
Home » Peptide drugs (Page 2)
Category Archives: Peptide drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ropeginterferon alfa-2b
PCDLPQTHSL GSRRTLMLLA QMRRISLFSC LKDRHDFGFP QEEFGNQFQK AETIPVLHEM
IQQIFNLFST KDSSAAWDET LLDKFYTELY QQLNDLEACV IQGVGVTETP LMKEDSILAV
RKYFQRITLY LKEKKYSPCA WEVVRAEIMR SFSLSTNLQE SLRSKE
(Disulfide bridge: 2-99, 30-139)
Ropeginterferon alfa-2b
- AOP2014
CAS 1335098-50-4
UNII981TME683S
FDA APPROVED, 2021/11/12, BESREMI
PEPTIDE, Antineoplastic, Antiviral
Polycythemia vera (PV) is the most common Philadelphia chromosome-negative myeloproliferative neoplasm (MPN), characterized by increased hematocrit and platelet/leukocyte counts, an increased risk for hemorrhage and thromboembolic events, and a long-term propensity for myelofibrosis and leukemia.1,2 Interferon alfa-2b has been used for decades to treat PV but requires frequent dosing and is not tolerated by all patients.2 Ropeginterferon alfa-2b is a next-generation mono-pegylated type I interferon produced from proline-IFN-α-2b in Escherichia coli that has high tolerability and a long half-life.4,6 Ropeginterferon alfa-2b has shown efficacy in PV in in vitro and in vivo models and clinical trials.3,4
Ropeginterferon alfa-2b was approved by the FDA on November 12, 2021, and is currently marketed under the trademark BESREMi by PharmaEssentia Corporation.6
Ropeginterferon alfa-2b, sold under the brand name Besremi, is a medication used to treat polycythemia vera.[1][2][3][4] It is an interferon.[1][3] It is given by injection.[1][3]
The most common side effects include low levels of white blood cells and platelets (blood components that help the blood to clot), muscle and joint pain, tiredness, flu-like symptoms and increased blood levels of gamma-glutamyl transferase (a sign of liver problems).[3] Ropeginterferon alfa-2b can cause liver enzyme elevations, low levels of white blood cells, low levels of platelets, joint pain, fatigue, itching, upper airway infection, muscle pain and flu-like illness.[2] Side effects may also include urinary tract infection, depression and transient ischemic attacks (stroke-like attacks).[2]
It was approved for medical use in the European Union in February 2019,[3] and in the United States in November 2021.[2][5] Ropeginterferon alfa-2b is the first medication approved by the U.S. Food and Drug Administration (FDA) to treat polycythemia vera that people can take regardless of their treatment history, and the first interferon therapy specifically approved for polycythemia vera.[2]
https://www.fda.gov/news-events/press-announcements/fda-approves-treatment-rare-blood-disease#:~:text=FDA%20NEWS%20RELEASE-,FDA%20Approves%20Treatment%20for%20Rare%20Blood%20Disease,FDA%2DApproved%20Option%20Patients%20Can%20Take%20Regardless%20of%20Previous%20Therapies,-ShareFor Immediate Release:November 12, 2021
Today, the U.S. Food and Drug Administration approved Besremi (ropeginterferon alfa-2b-njft) injection to treat adults with polycythemia vera, a blood disease that causes the overproduction of red blood cells. The excess cells thicken the blood, slowing blood flow and increasing the chance of blood clots.
“Over 7,000 rare diseases affect more than 30 million people in the United States. Polycythemia vera affects approximately 6,200 Americans each year,” said Ann Farrell, M.D., director of the Division of Non-Malignant Hematology in the FDA’s Center for Drug Evaluation and Research. “This action highlights the FDA’s commitment to helping make new treatments available to patients with rare diseases.”
Besremi is the first FDA-approved medication for polycythemia vera that patients can take regardless of their treatment history, and the first interferon therapy specifically approved for polycythemia vera.
Treatment for polycythemia vera includes phlebotomies (a procedure that removes excess blood cells though a needle in a vein) as well as medicines to reduce the number of blood cells; Besremi is one of these medicines. Besremi is believed to work by attaching to certain receptors in the body, setting off a chain reaction that makes the bone marrow reduce blood cell production. Besremi is a long-acting drug that patients take by injection under the skin once every two weeks. If Besremi can reduce excess blood cells and maintain normal levels for at least one year, then dosing frequency may be reduced to once every four weeks.
The effectiveness and safety of Besremi were evaluated in a multicenter, single-arm trial that lasted 7.5 years. In this trial, 51 adults with polycythemia vera received Besremi for an average of about five years. Besremi’s effectiveness was assessed by looking at how many patients achieved complete hematological response, which meant that patients had a red blood cell volume of less than 45% without a recent phlebotomy, normal white cell counts and platelet counts, a normal spleen size, and no blood clots. Overall, 61% of patients had a complete hematological response.
Besremi can cause liver enzyme elevations, low levels of white blood cells, low levels of platelets, joint pain, fatigue, itching, upper airway infection, muscle pain and flu-like illness. Side effects may also include urinary tract infection, depression and transient ischemic attacks (stroke-like attacks).
Interferon alfa products like Besremi may cause or worsen neuropsychiatric, autoimmune, ischemic (not enough blood flow to a part of the body) and infectious diseases, which could lead to life-threatening or fatal complications. Patients who must not take Besremi include those who are allergic to the drug, those with a severe psychiatric disorder or a history of a severe psychiatric disorder, immunosuppressed transplant recipients, certain patients with autoimmune disease or a history of autoimmune disease, and patients with liver disease.
People who could be pregnant should be tested for pregnancy before using Besremi due to the risk of fetal harm.
Besremi received orphan drug designation for this indication. Orphan drug designation provides incentives to assist and encourage drug development for rare diseases.
The FDA granted the approval of Besremi to PharmaEssentia Corporation.
Medical uses
In the European Union, ropeginterferon alfa-2b is indicated as monotherapy in adults for the treatment of polycythemia vera without symptomatic splenomegaly.[3] In the United States it is indicated for the treatment of polycythemia vera.[1][2][5]
History
The effectiveness and safety of ropeginterferon alfa-2b were evaluated in a multicenter, single-arm trial that lasted 7.5 years.[2] In this trial, 51 adults with polycythemia vera received ropeginterferon alfa-2b for an average of about five years.[2] The effectiveness of ropeginterferon alfa-2b was assessed by looking at how many participants achieved complete hematological response, which meant that participants had a red blood cell volume of less than 45% without a recent phlebotomy, normal white cell counts and platelet counts, a normal spleen size, and no blood clots.[2] Overall, 61% of participants had a complete hematological response.[2] The U.S. Food and Drug Administration (FDA) granted the application for Ropeginterferon_alfa-2b orphan drug designation and granted the approval of Besremi to PharmaEssentia Corporation[2]
REF
- Bartalucci N, Guglielmelli P, Vannucchi AM: Polycythemia vera: the current status of preclinical models and therapeutic targets. Expert Opin Ther Targets. 2020 Jul;24(7):615-628. doi: 10.1080/14728222.2020.1762176. Epub 2020 May 18. [Article]
- How J, Hobbs G: Use of Interferon Alfa in the Treatment of Myeloproliferative Neoplasms: Perspectives and Review of the Literature. Cancers (Basel). 2020 Jul 18;12(7). pii: cancers12071954. doi: 10.3390/cancers12071954. [Article]
- Verger E, Soret-Dulphy J, Maslah N, Roy L, Rey J, Ghrieb Z, Kralovics R, Gisslinger H, Grohmann-Izay B, Klade C, Chomienne C, Giraudier S, Cassinat B, Kiladjian JJ: Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells in vitro and in vivo. Blood Cancer J. 2018 Oct 4;8(10):94. doi: 10.1038/s41408-018-0133-0. [Article]
- Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, Wolf D, Kralovics R, Gisslinger B, Strecker K, Egle A, Melchardt T, Burgstaller S, Willenbacher E, Schalling M, Them NC, Kadlecova P, Klade C, Greil R: Ropeginterferon alfa-2b, a novel IFNalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood. 2015 Oct 8;126(15):1762-9. doi: 10.1182/blood-2015-04-637280. Epub 2015 Aug 10. [Article]
- EMA Approved Products: Besremi (ropeginterferon alfa-2b ) solution for injection [Link]
- FDA Approved Drug Products: BESREMi (ropeginterferon alfa-2b-njft) injection [Link]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b c d e https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761166s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA Approves Treatment for Rare Blood Disease”. U.S. Food and Drug Administration (FDA) (Press release). 12 November 2021. Retrieved 12 November 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g “Besremi EPAR”. European Medicines Agency (EMA). Retrieved 14 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Wagner SM, Melchardt T, Greil R (March 2020). “Ropeginterferon alfa-2b for the treatment of patients with polycythemia vera”. Drugs of Today. Barcelona, Spain. 56 (3): 195–202. doi:10.1358/dot.2020.56.3.3107706. PMID 32282866. S2CID 215758794.
- ^ Jump up to:a b “U.S. FDA Approves Besremi (ropeginterferon alfa-2b-njft) as the Only Interferon for Adults With Polycythemia Vera” (Press release). PharmaEssentia. 12 November 2021. Retrieved 14 November 2021 – via Business Wire.
External links
- “Ropeginterferon alfa-2b”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01193699 for “Safety Study of Pegylated Interferon Alpha 2b to Treat Polycythemia Vera (PEGINVERA)” at ClinicalTrials.gov
- Clinical trial number NCT02218047 for “AOP2014 vs. BAT in Patients With Polycythemia Vera Who Previously Participated in the PROUD-PV Study. (CONTI-PV)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Besremi |
| Other names | AOP2014, ropeginterferon alfa-2b-njft |
| License data | EU EMA: by INNUS DailyMed: Ropeginterferon_alfa |
| Pregnancy category | Contraindicated |
| Routes of administration | Subcutaneous |
| Drug class | Interferon |
| ATC code | L03AB15 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2]EU: Rx-only [3] |
| Identifiers | |
| CAS Number | 1335098-50-4 |
| DrugBank | DB15119 |
| UNII | 981TME683S |
| KEGG | D11027 |
/////////Ropeginterferon alfa-2b, FDA 2021, APPROVALS 2021, BESREMI, PEPTIDE, Antineoplastic, Antiviral, AOP 2014, PharmaEssentia

NEW DRUG APPROVALS
ONE TIME
$10.00
Vosoritide
| PGQEHPNARK YKGANKKGLS KGCFGLKLDR IGSMSGLGC (Disulfide bridge: 23-39) |

H-Pro-Gly-Gln-Glu-His-Pro-Asn-Ala-Arg-Lys-Tyr-Lys-Gly-Ala-Asn-Lys-Lys-Gly-Leu-Ser-Lys-Gly-Cys(1)-Phe-Gly-Leu-Lys-Leu-Asp-Arg-Ile-Gly-Ser-Met-Ser-Gly-Leu-Gly-Cys(1)-OH
PGQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC
H-PGQEHPNARKYKGANKKGLSKGC(1)FGLKLDRIGSMSGLGC(1)-OH
PEPTIDE1{P.G.Q.E.H.P.N.A.R.K.Y.K.G.A.N.K.K.G.L.S.K.G.C.F.G.L.K.L.D.R.I.G.S.M.S.G.L.G.C}$PEPTIDE1,PEPTIDE1,23:R3-39:R3$$$
L-prolyl-glycyl-L-glutaminyl-L-alpha-glutamyl-L-histidyl-L-prolyl-L-asparagyl-L-alanyl-L-arginyl-L-lysyl-L-tyrosyl-L-lysyl-glycyl-L-alanyl-L-asparagyl-L-lysyl-L-lysyl-glycyl-L-leucyl-L-seryl-L-lysyl-glycyl-L-cysteinyl-L-phenylalanyl-glycyl-L-leucyl-L-lysyl-L-leucyl-L-alpha-aspartyl-L-arginyl-L-isoleucyl-glycyl-L-seryl-L-methionyl-L-seryl-glycyl-L-leucyl-glycyl-L-cysteine (23->39)-disulfide
(4R,10S,16S,19S,22S,28S,31S,34S,37S,40S,43S,49S,52R)-52-[[2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-6-amino-2-[[(2S)-6-amino-2-[[(2S)-4-amino-2-[[(2S)-2-[[2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-5-amino-5-oxo-2-[[2-[[(2S)-pyrrolidine-2-carbonyl]amino]acetyl]amino]pentanoyl]amino]-4-carboxybutanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]pyrrolidine-2-carbonyl]amino]-4-oxobutanoyl]amino]propanoyl]amino]-5-carbamimidamidopentanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]hexanoyl]amino]acetyl]amino]propanoyl]amino]-4-oxobutanoyl]amino]hexanoyl]amino]hexanoyl]amino]acetyl]amino]-4-methylpentanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]acetyl]amino]-40-(4-aminobutyl)-49-benzyl-28-[(2S)-butan-2-yl]-31-(3-carbamimidamidopropyl)-34-(carboxymethyl)-16,22-bis(hydroxymethyl)-10,37,43-tris(2-methylpropyl)-19-(2-methylsulfanylethyl)-6,9,12,15,18,21,24,27,30,33,36,39,42,45,48,51-hexadecaoxo-1,2-dithia-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50-hexadecazacyclotripentacontane-4-carboxylic acid
Vosoritide
| Formula | C176H290N56O51S3 |
|---|---|
| CAS | 1480724-61-5 |
| Mol weight | 4102.7254 |
1480724-61-5[RN]BMN 111L-Cysteine, L-prolylglycyl-L-glutaminyl-L-α-glutamyl-L-histidyl-L-prolyl-L-asparaginyl-L-alanyl-L-arginyl-L-lysyl-L-tyrosyl-L-lysylglycyl-L-alanyl-L-asparaginyl-L-lysyl-L-lysylglycyl-L-leucyl-L-seryl-L-lysylglycyl-L-cysteinyl-L-phenylalanylglycyl-L-leucyl-L-lysyl-L-leucyl-L-α-aspartyl-L-arginyl-L-isoleucylglycyl-L-seryl-L-methionyl-L-serylglycyl-L-leucylglycyl-, cyclic (23→39)-disulfideL-prolylglycyl-(human C-type natriuretic peptide-(17-53)-peptide (CNP-37)), cyclic-(23-39)-disulfideUNII:7SE5582Q2Pвосоритид [Russian] [INN]فوسوريتيد [Arabic] [INN]伏索利肽 [Chinese] [INN]
Voxzogo, 2021/8/26 EU APPROVED
| Product details | |
|---|---|
| Name | Voxzogo |
| Agency product number | EMEA/H/C/005475 |
| Active substance | Vosoritide |
| International non-proprietary name (INN) or common name | vosoritide |
| Therapeutic area (MeSH) | Achondroplasia |
| Anatomical therapeutic chemical (ATC) code | M05BX |
| Orphan |
This medicine was designated an orphan medicine. This means that it was developed for use against a rare, life-threatening or chronically debilitating condition or, for economic reasons, it would be unlikely to have been developed without incentives. For more information, see Orphan designation. |
| Publication details | |
|---|---|
| Marketing-authorisation holder | BioMarin International Limited |
| Date of issue of marketing authorisation valid throughout the European Union | 26/08/2021 |
On 24 January 2013, orphan designation (EU/3/12/1094) was granted by the European Commission to BioMarin Europe Ltd, United Kingdom, for modified recombinant human C-type natriuretic peptide for the treatment of achondroplasia.
The sponsorship was transferred to BioMarin International Limited, Ireland, in February 2019.
This medicine is now known as Vosoritide.
The medicinal product has been authorised in the EU as Voxzogo since 26 August 2021.
PEPTIDE
| Treatment of Achondroplasia modified recombinant human C-type natriuretic peptide (CNP) |
Vosoritide, sold under the brand name Voxzogo, is a medication used for the treatment of achondroplasia.[1]
The most common side effects include injection site reactions (such as swelling, redness, itching or pain), vomiting and decreased blood pressure.[1]
Vosoritide was approved for medical use in the European Union in August 2021.[1][2]
Voxzogo is a medicine for treating achondroplasia in patients aged 2 years and older whose bones are still growing.
Achondroplasia is an inherited disease caused by a mutation (change) in a gene called fibroblast growth-factor receptor 3 (FGFR3). The mutation affects growth of almost all bones in the body including the skull, spine, arms and legs resulting in very short stature with a characteristic appearance.
Achondroplasia is rare, and Voxzogo was designated an ‘orphan medicine’ (a medicine used in rare diseases) on 24 January 2013. Further information on the orphan designation can be found here: ema.europa.eu/medicines/human/orphan-designations/EU3121094.
Voxzogo contains the active substance vosoritide.
Medical uses
Vosoritide is indicated for the treatment of achondroplasia in people two years of age and older whose epiphyses are not closed.[1]
Mechanism of action

A: Chondrocyte with constitutionally active FGFR3 that down-regulates its development via the MAPK/ERK pathway
B: Vosoritide (BMN 111) blocks this mechanism by binding to the atrial natriuretic peptide receptor B (NPR-B), which subsequently inhibits the MAPK/ERK pathway at the RAF-1 protein.[3]
Vosoritide works by binding to a receptor (target) called natriuretic peptide receptor type B (NPR-B), which reduces the activity of fibroblast growth factor receptor 3 (FGFR3).[1] FGFR3 is a receptor that normally down-regulates cartilage and bone growth when activated by one of the proteins known as acidic and basic fibroblast growth factor. It does so by inhibiting the development (cell proliferation and differentiation) of chondrocytes, the cells that produce and maintain the cartilaginous matrix which is also necessary for bone growth. Children with achondroplasia have one of several possible FGFR3 mutations resulting in constitutive (permanent) activity of this receptor, resulting in overall reduced chondrocyte activity and thus bone growth.[3]
The protein C-type natriuretic peptide (CNP), naturally found in humans, reduces the effects of over-active FGFR3. Vosoritide is a CNP analogue with the same effect but prolonged half-life,[3] allowing for once-daily administration.[4]
Chemistry
Vosoritide is an analogue of CNP. It is a peptide consisting of the amino acids proline and glycine plus the 37 C-terminal amino acids from natural human CNP. The complete peptide sequence isPGQEHPNARKYKGANKKGLS KGCFGLKLDR IGSMSGLGC
with a disulfide bridge between positions 23 and 39 (underlined).[5] The drug must be administered by injection as it would be rendered ineffective by the digestive system if taken by mouth.
History
Vosoritide is being developed by BioMarin Pharmaceutical and, being the only available causal treatment for this condition, has orphan drug status in the US as well as the European Union.[1][2][6] As of September 2015, it is in Phase II clinical trials.[7][4]
Society and culture
Controversy
Some people with achondroplasia, as well as parents of children with this condition, have reacted to vosoritide’s study results by saying that dwarfism is not a disease and consequently does not need treatment.[8]
Research
Vosoritide has resulted in increased growth in a clinical trial with 26 children. The ten children receiving the highest dose grew 6.1 centimetres (2.4 in) in six months, compared to 4.0 centimetres (1.6 in) in the six months before the treatment (p=0.01).[9] The body proportions, more specifically the ratio of leg length to upper body length – which is lower in achondroplasia patients than in the average population – was not improved by vosoritide, but not worsened either.[7][10]
As of September 2015, it is not known whether the effect of the drug will last long enough to result in normal body heights,[10] or whether it will reduce the occurrence of achondroplasia associated problems such as ear infections, sleep apnea or hydrocephalus. This, together with the safety of higher doses, is to be determined in further studies.[4]
References
- ^ Jump up to:a b c d e f g “Voxzogo EPAR”. European Medicines Agency. 23 June 2021. Retrieved 9 September 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “European Commission Approves BioMarin’s Voxzogo (vosoritide) for the Treatment of Children with Achondroplasia from Age 2 Until Growth Plates Close”. BioMarin Pharmaceutical Inc. (Press release). 27 August 2021. Retrieved 9 September 2021.
- ^ Jump up to:a b c Lorget F, Kaci N, Peng J, Benoist-Lasselin C, Mugniery E, Oppeneer T, et al. (December 2012). “Evaluation of the therapeutic potential of a CNP analog in a Fgfr3 mouse model recapitulating achondroplasia”. American Journal of Human Genetics. 91 (6): 1108–14. doi:10.1016/j.ajhg.2012.10.014. PMC 3516592. PMID 23200862.
- ^ Jump up to:a b c Clinical trial number NCT02055157 for “A Phase 2 Study of BMN 111 to Evaluate Safety, Tolerability, and Efficacy in Children With Achondroplasia (ACH)” at ClinicalTrials.gov
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN): List 112” (PDF). WHO Drug Information. 28 (4): 539. 2014.
- ^ “Food and Drug Administration Accepts BioMarin’s New Drug Application for Vosoritide to Treat Children with Achondroplasia” (Press release). BioMarin Pharmaceutical. 2 November 2020. Retrieved 9 September 2021 – via PR Newswire.
- ^ Jump up to:a b Spreitzer H (6 July 2015). “Neue Wirkstoffe – Vosoritid”. Österreichische Apothekerzeitung (in German) (14/2015): 28.
- ^ Pollack A (17 June 2015). “Drug Accelerated Growth in Children With Dwarfism, Pharmaceutical Firm Says”. The New York Times.
- ^ “BMN 111 (vosoritide) Improves Growth Velocity in Children With Achondroplasia in Phase 2 Study”. BioMarin. 17 June 2015.
- ^ Jump up to:a b “Vosoritid” (in German). Arznei-News.de. 20 June 2015.
External links
- “Vosoritide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Voxzogo |
| Other names | BMN-111 |
| Routes of administration |
Subcutaneous injection |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 1480724-61-5 |
| DrugBank | DB11928 |
| ChemSpider | 44210446 |
| UNII | 7SE5582Q2P |
| KEGG | D11190 |
| Chemical and physical data | |
| Formula | C176H290N56O51S3 |
| Molar mass | 4102.78 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////Vosoritide, Voxzogo, PEPTIDE, ボソリチド (遺伝子組換え) , восоритид , فوسوريتيد , 伏索利肽 , APPROVALS 2021, EU 2021, BMN 111, ORPHAN DRUG
CCC(C)C1C(=O)NCC(=O)NC(C(=O)NC(C(=O)NC(C(=O)NCC(=O)NC(C(=O)NCC(=O)NC(CSSCC(C(=O)NC(C(=O)NCC(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)N1)CCCNC(=N)N)CC(=O)O)CC(C)C)CCCCN)CC(C)C)CC2=CC=CC=C2)NC(=O)CNC(=O)C(CCCCN)NC(=O)C(CO)NC(=O)C(CC(C)C)NC(=O)CNC(=O)C(CCCCN)NC(=O)C(CCCCN)NC(=O)C(CC(=O)N)NC(=O)C(C)NC(=O)CNC(=O)C(CCCCN)NC(=O)C(CC3=CC=C(C=C3)O)NC(=O)C(CCCCN)NC(=O)C(CCCNC(=N)N)NC(=O)C(C)NC(=O)C(CC(=O)N)NC(=O)C4CCCN4C(=O)C(CC5=CN=CN5)NC(=O)C(CCC(=O)O)NC(=O)C(CCC(=O)N)NC(=O)CNC(=O)C6CCCN6)C(=O)O)CC(C)C)CO)CCSC)CO
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn

join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Difelikefalin acetate
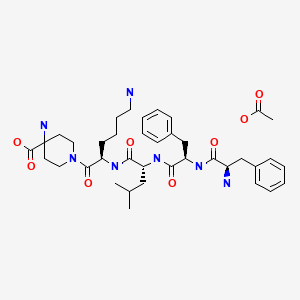
Difelikefalin acetate
ジフェリケファリン酢酸塩
CAS 1024829-44-4
| Formula | C36H53N7O6. (C2H4O2)x |
|---|
D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4- carboxylic acid)]-OH
FDA APPROVED, 2021/8/23, FORSUVA
Analgesic, Antipruritic, Opioid receptor agonist
Treatment of moderate-to-severe pruritus associated with chronic kidney disease in adults undergoing hemodialysis
Difelikefalin, CR-845; MR-13A-9; MR-13A9
4-amino-1- (D-phenylalanyl-D-phenylalanyl-D-leucyl-D-lysyl) piperidine-4-carboxylic acid
C36H53N7O6, 679.40573
| ORIGINATOR | Ferring Pharmaceuticals |
|---|---|
| DEVELOPER | Cara Therapeutics |
| CLASS | Analgesic drugs (peptides) |
| MECHANISM OF ACTION | Opioid kappa receptor agonists |
| WHO ATC CODES | D04A-X (Other antipruritics), N02A (Opioids) |
| EPHMRA CODES | D4A (Anti-Pruritics, Including Topical Antihistamines, Anaesthetics, etc), N2A (Narcotics) |
| INDICATION | Pain, Osteoarthritis, Pruritus |
Difelikefalin, sold under the brand name Korsuva , is an analgesic opioid peptide used for the treatment of moderate-to-severe pruritus. It acts as a peripherally specific, highly selective agonist of the κ-opioid receptor (KOR).[3][4][5][6]
Difelikefalin was approved for medical use in the United States in August 2021.[2][7][8]
Difelikefalin acts as an analgesic by activating KORs on peripheral nerve terminals and KORs expressed by certain immune system cells.[3] Activation of KORs on peripheral nerve terminals results in the inhibition of ion channels responsible for afferent nerve activity, causing reduced transmission of pain signals, while activation of KORs expressed by immune system cells results in reduced release of proinflammatory, nerve-sensitizing mediators (e.g., prostaglandins).[3]

NEW DRUG APPROVALS
ONE TIME
$10.00
Research
It is under development by Cara Therapeutics as an intravenous agent for the treatment of postoperative pain.[3][4][6] An oral formulation has also been developed.[6] Due to its peripheral selectivity, difelikefalin lacks the central side effects like sedation, dysphoria, and hallucinations of previous KOR-acting analgesics such as pentazocine and phenazocine.[3][4] In addition to use as an analgesic, difelikefalin is also being investigated for the treatment of pruritus (itching).[3][4][5] Difelikefalin has completed phase II clinical trials for postoperative pain and has demonstrated significant and “robust” clinical efficacy, along with being safe and well tolerated.[4][6] It has also completed a phase III clinical trial for uremic pruritus in hemodialysis patients.[9]Kappa opioid receptors have been suggested as targets for intervention for treatment or prevention of a wide array of diseases and conditions by administration of kappa opioid receptor agonists. See for example, Jolivalt et al., Diabetologia, 49(11):2775-85; Epub Aug. 19, 2006), describing efficacy of asimadoline, a kappa receptor agonist in rodent diabetic neuropathy; and Bileviciute-Ljungar et al., Eur. J. Pharm. 494:139-46 (2004) describing the efficacy of kappa agonist U-50,488 in the rat chronic constriction injury (CCI) model of neuropathic pain and the blocking of its effects by the opioid antagonist, naloxone. These observations support the use of kappa opioid receptor agonists for treatment of diabetic, viral and chemotherapy- induced neuropathic pain. The use of kappa receptor agonists for treatment or prevention of visceral pain including gynecological conditions such as dysmenorrheal cramps and endometriosis has also been reviewed. See for instance, Riviere, Br. J. Pharmacol. 141:1331-4 (2004).[0004] Kappa opioid receptor agonists have also been proposed for the treatment of pain, including hyperalgesia. Hyperalgesia is believed to be caused by changes in the milieu of the peripheral sensory terminal occur secondary to local tissue damage. Tissue damage (e.g., abrasions, burns) and inflammation can produce significant increases in the excitability of polymodal nociceptors (C fibers) and high threshold mechanoreceptors (Handwerker et al. (1991) Proceeding of the VIth World Congress on Pain, Bond et al., eds., Elsevier Science Publishers BV, pp. 59-70; Schaible et al. (1993) Pain 55:5-54). This increased excitability and exaggerated responses of sensory afferents is believed to underlie hyperalgesia, where the pain response is the result of an exaggerated response to a stimulus. The importance of the hyperalgesic state in the post-injury pain state has been repeatedly demonstrated and appears to account for a major proportion of the post-injury/inflammatory pain state. See for example, Woold et al. (1993) Anesthesia and Analgesia 77:362-79; Dubner et al.(1994) In, Textbook of Pain, Melzack et al., eds., Churchill-Livingstone, London, pp. 225-242.[0005] Kappa opioid receptors have been suggested as targets for the prevention and treatment of cardiovascular disease. See for example, Wu et al. “Cardioprotection of Preconditioning by Metabolic Inhibition in the Rat Ventricular Myocyte – Involvement of kappa Opioid Receptor” (1999) Circulation Res vol. 84: pp. 1388-1395. See also Yu et al. “Anti-Arrhythmic Effect of kappa Opioid Receptor Stimulation in the Perfused Rat Heart: Involvement of a cAMP-Dependent Pathway”(1999) JMoI Cell Cardiol, vol. 31(10): pp. 1809-1819.[0006] It has also been found that development or progression of these diseases and conditions involving neurodegeneration or neuronal cell death can be prevented, or at least slowed, by treatment with kappa opioid receptor agonists. This improved outcome is believed to be due to neuroprotection by the kappa opioid receptor agonists. See for instance, Kaushik et al. “Neuroprotection in Glaucoma” (2003) J. Postgraduate Medicine vol. 49 (1): pp. 90-95. [0007] The presence of kappa opioid receptors on immune cells (Bidlak et al.,(2000) Clin. Diag. Lab. Immunol. 7(5):719-723) has been implicated in the inhibitory • action of a kappa opioid receptor agonist, which has been shown to suppress HIV-I expression. See Peterson PK et al, Biochem Pharmacol 2001, 61(19):1145-51. [0008] Walker, Adv. Exp. Med. Biol. 521: 148-60 (2003) appraised the antiinflammatory properties of kappa agonists for treatment of osteoarthritis, rheumatoid arthritis, inflammatory bowel disease and eczema. Bileviciute-Ljungar et al., Rheumatology 45:295-302 (2006) describe the reduction of pain and degeneration in Freund’s adjuvant-induced arthritis by the kappa agonist U-50,488.[0009] Wikstrom et al, J. Am. Soc. Nephrol. 16:3742-7 (2005) describes the use of the kappa agonist, TRK-820 for treatment of uremic and opiate-induced pruritis, and Ko et al., J. Pharmacol. Exp. Ther. 305: 173-9 (2003) describe the efficacy of U- 50,488 in morphine-induced pruritis in the monkey. [0010] Application of peripheral opioids including kappa agonists for treatment of gastrointestinal diseases has also been extensively reviewed. See for example, Lembo, Diges. Dis. 24:91-8 (2006) for a discussion of use of opioids in treatment of digestive disorders, including irritable bowel syndrome (IBS), ileus, and functional dyspepsia.[0011] Ophthalmic disorders, including ocular inflammation and glaucoma have also been shown to be addressable by kappa opioids. See Potter et ah, J. Pharmacol. Exp. Ther. 309:548-53 (2004), describing the role of the potent kappa opioid receptor agonist, bremazocine, in reduction of intraocular pressure and blocking of this effect by norbinaltorphimine (norBNI), the prototypical kappa opioid receptor antagonist; and Dortch-Carnes et al, CNS Drug Rev. 11(2): 195-212 (2005). U.S. Patent 6,191,126 to Gamache discloses the use of kappa opioid agonists to treat ocular pain. Otic pain has also been shown to be treatable by administration of kappa opioid agonists. See U.S. Patent 6,174,878 also to Gamache.[0012] Kappa opioid agonists increase the renal excretion of water and decrease urinary sodium excretion (i.e., produces a selective water diuresis, also referred to as aquaresis). Many, but not all, investigators attribute this effect to a suppression of vasopressin secretion from the pituitary. Studies comparing centrally acting and purportedly peripherally selective kappa opioids have led to the conclusion that kappa opioid receptors within the blood-brain barrier are responsible for mediating this effect. Other investigators have proposed to treat hyponatremia with nociceptin peptides or charged peptide conjugates that act peripherally at the nociceptin receptor, which is related to but distinct from the kappa opioid receptor (D. R. Kapusta, Life ScL, 60: 15-21, 1997) (U.S. Pat. No. 5,840,696). U.S. Pat Appl. 20060052284.
PATENTJpn. Tokkyo Koho, 5807140US 20090156508WO 2008057608
PATENTUS 20100075910https://patents.google.com/patent/US8236766B2/en


Example 2Synthesis of Compound (2): D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OHSee the scheme of FIG. 3 and Biron et al., Optimized selective N-methylation of peptides on solid support. J. Peptide Science 12: 213-219 (2006). The amino acid derivatives used were Boc-D-Phe-OH, Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Dde)-OH, and N-Boc-amino-(4-N-Fmoc-piperidinyl)carboxylic acid. HPLC and MS analyses were performed as described in the synthesis of compound (1) described above.The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (1.8 g, 0.9 mmol; Peptide International). Attachment of N-Boc-amino-(4-N-Fmoc-piperidinyl)carboxylic acid followed by peptide chain elongation and deprotection of Dde in D-Lys(Dde) at Xaa4 was carried out according to the procedure described in the synthesis of compound (1). See above. The resulting peptide resin (0.9 mmol; Boc-D-Phe-D-Phe-D-Leu-D-Lys-(N-Boc-amino-4-piperidinylcarboxylic acid)-[2-Cl-Trt resin]) was split and a portion of 0.3 mmol was used for subsequent cleavage. The peptide resin (0.3 mmol) was then treated with a mixture of TFA/TIS/H2O (15 ml, v/v/v=95:2.5:2.5) at room temperature for 90 minutes. The resin was then filtered and washed with TFA. The filtrate was evaporated in vacuo and the crude synthetic peptide amide (0.3 mmol; D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OH) was precipitated from diethyl ether.For purification, the crude synthetic peptide amide (0.3 mmol) was dissolved in 2% acetic acid in H2O (50 ml) and the solution was loaded onto an HPLC column and purified using TEAP buffer system with a pH 5.2 (buffers A=TEAP 5.2 and B=20% TEAP 5.2 in 80% ACN). The compound was eluted with a linear gradient of buffer B, 7% B to 37% B over 60 minutes. Fractions with purity exceeding 95% were pooled and the resulting solution was diluted with two volumes of water. The diluted solution was then loaded onto an HPLC column for salt exchange and further purification with a TFA buffer system (buffers A=0.1% TFA in H2O and B=0.1% TFA in 80% ACN/20% H2O) and a linear gradient of buffer B, 2% B to 75% B over 25 minutes. Fractions with purity exceeding 97% were pooled, frozen, and dried on a lyophilizer to yield the purified synthetic peptide amide as white amorphous powder (93 mg). HPLC analysis: tR=16.43 min, purity 99.2%, gradient 5% B to 25% B over 20 min; MS (MH+): expected molecular ion mass 680.4, observed 680.3.Compound (2) was also prepared using a reaction scheme analogous to that shown in FIG. 3 with the following amino acid derivatives: Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Boc)-OH, and Boc-4-amino-1-Fmoc-(piperidine)-4-carboxylic acid.The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (PS 1% DVB, 500 g, 1 meq/g). The resin was treated with Boc-4-amino-1-Fmoc-4-(piperidine)-4-carboxylic acid (280 g, 600 mmol) in a mixture of DMF, DCM and DIEA (260 mL of each) was added. The mixture was stirred for 4 hours and then the resin was capped for 1 h by the addition of MeOH (258 mL) and DIEA (258 mL).The resin was isolated and washed with DMF (3×3 L). The resin containing the first amino acid was treated with piperidine in DMF (3×3 L of 35%), washed with DMF (9×3 L) and Fmoc-D-Lys(Boc)-OH (472 g) was coupled using PyBOP (519 g) in the presence of HOBt (153 g) and DIEA (516 mL) and in DCM/DMF (500 mL/500 mL) with stiffing for 2.25 hours. The dipeptide containing resin was isolated and washed with DMF (3×3.6 L). The Fmoc group was removed by treatment with piperidine in DMF(3×3.6 L of 35%) and the resin was washed with DMF (9×3.6 L) and treated with Fmoc-D-Leu-OH (354 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL/500 mL) and stirred for 1 hour. Subsequent washing with DMF (3×4.1 L) followed by cleavage of the Fmoc group with piperidine in DMF (3×4.2 L of 35%) and then washing of the resin with DMF (9×4.2 L) provided the resin bound tripeptide. This material was treated with Fmoc-D-Phe-OH (387 g), DIC (157 mL) and HOBt (153 g) in DCM/DMF (500 mL/500 mL) and stirred overnight. The resin was isolated, washed with DMF (3×4.7 L) and then treated with piperidine in DMF (3×4.7 L of 35%) to cleave the Fmoc group and then washed again with DMF (9×4.7 L). The tetrapeptide loaded resin was treated with Fmoc-D-Phe-OH (389 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL/500 mL) and stirred for 2.25 hours. The resin was isolated, washed with DMF (3×5.2 L) and then treated piperidine (3×5.2 L of 35%) in DMF. The resin was isolated, and washed sequentially with DMF (9×5.2 L) then DCM (5×5.2 L). It was dried to provide a 90.4% yield of protected peptide bound to the resin. The peptide was cleaved from the resin using TFA/water (4.5 L, 95/5), which also served to remove the Boc protecting groups. The mixture was filtered, concentrated (⅓) and then precipitated by addition to MTBE (42 L). The solid was collected by filtration and dried under reduced pressure to give crude synthetic peptide amide.For purification, the crude synthetic peptide amide was dissolved in 0.1% TFA in H2O and purified by preparative reverse phase HPLC (C18) using 0.1% TFA/water—ACN gradient as the mobile phase. Fractions with purity exceeding 95% were pooled, concentrated and lyophilized to provide pure synthetic peptide amide (>95.5% pure). Ion exchange was conducted using a Dowex ion exchange resin, eluting with water. The aqueous phase was filtered (0.22 μm filter capsule) and freeze-dried to give the acetate salt of the synthetic peptide amide (2) with overall yield, 71.3%, >99% purity.Hydrochloride, hydrobromide and fumarate counterions were evaluated for their ability to form crystalline salts of synthetic peptide amide (2). Approximately 1 or 2 equivalents (depending on desired stoichiometry) of hydrochloric acid, hydrobromic acid or fumaric acid, as a dilute solution in methanol (0.2-0.3 g) was added to synthetic peptide amide (2) (50-70 mg) dissolved in methanol (0.2-0.3 g). Each individual salt solution was added to isopropyl acetate (3-5 mL) and the resulting amorphous precipitate was collected by filtration and dried at ambient temperature and pressure. Crystallization experiments were carried out by dissolving the 10-20 mg of the specific amorphous salt obtained above in 70:30 ethanol-water mixture (0.1-0.2 g) followed by the addition of ethanol to adjust the ratio to 90:10 (˜0.6-0.8 mL). Each solution was then seeded with solid particles of the respective precipitated salt. Each sample tube was equipped with a magnetic stir bar and the sample was gently stirred at ambient temperature. The samples were periodically examined by plane-polarized light microscopy. Under these conditions, the mono- and di-hydrochloride salts, the di-hydrobromide salt and the mono-fumarate salt crystallized as needles of 20 to 50 μm in length with a thickness of about 1 μm.PATENT
WO 2008057608
https://patents.google.com/patent/WO2008057608A2/en Compound (2): D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4- carboxylic acid)]-OH (SEQ ID NO: 2):

EXAMPLE 2: Synthesis of compound (2)[00288] D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OH (SEQ ID NO: 2):[00289] See the scheme of Figure 2 and B iron et al., Optimized selective N- methylation of peptides on solid support. J. Peptide Science 12: 213-219 (2006). The amino acid derivatives used were Boc-D-Phe-OH, Fmoc-D-Phe-OH, Fmoc-D-Leu- OH, Fmoc-D-Lys(Dde)-OH, and N-Boc-amino-(4-N-Fmoc-piperidinyl) carboxylic acid. HPLC and MS analyses were performed as described in the synthesis of compound (1) described above.[00290] The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (1.8 g, 0.9 mmol; Peptide International). Attachment of N-Boc-amino-(4-N-Fmoc-piperidinyl) carboxylic acid followed by peptide chain elongation and deprotection of Dde in D-Lys(Dde) at Xa^ was carried out according to the procedure described in the synthesis of compound (1). See above. The resulting peptide resin (0.9 mmol; Boc-D-Phe-D-Phe-D-Leu-D-Lys-(N- Boc-amino-4-piperidinylcarboxylic acid)-[2-Cl-Trt resin]) was split and a portion of 0.3 mmol was used for subsequent cleavage. The peptide resin (0.3 mmol) was then treated with a mixture of TFA/TIS/H2O (15 ml, v/v/v = 95:2.5:2.5) at room temperature for 90 min. The resin was then filtered and washed with TFA. The filtrate was evaporated in vacuo and the crude peptide (0.3 mmol; D-Phe-D-Phe-D- Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OH) was precipitated from diethyl ether.[00291] For purification, the crude peptide (0.3 mmol) was dissolved in 2% acetic acid in H2O (50 ml) and the solution was loaded onto an HPLC column and purified using TEAP buffer system with a pH 5.2 (buffers A = TEAP 5.2 and B = 20% TEAP 5.2 in 80% ACN). The compound was eluted with a linear gradient of buffer B, 7%B to 37%B over 60 min. Fractions with purity exceeding 95% were pooled and the resulting solution was diluted with two volumes of water. The diluted solution was then loaded onto an HPLC column for salt exchange and further purification with a TFA buffer system (buffers A = 0.1% TFA in H2O and B = 0.1% TFA in 80% ACN/20% H2O) and a linear gradient of buffer B, 2%B to 75%B over 25 min. Fractions with purity exceeding 97% were pooled, frozen, and dried on a lyophilizer to yield the purified peptide as white amorphous powder (93 mg). HPLC analysis: tR = 16.43 min, purity 99.2%, gradient 5%B to 25%B over 20 min; MS (M+H+): expected molecular ion mass 680.4, observed 680.3.[00292] Compound (2) was also prepared using a reaction scheme analogous to that shown in figure 2 with the following amino acid derivatives: Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Boc)-OH, and Boc-4-amino-l-Fmoc-(piperidine)-4- carboxylic acid.[00293] The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (PS 1%DVB, 500 g, 1 meq/g). The resin was treated with Boc-4-amino-l-Fmoc-4-(piperidine)-4-carboxylic acid (280 g, 600 mmol) in a mixture of DMF, DCM and DIEA (260 mL of each) was added. The mixture was stirred for 4 hours and then the resin was capped for Ih by the addition of MeOH (258 mL) and DIEA[00294] (258 mL). The resin was isolated and washed with DMF (3 x 3 L). The resin containing the first amino acid was treated with piperidine in DMF (3 x 3 L of 35%), washed with DMF (9 x 3 L) and Fmoc-D-Lys(Boc)-OH (472 g) was coupled using PyBOP (519 g) in the presence of HOBt (153 g) and DIEA (516 mL) and in DCM/DMF (500 mL/ 500 mL) with stirring for 2.25 hours. The dipeptide containing resin was isolated and washed with DMF (3 x 3.6 L). The Fmoc group was removed by treatment with piperidine in DMF [00295] , (3 x 3.6 L of 35%) and the resin was washed with DMF (9 x 3.6 L) and treated with Fmoc-D-Leu-OH (354 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL / 500 mL) and stirred for 1 hour. Subsequent washing with DMF (3 x 4.1 L) followed by cleavage of the Fmoc group with piperidine in DMF (3 x 4.2 L of 35%) and then washing of the resin with DMF (9 x 4.2 L) provided the resin bound tripeptide. This material was treated with Fmoc-D-Phe-OH (387 g), DIC (157 mL) and HOBt (153 g) in DCM/DMF (500 mL / 500 mL) and stirred overnight. The resin was isolated, washed with DMF (3 x 4.7 L) and then treated with piperidine in DMF (3 x 4.7 L of 35%) to cleave the Fmoc group and then washed again with DMF (9 x 4.7 L). The tetrapeptide loaded resin was treated with Fmoc-D-Phe-OH (389 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL / 500 mL) and stirred for 2.25 hours. The resin was isolated, washed with DMF (3 x 5.2 L) and then treated piperidine (3 x 5.2 L of 35%) in DMF. The resin was isolated, and washed sequentially with DMF (9 x 5.2 L) then DCM (5 x 5.2 L). It was dried to provide a 90.4% yield of protected peptide bound to the resin. The peptide was cleaved from the resin using TFA/ water (4.5 L, 95/5), which also served to remove the Boc protecting groups. The mixture was filtered, concentrated (1/3) and then precipitated by addition to MTBE (42 L). The solid was collected by filtration and dried under reduced pressure to give crude peptide.[00296] For purification, the crude peptide was dissolved in 0.1% TFA in H2O and purified by preparative reverse phase HPLC (C 18) using 0.1% TF A/water – ACN gradient as the mobile phase. Fractions with purity exceeding 95% were pooled, concentrated and lyophilized to provide pure peptide (> 95.5% pure). Ion exchange was conducted using a Dowex ion exchange resin, eluting with water. The aqueous phase was filtered (0.22 μm filter capsule) and freeze-dried to give the acetate salt of the peptide (overall yield, 71.3%, >99% pure).
PATENT
κ opioid receptor agonists are known to be useful as therapeutic agents for various pain. Among, kappa opioid receptor agonist with high selectivity for peripheral kappa opioid receptors, are expected as a medicament which does not cause the central side effects. Such as peripherally selective κ opioid receptor agonist, a synthetic pentapeptide has been reported (Patent Documents 1 and 2). The following formula among the synthetic pentapeptide (A)
[Formula 1] Being Represented By Compounds Are Useful As Pain Therapeutics. The Preparation Of This Compound, Solid Phase Peptide Synthesis Methods In Patent Documents 1 And 2 Have Been Described.Document 1 Patent: Kohyo 2010-510966 JP
Patent Document 2: Japanese Unexamined Patent Publication No. 2013-241447 Compound (1) or a salt thereof and compound (A), for example as shown in the following reaction formula, 4-aminopiperidine-4-carboxylic acid, D- lysine (D-Lys), D- leucine (D-Leu) , it can be prepared by D- phenylalanine (D-Phe) and D- phenylalanine (D-Phe) sequentially solution phase peptide synthesis methods condensation.[Of 4]The present invention will next to examples will be described in further detail.Example
1 (1) Synthesis of Cbz-D-Lys (Boc) -α-Boc-Pic-OMe (3)
to the four-necked flask of 2L, α-Boc-Pic- OMe · HCl [α-Boc-4 – aminopiperidine-4-carboxylic acid methyl hydrochloride] were charged (2) 43.7g (148mmol), was suspended in EtOAc 656mL (15v / w). To the suspension of 1-hydroxybenzotriazole (HOBt) 27.2g (178mmol), while cooling with Cbz-D-Lys (Boc) -OH 59.2g (156mmol) was added an ice-bath 1-ethyl -3 – (3-dimethylcarbamoyl amino propyl) was added to the carbodiimide · HCl (EDC · HCl) 34.1g (178mmol). After 20 minutes, stirring was heated 12 hours at room temperature. After completion of the reaction, it was added and the organic layer was 1 N HCl 218 mL of (5.0v / w). NaHCO to the resulting organic layer 3 Aq. 218ML (5.0V / W), Et 3 N 33.0 g of (326Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 218ML 1N (5.0V / W), NaHCO 3 Aq. 218mL (5.0v / w), NaClaq . Was washed successively with 218ML (5.0V / W), Na 2 SO 4 dried addition of 8.74g (0.2w / w). Subjected to vacuum filtration, was concentrated under reduced pressure resulting filtrate by an evaporator, and pump up in the vacuum pump, the Cbz-D-Lys (Boc) -α-Boc-Pic-OMe (3) 88.9g as a white solid obtained (96.5% yield, HPLC purity 96.5%).[0033](2) D-Lys (Boc) Synthesis Of -Arufa-Boc-Pic-OMe (4)
In An Eggplant-Shaped Flask Of 2L, Cbz-D-Lys (Boc) -Arufa-Boc-Pic-OMe (3) 88.3g (142mmol) were charged, it was added and dissolved 441mL (5.0v / w) the EtOAc. The 5% Pd / C to the reaction solution 17.7g (0.2w / w) was added, After three nitrogen substitution reduced pressure Atmosphere, Was Performed Three Times A Hydrogen Substituent. The Reaction Solution Was 18 Hours With Vigorous Stirring At Room Temperature To Remove The Pd / C And After The Completion Of The Reaction Vacuum Filtration. NaHCO The Resulting Filtrate 3 Aq. 441ML And (5.0V / W) Were Added For Liquid Separation, And The Organic Layer Was Extracted By The Addition Of EtOAc 200ML (2.3V / W) In The Aqueous Layer. NaHCO The Combined Organic Layer 3 Aq. 441ML And (5.0V / W) Were Added for liquid separation, and the organic layer was extracted addition of EtOAc 200mL (2.3v / w) in the aqueous layer. NaClaq the combined organic layers. 441mL and (5.0v / w) is added to liquid separation, was extracted by the addition EtOAc 200ML Of (2.3V / W) In The Aqueous Layer. The Combined Organic Layer On The Na 2 SO 4 Dried Addition Of 17.7 g of (0.2W / W), Then The Filtrate Was Concentrated Under Reduced Pressure Obtained Subjected To Vacuum Filtration By an evaporator, and pump up in the vacuum pump, D-Lys (Boc) -α-Boc-Pic- OMe (4) to give 62.7g (90.5% yield, HPLC purity 93.6%).(3) Cbz-D-Leu -D-Lys (Boc) -α-Boc-Pic-OMe synthesis of (5)
in the four-necked flask of 2L, D-Lys (Boc) -α-Boc-Pic-OMe (4) was charged 57.7 g (120 mmol), was suspended in EtOAc 576mL (10v / w). HOBt 19.3g (126mmol) to this suspension, was added EDC · HCl 24.2g (126mmol) while cooling in an ice bath added Cbz-D-Leu-OH 33.4g (126mmol). After 20 minutes, after stirring the temperature was raised 5 hours at room temperature, further the EDC · HCl and stirred 1.15 g (6.00 mmol) was added 16 h. After completion of the reaction, it was added liquid separation 1N HCl 576mL (10v / w) . NaHCO to the resulting organic layer 3 Aq. 576ML (10V / W), Et 3 N 24.3 g of (240Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 576ML 1N (10V / W), NaHCO 3 Aq. 576mL (10v / w), NaClaq . Was washed successively with 576ML (10V / W), Na 2 SO 4 dried addition of 11.5g (0.2w / w). After the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and pump up in the vacuum pump, the Cbz-D-Leu-D- Lys (Boc) -α-Boc-Pic-OMe (5) 85.8g It was obtained as a white solid (98.7% yield, HPLC purity 96.9%).(4) D-Leu-D -Lys (Boc) -α-Boc-Pic-OMe synthesis of (6)
in an eggplant-shaped flask of 1L, Cbz-D-Leu- D-Lys (Boc) -α-Boc-Pic -OMe the (5) 91.9g (125mmol) were charged, was added and dissolved 459mL (5.0v / w) the EtOAc. The 5% Pd / C to the reaction solution 18.4g (0.2w / w) was added, After three nitrogen substitution reduced pressure atmosphere, was performed three times a hydrogen substituent. The reaction solution was subjected to 8 hours with vigorous stirring at room temperature to remove the Pd / C and after the completion of the reaction vacuum filtration. NaHCO the resulting filtrate 3 Aq. 200mL (2.2v / w) were added to separate liquid, NaHCO to the organic layer 3 Aq. 200mL (2.2v / w), NaClaq . It was sequentially added washed 200mL (2.2v / w). To the resulting organic layer Na 2 SO 4 dried added 18.4g (0.2w / w), to the filtrate concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and a pump-up with a vacuum pump. The resulting amorphous solid was dissolved adding EtOAc 200mL (2.2v / w), was crystallized by the addition of heptane 50mL (1.8v / w). Was filtered off precipitated crystals by vacuum filtration, the crystals were washed with a mixed solvent of EtOAc 120mL (1.3v / w), heptane 50mL (0.3v / w). The resulting crystal 46.1g to added to and dissolved EtOAc 480mL (5.2v / w), was crystallized added to the cyclohexane 660mL (7.2v / w). Was filtered off under reduced pressure filtered to precipitate crystals, cyclohexane 120mL (1.3v / w), and washed with a mixed solvent of EtOAc 20mL (0.2v / w), and 30 ° C. vacuum dried, D-Leu- as a white solid D-Lys (Boc) -α- Boc-Pic-OMe (6) to give 36.6 g (48.7% yield, HPLC purity 99.9%).(5) Synthesis of Cbz-D-Phe-D- Leu-D-Lys (Boc) -α-Boc-Pic-OMe (7)
to the four-necked flask of 1L, D-Leu-D- Lys (Boc) -α-Boc-Pic-OMe with (6) 35.8g (59.6mmol) was charged, it was suspended in EtOAc 358mL (10v / w). To this suspension HOBt 9.59g (62.6mmol), Cbz- D-Phe-OH 18.7g was cooled in an ice bath is added (62.6mmol) while EDC · HCl 12.0g (62.6mmol) It was added. After 20 minutes, a further EDC · HCl After stirring the temperature was raised 16 hours was added 3.09 g (16.1 mmol) to room temperature. After completion of the reaction, it was added and the organic layer was 1N HCl 358mL of (10v / w). NaHCO to the resulting organic layer 3 Aq. 358ML (10V / W), Et 3 N 12.1 g of (119Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 358ML 1N (10V / W), NaHCO 3 Aq. 358mL (10v / w), NaClaq . Was washed successively with 358ML (10V / W), Na 2 SO 4 dried addition of 7.16g (0.2w / w). After the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and pump up in the vacuum pump, Cbz-D-Phe-D -Leu-D-Lys (Boc) -α-Boc-Pic-OMe (7) was obtained 52.5g as a white solid (yield quant, HPLC purity 97.6%).(6) D-Phe-D -Leu-D-Lys (Boc) synthesis of -α-Boc-Pic-OMe ( 8)
in an eggplant-shaped flask of 2L, Cbz-D-Phe- D-Leu-D-Lys ( Boc) -α-Boc-Pic- OMe (7) the 46.9g (53.3mmol) were charged, the 840ML EtOAc (18V / W), H 2 added to and dissolved O 93.8mL (2.0v / w) It was. The 5% Pd / C to the reaction mixture 9.38g (0.2w / w) was added, After three nitrogen substitution reduced pressure atmosphere, was performed three times a hydrogen substituent. The reaction solution was subjected to 10 hours with vigorous stirring at room temperature to remove the Pd / C and after the completion of the reaction vacuum filtration. NaHCO the resulting filtrate 3 Aq. 235mL (5.0v / w) were added to separate liquid, NaHCO to the organic layer 3 Aq. 235mL (5.0v / w), NaClaq . It was added sequentially cleaning 235mL (5.0v / w). To the resulting organic layer Na 2 SO 4 dried addition of 9.38g (0.2w / w), then the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, pump up with a vacuum pump to D-Phe -D-Leu-D-Lys ( Boc) -α-Boc-Pic-OMe (7) was obtained 39.7g (yield quant, HPLC purity 97.3%).351mL was suspended in (10v / w). To this suspension HOBt 7.92g (51.7mmol), Boc-D-Phe-OH HCl HCl(8) D-Phe-D -Phe-D-Leu-D-Lys-Pic-OMe Synthesis Of Hydrochloric Acid Salt (1)
In An Eggplant-Shaped Flask Of 20ML Boc-D-Phe-D -Phe-D- Leu-D- lys (Boc) -α -Boc- Pic-OMe (9) and 2.00gg, IPA 3.3mL (1.65v / w), was suspended by addition of PhMe 10mL (5v / w). It was stirred at room temperature for 19 hours by addition of 6N HCl / IPA 6.7mL (3.35v / w). The precipitated solid was filtered off by vacuum filtration and dried under reduced pressure to a white solid of D-Phe-D-Phe- D- Leu-D-Lys-Pic- OMe 1.59ghydrochloride (1) (yield: 99 .0%, HPLC purity 98.2%) was obtained.(9) D-Phe-D -Phe-D-Leu-D-Lys-Pic-OMe Purification Of The Hydrochloric Acid Salt (1)
In An Eggplant-Shaped Flask Of 20ML-D-Phe-D- Phe D-Leu -D-Lys- pic-OMe hydrochloride crude crystals (1) were charged 200mg, EtOH: MeCN = 1: after stirring for 1 hour then heated in a mixed solvent 4.0 mL (20v / w) was added 40 ° C. of 5 , further at room temperature for 2 was time stirring slurry. Was filtered off by vacuum filtration, the resulting solid was dried under reduced pressure a white solid ((1) Purification crystals) was obtained 161 mg (80% yield, HPLC purity 99.2% ).(10) D-Phe-D -Phe-D-Leu-D-Lys-Pic Synthesis (Using Purified
(1)) Of (A) To A Round-Bottomed Flask Of 10ML D-Phe-D-Phe-D- -D-Lys Leu-Pic-OMe Hydrochloride Salt (1) Was Charged With Purified Crystal 38.5Mg (0.0488Mmol), H 2 Was Added And Dissolved O 0.2ML (5.2V / W). 1.5H Was Stirred Dropwise 1N NaOH 197MyuL (0.197mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 48.8μL (0.0488mmol), to obtain a D-Phe-D-Phe- D-Leu-D-Lys- Pic (A) (yield: quant , HPLC purity 99.7%).
D-Phe-D-Phe- D-Leu-D-Lys-Pic-OMe (1) physical properties 1 H NMR (400 MHz, 1M DCl) [delta] ppm by: 0.85-1.02 (yd,. 6 H), 1.34-1.63 ( m, 5 H), 1.65-2.12 ( m, 5 H), 2.23-2.45 (m, 2 H), 2.96-3.12 (m, 4 H), 3.19 (ddt, J = 5.0 & 5.0 & 10.0 Hz), 3.33-3.62 (m, 1 H), 3.68-3.82 (m, 1 H), 3.82-3.95 (m, 4 H), 3.95-4.18 (m, 1 H), 4.25-4.37 (m, 2 H), 4.61-4.77 (M, 2 H), 7.21-7.44 (M, 10 H) 13 C NMR (400MHz, 1M DCl) Deruta Ppm: 21.8, 22.5, 24.8, 27.0, 30.5, 30.8, 31.0, 31.2, 31.7, 37.2 , 37.8, 38.4, 39.0, 39.8, 40.4, 40.6, 41.8, 42.3, 49.8, 50.2, 52.2, 52.6, 54.6, 55.2, 57.7, 57.9, 127.6, 128.4, 129.2, 129.6, 129.7, 129.8 dp 209.5 ℃Example 2
(Trifluoroacetic Acid (TFA)
Use) (1) D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe TFA Synthesis Of Salt (1)
TFA 18ML Eggplant Flask Of 50ML (18V / W) , 1- Dodecanethiol 1.6ML (1.6V / W), Triisopropylsilane 0.2ML (0.2V / W), H 2 Sequentially Added Stirring The O 0.2ML (0.2V / W) Did. The Solution To The Boc-D-Phe- D- Phe-D-Leu-D -Lys (Boc) -α-Boc-Pic-OMe the (9) 1.00g (1.01mmol) was added in small portions with a spatula. After completion of the reaction, concentrated under reduced pressure by an evaporator, it was added dropwise the resulting residue in IPE 20mL (20v / w). The precipitated solid was filtered off, the resulting solid was obtained and dried under reduced pressure to D-Phe-D-Phe- D-Leu -D-Lys-Pic-OMe · TFA salt as a white solid (1) (Osamu rate 93.0%, HPLC purity 95.2%).(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic synthesis of (A)
to a round-bottomed flask of 10mL D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe TFA were charged salt (1) 83mg (0.0843mmol), was added and dissolved H2O 431μL (5.2v / w). Was 12h stirring dropwise 1N NaOH 345μL (0.345mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 84.3μL (0.0843mmol), to obtain a D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) ( yield: quant, HPLC purity 95.4%).Example
3 (HCl / EtOAc
Use) (1) In An Eggplant-Shaped Flask Of 30ML Boc-D-Phe-D -Phe-D-Leu-D-Lys (Boc) -Arufa-Boc-Pic-OMe (9) 1. It was charged with 00g (1.01mmol ), was added and dissolved EtOAc7.0mL (7.0v / w). 4N HCl / EtOAc 5.0mL (5.0v / w) was added after 24h stirring at room temperature, the precipitated solid was filtered off by vacuum filtration, washed with EtOAc 2mL (2.0v / w). The resulting solid D-Phe-D-Phe- D-Leu-D-Lys-Pic-OMe hydrochloride (1) was obtained 781mg of a white solid was dried under reduced pressure (the 96.7% yield, HPLC purity 95.4%).(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic (A) Synthesis of
eggplant flask of 10mL D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe hydrochloride were charged salt (1) 90 mg (0.112 mmol), H 2 was added and dissolved O 0.47mL (5.2v / w). Was 12h stirring dropwise 1N NaOH 459μL (0.459mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 0.112μL (0.112mmol), was obtained D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) ( yield: quant, HPLC purity 93.1%).4 Example
Compound (1) Of The Compound By Hydrolysis Synthesis Of (The A) (Compound (1) Without
Purification) Eggplant Flask 10ML D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe (1) Charged Hydrochloride Were (Without Pre-Step Purification) 114.5Mg (0.142Mmol), H 2 Was Added And Dissolved O 595MyuL (5.2V / W). Was 14H Stirring Dropwise 1N NaOH 586MyuL (0.586Mmol) At Room Temperature. After Completion Of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 0.15μL (0.150mmol), was obtained D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) (yield: quant, HPLC purity 95.2 %).Example 1 Comparative
Path Not Via The Compound (1) (Using Whole Guard Boc-D-Phe-D-Phe-D-Leu-D-Lys (Boc) -Alpha-Boc-Pic-OMe
(A)) (1) D–Boc Phe- D-Phe-D-Leu-D-Lys (Boc) -Arufa-Boc-Pic-OH Synthesis Of
Eggplant Flask Of 30ML Boc-D-Phe-D -Phe-D-Leu-D- Lys (Boc) -α- Boc-Pic -OMe (9) were charged 1.00g (1.00mmol), was added and dissolved MeOH 5.0mL (5.0v / w). After stirring for four days by the addition of 1N NaOH 1.1 mL (1.10mmol) at room temperature, further MeOH 5.0mL (5.0v / w), 1N NaOH 2.0mL the (2.0mmol) at 35 ℃ in addition 3h and the mixture was stirred. After completion of the reaction, 1 N HCl 6.1 mL was added, After distilling off the solvent was concentrated under reduced pressure was separated and the organic layer was added EtOAc 5.0mL (5.0mL) .NaClaq. 5.0mL (5.0v / w) Wash the organic layer was added, the organic layer as a white solid was concentrated under reduced pressure to Boc-D-Phe-D- Phe-D-Leu-D-Lys (Boc) – α-Boc-Pic-OH 975.1mg (99.3% yield, HPLC purity 80.8% )(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic synthesis of (A)
to a round-bottomed flask of 20mL Boc-D-Phe-D -Phe-D-Leu-D-Lys (Boc) It was charged -α-Boc-Pic-OH ( 10) 959mg (0.978mmol), was added and dissolved EtOAc 4.9mL (5.0v / w). And 4h stirring at room temperature was added dropwise 4N HCl / EtOAc 4.9mL (5.0mL) at room temperature. After completion of the reaction, it was filtered under reduced pressure, a white solid as to give D-Phe-D-Phe- D-Leu-D-Lys-Pic the (A) (96.4% yield, HPLC purity 79.2%) . If not via the compound of the present invention (1), the purity of the compound obtained (A) was less than 80%.
PATENThttp://www.google.com/patents/US20110212882
References
- ^ Janecka A, Perlikowska R, Gach K, Wyrebska A, Fichna J (2010). “Development of opioid peptide analogs for pain relief”. Curr. Pharm. Des. 16 (9): 1126–35. doi:10.2174/138161210790963869. PMID 20030621.
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214916s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j Raymond S. Sinatra; Jonathan S. Jahr; J. Michael Watkins-Pitchford (14 October 2010). The Essence of Analgesia and Analgesics. Cambridge University Press. pp. 490–491. ISBN 978-1-139-49198-3.
- ^ Jump up to:a b c d e Jeffrey Apfelbaum (8 September 2014). Ambulatory Anesthesia, An Issue of Anesthesiology Clinics. Elsevier Health Sciences. pp. 190–. ISBN 978-0-323-29934-3.
- ^ Jump up to:a b Alan Cowan; Gil Yosipovitch (10 April 2015). Pharmacology of Itch. Springer. pp. 307–. ISBN 978-3-662-44605-8.
- ^ Jump up to:a b c d Charlotte Allerton (2013). Pain Therapeutics: Current and Future Treatment Paradigms. Royal Society of Chemistry. pp. 56–. ISBN 978-1-84973-645-9.
- ^ “Korsuva: FDA-Approved Drugs”. U.S. Food and Drug Administration. Retrieved 24 August 2021.
- ^ “Vifor Pharma and Cara Therapeutics announce U.S. FDA approval of Korsuva injection for the treatment of moderate-to-severe pruritus in hemodialysis patients” (Press release). Vifor Pharma. 24 August 2021. Retrieved 24 August 2021 – via Business Wire.
- ^ Fishbane S, Jamal A, Munera C, Wen W, Menzaghi F (2020). “A phase 3 trial of difelikefalin in hemodialysis patients with pruritus”. N Engl J Med. 382 (3): 222–232. doi:10.1056/NEJMoa1912770. PMID 31702883.
External links
- “Difelikefalin”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03422653 for “A Study to Evaluate the Safety and Efficacy of CR845 in Hemodialysis Patients With Moderate-to-Severe Pruritus (KALM-1)” at ClinicalTrials.gov
- Clinical trial number NCT03636269 for “CR845-CLIN3103: A Global Study to Evaluate the Safety and Efficacy of CR845 in Hemodialysis Patients With Moderate-to-Severe Pruritus (KALM-2)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Korsuva |
| Other names | CR845, FE-202845, D-Phe-D-Phe-D-Leu-D-Lys-[γ-(4-N-piperidinyl)amino carboxylic acid][1] |
| License data | US DailyMed: Difelikefalin |
| Routes of administration | Intravenous |
| Drug class | Kappa opioid receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2] |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV)[3] |
| Metabolism | Not metabolized[3] |
| Elimination half-life | 2 hours[3] |
| Excretion | Excreted as unchanged drug via bile and urine[3] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1024828-77-0 |
| PubChem CID | 24794466 |
| ChemSpider | 44208824 |
| UNII | NA1U919MRO |
| KEGG | D11111 |
| Chemical and physical data | |
| Formula | C36H53N7O6 |
| Molar mass | 679.863 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
//////////Difelikefalin acetate, FDA 2021, APPROVALS 2021, FORSUVA, ジフェリケファリン酢酸塩 , Difelikefalin, CR 845, MR 13A-9, MR-13A9, PEPTIDE
Lonapegsomatropin
FPTIPLSRLF DNAMLRAHRL HQLAFDTYQE FEEAYIPKEQ KYSFLQNPQT SLCFSESIPT
PSNREETQQK SNLELLRISL LLIQSWLEPV QFLRSVFANS LVYGASDSNV YDLLKDLEEG
IQTLMGRLED GSPRTGQIFK QTYSKFDTNS HNDDALLKNY GLLYCFRKDM DKVETFLRIV
QCRSVEGSCG F
(Disulfide bridge: 53-165, 182-189)

Lonapegsomatropin, ロナペグソマトロピン
FDA APPROVED, 25/8/21, Skytrofa, Treatment of growth hormone deficiency
To treat short stature due to inadequate secretion of endogenous growth hormone
1934255-39-6 CAS, UNII: OP35X9610Y
Molecular Formula, C1051-H1627-N269-O317-S9[-C2-H4-O]4n
ACP 001; ACP 011; lonapegsomatropin-tcgd; SKYTROFA; TransCon; TransCon growth hormone; TransCon hGH; TransCon PEG growth hormone; TransCon PEG hGH; TransCon PEG somatropin,
WHO 10598
PEPTIDE
Biologic License Application (BLA): 761177
Company: ACENDIS PHARMA ENDOCRINOLOGY DIV A/S
SKYTROFA is a human growth hormone indicated for the treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH) (1).
- OriginatorAscendis Pharma
- DeveloperAscendis Pharma; VISEN Pharmaceuticals
- ClassGrowth hormones; Hormonal replacements; Polyethylene glycols
- Mechanism of ActionSomatotropin receptor agonists
- Orphan Drug StatusYes – Somatotropin deficiency
- RegisteredSomatotropin deficiency
- 25 Aug 2021Registered for Somatotropin deficiency (In children, In infants) in USA (SC)
- 27 May 2021Ascendis Pharma expects European Commission decision on the Marketing Authorisation Application (MAA) for Somatotropin deficiency (In children, In infants, In neonates) in fourth quarter of 2021
- 27 May 2021Phase-III clinical trials in Somatotropin deficiency (In children, Treatment-naive) in Japan (SC)
Ascendis Pharma A/S Announces U.S. Food and Drug Administration Approval of SKYTROFA® (lonapegsomatropin-tcgd), the First Once-weekly Treatment for Pediatric Growth Hormone Deficiency
SKYTROFA, the first FDA approved treatment utilizing TransCon™ technology, is a long-acting prodrug of somatropin that releases the same somatropin used in daily therapies –
– Once weekly SKYTROFA demonstrated higher annualized height velocity (AHV) at week 52 compared to a daily growth hormone with similar safety and tolerability –
– Availability in the U.S. expected shortly supported by a full suite of patient support programs –
– Ascendis Pharma to host investor conference call today, Wednesday, August 25 at 4:30 p.m. E.T. –
COPENHAGEN, Denmark, Aug. 25, 2021 (GLOBE NEWSWIRE) — Ascendis Pharma A/S (Nasdaq: ASND), a biopharmaceutical company that utilizes its innovative TransCon technologies to potentially create new treatments that make a meaningful difference in patients’ lives, today announced that the U.S. Food and Drug Administration (FDA) has approved SKYTROFA (lonapegsomatropin-tcgd) for the treatment of pediatric patients one year and older who weigh at least 11.5 kg (25.4 lb) and have growth failure due to inadequate secretion of endogenous growth hormone (GH).
As a once-weekly injection, SKYTROFA is the first FDA approved product that delivers somatropin (growth hormone) by sustained release over one week.
“Today’s approval represents an important new choice for children with GHD and their families, who will now have a once-weekly treatment option. In the pivotal head-to-head clinical trial, once-weekly SKYTROFA demonstrated higher annualized height velocity at week 52 compared to somatropini,” said Paul Thornton, M.B. B.Ch., MRCPI, a clinical investigator and pediatric endocrinologist in Fort Worth, Texas. “This once-weekly treatment could reduce treatment burden and potentially replace the daily somatropin therapies, which have been the standard of care for over 30 years.”
Growth hormone deficiency is a serious orphan disease characterized by short stature and metabolic complications. In GHD, the pituitary gland does not produce sufficient growth hormone, which is important not only for height but also for a child’s overall endocrine health and development.
The approval includes the new SKYTROFA® Auto-Injector and cartridges which, after first removed from a refrigerator, allow families to store the medicine at room temperature for up to six months. With a weekly injection, patients switching from injections every day can experience up to 86 percent fewer injection days per year.
“SKYTROFA is the first product using our innovative TransCon technology platform that we have developed from design phase through non-clinical and clinical development, manufacturing and device optimization, and out to the patients. It reflects our commitment and dedication to addressing unmet medical needs by developing a pipeline of highly differentiated proprietary products across multiple therapeutic areas,” said Jan Mikkelsen, Ascendis Pharma’s President and Chief Executive Officer. “We are grateful to the patients, caregivers, clinicians, clinical investigators, and our employees, who have all contributed to bringing this new treatment option to children in the U.S. with GHD.”
In connection with the commercialization of SKYTROFA, the company is committed to offering a full suite of patient support programs, including educating families on proper injection procedures for SKYTROFA as the first once-weekly treatment for children with GHD.
“It is wonderful that patients and their families now have the option of a once-weekly growth hormone therapy,” said Mary Andrews, Chief Executive Officer and co-founder of the MAGIC Foundation, a global leader in endocrine health, advocacy, education, and support. “GHD is often overlooked and undertreated in our children and managing it can be challenging for families. We are excited about this news as treating GHD is important, and children have a short time to grow.”
The FDA approval of SKYTROFA was based on results from the phase 3 heiGHt Trial, a 52-week, global, randomized, open-label, active-controlled, parallel-group trial that compared once-weekly SKYTROFA to daily somatropin (Genotropin®) in 161 treatment-naïve children with GHDii. The primary endpoint was, AHV at 52 weeks for weekly SKYTROFA and daily hGH treatment groups. Other endpoints included adverse events, injection-site reactions, incidence of anti-hGH antibodies, annualized height velocity, change in height SDS, proportion of subjects with IGF-1 SDS (0.0 to +2.0), PK/PD in subjects < 3 years, and preference for and satisfaction with SKYTROFA.
At week 52, the treatment difference in AHV was 0.9 cm/year (11.2 cm/year for SKYTROFA compared with 10.3 cm/year for daily somatropin) with a 95 percent confidence interval [0.2, 1.5] cm/year. The primary objective of non-inferiority in AHV was met for SKYTROFA in this trial and further demonstrated a higher AHV at week 52 for lonapegsomatropin compared to daily somatropin, with similar safety, in treatment-naïve children with GHD.
No serious adverse events or discontinuations related to SKYTROFA were reported. Most common adverse reactions (≥ 5%) in pediatric patients include: infection, viral (15%), pyrexia (15%), cough (11%), nausea and vomiting (11%), hemorrhage (7%), diarrhea (6%), abdominal pain (6%), and arthralgia and arthritis (6%)ii. In addition, both arms of the study reported low incidences of transient, non-neutralizing anti-hGH binding antibodies and no cases of persistent antibodies.
Conference Call and Webcast Information
| Date | Wednesday, August 25, 2021 |
| Time | 4:30 p.m. ET/1:30 p.m. Pacific Time |
| Dial In (U.S.) | 844-290-3904 |
| Dial In (International) | 574-990-1036 |
| Access Code | 8553236 |
A live webcast of the conference call will be available on the Investors and News section of the Ascendis Pharma website at www.ascendispharma.com. A webcast replay will be available on this website shortly after conclusion of the event for 30 days.
The Following Information is Intended for the U.S. Audience Only
INDICATION
SKYTROFA® is a human growth hormone indicated for the treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH).
IMPORTANT SAFETY INFORMATION
- SKYTROFA is contraindicated in patients with:
- Acute critical illness after open heart surgery, abdominal surgery or multiple accidental trauma, or if you have acute respiratory failure due to the risk of increased mortality with use of pharmacologic doses of somatropin.
- Hypersensitivity to somatropin or any of the excipients in SKYTROFA. Systemic hypersensitivity reactions have been reported with post-marketing use of somatropin products.
- Closed epiphyses for growth promotion.
- Active malignancy.
- Active proliferative or severe non-proliferative diabetic retinopathy.
- Prader-Willi syndrome who are severely obese, have a history of upper airway obstruction or sleep apnea or have severe respiratory impairment due to the risk of sudden death.
- Increased mortality in patients with acute critical illness due to complications following open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure has been reported after treatment with pharmacologic doses of somatropin. Safety of continuing SKYTROFA treatment in patients receiving replacement doses for the approved indication who concurrently develop these illnesses has not been established.
- Serious systemic hypersensitivity reactions including anaphylactic reactions and angioedema have been reported with post-marketing use of somatropin products. Do not use SKYTROFA in patients with known hypersensitivity to somatropin or any of the excipients in SKYTROFA.
- There is an increased risk of malignancy progression with somatropin treatment in patients with active malignancy. Preexisting malignancy should be inactive with treatment completed prior to starting SKYTROFA. Discontinue SKYTROFA if there is evidence of recurrent activity.
- In childhood cancer survivors who were treated with radiation to the brain/head for their first neoplasm and who developed subsequent growth hormone deficiency (GHD) and were treated with somatropin, an increased risk of a second neoplasm has been reported. Intracranial tumors, in particular meningiomas, were the most common of these second neoplasms. Monitor all patients with a history of GHD secondary to an intracranial neoplasm routinely while on somatropin therapy for progression or recurrence of the tumor.
- Because children with certain rare genetic causes of short stature have an increased risk of developing malignancies, practitioners should thoroughly consider the risks and benefits of starting somatropin in these patients. If treatment with somatropin is initiated, carefully monitor these patients for development of neoplasms. Monitor patients on somatropin therapy carefully for increased growth, or potential malignant changes of preexisting nevi. Advise patients/caregivers to report marked changes in behavior, onset of headaches, vision disturbances and/or changes in skin pigmentation or changes in the appearance of preexisting nevi.
- Treatment with somatropin may decrease insulin sensitivity, particularly at higher doses. New onset type 2 diabetes mellitus has been reported in patients taking somatropin. Undiagnosed impaired glucose tolerance and overt diabetes mellitus may be unmasked. Monitor glucose levels periodically in all patients receiving SKYTROFA. Adjust the doses of antihyperglycemic drugs as needed when SKYTROFA is initiated in patients.
- Intracranial hypertension (IH) with papilledema, visual changes, headache, nausea, and/or vomiting has been reported in a small number of patients treated with somatropin. Symptoms usually occurred within the first 8 weeks after the initiation of somatropin and resolved rapidly after cessation or reduction in dose in all reported cases. Fundoscopic exam should be performed before initiation of therapy and periodically thereafter. If somatropin-induced IH is diagnosed, restart treatment with SKYTROFA at a lower dose after IH-associated signs and symptoms have resolved.
- Fluid retention during somatropin therapy may occur and is usually transient and dose dependent.
- Patients receiving somatropin therapy who have or are at risk for pituitary hormone deficiency(s) may be at risk for reduced serum cortisol levels and/or unmasking of central (secondary) hypoadrenalism. Patients treated with glucocorticoid replacement for previously diagnosed hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SKYTROFA therapy. Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in those with known hypoadrenalism.
- Undiagnosed or untreated hypothyroidism may prevent response to SKYTROFA. In patients with GHD, central (secondary) hypothyroidism may first become evident or worsen during SKYTROFA treatment. Perform thyroid function tests periodically and consider thyroid hormone replacement.
- Slipped capital femoral epiphysis may occur more frequently in patients undergoing rapid growth. Evaluate pediatric patients with the onset of a limp or complaints of persistent hip or knee pain.
- Somatropin increases the growth rate and progression of existing scoliosis can occur in patients who experience rapid growth. Somatropin has not been shown to increase the occurrence of scoliosis. Monitor patients with a history of scoliosis for disease progression.
- Cases of pancreatitis have been reported in pediatric patients receiving somatropin. The risk may be greater in pediatric patients compared with adults. Consider pancreatitis in patients who develop persistent severe abdominal pain.
- When SKYTROFA is administered subcutaneously at the same site over a long period of time, lipoatrophy may result. Rotate injection sites when administering SKYTROFA to reduce this risk.
- There have been reports of fatalities after initiating therapy with somatropin in pediatric patients with Prader-Willi syndrome who had one or more of the following risk factors: severe obesity, history of upper airway obstruction or sleep apnea, or unidentified respiratory infection. Male patients with one or more of these factors may be at greater risk than females. SKYTROFA is not indicated for the treatment of pediatric patients who have growth failure due to genetically confirmed Prader-Willi syndrome.
- Serum levels of inorganic phosphorus, alkaline phosphatase, and parathyroid hormone may increase after somatropin treatment.
- The most common adverse reactions (≥5%) in patients treated with SKYTROFA were: viral infection (15%), pyrexia (15%), cough (11%), nausea and vomiting (11%), hemorrhage (7%), diarrhea (6%), abdominal pain (6%), and arthralgia and arthritis (6%).
- SKYTROFA can interact with the following drugs:
- Glucocorticoids: SKYTROFA may reduce serum cortisol concentrations which may require an increase in the dose of glucocorticoids.
- Oral Estrogen: Oral estrogens may reduce the response to SKYTROFA. Higher doses of SKYTROFA may be required.
- Insulin and/or Other Hypoglycemic Agents: SKYTROFA may decrease insulin sensitivity. Patients with diabetes mellitus may require adjustment of insulin or hypoglycemic agents.
- Cytochrome P450-Metabolized Drugs: Somatropin may increase cytochrome P450 (CYP450)-mediated antipyrine clearance. Carefully monitor patients using drugs metabolized by CYP450 liver enzymes in combination with SKYTROFA.
You are encouraged to report side effects to FDA at (800) FDA-1088 or www.fda.gov/medwatch. You may also report side effects to Ascendis Pharma at 1-844-442-7236.
Please click here for full Prescribing Information for SKYTROFA.
About SKYTROFA® (lonapegsomatropin-tcgd)
SKYTROFA® is a once-weekly prodrug designed to deliver somatropin over a one-week period. The released somatropin has the same 191 amino acid sequence as daily somatropin.
SKYTROFA single-use, prefilled cartridges are available in nine dosage strengths, allowing for convenient dosing flexibility. They are designed for use only with the SKYTROFA® Auto-Injector and may be stored at room temperature for up to six months. The recommended dose of SKYTROFA for treatment-naïve patients and patients switching from daily somatropin is 0.24 mg/kg body weight, administered once weekly. The dose may be adjusted based on the child’s weight and insulin-like growth factor-1 (IGF-1) SDS.
SKYTROFA has been studied in over 300 children with GHD across the Phase 3 program which consists of the heiGHt Trial (for treatment-naïve patients), the fliGHt Trial (for treatment-experienced patients), and the enliGHten Trial (an ongoing long-term extension trial). Patients who completed the heiGHt Trial or the fliGHt Trial were able to continue into the enliGHten Trial and some have been on SKYTROFA for over four years.
SKYTROFA is being evaluated for pediatric GHD in Phase 3 trials in Japan and Greater China, including the People’s Republic of China, Hong Kong, Macau and Taiwan. Ascendis Pharma is also conducting the global Phase 3 foresiGHt Trial in adults with GHD. SKYTROFA has been granted orphan designation for GHD in both the U.S. and Europe.
About TransCon™ Technologies
TransCon refers to “transient conjugation.” The proprietary TransCon platform is an innovative technology to create new therapies that are designed to potentially optimize therapeutic effect, including efficacy, safety and dosing frequency. TransCon molecules have three components: an unmodified parent drug, an inert carrier that protects it, and a linker that temporarily binds the two. When bound, the carrier inactivates and shields the parent drug from clearance. When injected into the body, physiologic conditions (e.g., pH and temperature) initiate the release of the active, unmodified parent drug in a predictable manner. Because the parent drug is unmodified, its original mode of action is expected to be maintained. TransCon technology can be applied broadly to a protein, peptide or small molecule in multiple therapeutic areas, and can be used systemically or locally.
About Ascendis Pharma A/S
Ascendis Pharma is applying its innovative platform technology to build a leading, fully integrated biopharma company focused on making a meaningful difference in patients’ lives. Guided by its core values of patients, science and passion, the company utilizes its TransCon technologies to create new and potentially best-in-class therapies.
Ascendis Pharma currently has a pipeline of multiple independent endocrinology rare disease and oncology product candidates in development. The company continues to expand into additional therapeutic areas to address unmet patient needs.
Ascendis is headquartered in Copenhagen, Denmark, with additional facilities in Heidelberg and Berlin, Germany, in Palo Alto and Redwood City, California, and in Princeton, New Jersey.
Please visit www.ascendispharma.com (for global information) or www.ascendispharma.us (for U.S. information).

NEW DRUG APPROVALS
ONE TIME
$10.00
///////////Lonapegsomatropin, Skytrofa, APPROVALS 2021, FDA 2021, PEPTIDE, ロナペグソマトロピン , ACP 00, ACP 011, lonapegsomatropin-tcgd, TransCon, TransCon growth hormone, TransCon hGH, TransCon PEG growth hormone, TransCon PEG hGH, TransCon PEG somatropin, ORPHAN DRUG
Pepinemab, VX 15
(Heavy chain)
QVQLVQSGAE VKKPGSSVKV SCKASGYSFS DYYMHWVRQA PGQGLEWMGQ INPTTGGASY
NQKFKGKATI TVDKSTSTAY MELSSLRSED TAVYYCARYY YGRHFDVWGQ GTTVTVSSAS
TKGPSVFPLA PCSRSTSEST AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL
YSLSSVVTVP SSSLGTKTYT CNVDHKPSNT KVDKRVESKY GPPCPPCPAP EFLGGPSVFL
FPPKPKDTLM ISRTPEVTCV VVDVSQEDPE VQFNWYVDGV EVHNAKTKPR EEQFNSTYRV
VSVLTVLHQD WLNGKEYKCK VSNKGLPSSI EKTISKAKGQ PREPQVYTLP PSQEEMTKNQ
VSLTCLVKGF YPSDIAVEWE SNGQPENNYK TTPPVLDSDG SFFLYSRLTV DKSRWQEGNV
FSCSVMHEAL HNHYTQKSLS LSLGK
(Light chain)
DIVMTQSPDS LAVSLGERAT INCKASQSVD YDGDSYMNWY QQKPGQPPKL LIYAASNLES
GVPDRFSGSG SGTDFTLTIS SLQAEDVAVY YCQQSNEDPY TFGQGTKLEI KRTVAAPSVF
IFPPSDEQLK SGTASVVCLL NNFYPREAKV QWKVDNALQS GNSQESVTEQ DSKDSTYSLS
STLTLSKADY EKHKVYACEV THQGLSSPVT KSFNRGEC
(Disulfide bridge: H22-H96, H132-L218, H145-H201, H224-H’224, H227-H’227, H259-H319, H365-H423, H’22-H’96, H’132-L’218, H’145-H’201, H’259-H’319, H’365-H’423, L23-L92, L138-L198, L’23-L’92, L’138-L’198)
Pepinemab
VX15/2503
Antineoplastic, Anti-human semaphorin 4D antibody
Monoclonal antibody
Treatment of solid tumors, multiple sclerosis and Huntington’s disease
| Formula | C6442H9910N1702O2052S48 |
|---|---|
| MOL WGT | 145481.0022 |
- Moab VX15/2503
- Pepinemab
- UNII-BPZ4A29SYE
- VX-15
- VX15
- VX15/2503
| Product name | Pepinemab Biosimilar – Anti-SEMA4D mAb – Research Grade |
|---|---|
| Source | CAS 2097151-87-4 |
| Species | Chimeric,Humanized |
| Expression system | Mammalian cells |
- OriginatorVaccinex
- DeveloperBristol-Myers Squibb; Children’s Oncology Group; Emory University; Merck KGaA; National Cancer Institute (USA); Teva Pharmaceutical Industries; UCLAs Jonsson Comprehensive Cancer Center; Vaccinex
- ClassAntibodies; Antidementias; Antineoplastics; Immunotherapies; Monoclonal antibodies
- Mechanism of ActionCD100 antigen inhibitors
- Orphan Drug StatusYes – Huntington’s disease
- New Molecular EntityYes
- Phase IIHuntington’s disease
- Phase I/IIAlzheimer’s disease; Non-small cell lung cancer; Osteosarcoma; Solid tumours; Squamous cell cancer
- Phase IColorectal cancer; Malignant melanoma; Pancreatic cancer
- No development reportedMultiple sclerosis
- 22 May 2021Pepinemab is still in phase I trials for Colorectal cancer and Pancreatic cancer in USA (NCT03373188)
- 17 May 2021Phase-I/II clinical trials in Squamous cell cancer (Combination therapy, Late-stage disease, Metastatic disease, Recurrent, Second-line therapy or greater) in USA (IV) (NCT04815720)
- 17 May 2021Vaccinex plans a phase I/II trial for Alzheimer’s disease (In volunteers), in H2 2021
Semaphorin 4D (SEMA4D) plays a role in multiple cellular processes that contribute to the pathophysiology of neuroinflammatory/neurodegenerative diseases. SEMA4D is, therefore, a uniquely promising target for therapeutic development.
Pepinemab is a novel monoclonal antibody that blocks the activity of SEMA4D, and preclinical testing has demonstrated the beneficial effects of anti-SEMA4D treatment in a variety of neurodegenerative disease models. Vaccinex is committed to the development of this potentially important antibody that has the potential to help people with different neurodegenerative disorders that share common mechanisms of pathology.
Note: Pepinemab (VX15/2503) is an investigational drug currently in clinical studies. It has not been demonstrated to be safe and effective for any disease indication. There is no guarantee that pepinemab (VX15/2503) will be approved for the treatment of any disease by the U.S. Food and Drug Administration or by any other health authority worldwide.
////////////////////Pepinemab, VX15/2503, vx 15, Antineoplastic, Anti-human semaphorin 4D antibody, Monoclonal antibody, solid tumors, multiple sclerosis, Huntington’s disease, PEPTIDES
NEW DRUG APPROVALS
ONE TIME
$10.00
Anifrolumab
(Heavy chain)
EVQLVQSGAE VKKPGESLKI SCKGSGYIFT NYWIAWVRQM PGKGLESMGI IYPGDSDIRY
SPSFQGQVTI SADKSITTAY LQWSSLKASD TAMYYCARHD IEGFDYWGRG TLVTVSSAST
KGPSVFPLAP SSKSTSGGTA ALGCLVKDYF PEPVTVSWNS GALTSGVHTF PAVLQSSGLY
SLSSVVTVPS SSLGTQTYIC NVNHKPSNTK VDKRVEPKSC DKTHTCPPCP APEFEGGPSV
FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY
RVVSVLTVLH QDWLNGKEYK CKVSNKALPA SIEKTISKAK GQPREPQVYT LPPSREEMTK
NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG
NVFSCSVMHE ALHNHYTQKS LSLSPGK
(Lihgt chain)
EIVLTQSPGT LSLSPGERAT LSCRASQSVS SSFFAWYQQK PGQAPRLLIY GASSRATGIP
DRLSGSGSGT DFTLTITRLE PEDFAVYYCQ QYDSSAITFG QGTRLEIKRT VAAPSVFIFP
PSDEQLKSGT ASVVCLLNNF YPREAKVQWK VDNALQSGNS QESVTEQDSK DSTYSLSSTL
TLSKADYEKH KVYACEVTHQ GLSSPVTKSF NRGEC
(Disulfide bridge: H22-96, H144-H200, H220-L215, H226-H’226, H229-H’229, H261-H321, H367-H425, H’22-H’96, H’144-H’200, H’220-L’215, H’261-H’321, H’367-H’425, L23-L89, L135-L195, L’23-L’89, L’135-L’195)
Anifrolumab
| アニフロルマブ (遺伝子組換え) |
FDA APPROVED 2021/7/30, Saphnelo
- MEDI 546
| Formula | C6444H9964N1712O2018S44 |
|---|---|
| Cas | 1326232-46-5 |
| Mol weight | 145117.1846 |
| Immunomodulator, Anti-IFN-type 1 receptor antibody | |
| Disease | Systemic lupus erythematosus |
|---|
Monoclonal antibody
Treatment of systemic lupus erythematosus (SLE)
- OriginatorMedarex
- DeveloperAstraZeneca; Medarex; MedImmune
- ClassAntirheumatics; Monoclonal antibodies; Skin disorder therapies
- Mechanism of ActionInterferon alpha beta receptor antagonists
- RegisteredSystemic lupus erythematosus
- Phase IILupus nephritis
- DiscontinuedRheumatoid arthritis; Scleroderma
- 02 Jul 2021Phase-III clinical trials in Systemic lupus erythematosus in USA (SC) (NCT04877691)
- 25 Jun 2021AstraZeneca plans a phase III trial in Systemic lupus erythematosus (Adjunctive treatment) in the China, Hong Kong, South Korea, Philipines, Taiwan and Thailand (IV, Infusion), in July 2021 (NCT04931563)
- 02 Jun 2021Pharmacokinetic, efficacy and adverse events data from a phase II TULIP-LN1 trial in Lupus nephritis presented at the 22nd Annual Congress of the European League Against Rheumatism (EULAR-2021)
Anifrolumab, sold under the brand name Saphnelo, is a monoclonal antibody used for the treatment of systemic lupus erythematosus (SLE).[1][2] It binds to the type I interferon receptor, blocking the activity of type I interferons such as interferon-α and interferon-β.[medical citation needed]
Anifrolumab was approved for medical use in the United States in August 2021.[1][3][4][5]
Anifrolumab is a monoclonal antibody that inhibits type 1 interferon receptors, indicated in the treatment of moderate to severe systemic lupus erythematosus.
Anifrolumab, or MEDI-546, is a type 1 interferon receptor (IFNAR) inhibiting IgG1κ monoclonal antibody indicated in the treatment of adults with moderate to severe systemic lupus erythematosus.7,11 The standard therapy for systemic lupus erythematosus consists of antimalarials like hydroxychloroquine, glucocorticoids like dexamethasone, and disease modifying antirheumatic drugs like methotrexate.8,11
Three monoclonal antibodies (anifrolumab, rontalizumab, and sifalimumab) that target the type 1 interferon pathway entered clinical trials as potential treatments for systemic lupus erythematosus, but so far only anifrolumab has been approved.3
The design of early clinical trials of anti-interferon treatments such as anifrolumab, rontalizumab, and sifalimumab have come under criticism.3 The design of the clinical trials use different definitions of autoantibody positivity, making comparison between trials difficult; all trials involve large portions of patients also using corticosteroids, which may alter patient responses in the experimental and placebo groups; and patient populations were largely homogenous, which may have increased the odds of success of the trial.3
Anifrolumab has also been investigated for the treatment of Scleroderma.1
Anifrolumab was granted FDA approval on 30 July 2021.11
Adverse effects
The most common adverse effect was shingles, which occurred in 5% of patients in the low-dose group, to 10% in the high-dose group, and to 2% in the placebo group. Overall adverse effect rates were comparable in all groups.[6]
History
The drug was developed by MedImmune, a unit of AstraZeneca, which chose to move anifrolumab instead of sifalimumab into phase III trials for lupus in 2015.[7][8][9]
Clinical trial results
Anifrolumab failed to meet its endpoint of significant reduction in disease as assessed by the SLE Responder Index 4 instrument in the TULIP 1 phase III trial.[10] This multi-center, double-blind, placebo-controlled study followed adults with moderate to severe SLE over the course of one year. Preliminary results were announced on 31 August 2018.
Names
Anifrolumab is the international nonproprietary name (INN).[11]
References
- ^ Jump up to:a b chttps://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761123s000lbl.pdf
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Anifrolumab, American Medical Association.
- ^https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/761123Orig1s000ltr.pdf
- ^ https://www.astrazeneca.com/media-centre/press-releases/2021/saphnelo-approved-in-the-us-for-sle.html
- ^ “Saphnelo (anifrolumab) Approved in the US for Moderate to Severe Systemic Lupus Erythematosus” (Press release). AstraZeneca. 2 August 2021. Retrieved 2 August 2021 – via Business Wire.
- ^ Spreitzer H (29 August 2016). “Neue Wirkstoffe – Anifrolumab”. Österreichische Apothekerzeitung (in German) (18/2016).
- ^ “Press release: New Hope for Lupus Patients”. MedImmune. 11 August 2015. Archived from the original on 31 July 2017.
- ^ “Anifrolumab”. NHS Specialist Pharmacy Service. Retrieved 31 July 2017.
- ^ “Anifrolumab”. AdisInsight. Retrieved 31 July 2017.
- ^ “Update on TULIP 1 Phase III trial for anifrolumab in systemic lupus erythematosus”. http://www.astrazeneca.com. Retrieved 2019-02-05.
- ^ World Health Organization (2014). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 71”. WHO Drug Information. 28 (1). hdl:10665/331151.
Further reading
- Anderson E, Furie R (April 2020). “Anifrolumab in systemic lupus erythematosus: current knowledge and future considerations”. Immunotherapy. 12 (5): 275–86. doi:10.2217/imt-2020-0017. PMID 32237942.
External links
- “Anifrolumab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01438489 for “A Study of the Efficacy and Safety of MEDI-546 in Systemic Lupus Erythematosus” at ClinicalTrials.gov
- Clinical trial number NCT02446912 for “Efficacy and Safety of Two Doses of Anifrolumab Compared to Placebo in Adult Subjects With Active Systemic Lupus Erythematosus” at ClinicalTrials.gov
- Clinical trial number NCT02446899 for “Efficacy and Safety of Anifrolumab Compared to Placebo in Adult Subjects With Active Systemic Lupus Erythematosus” at ClinicalTrials.gov
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Interferon α/β receptor |
| Clinical data | |
| Trade names | Saphnelo |
| Other names | MEDI-546, anifrolumab-fnia |
| License data | US DailyMed: Anifrolumab |
| Routes of administration | Intravenous |
| Drug class | type I interferon receptor antagonist (IFN) |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1326232-46-5 |
| DrugBank | DB11976 |
| ChemSpider | none |
| UNII | 38RL9AE51Q |
| KEGG | D11082 |
| Chemical and physical data | |
| Formula | C6444H9964N1712O2018S44 |
| Molar mass | 145119.20 g·mol−1 |
- Goldberg A, Geppert T, Schiopu E, Frech T, Hsu V, Simms RW, Peng SL, Yao Y, Elgeioushi N, Chang L, Wang B, Yoo S: Dose-escalation of human anti-interferon-alpha receptor monoclonal antibody MEDI-546 in subjects with systemic sclerosis: a phase 1, multicenter, open label study. Arthritis Res Ther. 2014 Feb 24;16(1):R57. doi: 10.1186/ar4492. [Article]
- Peng L, Oganesyan V, Wu H, Dall’Acqua WF, Damschroder MM: Molecular basis for antagonistic activity of anifrolumab, an anti-interferon-alpha receptor 1 antibody. MAbs. 2015;7(2):428-39. doi: 10.1080/19420862.2015.1007810. [Article]
- Massarotti EM, Allore HG, Costenbader K: Editorial: Interferon-Targeted Therapy for Systemic Lupus Erythematosus: Are the Trials on Target? Arthritis Rheumatol. 2017 Feb;69(2):245-248. doi: 10.1002/art.39985. [Article]
- Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, Illei GG, Drappa J, Wang L, Yoo S: Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017 Feb;69(2):376-386. doi: 10.1002/art.39962. [Article]
- Tummala R, Rouse T, Berglind A, Santiago L: Safety, tolerability and pharmacokinetics of subcutaneous and intravenous anifrolumab in healthy volunteers. Lupus Sci Med. 2018 Mar 23;5(1):e000252. doi: 10.1136/lupus-2017-000252. eCollection 2018. [Article]
- Riggs JM, Hanna RN, Rajan B, Zerrouki K, Karnell JL, Sagar D, Vainshtein I, Farmer E, Rosenthal K, Morehouse C, de Los Reyes M, Schifferli K, Liang M, Sanjuan MA, Sims GP, Kolbeck R: Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci Med. 2018 Apr 5;5(1):e000261. doi: 10.1136/lupus-2018-000261. eCollection 2018. [Article]
- Bui A, Sanghavi D: Anifrolumab . [Article]
- Trindade VC, Carneiro-Sampaio M, Bonfa E, Silva CA: An Update on the Management of Childhood-Onset Systemic Lupus Erythematosus. Paediatr Drugs. 2021 Jul;23(4):331-347. doi: 10.1007/s40272-021-00457-z. Epub 2021 Jul 10. [Article]
- Ryman JT, Meibohm B: Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacometrics Syst Pharmacol. 2017 Sep;6(9):576-588. doi: 10.1002/psp4.12224. Epub 2017 Jul 29. [Article]
- Koh JWH, Ng CH, Tay SH: Biologics targeting type I interferons in SLE: A meta-analysis and systematic review of randomised controlled trials. Lupus. 2020 Dec;29(14):1845-1853. doi: 10.1177/0961203320959702. Epub 2020 Sep 22. [Article]
- FDA Approved Drug Products: Saphnelo (Anifrolumab-fnia) Intravenous Injection [Link]
 //////////Anifrolumab, Saphnelo, FDA 2021, APPROVALS 2021, peptide, Monoclonal antibody, アニフロルマブ (遺伝子組換え) , MEDI 546, AstraZeneca, Medarex, MedImmune
//////////Anifrolumab, Saphnelo, FDA 2021, APPROVALS 2021, peptide, Monoclonal antibody, アニフロルマブ (遺伝子組換え) , MEDI 546, AstraZeneca, Medarex, MedImmune
NEW DRUG APPROVALS
one time
$10.00
Nangibotide


Nangibotide
LQEEDAGEYGCM-amide
CAS 2014384-91-7
- Molecular FormulaC54H82N14O22S2
- Average mass1343.439 Da
- 2014384‐91‐7
- L-Leucyl-L-glutaminyl-L-α-glutamyl-L-α-glutamyl-L-α-aspartyl-L-alanylglycyl-L-α-glutamyl-L-tyrosylglycyl-L-cysteinyl-L-methioninamide
- LR 12 peptide
- LQEEDAGEYG CM
L-Leucyl-L-glutaminyl-L-glutaminyl-L-α-glutamyl-L-α-aspartyl-L-alanylglycyl-L-α-glutamyl-L-tyrosylglycyl-L-cysteinyl-L-methionine
L-Methionine, L-leucyl-L-glutaminyl-L-glutaminyl-L-α-glutamyl-L-α-aspartyl-L-alanylglycyl-L-α-glutamyl-L-tyrosylglycyl-L-cysteinyl-нангиботидمانغيبوتيد南吉博肽
| Sequence (one letter code) | LQEEDAGEYGCM-amide |
|---|---|
| Sequence (three letter code) | H-Leu-Gln-Glu-Glu-Asp-Ala-Gly-Glu-Tyr-Gly-Cys-Met-NH2 |
- OriginatorInotrem
- ClassAnti-infectives; Anti-inflammatories; Anti-ischaemics; Antivirals; Peptides
- Mechanism of ActionTREML1 protein inhibitors
- Phase II/IIICOVID 2019 infections
- Phase IISeptic shock
- Phase IMyocardial infarction
- 12 Jul 2021Inotrem has patents pending for nangibotide use in severe forms of COVID-19
- 12 Jul 2021Inotrem receives funding from French government by Bpifrance for nangibotide development in COVID-2019 infections
- 12 Jul 2021Inotrem receives authorization from both the French and Belgian authorities to proceed with clinical development of nangibotide up to registration in COVID-2019 infections
Nangibotide, also referred as LR12, is an antagonist of triggering receptor expressed on myeloid cells (TREM)-1, and was derived from residues 94 to 105 of TREM-like transcript-1 (TLT-1).
TREM-1 plays a crucial role in the onset of sepsis by amplifying the host immune response. TLT-1– and TLT-1–derived peptides therefore exhibit anti-inflammatory properties by dampening TREM-1 signalling. LR12 blocks TREM-1 by binding to the TREM-1 ligand and provides protective effects during sepsis such as inhibiting hyper-responsiveness, organ damage, and death, without causing deleterious effects. The protective effects of modulating TREM-1 signalling are also evident in other models of inflammation such as: pancreatitis; haemorrhagic shock; inflammatory bowel diseases and inflammatory arthritis
Inotrem is developing the peptide nangibotide, a triggering receptor expressed on myeloid cells 1 inhibitor, for treating sepsis and septic shock. In July 2021, this drug was reported to be in phase 3 clinical development.
Nangibotide is an inhibitor of TREM-1, a receptor found on certain white blood cells. Activation of TREM-1 stimulates inflammation. Nangibotide is therefore being investigated as a treatment for the overwhelming inflammation typically seen in severe sepsis.
Mode of action
TREM-1 is a receptor found on neutrophils, macrophages and monocytes, key elements of the immune system. Activation of TREM-1 results in expression of NF-κB, which promotes systemic inflammation. Nangibotide inhibits TREM-1, thereby preventing the inflammatory activation. Absence of TREM-1 results in vastly reduced inflammation without impairing the ability to fight infection.[2]
Animal models
LR17, a mouse equivalent of nangibotide, improves survival in mouse models of severe sepsis.[3] In a pig model of sepsis, LR12 – another animal equivalent of nangibotide – resulted in significantly improved haemodynamics and less organ failure.[4] In monkeys, LR12 also reduced the inflammatory and hypotensive effects of sepsis.[5]
Human studies
Nangibotide has demonstrated safety in Phase 1 (healthy volunteers)[6] and Phase 2 (sick patients with septic shock)[7] studies. The ASTONISH trial will examine clinical efficacy in 450 patients with septic shock.[8]
Inotrem Receives Approval to Expand Nangibotide Clinical Trial in Critically Ill COVID-19 Patients and Receives Additional Public Funding of €45 Million
- Inotrem’s phase 2/3 clinical trial “ESSENTIAL” will enroll up to 730 patients in Europe to demonstrate the safety and efficacy of nangibotide to treat critically ill COVID-19 patients with respiratory failure.
- Recent preclinical studies have strengthened the body of evidence for targeting the TREM-1 pathway which is activated in a subset of patients suffering from severe COVID-19.
July 12, 2021 03:00 AM Eastern Daylight Time
PARIS–(BUSINESS WIRE)–Inotrem S.A., a biotechnology company specializing in the development of immunotherapies targeting the TREM-1 pathway, announces that it has obtained authorization to pursue the clinical development of nangibotide up to registration in COVID-19 patients from both the French and Belgian competent authorities.
As part of this program, Inotrem receives additional 45 million euros in public funding under the “Capacity Building” Call for Expression of Interest, operated on behalf of the French government by Bpifrance, the French national investment bank, as part of the Programme d’investissements d’avenir (PIA) and the France Recovery Plan, bringing French state support for the project to a total of 52,5 million euros. This public funding will support Inotrem’s clinical program including the phase 2/3 study “ESSENTIAL” which aims to demonstrate the efficacy and safety of nangibotide in treating patients in respiratory distress with severe forms of COVID-19.
The primary endpoint is evaluation of the impact of nangibotide on the progression of disease in patients receiving ventilatory support due to COVID-19 as well as on the severity of the respiratory failure, duration of mechanical ventilation, length of stay in intensive care and mortality. In “ESSENTIAL”, a Phase 2/3 clinical program, up to 730 patients will be enrolled initially in France and Belgium and, possibly in other European countries. Pre-defined interim analyses will be conducted by an independent Data Monitoring Board to test futility and to allow for the study design to be adapted as necessary. “ESSNTIAL” is the continuation of a 60 patients phase 2a evaluating the safety and efficacy of nangibotide in patients suffering from severe COVID-19. In July 2020, the CoviTREM-1 consortium, which includes the Nancy and Limoges university hospitals and Inotrem, obtained public funding of 7,5 million euros under the “PSPC-COVID” call for projects, operated on behalf of the French government by Bpifrance
New pre-clinical studies with nangibotide have demonstrated that the administration of nangibotide in murine models infected with SARS-CoV-2 was associated with a decrease in inflammatory mediators and an improvement of clinical signs, in particular respiratory function, and survival. Inotrem also confirmed in 3 different and independent cohorts that sTREM-1, a marker of the activation of the TREM-1 biological pathway, is associated with both severity and mortality in critically ill COVID-19 patients.
Leveraging the results of these preclinical studies and the implications for the role of the TREM-1 pathway in COVID-19, Inotrem has filed additional patents to cover nangibotide use in severe forms of COVID-19 as well as the use of sTREM-1 as a biomarker and companion diagnostic. This significantly strengthens Inotrem’s already broad patent estate.
Jean-Jacques Garaud, Executive Vice-President, Head of Scientific and Medical Affairs and Inotrem’s co-founder said :“We are eager to pursue the development of nangibotide in these severe forms of COVID-19. Nangibotide is a TREM-1 inhibitor which has already demonstrated a trend towards efficacy in septic shock patients and has the potential to modulate the dysregulated immune response in critically ill COVID-19 patients. With this large clinical study, we can demonstrate efficacy for nangibotide in a further indication with the goals of reducing the duration of hospitalization and mortality.”
Sven Zimmerman, CEO of Inotrem, also declared: “The size of the financial support awarded to us as part of the French government’s initiative against COVID-19 is a testimony to the relevance of targeting the TREM-1 pathway with nangibotide in these severely ill patients. We are delighted by the confidence placed in our technology and our team. Everyone at Inotrem is fully committed to deliver on this ambitious program alongside nangibotide’s ongoing Phase 2b trial in septic shock patients.”
About Inotrem
Inotrem S.A. is a biotechnology company specialized in immunotherapy for acute and chronic inflammatory syndromes. The company has developed a new concept of immunomodulation that targets the TREM-1 pathway to control unbalanced inflammatory responses. Through its proprietary technology platform, Inotrem has developed the first-in-class TREM-1 inhibitor, LR12 (nangibotide), with potential applications in a number of therapeutic indications such as septic shock and myocardial infarction. In parallel, Inotrem has also launched another program to develop a new therapeutic modality targeting chronic inflammatory diseases. The company was founded in 2013 by Dr. Jean-Jacques Garaud, a former head of research and early development at the Roche Group, Prof. Sébastien Gibot and Dr. Marc Derive. Inotrem is supported by leading European and North American investors.
About TREM-1 pathway
TREM-1 pathway is an amplification loop of the immune response that triggers an exuberant and hyperactivated immune state which is known to play a crucial role in the pathophysiology of septic shock and acute myocardial infarction.
About Nangibotide
Nangibotide is the formulation of the active ingredient LR12, which is a 12 amino-acid peptide prepared by chemical synthesis. LR12 is a specific TREM-1 inhibitor, acting as a decoy receptor and interfering in the binding of TREM-1 and its ligand. In preclinical septic shock models, nangibotide was able to restore appropriate inflammatory response, vascular function, and improved animals’ survival post septic shock.
About ESSENTIAL study:
The Efficacy and Safety Study Exploring Nangibotide Treatment in COVID-19 pAtients with ventiLatory support, is a randomized, double-blind, placebo-controlled confirmatory study with adaptive features that will be performed in Europe. This is a pivotal study and it is expected that based on its results, nangibotide could be registered in this indication. The first part of the study (i.e.: 60 patients) has been already finalized and assessed by an independent data monitoring committee with excellent safety results. The study will recruit up to 730 patients in up to 40 sites. Several interim and futility analyses are foreseen as part of the adaptive design of the study.
About Bpifrance
Bpifrance is the French national investment bank: it finances businesses – at every stage of their development – through loans, guarantees, equity investments and export insurances. Bpifrance also provides extra-financial services (training, consultancy.). to help entrepreneurs meet their challenges (innovation, export…).
PATENT
WO-2021144388
Process for preparing nangibotide by solid phase synthesis, useful for treating acute inflammatory disorders such as septic shock. Also claims novel peptide fragments, useful in the synthesis of nangibotide.
Example 1
Preparation of nangibotide by full SPPS (Reference)
Step 1 : Loading of the first amino acid onto the Rink Amide Resin
2 g of MBHA resin (1.0-1.3 mmol/g) was swelled using 16 mL of DMF for 30 min. 2 eq Fmoc-Met-OH (2.4 mmol, 2.67 g), 2 eq DIC (2.4 mmol, 1.136 mL) and 2 eq OxymaPure (2.4 mmol, 1.023 g) were dissolved in 8 mL of DMF at 0.3 M cone, and added to the resin after 5 min. All the coupling steps were conducted in this way unless described differently. The loading step was carried out for 1.5 hour. After the loading, the resin was filtered and washed 3 times with 12 mL of DMF. The Fmoc deprotection step was carried out by addition of 12 mL of 20% piperidine solution in DMF for two 10 min cycles. This step was performed analogously for all the amino acid residues. The loading, calculated by UV absorption for the peptidyl resin, was 0.8 mmol/g.
Step 2: peptide elongation
For the coupling of all the amino acids involved in the synthesis of nangibotide, 3 eq of each amino acid were activated by 3 eq of DIC and OxymaPure dissolved in DMF at 0.3 M cone. At the end of the peptide elongation, a final Fmoc deprotection, as already described, was performed before moving to the cleavage step.
Step 3: Cleavage and precipitation of crude nangibotide
The cleavage of nangibotide off the resin was carried out using a solution of 16 mL of TFA/DODT/TIPS/water in 90/4/3/3 ratio cooled at 0°C. The peptidyl resin was added portionwise in 30 min keeping the internal temperature under 25°C. The cleavage was run for 3.5 hours, then the resin was filtered and washed by 10 mL of TFA for 10 min.
DIPE was used for the precipitation of the peptide, adding 12 volumes (300 mL) dropwise to the peptide TFA solution, keeping the temperature under 20°C. The suspension with nangibotide was filtered on a gooch funnel, the peptide washed again with 100 mL of DIPE and then dried under vacuum overnight. Molar yield 40%. Purity 61%.
Example 2
Preparation of nangibotide by three-fragment condensation
In the approach using three fragments, only the cysteine residue was coupled to the methionine on rink amide resin to prepare fragment 11-12, whereas protected peptide fragments 1-7 and 8-10 were synthesized using 2-CTC resin.
Step 1: Synthesis of fragment 11-12
2 g of MBHA resin (1.0-1.3 mmol/g) was swelled using 16 mL of DMF for 30 min 2 eq of Fmoc-Met-OH (2.4 mmol, 2.67 g), 2 eq DIC (2.4 mmol, 1.136 mL) and 2 eq OxymaPure (2.4 mmol, 1.023 g) were dissolved in 8 mL of DMF at 0.3 M cone, and added to the resin. The loading step was carried out for 1 and half hour. After the loading, the resin was filtered and washed 3 times with 12 mL of DMF. The Fmoc deprotection step was carried out by
addition of 12 mL of a 20% piperidine solution in DMF for two 10 min cycles. Same procedure was repeated for the coupling of Fmoc-Cys(Trt)-OH to obtain resin-attached Fmoc-deprotected fragment 11-12. The loading, calculated by UV absorption for the peptidyl resin relative to the first amino acid inserted, was 0.8 mmol/g.
Step 2: Synthesis of fragments 1-7 and 8-10
For the synthesis of both fragments the loading of 2-chloro trityl chloride resin was performed on 5 g (1.6 mmol/g) using 0.8 eq Fmoc-Gly-OH (6.40 mmol, 1.90 g) dissolved in 30 mL of DCM and addition of 3 eq DIPEA (24 mmol, 4.19 mL). The loading step was carried out for 1 hour, then the resin was washed by 30 mL DCM for three times and eventual Cl-groups were capped by two different capping solutions: first by 30 mL of methanol/DIPEA/DCM (1:2:7) and then by 30 mL AC2O/DIPEA/DCM in the same ratio. After the treatment with these solutions for 15 min and subsequent washing with DCM, the resin was washed three times with DMF, before deprotection of Fmoc and evaluation of the resin loading. Generally, this protocol gave a resin loaded with 1.1 mmol/g Fmoc-Gly-OH. The Fmoc deprotection and coupling step protocols were equally performed with all the amino acids in the respective sequences: Fmoc-Tyr(tBu)-OH and Fmoc-Glu(tBu)-OH for fragment 8-10, and Fmoc-Ala-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Glu(OtBu)-OH twice, Fmoc-Gln(Trt)-OH and Fmoc-Leu-OH for fragment 1-7.
For each coupling, 3 eq amino acid were activated by 3 eq DIC and 3 eq OxymaPure dissolved in DMF at 0.3 M cone.
Fragment Fmoc-Glu(tBu)-Tyr(tBu)-Gly-OH (8-10) was obtained by cleavage off the resin using 6 volumes (30 mL) of a TFA 1.5 % solution in DCM, 5 times for 2 min. The final TFA solution was neutralized by 1.2 eq pyridine (15.89 mmol, 1.3 mL) diluted in 30 mL methanol. The final solution was concentrated to 50 mL under vacuum then washed by water and brine. The organic layer was dried by anhydrous sodium sulphate, filtered and further concentrated before crystallization of the tripeptide with 5 volumes of petroleum ether at 0°C. The peptide was filtered, washed by petroleum ether and dried overnight in a vacuum oven at 37°C. Molar yield 65%. Purity 90%.
Fragment Fmoc-Leu-Gln(Trt)-Glu(OtBu)-Glu(OtBu)-Asp(OtBu)-Ala-Gly-OH (1-7) was obtained by cleavage off the resin using 6 volumes (30 mL) of a TFA 1.5 % solution in DCM, 5 times for 2 min. The final TFA solution was neutralized by 1.2 eq pyridine (15.89 mmol, 1.3 mL) diluted in 30 mL methanol. The DCM was evaporated and replaced by methanol, adding and evaporating 30 mL methanol a couple of times till one third of the volume. The peptide fragment was precipitated by adding 5 volumes (150 mL) water to the methanol solution at 0°C and filtered after stirring for 30 min. The full protected heptapeptide was washed by water and dried overnight in a vacuum oven at 37°C. Molar yield 85%. Purity 89%.
Step 3: Synthesis of fragment 8-12 (Fragment condensation 1)
The fragment condensation between Fmoc-Glu(tBu)-Tyr(tBu)-Gly-OH (8-10) and H-Cys(Trt)-Met-MBHA resin (11-12) was carried out activating 2 eq (1.6 mmol, 1.12 g) of fragment 8-10 dissolved in 6 mL of DMF at 40°C by using 2 eq OxymaPure (1.6 mmol, 0.22 g) and 2 eq DIC (1.6 mmol, 0.25 mL) for 10 min. The activated ester of tripeptide 8-10 was added to the resin-attached fragment 11-12 and stirred for 3 hours at 40°C. After filtration, the resin was washed three times by 15 mL DMF and then capped by 12 mL of AC2O 10% in DMF for 15 min. The resin was washed three timed by 12 mL DMF before deprotection of Fmoc to finally obtain resin-attached Fmoc-protected fragment 8-12. Molar yield 91%. Purity 89%.
Step 4: Synthesis of nanaibotide (Fragment condensation 2)
The fragment condensation between fragment 1-7 and H-Glu(OtBu)-Tyr(tBu)-Gly-Cys(Trt)-Met-MBHA resin (8-12) was carried out activating 1.5 eq (2.25 mmol, 2.64 g) of fragment 1-7 dissolved in 25 mL DMF at 40°C by using 2 eq OxymaPure (2.25 mmol, 0.32 g) and 2 eq DIC (2.25 mmol, 0.35 mL) for 15 min. The activated ester of fragment 1-7 was added to the resin-attached fragment 8-12 and stirred for 3.5 hours at 40°C. After filtration, the resin was washed three times by 12 mL DMF before deprotection of Fmoc with the standard procedure described above. After Fmoc deprotection, the resin was washed again by DMF and DCM and then dried at vacuum pump.
Step 5: Cleavage and precipitation of crude nanaibotide
The cleavage of nangibotide off the resin was carried out using a solution of 16 mL of TFA/DODT/TIPS/water in 90/4/3/3 ratio cooled at 0°C. The peptidyl resin was added portionwise in 30 min keeping the internal temperature under 25°C. The cleavage was run for 3.5 hours, then the resin filtered and washed by 10 mL of TFA for 10 min.
DIPE was used to precipitate the peptide, adding 12 volumes (300 mL) dropwise to the peptide TFA solution, keeping the temperature under 20°C. The suspension with nangibotide was filtered on a gooch funnel, the peptide washed again with 100 mL of DIPE and then dried at vacuum pump overnight. Molar yield 61%. Purity 73%.
Example 3
Preparation of nangibotide by two-fragment condensation
In the approach using two fragments, the SPPS elongation onto MBHA resin, as described in Example 2, step 1, was continued until Glu8 was attached to provide fragment 8-12, then fragment 1-7, synthesized on 2-CTC resin as described in example 2, step 2, was coupled to the resin-attached fragment 8-12 as described in example 2, step 4.
Step 1: Synthesis of fragment 8-12
2 g of MBHA resin (1.0-1.3 mmol/g) was swelled using 16 mL of DMF for 30 min 2 eq of Fmoc-Met-OH (2.4 mmol, 2.67 g), 2 eq DIC (2.4 mmol, 1.136 mL) and 2 eq OxymaPure (2.4 mmol, 1.023 g) were dissolved in 8 mL of DMF at 0.3 M cone, and added to the resin. The loading step was carried out for 1 and half hour. After the loading, the resin was filtered and washed 3 times with 12 mL of DMF. The Fmoc deprotection step was carried out by addition of 12 mL of a 20% piperidine solution in DMF for two 10 min cycles. Same procedure was repeated for the coupling of Fmoc-Cys(Trt)-OH; Fmoc-Glu(OtBu)-OH; Fmoc-Tyr(tBu)-OH; Fmoc-Gly-OH to obtain fragment 8-12. The loading, calculated by UV absorption for the peptidyl resin relative to the first amino acid inserted, was 0.8 mmol/g. Molar yield 88%. Purity 83%.
Step 2: Synthesis of nanaibotide (Fragment condensation 2)
The final fragment condensation was performed as described in example 2, step 4.
Step 3: Cleavage and precipitation of crude nanaibotide
The cleavage of nangibotide off the resin was carried out as described in example 2, step 5. Molar yield 60%. Purity 70%.
PAPER
Methods in enzymology (2000), 312, 293-304
Journal of the American College of Cardiology (2016), 68(25), 2776-2793
PATENT
https://patents.google.com/patent/WO2011124685A1/en
Product pat, WO2011124685 ,protection in the EU states and the US April 2031
References
- ^ Cuvier V, Lorch U, Witte S, Olivier A, Gibot S, Delor I, Garaud JJ, Derive M, Salcedo-Magguilli M (2018). “A first-in-man safety and pharmacokinetics study of nangibotide, a new modulator of innate immune response through TREM-1 receptor inhibition”. Br J Clin Pharmacol. 84 (10): 2270–2279. doi:10.1111/bcp.13668. PMC 6138490. PMID 29885068.
- ^ Weber B, Schuster S, Zysset D, Rihs S, Dickgreber N, Schürch C, Riether C, Siegrist M, Schneider C, Pawelski H, Gurzeler U, Ziltener P, Genitsch V, Tacchini-Cottier F, Ochsenbein A, Hofstetter W, Kopf M, Kaufmann T, Oxenius A, Reith W, Saurer L, Mueller C (2014). “TREM-1 deficiency can attenuate disease severity without affecting pathogen clearance”. PLOS Pathog. 10 (1): e1003900. doi:10.1371/journal.ppat.1003900. PMC 3894224. PMID 24453980.
- ^ Derive M, Bouazza Y, Sennoun N, Marchionni S, Quigley L, Washington V, Massin F, Max JP, Ford J, Alauzet C, Levy B, McVicar DW, Gibot S (1 June 2012). “Soluble TREM-like transcript-1 regulates leukocyte activation and controls microbial sepsis”. Journal of Immunology. 188 (11): 5585–5592. doi:10.4049/jimmunol.1102674. PMC 6382278. PMID 22551551.
- ^ Derive M, Boufenzer A, Bouazza Y, Groubatch F, Alauzet C, Barraud D, Lozniewski A, Leroy P, Tran N, Gibot S (Feb 2013). “Effects of a TREM-like transcript 1-derived peptide during hypodynamic septic shock in pigs”. Shock. 39 (2): 176–182. doi:10.1097/SHK.0b013e31827bcdfb. PMID 23324887. S2CID 23583753.
- ^ Derive M, Boufenzer A, Gibot S (April 2014). “Attenuation of responses to endotoxin by the triggering receptor expressed on myeloid cells-1 inhibitor LR12 in nonhuman primate”. Anaesthesiology. 120 (4): 935–942. doi:10.1097/ALN.0000000000000078. PMID 24270127. S2CID 10347527.
- ^ Cuvier V, Lorch U, Witte S, Olivier A, Gibot S, Delor I, Garaud JJ, Derive M, Salcedo-Magguilli M (2018). “A first-in-man safety and pharmacokinetics study of nangibotide, a new modulator of innate immune response through TREM-1 receptor inhibition”. Br J Clin Pharmacol. 84 (10): 2270–2279. doi:10.1111/bcp.13668. PMC 6138490. PMID 29885068.
- ^ François B, Wittebole X, Ferrer R, Mira JP, Dugernier T, Gibot S, Derive M, Olivier A, Cuvier V, Witte S, Pickkers P, Vandenhende F, Garaud JJ, Sánchez M, Salcedo-Magguilli M, Laterre PF (July 2020). “Nangibotide in patients with septic shock: a Phase 2a randomized controlled clinical trial”. Intensive Care Medicine. 46 (7): 1425–1437. doi:10.1007/s00134-020-06109-z. PMID 32468087. S2CID 218912723.
- ^ “Efficacy, Safety and Tolerability of Nangibotide in Patients With Septic Shock (ASTONISH)”. ClinicalTrials.gov. US National Library of Medicine. Retrieved 13 July 2020.
Derive et al (2013) Effects of a TREM-Like Transcript 1–Derived Peptide During Hypodynamic Septic Shock in Pigs. Shock39(2) 176 PMID: 23324887
Derive et al (2014) Attenuation of Responses to Endotoxin by the Triggering Receptor Expressed on Myeloid Cells-1 Inhibitor LR12 in Nonhuman Primate. Anesthesiology120(4) 935 PMID: 24270127
Derive et al (2012) Soluble Trem-like Transcript-1 Regulates Leukocyte Activation and Controls Microbial Sepsis. J. Immunol.188(11) 5585 PMID: 22551551
| Clinical data | |
|---|---|
| Routes of administration | Intravenous; intraperitoneal |
| Physiological data | |
| Receptors | TREM-1 |
| Metabolism | Enzymatic in bloodstream |
| Pharmacokinetic data | |
| Metabolism | Enzymatic in bloodstream |
| Elimination half-life | 3 minutes |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2014384‐91‐7 |
| ChemSpider | 64835227 |
| UNII | 59HD7BLX9H |
| ChEMBL | ChEMBL4297793 |
| Chemical and physical data | |
| Formula | C54H82N14O22S2 |
| Molar mass | 1343.439 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
//////////////Nangibotide, phase 3, нангиботид , مانغيبوتيد , 南吉博肽 , INOTREM, SEPTIC SHOCK, PEPTIDE
NEW DRUG APPROVALS
one time
$10.00
Tralokinumab
(Heavy chain)
QVQLVQSGAE VKKPGASVKV SCKASGYTFT NYGLSWVRQA PGQGLEWMGW ISANNGDTNY
GQEFQGRVTM TTDTSTSTAY MELRSLRSDD TAVYYCARDS SSSWARWFFD LWGRGTLVTV
SSASTKGPSV FPLAPCSRST SESTAALGCL VKDYFPEPVT VSWNSGALTS GVHTFPAVLQ
SSGLYSLSSV VTVPSSSLGT KTYTCNVDHK PSNTKVDKRV ESKYGPPCPS CPAPEFLGGP
SVFLFPPKPK DTLMISRTPE VTCVVVDVSQ EDPEVQFNWY VDGVEVHNAK TKPREEQFNS
TYRVVSVLTV LHQDWLNGKE YKCKVSNKGL PSSIEKTISK AKGQPREPQV YTLPPSQEEM
TKNQVSLTCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS RLTVDKSRWQ
EGNVFSCSVM HEALHNHYTQ KSLSLSLGK
(Light chain)
SYVLTQPPSV SVAPGKTARI TCGGNIIGSK LVHWYQQKPG QAPVLVIYDD GDRPSGIPER
FSGSNSGNTA TLTISRVEAG DEADYYCQVW DTGSDPVVFG GGTKLTVLGQ PKAAPSVTLF
PPSSEELQAN KATLVCLISD FYPGAVTVAW KADSSPVKAG VETTTPSKQS NNKYAASSYL
SLTPEQWKSH RSYSCQVTHE GSTVEKTVAP TECS
(Disulfide bridge: H22-H96, H149-H205, H263-H323, H369-H427, H228-H’228, H231-H’231, L22-L87, L136-L195, H136-L213)
Tralokinumab
トラロキヌマブ (遺伝子組換え)
| Formula | C6374H9822N1698O2014S44 |
|---|---|
| CAS | 1044515-88-9 |
| Mol weight | 143873.2167 |
EU APPROVED, Adtralza, 2021/6/17
Antiasthmatic, Anti-inflammatory, Anti-IL-13 antibody
Tralokinumab is a human monoclonal antibody which targets the cytokine interleukin 13,[1] and is designed for the treatment of asthma and other inflammatory diseases.[2] Tralokinumab was discovered by Cambridge Antibody Technology scientists, using Ribosome Display, as CAT-354[3] and taken through pre-clinical and early clinical development.[4] After 2007 it has been developed by MedImmune, a member of the AstraZeneca group, where it is currently in Ph3 testing for asthma and Ph2b testing for atopic dermatitis.[5][6] This makes it one of the few fully internally discovered and developed drug candidates in AstraZeneca’s late stage development pipeline.
Discovery and development
Tralokinumab (CAT-354) was discovered by Cambridge Antibody Technology scientists[7] using protein optimization based on Ribosome Display.[8] They used the extensive data sets from ribosome display to patent protect CAT-354 in a world-first of sequence-activity-relationship claims.[7] In 2004, clinical development of CAT-354 was initiated with this first study completing in 2005.[9] On 21 July 2011, MedImmune LLC initiated a Ph2b, randomized, double-blind study to evaluate the efficacy of tralokinumab in adults with asthma.[10]
In 2016, MedImmune and AstraZeneca were developing tralokinumab for asthma (Ph3) and atopic dermatitis (Ph2b) while clinical development for moderate-to-severe ulcerative colitis and idiopathic pulmonary fibrosis (IPF) have been discontinued.[9] In July of that year AstraZeneca licensed Tralokinumab to LEO Pharma for skin diseases.[11]
A phase IIb study of Tralokinumab found that treatment was associated with early and sustained improvements in atopic dermatitis symptoms and tralokinumab had an acceptable safety and tolerability profile, thereby providing evidence for targeting IL-13 in patients with atopic dermatitis.[12]
On 15 June 2017, Leo Pharma announced that they were starting phase III clinical trials with tralokinumab in atopic dermatitis.[13]
Society and culture
Legal status
On 22 April 2021, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Adtralza, intended for the treatment of moderate‑to‑severe atopic dermatitis.[14]
The applicant for this medicinal product is LEO Pharma A/S.
References
- ^ Kopf M, Bachmann MF, Marsland BJ (September 2010). “Averting inflammation by targeting the cytokine environment”. Nature Reviews. Drug Discovery. 9 (9): 703–18. doi:10.1038/nrd2805. PMID 20811382. S2CID 23769909.
- ^ “Statement On A Nonproprietary Name Adopted By The USAN Council: Tralokinumab” (PDF). American Medical Association.
- ^ Thom G, Cockroft AC, Buchanan AG, Candotti CJ, Cohen ES, Lowne D, et al. (May 2006). “Probing a protein-protein interaction by in vitro evolution” [P]. Proceedings of the National Academy of Sciences of the United States of America. 103 (20): 7619–24. Bibcode:2006PNAS..103.7619T. doi:10.1073/pnas.0602341103. PMC 1458619. PMID 16684878.
- ^ May RD, Monk PD, Cohen ES, Manuel D, Dempsey F, Davis NH, et al. (May 2012). “Preclinical development of CAT-354, an IL-13 neutralizing antibody, for the treatment of severe uncontrolled asthma”. British Journal of Pharmacology. 166 (1): 177–93. doi:10.1111/j.1476-5381.2011.01659.x. PMC 3415647. PMID 21895629.
- ^ “Pipeline”. MedImmune. Retrieved 11 June 2013.
- ^ “Studies found for CAT-354”. ClinicalTrials.gov. Retrieved 11 June 2013.
- ^ Jump up to:a b Human Antibody Molecules for Il-13, retrieved 2015-07-26
- ^ Jermutus L, Honegger A, Schwesinger F, Hanes J, Plückthun A (January 2001). “Tailoring in vitro evolution for protein affinity or stability”. Proceedings of the National Academy of Sciences of the United States of America. 98 (1): 75–80. Bibcode:2001PNAS…98…75J. doi:10.1073/pnas.98.1.75. PMC 14547. PMID 11134506.
- ^ Jump up to:a b “Tralokinumab”. Adis Insight. Springer Nature Switzerland AG.
- ^ Clinical trial number NCT01402986 for “A Phase 2b, Randomized, Double-blind Study to Evaluate the Efficacy of Tralokinumab in Adults With Asthma” at ClinicalTrials.gov
- ^ “AstraZeneca enters licensing agreements with LEO Pharma in skin diseases”.
- ^ Wollenberg A, Howell MD, Guttman-Yassky E, Silverberg JI, Kell C, Ranade K, et al. (January 2019). “Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb”. The Journal of Allergy and Clinical Immunology. 143 (1): 135–141. doi:10.1016/j.jaci.2018.05.029. PMID 29906525.
- ^ “LEO Pharma starts phase 3 clinical study for tralokinumab in atopic dermatitis”. leo-pharma.com. AstraZeneca. 1 July 2016.
- ^ “Adtralza: Pending EC decision”. European Medicines Agency. 23 April 2021. Retrieved 23 April 2021.
| Tralokinumab Fab fragment bound to IL-13. From PDB 5L6Y. | |
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL-13 |
| Clinical data | |
| ATC code | D11AH07 (WHO) |
| Identifiers | |
| CAS Number | 1044515-88-9 |
| ChemSpider | none |
| UNII | GK1LYB375A |
| KEGG | D09979 |
| Chemical and physical data | |
| Formula | C6374H9822N1698O2014S44 |
| Molar mass | 143875.20 g·mol−1 |
| (what is this?) (verify) |
/////////Tralokinumab, Adtralza, EU 2021, APPROVALS 2021, Antiasthmatic, Anti-inflammatory, Anti-IL-13 antibody, MONOCLONAL ANTIBODY, PEPTIDE, トラロキヌマブ (遺伝子組換え) ,
NEW DRUG APPROVALS
ONE TIME
$10.00
EpiVacCorona


Origin of EpiVacCorona antigenes
- MKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNSSSNNNNNNNNNNLGDNGPQNQRNAPRITFGGPSDSTGSNQNGERSGARSKQRRPQGLPNNTASWFTALTQHGKEDLKFPRGQGVPINTNSSPDDQIGYYRRATRRIRGGDGKMKDLSPRWYFYYLGTGPEAGLPYGANKDGIIWVATEGALNTPKDHIGTRNPANNAAIVLQLPQGTTLPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPARMAGNGGDAALALLLLDRLNQLESKMSGKGQQQQGQTVTKKSAAEASKKPRQKRTATKAYNVTQAFGRRGPEQTQGNFGDQELIRQGTDYKHWPQIAQFAPSASAFFGMSRIGMEVTPSGTWLTYTGAIKLDDKDPNFKDQVILLNKHIDAYKTFPPTEPKKDKKKKADETQALPQRQKKQQTVTLLPAADLDDLSKQLQQSMSSADSTQA. “Carrier protein sequence”.
EpiVacCorona
Federal Budgetary Research Institution State Research Center of Virology and Biotechnology
peptide, russia
PATENT https://www.fips.ru/registers-doc-view/fips_servlet?DB=RUPAT&DocNumber=2743594&TypeFile=htmlRU 2 743 594 RU 2 743 593RU 2 743 595 RU 2 738 081 Science (Washington, DC, United States) (2021), 372(6538), 116-117.
EpiVacCorona (Russian: ЭпиВакКорона, tr. EpiVakKorona) is a peptide-based vaccine against COVID-19 developed by the VECTOR center of Virology.[1][2][3] It consists of three chemically synthesized peptides (short fragments of a viral spike protein) that are conjugated to a large carrier protein. This protein is a fusion product of a viral nucleocapsid protein and a bacterial MBP protein.The third phase of a clinical trial, which should show whether the vaccine is able to protect people from COVID-19 or not, was launched in November 2020 with more than three thousand participants.[2] It is assumed it will be completed in August 2021.[2] According to the vaccine developers, the peptides and the viral part of the chimeric protein should immunize people who received this vaccine against SARS-CoV-2 and trigger the production of protective antibodies. However, some experts in the field have expressed concerns about the selection of peptides for use as vaccine antigens.[3][4] In addition, there are also serious concerns about the vaccine immunogenicity data, which have fueled independent civic research efforts[5][6][7] and criticism by some experts.[3][8][4][9][10] Meanwhile, the EpiVacCorona has received vaccine emergency authorization in a form of government registration and is available for vaccination outside the clinical trials.[11] The vaccine delivered via intramuscular route and aluminum hydroxide serves as an immunological adjuvant.
Description[edit]
Origin of EpiVacCorona antigenes
Composition
The vaccine includes three chemically synthesized short fragments of the viral spike protein – peptides, which, according to the developers of EpiVacCorona represent the protein regions containing B-cell epitopes that should be recognized by the human immune system.
These peptides are represented by following amino acid sequences:
1) CRLFRKSNLKPFERDISTEIYQAGS, 2) CKEIDRLNEVAKNLNESLIDLQE, 3) CKNLNESLIDLQELGKYEQYIK.[1][12][13]
In the vaccine all peptides are conjugated to a carrier protein, which is an expression product of the chimeric gene. This chimeric gene was created by fusion of two genes originating from different organisms, namely a gene encoding a viral nucleocapsid protein and a gene encoding a bacterial maltose-binding protein (MBP). The fusion chimeric gene expressed in Escherichia coli. The sequence of the chimeric protein is available from the patent.[4] The genetic construct of the chimeric gene also includes a short genetic fragment encoding a polyhistidine-tag, which is used to purify the chimeric protein from E. coli lysate. After the purification, the protein is conjugated with three peptides in a way that only one variant of the peptide molecule is attached to each protein molecule. As a result, three types of conjugated molecules are created: chimeric protein with attached peptide number 1, the same protein with peptide number 2, and finally the same protein with peptide number 3. All three types of conjugated molecules are included in the vaccine.[citation needed]

EpiVacCorona: antigens origin and composition
Vaccine antigens and antibodies
According to the developers’ publications,[14][5][6] vaccine antigens are three peptides of the spike protein and a chimeric protein consisting of two parts (viral nucleocapsid protein and bacterial maltose-binding protein). In addition, the polyhistidine-tag – a short peptide that is introduced into a vaccine composition to purify a chimeric protein from a bacterial lysate – is also a vaccine antigen against which antibodies can form in those who have received the vaccine. A person vaccinated with EpiVacCorona can develop antibodies not only to the peptides of the spike protein, but also to other antigens present in the vaccine. According to Anna Popova who is a head of the Federal Service for Supervision of Consumer Rights Protection and Human Welfare, it takes 42 days for those vaccinated with EpiVacCorona to develop immunity.[15]
Development

Immunogenic peptide screening in rabbits for EpiVacCorona design
Preclinical studies
The primary screening of peptides for the search for the most immunogenic ones was carried out in animals. The level of antibodies that was triggered by each tested peptide after administration to rabbits was measured. In the test, hemocyanin protein was used as a carrier protein for the studied peptides. Further, on six species of animals (mice, rats, rabbits, African green monkeys, rhesus monkeys, guinea pigs), the vaccine was shown to be harmless in terms of such parameters as general toxicity, allergic properties, and mutagenic activity. In four species of animals (hamsters, ferrets, African green monkeys, rhesus monkeys), specific activity was shown: immunogenicity and protective properties against SARS-CoV-2. The main results of preclinical studies are published in the “Bulletin of the Russian Academy of Medical Sciences”.[12][13]
Clinical studies
The studies development timeline was reported in Russian media in January 2021.[16] There are currently two clinical trials of EpiVacCorona registered in the ClinicalTrials.gov database.[17][18][2]
Phase I-II
The trial “Study of the Safety, Reactogenicity and Immunogenicity of “EpiVacCorona” Vaccine for the Prevention of COVID-19 (EpiVacCorona)”[18] was registered in clinical trial database with ClinicalTrials.gov identifier: NCT04780035. Another trial with the same title was registered with ClinicalTrials.gov Identifier: NCT04527575. Results of the trial that included data on 86 participants were published in Russian Journal of Infection and Immunity, indicating preliminary evidence of safety and an immune response.[1] The publication reports preliminary results of the first two phases of clinical trials of the vaccine in volunteers, of which 14 people aged 18-30 years participated in the first phase, and 86 volunteers aged 18-60 years in the second phase. It is claimed that antibodies were formed in 100% of the volunteers, and the vaccine is also claimed to be safe.[1]

EpiVacCorona Vaccine Development Timeline
Phase III
The third phase of a clinical trial, which should show whether the vaccine is able to protect people from COVID-19 or not, was launched in November 2020 with more than three thousand participants planned. It is expected to be completed in September 2021.[2] In the clinical trials database the phase III trial etitled “Study of the Tolerability, Safety, Immunogenicity and Preventive Efficacy of the EpiVacCorona Vaccine for the Prevention of COVID-19[2]” was registered only in March 2021 with ClinicalTrials.gov Identifier: NCT04780035. Phase 3-4 trial was registered in Russia at 18.11.2020 with 4991 participants planned.[19]
Intellectual property
The following patents of the Russian Federation for invention have been published, which protect the EpiVacCorona vaccine:
“Peptide immunogens and vaccine composition against coronavirus infection COVID-19 using peptide immunogens” (No. 2738081). There are 7 peptides in patented vaccine compositions.
“Peptide immunogens and vaccine composition against coronavirus infection COVID-19 using peptide immunogens” (No. 2743593). The patented vaccine composition contains 2 peptides.
“Peptide immunogens used as a component of a vaccine composition against coronavirus infection COVID-19″ (No. 2743594). The patented vaccine composition contains 3 peptides.
“Vaccine composition against coronavirus infection COVID-19″ (No. 2743595). The patented vaccine composition contains 3 peptides.
In all of these patents, the carrier protein is referred to as a chimeric fusion protein with an amino acid sequence derived from two parts, a bacterial maltose binding protein and a viral nucleocapsid protein.[20]

EpiVacCorona vaccine registration certificate
Authorization
| Full authorization Emergency authorization |
See also: List of COVID-19 vaccine authorizations § EpiVacCorona
The VECTOR has received vaccine emergency authorization in a form of government registration in October 2020.[21]
In Russia phase III clinical study is called post-registration study. Therefore, government registration of the vaccine means permission to perform phase III clinical research and public vaccination outside of clinical trials as well.[21] Since December 2020, the vaccine has been released for public vaccination in Russia.[22]
As of March 2021, Turkmenistan is the only foreign state to register EpiVacCorona with full authorization.[23][24]
Russia’s Chief Health Officer Anna Popova said: “In December 2020 the EpiVacCorona documents were presented to the World Health Organization, and we are expecting a decision from WHO.”[25] However, Deutsche Welle reports “As of March 1, the WHO had yet to receive an Expression of Interest (EOI) from EpiVacCorona’s developers, “VECTOR,” to enable WHO experts to evaluate their vaccine.”[26]
Export
The Deputy Director-General of the World Health Organization (WHO) Dr. Soumya Swaminathan during news conference in Geneva that took place in October 2020, told: “We will only be able to have a position on a vaccine when we see results of the phase III clinical trials.”[27] According to the center’s director Rinat Maksyutov, many government and non-government organizations want to test or be involved in the production of the vaccine.[28] As of March 30, Venezuela obtained 1000 doses of the Russian EpiVacCorona vaccine for a trial.[29] Venezuela also has reached a deal to purchase doses of the vaccine, as well as manufacture it locally, Vice President Delcy Rodriguez provided this information on June 4, 2021.[30] Turkmenistan expects to receive EpiVacCorona, as the vaccine has already been approved for use in that country.[31]
Controversy
Independent study of clinical trial participants

Ministry of Health’s response to a request from trial participants to perform independent antibody screening tests

English translation of Ministry of Health’s response to a request from trial participants to perform independent antibody screening tests.
At the start of the Phase III, trial participants and those vaccinated outside the trial began to form a community through the Telegram messenger network. On January 18, 2021, the members of the community turned to the Ministry of Health of the Russian Federation with an open letter, in which they stated that the production of antibodies after vaccination among them is much lower than declared by vaccine developers. Study participants claimed that antibodies were not found in more than 50% of those who documented their participation in the study, although only 25% of the participants should have had a placebo according to the study design. The trial participants also claimed that negative results were obtained using the a special ELISA test developed and recommended by VECTOR for EpiVacCorona detection.[5][6][4] More questions about the quality and protectiveness of antibodies induced by EpiVacCorona appeared along with the first results of a special antibody VECTOR’s test, when, with a positive special test, negative results of all other commercially available tests were otained: LIAISON SARS-CoV-2 S1 / S2 IgG – DiaSorin, IgM / IgG – Mindray, SARS-CoV-2 IgG – Abbott Architect, Anti-SARS-CoV-2 ELISA (IgG) – Euroimmun, Access SARS-CoV-2 IgG (RBD) – Beckman Coulter, “SARS-CoV-2-IgG-ELISA -BEST “-” Vector-Best “,” Anti-RBD IgG “- Gamaleya Research Center.[5][6][4][8] Clinical trial participants conducted their own antibody mini-study that was performed in independent Russian laboratory. The study participants asked Dr. Alexander Chepurnov, the former head of the infectious diseases department at VECTOR, who now works at another medical institute, to check neutralizing antibodies presence in their serum samples.[3] They also sent to Dr. Chepurnov control serum samples from former COVID-19 patients or people vaccinated with another Russian vaccine, Sputnik V, which is known to trigger the production of neutralizing antibodies.[32] All serum samples were blinded before antibody tests. On 23 March 2021, the participants reported the results of their mini-study in an open letter to the Ministry of Health of the Russian Federation.[6][7] According to the letter, even with the help of the VECTOR antibody detection system, antibodies were detected only in 70-75% of those vaccinated with EpiVacCorona. However, the level of antibodies was very low. Moreover, according to the letter, virus-neutralizing antibodies were not detected in the independent research Dr. Alexander Chepurnov laboratory at all.[3][6][7] The trial participants asked Ministry of Health in their open letter to perform independent study for the verification of their findings.[3][6][7] In addition, the letter reports 18 cases of COVID-19 cases as of March 22, 2021 among those who received the vaccine and became ill (sometimes severe) three weeks or later after the second dose of EpiVacCorona.[33][6][7] April 20, 2021 the study participants got a reply, with refusal of performing any additional verification antibody tests or investigation of sever COVID-19 cases among vaccinated individuals. The reply include the following text: “Considering that the listed immunobiological preparations (vaccines) for the prevention of COVID-19 are registered in the prescribed manner, their effectiveness and safety have been confirmed.”
Vaccine criticism by independent experts
Some independent experts criticized the vaccine design[3][4] and clinical data presentation in the publication.[8][9][10] The experts are saying that peptide selection is “crucial” for the innovative peptide approach, which VECTOR uses for EpiVacCorona design. However, some researchers are not convinced that the viral spike protein peptides selected for the vaccine are actually “visible” by human immune system.[3][4][34] They stated that these peptides do not overlap[35] with peptides that have been shown in several publications to contain human linear B cell epitopes in spike protein of SARS-CoV-2.[36][37][38][39][40] Moreover, the study was criticized for the lack of positive control of convalescent plasma samples in reports related to neutralizing antibody titers in vaccinated individuals.[1][10] The same study was also criticized for presence of detectable antibodies in negative controls samples that were not discussed by authors.[1][10] In addition, vaccine developers have been criticized for aggressively advertising their vaccine efficacy prior to the completion of phase III clinical trial. The most substantial criticism came from Dr. Konstantin Chumakov, who currently serves as the Associate Director for Research at the FDA Office of Vaccines Research and Review. Dr. Chumakov said: “I would not be in a hurry to call this peptide formulation a vaccine yet, because its effectiveness has not yet been proven…For the introduction of such a vaccine, the level of evidence must be much higher, and therefore the developers of EpiVacCorona, before launching their vaccine on the market, had to conduct clinical trials and prove that their vaccine actually protects against the disease. However, such tests were not carried out, which is absolutely unacceptable.”[41]

The title page of the “EpiVacCorona” patent with Anna’s Popova name among inventors
Conflict of interest
The vaccine design was protected by several already issued patents (see section above). In each patent one of its co-authors is a namesake of Anna Popova who is a head of the Federal Service for Supervision of Consumer Rights Protection and Human Welfare. This patent authorship represents an issue as far as Anna Popova is a head of the Russian agency that is charged with overseeing vaccine safety and efficacy. As a co-author of these patents, she might have an interest in promoting the vaccine despite its shortcomings.
References
- ^ Jump up to:a b c d e f Ryzhikov AB, Ryzhikov EA, Bogryantseva MP, Usova SV, Danilenko ED, Nechaeva EA, Pyankov OV, Pyankova OG, Gudymo AS, Bodnev SA, Onkhonova GS, Sleptsova ES, Kuzubov VI, Ryndyuk NN, Ginko ZI, Petrov VN, Moiseeva AA, Torzhkova PY, Pyankov SA, Tregubchak TV, Antonec DV, Gavrilova EV, Maksyutov RA (2021). “A single blind, placebo-controlled randomized study of the safety, reactogenicity and immunogenicity of the “EpiVacCorona” Vaccine for the prevention of COVID-19, in volunteers aged 18–60 years (phase I–II)”. Russian Journal of Infection and Immunity. 11 (2): 283–296. doi:10.15789/2220-7619-ASB-1699.
- ^ Jump up to:a b c d e Federal Budgetary Research Institution State Research Center of Virology and Biotechnology “Vector” (2 March 2021). “Multicenter Double-blind Placebo-controlled Comparative Randomized Study of the Tolerability, Safety, Immunogenicity and Prophylactic Efficacy of the EpiVacCorona Peptide Antigen-based Vaccine for the Prevention of COVID-19, With the Participation of 3000 Volunteers Aged 18 Years and Above (Phase III-IV)”.
- ^ Jump up to:a b c d e f g DobrovidovaApr. 6, Olga; 2021; Am, 11:05 (6 April 2021). “Russia’s COVID-19 defense may depend on mystery vaccine from former bioweapons lab—but does it work?”. Science | AAAS. Retrieved 24 April 2021.
- ^ Jump up to:a b c d e f Dobrovidova, Olga (9 April 2021). “Latest Russian vaccine comes with a big dose of mystery”. Science. 372 (6538): 116–117. doi:10.1126/science.372.6538.116. ISSN 0036-8075. PMID 33833104. S2CID 233191522.
- ^ Jump up to:a b c Staff, Reuters (26 March 2021). “Volunteers break rank to raise doubts in trial of Russia’s second COVID-19 vaccine”. Reuters. Retrieved 23 April 2021.
- ^ Jump up to:a b c d e f g “”ЭпиВакКорона” глазами участников клинических испытаний и ученых-биологов”. Троицкий вариант — Наука (in Russian). 23 March 2021. Retrieved 23 April 2021.
- ^ Jump up to:a b c d e https://epivakorona.com/openletter.htm
- ^ Jump up to:a b c “EpiVacCorona’s race to the finish line Meduza speaks to the developer and manufacturer about concerns surrounding Russia’s latest coronavirus vaccine”. meduza.io. Retrieved 23 April2021.
- ^ Jump up to:a b “Нет антител, вопросы к составу, непрозрачность данных. Что не так с вакциной “ЭпиВакКорона””. BBC News Русская служба (in Russian). Retrieved 23 April 2021.
- ^ Jump up to:a b c d “Sputnik V’s ugly cousin Clinical results for Russia’s EpiVacCorona vaccine are finally here, but developers published in an obscure local journal, raising questions and concerns”. meduza.io. Retrieved 23 April 2021.
- ^ “About 200,000 EpiVacCorona vaccine doses go into civil circulation in Russia”. TASS. Retrieved 25 April 2021.
- ^ Jump up to:a bhttps://www.researchgate.net/publication/350822775_Immunogenicity_and_protectivity_of_the_peptide_candidate_vaccine_against_SARS-CoV-2
- ^ Jump up to:a b Ryzhikov AB, Ryzhikov EA, Bogryantseva MP, Usova SV, Danilenko ED, Imatdinov IR, Nechaeva EA, Pyankov OV, Pyankova OG, Gudymo AS, Bodnev SA, Onkhonova GS, Sleptsova ES, Kuzubov VI, Ryndyuk NN, Ginko ZI, Petrov VN, Moiseeva AA, Torzhkova PY, Pyankov SA, Tregubchak TV, Antonec DV, Sleptsova ES, Gavrilova EV, Maksyutov RA (2021). “Immunogenicity and Protectivityof the Peptide Vaccine againstSARS-CoV-2”. Annals of the Russian Academy of Medical Sciences. 76 (1): 5–19. doi:10.15690/vramn1528.
- ^ Ryzhikov, A. B.; Ryzhikov, Е. А.; Bogryantseva, M. P.; Usova, S. V.; Danilenko, E. D.; Nechaeva, E. A.; Pyankov, O. V.; Pyankova, O. G.; Gudymo, A. S. (24 March 2021). “A single blind, placebo-controlled randomized study of the safety, reactogenicity and immunogenicity of the “EpiVacCorona” Vaccine for the prevention of COVID-19, in volunteers aged 18–60 years (phase I–II)”. Russian Journal of Infection and Immunity. Retrieved 23 April 2021.
- ^ “People vaccinated with Russia’s EpiVacCorona need 42 days to develop immunity – watchdog”. TASS. Retrieved 25 April 2021.
- ^ “Что ждать от “ЭпиВакКороны”. Все о пептидной вакцине против COVID-19″. РИА Новости(in Russian). 1 January 2021. Retrieved 24 April 2021.
- ^ s.r.o, Direct Impact. “AIM database substance – EpiVacCorona”. AIM. Retrieved 25 April 2021.
- ^ Jump up to:a b Federal Budgetary Research Institution State Research Center of Virology and Biotechnology “Vector” (20 February 2021). “Simple, Blind, Placebo-controlled, Randomized Study of the Safety, Reactogenicity and Immunogenicity of Vaccine Based on Peptide Antigens for the Prevention of COVID-19 (EpiVacCorona), in Volunteers Aged 18-60 Years (I-II Phase)”.
- ^ Реестр Клинических исследований COV/pept-03/20; [1]
- ^MKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQTNSSSNNNNNNNNNNLGDNGPQNQRNAPRITFGGPSDSTGSNQNGERSGARSKQRRPQGLPNNTASWFTALTQHGKEDLKFPRGQGVPINTNSSPDDQIGYYRRATRRIRGGDGKMKDLSPRWYFYYLGTGPEAGLPYGANKDGIIWVATEGALNTPKDHIGTRNPANNAAIVLQLPQGTTLPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPARMAGNGGDAALALLLLDRLNQLESKMSGKGQQQQGQTVTKKSAAEASKKPRQKRTATKAYNVTQAFGRRGPEQTQGNFGDQELIRQGTDYKHWPQIAQFAPSASAFFGMSRIGMEVTPSGTWLTYTGAIKLDDKDPNFKDQVILLNKHIDAYKTFPPTEPKKDKKKKADETQALPQRQKKQQTVTLLPAADLDDLSKQLQQSMSSADSTQA. “Carrier protein sequence”.
- ^ Jump up to:a b “Russia begins post-registration trials of EpiVacCorona Covid-19 vaccine”. http://www.clinicaltrialsarena.com. Retrieved 25 April 2021.
- ^ “Вакцина “ЭпиВакКорона” поступила в гражданский оборот”. РИА Новости (in Russian). 11 December 2020. Retrieved 23 April 2021.
- ^ “Turkmenistan registers vaccines for the prevention of infectious diseases”. Turkmenistan Today. 29 January 2021.
- ^ “Turkmenistan: Master Berdymukhamedov goes to Moscow | Eurasianet”. eurasianet.org. Retrieved 25 April 2021.
- ^ “Russia submits EpiVacCorona vaccine documents to WHO – Rospotrebnadzor head Popova”. interfax.com. Retrieved 23 April 2021.
- ^ Welle (www.dw.com), Deutsche. “Two more Russian vaccines: What we do and don’t know | DW | 09.03.2021”. DW.COM. Retrieved 23 April 2021.
- ^ “COVID-19 vaccine: WHO in talks with Russia on its second vaccine EpiVacCorona”. mint. 16 October 2020. Retrieved 9 June 2021.
- ^ “Vector Center says has over 45 inquiries from abroad about its EpiVacCorona vaccine”. TASS. Retrieved 25 April 2021.
- ^ Foundation, Thomson Reuters. “Venezuela receives doses of Russian EpiVacCorona vaccine for trials”. news.trust.org. Retrieved 25 April 2021.
- ^ “Venezuela to purchase and manufacture Russia’s EpiVacCorona vaccine”. Reuters. 5 June 2021. Retrieved 13 June 2021.
- ^ turkmenportal. “Turkmenistan Approves Use of Russia’s EpiVacCorona Vaccine | Society”. Business Turkmenistan Information Center. Retrieved 25 April 2021.
- ^ Jones, Ian; Roy, Polly (20 February 2021). “Sputnik V COVID-19 vaccine candidate appears safe and effective”. The Lancet. 397 (10275): 642–643. doi:10.1016/S0140-6736(21)00191-4. ISSN 0140-6736. PMC 7906719. PMID 33545098.
- ^ “Участники КИ “ЭпиВакКороны” продолжают исследовать эффективность вакцины”. pcr.news. Retrieved 24 April 2021.
- ^ Li, Yang; Ma, Ming-Liang; Lei, Qing; Wang, Feng; Hong, Wei; Lai, Dan-Yun; Hou, Hongyan; Xu, Zhao-Wei; Zhang, Bo; Chen, Hong; Yu, Caizheng (30 March 2021). “Linear epitope landscape of the SARS-CoV-2 Spike protein constructed from 1,051 COVID-19 patients”. Cell Reports. 34 (13): 108915. doi:10.1016/j.celrep.2021.108915. ISSN 2211-1247. PMC 7953450. PMID 33761319.
- ^ “Вакцина “ЭпиВакКорона” в иллюстрациях”. Троицкий вариант — Наука (in Russian). 23 March 2021. Retrieved 24 April 2021.
- ^ Yi, Zhigang; Ling, Yun; Zhang, Xiaonan; Chen, Jieliang; Hu, Kongying; Wang, Yuyan; Song, Wuhui; Ying, Tianlei; Zhang, Rong; Lu, HongZhou; Yuan, Zhenghong (December 2020). “Functional mapping of B-cell linear epitopes of SARS-CoV-2 in COVID-19 convalescent population”. Emerging Microbes & Infections. 9 (1): 1988–1996. doi:10.1080/22221751.2020.1815591. ISSN 2222-1751. PMC 7534331. PMID 32844713.
- ^ Poh, Chek Meng; Carissimo, Guillaume; Wang, Bei; Amrun, Siti Naqiah; Lee, Cheryl Yi-Pin; Chee, Rhonda Sin-Ling; Fong, Siew-Wai; Yeo, Nicholas Kim-Wah; Lee, Wen-Hsin; Torres-Ruesta, Anthony; Leo, Yee-Sin (1 June 2020). “Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients”. Nature Communications. 11 (1): 2806. doi:10.1038/s41467-020-16638-2. ISSN 2041-1723. PMC 7264175. PMID 32483236.
- ^ Li, Yang; Lai, Dan-Yun; Zhang, Hai-Nan; Jiang, He-Wei; Tian, Xiaolong; Ma, Ming-Liang; Qi, Huan; Meng, Qing-Feng; Guo, Shu-Juan; Wu, Yanling; Wang, Wei (October 2020). “Linear epitopes of SARS-CoV-2 spike protein elicit neutralizing antibodies in COVID-19 patients”. Cellular & Molecular Immunology. 17 (10): 1095–1097. doi:10.1038/s41423-020-00523-5. ISSN 2042-0226. PMC 7475724. PMID 32895485.
- ^ Farrera-Soler, Lluc; Daguer, Jean-Pierre; Barluenga, Sofia; Vadas, Oscar; Cohen, Patrick; Pagano, Sabrina; Yerly, Sabine; Kaiser, Laurent; Vuilleumier, Nicolas; Winssinger, Nicolas (2020). “Identification of immunodominant linear epitopes from SARS-CoV-2 patient plasma”. PLOS ONE. 15 (9): e0238089. doi:10.1371/journal.pone.0238089. ISSN 1932-6203. PMC 7480855. PMID 32903266.
- ^ Shrock, Ellen; Fujimura, Eric; Kula, Tomasz; Timms, Richard T.; Lee, I.-Hsiu; Leng, Yumei; Robinson, Matthew L.; Sie, Brandon M.; Li, Mamie Z.; Chen, Yuezhou; Logue, Jennifer (27 November 2020). “Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity”. Science. 370 (6520): eabd4250. doi:10.1126/science.abd4250. ISSN 1095-9203. PMC 7857405. PMID 32994364.
- ^ “Константин Чумаков: “Даже если человек переболел COVID-19, ему все равно нужно привиться. Иммунный ответ на прививку лучше и долговечнее, чем на саму болезнь””. republic.ru (in Russian). Retrieved 24 April 2021.
External links
- Margarita Romanenko’s Lecture about Russian Covid-vaccines
- Meduza – Interview with EpiVacCorona developers, 23 March 2021
- Infection and Immunity – Study of the safety, reactogenicity and immunogenecity of the “EpiVacCorona” vaccine (PHASE I–II)
| EpiVacCorona vaccine | |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Peptide subunit |
| Clinical data | |
| Trade names | EpiVacCorona |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | Registered in Russia on 14 October 2020 RU Registered.TU approved.Full list : List of EpiVacCorona COVID-19 vaccine authorizations |
| Identifiers | |
| DrugBank | DB16439 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| COVID-19 (disease)SARS-CoV-2 (virus) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
EpiVacCorona Vaccine, developed by the Vektor State Research Center of Virology and Biotechnology in Russia, is based on peptide-antigens that facilitate immunity to the SARS-CoV-2 virus1. It is currently being tested in Phase I/II clinical trials for safety and immunogenicity (NCT04527575)1,2.
- Precision Vaccinations: VACCINE INFO EpiVacCorona Vaccine [Link]
- The Pharma Letter: Russia’s EpiVacCorona vaccine post-registration trials started [Link]
//////EpiVacCorona, SARS-CoV-2, RUSSIA, CORONA VIRUS, COVID 19, VACCINE, PEPTIDE
NEW DRUG APPROVALS
ONE TIME
$10.00
Pegcetacoplan
Sequence:
1ICVWQDWGAH RCTXK
Sequence:
1ICVWQDWGAH RCTXK
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| terminal mod. | Lys-15 | C-terminal amide |
| terminal mod. | Lys-15′ | C-terminal amide |
| bridge | Cys-2 – Cys-12 | disulfide bridge, dimer |
| bridge | Lys-15 – Lys-15′ | covalent bridge, dimer |
| bridge | Cys-2′ – Cys-12′ | disulfide bridge, dimer |
| uncommon | Oaa-14 | – |
| uncommon | Oaa-14′ | – |
Pegcetacoplan
ペグセタコプラン;
FDA APPROVED Empaveli, 2021/5/14
Protein Sequence
Sequence Length: 30, 15, 15multichain; modifiedPoly(oxy-1,2-ethanediyl), α-hydro-ω-hydroxy-, 15,15′-diester with N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-α-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-[2-(2-aminoethoxy)ethoxy]acetyl-N6-carboxy-L-lysinamide cyclic (2→12)-(disulfide)Polymer
Poly(oxy-1,2-ethanediyl), alpha-hydro-omega-hydroxy-, 15,15′-diester with N-acetyl-Lisoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-alpha-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-(2-(2-aminoethoxy)ethoxy)acetyl-N6-carboxy-L-lysinamide cyclic (2�->12)-(disulfide)
O,O’-bis((S2,S12-cyclo(N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-Ltryptophyl-L-glutaminyl-L-alpha-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-(2-(2-aminoethoxy)ethoxy)acetyl-L-lysinamide))-N6.15-carbonyl)polyethylene glycol(n = 800-1100)
- APL-2
- WHO 10743
| Formula | C170H248N50O47S4. (C2H4O)n3872.40 g·mol−1 |
|---|---|
| EfficacyDisease | Complement inhibitorParoxysmal nocturnal hemoglobinuria |
| CAS | 2019171-69-6 |
| Comment | Treatment of paroxysmal nocturnal hemoglobinuria (PNH), complement-mediated nephropathies, and age-related macular degeneration (AMD) |
- OriginatorApellis Pharmaceuticals
- ClassAnti-inflammatories; Anti-ischaemics; Antianaemics; Cyclic peptides; Eye disorder therapies; Polyethylene glycols; Urologics
- Mechanism of ActionComplement C3 inhibitors
- Orphan Drug StatusYes – Paroxysmal nocturnal haemoglobinuria; Autoimmune haemolytic anaemia; Glomerulonephritis
- RegisteredParoxysmal nocturnal haemoglobinuria
- Phase IIIAge-related macular degeneration
- Phase IIAmyotrophic lateral sclerosis; Autoimmune haemolytic anaemia; Glomerulonephritis; IgA nephropathy; Lupus nephritis; Membranous glomerulonephritis
- Phase I/IIWet age-related macular degeneration
- DiscontinuedIschaemia
- 02 Jun 2021Apellis Pharmaceuticals plans a phase III trial for Glomerulonephritis in the second half of 2021
- 25 May 2021Top-line efficacy and safety results from the phase III PRINCE trial for Paroxysmal nocturnal haemoglobinuria released by Apellis Pharmaceuticals
- 18 May 2021Registered for Paroxysmal nocturnal haemoglobinuria in USA (SC) – First global approval
Pegcetacoplan, sold under the brand name Empaveli, is a medication used to treat paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
The most common side effects include injection-site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.[2]
Paroxysmal nocturnal hemoglobinuria is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells).[1]
Pegcetacoplan is the first treatment for paroxysmal nocturnal hemoglobinuria that binds to complement protein C3.[1] Pegcetacoplan was approved for medical use in the United States in May 2021.[1][3]
Pegcetacoplan is a complement inhibitor indicated in the treatment of paroxysmal nocturnal hemoglobinuria (PNH).5,7 Prior to its FDA approval, patients with PNH were typically treated with the C5 inhibiting monoclonal antibody eculizumab.5 Patients given eculizumab experienced less hemolysis caused by the membrane attack complex, but were still somewhat susceptible to hemolysis caused by C3b opsonization.5,6 Pegcetacoplan was developed out of a need for an inhibitor of complement mediated hemolysis further upstream of C5.5,6 Pegcetacoplan is a pegylated C3 inhibitor that can disrupt the processes leading to both forms of hemolysis that threaten patients with PNH.5
Pegcetacoplan was granted FDA approval on 14 May 2021.7
Medical uses
Pegcetacoplan is indicated to treat adults with paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
EMPAVELI contains pegcetacoplan, a complement inhibitor. Pegcetacoplan is a symmetrical molecule comprised of two identical pentadecapeptides covalently bound to the ends of a linear 40-kiloDalton (kDa) PEG molecule. The peptide portions of pegcetacoplan contain 1-methyl-L-tryptophan (Trp(Me)) in position 4 and amino(ethoxyethoxy)acetic acid (AEEA) in position 14.
The molecular weight of pegcetacoplan is approximately 43.5 kDa. The molecular formula is C1970H3848N50O947S4. The structure of pegcetacoplan is shown below.
 |
EMPAVELI injection is a sterile, clear, colorless to slightly yellowish aqueous solution for subcutaneous use and is supplied in a 20-mL single-dose vial. Each 1 mL of solution contains 54 mg of pegcetacoplan, 41 mg of sorbitol, 0.384 mg of glacial acetic acid, 0.490 mg of sodium acetate trihydrate, and Water for Injection USP. EMPAVELI may also contain sodium hydroxide and/or additional glacial acetic acid for adjustment to a target pH of 5.0.
FDA approves new treatment for adults with serious rare blood disease..
FDA has approved Empaveli (pegcetacoplan) injection to treat adults with paroxysmal nocturnal hemoglobinuria (PNH), a rare, life-threatening blood disease. Empaveli is the first PNH treatment that binds to compliment protein C3.
PNH is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells). The disease affects 1-1.5 people per million. Individuals are typically diagnosed around ages 35 to 40. PNH can be serious, with median survival of 10 years after diagnosis. However, some patients live for decades with only minor symptoms.
PNH is caused by gene mutations that affect red blood cells. Red blood cells in people with these mutations are defective and can be destroyed by the immune system, which causes anemia.
The effectiveness of Empaveli was evaluated in a study enrolling 80 patients with PNH and anemia who had been taking eculizumab, a treatment previously approved for PNH. Patients first completed a four-week period during which they received Empaveli 1,080 mg twice weekly in addition to eculizumab at their previous dose. After the first four weeks, patients were randomly assigned to receive either Empaveli or their current dose of eculizumab for 16 weeks.
After 16 weeks, the severity of anemia was compared in the two treatment groups on the basis of hemoglobin concentration (a laboratory measure of anemia). In both treatment groups, the average hemoglobin was 8.7 g/dL at baseline, indicating severe anemia. (Normal hemoglobin values in adult men are 14 g/dL or above; normal values in adult women are 12 g/dL or above.) During the 16 weeks of treatment, patients in the Empaveli group had an average increase in their hemoglobin of 2.4 g/dL. Meanwhile, patients in the eculizumab group had an average decrease in their hemoglobin of 1.5 g/dL.
Empaveli is available only through a restricted program under a risk evaluation and mitigation strategy. Meningococcal (a type of bacteria) infections can occur in patients taking Empaveli and can become life-threatening or fatal if not treated early. Empaveli may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria. Patients should be monitored for infusion-related reactions. Empaveli can interfere with certain laboratory tests. The most common side effects are injection site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.
Empaveli received priority review, fast track and orphan drug designations for this indication.
FDA granted the approval of Empaveli to Apellis Pharmaceuticals.
Adverse effects
Meningococcal (a type of bacteria) infections can occur in people taking pegcetacoplan and can become life-threatening or fatal if not treated early.[1] Pegcetacoplan may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria.[1]
History
The effectiveness of pegcetacoplan was evaluated in a study enrolling 80 participants with paroxysmal nocturnal hemoglobinuria and anemia who had been taking eculizumab, a treatment previously approved for paroxysmal nocturnal hemoglobinuria.[1]
References
- ^ Jump up to:a b c d e f g h i “FDA approves new treatment for adults with serious rare blood disease”. U.S. Food and Drug Administration (FDA). 14 May 2021. Retrieved 14 May 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d https://pi.apellis.com/files/PI_Empaveli.pdf
- ^ “Apellis Announces U.S. Food and Drug Administration (FDA) Approval of Empaveli (pegcetacoplan) for Adults with Paroxysmal Nocturnal Hemoglobinuria (PNH)” (Press release). Apellis Pharmaceuticals. 14 May 2021. Retrieved 14 May 2021 – via GlobeNewswire.
External links
- “Pegcetacoplan”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03500549 for “Study to Evaluate the Efficacy and Safety of APL-2 in Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Empaveli |
| Other names | APL-2 |
| License data | US DailyMed: Pegcetacoplan |
| Routes of administration | Subcutaneous infusion |
| Drug class | Complement inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 2019171-69-6 |
| UNII | TO3JYR3BOU |
| KEGG | D11613 |
| ChEMBL | ChEMBL4298211 |
| Chemical and physical data | |
| Formula | C170H248N50O47S4 |
| Molar mass | 3872.40 g·mol−1 |
/////////Pegcetacoplan, ペグセタコプラン , FDA 2021, APPROVALS 2021, APL-2, WHO 10743, Apellis Pharmaceuticals, Empaveli, priority review, fast track, orphan drug
https://www.sec.gov/Archives/edgar/data/1492422/000156459020007350/apls-10k_20191231.htm
NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}