
Home » Antineoplastic (Page 2)
Category Archives: Antineoplastic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Gozanertinib
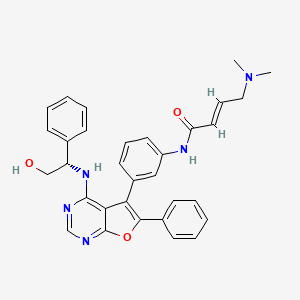
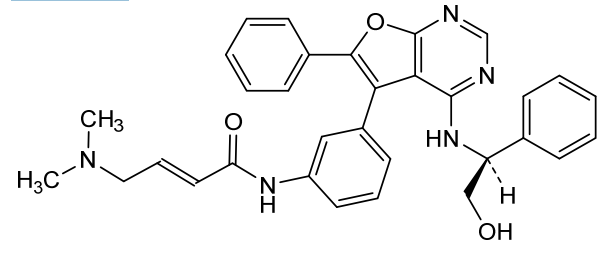
Gozanertinib
CAS 1226549-49-0
MF C32H31N5O3 MW533.6 g/mol
(E)-4-(dimethylamino)-N-[3-[4-[[(1S)-2-hydroxy-1-phenylethyl]amino]-6-phenylfuro[2,3-d]pyrimidin-5-yl]phenyl]but-2-enamide
(2E)-4-(dimethylamino)-N-[3-(4-{[(1S)-2-hydroxy-1-phenylethyl]amino}-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl]but-2-
enamide
epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, DBPR 112, ABT 101, 6G0COS33K4
Gozanertinib (also known as DBPR112 or ABT-101) is an orally bioavailable, advanced small-molecule dual kinase inhibitor designed to treat advanced non-small cell lung cancer (NSCLC). It targets alterations in the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) families.
Mechanism of Action
Gozanertinib is a furanopyrimidine-based tyrosine kinase inhibitor. It functions by entering the ATP-binding pocket of the receptor and forming an irreversible covalent bond with a specific cysteine residue (Cys797). By permanently blocking these receptors, it halts downstream oncogenic signaling pathways—specifically the RAS/RAF/MEK/ERK and PI3K/AKT cascades—thereby inducing cancer cell death and suppressing tumor expansion.
Target Profile and Key Mutations
Unlike earlier generations of tyrosine kinase inhibitors that only target standard configurations, gozanertinib is optimized to combat specific treatment-resistant mutations:
- EGFR Mutations: It effectively targets wild-type EGFR as well as the dual L858R/T790M resistance mutations.
- Exon 20 Insertions: A standout feature of gozanertinib is its preclinical potency against EGFR and HER2 exon 20 insertion (Ex20ins) mutations. According to chemical development findings published in the Journal of Medicinal Chemistry, it demonstrated ten times better potency against these specific insertions than the widely used third-generation inhibitor, osimertinib.
Development and Status
The drug was initially discovered through scaffold optimization by the National Health Research Institutes (NHRI) and is being co-developed with Anbogen Therapeutics. The International Nonproprietary Name (INN) “gozanertinib” was formally proposed for the compound in early 2025. Preclinical evaluations indicated favorable oral bioavailability and strong anti-tumor efficacy compared to older inhibitors like afatinib, advancing the compound into early-phase clinical trials
Gozanertinib is an orally bioavailable dual kinase inhibitor of epidermal growth factor receptor (EGFR; ErbB1) and human epidermal growth factor receptor 2 (HER2; EGFR2; ErbB2), including EGFR L858R, EGFR T790M and HER2 exon 20 insertion (Ex20ins) mutations, with potential antineoplastic activity. Upon oral administration, gozanertinib targets, binds to and inhibits the activity of EGFR or HER2 insertions or mutations. This prevents EGFR/HER2-mediated signaling, which may induce cell death and inhibit tumor growth in EGFR/HER2-overexpressing tumor cells. The ErbB receptor tyrosine kinase family is involved in key cellular functions, including cell growth and survival. EGFR and HER2 alterations constitutively upregulate kinase activity.
- Phase 1b/2 Study to Evaluate ABT-101 in Solid Tumor and NSCLC PatientsCTID: NCT05532696Phase: Phase 1/Phase 2Status: RecruitingDate: 2024-06-24
- A Study of DBPR112 in Patients With Head and Neck Cancer and EGFR Mutated Lung CancerCTID: NCT03246854Phase: Phase 1Status: TerminatedDate: 2020-12-17
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US43249513&_cid=P11-MQ1QG3-86325-1
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Development of Furanopyrimidine-Based Orally Active Third-Generation EGFR Inhibitors for the Treatment of Non-Small Cell Lung CancerPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-02-07PMCID: PMC9969398PMID: 36749735DOI: 10.1021/acs.jmedchem.2c01434
- Discovery of a Furanopyrimidine-Based Epidermal Growth Factor Receptor Inhibitor (DBPR112) as a Clinical Candidate for the Treatment of Non-Small Cell Lung CancerPublication Name: Journal of Medicinal ChemistryPublication Date: 2019-09-27PMID: 31560541DOI: 10.1021/acs.jmedchem.9b00722
PAT
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: EP-4248214-A1Priority Date: 2020-11-19
- Fused Bicyclic and Tricyclic Pyrimidine Compounds as Tyrosine Kinase InhibitorsPublication Number: US-2010120805-A1Priority Date: 2008-11-10
- Fused bicyclic and tricyclic pyrimidine compounds as tyrosine kinase inhibitorsPublication Number: US-8507502-B2Priority Date: 2008-11-10Grant Date: 2013-08-13
- Fused bicyclic and tricyclic pyrimidine compounds as tyrosine kinase inhibitorsPublication Number: WO-2010054285-A2Priority Date: 2008-11-10
- Fused bicyclic and polycyclic pyrimidine compounds as tyrosine kinase inhibitorsPublication Number: CN-102264745-APriority Date: 2008-11-10
- Active cancer immunotherapy through immune modulation via GLOBO series antigensPublication Number: CN-116847875-APriority Date: 2020-11-19
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: CA-3200572-A1Priority Date: 2020-11-19
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: WO-2022109601-A1Priority Date: 2020-11-19
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: IL-302947-APriority Date: 2020-11-19
- Active cancer immunotherapy by immunomodulation through GLOBO family antigensPublication Number: KR-20230110529-APriority Date: 2020-11-19
- Crystalline forms of (s, e)-4-(dimethylamino)-n-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: TW-I809967-BPriority Date: 2021-07-06Grant Date: 2023-07-21
- Crystalline forms of (S, E)-4-(dimethylamino)-N-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2- enamide free basePublication Number: US-12240858-B2Priority Date: 2021-07-06Grant Date: 2025-03-04
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: TW-202237177-APriority Date: 2020-11-19
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: US-2024139301-A1Priority Date: 2020-11-19
- Active cancer immunotherapy by immune modulation via globo series antigensPublication Number: AU-2021382807-A1Priority Date: 2020-11-19
- Crystalline forms of (s, e)-4-(dimethylamino)-n-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: US-2023021909-A1Priority Date: 2021-07-06
- Crystalline forms of (s, e)-4-(dimethylamino)-n-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: WO-2023283269-A1Priority Date: 2021-07-06
- Crystalline forms of (s, e)-4-(dimethylamino)-n-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: US-2024368175-A1Priority Date: 2021-07-06
- Crystalline forms of (s, e)-4-(dimethylamino)-n-(3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: TW-202309041-APriority Date: 2021-07-06
- Crystalline forms of (s, e)-4-(dimethylamino)-n- (3-(4-(2-hydroxy-1-phenylethylamino)-6-phenylfuro[2,3-d]pyrimidin-5-yl)phenyl)but-2-enamide free basePublication Number: EP-4330259-A1Priority Date: 2021-07-06
//////gozanertinib, ANAX LABS, epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, DBPR 112, ABT 101, 6G0COS33K4
Gintemetostat
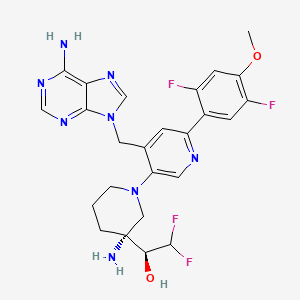
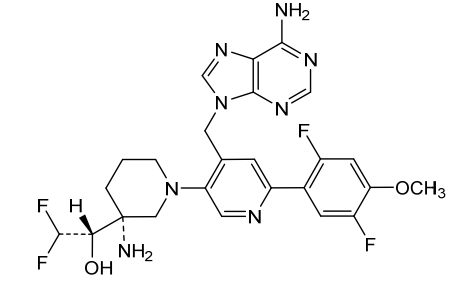
Gintemetostat
(1S)-1-[(3R)-3-amino-4′-[(6-amino-9H-purin-9-yl)methyl]-6′-(2,5-difluoro-4-methoxyphenyl)-3,4,5,6-tetrahydro-2H-[1,3′-bipyridin]-3-yl]-2,2-difluoroethan1-ol
antineoplastic, KTX 1001, NSD2 inhibitor 161, A48CGJ5UQM
CAS 2604513-16-6
MF C25H26F4N8O2 MW 546.5 g/mol
(S)-1-((R)-3-Amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6-(2,5-difluoro-4-methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol
Gintemetostat (also known as KTX-1001) is a first-in-class, orally administered small molecule being developed to treat relapsed and refractory multiple myeloma. It works as a selective inhibitor of NSD2 (also known as MMSET), targeting the epigenetic drivers of high-risk cancers.
How it Works
- Mechanism: Gintemetostat selectively binds to the catalytic SET domain of the NSD2 enzyme.
- Effect: By blocking this enzyme, it downregulates oncogenic signaling, decreases cancer cell growth, and can enhance T-cell activation against the tumor.
Target Patient Population
- High-Risk Myeloma: The drug focuses heavily on patients harboring the t(4;14) translocation, a genetic alteration found in 10-15% of patients that often causes aggressive relapses.
- Refractory Cases: It has shown notable single-agent activity in heavily pretreated patients who have exhausted standard-of-care, triple-class refractory treatment options.
Current Clinical Status
- Phase 1 Trial: Early data from phase 1 trials (such as NCT05651932) showed the drug has manageable safety profiles and offers clinical benefit (ranging from stable disease to very good partial response) in patients with aggressive, hard-to-treat multiple myeloma.
- Future Developments: Researchers are expanding studies to pair gintemetostat with other standard myeloma treatments, such as proteasome inhibitors and CELMoDs, to create stronger synergistic anti-cancer effects.
Gintemetostat is an orally available small molecule inhibitor of the histone-lysine N-methyltransferase nuclear receptor-binding SET domain protein 2 (NSD2; MMSET; WHSC1), with potential antineoplastic activity. Upon oral administration, gintemetostat selectively targets and binds to NSD2, and inhibits its catalytic activity and the mono- and di-methylation of histone H3 lysine 36 (H3K36). This modulates the expression of genes involved in cellular processes including cellular proliferation, which may lead to decreased growth of cancer cells. NSD2, a member of the NSD family of histone lysine methyltransferase enzymes that catalyzes the mono- and di-methylation of H3K36, is overexpressed and dysregulated in many types of cancers.
SYN
Discovery of a Highly Potent and Selective Inhibitor Targeting Protein Lysine Methyltransferase NSD2
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2024-09-04
PMID: 39230932
DOI: 10.1021/acs.jmedchem.4c00639
SYN
PAT
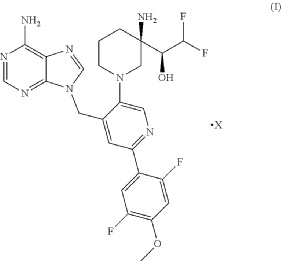
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021028854&_cid=P12-MQ0AZT-13511-1
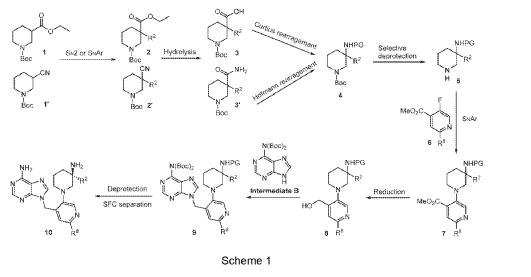
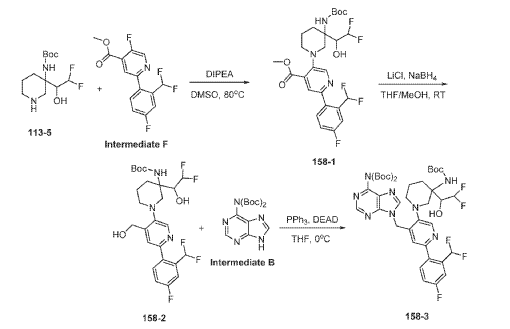
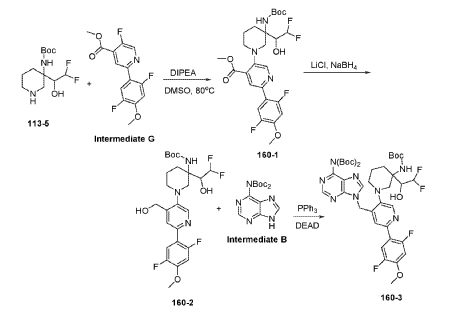
Example 160 and Example 161: (R)-1-((R)-3-amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6- (2,5-difluoro-4-methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol and (S)-1-((R)-3- amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6-(2,5-difluoro-4-methoxyphenyl)pyridin-3- yl)piperidin-3-yl)-2,2-difluoroethan-1-ol
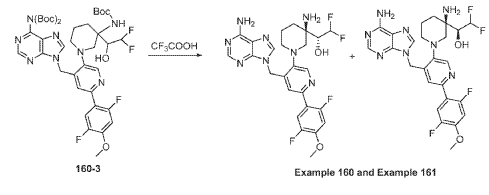
To a solution of tert-butyl (tert-butoxycarbonyl)(9-((5-(3-((tert-butoxycarbonyl)amino)-3-(2,2- difluoro-1-hydroxyethyl)piperidin-1-yl)-2-(2,5-difluoro-4-methoxyphenyl)pyridin-4-yl)methyl)-9H- purin-6-yl)carbamate (Intermediate 160-3) (200 mg, 0.237 mmol) in DCM (18 mL), was added TFA (36 mL), and the reaction mixrture was stirred at rt for 30 min under N2 atmosphere. The reaction mixture was concentrated in vacuo to give the crude product. The crude product was purifed by Pre-HPLC and SFC to afford (R)-1-((R)-3-amino-1-(4-((6-amino-9H-purin-9- yl)methyl)-6-(2,5-difluoro-4-methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol (Example 160) and (S)-1-((R)-3-amino-1-(4-((6-amino-9H-purin-9-yl)methyl)-6-(2,5-difluoro-4- methoxyphenyl)pyridin-3-yl)piperidin-3-yl)-2,2-difluoroethan-1-ol (Example 161).
Example 160: 1H NMR (400 MHz, CD3OD) d ppm 8.48 (s, 1H), 8.20 (d, J = 1.6 Hz, 2H), 7.58 (dd, J = 12.2, 7.3 Hz, 1H), 7.11 (d, J = 1.3 Hz, 1H), 6.90 (dd, J = 12.6, 7.1 Hz, 1H), 6.06 (td, J = 55.1, 3.9 Hz, 1H), 5.67 (s, 2H), 3.87 (s, 3H), 3.75 – 3.58 (m, 1H), 3.25 – 2.75 (m, 4H), 2.26 – 1.60 (m, 4H). LC-MS: [M+H]+ = 547.2, 548.2.
Example 161: 1H NMR (400MHz, CD3OD) d = 8.51 – 8.44 (m, 1H), 8.24 – 8.16 (m, 2H), 7.62 – 7.48 (m, 1H), 7.03 (s, 1H), 6.93 – 6.79 (m, 1H), 6.25 – 5.86 (m, 1H), 5.71 – 5.59 (m, 2H), 4.00 (m, 1H), 3.88 – 3.80 (m, 3H), 3.28 – 2.87 (m, 4H), 1.99 – 1.56 (m, 4H). LC-MS: [M+H]+ =547.4.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: EP-4559915-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: EP-4013755-B1Priority Date: 2019-08-14Grant Date: 2025-01-08
- Piperidinyl-methyl-purinamines as NSD2 inhibitors and anticancer agentsPublication Number: CN-114585622-APriority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as NSD2 inhibitors and anti-cancer agentsPublication Number: US-12312353-B2Priority Date: 2019-08-14Grant Date: 2025-05-27
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: WO-2021026803-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: EP-4013755-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: WO-2021028854-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purineamine D-tartrate, crystalline forms, and use thereof in the treatment of medical diseases and conditionsPublication Number: CN-119744262-APriority Date: 2022-05-18
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: EP-4526305-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purineamines as nsd2 inhibitors and anti-cancer agentsPublication Number: US-2023002388-A1Priority Date: 2019-08-14
- Piperidinyl-methyl-purinamines as NSD2 inhibitors and anticancer agentsPublication Number: CN-114585622-BPriority Date: 2019-08-14Grant Date: 2024-08-09
- Piperidinyl-methyl-purineamines as NSD2 inhibitors and anti-cancer agentsPublication Number: US-11420970-B1
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: AU-2023273656-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225144-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine fumaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225150-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: US-2025326752-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purinic amine D-tartrate salt, crystalline form and their use in the treatment of medical diseases and conditionsPublication Number: KR-20250012083-APriority Date: 2022-05-18
- Pharmaceutical compositions containing a piperidinyl-methyl-purine amine and their use in treating diseases and conditionsPublication Number: US-2024207192-A1Priority Date: 2022-12-12
- Pharmaceutical compositions containing a piperidinyl-methyl-purine amine and their use in treating diseases and conditionsPublication Number: WO-2024129670-A1Priority Date: 2022-12-12
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: US-2024002385-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225154-A1Priority Date: 2022-05-18
- Piperidinyl-methyl-purine amine d-tartaric acid salts, crystalline forms, and their use in treating medical diseases and conditionsPublication Number: WO-2023225141-A1Priority Date: 2022-05-18
////////////gintemetostat, ANAX LABS, antineoplastic, KTX 1001, NSD2 inhibitor 161, A48CGJ5UQM
Epaldeudomide
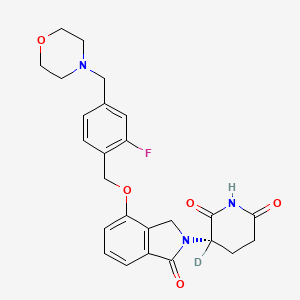
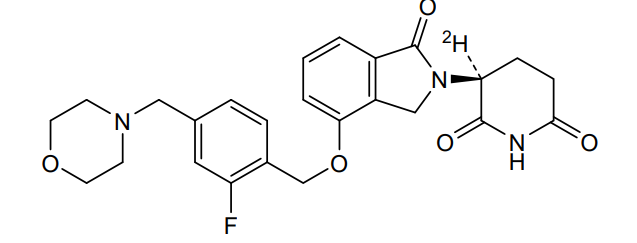
Epaldeudomide
CAS 1918159-31-5
MF C25H252HFN3O5, MW 468.5 g/mol
(3S)-3-deuterio-3-[7-[[2-fluoro-4-(morpholin-4-ylmethyl)phenyl]methoxy]-3-oxo-1H-isoindol-2-yl]piperidine-2,6-dione

KPG-818, KPG 818, ANTINEOPLASTIC, KV0TBL8MUS
Epaldeudomide (also known as KPG-818) is an investigational, next-generation immunomodulatory drug and “molecular glue” developed by Kangpu Biopharmaceuticals. Designed as a targeted therapy, it works by binding to the CRL4-CRBN E3 ubiquitin ligase complex to degrade specific proteins, showing promise in treating blood cancers, solid tumors, and autoimmune diseases.
Mechanism of Action
- Molecular Glue: It is a small molecule that acts as a modulator of the cereblon (CRBN) E3 ligase.
- Protein Degradation: It targets and induces the rapid degradation of two Ikaros zinc-finger transcription factors: IKZF3 (Aiolos) and IKZF1 (Ikaros).
- Immunomodulation: By degrading these targets, epaldeudomide triggers broad-spectrum immune responses, reduces tumor proliferation, and suppresses inflammation (such as the production of TNF-\(\alpha \)).
Therapeutic Pipeline and Research
Epaldeudomide is currently undergoing clinical evaluation to assess its safety, tolerability, and efficacy.
- Hematology/Oncology: It is being studied for the treatment of hematologic malignancies (such as multiple myeloma and lymphomas). It demonstrates potent anti-tumor and anti-angiogenic activity without several severe side effects typically associated with earlier immunomodulatory drugs.
- Autoimmune and Inflammatory Disorders: Because of its broad anti-inflammatory effects and ability to inhibit TNF-\(\alpha \), it is being explored for use against autoimmune conditions and inflammatory arthritis.
- OriginatorKangpu Biopharmaceuticals
- ClassAnti-inflammatories; Antineoplastics; Small molecules
- Mechanism of ActionCRBN protein modulators; Ubiquitin protein ligase complex modulators
- Phase IIInflammatory bowel diseases
- Phase I/IISystemic lupus erythematosus
- Phase IHaematological malignancies
- PreclinicalBehcet’s syndrome; Crohn’s disease; Multiple myeloma
- 06 Dec 2025Efficacy, pharmacokinetics and adverse events data from a phase I trial in Haematological malignancies presented at 67th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2025)
- 26 Nov 2025Epaldeudomide is still in phase I trials for Haematological malignancies (Second-line therapy or greater) in USA (PO, Capsule) (NCT04283097)
- 18 Nov 2025Efficacy and adverse events data from a phase I trial in Haematological malignancies released by Kangpu Biopharmaceuticals
SYN
US-10017492-B2
US-20170313676-A1
SYN
EP-3643709-A1
EP-3643709-B1
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP293972088&_cid=P21-MPEVL7-37300-1
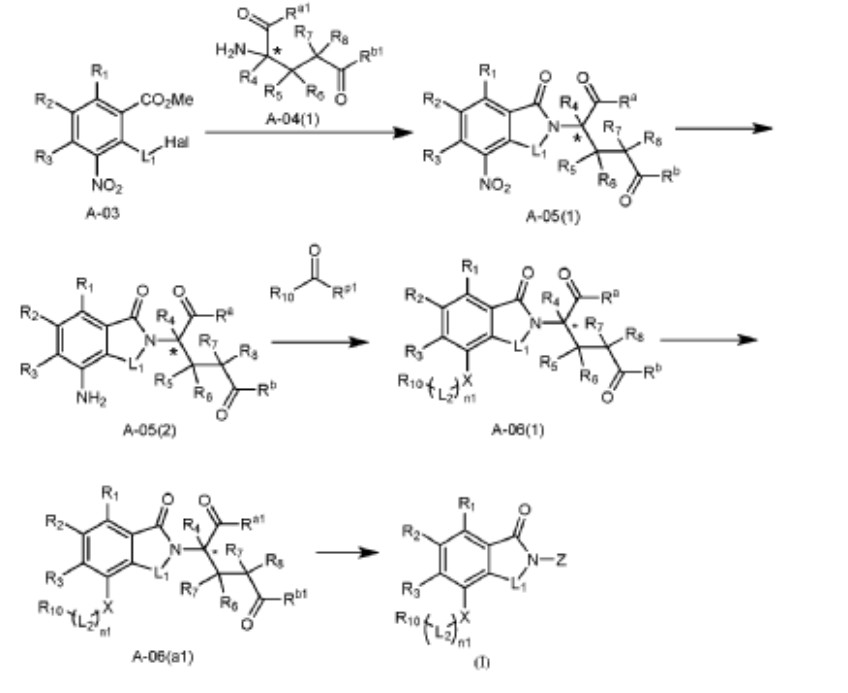
Example 37: Compound A382
[0196] 3-(4-((2-fluoro-5-(3-morpholinopropoxy)benzyl)amino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione, A382.
[0197] 1H NMR (DMSO- d 6, 300 MHz): δ 11.00 (s, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 7.05-7.13 (m, 2H), 6.93 (d, J = 7.5 Hz, 1H), 6.64 (d, J = 7.8 Hz, 1H), 6.28 (t, J = 6.3 Hz, 1H), 5.07-5.13 (m, 1H), 4.38 (d, J= 5.7 Hz, 2H), 4.28 (d, J= 17.4 Hz, 1H), 4.16 (d, J= 17.4 Hz, 1H), 3.54 (t, J= 4.5 Hz, 4H), 3.42 (s, 2H), 2.85-2.97 (m, 1H), 2.57-2.63 (m, 1H), 2.26-2.38 (m, 5H), 2.00-2.09 (m, 1H). LCMS: 467.2 ([M+1] +).
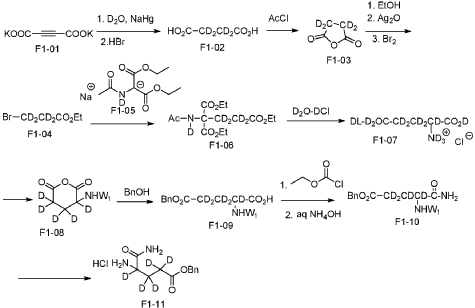
Example 69: Compound A406
[0296] ( S)-3-deuterium-3-(4-((2-fluoro-4-(morpholinomethyl)benzyl)oxy)-1-oxoisoindolin-2-yl)piperidine-2,6-dione, A406.
[0297] 1H NMR (DMSO- d 6, 300 MHz): δ 10.98 (s, 1H),7.47-7.55 (m, 2H), 7.31-7.38 (m, 2H), 7.16-7.20 (m, 2H), 5.24 (s, 2H), 5.06-5.12 (m, 0.04H), 4.35 (d, J = 18.0 Hz, 1H), 4.19 (d, J = 18.0 Hz, 1H), 3.55 (br, 4H), 3.47 (s, 2H), 2.82-2.94 (m, 1H), 2.48-2.57 (m, 1H), 2.33-2.42 (m, 5H), 1.91-1.96 (m, 1H). LCMS: 469.2 ([M+1] +).
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Isoindoline derivative, intermediate, preparation method, pharmaceutical composition and use thereofPublication Number: US-10017492-B2Priority Date: 2014-10-30Grant Date: 2018-07-10
- Isoindoline derivative, intermediate, preparation method, pharmaceutical composition and use thereofPublication Number: EP-3643709-A1Priority Date: 2014-10-30
- Isoindoline derivative, intermediate, preparation method, pharmaceutical composition and use thereofPublication Number: US-2017313676-A1Priority Date: 2014-10-30
- Isoindoline derivative, intermediate, preparation method, pharmaceutical composition and use thereofPublication Number: EP-3643709-B1Priority Date: 2014-10-30Grant Date: 2021-10-20
/////////epaldeudomide, ANAX LABS, KPG-818, KPG 818, ANTINEOPLASTIC, KV0TBL8MUS
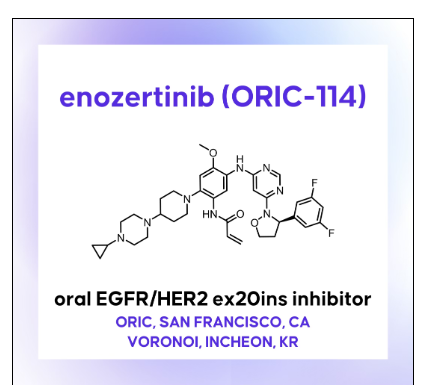
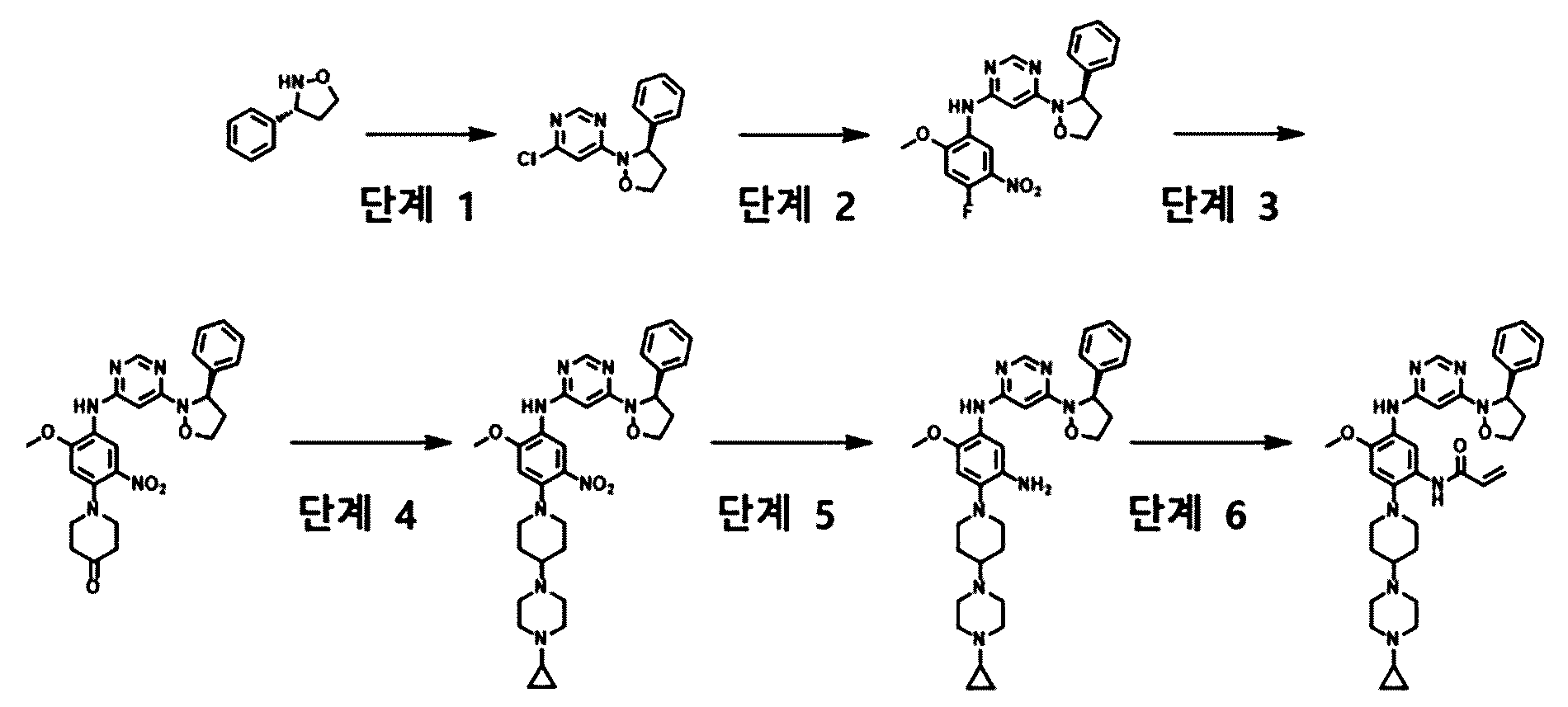
Enozertinib
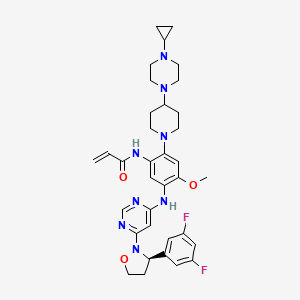
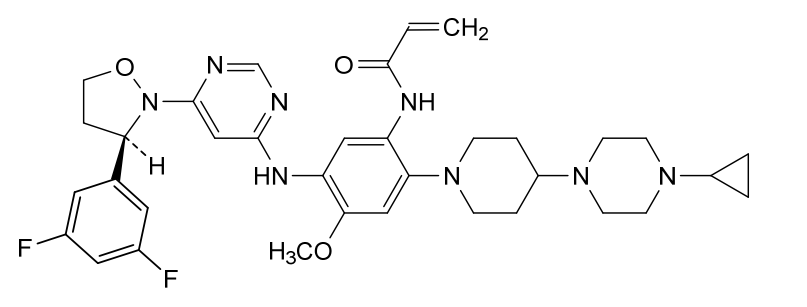
Enozertinib
CAS 2489185-38-6
MF C35H42F2N8O3 MW660.8
N-[2-[4-(4-cyclopropylpiperazin-1-yl)piperidin-1-yl]-5-[[6-[(3R)-3-(3,5-difluorophenyl)-1,2-oxazolidin-2-yl]pyrimidin-4-yl]amino]-4-methoxyphenyl]prop-2-enamide
- N-(2-(4-(4-cyclopropylpiperazine-1-yl)piperidine-1-yl)-5-((6-((R)-3-(3,5-difluorophenyl)isoxazolidine-2-yl)pyrimidine-4-yl)amino)-4-methoxyphenyl)acrylamide
- N-[2-[4-(4-Cyclopropyl-1-piperazinyl)-1-piperidinyl]-5-[[6-[(3R)-3-(3,5-difluorophenyl)-2-isoxazolidinyl]-4-pyrimidinyl]amino]-4-methoxyphenyl]-2-propenamide
- N-[2-[4-(4-cyclopropylpiperazin-1-yl)piperidin-1-yl]-5-[[6-[(3R)-3-(3,5-difluorophenyl)-1,2-oxazolidin-2-yl]pyrimidin-4-yl]amino]-4-methoxyphenyl]prop-2-enamide
epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, ORIC-114, ORIC 114, DU24UP8R94
Enozertinib (formerly ORIC-114) is an investigational, orally bioavailable, and brain-penetrant dual EGFR/HER2 inhibitor developed by ORIC Pharmaceuticals. It targets cancers with exon 20 insertion and atypical EGFR mutations. Its core profile highlights its ability to cross the blood-brain barrier.
How it Works
Enozertinib acts as an irreversible, mutant-selective covalent inhibitor. By blocking overactive EGFR and HER2 signaling, it induces cell death and inhibits tumor growth. Because it penetrates the central nervous system (CNS), it is uniquely suited to treat both primary brain tumors and brain metastases—a common complication in non-small cell lung cancer (NSCLC).
Enozertinib is an orally bioavailable, central nervous system (CNS) penetrating, mutant-selective covalent inhibitor of epidermal growth factor receptor (EGFR; ErbB1) and human epidermal growth factor receptor 2 (HER2; EGFR2; ErbB2) alterations, including exon 20 insertion (Ex20ins) mutations, with potential antineoplastic activity. Upon oral administration, enozertinib selectively targets, irreversibly binds to and inhibits the activity of EGFR or HER2 insertions or mutations. This prevents EGFR/HER2-mediated signaling. This may induce cell death and inhibit tumor growth in EGFR/HER2-overexpressing tumor cells. Enozertinib is able to penetrate the blood-brain-barrier (BBB) and may therefore exert its activity against EGFR Ex20ins-driven CNS primary tumors and CNS metastases. The ErbB receptor tyrosine kinase family is involved in key cellular functions, including cell growth and survival. EGFR and HER2 alterations constitutively upregulate kinase activity.
SYN
https://drughunter.com/molecule/enozertinib-oric-114
SYN

PAT

SIMILAR SYNTHESIS
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Egfr inhibitor for treating cancers comprising atypical egfr mutationsPublication Number: WO-2024233313-A1Priority Date: 2023-05-05
- Fumarate, tartrate, malate, and citrate salts of an egfr inhibitorPublication Number: WO-2024096624-A1Priority Date: 2022-11-03
- Malonate and glycolate salts of an egfr inhibitorPublication Number: WO-2024097848-A1Priority Date: 2022-11-03
- Malonate and glycolate salts of an egfr inhibitorPublication Number: EP-4611902-A1Priority Date: 2022-11-03
- Fumarate, tartrate, malate, and citrate salts of an egfr inhibitorPublication Number: EP-4612147-A1Priority Date: 2022-11-03
- Fumarate, tartrate, malate and citrate salts of EGFR inhibitorsPublication Number: CN-120035590-APriority Date: 2022-11-03
- Heteroaryl derivative, method for producing same, and pharmaceutical composition comprising same as effective componentPublication Number: US-11466000-B2Priority Date: 2019-03-19Grant Date: 2022-10-11
- Heteroaryl derivatives, their preparation methods, and pharmaceutical compositions containing them as active ingredientsPublication Number: CN-115838369-APriority Date: 2019-03-19
- Heteroaryl derivatives, methods for their preparation, and pharmaceutical compositions containing them as active ingredientsPublication Number: CN-114605400-APriority Date: 2019-03-19
- HETEROARYL DERIVATIVE, METHOD FOR PRODUCING THE SAME, AND PHARMACEUTICAL COMPOSITION INCLUDING THE SAME AS AN EFFECTIVE COMPONENTPublication Number: BR-112021018704-B1Priority Date: 2019-03-19
- Heteroaryl derivative, method for producing same, and pharmaceutical composition comprising same as effective componentPublication Number: US-2022289733-A1Priority Date: 2019-03-19
- Heteroaryl derivatives, methods for producing heteroaryl derivatives, and pharmaceutical compositions containing heteroaryl derivatives as active ingredientsPublication Number: JP-7394298-B2Priority Date: 2019-03-19Grant Date: 2023-12-08
- Heteroaryl derivatives, methods for their preparation, and pharmaceutical compositions containing them as active ingredientsPublication Number: CN-113993866-APriority Date: 2019-03-19
//////////enozertinib, anax labs, epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, ORIC-114, ORIC 114, DU24UP8R94
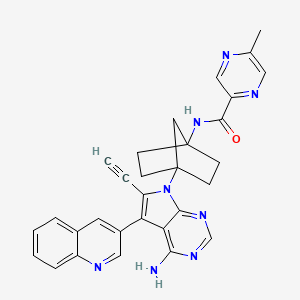
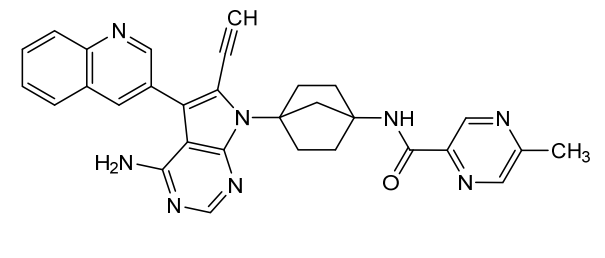
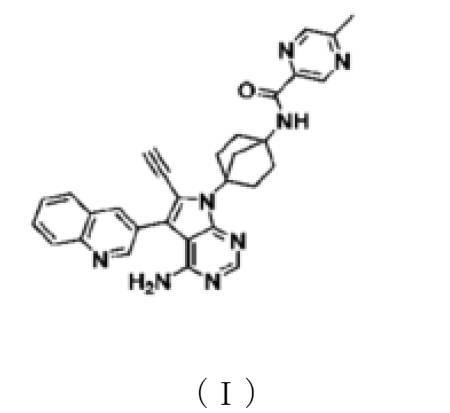
Emupertinib
Emupertinib
CAS 2472802-77-8
MFC30H26N8O MW514.6 g/mol
2-Pyrazinecarboxamide, N-[4-[4-amino-6-ethynyl-5-(3-quinolinyl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]bicyclo[2.2.1]hept-1-yl]-5-methyl-
N-{4-[4-amino-6-ethynyl-5-(quinolin-3-yl)-7Hpyrrolo[2,3-d]pyrimidin-7-yl]bicyclo[2.2.1]heptan-1-yl}-
5-methylpyrazine-2-carboxamide
epidermal growth factor receptor tyrosine kinase, inhibitor, antineoplastic, TAS3351, TAS 3351, CU9YW8A5TP
Emupertinib is a potent, small-molecule epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor. It possesses selective antineoplastic potential for targeting specific mutant profiles of cancer cells. The compound was originally developed by Taiho Pharmaceutical Co., Ltd. under the developmental code TAS3351
Development Profile
The International Nonproprietary Name (INN) for this therapeutic chemical structure was formally proposed under the World Health Organisation (WHO) proposed INN list 132 in early 2025. Global research pipelines list the compound’s structural classification profile within non-small cell lung cancer (NSCLC) primary discovery programs. The drug currently remains a specialized compound designated for global laboratory research use only, rather than standard human prescription or veterinary clinical treatments
SYN
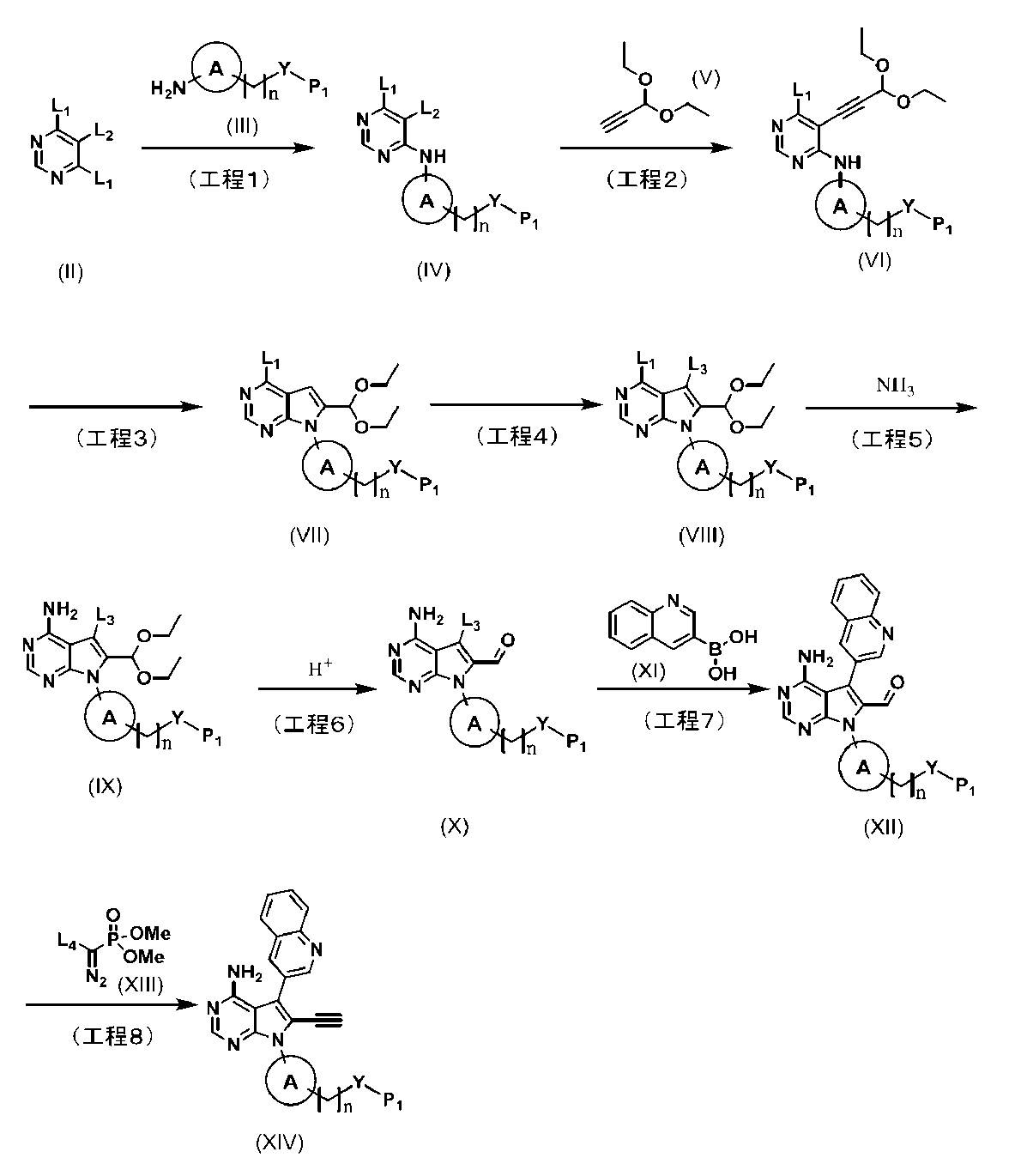
[0184][Example 37]
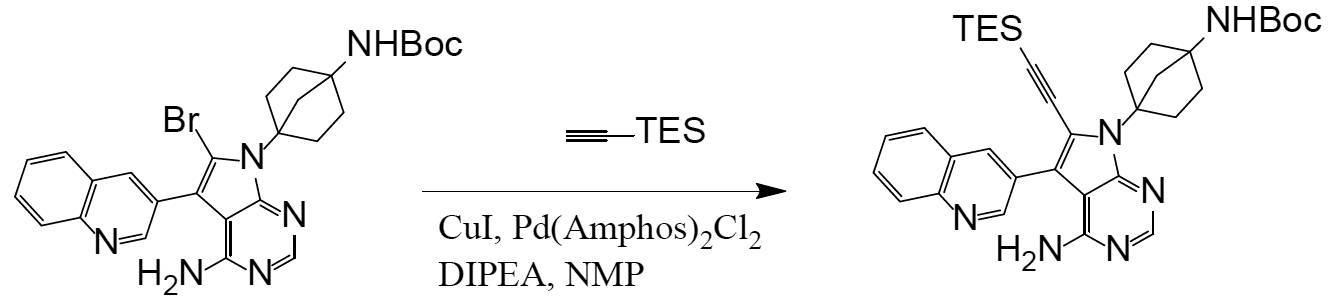
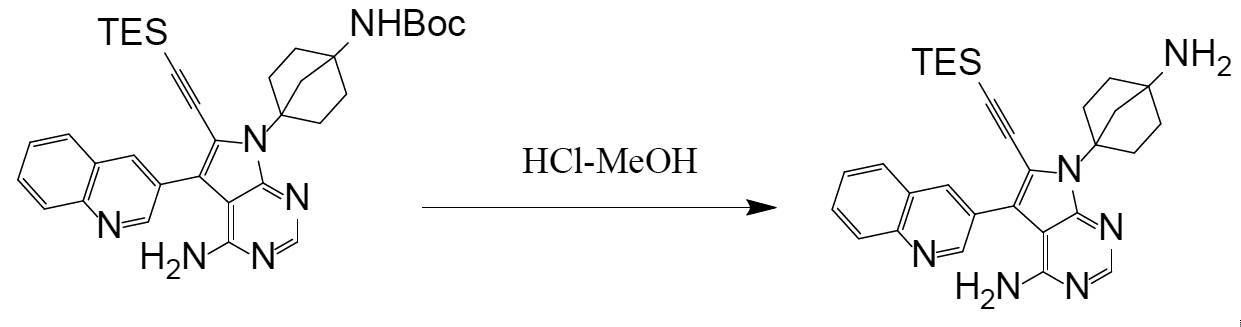
N-(4-(4-amino-6-ethynyl-5-(quinoline-3-yl)-7H-pyrrolo[2,3-d]pyrimidine-7-yl)bicyclo[2.2.1]heptan-1-yl)
-5-methylpyrazine-2-carboxamide The title compound was obtained by following the same method as in Example 29 (step 6), except that 5-methylpyrazine-2-carboxylic acid was used instead of 5-(fluoromethyl)-2-methylpyrazole-3-carboxylic acid used in Example 29.
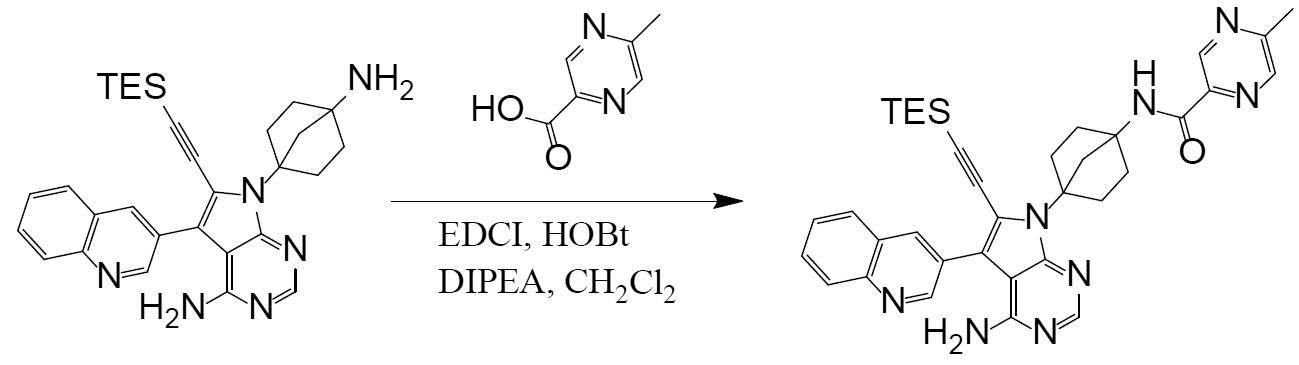
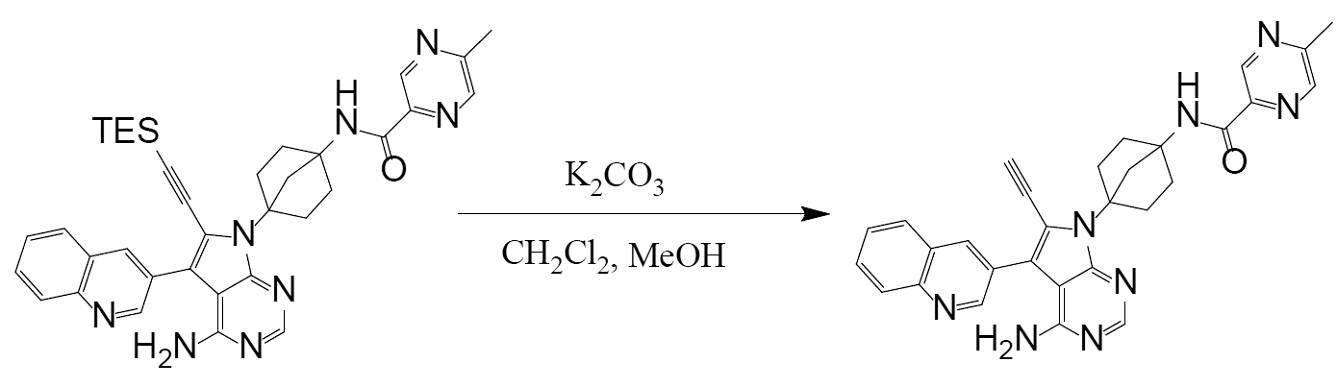
(Step 4)
Synthesis of N-(4-(4-amino-6-ethynyl-5-(quinoline-3-yl)-7H-pyrrolo[2,3-d]pyrimidine-7-yl)bicyclo[2.2.1]heptan-1-yl)-5-methylpyrazine-2-carboxamide (compound (1))
[Chemical Formula 7]
It can be obtained by deprotecting the acetylene protecting group TES of N-(4-(4-amino-5-(quinoline-3-yl)-6-((triethylsilyl)ethynyl)-7H-pyrrolo[2,3-d]pyrimidine-7-yl)bicyclo[2.2.1]heptan-1-yl)-5-methylpyrazine-2-carboxamide obtained in Step 3 under basic conditions.
The reagents used to create basic conditions are not particularly limited as long as the reaction proceeds, but examples of inorganic bases include metal hydroxides (sodium hydroxide, calcium hydroxide, etc.), metal hydrides (lithium hydride, sodium hydride, etc.), and metal carbonates (sodium carbonate, potassium carbonate, cesium carbonate, calcium carbonate, lithium carbonate, magnesium carbonate, sodium bicarbonate, etc.). Examples of organic bases include metal alkoxides (sodium methoxide, potassium tert-butoxide, etc.), metal amides (sodium amide, lithium diisopropylamide, etc.), alkyl metal compounds (n-butyllithium, trimethylaluminum, etc.), alkylamines (triethylamine, tetramethylethylenediamine, piperidine, 1,4-diazabicyclo[2.2.2]octane, etc.), heterocyclic amines (diazabicycloundecene, pyridine, imidazole, etc.), and quaternary ammonium fluorides (tetra-n-butylammonium fluoride). Preferably, the reagent used to create basic conditions is a reagent that does not contain fluoride ions, more preferably a metal carbonate, and even more preferably potassium carbonate. These can be used alone or in combination to adjust the pH to the desired level.
The amount of reagent used is not particularly limited as long as the reaction proceeds, but for example, 0.1 to 50 moles can be used per mole of the starting compound (the compound represented by formula (II)). Preferably, 0.1 to 10 moles, and more preferably 0.1 to 2 moles.
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
Substituted pyrrolo[2,3-d]pyrimidines as EGFR inhibitors
Publication Number: US-11786534-B2
Priority Date: 2019-02-15
Grant Date: 2023-10-17
- Crystal of 7h-pyrrolo[2,3-d]pyrimidine-4-amine derivativePublication Number: EP-4512808-A1Priority Date: 2022-04-22
- 7H-pyrrolo[2,3-d]pyrimidin-4-amine derivativePublication Number: KR-102645237-B1Priority Date: 2019-02-15Grant Date: 2024-03-07
- 7h-pyrrolo[2,3-d]pyrimidine-4-amine derivativePublication Number: WO-2020166680-A1Priority Date: 2019-02-15
- 7h-pyrrolo[2,3-d]pyrimidine-4-amine derivativePublication Number: US-2022160719-A1Priority Date: 2019-02-15
- 7H-Pyrrolo[2,3-d]pyrimidin-4-amine derivativesPublication Number: CN-113453764-BPriority Date: 2019-02-15Grant Date: 2024-04-16
- Brain-migrating tumor treatment agent containing, as active ingredient, n-(4-(4-amino-6-ethynyl-5-(quinolin-3-yl)-7h-pyrrolo[2,3-d]pyrimidin-7-yl) bicyclo[2.2.1]heptan-1-yl)-5-methylpyrazine-2-carboxamide or salt thereofPublication Number: WO-2025127108-A1Priority Date: 2023-12-13
- Crystals of 7h-pyrrolo[2,3-d]pyrimidin-4-amine derivativesPublication Number: WO-2025072720-A1Priority Date: 2023-09-29
- Crystal of 7h-pyrrolo[2,3-d]pyrimidine-4-amine derivativePublication Number: WO-2023204303-A1Priority Date: 2022-04-22
- Method for producing 7h-pyrrolo[2,3-d]pyrimidine-4-amine derivativePublication Number: WO-2023204304-A1Priority Date: 2022-04-22
- Method for producing 7h-pyrrolo[2,3-d]pyrimidine-4-amine derivativePublication Number: US-2025270214-A1Priority Date: 2022-04-22
///////emupertinib, anax labs, epidermal growth factor receptor tyrosine kinase, inhibitor, antineoplastic, TAS3351, TAS 3351, CU9YW8A5TP
Elisrasib
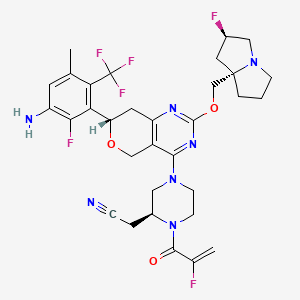
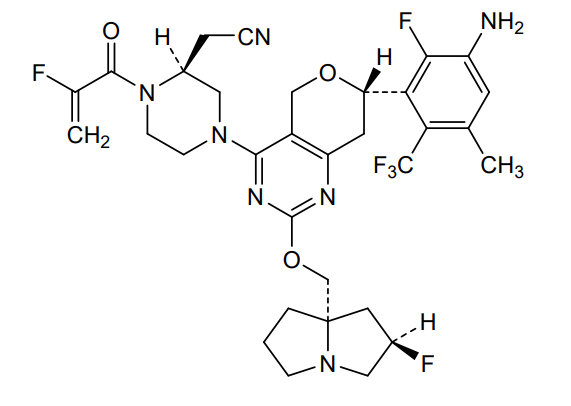
Elisrasib
CAS2914919-85-8
MFC32H35F6N7O3. MW 679.7 g/mol
2-[(2S)-4-[(7S)-7-[3-amino-2-fluoro-5-methyl-6-(trifluoromethyl)phenyl]-2-[[(2R,8S)-2-fluoro-1,2,3,5,6,7-hexahydropyrrolizin-8-yl]methoxy]-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile
[(2S)-4-[(7S)-7-[3-amino-2-fluoro-5-methyl-6-(trifluoromethyl)phenyl]-2-{[(2R,7aS)-2-fluorotetrahydro-1Hpyrrolizin-7a(5H)-yl]methoxy}-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile
Kirsten rat sarcoma viral oncogene homolog inhibitor, antineoplastic, D3S 001, PFW9YLB86H
Elisrasib (D3S-001) is a next-generation, orally available KRAS G12C inhibitor developed by D3 Bio that demonstrates high potency, sustained target engagement, and strong clinical activity in advanced solid tumors, including those resistant to first-generation inhibitors. As of April 2026, clinical trials show it has a 52% objective response rate (ORR) in G12C inhibitor-naive patients and a 30% ORR in refractory populations.
Key Aspects of Elisrasib (D3S-001):
- Mechanism of Action: It is a highly potent, covalent inhibitor that selectively binds the GDP-bound (inactive) form of the KRAS G12C mutant, effectively halting tumor cell proliferation and metastasis.
- Superior Efficacy: Preliminary data suggests elisrasib may be more potent than earlier inhibitors like sotorasib and adagrasib, providing higher target occupancy at lower doses.
- Clinical Performance (AACR 2026 Data):
- Naive Patients: 52% ORR, with a median duration of response (mDOR) of 16.5 months and median progression-free survival (mPFS) of 12.2 months at the 600 mg dose.
- Refractory Patients: 32% ORR, with a mDOR of 15.6 months and mPFS of 8.1 months.
- Targeted Cancers: Clinical trials are focused on KRAS G12C-mutant tumors, specifically non-small cell lung cancer (NSCLC), colorectal cancer (CRC), and other solid tumors.
- Safety Profile: The drug has shown good tolerability and a safe profile in early studies.
Elisrasib is in Phase 1/2 development and was highlighted for its promising results in treating patients with KRAS G12C-mutant tumors
Elisrasib is an orally bioavailable inhibitor of the oncogenic KRAS substitution mutation G12C, with potential antineoplastic activity. Upon oral administration, elisrasib selectively targets the KRAS G12C mutant and inhibits KRAS G12C-mediated signaling. This may halt proliferation and metastasis in susceptible tumor cells. KRAS, a member of the RAS family of oncogenes, serves an important role in cell signaling, division and differentiation. Mutations of KRAS may induce constitutive signal transduction leading to tumor cell proliferation, invasion, and metastasis.
- A Phase 1 Study to Assess Food Effect on the Pharmacokinetics of D3S-001 in Healthy Adult ParticipantsCTID: NCT07093398Phase: Phase 1Status: CompletedDate: 2026-03-25
- A Phase 1/2 Study of D3S-002 as Monotherapy or Combination Therapy in Adult Subjects With Advanced Solid Tumors With MAPK Pathway MutationsCTID: NCT05886920Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2026-03-23
- A Study of D3S-001 Monotherapy or Combination Therapy in Subjects With Advanced Solid Tumors With a KRAS p.G12C MutationCTID: NCT05410145Phase: Phase 1/Phase 2Status: RecruitingDate: 2026-03-12
PAT
SYN
Example 17
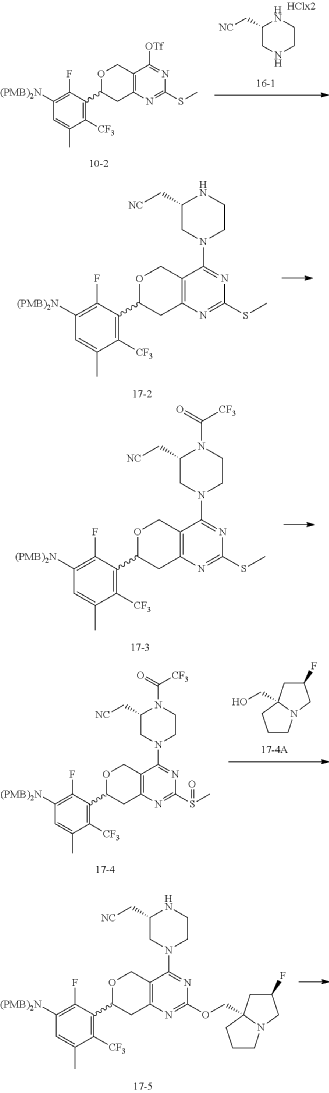
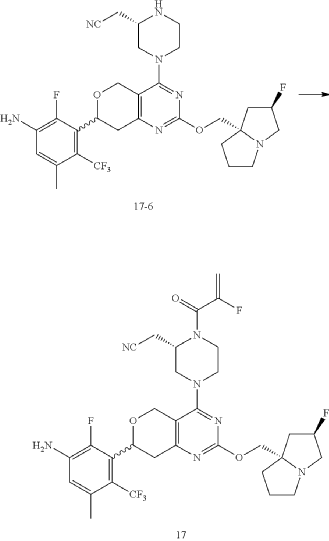
Step 6: Synthesis of Compound 17
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Pyrimidoheterocyclic compounds and application thereofPublication Number: EP-4105211-A1Priority Date: 2020-03-12
- Pyrimidoheterocyclic compounds and application thereofPublication Number: US-2023151004-A1Priority Date: 2020-03-12
//////////elisrasib, anax labs, Kirsten rat sarcoma viral oncogene homolog inhibitor, antineoplastic, D3S 001, PFW9YLB86H
Elironrasib
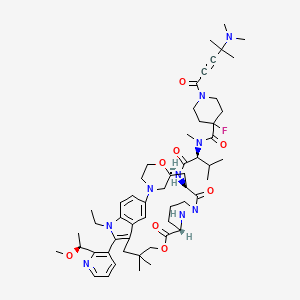
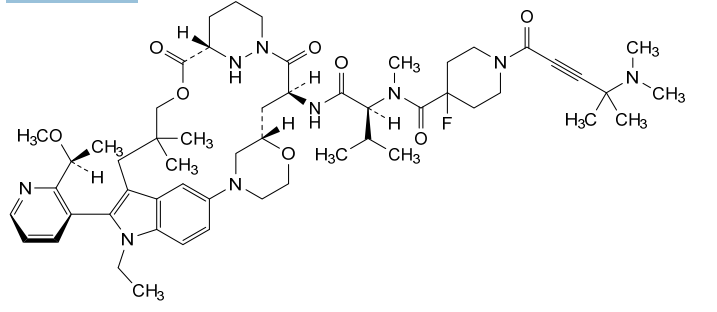
Elironrasib
CAS 2641998-63-0
MFC55H78FN9O8 MW 1012.3 g/mol
1-[4-(dimethylamino)-4-methylpent-2-ynoyl]-N-[(2S)-1-[[(6S,8S,14S)-22-ethyl-21-[2-[(1S)-1-methoxyethyl]-3-pyridinyl]-18,18-dimethyl-9,15-dioxo-5,16-dioxa-2,10,22,28-tetrazapentacyclo[18.5.2.12,6.110,14.023,27]nonacosa-1(26),20,23(27),24-tetraen-8-yl]amino]-3-methyl-1-oxobutan-2-yl]-4-fluoro-N-methylpiperidine-4-carboxamide
- 1-[4-(dimethylamino)-4-methylpent-2-ynoyl]-N-[(2S)-1-[[(6S,8S,14S)-22-ethyl-21-[2-[(1S)-1-methoxyethyl]pyridin-3-yl]-18,18-dimethyl-9,15-dioxo-5,16-dioxa-2,10,22,28-tetrazapentacyclo[18.5.2.12,6.110,14.023,27]nonacosa-1(26),20,23(27),24-tetraen-8-yl]amino]-3-methyl-1-oxobutan-2-yl]-4-fluoro-N-methylpiperidine-4-carboxamide
- 3-Pyridazinecarboxylic acid, N1-[N-[[1-[4-(dimethylamino)-4-methyl-1-oxo-2-pentyn-1-yl]-4-fluoro-4-piperidinyl]carbonyl]-N-methyl-L-valyl-3-[4-[(2R)-1-ethyl-3-(3-hydroxy-2,2-dimethylpropyl)-2-[2-[(1S)-1-methoxyethyl]-3-pyridinyl]-1H-indol-5-yl]-2-morpholinyl]-L-alanyl]hexahydro-, (3–>2)-lactone, (3S)-
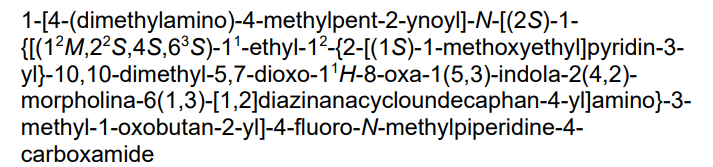
Kirsten rat sarcoma viral oncogene homolog inhibitor, antineoplastic, RMC-6291, RMC 6291, 942KVV5CJP
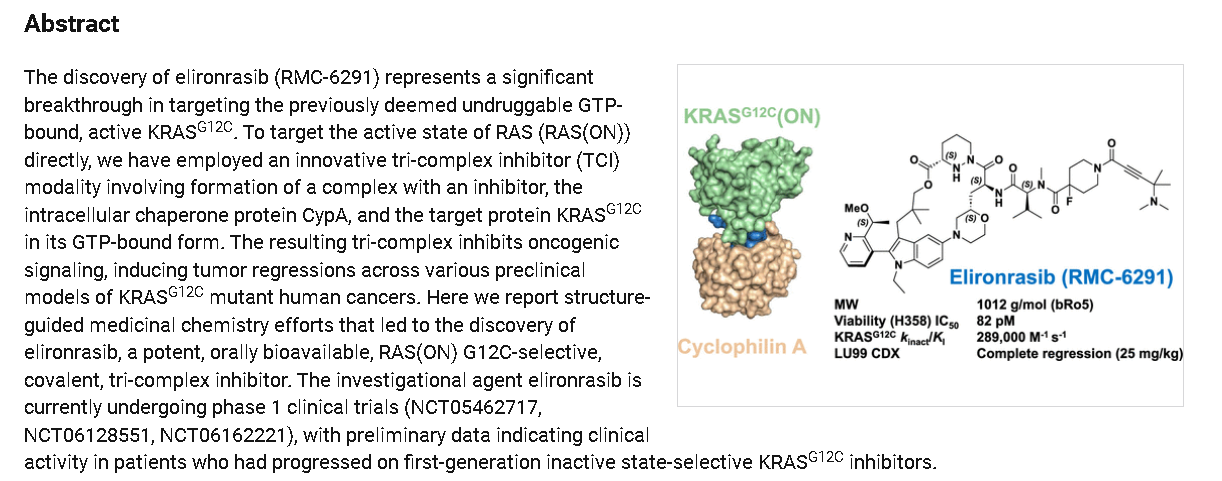
Elironrasib (RMC-6291) is an investigational, orally bioavailable, RAS(ON) G12C-selective inhibitor developed by Revolution Medicines that targets the active GTP-bound form of KRAS G12C. In Phase 1 trials, it showed significant promise in treating advanced KRAS G12C-mutated solid tumors, including non-small cell lung cancer (NSCLC). [1, 2, 3]
Key Clinical Trial Results (as of Oct 2025):
- Response Rate: 42% objective response rate (ORR).
- Disease Control: 79% disease control rate (DCR).
- Durability: Median duration of response was 11.2 months.
- Survival: Median progression-free survival was 6.2 months.
- Overcoming Resistance: Demonstrated efficacy in patients who had previously progressed on first-generation KRAS G12C(OFF) inhibitors.
Mechanism of Action:
Elironrasib acts as a covalent tri-complex inhibitor (TCI). It forms a complex with the intracellular chaperone protein cyclophilin A (CypA) and the active KRAS G12C(GTP) protein, effectively shutting down oncogenic signaling.
Development Status:
- Designation: It has received FDA breakthrough therapy designation for KRAS G12C-mutant NSCLC.
- Trials: Currently in Phase 1 clinical trials (e.g., NCT05462717) to evaluate safety, tolerability, and efficacy, both as a monotherapy and in combination.
- Target Population: Patients with KRAS G12C-addicted solid tumors.
Discovery of Elironrasib (RMC-6291), a Potent and Orally Bioavailable, RAS(ON) G12C-Selective, Covalent Tricomplex Inhibitor for the Treatment of Patients with RAS G12C-Addicted Cancers – PubMed27 Mar 2025 — This information does not constitute medical advice or diagnosis. Elirronrasib (RMC-6291) is a potent, orally bioavailable,
- Revolution Medicines to Present Updated Elironrasib Safety and Efficacy Data in Patients with KRAS G12C Non-Small Cell Lung Cancer Following Treatment with a KRAS(OFF) G12C Inhibitor22 Oct 2025 — This information does not constitute medical advice or diagnosis. Elirronrasib is a RAS(ON) G12C-selective inhibitor being develop…
Revolution Medicines
- Elironrasib May Overcome Resistance to Prior KRAS G12C Inhibition in Non-small Cell Lung Cancer
- OriginatorREVOLUTION Medicines
- ClassAntineoplastics; Morpholines; Piperidines; Pyridazines; Small molecules
- Mechanism of ActionKRAS protein inhibitors
- Phase I/IISolid tumours
- Clinical Phase UnknownNon-small cell lung cancer
- 30 Jan 2026Phase-I/II clinical trials in Solid tumours (Combination therapy, Late-stage disease, Metastatic disease) in USA (PO) (NCT07397338)
- 31 Oct 2025Elironrasib is still in phase I trial in Solid tumours (Late-stage disease, Metastatic disease, Monotherapy) in Australia, Italy, South Korea, Malaysia, Singapore, Spain, Czech Republic, Thailand and USA (PO, Tablet) (NCT05462717)
- 28 Oct 2025No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Metastatic disease, Monotherapy) in Australia, Italy, South Korea, Malaysia, Singapore, Spain, Czech Republic, Thailand (PO, Tablet)
Elironrasib is an orally bioavailable, covalent inhibitor of the active, guanosine triphosphate (GTP)-bound form of the oncogenic KRAS substitution mutation G12C, KRAS G12C(ON), with potential antineoplastic activity. Upon oral administration, elironrasib forms a tri-complex with the intracellular chaperone protein and immunophilin cyclophilin A (CypA) and KRAS G12C(ON). This tri-complex inhibits KRAS G12C(ON)-mediated signaling, which may inhibit tumor cell proliferation. KRAS, a member of the RAS family of oncogenes, serves an important role in cell signaling, division and differentiation. Mutations of KRAS may induce constitutive signal transduction leading to tumor cell growth, proliferation, invasion, and metastasis.
- Study of Elironrasib and Daraxonrasib as Monotherapies and Combination Therapy in Participants With Advanced KRAS G12C Mutant Solid TumorsCTID: NCT06128551Phase: Phase 1/Phase 2Status: RecruitingDate: 2026-04-23
- Dose Escalation and Dose Expansion Study of RMC-6291 Monotherapy in Subjects With Advanced KRASG12C Mutant Solid TumorsCTID: NCT05462717Phase: Phase 1Status: Active, not recruitingDate: 2026-04-08
- Study of RAS(ON) Inhibitors in Combination With Ivonescimab in Patients With Solid TumorsCTID: NCT07397338Phase: Phase 1/Phase 2Status: RecruitingDate: 2026-03-30
- Study of RAS(ON) Inhibitors in Patients With Advanced RAS-mutated NSCLCCTID: NCT06162221Phase: Phase 1/Phase 2Status: RecruitingDate: 2026-03-09
SYN
https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c02313
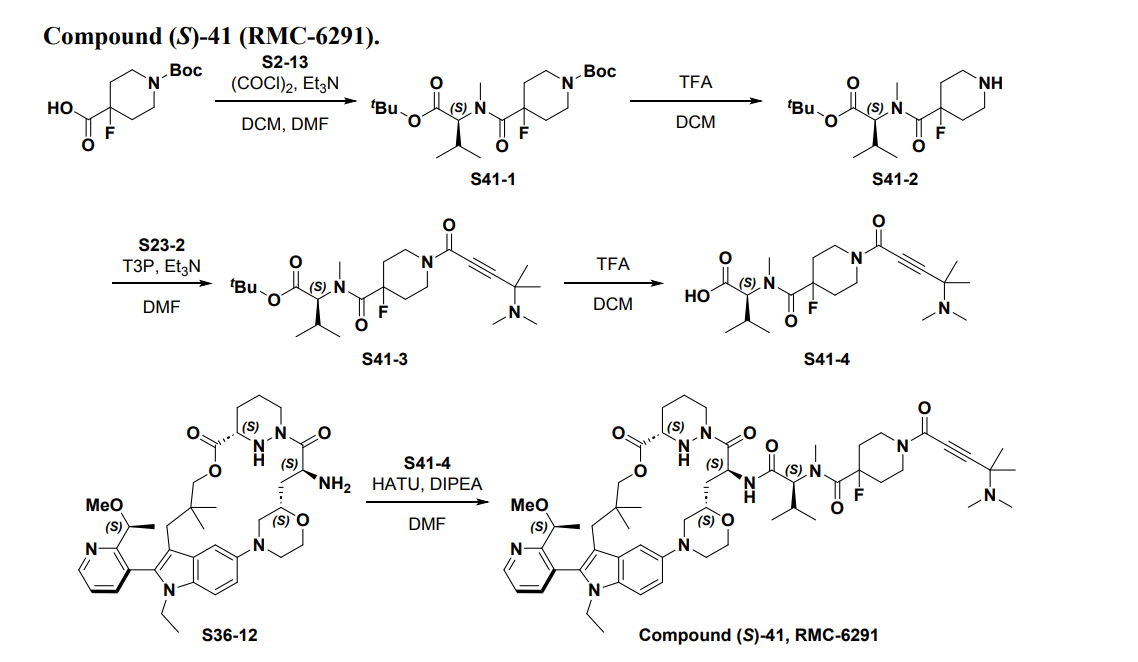

SYN
PAT’
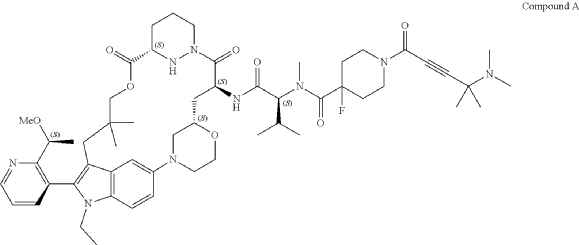
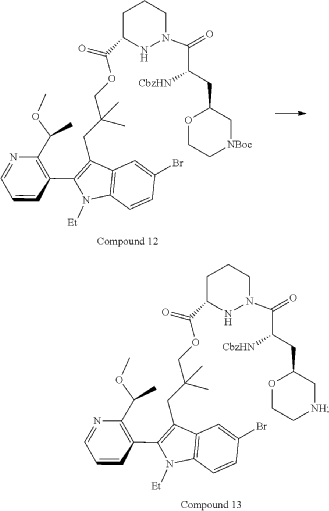
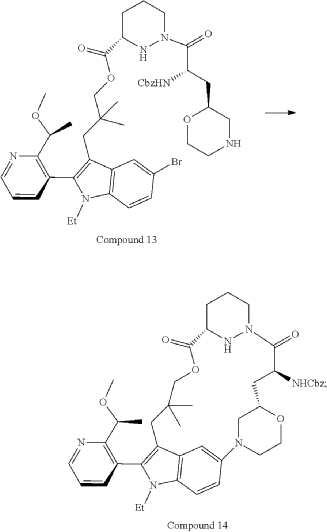
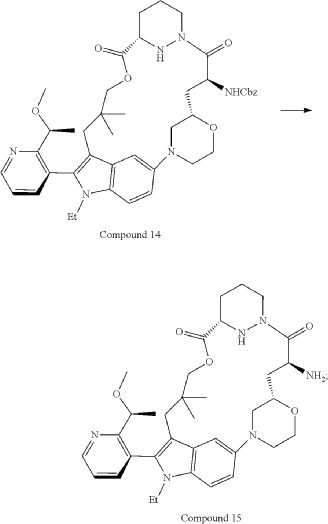
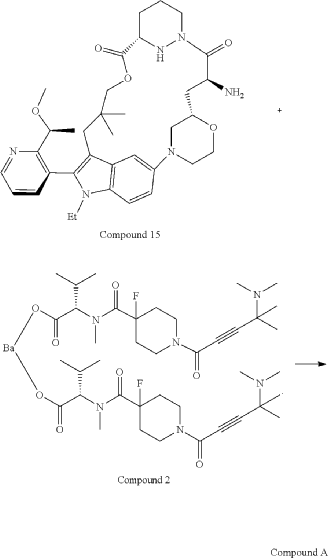
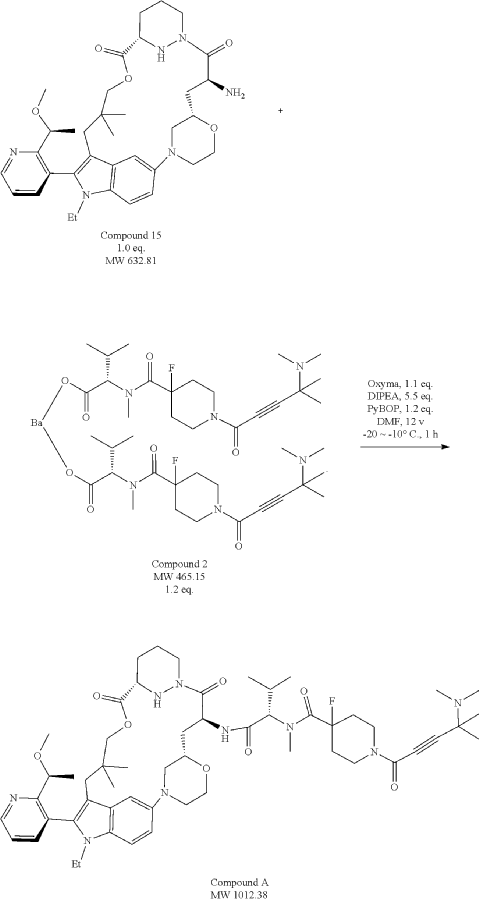
Part 5—Synthesis of Compound A—(12M)-1-(4-(dimethylamino)-4-methylpent-2 ynoyl)-N-((2S)-1-(((22S,63S,4S)-11-ethyl-12-(2-((S)-1-methoxyethyl)pyridin-3-yl)-10,10-dimethyl-5,7-dioxo-61,62,63,64,65,66-hexahydro-11H-8-oxa-2(4,2)-morpholina-1(5,3)-indola-6(1,3) pyridazinacycloundecaphane-4-yl)amino)-3-methyl-1-oxobutan-2 yl)-4-fluoro-N-methylpiperidine-4-carboxamide
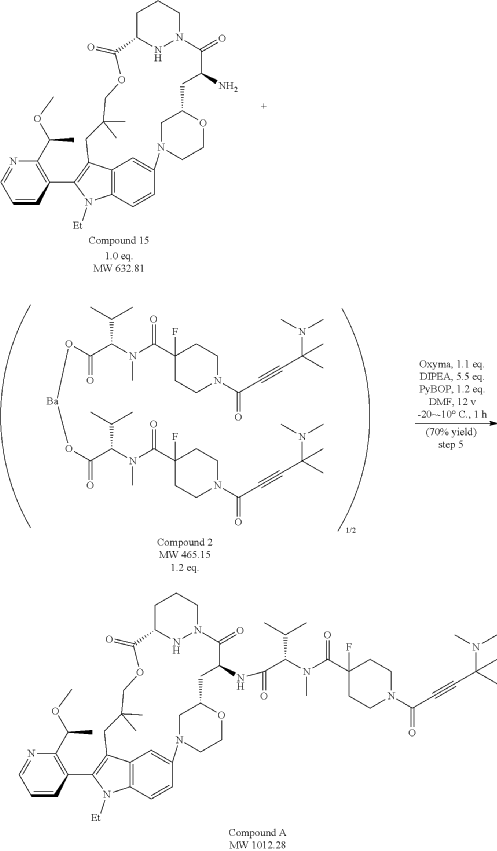
| To a 50 L glass reactor was charged Compound 15 (1.91 kg, 1.0 eq) and DMF (13.9 kg). The mixture was agitated at 20-30° C. until all of the solids were dissolved. Compound 2 (1.70 kg, 1.2 eq) and DMF (3.8 kg) were charged. The mixture was agitated at 20-30° C. until all of the solids were dissolved. DIPEA (2.20 kg, 5.50 eq) was charged at 20-30° C. and the mixture was cooled to −20-−10° C. under agitation. Ethyl cyanoglyoxylate-2-oxime (Oxyma) (0.48 kg, 1.1 eq) was charged to the reactor and the reaction mixture was agitated at −20 to −10° C. for 30 min. PyBOP was charged as a DMF solution (1.89 kg dissolved in 3.62 kg DMF, 1.2 eq) to the reactor at −20 to −10° C. in </=1 h. The reaction mixture was agitated at −20 to −10° C. for 1-3 h. Reaction monitoring by HPLC showed the reaction was complete. |
| The crude product was then further purified by recrystallization with a mixture of EtOAc and n-Heptane to give purified Compound A as a white solid. |
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
References
- Macrocyclic heterocycles and uses thereofPublication Number: US-2023339952-A1Priority Date: 2022-04-20
- Macrocyclic heterocycles and uses thereofPublication Number: WO-2023205701-A1Priority Date: 2022-04-20
- Methods for delaying, preventing, and treating acquired resistance to ras inhibitorsPublication Number: US-2023233569-A1Priority Date: 2020-06-18
- Ras inhibitorsPublication Number: US-2021130303-A1Priority Date: 2019-11-04
- Ras inhibitorsPublication Number: US-2023234929-A1Priority Date: 2019-11-04
- Ras inhibitors
- Publication Number: US-11566007-B2
- Priority Date: 2019-11-04
- Grant Date: 2023-01-31
////////elironrasib, ANAX LABS, Kirsten rat sarcoma viral oncogene homolog inhibitor, antineoplastic, RMC-6291, RMC 6291, 942KVV5CJP
Dirozalkib
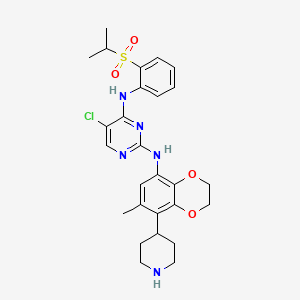
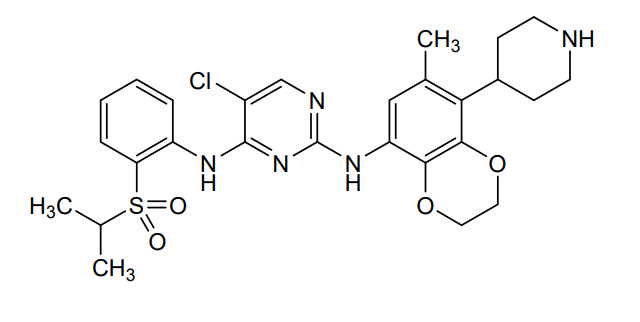
Dirozalkib
CAS 1893419-37-8
MF C27H32ClN5O4S MW558.1 g/mol
5-chloro-2-N-(6-methyl-5-piperidin-4-yl-2,3-dihydro-1,4-benzodioxin-8-yl)-4-N-(2-propan-2-ylsulfonylphenyl)pyrimidine-2,4-diamine

anaplastic lymphoma kinase (ALK) inhibitor, antineoplastic, XZP-3621, XZP 3621, Xuanzhu Biopharmaceutical, 2FH56C28YT
Dirozalkib (XZP-3621) is a novel, potent, and highly selective ALK/ROS1 tyrosine kinase inhibitor developed by Xuanzhu Biopharmaceutical to treat advanced ALK-positive non-small cell lung cancer (NSCLC). It demonstrated high efficacy (47.4% ORR, up to 89.3% in naive patients) in clinical trials and is designed to overcome resistance to earlier inhibitors.
Key Aspects of Dirozalkib
- Indication: Treatment of adult patients with ALK-positive locally advanced or metastatic non-small cell lung cancer (NSCLC).
- Mechanism: Acts as a dual-target ALK/ROS1 tyrosine kinase inhibitor (TKI), effective against ALK fusion-positive cells and various resistance mutations.
- Clinical Efficacy (Phase I/II): In studies, the drug showed significant antitumor activity with an Objective Response Rate (ORR) of 47.4% and an 89.3% ORR in ALK inhibitor-naive patients at 500 mg/day.
- Safety Profile: No dose-limiting toxicities occurred; the maximum tolerated dose was 600 mg/day, with a recommended dose of 500 mg/day. Common adverse events included diarrhea.
- Status: As of early 2026, the NDA (New Drug Application) for Dexitinib (Dirozalkib) was accepted by China’s NMPA, with potential for further market expansion.
- OriginatorXuanzhu Biopharmaceutical
- Class2 ring heterocyclic compounds; Amines; Aniline compounds; Antineoplastics; Chlorinated hydrocarbons; Piperidines; Pyrimidines; Small molecules; Sulfones
- Mechanism of ActionAnaplastic lymphoma kinase inhibitors
- RegisteredNon-small cell lung cancer
- 26 Aug 2025Chemical structure information added.
- 22 Aug 2025Registered for Non-small cell lung cancer (Late-stage disease) in China (PO) – First global approval
- 22 Aug 2025Efficacy and adverse events data from a phase III trial in Non-small cell lung cancer released by Xuanzhu Biopharmaceutical
- A Phase I Study of XZP-3621 in Chinese Patients With ALK or ROS1 Rearrangement Non-small Cell Lung CancerCTID: NCT05055232Phase: Phase 1Status: CompletedDate: 2025-07-24
- Food Effect and Mass Balance Study of XZP-3621 TabletsCTID: NCT05034120Phase: Phase 1Status: CompletedDate: 2025-05-25
- A Study of XZP-3621 in Chinese Patients With ALK Positive NSCLCCTID: NCT05482087Phase: Phase 2Status: Unknown statusDate: 2022-08-01
- A Study to Evaluate and Compare the Efficacy and Safety of XZP-3621 Versus CrizotinibCTID: NCT05204628Phase: Phase 3Status: Unknown statusDate: 2022-01-24
PAT
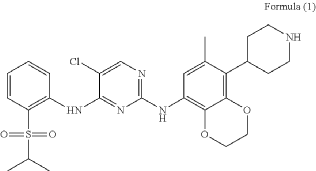
PAT
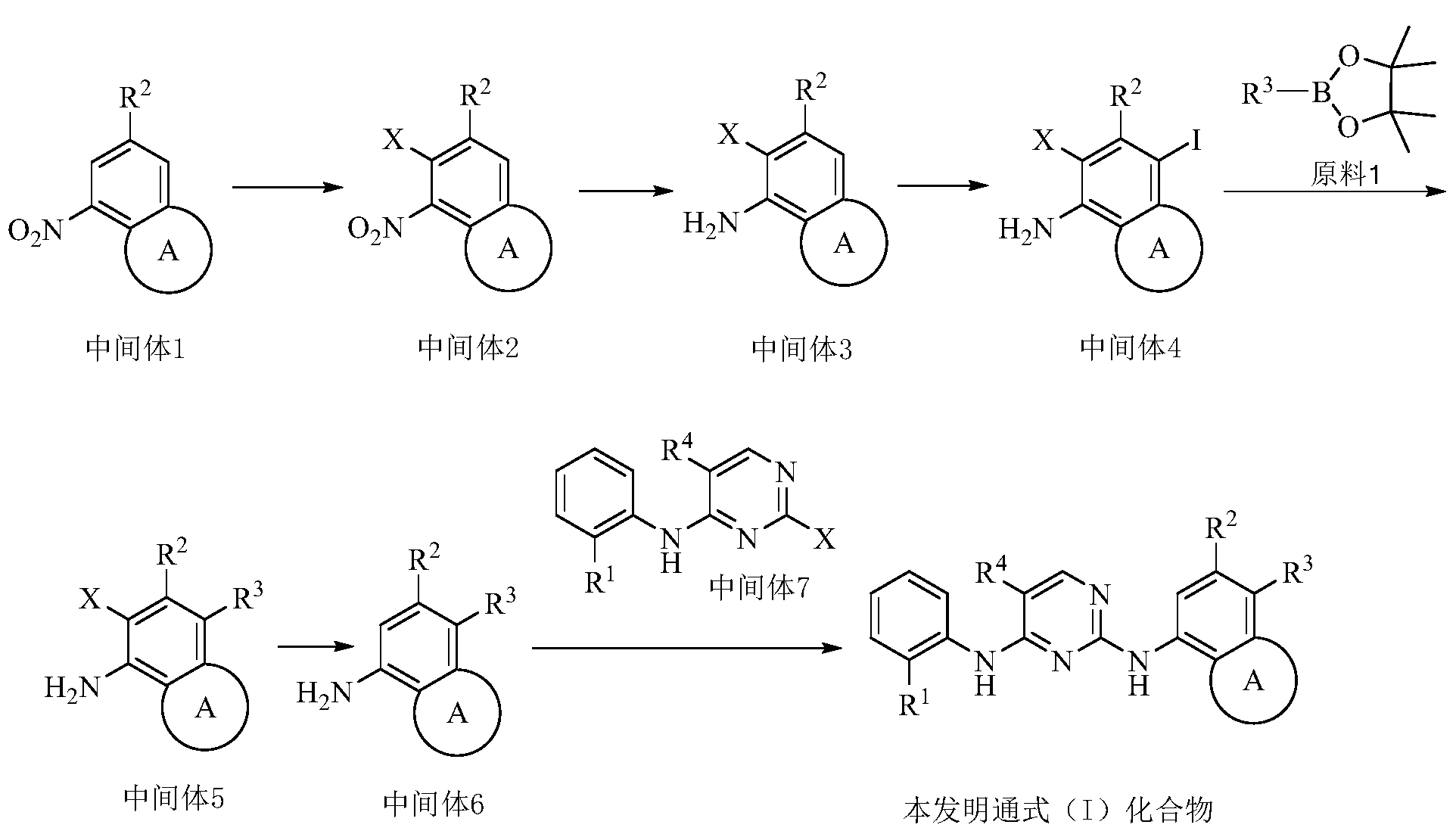
Example 3 Preparation of 2-((5-chloro-2-((7-methyl-8-(piperidin-4-yl)-2,3-dihydrobenzo[b][1,4]dioxin- 5-yl)amino)pyrimidin-4-yl)amino)-N,N-dimethylbenzenesulfonamide (compound 3)
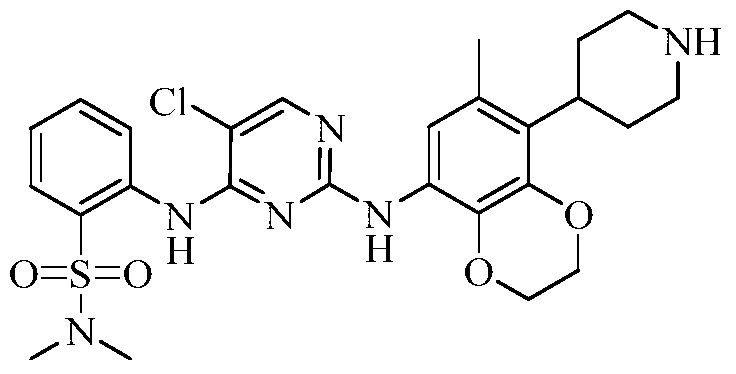
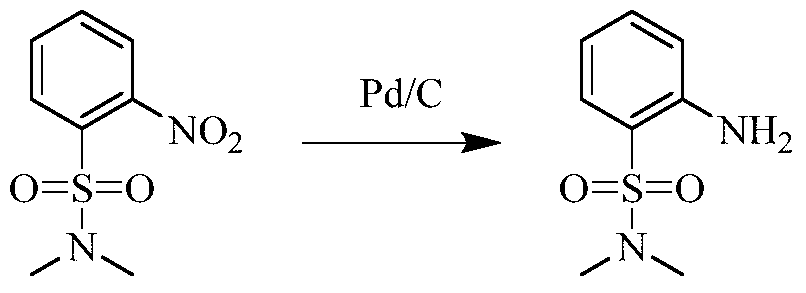
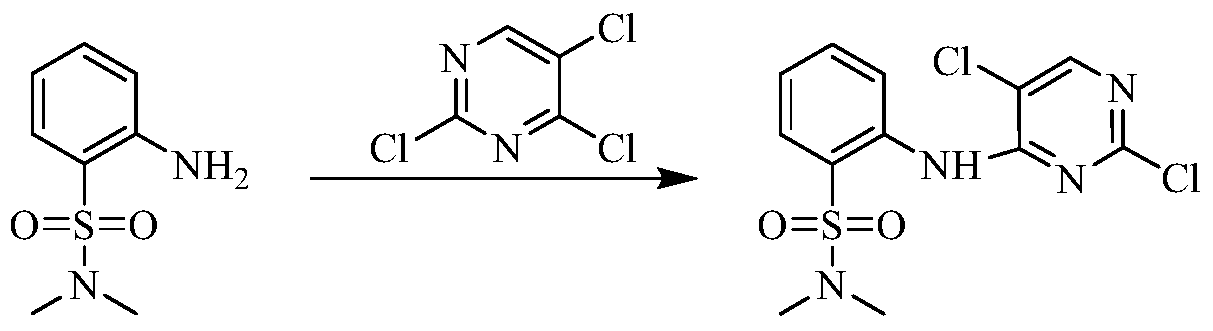
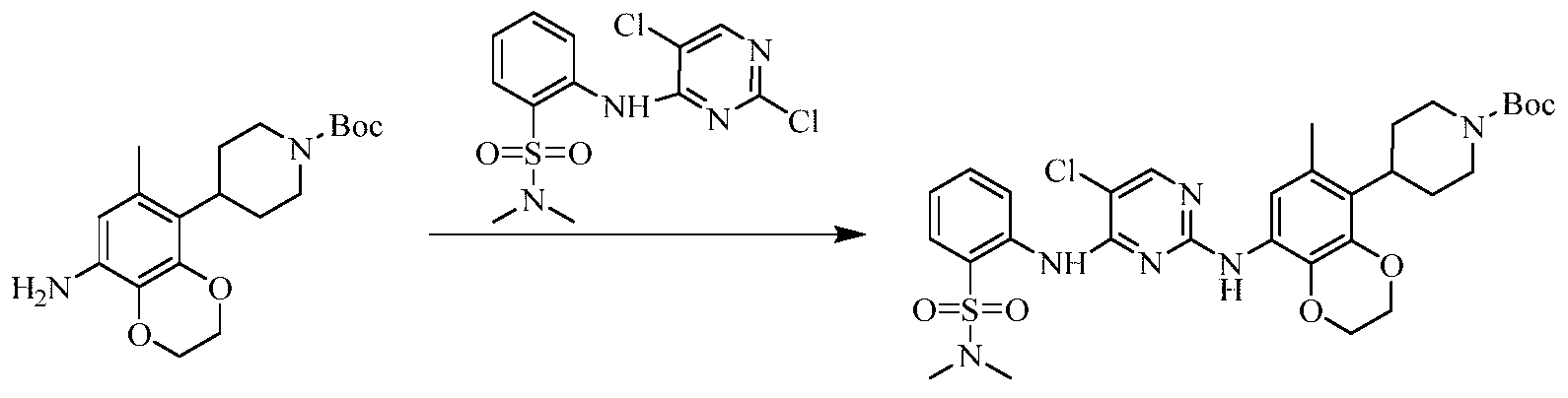
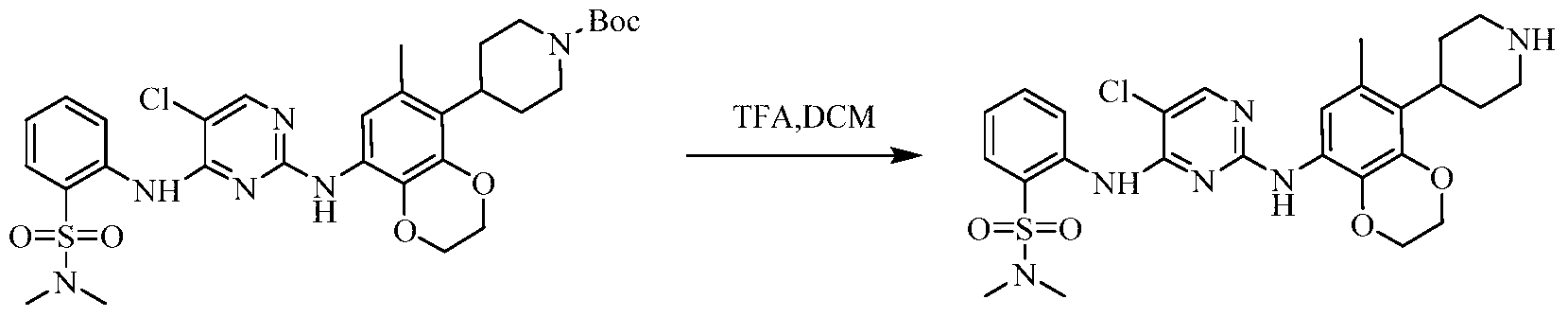
(5) Preparation of 2-((5-chloro-2-((7-methyl-8-(piperidin-4-yl)-2,3-dihydrobenzo[b][1,4]dioxin-5-yl)amino)pyrimidin-4-yl)amino)-N,N-dimethylbenzenesulfonamide
75 mg (0.114 mmol) of tert-butyl 4-(8-((5-chloro-4-((2-(N,N-dimethylaminosulfonyl)phenyl)amino)pyrimidin-2-yl)amino)-6-methyl-2,3-dihydrobenzo[b][1,4]dioxin-5-yl)piperidine-1-carboxylic acid ester was dissolved in dichloromethane (10 mL), and trifluoroacetic acid (1 mL) was added. The mixture was stirred at room temperature for 12 hours. The starting material disappeared as detected by TLC. Water (20 mL) was added, and the mixture was separated. The aqueous phase was extracted twice with dichloromethane (20 mL × 2). The organic phases were combined, dried over anhydrous sodium sulfate, and the solvent was removed by rotary evaporation. The crude product was purified by silica gel column chromatography (methanol:dichloromethane = 1:50) to obtain the final product (30 mg, yield 47.2%).
[0415]Molecular formula:
C26H31ClN6O4S Molecular weight: 559.08 LC-MS (m / z): 280.2 [ M /2+H ] +
[0416]
1H-NMR(400MHz,MeOD)δ:8.44(d,1H,J=1.2),8.11(s,1H),7.86(d,1H,J=1.2),7.56-7.60(m,1H),7.28-7.35(m,2H),4.26(s,4H),3.45-3.48(m,2H),3.06-3.15(m,3H),2.56-2.74(m,8H),2.17(s,3H),1.76-1.80(m,2H).
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Polycyclic inhibitor of anaplastic lymphoma kinasePublication Number: US-10011592-B2Priority Date: 2014-09-29Grant Date: 2018-07-03
- Polycyclic inhibitor of anaplastic lymphoma kinasePublication Number: US-2018086745-A9Priority Date: 2014-09-29
- Polycyclic anaplastic lymphoma kinase inhibitorPublication Number: EP-3202765-A1Priority Date: 2014-09-29
- Polycyclic inhibitor of anaplastic lymphoma kinasePublication Number: KR-20170055555-APriority Date: 2014-09-29
- Crystal form of polycyclic anaplastic lymphoma kinase inhibitorPublication Number: US-2023348443-A1Priority Date: 2020-01-17
- Crystal form of polycyclic anaplastic lymphoma kinase inhibitorPublication Number: US-12441717-B2Priority Date: 2020-01-17Grant Date: 2025-10-14
- Polycyclic inhibitor of anaplastic lymphoma kinasePublication Number: US-2017240534-A1Priority Date: 2014-09-29
- Polycyclic inhibitor of anaplastic lymphoma kinasePublication Number: KR-101909404-B1Priority Date: 2014-09-29Grant Date: 2018-10-17
- Polycyclic anaplastic lymphoma kinase inhibitorPublication Number: WO-2016050171-A1Priority Date: 2014-09-29
- Pharmaceutical composition of anaplastic lymphoma kinase inhibitor and preparation method thereforPublication Number: WO-2025140560-A1Priority Date: 2023-12-28
- CRYSTALLINE FORM OF A POLYCYCLIC ANAPLASIC LYMPHOMA KINASE INHIBITORPublication Number: EP-4092021-A4Priority Date: 2020-01-17
- Polycyclic anaplastic lymphoma kinase inhibitor crystalline formPublication Number: CN-113135905-BPriority Date: 2020-01-17Grant Date: 2023-11-21
- Crystal form of polycyclic anaplastic lymphoma kinase inhibitorPublication Number: WO-2021143819-A1Priority Date: 2020-01-17
- Crystal form of polycyclic anaplastic lymphoma kinase inhibitorPublication Number: EP-4092021-A1Priority Date: 2020-01-17
/////////dirozalkib, anax labs, anaplastic lymphoma kinase (ALK) inhibitor, antineoplastic, XZP-3621, XZP 3621, Xuanzhu Biopharmaceutical, 2FH56C28YT
Darlifarnib
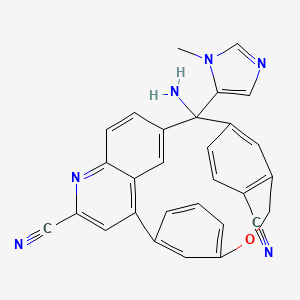
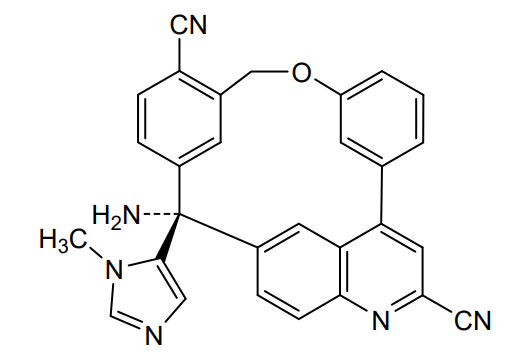
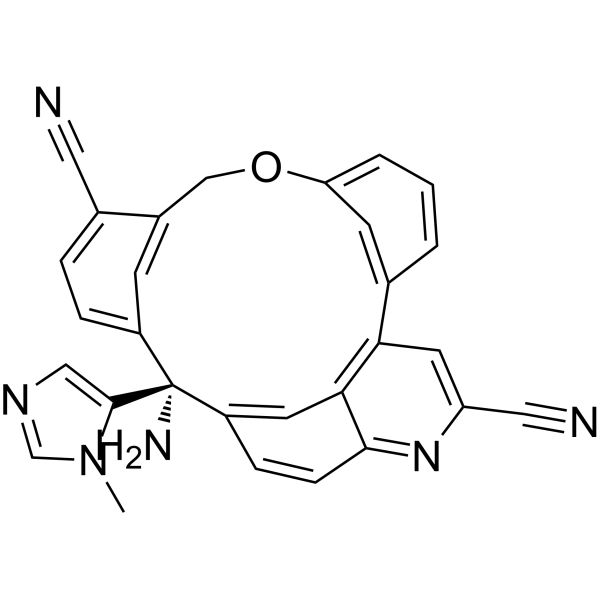
Darlifarnib
CAS 2939824-30-1
MF C29H20N6O MW 468.51
14-amino-14-(3-methylimidazol-4-yl)-7-oxa-19-azapentacyclo[13.6.2.12,6.19,13.018,22]pentacosa-1(22),2(25),3,5,9,11,13(24),15(23),16,18,20-undecaene-10,20-dicarbonitrile

farnesyl transferase inhibitor, antineoplastic, KO-2806, KO 2806, T206317
Darlifarnib (KO-2806) is an investigational, orally active next-generation farnesyl transferase inhibitor (FTI) being developed by Kura Oncology to treat solid tumors, such as clear cell renal cell carcinoma (ccRCC). It inhibits the enzyme farnesyl transferase, blocking KRAS and mTORC1 signaling to induce tumor regression. It is often combined with other agents to overcome resistance.
Key Details About Darlifarnib
- Mechanism of Action: As a FTI, darlifarnib binds to and inhibits farnesyl transferase, which prevents the activation of RAS oncogenes and inhibits downstream mTORC1 signaling, leading to tumor cell death.
- Target Indications: Preclinical and early clinical data show potential in treating KRAS-mutant cancers, including non-small cell lung cancer (NSCLC), colorectal cancer (CRC), and clear cell renal cell carcinoma (ccRCC).
- Combination Therapy: Data from the Phase 1 FIT-001 trial (presented in April 2026) showed that combining darlifarnib with the TKI cabozantinib demonstrated robust activity in patients with pretreated, advanced ccRCC.
- Overcoming Resistance: Darlifarnib is designed to re-sensitize tumors that have become resistant to prior therapies, such as RAS inhibitors and tyrosine kinase inhibitors (TKIs).
- Status: It is an investigational drug and not yet FDA-approved.
- OriginatorKura Oncology
- ClassAntineoplastics; Small molecules
- Mechanism of ActionFarnesyltranstransferase inhibitors
- Phase IAdenocarcinoma; Colorectal cancer; Non-small cell lung cancer; Renal cell carcinoma; Solid tumours
- 12 Jan 2026Kura Oncology plans the one or more expansion cohorts of KO 2806 and cabozantinib in patients with advanced renal cell carcinoma in the first half of 2026
- 22 Oct 2025Pharmacodynamics data from a preclinical trial in Cancer presented at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics 2025 (AACR-NCI-EORTC-2025)
- 18 Oct 2025Adverse events and efficacy data from a phase I trial in Non-small cell lung cancer, Renal cell carcinoma, Adenocarcinoma released by Kura Oncology
PAT
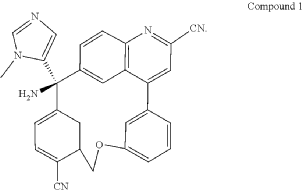
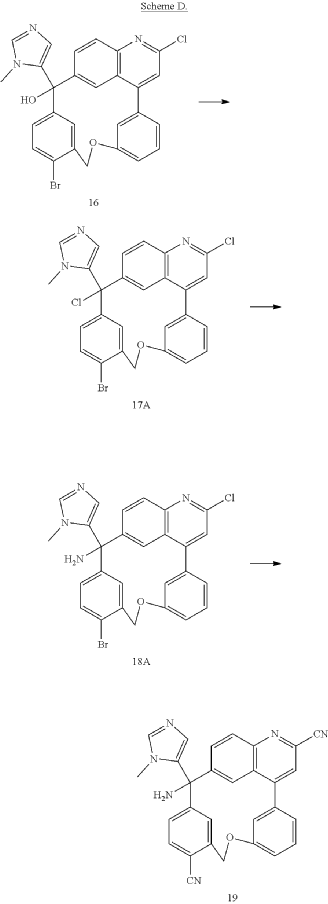
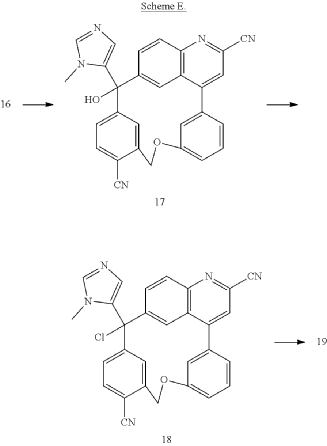
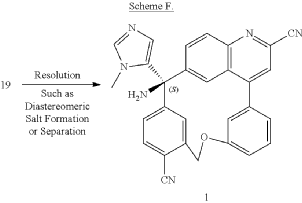
PAT
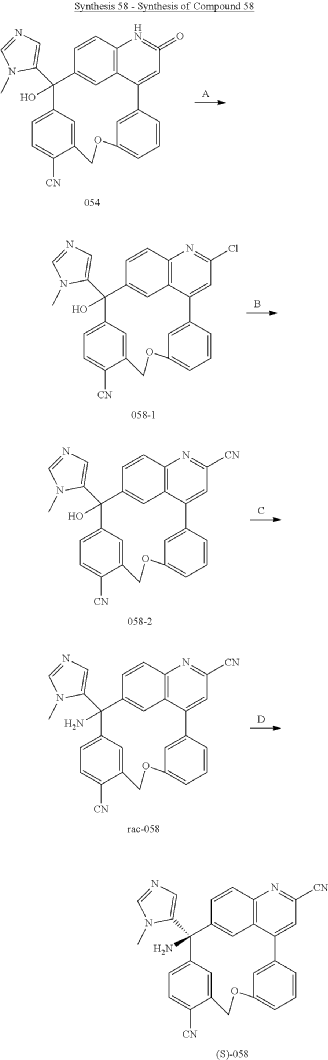
Step A: Preparation of (058-1)
Step B: Preparation of (058-2)
Step C: Preparation of (rac)-3-amino-3-(1-methyl-1H-imidazol-5-yl)-6-oxa-2(4,6)-quinolina-1,4(1,3)-dibenzenacyclohexaphane-22,44-dicarbonitrile (rac-058)
Step D: Preparation of (S)-3-amino-3-(1-methyl-1H-imidazol-5-yl)-6-oxa-2(4,6)-quinolina-1,4(1,3)-dibenzenacyclohexaphane-22,44-dicarbonitrile ((S)-058)
PAT
- Macrocyclic compounds and compositions, and methods of preparing and using the samePublication Number: US-2023322711-A1Priority Date: 2021-11-30
- Macrocyclic compounds and compositions, and methods of preparing and using the samePublication Number: US-12018011-B2Priority Date: 2021-11-30Grant Date: 2024-06-25
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
/////////////darlifarnib, ANAX LAB, farnesyl transferase inhibitor, antineoplastic, KO-2806, KO 2806, T206317
Daraxonrasib
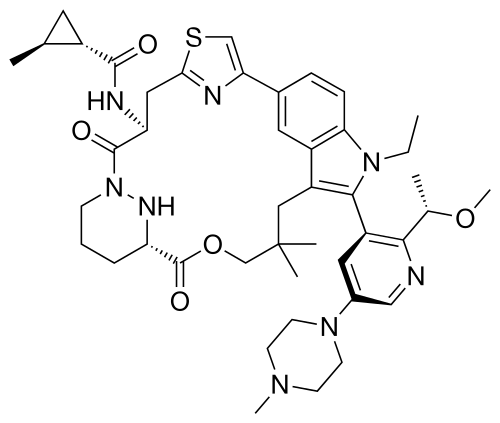
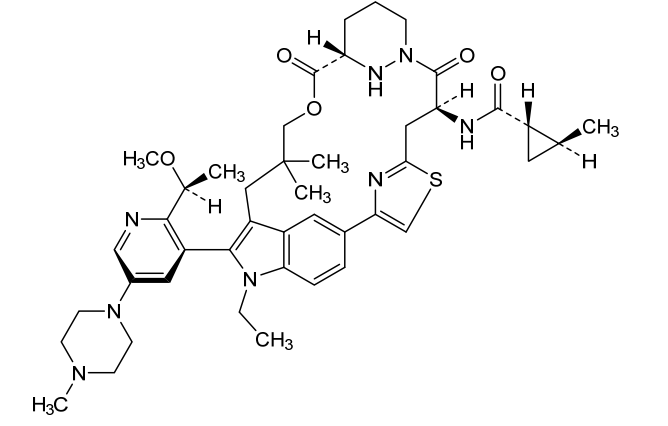
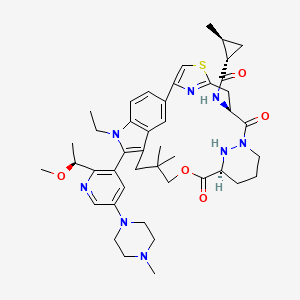
Daraxonrasib
CAS 2765081-21-6
MFC44H58N8O5S MW811.0 g/mol
trans-(1S,2S)-N-[(7S,13S)-21-ethyl-20-[2-[(1S)-1-methoxyethyl]-5-(4-methylpiperazin-1-yl)-3-pyridinyl]-17,17-dimethyl-8,14-dioxo-15-oxa-4-thia-9,21,27,28-tetrazapentacyclo[17.5.2.12,5.19,13.022,26]octacosa-1(25),2,5(28),19,22(26),23-hexaen-7-yl]-2-methylcyclopropane-1-carboxamide
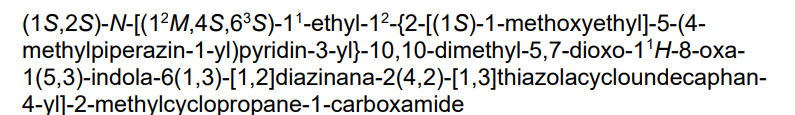
Kirsten rat sarcoma viral oncogene homolog inhibitor, antineoplastic, RMC-6236, RMC 6236, B6T47Y2UAP, RAS-IN-2,
Daraxonrasib (formerly RMC-6236) is an investigational, orally administered “molecular glue” RAS inhibitor developed by Revolution Medicines for treating advanced solid tumors with RAS mutations, particularly metastatic pancreatic cancer. April 2026 Phase 3 trials showed it significantly improves survival, demonstrating high potential as a first-line treatment.
Key Clinical Findings and Updates (as of April 2026):
- Mechanism: It acts as a RAS(ON) inhibitor, targeting mutated and wild-type RAS proteins (
) to disrupt cancer signaling.
- Breakthrough Results: Data from the RASolute 302 trial showed a substantial survival benefit in patients with previously treated metastatic pancreatic ductal adenocarcinoma (PDAC).
- High Response Rates: In trials, daraxonrasib combined with chemotherapy showed a 58% confirmed objective response rate (ORR) and 84% progression-free survival (PFS) at 6 months in untreated RAS-mutant metastatic pancreatic cancer.
- Safety Profile: Generally well-tolerated, with side effects including rash, diarrhea, stomatitis, and nausea.
- Recognition: Named the “2025 Molecule of the Year” by Drug Hunter for its, novel mechanism and clinical potential.


Daraxonrasib is currently being studied in the Phase 3 RASolute 303 trial for first-line treatment of pancreatic cancer.
Daraxonrasib (RMC-6236) is a RAS inhibitor drug. It is undergoing testing by Revolution Medicines to treat advanced solid tumors with RAS mutations, especially metastatic pancreatic ductal adenocarcinoma (PDAC) containing KRAS G12X mutations.[1] It received a breakthrough therapy designation from the U.S. Food and Drug Administration.[2]
Daraxonrasib is orally active and multi-selective RAS inhibitor. It uses a tri-complex mechanism to target the active, GTP-bound form of RAS proteins, including mutant and wild-type forms. Unlike conventional RAS inhibitors, it first binds to the chaperone-like protein cyclophilin A to form a complex, which then attaches to active RAS. This interaction blocks downstream effector binding and inhibits oncogenic signaling.[3]
In 2026, Daraxonrasib clinical trial completed a phase 3 clinical trial (RASolute 302) to assess efficacy compared to standard-of-care chemotherapy.[4] The trial met all primary and key secondary endpoints, including progression-free survival (PFS). The company reported median survival of 13.2 months with daraxonrasib vs. 6.7 months with standard chemotherapy. The hazard ratio for death was 0.40 (a 60% reduction in risk of death; p < 0.0001). Daraxonrasib was generally well tolerated with a manageable safety profile and no new safety signals.[5]
PAT
PAT
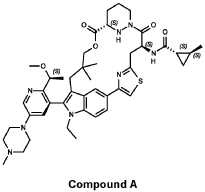
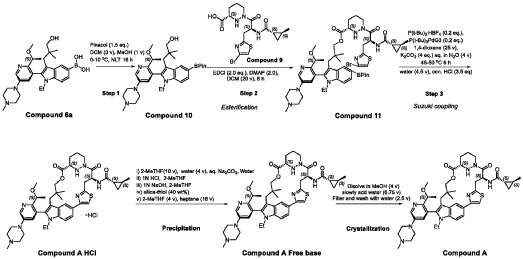
PATENT ATTORNEY DOCKET: 51432-038WO2 Part 4 – Purification of Compound A – (1S,2S)-N-[(7S,13S)-21-ethyl-20-{2-[(1S)-1- methoxyethyl]-5-(4-
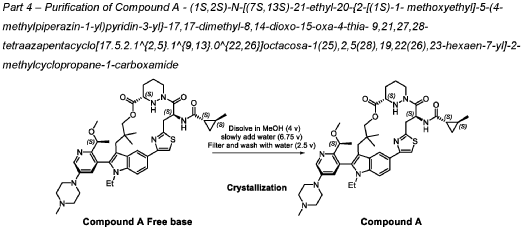
1.0equiv) at 25°C. The resulting suspension was stirred until solids were completely dissolved. The resulting methanol solution was filtered through microporous filter and transferred to another reactor. Then the reactor temperature was maintained at 25°C and slowly water (2.41kg, 1.0 V) water was added over a period of 30 minutes. The resulting cloudy solution was stirred for another 30 minutes at 25°C. Then a solution of methanol and water (3.42kg, 1:2, v/v) slowly over 1 hour. The resulting suspension was stirred for 2 hours at 25°C. Again, to the suspension additional water (2.48kg) slowly added over 1 hour. The final, suspension was stirred for additional 1 hour. Water (9.29kg, 3.75 V) was added to the suspension slowly over 2 hours and the mixture was stirred for at least for 16 hours at 25°C. The resulting suspension was filtered and washed with mixed solvent water: MeOH (3:2, v/v) twice (2x 2.2 kg), followed by water (4.91kg) washing. The wet cake was dried under reduced pressure and controlled humidity (temperature: 25 ± 5 ˚C, vacuum ≥ -0.085 MPa, humidity: 10%~20%) for 37 hours to afford Compound A as a white solid (2.68 kg, 99.4% a/a purity, 93.0% w/w assay, KF: 6.7%, 3.07 mol, 92% yield, Table 27).
PAT
- Synthesis of ras inhibitorsPublication Number: WO-2024216017-A2Priority Date: 2023-04-14
- Macrocycle compounds useful as kras inhibitorsPublication Number: WO-2024008834-A1Priority Date: 2022-07-08
- Ras inhibitorsPublication Number: US-11690915-B2Priority Date: 2020-09-15Grant Date: 2023-07-04
- Ras inhibitorsPublication Number: US-2023226186-A1Priority Date: 2020-09-15
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- Cregg J, Edwards AV, Chang S, Lee BJ, Knox JE, Tomlinson AC, et al. (March 2025). “Discovery of Daraxonrasib (RMC-6236), a Potent and Orally Bioavailable RAS(ON) Multi-selective, Noncovalent Tri-complex Inhibitor for the Treatment of Patients with Multiple RAS-Addicted Cancers”. Journal of Medicinal Chemistry. 68 (6): 6064–6083. doi:10.1021/acs.jmedchem.4c02314. PMID 40056080.
- Sava J (July 1, 2025). “Daraxonrasib Earns FDA Breakthrough Status in Pancreatic Cancer”. Targeted Oncology. Retrieved October 12, 2025.
- Jiang J, Jiang L, Maldonato BJ, Wang Y, Holderfield M, Aronchik I, et al. (June 2024). “Translational and Therapeutic Evaluation of RAS-GTP Inhibition by RMC-6236 in RAS-Driven Cancers”. Cancer Discovery. 14 (6): 994–1017. doi:10.1158/2159-8290.CD-24-0027. PMC 11149917. PMID 38593348.
- Clinical trial number NCT05379985 at ClinicalTrials.gov
- Mast J (2026-04-13). “Revolution Medicines touts ‘unprecedented’ data for pancreatic cancer pill”. STAT. Retrieved 2026-04-13.
| Clinical data | |
|---|---|
| Other names | RMC-6236 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2765081-21-6 |
| PubChem CID | 164726578 |
| IUPHAR/BPS | 13368 |
| ChemSpider | 115275938 |
| UNII | B6T47Y2UAP |
| KEGG | D13265 |
| ChEBI | CHEBI:746946 |
| Chemical and physical data | |
| Formula | C44H58N8O5S |
| Molar mass | 811.06 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////daraxonrasib, anax labs, Kirsten rat sarcoma viral oncogene homolog inhibitor, antineoplastic, RMC-6236, RMC 6236, B6T47Y2UAP, RAS-IN-2,

{kind=link}
{kind=link}