WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Remlifanserin is a small molecule drug. The usage of the INN stem ‘-anserin’ in the name indicates that Remlifanserin is a serotonin receptor antagonist. Remlifanserin has a monoisotopic molecular weight of 429.22 Da.
The drug is an improved follow-up compound to its developer’s earlier drug pimavanserin (Nuplaizid; ACP-103).[6] It is more potent and selective than pimavanserin as a serotonin 5-HT2A receptor inverse agonist.[10] Remlifanserin shows 32- to 123-fold selectivity for antagonism and inverse agonism of the serotonin 5-HT2A receptor over the serotonin 5-HT2C receptor depending on the bioassay.[10] For comparison, pimavanserin’s selectivity was 8- to 37-fold depending on the assay.[10] Remlifanserin shows very low affinity for the serotonin 5-HT2B receptor compared to the serotonin 5-HT2A and 5-HT2C receptors.[10] It is expected to have less QT prolongation than pimavanserin.[10] The drug blocks the head-twitch response induced by the serotonergic psychedelicDOI and the hyperlocomotion induced by the NMDA receptor antagonistdizocilpine (MK-801) in rodents.[10]
Remlifanserin is under development by Acadia Pharmaceuticals.[1][5] As of January 2025, it is in phase 3clinical trials.[1][5] Its clinicaltrials.gov identifier (nct number) is NCT06159673.[11]
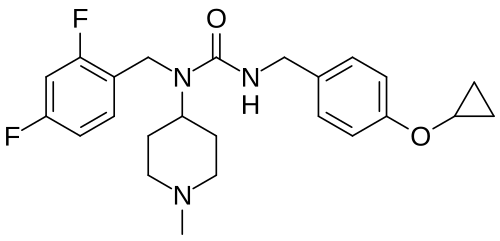
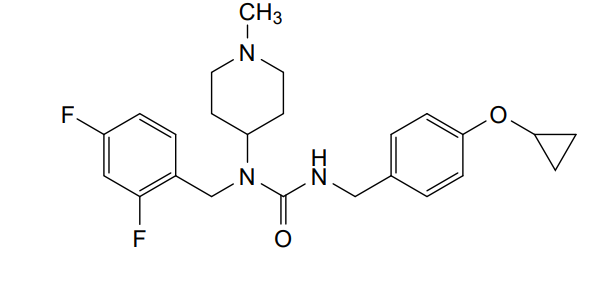
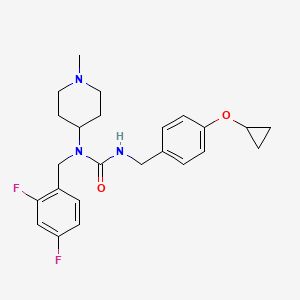
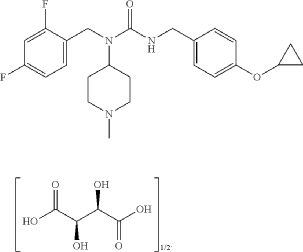
N-[(2,4-difluorophenyl)methyl]-1-methylpiperidin-4-amine (1.89 mmol, 478 mg), phenyl N-[(4-cyclopropoxyphenyl)methyl]carbamate (97%, 585 mg, 2.0 mmol) and potassium carbonate (2.5 mmol, 350 mg) were suspended in toluene (5.0 ml). The mixture was stirred at 70° C. for 16 hours, then partitioned between toluene and sodium hydroxide (aqueous, 0.5 M). The organic phase was separated and concentrated. The material was purified by silica gel chromatography, eluting with 0-50% methanol in ethyl acetate. Fractions containing the desired product were pooled and concentrated. Diethyl ether (10 ml) was added. The mixture was filtered and concentrated to give 3-[(4-cyclopropoxyphenyl)methyl]-1-[(2,4-difluorophenyl)methyl]-1-(1-methylpiperidin-4-yl)urea (725 mg, 1.688 mmol, 89% yield). This material (725 mg, 1.688 mmol) and L-(+)-tartaric acid (0.844 mmol, 127.3 mg) were dissolved in ethanol (5.0 ml) using an ultrasonication bath. The solvents were then evaporated to give the title compound as the hemitartrate salt (glassy foam, 906 mg). 1H NMR (400 MHz, Chloroform-d) δ 7.16 (q, 1H), 7.05 (d, 2H), 6.94 (d, 2H), 6.84-6.73 (m, 2H), 4.70 (bt, 1H), 4.62-4.48 (m, 1H), 4.41 (s, 2H), 4.33 (s, 1H), 4.28 (d, 2H), 3.70 (m, 1H), 3.42 (t, 2H), 2.72-2.56 (m, 2H), 2.63 (s, 3H), 2.18 (m, 2H), 1.81 (d, 2H), 0.76 (m, 4H); LC-MS: 430.3 [M+H] +.
Relicpixant is a small molecule drug. The usage of the INN stem ‘-pixant’ in the name indicates that Relicpixant is a purinoreceptor (P2X) antagonist. Relicpixant has a monoisotopic molecular weight of 500.07 Da.
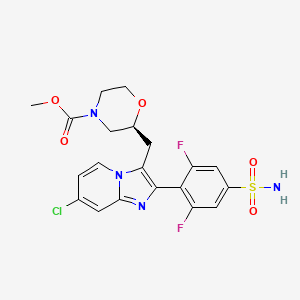
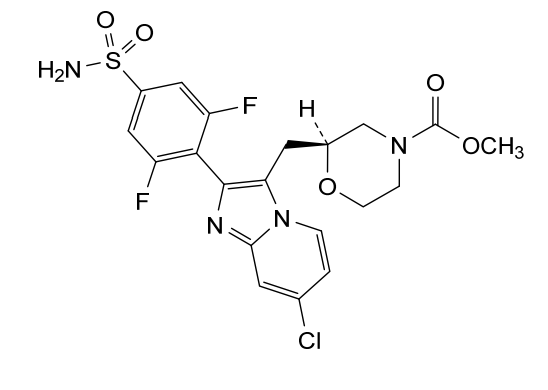
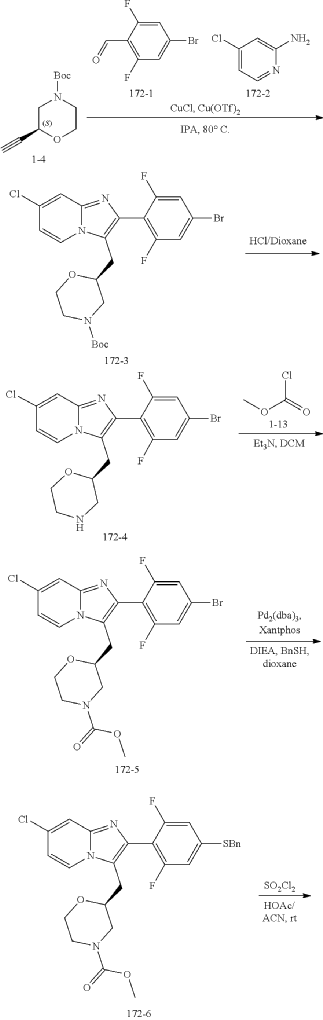
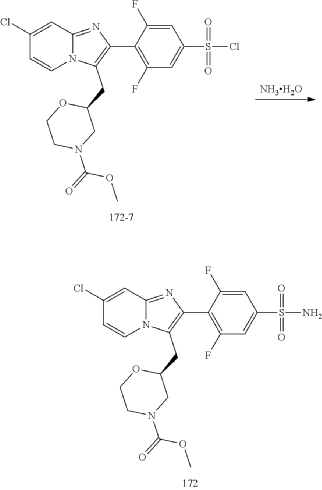
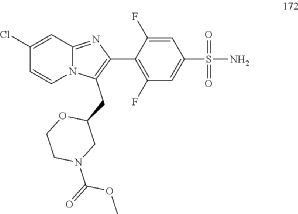
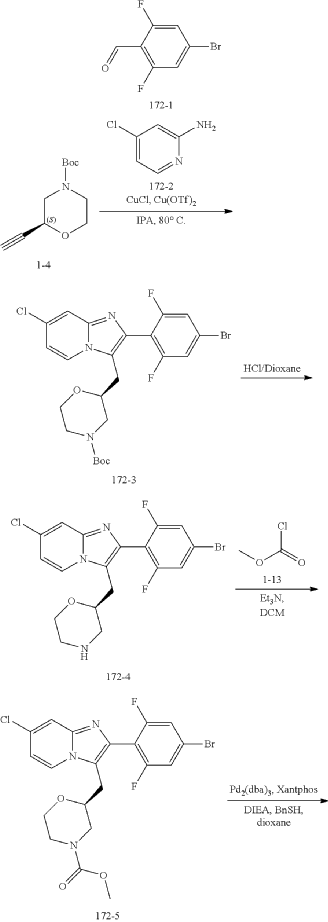
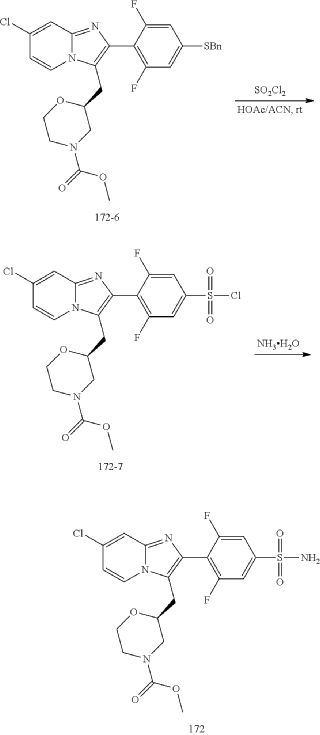
Step (8) Preparation of methyl (S)-2-((7-chloro-2-(2,6-difluoro-4-sulfamoylphenyl)imidazo[1,2-a]pyridin-3-yl)methyl)morpholine-4-carboxylate
Aqueous ammonia (2 mL) was diluted with acetonitrile (1 mL) and added dropwise to the above reaction system at 0° C. The reaction system was reacted at room temperature for 0.5 h. The starting material was consumed completely, and a target product was generated as detected by LCMS. The reaction system was extracted with water and ethyl acetate twice, washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by preparative chromatography to give compound 172 (185 mg, 99.74% purity) in the form of a white solid. LC-MS: [M+H] +=501.1.
To the above reaction mixture was added dropwise ammonia water (2 mL) diluted with acetonitrile (1 mL) at 0° C., and reaction mixture was reacted at room temperature for 0.5 hours. The raw material completely disappeared, and a target product was generated, as shown by LC-MS. The reaction mixture was extracted twice with water and ethyl acetate, washed with brine solution, dried over anhydrous sodium sulfate, concentrated, and purified by C18 chromatography column (water/acetonitrile, RRt=22.5 min). Compound A in an amorphous state (compound A is the compound of formula A) (185 mg, purity: 99.74%) was obtained as a white solid. LC-MS: [M+H]=501.1.
Mechanism of ActionIsocitrate dehydrogenase 1 inhibitors; Isocitrate dehydrogenase 2 inhibitors
Phase IIIAcute myeloid leukaemia
No development reportedHaematological malignancies; Solid tumours
28 Sep 2025No recent reports of development identified for phase-I development in Haematological-malignancies(Late-stage disease, Second-line therapy or greater) in Spain (PO)
28 Sep 2025No recent reports of development identified for phase-I development in Haematological-malignancies(Late-stage disease, Second-line therapy or greater) in USA (PO)
19 Sep 2025No development reported – Phase-I for Solid tumours (Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA (PO)
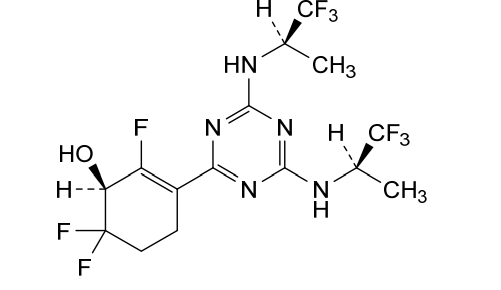
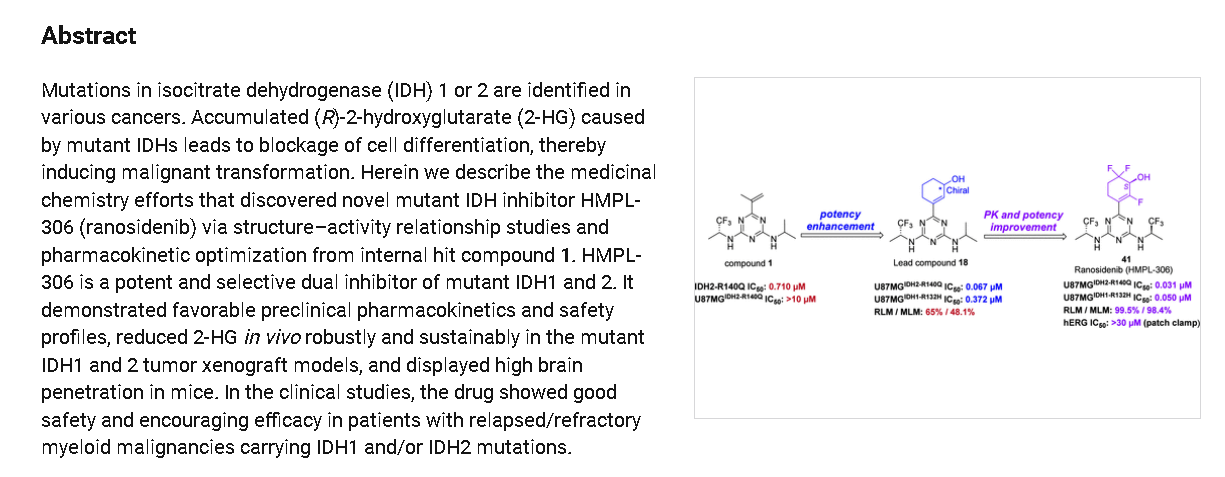
Ranosidenib is a small molecule drug. Ranosidenib is under investigation in clinical trial NCT06387069 (A Study to Evaluate HMPL-306 in Patients With IDH1- and IDH2-mutated Acute Myeloid Leukemia). Ranosidenib has a monoisotopic molecular weight of 453.12 Da.
Ranosidenib is an orally bioavailable inhibitor of mutated forms of both isocitrate dehydrogenase type 1 (IDH1, IDH1 [NADP+] soluble) in the cytoplasm and type 2 (IDH2, isocitrate dehydrogenase [NADP+], mitochondrial) in the mitochondria, with potential antineoplastic activity. Upon administration, ranosidenib specifically targets and inhibits mutant forms of IDH1 and IDH2, thereby inhibiting the formation of the oncometabolite 2-hydroxyglutarate (2HG) from alpha-ketoglutarate (a-KG). This prevents 2HG-mediated signaling and leads to both an induction of cellular differentiation and an inhibition of cellular proliferation in tumor cells expressing IDH mutations. IDH1 and 2, metabolic enzymes that catalyze the conversion of isocitrate into a-KG, play key roles in energy production and are mutated in a variety of cancer cell types. Mutant forms of IDH1 and 2 catalyze the formation of 2HG and drive cancer growth by blocking cellular differentiation and inducing cellular proliferation.
A Study of HMPL-306 in Advanced Hematological Malignancies With mIDHCTID: NCT04764474Phase: Phase 1Status: TerminatedDate: 2026-01-29
A Study of HMPL-306 in Advanced Solid Tumors With IDH MutationsCTID: NCT04762602Phase: Phase 1Status: TerminatedDate: 2025-09-16
A Study to Evaluate HMPL-306 in Patients With IDH1or IDH2-mutated Acute Myeloid LeukemiaCTID: NCT06387069Phase: Phase 3Status: RecruitingDate: 2025-08-14
Phase I Study of HMPL-306 for the Treatment of Gliomas With IDH1 and/or IDH2 MutationsCTID: NCT07025018Phase: Phase 1Status: RecruitingDate: 2025-08-01
A Study of HMPL-306 in Patients With IDH1 and/or IDH2 Mutation of Relapsed/Refractory Myeloid Leukemia/NeoplasmsCTID: NCT04272957Phase: Phase 1Status: Unknown statusDate: 2020-06-16
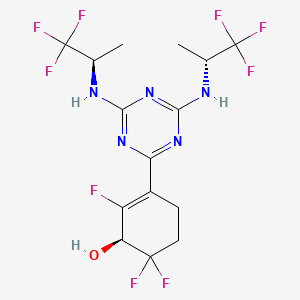
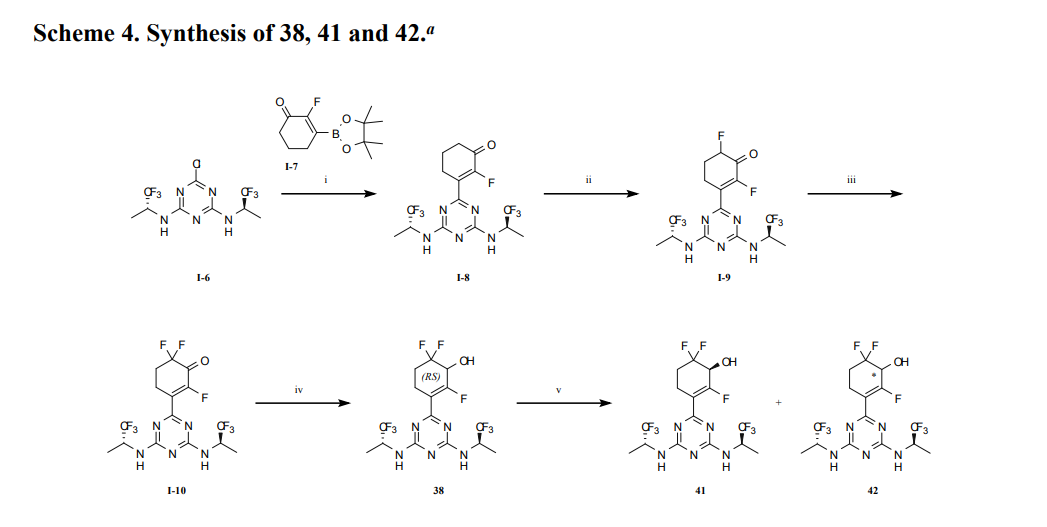
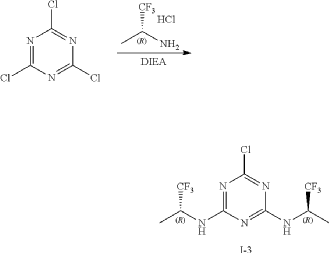
At 0° C., to a flask were added 1,4-dioxane (50 mL), 2,4,6-trichloro-1,3,5-triazine (1.84 g, 10 mmo), (R)-1,1,1-trifluoropropan-2-amine hydrochloride (2.99 g, 20 mmol) and DIEA (5.17 g, 40 mmol). The reaction was heated to 60° C. and stirred for 4 hours. After the reaction was completed, the mixture was condensed and purified by flash column chromatography (eluting with gradient water/MeOH=100:0-0:100) to give Intermediate I-3 as yellow solid (2.50 g, yield: 74%). MS (m/z): 338.0 [M+H] +
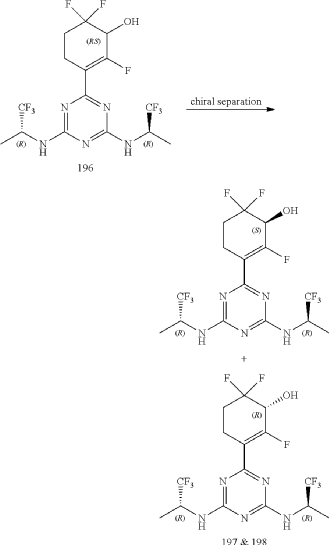
Compounds 197 and 198
3-(4,6-Bis(((R)-1,1,1-trifluoropropan-2-yl)amino)-1,3,5-triazin-2-yl)-2,6,6-trifluorocyclohex-2-en-1-ol, optically pure diastereoisomers
The Compound 196 was resolved by chiral HPLC to provide a pair of optically pure diastereoisomers, Compounds 197 and 198 (Chiral HPLC conditions: Preparation instrument: Shimadzu LC-10AD vp; Column: Daicel AD-H(250 mm*30 mm, 5 um); mobile phase: n-heptane/isopropanol=90/10; flow rate: 40 mL/min; column temperature: 40° C.). The first eluent (RT=4.203 min) was concentrated and purified by flash column chromatography (eluting with gradient PE/EA=100:0-0:100) to give a compound named as Compound 197, de %=99.27%, MS (m/z): 454.1 [M+1] +. The second eluent (RT=5.906 min) was concentrated and purified by flash column chromatography (eluting with gradient PE/EA=100:0-0:100) to give a compound named as Compound 198, de %=97.82%, MS (m/z): 454.2 [M+1] +.
Progerinin (SLC-D011) is an orally active, targeted inhibitor designed to reduce the toxic, premature-aging protein “progerin” in Hutchinson-Gilford Progeria Syndrome (HGPS). It binds to progerin, disrupting its interaction with lamin A and promoting its degradation. Studies show it improves cardiac function, increases lifespan in mouse models, and is currently in clinical trials
Key Aspects of Progerinin:
Mechanism of Action: Progerinin is an optimized progerin-lamin A binding inhibitor that selectively reduces progerin levels while sparing wild-type lamin A, B, and C.
Disease Application: It targets HGPS, a rare genetic disease that causes premature, rapid aging and death due to cardiac issues.
Preclinical Results: In Lmna mouse models, progerinin demonstrated improved physical conditions (hair morphology, body weight) and significantly extended lifespan (up to 14–21 weeks).
Cardiac Benefits: It alleviates cardiovascular abnormalities, such as reducing cardiac muscle weakness, which is a major cause of death in HGPS patients.
Clinical Status: A Phase I clinical trial was conducted for safety in healthy volunteers. As of 2025, trials are examining its efficacy, sometimes in combination with lonafarnib (Zokinvy).
Administration: It is developed as a nanosuspension for oral administration. National Institutes of Health (NIH) | (.gov) +7
Progerinin was developed by Korean-based biotech company PRG Science & Technology Co., Ltd. (PRG S&T)
Progerinin (SLC-D011) is an orally active progerin-lamin A binding inhibitor. Progerinin selectively binds to the C-terminal region of progerin, disrupting its interaction with lamin A and promoting progerin degradation while sparing wild-type lamin A, B, and C. Progerinin ameliorates nuclear deformation, increases H3K9me3 levels, and reduces progerin expression in HGPS patient-derived fibroblasts. Progerinin extends lifespan in LmnaG609G/G609G mice and LmnaG609G/+ mice, improves body weight, hair morphology, cardiac function, and histological phenotypes. Progerinin can be used for the study of Hutchinson-Gilford progeria syndrome (HGPS).
Study to Determine Optimal Dose and Evaluate Safety, Tolerability, and Pharmacokinetics of Progerinin in Patients With Hutchinson-Gilford Progeria Syndrome (HGPS)CTID: NCT06775041Phase: Phase 2Status: Active, not recruitingDate: 2026-02-09
Phase 2, Open-Label Study to Evaluate the Safety and Tolerability of Progerinin in Werner SyndromeCTID: NCT05847179Phase: Phase 2Status: Not yet recruitingDate: 2026-01-23
Phase I Study of Progerinin in Healthy VolunteersCTID: NCT04512963Phase: Phase 1Status: CompletedDate: 2021-09-22
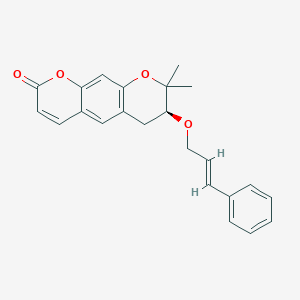
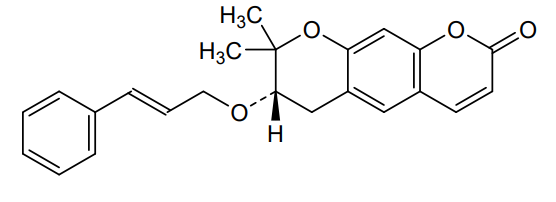
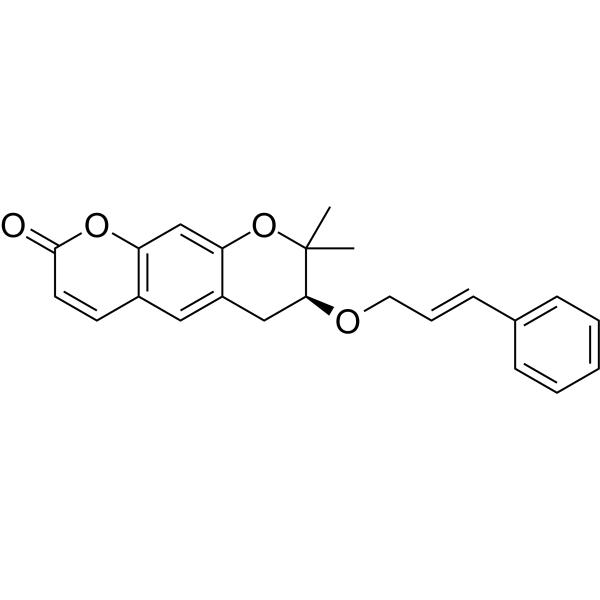
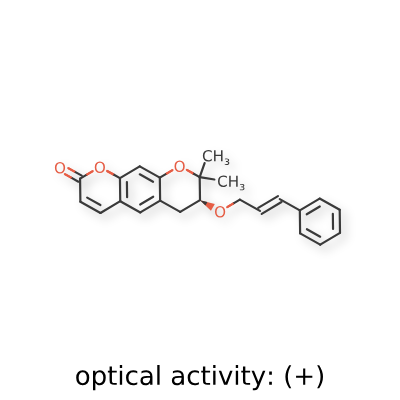
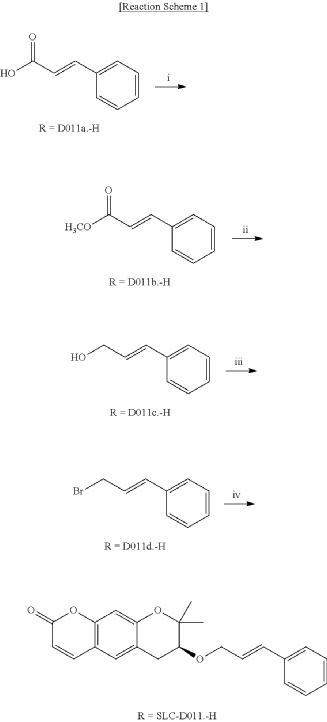
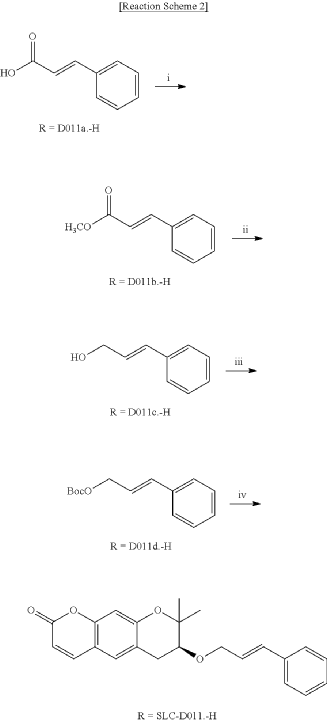
<Example 1> Synthesis of Ether-Form (+)-Decursin Derivative (SLC-D011)
(7S)-(+)-8,8-dimethyl-7-(3-phenyl-allyloxy)-7,8-dihydro-6H-pyrano[3,2-g]chromen-2-one (SLC-D011) was synthesized through the manner as in the to following Reaction Schemes 1 and 2.
1. Synthesis Process I
Step (I): After dissolving trans-cinnamic acid (D0111a, 5 g, 33.7 mmol) in methanol (50 ml) in a 100 ml round bottom flask, 5 drops of concentrated H 2SO 4 was added and the mixture was refluxed by heating at 80° C. for 24 hours and was cooled to room temperature and then concentrated under reduced pressure.
Then, the mixture was separated with dichloromethane (300 ml) and distilled water (300 ml) to collect the organic layer and dehydrated with sodium sulfate and filtered.
After filtration, the filtrate was concentrated under reduced pressure to obtain 3-phenyl-acrylic acid, methyl ester (D011b, 5.39 g, yield=98.5%) as a pure product to apply to the next step.
Step (II): 3-phenyl-acrylic acid, methyl ester (D011b, 4 g, 24.7 mmol, 1 eq) was added into a 500 ml round bottom flask filled with N 2 gas and was dissolved in anhydrous dichloromethane and then placed in a low-temperature reactor set at −78° C.
Diisobutylaluminium hydride 1M solution (DIBAL-H; 1M solution in hexane, 74 ml, 74.0 mmol, 3 eq) was slowly added dropwise over 30 minutes to the reaction solution and methanol (22 ml) was slowly added dropwise while the reaction temperature was raised to 0° C. and stirring was carried out for 1 hour.
The reaction solution was transferred to room temperature, stirred for 30 minutes and then a saturated aqueous solution of Rochelle’s salt (88 ml) was added thereto.
The reaction mixture was vigorously stirred at room temperature for 2 hours, and the mixture was partitioned twice with dichloromethane (300 ml) and distilled water (300 ml) to collect the organic layers and was dehydrated with sodium sulfate, filtered and the resulting filtrate was concentrated under reduced pressure.
The concentrate was purified by silica gel column chromatography (ethyl acetate:n-hexane=3:1) to obtain the pure product 3-phenyl-pro-2-pen-1-ol (D011c, 3.1 g, yield=93.9%, Rf=0.37 (2:1 n-hexane-ethyl acetate) to apply to the next step.
Step (III): To a 100 ml round bottom flask was added 3-phenyl-pro-2-pen-1-ol (D011c, 1 g, 7.45 mmol, 1 eq), was dissolved in anhydrous dichloromethane, PBr 3 (phosphoric tribromide, 253.6 μl, 2.608 mmol, 0.35 eq) was added on the steam bath and stirred for 1 hour.
The reaction mixture was concentrated and purified by silica gel column chromatography (ethyl acetate:n-hexane=1:8) to obtain the pure product, 3-bromo-propenyl)-benzene (D005d, 1.42 g, yield=96.2%, Rf=0.34 (5:1 n-hexane-ethyl acetate) which was applied to the next step.
Step (IV): (S)-(+)-decursinol (SLC-B001, 2.33 g, 9.47 mmol, 1 eq) was dissolved in anhydrous N,N-dimethylformamide (DMF, 10 ml) in a 100 ml round bottom flask under an N 2 gas and was placed in a low temperature reactor set at −20° C.
(3-bromo-propenyl)-benzene (D005d, 2.8 g, 14.2 mmol, 1.5 eq) and sodium sulfate (NaH 60%, 757 mg, 18.9 mmol) were added to the reaction mixture and stirred for 4 hours. 3 ml of distilled water was added and after 10 minutes, it was taken out from the low-temperature reactor. Then, it was separated into twice with dichloromethane (200 ml) and distilled water (200 ml) to collect the organic layer and dehydrated with sodium sulfate, filtered and the resulting filtrate was concentrated under reduced pressure.
Step (I): After dissolving trans-cinnamic acid (D011a, 5 g, 33.7 mmol) in methanol (50 ml) in a 100 ml round bottom flask, 5 drops of concentrated H 2SO 4 was added and the mixture was refluxed by heating at 80° C. for 24 hours. The reaction mixture was cooled to room temperature and then concentrated under reduced pressure and was separated with dichloromethane (300 ml) and distilled water (300 ml) to collect the organic layer and dehydrated with sodium sulfate and filtered.
After filtration, the obtained filtrate was concentrated under reduced pressure to obtain 3-phenyl-acrylic acid, methyl ester (D011b, 5.39 g, yield=98.5%) as a pure product which was applied to the next step.
Step (II): 3-phenyl-acrylic acid, methyl ester (D011b, 4 g, 24.7 mmol, 1 eq) was added into a 500 ml round bottom flask filled with N 2 gas and was dissolved in anhydrous dissolved in anhydrous dichloromethane and then placed in a low-temperature reactor set at −78° C.
Diisobutylaluminium hydride 1M solution (DIBAL-H; 1M solution in hexane, 74 ml, 74.0 mmol, 3 eq) was slowly added dropwise over 30 minutes to the reaction solution and methanol (22 ml) was slowly added dropwise while the reaction temperature was raised to 0° C. and stirring was carried out for 1 hour.
The reaction solution was transferred to room temperature, stirred for 30 minutes and then a saturated aqueous solution of Rochelle’s salt (88 ml) was added thereto. The reaction mixture was vigorously stirred at room temperature for 2 hours, and the mixture was partitioned twice with dichloromethane (300 ml) and distilled water (300 ml).
The organic layers were collected and were dehydrated with sodium sulfate, filtered and the resulting filtrate was concentrated under reduced pressure.
The concentrate was separated by silica gel column chromatography (ethyl acetate:n-hexane=3:1) to obtain the pure product 3-phenyl-pro-2-pen-1-ol (D011c, 3.1 g, yield=93.9%, Rf=0.37 (2:1 n-hexane-ethyl acetate) which was applied to the next step.
Step (III): 3-phenyl-prop-2-pen-1-ol (D011c, 1 g, 7.45 mmol, 1 eq) was added to a 100 ml round bottom flask filled with N 2 gas and was dissolved in anhydrous dichloromethane, and trimethylamine (Et 3N, 1.04 ml, 7.45 mmol, 1 eq), 4-dimethylaminopyridine (4-DMAP, 92 mg, 0.75 mmol, 0.1 eq), di-tert-butyl-dicarbonate (0.13 ml, 7.45 mmol, 1 eq), 4-dimethylaminopyridine (4-DMAP, 92 mg, 0.75 mmol, tert-butyl-dicarbonate (Boc2O, 2.57 ml, 11.18 mmol, 1.5 eq) were sequentially added, and the reaction solution was stirred at room temperature for 2 hours.
The reaction mixture was concentrated and separated by silica gel column chromatography (ethyl acetate:n-hexane=1:30) to obtain the pure product, tert-butyl cinnamyl carbonate (D011d, 1.30 g, yield=74.7%, Rf=0.32 (20:1 n-hexane-ethyl acetate)) which was applied to the next step.
Step (IV): Tert-butyl cinnamyl carbonate (D011d, 1.43 g, 6.09 mmol, 1.5 eq), (S)-(+)-decursinol (SLC—4.06 mmol, 1 eq) were added into a 100 ml round bottom flask and the mixture was dried under vacuum for 1 hour.
The dried mixture was dissolved in anhydrous tetrahydrofuran under N 2 gas and after bubbling the solution for 1 hour using N 2 gas, tetrakis(triphenylphosphine) palladium (Pd (PPh 3) 4, 188 mg, 0.162 mmol, 0.04 eq) was added to the reaction mixture and was refluxed overnight. The mixture liquid was concentrated under reduced pressure, and was separated by silica gel column chromatography (ethyl acetate:n-hexane=gradient elution to 1:4 from 1:8) to obtain compound (7S)-(+)-8,8-dimethyl-7-(3-phenyl-allyloxy)-7,8-dihydro-6H-pyrano[3,2-g]chromen-2-one (SLC-D011) of 1.20 g (81.3%). Yield 81.3%, white solid, mp: 143° C., R f=0.39 (2:1 n-hexane-ethyl acetate); [α] 25D+117.6 (c=1, CHCl 3); 1H NMR (400 MHz, CDCl 3): δ H 7.56 (1H, d, J=9.6 Hz, H-4), 7.38-7.23 (5H, m, H-5′, H-6′, H-7′, H-8′, H-9′), 7.15 (1H, s, H-5), 6.76 (1H, s, H-10), 6.59 (1H, d, J=16.0 Hz, H-3′), 6.30-6.23 (1H, m, H-2′), 6.20 (1H, d, J=9.6 Hz, H-3), 4.34 (1H, dd, J=6.0, 12.8 Hz, H-1a′), 4.21 (1H, dd, J=6.0, 12.4 Hz, H-1b′), 3.59 (1H, dd, J=5.2, 7.6 Hz, H-7), 3.07 (1H, dd, J=4.8, 16.0 Hz, H-6a), 2.85 (1H, dd, J=7.2, 16.4 Hz, H-6b), 1.41 (3H, s CH 3-8), 1.36 (3H, s, CH 3-8); 13C NMR (100 MHz, acetone-d 6) δ C 161.2 (C-2), 157.8 (C-9a), 155.3 (C-10a), 144.5 (C-4), 137.9 (C-4′), 132.9 (C-3′), 130.4 (C-5), 129.6 (C-6′, C-8′), 128.6 (C-7′), 127.5 (C-2′), 127.4 (C-5′, C-9′), 118.3 (C-5a), 113.7 (C-3), 113.6 (C-4a), 104.5 (C-10), 78.8 (C-7), 76.4 (C-8), 70.8 (C-1′), 27.8 (C-6), 26.1 (CH 3-8), 22.2 (CH 3-8); ESI-MS: m/z=363 [M+H]+. Anal. Calc. for C 23H 22O 4: C, 76.22; H, 6.12; Found: C, 76.20; H, 6.10.
Mechanism of ActionEndonuclease inhibitors; Virus replication inhibitors
MarketedInfluenza virus infections
27 Feb 2026Launched for Influenza virus infections (In adults, In adolescents) in China (PO), prior to February 2026 (TaiGen Biotechnology pipeline, February 2026)
26 Jan 2026Pixavir marboxil licensed to Boryung Biopharma for commercialization in South Korea
16 Dec 2025Chemical structure information added.
Pixavir marboxil (also known as TG-1000) is an investigational antiviral drug designed to treat and inhibit influenza virus infections. It belongs to a class of compounds known as cap-dependent endonuclease (CEN) inhibitors, which target a key viral enzyme necessary for influenza virus replication.
Mechanism of Action
Blocks viral replication: Pixavir marboxil works by inhibiting the influenza virus’s cap-dependent endonuclease, a part of the viral RNA polymerase complex the virus needs to “snatch” capped RNA fragments from host cell mRNA. Without this process, the virus cannot efficiently produce its own viral proteins or replicate.
What Viruses It Targets
Pixavir marboxil has shown activity against:
Influenza A viruses
Influenza B viruses
Certain drug-resistant influenza strains
This broad spectrum makes it useful for seasonal flu and potentially strains less responsive to older antiviral drugs
Clinical Development & Approval Status
Phase Trials & Results
Completed Phase III: Clinical trials in adults and adolescents (age ≥12) showed that a single dose shortened time to symptom relief compared to placebo (e.g., median ~60.9 h vs ~87.9 h).
Symptom relief benefits: The data indicated statistically significant improvement in flu symptoms and faster viral inactivation in treated patients versus placebo.
Pediatric Formulation: China’s health authority approved pediatric Phase III studies for Pixavir (children <12), indicating further development for younger patients.
Regulatory Filings
NDA (New Drug Application): Pixavir marboxil has been submitted for approval to the National Medical Products Administration (NMPA) in mainland China based on Phase III results.
Generic Name Approved: The drug has been officially recognized with the generic name “Pixavir marboxil,” moving it closer to commercialization.
Pixavir marboxil is a small molecule drug. The usage of the INN stem ‘-xavir’ in the name indicates that Pixavir marboxil is a influenza CAP-dependent endonuclease inhibitor. Pixavir marboxil has a monoisotopic molecular weight of 540.12 Da.
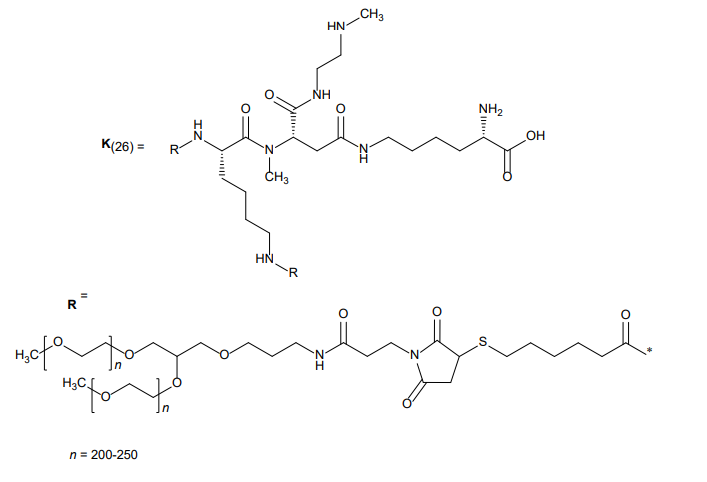
MOLECULAR FORMULA C231H386N64O67S5 + (C2H4O)4n MOLECULAR WEIGHT approx. 45 kDa
The structure of navepegritide (YUVIWEL®) is built using a “prodrug” design. It is not a simple small molecule, but rather a complex conjugate consisting of three distinct components designed to release the active drug slowly over time.
1. The Active Part: C-Type Natriuretic Peptide (CNP)
The core of the molecule is a synthetic 38-amino acid peptide (CNP-38).
Sequence: This peptide mimics the natural human C-type natriuretic peptide, which is essential for bone growth.
Function: Once released, this peptide binds to the natriuretic peptide receptor B (NPR-B) on the surface of chondrocytes (cartilage cells) in the growth plates, stimulating bone formation.
2. The Carrier: Polyethylene Glycol (PEG)
To prevent the body from clearing the small peptide too quickly, it is attached to a large, inert carrier.
Type: It uses a multi-arm, branched 40 kDa Polyethylene Glycol (PEG) molecule.
Purpose: The PEG carrier acts as a shield and a “weight,” making the molecule too large to be filtered out rapidly by the kidneys. This is what allows for once-weekly dosing instead of daily injections.
3. The Linker: TransCon™ Technology
This is the most critical part of the structure. The peptide is attached to the PEG carrier via a cleavable linker.
Mechanism: This linker is designed to break down spontaneously at a predictable rate under physiological conditions (neutral pH and body temperature).
The Result: As the linker slowly breaks, it releases the unmodified, active CNP-38 into the bloodstream. Because the peptide is released in its natural state, it retains its full biological activity.
Summary Table: Structural Components
Component
Description
Role
Peptide
CNP-38 (38 amino acids)
The “payload” that stimulates bone growth.
Linker
pH-sensitive cleavable bond
Controls the slow release of the peptide.
Carrier
40 kDa PEG
Increases the half-life and prevents rapid clearance.
Note: This structure is technically a prodrug because the large PEG-bound version is inactive; only the released CNP-38 peptide performs the therapeutic work.
C-Type natriuretic peptide (CNP), human, (89-126)-fragment (1-38) (CNP-38), conjugated at N6 of Lys26 with four O-methylpoly(ethylene glycol) chains (approx. 10 kDa each) via a cleavable tetra-antennary linker; L-leucyl-L-glutaminyl-L-?-glutamyl-L-histid
Poly(oxy-1,2-ethanediyl), ?-hydro-?-methoxy-, 26,26,26,26-tetraether with L-leucyl-L-glutaminyl-L-?-glutamyl-L-histidyl-L-prolyl-L-asparaginyl-L-alanyl-L-arginyl-L-lysyl-L-tyrosyl-L-lysylglycyl-L-alanyl-L-asparaginyl-L-lysyl-L-lysylglycyl-L-leucyl-L-sery
FDA 2026, APPROVALS 2026, 2/27/2026, Yuviwel, Y3BH8M899D, MN-266, TRANSCON CNP, PA (224-233), Influenza, DA-66438, ACP-015, WHO 11981,
To increase linear growth in pediatric patients 2 years and older with achondroplasia with open epiphyses
Navepegritide is a prodrug consisting of a 38-amino acid C-type natriuretic peptide (CNP) moiety conjugated to a multi-arm polyethylene glycol (PEG) carrier via a cleavable linker. This structure allows for the once-weekly dosing approved by the FDA for children with achondroplasia.
Key Details
Purpose: It is designed to increase linear growth by providing continuous exposure to C-type natriuretic peptide (CNP), a protein that helps regulate bone growth.
Mechanism: As a prodrug, it uses Ascendis Pharma’s TransCon technology to release active CNP slowly into the body over a week, maintaining steady levels and avoiding high peaks.
Clinical Benefits: In the pivotal ApproaCH trial, patients treated with navepegritide showed a significant improvement in annualized growth velocity (AGV) compared to those on a placebo. It also showed potential improvements in body proportionality and lower-limb alignment.
Administration: It is administered via a once-weekly subcutaneous injection, offering a less frequent alternative to daily treatments like vosoritide.
Safety: Most common side effects include injection site reactions (redness, itching, or swelling) and a risk of low blood pressure (hypotension).
Pebezertinib is a small molecule drug. The usage of the INN stem ‘-ertinib’ in the name indicates that Pebezertinib is a epidermal growth factor receptor (EGFR) inhibitor. Pebezertinib is under investigation in clinical trial NCT05241873 ((Concerto) Study of BLU-451 in Advanced Cancers With EGFR Exon 20 Insertion Mutations). Pebezertinib has a monoisotopic molecular weight of 497.16 Da.
Pebezertinib is an orally bioavailable, central nervous system (CNS) penetrating, mutant-selective covalent inhibitor of epidermal growth factor receptor (EGFR) exon 20 insertion (Ex20ins) activating mutations, with potential antineoplastic activity. Upon oral administration, pebezertinib selectively targets, irreversibly binds to and inhibits the activity of EGFR Ex20ins and some other oncogenic point mutations. This prevents EGFR Ex20ins-mediated signaling. This may induce cell death and inhibit tumor growth in EGFR Ex20ins-overexpressing tumor cells. EGFR, a receptor tyrosine kinase mutated in many tumors, plays a key role in tumor cell proliferation and tumor vascularization. Pebezertinib is able to penetrate the blood-brain-barrier (BBB) and may therefore exert its activity against EGFR Ex20ins-driven CNS primary tumors and CNS metastases. Pebezertinib does not inhibit the activity of wild-type (WT) EGFR. EGFR Ex20ins are oncogenic driver mutations that constitutively upregulate kinase activity.
(Concerto) Study of BLU-451 in Advanced Cancers With EGFR Exon 20 Insertion Mutations
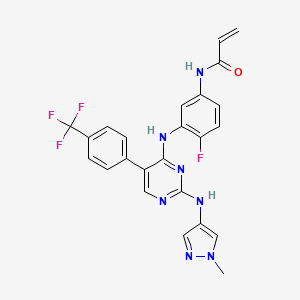
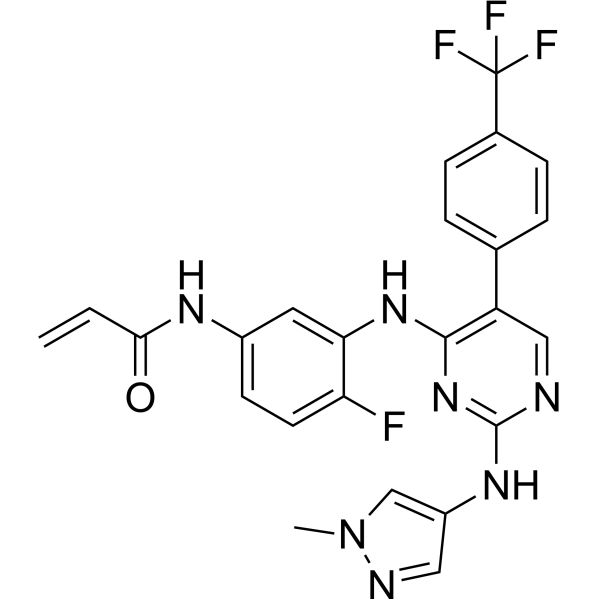
Scheme 21: Synthesis of N-(4-fluoro-3-((2-((1-methyl-1H-pyrazol-4-yl)amino)-5-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)amino)phenyl)acrylamide (Compound 37):
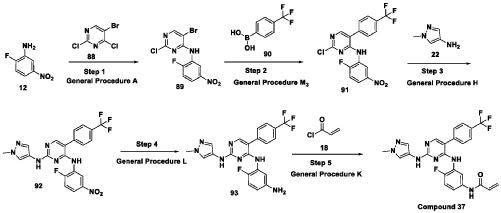
Step 1: Synthesis of 5-bromo-2-chloro-N-(2-fluoro-5-nitrophenyl)pyrimidin-4-amine (89):
[0286] Title compound was prepared in a manner substantially similar to procedure mentioned in General Procedure A. The crude was purified by combiflash eluted with 40% ethyl acetate in hexane to get (89) as pale yellow solid (1.3 g, Yield: 44.24 %). MS: [M+H]+ 346.97.
Step 2: Synthesis of 2-chloro-N-(2-fluoro-5-nitrophenyl)-5-(4-(trifluoromethyl)phenyl)pyrimidin-4-amine (91):
[0287] Title compound was prepared in a manner substantially similar to procedure mentioned in General Procedure M3. The crude was purified by combiflash eluted with 35% ethyl acetate in hexane to get desired product (91) as light yellow solid (700 mg; Yield: 50.12%). MS:
[M+H]+ 413.10
Step 3: Synthesis of N4-(2-fluoro-5-nitrophenyl)-N2-(1-methyl-1H-pyrazol-4-yl)-5-(4-(trifluoromethyl)phenyl)pyrimidine-2,4-diamine (92):
[0288] Title compound was prepared in a manner substantially similar to procedure mentioned in General Procedure H. The crude was purified by combiflash eluted with 1% methanol in dichloromethane to get desired product (92) as pale yellow solid (500 mg; Yield: 70.24%). MS:
[M+H]+ 474.09
Step 4: Synthesis of N4-(5-amino-2-fluorophenyl)-N2-(1-methyl-1H-pyrazol-4-yl)-5-(4-(trifluoromethyl)phenyl)pyrimidine-2,4-diamine (93):
[0289] Title compound was prepared in a manner substantially similar to procedure mentioned in General Procedure L to get (93) as semi solid (350 mg; Yield: 74.78%). MS: [M+H]+ 444.11
Step 5: Synthesis of N-(4-fluoro-3-((2-((1-methyl-1H-pyrazol-4-yl)amino)-5-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)amino)phenyl)acrylamide (Compound 37):
[0290] Title compound was prepared in a manner substantially similar to procedure mentioned in General Procedure K. The crude was purified by Prep HPLC to get Compound 37 as off white solid (30 mg, Yield: 13.33%).1H NMR (400 MHz, DMSO-d6): δ 10.21 (bs, 1H), 9.24 (bs, 1H), 8.53 (bs, 1H), 7.99 (s, 1H), 7.71-7.81 (m, 5H), 7.57 (s, 1H), 7.08-7.16 (m, 3H), 6.37-6.44 (m, 1H), 6.21-6.26 (m, 1H), 5.74 (d, J = 8.4 Hz, 1H), 3.54 (s, 3H). LCMS: [M+H]+ 498.35.
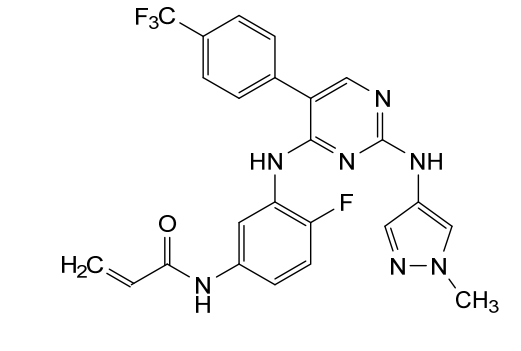
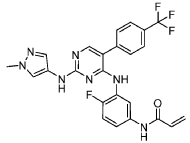
International Patent Application No. PCT/US2021/057472, the entire teachings of which are incorporated herein by reference, discloses selective inhibitors of EGFR, including exon 20 mutant proteins, which can be used to treat various cancers. The structure of one of the inhibitors disclosed in PCT Patent Application No. PCT/US2021/057472, referred to
herein as “Compound (I)” is shown below:
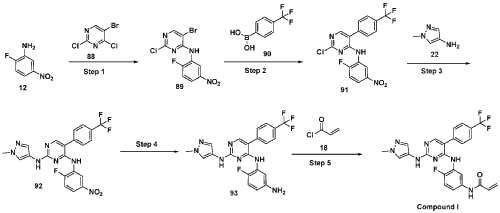
Example 1 : Preparation of Compound (I)
Synthesis of N-(4-fluoro-3-((2-((l-methyl-lH-pyrazol-4-yl)amino)-5-(4-(trifluoro methyl)phenyl)pyrimidin-4-yl)amino)phenyl)acrylamide (Compound I):
Step 1 : Synthesis of 5-bromo-2-chloro-N-(2-fluoro-5-nitrophenyl)pyrimidin-4-amine (89):
To an ice cold solution of 2-fluoro-5-nitroaniline (12) (1.0 eq) in tetrahydrofuran was added sodium hydride (60% dispersion in mineral oil, 3.0 eq) portion-wise. The resulting reaction mixture was stirred at room temperature for 30 minutes and followed by the addition of 2, 4-di chi oro-5 -bromopyrimidine (88) (1.0 eq). The resulting reaction mixture was heated at 60 °C for 16 hours. After completion (TLC monitoring), quenched with ice, extracted with ethyl acetate (3 times). The combined organic layers were washed with water, brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude was purified by combiflash eluted with 40% ethyl acetate in hexane to get (89) as pale yellow solid (1.3 g, Yield: 44.24 %). MS: [M+H]+ 346.97.
Step 2: Synthesis of 2-chloro-N-(2-fluoro-5-nitrophenyl)-5-(4-(trifluoromethyl)phenyl) pyrimidin-4-amine (91):
To a solution of halo derivative (89) (1.0 eq) and respective boronate acid/ester derivative (90) (1.1 eq) in A A i methyl form am ide: water (4: 1) was added sodium carbonate or sodium bicarbonate (2.0 eq). The resulting reaction mixture was degassed under argon atmosphere for 15 minutes, followed by addition of tetrakis(triphenylphosphine)palladium(0) (0.1 eq). The resulting reaction mixture was heated at 90 °C for 16 hours. After completion of reaction (TLC monitoring), the reaction mixture was cooled to room temperature, water was added and extracted with ethyl acetate (3 times). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude was purified by combiflash eluted with 35% ethyl acetate in hexane to get desired product (91) as light yellow solid (700 mg; Yield: 50.12%). MS: [M+H]+413.10.
Step 3 : Synthesis of N4-(2-fluoro-5-nitrophenyl)-N2-(l-methyl-lH-pyrazol-4-yl)-5-(4-(trifluoromethyl)phenyl)pyrimidine-2,4-diamine (92):
To an ice-cold solution of chloro compound (91) (1.0 eq) in isopropanol was added amine (22) (1.2 eq) and trifluoroacetic acid (2.0 eq). The reaction mixture was heated at 110 °C for 16 hours. After completion of the reaction (TLC monitoring), the reaction mixture was concentrated under reduced pressure, added saturated solution of sodium bicarbonate and extracted with dichloromethane (3 times). The combined organic layers were washed with brine solution, dried over anhydrous sodium sulfate and evaporated under reduced pressure. The crude was purified by combiflash eluted with 1% methanol in di chloromethane to get desired product (92) as pale yellow solid (500 mg; Yield: 70.24%). MS: [M+H]+ 474.09.
Step 4: Synthesis of N4-(5-amino-2-fluorophenyl)-N2-(l-methyl-lH-pyrazol-4-yl)-5-(4-(trifluoromethyl)phenyl)pyrimidine-2,4-diamine (93):
To an ice cold solution of nitro derivative (92) (1.0 eq) in methanol: tetrahydrofuran: water (2:2: 1) were added zinc-dust or iron powder (5 eq) and ammonium chloride (5 eq). The resultant reaction mixture was stirred at room temperature for 2 hours. After completion of reaction (TLC monitoring), reaction mixture passed through celite bed washed with 5% methanol in dichloromethane. The filtrate was washed with water, brine, dried over anhydrous sodium sulfate, filtered and concentrated to dryness to get the desired product (93) as semi solid (350 mg; Yield: 74.78%). MS: [M+H]+ 444.11.
Step 5 : Synthesis of N-(4-fluoro-3-((2-((l-methyl-lH-pyrazol-4-yl)amino)-5-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)amino)phenyl)acrylamide (Compound I):
To a solution of amino compound (93) (1.0 eq) in dichloromethane: tetrahydrofuran (1 :1) was cooled to -40 °C followed by triethylamine (3-5 eq) and acryloyl chloride (1.0 eq) were added. The mixture was stirred at the same temperature for 2 hours. After completion of reaction (monitored by TLC), added water and extracted with dichloromethane (3 times). The combined organic layers washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crudes were purified by Prep-HPLC purification to to obtain Compound I as off white solid (30 mg, Yield: 13.33%). ‘H NMR (400 MHz, DMSO-de): 8 10.21 (bs, 1H), 9.24 (bs, 1H), 8.53 (bs, 1H), 7.99 (s, 1H), 7.71-7.81 (m, 5H), 7.57 (s, 1H), 7.08-7.16 (m, 3H), 6.37-6.44 (m, 1H), 6.21-6.26 (m, 1H), 5.74 (d, J= 8.4 Hz, 1H), 3.54 (s, 3H). LCMS: [M+H]+ 498.35.
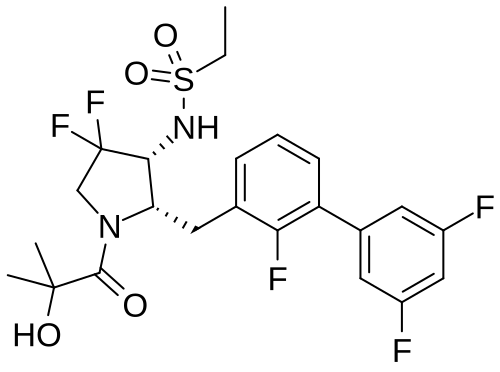
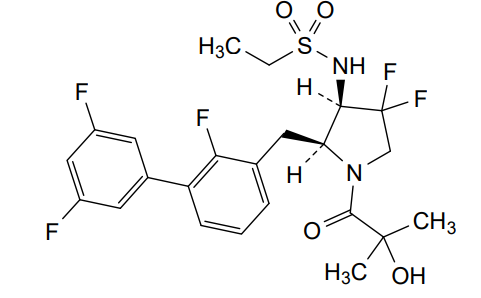
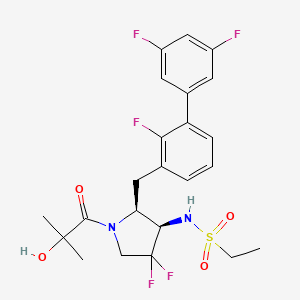
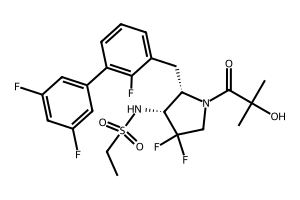
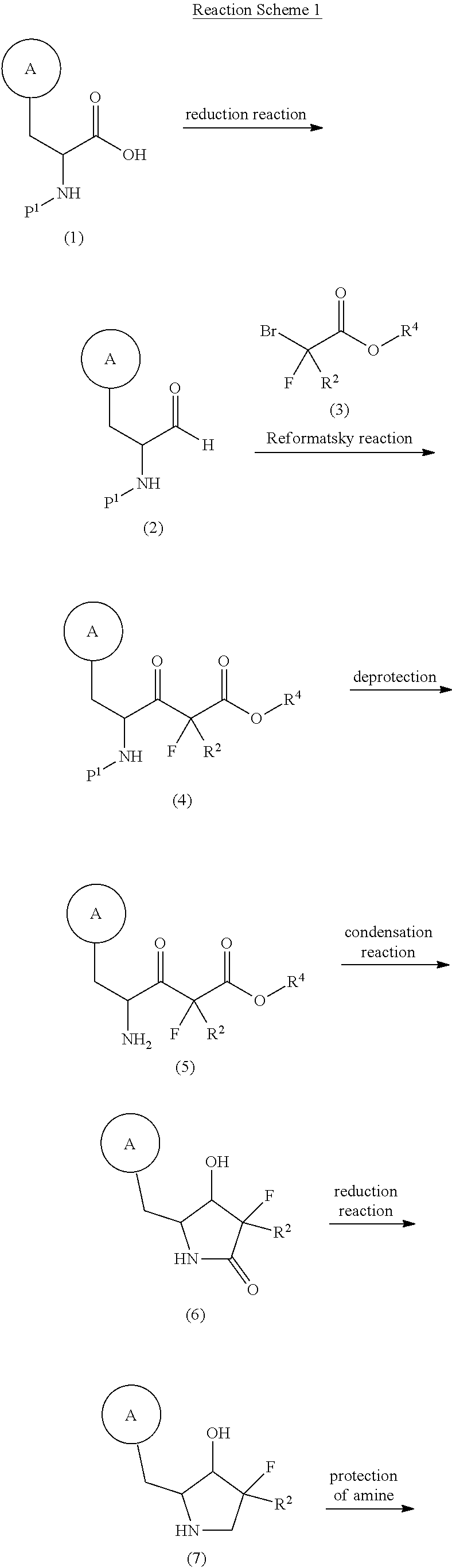
N-{(2S,3R)-4,4-difluoro-1-(2-hydroxy-2-methylpropanoyl)-2-[(2,3′,5′-trifluoro-[1,1′-biphenyl]-3-yl)methyl]pyrrolidin-3-yl}ethane-1-sulfonamide orexin type 2 receptor agonist, TAK-861, TAK 861, 59MF6P2ATF
Oveporexton is a small molecule drug. The usage of the INN stem ‘-orexton’ in the name indicates that Oveporexton is a orexin receptor agonist. Oveporexton has a monoisotopic molecular weight of 520.15 Da.
Oveporexton is being developed by Takeda.[1] As of July 2025, it has completed phase 3 clinical trials for treatment of narcolepsy, whereas no recent development has been reported for treatment of idiopathic hypersomnia.[1][5][10] Takeda plans to submit a New Drug Application (NDA) of oveporexton for the treatment of narcolepsy to the United StatesFood and Drug Administration (FDA) in 2025.[5] Oveporexton is a follow-on and replacement compound for Takeda’s earlier lead drug danavorexton (TAK-925), which is administered intravenously and stopped being developed due to unexpected liver toxicity findings.[10]
A Trial of TAK-861 for the Treatment of Narcolepsy With CataplexyCTID: NCT07363720Phase: Phase 3Status: Not yet recruitingDate: 2026-01-23Conditions: Narcolepsy Type 1 (NT1); Narcolepsy With CataplexyInterventions: PlaceboLinked Compound CID: 154617563
A Study of TAK-861 for the Treatment of Selected Central Hypersomnia ConditionsCTID: NCT05816382Phase: Phase 2/Phase 3Status: RecruitingDate: 2025-12-01Conditions: Narcolepsy Type 1Interventions: TAK-861Linked Compound CID: 154617563
A Study of TAK-861 in People With Narcolepsy Type 1CTID: NCT06505031Phase: Phase 3Status: CompletedDate: 2025-09-15Conditions: Narcolepsy Type 1Interventions: PlaceboLinked Compound CID: 154617563
A Study of TAK-861 for the Treatment of Narcolepsy Type 1CTID: NCT06470828Phase: Phase 3Status: CompletedDate: 2025-07-01Conditions: Narcolepsy Type 1Interventions: PlaceboLinked Compound CID: 154617563
A Study of TAK-861 in Participants With Narcolepsy Type 1CTID: NCT05687903Phase: Phase 2Status: CompletedDate: 2025-01-09Conditions: Narcolepsy Type 1Interventions: PlaceboLinked Compound CID: 154617563
A Randomized, Double-blind, Placebo-Controlled Study to Evaluate the Efficacy, Safety, and Tolerability of TAK-861 for the Treatment of Narcolepsy With Cataplexy (Narcolepsy Type 1)EudraCT: 2022-001654-38Phase: Phase 2Status: CompletedDate: 2023-05-26Linked Compound CID: 154617563
A Long-term Extension Study to Evaluate the Safety and Tolerability of TAK-861 in Participants With Selected Central Hypersomnia ConditionsEudraCT: 2022-002965-13Phase: Phase 2, Phase 3Status: Trial now transitionedDate: 2023-04-11Linked Compound CID: 154617563
A Randomized, Double-blind, Placebo-Controlled Study to Evaluate the Efficacy, Safety, and Tolerability of TAK-861 for the Treatment of Narcolepsy Without Cataplexy (Narcolepsy Type 2)EudraCT: 2022-002966-34Phase: Phase 2Status: CompletedDate: 2023-03-20Linked Compound CID: 154617563
A) tert-butyl (2S,3R)-3-(ethylsulfonamido)-4,4-difluoro-2-((2,3′,5′-trifluoro-[1,1′-biphenyl]-3-yl)methyl)pyrrolidine-1-carboxylate
To a mixture of tert-butyl (2S,3R)-2-(3-chloro-2-fluorobenzyl)-3-(ethylsulfonamido)-4,4-difluoropyrrolidine-1-carboxylate (3.70 g), (3,5-difluorophenyl)boronic acid (2.56 g) and 1 M aqueous potassium phosphate solution (24.3 mL) in DME (50 mL) was added XPhos Pd G3 (0.343 g) at room temperature. The mixture was stirred at 90° C. under nitrogen atmosphere for 15 h. The reaction mixture was poured into water and extracted with EtOAc. The organic layer was washed with saturated brine, dried over magnesium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, EOAc/hexane) to give the title compound (3.30 g).
MS: [M−H] − 533.2.
B) N-((2S,3R)-4,4-difluoro-2-((2,3′,5′-trifluoro-[1,1′-biphenyl]-3-yl)methyl)pyrrolidin-3-yl)ethanesulfonamide hydrochloride
A mixture of tert-butyl (2S,3R)-3-(ethylsulfonamido)-4,4-difluoro-2-((2,3′,5′-trifluoro-[1,1′-biphenyl]-3-yl)methyl)pyrrolidine-1-carboxylate (3.30 g) and 4 M HCl/CPME solution (30 mL) was stirred overnight at room temperature. By filtration, the title compound (2.86 g) was obtained.
MS: [M+H] + 435.1.
C) N-{(2S,3R)-4,4-difluoro-1-(2-hydroxy-2-methylpropanoyl)-2-[(2,3′,5′-trifluoro[1,1′-biphenyl]-3-yl)methyl]pyrrolidin-3-yl}ethanesulfonamide
To a mixture of N-((2S,3R)-4,4-difluoro-2-((2,3′,5′-trifluoro-[1,1′-biphenyl]-3-yl)methyl)pyrrolidin-3-yl)ethanesulfonamide hydrochloride (200 mg) and DIPEA (0.367 ml) in THF (3 mL) was added alpha-acetoxy-isobutyryl chloride (0.074 ml) at 0° C., and the mixture was stirred at same temperature for 10 min. To the mixture were added water (1 ml) and 4 M lithium hydroxide solution (1.06 ml), and the mixture was stirred overnight at room temperature. The mixture was diluted with saturated brine and extracted with EtOAc. The extract was dried over magnesium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, EtOAc/hexane) and recrystallized from EtOAc/hexane to give the title compound (154 mg).
1H NMR (400 MHz, CDCl 3) δ 1.32-1.40 (9H, m), 2.27-2.54 (1H, m), 2.88-3.16 (4H, m), 4.02-4.49 (3H, m), 4.86-5.20 (2H, m), 6.78-6.86 (1H, m), 7.02-7.10 (2H, m), 7.16-7.22 (1H, m), 7.27-7.31 (1H, m), 7.35-7.43 (1H, m).
N-{(2S,3R)-4,4-difluoro-l-(2-hydroxy-2-methylpropanoyl)-2-[(2,3′,5′-trifluoro[l,l’-biphenyl]-3-yl)methyl]pyrrolidin-3-yl}ethanesulfonamide, (hereafter referred as to “Compound A”) is described in U.S. Patent No. 11,028,048.
Dauvilliers Y, Plazzi G, Mignot E, Lammers GJ, Del Río Villegas R, Khatami R, et al. (May 2025). “Oveporexton, an Oral Orexin Receptor 2-Selective Agonist, in Narcolepsy Type 1”. The New England Journal of Medicine. 392 (19): 1905–1916. doi:10.1056/NEJMoa2405847. PMID40367374.
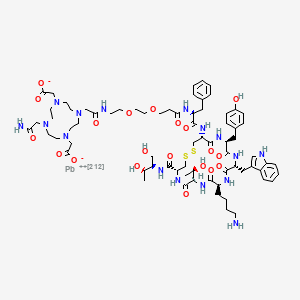
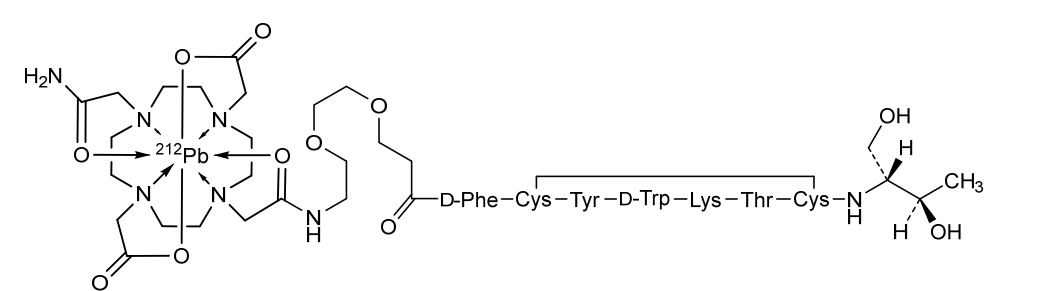
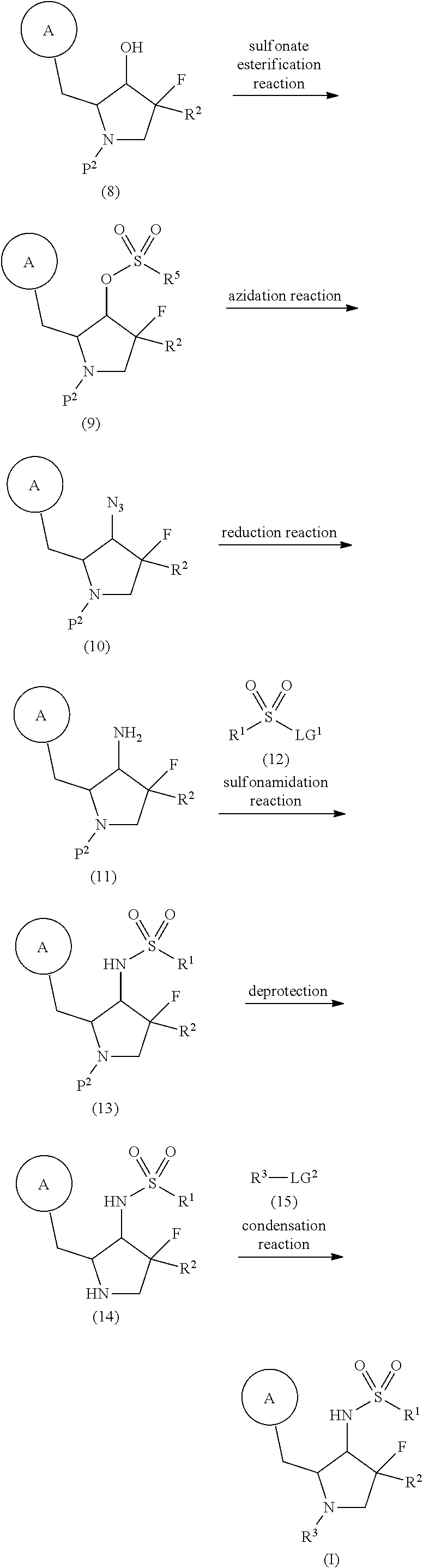
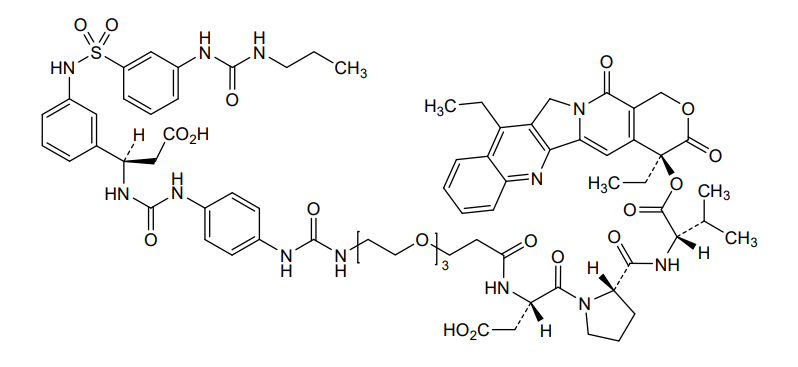
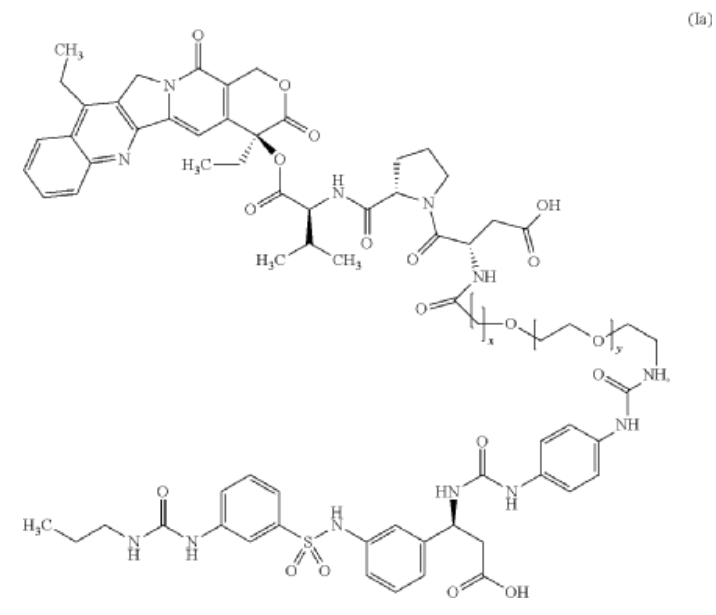
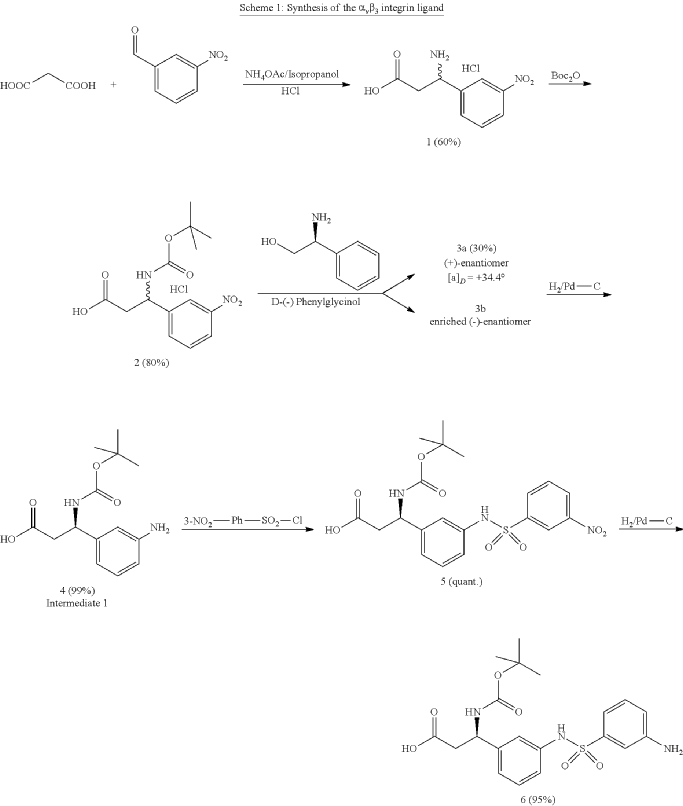
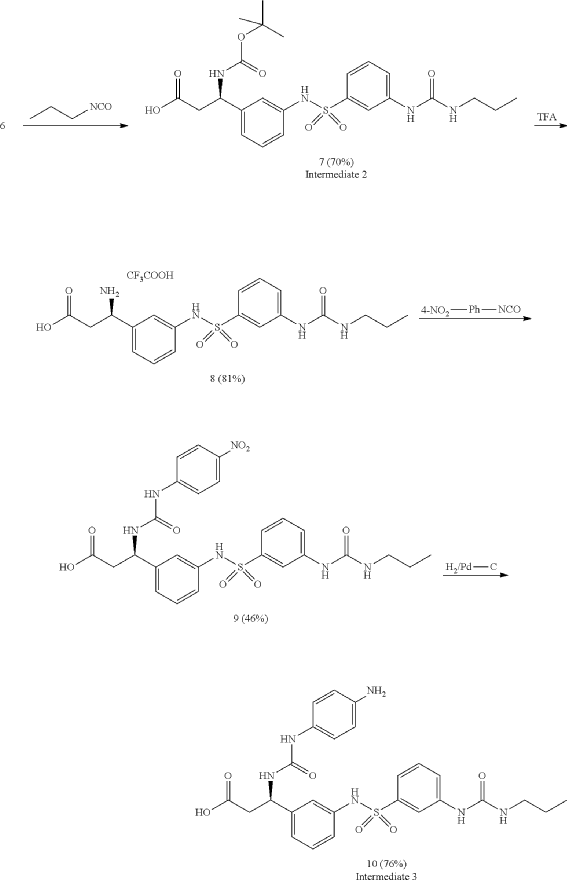
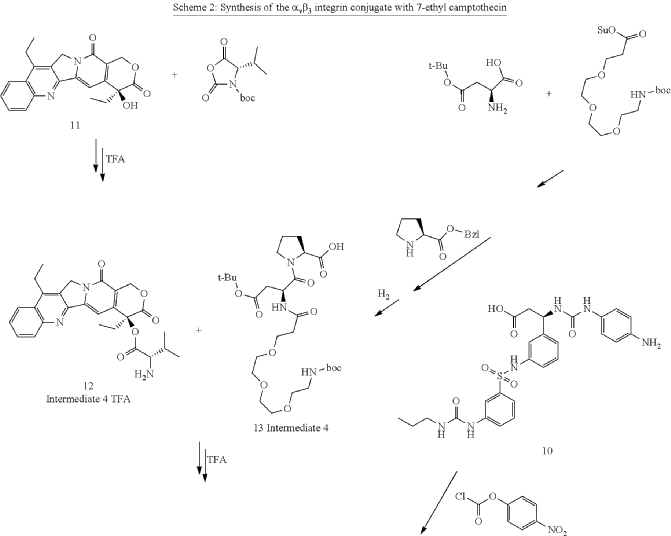
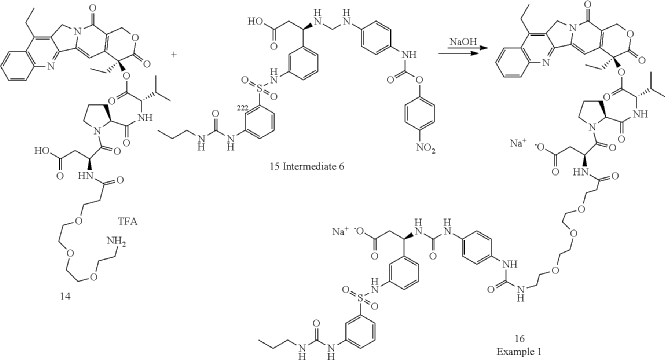
Orenasitecan is a small molecule drug. The usage of the INN stem ‘-tecan’ in the name indicates that Orenasitecan is a antineoplastic, topoisomerase I inhibitor. Orenasitecan has a monoisotopic molecular weight of 1470.58 Da.
ORENASITECAN is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader























{kind=link}
{kind=link}
{kind=link}