
Home » NEW DRUGS (Page 3)
Category Archives: NEW DRUGS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Actelion wins crucial FDA approval for next-gen lung disease drug Opsumit

MACITENTAN
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide, CAS NO 441798-33-0
Late on Friday the FDA came through with an approval for Actelion’s pulmonary arterial hypertension (PAH) drug Opsumit (macitentan), its next-gen successor to the franchise drug Tracleer.
Read more: Actelion wins crucial FDA approval for next-gen lung disease drug Opsumit – FierceBiotech http://www.fiercebiotech.com/story/actelion-wins-crucial-fda-approval-next-gen-lung-disease-drug-opsumit/2013-10-18#ixzz2i7tDhpZT
Subscribe at FierceBiotech
Macitentan (Opsumit® )is a novel dual endothelin receptor antagonist that resulted from a tailored drug discovery process. Macitentan has a number of potentially key beneficial characteristics – i.e., increased in vivo preclinical efficacy vs. existing ERAs resulting from sustained receptor binding and tissue penetration properties. A clinical pharmacology program indicated a low propensity of macitentan for drug-drug interactions.
Macitentan is an investigational drug being studied for the treatment of pulmonary arterial hypertension. It acts as a dualendothelin receptor antagonist and is being developed by Actelion.[1] A Phase III clinical trial was successfully completed in 2012.[2]
on 22 October 2012 – Actelion (SIX: ATLN) announced that it has submitted a New Drug Application (NDA) to the US Food and Drug Administration (FDA) seeking approval for macitentan (Opsumit®) for the treatment of patients with pulmonary arterial hypertension
Actelion’s experimental lung drug macitentan prolonged overall survival by more than a third according to detailed study data, which the company hopes will convince investors it has a viable follow-up product to secure its commercial future.
Europe’s largest standalone biotech company wants the drug, which treats pulmonary arterial hypertension — a disease that causes high blood pressure in the arteries of the lungs — to replace blockbuster Tracleer.
Tracleer currently makes up 87 percent of sales but loses patent protection in 2015 and has also seen its market share eroded by Gilead’s Letairis.
Pharmacokinetics
Macitentan has an active metabolite, ACT-132577, which is an oxidative depropylation product. Both macitentan and ACT-132577 are mainly excreted in form of hydrolysis products via urine (about 2/3 of all metabolites) and faeces (1/3).[3]
Co-administration of ciclosporin has only a slight effect on the concentrations of macitentan and its active metabolite, whilerifampicin decreases the area under the curve (AUC) of the drug’s blood plasma concentration by 79%, and ketoconazoleapproximately doubles it. This corresponds to the finding that macitentan is mainly metabolised via the liver enzyme CYP3A4.[4]

- ^ Bolli, M. H.; Boss, C.; Binkert, C.; Buchmann, S.; Bur, D.; Hess, P.; Iglarz, M.; Meyer, S.; Rein, J.; Rey, M.; Treiber, A.; Clozel, M.; Fischli, W.; Weller, T. (2012). “The Discovery of N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (Macitentan), an Orally Active, Potent Dual Endothelin Receptor Antagonist”. Journal of Medicinal Chemistry 55 (17): 7849–7861. doi:10.1021/jm3009103. PMID 22862294. edit
- ^ “Macitentan”. Actelion. Retrieved 22 August 2012.
- ^ Bruderer, S.; Hopfgartner, G. R.; Seiberling, M.; Wank, J.; Sidharta, P. N.; Treiber, A.; Dingemanse, J. (2012). “Absorption, distribution, metabolism, and excretion of macitentan, a dual endothelin receptor antagonist, in humans”. Xenobiotica 42 (9): 901–910.doi:10.3109/00498254.2012.664665. PMID 22458347. edit
- ^ Bruderer, S.; Äänismaa, P. I.; Homery, M. C.; Häusler, S.; Landskroner, K.; Sidharta, P. N.; Treiber, A.; Dingemanse, J. (2011).“Effect of Cyclosporine and Rifampin on the Pharmacokinetics of Macitentan, a Tissue-Targeting Dual Endothelin Receptor Antagonist”. The AAPS Journal 14 (1): 68–78. doi:10.1208/s12248-011-9316-3. PMC 3282010. PMID 22189899. edit
External links
Actelion Ltd
Actelion Ltd is a biopharmaceutical company with its corporate headquarters in Allschwil/Basel, Switzerland. Actelion’s first drug Tracleer®, an orally available dual endothelin receptor antagonist, has been approved as a therapy for pulmonary arterial hypertension. Actelion markets Tracleer through its own subsidiaries in key markets worldwide, including the United States (based in South San Francisco), the European Union, Japan, Canada, Australia and Switzerland. Actelion, founded in late 1997, is a leading player in innovative science related to the endothelium – the single layer of cells separating every blood vessel from the blood stream. Actelion’s over 2,400 employees focus on the discovery, development and marketing of innovative drugs for significant unmet medical needs. Actelion shares are traded on the SIX Swiss Exchange (ticker symbol: ATLN) as part of the Swiss blue-chip index SMI (Swiss Market Index SMI®).
NovoEight (turoctocog alfa) Receives Approval from the FDA

Bagsværd, Denmark, 16 October 2013 – Today, Novo Nordisk announced that the U.S. Food and Drug Administration (FDA) has approved its Biologics License Application (BLA) for recombinant coagulation factor VIII, Novoeight.
The FDA approved Novoeight for use in adults and children with hemophilia A for:
- Control and prevention of bleeding
- Perioperative management
- Routine prophylaxis to prevent or reduce the frequency of bleeding episodes.
http://www.drugs.com/newdrugs/novoeight-turoctocog-alfa-receives-approval-fda-3931.html

turoctocog alfa (NN7008)
old clip
Novo Nordisk recently announced the company has submitted the regulatory application to the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for its turoctocog alfa (NN7008) for prevention and treatment of bleeding in people with hemophilia A.
The decision for submission was based on results from the guardian trials consisting of over 200 people with hemophilia A, making guardian the largest pre-registration clinical trial program for hemophilia A. The trials contained previously treated adults and children with severe hemophilia A.
Turoctocog alfa is a third-generation recombinant coagulation factor VIII drug, designed to increase reliability, safety and portability for patients with hemophilia A.
“We are very excited about having reached this goal. Turoctocog alfa represents a new treatment alternative for people with hemophilia A and is one of the first important outcomes of the hemophilia research strategy we embarked upon in 2006,” Mads Krogsgaard Thomsen, executive vice president and chief science officer of Novo Nordisk said in a news release.
The company said that in the next few months, it plans to submit applications for regulatory approval in other countries as well.
Hemophilia A is estimated to affect 500,000 people worldwide, and is extremely under-diagnosed in developing countries.
ema clip
On 19 September 2013, the Committee for Medicinal Products for Human Use(CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the medicinal product NovoEight 250, 500, 100, 1500, 2000 or 3000 IU, powder and solvent for solution for injection, intended for the treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor-VIII deficiency).The applicant for this medicinal product is Novonordisk. It may request are-examination of the CHMP opinion, provided it notifies the European Medicines Agency in writing of its intention within 15 days of receipt of the opinion.
The active substance of NovoEight is turoctocog alfa, human recombinant factor VIII that enables the temporary substitution of the endogenous coagulation factor VIII in haemophilia A patients. The benefits with NovoEight are its ability to prevent and treat the bleeds in previously treated patients with severe haemophilia A. The most common side effects are increase in hepatic enzymes and injection-site reaction.
A pharmacovigilance plan for NovoEight will be implemented as part of themarketing authorisation.
The approved indication is:
Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor-VIII deficiency).
It is proposed that NovoEight be prescribed by physicians experienced in the treatment of haemophilia A. It is proposed that treatment should be initiated under the supervision of a doctor experienced in the treatment of haemophilia.
Detailed recommendations for the use of this product will be described in thesummary of product characteristics (SmPC), which will be published in the European public assessment report (EPAR) and made available in all official European Union languages after the marketing authorisation has been granted by the European Commission.
The CHMP, on the basis of quality, safety and efficacy data submitted, considers there to be a favourable benefit-to-risk balance for NovoEight and therefore recommends the granting of the marketing authorisation.
The U.S. Food and Drug Administration approved Adempas (riociguat) to treat adults with two forms of pulmonary hypertension.

October 8, 2013 — The U.S. Food and Drug Administration today approved Adempas (riociguat) to treat adults with two forms of pulmonary hypertension.
Pulmonary hypertension is caused by abnormally high blood pressure in the arteries of the lungs. It makes the right side of the heart work harder than normal. In its various forms, pulmonary hypertension is a chronic, progressive, debilitating disease, often leading to death or need for lung transplantation
read all at
http://www.drugs.com/newdrugs/fda-approves-adempas-pulmonary-hypertension-3927.html
In the area of pulmonary hypertension Adempas (Riociguat) is the first member of a novel class of compounds – so-called ‘soluble guanylate cyclase (sGC) stimulators’ – being investigated as a new and specific approach to treating different types of pulmonary hypertension (PH). Adempas has the potential to overcome a number of limitations of currently approved treatments for pulmonary arterial hypertension (PAH) and addresses the unmet medical need in patients with chronic thromboembolic pulmonary hypertension (CTEPH). It was approved for the treatment of CTEPH in Canada in September 2013, making it the world’s first drug approved in this deadly disease.
Riociguat has already shown promise as a potential treatment option beyond these two PH indications. An early clinical study was conducted in PH-ILD (interstitial lung disease), a disease characterized by lung tissue scarring (fibrosis) or lung inflammation which can lead to pulmonary hypertension, and, based on positive data, the decision was taken to initiate Phase IIb studies in PH-IIP (idiopathic pulmonary fibrosis), a subgroup of PH-ILD. Moreover, scientific evidence was demonstrated in preclinical models that the activity may even go beyond vascular relaxation. To prove the hypothesis Bayer is initiating clinical studies in the indication of systemic sclerosis (SSc), an orphan chronic autoimmune disease of the connective tissue affecting several organs and associated with high morbidity and mortality. If successful, Riociguat has the potential to become the first approved treatment for this devastating disease.
synthesis
Generic Name: Riociguat
Trade Name: Adempas
Synonym: BAY 63-2521
CAS number: 625115-55-1
Chemical Name: Methyl N-[4,6-Diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate
Mechanism of Action: soluble guanylyl cyclase (sGC) stimulator
Date of Approval: October 8, 2013(US)
Indication: Pulmonary Hypertension
Company: Bayer AG

1)J. Mittendorf.; S. Weigand.; C. Alonso-Alija.; E. Bischoff.; A. Feurer.; M. Gerisch.; A. Kern.; A. Knorr.; D. Lang.; K. Muenter.; M. Radtke.; H. Schirok.; K.-H. Schlemmer.; E. Stahl.; A. Straub.; F. Wunder.; J.-P. Stasch. Discovery of Riociguat (BAY 63-2521): A Potent, Oral Stimulator of Soluble Guanylate Cyclase for the Treatment of Pulmonary Hypertension, ChemMedChem. 2009, 4, 853-865.
2)Cristina Alonso-Alija, Bayer Ag, Erwin Bischoff, Achim Feurer, Klaus Muenter, Elke Stahl, Johannes-Peter Stasch, Stefan Weigand, Carbamate-substituted pyrazolopyridines, WO2003095451 A1
3)Franz-Josef Mais, Joachim Rehse, Winfried Joentgen, Konrad SIEGEL, Process for preparing methyl methylcarbamate and its purification for use as pharmaceutically active compound,US20110130410
4)Claudia Hirth-Dietrich, Peter Sandner, Johannes-Peter Stasch, Andreas Knorr, Degenfeld Georges Von, Michael Hahn, Markus Follmann, The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc), WO 2011147810A1
5)Li Liang, Li Xing-zhou, Liu Ya-dan, Zheng Zhi-bing, Li Song, Synthesis of riociguat in treatment of pulmonary hypertension, Chinese Journal of Medicinal Chemistry(Zhongguo Yaowu Huaxue Zazhi), 21(2),120-125; 2011

Jens Ackerstaff, Lars BÄRFACKER, Markus Follmann, Nils Griebenow, Andreas Knorr, Volkhart Min-Jian Li, Gorden Redlich, Johannes-Peter Stasch, Stefan Weigand, Frank Wunder, Bicyclic aza heterocycles, and use thereof, WO2012028647 A1
2)Claudia Hirth-Dietrich, Peter Sandner, Johannes-Peter Stasch, Andreas Knorr, Degenfeld Georges Von, Michael Hahn, Markus Follmann, The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc), WO 2011147810A1

Jin Li, Xiaoyu Yang, Jingwei ZHU, Minmin Yang, Xihan Wu, Method for synthesizing 1-(2-fluorobenzyl)-1H -pyrazolo[3,4-b]pyridin -3-formamidine hydrochloride, WO2013086935 A1

veerareddy Arava, Surendrareddy Gogireddy, An expeditious synthesis of riociguat, A pulmonary hypertension drug, Der Pharma Chemica, 2013, 5(4):232-239
cut paste from my earlier post

RIOCIQUAT
CAS NO 625115-55-1
Methyl N-[4,6-Diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate
9 APRIL2013
Bayer has been boosted by the news that regulators in the USA are fast-tracking the German group’s investigational pulmonary arterial hypertension riociguat.
The US Food and Drug Administration has granted priority review to the New Drug Application for riociguat, which Bayer filed in February on both sides of the Atlantic for PAH and a related condition, inoperable chronic thromboembolic pulmonary hypertension (CTEPH). The FDA bestows a priority review on medicines that offer major advances in care or that provide a treatment where no adequate therapy exists. The agency aims to complete its assessment within eight months from the submission of the NDA, rather than the standard 12 months.
Riociguat (BAY 63-2521) is a novel drug that is currently in clinical development by Bayer. It is a stimulator of soluble guanylate cyclase (sGC). At the moment Phase III clinical trialsinvestigate the use of riociguat as a new approach to treat two forms of pulmonary hypertension (PH): chronic thromboembolic pulmonary hypertension (CTEPH) andpulmonary arterial hypertension (PAH). Riociguat constitutes the first drug of a novel class of sGC stimulators
The submissions are based on two Phase III studies and riociguat, the first member of a novel class of compounds called stimulators of soluble guanylate cyclase (sGC), met its primary endpoint in both trials, a change in exercise capacity after 12- or 16 weeks respectively. The drug was generally well tolerated, with a good safety profile.

If approved, riociguat would be going up against Actelion’s Tracleer (bosentan) and Gilead Sciences/GlaxoSmithKline’s Letairis/Volibris (ambrisentan). Actelion, which has dominated the PAH market, has already filed its follow-up to Tracleer, Opsumit (macitentan).

Orexigen files obesity drug Contrave for approval in Europe

Orexigen files obesity drug Contrave for approval in Europe
Orexigen Therapeutics has submitted the Marketing Authorization Application (MAA) for Contrave, an investigational weight-loss drug to the European Medicines Agency (EMA).
 The La Jolla, CA-based drug firm is using the EMA’s centralised procedure to seek approval for Contrave Orexigen (32 mg naltrexone sustained release (SR) / 360 mg bupropion SR) for the management of obesity, including weight loss and maintenance of weight loss, in conjunction with lifestyle modification. The company filed the application after meeting with the European agency to discuss the filing strategy and “both were supportive” of the company’s plan to file in advance of an eagerly-anticipated interim analysis of a cardiovascular outcomes trial called the Light study. Orexigen and the European regulator have also agreed upon an investigation plan in children and adolescents.
The La Jolla, CA-based drug firm is using the EMA’s centralised procedure to seek approval for Contrave Orexigen (32 mg naltrexone sustained release (SR) / 360 mg bupropion SR) for the management of obesity, including weight loss and maintenance of weight loss, in conjunction with lifestyle modification. The company filed the application after meeting with the European agency to discuss the filing strategy and “both were supportive” of the company’s plan to file in advance of an eagerly-anticipated interim analysis of a cardiovascular outcomes trial called the Light study. Orexigen and the European regulator have also agreed upon an investigation plan in children and adolescents.
read all at
http://www.pharmatopics.com/2013/10/orexigen-files-obesity-drug-contrave-approval-europe/
FDA Approves Pfizer’s Duavee

Pfizer Inc. Announces FDA Approval Of DUAVEETM (conjugated estrogens/ bazedoxifene) For The Treatment Of Moderate-To-Severe Vasomotor Symptoms (Hot Flashes) Associated With Menopause And The Prevention Of Postmenopausal Osteoporosis
DUAVEE is the first and only therapy to pair conjugated estrogens with an estrogen agonist/antagonist, also known as a selective estrogen receptor modulator (SERM) [2]
Thursday, October 3, 2013 – 4:14pm EDT
NEW YORK, N.Y., October 3 – Pfizer Inc. (NYSE: PFE), a leader in the development of treatments for menopausal symptoms, is pleased to announce that the United States Food and Drug Administration (FDA) has approved DUAVEETM (conjugated estrogens/bazedoxifene) 0.45mg / 20mg tablets, a novel therapy for women with a uterus, for the treatment of moderate-to-severe vasomotor symptoms associated with menopause and the prevention of postmenopausal osteoporosis
read all http://www.pharmalive.com/fda-approves-pfizer-s-duavee
Vortioxetine ボルチオキセチン 臭化水素酸塩 – FDA Approves Brintellix to Treat Major Depressive Disorder

Vortioxetine
ボルチオキセチン 臭化水素酸塩
1-[2-(2,4-dimethylphenyl)sulfanylphenyl]piperazine
Lu AA21004
| VORTIOXETINE; CAS 508233-74-7;
1-(2-((2,4-Dimethylphenyl)thio)phenyl)piperazine; Lu AA21004; UNII-3O2K1S3WQV; C18H22N2S; |
|
| Molecular Formula: | C18H22N2S |
|---|---|
| Molecular Weight: | 298.44568 g/mol |
Vortioxetine Hydrobromide

C18H22N2S.HBr : 379.36
[960203-27-4] HYDROBROMIDE
Vortioxetine is an atypical antipsychotic and antidepressant indicated for the treatment of major depressive disorder (MDD). It is classified as a serotonin modulator and simulator (SMS) as it has a multimodal mechanism of action towards the serotonin neurotransmitter system whereby it simultaneously modulates one or more serotonin receptors and inhibits the reuptake of serotonin. More specifically, vortioxetine acts via the following biological mechanisms: as a serotonin reuptake inhibitor (SRI) through inhibition of the serotonintransporter, as a partial agonist of the 5-HT1B receptor, an agonist of 5-HT1A, and an antagonist of the 5-HT3, 5-HT1D, and 5-HT7 receptors. SMSs were developed because there are many different subtypes of serotonin receptors, however, not all of these receptors appear to be involved in the antidepressant effects of SRIs. Some serotonin receptors seem to play a relatively neutral or insignificant role in the regulation of mood, but others, such as 5-HT1A autoreceptors and 5-HT7 receptors, appear to play an oppositional role in the efficacy of SRIs in treating depression.
Sept. 30, 2013 — The U.S. Food and Drug Administration today approved Brintellix (vortioxetine) to treat adults with major depressive disorder.
Major depressive disorder (MDD),
 Commonly referred to as depression, is a mental disorder characterized by mood changes and other symptoms that interfere with a person’s ability to work, sleep, study, eat and enjoy once-pleasurable activities. Episodes of depression often recur throughout a person’s lifetime, although some may experience a single occurrence.
Commonly referred to as depression, is a mental disorder characterized by mood changes and other symptoms that interfere with a person’s ability to work, sleep, study, eat and enjoy once-pleasurable activities. Episodes of depression often recur throughout a person’s lifetime, although some may experience a single occurrence.
READ ALL AT
http://www.drugs.com/newdrugs/fda-approves-brintellix-major-depressive-disorder-3918.html
The disease: Major depression
The developers: Lundbeck, Takeda
Vortioxetine (vor-tye-OX-e-teen, code name Lu AA21004) is an experimental drug currently under development by Lundbeck and Takeda for the treatment of major depressive disorder (MDD) and generalized anxiety disorder (GAD).Commercial names chosen are Brintellix and Rexulti.
Regulatory approval for the treatment of MDD for the European market has been filed in September 2012, for the United States in October 2012, and filing for Canada should follow. Filing for the Japanese market is expected in 2013.

Depression
In May 22 2011, Lundbeck presented the results of four phase III trials on vortioxetine at the 2011 Annual Meeting of the American Psychiatric Association. A statistically significant effect was shown in two of the studies (one for active treatment using the Hamilton Depression Rating Scale (HAM-D), the second as a maintenance treatment), vortioxetine failed to prove superiority over placebo in a third (again using the HAM-D) and the fourth was nullified by an exceptionally high placebo response (according to the Montgomery-Åsberg Depression Rating Scale (MADRS)).
In July 2011, Lundbeck published the results of a double-blind, randomized, placebo-controlled clinical trial with venlafaxine as an active reference. It was found to be superior to placebo in treating MDD while having fewer side effects than venlafaxine. Similarly, in May 2012, Lundbeck published the results of a double-blind, randomized, placebo-controlled clinical trial with duloxetine evaluating vortioxetine in elderly depressed patients, and it was found superior to placebo, with fewer side effects than duloxetine.
In May 2012, Lundbeck disclosed the results of three phase III clinical trials, showing vortioxetine’s superiority over placebo according to the MADRS.
In August 2012, a randomized, double-blind trial confirms the superiority of vortioxetine over placebo according to all measures, excepted the Sheehan Disability scale.
In September 2012, a randomised, double-blind trial reveals that a dose of 5mg shows superiority over placebo only in patients that suffer from comorbid anxiety.This is consistent with results from another trial published in December 2012, demonstrating that 2.5 mg and 5 mg doses are ineffective.
Anxiety
August 2012, contradictory results of two randomized, double-blind trial were published. While the first demonstrated vortioxetine’s superiority over the placebo, the second showed that the drug had no efficacy, leading the authors to question the designs of the different trials.

United States Patent Number: 7,144,884 , 8,476,279
related to Chinese patent: CN1319958 C , CN1561336 A; CN1319958C, CN1561336A
patent validity: January 9, 2023 (U.S. Patent Number: 7,144,884), October 2, 2022 (U.S. Patent No.: 8,476,279)
peak annual sales (estimated): $ 2 billion
drug companies: Lundbeck (Lundbeck), Takeda (Takeda)
Wal antidepressant drug Paxil (Brintellix, Vortioxetine) for – 1 – Preparation – [2 – (2,4 methyl) phenyl] piperazine process
Method II:
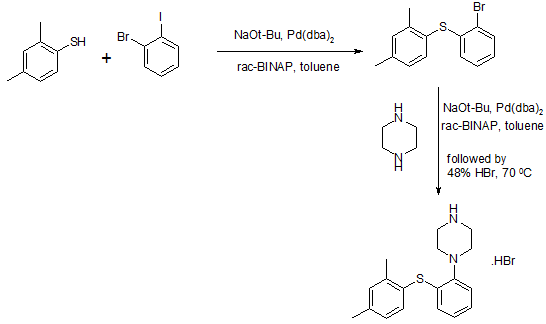
815g of the NaOBut (8,48 mo1), 844 g of piperazine Qin (9,8 mol), 6,6 g of Pd (dba) 2 (11,48 mmol) and 13,6 g of rac-BINAP (21, 84 mmol) was stirred for 50 minutes with 4L ofbenzene. Then, 840 g 2 – bromo – iodobenzene (2,97 mol) and 1.5L of Yue added to the mixture with benzene, and the stirring was continued for 30 minutes. Finally, 390.8g of 2,4 -thiophenol (2,83 mol) was added together with 1.5L toluene. The resulting suspension was heated to reflux and reflux was continued for 5 hours. The reaction mixture was cooled overnight. 2L of water was added and stirred for l hour and then filtered through a filter aid, the resulting mixture. Then, the filtrate was washed with brine 3xlL. Subsequently, the combined aqueous phase extracted with 600ml of benzene. Then, Yue The combined benzene phase was heated to 70 ° C, then adding 329.2ml 48-wt. / HBr (aq.) and 164.6ml water o’s. The mixture was cooled to room temperature overnight. Final product was collected by filtration (l-[2 – (2,4 – di曱group – phenylsulfanyl) – phenyl] – piperazine hydrobromide Qin), and dried under vacuum (60 0 C), to give 895g of product (84% yield).
Method III:
The benzene is placed 500ml three-necked 1L round bottom flask equipped with a mechanical stirrer and add 809mg Pd2dba3 (0.88mmol; 0.5 mol%) and 952 mg DPEPhos (1.77 mmol; 0.5mol-%). The deep red solution was purged with nitrogen for 5 minutes, then add 100g2-bromo-iodobenzene (353 mmol) and 48.9 g 2,4 – bis thiophenol (353 mmol). Add 43.6g KOBut (389 mmol) caused an exothermic reaction, so that the temperature rise of 20 ° C 42 ° C, while forming a non-uniform mixture, and the color changed from deep red to orange / brown. The force of the suspension under nitrogen was heated to port 100 ° C. After only 20 minutes, HPLC showed complete conversion to have l-(2 – bromo – phenylsulfanyl) -2,4 – Yue group – benzene. The mixture was cooled to 40 ° C, was added to 600ml 15-wt% NaCl, and stirred for 5 minutes. The organic phase was separated, and the aqueous phase was washed 2xl00mwith benzene. The combined organic phase was washed with HCl (aq) NaCl and washed with 100ml 2M 100ml 15-wt%, and then Na 2 S04 dried by activated charcoal (10 g) at reflux for 15 minutes, filtered twice and evaporated to 107.3 g of orange-red oil (103%), the oil was found by HPLC purity of 98%.
To 90 g of the orange-red oil (307 mmol) in 500ml of anhydrous toluene was added 57 g boc-piperazine Qin (307 mmol), degassed with nitrogen for 5 minutes, was added 1.4g Pd2dba3 (1.53 mmol- %; 0.5 mol%) and 2.9g mc-BINAP (4.6 mmol; 1.5 mol-%), degassed and then another 2 minutes, then add 35.4 g of NaOtBu (368 mmol), and heated to 80 ° C for 18 hours. HPLC showed complete conversion to have the reaction mixture was cooled to RT, filtered, and the filter cake was washed with 2 x 100ml of曱benzene. % NaCl, washed twice in Na2S04 dried, added charcoal, refluxed for 30 minutes, filtered twice and evaporated to 140.7 g of a brown oil (4 – – The combined filtrates with 2 x 150ml 15 [2 – (2, 4 – di曱group – phenylsulfanyl) -. phenyl]-BOC-piperazine Qin). The resulting crude oil was dissolved in 300ml MeOH and 200ml 6MHCl (aq.) and refluxed for l hour, after which HPLC showed complete deprotection. After cooling to RT, the vacuum on a rotary evaporator to remove曱alcohol was added 20ml of concentrated NaOH (pH was measured to 13-14), after which the mixture with 1000ml EtOAc – 15 minutes from stirring. The organic phase was collected and dried 300ml 15wtQ /. Saline extraction in Na2S04 dried, and added 46.3 g of fumaric acid in 300mlMeOH (399 mmol) was added. The mixture was heated to reflux, cooled to room temperature and then placed in the tank (-18. C) overnight. The precipitate was collected, washed with 100ml and 100ml of acetone with EtOAc, and dried in vacuo (50 ° C), to give 103.2g of l-[2 – (2,4 – di group – phenylsulfanyl) – phenyl] – piperazine. Qin fumarate (249mmo1), as a white powder, overall yield 81%, determined by LC-MS and the purity was 99% fumarate. Use EtOAc/H20 / concentrated NaOH to the fumarate salt into the free base (l-[2 – (2,4 – dimethyl – phenylsulfanyl) – phenyl] – piperazine Qin), The organic phase was washed with brine, dried over Na 2 S04 sulfate, filtered and to the filtrate was added 34ml48-wto / o of HBr (aq.), to form a white solid precipitated. The solid was collected, and the solid was washed with 1000ml H20 boiling process, the resultant was cooled to room temperature and purified by forming a slurry. The final product was collected by filtration (l-[2 – (2,4 – digroup – phenylsulfanyl) – phenyl] – piperazine hydrobromide Qin Kr), and dried in vacuo (50 ° C), to produce 83g of white powder (total yield 71%).
Source:
1) Bang-Andersen B, Ruhland T, Jørgensen M, Smith G, Frederiksen K, Jensen KG, Zhong H, Nielsen SM, Hogg S, Mørk A, Stensbøl TB “Discovery of 1 -. [2 – (2,4 – dimethylphenylsulfanyl) phenyl] PIPERAZINE (Lu AA21004): a novel multimodal Major Compound for the treatment of depressive disorder. ” Journal of Medicinal Chemistry 54 (9): 3206-21.
2) Thomas Ruhland, Garrick Paul Smith, Benny Bang-Andersen, Ask Puschl, Ejner Knud Moltzen, Kim Andersen,; Phenyl-piperazine derivatives as serotonin reuptake inhibitors; US patent number 7144884 ; also published as CA2462110A1, CA2462110C , CN1319958C, CN1561336A, DE60225162D1, DE60225162T2, DE60233608D1, EP1436271A1, EP1436271B1, EP1749818A2, EP1749818A3, EP1749818B1, US7138407, US7148238, US7683053, US8110567, US8476279, US20050014740, US20060084662, US20060089368, US20070060574, US20110009423, US20120302553, WO2003029232A1; H. Lundbeck A / S;
T · Rouland, G · P · Smith, B · Bang – Anderson, A · Pi Shier, E · K · Moore Cen, K · Anderson; as serotonin reuptake inhibitors phenyl piperazine derivatives matter; CN 1319958 C
T · Rouland, G · P · Smith, B · Bang – Anderson, A · Pi Shier, E · K · Moore Cen, K · Anderson; as serotonin reuptake inhibitors phenyl piperazine derivatives; CN 1561336 A
3) Benny Bang-Andersen; Phenyl-piperazine derivatives as serotonin reuptake inhibitors; US patent number 8476279 B2 ; Also published as CA2462110A1, CA2462110C, CN1319958C, CN1561336A, DE60225162D1, DE60225162T2, DE60233608D1, EP1436271A1, EP1436271B1, EP1749818A2, EP1749818A3, EP1749818B1, US7138407, US7144884, US7148238, US7683053, US8110567, US20050014740, US20060084662, US20060089368, US20070060574, US20110009423, US20120302553, WO2003029232A1; H. Lundbeck A / S;
4) Kim Lasse Christensen; Process for the manufacture of 1 – [2 – (2,4-dimethyl-phenylsulfanyl)-phenyl]-piperazine; PCT application, WO2013102573 A1
5) Benny Bang-Andersen, Joergen Brodersen, Andre Faldt, Rene Holm, Morten Joergensen, De Diego Heidi Lopez, Michael J Mealy, Arne Moerk, Nicholas Moore, Lone Munch Ringgaard, Michael Harold Rock, Tine Bryan Stensboel; 1 – [2 – (2, 4-dimethylphenylsulfanyl)-phenyl] piperazine as a compound with combined serotonin reuptake, 5-ht3 and 5-ht1a activity for the treatment of cognitive impairment; WO2007144005 A1
Updated oct 2015…………….

Vortioxetine (vor-tye-oks-e-teen, trade name Trintellix) is an atypical antidepressant (a serotonin modulator and stimulator) made by Lundbeck and Takeda.[1]
Vortioxetine [1-[2-(2,4-Dimethylphenyl-sulfanyl)-phenyl]-piperazine] is an orally administered small molecule developed as once-daily treatment of major depressive disorder (MDD) and generalized anxiety disorder (GAD). As a drug, Vortioxetine is a bis-aryl-sulphanyl amine compound that combines serotonin (5-HT) reuptake inhibition with other characteristics, including receptor activity modulation.
Animal and in vitro studies indicate that several neurotransmitter systems may be impacted by vortioxetine, with the drug enhancing levels of 5-HT, noradrenaline, dopamine, acetylcholine and histamine in certain areas of the brain, as well as modulating γ-aminobutyric acid and glutamate neurotransmission. Results from additional animal models suggest vortioxetine may also improve measures of cognitive function, such as memory. In healthy volunteers, single or repeated administration of vortioxetine (10 mg) did not impair cognitive function, psychomotor performance or driving ability in a placebo-controlled study.
Medical use
Vortioxetine is used as first-line treatment for major depressive disorder.[1][2][3][4][5]
Pharmacokinetics
Vortioxetine reaches peak plasma concentration (Cmax) within 7 to 11 hours post-administration (Tmax), and its mean terminal half-life (t½) is ≈ 66 hours. Steady-state plasma concentrations are typically reached within two weeks.[1] It has no active metabolites (i.e. it is not a prodrug).[1]
Research
Vortioxetine has been studied in several clinical trials as a potential treatment for general anxiety disorder but results were inconsistent.[9][10]
History
Vortioxetine was discovered by scientists at Lundbeck who reported the rationale and synthesis for the drug (then called Lu AA21004) in a 2011 paper.[7][11]
In 2007, the compound was in Phase II clinical trials, and Lundbeck and Takeda entered into a partnership in which Takeda paid Lundbeck $40 million upfront, with promises of up to $345 million in milestone payments, and Takeda agreed to pay most of the remaining cost of developing the drug. The companies agreed to co-promote the drug in the US and Japan, and that Lundbeck would receive a royalty on all such sales. The deal included another drug candidate, tedatioxetine (Lu AA24530), and could be expanded to include two other Lundbeck compounds.[12]
Vortioxetine was approved by the U.S. FDA for the treatment of major depressive disorder (MDD) in adults in September, 2013,[13] and it was approved in Europe later that year.[14]
Vortioxetine was previously trademarked as Brintellix in the United States, but on May 2, 2016, the US FDA approved a name change to Trintellix in order to avoid confusion with the blood-thinning medication ticagrelor (Brilinta).[15]
WO2015155153, SYNTHESIS OF VORTIOXETINE VIA (2,4-DIMETHYLPHENYL)(2-IODOPHENYL)SULFANE INTERMEDIATE
| LEK PHARMACEUTICALS D.D. [SI/SI]; Verovskova 57 1526 Ljubljana (SI) | |
| Inventors:ZUPANCIC, Borut; (SI) |
Vortioxetine is disclosed as Example 1 e in WO 2003/029232 A1 and is described as being prepared analogously to Example 1 . The process used to prepare Example 1 involves the preparation of 1 -(2-((2-(trifluoromethyl)phenyl)thio)phenyl)piperazine on a solid polystyrene support, followed by decomplexation using visible light irradiation, and purification by preparative LC-MS and ion-exchange chromatography. The overall yield for the preparation of vortioxetine is described as 17%.
Several alternative palladium catalyzed processes for the preparation of vortioxetine are described in Examples 17 to 25 of WO 2007/144005 A1 . These processes describe the preparation of vortioxetine from 2,4-dimethylthiophenol and 2-bromoiodobenzene (or 1 ,2-dibromobenzene) starting materials via a 1 -(2-bromo-phenylsulfanyl)-2,4-dimethyl-benzene intermediate. Each of these processes involves the use of a palladium catalyst and a phosphine ligand.
The preparation of vortioxetine is also described by Bang-Andersen et al. in J. Med. Chem. (201 1 ), Vol. 54, 3206-3221 . Here, in a first step, te/t-butyl 4-(2-bromophenyl)piperazine-1 -carboxylate intermediate is prepared from Boc-piperazine and 2-bromoiodobenzene in a palladium catalyzed coupling reaction. te/t-Butyl 4-(2-bromophenyl)piperazine-1 -carboxylate is then reacted with 2,4-dimethylthiophenol, again in the presence of palladium catalyst and a phosphine ligand, to provide Boc-protected vortioxetine. In the final step, vortioxetine is deprotected using hydrochloric acid to give vortioxetine hydrochloride.
WO 2013/102573 A1 describes a reaction between 1 -halogen-2,4-dimethyl-phenyl, 2-halogen-thiophenol and an optionally protected piperazine in the presence of a base and a palladium catalyst consisting of a palladium source and a phosphine ligand.
Each of the above processes has disadvantages. The process described in WO 2003/029232 is low yielding and unsuitable for the large scale production of vortioxetine, whereas the processes described in WO 2007/144005 A1 , WO 2013/102573 A1 and by Bang-Andersen et al. require the use of expensive starting materials, palladium catalyst and phosphine ligand. In addition, the toxicity of palladium is well known, Liu et al. Toxicity of Palladium, Toxicology Letters, 4 (1979) 469-473, and the European Medicines Agency’ s Guideline on the Specification for Residues of Metal Catalysts sets clear limits on the permitted daily exposure to palladium arising from palladium residue within drug substances, http://www.ema.europa.eu. Thus it would be desirable to avoid the use of a palladium catalyst in the synthesis of vortioxetine and the subsequent purification steps required to remove palladium residue from the final pharmaceutical product.
The invention is described below in further detail by embodiments, without being limited thereto.
A general concept of the process of the present invention may be represented in Scheme 1 .

Scheme 1 : General representation of the basic synthetic concept of the present invention.
Scheme 2.

X = NH2: lb
Scheme 2: Representation of a particular synthetic embodiment of the present invention.
Compound III can also be prepared from 2,4-dimethylbenzenethiol (II) and 1 -fluoro-2-nitrobenzene (l”‘a) or 1 -chloro-2-nitrobenzene (l'”b). In the first step (2,4-dimethylphenyl)(2- nitrophenyl)sulfane (III’) is formed and in the second reaction step nitro group is reduced to ami

Z = F: l”‘a
Z = CI: l”‘b
Scheme 3: Representation of a particular synthetic embodiment of the present invention.
Example 7: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine vortioxetine, VII)

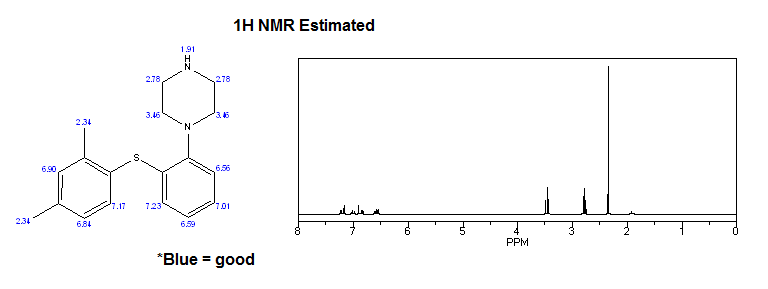
Mixture of (2,4-dimethylphenyl)(2-iodophenyl)sulfane V (0.34 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and 2-phenylphenol (68 mg, 0.4 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120°C for 20 h. Water (10 mL) is then added and product is extracted to EtOAc (3 x 10 mL). Combined organic layers were washed with water (3 x 10 mL) and brine (2 x 10 mL) and dried over Na2S04. After evaporation of the solvent crude product is purified by chromatography to afford title compound: H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H), 3.02-
3.09 (m, 8H), 6.52 (m, 1 H), 6.87 (m, 1 H), 7.04 (m, 1 H), 7.06-7.10 (m, 2H), 7.16 (m, 1 H), 7.39 (d, J= 7.8 Hz, 1 H); MS (ESI) m/z: 299 [MH]+.
Example 8: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine (vortioxetine, VII)

Mixture of (2,4-dimethylphenyl)(2-iodophenyl)sulfane V (0.34 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and N,N-diethyl-2-hydroxybenzamide (39 mg, 0.2 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120 ^ for 20 h. Water (10 mL) is then added and product is extracted to EtOAc (3 x 10 mL). Combined organic layers were washed with water (3 x 10 mL) and brine (2 x 10 mL) and dried over Na2S04. After evaporation of the solvent crude product is purified by chromatography to afford title compound: H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H), 3.02-3.09 (m, 8H), 6.52 (m, 1 H), 6.87 (m, 1 H), 7.04 (m, 1 H), 7.06-7.10 (m, 2H), 7.16 (m, 1 H), 7.39 (d, J= 7.8 Hz, 1 H); MS (ESI) m/z: 299 [MH]+.
Example 9: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine hydrobromide
(vortioxetine HBr, VII.HBr)

To a solution of vortioxetine VII (1 .80 g, 6.03 mmol) in iPrOAc (20 mL) at room temperature 48% HBr (0.68 mL, 6.03 mmol) was slowly added. Obtained mixture was stirred at room temperature for 1 h, white precipitate was then filtered off, washed with acetone (2 x 20 mL), and dried to afford title compound VII.HBr as a white powder (2.15 g, 94% yield): H NMR (DMSO-d6, 500 MHz) δ 2.23 (s, 3H), 2.32 (s, 3H), 3.15-3.27 (m, 8H), 6.40 (m, 1 H), 6.96 (m, 1 H), 7.08-7.17 (m, 3H), 7.24 (m, 1 H), 7.32 (d, J= 7.8 Hz, 1 H), 8.85 (br, 2H).
Reference Example 1 : Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of piperazine (1 .0 g, 1 1 .6 mmol), NaOtBu (1 .37 g, 13.8 mmol), Pddba2 (40 mg, 0.07 mmol), and 1 ,3-bis(2,6-di-i-propylphenyl)imidazolium chloride (24 mg, 0,07 mmol) in dry and degassed toluene (10 mL) is stirred at room temperature for 1 h. (2,4-Dimethylphenyl)(2-iodophenyl)sulfane V (1 .32 g, 3.86 mmol) is then added, reaction mixture is heated to l OO’C and stirred for 24 h. After cooling to room temperature to the reaction mixture water (5 mL) and Celite (0.4 g) is added. After stirring for 20 min salts are filtered off, organic layer is separated, washed with brine (2 x 10 mL), dried over Na2S04 and solvent is evaporated to afford crude product, which is then purified by chromatography to afford title compound as yellowish crystals: H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H),
Reference Example 2: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of piperazine (1 .29 g, 15.0 mmol), NaOtBu (1 .77 g, 17.8 mmol), Pddba2 (52 mg, 0.09 mmol), and rac-BINAP (93 mg, 0,15 mmol) in dry and degassed toluene (10 mL) was stirred at room temperature for 1 h. (2,4-Dimethylphenyl)(2-iodophenyl)sulfane V (1 .70 g, 5.0 mmol) was then added, reaction mixture was heated to 100°C and stirred for 24 h. After cooled to room temperature to the reaction mixture water (5 mL) and Celite (0.4 g) were added. After stirring for 20 min salts were filtered off, organic layer was separated, washed with brine (2 x 10 mL), dried over Na2S04 and solvent was evaporated to afford product as an orange oil (1 .41 g, 95% yield): H NMR (CDCI3, 500 MHz) δ 1 .63 (br s, 1 H), 2.33 (s, 3H), 2.37 (s, 3H), 3.02-3.09 (m, 8H), 6.52 (m, 1 H), 6.87 (m, 1 H), 7.04 (m, 1 H), 7.06-7.10 (m, 2H), 7.16 (m, 1 H), 7.39 (d, J = 7.8 Hz, 1 H); MS (ESI) m/z: 299 [MH]+.
Comparative Example 1 : Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of (2,4-dimethylphenyl)(2-bromohenyl)sulfane V” (0.29 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and 2-phenylphenol (68 mg, 0.4 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120°C for 20 h. Vortioxetine VII was not formed.
Comparative Example 2: Preparation of 1 -(2-((2,4-dimethylphenyl)thio)phenyl)piperazine
(vortioxetine, VII)

Mixture of (2,4-dimethylphenyl)(2-bromophenyl)sulfane V (0.29 g, 1 .0 mmol), piperazine VI (0.13 g, 1 .5 mmol), K3P03 (0.42 g, 2.0 mmol), Cul (19 mg, 0.1 mmol), and N,N-diethyl-2-hydroxybenzamide (39 mg, 0.2 mmol) in dry and degassed DMSO (2 mL) was heated under nitrogen atmosphere at 120 ^ for 20 h. Vortioxetine VII was not formed.
WO2007144005A1: Industrial process

Discovery of 1-[2-(2,4-Dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): A Novel Multimodal Compound for the Treatment of Major Depressive Disorder

The synthesis and structure−activity relationship of a novel series of compounds with combined effects on 5-HT3A and 5-HT1A receptors and on the serotonin (5-HT) transporter (SERT) are described. Compound 5m (Lu AA21004) was the lead compound, displaying high affinity for recombinant human 5-HT1A (Ki = 15 nM), 5-HT1B (Ki = 33 nM), 5-HT3A (Ki = 3.7 nM), 5-HT7 (Ki = 19 nM), and noradrenergic β1 (Ki = 46 nM) receptors, and SERT (Ki = 1.6 nM). Compound 5mdisplayed antagonistic properties at 5-HT3A and 5-HT7 receptors, partial agonist properties at 5-HT1B receptors, agonistic properties at 5-HT1A receptors, and potent inhibition of SERT. In conscious rats, 5m significantly increased extracellular 5-HT levels in the brain after acute and 3 days of treatment. Following the 3-day treatment (5 or 10 (mg/kg)/day) SERT occupancies were only 43% and 57%, respectively. These characteristics indicate that 5m is a novel multimodal serotonergic compound, and 5m is currently in clinical development for major depressive disorder.
1-[2-(2,4-Dimethylphenylsulfanyl)phenyl]piperazine Hydrochloride (5m)
ALERT HYDROCHLORIDE DATA
References
- US Label Last updated July 2014 after review in September, 2014. Versions of label are available at FDA index page Page accessed January 19, 2016
- [No authors listed] Vortioxetine. Aust Prescr. 2015 Jun;38(3):101-2. PMID 26648632Free full text
- “Relative efficacy and tolerability of vortioxetine versus selected antidepressants by indirect comparisons of similar clinical studies.”. Curr Med Res Opin 30: 2589–606. Oct 10, 2014. doi:10.1185/03007995.2014.969566. PMID 25249164.
- Köhler S, Cierpinsky K, Kronenberg G, Adli M. The serotonergic system in the neurobiology of depression: Relevance for novel antidepressants. J Psychopharmacol. 2016 Jan;30(1):13-22. PMID 26464458
- Kelliny M, Croarkin PE, Moore KM, Bobo WV. Profile of vortioxetine in the treatment of major depressive disorder: an overview of the primary and secondary literature. Ther Clin Risk Manag. 2015 Aug 12;11:1193-212. PMID 26316764 Free full text
- “Lundbeck’s “Serotonin Modulator and Stimulator” Lu AA21004: How Novel? How Good? – GLG News”.
- ^ Jump up to:a b c Bang-Andersen B, Ruhland T, Jørgensen M, et al. (May 2011). “Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder”. Journal of Medicinal Chemistry 54 (9): 3206–21. doi:10.1021/jm101459g. PMID 21486038.
- N. Moore; B. Bang-Andersen; L. Brennum; K. Fredriksen; S. Hogg; A. Mork; T. Stensbol; H. Zhong; C. Sanchez; D. Smith (August 2008). “Lu AA21004: a novel potential treatment for mood disorders”. European Neuropsychopharmacology 18 (Supplement 4): S321.doi:10.1016/S0924-977X(08)70440-1.
- Pae CU et al. Vortioxetine, a multimodal antidepressant for generalized anxiety disorder: a systematic review and meta-analysis. J Psychiatr Res. 2015 May;64:88-98. PMID 25851751
- Reinhold JA, Rickels K. Pharmacological treatment for generalized anxiety disorder in adults: an update. Expert Opin Pharmacother. 2015;16(11):1669-81. PMID 26159446
- Sanchez C, Asin KE, Artigas F Vortioxetine, a novel antidepressant with multimodal activity: review of preclinical and clinical data. Pharmacol Ther. 2015 Jan;145:43-57. PMID 25016186 Free full text
- Daniel Beaulieu for First Word Pharma. September 5th, 2007 Lundbeck, Takeda enter strategic alliance for mood disorder, anxiety drugs
- FDA approves new drug to treat major depressive disorder, U.S. Food and Drug Administration Press Announcement.
- EMA Brintellix page at EMA site Page accessed January 19, 2016
- Commissioner, Office of the. “Safety Alerts for Human Medical Products – Brintellix (vortioxetine): Drug Safety Communication – Brand Name Change to Trintellix, to Avoid Confusion With Antiplatelet Drug Brilinta (ticagrelor)”. http://www.fda.gov. Retrieved2016-05-02.
| Patent ID | Date | Patent Title |
|---|---|---|
| US7683053 | 2010-03-23 | PHENYL-PIPERAZINE DERIVATIVES AS SEROTONIN REUPTAKE INHIBITORS |
| US7148238 | 2006-12-12 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| US7144884 | 2006-12-05 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| US7138407 | 2006-11-21 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2013184291 | 2013-07-18 | THERAPEUTIC USES OF 1-[2-(2,4-DIMETHYL-PHENYLSUFLANYL)PHENYL]PIPERAZINE |
| US8476279 | 2013-07-02 | Phenyl-piperazine derivatives as serotonin reuptake inhibitors |
| US2013115292 | 2013-05-09 | ENTERIC TABLET |
| US2012189697 | 2012-07-26 | COMPOSITIONS OF 1-[2-(2,4-DIMETHYL-PHENYLSULFANYL)-PHENYL]PIPERAZINE |
| US2012035188 | 2012-02-09 | LIQUID FORMULATIONS OF SALTS OF 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)PHENYL]-PIPERAZINE |
| US8110567 | 2012-02-07 | PHENYL-PIPERAZINE DERIVATIVES AS SEROTONIN REUPTAKE INHIBITORS |
| US2012004409 | 2012-01-05 | Purification of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine |
| US2011201617 | 2011-08-18 | Therapeutic Uses Of Compounds Having Combined SERT, 5-HT3 And 5-HT1A Activity |
| US2011009422 | 2011-01-13 | 1- [2-(2,4-DIMETHYLPHENYLSULFANYL)-PHENYL] PIPERAZINE AS A COMPOUND WITH COMBINED SEROTONIN REUPTAKE, 5-HT3 AND 5-HT1A ACTIVITY FOR THE TREATMENT OF PAIN OR RESIDUAL SYMPTOMS IN DEPRESSION RELATING TO SLEEP AND COGNITION |
| US2010297240 | 2010-11-25 | 1- [2- (2,4-DIMETHYLPHENYLSULFANYL)-PHENYL] PIPERAZINE AS A COMPOUND WITH COMBINED SEROTONIN REUPTAKE, 5-HT3 AND 5-HT1A ACTIVITY FOR THE TREATMENT OF COGNITIVE IMPAIRMENT |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015094316 | 2015-04-02 | LIQUID FORMULATIONS OF SALTS OF 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)PHENYL]PIPERAZINE |
| US8969355 | 2015-03-03 | 1-[2-(2,4 dimethylphenylsulfanyl)-phenyl]piperazine as a compound with combined serotonin reuptake, 5-HT3 and 5-HT1a activity for the treatment of cognitive impairment |
| US2015005318 | 2015-01-01 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014371453 | 2014-12-18 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014343287 | 2014-11-20 | Process for the Manufacture of 1-[2-(2,4-Dimethyl-Phenylsulfanyl)-Phenyl]-Piperazine |
| US2014315921 | 2014-10-23 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014256943 | 2014-09-11 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014248355 | 2014-09-04 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014248356 | 2014-09-04 | 1-[2-(2,4-Dimethylphenylsulfanyl)-Phenyl]Piperazine As A Compound With Combined Serotonin Reuptake, 5-HT3 And 5-HT1a Activity For The Treatment Of Cognitive Impairment |
| US2014163043 | 2014-06-12 | PHENYL-PIPERAZINE DERIVATIVES AS SEROTONIN REUPTAKE INHIBITORS |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016083359 | 2016-03-24 | 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)-PHENYL]PIPERAZINE AS A COMPOUND With COMBINED SEROTONIN REUPTAKE, 5-HT3 AND 5-HT1A ACTIVITY FOR THE TREATMENT OF COGNITIVE IMPAIRMENT |
| US2016060215 | 2016-03-03 | New Process For The Synthesis Of 1-(2-((2,4-Dimethylphenyl)Thio)Phenyl)Piperazine |
| US2016015706 | 2016-01-21 | CRYSTALLINE FORMS OF AN ANTIDEPRESSANT COMPOUND |
| US2016009670 | 2016-01-14 | VORTIOXETINE MANUFACTURING PROCESS |
| US9211288 | 2015-12-15 | Compositions comprising vortioxetine and donepezil |
| US2015297585 | 2015-10-22 | COMPOSITIONS COMPRISING VORTIOXETINE AND DONEPEZIL |
| US2015266841 | 2015-09-24 | Novel Crystalline Form Of Vortioxetine Hydrobromide |
| US2015150867 | 2015-06-04 | COMPOSITIONS OF 1-[2-(2,4-DIMETHYL-PHENYLSULFANYL)-PHENYL]PIPERAZINE |
| US2015110873 | 2015-04-23 | ENTERIC TABLET |
| US2015094316 | 2015-04-02 | LIQUID FORMULATIONS OF SALTS OF 1-[2-(2,4-DIMETHYLPHENYLSULFANYL)PHENYL]PIPERAZINE |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
1-[2-(2,4-Dimethyl-phenylsulfanyl)-phenyl]piperazine
|
|
| Clinical data | |
| Trade names | Trintellix, Brintellix |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 75% (peak at 7–11 hours) |
| Protein binding | 98% |
| Metabolism | extensive hepatic, primarilyCYP2D6-mediated oxidation |
| Biological half-life | 66 hours |
| Excretion | 59% in urine, 26% in feces |
| Identifiers | |
| CAS Number | 508233-74-7 |
| ATC code | N06AX26 (WHO) |
| PubChem | CID 9966051 |
| IUPHAR/BPS | 7351 |
| ChemSpider | 8141643 |
| KEGG | D10184 |
| ChEBI | CHEBI:76016 |
| Synonyms | Lu AA21004 |
| Chemical data | |
| Formula | C18H22N2S |
| Molar mass | 298.45 g/mol (379.36 as hydrobromide) |
/////////////
-
CC(C=C(C)C=C1)=C1SC2=C(N3CCNCC3)C=CC=C2
FDA Approves Perjeta for Neoadjuvant Breast Cancer Treatment

The structure of HER2 and pertuzumab
pertuzumab
Sept. 30, 2013 — The U.S. Food and Drug Administration today granted accelerated approval to Perjeta (pertuzumab) as part of a complete treatment regimen for patients with early stage breast cancer before surgery (neoadjuvant setting). Perjeta is the first FDA-approved drug for the neoadjuvant treatment of breast cancer.
Perjeta was approved in 2012 for the treatment of patients with advanced or late-stage (metastatic) HER2-positive breast cancer. HER2-positive breast cancers have increased amounts of the HER2 protein that contributes to cancer cell growth and survival
cut paste of my old article
he European Medicines Agency (EMA) has approved Roche’s PERJETA (pertuzumab) for patients with previously untreated HER2-positive metastatic breast cancer (mBC)
MARCH 5, 2013 8:59 AM / 4 COMMENTS /
The structure of HER2 and pertuzumab
march 4, 2013
Pertuzumab (also called 2C4, trade name Perjeta) is a monoclonal antibody. The first of its class in a line of agents called “HER dimerization inhibitors”. By binding to HER2, it inhibits the dimerization of HER2 with other HER receptors, which is hypothesized to result in slowed tumor growth.[1] Pertuzumab received US FDA approval for the treatment of HER2-positive metastatic breast cancer on June 8, 2012.[2] Pertuzumab was developed at Genentech and is now owned by Roche which acquired Genentech in 2009.
Clinical trials
Early clinical trials of pertuzumab in prostate, breast, and ovarian cancers have been met with limited success.[3]
The dosage of pertuzumab used in the pivotal phase III CLEOPATRA (Clinical Evaluation of Pertuzumab and Trastuzumab) trial was as follows: IV 840 mg loading dose followed by IV 420 mg every three weeks.[4]
The pharmacokinetics of intravenous pertuzumab appear to be unaffected by age and no drug-drug interaction has been reported with docetaxel. The pharmacokinetics and pharmacodynamics of pertuzumab were summarized in a Feb 2012 review by Gillian Keating.[4]
The combination of pertuzumab plus trastuzumab plus docetaxel, as compared with placebo plus trastuzumab plus docetaxel, when used as first-line treatment for HER2-positive metastatic breast cancer, significantly prolonged progression-free survival, with no increase in cardiac toxic effects in the randomized, double-blind, multinational, phase III CLEOPATRA trial.[5]
Intravenous pertuzumab is currently being evaluated in patients with breast cancer in the following trials: MARIANNE (advanced breast cancer), NEOSPHERE (early breast cancer), TRYPHAENA (HER2-positive stage II/III breast cancer) and APHINITY (HER2-positive nonmetastatic breast cancer).[4]
References
- de Bono, Johann S.; Bellmunt, J; Attard, G; Droz, JP; Miller, K; Flechon, A; Sternberg, C; Parker, C et al. (20 January 2007). “Open-Label Phase II Study Evaluating the Efficacy and Safety of Two Doses of Pertuzumab in Castrate Chemotherapy-Naive Patients With Hormone-Refractory Prostate Cancer”. Journal of Clinical Oncology 25 (3): 257–262.doi:10.1200/JCO.2006.07.0888. PMID 17235043.
- “FDA Approves Perjeta (Pertuzumab) for People With HER2-Positive Metastatic Breast Cancer” (Press release). Genentech. Retrieved 2012-06-09.
- Genentech press release – May 15, 2005
- Keating GM. Pertuzumab: in the first-line treatment of HER2-positive metastatic breast cancer. Drugs 2012 Feb 12; 72 (3): 353-60.Link text
- Baselga J, Cortés J, Kim SB, and the CLEOPATRA Study Group. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 2012 Jan 12; 366 (2): 109-19. Link text
Stelara (ustekinumab) Receives FDA Approval to Treat Active Psoriatic Arthritis

Ustekinumab
| CAS No: | 815610-63-0 |
|---|---|
| Molecular Weight: | 145.64 g/mol |
| Chemical Formula: | C9H18N2O2 |
| IUPAC Name: | Immunoglobulin G1, anti-(human interleukin 12 p40 subunit) (human monoclonal CNTO 1275 gamma1-chain), disulfide with human monoclonal CNTO 1275 kappa-chain, dimer |
HORSHAM, Pa., Sept. 23, 2013 /PRNewswire/ — Janssen Biotech, Inc., announced today that the U.S. Food and Drug Administration (FDA) has approved Stelara (ustekinumab) alone or in combination with methotrexate for the treatment of adult patients (18 years or older) with active psoriatic arthritis. It is estimated that more than two million people in the U.S. are living with psoriatic arthritis, a chronic autoimmune disease characterized by both joint inflammation and psoriasis skin lesions
read all at
Ustekinumab (INN, experimental name CNTO 1275, proprietary commercial name Stelara, Centocor) is a human monoclonal antibody. It is directed against interleukin 12 and interleukin 23, naturally occurring proteins that regulate the immune system and immune-mediated inflammatory disorders.
Ustekinumab is a fully human monoclonal antibody (mAb) targeting the interleukin (IL)-12/23p40 subunit.
Interleukins are small soluble proteins that communicate between white blood cells (leukocytes), such as T cells. Interleukins mediate the differentiation, proliferation and many other processes of these cells. IL-12 and IL-23 are involved in the differentiation of naive T cells into T helper (Th) 1 and Th17 cells respectively.
Th1 and Th17 cells have been implicated in several autoimmune disorders, such as psoriasis. Ustekinumab targets the common p40 subunit of IL-12 and IL-23 to stop these cytokines from binding to their receptors and consequently preventing the development of Th1 and Th17 cells in an immune response.
In two Phase III trials for moderate to severe psoriasis, the longest >76 weeks, ustekinumab was safe and effective.
A third Phase III trial, ACCEPT, compared the efficacy and safety of ustekinumab with etanercept in the treatment of moderate to severe plaque psoriasis. This trial found a significantly higher clinical response with ustekinumab over the 12-week study period compared to high-dose etanercept. It also demonstrated the clinical benefit of ustekinumab among patients who failed to respond to etanercept.
Ustekinumab is approved in Canada, Europe and the United States to treat moderate to severe plaque psoriasis.
As of November 2009, the drug is being investigated for the treatment of psoriatic arthritis. It has also been tested in Phase II studies for multiple sclerosis and sarcoidosis, the latter versus golimumab (Simponi).
The US Food and Drug Administration (FDA) and European Union (EU) have approved the interleukin (IL) 12/23 inhibitor ustekinumab (Stelara, Janssen Biotech) for adults with active psoriatic arthritis who have not responded adequately to previous nonbiological disease-modifying antirheumatic drug therapy, the company announced today.
Approval of ustekinumab for psoriatic arthritis is “significant for patients and physicians as it marks the first treatment approved for this devastating and complex disease since the introduction of anti-TNF biologic medicines more than a decade ago,” Jerome A. Boscia, MD, vice president and head of immunology development, Janssen Research & Development, LLC, said in a statement.
The European Medicine Agency’s Committee for Medicinal Products for Human Use (CHMP) recommended approval of ustekinumab for active psoriatic arthritis in June, as reported by Medscape Medical News.
Ustekinumab is already approved in the US and EU for treatment of moderate to severe psoriatic plaques in adults. The drug, which can be used alone or in combination with methotrexate, is novel in that it targets both IL-12 and IL-23.
Image source: Crystal structure of human IL-12, Wikipedia, public domain

Ustekinumab binding to IL-12/23p40
FDA OKs Teva’s Injectable Treanda

FDA OKs Teva’s Injectable Treanda
FDA Approves Teva’s Injectable Treanda

bendamustine
Sept. 17, 2013 (GLOBES)–Teva Pharmaceutical Industries Ltd. (NYSE: TEVA; TASE: TEVA) has announced that the US Food and Drug Administration (FDA) has approved a new injectable version Treanda for treatment of indolent B-cell non-Hodgkin lymphoma that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen, and chronic lymphocytic leukemia. read all at
http://www.pharmalive.com/fda-oks-tevas-injectable-treanda
Bendamustine (INN, trade names Treakisym, Ribomustin, Levact and Treanda; also known as SDX-105) is a nitrogen mustard used in the treatment of chronic lymphocytic leukemia[1] and lymphomas. It belongs to the family of drugs called alkylating agents. It is also being studied for the treatment of sarcoma.[2]
History
Bendamustine was first synthesized in 1963 by Ozegowski and Krebs in East Germany(the former German Democratic Republic). Until 1990 it was available only in East Germany. East German investigators found that it was useful for treating chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myelomaand lung cancer.
Bendamustine received its first marketing approval in Germany, where it is marketed under the tradename Ribomustin, by Astellas Pharma GmbH’s licensee, Mundipharma International Corporation Limited. It is indicated as a single-agent or in combination with other anti-cancer agents for indolent non-Hodgkin’s lymphoma, multiple myeloma, and chronic lymphocytic leukemia. SymBio Pharmaceuticals Ltd holds exclusive rights to develop and market bendamustine HCl in Japan and selected Asia Pacific Rim countries.
In March 2008, Cephalon received approval from the United States Food and Drug Administration to market bendamustine in the US, where it is sold under the tradename Treanda, for treatment of chronic lymphocytic leukemia.[3]
In October 2008, the FDA granted further approval to market Treanda for the treatment of indolent B-cell non-Hodgkin’s lymphoma that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen. [4]
Bendamustine, 4-{5-[Bis(2-chloroethyl)amino]-1-methyl-2-benzimidazolyl}butyric acid:

was initially synthesized in 1963 in the German Democratic Republic (GDR) and was available from 1971 to 1992 there under the tradename Cytostasan®. See, e.g., W. Ozegowski and D. Krebs, IMET 3393 γ-[1-methyl-5-bis-(β-chloroethyl)-aminobenzimidazolo-(2)]-butyryl chloride, a new cytostatic agent of the group of benzimidazole nitrogen mustards. Zbl. Pharm. 110, (1971) Heft 10, 1013-1019, describing the synthesis of bendamustine hydrochloride monohydrate. Since that time, it has been marketed in Germany under the tradename Ribomustin®. Bendamustine is an alkylating agent that has been shown to have therapeutic utility in treating diseases such as chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer.
While bendamustine has been demonstrated as efficacious, it is known to be unstable, especially in aqueous solutions, leading to technical difficulties in its preparation and administration. Researchers, therefore, have investigated methods of improving the preparation and stability of bendamustine and its formulations. For example, German (GDR) Patent No. 159877 discloses a method for preparing bendamustine free base by reaction of the bis-hydroxyl precursor with thionyl chloride followed by recrystallization from water.
German (GDR) Patent No. 34727 discloses a method of preparing derivatives of bendamustine. The described derivatives differ from bendamustine in the substitution at the 1-position.
German (GDR) Patent No. 80967 discloses an injectable preparation of bendamustine hydrochloride monohydrate, ascorbic acid, and water. GDR 80967 describes that lyophilization of compounds such as bendamustine is only possible if the compound is of sufficient stability that it can withstand the processing conditions. The preparation described in GDR 80967 is not lyophilized.
German (GDR) Patent No. 159289 discloses a ready-to use, injectable solution of bendamustine hydrochloride that avoids lyophilization. GDR 159289 describes an anhydrous solution of bendamustine hydrochloride in 1,2-propylene glycol or ethanol.
U.S. application Ser. No. 11/330,868, filed Jan. 12, 2006, assigned to Cephalon, Inc., Frazer, P A, discloses methods of preparing lyophilized pharmaceutical compositions comprising bendamustine hydrochloride.
Chemotherapeutic uses
Bendamustine has been used both as sole therapy and in combination with other agents including etoposide, fludarabine, mitoxantrone,methotrexate, prednisone, rituximab, vincristine and 90Y-ibritumomab tiuxetan.
One combination for stage III/IV relapsed or refractory indolent lymphomas and mantle cell lymphoma (MCL), with or without prior rituximab-containing chemoimmunotherapy treatment, is bendamustine with mitoxantrone and rituximab.[5] In Germany in 2012 it has become the first line treatment of choice for indolent lymphoma.[6] after Trial results released in June 2012 showed that it more than doubled disease progression-free survival when given along with rituximab. The combination also left patients with fewer side effects than the older R-CHOP treatment.[7]
Common adverse reactions are typical for the class of nitrogen mustards, and include nausea, fatigue, vomiting, diarrhea, fever, constipation, loss of appetite, cough, headache, unintentional weight loss, difficulty breathing, rashes, and stomatitis, as well as immunosuppression, anemia, and low platelet counts. Notably, this drug has a low incidence of hair loss (alopecia) unlike most other chemotherapy drugs.[8]
References
- Kath R, Blumenstengel K, Fricke HJ, Höffken K (January 2001). “Bendamustine monotherapy in advanced and refractory chronic lymphocytic leukemia”. J. Cancer Res. Clin. Oncol. 127 (1): 48–54. doi:10.1007/s004320000180. PMID 11206271.
- Bagchi S (August 2007). “Bendamustine for advanced sarcoma”. Lancet Oncol. 8 (8): 674. doi:10.1016/S1470-2045(07)70225-5.PMID 17726779.
- “Cephalon press release – Cephalon Receives FDA Approval for TREANDA, a Novel Chemotherapy for Chronic Lymphocytic Leukemia”. Retrieved 2008-03-23.
- “Cephalon press release -Cephalon Receives FDA Approval for TREANDA to Treat Patients with Relapsed Indolent Non-Hodgkin’s Lymphoma”. Retrieved 2008-11-03.
- Weide R, Hess G, Köppler H, et al. (2007). “High anti–lymphoma activity of bendamustine/mitoxantrone/rituximab in rituximab pretreated relapsed or refractory indolent lymphomas and mantle cell lymphomas. A muticenter phase II study of the German Low Grade Lymphoma Study Group (GLSG)”. Leuk. Lymphoma. 48 (7): 1299–1306. doi:10.1080/10428190701361828. PMID 17613757.
- New Combo Replaces CHOP for Lymphoma. Dec 2012
- “‘Rediscovered’ Lymphoma Drug Helps Double Survival: Study”. June 3, 2012.
- Tageja, Nishant; Nagi, Jasdeepa; “Bendamustine: something old, something new”; Cancer Chemotherapy and Pharmacology, 2010 Aug;66(3):413-23. doi: 10.1007/s00280-010-1317-x.
External links
- Manufacturer’s official website intended for US patients
more info
Bendamustine hydrochloride, 4-{5-[Bis(2-chloroethyl) amino]- l-methyl-2- benzimidazolyl} butyric acid hydrochloride, of the formula (VI) :

was initially synthesized in 1963 in the German Democratic Republic (GDR) and was available from 1971 to 1992 there, as the hydrochloride salt, under the trade name Cytostasan®. Since that time, it has been marketed in Germany under the trade name Ribomustin®. Bendamustine Hydrochloride as injection is available in the United States under the tradename Treanda®. Bendamustine hydrochloride is an alkylating agent that is approved for the treatment of non-Hodgkin’s lymphoma, multiple myeloma and chronic lymphocytic leukemia.
Bendamustine hydrochloride is a benzimidazole analog. While bendamustine has been demonstrated as efficacious, it is known to be unstable, especially in aqueous solutions, leading to formation of non-bendamustine products (i.e. “degradation impurities”) which leads to technical difficulties in its preparation and administration. In light of its instability in aqueous solution, bendamustine is supplied as a lyophilized cake of bendamustine hydrochloride salt. US2006/159713, US 2006/128777 and WO2010/036702 disclose various impurities of Bendamustine hydrochloride which are as follows:

PC-1 PC-2
Jena et al. were the first to disclose the synthesis of Bendamustine hydrochloride in German (GDR) Patent No. 34727. Krueger et al. in German (GDR) Patent No. 159877 recite a method as summarized in scheme-1, for the synthesis of bendamustine hydrochloride comprising the reaction of the 4-[l-methyl-5-bis-(2- hydroxyethyl)-benzimidazolyl-2]butyric acid ethyl ester (4) (or the corresponding methyl, propyl or butyl ester) with thionyl chloride in chloroform at 0-5°C to form 4-[l- methyl-5-bis-(2-chloroethyl)-benzimidazolyl-2]butyric acid ethyl ester (5). Excess of thionyl chloride is destroyed by stirring the reaction mixture in aqueous HCl. Finally chloroform is distilled off and stirred at 95°C for 3 hours. The reaction mixture is partially concentrated and the residue is diluted with water and stirred upto crystallization. Further purification is done by recrystallization from water.
Scheme-1: Method disclosed by Krueger et al. in DD159877 for the synthesis of Bendamustine hydrochloride

Bendamustine hydrochloride (6)
Ozegowski et al in Zentralblatt fuer Pharmazie, Pharmakotherapie und Laboratoriumsdiagnostik 1 10 (10), 1013-1019 (1971) discloses a process for the preparation of bendamustine hydrochloride monohydrate. The Chinese journal “Chinese journal of New Drugs “, 2007, No. 23, Vol. 16, 1960-61 and J. Prakt. Chem. 20, 178-186 (1963) disclose another method for the synthesis of Bendamustine hydrochloride monohydrate starting from 2,4-dinitrochlorobenzene as summarized in scheme-2.

The crucial conversions are reaction of l-methyl-2-(4′-ethyl butyrate)-5- amino]-lH-benzimidazole 6 with ethylene oxide in the presence of water, sodium acetate and acetic acid, by maintaining at 5°C for 5 hours and overnight at 20°C to give 4-{5-[bis-(2-hydroxy-ethyl)-amino]-l-methyl-lH-benzimidazol-2-yl}-butyric acid ethyl ester (dihydroxy ester) 7 as a jelly mass, which on chlorination using thionyl chloride in chloroform and subsequent in situ hydrolysis with concentrated HCI gave bendamustine hydrochloride. It also discloses a process for the recrystallization of bendamustine hydrochloride from water and the product obtained is a monohydrate with a melting point of 148-151°C.
IP.com Journal 2009, 9(7B), 21 discloses another process as shown below for the preparation of ethyl-4-[5-[bis(2-hydroxyethyl) amino]- l-methylbenzimidazol-2- yl]butanoate (III) wherein ethyl-4-(5 -amino- 1 -methyl- lH-benzo[d]imidazol-2-yl) butanoate (II) is reacted with 2-halo ethanol in the presence of an inorganic base selected from the group consisting potassium carbonate, potassium bicarbonate, sodium

The PCT application WO 2010/042568 assigned to Cephalon discloses the synthesis of Bendamustine hydrochloride as summarized in schem-3 starting from 2,4- dintroaniline in six steps. The crucial step is reductive alkylation of Il-a, using borane- tetrahydrofuran and chloroacetic acid at ambient temperature, producing compound of formula I-a. Acid mediated hydrolysis of I-a using concentrated hydrochloric acid at reflux produced bendamustine hydrochloride which has a purity of 99.1%. The above PCT Patent application also discloses a method of purification of Bendamustine hydrochloride by agitating the Bendamustine hydrochloride in a mixture of DMF and THF at 75°C for about 30 minutes followed by cooling to ambient temperature and isolating the solid by filtration.
Scheme-3:


iil-a


Bemdamuatine hydrochloride
The PCT application WO 2011/079193 assigned to Dr. Reddy’s Laboratories discloses the synthesis of Bendamustine hydrochloride as summarized in schem-4 starting from compound of formula (II). The crucial step is alkylation of compound of formula II with 2-haloethanol in the presence of an organic base to give a compound of formula (III) which on chlorination with a chlorinating agent affords a compound of formula (IV). Compound of formula (IV) on hydrolysis in acidic medium gives bendamustine hydrochloride. It further discloses purification of bendamustine hydrochloride using aqueous hydrochloric acid and acetonitrile.
Scheme-4:

Bendamustine hydrochloride (Pure)
The most of the prior art processes described above involve
• The use of ethylene oxide for the preparation of bendamustine hydrochloride, which is often not suitable for industrial scale processes due to difficulty in handling ethylene oxide, since it is shipped as a refrigerated liquid.
• Further, the known processes involve the use of strongly acidic conditions and high temperatures for the hydrolysis of ethyl ester of bendamustine and subsequent in-situ formation of bendamustine hydrochloride, thereby resulting in increased levels of various process-related impurities IMP. -A (RRT-0.46), IMP. -B (RRT-1.27) and IMP. -C (RRT-1.31) whose removal is quite difficult and make the process less economically viable.

IMP.-B
International Application Publication No. WO 2009/120386 describes various solid forms of bendamustine hydrochloride designated as bendamustine hydrochloride Form 1, bendamustine hydrochloride Form 2, bendamustine hydrochloride Form 3, bendamustine hydrochloride Form 4, amorphous bendamustine hydrochloride or a mixture thereof, processes for their preparation and lyophilized composition comprising the solid forms. According to the disclosure, monohydrate of bendamustine hydrochloride has been prepared previously. The monohydrate has a reported melting point of 152-156°C which is similar to that of the observed melting point of bendamustine hydrochloride Form 2.
It is known that synthetic compounds can contain extraneous compounds or impurities resulting from their synthesis or degradation. The impurities can be unreacted starting materials, by-products of the reaction, products of side reactions, or degradation products. Generally, impurities in an active pharmaceutical ingredient (API) may arise from degradation of the API itself, or during the preparation of the API. Impurities in Bendamustine hydrochloride or any active pharmaceutical ingredient (API) are undesirable and might be harmful.
Regulatory authorities worldwide require that drug manufacturers isolate, identify and characterize the impurities in their products. Furthermore, it is required to control the levels of these impurities in the final drug compound obtained by the manufacturing process and to ensure that the impurity is present in the lowest possible levels, even if structural determination is not possible. The product mixture of a chemical reaction is rarely a single compound with sufficient purity to comply with pharmaceutical standards. Side products and byproducts of the reaction and adjunct reagents used in the reaction will, in most cases, also be present in the product mixture. At certain stages during processing of the active pharmaceutical ingredient, the product is analyzed for purity, typically, by HPLC, TLC. or GC analysis, to determine if it is suitable for continued processing and, ultimately, for use in a pharmaceutical product. Purity standards are set with the intention of ensuring that an API is as free of impurities as possible, and, thus, are as safe as possible for clinical use. The United States Food and Drug Administration guidelines recommend that the amounts of some impurities are limited to less than 0.1 percent.
Generally, impurities are identified spectroscopically and by other physical methods, and then the impurities are associated with a peak position in a chromatogram (or a spot on a TLC plate). Thereafter, the impurity can be identified by its position in the chromatogram, which is conventionally measured in minutes between injection of the sample on the column and elution of the particular component through the detector, known as the “retention time” (“RT”). This time period varies daily based upon the condition of the instrumentation and many other factors. To mitigate the effect that such variations have upon accurate identification of an impurity, practitioners use “relative retention time” (“RRT”) to identify impurities. The RRT of an impurity is its retention time divided by the retention time of a reference marker.
It is known by those skilled in the art, the management of process impurities is greatly enhanced by understanding their chemical structures and synthetic pathways, and by identifying the parameters that influence the amount of impurities in the final product.
Therefore, there remains a need for improved process for the preparation of bendamustine hydrochloride, producing high yield and purity, and well-suited for use on an industrial scale. Despite the existence of various polymorphic forms of bendamustine hydrochloride, there exists a need for a simple process for the preparation of the stable form of bendamustine hydrochloride which is amenable to scale up and results in high yield and purity.
Bendamustine, (4-{5-[bis(2-chloroethyl)amino]-1-methyl-2-benzimidazolyl}butyric

Bendamustine
is an atypical structure with a benzimidazole ring, which structure includes an active nitrogen mustard. Bendamustine was initially synthesized in 1963 in the German Democratic Republic and was available from 1971 to 1992 in that location under the name Cytostasan®. Since that time, it has been marketed in Germany under the tradename Ribomustin®. It is currently available for use in the United States under the tradename Treanda® (Cephalon, Inc., Frazer, Pa.). It has been widely used to treat chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer.
Like other nitrogen mustards, bendamustine hydrolyzes in aqueous solution, with the major degradant being the primary alcohol HP1 (See U.S. application Ser. No. 11/330,868, the entirety of which is incorporated herein):
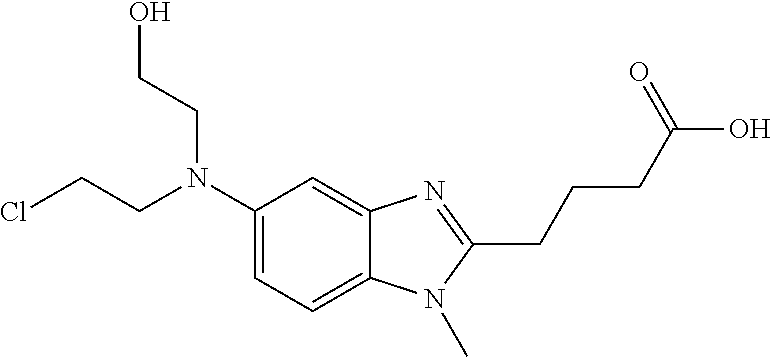
In light of its instability in aqueous solution, bendamustine is currently supplied as a lyophilized powder for injection. Just prior to its infusion, the medical practitioner reconstitutes the powder with Sterile Water for Injection. Reconstitution should yield a clear, colorless to pale yellow solution and the powder should completely dissolve in about 5 minutes. If particulate matter is observed, the reconstituted product should not be used and should be discarded. The reconstituted product is then transferred to a 0.9% Sodium Chloride Injection infusion bag within 30 minutes of reconstitution. This admixture should be a clear and colorless to slightly yellow solution. If the admixture comprises particulate matter or is discolored, it should be discarded and a fresh sample prepared.
The salt bendamustine hydrochloride is an alkylating agent, originally synthesized in 1963 at the Institute for Microbiology & Experimental Therapy in Jena, German Democratic Republic, with the intent to produce an agent with both alkylating and antimetabolite properties. Jenapharm (now Schering AG) formerly marketed it in Germany under the trade name Cytostasan from 1971 to 1992. Cytostasan was a lyophilised powder for solution for injection (vials) conatining 25 mg of Bendamustine HCI. It was widely used but never studied systematically in patients until the 1990s, then German investigators demonstrated its clinical activity in a number of malignancies. Since 1993, Ribosepharm was marketing bendamustine in Germany under the brand name Ribomustin RBO. Ribomustin is available as a lyophilized powder for injection, containing 100 mg of drug in each 50 ml_ vial, or 25 mg of drug in each 20 ml_ vial, also comprising mannitol, and indicated for the treatment of chronic lymphocytic leukemia. The lyophilized powder is reconstituted as close to the time of patient administration as possible with 40 ml_ (for a 100 mg product) or 10 mL (for a 25 mg product) of sterile water for injection. The reconstituted product then is further diluted to 500 mL with 0.9% sodium chloride for injection. The route of administration is by intravenous infusion over 30 to 60 minutes.
Another bendamustine product is sold in the United States by Cephalon, Inc. as TREANDA® for Injection, a lyophilized powder in a single-use vial indicated for the treatment of patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin’s lymphoma. A 25 mg dose vial contains 25 mg of bendamustine hydrochloride and 42.5 mg of mannitol, and a 100 mg dose vial contains 100 mg of bendamustine hydrochloride and 170 mg of mannitol.
TREANDA is intended for intravenous infusion only after reconstitution with Sterile Water for Injection USP, and then further dilution with either 0.9% Sodium
Chloride Inj.ection, USP, or 2.5% Dextrose/0.45% Sodium Chloride Inj.ection, USP. The pH of the reconstituted solution is 2.5-3.5. TREANDA is supplied as a sterile non-pyrogenic white to off-white lyophilized powder, in a single-use vial.
Bendamustine hydrochloride is very unstable in an aqueous solution. The bis-2-chlorethylamino bond is hydrolyzed in weak acid, neutral, or alkaline solution. Monohydroxybendamustine [HP-1 ] is formed rapidly in the presence of water. Bendamustine ethyl ester [BM1 EE] is formed when bendamustine reacts with ethyl alcohol. BM1 EE can be formed during drug substance manufacturing, e.g., during recrystalization and/or purification processes. BM1 EE is a more potent cytotoxic drug than bendamustine.
FDA Advisory Committee Recommends Approval in U.S. of Umeclidinium/Vilanterol for the Treatment of COPD
umeclidinium

vilanterol
09/10/13 — GlaxoSmithKline plc (LSE: GSK) and Theravance, Inc. (NASDAQ: THRX) today announced that the Pulmonary-Allergy Drugs Advisory Committee (PADAC) to the US Food and Drug Administration (FDA) voted 11 yes to 2 no that the efficacy and safety data provide substantial evidence to support approval of umeclidinium/vilanterolumeclidinium (UMEC/VI, 62.5/25mcg dose) for the long-term, once-daily, maintenance bronchodilator treatment of airflow obstruction in patients with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema.
Anoro Ellipta is the proposed proprietary name for UMEC/VI, a combination of two investigational bronchodilator molecules — GSK573719 or umeclidinium bromide (UMEC), a long-acting muscarinic antagonist (LAMA) and vilanterol (VI), a long-acting beta2 agonist (LABA), administered using the Ellipta inhaler.
The FDA Advisory Committee also voted that the safety of the investigational medicine has been adequately demonstrated at the 62.5/25mcg dose for the proposed indication (10 yes, 3 no), and the efficacy data provided substantial evidence of a clinically meaningful benefit for UMEC/VI 62.5/25mcg once daily for the long-term, maintenance treatment of airflow obstruction in COPD (13 yes, 0 no).
Patrick Vallance, GSK’s President of Pharmaceuticals R&D, said: “Today’s recommendation is good news and a reflection of our commitment to giving an alternative treatment option for patients living with COPD — a disease that affects millions of Americans. If approved, Anoro Ellipta will be the first, once-daily dual bronchodilator available in the US, marking another significant milestone for GSK’s portfolio of medicines to treat respiratory disease. We will continue to work with the FDA as they complete their review.”
“We are pleased with the Advisory Committee’s support of UMEC/VI,” said Rick E Winningham, Chief Executive Officer of Theravance. “This is a transformative year for Theravance and today’s positive recommendation brings the second major respiratory medicine in our GSK collaboration closer to approval and becoming an important therapeutic option for COPD patients.”
In December 2012, a New Drug Application (NDA) was submitted to the FDA for the use of UMEC/VI administered by the Ellipta™ inhaler for the long-term once-daily maintenance bronchodilator treatment of airflow obstruction in patients with COPD, including chronic bronchitis and/or emphysema. UMEC/VI is not proposed for the relief of acute bronchospasm or for the treatment of asthma in any of the regulatory applications.
The FDA Advisory Committee provides non-binding recommendations for consideration by the FDA, with the final decision on approval made by the FDA. The Prescription Drug User Fee Act (PDUFA) goal date for UMEC/VI is 18 December 2013.
UMEC/VI is an investigational medicine and is not currently approved anywhere in the world.
Safety Information
Across the four pivotal COPD studies for UMEC/VI, the most frequently reported adverse events across all treatment arms, including placebo, were headache, nasopharyngitis, cough, upper respiratory tract infection, and back pain. COPD exacerbation was the most common serious adverse event reported. In addition, in the four pivotal COPD studies, a small imbalance was observed in cardiac ischemia which was not observed in the long term safety study.
The UMEC/VI clinical development programme involved over 6,000 COPD patients.
About COPD
Chronic obstructive pulmonary disease (COPD) is a term referring to two lung diseases, chronic bronchitis and emphysema, that are characterized by obstruction to airflow that interferes with normal breathing. COPD is the third most common cause of death in the US and The National Heart, Lung and Blood Institute (NHLBI) estimates that nearly 15 million US adults have COPD and another 12 million are undiagnosed or developing COPD(1).
According to the NHLI, long-term exposure to lung irritants that damage the lungs and the airways are usually the cause of COPD and in the United States, the most common irritant that causes COPD is cigarette smoke. Breathing in second hand smoke, air pollution, or chemical fumes or dust from the environment or workplace also can contribute to COPD. Most people who have COPD are at least 40 years old when symptoms begin.

{kind=link}