PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Telapristone (proposed trade names Proellex and Progenta) is an investigational selective progesterone receptor modulator, tested for treatment of progesterone sensitive myomata.[1] CDB-4124 was originally developed the National Institutes of Health, and as of 2012 is in Phase II clinical trials for uterine fibroids and endometriosis.[2] It also has some antiglucocorticoidactivity
17α-acetoxy-21-methoxy-11β-[4-N,N-dimethylaminophenyl]-19-norpregna-4,9-diene-3,20-dione, (also known as CDB-4124)
17α-acetoxy-21-methoxy-11β-[4-N,N-dimethylaminophenyl]-19-norpregna-4,9-diene-3,20-dione) is a selective progesterone receptor modulator, it is being tested for treatment of progesterone sensitive myomata.
International patent application WO 97/41145disclosed for the first time the preparation of 17α-acetoxy-21-methoxy-11β-[4-N,N-dimethylaminophenyl]-19-norpregna-4,9-diene-3,20-dione). In example 9 it is characterized as light-yellow powder with a melting point of 116° C. (purity: 98.06%, characteristic FT-IR absorption bands at: 1124, 1235, 1370, 1446, 1518, 1612, 1663, 1734, 2940 cm−1).
According to the published international patent applications of WO 01/47945 and WO 01/74840 the obtained 17α-acetoxy-21-methoxy-11β-[4-N,N-dimethylaminophenyl]-19-norpregna-4,9-diene-3,20-dione) was light-yellow powder as well having a melting point of 116° C. (purity: 98.87%, 98.06%, characteristic FT-IR absorption bands at: 1124, 1235, 1370, 1446, 1518, 1612, 1662, 1734, 2940 cm−1)
Preparation of 17α-hydroxy-llβ-[4-(N,N-dimethylamino)phenyl]-21-methoxy- 19-norpregna-4,9-diene-3,20-dione (10) :
A suspension of 2-iodoxybenzoic acid (IBX, 599 mg, 2.14 mmol) in anhydrous dimethylsulfoxide (DMSO) (5.0 mL; Aldrich, Sure-Seal) was stirred magnetically under nitrogen and warmed in an oil bath at 55 – 60°C. After several minutes, all of the IBX was solubilized. To the IBX solution was added a solution of the 20-alcohol (18, 500 mg, 1.07 mmol) in DMSO (5 mL). Additional DMSO (3 mL) was used to rinse in residual 18. After a period V2 hr of reaction, approximately 70% of the 20-alcohol (18) had been converted to the 20-ketone (10), as evidenced by TLC (15% acetone in methylene chloride; aliquot was diluted in water and extracted by EtOAc). After 3 hr, there was no observable change in the conversion. The reaction mixture was transferred to a separatory funnel, diluted with water, and extracted by EtOAc (3x). The EtOAc extracts were washed with additional water (2x) and brine (lx). The combined extracts were dried by filtration through sodium sulfate, evaporated in vacuo, and dried overnight under high vacuum to recover 600 mg of a brown film. The film product was taken up in EtOAc and filtered through silica on a sintered glass funnel to remove residual DMSO and highly polar impurities. Evaporation of EtOAc afforded 450 mg of a yellow film. Repeated trituration with hexane, with scratching and sonicating, produced a solid. The solid was dried overnight under high vacuum to give 349 mg of 10 as a yellow powder in 70.1% yield. The product was carried directly to the next reaction without further purification. NMR (300 MHz, CDCI3) : δ 0.408 (s, 3 H, C18-CH3),2.906 (s, 6 H, -N(CH3)2), 3.454 (s, 3 H, C21-OCH3), 4.245 and 4.388 (AB, 2 H, C21-CH2, JAB = 17.41 Hz), 4.378 (d, 1 H, Cllβ-CH, J = 7.50), 5.758 (s, 1 H, C4-CH), 6.638 (d, 2 H, 3′,5′-aromatic CH, J = 8.55 Hz) and 6.975 (d, 2 H, 2′,6′-aromatic CH, J = 8.55 Hz).
Preparation of 17α-acetoxy-llβ-[4-(N,N-dimethylamino)phenyl]-21-methoxy- 19-norpregna-4, 9-diene-3,20-dione(11) :
A mixture of trifluoroacetic anhydride (47 mL) and glacial acetic acid (19.1 mL) in methylene chloride (300 mL) was allowed to stir at room temperature under nitrogen. After 1/2 hr of stirring, the mixture was cooled to 0°C in an ice water bath and tosic acid (2.85 g, 14.98 mmol) was added. A solution of the 17α-hydroxy compound (10, 6.18 g, 13.33 mmol) was added in 50 mL of methylene chloride and rinsed in with additional CH2CI2 (50 mL). After stirring for a period of 2 hr at 0°C, examination by TLC (silica; 10% acetone in methylene chloride; neutralized with NH4OH before developing) indicated that the reaction was >95% complete. The reaction mixture was diluted with water (300 mL) and neutralized by careful addition of concentrated NH4OH (75 mL).
More NH4OH was added to a pH of 7 as indicated by a pH paper. The product obtained was extracted by CH2CI2 (3x) and the organic extracts were washed with water (2x) and brine (lx). The combined organic extracts were dried by filtration through Na2SO4 and evaporated in vacuo to give 7.13 g of the crude product (11). A pure material was obtained by flash column chromatography (silica; 10% acetone in methylene chloride). The impure fractions were combined and chromatographed a second time. The pure fractions from both chromatographic runs were combined and evaporated in vacuo, then evaporated from ether, and further dried under high vacuum to produce a pale yellow foam. Treatment with pentane followed by scratching and sonicating produced 4.13 g of 11 as a fine yellow powder in 61.3% yield; m.p. softens at 116°C.
Analysis by a reverse phase HPLC on a NOVAPAK™ Cι8 column eluted with 70% CH3OH in water with 0.03% Et3N at a flow rate of 1 mL per min and at λ = 302 indicated 98.87 % purity of 11 with retention time tR = 6.45 min.
According to the above mentioned facts, there is no such known process, which is suitable for the realization of the synthesis of CDB-4124 on industrial scale using simple reaction conditions. Our aim was to elaborate a process, which is easy to scale-up, the industrial realization of which is safe, economical and the purity of the active ingredient fulfils the requirements of the pharmacopoeia.
Surprisingly it was found, that the following process fulfils the above mentioned requirements: i) epoxide formation on the double bond in position 5(10) of 3,3-[l,2-ethandiyl- bis(oxy)]-oestr-5(10),9(l l)-dien-17-one of formula (II)
with hydrogen peroxide; ii) addition of hydrogen cyanide formed in situ on position 17 of the obtained 5,1 Oa- epoxy-3,3-[l,2-ethandiyl-bis(oxy)]-5α-oestr-9(l l)-en-17-one of formula (III)
iii) silylation of the hydroxyl group in position 17 of the formed 5,10α-epoxy-3,3-[l,2- ethandiyl-bis(oxy)]-17α-hydroxy-5α-oestr-9(l l)-en-17β-carbonitrile of formula (IV)
with trimethyl chlorosilane; iv) reacting the obtained 5,10α-epoxy-3,3-[l,2-ethandiyl-bis(oxy)]-17-[trimethyl-silyl- oxy]-5α-oestr-9(ll)-en-17β-carbonitrile of formula (V)
with 4-(dimethylamino)-phenyl magnesium bromide Grignard reagent in the presence of CuCl
(Teutsch reaction); v) silylation of the hydroxyl group in position 5 of the formed 1 lβ-[4-(dimethyl-amino)- phenyl]-3 ,3-[ 1 ,2-ethandiyl-bis(oxy)] -5-hydroxy- 17α-[trimethylsilyl-(oxy)] -5α-oestr-9-en- 17β- carbonitrile of formula (VI)
with trimethyl chlorosilane; vi) reacting the obtained llβ-[4-(dimethylamino)-phenyl]-3,3-[l,2-ethandiyl-bis(oxy)]- 5,17α-bis-[trimethyl-silyl-(oxy)]-5α-oestr-9-en-l 7β-carbonitrile of formula (VII)
with diisobutyl aluminum hydride and after addition of acid to the reaction mixture vii) methoxy-methylation of the obtained llβ-[4-(dimethylamino)-phenyl]-3,3-[l,2- ethandiyl-bis(oxy)]-5,17α-bis-[trimethyl-silyl-(oxy)]-5α-oestr-9-en-17β-carbaldehide of formula (VIII)
with methoxy-methyl Grignard reagent formed in situ, while hydrolyzing the trimethylsilyl protective groups; viii) oxidation of the hydroxy! group in position 20 of the obtained 17,20ξ-dihydroxy-
3-[4-(dimethylamino)-phenyl]-21 -methoxy- 19-norpregna-4,9-dien-3-one of formula (IX)
with dicyclohexyl carbodiimide in the presence of dimethyl sulfoxide and a strong organic acid (Swern oxidation), and in given case after purification by chromatography ix) acetylation of the hydroxyl group in position 17 of the obtained l lβ-[4- (dimethylamino)-phenyl]- 17-hydroxy-21 -methoxy- 19-norpregna-4,9-dien-3 ,20-dione of formula (X)
with acetic anhydride in the presence of perchloric acid, and in given case the obtained 7- acetoxy-11 β-[4-(dimethylamino)-phenyl)]-21-methoxy-19-norpregna-4,9-dien-3 ,20-dione of formula (I) is purified by chromatography.
Example 11
17-Acetoxy-llβ-f4-(dimethylamino)-phenyl)1-21-methoxy-19-norpregna-4,9-dien-3,20- dione [compound of formula (Dl 70 % Perchloric acid (6 ml) was added to stirred and cooled ((-20) – (-25) 0C) acetic anhydride (45 ml) at such a rate to keep the temperature below (-15) °C. Then a solution of l lβ-[4-(dimethylamino)-phenyl)]-17-hydroxy-21-methoxy-19-norpregna-4,9-dien-3,20-dione (15.5 g) in dichloromethane (60 ml) was added at (-20) – (-25) 0C. After completion of the reaction – followed by thin layer chromatography – the reaction mixture was diluted with dichloromethane (50 ml), cooled to (-10) 0C and ion exchanged water (52 ml) was added to decompose the acetic anhydride. After stirring for 10 min 25 % ammonium hydroxide solution (77 ml) was added at such rate to keep the temperature below 25 0C (pH=7-8). Then the precipitated carbamide by-product was filtered off, the aqueous phase was separated, extracted with dichloromethane (2×30 ml) and the combined organic layers were concentrated to yield 16.2 g (95.8 %) of the title compound, which was purified by HPLC according to method described in the next example. NMR: 1H NMR C500 MHz. CDCl1 (TMS), δ (ppmT): 0.40 (3H, s, 18-CH3); 2.10 (3H5 s, O-CO- CH3); 2.90 (6H, s, N-CH3); 3.41 (3H, s, 0-CH3); 4.09 (IH, d, Hx-21); 4.38 (IH, m, H-Il); 4.29 (IH, d, Hy-21); 5.77 (IH, br, H-4); 6.62 (2H5 m, H-3′ & H-5′); 6.96 (2H, m, H-2′ & H-6′) 13C NMR (125 MHz. CDCU (TMS), δ fppmϊ): 15.6 (C-18); 21.1 (0-CO-CH3); (39.3 (C-Il); 40.6 (N-CH3); 59.4 (0-CH3); 76.0 (C-21); 93.9 (C-17); 112.8 (C-3′ & C-5′); 123.0 (C-4); 127.3 (C-2′ & C-6′); 129.4 (C-IO); 131.3 (C-I’); 145.5 (C-9); 148.7 (C-4′); 156.4 (C-5); 170.7 (0-CO-CH3); 199.4 (C-3); 202.7 (C-20)
Example 12 Purification of crude CDB-4124 by HPLC (eluent: cyclohexanermethyl-tert-butyl- ether;acetone = 60:30:10) (laboratory scale) [compound of formula (DI
Silicagel (51O g, ZEOPREP C-GEL C-490L, 15-35 μm of particle size; bed length about 60 cm) was filled to an axial bed compression HPLC column of 5 cm of diameter with slurry packing method and the column was equilibrated with a 60:30:10 mixture of cyclohexane – methyl-tert-butyl ether – acetone eluent. 5.1 g of the crude compound of formula (I) (CDB-4124) obtained in the previous example (content of impurities: less than 4 %) was dissolved in the eluent (100 ml), filtered and injected on the column. The product was eluted with 85 ml/min flow rate and UV detection was used. The first fraction was about 40 ml, the main fraction containing the pure CDB-4124 was about 560 ml. The solid title compound was obtained by concentration of the eluted main fraction. Yield: 4.25 g (83.33 %), content of impurities: less than 0.5 %. Melting point: 1180C.
Attardi BJ, Burgenson J, Hild SA, Reel JR (2004). “In vitro antiprogestational/antiglucocorticoid activity and progestin and glucocorticoid receptor binding of the putative metabolites and synthetic derivatives of CDB-2914, CDB-4124, and mifepristone”. J Steroid Biochem Mol Biol88 (3): 277–88. doi:10.1016/j.jsbmb.2003.12.004. PMID15120421.
Industrial method for the synthesis of 17-acetoxy-11[beta][4-(dimethylamino)-phenyl]-21-methoxy-19-norpregna-4,9-dien-3,20-dione and the key intermediates of the process
Onapristone is a progesterone receptor antagonist in phase II clinical trials at Arno Therapeutics for the treatment of breast cancer and for the treatment of men with advanced castration-resistant prostate cancer (CRPC) after failure of abiraterone or enzalutamide. Early clinical studies are underway for the treatment of post-menopausal women with progesterone receptor (PR) positive tumors. In 2012, the product was licensed to Arno Therapeutics by Invivis Pharmaceuticals on an exclusive worldwide basis.
Mifepristone Onapristone Asoprisnil(ZK-98299)
Proellex ORG-33628 Lonaprisan
……………..syn 1
The Grignard reaction of the protected epoxide (I) with 4-(dimethylamino)phenylmagnesium bromide (II) gives the 11-substituted compound (III), which is submitted to an Oppenhauer oxidation with cyclohexanone and aluminum isopropoxide yielding the 17-keto derivative (IV). The photochemical epimerization of the 13beta-methyl of (IV) with a high pressure mercury lamp in dioxane affords the epimer (V), which is condensed with 1-tetrahydropyranyloxy-2-propyne (VI) by means of butyllithium in THF to give the acetylenic alcohol (VII). Finally, this compound is reduced with hydrogen over Pd/C in ethanol and deprotected and dehydrated by treatment with hot aqueous acetic acid.
…………………..
Image: Micrograph of prostate adenocarcinoma, acinar type, the most common type of prostate cancer. Photo: courtesy of Nephron.
US-based clinical stage biopharmaceutical firm Arno Therapeutics (ARNI) has started enrolling patients in a Phase I/II trial (NCT02049190) assessing its oral, anti-progestin hormone blocker ‘onapristone’ in men with advanced castration-resistant prostate cancer (CRPC) after failure of abiraterone or enzalutamide.
In previous Phase II clinical trials, onapristone has shown to exhibit anti-tumour activity in patients with breast cancer.
The pre-clinical testing has showed that onapristone had blocked the activation of the progesterone receptor (PR), which is believed to be a mechanism that inhibits the growth of APR-driven breast, endometrial and other tumours.
The company said that tests for the activated form of the progesterone receptor (APR) have the potential to function as a biomarker of anti-progestin activity, as detected by a companion diagnostic under development.
Enrolment of patients in the randomised, open-label Phase I/II trial follows approval of an Investigational Medicinal Product Dossier from the UK Health Authority, Medicines and Healthcare products Regulatory Agency (MHRA), ethics committee authorisation and subsequent site authorisation.
Arno Therapeutics president and chief executive officer Glenn Mattes said globally, prostate cancer is the second most common cancer in men, and the fifth leading cause of death from cancer in men, with an estimated 1.1 million new cases diagnosed and 307,000 deaths during 2012 alone, according to the International Agency for Research on Cancer.
“These numbers are staggering, and our ultimate goal is to evaluate onapristone in the subset of advanced CRPC patients who are more likely to respond to this personalised treatment, for which there is an immense unmet medical need,” Mattes said.
“The trial marks Arno’s second Phase I study actively enrolling this year and we are excited by the momentum generated thus far.”
The Phase I/II trial, designed to assess the safety and anti-cancer activity of onapristone in the select patient population, is being carried out at the Institute of Cancer Research, London, and the Royal Marsden NHS Foundation Trust in the UK.
A total of 60 patients will be enrolled in the trial, which additional sites are planned for in the UK.
The company has engaged Biotrial, a drug evaluation and pharmacology research company, as its contract research organisation (CRO) for the Phase I/II trial.
The trial will evaluate onapristone in extended-release tablet formulations in up to five dose levels (10mg-50mg BID) in patients with advanced CRPC where PR may be contributing to tumour progression.
Patients in the trial will be evaluated for whether their tumours express APR, which may help identify patients who are more likely to respond to onapristone.
A second group of patients will be included at the recommended Phase II dose to gain additional understanding of the onapristone safety profile and potential anti-cancer activity.
MK-8742 is in phase II clinical development at Merck & Co. for the oral treatment of chronic hepatitis C infection in combination with MK-5172 and ribavirin. Phase I clinical trials are uongoing for the treatment of hepatitis C infected males. In 2013, breakthrough therapy designation was assigned to the compound.
MK-8742 is an inhibitor of Hepatitis C Virus (HCV) non-structural protein 5A (NS5A) that is being developed for the treatment of HCV infection. MK-8742 has broad, potent HCV genotypic activity in vitro against viral variants that are resistant to other NS5A inhibitors. MK-8742 exhibits potent antiviral activity during 5 days of monotherapy in patients with GT1 and GT3 chronic HCV infection. MK-8742 is currently in Phase IIB development.
ELBASVIR
MK-8742 is an inhibitor of Hepatitis C Virus (HCV) non-structural protein 5A (NS5A) that is being developed for the treatment of HCV infection. MK-8742 has broad, potent HCV genotypic activity in vitro against viral variants that are resistant to other NS5A inhibitors. MK-8742 exhibits potent antiviral activity during 5 days of monotherapy in patients with GT1 and GT3 chronic HCV infection. MK-8742 is currently in Phase IIB development.
A mixture of Compound Int-19b (1.1 g, 3 mmol), (dibromomethyl)benzene (2.25 g, 9 mmol) and K2C03 (1.2 g, 9 mmol) in 15 mL of DMF was heated to 100 °C and allowed to stir at this temperature for 3 hours. The reaction mixture was cooled to room temperature, concentrated in vacuo and the residue obtained was dissolved with
dichloromethane and water. The aqueous phase was extracted with dichloromethane. The combined organic extracts were washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using flash column
chromatography on silica gel to provide Compound Int-23a (380 mg, 28 %) as a white solid. 1H MR (CDCI3): δ 7.72 (bs, 1 H), 7.44 – 7.46 (d, J= 8.4 Hz, 1 H), 7.21 – 7.28 (m, 3 H), 7.09 – 7.12 (m, 3 H), 7.04 (s, 1 H), 6.99 – 7.01 (bs, J= 6.8 Hz, 2 H), 6.78 (s, 1 H), 6.63 – 6.65 (d, J = 8.4 Hz, 1 H). MS (ESI)
m/e (M+H+): 456. Step B – Pre aration of Compound Int-23b
lnt-23a lnt-23b
To a solution of Int-23a (456 mg, 1.0 mmol) in 1,4-dioxane was added bis pinacol borate (2.2 mmol) , Pd(dppf)Cl2 (0.04 mmol) and KOAc (4 mmol). The reaction mixture was put under N¾ heated to 110°C and allowed to stir at this temperature for 3 hours. The reaction mixture was cooled to room temperature, concentrated in vacuo, and the residue obtained was purified using column chromatography on silica gel to provide Compound Int- 23b (590 mg, 87 % yield). 1H MR (CDC13): δ 8.13 (s, 1 H), 7.60 (d, J= 7.6 Hz, 1 H), 7.52 (d, J= 8.0 Hz, 1H), 7.36 – 7.39 (m, 1 H), 7.14 -7.19 (m, 4 H), 6.93 – 6.95 (m, 3 H), 6.90 (s, 1 H), 1.26 – 1.29 (s, 24 H). MS (ESI) m / e (M+H+): 550.
– Pre aration of Compound Int-23c
lnt-23b lnt-23c
A suspension of Int-23b (550 mg, 1.0 mmol), tert-butyl 2-(2-bromo-lH- imidazol-5-yl) pyrrolidine- 1-carboxylate (2.4 mmol), Pd(dppf) Cl2 (200 mg), Na2C03 (3 mmol) and in THF/H20 (10: 1, 33 mL) was allowed to stir at reflux for about 15 hours under N2. The reaction mixture was cooled to room temperature and filtered, and the filtrate was washed with water (50 mL) and extracted with EtOAc (100 mL). The organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified using column chromatography on silica gel to provide Compound Int-23c (160 mg). MS (ESI) m / e (M+H+): 768.
Preparation of Compound Int-23d
Int-23c (0.10 g, 0.13 mmol) was added to HCl/CH3OH (5 mL, 3M) and the resulting reaction was allowed to stir at room temperature for about 3 hours. The reaction mixture was then concentrated in vacuo to provide Compound Int-23d, which was used without further purification. MS (ESI) m / e (M+H+): 568.
– Preparation of Compound A
To a solution of Int-23d (56.8 mg, 0.10 mmol), (S)-2- (methoxycarbonylamino)-3-methylbutanoic acid (35.0 mg, 0.20 mmol) and DIPEA (0.8 mmol) in CH3CN (1 mL) was added BOP (98 mg, 0.22 mmol). The resulting reaction was allowed to stir at room temperature and monitored using LCMS. After LCMS showed the starting material to be consumed, the reactionmixture was filtered, and the filtrate was purified using HPLC to provide Compound A as a white solid. 1H MR (MeOD): δ 7.94 (s,
The NS5A protein plays a critical role in the replication of HCV and has been the focus of numerous research efforts over the past few years. NS5A inhibitors have shown impressive in vitro potency profiles in HCV replicon assays, making them attractive components for inclusion in all oral combination regimens. Early work in the NS5A arena led to the discovery of our first clinical candidate, MK-4882 [2-((S)-pyrrolidin-2-yl)-5-(2-(4-(5-((S)-pyrrolidin-2-yl)-1H-imidazol-2-yl)phenyl)benzofuran-5-yl)-1H-imidazole]. While preclinical proof-of-concept studies in HCV-infected chimpanzees harboring chronic genotype 1 infections resulted in significant decreases in viral load after both single- and multiple-dose treatments, viral breakthrough proved to be a concern, thus necessitating the development of compounds with increased potency against a number of genotypes and NS5A resistance mutations. Modification of the MK-4882 core scaffold by introduction of a cyclic constraint afforded a series of tetracyclic inhibitors, which showed improved virologic profiles. Herein we describe the research efforts that led to the discovery of MK-8742, a tetracyclic indole-based NS5A inhibitor, which is currently in phase 2b clinical trials as part of an all-oral, interferon-free regimen for the treatment of HCV infection.
see
Journal of Medicinal Chemistry (2014), 57(5), 1643-1672.
Factor Xa (FXa) is an essential blood coagulation factor[2] that is responsible for the initiation of the coagulation cascade. FXa cleaves prothrombin to its active form thrombin, which then acts to convert soluble fibrinogen to insoluble fibrin and to activateplatelets. Stabilization of the platelet aggregation by fibrin mesh ultimately leads to clot formation.[4]
Metabolism
Darexaban is rapidly absorbed and extensively metabolized in the liver to its active metabolite, darexaban glucuronide (YM-222714) during first pass metabolism via glucuronidation.[5] The metabolism of darexaban also occurs in the small intestine but to a much lesser extent.[2] Glucuronidation of darexaban occurs quickly, thus the half life of darexaban itself is short. However, the resultant darexaban glucuronide metabolite has a long half life of approximately 14-18 hours, reaching its maximum levels in the blood 1-1.5 hour post dose.[2] As a result, darexaban glucuronide is the main determinant of the antithrombotic effects.[3] Darexaban shows minimal interaction with food and is excreted through the kidneys (urine) and feces.[6]
Mechanism of action
Darexaban and darexaban glucuronide selectively and competitively inhibit FXa, suppressing prothrombin activity at the sites of blood clot (thrombus) formation. This leads to a decrease in blood clot formation in a dose dependent manner.[2] Reducing blood clot formation will decrease blood flow blockages, thus possibly lowering the risk of myocardial infarction, unstable angina, venous thrombosis, and ischemic stroke.[7]
Clinical uses
Atrial fibrillation
Atrial fibrillation is an abnormal heart rhythm that causes a reduction in the cardiac output and blood flow to the brain. It also promotes the formation of blood clots in the atria.[4]Atrial fibrillation is associated with an increased risk of embolic stroke due to the increased risk of blood clot development.[8] Oral anticoagulant drugs such as Darexaban decrease the incidence and severity of stroke in patients with atrial fibrillation by preventing the formation of blood clots.[9]
Contraindictions
The RUBY-1 phase II trial results show that oral administration of darexaban in combination with the standard dual antiplatelet therapy used for ACS patients caused a two- to four-fold increase in bleeding rates and no effect on ACS.[6] Though there were no cases of fatal bleeding or intracranial haemorrhage, the results of this study questions the concept of adding an oral anticoagulant to standard of care dual antiplatelet therapy in order to prevent recurrent ischemic events after ACS. The developpement of darexaban was discontinued in september 2011.
References
Eriksson, B., et al. “A dose escalation study of YM150, an oral direct factor Xa inhibitor, in the prevention of venous thromboembolism in elective primary hip replacement surgery.” Journal of Thrombosis and Haemostasis (2007): 1660-1665
Yoshiyuki, I., et al. “Biochemical and pharmalogical profile of darexaban, an oral direct Xa inhibitor.” European Journal of Pharmacology (2011): 49-55
Toshifumi, S., et al. “Identification of UDP-Glucuronosyltransferases Responsible for the Glucuronidation of Darexaban, an Oral Factor Xa Inhibitor, in Human Liver anD Intestine.” The American Society for Pharmacology and Experimental Therapeutics (2011): 278-282
Katsung, B., S. Masters and A. Trevor. Basic and Clinical Pharmacology 11th Edition. United States of America: McGraw-Hill, 2009
Turpie, A., et al. “Prevention of venous thromboembolism with an oral factor Xa inhibitor, YM150, after total hip arthoplasty. A dose finding study (ONYX-2).” Journal of Thrombosis and Haemostasis (2010): 714-721
Hirayama, F., et al. “Discovery of N-[2-Hydroxy-6-(4-methoxybenamido)phenyl]-4-(4-methyl-1,4-diazepan-1-yl)benzamide (Darexaban, YM150) as a Potent and Orally Available Factor Xa Inhibitor.” Journal of Medicinal Chemistry (2011): 8051-8065
Zhong, Y., et al. “Atrial Fibrillation as a Risk Factor for Stroke: A Retrospective Cohort Study of Hospitalized Medicare Beneficiaries.” American Journal of Public Health (1998): 395-400
Hylek, E., et al. “Effect of intesity of oral anticoagulation on stroke severity and mortality in atrial fibrillation.” The New England Journal of Medicine (2003): 1019-26
In December 2021, the U.S. Food and Drug Administration approved cabotegravir for pre-exposure prophylaxis (PrEP) in at-risk people under the brand name Apretude.[11]
GSK744 (also known as S/GSK1265744) is an investigational new drug under development for the treatment of HIV infection. It is anintegrase inhibitor, with a carbamoyl pyridone structure similar to dolutegravir. In investigational studies, the agent has been packaged into nanoparticles (GSK744LAP) conferring an exceptionally long half-life of 21–50 days following a single dose. In theory, this would make possible suppression of HIV with dosing as infrequently as once every three months.[1]
S-265744 LAP is in phase II clinical development at Shionogi-GlaxoSmithKline for the treatment of HIV infection. Phase III clinical trials had been ongoing for this indication; however, no recent development has been reported for this study.
Cabotegravir, or GSK1265744, is an HIV-1 integrase inhibitor that is prescribed with the non-nucleoside reverse transcriptase inhibitor, rilpivirine.4,6,7 Early research into cabotegravir showed it had lower oral bioavailability than dolutegravir,4 which resulted in the development of long acting monthly intramuscular injection formulation for cabotegravir.4,7
Cabotegravir was granted FDA approval on 21 January 2021 in combination with rilpivirine to treat HIV-1 infection in virologically suppressed individuals.8 While previously administered once monthly only, this combination product was granted FDA approval for dosing every two months on February 01, 2022 11 and without the need for an oral lead-in period prior.7
The human immunodeficiency virus (“HIV”) is the causative agent for acquired immunodeficiency syndrome (“AIDS”), a disease characterized by the destruction of the immune system, particularly of CD4+ T-cells, with attendant susceptibility to opportunistic infections, and its precursor Al DS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. HIV is a retrovirus; the conversion of its RNA to DNA is accomplished through the action of the enzyme reverse transcriptase. Compounds that inhibit the function of reverse transcriptase inhibit replication of HIV in infected cells. Such compounds are useful in the prevention or treatment of HIV infection in humans.
A required step in HIV replication in human T-cells is the insertion by virally-encoded integrase of proviral DNA into the host cell genome. Integration is believed to be mediated by integrase in a process involving assembly of a stable nucleoprotein complex with viral DNA sequences, cleavage of two nucleotides from the 3′ termini of the linear proviral DNA and covalent joining of the recessed 3′ OH termini of the proviral DNA at a staggered cut made at the host target site. The repair synthesis of the resultant gap may be accomplished by cellular enzymes. There is continued need to find new therapeutic agents to treat human diseases. HIV integrase is an attractive target for the discovery of new therapeutics due to its important role in viral infections, particularly HIV infections. Integrase inhibitors are disclosed in WO2006/116724.
(3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, a compound of formula (I), also referred to as compound (I), has proven antiviral activity against human immunodeficiency virus (HIV).
The present invention features pharmaceutical compositions comprising the active ingredient (3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, or a pharmaceutically acceptable salt thereof, suitable for administration once monthly or longer.
Medical uses
Cabotegravir in combination with rilpivirine is indicated for the treatment of human immunodeficiency virus type-1 (HIV-1) in adults.[1][5] The combination injection is intended for maintenance treatment of adults who have undetectable HIV levels in the blood (viral load less than 50 copies/mL) with their current antiretroviral treatment, and when the virus has not developed resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs) and integrase strand transfer inhibitors.[5] The tablets are used to check whether a person tolerates the treatment before the injection therapy is started.[12][5]
The two medicines are the first antiretroviral drugs that come in a long-acting injectable formulation.[12]
Cabotegravir (Apretude) is indicated for use in at-risk people weighing at least 35 kilograms (77 lb) for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV.[11]
Contraindications and interactions
Cabotegravir must not be combined with the drugs rifampicin, rifapentine, carbamazepine, oxcarbazepine, phenytoin or phenobarbital, which induce the enzyme UGT1A1.[5] These drugs significantly decrease cabotegravir concentrations in the body and thus may reduce its effectiveness.[9][5] Additionally, they induce the enzyme CYP3A4, which leads to reduced rilpivirine concentrations in the body.[5][13][14][15] Additionally, patients who are breastfeeding or plan to breastfeed should not take Cabotegravir because it is not known if it will pass within the breast milk.[16]
Adverse effects
The most common side effects of the injectable combination therapy with rilpivirine are reactions at the injection site (in up to 84% of patients) such as pain and swelling, as well as headache (up to 12%) and fever or feeling hot (in 10%). For the tablets, headache and a hot feeling were slightly less frequent. Less common side effects (under 10%) for both formulations are depressive disorders, insomnia, and rashes.[9]
Pharmacology
Mechanism of action
Cabotegravir is an integrase strand transfer inhibitor. This means it blocks the HIV’s enzyme integrase, thereby preventing its genome from being integrated into the human cells’ DNA.[9] As this is a necessary step for the virus to replicate, its further spread is hampered.[9]
When taken by mouth, cabotegravir reaches highest blood plasma levels after three hours. Taking the drug together with food slightly increases its concentrations in the blood, but this is not clinically relevant. After injection into the muscle, cabotegravir is slowly absorbed into the bloodstream, reaching its highest blood plasma levels after about seven days.[9]
Over 99% of the substance are bound to plasma proteins. The drug is inactivated in the body by glucuronidation, mainly by the enzyme UGT1A1, and to a much lesser extent by UGT1A9. More than 90% of the circulating substance are the unchanged cabotegravir, however. The biological half-life is 41 hours for the tablets and 5.6 to 11.5 weeks for the injection.[9]
Elimination has only been studied for oral administration: Most of the drug is eliminated via the faeces in unchanged form (47%). It is not known how much of this amount comes from the bile, and how much was not absorbed in the first place. (The bile actually contains the glucuronide, but this could be broken up again in the gut lumen to give the parent substance that is observed in the faeces.) To a lesser extent it is excreted via the urine (27%), almost exclusively as the glucuronide.[9]
Pharmacogenomics
UGT1A1 poor metabolizers have 1.3- to 1.5-fold increased cabotegravir concentrations in the body. This is not considered clinically significant.[9]
Chemistry
Cabotegravir is a white to off-white, crystalline powder that is practically insoluble in aqueous solutions under pH 9, and slightly soluble above pH 10. It is slightly acidic with a pKa of 7.7 for the enolic acid and 1.1 (calculated) for the carboxamide. The molecule has two asymmetric carbon atoms; only one of the four possible configurations is present in the medication.[18]
Formulation
In studies, the agent was packaged into nanoparticles (GSK744LAP) conferring a biological half-life of 21 to 50 days[citation needed] following a single dose. The marketed injection achieves its long half-life not via nanoparticles but with a suspension of the free cabotegravir acid. The tablets contain cabotegravir sodium salt.[18]
History
Cabotegravir was examined in the clinical trials HPTN 083 and HPTN 084.[19][20] In 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vocabria intended for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in combination with rilpivirine injection.[21] The EMA also recommended marketing authorization be given for rilpivirine and cabotegravir injections to be used together for the treatment of people with HIV-1 infection.[12] Cabotegravir was approved for medical use in the European Union in December 2020.[8]
In 2020, results for some studies were released showing success in using injectable cabotegravir for long-acting pre-exposure prophylaxis (PrEP) with greater efficacy than the emtricitabine/tenofovir combination being widely used for PrEP at the time.[24][25]
The safety and efficacy of cabotegravir to reduce the risk of acquiring HIV were evaluated in two randomized, double-blind trials that compared cabotegravir to emtricitabine/tenofovir, a once daily oral medication for HIV PrEP.[11] Trial 1 included HIV-uninfected men and transgender women who have sex with men and have high-risk behavior for HIV infection.[11] Trial 2 included uninfected cisgender women at risk of acquiring HIV.[11]
In Trial 1, 4,566 cisgender men and transgender women who have sex with men received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections among trial participants taking daily cabotegravir followed by cabotegravir injections every two months compared to daily oral emtricitabine/tenofovir.[11] The trial showed participants who took cabotegravir had 69% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In Trial 2, 3,224 cisgender women received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections in participants who took oral cabotegravir and injections of cabotegravir compared to those who took emtricitabine/tenofovir orally.[11] The trial showed participants who took cabotegravir had 90% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In December 2021, the U.S. Food and Drug Administration (FDA) approved cabotegravir for pre-exposure prophylaxis.[11] The FDA granted the approval of Apretude to Viiv.[11]
Methods for the preparation of a compound of formula (I) are described in WO 2006/1 16764, WO2010/01 1814, WO2010/068262, and WO2010/068253
3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0,14 mL, 1.74 mmol) and 10 drops of glacial acetic acid.
The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added
to the mixture and the solvents removed in vacuo and the material was purified via
silica gel chromatography (2% CH3OH/CH2CI2 gradient elution) to give
The title compound was made in two steps using a similar process to that described
in example Z-I. 16a (510 mg, 1.08 mmol) and (2«5)-2-amino-1-propanol (0.17 mL, 2,17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give
The starting material of Example A is compound 8, which is identical to formula (Ia). Thus, Example A depicts a process in providing an intermediate for the compound of formula 17 below which is isomeric to the compound ZZ-2 at page 237 of WO 2006/116764 to Brian Johns et al.
14
Example Aa After dissolution of mixture of 320 g of compound 8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 mL of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 °C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 mL of cold 2-propanol and drying provided 167 g of compound 14 (52% yield) as a crystal. 1H NMR(300 MHz1 CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1H), 5.46 (d, J = 10.5 Hz, 1H), 5.23 (d, J = 10.2 Hz, 1H), 5.20 (dd, J = 3.9, 9.6 Hz, 1H), 4.46- 4.34 (m, 1H)1 4.31 (dd, J = 6.6, 8.7 Hz, 1H)1 4.14 (dd, J = 3.9, 12.3 Hz1 1H)1 3.79 (dd, J = 9.9, 12.3 Hz1 1 H), 3.62 (dd, J = 6.9, 8.7 Hz1 1 H), 1.38 (d, J = 6.3 Hz1 3H).
Example Ab
To slurry of 156 g of compound 14 (1.0 eq.) in 780 ml_ of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound 15 (84% yield) as a crystal.
Under carbon mono-oxide atmosphere, a mixture of 163 g of compound 15 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 ml_ of 2,4-difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 11.3 g of
Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O1 the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt. The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound 16 (96% yield) as foam.
Under hydrogen atmosphere, a mixture of 184 g of compound 16 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1), the filtrate was concentrated. After 200 ml_ of AcOEt was added to the residue, filtration afforded crude solid of compound 17. The precipitates were combined and extracted with 4.0 L of CHCl3/MeOH(5/1). After concentration of the CHCI3ZMeOH solution and addition of 250 ml_ of AcOEt to the residue, filtration afforded crude solid of compound 17. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated (three times). After cooling of the residue, filtration and drying provided 132 g of compound 17 (88% yield) as a crystal. 1H NMR(300 MHz, DMSO-cfe) δ 11.47 (brs, 1H), 10.31 (t, J = 6.0 Hz, 1H), 8.46 (s, 1H), 7.40 (td, J = 8.6, 6.9 Hz, 1H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1H), 7.11-7.01 (m, 1H), 5.39 (dd, J = 4.1, 10.4 Hz, 1H), 4.89 (dd, J = 4.2, 12.3 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1H), 4.36-^.22 (m, 1H)1 4.00 (dd, J = 10.2, 12.3 Hz, 1H), 3.67 (dd, J = 6.7, 8.6 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H).
Example Ae
After dissolution of 16.0 g of compound 17 (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml_ of 1N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml_ of EtOH and drying provided 13.5 g of compound 18 (80% yield) as a crystal.
The following examples are intended for illustratation only and are not intended to limit the scope of the invention in any way. Preparation 1 : (3S.11 af?VΛ/-r(2.4-DifluoroDhenvnmethyll-6-hvdroxy-3-methyl-5.7-dioxo- 2,3,5,7, 11 ,11 a-hexahydroM ,31oxazolor3,2-alpyridori ,2-c/1pyrazine-8-carboxamide sodium salt (compound 1 b, scheme 2).
I) MsCI, Et3N
2) DBU
P-1 P-2 P-3
a) Synthesis of 2-methyl-3-[(phenylmethvl)oxvl-4/-/-pvran-4-one (compound P-2). To a slurry of 2000 g of compound P-1(1.0 eq.) in 14.0 L of MeCN were added 2848 g of benzyl bromide(1.05 eq.) and 2630 g of K2CO3(1.2 eq.). The mixture was stirred at 80 0C for 5 h and cooled to 13°C. Precipitate was filtered and washed with 5.0 L of MeCN. The filtrate was concentrated and 3.0 L of THF was added to the residue. The THF solution was concentrated to give 3585 g of crude compound P-2 as oil. Without further purification, compound P-2 was used in the next step. 1H NMR(300 MHz, CDCI3) δ 7.60 (d, J = 5.7 Hz, 1 H), 7.4-7.3 (m, 5H), 6.37 (d, J = 5.7 Hz, 1 H), 5.17 (s, 2H), 2.09 (s, 3H).
b) Synthesis of 2-(2-hydroxy-2-phenylethyl)-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound P-3). To 904 g of the crude compound P-2 was added 5.88 L of THF and the solution was cooled to -60 0C. 5.00 L of 1.0 M of Lithium bis(trimethylsilylamide) in THF(1.25 eq.) was added dropwise for 2 h to the solution of compound 2 at -60 0C. Then, a solution of 509 g of benzaldehyde(1.2 eq.) in 800 ml. of THF was added at -60 0C and the reaction mixture was aged at -60 0C for 1 h. The THF solution was poured into a mixture of 1.21 L of conc.HCI, 8.14 L of ice water and 4.52 L of EtOAc at less than 2 0C.
The organic layer was washed with 2.71 L of brine (twice) and the aqueous layer was extracted with 3.98 L of EtOAc. The combined organic layers were concentrated. To the mixture, 1.63 L of toluene was added and concentrated (twice) to provide toluene slurry of compound P-3. Filtration, washing with 0.90 L of cold toluene and drying afforded 955 g of compound P-3 (74% yield from compound P-1 ) as a solid. 1H NMR(300 MHz, CDCI3) δ
c) Synthesis of 2-[(£)-2-phenylethenyl]-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound
P-4). To a solution of 882 g of compound P-3 (1.0 eq.) in 8.82 L of THF were added 416 g of Et3N(1.5 eq.) and 408 g of methanesulfonyl chloride(1.3 eq.) at less than 30 0C. After confirmation of disappearance of compound P-3, 440 ml. of NMP and 1167 g of DBU(2.8 eq.) were added to the reaction mixture at less than 30 0C and the reaction mixture was aged for 30 min. The mixture was neutralized with 1.76 L of 16% sulfuric acid and the organic layer was washed with 1.76 L of 2% Na2S03aq. After concentration of the organic layer, 4.41 L of toluene was added and the mixture was concentrated (tree times). After addition of 4.67 L of hexane, the mixture was cooled with ice bath. Filtration, washing with 1.77 L of hexane and drying provided 780 g of compound P-4 (94% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.69 (d, J = 5.7 Hz, 1 H), 7.50-7.25 (m, 10H), 7.22 (d, J = 16.2
Hz, 1 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.41 (d, J = 5.7 Hz, 1 H), 5.27 (s, 2H). d) Synthesis of 4-oxo-3-[(phenylmethyl)oxy]-4H-pyran-2-carboxylic acid (compound P-5). To a mixture of 822 g of compound P-4 (1.0 eq.) and 1 1.2 g of RuCI3-nH2O(0.02 eq.) in 2.47 L of MeCN, 2.47 L of EtOAc and 2.47 L of H2O was added 2310 g of NalO4(4.0 eq.) at less than 25 0C. After aging for 1 h, 733 g of NaCIO2(S-O eq.) was added to the mixture at less than 25 0C. After aging for 1 h, precipitate was filtered and washed with 8.22 L of
EtOAc. To the filtrate, 1.64 L of 50% Na2S203aq, 822 ml. of H2O and 630 ml. of coc.HCI were added. The aqueous layer was extracted with 4.11 L of EtOAc and the organic layers were combined and concentrated. To the residue, 4 L of toluene was added and the mixture was concentrated and cooled with ice bath. Filtration, washing with 1 L of toluene and drying provided 372 g of compound P-5 (56% yield) as a solid. 1H NMR(300 MHz,
e) Synthesis of 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylic acid (compound P-6). A mixture of 509 g of compound P-5 (1.0 eq.) and
407 g of 3-amino-propane-1 ,2-diol(2.5 eq.) in 1.53 L of EtOH was stirred at 65 0C for 1 h and at 80 0C for 6 h. After addition of 18.8 g of 3-Amino-propane-1 ,2-diol(0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 1 h. After addition of 18.8 g of 3-amino- propane-1 ,2-diol (0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 30 min. After cooling and addition of 509 ml. of H2O, the mixture was concentrated. To the residue,
2.54 L of H2O and 2.54 L of AcOEt were added. After separation, the aqueous layer was washed with 1.02 L of EtOAc. To the aqueous layer, 2.03 L of 12% sulfuric acid was added at less than 12 0C to give crystal of compound P-6. Filtration, washing with 1.53 L of cold H2O and drying provided 576 g of compound P-6 (83% yield) as a solid. 1H NMR(300 MHz, DMSO-de) δ 7.67 (d, J = 7.5 Hz, 1 H), 7.5-7.2 (m, 5H), 6.40 (d, J = 7.5 Hz, 1 H), 5.07
f) Synthesis of methyl 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-7). To a slurry of 576 g of compound P-6 (1.0 eq.: 5.8% of H2O was contained) in 2.88 L of NMP were added 431 g of NaHCO3(3.0 eq.) and 160 ml. of methyl iodide(1.5 eq.) and the mixture was stirred at room temperature for 4 h. After cooling to 5 0C, 1.71 L of 2N HCI and 1.15 L of 20% NaClaq were added to the mixture at less than 10 0C to give crystal of compound 7. Filtration, washing with 1.73 L of H2O and drying provided 507 g of compound P-7 (89% yield) as a solid. 1H NMR(300 MHz, DMSO- cfe) δ 7.59 (d, J = 7.5 Hz, 1 H), 7.40-7.28 (m, 5H), 6.28 (d, J = 7.5 Hz, 1 H), 5.21 (d, J = 5.4 Hz, 1 H), 5.12 (d, J = 10.8 Hz, 1 H), 5.07 (d, J = 10.8 Hz, 1 H), 4.83 (t, J = 5.7 Hz, 1 H), 3.97 (dd, J = 2.4, 14.1 Hz, 1 H), 3.79 (s, 3H), 3.70 (dd, J = 9.0, 14.4 Hz, 1 H), 3.65-3.50 (m, 1 H), 3.40-3.28 (m, 1 H), 3.26-3.14 (m, 1 H).
g) Synthesis of methyl 1-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-8). To a mixture of 507 g of compound P -7 (1.0 eq.) in
5.07 L of MeCN, 5.07 L of H2O and 9.13 g of AcOH(0.1 eq.) was added 390 g of NaIO4(1.2 eq.) and the mixture was stirred at room temperature for 2 h. After addition of 1.52 L of 10% Na2S2OsBq., the mixture was concentrated and cooled to 10 0C. Filtration, washing with H2O and drying provided 386 g of compound P-8 (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.42-7.30 (m, 5H), 6.33 (d, J = 6.0 Hz, 2H),
h) Synthesis of (3S, 11 aR)-3-methyl-6-[(phenylmethyl)oxy]-2,3, 1 1 ,1 1a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-9). After dissolution of mixture of 320 g of compound P-8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 ml. of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 0C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 ml. of cold 2-propanol and drying provided 167 g of compound P-9 (52% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1 H), 5.46 (d, J = 10.5 Hz, 1 H), 5.23 (d, J = 10.2 Hz, 1 H), 5.20 (dd, J = 3.9, 9.6 Hz, 1 H), 4.46-4.34 (m, 1 H), 4.31 (dd, J = 6.6, 8.7 Hz, 1 H), 4.14 (dd, J = 3.9, 12.3 Hz, 1 H), 3.79 (dd, J = 9.9, 12.3 Hz, 1 H), 3.62 (dd, J = 6.9, 8.7 Hz, 1 H), 1.38 (d, J = 6.3 Hz, 3H).
i) Synthesis of (3 S, 1 1 aR)-8-bromo-3-methyl-6-[(phenylmethyl)oxy]-2,3, 11 ,11a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-10). To slurry of 156 g of compound P-9 (1.0 eq.) in 780 ml. of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound P-10 (84% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 8.37 (s, 1 H), 7.55- 7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1 H), 5.18 (d, J = 10.8 Hz, 1 H), 5.03 (d, J = 10.5 Hz, 1 H), 4.53 (dd, J = 3.6, 12.0 Hz, 1 H), 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 1 1.7 Hz, 1 H), 3.64 (dd, J = 5.7, 8.1 Hz, 1 H), 1.27 (d, J = 6.3 Hz, 3H). j) Synthesis of (3S,1 1aS)-Λ/-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,3,5,7, 11 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8- carboxamide (compound P-11). Under carbon mono-oxide atmosphere, a mixture of 163 g of compound P-10 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 mL of 2,4- difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 1 1.3 g of Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O, the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt.
k) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide (compound 1a). Under hydrogen atmosphere, a mixture of 184 g of compound P-11 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1 ), the filtrate was concentrated. After 200 mL of AcOEt was added to the residue, filtration afforded crude solid of compound 1 a.
The precipitates were combined and extracted with 4.0 L of CHCI3/Me0H(5/1 ). After concentration of the CHCI3/MeOH solution and addition of 250 mL of AcOEt to the residue, filtration afforded crude solid of compound 1a. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated
†Global API Chemistry, ‡MDR Chemical Science,§Analytical Sciences, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
A novel synthesis of GSK1265744, a potent HIV integrase inhibitor, is described. The synthesis is highlighted by an efficient construction of the densely functionalized pyridinone core as well as a highly diastereoselective formation of the acyl oxazolidine moiety. The latter exploits the target molecule’s ability to chelate to Mg2+, a key feature in the integrase inhibitor’s mechanism of action.
Bictegravir and dolutegravir are two recently approved integrase inhibitors for the treatment of HIV. A third inhibitor, cabotegravir, is in Phase 3 development. As a continuation of a series of articles on synthetic routes to newly approved drugs, the current article reviews the patent and journal literature regarding synthetic routes and final forms of these drug
^“Adopted USANs”(PDF). American Medical Association. Retrieved 19 September 2014.
^World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1): 70–1. hdl:10665/331088.
Ziegler, Robert E.; Desai, Bimbisar K.; Jee, Jo-Ann; Gupton, B. Frank; Roper, Thomas D.; Jamison, Timothy F. 7-Step Flow Synthesis of the HIV Integrase Inhibitor Dolutegravir. Angewandte Chemie, International Edition. Volume 57. Issue 24. Pages 7181-7185. Journal; Online Computer File. (2018).
SYN 4
Synthetic Reference
Rajan, Srinivasan Thirumalai; Eswaraiah, Sajja; Reddy, Ghojala Venkat; Reddy, Sagyam Rajeshwar; Markandeya, Bekkam; Rajesham, Boge. Novel crystalline polymorph of sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate and process for preparation thereof. Assignee MSN Research & Development Center, India. IN 201641037221. (2018).
Synthetic Reference 5
Sharma, Pramodkumar; Rao, Bhatraju Srinivasa; Deo, Keshav. A process for the preparation of Dolutegravir or its pharmaceutical acceptable salts thereof. Assignee Wockhardt Limited, India. IN 2015MU01007. (2016).
Synthetic Reference 6
Weaver, Jimmie Dean. Preparation of fluoroarenes via hydrogen bond directed photocatalytic hydrodefluorination of perfluoroarenes. Assignee The Board of Regents for Oklahoma State University, USA. WO 2018187336. (2018).
Vellanki, Sivaram Prasad; Nadella, Madumurthy; Bhalme, Mitali; Ramabhotla, Revathi Srinivas. Process for the preparation of dolutegravir, an integrase inhibitor for HIV-1 infection therapy. Assignee Mylan Laboratories Ltd., India. IN 2015CH00588. (2016).
SYN 9
Synthetic Reference
Sankareswaran, Srimurugan; Mannam, Madhavarao; Chakka, Veerababu; Mandapati, Srirami Reddy; Kumar, Pramod. Identification and Control of Critical Process Impurities: An Improved Process for the Preparation of Dolutegravir Sodium. Organic Process Research & Development. Volume 20. Issue 8. Pages 1461-1468. Journal; Online Computer File. (2016).
LM11A-31 is a non-peptide ligand of the p75 neurotrophin receptor (p75NTR). LM11A-31 blocks pro-NGF induced cell death in neuronal cultures, and protects neuronal cells from the the cytotoxic effects of cisplatin or methotrexate. Oral administration of LM11A-31 promotes the survival of oligodendrocytes and myelinated axons in a mouse spinal cord injury model and improves function in both weight-bearing and non-weight bearing tests.Inhibits death of hippocampal neurons at 100–1,000 pM
LM11A-31, C12 H25 N3 O2, Pentanamide, 2-amino-3-methyl-N-[2-(4-morpholinyl)ethyl]- WO 2010102212 TO LONGO FRANK, PUB 10.09.2010 THE UNIVERSITY OF NORTH CAROLINA AT CHAPEL HILL
Scientists have developed a pill which they claim could help paralyzed people walk again.
The new drug allowed mice with no movement in their lower limbs to walk with ‘well-coordinated steps’ and even to replicate swimming motions, researchers said.
The experimental drug, called LM11A-31, was developed by Professor Frank Longo, of Stanford University, California.
The researchers gave three different oral doses of LM11A-31, as well as a placebo, to different groups of mice beginning four hours after injury and then twice daily for a 42 day experimental period, the ‘Daily Mail’ reported.
In tests, the experimental medication did not increase pain in the mice and showed no toxic effects on the animals.
It also efficiently crossed the blood brain barrier, which protects the central nervous system from potentially harmful chemicals carried around in the rest of the bloodstream.
An injury to the spinal cord stops the brain controlling the body and this is the first time an oral drug has been shown to provide an effective therapy.
“This is a first to have a drug that can be taken orally to produce functional improvement with no toxicity in a rodent model,” Professor Sung Ok Yoon, of Ohio State University, Columbus, said.
“So far, in the spinal cord injury field with rodent models, effective treatments have included more than one therapy, often involving invasive means. Here, with a single agent, we were able to obtain functional improvement,” Yoon said.
The small molecule in the study was tested for its ability to prevent the death of cells called oligodendrocytes.
These cells surround and protect axons, long projections of a nerve cell, by wrapping them in a myelin sheath that protect the fibres.
In addition to functioning as axon insulation, myelin allows for the rapid transmission of signals between nerve cells.
The drug preserved oligodendrocytes by inhibiting the activation of a protein called p75. Yoon’s lab previously found p75 is linked to the death of these specialised cells after a spinal cord injury. When they die, axons that are supported by them degenerate.
“Because we know oligodendrocytes continue to die for a long period of time after an injury, we took the approach that if we could put a brake on that cell death, we could prevent continued degeneration of axons,” she said.
Small, Nonpeptide p75NTR Ligands Induce Survival Signaling and Inhibit proNGF-Induced Death in Journal of neuroscience, 26(20): 5288-5300; doi: 10.1523/JNEUROSCI.3547-05.2006 by SM Massa – 2006 – Cited by 51 – Related articles
17 May 2006 – At 5 nm, LM11A-24 and -31 inhibit TUNEL staining to a degree … We further prioritized LM11A–31, because preliminary studies
Small, Nonpeptide p75NTR Ligands Induce Survival Signaling and Inhibit proNGF-Induced Death
Example 32: Preparation of enantiomerically pure 2-amino-3-methyl-N-(2- morpholino-ethyϊ)-pentanamide
[00332] 2-amino-3-methyl-N-(2-morpholinoethyl)-pentanamide can be prepared by a method shown in Scheme 4 below. First, 2-aminoethanol (Compound IE) is transformed to its derivative with a leaving group (Compound 2E). Examples of the leaving group include halides and alkoxy or other activated hydroxyl group. Second, Compound 2E reacts with morpholine at a neutral or basic condition to yield 2-morpholinoethanamine (Compound 3E). The aforementioned two steps may also be performed continuously as one step with Compound 2E being generated in situ. For example, Compound 3 E can be prepared from Compound IE directly through a Mitsunobu reaction wherein the hydroxyl group of Compound IE is activated by diethyl azodicarboxylate (DEAD) before morpholine is added. The final product, 2-amino-3-methyl-N-(2-moipholinoethyl)-pentanamide (Compound 5E), can be obtained by coupling 2-morpholinoethanamine with 2-amino-3- methylpentanoic acid (Compound 4E) via a peptide coupling agent. Examples of the peptide coupling agent include l,r-carbonyldiimidazole (CDI), hydroxybenzotriazole (HOBT), 1,3-dicyclohexylcarbodiimide (DCC), 1- hydroxybenzo-7-azatriazole (HOAt), and the like. Scheme 4:
H2N^0H — H2N^ / LG , p , .
1Ot= LG: a leaving group
1E zt
[00333] A chiral 2-amino-3-methyl-N-(2-moφholinoethyl)-pentanamide (Compound 5E) can be obtained by using the corresponding chiral 2-amino-3- methylpentanoic acid (Compound 4E) in the above coupling step. For example, (2S,3S)-2-amino-3-methyl-N-(2-moφholinoethyl)-pentanamide; (2R,3R)-2-amino- 3 -methyl-N-(2-morpholinoethyl)-pentanamide; (2R,3 S)-2-amino-3 -methyl-N-(2- moφholinoethyl)-pentanamide; and (2S,3R)-2-ammo-3-methyl-N-(2- morpholinoethyl)-pentanamide can be obtained by using (2S,3S)-2-amino-3- methylpentanoic acid, i.e., L-isoleucine; (2R,3R)-2-amino-3-methylpentanoic acid, i.e., D-isoleucine; (2R,3S)-2-amino-3-methylpentanoic acid, i.e., D-alloisoleucine; and (2S,3R)-2-amino-3-methylpentanoic acid, i.e., L-alloisoleucine, respectively. [00334] The chiral purity, also known as, enantiomeric excess or EE, of a chiral Compound 5E can be determined by any method known to one skilled in the art. For example, a chiral Compound 5E can be hydrolyzed to Compound 3E and the corresponding chiral Compound 4E. Then, the chiral Compound 4E obtained through hydrolysis can be compared with a standard chiral sample of Compound 4E to determine the chiral purity of the chiral Compound 5E. The determination can be conducted by using a chiral HPLC.
The free base compound of 2-amino-3-niethyl- -(2-morpholinoethyl)-pentanamide can be prepared from isoleucine by synthetic methods known to one skilled in the art.
Standard procedures and chemical transformation and related methods are well known to one skilled in the art, and such methods and procedures have been described, for example, in standard references such as Fiesers’ Reagents for Organic Synthesis, John Wiley and Sons, New York, NY, 2002: Organic Reactions, vols, 1-83, John Wiley and Sons, New York, NY, 2006; March J, and Smith M,, Advanced Organic Chemistry, 6th ed., John Wiley and Sons, New York, NY; and Larock R.C., Comprehensive Organic Transformations, Wiley-VCH Publishers, New York, 1999. All texts and references cited herein are incorporated by reference in their entirety. Other related synthetic methods can be found in U.S. Patent Application Publication Nos. 2006/024072 and 2007/0060526, the contents of which are herein incorporated by reference in their entirety for all purposes. The amorphous dihydrochloride (di-HCl) salt of 2-amino-3-methyl-N-(2-morpholinoethyl)-pentanamide can be prepared by mixing two molar ecjuivalents of HC1 with one molar equivalent of 2-amino- 3-methyl-N-(2-morpholinoethyl)~pentanamide in appropriate solvent(s) and then separating the di-HCl salt from the solvent(s) mixture.
The amorphous di-HCl salt of 2-aniino-3-methyl-N-(2-moi holinoethyl)-pentariamide was analyzed via the methods as described above. The XRD analysis indicated it was amorphous/low ordered as shown in Figure 1 , The DSC thermogram exhibited a broad endotherm with onset temperature 37 °C and peak temperature 74 °C and an enthalpy value of ΔΗ = 80 J/g. The TGA thermogram indicated the di-HCl salt is anhydrous and starts to decompose after about 200°C. An overlay of DSC and TGA thermograms are shown in Figure 2. The moisture sorption-desorpiion isotherm of the di-HC! salt (Figures 3 A and 3 B ) was collected using dynamic vapor sorption (DVS) analysis. The material did not adsorb much moisture from 0% to 20% RH, then it showed steady sorption up to 140 wt% moisture at 95% RH (likely deliquescence). This sample showed rapid desorption from 95% to 70% RH and then continues desorbing at a relatively slower pace to a mass about 5 wt% greater than the original value at 0% RH. This sample shows a small hysteresis between the sorption and desorption phase. O verall this material is quite hygroscopic. The crude solubility of the di-HCl salt in water was >30 mg/niL. The proton N MR spectrum of the amorphous di-HCl salt is shown in Figure 4. Example 2. Preparation of 2-amino-3-methyl- -(2-morpholinoethy[)-pentanamide (free base):
Five grams of 2-amino-3-methyl-N-(2-morpholinoethyl)-pentanamide di-HCl salt was dissolved in 150 mL of ethanol. Sodium bicarbonate (5.3 g), dissolved in 100 mL of HPLC water, was added to this solution. The mixed solution was sonicated for ~10 minutes. This solution was concentrated using a rotovap, and the residue was dissolved in 300 mL of methylene chloride. This solution was passed through a short plug of carbonate bonded silica gel. This solution was concentrated using rotovap and the residue was lyophilized to dry, resulting in 3.6 g of the free base as a white solid. Proton NMR, C-13 NMR and LC/MS confirmed the structure of this material as the free base of 2-amino-3-methyl-N-(2- morpholmoethyl)-pentanamide.
In the process of converting the di-HCl salt to free base, the sample was lyophilized to avoid formation of oil. XRD analysis of the lyophilized free base surprisingly re vealed it was crystalline, as shown in Figure 5. The DSC thermogram exhibited an endotherm with extrapolated onset temperature 51 °C and peak temperature 53 °C and an enthalpy value of Δ¾= 104 J/g. The TGA thermogram shows less than 0.6 wt% loss at 105 °C, suggesting it was solvent free. An overlay of the DSC and TGA thermograms can be seen in Figure 6. The crude solubility of free base in water was >30 mg/mL. The proton NMR was consistent with the free base. The NMR and Raman spectra are shown in Figures 7 and 8A and 8B, respectively. The moisture sorption-desorption isotherm (Figures 9 A and 9B) was collected using dynamic vapor sorption (DVS) analysis. The sample did not adsorb much moisture content from 0% to 45% RH under the experimental conditions. Above 45 %RH the sample appears to adsorb moisture of – 10 wt% from 45% to 50% RH followed by rapid sorption up to 96 wt% moisture at 95% RH. In the desorption phase, the free base shows a rapid desorption from 95% to 80°/» RH, then the sample desorbs at a relatively slow pace to the original weight at 0% RH. The sample may form a hydrate near 45 %>RH, The putative hydrate appears to deliquesce resulting in an amorphous glass by the end of the scan.
Crystalline forms of neurotrophin mimetic compounds and their salts
Type II TNF receptor agonist; NGF receptor modulator
Crystalline forms of (2S,3S)-2-amino-3-methyl-N-(2-morpholinoethyl)-pentanamide (LM11A-31-BHS), useful for the treatment of neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease and multiple sclerosis. See WO2011066544 claiming deuterated compounds of LM11A-31-BHS, useful for treating neurodegenerative diseases. PharmatrophiX is investigating the p75 neutrophin receptor ligand, LM11A-31-BHS, for the oral treatment of AD. By March 2013, a phase I trial was planned. The drug was formerly being investigated in collaboration with Elan Corp and the deal was terminated by the fourth quarter of 2010.
TAK-385 is a luteinizing hormone-releasing hormone (LH-RH) receptor antagonist administered orally. By preventing LH-RH from binding with the LH-RH receptor in the anterior pituitary gland and suppressing the secretion of luteinizing hormone (LH) and follicle stimulation hormone (FSH) from the anterior pituitary gland, TAK-385 controls the effect of LH and FSH on the ovary, reduces the level of estrogen in blood, which is known to be associated with the development of endometriosis and uterine fibroids, and is expected to improve the symptoms of these disorders.
TAK-385 in Japan for the treatment of endometriosis and uterine fibroids. TAK-385 is a luteinizing hormone-releasing hormone (LH-RH) *1 receptor antagonist administered orally. By preventing LH-RH from binding with the LH-RH receptor in the anterior pituitary gland and suppressing the secretion of luteinizing hormone (LH) *2 and follicle stimulation hormone (FSH) *3 from the anterior pituitary gland, TAK-385 controls the effect of LH and FSH on the ovary, reduces the level of estrogen in blood, which is known to be associated with the development of endometriosis and uterine fibroids, and is expected to improve the symptoms of these disorders. The safety and efficacy of TAK-385 in subjects with endometriosis and uterine fibroids will be evaluated in two individual phase 2, double-blind, comparative studies. There are medical needs which cannot be met by the current therapies in the treatment of endometriosis and uterine fibroids. We are committed to the rapid development to deliver the oral LH-RH antagonist TAK-385, which could become a new treatment option for patients with these conditions.
*1 The hormone that controls the secretion of LH and FSH, gonadotropic hormones, secreted from the anterior pituitary gland.
*2 A hormone that is secreted from the anterior pituitary gland by the action of LH-RH and encourages follicular maturation, ovulation and luteinization by acting on the ovaries.
*3 A hormone that is secreted from the anterior pituitary gland by the action of LH-RH and encourages follicular maturation by stimulating the ovaries.
TAK-385, an oral antagonist of gonadotropin-releasing hormone (GnRH), was originated by Takeda. It is in phase II clinical trials for the treatment of endometriosis and for the treatment of uterine fibroids (myoma). Phase I clinical trials are also underway for the treatment of prostate cancer.
TAK-385 (relugolix) is a novel, non-peptide, orally active gonadotropin-releasing hormone (GnRH) antagonist, which builds on previous work with non-peptide GnRH antagonist TAK-013. TAK-385 possesses higher affinity and more potent antagonistic activity for human and monkey GnRH receptors compared with TAK-013. Both TAK-385 and TAK-013 have low affinity for the rat GnRH receptor, making them difficult to evaluate in rodent models. Here we report the human GnRH receptor knock-in mouse as a humanized model to investigate pharmacological properties of these compounds on gonadal function. Twice-daily oral administration of TAK-013 (10 mg/kg) for 4 weeks decreased the weights of testes and ventral prostate in male knock-in mice but not in male wild-type mice, demonstrating the validity of this model to evaluate antagonists for the human GnRH receptor.
The same dose of TAK-385 also reduced the prostate weight to castrate levels in male knock-in mice. In female knock-in mice, twice-daily oral administration of TAK-385 (100 mg/kg) induced constant diestrous phases within the first week, decreased the uterus weight to ovariectomized levels and downregulated GnRH receptor mRNA in the pituitary after 4 weeks. Gonadal function of TAK-385-treated knock-in mice began to recover after 5 days and almost completely recovered within 14 days after drug withdrawal in both sexes. Our findings demonstrate that TAK-385 acts as an antagonist for human GnRH receptor in vivo and daily oral administration potently, continuously and reversibly suppresses the hypothalamic–pituitary–gonadal axis. TAK-385 may provide useful therapeutic interventions in hormone-dependent diseases including endometriosis, uterine fibroids and prostate cancer.
Production of N-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxy-3-pyridazinyl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-N’-methoxyurea
The similar reaction as described in Example 4 by using the compound (100 mg, 0.164 mmol) obtained in Reference Example 54 and methyl iodide (0.010 ml, 0.164 mmol) gave the title compound (17.3 mg, 17 %) as colorless crystals. 1 H-NMR(CDCl3) δ: 2.15 (6H, s), 3.6-3.8 (2H, m), 3.82 (3H, s), 4.18 (3H, s), 5.35 (2H, s), 6.92 (2H, t, J = 8.2 Hz), 7.12 (1H, d, J = 8.8 Hz), 7.2-7.65 (7H, m), 7.69 (1H, s).
……………
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
Method for the production of TAK-385 or its salt and crystals starting from 6-(4-aminophenyl)-1-(2,6-difluorobenzyl)-5-dimethylaminomethyl-3-(6-methoxypyridazin-3-yl) thieno[2,3-d] pyrimidine-2,4 (1H,3H)-dione or its salt. Takeda Pharmaceutical is developing relugolix (TAK-385), an oral LHRH receptor antagonist analog of sufugolix, for the treatment of endometriosis and uterine fibroids. As of April 2014, the drug is in Phase 2 trails. See WO2010026993 claiming method for improving the oral absorption and stability of tetrahydro-thieno[2,3-d]pyrimidin-6-yl]-phenyl)-N’-methoxy urea derivatives.
references
Discovery of TAK-385, a thieno[2,3-d]pyrimidine-2,4-dione derivative, as a potent and orally bioavailable nonpeptide antagonist of gonadotropin releasing hormone (GnRH) receptor
238th ACS Natl Meet (August 16-20, Washington) 2009, Abst MEDI 386
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
Condensation of ethyl 2,4,5-trifluorobenzoylacetate (I) with triethyl orthoformate in refluxing Ac2O produced the benzoyl ethoxyacrylate (II), which was further condensed with 2-amino-5-fluoropyridine (III) to afford enamine (IV). Cyclization of (IV) in the presence of K2CO3 gave rise to the quinolone (V). The 7-fluoride group of (V) was then displaced by N-methylpiperazine (VI) in cold pyridine to furnish the piperazinyl quinolone (VII). Finally, ester hydrolysis in (VII) under acidic conditions yielded the target compound. In a closely related procedure, ester (V) was hydrolyzed to acid (VIII) using HCl. Subsequent displacement of the 7-fluoride of (VIII) with N-methylpiperazine (VI) provided the desired piperazinyl quinolone.
synthesis 2
Condensation of ethyl 2,4,5-trifluorobenzoylacetate (I) with triethyl orthoformate in refluxing Ac2O produced the benzoyl ethoxyacrylate (II), which was further condensed with 2-amino-5-fluoropyridine (III) to afford enamine (IV). Cyclization of (IV) in the presence of K2CO3 gave rise to the quinolone (V). The 7-fluoride group of (V) was then displaced by N-methylpiperazine (VI) in cold pyridine to furnish the piperazinyl quinolone (VII). Finally, ester hydrolysis in (VII) under acidic conditions yielded the target compound. In a closely related procedure, ester (V) was hydrolyzed to acid (VIII) using HCl. Subsequent displacement of the 7-fluoride of (VIII) with N-methylpiperazine (VI) provided the desired piperazinyl quinolone.
Synthesis, pharmacokinetics, and biological activity of a series of new pyridonecarboxylic acid antibacterial agents bearing a 5-fluoro-2-pyridyl group or a 3-fluoro-4-pyridyl group at N-1
J Heterocycl Chem 1997, 34(3): 1021
TBR-652 (formerly TAK-652) is a highly potent and orally active CCR5 antagonist in phase II clinical trials at Takeda for the treatment of HIV infection. Tobira Therapeutics is evaluating the compound in preclinical studies for the treatment of rheumatoid arthritis.
TBR-652 binds CCR5 receptors to interfere with the entry of the HIV-1 virus into macrophages and activated T-cells by inhibiting fusion between viral and cellular membranes. This mechanism of action differs from currently used HIV treatments such as nucleoside reverse transcriptase inhibitors and protease inhibitors.
In 2007, Takeda entered into an agreement with Tobira pursuant to which Tobira obtained exclusive worldwide rights to develop, manufacture and commercialize TBR-652 for the treatment of HIV infection.
Cenicriviroc is an inhibitor of CCR2 and CCR5 receptors,[2] allowing it to function as an entry inhibitor which prevents the virus from entering into a human cell. Inhibition of CCR2 may have an anti-inflammatory effect.
A double-blind, randomized, placebo-controlled clinical study to assess the antiviral activity, safety, and tolerability of cenicriviroc was conducted in 2010. HIV-infected patients taking cenicriviroc had significant reductions in viral load, with the effect persisting up to two weeks after discontinuation of treatment.[3] Additional Phase II clinical trials are underway.[4]
Phase IIb data presented at the 20th Conference on Retroviruses and Opportunistic Infections (CROI) in March 2013 showed similar viral suppression rates of 76% for patients taking 100 mg cenicriviroc, 73% with 200 mg cenicriviroc, and 71% with efavirenz. Non-response rates were higher with cenicriviroc, however, largely due to greater drop-out of patients. A new tablet formulation with lower pill burden may improve adherence. Looking at immune and inflammatory biomarkers, levels of MCP-1 increased and soluble CD14 decreased in the cenicriviroc arms.[5]
Although HIV has been largely rendered a chronic infection, there remains a need for new drugs because of the virus’s propensity to develop resistance to the drugs used to keep it at bay.
Pfizer’s maraviroc was the first drug that acted on the cells to prevent viral entry by antagonising the CCR5 co-receptor. Several others have been investigated and have failed; another that is undergoing clinical trials is Takeda’s cenicriviroc, which has been licensed to Tobira Therapeutics. Unlike maraviroc, the new agent also acts at the CCR2 co-receptor, which is implicated in cardiovascular and metabolic diseases.
In a Phase I double blind, placebo controlled trial designed to study safety, efficacy and pharmacokinetics, treatment-experienced but CCR5 antagonist-naïve patients with HIV-1 were given doses of 25, 50, 75, 100 or 150mg of the drug, or placebo once a day for 10 days.2 The maximum median reductions in HIV-1 RNA values were 0.7, 1.6, 1.8 and 1.7 log10 copies/ml for the respective doses, with a median time to nadir of 10 to 11 days. The effect on CD-4 cell counts was negligible. There was also a significant reduction in levels of monocyte chemotactic protein 1, suggesting that CCR2 was also being blocked. The drug was both generally safe and well tolerated, and no patients withdrew from the trial due to adverse events.
In another Phase I trial, designed to look at pharmacokinetics and pharmacodynamics and carried out in a similar patient population, subjects were given the drug as oral monotherapy for 10 days, again in doses of 25, 50, 75, 100 and 150mg, or placebo.3 The drug was well absorbed into the systemic circulation, and the concentration levels declined slowly, with meant elimination half-lives of one to two days. Potent, dose-dependent reductions in viral load were seen, and again it was generally safe and well tolerated across all levels.
In June 2011, Tobira initiated a multi-centre, double blind, double dummy, 48-week comparative Phase IIb trial in 150 patients with HIV-1 infection. Subjects are being given 100 or 200mg once-daily doses of the drug to evaluate its efficacy, safety and tolerability.
ancriviroc (formerly known as SCH-C), vicroviroc which has the chemical name (4,6-dimethylprymidine-5-yl){4- [(3S)-4-{(1 R)-2-methoxy-1 -[4-(trifluoromethyl)phenyl]ethyl}-3-methylpiperazin-1 -yl]-4-methylpiperidin-1 – yljmethanone, PRO-140, apliviroc (formerly known as GW-873140, Ono-4128, AK-602), AMD-887, INC- B9471 , CMPD-167 which has the chemical name N-methyl-N-((1R,3S,4S)-3-[4-(3-benzyl-1-ethyl-1H- pyrazol-δ-yOpiperidin-i-ylmethylH-IS-fluorophenyllcyclopent-i-yll-D-valine), methyl1-endo-{8-[(3S)-3- (acetylamino)-3-(3-fluorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl}-2-methyl-4,5,6,7-tetrahydro-1 H- imidazo[4,5-c]pyridine-5-carboxylate, methyl 3-endo-{8-[(3S)-3-(acetamido)-3-(3-fluorophenyl)propyl]-8- azabicyclo[3.2.1]oct-3-yi}-2-methyl-4,5,6,7-tetrahydro-3H-imidazo[4,5-c]pyridine-5-carboxylate, ethyl 1- endo-{8-[(3S)-3-(acetylamino)-3-(3-fiuorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl}-2-methyl-4,5,6,7- tetrahydro-1 H-imidazo[4,5-c]pyridine-5-carboxylate and N-{(1S)-3-[3-endo-(5-lsobutyryl-2-methyl-4,5,6,7- tetrahydro-1H-imidazo[4,5-c]pyridin-1-yl)-8-azabicyclo[3.2.1]oct-8-yl]-1-(3-fluorophenyl)propyl}acetamide) and pharmaceutically acceptable salts, solvates or derivatives of the above. The last four compounds are disclosed in WO 03/084954 and WO 05/033107.
Compound (S)-(−)-5b (TAK-652) also inhibited the replication of six macrophage-tropic (CCR5-using or R5) HIV-1 clinical isolates in peripheral blood mononuclear cells (PBMCs) (mean IC90 = 0.25 nM).
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-propyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide ((S)–(−)-5a). The 1 N HCl (160 mL) was added to 1931 (35.68 g, 53.4 mmol), and the mixture was extracted with EtOAc. To the aqueous layer was added 25% aqueous K2CO3 (160 mL), and the mixture was extracted with a mixture of EtOAc and i-PrOH (4:1). The organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo to give (S)-18. To a solution of 16a (18.0 g, 41.1 mmol) and DMF (0.5 mL) in THF (180 mL) was added thionyl chloride (SOCl2) (4.50 mL, 61.7 mmol) at room temperature. After being stirred at room temperature for 1.5 h, the reaction mixture was concentrated in vacuo. A solution of the residue in THF (200 mL) was added dropwise to a mixture of (S)-18 and triethylamine (Et3N) (35.0 mL, 251 mmol) in THF (150 mL) under ice cooling. After being stirred at room temperature for 4 h, water was added to the reaction mixture. The mixture was washed with 10% aqueous AcOH, saturated aqueous NaHCO3, and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on a NH silica gel (hexane/EtOAc = 1:5 → 1:8 → 1:9) to give 21.14 g (75%) of (S)-(−)-5a as a yellow amorphous powder, [α]D = −132.5° (C = 0.507%, EtOH). 1H NMR (300 MHz, CDCl3) δ 0.87−1.03 (9H, m), 1.34−1.49 (2H, m), 1.50−1.85 (8H, m), 2.55−2.65 (2H, m), 3.15−3.25 (2H, m), 3.52−3.58 (4H, m), 3.75−3.83 (4H. m), 4.02 (1H, d, J = 13.8 Hz), 4.08−4.17 (3H, m), 6.56 (1H, d, J = 1.0 Hz), 6.80 (1H, d, J = 8.8 Hz), 6.96 (2H, d, J = 8.8 Hz), 7.31−7.46 (7H, m), 7.55 (1H, s), 7.76 (2H, d, J = 8.8 Hz), 7.98 (1H, s). Anal. (C40H50N4O4S·0.25H2O) C, H, N.
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide methanesulfonate ((S)–(−)-5b). The free base of (S)-(−)-5b was prepared in 80% yield from 16band 19 by a method similar to that described for (S)-(−)-5a. To a solution of the free base of (S)-(−)-5b (64.91 g, 93.1 mmol) in EtOAc (600 mL) was added dropwise a solution of methanesulfonic acid (8.95 g, 93.1 mmol) in EtOAc (160 mL) at room temperature. After being stirred at room temperature for 4 h, the crystals were collected by filtration and washed with EtOAc to give 69.09 g (94%) of (S)-(−)-5b as yellow crystals. The crystals (68.0 g) were purified by recrystallization from 2-butanone to give 58.9 g (85%) of (S)-(−)-5b as yellow crystals, mp 145.5−147.5 °C, [α]D = −191.2° (c = 0.508%, EtOH). 1H NMR (300 MHz, DMSO-d6) δ 0.82−0.97 (12H, m), 1.29−1.39 (2H, m), 1.40−1.55 (4H, m), 1.65−1.85 (2H, m), 2.00−2.25 (1H, m), 2.29 (3H,s), 2.38−2.60 (2H, m), 3.10 (2H, d, J = 7.8 Hz), 3.30−3.60 (4H, m), 3.70 (2H, t, J = 4.8 Hz), 3.98 (2H, t,J = 6.6 Hz), 4.10 (2H, t, J = 4.8 Hz), 4.34 (1H, d, J = 15.0 Hz), 4.68 (1H, d, J = 15.0 Hz), 6.87 (1H, d, J = 8.7 Hz), 6.99 (2H, d, J = 8.7 Hz), 7.16 (1H, s), 7.42−7.60 (8H, m), 7.93 (2H, d, J = 8.7 Hz), 9.05 (1H, s), 10.18 (1H, s). Anal. (C42H56N4O7S2) C, H, N.
8-[4-(2-Butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propyl-1H-imidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzazocin-5-carboxamide (317 mg) was resolved by using CHIRAKCEL OJ 50 mm ID×500 mL (hexane/ethanol) to give (−)-8-[4-(2-butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propylimidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzoazocine-5-carboxamide (142 mg) (Compound 9) and (+)-8-[4-(2-butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propylimidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzoazocine-5-carboxamide (143 mg) (Compound 10).
Example 21 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide
To a solution of 8-[4-(2-butoxyethoxy)phenyl]-1-isobutyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid (45 g) in tetrahydrofuran (135 ml) was added N,N-dimethylformamide (230 mg) and added dropwise thionyl chloride (12.45 g) at 10 to 15° C., and the resulting solution was stirred at the same temperature for 40 minutes to prepare an acid chloride.
Separately, to a solution of (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine in tetrahydrofuran (270 ml) was added pyridine (27.59 g), the resulting mixture was adjusted to 5° C. or lower, and then thereto was added dropwise the acid chloride solution at 5° C. or less, and the resulting mixture was stirred at the same temperature for 2 hours. To the mixture were added water (270 ml) and 20% aqueous citric acid solution (180 ml), tetrahydrofuran was distilled off under reduced pressure and the residue was extracted with ethyl acetate. The extract was sequentially washed with water, saturated sodium bicarbonate solution and water, and then the solvent was distilled off. To the residue was added ethyl acetate (360 ml), added heptane (360 ml) at 40° C. and added seed crystals of (−)-8-[4-(2-butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide (10 mg), and the mixture was stirred at 25° C. for 2 hours and stirred at 5° C. for 1 hour. The precipitated crystals were collected by filtration to obtain 63.97 g (yield: 92.1%) of the title compound. Melting point: 120-122° C.
Elemental analysis value: in terms of C41H52N4O4S
Calcd. value: C, 70.66; H, 7.52; N, 8.04.
Analytical value: C, 70.42; H, 7.52; N, 8.01
Industrial Applicability
According to the present invention, an optically active sulfoxide derivative having CCR5 antagonism or an intermediate compound thereof can be prepared without causing side reactions such as racemization and Pummerer rearrangement. In particular, Process 7 is industrially advantageous since it is possible to prepare an optically active Compound (II) by asymmetric oxidization in the presence of an optically active acid.
Example 20 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-propyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide.methanesulfonate
According to the same method as that described in Example 15, the title compound was produced from 8-[4-(2-butoxyethoxy)phenyl]-1-propyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid and (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine.
Elemental analysis value: in terms of C41H54N4O7S2
Calcd. value: C, 63.21; H, 6.99; N, 7.19; S, 8.23.
Analytical value: C, 63.00; H, 7.09; N, 7.41; S, 8.25
Example 15 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide.methanesulfonate
8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid (986 mg) was dissolved in tetrahydrofuran (3 ml) and thereto was added N,N-dimethylformamide (one drop). Subsequently, to the resulting solution was added dropwise oxalyl chloride (0.2 ml, 2.29 mmol) under ice-cooling and the mixture was stirred for 80 minutes under ice-cooling to prepare an acid chloride.
Separately, (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine (689 mg) was added to tetrahydrofuran (7 ml) and the resulting solution was cooled to 5° C. To the solution was added dropwise pyridine (0.62 ml) and added dropwise the acid chloride solution at 3 to 5° C., and the mixture was stirred for 2 hours under ice-cooling. To the mixture was added water (20 ml) at 10° C. or lower and the mixture was extracted with ethyl acetate. The organic layer was sequentially washed with water, saturated sodium bicarbonate solution and water, and concentrated under reduced pressure. Thereto was added toluene and the mixture was concentrated under reduced pressure. Thereto was added acetonitrile and the mixture was concentrated under reduced pressure. The residue was dissolved in acetonitrile (7 ml) and acetone (7 ml), thereto was added dropwise methanesulfonic acid (209 mg), and added seed crystals and the mixture was stirred at room temperature for 100 minutes. Subsequently, to the mixture was added acetone-acetonitrile (1:1, 5 ml). After stirring at room temperature overnight, the mixture was stirred for 2.5 hours under ice-cooling. The precipitated crystals were collected by filtration and washed with the ice-cooled acetone (9 ml). The crystals were dried at 40° C. under reduced pressure to obtain 1.51 g (yield: 87%) of the title compound as yellow crystals.
1H-NMR (300 MHz, DMSO-d6, δ): 0.78-0.96 (12H, m), 1.25-1.40 (2H, m), 1.41-1.51 (4H, m), 1.65-1.85 (2H, m), 2.05-2.15 (1H, m), 2.30 (3H, s), 2.35-2.50 (2H, m), 3.05-3.15 (2H, m), 3.30-3.55 (4H, m), 3.65-3.70 (2H, m), 3.90-4.05 (2H, m), 4.05-4.10 (2H, m), 4.30 (1H, d, J=14.73 Hz), 4.65 (1H, d, J=14.73 Hz), 6.85 (1H, d, J=8.97 Hz), 6.97 (1H, d, J=8.79 Hz), 7.17 (1H, s), 7.35-7.75 (6H, m), 7.92 (2H, d, J=8.79 Hz), 9.08 (1H, s), 10.15 (1H, s).
Elemental analysis value: in terms of C41H52N4O4S.CH4SO3
Calcd. value: C, 63.61; H, 7.12; N, 7.06; S, 8.09.
Found value: C, 63.65; H, 7.23; N, 7.05; S, 8.08.
………………………….
References
Klibanov, Olga M.; Williams, Shannon H.; Iler, Cameron A (2010). “Cenicriviroc, an orally active CCR5 antagonist for the potential treatment of HIV infection”. Current Opinion in Investigational Drugs11 (8): 940–950. PMID20721836.
The relative activity of “function sparing” HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property?
Highly potent and orally active CCR5 antagonists as anti-HIV-1 agents: synthesis and biological activities of 1-benzazocine derivatives containing a sulfoxide moiety.
Stereoselective synthesis of [L-Arg-L/D-3-(2-naphthyl)alanine]-type (E)-alkene dipeptide isosteres and its application to the synthesis and biological evaluation of pseudopeptide analogues of the CXCR4 antagonist FC131.
TAK-652, a novel CCR5 inhibitor, has favourable drug interactions with other antiretrovirals in vitro.
Antiviral therapy
……………….
Chemical structures of selected small molecule CCR5 inhibitors. A. Maraviroc (MVC, Selzentry), B. Vicriviroc (VCV), C. Cenicriviroc (TBR-652), D. PF-232798.
Infections due to Gram-positive bacteria such as methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus faecium (VRE), and penicillin-resistant Streptococcus pneumoniae(PRSP) are the leading cause of morbidity and mortality in hospital settings and community today. Oxazolidinones are a new class of totally synthetic antibacterial agents active against Gram-positive infections. Linezolid (Zyvox™, Pharmacia/Pfizer, is a drug in this class, approved in the United States and Europe for treatment of Gram-positive nosocomial and community-acquired pneumoniae and skin infections. Oxazolidinones inhibit the bacterial protein synthesis prior to the chain initiation step, by binding to the 23S rRNA of 50S ribosomal subunit, and interfering with the initiator fMet–tRNA binding to the P-site of the ribosomal peptidyltransferase centre
Plymorphic forms of phenyl oxazolidinone derivatives
The title compound is prepared by reductive alkylation of the known piperazinyl oxazolidinone derivative (I) with 5-nitro-2-furfural (II) in the presence of NaBH(OAc)3, followed by conversion to the corresponding hydrochloride salt.
EP 1303511; US 2002103186; WO 0206278; WO 0307870; WO 0308389
…………….
synthesis
The antibacterial activity of RBx-7644 is due to the 5(S)-acetamidomethyl configuration at the oxazolidinone ring, and thus, asymmetric synthesis of only the 5(S)-enantiomer was desirable: 3,4-Difluoronitrobenzene (I) is condensed with piperazine in acetonitrile to give 4-(2-fluoro-4-nitrophenyl)-piperazine (II) as a light yellow compound. Compound (II) is dissolved in dichloromethane and triethylamine, followed by the addition of Boc-anhydride, to provide compound (III). 4-(tert-Butoxycarbonyl)-1-(2-fluoro-4-nitrophenyl)piperazine (III), upon hydrogenation with H2 over Pd/C in methanol at 50 psi, yields 4-(tert-butoxycarbonyl)-1-(2-fluoro-4-aminophenyl)piperazine (IV) as a dark solid. Compound (IV) reacts with benzylchloroformate in dry THF in the presence of solid sodium bicarbonate to afford the desired compound (V). 4-(tert-Butoxycarbonyl)-1-[2-fluoro-4-(benzyloxycarbonylamino)phenyl]piperazine (V), upon treatment with n-BuLi and (R)-glycidyl butyrate at -78 癈, gives the desired (R)-(-)-3-[3-fluoro-4-[4-(tert-butoxycarbonyl)piperazin-1-yl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone (VI). The hydroxymethyl compound (VI) is treated with methanesulfonyl chloride in dichloromethane in the presence of triethylamine to give (R)-(-)-3-[3-fluoro-4-[4-(tert-butoxycarbonyl)piperazin-1-yl]phenyl]-5-(methylsulfonyloxymethyl)-2-oxazolidinone (VII). The sulfonyl derivative (VII) is treated with sodium azide in dimethylformamide to provide the azide (VIII) as a white solid. (R)-(-)-3-[3-Fluoro-4-[4-(tert-butoxycarbonyl)piperazin-1-yl)phenyl]-5-(azidomethyl)-2-oxazolidinone (VIII), upon hydrogenation with H2 over Pd/C at 45 psi, gives (S)-(-)-3-[3-fluoro-4-[4-(tert-butoxycarbonyl)-piperazin-1-yl]phenyl]-5-(aminomethyl)-2-oxazolidinone (IX). The aminomethyl compound (IX), upon treatment with acetic anhydride in dichloromethane in the presence of triethylamine, affords the acetamide derivative (X). The acetamidomethyl-oxazolidinone derivative (X), upon treatment with trifluoroacetic acid, gives (S)-(-)-3-[3-fluoro-4-(1-piperazinyl)phenyl]-5-(acetamidomethyl)-2-oxazolidinone, which, without isolation, is treated with 5-nitro-2-furaldehyde in the presence of sodium triacetoxy borohydride to provide compound (XI). Compound (XI), upon treatment with ethanolic HCl, affords RBx-7644 as a light yellow crystalline solid.
(S)-N-[[3-fluoro-4-[N-1[4-{2-furyl-(5-nitro)methyl}]piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]-methyl]acetamidehydrochloride having the Formula I.
The compound of Formula I, namely, (S)-N-[[3-fluoro-4-[N-1 [4-{2-furyl-(5-nitro)methyl}] piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]-methyl]acetamide hydrochloride is a phenyl oxazolidinone derivative, as disclosed in PCT application WO 02/06278. It is said to be useful as antimicrobial agent, effective against a number of human and veterinary pathogens, including gram-positive aerobic bacteria, such as multiply resistant staphylococci, streptococci and enterococci as well as anaerobic organisms such as Bacterioides spp. andClostridia spp. species, and acid fast organisms such as Mycobacterium tuberculosis, Mycobacterium avium and Mycobacterium spp.
The PCT application WO 02/06278 describes the preparation of compounds of Formula I. The products of Formula I obtained by following the cited methods tend to be hygroscopic and difficult to filter. These types of disadvantageous properties have proven to be serious obstacles to the large-scale manufacture of a compound. Further, handling problems are encountered during the preparation of pharmaceutical compositions comprising the hygroscopic compound of Formula I obtained by following the method disclosed in WO 02/06278.
EXAMPLE 1 Preparation of Polymorphic ‘Form A’ of the Compound of Formula I
50 gm of free base of Formula I was dissolved in ethanol (750 ml) by heating at about 60° C. and to this solution was added ethanolic HCl (13.36 ml, 8.9 N) at about 45-50° C. The reaction mixture was cooled to about 10° C., and stirred for about 4 hours. The separated solid was filtered off and dried under vacuum at 60° C. The solid was then digested in ethanol (150 ml) at 70-80° C. for about 4 hours. It was then cooled to about 10° C., the solid was filtered and dried under vacuum at 60-65° C. to give 30 gm of the pure polymorphic ‘Form A’ of compound of Formula I.
………………
Synthesis and SAR of novel oxazolidinones: Discovery of ranbezolid
Novel oxazolidinones were synthesized containing a number of substituted five-membered heterocycles attached to the ‘piperazinyl–phenyl–oxazolidinone’ core of eperezolid. Further, the piperazine ring of the core was replaced by other diamino-heterocycles. These modifications led to several compounds with potent activity against a spectrum of resistant and susceptible Gram-positive organisms, along with the identification of ranbezolid (RBx 7644) as a clinical candidate.
Substitution of five-membered heterocycles on to the ‘piperazinyl–phenyl–oxazolidinone’ core structure led to the identification of ranbezolid as a clinical candidate. Further replacement of piperazine ring with other diamino-heterocycles led to compounds with potent antibacterial activity.
Scheme 5.
Reagents and conditions: (a) Method A: TFA, CH2Cl2, 0 °C → rt; 5-chloromethyl-2-furaldehyde, potassium carbonate, DMF, rt; or (b) Method B: TFA, CH2Cl2, 0 °C → rt; 5-nitrofuran-2-carboxaldehyde, sodiumtriacetoxyborohydride, THF, molecular sieves 3 Å, rt. 7 = ranbezolid
Synthesis of compound 7: (S)-N-[[3-[3-Fluoro-4-(N-4-tert-butoxycarbonyl-piperazin-1-yl)phenyl]-2-oxo-5-oxa-zolidinyl]-methyl]acetamide (28a, 3.65 kg, 8.37 mol) was dissolved in dichloromethane (30.86 L) and cooled to 5 °C. To it trifluoroacetic acid (6.17 L) added dropwise and stirred for 14 h allowing the reaction mixture to warm to rt. The reaction mixture was evaporated in vacuo and the residue dissolved in tetrahydrofuran (58 L) followed by addition of molecular sieves 4 Å (4.2 kg). To the resulting mixture 5-nitro-2-furaldehyde (1.5 kg, 10.77 mol) was added followed by sodium triacetoxyborohydride (5.32 kg, 25.1 mol) and stirred for 14 h. The reaction mixture was filtered over Celite and filtrate evaporated in vacuo. The residue was dissolved in ethylacetate (85.6 L) and washed with satd sodium bicarbonate solution (36 L) and water (36 L). The organic layer was dried over anhyd sodium sulfate (3 kg) and evaporated in vacuo. The crude residue was purified by column chromatography (1–3% methanol in ethylacetate) to obtain (S)-N-[[3-[3-fluoro-4-[N-4-(5-nitro-2-furylmethyl)-piperazin-1-yl]phenyl]-2-oxo-5-oxa-zolidinyl]methyl]acetamide (39, 2.6 kg, yield 67%). Mp: 136 °C. 1H NMR (CDCl3): δ 7.42 (dd, 1H, phenyl–H), 7.29 (m, 2H, furyl–H), 7.07 (d, 1H, phenyl–H), 6.92 (t, 1H, phenyl–H), 6.51 (d, 1H, furyl–H), 6.11 (t, 1H, –NHCO–), 4.77 (m, 1H, oxazolidinone ring C5–H), 4.01 (t, 1H), 3.85–3.45 (m, 5H), 3.09 (m, 4H, piperazine–H), 2.72 (m, 4H, piperazine–H), 2.02 (s, 3H, –COCH3). MS m/z (rel. int.): 462.1 [(M+H)+, 100%], 484 [(M+Na)+, 25%], 500.2 [(M+K)+, 20%]. HPLC purity: 98%.
Compound 39(3.6 kg, 7.81 mol) was dissolved in abs ethanol (53.8 L) by heating to 60 °C. The resulting solution was cooled to 45 °C and ethanolic hydrochloride (1.48 L, 7.9 N) was added dropwise in 10 min. The mixture was then cooled to 10 °C and stirred for 4 h and the precipitate formed was filtered and washed with ethanol and dried to obtain (S)-N-[[3-[3-fluoro-4-[N-4-(5-nitro-2-furylmethyl)-piperazin-1-yl]phenyl]-2-oxo-5-oxazolidinyl]-methyl]acetamide hydrochloride, ranbezolid (7, 3.2 kg, yield from 39: 82%, yield from 28a: 55%).
General Manager, R&D at Calyx Chemicals & Pharmaceuticals Ltd.
in.linkedin.com/pub/vijay-kaul/b/aa9/962
DR VIJAY KAUL QUOTE……………Kindly go through my patent describing a two step cost effective environmentally benign process for the key intermediate of Ranbezolid.
Process for the preparation of 4-(4-benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-1-carboxylic acid tert-butyl ester W02005051933 ,……………….UNQUOTE
novel methods for the synthesis of the 4-(4- benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-l-carboxylic acid tert-butyl ester of Formula I, which provides improvements over prior methods of synthesis. In one aspect, there is provided a process for the synthesis of highly pure 4-(4- benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-l-carboxylic acid tert-butyl ester of Formula I,
Formula I comprising the steps of: condensing piperazine with l,2-difluoro-4-nitrobenzene to form l-(2-fluoro-4-nitro-phenyl)- piperazine of Formula II,
contacting the compound of Formula II with di-tert-butoxycarbonyl anhydride to form 4- (2- fluoro-4-nitrophenyl)-piperazine 1-carboxylic acid tert-butyl ester of Formula III,
reducing the compound of Formula III to form 4-(4-amino-2-fluorophenyl)-piperazin-l- carboxylic acid tert-butyl ester of Formula IV,
Formula IV and reacting the compound of Formula IN with benzylchloroformate to form 4-(4-benzyloxy- carbonylamino-2-fluorophenyl)-piperazine- 1-carboxylic acid tert-butyl ester of Formula I. In one aspect, the step of condensing piperazine with l,2-difluoro-4-nitrobenzene is carried out in an aromatic hydrocarbon, such as toluene, xylene and the like, or mixtures thereof, and at a temperature of, for example, about 40 °C to about 90 °C, or from about 80 °C to about 90 °C.
Oxazolidinone compounds can be prepared from compounds of Formula I using, for example, using methods disclosed in U.S. Patent No. 6,734,307 and PCT Publication Nos. WO 02/06278, WO 03/007870, WO 03/097059, WO04/089944 and WO04/14392, which are incorporated herein by reference. Scheme I below shows a synthetic route starting from a compound of Formula I to oxazolidinone compounds.
Formula lb
Formula lc
Formula Id Scheme I A compound of Formula I
Formula I can be reacted with a base, e.g., butyl lithium, and glycidyl butyrate to form a compound of
Formula la.
Formula la
The compound of Formula la can be reacted with methane sulphonyl chloride, followed by ammonium hydroxide, and finally acetyl halide of Formula CH3CO-hal (wherein hal is Br, CI or I) to form a compound of Formula lb.
Formula lb
The compound of Formula lb can be deprotected to form a compound of Formula Ic.
The compound of Formula Ic can be reacted with R-T-(W)0-ι-R12 to form a compound of Formula Id
EXAMPLE Preparation of 4-(4-benzyloxy-carbonylamino-2-fluorophenyl -piperazine- 1 – carboxylic acid tert-butyl ester of Formula I
Piperazine (0.77 mol, 66.2 g) was mixed with toluene (500 mL) and stirred at room temperature and subsequently stirred at 50 °C until a homogenous solution was obtained. 1,2- difluoro-4-nitrobenzene (0.314 mol, 50 g) was added to the piperazine/toluene solution and the reaction mixture was stirred at 80-90 °C for 3-6 hours.
The reaction mixture then was cooled to 40-45 °C and diluted with deionized water. The organic layer was separated and about 250-350 mL of toluene was evaporated off under reduced pressure at 40 °C. Di-tert- butoxycarbonyl anhydride (0.334mol, 75 g) was then added dropwise to the reaction mixture at room temperature. The resulting reaction mixture was stirred at room temperature for 1-2 hours and then further diluted with hexane (200 mL) and stirred for 15-20 minutes at room temperature.
The solid product formed in the reaction mixture was filtered, washed with hexane (150 mL), and dried under reduced pressure at 60-70°C to yield 4-(2-fluoro-4- nitrophenyl)-piperazine- 1-carboxylic acid tert butyl ester of Formula III. Yield = 1.8-1.9 (w\w); Purity = 96-98% by HPLC.
The compound of Formula III (0.246 mol, 80 g) was added to toluene (800 mL) followed by the addition of palladium on carbon (4 g) at room temperature with continuous stirring. Hydrogen gas was bubbled into the resulting reaction mixture at a pressure of 72 psi. The reaction mixture was stirred for 12-16 hours and then diluted with toluene (150 mL). The reaction mixture was filtered through a celite pad and washed with toluene (200 mL).
Sodium bicarbonate solution was added to the reaction mixture at room temperature with continuous stirring. Benzyl chloroformate (0.310 mol, 103 g) was added dropwise to the reaction mixture with continuous stirring for 2-3 hours. Ethyl acetate (1600 mL) was added to the reaction mixture and stirred for about 30 minutes followed by addition of deionized water (400 mL). The organic layer was separated and the solvent was removed under reduced pressure. The semi-solid product was washed with hexane (350 mL) to obtain 4-(4-benzyloxy- carbonylamino-2-fluorophenyl)-piperazine- 1-carboxylic acid tert-butyl ester of Formula I as a solid. Yield = 1.16-1.23 (w/w); Purity = 97-99% by HPLC.
References
European Journal of Pharmacology. 2006. 545, 167–172
US2005209248, 9-23-2005
Plymorphic forms of phenyl oxazolidinone derivatives

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



























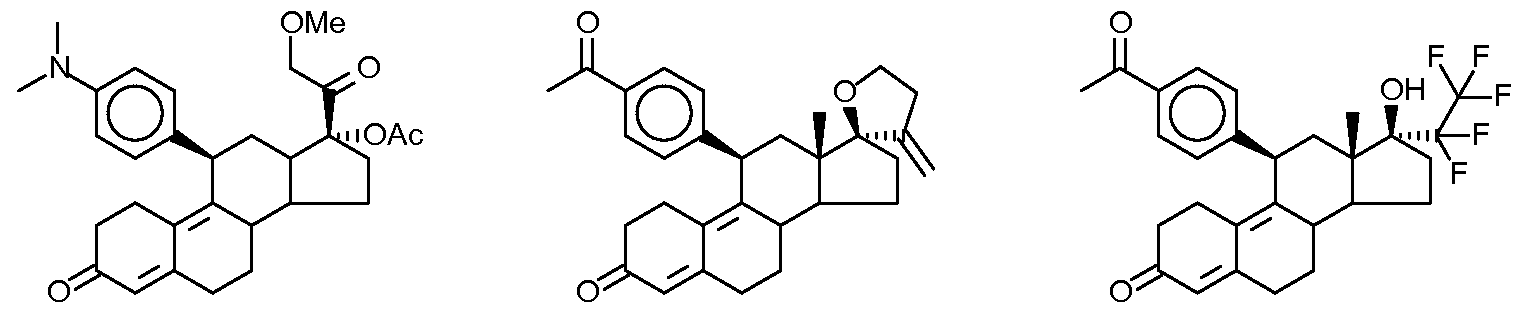




 ELBASVIR
ELBASVIR




































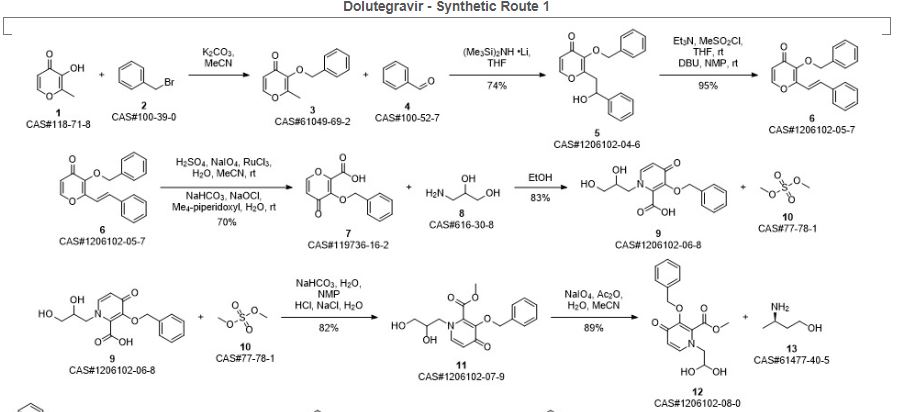
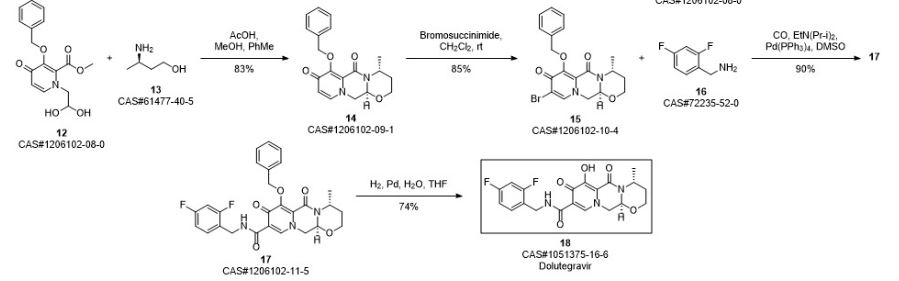







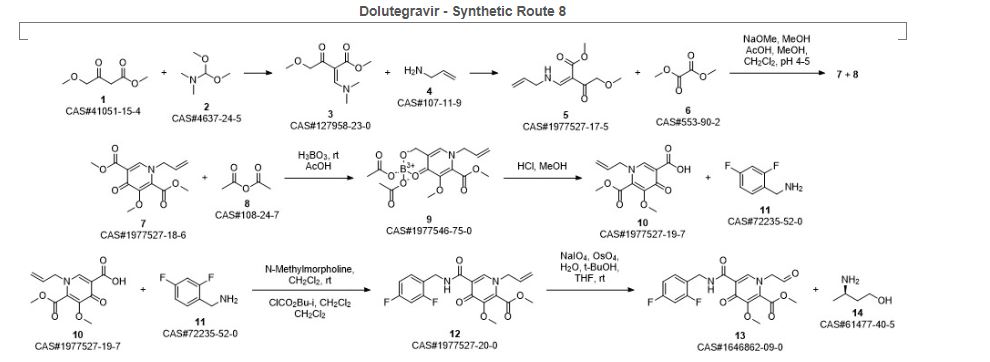

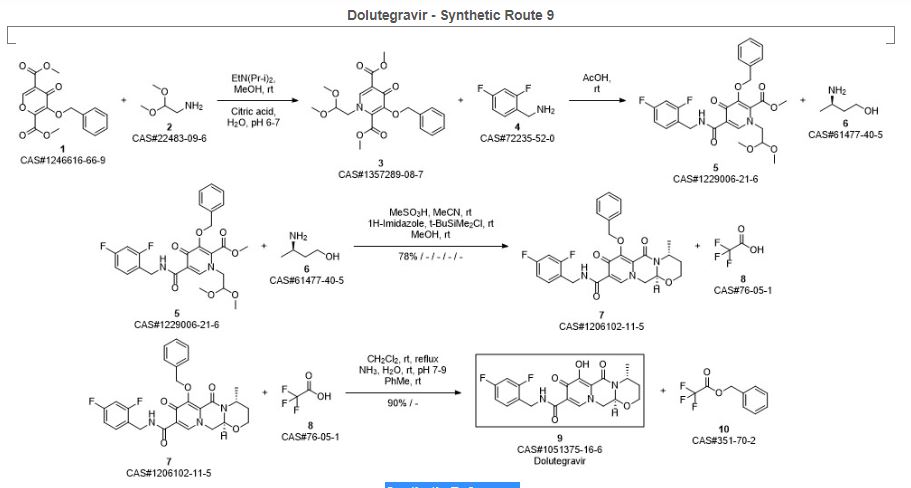


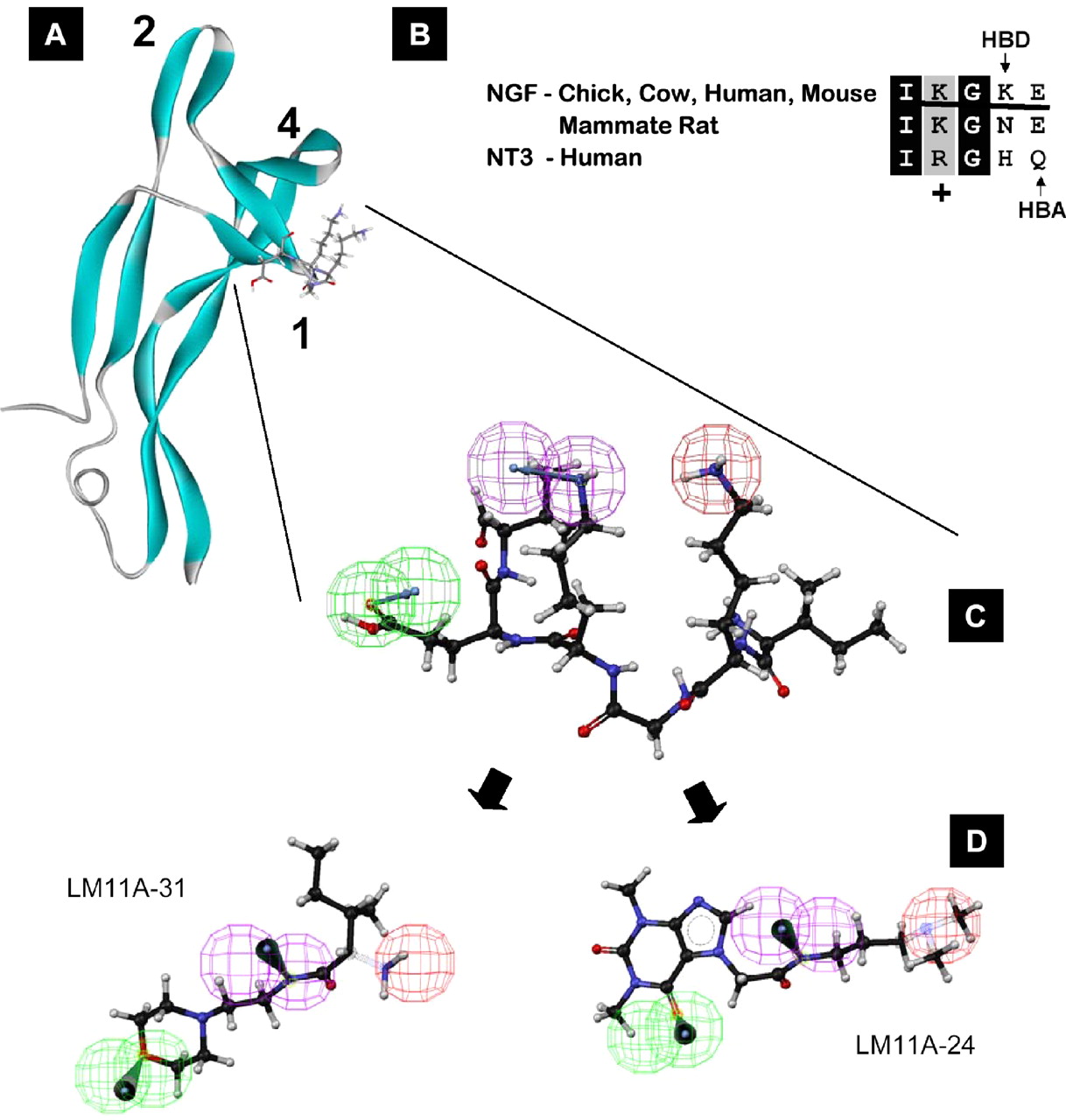
 The experimental drug was developed by Prof Frank Longo from Stanford University
The experimental drug was developed by Prof Frank Longo from Stanford University













 Relugolix (TAK-385)
Relugolix (TAK-385)


 tak 385
tak 385










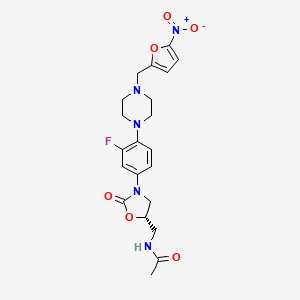





















.svg){kind=link}
{kind=link}