
Home » Phase2 drugs (Page 20)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves Alcobra’s protocol for Phase IIb study of metadoxine drug candidate

Metadoxine
Israel-based Alcobra has received approval from the US Food and Drug Administration (FDA) for its protocol for planned Phase IIb clinical study of Metadoxine Extended Release (MDX) drug candidate for the treatment of Fragile X Syndrome.

The multi-center, randomized, placebo-controlled, Phase IIb study, will be conducted primarily in the US and patient enrollment is expected to begin in the near future.
The study is supported by data collected from multiple earlier pre-clinical studies which demonstrated significant improvement in behavioral and cognitive outcomes based on evaluations of memory, learning, and social interaction.
Metadoxine, or Pyridoxol L-2-pyrrolidone-5-carboxylate, whose structure formula is reported hereinbelow

is known for its effectiveness in acute and chronic alcoholism and for the prevention of alcohol related pathology
 |
|
| Systematic (IUPAC) name | |
|---|---|
| L-Proline, 5-oxo-, compd. with 5-hydroxy-6-methylpyridine-3,4-dimethanol (1:1) | |
| Clinical data | |
| Legal status | PHASE 2 |
| Routes | Oral, IV |
| Identifiers | |
| CAS number | 0074536-44-0 |
| ATC code | N07BB |
| Chemical data | |
| Formula | C13H18N2O6 |
| Mol. mass | 298 g/mol |
………..
Metadoxine, also known as pyridoxine-pyrrolidone carboxylate, is a drug used to treat chronic and acute alcohol abuse.[1] Metadoxine improved the clinical signs of acute alcohol intoxication and accelerated alcohol clearance from the blood [2]It is presently in human clinical trials as an attention-deficit/hyperactivity disorder predominantly inattentive treatment.[3]
Pyridoxine is one form of vitamin B6 and a precursor to the metabolically active pyridoxal phosphate. Pyridoxal phosphate is a coenzyme to many enzymes: see vitamin B6 metabolic functions.
Pyrrolidone carboxylate is involved in amino acid metabolism through the glutathione pathway.[4] Glutathione is an important antioxidant and combats redox imbalance. It also supports de novo ATP synthesis.[5]
Alcohol-induced liver diseases are a common disorder in modern communities and societies. For example, in Europe there are more than 45 million individuals showing signs of alcohol-related damage such as liver disease and myopathies. Chronic alcohol consumption increases hepatic accumulation of triglycerides and leads to hepatic steatosis, which is the earliest and most common response to severe alcohol intoxication.
Thus, severe alcohol intoxication is a serious disease that should be treated with medication in order to reduce the damage to the human body of the alcohol intoxicated individual. For example, alcohol intoxication can be treated with metadoxine (pyridoxine L-2-pyrrolidone-5-carboxylate). Metadoxine is a salt of the corresponding anion of L-2-pyrrolidone-5-carboxylic acid (L-2-pyroglutamic acid) (1) and the protonated derivative of pyridoxine (vitamin B6) (2), having the following structures:

(1) (2)
WO 2008/066353 discloses the use of Metadoxine in the treatment of alcohol intoxication either alone or in combination with other active agents. WO 2008/066353 mentions that metadoxine does not inhibit the expression and activation of an alcohol-induced cytochrome P450 2El, which is the key enzyme involved in alcohol-induced toxicity. Thus, the use of metadoxine may be limited.
Several studies have shown that in order to effectively treat alcohol intoxication, there is a need for a relatively high daily dose (ca. 900 mg) administered intravenously (see, e.g., Lu et al. Chin. Med. J. 2007, 120 (2), 155-168 and Shpilenya et al. Alcohol Clin. Exp. Res. 2002, 26 (3), 340-346). These studies disclose side effects associated with the use of metadoxine, including nausea and vomiting.
Thus, there exists a need in the art for effective and safe drugs for treating alcohol intoxication and other associated diseases.
History
Metadoxine is predominantly used in developing nations for acute alcohol intoxication. Alternate names include: Abrixone (Eurodrug, Mexico), Alcotel (Il Yang, South Korea), Ganxin (Qidu Pharmaceutical, China), Metadoxil (Baldacci, Georgia; Baldacci, Italy; Baldacci, Lithuania; CSC, Russian Federation; Eurodrug, Colombia; Eurodrug, Hungary; Eurodrug, Thailand; Micro HC, India), Viboliv (Dr. Reddy’s, India), and Xin Li De (Zhenyuan Pharm, China).[6]
Fatty liver refers to a pathogenic condition where fat comprises more than 5% of the total weight of the liver. Liver diseases including the fatty liver, hepatitis, fibrosis and cirrhosis are known to be the most serious disease next to cancer causing death in people with ages 40 to 50, in the advanced countries. In advanced countries, nearly about 30% of the population is with fatty liver, and about 20% of people with fatty liver progresses to cirrhosis. About half of the cirrhosis patients die of liver diseases within 10 years after the diagnosis. Fatty liver and steatohepatitis are frequently found in people who intake excessive alcohols and who have obesity, diabetes, hyperlipemia, etc. Among them, alcoholic steatohepatitis (ASH), which is caused by excessive alcohol intake, is at high risk of progressing to hepatitis, cirrhosis and hepatoma, along with non-alcoholic steatohepatitis (NASH).
When taken in, alcohol is carried to the liver and oxidized to acetaldehyde by such enzymes as alcohol dehydrogenase, catalase, etc. The acetaldehyde is metabolized and converted into acetate and is used as energy source. Repeated alcohol intake induces the increase of NADH and NADP+ during the metabolism and acetaldehyde which as the metabolite product of alcohol depletes GSH, thereby changing intracellular oxidation-reduction homeostasis and inducing oxidative stress. Oxidative stress may cause mitochondrial dysfunction, lipid peroxidation and protein modification, thereby leading to death of hepatocytes, inflammation, activation of astrocytes, and the like. In addition, the increase of NADH promotes lipid synthesis, thereby inducing fatty liver.
At present, there are few therapeutically effective drugs for treating fatty liver. Exercise and controlled diet are recommended, but these are not so effective in treating fatty liver. The development of an effective treatment drug is in desperate need. As it is known that fatty liver is related with insulin resistance which is found in diabetes and obesity, the therapeutic effect of some anti-diabetic drugs, e.g., metformin, on fatty liver has been reported. But, the drug has the problem that it may induce adverse reactions such as hepatotoxicity or lactic acidosis. Betaine, glucuronate, methionine, choline and lipotrophic agents are often used as alternative supplementary drug therapy, but they are not fully proven on medical or pharmaceutical basis. Accordingly, development of a fatty liver treatment having superior effect and safety with no adverse reactions is in need.
Metadoxine (pyridoxol 1-2-pyrrolidone-5-carboxylate) is a complex compound of pyridoxine and pyrrolidone carboxylate represented by the formula (1) below:

Metadoxine is a drug used to treat alcoholic liver disease. It is used to treat liver fibrosis and fatty liver through increasing alcohol metabolism and turnover, reducing toxicity of free radicals and restoring the level of ATP and glutathione (Arosio, et al., Pharmacol. Toxicol. 73: 301-304, 1993; Calabrese, et al., Int. J. Tissue React. 17: 101-108, 1995; Calabrese, et al., Drugs Exp. Clin. Res. 24: 85-91, 1998; Caballeria, et al., J. Hepatol. 28: 54-60, 1998; and Muriel, et al., Liver Int. 23: 262-268, 2003).
However, metadoxine is unable to inhibit the expression and activation of alcohol-induced cytochrome P4502E1 (CYP2E1), which is a key enzyme involved in alcohol-induced toxicity, and thus unable to control the augmentation of inflammation mediated by CYP2E1. Therefore, the treatment of alcohol-induced fatty liver using metadoxine is very limited. Further, the expression of CYP2E1 is related with insulin resistance, thus metadoxine cannot not overcome insulin resistance.
Garlic oil is a liquid including about 1% of allicin along with reduced allicin and other sulfur-containing substances. Upon binding to vitamin B1, allicin is turned into allithiamin, which is chemically stable, acts swiftly, and is easily absorbed by the digestive organs. The substance inhibits carcinogenesis induced by chemicals in white rats (Brady, et al., Cancer Res. 48: 5937-5940, 1988; and Reddy, et al., Cancer Res. 53: 3493-3498, 1993), induces phase II enzyme (Hayes, et al., Carcinogenesis 8: 1155-1157, 1987; and Sparnins, et al., Carcinogenesis 9: 131-134, 1988), and inactivates CYP2E1 (Brady, et al., Chem. Res. Toxicol. 4:642-647, 1991). In addition, garlic oil is reported to have antithrombotic, anti-atherosclerotic, antimutagenic, anticancer and antibacterial activities (Agarwal, Med. Res. Rev. 16: 111-124, 1996; and Augusti, Indian J. Exp. Biol. 34: 634-660, 1996).
Pharmacology
Treatment for acute alcohol abuse
In an animal model, metadoxine treatment increased the clearance of alcohol and acetaldehyde, reduced the damaging effect of free radicals, and enabled cells to restore cellular ATP and glutathione levels. [7][8] It increases the urinary elimination of ketones, which are formed when the oxidation rate of acetaldehyde into acetate is exceeded on massive alcohol intoxication.[8][4]
As a medical treatment, it is typically given intravenously.
Treatment for AD/HD-PI
Metadoxine is a selective antagonist to the 5-HT2B receptor, a member of the serotonin receptor family.[3] Electrophysiological studies also showed that Metadoxine caused a dose-dependent, reversible reduction in glutamatergic excitatory transmission and enhancement of GABAergic inhibitory transmission, changes that may be associated with cognitive regulation.[3] It is given orally in an extended release pill, which differs from the instant release alcohol treatment.
Treatment for liver disease
Metadoxine may block the differentiation step of preadipocytes by inhibiting CREB phosphorylation and binding to the cAMP response element, thereby repressing CCAAT/enhancer-binding protein b during hormone-induced adipogenesis.[7]
Treatment for Fragile X Syndrome
Metadoxine treatment led to significant improvement in blood and brain biological markers (AKT and ERK), which may have a role in learning and memory.[3] The study also demonstrated a reduction in the amount of immature neurons and abnormally increased protein levels.[3]
…………………..
PATENT
http://www.google.com/patents/WO2010013242A1?cl=en
Scheme 1

[0054] In another aspect, the invention provides methods of synthetically preparing, e.g., carboxylated lactam ring of formula (II) (e.g. wherein n=2 for a reactant of formula (IVb) in Scheme 2), carboxylated lactam ring of (III) (e.g. wherein n=3 for a reactant of formula (IVb) in Scheme 2) and carboxylated lactam ring of formula (IV) (e.g. wherein n=4 for a reactant of formula (IVb) in Scheme 2), as depicted in Scheme 2.
Scheme 2
pyridoxine

Compound IVb, n=2,3,4

[0055] In another aspect, the invention provides methods of preparing a salt adduct including a positively charged pyridoxine moiety, or a derivative thereof, and a carboxylated 5- to 7-membered lactam ring, including the steps of:
(a) suspending an optionally substituted amino dioic acid in water and heating for a sufficient period of time to allow completing lactamization reaction;
(b) optionally decolorizing the reaction mixture to eliminate impurities;
(c) isolating the lactam carboxylate;
(d) optionally purifying the obtained lactam carboxylate by crystallization;
(e) admixing the obtained lactam carboxylate and a pyridoxine base or a derivative thereof in a solvent mixture optionally under heating; and
(f) isolating the product.
In certain embodiments, a solvent mixture of step (e) includes a mixture of an alcohol such as methanol, ethanol, isopropanol and the like, and water. [0059] According to yet another embodiment, there is provided methods of preparing N-substituted L-pyroglutamic acid and the carboxylate thereof, such as, for example, N-methyl-L-pyroglutamic acid (1-methyl-L-pyroglutamic acid), starting from L-pyroglutamic acid ethyl ester, as depicted in Scheme 3 below. Scheme 3


1-methyl-L-pyroglutamic acid
[0060] The invention further provides methods of preparing a salt adduct of the invention, wherein said positively charged moiety is a substituted pyridoxine, as depicted in Scheme 4 below. The starting reagent is 2-methyl-3-hydroxy-4- methoxymethyl-5-hydroxymethyl-pyridine hydrochloride (Compound (V)). The preparation of the corresponding salt is described in Example 1. Scheme 4
HCI NH 3 / MeOH 2 L-pyroglutamic acid


Compound V Compound Vl

Salt lid
[0061] The invention further provides methods of preparing a salt adduct of the invention, wherein said positively charged moiety is a substituted pyridoxine, as depicted in Scheme 5 below. The starting reagent in scheme 5 is 2-methyl-3-hydroxy- 4-methoxymethyl-5-hydroxymethyl-pyridine hydrochloride (Compound V). The preparation of the corresponding salt is described in Example 2. Scheme 5

Compound V
HCI

Compound VIII IX Compound L-pyroglutamic acid

, SaIt IIe
| WO2010150261A1 * | June 24, 2010 | Dec 29, 2010 | Alcobra Ltd. | A method for the treatment, alleviation of symptoms of, relieving, improving and preventing a cognitive disease, disorder or condition |
| WO2011061743A1 * | Nov 18, 2010 | May 26, 2011 | Alcobra Ltd. | Metadoxine and derivatives thereof for use in the treatment of inflammation and immune-related disorders |
| US8476304 | Jul 3, 2012 | Jul 2, 2013 | Alcobra Ltd. | Method for decreasing symptoms of alcohol consumption |
| US8710067 | Jul 3, 2012 | Apr 29, 2014 | Alcobra Ltd. | Method for the treatment, alleviation of symptoms of, relieving, improving and preventing a cognitive disease, disorder or condition |
| WO2008066353A1 * | Nov 30, 2007 | June 5, 2008 | Jae Hoon Choi | Pharmaceutical composition comprising metadoxine and garlic oil for preventing and treating alcohol-induced fatty liver and steatohepatitis |
| WO2009004629A2 * | Jul 3, 2008 | Jan 8, 2009 | Alcobra Ltd | A method for decreasing symptoms of alcohol consumption |
| FR2172906A1 * | Title not available | |||
| US4313952 * | Dec 8, 1980 | Feb 2, 1982 | Maximum Baldacci | Method of treating acute alcoholic intoxication with pyridoxine P.C.A. |
References
- Addolorato, G; Ancona C, Capristo E, Gasbarrini G (2003). “Metadoxine in the treatment of acute and chronic alcoholism: a review”. International Journal of Immunopathology and Pharmacology.
- Martinez, Diaz; Villamil Salcedo; Cruz Fuentes (2001). “Efficacy of Metadoxine in the Management of Acute Alcohol Intoxication”. Journal of International Medical Research.
- “Metadoxine extended release (MDX) for adult ADHD” (in English). Alcobra Ltd. 2014. Retrieved 2014-05-07.
- Shpilenya, Leonid S.; Alexander P. Muzychenko; Giovanni Gasbarrini; Giovanni Addolorato (2002). “Metadoxine in Acute Alcohol Intoxication: A Double-Blind, Randomized, Placebo-Controlled Study”. Alcoholism:Clinical and Experimental Research.
- Shull, Kenneth H.; Robert Kisilevsky (1996). “Effects of Metadoxine on cellular status of glutathione and on enzymetric defence system following acute ethanol intoxication in rats”. Drugs Exp Clin Res.
- “Metadoxine – Drugs.com” (in English). Drugs.com. 2014. Retrieved 2014-05-08.
- Yang, YM; HE Kim; SH Ki; SG Kim (2009). “Metadoxine, an ion-pair of pyridoxine and L-2-pyrrolidone-5-carboxylate, blocks adipocyte differentiation in association with inhibition of the PKA-CREB pathway.”. Archives of Biochemistry and Biophysics.
- Calabrese, V; A Calderone; N Ragusa; V Rizza (1971). “Effects of l-2-pyrrolidone-5-carboxylate on hepatic adenosine triphosphate levels in the ethionine-treated rat”. Biochemical Pharmacology.
XenoPort begins phase II trial of XP-23829 in patients with psoriasis
![]()
XP 23829 from Xenoport is an interesting molecule and as on 27 July 2014, I did not find conclusive evidence
See some structures below
 Not sure about the structure of XP 23829
Not sure about the structure of XP 23829
OR
 Best fit
Best fit
OR
 Not sure?
Not sure?(N,N-dimethylcarbamoyl)methyl methyl(2E)but-2-ene-1,4-dioate.
OR

I AM NOT SURE ABOUT THIS ONE ALSO????????
As Football worldcup2014 goes on in Brazil

A thought for it is due…………
……………………………………………………
Best fit is probably is as shown below, and there are reasons
(N,N- Diethylcarbamoyl)methyl methyl (2E)but-2-ene-l,4-dioate
Introduction
(N,N-Diethylcarbamoyl)methyl methyl (2E)-but-2-ene-1,4-dioate


C11 H17 N O5, mw 243.13
M.p.: 53-56 °C.
1 H NMR (CDCI3, 400 MHz): δ 6.99-6.90 (m, 2H), 4.83 (s, 2H), 3.80 (s, 3H), 3.39 (q, J = 1.1 Hz, 2H), 3.26 (q, J = 7.2 Hz, 2H), 1 .24 (t, J = 7.2 Hz, 3H), 1 .14 (t, J = 7.2 Hz, 3H). MS (ESI): m/z 244.13 (M+H)+.
Cas…….1208229-58-6
XP-23829 PROBABLE
For the treatment of moderate-to-severe chronic plaque-type psoriasis.
XP-H-093
US8148414 Basic patent
Xenoport, Inc. Innovator
XenoPort has initiated a phase II trial of XP-23829, a proprietary investigational next-generation fumaric acid product candidate (ClinicalTrials.gov Identifier NCT02173301). The multicenter, randomized, double-blind, placebo-controlled study is designed to assess the efficacy and safety of XP-23829 as a potential treatment of patients with moderate to severe chronic plaque-type psoriasis. XenoPort expects to enroll approximately 200 subjects in this trial, which is being conducted in the U.S. The study will include a screening and washout phase of up to 4 weeks, a 12-week treatment phase and a 4-week post-treatment phase. Eligible study subjects will be randomized to placebo or one of three treatment arms of XP-23829: 400 or 800 mg once daily or 400 mg twice daily. The primary endpoint will examine the percent change in Psoriasis Area and Severity Index (PASI) score from baseline at the end of week 12. Secondary endpoints will include the proportion of subjects who achieve a reduction of 75% or greater from baseline in PASI (PASI75) score and subjects who achieve a Static Physicians Global Assessment score of “clear” or “almost clear.” Topline results are expected in the third quarter of 2015 (XenoPort News Release).
XP23829 — A Prodrug of Monomethyl Fumarate
Our third product candidate, XP23829, is in Phase 1 clinical development. Provided we are able to demonstrate the safety and desired pharmacokinetic, or PK, profile of XP23829 in our Phase 1 trials, we believe that XP23829 could be a potential treatment of patients with RRMS, psoriasis and/or certain other disorders where the mechanism of action of XP23829 may be relevant. For example, we are exploring the potential of XP23829 to protect against neurodegeneration in experimental preclinical models of Parkinson’s disease through a grant from The Michael J. Fox Foundation. We hold a composition-of-matter patent and a formulation patent in the United States on XP23829 and hold patents or pending patent applications directed to the XP23829 methods of synthesis and use in the United States. We have also filed applications directed to the XP23829 composition of matter and methods of synthesis and use in other jurisdictions.
Prodrug Background
XP23829 is a fumaric acid ester compound and a patented prodrug of MMF. Fumaric acid ester compounds have shown immuno-modulatory and neuroprotective effects in cell-based systems and preclinical models of disease. A product containing a combination of fumaric acid ester compounds, known as Fumaderm, is approved in Germany for the treatment of psoriasis. Tecfidera (a formulation of DMF, also known as BG-12) from Biogen Idec Inc. is another fumaric acid ester prodrug that converts to MMF in the body. Phase 3 clinical trials of Tecfidera as a potential treatment for RRMS showed statistically significant benefits of Tecfidera versus placebo. Tecfidera is currently under U.S. regulatory review as a potential treatment for RRMS.
Our Prodrug
XP23829 is a novel prodrug of MMF that we believe may provide improved tolerability and efficacy compared to DMF. In preclinical studies that compared molar equivalent doses of XP23829 to DMF, XP23829 provided higher blood levels of the biologically active molecule MMF and a similar or greater degree of efficacy in MS and psoriasis animal models. Toxicology studies conducted in two species showed that XP23829 caused less stomach irritation when compared to DMF.
Phase 1 Clinical Trial in Healthy Volunteers
In October 2012, we reported favorable preliminary results from our first Phase 1 clinical trial in healthy adults designed to assess the pharmacokinetics, safety and tolerability of single doses of four different formulations of XP23829. The trial was a randomized, double-blind, two-period crossover, food effect comparison clinical trial of XP23829. Sixty subjects were assigned to five cohorts of 12, with each cohort receiving one of four different formulations of XP23829 or placebo. The trial demonstrated that administration of XP23829 resulted in the expected levels of MMF in the blood. As anticipated, the four formulations produced

April 4, 2012
http://investor.xenoport.com/releasedetail.cfm?ReleaseID=708145
XenoPort Awarded U.S. Patent Directed to Composition and Formulations of XP23829, a Novel Fumarate Analog for the Potential Treatment of Relapsing-Remitting Multiple Sclerosis and Psoriasis
SANTA CLARA, Calif.–(BUSINESS WIRE)–Apr. 4, 2012– XenoPort, Inc. (Nasdaq: XNPT) announced today that it was awarded U.S. Patent 8,148,414 for “Prodrugs of Methyl Hydrogen Fumarate, Pharmaceutical Compositions Thereof, and Methods of Use.” The term of the patent extends until 2029, subject to potential Hatch-Waxman patent term extensions.
The patent is directed to the XP23829 compound, analogs thereof and formulations thereof. A related U.S. patent application directed to therapeutic uses of XP23829 is now pending.
XP23829 is a prodrug of methyl hydrogen fumarate, also known as monomethyl fumarate (MMF). In cell- and animal-based models, MMF has been shown to exhibit immuno-modulatory properties and inhibit damage from oxidative stress.
In XenoPort’s preclinical animal studies that compared molar equivalent doses of XP23829 to dimethyl fumarate (DMF), another prodrug of MMF, XP23829 demonstrated a greater degree of efficacy in animal models of both multiple sclerosis (MS) and psoriasis. Toxicology studies conducted in two species showed that XP23829 caused less stomach irritation compared to DMF.
XenoPort intends to file an Investigational New Drug Application (IND) for XP23829 for the treatment of relapsing remitting MS with the U.S. Food and Drug Administration (FDA) in the second quarter of 2012 and expects to initiate human clinical trials later this year.
XenoPort owns all rights to XP23829.
About XenoPort
XenoPort is a biopharmaceutical company focused on developing and commercializing a portfolio of internally discovered product candidates for the potential treatment of neurological disorders. Horizant® (gabapentin enacarbil) Extended-Release Tablets is XenoPort’s first FDA-approved product. GlaxoSmithKline holds commercialization rights and certain development rights for Horizant in the United States. Regnite® (gabapentin enacarbil) is approved for the treatment of moderate-to-severe primary restless legs syndrome in Japan. Astellas Pharma Inc. holds all development and commercialization rights for Regnite in Japan and five Asian countries. XenoPort holds all other world-wide rights and has co-promotion and certain development rights to gabapentin enacarbil in the United States. XenoPort’s pipeline of product candidates includes potential treatments for patients with postherpetic neuralgia, spasticity and Parkinson’s disease.
To learn more about XenoPort, please visit the company Website at http://www.XenoPort.com.
More info about this drug
SEE a patent
WO 2010022177
……………………………………….
WO 2013181451
http://www.google.com/patents/WO2013181451A1?cl=en
Scheme 5:

ONE OUT OF THESE
Example 6: (/V,/V-Diethylcarbamoyl)methyl methyl (2£)but-2-ene-1 ,4-dioate

[0138] Following general procedure A, methyl hydrogen fumarate (MHF) (0.39 g, 3.00 mmol) dissolved in NMP was reacted at about 55 °C with 2-chloro-/V,/V-diethylacetamide (0.44 g, 3.00 mmol) in the presence of CsHC03 (0.69 g, 3.60 mmol) to afford 0.37 g (51 % yield) of the title compound after purification by silica gel column chromatography (Biotage) using a mixture of ethyl acetate (EtOAc) and hexanes (1 :1 ) as eluent. M.p.: 53-56 °C. 1 H NMR (CDCI3, 400 MHz): δ 6.99-6.90 (m, 2H), 4.83 (s, 2H), 3.80 (s, 3H), 3.39 (q, J = 1.1 Hz, 2H), 3.26 (q, J = 7.2 Hz, 2H), 1 .24 (t, J = 7.2 Hz, 3H), 1 .14 (t, J = 7.2 Hz, 3H). MS (ESI): m/z 244.13 (M+H)+.
Example 7: Methyl 2-morpholin-4-yl-2-oxoethyl (2 £)but-2-ene-1 ,4-dioate
[0139] Following general procedure A, methyl hydrogen fumarate (MHF) (0.50 g, 3.84 mmol) dissolved in NMP was reacted at about 55 °C with 4-(chloroacetyl) morpholine (0.75 g, 4.61 mmol) in the presence of CsHC03 (0.89 g, 4.61 mmol) to afford 0.34 g (35% yield) of the title compound as a white solid after purification by mass-guided preparative HPLC and lyophilization. M.p.: 124 to 126°C; 1 H NMR (CDCI3, 400 MHz): δ 6.97-6.91 (m, 2H), 4.84 (s, 2H), 3.82 (s, 3H), 3.72-3.70 (m, 4H), 3.64-3.62 (m, 2H), 3.46-3.41 (m, 2H). MS (ESI): m/z 258.04 (M+H)+. Example 8: A/,A/-Dimethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4-dioate

[0140] Following general procedure A, methyl hydrogen fumarate (MHF) (0.50 g, 3.84 mmol) dissolved in NMP was reacted at about 55 °C with /V,/V-dimethyl chloroacetamide (0.56 g, 4.61 mmol) in the presence of CsHC03 (0.89 g, 4.61 mmol). The crude material was precipitated out from a mixture of ethyl acetate (EtOAc) and hexanes (Hxn) (1 :1 ) to provide a white solid. This solid was further dissolved in dichloromethane (DCM) and the organic layer washed with water. After removal of the solvents 0.55 g (67% yield) of the title compound was obtained as a white solid. 1 H NMR (CDCI3, 400 MHz): δ 6.98- 6.90 (m, 2H), 4.84 (s, 2H), 3.80 (s, 3H), 2.99-2.97 (2s, 6H). MS (ESI): m/z 216 (M+H)+.
Example 9: Methyl (2-morpholino-4-ylethyl) fumarate

[0141] Following general Procedure A, methyl hydrogen fumarate (MHF) dissolved in NMP is reacted at about 55 °C with 4-(chloroethyl) morpholine (0.75 g, 4.61 mmol) in the presence of CsHC03 to afford the title compound after purification by mass-guided preparative HPLC and lyophilization. Example 10: Methyl (3-mor holino-4-ylpropyl) fumarate

[0142] Following the procedure of Methyl (2-morpholino-4-ylethyl) fumarate, and replacing 4-(chloroethyl) morpholine with 4-(chloropropyl) morpholine provides the title compound.
Example 11 : Methyl (4-morpholino-4-ylbutyl) fumarate

[0143] Following the procedure of Methyl (2-morpholino-4-ylethyl) fumarate, and replacing 4-(chloroethyl) morpholine with 4-(chlorobutyl) morpholine provides the title compound. Example 12: Methyl 5-morpholino-4-ylpentyl) fumarate

[0144] Following the procedure of Methyl (2-morpholino-4-ylethyl) fumarate, and replacing 4-(chloroethyl) morpholine with 4-(chloropentyl) morpholine provides the title compound. Example 13: (A/-cyclopropyl-W-ethylcarbamoyl)methyl methyl 2(E)but-2-ene-1 ,4-dioate

[0145] Following the general procedure A, methyl hydrogen fumarate (MHF) (38.7 g, 0.297 mol) suspended in toluene (100 mL) was reacted at about 80 °C with 2-chloro-/V-cyclopropyl- N-ethylacetamide (48 g, 0.297 mol) in the presence of W,/V-diisopropylethylamine (DIEA; 42.3 g, 57 mL, 0.327 mol) to afford 50 g (63.3%) of the title compound after recrystallization using methyl ferf-butyl ether. The crystalline compound had a melting point of 92.1 °C. 1 H NMR (CDCI3, 400 MHz): δ 7.01 -6.92 (m, 2H), 4.99 (s, 2H), 3.81 (s, 3H), 3.44 (q, J = 7.2 Hz, 2H), 2.69-2.66 (m, 1 H), 1 .14 (t, J = 7.2 Hz, 3H), 0.94-0.91 (m, 2H), 0.83-0.81 (m, 2H). MS (ESI): m/z 256.2 (M+H)+.
Example 14: (/V-cyclopropyl-/V-methylcarbamoyl)methyl methyl 2(E)but-2-ene-1 , 4- dioate

[0146] Following general procedure A, methyl hydrogen fumarate (MHF) (38.7 g, 0.40 mol) suspended in toluene (100 mL) was reacted at about 80 °C with 2-chloro-/V-cyclopropyl-/V- methylacetamide (60 g, 0.40 mol) in the presence of Ν,Ν-diisopropylethylamine (DIEA; 57.8 g, 78 mL, 0.44 mol) to afford 50 g (50.86%) of the title compound after recrystallization using methyl fe/t-butyl ether. The crystalline compound had a melting point of 93.6 °C. 1 H NMR (CDCI3, 400 MHz): δ 7.01 -6.91 (m, 2H), 5.01 (s, 2H), 3.82 (s, 3H), 2.94 (s, 3H), 2.73-2.68 (m, 1 H), 0.94-0.86 (m, 2H), 0.83-0.78 (m, 2H). MS (ESI): m/z 242.2 (M+H)+.
Example 15: Methyl 2-oxo-2-pyrrolidinylethyl 2(E)but-2-ene-1 ,4-dioate

[0147] Following general procedure A, methyl hydrogen fumarate (MHF) (20.78 g, 0.159 mol) suspended in toluene (60 mL) was reacted at about 80 °C with 2-chloro-1 -pyrrolidin-1 -yl- ethanone (23.5 g, 0.159 mol) in the presence of N,N-diisopropylethylamine (DIEA; 22.69 g, 31 .5 mL, 0.175 mol) to afford 24 g (62.3%) of the title compound after recrystallization using methyl fe/t-butyl ether. The crystalline compound had a melting point of 102.1 °C. 1 H NMR (CDCI3, 400 MHz): δ 7.00-6.92 (m, 2H), 4.75 (s, 2H), 3.81 (s, 3H), 3.53-3.49 (t, J = 6.8 Hz, 2H), 3.42-3.39 (t, J = 6.8 Hz, 2H), 2.20-1 .97 (m, 2H), 1 .91 -1 .82 (m, 2H). MS (ESI): m/z 242 (M+H)+.
………………………………
Patent
http://www.google.co.in/patents/US8148414
(I):

Following general procedure A, methyl hydrogen fumarate (MHF) (0.39 g, 3.00 mmol) dissolved in NMP was reacted at ca. 55° C. with 2-chloro-N,N-diethylacetamide (0.44 g, 3.00 mmol) in the presence of CsHCO3 (0.69 g, 3.60 mmol) to afford 0.37 g (51% yield) of the title compound (1) after purification by silica gel column chromatography (Biotage) using a mixture of ethyl acetate (EtOAc) and hexanes (1:1) as eluent. M.p.: 53-56° C. 1H NMR (CDCl3, 400 MHz): δ 6.99-6.90 (m, 2H), 4.83 (s, 2H), 3.80 (s, 3H), 3.39 (q, J=7.2 Hz, 2H), 3.26 (q, J=7.2 Hz, 2H), 1.24 (t, J=7.2 Hz, 3H), 1.14 (t, J=7.2 Hz, 3H). MS (ESI): m/z 244.13 (M+H)+.

Methyl 2-oxo-2-piperazinylethyl(2E)but-2-ene-1,4-dioate hydrochloride (14) (0.20 g, 0.68 mmol) was reacted with acetyl chloride (AcCl) (0.60 mL, 0.66 g, 0.84 mmol) and diisopropylethylamine (0.70 mL, 0.52 g, 4.0 mmol) in dichloromethane (DCM). Following aqueous work-up, the crude product was purified by silica gel flash chromatography to afford 0.12 g (54% yield) of the title compound (16) as a white solid. 1H NMR (CDCl3, 400 MHz): δ 6.98-6.93 (m, 2H), 4.86 (s, 2H), 3.83 (s, 3H), 3.66 3.63 (m, 4H), 3.50-3.40 (m, 4H), 2.14 (s, 3H). MS (ESI): m/z 299.12 (M+H)+.
Example 9N,N-Dimethylcarbamoyl)methyl methyl(2E)but-2-ene-1,4-dioate (9)
Following general procedure A, methyl hydrogen fumarate (MHF) (0.50 g, 3.84 mmol) dissolved in NMP was reacted at ca. 55° C. with N,N-dimethyl chloroacetamide (0.56 g, 4.61 mmol) in the presence of CsHCO3 (0.89 g, 4.61 mmol). The crude material was precipitated out from a mixture of ethyl acetate (EtOAc) and hexanes (Hxn) (1:1) to provide a white solid. This solid was further dissolved in dichloromethane (DCM) and the organic layer washed with water. After removal of the solvents 0.55 g (67% yield) of the title compound (9) was obtained as a white solid. 1H NMR (CDCl3, 400 MHz): δ 6.98-6.90 (m, 2H), 4.84 (s, 2H), 3.80 (s, 3H), 2.99-2.97 (2s, 6H). MS (ESI): m/z 216 (M+H)+.
………………………..
http://www.google.com/patents/WO2014031844A1?cl=en
Compound (1).
Table 1 : Flushing Incidence as a Function of MMF Cmax


*Formulation 2 is the dosage form described in Example 10; Formulation 3 is the dosage form described in Example 3 ; Formulation 4 is the dosage form described in Example 5 ;
** maximum average Concentration; ***average Cmax; Poster (see above); Compound (1) referred to in the above table is an MMF prodrug of Formula (II); (N,N- Diethylcarbamoyl)methyl methyl (2£)but-2-ene-l,4-dioate having the following chemical structure:
Compound (1).
The maximum slope values ( dose and ng) for different dosage treatments are given in Table 2. The Figures 15-16 show plots of maximum MMF slope vs flushing incidence. The curves in the figures were fitted using a Hill Emax model. Table 2

Compound, Flushing
Table 3: Composition of Enteric Coated Sustained Release Tablet (15% HPMC in Core)

Quantity Quantity
Component Manufacturer Role
(mg tablet) (%w/w)
Vertellus (Greensboro,
Triethyl Citrate Plasticizer 1.25 0.42
NC)
Emerson Resources Anti- tacking
PlasAC YL™ T20 2.41 0.80
(Norristown, PA) agent
Total Enteric
27.87 9.30 Coating
Total Tablet 334.69 111.68
[00191] The tablets were made according to the following steps. The core tablets were prepared using a wet granulation process. The granulation was performed in two batches at 456 g per batch. Compound (1) and hydroxypropyl cellulose were passed through a conical mill with a 610 micron round holed screen. Compound (1) and hydroxypropyl cellulose were then combined in a Key KG- 5 granulator bowl and mixed with water addition for approximately 7 minutes. The wet granules were dried in a Glatt GPCG-1 fluid bed dryer at 40 °C. The two portions of dried granules were sized by passing through a conical mill with an approximately 1300 micron grater type screen. The milled granules were blended with the hypromellose 2208, silicon dioxide, and lactose monohydrate for 10 minutes in an 8 quart (7.6 1) V-blender. This blend was passed through an 850 micron mesh screen. The magnesium stearate was passed through a 600 micron mesh screen and blended with the additional core materials in the V-blender for 5 minutes. Core tablets (299.69 mg) were compressed using a GlobePharma Minipress II rotary tablet press with 8.6 mm round concave tooling. The core tablets had a final mean hardness of approximately 12 kp. For the coating, an aqueous suspension was prepared by mixing with an impeller 63.8 g Opadry 03019184 with 770.7 g of purified water. The water contained in the suspension is removed during the film coating process and therefore not included in the final formulation in Table 3. The tablets were coated with the aqueous suspension in an O’ Hara Technologies Labcoat M coater with a 12″ (30.5 cm) diameter perforated pan until the desired weight gain of barrier coat was achieved. The coating process occurred at an inlet temperature of approximately 52 °C and an outlet temperature of 36 °C. After coating, the tablets were dried for 2 hours at 40 °C. An aqueous suspension was prepared by mixing with an impeller 405.1 g methacrylic acid copolymer dispersion, 6.3 g triethyl citrate, 60.6 g PlasACRYL™ T20 with 228.1 g water. The water contained in the methacrylic acid copolymer dispersion and the
PlasACRYL™ T20 is removed during the film coating process and therefore not included in the final formulation in Table 3. The tablets were coated with the aqueous suspension in the O’ Hara Technologies Labcoat M coater until the desired weight gain of enteric film was achieved. The coating process occurred at an inlet temperature of approximately 40 °C and an outlet temperature of 30 °C. After coating, the tablets were dried for 2 hours at 40 °C.
Example 2
In Vitro Dissolution Profile of Example 1 Dosage Form
[00192] A two-stage dissolution method was used to determine the in vitro dissolution profile of dosage forms prepared according to Example 1. The 2-stage dissolution test was used to better approximate the pH conditions experienced by a dosage form after swallowing by a patient, i.e., low pH of the stomach followed by near neutral pH of the intestines. The dosage forms were first placed into a dissolution vessel (USP, Type I, basket) containing 750 mL of 0.1 N hydrochloric acid (pH 1.2). After 2 hours, 250 mL of 200 mM tribasic sodium phosphate was added to the vessel resulting in a pH adjustment from 1.2 to 6.8. The dissolution medium was kept at 37 °C and was agitated at 100 rpm.
[00193] For the Example 1 dosage forms, samples of the dissolution medium were withdrawn after 1 and 2 hours in the low pH stage, and at 0.5, 2, 4, 7, 10, and 14 hours following buffer addition. The released amount of the MMF prodrug in the samples was determined by reverse phase HPLC using a C18 column and a 7 minute gradient method according to Table 4 where Mobile Phase A is water/0.1 ]¾Ρθ4 and Mobile Phase B is water/acetonitrile/H3PC>4 (10/90/0.1 by volume) with UV detection at 210 nm.
Table 4: HPLC Gradient Conditions

[00194] As shown in FIG. 1, for dosage forms prepared according to Example 1, drug release is delayed for approximately 2 hours, followed by sustained release reaching >90 at 12 hours.
Example 3
Preparation of Delayed Sustained Release Dosage Form (Enteric Coated, 15% HPMC in Core, without Barrier Layer) [00195] Delayed sustained release tablets containing compound (1) were made having the ingredients shown in Table 5:
Table 5: Composition of Enteric Coated Sustained Release Tablet (15% HPMC in Core, without Barrier Layer)

[00196] The tablets were made according to the following steps. The core tablets were prepared using a wet granulation process. The granulation was performed in two batches at 463.9 g per batch. Compound (1) and hydroxypropyl cellulose were passed through a conical mill with a 610 micron round holed screen. Compound (1) and hydroxypropyl cellulose were then combined in a Key KG- 5 granulator bowl and mixed with water addition for approximately 10 minutes. The wet granules were dried in a Glatt GPCG-1 fluid bed dryer at 40 °C. The two portions of dried granules were blended with silicon dioxide and sized by passing through a conical mill with an approximately 1300 micron grater type screen. The milled granules were blended with the hypromellose 2208 and lactose monohydrate for 10 minutes in an 8 quart (7.6 1) V-blender. This blend was passed through an 850 micron mesh screen. The magnesium stearate was passed through a 600 micron mesh screen and blended with the additional core materials in the V-blender for 5 minutes. Core tablets (299.68 mg) were compressed using a GlobePharma Minipress II rotary tablet press with 11/32″ round concave tooling. The core tablets had a final mean hardness of approximately 11 kp. For the coating, an aqueous suspension was prepared by mixing with an impeller 578.7 g methacrylic acid copolymer dispersion, 9.0 g triethyl citrate, 86.5 g PlasACRYL™ T20 with 325.8 g water. The water contained in the methacrylic acid copolymer dispersion and the
PlasACRYL™ T20 is removed during the film coating process and therefore not included in the final formulation in Table 4. The tablets were coated with the aqueous suspension in the O’ Hara Technologies Labcoat M coater until the desired weight gain of enteric film was achieved. The coating process occurred at an inlet temperature of approximately 41 °C and an outlet temperature of 31 °C. After coating, the tablets were dried for 2 hours at 40 °C.
…………………………
WO 2014071371
http://www.google.com/patents/WO2014071371A1?cl=en
(N,N-Diethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4-dioate has the following chemical structure:

This compound was synthesized in Example 1 of Gangakhedkar et al., U.S. Patent No. 8,148,414. The compound is a prodrug of methyl hydrogen fumarate (MHF) and has a disclosed melting point of between 53 °C and 56 °C.
Cocrystals are crystals that contain two or more non-identical molecules that form a crystalline structure. The intermolecular interactions between the non-identical molecules in the resulting crystal structures can result in physical and chemical properties that differ from the properties of the individual components. Such properties can include, for example, melting point, solubility, chemical stability, mechanical properties and others. Examples of cocrystals may be found in the Cambridge Structural Database and in Etter, et al.,
“The use of cocrystallization as a method of studying hydrogen bond preferences of 2-aminopyridine” J. Chem. Soc, Chem. Commun. (1990), 589-591 ; Etter, et al., “Graph-set analysis of hydrogen-bond patterns in organic crystals” Acta Crystallogr., Sect. B, Struct. Sci. (1990), B46: 256-262; and Etter, et al., “Hydrogen bond directed cocrystallization and molecular recognition properties of diarylureas” J. Am. Chem. Soc. (1990), 1 12: 8415-8426. Additional information relating to cocrystals can be found in: Carl Henrik Gorbotz and Hans-Petter Hersleth,
“On the inclusion of solvent molecules in the crystal structures of organic compounds”; Acta Cryst. (2000), B56: 625-534; and Senthil Kumar, et al., “Molecular Complexes of Some Mono- and Dicarboxylic Acids with trans-1 ,4,-Dithiane-1 ,4-dioxide” American Chemical Society, Crystal Growth & Design (2002) , 2(4) : 313-318.
(N,N-Diethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4-dioate is a prodrug of methyl hydrogen fumarate. Once administered, the compound is metabolized in vivo into an active metabolite, namely, methyl hydrogen fumarate (MHF) which is also referred to herein as monomethyl fumarate (MMF). The in vivo metabolism of (N,N-Diethylcarbamoyl)methyl

(N,N-Diethylcarbamoyl)methyl methyl Methyl hydrogen fumarate N ^ diethyl glycolamide
(2E)but-2-ene-1 ,4-dioate
Table 1

As can be seen from the data in Table 1 , the six cocrystals disclosed herein each exhibit a higher melting point than crystalline (N,N-Diethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4- dioate.

…………………………….
Steady state pharmacokinetics of formulations of XP23829, a novel prodrug of monomethyl fumarate (MMF), in healthy subjects
66th Annu Meet Am Acad Neurol (AAN) (April 26-May 3, Philadelphia) 2014, Abst P1.188
………………………………….
Lymphocyte and eosinophil responses in healthy subjects dosed with Tecfidera and XP23829, a novel fumaric acid ester (FAE)
66th Annu Meet Am Acad Neurol (AAN) (April 26-May 3, Philadelphia) 2014, Abst P1.201
………………………..
A comparison of XP23829 with DMF, the active ingredient of BG-12
4th Cooperative Meet Consorti Mult Scler Cent (CMSC) Am Comm Treat Res Mult Scler (ACTRIMS) (May 30-June 2, San Diego) 2012, Abst SC03
http://annualmeeting.mscare.org/index.php?option=com_content&view=article&id=174&Itemid=101
…………………………..
Favorable metabolism and pharmacokinetics of formulations of XP23829, a novel fumaric acid ester, in healthy subjects
65th Annu Meet Am Acad Neurol (AAN) (March 16-23, San Diego) 2013, Abst P05.189
…………………………………..
Comparison of the efficacy and tolerability of a novel methyl hydrogenfumarate prodrug with dimethyl fumarate in rodent EAE and GI irritation models
Neurology 2011, 76(9): Abst P05.040
| WO2013119791A1 * | Feb 7, 2013 | Aug 15, 2013 | Xenoport, Inc. | Morpholinoalkyl fumarate compounds, pharmaceutical compositions, and methods of use |
| US20120034303 * | Jan 8, 2010 | Feb 9, 2012 | Forward Pharma A/S | Pharmaceutical formulation comprising one or more fumaric acid esters in an erosion matrix |
| US20120095003 * | Oct 14, 2011 | Apr 19, 2012 | Xenoport, Inc. | Methods of using prodrugs of methyl hydrogen fumarate and pharmaceutical compositions thereof |
| US20120157523 * | Oct 14, 2011 | Jun 21, 2012 | Xenoport, Inc. | Prodrugs of methyl hydrogen fumarate, pharmaceutical compositions thereof, and methods of use |
| K Gogas ET AL: “Comparison of the efficacy and tolerability of a novel methylhydrogenfumarate prodrug with dimethylfumarate in rodent experimental autoimmune encephalomyelitis and GI irritation models“, 26th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) & 15th Annual Conference of Rehabilitation in MS (RIMS), 15 October 2010 (2010-10-15), XP055076728, Retrieved from the Internet: URL:http://registration.akm.ch/einsicht.php?XNABSTRACT_ID=115706&XNSPRACHE_ID=2&XNKONGRESS_ID=126&XNMASKEN_ID=900 [retrieved on 2013-08-27] |
![]()
| WO2013119791A1 * | Feb 7, 2013 | Aug 15, 2013 | Xenoport, Inc. | Morpholinoalkyl fumarate compounds, pharmaceutical compositions, and methods of use |
| US20100048651 * | Aug 19, 2009 | Feb 25, 2010 | Xenoport, Inc. | Prodrugs of methyl hydrogen fumarate, pharmaceutical compositions thereof, and methods of use |
| US8669281 | 20 Sep 2013 | 11 Mar 2014 | Alkermes Pharma Ireland Limited | Prodrugs of fumarates and their use in treating various diseases |
| WO2014031894A1 | 22 Aug 2013 | 27 Feb 2014 | Xenoport, Inc. | Oral dosage forms of methyl hydrogen fumarate and prodrugs thereof |
| WO2014071371A1 | 5 Nov 2013 | 8 May 2014 | Xenoport, Inc. | Cocrystals of (n,n-diethylcarbamoyl)methyl methyl (2e)but-2-ene-1,4-dioate |
Inovio Kicks Off Study of Cervical Cancer Immunotherapy INO 3112

Inovio Kicks Off Study of Cervical Cancer Immunotherapy
Inovio Pharmaceuticals Inc. announced it has initiated a Phase 1/2a clinical trial to evaluate safety, immunogenicity, clinical responses and disease-free survival of its DNA immunotherapy product, INO-3112, in treating human papillomavirus (HPV)-associated cervical cancer. Read more…
Inovio Pharmaceuticals Inc. announced it has initiated a Phase 1/2a clinical trial to evaluate safety, immunogenicity, clinical responses and disease-free survival of its DNA immunotherapy product, INO-3112, in treating human papillomavirus (HPV)-associated cervical cancer. INO-3112 is a combination of Inovio’s lead active immunotherapy product, VGX-3100, and its proprietary immune activator expressing interleukin-12 (IL-12). VGX-3100 is currently being evaluated in a randomized Phase 2 efficacy trial for the treatment of high grade cervical dysplasia (pre-cancer).
DARA BioSciences receives FDA orphan drug designation for KRN5500 (SPK 241) …..Antitumor agent

KRN5500
Antitumor agent
151276-95-8 cas
IUPAC/Chemical name:
(2E,4E)-N-(2-(((2R,3R,4R,5R,6S)-6-((7H-purin-6-yl)amino)-2-((S)-1,2-dihydroxyethyl)-4,5-dihydroxytetrahydro-2H-pyran-3-yl)amino)-2-oxoethyl)tetradeca-2,4-dienamide
C28H43N7O7
Exact Mass: 589.32240
L-glycero-beta-L-manno-Heptopyranosylamine, 4-deoxy-4-((((1-oxo-2,4-tetradecadienyl)amino)acetyl)amino)-N-1H-purin-6-yl-, (E,E)-
L-glycero-beta-L-manno-Heptopyranosylamine, 4-deoxy-4-(((((2E,4E)-1-oxo-2,4-tetradecadienyl)amino)acetyl)amino)-N-1H-purin-6-yl-

-
- (1) Melting point: 182-183 °C,
- (2) Specific rotation [a]0 2S = 0 (c = 0.1, in methanol),
- (3) Elementary analysis:

- (4) FD mass spectrum (m/z): 590 (M + H) , C28 H4 3 N707
- (5) Infrared spectrum (KBr disc): 3250 cm-1, 1650 cm-1, 1620 cm-1,
- (6) Proton nuclear magnetic resonance spectrum (500 MHz, in CD30D) δH: 0.89 (3H, t, J = 7.3 Hz), 1.20-1.50 (14H, m), 2.18 (2H, dt, J = 7.3, 7.3 Hz), 3.6-3.8 (5H, m), 3.95 (1 H, d, J = 16.3 Hz), 3.98 (1H, d, J = 16.3 Hz), 4.00 (1H, dd, J = <1, 2.9 Hz), 4.15 (1H, dd, J = 10.8, 10.8 Hz), 5.66 (1 H, brs), 5.98 (1 H, d, J = 15.7 Hz), 6.12 (1 H, dt, J = 7.3, 15.7 Hz), 6.22 (1 H, dd, J = 10.0, 15.7 Hz), 7.17 (1 H, dd, J = 10.0, 15.7 Hz), 8.15 (1 H, s), 8.30 (1 H, s).
- EP 0525479; JP 1993186494; US 5461036; US 5631238

DARA BioSciences receives FDA orphan drug designation for KRN5500
DARA BioSciences has received orphan drug designation from the US Food and Drug Administration’s (FDA) Office of Orphan Products Development for KRN5500, for treating multiple myeloma
Multiple myeloma is a hematologic cancer or cancer of the blood.
KRN5500 is a non-opioid, non-narcotic compound that is currently being tested in Phase I clinical trial.
Earlier this year, KRN5500 received orphan status to be developed for the parenteral treatment of painful, chronic, chemotherapy-induced peripheral neuropathy (CCIPN) that is refractory to conventional analgesics in patients with cancer.
“We believe this myeloma-specific orphan designation enhances both the viability and the future market opportunity for this valuable pipeline product.”
DARA BioSciences MD, CEO and chief medical officer David J Drutz said: “It is noteworthy in this regard that up to 20% of myeloma patients have intrinsic peripheral neuropathy, an incidence that increases to the range of 75% in patients treated with neurotoxic drugs such as thalidomide or bortezomib.
KRN5500 is a semisynthetic derivative of the nucleoside-like antineoplastic antibiotic spicamycin, originally isolated from the bacterium Streptomyces alanosinicus. KRN 5500 inhibits protein synthesis by interfering with endoplasmic reticulum and Golgi apparatus functions. This agent also induces cell differentiation and caspase-dependent apoptosis.
KRN5500 is available as a solution for intravenous (IV) administration. KRN5500 was discovered in an effort to identify new agents that induced differentiation of myeloid leukemia cells.
Safety and efficacy data from Phase I trials have been leveraged to support DARA Therapeutics’ active IND and ongoing Phase 2a clinical trial. The objective of this Phase 2a feasibility study is to determine the potential of KRN5500 (a spicamycin analogue) to be a breakthrough medicine for the treatment of neuropathic pain in cancer patients.
Four clinical trials have been conducted in cancer patients, including one in Japan and 3 in the United States. Three of these studies are complete; the fourth was closed to patient accrual and treatment in December 2004.
A total of 91 patients with solid tumors have been treated with single IV KRN5500 doses of up to 21 mg/m2 and weekly doses of up to 42 mg/m2. While KRN5500 has not shown anti-cancer efficacy in any trial, its use in pain elimination is encouraging. (source: http://www.darabiosciences.com/krn5500.htm).

Chemical structures of KRN5500 and its known metabolites.
………………..
http://www.google.com/patents/EP0525479A1?cl=en
spk 241
- 6-[4′-N-(N’-trans,trans-2,4-tridecadienylglycyl)spicamynyl-amino]purine,
- (20) SPK241:


Example 52: Preparation of SPK241
-
[0214]To trans-2-dodecenal (4.5 g) dissolved in methylene chloride (80 ml) was added (carbomethoxymethylene)triphenylphosphorane (8.3 g), and the mixture was stirred for 2 hours. The reaction mixture was subjected to chromatography on a silica gel column with eluent systems of n-hexane- ethyl acetate (from 100:1 to 20:1) to give the methyl ester of trans,trans-2,4-tetradecadienoic acid (5.4 g). Potassium hydroxide (6.5 g) was dissolved in a mixed solvent of ethanol-water (1:1) (100 ml). The methyl ester of trans,trans-2,4-tetradecadienoic acid (5.4 g) was added to the mixture, and the resulting mixture was stirred at 60 °C for 40 minutes. After the reaction mixture was cooled, it was adjusted to the weak acidic range of pH with citric acid and extracted with ethyl acetate. The ethyl acetate layer was dried over anhydrous sodium sulfate and concentrated to give trans,trans-2,4-tetradecadienoic acid (4.4 g). Hereafter, the title compound can be synthesized by the two methods described below.
-
[0215]In the first method, trans,trans-2,4-tetradecadienoic acid (4.3 g) is first dissolved in N,N-dimethylformamide (DMF, 50 ml). Para-nitrophenol (2.67 g) and N,N’-dicyclohexylcarbodiimide (3.9 g) were added to trans,trans-2,4-tetradecadienoic acid solution, and the mixture was stirred for 12 hours. After precipitates produced were removed by filtration and the solvent (DMF) was removed by distillation, the residue was subjected to chromatography on a silica gel column with eluent systems of n-hexane-ethyl acetate (from 200:1 to 50:1) to give the active ester of trans,trans-2,4-tetradecadienoic acid (5.1 g). To the active ester (500 mg) dissolved in DMF (30 ml) were added 6-(4′-N-glycyl-spicamynyl-amino)purine hydrochloride (556 mg) and triethylamine (1.2 ml). The mixture was stirred for 12 hours. After the solvent was removed by distillation, the residue was subjected to chromatography on a silica gel column with eluent systems of chloroform-methanol (from 7:1 to 5:1) to give SPK241 in the yield of 398 mg.
-
[0216]In the second method, trans,trans-2,4-tetradecadienoic acid (99.6 g) was dissolved in thionyl chloride (87 ml), and the mixture was stirred at room temperature. The excessive thionyl chloride was removed by distillation to give trans,trans-2,4-tetradecadienoic acid chloride (102.0 g). To glycine (66.8 g) dissolved in an aqueous 2N sodium hydroxide solution (540 ml) were added at the same time trans,trans-2,4-tetradecadienoic acid chloride (102.0 g) and 2N sodium hydroxide (270 ml) with 1/10 portions at a 3 minute interval. After the addition was completed, the mixture was warmed to room temperature, stirred for 15 minutes and acidified with concentrated hydrochloric acid (140 ml) under ice-cooling. Precipitates thus produced were collected by filtration and desiccated to give trans,trans-2,4-tetradecadienoyl glycine (75.0 g). To the solution of trans,trans-2,4-tetradecadienoyl glycine (4.7 g) and 6-(4′-N-glycyl-spicamynyl-amino)-purine (5.1 g) in N,N-dimethylformamide (DMF, 60 ml) was added N-hydroxysuccinimide (2.1 g), and the mixture was ice-cooled. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (3.4 g) dissolved in DMF (100 ml) was added dropwise to the mixture. After the addition was completed, the mixture was heated to room temperature and stirred for 12 hours. Water (500 ml) was added to the reaction mixture, and precipitates produced were collected by filtration and desiccated. Sodium methoxide (3.1 g) was added to a suspension of the precipitates in methanol (100 ml), and the mixture was stirred at room temperature, then ice-cooled and acidified by adding dropwise thereto a 10% methanolic hydrochloric acid solution. Precipitates produced were filtered, dried and subjected to chromatography on a silica gel column with eluent systems of chloroform-methanol (from 7:1 to 5:1) to give SPK241 in the yield of 5.00 g.
Physicochemical properties of SPK241
-
[0217]
- (1) Melting point: 182-183 °C,
- (2) Specific rotation [a]0 2S = 0 (c = 0.1, in methanol),
- (3) Elementary analysis:
- (4) FD mass spectrum (m/z): 590 (M + H) , C28 H4 3 N707
- (5) Infrared spectrum (KBr disc): 3250 cm-1, 1650 cm-1, 1620 cm-1,
- (6) Proton nuclear magnetic resonance spectrum (500 MHz, in CD30D) δH: 0.89 (3H, t, J = 7.3 Hz), 1.20-1.50 (14H, m), 2.18 (2H, dt, J = 7.3, 7.3 Hz), 3.6-3.8 (5H, m), 3.95 (1 H, d, J = 16.3 Hz), 3.98 (1H, d, J = 16.3 Hz), 4.00 (1H, dd, J = <1, 2.9 Hz), 4.15 (1H, dd, J = 10.8, 10.8 Hz), 5.66 (1 H, brs), 5.98 (1 H, d, J = 15.7 Hz), 6.12 (1 H, dt, J = 7.3, 15.7 Hz), 6.22 (1 H, dd, J = 10.0, 15.7 Hz), 7.17 (1 H, dd, J = 10.0, 15.7 Hz), 8.15 (1 H, s), 8.30 (1 H, s).

| DE3407979A1 * | Mar 3, 1984 | Sep 6, 1984 | Kirin Brewery | Spicamycin sowie verfahren zu seiner herstellung |
| JPS59161396A | Title not available | |||
| US3155647 | Jul 24, 1963 | Nov 3, 1964 | Olin Mathieson | Septaciding and derivatives thereof |
| WO1990015811A1 | Jun 14, 1990 | Dec 27, 1990 | Kirin Brewery | Spicamycin x and its use |
| EP1328236A2 * | Sep 20, 2001 | Jul 23, 2003 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
| EP2305264A1 * | Sep 20, 2001 | Apr 6, 2011 | The General Hospital Corporation | Spicamycin derivatives for use in decreasing or preventing pain |
| EP2349285A2 * | Oct 9, 2009 | Aug 3, 2011 | Dara Biosciences, Inc. | Methods for treating or preventing pain using spicamycin derivatives |
| EP2597082A1 | Nov 24, 2011 | May 29, 2013 | Symrise AG | Compounds for masking an unpleasant taste |
| US5905069 * | Jan 26, 1998 | May 18, 1999 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin or derivatives thereof |
| US7196071 | Sep 20, 2001 | Mar 27, 2007 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
| US7375094 | Mar 15, 2007 | May 20, 2008 | The General Hospital Corporation | Produced via Streptomyces; antitumor agents; time-release agents; for opiod-resistant pain; drug screening |
| US7632825 | Apr 30, 2008 | Dec 15, 2009 | Bayer Pharmaceuticals Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
|
References 1: Mizumura Y. [Spicamycin derivative]. Nippon Rinsho. 2006 Feb;64(2):322-8. Review. Japanese. PubMed PMID: 16454188. 2: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2004 Apr;26(3):211-44. PubMed PMID: 15148527. 3: Borsook D, Edwards AD. Antineuropathic effects of the antibiotic derivative spicamycin KRN5500. Pain Med. 2004 Mar;5(1):104-8. PubMed PMID: 14996243. 4: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2003 Dec;25(10):831-55. PubMed PMID: 14735233. 5: Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2003 Nov;25(9):747-71. PubMed PMID: 14685303. 6: Supko JG, Eder JP Jr, Ryan DP, Seiden MV, Lynch TJ, Amrein PC, Kufe DW, Clark JW. Phase I clinical trial and pharmacokinetic study of the spicamycin analog KRN5500 administered as a 1-hour intravenous infusion for five consecutive days to patients with refractory solid tumors. Clin Cancer Res. 2003 Nov 1;9(14):5178-86. PubMed PMID: 14613997. 7: Yamamoto N, Tamura T, Kamiya Y, Ono H, Kondoh H, Shirao K, Matsumura Y, Tanigawara Y, Shimada Y. Phase I and pharmacokinetic study of KRN5500, a spicamycin derivative, for patients with advanced solid tumors. Jpn J Clin Oncol. 2003 Jun;33(6):302-8. PubMed PMID: 12913085. 8: Kobierski LA, Abdi S, DiLorenzo L, Feroz N, Borsook D. A single intravenous injection of KRN5500 (antibiotic spicamycin) produces long-term decreases in multiple sensory hypersensitivities in neuropathic pain. Anesth Analg. 2003 Jul;97(1):174-82, table of contents. PubMed PMID: 12818962. 9: Gadgeel SM, Boinpally RR, Heilbrun LK, Wozniak A, Jain V, Redman B, Zalupski M, Wiegand R, Parchment R, LoRusso PM. A phase I clinical trial of spicamycin derivative KRN5500 (NSC 650426) using a phase I accelerated titration “2B” design. Invest New Drugs. 2003 Feb;21(1):63-74. PubMed PMID: 12795531. 10: Byrd JC, Lucas DM, Mone AP, Kitner JB, Drabick JJ, Grever MR. KRN5500: a novel therapeutic agent with in vitro activity against human B-cell chronic lymphocytic leukemia cells mediates cytotoxicity via the intrinsic pathway of apoptosis. Blood. 2003 Jun 1;101(11):4547-50. Epub 2003 Feb 20. PubMed PMID: 12595316. |
BMS-791325, Beclabuvir In Phase 2 for Hepatitis C (HCV)
BMS-791325, Beclabuvir
IN PHASE 2 for Hepatitis C (HCV)
An NS5B inhibitor.

BMS-791325 preferably is

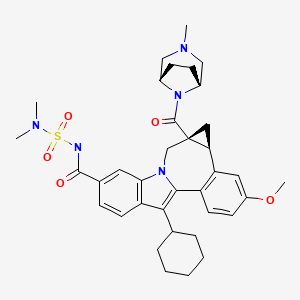

958002-33-0
958002-36-3 (as hydrochloride)
C36 H45 N5 O5 S, 659.838
Cycloprop(d)indolo(2,1-a)(2)benzazepine-9-carboxamide, 12-cyclohexyl-N-((dimethylamino)sulfonyl)-4b,5,5a,6-tetrahydro-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-, (4bS,5aR)-
(4bS,5aR)-12-Cyclohexyl-N-(dimethylsulfamoyl)-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-4b,5,5a,6-tetrahydrocyclopropa(d)indolo(2,1-a)(2)benzazepine-9-carboxamide
(4bS,5aR)-12-Cyclohexyl-N-(dimethylsulfamoyl)-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-4b,5,5a,6-tetrahydrocyclopropa(d)indolo(2,1-a)(2)benzazepine-9-carboxamide
(1aR,12bS)-8-Cyclohexyl-N-(dimethylsulfamoyl)-11-methoxy-1a-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8-yl)carbonyl]-1,1a,2,12b-tetrahydrocyclopropa[d]indolo[2,1-a][2]benzazepine-5-carboxamide
Cycloprop [d] indolo [2, 1 -a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (laR,12bS)-
Bristol-Myers Squibb (Originator)
RNA-Directed RNA Polymerase (NS5B) Inhibitors
UNII-MYW1X5CO9S
BMS-791325 is in phase II clinical studies at Bristol-Myers Squibb for the treatment of chronic hepatitis C. In 2013, the company received breakthrough therapy designation in the U.S. for the treatment of chronic hepatitis C in combination with daclatasvir and asunaprevir.
| Patent | WO 2007136982 |
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
http://www.google.com/patents/WO2007136982A1?cl=en
Scheme 1.

N-protected piperazines can also be coupled to the intermediate indolobenzazepine acids and the resultant piperazine carboxamides can be deprotected using methods known in the art and derivatized using a variety of synthetic protocols, some illustrative examples of which are shown below (See Scheme 2).
Scheme 2.

An intermediate useful for the synthesis of some compounds of the invention involves the preparation of the tert-butyl ester indolobenzazepine shown in Scheme 3. Scheme 3.

t-Butylation either:

This methodology involves base catalyzed hydrolysis of the indole methyl ester shown, followed by its reaction with either thionyl chloride and potassium tertiary butoxide, or alkylation with silver carbonate and tertiary butyl bromides. The resultant compound can be transformed using chemistry analogous to that outlined previously to provide the mixed ester indolobenzazepines shown above.
Scheme 4.

Some examples exist as stereoisomeric mixtures. The invention encompasses all stereoisomers of the compounds. Methods of fractionating stereoisomeric mixtures are well known in the art, and include but are not limited to; preparative chiral supercritical fluid chromatography (SFC) and chiral high performance liquid chromatography (HPLC). An example using this approach is shown in scheme 5. Scheme 5.

An additional method to achieve such separations involves the preparation of mixtures of diastereomers which can be separated using a variety of methods known in the art. One example of this approach is shown below (Scheme 6).
Scheme 6.

Diastereomers separated by reverse phase HPLC
Some diastereomeric amides can be separated using reverse phase HPLC. After hydroysis, the resultant optically active acids can be coupled with bridged piperazine derivatives (Scheme 6). For example, O-(lH-benzotriazol-l-yl)-N,N, N’,N’-tetramethyluronium tetrafluoroborate and diisopropyl ethyl amine in DMSO can be used to give the alkyl bridged piperazine carboxamides. Other standard acid amine coupling methods can also be used to give optically active carboxamides.

Schemes 7-9 illustrate other methods of making intermediates and compounds.

Scheme 8.

Scheme 9.


Biological Methods
The compounds demonstrated activity against HCV NS5B as determined in the following HCV RdRp assays.
DESCRIPTION OF SPECIFIC EMBODIMENTS
Unless otherwise specified, analytical LCMS data on the following intermediates and examples were acquired using the following columns and conditions. Stop time: Gradient time + 1 minute; Starting cone: 0% B unless otherwise noted; Eluent A: 5% CH3CN / 95% H2O with 10 mM NH4OAc (for columns A, D and E); 10 % MeOH / 90 % H2O with 0.1% TFA (for columns B and C); Eluent B: 95% CH3CN / 5% H2O with 10 mM NH4OAc (for columns A, D and E); 90 % MeOH / 10 % H2O with 0.1% TFA (for columns B and C); Column A:
Phenomenex lOμ 4.6 x 50 mm C18; Column B: Phenomenex C18 lOμ 3.0 x 50 mm; Column C: Phenomenex 4.6 x 50 mm C18 lOμ; Column D: Phenomenex Lina C18 5μ 3.0 x 50 mm; Column E: Phenomenex 5μ 4.6 x 50 mm Cl 8.
Intermediate 1

lH-Indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-, methyl ester. Freshly recrystallized pyridinium tribromide (recrystallization from hot AcOH (5 mL per 1 g), rinsed with cold AcOH and dried under high vacuum over KOH) was added in portions (over 10 min.) to a stirring solution of methyl 3-cyclohexyl-lH-indole-6- carboxylate (60 g, 233 mmol) (prepared using procedures describe in WO2004/065367) in CHC1/THF (1: 1, 1.25 L) at 2o C. The reaction solution was stirred at 0-5 °C for 2.5h, and washed with sat. aq. NaHSO3 (1 L), 1 N HCl (1 L) and brine (1 L). The organic layer was dried (MgSO4) and concentrated. The resulting red oil was diluted with Et2θ and concentrated. The resulting pink solid was dissolved into Et2θ (200 mL) treated with hexanes (300 mL) and partially concentrated. The solids were collected by filtration and rinsed with hexanes. The mother liquor was concentrated to dryness and the procedure repeated. The solids were combined to yield lH-indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-, methyl ester (64 g, 190 mmol, 82%) as a fluffy pink solid, which was used without further purification. IHNMR (300 MHz, CDCl3) δ 8.47 (br s, IH), 8.03 (d, J = 1.4 Hz, IH), 7.74 (dd, J = 1.4, 8.8 Hz, IH), 7.69 (d, J = 8.8 Hz, IH), 3.92 (s, 3H), 2.82 (tt, J = 3.7, 11.7 Hz, IH), 1.98 – 1.72 (m, 7H), 1.50 – 1.27 (m, 3H). 13CNMR (75 MHz, CDC13) δ 168.2, 135.6, 130.2, 123.1, 120.8, 120.3, 118.7, 112.8, 110.7, 52.1, 37.0, 32.2(2), 27.0(2), 26.1. LCMS: m/e 334 (M-H)“, ret time 3.34 min, column A, 4 minute gradient.
Intermediate 2

lH-Indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-. A solution of methyl 2- bromo-S-cyclohexyl-lH-indole-ό-carboxylate (20 g, 60 mmol) and LiOH (3.8 g, 160 mmol) in MeOΗ/TΗF/Η2O ( 1 : 1 : 1 , 300 mL) was heated at 90 °C for 2h. The reaction mixture was cooled in an ice/H2O bath, neutralized with IM HCl (-160 mL) diluted with H2O (250 mL) and stirred for Ih at rt. The precipitates were collected by filtration rinse with H2O and dried to yield lH-indole-6-carboxylic acid, 2-bromo-3- cyclohexyl- (quant.) which was used without further purification.
An alternative procedure that can by used to provide lH-indole-6-carboxylic acid, 2-bromo-3-cyclohexyl- is described below: A solution of methyl 2-bromo-3-cyclohexyl-lH-indole-6-carboxylate (117 g, 349 mmol) and LiOKH2O (26.4 g, 629 mmol) in MeOH/THF/H2O (1: 1: 1, 1.8 L) was heated at reflux for 3h. The reaction mixture was cooled in an ice/H2O bath to ~2 °C, neutralized with IM HCl (-650 mL) (added at such a rate that temperature did not exceed 5 °C), diluted with H2O (1 L) and stirred while warming to ambient temperature. The precipitates were collected by filtration rinsed with H2O and dried to yield the mono THF solvate of lH-indole-6-carboxylic acid, 2-bromo-3- cyclohexyl- (135.5 g, 345 mmol, 99%) as a yellow solid, which was used without further purification. IHNMR (300 MHz, CDCl3) δ 11.01 (br s, IH), 8.77 (s, IH), 8.07 (d, J = 1.5 Hz, IH), 7.82 (dd, J = 1.5, 8.8 Hz, IH), 7.72 (d, J = 8.8 Hz, IH), 3.84 – 3.74 (m, 4H), 2.89 (m, IH), 1.98 – 1.72 (m, HH), 1.50 – 1.24 (m, 3H). 13CNMR (75 MHz, CDC13) δ 172.7, 135.5, 130.7, 122.3, 120.9(2), 118.8, 113.3, 111.1, 67.9(2), 37.0, 32.2(2), 27.0(2), 26.1, 25.5(2). LCMS: m/e 320 (M-H)“, ret time 2.21 min, column A, 4 minute gradient.
Intermediate 3

lH-Indole-6-carboxamide, 2-bromo-3-cyclohexyl-N-
[(dimethylamino)sulfonyl]-. l,l’-Carbonyldiimidazole (1.17 g, 7.2 mmol) was added to a stirred solution of 2-bromo-3-cyclohexyl-lH-indole-6-carboxylic acid (2.03 g, 6.3 mmol) in THF (6 mL) at 22 °C. The evolution of CO2 was instantaneous and when it slowed the solution was heated at 50°C for 1 hr and then cooled to 220C. N,N-Dimethylsulfamide (0.94 g, 7.56 mmol) was added followed by the dropwise addition of a solution of DBU (1.34 g ,8.8 mmol) in THF (4 mL). Stirring was continued for 24 hr. The mixture was partitioned between ethyl acetate and dilute HCl. The ethyl acetate layer was washed with water followed by brine and dried over Na2SO4. The extract was concentrated to dryness to leave the title product as a pale yellow friable foam, (2.0 g, 74 %, >90 % purity , estimated from NMR). 1H NMR (300 MHz, DMSO-D6) δ ppm 1.28 – 1.49 (m, 3 H) 1.59 – 2.04 (m, 7 H) 2.74 – 2.82 (m, 1 H) 2.88 (s, 6 H) 7.57 (dd, J=8.42, 1.46 Hz, 1 H) 7.74 (d, J=8.78 Hz, 1 H) 7.91 (s, 1 H) 11.71 (s, 1 H) 12.08 (s, 1 H).
An alternative method for the preparation of lH-indole-6-carboxamide, 2- bromo-3-cyclohexyl-N-[(dimethylamino)sulfonyl]- is described below.
To a 1 L four necked round bottom flask equipped with a mechanical stirrer, a temperature controller, a N2 inlet , and a condenser, under N2, was added 2-bromo-3- cyclohexyl-lH-indole-6-carboxylic acid (102.0 g, 0.259 mol) and dry TΗF (300 mL). After stirring for 10 min, CDI (50.3 g, 0.31 mol) was added portion wise. The reaction mixture was then heated to 50 oC for 2 h. After cooling to 30 oC, N,N- dimethylaminosulfonamide (41.7 g, 0.336 mol) was added in one portion followed by addition of DBU (54.1 mL, 0.362 mol) drop wise over a period of 1 h. The reaction mixture was then stirred at rt for 20 h. The solvent was removed in vacuo and the residue was partitioned between EtOAc and 1 Ν HCl (1 : 1, 2 L). The organic layer was separated and the aqueous layer was extracted with EtOAc (500 mL). The combined organic layers were washed with brine (1.5 L) and dried over MgSO4. The solution was filtered and concentrated in vacuo to give the crude product (111.0 g). The crude product was suspended in EtOAc (400 mL) at 60 oC. To the suspension was added heptane (2 L) slowly. The resulting suspension was stirred and cooled to 0 oC. It was then filtered. The filter cake was rinsed with small amount of heptane and house vacuum air dried for 2 days. The product was collected as a white solid (92.0 g, 83%). 1H ΝMR (MeOD, 300 MHz) δ 7.89 (s, H), 7.77 (d, J= 8.4 Hz, IH), 7.55 (dd, J= 8.4 and 1.8 Hz, IH), 3.01 (s, 6H), 2.73-2.95 (m, IH), 1.81-2.05 (m, 8H), 1.39-1.50 (m, 2H); m/z 429 (M +H)+. Intermediate 4

lH-Indole-6-carboxamide, 3-cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2- formyl-4-methoxyphenyl)-. A mixture of the 2-Bromo-3-cyclohexyl- N- [(dimethylamino)sulfonyl]-lH-indole-6-carboxamide (4.28g, 0.01 mol), 4-methoxy- 2-formylphenyl boronic acid (2.1%, 0.015 mol), 2-dicyclohexylphosphino-2′,6′- dimethoxy-biphenyl (41 mg, 0.0001 mol), palladium acetate (11.2 mg), and finely ground potassium carbonate (4.24g, 0.02 mol) in toluene (30 mL) was stirred under reflux and under nitrogen for 30 min, at which time LC/MS analysis showed the reaction to be complete. The reaction mixture was then diluted with ethyl acetate and water, and then acidified with an excess of dilute HCl. The ethyl acetate layer was then collected and washed with dilute HCl, water and brine. The organic solution was then dried (magnesium sulfate), filtered and concentrated to give a gum. The gum was diluted with hexanes (250 ml) and ethyl acetate (25 mL), and the mixture was stirred for 20 hr at 22° C during which time the product was transformed into a bright yellow granular solid (4.8 g) which was used directly without further purification.
An alternative procedure for the preparation of lH-indole-6-carboxamide, 3- cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2-formyl-4-methoxyphenyl)- is provided below:
To a slurried solution of 2-bromo-3-cyclohexyl-N-[(dimethylamino)sulfonyl]- indole-6-carboxamide (54.0 g, 126 mmol), 4-methoxy-2-formylphenylboronic acid (29.5 g, 164 mmol) and LiCl (13.3 g, 315 mmol) in EtOH/toluene (1 : 1, 1 L) was added a solution of Na2CO3 (40.1 g, 379 mmol) in water (380 mL). The reaction mixture was stirred 10 min. and then Pd(PPh3)4 (11.3 g, 10.0 mmol) was added. The reaction solution was flushed with nitrogen and heated at 70 °C (internal monitoring) overnight and then cooled to rt. The reaction was diluted with EtOAc (1 L) and EtOH (100 mL), washed carefully with IN aqueous HCl (1 L) and brine (500 mL), dried (MgSO4), filtered and concentrated. The residual solids were stirred with Et20 (600 mL) for Ih and collected by filtration to yield lH-indole-6-carboxamide, 3- cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2-formyl-4-methoxyphenyl)- (52.8g, 109 mmol, 87%) as a yellow powder which was used without further purification. IHNMR (300 MHz, d6-DMSO) δ 11.66 (s, IH), 8.17 (s, IH), 7.75 (d, J = 8.4 Hz, IH), 7.74 (d, J = 8.4 Hz, IH), 7.59 (dd, J = 1.4, 8.4 Hz, IH), 7.23 – 7.16 (m, 2H), 7.08 (dd, J = 2.6, 8.4 Hz, IH), 6.54 (d, J = 8.8 Hz, IH), 3.86 (s, 3H), 3.22 – 3.08 (m, IH), 2.91 (s, 6H), 2.00 – 1.74 (m, 7H), 1.60 – 1.38 (m, 3H). 13CNMR (75 MHz, CDC13) δ 165.7, 158.8, 147.2, 139.1, 134.3, 132.0, 123.4, 122.0, 119.2, 118.2, 114.8, 112.3, 110.4, 109.8, 79.6, 45.9, 37.2(2), 34.7, 32.0(2), 25.9(2), 24.9. LCMS: m/e 482 (M- H)“, ret time 2.56 min, column A, 4 minute gradient.
Intermediate 5

6H-Isoindolo[2,l-a]indole-3-carboxamide, 11-cyclohexyl-N-
[(dimethylamino)sulfonyl]-6-ethoxy-8-methoxy-. To a 5 L four necked round bottom flask equipped with a temperature controller, a condenser, a N2 inlet and a mechanical stirrer, was charged toluene (900 mL), EtOH (900 mL), 2-bromo-3- cyclohexyl-N^NjN-dimethylsulfamoyiyiH-indole-ό-carboxamide (90 g, 0.21 mol), 2-formyl-4-methoxyphenylboronic acid (49.2 g, 0.273 mol) and LiCl (22.1 g, 0.525 mol). The resulting solution was bubbled with Ν2 for 15 mins. A solution of Na2CO3 (66.8 g, 0.63 mol) in Η2O (675 mL) was added and the reaction mixture was bubbled with N2 for another (10 mins). Pd(PPh3)4 (7.0 g, 6.3 mmol) was added and the reaction mixture was heated to 70 °C for 20 h. After cooling to 35 °C, a solution of 1 N HCl (1.5 L) was added slowly. The resulting mixture was transferred to a 6 L separatory funnel and extracted with EtOAc (2 X 1.5 L). The combined organic extracts were washed with brine (2 L), dried over MgSO4, filtered and concentrated in vacuo to give a yellow solid, which was triturated with 20% EtOAc in hexane (450 mL, 50 °C to 0 °C) to give 3-cyclohexyl-N-(N,N-dimethylsulfamoyl)-2-(2-formyl-4- methoxyphenyl)-lH-indole-6-carboxamide(65.9 g) as a yellow solid. HPLC purity, 98%.
The mother liquid from the trituration was concentrated in vacuo. The residue was refluxed with EtOH (50 mL) for 3 h. The solution was then cooled to 0 °C. The precipitates were filtered and washed with cooled TBME (5 °C) (20 mL). The filter cake was house vacuum air dried to give a further quantity of the title compound as a white solid (16.0 g). HPLC purity, 99%. 1H NMR (CDC13, 300 MHz) δ 8.75 (s, IH), 7.96 (s, IH), 7.73 (d, J= 8.4 Hz, IH), 7.67 (d, J= 8.4 Hz, IH), 7.45 (dd, J= 8.4 and 1.4 Hz, IH), 7.09 (d, J= 2.2 Hz, IH), 6.98 (dd, J= 8.4 and 2.2 Hz, IH), 6.50 (s, IH), 3.86 (s, 3H), 3.05 (s, 6H), 2.92-3.13 (m, 3H), 1.85-1.93 (m, 7 H), 1.40-1.42 (m, 3H), 1.05 (t, J= 7.1 Hz, 3H). m/z 512 (M + H)+.
Intermediate 6

lH-indole-6-carboxamide, 3-cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2- formyl-4-methoxyphenyl)-. 1 l-cyclohexyl-N-(N,N-dimethylsulfamoyl)-6-ethoxy-8- methoxy-6H-isoindolo[2,l-a]indole-3-carboxamide was dissolved in THF (75 mL). To the solution was added a solution of 2 N HCl (300 mL). The mixture was vigorously stirred under N2 at rt for 16 h. The resulting suspension was filtered and washed with cooled TBME (2 X 30 mL). the filer cake was vacuum air dried overnight to give the title compound as a yellow solid. HPLC purity, 99% 1H NMR (DMSO-d6, 300 MHz) δ 11.65 (s, IH), 8.16 (s, IH), 7.76 (d, J= 5.9 Hz, IH), 7.73 (d, J= 5.9 Hz, IH), 7.58 (dd, J= 8.5 and 1.5 Hz, IH), 7.17-7.20 (m, 2H), 7.08 (dd, J = 8.5 and 1.4 Hz, IH), 6.55 (d, J= 8.6 Hz, IH), 3.86 (s, 3H), 3.14-3.18 (m, IH), 2.91 (s, 6H), 1.75-1.99 (m, 7H), 1.48-1.60 (m, 3H); m/z 484 (M + H)+.
Intermediate 7

7H-Indolo[2, 1-a] ‘ [2] benzazepine-6-carboxylic acid, 13-cyclohexyl-10- [[[(dimethylamino)sulfonyl] amino] carbonyl]-3-methoxy-, methyl ester. A mixture of the 3-cyclohexyl-N-(N,N-dimethylsulfamoyl)-2-(2-formyl-4-methoxyphenyl)-lH- indole-6-carboxamide (4.8g, 0.01 mol), methyl 2-(dimethoxyphosphoryl)acrylate (9.7 g, 0.02 mol) and cesium carbonate (7.1g, 0.02 mol) in DMF (28mL) was stirred for 20 hr at an oil bath temperature of 55 ° C. The mixture was poured into ice-water and acidified with dilute HCl to precipitate the crude product. The solid was collected, dried and flash chromatographed on Siθ2 (11Og) using an ethyl acetate and methylene chloride (1: 10) solution containing 2% acetic acid. Homogeneous fractions were combined and evaporated to afford the title compound as a pale yellow solid (3.9g, 71 % yield). MS: 552 (M=H+).
An alternate procedure for the preparation of 7H-indolo[2,l- a] [2]benzazepine-6-carboxylic acid, 13-cyclohexyl-10- [[[(dimethylamino)sulfonyl]amino]carbonyl]-3-methoxy-, methyl ester is provided below. A solution of l l-cyclohexyl-N-[(dimethylamino)sulfonyl]-6-hydroxy-8- methoxy-6H-isoindolo[2,l-a]indole-3-carboxamide (cyclic hemiaminal) (63.0 g, 130 mmol), methyl 2-(dimethoxyphosphoryl)acrylate (60 g, 261 mmol), cesium carbonate (106 g, 326 mmol) in DMF (400 mL) was heated at 60 °C (bath temp) for 4.5h. Additional methyl 2-(dimethoxyphosphoryl)acrylate (15 g, 65 mmol) and cesium carbonate (21.2 g, 65 mmol) were added and the reaction was heated at 60 °C overnight then and cooled to rt. The stirring reaction mixture was diluted with H2O (1 L), slowly neutralized with IN aqueous HCl (800 mL), stirred 3h, and then the precipitates were collected by filtration. The solids were triturated with Et20 (800 mL) and dried to yield methyl 7H-indolo[2,l-a][2]benzazepine-6-carboxylic acid, 13- cyclohexyl-10-[[[(dimethylamino)sulfonyl]amino]carbonyl]-3-methoxy-, methyl ester (70.2 g, 127 mmol, 98%) as a yellow solid which was used without further purification. IHNMR (300 MHz, CDC13) δ 8.67 (s, IH), 8.09 (s, IH), 7.86 (d, J = 8.4 Hz, IH), 7.80 (s, IH), 7.50 (d, J = 8.4 Hz, IH), 7.42 (d, J = 8.8 Hz, IH), 7.08 (dd, J = 2.6, 8.8 Hz, IH), 6.98 (d, J = 2.6 Hz, IH), 5.75 – 5.51 (m, IH), 4.29 – 4.01 (m, IH), 3.89 (s, 3H), 3.82 (s, 3H), 3.05 (s, 6H), 2.87 – 2.73 (m, IH), 2.11 – 1.12 (m, 10H). LCMS: m/e 550 (M-H)-, ret time 3.21 min, column A, 4 minute gradient.
Example 1

Cycloprop[d]indolo[2,l-a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (+/-)-. TBTU (43.7 mg, 0.136mmol) and DIPEA (0.095 mL, 0.544 mmol) were added to a solution of (+/-) cycloprop[d]indolo[2,l-a][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (50 mg, 0.0906 mmol) in DMSO (2.0 mL). The reaction mixture was stirred at rt for 15 min. 3-Methyl-3,8-diaza-bicyclo[3.2. l]octane dihydrochloride {J & W PharmLab, LLC Morrisville, PA 19067-3620}. (27.1 mg, 0. 136 mmol) was then added and the reaction mixture was stirred at rt for 3 hr. It was then concentrated and the residue was purified by preparative reverse phase HPLC to give the final product as a yellow solid, (32 mg, 46% yield). MS m/z 660(MH+), Retention time: 2.445 min IH NMR (300 MHz, MeOD) δ ppm 0.20 (m, 0.23 H) 1.11 – 2.25 (m, 15.77 H) 2.58 (m, 0.23 H) 2.69 (m, 0.77 H) 2.75 – 3.11 (m, 10 H) 3.28 – 3.75 (m, 5 H) 3.91 (s, 2.31 H) 3.92 (s, 0.69 H) 4.15 – 4.37 (m, 1 H) 4.68 (m ,br, 1 H) 4.94 – 5.00 (m, 0.23 H) 5.16 (d, J=15.00 Hz, 0.77 H) 7.00 – 7.09 (m, 1 H) 7.18 (d, J=2.56 Hz, 0.23 H) 7.21 (d, J=2.56 Hz, 0.77 H) 7.33 (d, J=8.41 Hz, 0.77 H) 7.35 (d, J=8.42 Hz, 0.23 H) 7.57 (dd, J=8.42, 1.46 Hz, 0.77 H) 7.62 (dd, J=8.78, 1.46 Hz, 0.23 H) 7.91 (d, J=8.42 Hz, 0.77 H) 7.93 (d, J=8.42 Hz, 0.23 H) 8.00 (s, 0.77 H) 8.07 (s, 0.23 H).
Example 4

Cycloprop[d]indolo[2,l-a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonylj ‘- 1 , Ia, 2, 12b-tetrahydro-ll-methoxy-la-[(8-methyl-3, 8- diazabicyclo[3.2.1]oct-3-yl)carbonyl]-, (+/-)-. To a solution of (+/-) cycloprop[d]indolo[2,l-a][2]benzazepine-5-carboxamide, 8-cyclohexyl-la-(3,8- diazabicyclo[3.2.1]oct-3-ylcarbonyl)-N-[(dimethylamino)sulfonyl]-l,la,2,12b- tetrahydro-11-methoxy- (54 mg, 0.071 mmol) in methanol (3 mL), paraformaldehyde (6.4 mg, 0.213 mmol), ZnCl2 (29 mg, 0.213 mmol) and
Na(CN)BH3 (13.4 mg, 0.213 mmol) were added. The resultant mixture was heated at 60°C for 2hr, and then cooled to rt. The solid present was removed by filtration, and the filtrate was concentrated under vacuum and the residue purified by preparative reverse phase HPLC to give the title compound as a light yellow colored solid, (37 mg, 67% yield). MS ml 660(MH+), Retention time: 2.495 min. IH NMR (500 MHz, MeOD) δ ppm 0.21 (m, 0.3 H) 1.13 (m, 0.3 H) 1.18 – 2.22 (m, 15.4 H) 2.58 (m, 0.3 H) 2.68 (m, 0.7 H) 2.76 – 3.11 (m, 11 H) 3.32 – 3.37 (m, 1 H) 3.63 (d, J=15.56 Hz, 0.7 H) 3.82 – 4.32 (m, 7.3 H) 4.88 – 4.92 (m, 0.3 H) 5.08 (d, J=15.56 Hz, 0.7 H) 7.00 – 7.08 (m, 1 H) 7.18 (d, J=2.14 Hz, 0.3 H) 7.21 (d, J=2.14 Hz, 0.7 H) 7.32 (d, J=8.55 Hz, 0.7 H) 7.35 (d, J=8.55 Hz, 0.3H) 7.57 (d, J=7.93 Hz, 0.7 H) 7.62 (dd, J=8.39, 1.37 Hz, 0.3 H) 7.91 (d, J=8.55 Hz, 0.7 H) 7.93 – 7.99 (m, 1 H) 8.09 (s, 0.3 H).
Example 6

Cycloprop [d] indolo [2, 1 -a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (laR,12bS)-. To a solution of (-) cycloprop[d]indolo[2,l-a][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (204 mg, 0.37 mmol) in DMSO (8.0 mL), TBTU (178 mg, 0.555 mmol) and DIPEA (0.39 mL, 2.22 mmol) were added. The reaction mixture was stirred at rt for 15 min. Then 3- methyl-3,8-diaza-bicyclo[3.2.1]octane dihydrochloride (111 mg, 0. 555 mmol) was added and the reaction mixture was stirred at rt for 2 hr. It was then concentrated and the residue was purified by preparative reverse phase HPLC to give a yellow solid as final TFA salt. (265 mg, 92% yield). Average Specific Rotation: -53.56° Solvent, MeOH.; Wavelength 589 nm; 50 cm cell. MS m/z 660(MH+), Retention time: 3.035 min. 1H NMR (300 MHz, MeOD) δ ppm 0.20 (m, 0.23 H) 1.11 – 2.25 (m, 15.77 H) 2.58 (m, 0.23 H) 2.69 (m, 0.77 H) 2.75 – 3.11 (m, 10 H) 3.28 – 3.75 (m, 5 H) 3.91 (s, 2.31 H) 3.92 (s, 0.69 H) 4.15 – 4.37 (m, 1 H) 4.68 (m ,br, 1 H) 4.94 – 5.00 (m, 0.23 H) 5.16 (d, J=15.00 Hz, 0.77 H) 7.00 – 7.09 (m, 1 H) 7.18 (d, J=2.56 Hz, 0.23 H) 7.21 (d, J=2.56 Hz, 0.77 H) 7.33 (d, J=8.41 Hz, 0.77 H) 7.35 (d, J=8.42 Hz, 0.23 H) 7.57 (dd, J=8.42, 1.46 Hz, 0.77 H) 7.62 (dd, J=8.78, 1.46 Hz, 0.23 H) 7.91 (d, J=8.42 Hz, 0.77 H) 7.93 (d, J=8.42 Hz, 0.23 H) 8.00 (s, 0.77 H) 8.07 (s, 0.23 H). An alternate procedure for the synthesis of cycloprop[d]indolo[2,l- a][2]benzazepine-5-carboxamide, 8-cyclohexyl-N-[(dimethylamino)sulfonyl]- l,la,2,12b-tetrahydro-l l-methoxy-la-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8- yl)carbonyl]-, (laR,12bS)-rel-(-)-is provided below. To a mixture of (-) cycloprop[<i]indolo[2,l-α][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (25.2 g, 45.68 mmol) and 3-methyl-3,8-diazabicyclo-[3.2.1]octane dihydrochloride (10.0 g, 50.22 mmol) in anhydrous MeCN (300 mL) was added DIPEA (23.62 g, 182.72 mmol) under N2. After 15 min, TBTU (16.12 g, 50.22 mmol) was added. The reaction solution was stirred for 30 min under N2. The ΗPLC indicated the disappearance of starting material. The solvent in the solution was evaporated to give a foam. This was dissolved in EtOAc (2.5 L), washed with H2O (1.5 L), H2O/brine (8:2) (1.5 L), brine (1.5 L), dried over Na2SO4 and evaporated to give 28.8 g of crude product. This solid was pooled with 45.4 g of material obtained from five separated reactions to afford a total of 74.2 g of crude product. This was passed through a pad of silica gel (E. Merck 230-400 mesh, 1 kg), eluting with MeOH/CH2Cl2 (2.5:97.5). After evaporation, it gave a foam, which was treated with EtOAc and hexane to turn into a solid. After drying at 50 °C under vacuum for 7 h, the GC analysis indicated it has 1.4% each of EtOAc and hexane. After further drying at 61-64 °C, the GC analysis indicated it still has 1.0% of hexane and 1.4% of EtOAc. The product was dissolved in Et2O and slowly evaporated in vacuum three times, dried at 60 °C under vacuum for 3 h to give 68.3 g. This was washed with H2O (900 mL) and redried at 68 °C under vacuum for 7 h to give 67.1 g (77% yield) of the compound of example 6. The GC analysis indicated it has 0.97% Of Et2O. HPLC conditions column: Cadenza CD-C18 3 x 250 mm; UV: 257 and 220 nm; 25 °C; flow rate: 0.5 mL/min; gradient time: 38 min, 0 – 80% B (0 – 35 min) and 80% B (35 – 38 min); solvent A: 25 nM CH3COONH4 at pH 4.7 in water, solvent B: MeCN. HPLC purity 99.7% (Rt 26.54 min); Chiral HPLC conditions column: Regis (S5S) Whelk-Ol 250 x 4.6 mm; UV 258nm; 35 °C; flow rate 2.0 mL/min; mobile phase C02/Me0H; gradient time 20 min, 30% MeOH (0 – 1 min), 30 – 48% MeOH (1 – 19 min), 48% MeOH (19 – 20 min). Chiral HPLC purity > 99.8% (Rt 16.60 min); LC/MS (ES+) 660.36 (M+H, 100); HRMS: calcd. 660.3220, found 660.3197; [α]D 25 C – 79.66 ° (c 1.06, MeOH); Anal. Calcd for C36H45N5O5S-O-O H2O»0.09 Et2O: C, 64.53; H, 7.00; N, 10.35; S, 4.74; H2O, 1.51; Et2O, 0.97. Found: C, 64.50; H, 7.12; N, 10.41; S, 5.14; H2O, 1.52; Et2O, 0.97. The absolute stereochemistry of cycloprop[d]indolo[2,l- a][2]benzazepine-5-carboxamide, 8-cyclohexyl-N-[(dimethylamino)sulfonyl]- l,la,2,12b-tetrahydro-l l-methoxy-la-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8- yl)carbonyl]-, (laR,12bS)-rel-(-)- is as drawn above, and was determined from an x- ray crystal structure obtained on the (R)-camphorsulfonic acid salt.
Additionally, the following salts were prepared: hydrochloride, phosphate, acetate, sulfate, camsylate, sodium, calcium, and magnesium. The hydrochloride salt had the following characteristics. DSC: small, broad endotherm from 25°C to 75°C, and potential melt/degradation endotherm with peak at temperatures ranging between 253 °C and 258 °C; TGA: Early weight loss from 25°C to 75°C ranging between 0.003% and 1.5%, and degradation weight loss starting at approximately 200°C.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
| WO2006020082A1 * | Jul 15, 2005 | Feb 23, 2006 | Squibb Bristol Myers Co | Inhibitors of hcv replication |
| WO2006046030A2 * | Oct 25, 2005 | May 4, 2006 | Angeletti P Ist Richerche Bio | Tetracyclic indole derivatives as antiviral agents |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008111978A1 | Mar 13, 2007 | Sep 18, 2008 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2008112473A1 * | Mar 5, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112841A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112848A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112851A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv inhibitors |
| WO2008112863A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2009067108A1 * | Nov 20, 2007 | May 28, 2009 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2009067392A1 * | Nov 17, 2008 | May 28, 2009 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine derivatives for the treatment of hepatitis c |
| WO2009067481A1 * | Nov 19, 2008 | May 28, 2009 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2010080874A1 | Jan 7, 2010 | Jul 15, 2010 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| WO2013059265A1 * | Oct 17, 2012 | Apr 25, 2013 | Bristol-Myers Squibb Company | A compound for the treatment of hepatitis c |
| WO2014014885A1 * | Jul 16, 2013 | Jan 23, 2014 | Bristol-Myers Squibb Company | Novel methods and intermediates for the preparation of (4bs,5ar)-12-cyclohexyl-n-(n,n-dimethylsulfamoyl)-3-methoxy-5a-((1 r,5s) -3-methyl-3,8-diazabicyclo[3.2.1]octane-8-carbonyl)-4b,5,5a,6-tetrahydrobenzo [3,4]cyclopropa[5,6]azepino[1,2-a]indole-9-carboxamide |
| CN101679442B | Mar 13, 2008 | Feb 20, 2013 | 百时美施贵宝公司 | Compounds for the treatment of hepatitis c |
| EP2518073A1 * | Nov 19, 2008 | Oct 31, 2012 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
The First Kilogram Synthesis of Beclabuvir, an HCV NS5B Polymerase Inhibitor
 , Kenneth J. Fraunhoffer, Zhinong Gao, Chao Hang, Yi Hsiao, Wenhao Hu, Kishta Katipally, Adam Littke, Aghogho Pedro, Yuping Qiu, Maria Sandoval, Richard Schild, Michelle Soltani, Anthony Tedesco, Dale Vanyo, Purushotham Vemishetti, and Robert E. Waltermire
, Kenneth J. Fraunhoffer, Zhinong Gao, Chao Hang, Yi Hsiao, Wenhao Hu, Kishta Katipally, Adam Littke, Aghogho Pedro, Yuping Qiu, Maria Sandoval, Richard Schild, Michelle Soltani, Anthony Tedesco, Dale Vanyo, Purushotham Vemishetti, and Robert E. Waltermire
The process development and kilogram-scale synthesis of beclabuvir (BMS-791325, 1) is described. The convergent synthesis features the use of asymmetric catalysis to generate a chiral cyclopropane fragment and coupling with an indole fragment via an alkylation. Subsequent palladium-catalyzed intramolecular direct arylation efficiently builds the central seven-membered ring. The target was prepared in 12 linear steps with five isolations in an overall yield of 8%.
Preparation of (4bS,5aR)-12-Cyclohexyl-N-(N,N-dimethylsulfamoyl)-3-methoxy-5a-((1R,5S)-3-methyl-3,8-diazabicyclo[3.2.1]octane-8-carbonyl)-4b,5,5a,6-tetrahydrobenzo[3,4]cyclopropa[5,6]azepino[1,2-a]indole-9-carboxamide Hydrochloride (1·HCl)
BMS-791325·HCl (1·HCl) was isolated in 89.5% yield.
1H NMR (600 MHz, 10:1 v/v CD3CN/D2O): major rotamer: 7.91 (br s, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.55 (br d, J = 8.5 Hz, 1H), 7.29 (d, J = 8.5 Hz, 1H), 7.20 (d, J = 2.5 Hz, 1H), 7.00 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 5.03 (br d, J = 12.7 Hz, 1H), 4.58 (br d, J = 4.9 Hz, 2H), 3.87 (s, 3H), 3.56 (d, J = 15.5 Hz, 1H), 3.40 (br s, 3H), 3.32–3.28 (m, 4H), 2.96 (s, 6H), 2.92 (tt, J= 12.2, 3.6 Hz, 1H), 2.59 (br t, J = 7.0 Hz, 1H), 2.05–1.90 (m, 2H), 1.79–1.71 (m, 4H), 1.55 (br d, J= 12.2 Hz, 2H), 1.46–1.36 (m, 4H), 1.26 (t, J = 5.3 Hz, 2H), 1.23–1.15 (m, 2H);
minor rotamer: 8.05 (br s, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.58 (dd, J = 8.5, 1.4 Hz, 1H), 7.34 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 2.6 Hz, 1H), 6.98 (d, 1H, overlap with major rotamer), 4.91 (d, J = 15.0 Hz, 1H), 4.58 (br d, J = 4.9 Hz, 2H), 4.11 (d, J = 15.0 Hz, 1H), 3.89 (s, 3H), 3.46 (br d, J = 12.5 Hz, 2H), 3.17 (br d, J = 12.5 Hz, 2H), 2.97 (s, 6H), 2.85 (br s, 3H), 2.76 (tt, J = 12.1, 3.5 Hz, 1H), 2.49 (br s, 1H), 2.05–1.90 (m, 2H), 1.79–1.71 (m, 4H), 1.46–1.36 (m, 6H), 1.23–1.15 (m, 2H), 1.10 (m, 1H), 0.03 (t, J = 6.1 Hz, 1H).
13C NMR (125 MHz, 10:1 v/v CD3CN/D2O): major rotamer: 170.1, 167.7, 161.0, 140.4, 139.3, 135.9, 133.6, 131.1, 124.9, 123.0, 121.7, 120.8, 119.0, 118.6, 114.3, 110.7, 59.2, 56.2, 53.1, 48.3, 44.5, 38.9, 37.6, 34.8, 33.77, 33.72, 27.92, 27.77, 26.82, 26.5, 23.6, 18.5;
minor rotamer: 168.3, 168.0, 161.3, 138.4, 137.5, 135.8, 134.2, 130.0, 125.4, 121.9, 120.0, 119.64, 119.58, 117.9, 113.3, 111.3, 59.6, 56.3, 53.1, 44.6, 42.2, 38.9, 38.3, 37.4, 33.8, 33.6, 28.3, 27.74, 26.79, 26.5, 24.84, 11.9.
HRMS (ESI) calcd for C36H45N5O5S (free base) [M + H]+660.3214, found m/z 660.3220.
////////BMS-791325, Beclabuvir, Phase 2, Hepatitis C, HCV,
BI-836845 a fully human mAb targeting IGF-1 created using HuCAL technology from Morphosys, for the potential iv infusion treatment of cancer, including solid tumors and breast cancer.

BI-836845
Human monoclonal IgG1 lambda antibody against IGF-1 (insulin growth factor-1) and IGF-2
| IGF pathway modulator (iv, cancer), Boehringer Ingelheim; |
Phase 2 Clinical
Anticancer protein kinase inhibitor; Anticancer monoclonal antibody
WO-2008155387
Boehringer Ingelheim International Gmbh

Boehringer Ingelheim is developing BI-836845, a fully human mAb targeting IGF-1 created using HuCAL technology from Morphosys, for the potential iv infusion treatment of cancer, including solid tumors and breast cancer.
In April 2011, a phase I trial was initiated in the UK . In October 2011, another phase I trial was initiated in Taiwan. In February 2014, recruitment was ongoing. At that time, the trial was expected to be completed in March 2015 In June 2014, the drug was listed as being in phase I development for solid tumors in Japan and for breast cancer
In May 2014, an open-label, randomized, parallel-assigned, phase II trial (NCT02123823; 1280.4; 2013-001110-15) to evaluate the safety and efficacy of BI-836845 and everolimus in combination with exemestane in women with breast cancer (expected n = 198) was planned to be initiated in Belgium, France and the Netherlands. At that time, the trial was expected to complete in December 2017
In June 2014, an open-label, single-group assigned, phase I trial (NCT02145741; 1280.15) to evaluate BI-836845 in Japanese patients (expected n = 18) with advanced solid tumors was planned to be initiated in Japan. At that time, the trial was expected to complete in June 2015
In March 2011, a non-randomized, open-label, phase I study (NCT01317420; 1280.2; 2010-021714-29) was planned to begin later that month in patients with solid tumors (expected n = 70) in the UK, to assess the safety, efficacy, pharmacokinetics, pharmacodynamics and pharmacogenomics of BI-836845. The study began in April 2011; at that time, completion was expected in March 2013 .
In June 2012, preclinical data were presented at the 48th ASCO meeting in Chicago, IL. In the study, the combination of BI-836845 plus rapamycin was more effective than single agent therapy at inhibiting Ewing’s sarcoma cell proliferation in vitro and in a nude mouse xenograft model .

In November 2011, preclinical data were presented at the 23rd AACR-NCI-EORTC International Conference in San Francisco, CA. BI-836845 potently inhibited proliferation of the multiple myeloma cell line LP-1 with an EC50 of 0.4 nM.
BI-836845 is a human monoclonal IgG1 lambda antibody against IGF-1 (insulin growth factor-1) and IGF-2 (insulin growth factor-2). Phase II clinical trials are ongoing at Boehringer Ingelheim for the treatment of patients with breast cancer, and phase I clinical trials are ongoing with patients with advanced solid tumors.
Insulin-like growth factor-1 (IGF-1; a 70 amino-acid polypeptide) and insulin-like growth factor-2 (IGF-2; a 67 amino-acid polypeptide) are 7.5-kD soluble factors present in serum that can potently stimulate the growth of many mammalian cells (reviewed by Pollack et al., 2004). Although IGFs can be detectable in a number of tissues the main source of circulating IGFs is the liver which secretes the IGFs and IGF binding proteins (IGFBPs) in response to a complex signaling pathway that is initiated in the pituitary gland and transduced via growth hormone. On secretion into the bloodstream the IGFs form complexes with the IGFBPs which not only protects them from proteolytic degradation in the serum en route to their target tissues but also prevents their association with the IGF receptors. In addition to this endocrine source of IGFs they are also known to be secreted in an autocrine or paracrine manner in target tissues themselves. This is known to occur during normal fetal development where the IGFs play a key role in the growth of tissues, bone and organs. It is also seen in many cancer tissues where there is thought to be paracrine signaling between tumour cells and stromal cells or autocrine IGF production by the tumour cells themselves (reviewed by LeRoith D, 2003).
30 May 2014
MEDIA ALERT
ASCO 2014: Boehringer Ingelheim to present latest oncology research, including overall survival results
• Highly anticipated new overall survival data for Giotrif® (afatinib*) to be presented on June 2nd (3:00 – 6:00 PM, E Hall D2 [Abstract #8004 scheduled for 4:00 – 4:12 PM])
• 7 total abstracts accepted for Giotrif® (afatinib*), nintedanib** and BI 836845**: 1 for oral presentation and 6 posters
| BI 836845 (IGF ligand antibody)** | ||
| A Phase I dose escalation study of weekly BI 836845, a fully human, affinity-optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid cancers | Chia-Chi Lin, Kwang-Yu Chang, Dennis Chin-Lun Huang, Vicky Marriott, Ludy van Beijsterveldt, Li-Tzong Chen, Ann-Lii Cheng | Sunday, June 1 8:00 – 11:45 AM S Hall A2 (Abstract #2617 Poster #80) |
| Phase I dose escalation study of 3-weekly BI 836845, a fully human, affinity optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid tumours | Rihawi K, Ong M, Michalarea V, Bent L, Buschke S4, Bogenrieder T, Anthoney A, de Bono J, Twelves CJ | Sunday, June 1 8:00 – 11:45 AM S Hall A2 (Abstract #2622 Poster #85) |
The activity of the IGFs is thought to be regulated by a complex and relatively poorly understood interaction involving seven different IGFBPs and other serum proteins. Activation of the IGFs involves their release from this ternary complex after proteolytic release of the serum binding protein and IGFBPs, this is thought to occur in close proximity to cell surfaces where the IGFs are then free to bind to their receptors and transduce intracellular signals that ultimately leads to cellular proliferation and the inhibition of apoptosis. IGF-1 and IGF-2 are able to bind to the IGF-1 receptor (IGF-1R) expressed on many normal tissues, which functionally is a 460 kD heterotetramer consisting of a dimerised alpha- and beta-subunit, with similar affinities (Rubin et al., 1995). IGF-2 can also bind to the IGF-2 receptor (also know as the mannose-6-phosphate receptor) which does not have any known signaling function, rather it is thought to act as a sink for IGF-2 and prevent it from binding and signaling through the IGF-1R. In this respect the IGF-2R has been demonstrated to be a tumour suppressor protein. The IGF-1R is structurally similar to the insulin receptor which exists in two forms, IR-A and IR-B, which differ by an alternatively spliced 12 amino acid exon deletion in the extracellular domain of IR-A. IR-B is the predominant IR isoform expressed in most normal adult tissues where it acts to mediate the effects of insulin on metabolism. IR-A on the other hand is known to be highly expressed in developing fetal tissues but not in adult normal tissues. Recent studies have also shown that IR-A, but not IR-B, is highly expressed in some cancers. The exon deletion in IR-A has no impact on insulin binding but does cause a small conformational change that allows IGF-2 to bind with much higher affinity than for IR-B (Frasca et al., 1999; Pandini et al., 2002). Thus, because of it’s expression in cancer tissues and increase propensity for IGF-2 binding, IR-A may be as important as IGF1-R in mediating the mitogenic effects of IGF-2 in cancer.
Binding of the IGFs to IGF-1R triggers a complex intracellular signaling cascade which results in activation of proteins that stimulate growth and inhibit apoptosis (reviewed by Pollack et al., 2004). In terms of growth, upregulated translation is induced by the activation of p70 S6 kinase, which in turn phosphorylates the S6 ribosomal protein (Dufner and Thomas, 1999). Thus, IGF-stimulated cell growth can be measured by the rapid increase in phosphorylated S6 ribosomal protein.
Unlike the EGFR and Her2neu receptors there is no known amplification of the IGF1-R or IR-A receptors in cancers indicating that receptor activation is controlled by the presence of active ligand. There is a very large body of scientific, epidemiological and clinical literature implicating a role for the IGFs in the development, progression and metastasis of many different cancer types (reviewed by Jerome et al., 2003; and Pollack et al., 2004).
For example, in colorectal cancer the expression of IGF-2 mRNA and protein is elevated in clinical colorectal tumour specimens compared with adjacent normal tissue (Freier et al., 1999; Li et al., 2004). There is also a positive correlation of elevated IGF serum levels with proliferating cell index in patients with colorectal neoplasia (Zhao et al., 2005). In addition, elevated circulating levels of IGF-2 correlate with an increased risk of developing colorectal cancers and adenomas (Renehan et al., 2000a) and b); Hassan et al., 2000). Loss of parental imprinting (LOI) of the IGF-2 gene, an epigenetic alteration that results in elevated IGF-2 expression, is a heritable molecular trait that has recently been identified in patients with colorectal and other tumour types. Loss of IGF-2 imprinting has been shown to be associated with a five-fold risk of colorectal neoplasia (Cui et al., 2003; Cruz-Correa et al., 2004) and adenomas (Woodson et al., 2004). Antibodies targeting the alpha-subunit of the IGF-1R which block IGF binding and internalize the receptor have been shown to delay the growth of the xenografted colon cancer-derived cell lines such as COLO 205 (Burtrum et al., 2003).
Elevated levels of IGFs are associated with a poor prognosis in human pulmonary adenocarcinomas (Takanami et al., 1996) and IGFs are expressed and secreted by many SCLC— and NSCLC-derived cell lines (Quinn et al., 1996). Transgenic over-expression of IGF-2 induces spontaneous lung tumours in a murine model (Moorhead et al., 2003). In terms of hepatocellular carcinoma (HCC), human clinical specimens and animal models of HCC express higher levels of IGF mRNA and protein than corresponding normal tissues and this has been correlated with increased tumour growth (Wang et al., 2003; Ng et al., 1998). IGF-2 has also been shown to be a serological marker of HCC with elevated levels in the serum of HCC patients compared with controls (Tsai et al., 2005). An orthotopic xenograft tumour model of HCC was established using Hep 3B cells, and used to demonstrate that inhibition of IGF-2 expression using a methylated oligonucleotide enhances survival (Yao et al., 2003a) and b).
Many childhood solid tumours such as Ewing sarcoma and rhabdomyosarcoma appear to be particularly dependent on the IGF signaling pathway for their growth (Scotlandi et al., 1996). LOI of the IGF-2 gene has been implicated as a primary genetic event in the development for embryonal rhabdomyosarcoma (Fukuzawa et al., 1999). Autocrine IGF signaling is also thought to strongly influence the growth of Ewing sarcoma in cases where the type-1 EWS-FLI1 chimeric transcription factor is expressed through a chromosomal translocation resulting in elevated expression of target genes including the IGF ligands and IGF-1R, and reduced expression of IGFBP-3. Antibodies and small molecule compounds targeting the IGF-1R have been shown to reduce the growth of xenografted pediatric solid tumour derived cell lines (Kolb et al., 2008; Manara et al., 2007).
Using IGF ligand-specific antibodies it has been demonstrated that the growth of human prostate cancer cells in adult human bone implanted into SCID mice can be inhibited (Goya et al., 2004). In addition, it was demonstrated that the same IGF ligand antibodies could block the paracrine supply of IGF and suppress the liver metastasis of human colorectal cancer cells in a murine xenograft system (Miyamoto et al., 2005).
There is also considerable evidence suggesting that the IGF signaling system reduces the sensitivity of cancers to chemotherapeutic agents and radiation. One of the earliest findings in this respect was the demonstration that IGF-1R knock-out mouse embryos are refractory to transformation by viruses, oncogenes and over-expressed growth factor receptors (Sell et al., 1993; Sell et al., 1994) and that over-expression of IGF-1R protects cells from UV irradiation and gamma radiation-induced apoptosis (Kulik et al., 1997). Furthermore, using liver tumour cell lines that secrete large amounts of IGF-2, it was found that neutralization of IGF-2 significantly increased response to chemotherapeutic agents such as cisplatin and etoposide in vitro, especially at lower, cytostatic doses, suggesting that IGF-2 can reduce the susceptibility to chemotherapeutic agents (Lund et al., 2004). Consistent with these findings it has been demonstrated that antibodies targeting the IGF-1R increase the susceptibility of tumour xenografts to growth inhibition by chemotherapeutic drugs and radiation (Goetsch et al., 2005).
A number of antibodies that show cross-reactive binding to human IGF-1 and human IGF-2 have been reported. Antibody sm1. was raised against human IGF-1 and shows 40% cross-reactivity to human IGF-2 and was shown to inhibit the proliferation of a mouse fibroblast cell line BALB/c3T3 which was stimulated with 20 ng/ml human IGF-1 (Russell et al., 1984). In a study designed to functionally epitope map IGF-1 by raising monoclonal antibodies to whole IGF-1 protein and portions of the protein a number of antibodies where identified that cross reacted with IGF-2 (Manes et al., 1997). The percent cross-reactivity with IGF-2 ranged from 0 to 800% and several antibodies were identified which were equally IGF-1 and IGF-2 reactive. KM1486 is a rat monoclonal antibody that cross-reacts with human IGF-1 and IGF-2 and it was demonstrated that KM1486 can inhibit growth of human prostate cancer cells in human adult bone implanted into nonobese diabetic/severe combined immunodeficient mice (Goya et al., 2004). In addition, it was demonstrated that KM1486 suppresses the liver metastasis of human colorectal cancers (Miyamoto et al., 2005). KM1486 has also been described in WO 03/093317, JP 2003-310275, WO 2005/018671, WO 2005/028515, and WO 2005/027970.
For the treatment of human disease an antibody with a fully human sequence is highly desirable in order to minimize the risk of generating a human anti-antibody reaction and neutralizing antibodies that will rapidly eliminate the administered antibody from the body and thereby reduce the therapeutic effect. As such, and given the roles of IGF-1 and IGF-2 dependent signaling in the development and progression of cancers it would be desirable to obtain high affinity fully human antibodies that co-neutralise the mitogenic effects of both ligands.
In addition, to maximize the therapeutic potential of such an antibody, it is important to have a suitably long terminal half life (T1/2). Prior to terminal half life determination in human subjects, the most accurate estimation of an antibody’s human terminal half life can be obtained from administration to non-human primates such as cynomolgus monkeys. For example, bevacizumab, a registered humanized monoclonal antibody against vascular endothelial growth factor (VEGF) used for the treatment of several human cancers, has a terminal half-life in cynomolgus monkeys of 8.57±0.38 days (Lin et al., 1999), which translates to a terminal half life in humans of approximately 20 days allowing for a single administration once every two weeks (Lu et al., 2008).

It was a further object of the invention to obtain an antibody that does not affect binding of insulin to its receptor.
The clinical development of therapeutic agents is supported by pharmacodynamic biomarkers of drug activity. Clinical studies with antibodies targeting the IGF-1R have demonstrated that an increase in total serum IGF-1 levels may be a useful pharmacodynamic marker for these agents (Pollack et al., 2007). The reason for the increase in total serum IGF-1 levels is likely due to a feedback mechanism involving pituitary growth hormone (GH) secretion which releases both IGF-1 and IGFBPs from the liver. Indeed, in humans it has been demonstrated that free or bioactive IGF-1, which represents only around 1% of total IGF-1 levels, determines the feedback response (Chen et al., 2005). The inventors thus sought to confirm whether total serum IGF-1 levels are also a useful pharmacodynamic marker for the activity of a therapeutic anti-IGF antibody. In this case it would be desirable for such antibody to be cross-reactive with IGFs from a suitable animal species, e.g. mouse or rat, such that a pharmacodynamic effect can already be tested pre-clinically.

Boehringer Ingelheim
The Boehringer Ingelheim group is one of the world’s 20 leading pharmaceutical companies. Headquartered in Ingelheim, Germany, Boehringer Ingelheim operates globally with 142 affiliates and a total of more than 47,400 employees. The focus of the family-owned company, founded in 1885, is researching, developing, manufacturing and marketing new medications of high therapeutic value for human and veterinary medicine.
Taking social responsibility is an important element of the corporate culture at Boehringer Ingelheim. This includes worldwide involvement in social projects, such as the initiative “Making more Health” and caring for the employees. Respect, equal opportunities and reconciling career and family form the foundation of the mutual cooperation. In everything it does, the company focuses on environmental protection and sustainability.
In 2013, Boehringer Ingelheim achieved net sales of about 14.1 billion euros. R&D expenditure corresponds to 19.5% of its net sales.

Fig.1 Production of MAb
| Adam, P.J.; Friedbichler, K.; Hofmann, M.H.; Bogenrieder, T.; Borges, E.; Adolf, G.R. BI 836845, a fully human IGF ligand neutralizing antibody, to improve the efficacy of rapamycin by blocking rapamycin-induced AKT activation 48th Annu Meet Am Soc Clin Oncol (ASCO) (June 1-5, Chicago) 2012, Abst 3092 |
| Lin, C.-C.; Chang, K.-Y.; Huang, D.C.; Marriott, V.; Van Beijsterveldt, L.; Chen, L.-T.; Cheng, A.-L. A phase I dose escalation study of weekly BI 836845, a fully human, affinity-optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid cancers 50th Annu Meet Am Soc Clin Oncol (ASCO) (May 30-June 3, Chicago) 2014, Abst 2617 |
Adam, P.J.; Ostermann, E.; Lamche, H.R.; Hofmann, M.H.; Kroez, M.; Borges, E.; Adolf, G.R.
Pharmacodynamic properties and anti-tumor efficacy of BI 836845, a fully human IGF ligand neutralizing antibody
AACR-NCI-EORTC Int Conf Mol Targets Cancer Ther (November 12-16, San Francisco) 2011, Abst A208
| Rihawi, K.; Ong, M.; Michalarea, V.; et al. Phase I dose escalation study of 3-weekly BI 836845, a fully human, affinity optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid tumors 50th Annu Meet Am Soc Clin Oncol (ASCO) (May 30-June 3, Chicago) 2014, Abst 2622 |
Momenta Pharma receives FDA orphan drug designation for pancreatic cancer drug Necuparanib
heparan sulfate mimetic derived from unfractionated heparin with a molecular weight between 5500 and 6200 Da
Necuparanib
M-402
M-ONC-402
MONC 402
| Momenta Pharmaceuticals Inc |
Momenta Pharmaceuticals has received orphan drug designation from the US Food and Drug Administration (FDA) for its necuparanib, a heparan sulfate mimetic indicated for treatment of pancreatic cancer.
Momenta Pharmaceuticals chief medical officer Jim Roach said there is a great need for new medications for patients suffering from pancreatic cancer.
“We are encouraged by the progress of the programme to date, and in the next several months, we anticipate completing Part A of our ongoing Phase I/II study of necuparanib in combination with Abraxane and gemcitabine,” Roach said.
“In the next several months, we anticipate completing Part A of our ongoing Phase I/II study of necuparanib in combination with Abraxane and gemcitabine.”
“We look forward to sharing the results from Part A and advancing the product into the Phase II part of the study in the second half of 2014.”
Necuparanib has recently been adopted as the unique non-proprietary name for M402 by The United States Adopted Names.
The drug is derived from unfractionated heparin. It has been engineered to have significantly reduced anticoagulant activity while preserving the relevant antitumor properties of heparin.
Part A dose escalation component of the Phase I/II trial, which is evaluating necuparanib in combination with Abraxane (nab-paclitaxel) and gemcitabine in advanced metastatic pancreatic cancer patients, is expected to be completed in the next several months.
The company is expected to report the clinical data from Part A in the second half this year. The company also plans to begin Part B of the study by the year-end.
Part B will be a randomised, controlled, proof-of-concept study to assess the antitumor activity of necuparanib in combination with Abraxane plus gemcitabine, versus Abraxane plus gemcitabine alone.
Heparin, a highly sulfated heparin-like glycosaniinoglycan (HLGAG) produced by mast cells and isolated from natural sources, is a widely used clinical anticoagulant. However, the effects of natural, or unfractionated, heparin can be difficult to predict and patients must be monitored closely to prevent over- or under-anticoagulation. Low molecular weight heparins (LMWHs) obtained by various methods of fractionation or depolymerization of polymeric heparin have more predictable pharmacological action as anticoagulants, reduced side effects, sustained antithrombotic activity, and better bioavailability than unfractionated heparin (UFH). Several LMWHs are approved for outpatient treatment of thrombotic conditions.
There is increasing interest in the potential role of antithrombotic agents in the management of cancer patients. Results from several recent clinical trials have suggested a survival advantage for certain types of cancer patients treated with LMWHs (reviewed in Lemoine, 2005, Journal of Clinical Oncology, 23: 2119-20).
http://www.google.fm/patents/EP2207811A1?cl=en
The invention is based, in part, on the development of polysaccharide preparations, e.g., preparations of polysaccharides derived from heparin, that lack substantial anticoagulant activity (e.g., preparations of polysaccharides that have substantially no anticoagulant activity) but retain activity in other non-coagulation mediated biological processes, and methods to produce them. These compounds can have one or more of the following features: 1) an anti-Xa activity and an anti-IIa activity each less than 50 IU/mg, and 2) anti-metastatic, anti-angiogenic, anti-fibrotic and/or anti-inflammatory activity. The polysaccharides disclosed herein can also have structural characteristics that distinguish them from other polysaccharides, (e.g., from commercially available heparins). For example, a polysaccharide preparation provided herein can have one or more of the following characteristics: the preparation has less than 50% glycol split uronic acid residues; the preparation has no more than 3 glycol split uronic acid residues (UG) per polysaccharide chain; the preparation has greater than 40% U2SHNS>6S disaccharide residues; degree of desulfation of the preparation is less than 40%; one or more polysaccharide chains in the preparation have a 4,5-unsaturation of a non-reducing end uronic acid residue; one or more polysaccharide chains in the preparation have a 2,5-anhydromannitol residue at the reducing end; and the weight average molecular weight of the preparation is between 3,500 and 7,000 Da. This disclosure includes preparations having one or more of these properties and characteristics as well as methods of making and using such preparations. The disclosure also features methods of using such preparations.
Accordingly, in a first aspect, the invention features a polysaccharide preparation (e.g., a heparin-derived preparation) having the following characteristics: (a) a weight average chain molecular weight between 3,500 and 7,000 Da; (b) an anti-Xa activity and an anti-IIa activity each less than 50 IU/mg (e.g., an anti-Xa activity less than about 40 IU/mg, 30 IU/mg, 20 IU/mg, 15 IU/mg, or 10 IU/mg and an anti-IIa activity less than about 40 IU/mg, 30 IU/mg, 20 IU/mg, 10 IU/mg, 5 IU/mg, 4 IU/mg, or 3 IU/mg); and (c) less than 50% glycol split uronic acid residues (e.g., less than 40%, 30%, 25%, or 20% glycol split uronic acid residues) in the preparation. In some embodiments, the preparation contains between 5% and 50% glycol split uronic acid residues (e.g., between 5% and 40%, 5% and 30%, 10% and 50%, 10% and 40%, or 10% and 30% glycol split uronic acid residues).
In a second aspect, the invention features a polysaccharide preparation (e.g., a heparin- derived preparation) having the following characteristics: (a) a weight average chain molecular weight between 3,500 and 7,000 Da; (b) an anti-Xa activity and an anti-IIa activity each less than 50 IU/mg (e.g., an anti-Xa activity less than about 40 IU/mg, 30 IU/mg, 20 IU/mg, 15 IU/mg, or 10 IU/mg and an anti-IIa activity less than about 40 IU/mg, 30 IU/mg, 20 IU/mg, 10 IU/mg, 5 IU/mg, 4 IU/mg, or 3 IU/mg); and (c) the polysaccharide chains of the preparation have no more than 3 glycol split uronic acid residues (UQ) per polysaccharide chain (e.g., each polysaccharide chain has no more than 2 or no more than 1 glycol split uronic acid residue (UQ) per polysaccharide chain).
In a third aspect, the invention features a polysaccharide preparation (e.g., a heparin- derived preparation) having the following characteristics: (a) a weight average chain molecular weight between 3,500 and 7,000 Da; (b) an anti-Xa activity and an anti-IIa activity each less than 50 IU/mg (e.g., an anti-Xa activity less than about 40 IU/mg, 30 IU/mg, 20 IU/mg, 15 IU/mg, or 10 IU/mg and an anti-IIa activity less than about 40 IU/mg, 30 IU/mg, 20 IU/mg, 10 IU/mg, 5 IU/mg, 4 IU/mg, or 3 IU/mg); and (c) polysaccharide chains of the preparation have on average no more than 3 glycol split uronic acid residues (Uo) per polysaccharide chain (e.g., on average no more than 2.5, no more than 2, no more than 1.5, or no more than 1 glycol split uronic acid residues (UG) per polysaccharide chain.
In a fourth aspect, the invention features a polysaccharide preparation (e.g., a heparin- derived preparation) having the following characteristics: (a) a weight average chain molecular weight between 3,500 and 7,000 Da; (b) an anti-Xa activity and an anti-IIa activity each less than 50 IU/mg (e.g., an anti-Xa activity less than about 40 IU/mg, 30 IU/mg, 20 IU/mg, 15 IU/mg, or 10 IU/mg and an anti-IIa activity less than about 40 IU/mg, 30 IU/mg, 20 IU/mg, 10 IU/mg, 5 IU/mg, 4 IU/mg, or 3 IU/mg); and (c) the preparation has greater than 40% U2SHNS,6S disaccharide residues (e.g., greater than 50%, 60%, 70%, or 80% U2SHNS,6S disaccharide residues). In some embodiments, the preparation has a degree of desulfation less than 40% (e.g., less than 30%, 20%, or 10%).
In a fifth aspect, the invention features a polysaccharide preparation (e.g., a heparin- derived preparation) lacking substantial anticoagulant activity (e.g., having substantially no anticoagulant activity), wherein the preparatiorrmdudes-polv^accharides that include Formula I:
[Uw-HXjy)Z]m~[UG-HX5y5Z]n
wherein U indicates a uronic acid residue and H indicates a hexosamine residue; m and n are integers such that m = 4-16 (e.g., 4-8, 4-9, 4-10, 4-11, 4-12, 4-13, 4-14, or 4-15), and n = 1-4 (e.g., 1-2 or 1-3);
w = -2OS or -2OH; x = -NS or -NAc; y = -3OS or -3OH; z = -60S or -6OH;

wherein the symbol ~ indicates that the units marked m and n are distributed along the polysaccharide chain and are not necessarily in sequence, wherein w, x, y, and z are each the same or different on each unit marked m, and wherein x, y, and z are each the same or different on each unit marked n.
In a sixth aspect, the invention features a polysaccharide preparation (e.g., a heparin- derived preparation) lacking substantial anticoagulant activity (e.g., having substantially noanticoagulant activity) and having antimetastatic activity, wherein the preparation includes polysaccharides that include Formula II:
[Uw-HXjy;Z] m– [UG-HX)y;Z] n– [Uw-HX;y)Z] 0– [UG-HX^2] p– [Uw-HX;yjZ] q
wherein U indicates a uronic acid residue and H indicates a hexosamine residue; wherein m-r are integers such that: m = 0-10; n= 0- 3;
O = O-IO;
P = 0-3; q = 0-10;
w = -2OS or -2OH; x = -NS or -NAc; y = -3OS or -3OH; z = -60S or -6OH;

wherein w, x, y, and z are each the same or different on each unit marked m, n, o, p, or q. In some embodiments, the sum of n + p is less than or equal to 4 (e.g., less than or equal to 3, 2, 1, or 0). In some embodiments, the preparation has a weight average chain molecular weight between 3,500 and 7,000 Da.
Examples of such polysaccharide preparations include chains that include the following:
[Uw-HX;yjZ]m~[UG-Hx y z]n
wherein U indicates a uronic acid residue and H indicates a hexosamine residue, wherein m and n are integers such that m = 6-18, and n = 1 -4, w = -2OS or -2OH, x = -NS or -NAc, y = -3OS or -3OH, z = -60S or -6OH,

wherein the symbol ~ indicates that the units marked m and n are distributed along the polysaccharide chain and are not necessarily in sequence, wherein w, x, y, and z are each the same or different on each unit marked m, and wherein x, y, and z are each the same or different on each unit marked n; and
[Uw-HX)y)Z]m-[UG-HXiy)Z]n-[Uw-HXjyjZ]o-[UG-HX5y)Z]p-[Uw-HX!yiZ]q
wherein U indicates a uronic acid residue and H indicates a hexosamine residue, wherein m-r are integers such that: m = 0-10, n= 0- 3, o = 0-10, p = 0-3, q = 0-10, w = -2OS or -2OH, x = -NS or -NAc, y = -3OS or -3OH, z = -60S or -6OH,

wherein w, x, y, and z are each the same or different on each unit marked m, n, o, p, or q.
Anti-IIa Activity
Polysaccharide preparations are disclosed herein that provide substantially reduced anti- Ha activity, e.g., anti-IIa activity of about 0 to 50 IU/mg, about 0 to 40 IU/mg, about 0 to 30 IU/mg, about 0 to 25 IU/mg, about 0 to 20 IU/mg, about 0 to 10 IU/mg, about 0 to 5 IU/mg, about 5 to 10 IU/mg, about 5 to 15 IU/mg, about 5 to 20 IU/mg. Anti-IIa activity is calculated in International Units of anti- Ha activity per milligram using statistical methods for parallel line assays. The anti-IIa activity levels described herein are measured using the following principle.
Polysaccharide (PS) + ATIII→ [PS • ATIII]
Ha
PS • ATIII→[PS • ATIII • Ha] + Ha (Excess)
Ha (Excess) + Substrate -» Peptide + pNA (measured spectrophotometrically) Anti-factor Ha activity is determined by the sample potentiating effect on antithrombin (ATIII) in the inhibition of thrombin. Thrombin excess can be indirectly spectrophotometrically measured. The anti-factor Ha activity can be measured, e.g., on a Diagnostica Stago analyzer or on an ACL Futura3 Coagulation system, with reagents from Chromogenix (S-2238 substrate, Thrombin (53 nkat/vial), and Antithrombin), or on any equivalent system. Analyzer response is calibrated using the 2nd International Standard for Low Molecular Weight Heparin.
Karyopharm Announces Initiation of Phase 2 Study of Selinexor (KPT-330) an orphan drug
Selinexor (KPT-330)
1393477-72-9
WO2011109799A1
synthesis at http://www.allfordrugs.com/2014/06/10/karyopharm-announces-initiation-of-phase-2-study-of-selinexor-kpt-330/
- C17-H11-F6-N7-O
- 443.3099
Synonyms
- (Z)-3-(3-(3,5-Bis(trifluoromethyl)phenyl)-1H-1,2,4-triazol-1-yl)-N’-(pyrazin-2-yl)acrylohydrazide
- 2-Propenoic acid, 3-(3-(3,5-bis(trifluoromethyl)phenyl)-1H-1,2,4-triazol-1-yl)-, 2-(2-pyrazinyl)hydrazide, (2Z)-
- 3-[3-[3,5-Bis(trifluoromethyl)phenyl]-1H-1,2,4-triazol-1-yl]-N’-(pyrazin-2-yl)acrylohydrazide
- KPT-330
- Selinexor
Karyopharm Announces Initiation of Phase 2 Study of Selinexor (KPT-330) in Patients with …
MarketWatch
“These patients were treated in our Phase 1 clinical trial of Selinexor in … Additional Phase 1 and Phase 2 studies are ongoing or currently planned and … the discovery and development of novel first-in-class drugs directed against …
synthesis

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
amcrasto@gmail.com
web link
blogs are

New Drug Approvals
ALL ABOUT DRUGS
WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL
DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ
VIETNAM
http://me.zing.vn/u/amcrasto
ICELAND
http://amcrasto.bland.is/
RUSSIA
http://www.100zakladok.ru/amcrasto/
http://bobrdobr.ru/people/amcrasto/


Fexinidazole Hoe-239

Fexinidazole, Hoe-239
1-Methyl-2-{[4-(methylsulfanyl)phenoxy]methyl}-5-nitro-1H-imidazole
| cas59729-37-2 |
| Molecular formula | C12H13N3O3S |
| Molar mass | 279.31 g mol−1 |
Sanofi (Originator)
University of Dundee
Drugs for Neglected Diseases Initiative
Winkelmann, E.; Raether, W.
Chemotherapeutically active nitro compounds. 4,5-nitroimidazoles. Part III
Arzneim-Forsch Drug Res 1978, 28(5): 739
US 4042705, DE 2531303,
UPDATE 7/16/2021 FDA APPROVES
To treat human African trypanosomiasis caused by the parasite Trypanosoma brucei gambiense
600 MG TABLET ORAL, DRUGS FOR NEGLECTED DISEASES INITIATIVE
US FDA approves fexinidazole as the first all-oral treatment for sleeping sickness
POSTED ON JULY 19
The US Food and Drug Administration (FDA) has approved fexinidazole as the first all-oral treatment for both stages of the Trypanosoma brucei gambiense form of sleeping sickness (Human African trypanosomiasis) in patients 6 years of age and older and weighing at least 20 kg.
Fexinidazole was developed as part of an innovative partnership between the non-profit research and development organization Drugs for Neglected Diseases initiative (DNDi), which conducted the pivotal clinical trials for this treatment, in partnership with the National Sleeping Sickness Programs of the Democratic Republic of Congo (DRC) and Central African Republic (CAR), and Sanofi.
Sleeping sickness is a parasitic disease transmitted by the bite of an infected tse-tse fly. It affects mostly populations living in remote rural areas of sub-Saharan Africa, where about 65 million people are at risk of infection. Left untreated, sleeping sickness is almost always fatal. Through Sanofi’s collaboration the number of sleeping sickness cases reported to the WHO has been reduced by ~97% between 2001 and 2020. DNDi, Sanofi and partners are deeply committed to ensuring access to fexinidazole in all sleeping sickness-endemic countries.
Current treatment options for the disease are effective, but burdensome for patients and health workers due to the need for infusion or injection, requiring hospitalization, especially challenging for people living in remote areas.
“Having a simple, all-oral treatment for sleeping sickness is a dream come true for frontline clinicians,” said Dr Bernard Pécoul, DNDi Executive Director. “We are proud of this latest milestone in our long-term partnership with Sanofi, developed in close collaboration with researchers in countries hard-hit by sleeping sickness.”
Fexinidazole is indicated as a 10-day once-a-day treatment for Trypanosoma brucei gambiense sleeping sickness, the most common form of the disease found in West and Central Africa. Fexinidazole is the first all-oral treatment that works both for the first stage of the disease, as well as the second stage of the disease in which the parasites have crossed the blood-brain barrier, causing patients to suffer from neuropsychiatric symptoms.
“This FDA approval is a key milestone in Sanofi’s long-term commitment to fight sleeping sickness, started 20 years ago alongside the WHO through an ambitious partnership to combat Neglected Tropical Diseases” said Luc Kuykens, Senior Vice President, Sanofi Global Health unit. “Following the positive scientific opinion granted by the European Medicines Agency end 2018, the FDA approval is an important step to revitalize efforts to support the sustainable elimination of the disease”.
As a result of FDA approval, a Tropical Disease Priority Review Voucher (PRV) has been awarded to DNDi. The FDA Tropical Disease PRV Program was established in 2007 to incentivize development of new treatments for neglected tropical diseases, including sleeping sickness. Any benefits from the PRV will be shared between Sanofi and DNDi, which will enable continued investments in innovating for and ensuring access to new health tools for sleeping sickness and other neglected diseases. Sanofi commits to continue to provide the drug free-of-charge to the World Health Organization for distribution to affected countries, as part of a long-term collaboration with WHO.
About Sleeping sickness
Sleeping sickness, or human African trypanosomiasis (HAT), is usually fatal without treatment. Transmitted by the bite of an infected tse-tse fly, following a period with nonspecific symptoms, it evolves to cause neuropsychiatric symptoms, including abnormal behaviour, and a debilitating disruption of sleep patterns that have given this neglected disease its name. About 65 million people in sub-Saharan Africa are at moderate to very high risk of infection.
About DNDi
The Drugs for Neglected Diseases initiative (DNDi) is a collaborative, patient needs-driven, not-for-profit research and development (R&D) organization that develops safe, effective, and affordable treatments for sleeping sickness, leishmaniasis, Chagas disease, filarial infections, mycetoma, paediatric HIV, hepatitis C, and covid-19. Since its inception in 2003, DNDi has delivered eight new treatments, including nifurtimox-eflornithine combination therapy (NECT) for late-stage sleeping sickness, and fexinidazole, the first all-oral drug for sleeping sickness.
Fexinidazole is an antiparasitic agent.[1] It has activity against Trypanosoma cruzi, Tritrichomonas foetus, Trichomonas vaginalis,Entamoeba histolytica,[1] Trypanosoma brucei,[2] and Leishmania donovani.[3] The biologically relevant active metabolites in vivo are the sulfoxide and sulfone [3][4]
Fexinidazole was discovered by the German pharmaceutical company Hoechst AG, but its development as a pharmaceutical was halted in the 1980s.[5] Fexinidazole is now being studied through a collaboration between Sanofi and the Drugs for Neglected Diseases Initiative for the treatment of Chagas disease and human African trypanosomiasis (sleeping sickness).[6][7] Fexinidazole is the first drug candidate for the treatment of advanced-stage sleeping sickness in thirty years.[8]
Fexinidazole is currently in phase II/III clinical development at Drugs for Neglected Diseases Initiative for the oral treatment of African trypanosomiasis (sleeping sickness). In May 2009, Sanofi (formerly known as sanofi-aventis) licensed the drug candidate to Drugs for Neglected Diseases Initiative for the development, manufacturing and distribution as a treatment of human African trypanosomiasis. Once approved, the companies plan to make the drug available on a nonprofit basis.
Fexinidazole was originally developed by a German pharmaceutical company called Hoechst, now part of Sanofi; however, its development was abandoned in the 1980s when the company gave up its tropical disease programs. Fexinidazole is one of a class of drugs known as azoles, like fluconazole, that work against fungi and may work against cancer.
-
Onset of trypanosomiasis is caused by Trypanosoma protozoa and it is said that every year 200,000 to 300,000 of new patients of African sleeping sickness fall sick. At present the number of patients of African sleeping sickness cannot be confirmed due to the low reliability of the investigative data. According to the WHO, at least 150,000 people died of African sleeping sickness in 1996 and it is said that its aftereffect remains in not less than 100,000 people. Beyond that, enormous is the damage to domestic animals caused by a disease called as nagana, and several hundred thousands of cattle which are to be protein sources for people die every year. Further, in the area of about 10,000,000 km2of savanna equal to the United States of America, cattle-breeding is impossible due to Trypanosoma. Thus, African sleeping sickness remarkably damages the health and the economical development of African people, and this is the reason why the WHO adopts the trypanosomiasis as one of the infectious diseases that should be controlled.
-
African sleeping sickness is a protozoal infectious disease by Trypanosoma transmitted through tsetse flies and the protozoa appear in the blood stream in about 10 days after infection. In the initial period of infection the protozoa multiply in the blood stream and give fever, physical weakness, headache, a pain of muscles and joints and a feeling of itching to proceed. On entering the chromic period, the central nerve is affected to show symptoms such as mental confusion and systemic convulsion, and finally the patients lapse into lethargy and are led to death.
-
The trypanosomiasis of domestic animals has Trypanosoma brucei brucei, Trypanosoma evansi, Trypanosoma congolense and Trypanosoma vivax as pathogens and is a communicable disease which affects domestic animals such as horses, cattle, pigs and dogs and, in addition, mice, guinea pigs, rabbits and the like. Particularly, the loss of cattle and horses is greatest and almost fetal, and they are led to anemia, edema, weakening and the like and fall dead in one month after infection.
-
In treating trypanosomiasis, pentamidine, melarsoprol, eflornithine and the like are used and there was a feeling in the 1960s that its eradication might be possible. However, these drugs are old and are gradually losing their efficacy. Particularly, the resistance to melarsoprol of an arsenic agent causes a big problem and the situation is so dire that patients with no efficacy only await death and the development of novel antitrypanosoma agents are strongly desired.
-
Trypanosoma mainly lives in the blood stream of the human body. This bloodstream energy metabolism depends on the glycolytic pathway localized in the organelle characteristic of the protozoa which is called as glycosome and the so-called oxidative phosphorylation does not function. However, in order to efficiently drive this glycolytic pathway, the produced NADH has to be reoxidized, and the glycerol-3-phosphate oxidation system of mitochondria plays an important role in this reoxidation. The terminal oxidase of this oxidation system functions as a quinol oxidase having a reduced ubiquinone as an electron donor and has properties greatly different from those of cytochrome oxidase of an aerobic respiration system which the host has. Particularly, a remarkable point is that the terminal oxidase of the oxidation system is non-sensitive to the cyanide which quickly inhibits the cytochrome oxidase of the host. Then, many researchers centered around Western countries have tried to develop drugs targeting this cyanide resistant oxidase but effective drugs having a selective toxicity have not been obtained.
-
Under these circumstances the present inventors et al. found that isoprenoid based physiologically active substances of ascochlorin, ascofuranone and derivatives thereof, particularly ascofuranone specifically inhibits the glycerol-3-phosphate oxidation system of trypanosome at a very low concentration of the order of nM and filed a patent application (Japanese Patent Publication A No. : H09-165332). They also clarified that acofuranone exhibits a very strong multiplication inhibition effect in the copresence of glycerin (Molecular and Biochemical Parasitology, 81: 127-136, 1996).
In consideration of practical use of ascofuranone, it was found essential to discover agents which replace glycerin and exhibit an effect of the combined use in a small amount, and by using an alkaloid compound having an indole skeleton existing in a plant of the family Simaroubaceae together with ascofuranone, the prolongation of life and recovery effect in African seeping sickness was found and a patent application was filed (Japanese Patent Application No.: 2003-24643, Japanese Patent Publication A No.: 2004-23601).
Method for the preparation of fexinidazole, useful for the treatment of parasitic diseases, visceral leishmaniasis, chagas disease and human African trypanosomiasis. Family members of the product patent, WO2005037759, are expected to expire from October 2024. This to be the first application from Drugs for Neglected Diseases Initiative (DNDi) on this API. DNDi in collaboration with Sanofi, the Swiss Tropical & Public Health Institute and the University of Dundee, is developing fexinidazole, an antiparasitic agent, for treating human African trypanosomiasis (HAT) and visceral Leishmaniasis (VL). By June 2013, phase I clinical studies had been completed and at that time, DNDi was planning to initiate a phase II proof-of-concept study in VL patients in early 2013.

fexinidazole[inn], 59729-37-2, 1-Methyl-2-((4-(methylthio)phenoxy)methyl)-5-nitro-1H-imidazole, Fexinidazol, Fexinidazolum

………………..
http://www.google.com/patents/EP1681280A1?cl=en
…………..
US 4042705
http://www.google.co.in/patents/US4042705
…………
new patent june 2014
WO-2014079497
Process for preparing fexinidazole – comprising the reaction of 1-methyl-2-hydroxymethyl-5-nitro-imidazole with methanesulfonyl chloride, followed by reaction with 4-methylmercapto-phenol, and further manipulative steps.
1-Methyl-2-hydroxymethyl-5-nitro-imidazole is (I) and 1-methyl-2-(4-methylmercapto-phenyloxymethyl)-5-nitro-imidazole (fexinidazole) is (II) (claim 1, page 12).
The synthesis of (II) via intermediate (I) is described (example 1, pages 6-8).
A process for preparing fexinidazole comprising the reaction of 1-methyl-2-hydroxymethyl-5-nitro-imidazole with methanesulfonyl chloride in the presence of a suspension of powdered alkaline carbonate (eg potassium carbonate) in an anhydrous organic solvent (eg acetone), followed by reaction with 4-methylmercapto-phenol, removal of hydrochloride salt, and isolation and purification is claimed. Also claimed is their use for treating parasitic diseases, visceral leishmaniasis, chagas disease, and human African trypanosomiasis. Fexinidazole is known to be an antiparasitic agent.
|
2-1-1983
|
The activity of fexinidazole (HOE 239) against experimental infections with Trypanosoma cruzi, trichomonads and Entamoeba histolytica.
|
Annals of tropical medicine and parasitology
|
|
|
1-1-1983
|
The use of the 2 substituted 5-nitroimidazole, Fexinidazole (Hoe 239) in the treatment of chronic T. brucei infections in mice.
|
Zeitschrift für Parasitenkunde (Berlin, Germany)
|
|
5-1-2011
|
1-Aryl-4-nitro-1H-imidazoles, a new promising series for the treatment of human African trypanosomiasis.
|
European journal of medicinal chemistry
|
|
|
2-1-2011
|
Compounds containing 2-substituted imidazole ring for treatment against human African trypanosomiasis.
|
Bioorganic & medicinal chemistry letters
|
|
|
1-1-2011
|
Trypanocidal activity of nitroaromatic prodrugs: current treatments and future perspectives.
|
Current topics in medicinal chemistry
|
|
|
12-1-2010
|
Potential new drugs for human African trypanosomiasis: some progress at last.
|
Current opinion in infectious diseases
|
|
|
7-1-2010
|
Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis.
|
Antimicrobial agents and chemotherapy
|
|
|
1-1-2010
|
Fexinidazole–a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness.
|
PLoS neglected tropical diseases
|
|
|
1-1-1999
|
[Use of megazol for the treatment of trypanosomiasis].
|
Médecine tropicale : revue du Corps de santé colonial
|
|
|
11-1-1998
|
A method to assess invasion and intracellular replication of Trypanosoma cruzi based on differential uracil incorporation.
|
Journal of immunological methods
|
|
|
10-1-1996
|
Topical chemotherapy for experimental murine African CNS-trypanosomiasis: the successful use of the arsenical, melarsoprol, combined with the 5-nitroimidazoles, fexinidazole or MK-436.
|
Tropical medicine & international health : TM & IH
|
|
|
6-1-1991
|
Chemotherapy of CNS-trypanosomiasis: the combined use of the arsenicals and nitro-compounds.
|
|
11-15-2013
|
Targeting the human parasite Leishmania donovani: discovery of a new promising anti-infectious pharmacophore in 3-nitroimidazo[1,2-a]pyridine series.
|
Bioorganic & medicinal chemistry
|
|
|
10-1-2013
|
The R enantiomer of the antitubercular drug PA-824 as a potential oral treatment for visceral Leishmaniasis.
|
Antimicrobial agents and chemotherapy
|
|
|
2-1-2013
|
Assessing the essentiality of Leishmania donovani nitroreductase and its role in nitro drug activation.
|
Antimicrobial agents and chemotherapy
|
|
|
9-1-2012
|
Genotoxicity profile of fexinidazole–a drug candidate in clinical development for human African trypanomiasis (sleeping sickness).
|
Mutagenesis
|
|
|
7-15-2012
|
Discovery of nitroheterocycles active against African trypanosomes. In vitro screening and preliminary SAR studies.
|
Bioorganic & medicinal chemistry letters
|
|
|
2-1-2012
|
The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis.
|
Science translational medicine
|
|
|
1-1-2012
|
Fexinidazole: a potential new drug candidate for Chagas disease.
|
PLoS neglected tropical diseases
|
|
|
1-1-2012
|
Management of trypanosomiasis and leishmaniasis.
|
British medical bulletin
|
|
|
12-1-2011
|
Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness.
|
Antimicrobial agents and chemotherapy
|
|
|
6-1-2011
|
Development of novel drugs for human African trypanosomiasis.
|
Future microbiology
|
| US3682951 * | 2 Nov 1970 | 8 Aug 1972 | Searle & Co | 1-{8 {62 -(1-adamantyloxy)halophenethyl{9 {0 imidazoles and congeners |
| US3714179 * | 8 Sep 1970 | 30 Jan 1973 | Searle & Co | 1-alkyl-2-furfurylthioimidazoles and congeners |
| US3796704 * | 16 Aug 1971 | 12 Mar 1974 | Bayer Ag | Phenyl-imidazolylalkanyl derivatives |
| US3828065 * | 11 Dec 1972 | 6 Aug 1974 | Searle & Co | 2-methyl-5-nitro-1-(2-phenylthioethyl)imidazoles |
| US3842097 * | 22 Jan 1973 | 15 Oct 1974 | Searle & Co | 2-(phenoxyalkylthio)imidazoles and congeners |
| US3910925 * | 24 May 1974 | 7 Oct 1975 | Searle & Co | {8 2-(2-Methyl-5-nitro-1-imidazolyl)ethyl{9 benzo(b)pyridyloxy ethers |
| US3922277 * | 14 Nov 1974 | 25 Nov 1975 | Hoechst Ag | (1-Alkyl-5-nitro-imidazolyl-2-alkyl)-pyridyl compounds |
| DE2124103A1 * | 14 May 1971 | 25 Nov 1971 | Title not available |
References
- Raether, W; Seidenath, H (1983). “The activity of fexinidazole (HOE 239) against experimental infections with Trypanosoma cruzi, trichomonads and Entamoeba histolytica”. Annals of Tropical Medicine and Parasitology 77 (1): 13–26. PMID 6411009.
- Jennings, FW; Urquhart, GM (1983). “The use of the 2 substituted 5-nitroimidazole, Fexinidazole (Hoe 239) in the treatment of chronic T. brucei infections in mice”. Zeitschrift für Parasitenkunde 69 (5): 577–581. doi:10.1007/bf00926669. PMID 6636983.
- Wyllie, S; Patterson, S; Stojanovski, FRC; Norval, S; Kime, R; Read, RD; Fairlamb, AH (2012). “The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis”. Science Translational Medicine 4 (119): 119re1.doi:10.1126/scitranslmed.3003326. PMC 3457684. PMID 22301556.
- Sokolova, AY; Wyllie, S; Patterson, S; Oza, SL; Read, RD; Fairlamb, AH (2010). “Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis”. Antimicrobial Agents and Chemotherapy 54 (7): 2893–900. doi:10.1128/AAC.00332-10.PMID 20439607.
- “Jump-Start on Slow Trek to Treatment for a Disease”. New York Times. January 8, 2008.
- “Fexinidazole Progresses into Clinical Development”. DNDi Newsletter. November 2009.
- “Sanofi-aventis and DNDi enter into a Collaboration Agreement on a New Drug for Sleeping Sickness, Fexinidazole”. DNDi. May 18, 2009.
- Torreele, E; Bourdin Trunz, B; Tweats, D; Kaiser, M; Brun, R; Mazué, G; Bray, MA; Pécoul, B (2010). “Fexinidazole–a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness”. In Boelaert, Marleen. PLOS Neglected Tropical Diseases 4 (12): e923. doi:10.1371/journal.pntd.0000923. PMC 3006138. PMID 21200426.

AbbVie’S Investigational Oncology Compound ABT-199/GDC-0199, Venetoclax

ABT 199, RG 7601, GDC 0199
Venetoclax
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
SYNTHESIS UPDATED BELOW …………..

CAS 1257044-40-8 [RN]
2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)-4-(4-((2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-enyl)methyl)piperazin-1-yl)-N-(3-nitro-4-((tetrahydro-2H-pyran-4-yl)methylamino)phenylsulfonyl)benzamide
4-[4-[[2-(4-chlorophenyl)-4,4-dimethylcyclohexen-1-yl]methyl]piperazin-1-yl]-N-[3-nitro-4-(oxan-4-ylmethylamino)phenyl]sulfonyl-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
ABT 199
- Molecular Formula: C45H50ClN7O7S
- Average mass: 868.439209 Da
- Monoisotopic mass: 867.318115 Da
-
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
NORTH CHICAGO, Ill., May 31, 2014/NEWS.GNOM.ES/ — AbbVie (NYSE: ABBV) released interim results from a Phase Ib clinical trial of ABT-199/GDC-0199, an investigational B-cell lymphoma 2 (BCL-2) selective inhibitor, in combination with rituximab (Abstract 7013). Results showed anoverall response rate (ORR) of 84 percent, in patients with relapsed/refractory chronic lymphocytic leukemia(CLL), the most common leukemia in the UnitedStates. These results were presented at the 50thAnnual Meeting of the American Society of ClinicalOncology (ASCO), May 30 – June 3 in Chicago.
ABT-199 is a so-called BH3-mimetic drug, which is designed to block the function of the protein Bcl 2. In 1988, it was discovered that Bcl-2 allowed leukaemia cells to become long-lived, a discovery made at the Walter and Eliza Hall Institute by Professors David Vaux, Suzanne Cory and Jerry Adams. Subsequent research led by them and other institute scientists, including Professors Andreas Strasser, David Huang, Peter Colman and Keith Watson, has explained much about how Bcl-2 and related molecules function to determine if a cell lives or dies. These discoveries have contributed to the development of a new class of drugs called BH3-mimetics that kill, and thereby rapidly remove, leukaemic cells by blocking Bcl-2. (source:http://www.wehi.edu.au).
|
Highlights of recent research using this agent |
GDC-0199 (RG7601) is a novel small molecule Bcl-2 selective inhibitor designed to restore apoptosis, also known as programmed cell death, by blocking the function of a pro-survival Bcl-2 family protein. The Bcl-2 family proteins, which are expressed at high levels in many tumors, play a central role in regulating apoptosis and, consequently, are thought to impact tumor formation, tumor growth and resistance.
Venetoclax (previously: GDC-0199, ABT-199, RG7601 )[1] is a small molecule oral drug being investigated to treat chronic lymphocytic leukemia (CLL).[2][3]
In 2015, the FDA granted Breakthrough Therapy Designation to venetoclax for CLL in previously treated (relapsed/refractory) patients with the 17p deletion genetic mutation.[3]
Mechanism of action
Venetoclax (a BH3-mimetic[4]) acts as a Bcl-2 inhibitor, ie. it blocks the anti-apoptotic B-cell lymphoma-2 (BCL2) protein, leading toprogrammed cell death in CLL cells.[2]
Clinical trials
A phase 1 trial established a dose of 400mg/day.[2]
A trial of venetoclax in combination with rituximab had an encouraging complete response rate.[5]
A phase 2 trial met its primary endpoint which was overall response rate.[3] Interim results from a Phase 2b trial are encouraging, especially in patients with the 17p deletion.[2]
A phase 3 trial (NCT02005471)[1] has started.[3]
NOW IN PHASE 3 UPDATED…………
4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (hereafter, “Compound 1”) is a potent and selective Bcl-2 inhibitor having, inter alia, antitumor activity as an apoptosis-inducing agent. Compound 1 has the formula:

Compound 1 is currently the subject of ongoing clinical trials for the treatment of chronic lymphocytic leukemia. U.S. Patent Publication No. 2010/0305122 describes Compound 1, and other compounds which exhibit potent binding to a Bcl-2 family protein, and pharmaceutically acceptable salts thereof. U.S. Patent Publication Nos. 2012/0108590 and 2012/0277210 describe pharmaceutical compositions comprising such compounds, and methods for the treatment of neoplastic, immune or autoimmune diseases comprising these compounds. U.S. Patent Publication No. 2012/0157470 describes pharmaceutically acceptable salts and crystalline forms of Compound 1. The disclosures of U.S. 2010/0305122; 2012/0108590; 2012/0157470 and 2012/0277210 are hereby incorporated by reference in their entireties.



PATENT
US 2015183783
http://www.google.com/patents/US20150183783
PATENT
CN 104370905
http://www.google.com/patents/CN104370905A?cl=en

ABT-199 is developed AbbVie Bel-2 inhibitors, I trial (NCT01328626) enrolled 84 patients with relapsed type / refractory CLL / SLL patients and 44 cases of relapsing / refractory non-Hodgkin lymphoma patients. ABT-199 treatment response CLL / SLL rate of 79% (complete response rate of 22%), median duration of response time was 20.5 months; ABT-199 treatment of non-Hodgkin’s lymphoma response rate of 48% (complete response rate was 7.5%). The efficacy of ABT-199 is capable of obinutuzumab, idelalisib, ibrutinib rival, is expected to become the first listed Bcl_2 inhibitors, ABT-199 is currently ongoing Phase III clinical study.
ABT-199 compound CAS number 1257044-40-8, the compound is structured as follows:

Patent W02012058392, W02012071336, W02010138588 et al. Discloses the preparation of ABT-199 in order to -IH- 5-bromo-pyrrolo [2, 3-b] pyridine as raw material to protect hydroxylation, after replacing the compound 5, and reaction of compound 6, hydrolysis to give compound 9, compound 10 and compound 9 obtained by condensation of ABT-199, a specific line as follows:

use of 2-fluoro-4-nitrobenzoate (A) as a raw material, and substituted 5-hydroxy-7-aza-indole (B), reduction to produce compound ( D), the compound (D) with the compound by cyclization after (H) substitution, hydrolysis to yield compound (J), and then with the compound (K) to afford ABT-199.

Preparation of a compound of Example (F) of the
Example

First step: Synthesis of Compound (C)
2-fluoro-4-nitrobenzoate in IL three-necked flask 50. 0g, dissolved with dimethylformamide N’N- 250ml, was added successively 5-hydroxy-7-aza-indole indole 33. 6g, potassium carbonate 34. 7g, the reaction was heated to 50 degrees under nitrogen gas protection for 2 hours, poured into 2L of ice water was added and extracted three times with ethyl acetate, the organic phase was dried with saturated sodium chloride spin dry to give Compound (C) crude 82. 0g, crude without purification in the next reaction direct investment.
Step two: Synthesis of Compound (D)
The compound of the previous step (C) of the crude product was dissolved in methanol 400ml, was added 10% palladium on carbon 4. 0g, through the reaction of hydrogen at atmospheric pressure, after the end of the reaction by TLC spin solvent to give compound (D) The crude product 73. 2g, crude without purification in the next reaction direct investment.
The third step: Synthesis of compound (F)
Take the previous step the compound (D) crude 20. 0g, t-butanol were added 150ml, compound (E) 10. g, potassium carbonate 9. 7g, completion of the addition the reaction was refluxed for 48 hours the reaction solution was cooled, added acetic acid ethyl ester was diluted, washed with water three times, the combined aqueous phases extracted once with ethyl acetate, the combined ethyl acetate phases twice, dried over anhydrous sodium sulfate and the solvent was spin, the crude product obtained was purified by silica gel column chromatography to give 13. 9g, three-step overall yield of 57.4%.
Preparation Example II Compound (H),

[0029] Take compound (G) (prepared according to W02012058392 method) 5. 0g, dissolved with 50ml of dichloromethane, was added triethylamine 5. 6ml, the reaction solution was cooled to 0-5 ° with stirring, was added dropwise methanesulfonyl chloride 2. 7g, the addition was complete the reaction was warmed to room temperature overnight, after the end of the reaction by TLC the reaction was quenched with water, the organic phase was dried over anhydrous sodium sulfate and the solvent was spin, purified by silica gel column chromatography to give compound (H) 6. 5g , a yield of 99%.
Three ABT-199 Preparation of Example

First step: Synthesis of Compound (I)
In IOOml three-necked flask were added the compound (F) 2. 5g, compound ⑶2. 3g, potassium carbonate I. 9g, Ν ‘was added and reacted at 50 degrees N- dimethylformamide 15ml, nitrogen atmosphere, TLC detection After the reaction, the reaction solution was poured into ice-water, extracted with ethyl acetate twice added ethyl acetate phase was dried over anhydrous sodium sulfate spin, and purified by silica gel column chromatography to give compound (I) 3. 6g, yield 88 %.
Step two: Synthesis of Compound (J)
In IOml single jar Compound (I) I. 0g, followed by adding water 5ml, ethanol 5ml, tetrahydrofuran 5ml, sodium hydroxide 136mg, the reaction was stirred at room temperature the reaction, ethyl acetate was added after dilution of the reaction by TLC, adjusted with IN hydrochloric acid PH4-5, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate and spin dried to give compound (J) 907mg, 93% yield.
Step two: Synthesis of ABT-199
In a 25ml single neck flask was added the compound (J) 100mg, EDCI67mg, dichloromethane 10ml, the reaction was stirred for 30 minutes, was added the compound (K) (prepared in accordance with W02012058392) 55mg, finally added a catalytic amount of DMAP, the force After opening the reaction was stirred overnight, after the end of the reaction by TLC the solvent was spin, HPLC purified preparation obtained by pure ABT-199 ^ 9811, 65% yield.
PATENT
WO 2014165044
http://www.google.com/patents/WO2014165044A1?cl=en
PATENT
US 2014275540
http://www.google.com/patents/US20140275540

-

-
An exemplary reaction according to Scheme 2 is shown below.
 Scheme 3 below. Compound (E) is commercially available or may be prepared by techniques known in the art, e.g., as shown in U.S. Pat. No. 3,813,443 and Proceedings of the Chemical Society, London, 1907, 22, 302.
Scheme 3 below. Compound (E) is commercially available or may be prepared by techniques known in the art, e.g., as shown in U.S. Pat. No. 3,813,443 and Proceedings of the Chemical Society, London, 1907, 22, 302. -
 Scheme 4 below. Compound (M) is commercially available or may be prepared by techniques known in the art, e.g., as shown in GB 585940 and J. Am. Chem. Soc., 1950, 72, 1215-1218.
Scheme 4 below. Compound (M) is commercially available or may be prepared by techniques known in the art, e.g., as shown in GB 585940 and J. Am. Chem. Soc., 1950, 72, 1215-1218. -

-
In another embodiment, the compound of formula (1) is prepared from compound (D) and compound (I) as shown in Scheme 5 below. Compound (J) may be prepared by techniques known in the art, e.g., as shown in WO 2009/117626 and Organometallics, 2008, 27 (21), 5605-5611.
-

 Example 1 Synthesis of tert-butyl 4-bromo-2-fluorobenzoate (Compound (C))To a 100 ml jacketed reactor equipped with a mechanical stirrer was charged 4-bromo-2-fluoro1-iodobenzene, “Compound (A)” (5 g, 1.0 eq) and THF (25 ml). The solution was cooled to −5° C. 2 M isopropyl magnesium chloride in THF (10.8 ml, 1.3 eq) was slowly added maintaining the internal temperature below 0° C. The mixture was stirred at 0° C. for 1 h. Di-tert-butyl dicarbonate (5.44 g, 1.5 eq) in THF (10 ml) was added. After 1 h, the solution was quenched with 10% citric acid (10 ml), and then diluted with 25% NaCl (10 ml). The layers were separated and the organic layer was concentrated to near dryness and chased with THF (3×10 ml). The crude oil was diluted with THF (5 ml), filtered to remove inorganics, and concentrated to dryness. The crude oil (6.1 g, potency=67%, potency adjusted yield=88%) was taken to the next step without further purification. 1H NMR (DMSO-d6): δ 1.53 (s, 9H), 7.50-7.56 (m, 1H), 7.68 (dd, J=10.5, 1.9 Hz, 1H), 7.74 (t, J=8.2 Hz, 1H).
Example 1 Synthesis of tert-butyl 4-bromo-2-fluorobenzoate (Compound (C))To a 100 ml jacketed reactor equipped with a mechanical stirrer was charged 4-bromo-2-fluoro1-iodobenzene, “Compound (A)” (5 g, 1.0 eq) and THF (25 ml). The solution was cooled to −5° C. 2 M isopropyl magnesium chloride in THF (10.8 ml, 1.3 eq) was slowly added maintaining the internal temperature below 0° C. The mixture was stirred at 0° C. for 1 h. Di-tert-butyl dicarbonate (5.44 g, 1.5 eq) in THF (10 ml) was added. After 1 h, the solution was quenched with 10% citric acid (10 ml), and then diluted with 25% NaCl (10 ml). The layers were separated and the organic layer was concentrated to near dryness and chased with THF (3×10 ml). The crude oil was diluted with THF (5 ml), filtered to remove inorganics, and concentrated to dryness. The crude oil (6.1 g, potency=67%, potency adjusted yield=88%) was taken to the next step without further purification. 1H NMR (DMSO-d6): δ 1.53 (s, 9H), 7.50-7.56 (m, 1H), 7.68 (dd, J=10.5, 1.9 Hz, 1H), 7.74 (t, J=8.2 Hz, 1H). -
Example 2 Synthesis of tert-butyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-bromobenzoate (Compound (D))
-
To a 3 L three-neck Morton flask were charged 1H-pyrrolo[2,3-b]pyridin-5-ol (80.0 g, 1.00 eq.), tert-butyl 4-bromo-2-fluorobenzoate (193 g, 1.15 eq.), and anhydrous DMF (800 mL). The mixture was stirred at 20° C. for 15 min. The resulting solution was cooled to about zero to 5° C. A solution of sodium tert-butoxide (62.0 g) in DMF (420 mL) was added slowly over 30 min while maintaining the internal temperature at NMT 10° C., and rinsed with DMF (30 mL). The reaction mixture was stirred at 10° C. for 1 hour (an off-white slurry) and adjusted the internal temperature to ˜45° C. over 30 min. The reaction mixture was stirred at 45-50° C. for 7 hr and the reaction progress monitored by HPLC (IP samples: 92% conversion % by HPLC). The solution was cooled to ˜20° C. The solution was stirred at 20° C. overnight.
-
Water (1200 mL) was added slowly to the reaction mixture at <30° C. over 1 hour (slightly exothermic). The product slurry was adjusted to ˜20° C., and mixed for NLT 2 hours. The crude product was collected by filtration, and washed with water (400 mL). The wet-cake was washed with heptane (400 mL) and dried under vacuum at 50° C. overnight to give the crude product (236.7 g).
-
Re-crystallization or Re-slurry: 230.7 g of the crude product, (potency adjusted: 200.7 g) was charged back to a 3 L three-neck Morton flask. Ethyl acetate (700 mL) was added, and the slurry heated slowly to refluxing temperature over 1 hr (small amount of solids left). Heptane (1400 mL) was added slowly, and the mixture adjusted to refluxing temperature (78° C.). The slurry was mixed at refluxing temperature for 30 min., and cooled down slowly to down to ˜−10° C. at a rate of approximate 10° C./hour), and mixed for 2 hr. The product was collected by filtration, and rinsed with heptane (200 ml).
-
The solid was dried under vacuum at ˜50° C. overnight to give 194.8 g, 86% isolated yield of the product as an off-white solid. MS-ESI 389.0 (M+1); mp: 190-191° C. (uncorrected). 1H NMR (DMSO-d6): δ 1.40 (s, 9H), 6.41 (dd, J=3.4, 1.7 Hz, 1H), 7.06 (d, J=1.8 Hz, 1H), 7.40 (dd, J=8.3, 1.8 Hz, 1H), 7.51 (t, J=3.4 Hz, 1H), 7.58 (d, J=2.6 Hz, 1H), 7.66 (d, J=8.3 Hz, 1H), 8.03 (d, J=2.7 Hz, 1H), 11.72 (s, 1H, NH).
-
Example 3 Synthesis of 2-chloro-4,4-dimethylcyclohexanecarbaldehyde (Compound (F))
-
To a 500 mL RB flask were charged anhydrous DMF (33.4 g, 0.456 mol) and CH2Cl2 (80 mL). The solution was cooled down <−5° C., and POCl3 (64.7 g, 0.422 mol) added slowly over 20 min @<20° C. (exothermic), rinsed with CH2Cl2 (6 mL). The slightly brown solution was adjusted to 20° C. over 30 min, and mixed at 20° C. for 1 hour. The solution was cooled back to <5° C. 3,3-Dimethylcyclohexanone (41.0 g, 90%, ˜0.292 mol) was added, and rinsed with in CH2Cl2 (10 mL) (slightly exothermic) at <20° C. The solution was heated to refluxing temperature, and mixed overnight (21 hours).
-
To a 1000 mL three neck RB flask provided with a mechanical stirrer were charged 130 g of 13.6 wt % sodium acetate trihydrate aqueous solution, 130 g of 12% brine, and 130 mL of CH2Cl2. The mixture was stirred and cooled down to <5° C. The above reaction mixture (clear and brown) was transferred, quenched into it slowly while maintaining the internal temperature <10° C. The reaction vessel was rinsed with CH2Cl2 (10 mL). The quenched reaction mixture was stirred at <10° C. for 15 min. and allowed to rise to 20° C. The mixture was stirred 20° C. for 15 min and allowed to settle for 30 min. (some emulsion). The lower organic phase was separated. The upper aq. phase was back extracted with CH2Cl2 (50 mL). The combined organic was washed with a mixture of 12% brine (150 g)-20% K3PO4 aq. solution (40 g). The organic was dried over MgSO4, filtered and rinsed with CH2Cl2 (30 ml). The filtrate was concentrated to dryness under vacuum to give a brown oil (57.0 g, potency=90.9 wt % by qNMR, ˜100%). 1H NMR (CDCl3): δ 0.98 (s, 6H), 1.43 (t, J=6.4 Hz, 2H), 2.31 (tt, J=6.4, 2.2 Hz, 2H), 2.36 (t, J=2.2 Hz, 2H), 10.19 (s, 1H).
-
Example 4 Synthesis of 2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-enecarbaldehyde (Compound (G))
-
To a 250 mL pressure bottle were charged 2-chloro-4,4-dimethylcyclohex-1-enecarbaldehyde (10.00 g), tetrabutylammonium bromide (18.67 g), and acetonitrile (10 mL). The mixture was stirred at 20° C. for 5 min. 21.0 wt % K2CO3 aq. solution (76.0 g) was added. The mixture was stirred at room temperature (rt) for NLT 5 min. followed by addition of 4-chlorophenylboronic acid (9.53 g) all at once. The mixture was evacuated and purged with N2 for three times. Palladium acetate (66 mg, 0.5 mol %) was added all at once under N2. The reaction mixture was evacuated and purged with N2 for three times (an orange colored mixture). The bottle was back filled with N2 and heated to ˜35° C. in an oil bath (bath temp ˜35° C.). The mixture was stirred at 30° C. overnight (15 hours). The reaction mixture was cooled to RT, and pulled IP sample from the upper organic phase for reaction completion, typically starting material <2% (orange colored mixture). Toluene (100 mL) and 5% NaHCO3-2% L-Cysteine aq. solution (100 mL) were added. The mixture was stirred at 20° C. for 60 min. The mixture was filtered through a pad of Celite to remove black solid, rinsing the flask and pad with toluene (10 mL). The upper organic phase was washed with 5% NaHCO3 aq. solution-2% L-Cysteine (100 mL) once more. The upper organic phase was washed with 25% brine (100 mL). The organic layer (105.0 g) was assayed (118.8 mg/g, 12.47 g product assayed, 87% assayed yield), and concentrated to ˜1/3 volume (˜35 mL). The product solution was directly used in the next step without isolation. However, an analytical sample was obtained by removal of solvent to give a brown oil. 1HNMR (CDCl3): δ 1.00 (s, 6H), 1.49 (t, J=6.6 Hz, 2H), 2.28 (t, J=2.1 Hz, 2H), 2.38 (m, 2H), 7.13 (m, 2H), 7.34 (m, 2H), 9.47 (s, 1H).
-
Example 5 Synthesis of tert-butyl 4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazine-1-carboxylate (Compound (H))
-
To a 2 L three neck RB flask provided with a mechanical stirrer were charged a solution of 4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-carbaldehyde (50.0 g) in toluene (250 mL), BOC-piperazine (48.2 g) and anhydrous THF (250 mL). The yellow solution was stirred at 20° C. for 5 min. Sodium triacetoxyborohydride (52.7 g) was added in portion (note: the internal temperature rose to ˜29.5° C. in 15 min cooling may be needed). The yellow mixture was stirred at ˜25° C. for NLT 4 hrs. A conversion of starting material to product of 99.5% was observed by HPLC after a 3 hour reaction time.
-
12.5 wt % brine (500 g) was added slowly to quench the reaction. The mixture was stirred at 20° C. for NLT 30 min and allowed to settle for NLT 15 min. The lower aq. phase (˜560 mL) was separated (note: leave any emulsion in the upper organic phase). The organic phase was washed with 10% citric acid solution (500 g×2). 500 g of 5% NaHCO3 aq. solution was charged slowly into the flask. The mixture was stirred at 20° C. for NLT 30 min., and allowed to settle for NLT 15 min. The upper organic phase was separated. 500 g of 25% brine aq. solution was charged. The mixture was stirred at 20° C. for NLT 15 min., and allowed to settle for NLT 15 min. The upper organic phase was concentrated to ˜200 mL volume under vacuum. The solution was adjusted to −30° C., and filtered off the inorganic salt. Toluene (50 mL) was used as a rinse. The combined filtrate was concentrated to ˜100 mL volume. Acetonitrile (400 mL) was added, and the mixture heated to ˜80° C. to achieve a clear solution. The solution was cooled down slowly to 20° C. slowly at rate 10° C./hour, and mixed at 20° C. overnight (the product is crystallized out at ˜45-50° C., if needed, seed material may be added at 50° C.). The slurry was continued to cool down slowly to ˜−10° C. at rate of 10° C./hours. The slurry was mixed at ˜−10° C. for NLT 6 hours. The product was collected by filtration, and rinsed with pre-cooled acetonitrile (100 mL). The solid was dried under vacuum at 50° C. overnight (72.0 g, 85%). MS-ESI: 419 (M+1); mp: 109-110° C. (uncorrected); 1H NMR (CDCl3): δ 1.00 (s, 6H), 1.46 (s, 9H), 1.48 (t, J=6.5 Hz, 2H), 2.07 (s, br, 2H), 2.18 (m, 4H), 2.24 (t, J=6.4 Hz, 2H), 2.80 (s, 2H), 3.38 (m, 4H), 6.98 (m, 2H), 7.29 (m, 2H).
-
Example 6 Synthesis of 1-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazine dihydrochloride (Compound (I))
-
To a 2.0 L three-neck RB flask equipped with a mechanical stirrer were charged the Boc reductive amination product (Compound (H), 72.0 g) and IPA (720 mL). The mixture was stirred at rt for 5 min, and 59.3 g of concentrated hydrochloride aq. solution added to the slurry. The reaction mixture was adjusted to an internal temperature of ˜65° C. (a clear and colorless solution achieved). The reaction mixture was agitated at ˜65° C. for NLT 12 hours.
-
The product slurry was cooled down to −5° C. slowly (10° C./hour). The product slurry was mixed at ˜−5° C. for NLT 2 hours, collected by filtration. The wet cake was washed with IPA (72 mL) and dried at 50° C. under vacuum overnight to give 73.8 g (95%) of the desired product as a bis-hydrochloride IPA solvate (purity >99.5% peak area at 210 nm). MS-ESI: 319 (M+1); 1HNMR (CDCl3): δ 0.86 (s, 6H), 1.05 (d, J=6.0 Hz, 6H, IPA), 1.42 (t, J=6.1 Hz, 2H), 2.02 (s, br, 2H), 2.12 (m, 2H), 3.23 (m, 4H), 3.4 (s, br, 4H), 3.68 (s, 2H), 3.89 (septet, J=6.0 Hz, 1H, IPA), 7.11 (d, J=8.1 Hz, 2H), 7.41 (d, J=8.1 Hz, 2H).
-
Example 7 Synthesis of 3-nitro-4-(((tetrahydro-2H-pyran-4-yl)methyl)amino)-benzenesulfonamide (Compound (N))
-
To a 500 mL three-neck RB flask equipped with a mechanical stirrer were charged the 4-chloro-3-nitrobenzenesulfonamide, Compound M (10.0 g), diisopropylethylamine (17.5 g), (tetrahydro-2H-pyran-4-yl)methanamine (7.0 g) and acetonitrile (150 mL). The reaction mixture was adjusted to an internal temperature of 80° C. and agitated for no less than 12 hours.
-
The product solution was cooled down to 40° C. and agitated for no less than 1 hour until precipitation observed. The product slurry was further cooled to 20° C. Water (75 mL) was slowly charged over no less than 1 hour, and the mixture cooled to 10° C. and agitated for no less than 2 hours before collected by filtration. The wet cake was washed with 1:1 mix of acetonitrile:water (40 mL). The wet cake was then reslurried in water (80 mL) at 40° C. for no less than 1 hour before collected by filtration. The wet cake was rinsed with water (20 mL), and dried at 75° C. under vacuum to give 12.7 g of the desired product in 99.9% purity and in 91% weight-adjusted yield. 1H NMR (DMSO-d6): δ 1.25 (m, 2H), 1.60 (m, 2H), 1.89 (m, 1H), 3.25 (m, 2H), 3.33 (m, 2H), 3.83 (m, 2H), 7.27 (d, J=9.3 Hz, 1H), 7.32 (s, NH2, 2H), 7.81 (dd, J=9.1, 2.3 Hz, 1H), 8.45 (d, J=2.2 Hz, 1H), 8.54 (t, J=5.9 Hz, 1H, NH).
-
Example 8 Synthesis of tert-butyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoate (Compound (K))
-
General Considerations:
-
this chemistry is considered air and moisture sensitive. While the catalyst precursors in their solid, dry form can be handled and stored in air without special precautions, contact with even small amounts of solvent may render them susceptible to decomposition. As a result, traces of oxygen or other competent oxidants (e.g., solvent peroxides) must be removed prior to combination of the catalyst precursors with solvent and care must be used to prevent ingress of oxygen during the reaction. Also, care must be taken to use dry equipment, solvents, and reagents to prevent formation of undesirable byproducts. The sodium t-butoxide used in this reaction is hygroscopic and it should be properly handled and stored prior to or during use.
-
To a 2.0 L three-neck RB flask equipped with a mechanical stirrer were charged the bis-hydrochloride salt (Compound (I), 42.5 g) and toluene (285 ml). 20% K3PO4 (285 ml) was added and the biphasic mixture was stirred for 30 min. The layers were separated and the organic layer was washed with 25% NaCl (145 ml). The organic layer concentrated to 120 g and used in the coupling reaction without further purification.
-
NaOtBu (45.2 g) and Compound (I) in toluene solution (120 g solution −30 g potency adjusted) were combined in THF (180 ml) in a suitable reactor and sparged with nitrogen for NLT 45 min. Pd2dba3 (0.646 g), Compound (J) (0.399 g), and Compound (D) (40.3 g) were combined in a second suitable reactor and purged with nitrogen until oxygen level was NMT 40 ppm. Using nitrogen pressure, the solution containing Compound (I) and NaOtBu in toluene/THF was added through a 0.45 μm inline filter to the second reactor (catalyst, Compound (J) and Compound (D)) and rinsed with nitrogen sparged THF (30 ml).
-
The resulting mixture was heated to 55° C. with stirring for NLT 16 h, then cooled to 22° C. The mixture was diluted with 12% NaCl (300 g) followed by THF (300 ml). The layers were separated.
-
The organic layer was stirred with a freshly prepared solution of L-cysteine (15 g), NaHCO3 (23 g), and water (262 ml). After 1 h the layers were separated.
-
The organic layer was stirred with a second freshly prepared solution of L-cysteine (15 g), NaHCO3 (23 g), and water (262 ml). After 1 h the layers were separated. The organic layer was washed with 12% NaCl (300 g), then filtered through a 0.45 μm inline filter. The filtered solution was concentrated in vacuo to ˜300 mL, and chased three times with heptane (600 mL each) to remove THF.
-
The crude mixture was concentrated to 6 volumes and diluted with cyclohexane (720 ml). The mixture was heated to 75° C., held for 15 min, and then cooled to 65° C. over NLT 15 min. Seed material was charged and the mixture was held at 65° C. for 4 hours. The suspension was cooled to 25° C. over NLT 8 h, then held at 25° C. for 4 hours. The solids were filtered and washed with cyclohexane (90 ml) and dried at 50° C. under vacuum.
-
Isolated 52.5 g (88.9% yield) as a white solid. Melting point (uncorrected) 154-155° C. 1H NMR (DMSO-d6): δ 0.93 (s, 6H), 1.27 (s, 9H), 1.38 (t, J=6.4 Hz, 2H), 1.94 (s, 2H), 2.08-2.28 (m, 6H), 2.74 (s, 2H), 3.02-3.19 (m, 4H), 6.33 (dd, J=3.4, 1.9 Hz, 1H), 6.38 (d, J=2.4 Hz, 1H), 6.72 (dd, J=9.0, 2.4 Hz, 1H), 6.99-7.06 (m, 2H), 7.29 (d, J=2.7 Hz, 1H), 7.30-7.36 (m, 2H), 7.41-7.44 (m, 1H), 7.64 (t, J=6.7 Hz, 1H), 7.94 (d, J=2.7 Hz, 1H), 11.53 (s, 1H).
-
Example 9 Synthesis of 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoic acid (Compound (L))
-
Solution preparation: 10% KH2PO4 (aq): KH2PO4 (6 g) in water (56 g); 2:1 heptane/2-MeTHF:heptane (16 mL) in 2-MeTHF (8 mL).
-
Compound (K) (5.79 g), potassium tert-butoxide (4.89 g), 2-methyltetrahydrofuran (87 mL), and water (0.45 mL) were combined in a suitable reactor under nitrogen and heated to 55° C. until reaction completion. The reaction mixture was cooled to 22° C., washed with the 10% KH2PO4 solution (31 g) twice. The organic layer was then washed with water (30 g).
-
After removal of the aqueous layer, the organic layer was concentrated to 4 volumes (˜19 mL) and heated to no less than 50° C. Heptane (23 ml) was slowly added. The resulting suspension was cooled to 10° C. Solids were then collected by vacuum filtration with recirculation of the liquors and the filter cake washed with 2:1 heptane/2-MeTHF (24 ml). Drying of the solids at 80° C. under vacuum yielded 4.0 g of Compound (L) in approximately 85% weight-adjusted yield. 1H NMR (DMSO-d6): δ 0.91 (s, 6H), 1.37 (t, J=6.4 Hz, 2H), 1.94 (s, br, 2H), 2.15 (m, 6H), 2.71 (s, br, 2H), 3.09 (m, 4H), 6.31 (d, J=2.3 Hz, 1H), 6.34 (dd, J=3.4, 1.9 Hz, 1H), 6.7 (dd, J=9.0, 2.4 Hz, 1H), 7.02 (m, 2H), 7.32 (m, 2H), 7.37 (d, J=2.6 Hz, 1H), 7.44 (t, J=3.0 Hz, 1H), 7.72 (d, J=9.0 Hz, 1H), 7.96 (d, J=2.7 Hz, 1H) & 11.59 (m, 1H).
-
Example 10 Synthesis of 4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (Compound (I))
-
Solution preparation prior to reaction: 10% Acetic Acid:Acetic Acid (37 mL) in water (333 g); 5% NaHCO3:NaHCO3 (9 g) in water (176 g); 5% NaCl:NaCl (9 g) in water (176 g).
-
Compound (N) (13.5 g), DMAP (10.5 g), EDAC (10.7 g) and dichloromethane (300 mL) were combined in a suitable reactor and agitated at 25° C. In a second suitable reactor was charged the Acid (Compound (L), 25 g), Et3N (8.7 g) and dichloromethane (120 mL). The resulting Acid (Compound (L)) solution was slowly charged to the initial suspension of Compound (N) and agitated until reaction completion.








-
N,N-dimethylethylenediamine (9.4 g) was then charged to the reaction mixture with continued agitation. The reaction mixture was warmed to 35° C. and washed with 10% Acetic acid solution (185 mL) twice. The lower organic layer was diluted with more dichloromethane (75 mL) and methanol (12.5 mL). The organic, product layer was then washed with 5% NaHCO3 solution (185 mL) and then washed with 5% NaCl solution (185 mL) at 35° C. The lower, organic layer was separated and then concentrated to 8 vol (˜256 mL) diluted with methanol (26 mL) and warmed to 38° C. Ethyl Acetate (230 mL) was slowly charged. The resulting suspension was slowly cooled to 10° C. and then filtered. The wet cake was washed twice with a 1:1 mix of dichloromethane and ethyl acetate (˜2 vol, 64 mL). After drying the wet cake at 90° C., 32 g (84%) of Compound (I) was isolated.
-
1H NMR (DMSO-d6): δ 0.90 (s, 6H), 1.24 (m, 2H), 1.36 (t, J=6.4 Hz, 2H), 1.60 (m, 2H), 1.87 (m, 1H), 1.93 (s, br, 2H), 2.12 (m, 2H), 2.19 (m, 4H), 2.74 (s, br, 2H), 3.06 (m, 4H), 3.26 (m, 4H), 3.83 (m, 2H), 6.17 (d, J=2.1 Hz, 1H), 6.37 (dd, J=3.4, 1.9 Hz, 1H), 6.66 (dd, J=9.1, 2.2 Hz, 1H), 7.01 (m, 2H), 7.31 (m, 2H), 7.48 (m, 3H), 7.78 (dd, J=9.3, 2.3 Hz, 1H), 8.02 (d, J=2.61 Hz, 1H), 8.54 (d, J=2.33 Hz, 1H), 8.58 (t, J=5.9 Hz, 1H, NH), 11.65 (m, 1H).
PATENT

| Patent | Submitted | Granted |
|---|---|---|
| APOPTOSIS-INDUCING AGENTS FOR THE TREATMENT OF CANCER AND IMMUNE AND AUTOIMMUNE DISEASES [US2014275082] | 2014-02-10 | 2014-09-18 |
| Processes For The Preparation Of An Apoptosis-Inducing Agent [US2014275540] | 2014-03-12 | 2014-09-18 |
| APOPTOSIS INDUCING AGENTS FOR THE TREATMENT OF CANCER AND IMMUNE AND AUTOIMMUNE DISEASES [US2010305122] | 2010-12-02 | |
| Panel of micrornas that silence the MCL-1 gene and sensitize cancer cells to ABT-263 [US8742083] | 2010-12-23 | 2014-06-03 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
| METHODS OF TREATMENT USING SELECTIVE BCL-2 INHIBITORS [US2012129853] | 2011-11-22 | 2012-05-24 |
| INHIBITION OF MCL-1 AND/OR BFL-1/A1 [US2015051249] | 2013-03-14 | 2015-02-19 |
| COMBINATION THERAPY OF A TYPE II ANTI-CD20 ANTIBODY WITH A SELECTIVE BCL-2 INHIBITOR [US2014248262] | 2013-09-06 | 2014-09-04 |
References
- New Drugs Online Report for venetoclax
- Hard-to-Treat CLL Yields to Investigational Drug. ASH Dec 2015 refs: Roberts AW, et al “Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia” N Engl J Med 2015; DOI: 10.1056/NEJMoa1513257.
- Phase 2 Study of Venetoclax in Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia with 17p Deletion Meets Primary Endpoint
- ABT-199 BH-3 Mimetic Enters Phase Ia Trial For Chronic Lymphocytic Leukemia. 2011
- For Refractory CLL, Venetoclax’s Complete Response Rate Is Tops. 2015
External links
- ABT-199 inc formula and structure
|
References |
1: Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2
inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013 Jan 6. doi: 10.1038/nm.3048. [Epub ahead of print] PubMed PMID: 23291630.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
|
|
| Identifiers | |
| CAS Number | 1257044-40-8 |
| PubChem | CID: 49846579 |
| ChemSpider | 29315017 |
| Chemical data | |
| Formula | C45H50ClN7O7S |
| Molecular mass | 868.44 g/mol |
/////////
CC1(CCC(=C(C1)c2ccc(cc2)Cl)CN3CCN(CC3)c4ccc(c(c4)Oc5cc6cc[nH]c6nc5)C(=O)NS(=O)(=O)c7ccc(c(c7)[N+](=O)[O-])NCC8CCOCC8)C
OR
CC1(CCC(=C(C1)C2=CC=C(C=C2)Cl)CN3CCN(CC3)C4=CC(=C(C=C4)C(=O)NS(=O)(=O)C5=CC(=C(C=C5)NCC6CCOCC6)[N+](=O)[O-])OC7=CN=C8C(=C7)C=CN8)C