropyl]oxy]carbonyl]-3-methyl-L-valyl-(4R)-4-hydroxy-L-prolyl-1-
amino-N-(cyclopropylsulfonyl)-2-ethenyl-, cyclic (1→2)-ether, hydrate (1 :1) (1R,2S)-
ethenylcyclopropyl}-5-(1,1-dimethylethyl)-14-methoxy-3,6-dioxo-
1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide hydrate
MOLECULAR WEIGHT 784.92
9857
ACS Med Chem Lett 2012, 3(4): 332DOI: 10.1021/ml300017p
Org Lett 2013, 15(16): 4174
[1]. Steven Harper , John A. McCauley , Michael T. Discovery of MK-5172, a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. ACS Med. Chem. Lett., 2012, 3 (4), pp 332-336[2]. Summa V, Ludmerer SW, McCauley JA, MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012 Aug;56(8):4161-7.
IC50 Value: 7.4nM and 7nM for genotype1b and 1a respectively, in replicon system [1]
MK-5172 is a novel P2-P4 quinoxaline macrocyclic HCV NS3/4a protease inhibitor currently in clinical development.
in vitro: In biochemical assays, MK-5172 was effective against a panel of major genotypes and variants engineered with common resistant mutations observed in clinical studies with other NS3/4a protease inhibitors. In the replicon assay, MK-5172 demonstrated subnanomolar to low-nanomolar EC50s against genotypes 1a, 1b, and 2a [2].
in vivo: In rats, MK-5172 showed a plasma clearance of 28 ml/min/kg and plasma half-life of 1.4 hr. When dosed p.o. at 5 mg/kg, the plasma exposure of MK-5172 was good with an AUC of 0.7 uM.hr. The liver exposure of the compound was quite good (23 uM at 4 hr), and MK-5172 remained in liver 24 hr after a single p.o. 5 mg/kg dose. At 24 hr, the liver concentration of MK-5172 was 0.2 uM, which was over 25-fold higher than the IC50 in the replicon assay with 50% NHS. When dosed to dogs, MK-5172 showed low clearance of 5 ml/min/kg and a 3 hr half-life after i.v. 2 mg/kg dosing and had good plasma exposure (AUC=0.4 uM.hr) after a p.o. 1 mg/kg dose [1].
Clinical trial: Evaluation of Hepatic Pharmacokinetics for MK-5172 in Participants With Chronic Hepatitis C . Phase1


1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-
yl]carbonyl}amino)-2-ethenylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide (15 K-salt).
8.3 Hz, 1 H), 7.15 (m, 1 H), 7.04 (m, 1 H), 5.97 (m, 1 H), 5.73 (br s, 1 H), 4.96 (m, 1 H), 4.79 (apparent q, J = 9.3 Hz, 1 H), 4.26 (dd, J = 9.7, 7.7 Hz, 1 H), 4.20 (d, J = 11.3 Hz, 1 H), 4.14 (d, J = 8.8 Hz, 1 H), 3.90 (dd, J = 11.1, 3.2 Hz, 1 H), 3.86 (s, 3 H), 3.62 (m, 1 H), 2.86-2.60 (m, 3 H), 2.38 (m, 1 H), 2.21 (m, 1 H), 1.80-1.48 (m, 6 H), 1.42 (m, 5 H), 1.14 (m, 1 H), 0.95 (m, 10 H), 0.81 (m, 2 H), 0.72-0.50 (m, 3 H), 0.41 (m, 1 H) ppm.http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-
7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide (MK-5172, 15).
OD) δ 7.79 (dd, J = 9.6, 1.8 Hz, 1 H), 7.23 (s, 1 H), 7.22 (m, 1 H), 7.10 (d, J = 9.6 Hz, 1 H), 6.01 (apparent t, J = 3.6 Hz, 1 H), 5.74 (m, 1 H), 5.24 (dd, J = 17.0 Hz, 1.6 Hz, 1 H), 5.11 (dd, J = 10.4 Hz, 1.6 Hz, 1 H), 4.49 (d, J = 11.2 Hz, 1 H), 4.40 (m, 2 H), 4.13 (dd, J = 12.0 Hz, 4.0 Hz, 1 H), 3.92 (s, 3 H), 3.76 (m, 1 H), 2.92 (m, 2 H), 2.85 (m, 1 H), 2.55 (dd, J = 13.6 Hz, 6.4 Hz, 1 H), 2.28 (m, 1 H), 2.18 (apparent q, J =8.8 Hz, 1 H), 1.85 (dd, J = 8.0 Hz, 5.6 Hz, 1 H), 1.73 (m, 2 H), 1.5 (m, 2 H), 1.40 (dd, J = 9.6 Hz, 5.6 Hz, 1 H), 1.3 (m, 2 H), 1.23 (m, 4 H), 1.08 (s, 9 H), 0.99 (m, 2 H), 0.89 (m, 3 H), 0.73 (m, 1 H), 0.49 (m, 1 H) ppm; HRMS (ESI) m/z 767.3411 [(M+H)+; calcd for C38H51N6O9S: 767.3433].http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
| Intermediate # | Structure | Name | Lit. Reference |
| A1 |  |
(1R,2S)-1-Amino-N- (cyclopropylsulfonyl)-2- vinylcyclopropanecarboxamide hydrochloride | Wang et al., U.S. Pat. No. 6,995,174 |

















Step 1: Quinoxaline Hydroxyproline Methyl Ester HCl Salt

A 250-ml RB, equipped with magnetic stirrer and N2 bubbler, was charged with chloroquinoxaline BOC hydroxyproline adduct in MeOH (100 ml), and the mixture was cooled in an ice bath. Acetyl chloride (17.9 g) was then added, and the mixture was stirred at RT for 2 h. The batch was diluted with IP Ac (80 ml). Solids were filtered off and washed with IPAc (20 ml). The washed solids were dried under vacuum for 3 d, to provide 48.9 g (100% yield ). Part of this material was used in the next step.
Example 17: Preparation of Compound A, Method A

Macrocyclic acid hemihydrate, the product of Example 15 (10.16 g, 18.03 mmol) was dissolved in THF (50 mL to 90 mL). The solution was azetropically dried at a final volume of 100 mL. Sulfonamide pTSA salt (7.98 g, 1.983 mmol) followed by DMAc (15 mL) was added at RT. The batch was cooled to 0°C to 10°C, and pyridine (10 mL) was added dropwise. Then, EDC HCl (4.49 g, 23.44 mmol) was added in portions or one portion at 0°C to 10°C. The reaction mixture was aged at 0°C to 10°C for 1 h, and then warmed to 15°C to 20°C for 2 h to 4 h. MeOAc (100 mL) followed by 15 wt% citric acid in 5% NaCl in water (50 mL) was added, while the internal temperature was maintained to < 25°C with external cooling. The separated organic phase was washed with 15 wt% citric acid in 5% NaCl in water (50 mL) followed by 5% NaCl (50 mL). The organic phase was solvent-switched to acetone at a final volume of ~80 mL. Water (10 mL) was added dropwise at 35°C to 40°C. The batch was seeded with Compound A monohydrate form III (~10 mg) and aged for 0.5 h tol h at 35°C to 40°C. Additional water (22 mL) was added dropwise over 2 h to 4 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 60% acetone in water (2x 40 mL). Suction drying at RT gave Compound A monohydrate form III as a white solid.
XH NMR (400 MHz, CDC13) δ 9.95 (s, br, 1 H), 7.81 (d, J = 9.1 Hz, 1 H), 7.18 (dd, J = 9.1, 2.7 Hz, 1 H), 7.16 (s, br, 1 H), 7.13 (d, J = 2.7 Hz, 1 H), 5.96 (t, J = 3.8 Hz, 1 H), 5.72 (m, 1 H), 5.68 (d, J = 10.1 Hz, 1 H), 5.19 (d, J = 17.1 Hz, 1 H), 5.07 (d, J = 10.1 Hz, 1 H), 4.52 (d, J = 11.4 Hz, 1 H), 4.45 (d, J = 9.8 Hz, 1 H), 4.36 (d, J = 10.5, 6.9 Hz, 1 H), 4.05 (dd, J = 11.5, 3.9 Hz, 1 H), 3.93 (s, 3 H), 3.78 (m, 1 H), 2.90 (m, 1 H), 2.82 (tt, J = 8.0, 4.8 Hz, 1 H), 2.74 (dt, J = 13.2, 4.8 Hz, 1 H), 2.59 (dd, J = 14.0, 6.7 Hz, 1 H), 2.40 (ddd, J = 14.0, 10.6, 4.0 Hz, 1 H), 2.10 (dd, J = 17.7, 8.7 Hz, 1 H), 1.98 (2 H, mono hydrate H20), 1.88 (dd, J 8.2, 5.9 Hz, 1 HO, 1.74 (m, 3 H), 1.61 (m, 1 H), 1.50 (m, 3 H), 1.42 (dd, J = 9.6, 5.8 Hz, 1 H), 1.22 (m, 2 H), 1.07 (s, 9 H), 0.95 (m, 4 H), 0.69 (m, 1 H), 0.47 (m, 1 H).
1 C NMR (100 MHz, CDC13) δ 173.5, 172.1, 169.1, 160.4, 157.7, 154.9, 148.4, 141.0, 134.3, 132.7, 129.1, 118.8, 118.7, 106.5, 74.4, 59.6, 59.4, 55.8, 55.5, 54.9, 41.8, 35.4, 35.3, 35.2, 34.3,. 31.2, 30.7, 29.5, 28.6, 28.2, 26.6, 22.6, 18.7, 11.2, 6.31, 6.17.
HPLC conditions: Ascentis Express Column, 10 cm x 4.6 mm, 2.7 μηι; Column temperature of 40°C; Flow rate of 1.8 mL/min; and Wavelength of 215 nm.
Gradiant: mm 0.1% ¾PO4
0 20 80
5 55 45
15 55 45
25 95 5
27 95 5
27.1 20 80
32 20 80
Retention time: mm.
Compound A 14.50
Example 18: Preparation of Compound A, Method B

To a 50-L flask equipped with overhead stirring was added macrocyclic acid (1.06 kg crude, 1.00 eq), amine-pTSA (862 g crude, 1.12 eq) and MeCN (7.42 L) at 19°C. The slurry was cooled in a water bath, pyridine (2.12 L, 13.8 eq) was added, aged 15 min, and then added EDC (586 g, 1.60 eq) in one portion, aged 1.5 h, while it turned into a clear homogeneous solution.
The solution cooled in a water bath, then quenched with 2 N HC1 (1.7 L), and seeded (9.2 g), aged 15 min, and the rest of the aqueous HC1 was added over 2.5 h. A yellow slurry was formed. The slurry was aged overnight at RT, filtered, washed with MeCN/water (1 : 1 v/v, 8 L), to obtain Compound A (Hydrate II).
Compound A was dissolved in acetone (4 L) at RT, filtered and transferred to a
12-L round-bottom flask with overhead stirring, rinsed with extra acetone (1 L), heated to 50°C, water (0.9 L) was added, seeded with Compound A monohydrate form III (-10 mg), and aged 15 min, and then added water (0.8 L) over 2.5 h, extra water 3.3 v over 2.5 h was added, stopped heating, cooled to RT, aged at RT overnight, filtered, washed with water/acetone (1 : 1 v/v, 4 L), and dried in air under vacuum. Compound A Hydrate III, 670 g, was obtained as an off-white solid.
Example 19: Preparation of Compound A, Method C

Macrocyclic acid hemihydrate from Example 15 (10.16 g, 18.03 mmol) was dissolved in THF (50 ml to 90 mL). The solution was azetropically dried at a final volume of 100 mL. Sulfonamide pTSA salt (7.98 g, 19.83 mmol) was added, followed by DMAc (15 mL), at RT. The batch was cooled to 0° to 10°C, and pyridine (10 mL) was added dropwise. Then, EDC HC1 (4.49 g, 23.44 mmol) was added (in portions or one portion) at 0°C to 10°C. The reaction mixture was aged at 0°C to 10°C for 1 h, and then warmed to 15°C to 20°C for 2 h to 4 h. THF (50 mL) was added, followed by 15 wt% citric acid in 15 wt% aq. NaCl (50 mL), while the internal temperature was maintained at < 25°C with external cooling. The separated organic phase was washed with 15 wt% citric acid in 1 % aq. NaCl (40 mL), followed by 15% NaCl (40 mL). The organic phase was solvent-switched to acetone at a final volume of ~75 mL Water (1 1 mL to 12 mL) was added dropwise at 35°C to 40°C. The batch was seeded with Compound A monohydrate form III (~20 mg) and aged for 0.5 h to 1 h at 35°C to 40°C.
Additional water (22 mL) was added dropwise over 2 h to 4 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 60% acetone in water (40 mL x 2). Suction drying at RT or vacuum-oven drying at 45°C gave Compound A monohydrate form III as a white solid.
Example 20: Preparation of Compound A, Method D

Macrocyclic acid hemihydrate from Example 12 (10 g, 98.4wt%, 17.74 mmol) was dissolved in THF (70 mL). The solution was azetropically dried at a final volume of ~60 mL. Sulfonamide pTSA salt (7.53 g, 18.7 mmol) was added at RT. The batch was cooled to 0°C to 5°C, and pyridine (1 1.4 mL) was added dropwise. Then, EDC HC1 (4.26 g, 22.2 mmol) was added in portions at 0°C to 15°C. The reaction mixture was aged at 10°C to 15°C for 2 h to 4 h. 35 wt% Citric acid in 10 wt% aq. NaCl (80 mL) was added, while the internal temperature was maintained at < 25°C with external cooling. The separated organic phase was solvent-switched to acetone at a final volume of ~75 mL. Water (12 mL) was added dropwise at 50°C. The batch was seeded with Compound A monohydrate form III (-300 mg) and aged for 0.5 h to 1 h at 50°C. Additional water (25 mL) was added dropwise over 6 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 65%) acetone in water (40 mL). Suction drying at RT or vacuum-oven drying at 45°C gave Compound A monohydrate form III as a white solid.
Example 24: Ring Closing Metathesis

To a 50 mL 2-neck RB flask with reflux condenser and needle for N2 bubbling was charged the product of Example 20 (1.034 g, 0.869 mmol, 1.0 eq), toluene (20.68 ml, 20X), and the resulting solution was degassed with N2. Hoveyda-Grubbs 2nd generation catalyst (10.90 mg, 0.017 mmol) was charged to the pot, and the system was heated to 80°C with constant sparge of N2, with color change from green to reddish. The reaction was sampled (5 h) and assay by HPLC to be approximately 80% converted. The system was removed from the heat and allowed to stir at RT overnight under N2. The reaction was again assayed and deemed complete by HPLC. Toluene was removed by concentration and the resulting red oil was purified by gradient silica gel chromatography (50 g BlOTAGE SNAP Si gel column; loaded with DCM; eluted with 0 to 10% EtOAc in DCM over 10 column volumes; then 10 to 20% EtOAc in DCM over 3 column volumes; then hold; detect by TLC-UV) to yield a yellow solid, which was further slurried in EtOAc (3 mL) and hexanes (6 mL). The resulting slurry was filtered and washed with 25% EtOAc in hexanes (6 mL) to yield the product (445 mg, 0.754 mmol, 87% yield) as a white solid.


Merck reported interim data from the Phase 2 C-WORTHY study in April 2014 at the International Liver Congress (ILC) in London that evaluated the efficacy and safety of its two-drug regimen based on NS3/4A protease inhibitor MK-5172 and NS5A replication complex inhibitor MK-8742, given with or without ribavirin, in GT1 HCV patients with cirrhosis. The once-daily single pill (without ribavirin) showed a 98% SVR12 (12-week sustained virologic response) in genotype-1, treatment-naive patients. Merck will start the phase III clinical trials (NCT02105688, NCT02105662, NCT02105467 andNCT02105701) for the combination in June 2014.
Development of a Practical, Asymmetric Synthesis of the Hepatitis C Virus Protease Inhibitor MK-5172.Org. Lett. 2013;
15: 4174-4177

MK-5172 is a hepatitis C virus protease inhibitor. Key steps in the synthesis depicted are (1) the regioselective SNAr reaction of dichloroquinoxaline A with prolinol derivative B and (2) construction of the 18-membered macrocycle using a macrolactamization (F → G).
Comment
The medicinal chemistry route to MK-5172 is based on a ring-closing metathesis strategy (S. Harper et al.ACS Med. Chem. Lett. 2012, 3, 332). The best regioselectivity (20:1) and minimization of double substitution in the SNAr reaction of A with B was achieved using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the base in polar solvents such as DMSO, NMP, or DMAc.
SYNTHESIS, THESIS PROCEDURES, NMR see………..http://www.allfordrugs.com/2015/07/31/mk-5172-grazoprevir/
/////////////http://www.allfordrugs.com/2015/07/31/mk-5172-grazoprevir/
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html