
Home » 2021 (Page 4)
Yearly Archives: 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TNO 155
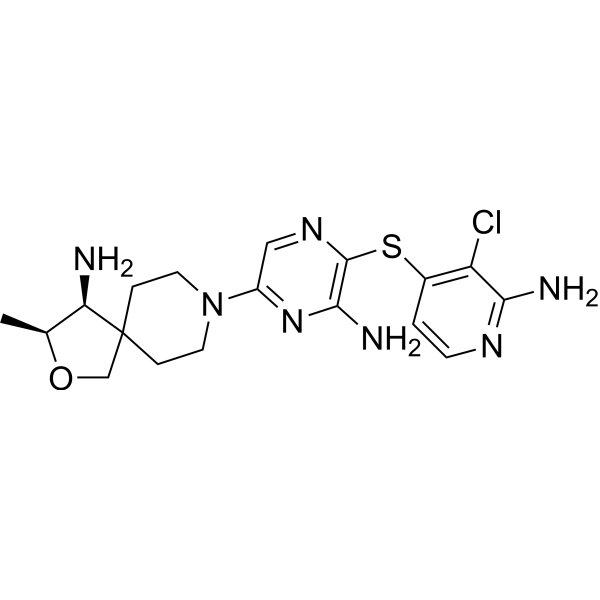
TNO 155
2-Oxa-8-azaspiro[4.5]decan-4-amine, 8-[6-amino-5-[(2-amino-3-chloro-4-pyridinyl)thio]-2-pyrazinyl]-3-methyl-, (3S,4S)-
- (3S,4S)-8-[6-Amino-5-[(2-amino-3-chloro-4-pyridinyl)thio]-2-pyrazinyl]-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine
- (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine
| Molecular Weight |
421.95 |
|---|---|
| Formula |
C₁₈H₂₄ClN₇OS |
| CAS No. |
- PTPN11 inhibitor TNO155
- SHP2 inhibitor TNO155
- TNO-155
- TNO155
- UNII-FPJWORQEGI
TNO155 is a potent selective and orally active allosteric inhibitor of wild-type SHP2 (IC50=0.011 µM). TNO155 has the potential for the study of RTK-dependent malignancies, especially advanced solid tumors.
- Originator Novartis
- Developer Mirati Therapeutics; Novartis
- Class Antineoplastics
- Mechanism of ActionProtein tyrosine phosphatase non receptor antagonists
- Phase I/IISolid tumours
- Phase IColorectal cancer
- 11 Jul 2021Phase I trial in Solid tumours is still ongoing in USA, Canada, Japan, South Korea, Netherlands, Singapore, Spain, Taiwan (NCT03114319)
- 04 Jun 2021Efficacy, safety and pharmacokinetics data from phase I trial in Solid tumours presented at 57th Annual Meeting of the American Society of Clinical Oncology (ASCO-2021)
- 08 Jan 2021Novartis plans a phase Ib/II trial for Solid tumours (Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in February 2021 (NCT04699188)
CLIP
Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling
Results: In EGFR-mutant lung cancer models, combination benefit of TNO155 and the EGFRi nazartinib was observed, coincident with sustained ERK inhibition. In BRAFV600E colorectal cancer models, TNO155 synergized with BRAF plus MEK inhibitors by blocking ERK feedback activation by different RTKs. In KRASG12C cancer cells, TNO155 effectively blocked the feedback activation of wild-type KRAS or other RAS isoforms induced by KRASG12Ci and greatly enhanced efficacy. In addition, TNO155 and the CDK4/6 inhibitor ribociclib showed combination benefit in a large panel of lung and colorectal cancer patient–derived xenografts, including those with KRAS mutations. Finally, TNO155 effectively inhibited RAS activation by colony-stimulating factor 1 receptor, which is critical for the maturation of immunosuppressive tumor-associated macrophages, and showed combination activity with anti–PD-1 antibody.
Conclusions: Our findings suggest TNO155 is an effective agent for blocking both tumor-promoting and immune-suppressive RTK signaling in RTK- and MAPK-driven cancers and their tumor microenvironment. Our data provide the rationale for evaluating these combinations clinically.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENT
WO 2015107495
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015107495
PATENT
WO 2020065453
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020065453
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine, which has the formula I,
WO/2015/107495 A1 describes a method for the manufacture of the compound of the formula I which can be characterized by the following reaction scheme 1:
[0008] The last compound resulting from step g above was then reacted as in the following scheme 2:
Scheme 2:
[0009] Thus the compound of formula I is obtained (last compound in the scheme 2, above). The synthesis requires at least the 9 steps shown and is rather appropriate for synthesis in laboratory amounts.
Scheme 1A:
[0016] Therefore, the process, though readily feasible on a laboratory scale, is not ideal for manufacture at a large scale.
[0017] The compound added in reaction b in Scheme 2 is obtained in WO
2015/107495 A1 as “Intermediate 10” follows:
Scheme 3:
[0018] An issue here is the relatively low yield of the amine resulting from reaction a in
Scheme 3.
[0019] In addition, while WO 2015/107495 A1 generically mentions that pharmaceutically acceptable salts of the compound of the formula I may be obtainable, no concrete reason for obtaining such salts and no specific examples of salts are described.
[0020] In addition, given the many potentially salt forming groups in formula I, it is not clear whether any salts with a clear stoichiometry can be formed at all.
Example 1
Method of synthesis of the compound of the formula I ((3S,4S)-8-(6-amino-5-((2-amino-3- chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine):
The overall synthesis can be described by the following Reaction Scheme A:
Scheme A:
Step a
[00293] To a solution of A1 (10.4 kg, 100 mol, 1.0 Eq) in CH2Cl2 (50 L) was added imidazole (8.16 kg, 120 mol, 1.2eq) and TBSCl (18 kg, 120 mol, 1.2 Eq) at 0 °C. After addition, the mixture was stirred at 0°C for 4 h . GC showed the reaction was finished. (A1/ (A1 + A2) < 1%). The reaction mixture was quenched with saturated NaHCO3 (14L) at 0-5°C. Phases were separated. The organic phase was washed with brine (14L). The organic layer was dried over Na2SO4, concentrated under vacuum at 40-45°C to afford A2 (23.3 kg, assay 88%, yield 94%) which was used for the next step directly. 1H NMR (400 MHz, CDC13) δ = 4.35 (d, J= 8.8 Hz, 1H), 3.74 (s, 3H), 2.48 (s, J= 8.8
Hz, 3H), 0.93 (s, 9H), 0.09 (s, 6H).
Step b
[00294] To a solution of A2 (7.5 kg, 34.3 mol, 1.0 Eq) and N,O-dimethylhydroxylamine hydrochloride (6.69 kg, 68.6mol, 2.0 Eq) in THF (20 L) was added drop-wise a solution
of chloro(isopropyl)magnesium (2 M, 51.45 L, 3.5 Eq) at 0 °C under N2 over 5-6 h. After addition, the reaction mixture was stirred at 0 °C for 1h, GC showed the reaction was finished (A2/(A2+A3) < 2 %). The mixture was quenched with NH4Cl (25 L) slowly by keeping the temperature at 0-5°C. After addition, the reaction mixture was stirred for 30min. Phase was separated. The aqueous layer was extracted with EA(2 x 20 L). The combined organic phase was washed with brine (25L), dried over Na2SO4, concentrated to give A3(9.4 kg, assay 86%, yield 95%) which was used for the next step directly. 1HNMR (400 MHz, CDCl3) δ = 4.67 (m, J= 6.6 Hz, 1H), 3.70 (s, 3H), 3.21 (s, 3H), 3.17 (d, 3H)2.48 (s , J= 6.6 Hz, 3H), 0.90 (s, 9H), 0.10 (s, 3H), 0.08 (s, 3H).
[00295] To a solution of A3 (7.1 kg, assay 86%, 24.65 mol, 1.0 Eq) in DCM (30 L) was added dropwise a solution of LiAlH4 (2.4 M, 11.3 L, 1.1 Eq) at -70 °C under N2. Then the reaction mixture was stirred at -70 °C for 3h, and TLC showed the reaction was finished (PSC-1). The mixture was warmed to 0 °C, and then quenched with sat. potassium sodium tartrate (35 L) at 0 °C. After addition, DCM (20L) was added and stirred for 2h at 20-25°C. Phases were separated. The aqueous layer was extracted with DCM (25 L). The combined organic phase was charged with sat. citric acid (45L) and stirred at 0°C for 8h. Phase was separated. The organic phase was washed with NaHCO3 (25L), brine (25 L), dried over Na2SO4, and the solvent was removed under vacuum at 25-30°C. n-Heptane (10 L) was added to the residue and concentrated under vacuum at 30-35°C. n-Heptane (10 L) was added to the residue again and concentrated under vacuum at 30-35°C to give A4 (4.2 kg, assay
60%, yield 54%) which was used for the next step directly.
Step d
[00296] To a solution of diisopropylamine (3.06 kg, 30.3 mol, 1.5 eq) in THF (20 L) cooled to approximately -10°C was added 2.5 M n-BuLi (12.12 L, 30.3 mol, 1.5 eq) under N2. The resulting mixture was stirred at approximately -10 °C for 30min, then a solution of A5 (5.2 kg, 20.20 mol, 1.0eq) in THF (10 L) was added slowly. After addition, the reaction mixture was stirred at -10°C for 30 min, and then cooled to -50°C. A4 (4.18 kg, 22.22 mol, 1.1eq) was added dropwise. After addition, the reaction mixture was stirred at -50°C for 30 min. The mixture was quenched with saturated aqueous NH4Cl (30L) and water (10L) at -50°C. The reaction mixture was warmed to 20-25°C. Phase was separated. The aqueous phase was extracted with EA (3 x 20 L). All organic phases were combined and washed with brine(20L), then concentrated to a yellow oil which was purified by column (silica gel, 100-200 mesh, eluted with n-heptane:EA from 50:1 to 10:1) to give A6 (5.5 kg, assay 90 %, yield 55%) as pale yellow oil. 1H NMR (400 MHz, CDCl3) δ = 4.35-4.15 (m, 2H), 3.95-3.74 (m, 3H), 3.52 (m, 2H), 2.67(m, 2H), 2.12-1.98 (m, 2H), 1.75-1.52 (m, 4H), 1.49 (s, 9H), 1.35-1.10 (m, 6H), 0.98 (s,
9H), 0.02 (s, 6H).
Step e
[00297] To a solution of A6 (11.4 kg, 25.58 mol, 1.0eq) in THF (60 L) was added LiBH4
(836 g, 38.37 mol, 1.5eq) in portions at 5-10 °C, and the reaction mixture was stirred at 20-25 °C for 18 h. HPLC showed the reaction was finished (A6/(A6+A7)<2%). The mixture was cooled to l0°C and slowly quenched with saturated NaHCO3 solution (15 L) and water (25L) with vigorously stirring. After gas formation stopped, vacuum filtration was applied to remove solids. The solid was washed with EA (2 x 15 L). Phase was separated; the aqueous phase was extracted with EA (3 x15L). All organic phases were combined and washed with brine (15L), and concentrated to obtain crude A7 (13.8 kg, assay 58%, yield 77%) which was used for the next step directly.
[00298] To a solution of A7 (8 kg, 19.82 mol, 1.0 eq) in THF (40 L) under nitrogen atmosphere was added TsCl (5.28 kg, 27.75 mol, 1.4 eq) at 10-15°C. After addition, the mixture was cooled to 0 °C, and 1M LiHMDS (29.7 L, 29.73 mol, 1.5 eq) was added dropwise during 2h. After addition, the mixture was stirred at 0°C for 3h. HPLC showed the reaction was finished (PSC-1 A7/ (A7+A8)<7%). TBAF (20.72 kg, 65.67 mol, 3.3 eq) was added into the mixture at 0 °C and the reaction mixture was stirred at 25-30 °C for 48h. HPLC showed the reaction was finished ( PSC-2, A9-intermedaite/(A9-intermediate+A9) < 2%). The mixture was quenched with saturated aqueous sodium bicarbonate solution (32L) and stirred for 30min at 0 °C. Phase was separated, and the aqueous phase was extracted with EA (3 x 20 L). The combined organic phase was washed with brine(20 L), dried over Na2SO4, and concentrated to a yellow oil which was purified by column (eluted with n-heptane:EA from 10:1 to 1:1) to give A9 (4.42 kg, assay 90%, yield 74 %) as pale yellow solid.
Step g
[00299] To a solution of A9 (4.0 kg, 14.74 mol, 1.0 eq) in DCM (40 L) cooled on an ice-bath was added DMP (9.36 kg, 23.58mol, 1.6eq) in portions, and it resulted in a suspension. After addition, the mixture stirred for 4 hours at 20-25°C. HPLC showed the reaction was finished (A9/(A9+A10)<2%). DCM (30L) was added at 0°C. After addition, the mixture was quenched with saturated aqueous Na2SO3 (20 L). The mixture was stirred for 30min at 0 °C, filtered and the white solid was washed with DCM (2 x15L). Phase was separated, and the organic phase was cooled to 0°C, to which was added saturated aqueous NaHCO3 (20L) and stirred for 1h. Phase was separated, and the organic phase was washed with brine(25L), dried over Na2SO4, and concentrated to a yellow oil which was purified by column (eluted with n-heptane:EA from 50:1 to 10:1) to give A10 (3.70 kg, assay 88%, ee value 95.3%, yield 82%) as white solid. 1H NMR (400 MHz, DMSO-d6) δ = 4.20 (d, J = 8.0 Hz,
1H), 3.98-3.67 (m, 4H), 3.08-2.90 (m, 2H), 1.54-1.39(m, 13H), 1.18 (d, J = 8.0 Hz, 3H).
Step h
[00300] To a solution of A10 (4.60 kg, 17.08 mol, 1.0 eq) in THF (40 L) was added
Ti(OEt)4 (15.58 kg, 68.32 mol, 4.0 eq) and (R)-t-Butyl sulfmamide (4.14 kg, 34.16 mol, 2.0 eq) at 25 °C. After addition, the mixture was heated to 70°C and stirred for 20h. HPLC showed the reaction was finished (PSC-l, A10/(A10+A12)<4%). The mixture was cooled to -30— 40°C, and MeOH (4 L) was added dropwise within 30 min and stirred for 1 h. 2M L1BH4 (8.1 L) solution was added dropwise to the reaction mixture at -40- -50°C and stirred for 1h. HPLC indicated all of imine was consumed (PSC-2, A12/(A12+A13)<1%). The mixture was warmed to -30 °C and stirred for 1h, then warmed to 0 °C within 2 h and stirred for 1h, then warmed to 20-25 °C and stirred for 30min. IP AC ( 25L) was added to above mixture, NaHCO3(5L) was added dropwise in about 1h at 25 °C and stirred for 30 min. The mixture was filtered under vacuum and the cake was washed with IP AC (8 x15L). The combined organic phase was washed with brine (25L), then evaporated under vacuum to get a solution of A13
(about 28kg) which was used for next step.
[00301] To a mixture of A13 in IPAC (about 28 kg, 17.08 mol, 1.0 eq) was added dropwise
4M HCl/IPA (8.54 L, 34.16 mol, 2.0 eq) at -5 °C and stirred for 5h at -5 °C. HPLC showed that A13 was consumed completely (A13/(A14+A13)<1%). MTBE (25 L) was added to above mixture within
30 min and stirred for 30 min at -5 °C .The solid was collected by vacuum filtration. The cake was washed with MTBE (2 x 2.5 L). The wet cake was used for next step directly.
Step j
[00302] The wet solid A14 (from 9.2 kg A10) was stirred in MTBE(76 L) at 25°C, then the
16% NaOH (9.84 kg) solution was added dropwise to the MTBE suspension while maintaining IT<10ºC. After addition, the mixture was stirred for 15 min and all solids were dissolved at 0°C. The organic phase was separated, and the aqueous phase was extracted with MTBE (2 x 20L). The combined organic phase was washed with brine (10 L) and evaporated under vacuum to remove all MTBE. ACN (24 L) was added to above residue, and the mixture was evaporated under vacuum to remove the organic solvents and yielded a crude A15 (5.42 kg, qnmr 90%, 18.04 mol, 1.0 eq). ACN (34.68 kg) was added to above residue and stirred for 10 min at 65°C. A solution of (-)-O-acetyl-D-mandelic acid (3.15kg,16.2 mol, 0.9 eq) in ACN(11.6 kg) was added drop-wise to the mixture (firstly added 1/3, stirred for 0.5 h, then added the others) over 3h. The mixture was stirred for 1 h at 65°C, then cooled to 25°C over 4h and stirred for l2h at 25°C . The solid was collected by vacuum filtration, and the cake was washed with pre-cooled ACN (2 x15kg) (PSC-1) and dried under vacuum to give
A16 (7.36 kg, yield 46% from A10 to A16). 1H NMR (400 MHz, DMSO-d6) δ = 7.43-7.29 (m, 5H),
5.58 (s, 2H), 4.12-4.07 (m, 1H), 3.75-3.65 (m, 3H), 3.51-3.49 (m, 1H), 3.18-3.17 (m, 1H), 2.84 (bs,
2H), 2.05 (s, 3H), 1.60-1.40 (m, 13H), 1.14-1.12 (d, J= 8.0 Hz, 3H).
Step k
[00303] To a solution of A16 (15 g) in MeOH (90 mL) was added dropwise 5N HC1/IPA
(45 mL) at room temperature within 15 minutes. After the addition, the mixture was stirred for 6 hours.
IP AC (180 mL) was added dropwise to above mixture within 1h at room temperature. The resulting mixture was stirred for another 30 minutes before it was cooled to 0-5 °C. The mixture was stirred at 0- 5 °C for another 2h and the precipitants were collected by filtration. The cake was washed with (45*2 mL) IP AC, dried under vacuum at 60 °C overnight to afford the product as a white solid. 1H NMR (400
MHz, DMSO-d6) δ = 9.37 (br s, 1H), 9.25 (br s, 1H), 8.42 (br s, 3H), 4.26 – 4.17 (m, 1H), 3.72 (ABq, J
= 9.1 Hz, 2H), 3.50 – 3.41 (m, 1H), 3.28 – 3.18 (m, 1H), 3.18 – 3.09 (m, 1H), 2.99 – 2.74 (m, 2H), 2.07 – 1.63 (m, 4H), 1.22 (d, J= 6.5 Hz, 3H).
Step l
[00304] To a mixture of A17 (10 g) and Z17a (9.5 g) in DMAC (60 mL) was added K2CO3
(22.5 g) and H2O (40 mL) at room temperature. The mixture was degassed with nitrogen and stirred at
90 °C overnight. The mixture was cooled to room temperature, diluted with Me-THF (500 mL) and
H2O (280 mL). The organic phase was separated and the aqueous phase was extracted with Me-THF
(300 mL*2). The combined organic phases were washed with brine (200 mL*3), concentrated under
vacuum to remove most of the solvent. The residue was diluted with IPA (60 mL) and H2O (20 mL), stirred at 50 °C for 1h, cooled to 5 °C within 3h, stirred at this temperature for 1h. The solid was collected by vacuum filtration, dried under vacuum to afford the product as a yellow solid (l2g,
87.4%). 1H NMR (400 MHz, DMSO-d6)δ = 7.64 (d, J= 6.2 Hz, 1H), 7.62 (s, 1H), 6.26 (s, 2H), 6.13 (s, 2H), 5.74 (d, J= 5.3 Hz, 1H), 4.12 – 4.02 (m, 1H), 3.90 – 3.78 (m, 2H), 3.67 (d, J= 8.4 Hz, 1H), 3.49 (d, J= 8.4 Hz, 1H), 3.33 (s, 2H), 2.91 (d, J= 5.1 Hz, 1H), 1.78 – 1.68 (m, 1H), 1.67 – 1.57 (m, 1H), 1.56 – 1.41 (m, 2H), 1.08 (d, J= 6.5 Hz, 3H).
Example 2
Formation of the succinate salt of the compound of the formula I:
[00305] The reaction is summarized by the following Reaction Scheme:
[00306] To a mixture of A18 (10 g) in MeOH (76 g) and H2O (24 g) was added succinic acid (2.94 g) at room temperature. The mixture was heated to 50 °C and stirred for 30 minutes to dissolve all solid. The solution was added to IPA (190 mL) at 60-65 °C. The resulting mixture was stirred at 60 °C >5 hours, cooled to -15 °C within 5 hours and stirred at this temperature >4 hours. The solid was collected by vacuum filtration, dried under vacuum to afford the product as an off-white solid(l0.8 g, 82.8%). 1H NMR (400 MHz, DMSO-d6)δ = 7.64 (d, J= 6.2 Hz, 1H), 7.63 (s, 1H), 6.26 (s, 2H), 6.16 (s, 2H), 5.74 (d, J= 5.3 Hz, 1H), 4.12 – 4.02 (m, 1H), 3.90 – 3.78 (m, 2H), 3.67 (d, J= 8.4 Hz, 1H), 3.49 (d, J= 8.4 Hz, 1H), 3.33 (s, 2H), 2.91 (d, J= 5.1 Hz, 1H), 2.34 (s, 4H), 1.71 – 1.60 (m, 4H), 1.13 (d, J = 6.5 Hz, 3H).
[00307] In a special variant, the reaction follows the following Reaction Scheme, also including an optional milling to yield the final product:
Example 3
Formation of the intermediate Z17a (3-((2-amino-3-chloropyridin-4-yl)thio)-6-chloropyrazin-2- amine). Variant 1:
[00308] The compound Z17a was obtained by reaction according to the following Reaction
Scheme:
[00309] In detail, the synthesis of Compound Z17a was carried out as follows:
Step a
[00310] Under nitrogen atmosphere, n-BuLi (2.5M, 7.6 L) was added dropwise to a solution of 3-chloro-2-fluoropyridine (2 kg) in THF (15 L) at -78°C. Then the resultant mixture was stirred for 1h. Then a solution of I2 (4.82 kg) in THF (6 L) was added dropwise. After addition, the reaction mixture was stirred for 30 min, and then quenched with sat. Na2SO3 (10 L), and warmed to 20- 25°C. Phase was separated. The aqueous phase was extracted with EA (2 x 10 L). The combined organic phase was washed with sat.Na2SO3 (2 x 8 L), brine (8 L), and dried over Na2SO4. The organic phase was concentrated under vacuum. The residue was slurried in MeOH (4 L), filtered, and dried to offer 3-chloro-2-fluoro-4-iodopyridine 1c (2.2 kg, yield 68%).
Step b
[00311] Into a solution of Compound 1c (8 kg) in DMSO (48 L) was passed through NH3
(gas) at 80 °C overnight. TLC showed the reaction was finished. The reaction mixture was cooled to RT. The reaction mixture was added to water (140 L). The solid was collected and washed with water (25 L), dried to afford Z17b (6.91 kg, yield 87%). 1H NMR (400 MHz, CDC13) δ = 7.61 (d, J= 6.8 Hz,
1H), 7.14 (s , J= 6.8 Hz, 1H), 5.09 (bs, 2H).
Step c
[00312] A solution of 2-amino-6-chloro-pyrazine la (1 kg, 7.69 mol) in DCM (15 L) was heated to reflux, to which was charged NBS (4l7g) in portions during 1 h. The reaction was cooled to room temperature. The reaction mixture was washed with water (3 L) and brine (3 L). The organic phase was evaporated, and the residue was purified by column chromatography to give product Z17f
(3-bromo-6-chloropyrazin-2-amine) (180 g, 11% yield).
[00313] To a solution of 3-bromo-6-chloropyrazin-2-amine Z17f (6.0 kg, 28.78 mol) in 1,4- Dioxane (40 L) was added Pd(OAc)2 (64.56 g, 287.6 mmol), Xantphos (333 g, 575.6 mmol), and DIPEA (7.44 kg, 57.56 mol) at room temperature under nitrogen. After another 30 minutes purging with nitrogen, methyl 3-mercaptopropanoate (3.81 kg, 31.70 mol) was added, resulting in darkening of the orange mixture. The mixture was heated to 90°C. HPLC showed complete conversion of the starting material. The mixture was allowed to cool to about room temperature, then diluted with EtOAc (40L). After aging for 30 min with stirring, the entire mixture was filtered and solids were washed with EtOAc (3 x 15L). The combined orange filtrate was concentrated to dryness and the solid residue was suspended in DCM (45 L). The mixture was heated to 35-40 °C and stirred for 1h until all solids were dissolved. Then n-heptane (45L) was added dropwise. Upon complete addition, the mixture was cooled to 15-20 °C with stirring for 1h. The solids were collected by vacuum filtration and solids were washed with cold 1:1 DCM/heptane (25 L), then heptane (25 L) (PSC-2). The solids were dried over the weekend to give Z17d (5.32 kg, yield 75%). 1H NMR (400 MHz, CDCl3) δ = 7.83 (s, 1H), 4.88 (bs,
2H), 3.73 (s, 3H), 3.47 (t, J= 9.2 Hz, 2H), 2.79 (t, J= 9.2 Hz, 2H).
Step e
[00314] To a solution of Z17d (8.0 kg, assay 95%, 30.68 mol) in THF (70 L) was added
EtONa (prepared from 776 g Na and 13.6 L EtOH) at room temperature and the mixture was stirred at
ambient temperature for 1 hour. The mixture was then concentrated to a wet yellow solid by rotary evaporation and the residue was suspended in DCM (40L). The mixture stirred under N2 for l6h. The solids were collected by vacuum filtration and the cake was washed with DCM (about 15 L) until the filtrate was colorless (PSC-2). The solids were then dried under vacuum to give Z17c (6.93 kg, qNMR
72 %, yield 88%). 1H NMR (400 MHz, D2O) δ = 7.37 (s, 1H).
Step f
[00315] To a mixture of Z17c (6.95 kg, assay 72%, 27.23 mol) in l,4-dioxane (72 L) was added Xantphos (233 g, 411 mmol, 0.015 eq), Pd2(dba)3 (186 g, 206 mmol, 0.0075 eq), Z17b (7.13 kg, 28.02 mol) and DIPEA (7.02 kg, 54.46 mol). The system was vacuated and purged with nitrogen gas three times. The mixture was stirred at 65 °C for 16 h under N2. The mixture was cooled to RT and water (50 L) was added, filtered. The cake was washed with EA (25 L). The filtrate was extracted with EA (4 x 20 L). The organic phase was concentrated in vacuum to offer the crude product which was combined with the cake. Then DCM (60 L) was added to the crude product and stirred at 25-30°C for l8h and then filtered. The filter cake was slurried with CH2Cl2 (30 L) for 4 hrs and filtered. The filter cake was slurred in CH2Cl2 (30 L) for 16 hrs and filtered. Then the filter cake was dried in vacuum to give Z17a (3-((2-amino-3-chloropyridin-4-yl)thio)-6-chloropyrazin-2-amine; 9.1 kg, 84 %) as light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 7.89 (s, 1H), 7.7 (d, J= 7.6 Hz, 1H), 7.18 (bs, 2H), 6.40 (bs, 2H), 5.97 (d, J= 7.6 Hz, 1H).
Example 4
Alternative formation of the intermediate Z17a (here also named Y7a)
[00316] By way of alternative and according to a preferred reaction method, the compound of the formula Z17a was obtained according to the following Reaction Scheme:
In detail, the synthesis of the compound of the formula Y7a = Z17a was carried out as follows:
Step a
[00317] 2, 3, 5-trichloropyrazine (70.50 g, 384.36 mmol, 1 equiv) and ammonia solution
(25% wt, 364.00 g, 400 mL, 2.68 mol, 6.14 equiv) were added to a 1-L sealed reactor. The mixture was heated to 80 °C and stirred for 24 h, and the reaction was completed. The reaction mixture was cooled to 30 °C and filtered to give a brown filter cake. The brown filter cake was dissolved in acetone
(50 mL), and filtered. To the filtrate was added petroleum ether (300 mL). The suspension was stirred for 4 h, and filtered to give the crude product. The crude product was slurried in combined solvents of petroleum ether and acetone (10/1, 200 mL) and filtered to give the product Y7d (51.00 g, 307.91 mmol, 80% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 7.63 (s, 1H).
[00318] To a 200 mL round bottom flask was added Na2S (10.816 g, 44wt% containing crystalline water, 60.978mmol) and toluene (100 mL). The mixture was heated to reflux, and water was removed with a Dean-Stark trap (about 5~6 mL water was distilled out). After cooling, the mixture was concentrated to dryness.
[00319] To above round bottom flask was added Y7d (5.000 g, 30.489mmol) and 2-methylbutan-2-ol (50 mL), the reaction was heated to reflux and stirred for 36 h. After cooling to 25 °C, the mixture was filtered. The solvent of the filtrate was exchanged with n-heptane (5 V, 3 times, based on Y7d), and finally concentrated to IV residue. THF (25 mL) was charged to the residue at 25 °C and stirred. The suspension was filtered and washed with THF/n-heptane (5 mL/5 mL) to give a brown solid (6.200 g).
[00320] To another 200 mL round bottom flask was added above brown solid (6.200 g),
10% brine (25 mL), Me-THF (30 mL) and n-Bu4NBr (9.829 g, 30.489 mmol). The mixture was stirred for 0.5 h at room temperature, and the phases were separated. The organic phase was washed with 20% brine (25 mL), and exchanged the solvent with iso-propanol (5 V *3 times, based on Y7d) to give the iso-propanol solution of Y7c (27.000g, 99.2% purity by HPLC area, 58.08% assay yield). 1H NMR (400 MHz, DMSO-d6) δ = 6.88 (s, 1H), 2.97 – 2.92 (m, 14H), 1.38 – 1.31 (m, 14H), 1.13 – 1.04 (m,
14H), 0.73 – 0.69 (t, 21H).
Step c
[00321] To a 25-mL round-bottom flask was added Y7c (4.7g, 23.27wt%, IPA solution from Step b, 2.723 mmol, 1.0 equiv), Y7b (1.052 g, 4.085 mmol, 1.5 equiv), l,lO-Phenanthroline (0.05 g, 0.272 mmol) and water (8 mL). The mixture was purged with nitrogen gas three times, and Cul (0.026 g, 0.136 mmol) was added under nitrogen atmosphere. The mixture was heated up to 65 °C and stirred for 3 h, and the reaction was completed. The reaction was cooled to room temperature and filtered, and the filter cake was washed with water (4 mL*3). The filter cake was slurried in MTBE (6 mL) for 30 min and filtered. The filter cake was washed with MTBE (6 mL) and dried to afford Y7a which is Z17a (565 mg, 72% yield).
[00322] Z17b is synthesized as described in Example 3 Step a and Step b.
Example 5
Alternative Synthesis of the intermediate Z17a:
[00323] According to another preferred method, the compound of the formula Z17a was obtained in accordance with the following Reaction Scheme:
[00324] The reactions were carried out as follows:
Step a
Y7d was synthesised as described in Example 4 step a.
[00325] To a three-necked round-bottle flask was added Y7d (200 mg, 1.22 mmol, 1 equiv), dioxane (4 mL). The solution was vacuated and purged with nitrogen gas three times. Xantphos (14mg, 0.024 mmol, 0.02 equiv), PdCl2(dppf) (8.9 mg, 0.012 mmol, 0.1 equiv), and DIPEA (0.32 g, 2.44 mmol, 2.0 equiv) were added under nitrogen atmosphere. The solution was heated to 85 °C for overnight. The reaction was cooled and evaporated. The residue was purified by column chromatography (eluent/ethyl acetate/heptane = 1/1) to give Z17d (259 mg, 0.99 mmol, 81%). 1H NMR (400 MHz, CDCl3) δ = 7.83 (s, 1H), 4.88 (bs, 2H), 3.73 (s, 3H), 3.47 (t, J= 9.2 Hz, 2H), 2.79 (t, J= 9.2 Hz, 2H).
[00326] The remaining steps were carried out as described in Example 4, Steps e and f, to yield Z17a. Z17b was synthesized as described in Example 3 Step a and Step b.
Example 6
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8- azaspiro[4.5]decan-4-amine. succinate (1:1) hemihydrate. modification (form) HA:Variant a)
[00327] 50 ml ethanol and 2.5 ml water were added to a 100ml flask containing 3.0 g of free base of 3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (obtained as A18 for example as described in Example 1) and 848.0 mg of succinic acid. The mixture was heated to 50°C to generate a clear solution. The temperature was lowered to 15°C during a period of 3 hours. The solution was kept stirring at 15°C overnight.
Precipitated solid was separated via suction filtration and 50 ml of acetone was added to produce a suspension. The suspension was stirred at 50°C for 3 hours. The solid was separated with suction filtration and dried at room temperature under vacuum for 3 hours. Yield was about 60%.
[00328] The succinate appeared as a highly crystalline solid, with a melting point onset of
94.4°C and an accompanying enthalpy of 96 J/g. The succinate salt crystals showed aggregates of broken drusy tabular particles.
[00329] Variant b)
[00330] 14.34 g of 3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)- 3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine free form (obtained as A18 for example as described in Example 1) and 4.053 g of succinic acid were equilibrated in 100 mL 95% EtOH at 50°C. Add 5 mL of water into the system and heat to 70-75 °C. Add 95 mL of pure EtOH and heat for 30 min more. Stir over night at 25 oC. Filter the mixture wash with EtOH and dry under vacuum in an oven at room temperature. Yield is 87.5%.
PATENT
WO 2020065452
PATENT
PHARMACEUTICAL COMBINATION COMPRISING TNO155 AND NAZARTINIB
PAPER
Journal of Medicinal Chemistry (2020), 63(22), 13578-13594.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01170
SHP2 is a nonreceptor protein tyrosine phosphatase encoded by the PTPN11 gene and is involved in cell growth and differentiation via the MAPK signaling pathway. SHP2 also plays an important role in the programed cell death pathway (PD-1/PD-L1). As an oncoprotein as well as a potential immunomodulator, controlling SHP2 activity is of high therapeutic interest. As part of our comprehensive program targeting SHP2, we identified multiple allosteric binding modes of inhibition and optimized numerous chemical scaffolds in parallel. In this drug annotation report, we detail the identification and optimization of the pyrazine class of allosteric SHP2 inhibitors. Structure and property based drug design enabled the identification of protein–ligand interactions, potent cellular inhibition, control of physicochemical, pharmaceutical and selectivity properties, and potent in vivo antitumor activity. These studies culminated in the discovery of TNO155, (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (1), a highly potent, selective, orally efficacious, and first-in-class SHP2 inhibitor currently in clinical trials for cancer.

file:///C:/Users/Inspiron/Downloads/jm0c01170_si_001.pdf
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8- azaspiro[4.5]decan-4-amine (1):

Step a: A mixture of (3S,4S)-tert-butyl 4-((R)-1,1-dimethylethylsulfinamido)-3-methyl-2-oxa-8- azaspiro[4.5]decane-8-carboxylate (51 mg, 0.136 mmol) and HCl (4 M in dioxane, 340 L, 1.362 mmol) in MeOH (5 mL) was stirred for 1 h at 40 °C. After cooling to RT, the volatiles were removed under reduced pressure to give (3S,4S)-3-methyl-2-oxa-8-azaspiro[4.5]decane-4-amine which was used in next step without further purification. MS m/z 171.1 (M+H)+. Step b: A mixture of (3S,4S)-3-methyl-2-oxa-8-azaspiro[4.5]decane-4-amine crude, 3-((2-amino3-chloropyridin-4-yl)thio)-6-chloropyrazin-2-amine (35.5 mg, 0.123 mmol), and DIPEA (193 L, 1.11 mmol) in DMSO (600 L) was stirred for 16 h at 100 °C. After cooling to RT, the volatiles were removed under reduced pressure and the resulting residue was purified by HPLC (gradient elution 15-40% acetonitrile in water, 5 mM NH4OH modifier) to give (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (11 mg, 0.026 mmol). 1 H NMR (400 MHz, METHANOL-d4) δ ppm 7.67-7.47 (m, 2 H), 5.91 (d, J=5.5 Hz, 1 H), 4.22 (qd, J=6.4, 4.8 Hz, 1 H), 4.03 (ddt, J=13.5, 8.9, 4.7 Hz, 2 H), 3.86 (d, J=8.7 Hz, 1 H), 3.71 (d, J=8.7 Hz, 1 H), 3.37 (td, J=9.9, 4.9 Hz, 1 H), 3.29-3.23 (m, 1 H), 3.00 (d, J=5.0 Hz, 1H) 1.91-1.56 (m, 4 H), 1.21 (d, J=6.4 Hz, 3 H). HRMS calcd for C18H25ClN7OS (M+H)+ 422.1530, found 422.1514.
//////////TNO 155, CANCER
CILENGITIDE
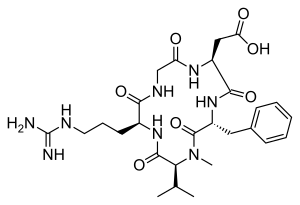
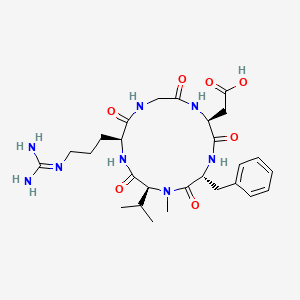
| IUPAC Condensed | cyclo[Arg-Gly-Asp-D-Phe-N(Me)Val] |
|---|---|
| HELM | PEPTIDE1{R.G.D.[dF].[meV]}$PEPTIDE1,PEPTIDE1,5:R2-1:R1$$$ |
| IUPAC | cyclo[L-arginyl-glycyl-L-alpha-aspartyl-D-phenylalanyl-N-methyl-L-valyl] |
CILENGITIDE
- Molecular FormulaC27H40N8O7
- Average mass588.656 Da
2-[(2S,5R,8S,11S)-5-benzyl-11-[3-(diaminomethylideneamino)propyl]-7-methyl-3,6,9,12,15-pentaoxo-8-propan-2-yl-1,4,7,10,13-pentazacyclopentadec-2-yl]acetic acid188968-51-6[RN]
4EDF46E4GI
7823
циленгитид
سيلانجيتيد
西仑吉肽
EMD 121974, EMD-121974, UNII-4EDF46E4GI
Cilengitide has been in phase III clinical trials by Merck Serono and NCI for the treatment of glioblastoma multiforme. However, this research has been discontinued.
Cilengitide was originally developed by Merck KGaA in collaboration with the Technical University of Munich, then received orphan drug designation from FDA for the treatment of glioma in 2005.
Cilengitide (EMD 121974) is a molecule designed and synthesized at the Technical University Munich in collaboration with Merck KGaA in Darmstadt. It is based on the cyclic peptide cyclo(-RGDfV-), which is selective for αv integrins, which are important in angiogenesis (forming new blood vessels), and other aspects of tumor biology. Hence, it is under investigation for the treatment of glioblastoma, where it may act by inhibiting angiogenesis, and influencing tumor invasion and proliferation.[1][2]
The European Medicines Agency has granted cilengitide orphan drug status.[3]
Cilengitide seems to function by inhibiting the FAK/src/AKT pathway and inducing apoptosis in endothelial cells.[4] Preclinical studies in mice of cilengitide were able to demonstrate efficacious tumor regression.[4]
In a rat xenograft model, cilengitide was able to potentiate the cytotoxic effects of radiation when cilengitide was administered prior to radiation therapy.[5] When combined with radiation, inhibition of integrin expression by cilengitide synergistically improves the cytotoxic effects of ionizing radiation for glioblastoma.[5]
Clinical trials
Phase II studies were able to demonstrate that cilengitide as a potential monotherapy in patients with recurrent glioblastoma[6] with high intratumor drug levels when 2000 mg of cilengitide is given twice weekly.[7]
Cilengitide is well tolerated, in combination with radiation and temozolomide, at a dose of 2000 mg in patients with newly diagnosed glioblastoma, regardless of MGMT promoter status.[8] In a phase I/IIa study, the addition of cilengitide to the standard of care for newly diagnosed glioblastoma (surgical resection followed by temozolomide and radiation therapy) improves progression-free survival and overall survival in patients with MGMT promoter methylation.[9]
However, in a subsequent study, cilengitide does not seem to alter the pattern of glioblastoma progression,[10]
and in an EORTC phase III randomized, controlled, multicenter clinical trial, consisting of over 500 patients in 23 countries, the addition of cilengitide to the standard of care did not improve overall survival in patients with newly diagnosed glioblastoma and methylated MGMT promoter status [11] A phase II study, the CORE trial, is currently being conducted in patients with newly diagnosed glioblastoma and unmethylated MGMT promoter status.[12]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
Angewandte Chemie, International Edition, 55(4), 1540-1543; 2016

SYN
Chemistry – A European Journal, 16(18), 5385-5390, S5385/1-S5385/36; 2010
Reference:1. WO0047228A1 / US7115261B1.
2. US6001961A.Route 2
Reference:1. CN102731627A.PATENTWO/2021/224234ANTIVIRAL USE OF CILENGITIDEhttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021224234&_cid=P20-KW0M52-85135-1
PATENThttps://patents.google.com/patent/CN102731627A/enEMD121974 (Cilengitide), the Chinese another name: ring (L-arginyl glycyl-L-aspartoyl-D-phenylalanyl-N-methyl-L-valyl) is an a kind of new classification cancer therapy drug of synthetic.Merkel company discovers that EMD121974 amalgamation radiotherapy (merging to reach assists TM to add radiotherapy) possibly prolong lifetime; Simultaneously integrate plain supressor antitumor drug as first; Got into the III clinical trial phase, its important mechanism is to grow targeting that the blood supply structure of nutrition, the growth of promotion cancer cell is provided in tumour and for tumour through line artery.The EMD121974 molecular formula is: C 27H 40N 8O 7, have following structure:
The preparation method of cyclic peptide mainly contains liquid phase synthesis process, solid phase synthesis precursor peptide cyclization process, process for solid phase synthesis in liquid phase at present; Wherein preceding two kinds of synthesis techniques all are the cyclisation in liquid phase of synthetic precursor peptide, and this method needs reactant in extremely rare solvent, to react (10 -3~10 -4Mol/L), and intermolecular be prone to react generation line style or cyclic polymer, greatly reduced the cyclisation yield, bring trouble for follow-up purifying, and in large-scale production, produce a large amount of waste liquids, be unfavorable for suitability for industrialized production.In conjunction with the structure of EMD121974, utilize the false rare principle of benefit of solid phase, developed a kind of efficient cyclization reaction, the cyclisation time shortens to 20%~30% of liquid phase cyclisation, and the 2%-8% of solvent as liquid phase used in reaction.Embodiment 1The preparation of Fmoc-L-Asp (OtBu)-Wang ResinThe Wang Resin that takes by weighing the 10g substitution degree and be 0.5mmol/g joins in the reactor drum, adds an amount of DCM, and swelling 30min takes out DCM; 6.17g Fmoc-L-Asp-OtBu, DIC 2.40ml, HOBT2.1g are dissolved among the 30ml DMF; At 0-5 ℃ of activation 15min, activation solution is joined in the reactor drum that contains Wang Resin, behind the reaction 10min; Add DMAP 0.18g again, at 0~30 ℃ of reaction 1~5h.After reaction finishes, add sealing Wang Resin unreacted hydroxylation reagent diacetyl oxide 1ml and pyridine 0.5ml, behind the capping 1h, DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min.Through detecting, obtain the Fmoc-L-Asp that substitution degree is 0.47mmol/g (OtBu)-Wang Resin.Embodiment 2The EMD121974 precursor:The preparation of A-Wang Resin (Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin)Fmoc-L-Asp (OtBu)-Wang Resin is joined in the reactor drum, behind DMF swelling 30min, take out solvent, the piperidines-DMF that adds 80ml 25% reacts 5min, and 80ml DMF washs 1 time (3min), and the piperidines-DMF that adds 80ml 25% reacts 15min; DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min; With 4.45g Fmoc-Gly-OH, 5.68g HBTU, 2.03g HOBt, be dissolved among the DMF of 30ml, dissolve the back and added DIEA 2.45ml; 0~5 ℃ of activation 15min; Activation solution is joined in the above-mentioned reactor drum, and behind reaction 1-3h under 0~30 ℃, reaction end detects with ninhydrin method.Adopt aforesaid method coupling Fmoc-L-Arg (Mtr)-OH, Fmoc-N-Me-L-Val, Fmoc-D-Phe-OH successively, finally obtain Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin.Embodiment 3EMD121974 precursor peptide: the preparation of B-Wang Resin (D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp-Wang Resin)With volume ratio is that piperidines-DMF of 25% is the Fmoc deprotection agent of Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin; Add piperidines-DMF 80ml of 25% first time; Reaction 5min, 80ml DMF washs 1 time (3min), adds piperidines-DMF 80ml of 25% for the second time; Behind the reaction 15min, DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min gets D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin after washing finishes.80% the PhOH-DCM solution that adds volume ratio and be 100ml takes off OtBu with the TFA of catalytic amount, reacts 8h; DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min gets D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp-Wang Resin.Embodiment 4The preparation of EMD121974-Wang Resin (Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp)-Wang Rsin)In above-mentioned reactor drum, add cyclization reagent 3.9g DPPA, 2.5ml DIEA (reactant cyclization reagent amount of substance ratio is 1: 3), at 10~40 ℃ of reaction 3h, the multiple cyclization reagent reaction 3~5h (reaction end detects with ninhydrin method) that throws once above-mentioned equivalent; DMF, DCM, the CH of 80ml used in washing successively 3OH washing 2,1,3 times, each 3min gets Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp)-Wang Rsin.Embodiment 5The preparation of EMD121974 (Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp))In above-mentioned reactor drum, add the TFA/H of lytic reagent 120ml again 2Behind O/TlS (volume ratio is 95: 2.5: 2.5) the reaction 3h, suction filtration is removed resin, and filtrating slowly joins in the no water-ice ether; Static 2-5h, high speed centrifugation obtain thick peptide, prepare through high-pressure liquid phase; Lyophilize gets smart EMD121974; Its purity>99.5%, single impurity<0.2%, total recovery reaches 63%.Choosing substitution degree in the present embodiment is the Wang Resin of 0.5mmol/g, and can also choose substitution degree is the arbitrary Wang Resin and Fmoc-L-Asp-OtBu prepared in reaction Fmoc-L-Asp (the OtBu)-Wang Resin of 0.4~0.9mmol/g scope.All can realize technical scheme of the present invention, and obtain technique effect of the present invention.Above content is an EMD121974 and become one of best preferred version of route; And to further explain that the present invention did; But can not assert that practical implementation of the present invention is only limited to these explanations; Under the prerequisite that does not break away from the present invention’s design, can also make some simple deductions and replacement, all should be regarded as protection domain of the present invention.
CLIPhttps://www.eurekaselect.net/article/2607Cilengitide, a cyclic RGD pentapeptide, is currently in clinical phase III for treatment of glioblastomas and in phase II for several other tumors. This drug is the first anti-angiogenic small molecule targeting the integrins αvβ3, αvβ5 and α5β1. It was developed by us in the early 90s by a novel procedure, the spatial screening. This strategy resulted in c(RGDfV), the first superactive αvβ3 inhibitor (100 to 1000 times increased activity over the linear reference peptides), which in addition exhibited high selectivity against the platelet receptor αIIbβ3. This cyclic peptide was later modified by N-methylation of one peptide bond to yield an even greater antagonistic activity in c(RGDf(NMe)V). This peptide was then dubbed Cilengitide and is currently developed as drug by the company Merck-Serono (Germany). This article describes the chemical development of Cilengitide, the biochemical background of its activity and a short review about the present clinical trials. The positive anti-angiogenic effects in cancer treatment can be further increased by combination with “classical” anti-cancer therapies. Several clinical trials in this direction are under investigation.
CLIPJournal of Protein Chemistry

Schematic of the one-step chemoenzymatic synthesis of cilengitide using wild-type Mcy TE. (1) The chemically synthesised (SPPS, solid-phase peptide synthesis) mimetic substrate was condensed with benzyl mercaptane to produce pentapeptide thioester (pentapeptide-BMT). (2) Models of the substrate-O-TE acyl enzyme intermediate are marked with brackets (protein data bank, 1JMK). (3) Mechanism of TE domain catalysis: a pentapeptide -O-TE acyl-enzyme intermediate is formed by transfer of the peptidyl chain from the phosphopantethiene of the terminal peptidyl carrier protein (PCP), which was substituted by benzyl mercaptane, to the active site serine of the TE domain. For hydrolyzing TE domains, the intermediate is captured by water, generating the linear peptide; for cyclizing TE domains, an intramolecular nucleophile captures the intermediate, resulting in “cilengitide”
PATENTWO 9745447
WO 9745137
DE 19534177
WO 2000053627
WO 2000047228
US 20040063790
WO 2009124754
WO 2011079015
WO 2011069629
WO 2011144756WO 2016059622
PATENTWO 2012062777https://patents.google.com/patent/WO2012062777A1/enSynthesis of cyclic peptidesCyclo[-Arg-Gly-Asp- 6 or 7 -Phe-Val-Ala-] (1 and 2). Resin loading. 2- chlorotrityl chloride-resin ( 1 50 m g , 1 .5m m ol/g ) was p laced i n a 20 m l polypropylene syringe fitted with a polyethylene filter disk. The resin was then washed with CH2CI2 (5 χ 0.5 min), and a solution of Fmoc-L-Gly-OH (334 mg, 1 .125 mmol, 5 equiv) and DIEA (239 μΙ_, 6.25 equiv) in CH2CI2 (2.5 ml_) was added. The mixture was then stirred for 15 min. Extra DIEA (239 μΙ_, total 12.5 mmol) was added, and the mixture was stirred for an additional 45 min. The reaction was stopped by adding 3 χ DCM/ MeOH/ DIEA (85: 10:5) and stirring for 1 0 m in. The Fmoc-L-Gly-O-resin product was subjected to the following washings/treatments with CH2CI2 (3 χ 0.5 min), DMF (3 χ 0.5 min), piperidine and DMF (5 χ 0.5 min). The loading was 0.50 mmol/g, as calculated by Fmoc determination.Peptide coupling. Fmoc-L-Arg(Pbf)-OH (243 mg, 0.375 mmol, 5 equiv), Fmoc- L-Ala-OH (1 17 mg, 0.375 mmol, 5 equiv), Fmoc-L-Val-OH ( 127 mg, 0.375 mmol, 5 equiv) and Fmoc- L-Phe-OH ( 145 mg, 0.375 mmol, 5 equiv) were added sequentially to the above obtained H-L-Gly-O-resin using HCTU (155 mg, 0.375 mmol, 5 equiv), HOBt (50 mg, 0.375 mmol, 5 equiv) and DIEA (127 μΙ_, 0.75 mmol, 10 equiv) in DMF (2.5 ml_). In all cases, after 90 min of coupling, the ninhydrin test was negative. Removal of Fmoc group and washings were performed as described in general procedures. /V-Alloc-thiazole 6 or 7 (92 mg, 0.375 mmol, 5 equiv) was coupled with HATU (143 mg, 0.375 mmol, 5 equiv), HOAt (51 mg, 0.375 mmol, 5 equiv) and DIEA (127 μΙ_, 0.75 mmol, 10 equiv) for 90 min. This coupling was repeated twice in the same conditions. The Alloc group of the peptide resin was removed with Pd (PPh3)4 (9 mg, 0.0075 mmol, 0.1 equiv) in the presence of PhSiH3 (92.5 μΙ_, 0.75 mmol, 10 equiv) in DCM for 20 min. This deprotection was repeated three times in the same conditions. After washing, the resin was treated with dry THF (2ml_) for 15 min. Meanwhile, Fmoc-L-Asp(tBu)-OH (154 mg, 0.375 mmol, 5 equiv) was added to a 68 mM solution of triphosgene in dry THF (1 .15 equiv). Sym-collidine (99.5 μΙ_, 0.75 mmol, 10 equiv) was added to the clear solution, upon which a precipitate of collidinium chloride was formed. DIEA (102 μΙ_, 0.6 mmol, 8 equiv) was added to the resin, immediately followed by addition of the suspension. This coupling was repeated four times in the same conditions. The reaction mixture was stirred at 50 °C during 48 h.Peptide cleavage. Following Fmoc deprotection, the peptidyl-resin was treated with TFA-CH2CI2 (1 :99) (5 χ 30 s). The filtrate was collected on H20 (4 ml_) and the H20 was partially removed under reduced pressure. MeCN was then added to dissolve solid that formed during the removal of H20, and the solution was lyophilized to give 12 mg and 10 mg of the linear compounds 28 and 29 respectively with a purity of > 91 % as checked by HPLC (Column A, Rt 7.43 min and Rt 7.38 min respectively, linear gradient 35%-40% ACN in 15 min.)], which was used without further purification. MALDI-TOF-MS calculated for C50H71 N11 O13S2 1098.29; found mlz 1099.29 [M + H]+, 1 121 .28 [M + Na]+, 1 137.39 [M + K]+.Synthesis in solution. Cyclization. The protected linear peptides 28 and 29 were dissolved in DMF (1 L, 10“4 M), and HOAt (9.6 mg, 0.07 mmol, 5 equiv), DIPEA (24 μΙ_, 0.14 mmol, 10 equiv), and PyAOP (36.6 mg, 0.07 mmol, 5 equiv) were added. The mixture was stirred for 24 h at room temperature, and the course of the cyclization step was then checked by HPLC (Column A, Rt 1 1 -67 min and Rt 10.70 min respectively, linear gradient 45%-55% ACN in 15 min.). The solvent was removed by evaporation under reduced pressure and the protected cycle 30 and 31 were used in the next step without further purification. MALDI-TOF-MS calculated for C50H69N11 O12S2 1080.28; found mlz 1081 .28 [M + H]+, 1 103.27 [M + Na]+, 1 1 19.38 [M + K]+.Side chain deprotection. The protected cyclopeptides 30 and 31 (14.7 mg, 19.04 pmol) were treated with TFA-H20 (95: 5) during 1 h. The solvent was removed by evaporation under reduced pressure.Peptide purification. The crude product was purified by HPLC (Symmetry C8 5 μη-Ί, 30 mm x 100 mm), gradient of MeCN (30% to 75% in 15 min) MeCN (+0.05% TFA) in water (+0.05% TFA), 20 mL/min, detection at 220 nm, to give the cyclopeptides 1 and 2 (4.5 mg, 5.8 pmol and 6.5 mg, 8.37 pmol, 7.7% and 12% yield respectively). The products were characterized by HPLC (Rt 8.99 min, and Rt 8.02 min Column A, respectively, linear gradient 0%-100% ACN in 1 5 min. ) and by MALDI-TOF-MS: calculated for C33H45N11 O9S 771 .84; found mlz 772.84 [M + H]+, 794.83 [M + Na]+, 810.94 [M + K]+.Cyc/o-[Arg-Gly-Asp-Thz1X-] (3). General procedure for cyclopeptide synthesis. Solid phase synthesis: The synthesis of the linear peptide H- Asp(tBu)-XX-Arg(Pbf)-Gly-OH was performed using Fmoc-based solid phase peptide synthesis with 2-chlorotrityl chloride resin (2.0 g, 3.2 mmol).Resin loading: Fmoc-Gly-OH (594 mg, 2.0 mmol) was attached to the resin with DIPEA in DCM at room temperature for 1 .5 h. The remaining trityl groups were capped adding 0.5 mL of MeOH for 30 min. After that, the resin was filtered and washed with DCM (2x), DMF (2x). The loading of the resin was determined by titration of the Fmoc group (Chan WC and White PD. Fmoc Solid Phase Peptide Synthesis. Oxford University Press: New York, 2000). The final loading was 2.0 mmol/g. The Fmoc group was eliminated by treatment with 20% piperidine in DMF (2X10 min). The resin was washed with DMF (3x), DCM (3x). Peptide coupling: Fmoc-Arg(Pbf)-OH (5.19 g, 8.0 mmol), DIPCDI (1.23 mL, 8.0 mmol) and HOBt (1.08 g, 8.0 mmol) were dissolved in DMF and added to the resin for 1 .5 h. The end of the coupling was monitored by ninhydrin test (free amine group) (Kaiser E et al. Anal Biochem 1970, 34:595-598). The resin was filtered and washed with DMF (3X) and DCM (3X). The Fmoc group was eliminated with 20 % piperidine in DMF (2X10 min).The coupling of the thiazole module was carried out with 8 (1 .14 g, 3.0 mmol), PyAOP (1 .56 g, 3.0 mmol) and DIPEA (1 .02 mL, 6.0 mmol) in DMF for 1 .5 h. The completion of the reaction was checked with the ninhydrin test. Finally the deprotection of the amine and coupling of the Fmoc-Asp(‘Bu)-OH were carried out under the same conditions of the second amino acid.Peptide cleavage: The resin bound peptide was treated with 2% TFA in DCM (6 x 30 sec.) The resin was washed with DCM and the combined solution was evaporated under vacuum with Et20 several times, furnishing the linear peptide 32 as a white solid. The peptide was used for the next step without purification.H PLC (gradient 20 to 80% of CH3CN in 1 5 m in): tR= 8.33 min. HPLC-MS (ES(+)): m/z 795.3.Synthesis in solution. Cyclization: The product 32 (200 mg, 0.251 mmol) was dissolved in anhydrous DMF (50 mL, 5 mM), PyAOP (262 mg, 0.503 mmol) and DIPEA (213 μί, 1 .255 mmol) were added. The reaction was monitored by HPLC. Once the reaction was finished, the DMF was evaporated under vacuum. The crude was dissolved in AcOEt and the solution was washed with NH4CISat and Na2CO3 sat. The organic layer was collected, dried over Na2SO4, filtered and concentrated under vacuum. The peptide was purified by flash chromatography (CHCIs/MeOH 8:2) furnishing the protected cyclic peptide 33 as a white solid (1 56 mg, XX%). HPLC (gradient 40 to 90% of CH3CN in 1 5 min): tR= 8.86 min. HPLC-MS (ES(+)): m/z 778.2Side chain deprotection: The protected peptide 33 (125 mg, XX mmol), was treated with 25 mL of a solution of TFA H2O (95:5). After 3 h, the solvent was evaporated under vacuum and the residue was precipitated with Et2O (4X). The Et2O solution was discarded and the white solid was lyophilized to afford 3 55 mg (XX%).
Peptide purification. The end product 3 was dissolved in 5 ml MilliQ water and it was filtered through a 0.2 pm filter. The cyclic peptide was purified by semipreparative RP-HPLC using acetronitrile (0.05% TFA)/water (0.1 % TFA). The HPLC sample was vacuum concentred and transformed into the hydrochloride salt lyophilized with water with 0.05% HCI.1H-NMR (500 MHz, H20:D20-d2 9: 1 , 278 K): δ = 9.29 (t, NH Gly), 9.20 (d, J = 7.24 Hz, NH Asp), 8.90 (t, J = 5.89/5.89 Hz, NH Thz), 8.46 (d, J = 8.93 Hz, NH Arg), 7.79 (s, CH Thz), 7.22 (t, J = 5.39/5.39 Hz, ΝΗε Arg), 4.75 (m, CHa Arg), 4.63 (m, CHa Asp), 4.04 (dd, J = 3.35/14.90 Hz, CHa Gly), 3.82 (dd, J = 6.69/14.96 Hz, CHa Gly), 3.17 (m, CH25 Arg), 2.89 (m, CH2p Asp), 1 .92 (m, CH p Arg), 1 .82 (m, CHP Arg), 1 .63 (m, CH2 Arg). HPLC (gradient 0 to 20% of CH3CN in 15 min): tR= 10.52 m in. HRMS (E IS) m/z calculated 468.1540

found 469.16099 (M+H)+.Cyc/o-[Arg-Gly-Asp-Thz2X-] (4). The cyclopeptide 4 was prepared according to the process followed for 3 and using bithiazole 9 (XX mg, YY mmol) instead of 8. The linear peptide 34: HPLC (gradient 0 to 100% CH3CN in 15 min.): tR = 10.34 min, HPLC-MS (ES(+)): m/z 877.81 . The protected peptide 35: HPLC (gradient 0 to 100% CH3CN in 15 min.): tR = 13.91 min, HPLC-MS (ES(+)): m/z 860.54. The final peptide 4: 1H-NMR (500 MHz, H20:D20-d2 9: 1 , 298 K): δ = 8.93 (sbroad, NH Gly), 8.82 (d, J = 7.62 Hz, NH Asp), 8.75 (t, J = 5.69/5.69 Hz, NH Thz), 8.51 (d, J = 7.62 Hz, NH Arg), 8.05 (s, CH Thz1), 7.50 (s, CH Thz2), 7.19 (t, J = 5.38/5.38 Hz, ΝΗε Arg), 4.13 (dd, J = 5.82/14.24 Hz, CH Gly), 3.87 (dd, J = 5.96/15.69 Hz, CH Gly), 3.21 (m , CH25 Arg), 2.94 (m, CH2p Asp), 1 .95 (m , CHP Arg), 1 .87 (m , CHP Arg), 1 .68 (m , CH2y Arg). HPLC (gradient 1 0 to 25% of CH3CN in 1 5 m in): tR = 8.73 min. HRMS (EIS) m/z calculated 551 .1369 (C2oH25N906S2) found 552.14392 (2M+2H)+.Cyc/o-[Arg-Gly-Asp-Thz3X-] (5). The cyclopeptide 5 was prepared according to the process for 3 and using trithiazole 10 (XX mg, YY mmol) instead of 8. The linear peptide 36: HPLC (gradient 20 to 80% of CH3CN in 15 min.): tR = 7.60 min, HPLC-MS (ES(+)): m/z 961 .23. The protected peptide 37: HPLC (gradient 20 to 80% of CH3CN in 15 m in. ): tR = 1 3.13 min, HPLC-MS (ES(+)): m/z 944.3. The final peptide 5: HPLC (gradient 10 to 30% CH3CN in 15 m in): tR = 8.26 m in. HRMS (E IS) m/z calculated 634.1 1 99 (C23H26N10O6S3) found 635.12683 (2M+2H)+. 1H-NMR (500 MHz, DMSO-d6 298 K): δ = 9.21 (t, J = 5.4, NH Gly), 8.72 (m, NH Asp + NH Thz), 8.37 (s, CH Thz1), 7.96 (d, J = 9.2, NHa Arg), 7.77 (s, CH Thz2), 7.68 (t, J = 6.0, ΝΗε Arg), 7.23 (s, CH Thz3), 4.83 (dd, J = 14.3, 8.5, CHa Arg), 4.72 (dd, J = 16.3, 6.6, CH Thz), 4.59 (m, CH Thz + CHa Asp), 3.89 (d, J = 1 1 .5, CH Gly), 3.59 (d, J = 9.7, CH Gly), 3.13 (dd, J = 12.6, 6.3, CH25 Arg), 2.81 (dd, J = 16.3, 4.3, CHP Asp), 2.58 (dd, J = 16.5, 8.7, CHP Asp), 1 .82 (m, CHP Arg), 1 .71 (m, CHP Arg), 1 .49 (m, CH2y Arg).Cilengitide. The cilengitide was prepared according to the method described in Dechantsreiter MA et al. (J Med Chem 1999, 42:3033-3040). 1H- NMR (500 MHz, H20:D20-d2 9: 1 , 298 K): δ = 8.55 (d, J = 8.06 Hz, NH Asp), 8.37 (d, J = 7.28 Hz, NH Arg), 8.13 ( d, J = 9.19 Hz, NH Phe), 7.97 (m, NH Gly), 7.34 (m, 2H, C6H5 Phe), 7.26 (m, 3H, C6H5 Phe), 7.22 (t, J = 5.53/5.53 Hz, ΝΗε Arg), 5.19 (dd, J = 8.58/16.02 Hz, CHa Phe), 4.56 (dd, J = 7.45/- Hz, CHa Asp), 4.34 (d, J = 10.89 Hz, CHa MeVal), 4.12 (dd, J = 7.80/14.63 Hz, CH Gly), 3.95 (dd, J = 6.84/15.33 Hz, CHa Arg), 3.54 (dd, J = 3.37/14.60 Hz, CH Gly), 3.20 (m , CH25 Arg), 3.02 (m, CH2p Phe), 2.88 (s, CH3 MeVal), 2.84 (dd, J = 7.26/16.68 Hz, CHP Asp), 2.63 (dd, J = 7.60/16.54 Hz, CHP Asp), 2.06 (m, CHP Val), 1 .91 (m, CH2p Arg), 1 .57 (m, CH2 Asp), 0.88 (d, J = 6.55 Hz, CH3 Val1), 0.56 (d, J = 6.49 Hz, CH3 Val2).
PAPERJournal of medicinal chemistry (1999), 42(16), 3033-40.Peptide Science (2001), Volume Date2000, 37th, 249-250. Current opinion in investigational drugs (London, England : 2000) (2003), 4(6), 741-5. Journal of medicinal chemistry (2005), 48(24), 7675-87.Peptide Science (2006), 43rd, 215-216Angewandte Chemie, International Edition (2010), 49(15), 2732-2737, S2732/1-S2732/53.Accounts of Chemical Research (2017), 50(7), 1541-1556.
References
- ^ Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL (August 2002). “Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts”. Cancer Research. 62 (15): 4263–72. PMID 12154028.
- ^ Goodman SL, Hölzemann G, Sulyok GA, Kessler H (February 2002). “Nanomolar small molecule inhibitors for alphav(beta)6, alphav(beta)5, and alphav(beta)3 integrins”. Journal of Medicinal Chemistry. 45 (5): 1045–51. doi:10.1021/jm0102598. PMID 11855984.
- ^ Spreitzer H (October 27, 2008). “Neue Wirkstoffe – Cilengitide”. Österreichische Apothekerzeitung (in German) (22/2008): 1136–7.
- ^ Jump up to:a b Yamada S, Bu XY, Khankaldyyan V, Gonzales-Gomez I, McComb JG, Laug WE (December 2006). “Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice”. Neurosurgery. 59 (6): 1304–12, discussion 1312. doi:10.1227/01.NEU.0000245622.70344.BE. PMID 17277694. S2CID 19861713.
- ^ Jump up to:a b Mikkelsen T, Brodie C, Finniss S, Berens ME, Rennert JL, Nelson K, Lemke N, Brown SL, Hahn D, Neuteboom B, Goodman SL (June 2009). “Radiation sensitization of glioblastoma by cilengitide has unanticipated schedule-dependency”. International Journal of Cancer. 124 (11): 2719–27. doi:10.1002/ijc.24240. PMID 19199360.
- ^ Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, et al. (December 2008). “Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme”. Journal of Clinical Oncology. 26 (34): 5610–7. CiteSeerX 10.1.1.688.8987. doi:10.1200/JCO.2008.16.7510. PMID 18981465.
- ^ Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, Cloughesy T, Lassman AB, Deangelis LM, Chang S, Prados M (January 2012). “Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery”. Journal of Neuro-Oncology. 106 (1): 147–53. doi:10.1007/s11060-011-0650-1. PMC 4351869. PMID 21739168.
- ^ Nabors LB, Mikkelsen T, Hegi ME, Ye X, Batchelor T, Lesser G, Peereboom D, Rosenfeld MR, Olsen J, Brem S, Fisher JD, Grossman SA (November 2012). “A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306)”. Cancer. 118 (22): 5601–7. doi:10.1002/cncr.27585. PMC 3423527. PMID 22517399.
- ^ Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, et al. (June 2010). “Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma” (PDF). Journal of Clinical Oncology. 28(16): 2712–8. doi:10.1200/JCO.2009.26.6650. PMID 20439646.
- ^ Eisele G, Wick A, Eisele AC, Clément PM, Tonn J, Tabatabai G, et al. (March 2014). “Cilengitide treatment of newly diagnosed glioblastoma patients does not alter patterns of progression”(PDF). Journal of Neuro-Oncology. 117 (1): 141–5. doi:10.1007/s11060-014-1365-x. PMID 24442484. S2CID 21636884.
- ^ Merck Group. “Phase III Trial of Cilengitide Did Not Meet Primary Endpoint in Patients With Newly Diagnosed Glioblastoma, Date accessed: 3/24/2014.”
- ^ ASCO Meeting Library. [1] “Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma and methylated O6-methylguanine-DNA methyltransferase (MGMT) gene promoter: Key results of the multicenter, randomized, open-label, controlled, phase III CENTRIC study, Date accessed: 3/24/2014
| Names | |
|---|---|
| IUPAC name2-[(2S,5R,8S,11S)-5-benzyl-11-{3-[(diaminomethylidene)amino]propyl}-7-methyl-3,6,9,12,15-pentaoxo-8-(propan-2-yl)-1,4,7,10,13-pentaazacyclopentadecan-2-yl]acetic acid | |
| Identifiers | |
| CAS Number | 188968-51-6 |
| 3D model (JSmol) | Interactive image |
| ChEMBL | ChEMBL429876 |
| ChemSpider | 154046 |
| IUPHAR/BPS | 6597 |
| KEGG | D03497 |
| MeSH | Cilengitide |
| PubChem CID | 176873 |
| UNII | 4EDF46E4GI |
| CompTox Dashboard (EPA) | DTXSID9044035 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C27H40N8O7 |
| Molar mass | 588.656 g/mol |
| Density | 1.417 g/mL |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
/////////CILENGITIDE, циленгитид , سيلانجيتيد ,西仑吉肽 , PHASE 3, EMD 121974, EMD-121974, UNII-4EDF46E4GI, orphan drug , MERCK, glioblastoma,
CC(C)C1C(=O)NC(C(=O)NCC(=O)NC(C(=O)NC(C(=O)N1C)CC2=CC=CC=C2)CC(=O)O)CCCN=C(N)N

NEW DRUG APPROVALS
ONE TIME
$10.00
Methotripremazine
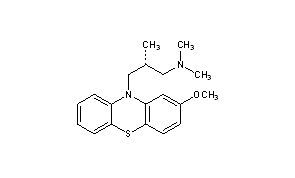
Methotripremazine
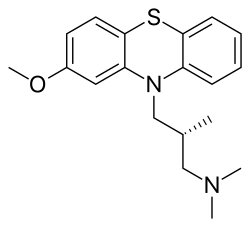
- CL 36467
- CL 39743
- N05AA02
- RP 7044
- RP-7044
- SK&F 5116
- XP-03
- XP03
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Methotrimeprazine hydrochloride | 42BB1Y2586 | 1236-99-3 | ODLGFPIWRAEFAN-PFEQFJNWSA-N |
| Methotrimeprazine maleate | 5KN5Y9V01K | 7104-38-3 | IFLZPECPTYCEBR-VIEYUMQNSA-N |
Methotrimeprazine
CAS Registry Number: 60-99-1
CAS Name: (bR)-2-Methoxy-N,N,b-trimethyl-10H-phenothiazine-10-propanamine
Additional Names: (-)-10-(3-dimethylamino-2-methylpropyl)-2-methoxyphenothiazine; levomepromazine; 2-methoxytrimeprazine; levomeprazine
Manufacturers’ Codes: RP-7044
Trademarks: Sinogan-Debil; Tisercin (EGYT); Neozine (Rh>e-Poulenc); Nirvan; Nozinan (Rh>e-Poulenc); Levoprome (Lederle)
Molecular Formula: C19H24N2OS
Molecular Weight: 328.47
Percent Composition: C 69.47%, H 7.36%, N 8.53%, O 4.87%, S 9.76%
Literature References: Prepn: Courvoisier et al.,C.R. Seances Soc. Biol. Ses Fil.151, 1378 (1957); Jacob, Robert, US2837518 (1958 to Rhône-Poulenc).Optical Rotatory Power, -17, Conc: 5 g/100mL; Solv: chloroform; Wavlen: 589.3 nm; Temp: 20 °C
Derivative Type: Maleate
CAS Registry Number: 7104-38-3
Trademarks: Minozinan; Milezin (Spofa); Neuractil; Neurocil (Bayer); Sofmin (Dainippon); Veractil
Molecular Formula: C19H24N2OS.C4H4O4
Molecular Weight: 444.54
Percent Composition: C 62.14%, H 6.35%, N 6.30%, O 18.00%, S 7.21%
Properties: Crystals, darkened by light. Dec about 190°. Sparingly sol in water (0.3% at 20°) and in ethanol (0.4%). pH of a 0.3% aq soln is 4.3. The free base is levorotatory: [a]D20 -17° (c = 5 in chloroform).
Optical Rotation: [a]D20 -17° (c = 5 in chloroform)
Therap-Cat: Analgesic.
Keywords: Analgesic (Non-Narcotic).
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Methotrimeprazine is a phenothiazine used in the management of psychosis, particular those of schizophrenia, and manic phases of bipolar disorder.
A phenothiazine with pharmacological activity similar to that of both chlorpromazine and promethazine. It has the histamine-antagonist properties of the antihistamines together with central nervous system effects resembling those of chlorpromazine. (From Martindale, The Extra Pharmacopoeia, 30th ed, p604)
Levomepromazine, also known as methotrimeprazine, is a phenothiazine neuroleptic drug. Brand names include Nozinan, Levoprome, Detenler, Hirnamin, Levotomin and Neurocil. It is a low-potency antipsychotic (approximately half as potent as chlorpromazine) with strong analgesic, hypnotic and antiemetic properties that are primarily used in palliative care.[1][2]
Serious side effects include tardive dyskinesia, akathisia, abnormalities in the electrical cycle of the heart, low blood pressure and the potentially fatal neuroleptic malignant syndrome.[1][2]
As is typical of phenothiazine antipsychotics, levomepromazine is a “dirty drug“, that is, it exerts its effects by blocking a variety of receptors, including adrenergic receptors, dopamine receptors, histamine receptors, muscarinic acetylcholine receptors and serotonin receptors.[1][2]
Medical uses
It can be used as an analgesic for moderate to severe pain in non-ambulant patients (the latter being because of its strong sedative effects).[3]
Levomepromazine is also used at lower doses for the treatment of nausea and insomnia.[1]
Levomepromazine is frequently prescribed and valued worldwide in palliative care medicine for its multimodal action, to treat intractable nausea or vomiting, and for severe delirium/agitation in the last days of life. Palliative care physicians will commonly prescribe it orally or via subcutaneous syringe drivers in combination with opioid analgesics such as hydromorphone.[1][2]
Levomepromazine is used for the treatment of psychosis, particularly those of schizophrenia, and manic phases of bipolar disorder. It should only be used with caution in the treatment of agitated depressions, as it can cause akathisia as a side effect, which could worsen the agitation.[1][2] A 2010 systematic review compared the efficacy of levomepromazine with atypical antipsychotic drugs:
Adverse effects
The most common side effect is akathisia.[2] Levomepromazine has prominent sedative and anticholinergic/sympatholytic effects (dry mouth, hypotension, sinus tachycardia, night sweats) and may cause weight gain.[2] These side effects normally preclude prescribing the drug in doses needed for full remission of schizophrenia, so it has to be combined with a more potent antipsychotic.[2] In any case, blood pressure and EKG should be monitored regularly.[2]
A rare but life-threatening side effect is neuroleptic malignant syndrome (NMS).[2] The symptoms of NMS include muscle stiffness, convulsions and fever.[2]
PAPER
Bulletin de la Societe de Pharmacie de Bordeaux (1964), 103(4), 224-30.
The authors define an extn. equil. const., pKe. When a basic mol., A, in an org. solvent (immiscible with water) is shaken with an aq. acid, part of A passes into the aq. phase in the equil. A + H+ .rdblhar. AH+, and Ke and pKe are defined by the equations Ke = [A]org[H+]H2O/[AH+]H2O and pKe = pKa -log ([A]org/[A]H2O), resp. Values of pKe are reported for levomepromazine, properidiazine, thioridazine, chlorpromazine, alimenazine, propiomazine, promethazine, and aminopromazine. Where 2 C atoms sep. the 2 N chain atoms, pKe is of the order of 5, and if 3, the value is near 4.3.
PATENT
JP 40009030
A soln. of 10.5 g. l-3-dimethylamino-2-methylpropanol in xylene is added a suspension of 2.5 g. Na in xylene and a soln. of 18 g. p-tosyl chloride in xylene is dropped in to give l-3-dimethylamino-2-methylpropanol tosylate (I), hydrochloride m. 98-100%. I is treated with 18 g. 2-methoxyphenothiazine and NaNH2 (prepd. from 1.85 g. Na) to give 80% l-3-(2-methoxy-10-phenothiazinyl)-2-methyl-1-dimethylaminopropane, m. 125-6° (hexane). Similarly are prepd. l-3-(3-ethyl-10-phenothiazinyl)-2-methyl-1-dimethylaminopropane (maleate m. 136°) and l-3-(10-phenothiazinyl)-2-methyl-1-dimethylaminopropane (maleate m. 174-5°). The products are tranquilizers.
PATENT
HU 152208
HU 157158
PL 66636
PAPER
Bulletin de la Societe Chimique de France (1968), (8), 3220-2.
Folia medica (1970), 12(1), 88-9
Journal of pharmaceutical sciences (1987), 76(7), 541-4.
SYN
| IN201203390 |
Deprotonation of 2-methoxyphenothiazine by means of KOH in refluxing touene/DMSO, followed by condensation of resulting pottasium salt with N-(3-chloro-2-methylpropyl)-N,N-dimethylamine in refluxing toluene leads to racemic levomepromazine , which upon finally resolution using (-)-dibenzoyl-L-tartaric acid in acetone or using di-p-toluoyl-L-tartaric acid and, optionally, HCOOH in EtOH at 60 °C affords the target levomepromazine

SYN

References
- ^ Jump up to:a b c d e f Brayfield A, ed. (13 December 2013). “Levomepromazine”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 12 May 2014.
- ^ Jump up to:a b c d e f g h i j k Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- ^ “Levomepromazine”. Farmacotherapeutisch Kompas (in Dutch). Retrieved 5 October 2016.
- ^ Jump up to:a b Sivaraman P, Rattehalli RD, Jayaram MB (October 2010). “Levomepromazine for schizophrenia”. The Cochrane Database of Systematic Reviews. 10 (10): CD007779. doi:10.1002/14651858.CD007779.pub2. PMC 3283151. PMID 20927765.
External links
- “Levomepromazine”. PubChem. National Center for Biotechnology Information.
- NOZINAN – Lévomépromazine Doctissimo Guides des Medicaments
- “Levomepromazine” (PDF). Grampians Palliative Care Team Publication. Victoria, Australia. May 2010. Archived from the original (PDF) on 2011-02-26.
- “Levomepromazine in Palliative Care” (PDF). Scotland, UK. August 2013. Archived from the original (PDF) on 2013-05-22.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Pregnancy category | Only if clearly needed |
| Routes of administration | Oral, seldom IM |
| Drug class | Typical antipsychotic |
| ATC code | N05AA02 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | ~50–60% |
| Metabolism | Hepatic |
| Elimination half-life | ~20 hours |
| Excretion | In feces and urine (metabolites), unchanged drug only 1% |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 60-99-1 7104-38-3 (maleate), 1236-99-3 HCl) |
| PubChem CID | 72287 |
| IUPHAR/BPS | 7603 |
| DrugBank | DB01403 |
| ChemSpider | 65239 |
| UNII | 9G0LAW7ATQ |
| KEGG | D00403 |
| ChEBI | CHEBI:6838 |
| ChEMBL | ChEMBL1764 |
| CompTox Dashboard (EPA) | DTXSID1023289 |
| ECHA InfoCard | 100.000.450 |
| Chemical and physical data | |
| Formula | C19H24N2OS |
| Molar mass | 328.47 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////methotripremazine, L 36467, CL 39743, N05AA02, RP 7044, RP-7044, SK&F 5116, XP-03, XP03
O(c2cc1N(c3c(Sc1cc2)cccc3)C[C@H](C)CN(C)C)C

NEW DRUG APPROVALS
one time
$10.00
Methiomeprazine
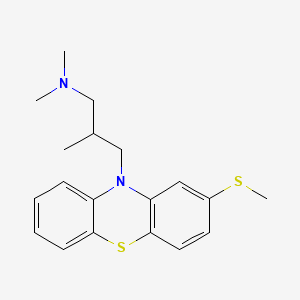
Methiomeprazine
N,N,2-trimethyl-3-(2-methylsulfanylphenothiazin-10-yl)propan-1-amine
CAS 7009-43-0
Molecular Formula, C19-H24-N2-S2, Molecular Weight, 344.5446,
- 10H-Phenothiazine-10-propanamine, N,N,β-trimethyl-2-(methylthio)-, (±)-
- Phenothiazine, 10-[3-(dimethylamino)-2-methylpropyl]-2-(methylthio)-, (±)- (8CI)
- N,N,β-Trimethyl-2-(methylthio)-10H-phenothiazine-10-propanamine
- (±)-10-(3-Dimethylamino-2-methylpropyl)-2-(methylthio)phenothiazine
- 10584-RP
- 2-Methylthio-10-(2-methyl-3-dimethylaminopropyl)phenothiazine
- Methiomeprazine
- SKF 6270
- (+-)-10-(3-Dimethylamino-2-methylpropyl)-2-(methylthio)phenothiazine
- Phenothiazine, 10-(3-(dimethylamino)-2-methylpropyl)-2-(methylthio)-, (+-)-
- 10584 RP
- EINECS 230-285-9
- Methiomeprazinum
- Methiomeprazinum [INN-Latin]
- Metiomeprazina
- Metiomeprazina [INN-Spanish]
- RP 10584
- SKF 6270
- UNII-X2R9QTF0OL
Methiomeprazine hydrochloride
14056-64-5
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////////////////////////////////////////////////////////////////////////////////////////////// Methiomeprazine is an antiemetic drug.
PATENTFR 2705 M 19640831.The title compd. and its derivs. are prepd. and can be used in the prepn. of antiemetic compns. A soln. of 2.280 g. 3-methylthio-10-(3-dimethylamino-2-methylpropyl)phenothiazine (I) in 12 l. EtOH is heated to 70° and added to a soln. (60°) of 969 g. d-tartaric acid in 27 l. EtOH, the soln. kept overnight and filtered, and the mother liquors from the 1st and 2nd crystns. combined and evapd. The residue (2.352 g.) is dissolved in H2O, the soln. made alk. with 700 ml. NaOH (d. 1.33) and extd. with 4 l. CH2Cl2, the org. phase sepd., the aq. phase extd. with 1 l. CH2Cl2, and the exts. combined and evapd. at ∼20 mm. The residue (1.183 g.) is taken up in 7 l. EtOH at 60°, the soln. added to 370 g. maleic acid in 1.7 l. EtOH (60°), and the mixt. kept overnight to give 1.192 g. I acid maleate (II), m. 176-7° (EtOH), [α]24D -21.2° ± 1.5° (c 2, CHCl3). II (300 g.) is added to a mixt. of 1 l. H2O and 2 l. CH2Cl2, 150 ml. NaOH (d. 1.33) added, and the org. phase sepd. and distd. to give 185 g. (-)-3-methylthio-10-(3-dimethylamino-2-methylpropyl)phenothiazine (III), m. 84-5° (iso-PrOH), [α]23D -45° ± 3° (c 2.5, C6H6).
PAPERJournal of Organic Chemistry (1960), 25, 944-7.https://pubs.acs.org/doi/abs/10.1021/jo01076a019cf. CA 54, 15391b. The prepn. of various 10-aminoalkyl derivs. of the following phenothiazines was described: 2-hydroxyphenothiazine (I), 2-methylthiophenothiazine (II), 2-methylsulfonylphenothiazine (III), 2-trifluoromethylsulfonylphenothiazine (IV), 2-trifluoromethylthiophenothiazine (V), 2-azaphenothiazine (VI), and 8-chloro-2-azaphenothiazine (VII). The direct alkylation of I was not attempted. Instead, 2-benzoyloxyphenothiazine was alkylated with NaNH2 in xylene and the ester group removed by basic hydrolysis during the workup. The alkylation of IV with 3-(4-methylpiperazinyl)propyl chloride required 48 hrs. VI (15 g.), 6.8 g. NaNH2, and 500 ml. PhMe refluxed 45 min. under N, treated with 21 g. 3-chloro-1-(1-formyl-4-piperazinyl)propane-HCl and 300 ml. PhMe, the mixt. cooled, 150 ml. H2O added, the PhMe layer extd. with dil. HCl, the acid exts. made alk., extd. with C6H6, and the solvent evapd. gave 21 g. oil. The oil dissolved in 250 ml. alc., 60 ml. H2O and 7 ml. 40% NaOH, the mixt. refluxed 2 hrs., the solvents removed, the residual oil dissolved in C6H6, the soln. extd. with HCl, made alk., extd. with C6H6, and the whole distd. gave 11 g. 10-[3-(1-piperazinyl)propyl]-2-azaphenothiazine. The distd. material was dissolved in 250 ml. MeOH and refluxed 1.5 hrs. with 1.8 g. ethylene oxide, the solvent evapd., the residue dissolved in 250 ml. C6H6, the soln. azeotropically distd. during 1 hr., cooled, and refluxed 1 hr. with 6.5 g. AcCl, the solvents evapd., the gum treated with 10% NaOH, and the C6H6 evapd. gave 4.3 g. 4-[3-(2-azaphenothiazin-10-yl)propyl]-1-piperazineëthanol; acetate dimaleate m. 147-8° (decompn.) (EtOAc). 1-Piperazinepropanol (57.6 g.) refluxed 1 hr. with 48 g. HCO2Me, the excess HCO2Me removed, and the residue distd. gave 65.3 g. oil, b1.1 174.5-7.0°, n24D 1.5072. This oil (42.8 g.) in 300 cc. CHCl3 treated with excess HCl, then 19 g. SOCl2, the mixt. refluxed 0.5 hr., 3 g. SOCl2 added, refluxing continued 2.5 hrs., and the solvents removed gave a cryst. HCl salt. Conversion of this to the free base gave 60% 1-formyl-4-(3-chloropropyl)piperazine, yellow oil, b0.4 144.5-8.5°, n25D 1.5053. By starting with I-VII the following 2,10-disubstituted phenothiazines were obtained (substituents at 2, 10, b.p./mm., and % yield given); SMe, (CH2)3NMe2, 220-3°/0.7 (HCl salt m. 149-50°), 88; SMe, CH2CHMeCH2NMe2, 218-21°/0.1 (HCl salt m. 173-4°), 93; SMe, (CH2)3N.(CH2)2.NMe.CH2.CH2, 239-42°/0.1 (di-HCl salt m. 224-5°), 92; SMe, CH2CHMeCH2N.(CH2)2. NMe.CH2.CH2, 200-20°/0.03 (dimaleate m. 174-5°), 44; SMe, (CH2)3N.(CH2)2.N[(CH2)2OAc].CH2.CH2 – (dimaleate m. 165-6°), 33; SO2Me, (CH2)3NMe2, 115-16° (HCl salt m. 112-15°), 62; SO2Me, CH2CHMeCH2NMe2, 255-60°/0.2 (HCl salt m. 234-5°), 60; SCF3, (CH2)3NMe2, 153-7°/0.1, 64; SCF3, CH2CHMeCH2NMe2, 153-7°/0.1 (picrate m. 158.5-9.5°), 54; SCF3, I (CH2)3N.(CH2)2.NMe.CH2.CH2, 220-3°/0.3 (dimaleate m. 182-3°), 63; SO2CF3, (CH2)3NMe2, 235-40°/0.04 (HCl salt m. 174-5°), 15; SO2CF3, CH2CHMeCH2NMe2, 182-4°/0.2 (picrate m. 203-4°), 19; SO2CF3, (CH2)3N.(CH2)2.NMe.CH2.CH2, – [di-HCl salt m. 249.5° (decompn.)], 16; OH, (CH2)3NMe2, 220-5°/0.05, m. 90-1° (dimaleate m. 132-3°), 49. The following 8,10-substituted 2-azaphenothiazines were similarly prepd. (8,10 substituents, m.p. or b.p., % yield given): H, (CH2)3NMe2, 165-70°/0.007 [di-HCl salt m. 240.5-4.5° (decompn.)], 63; H, CH2CHMeCH2NMe2, 190-5°/0.6 (di-HCl salt m. 234-5°), 82; H, (CH2)3N.(CH2)2.N[(CH2)2OAc].CH2.CH2, – (dimaleate m. 147-8° (decompn.), 9; Cl, (CH2)3NMe2, 215-20°/1 (di-HCl salt m. 249-50°), 66.
PATENTGB 802725N-Aminoalkyl derivs. of I, where the alkyl is a straight or branched 2-5 C atom chain and the amino may be mono- or dialkylated or may be substituted by a pyrrolidino, piperidino, morpholino, or 4-alkyl-1-piperazinyl group, are prepd. by condensing I with the appropriate halo amine or by decompg. a phenothiazine-10-carboxylate of the appropriate amino alcohol. I (4.9 g.) was heated in 50 cc. boiling anhyd. xylene with 0.88 g. sodamide 1 hr., 2.71 g. 3-dimethylamino-1-chloropropane added, the soln. boiled 6 hrs., treated with H2O, then with dil. HCl, made alk. with NaOH, extd. with ether, and the solvent was evapd. in vacuo to give 4.5 g. 3-methylthio-10-(3-dimethylaminopropyl)phenothiazine (III), b0.2 206-18°; III.2HCl m. 160° (acetone-ether); picrate m. 135° (acetone). 3-Methylthio-10-(3-dimethylamino-2-methylpropyl)phenothiazine, m. 88-9°, was prepd. from I and 3-dimethylamino-2-methyl-1-chloropropane; picrate m. 145° (EtOH). The following were similarly prepd.: 3-methylthio-10-[3-(4-methyl-1-piperazinyl)propyl]phenothiazine, b0.1 250-6° [dihydrochloride m. 220° (decompn.) (acetone-ether); dipicrate m. 252-3° (acetone-iso-PrOH); 3-methylthio-10 – (2 – dimethylaminopropyl)phenothiazine, b0.2 202-6° (hydrochloride m. 205-6°; picrate m. 190°); 3-methylthio-10- (3-pyrrolidinopropyl)phenothiazine, b0.9 261° (hydrochloride m. 161°). I was phosgenated in toluene in the presence of pyridine to the 3-methylthiophenothiazine-10-carbonyl chloride (IV), m. 125°; IV heated in toluene with 3-(4- methyl-1-piperazinyl)-2-methylpropanol gave 3-(4-methyl-1- piperazinyl)-2-methylpropyl 3-methylthiophenothiazine-10- carboxylate (V) (dihydrochloride m. 225°). A soln. of 13 7 g. V in 60 cc. ο-Cl2C6H4 was boiled for 5 hrs. till CO2 evolution ceased, the soln. cooled, 60 cc. ether added and the mixt. H2O-washed, extd. with 10% HCl, made alk. with NaOH, and extd. with ether. The ether soln. was dried over anhyd. Na2SO4 and distd. in vacuo to yield 11.25 g. crude base which gave, with an EtOH soln. of maleic acid, 12.7 g. 3-methylthio-10-[3-(4-methyl-1-piperazinyl)-2-methyl-propyl]phenothiazinecarboxylic acid dimaleate, m. 199°. 3-Methylthio-10- [2,3-bis(dimethylamino)propyl] phenothiazine neutral fumarate, m. 198°, was similarly obtained by decarboxylating 1,3-bis(dimethylamino)-2-propyl 3-methylthiophenothiazine-10-carboxylate and treating with fumaric acid. 3-Methylthio-10-(3-diethylaminopropyl)phenothiazine-HCl, m. 172°, was prepd. from 3-methylthio-10-[3-(p-toluenesulfonyloxy)propyl]phenothiazine (VI) and Et2NH; 3-methylthio-10-(3-methylaminopropyl)phenothiazine (H oxalate m. 186°), from VI and MeNH2. VI heated with excess NH3 in toluene gave 3-methylthio-10-(3-aminopropyl)phenothiazine (VII) (oxalate m. 198°). VII in dioxane was neutralized with N HCl and treated with 30% aq. HCHO and PtO2 to give III. These compds. are antiemetics and potentiators of general anasthetics or neuroleptics.
SYN

///////////Methiomeprazine , antiemetic, Metiomeprazina, RP 10584, RP-10584, RP10584, RP 10584, SKF 6270
Systematic name (3):
- 10-[3-(ジメチルアミノ)-2-メチルプロピル]-2-(メチルチオ)-10H-フェノチアジン
- N,N,β-トリメチル-2-(メチルチオ)-10H-フェノチアジン-10-プロパン-1-アミン
- N,N,β-トリメチル-2-メチルチオ-10H-フェノチアジン-10-プロパン-1-アミン
Other name (6):
- メチオメプラジン
- Methiomeprazine
- 10-[3-(Dimethylamino)-2-methylpropyl]-2-(methylthio)-10H-phenothiazine
- SKF-6270
- N,N,β-Trimethyl-2-(methylthio)-10H-phenothiazine-10-propan-1-amine
CSc1ccc2Sc3ccccc3N(CC(C)CN(C)C)c2c1

NEW DRUG APPROVALS
ONE TIME
$10.00
Dichlorquinazine

CORRECT STR OF Dichlorquinazine
7-chloro-N-[1-[4-[2-[(7-chloroquinolin-4-yl)amino]propyl]piperazin-1-yl]propan-2-yl]quinolin-4-amine;methanesulfonic acid
- 1,4-Piperazinediethanamine, N,N’-bis(7-chloro-4-quinolinyl)-α,α’-dimethyl- (9CI)
- Quinoline, 4,4-[1,4-piperazinediylbis[(1-methylethylene)imino]]bis[7-chloro- (7CI)
- Quinoline, 4,4′-[1,4-piperazinediylbis[(1-methylethylene)imino]]bis[7-chloro- (8CI)
- N1,N4-Bis(7-chloro-4-quinolinyl)-α1,α4-dimethyl-1,4-piperazinediethanamine
- 1,4-Bis[2-(7-chloro-4-quinolylamino)propyl]piperazine
- Bis[(chloro-7”-quinolyl-4”)amino-2′-propyl]-1,4-piperazine
- Dichlorquinazine
- N,N’-Bis(7-chloro-4-quinolyl)-α,α’-dimethylpiperazine-1,4-diethylamine
- NSC 129790
- RP 12278
- WR 3863
WRONG STRUCTURE
WRONG STRUCTURE
Dichlorquinazine
- BRN 0867697
- Dichlorquinazine
- EINECS 234-130-6
- NSC 129790
- RP 12278
- UNII-HT3GAD2SCM
- WR 3863
cas 10547-40-7
C28H32Cl2N6, mw
| 523.5 |
7-chloro-N-[2-[4-[2-[(7-chloroquinolin-4-yl)amino]propan-2-yl]piperazin-1-yl]propan-2-yl]quinolin-4-amine
VARIANT
RN: 23256-65-7
Molecular Formula, C28-H32-Cl2-N6.C-H4-O3-S, Molecular Weight, 619.6144
- RP-12278 mesylate
- WR-3863 mesylate
- Quinoline, 4,4′-(1,4-piperazinediylbis((1-methylethylene)imino))bis(7-chloro-, tetramethanesulfonate bis((7-chloro-4”-quinolyl)-2′-aminopropyl)-1,4-piperazine methanesulfonate
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENTS
BE 626239
4-(Chloro or alkoxy)quinolines are treated with a 1,4-bis(aminoalkyl)piperazine to give the title compds. which can be used as antiinflammatory agents and as amebicides. Thus, a mixt. of 16.3 g. 4-chloroquinoline, 10 g. 1,4-bis(3-aminopropyl)piperazine, 55 g. PhOH, and 0.2 g. NH4Cl is heated 5 hrs. at 175°, poured into a mixt. of 500 ml. H2O and 100 ml. NaOH (d. 1.33), filtered, the ppt. is treated with a mixt. of 80 ml. H2O and 20 ml. NaOH, the mixt. filtered, and the ppt. washed with 500 ml. H2O and dried to give 15.9 g. 1,4-bis[3-(4-quinolyl)aminopropyl]piperazine, m. 210°(MeOH-H2O). Similarly prepd. are the following I: n, R, R1, R2, X, Y, m.p.; 2, H, H, H, MeO, H, 245° (HCONMe2); 2, H, H, H, H, SO2NMe2, 271° (HCONMe2); 2, H, H, H, H, CF3, 293° (HCONMe2); 3, Me, H, H, H, H, ∼100°; 3, Me, Ac, H, H, H, -(1); 3, Me, H, H, MeOH, 180° and 190°; 3, Me, Ac, H, MeO, H, -(2); 1, Me, H, Me, H, Cl, 264°; 2, H, H, H, Cl, H, 264° (BuOH); 1, Me, H, H, H, CF3, 240° (MeCOEt); 2, H, H, H, H, MeO, 200° (EtOH); 2, H, H, Me, H, MeO, 216° (EtOH); 3, Me, H, H, H, MeO, 218° (CH2Cl2); (1) bis(acid maleate) m. 155° (iso-PrOH), (2) bis(acid maleate) m. 155° The following II were also prepd.: n, R, R1, R2, m.p.; 1, Me, A(R = R1 = X = Y = H,Z =Cl), A(R = R1 = X = Z = H,Y = Cl), 208-10° (HCONMe2); 1, Me, A(R = R1 = X = Y= H, Z = Cl), A(R = R1 = X = Y = H,Z = MeO), 206-8° (HCONMe2); 1, Me, A(R1 = X = Y = H, R = 4-ClC6H4, Z = Cl), A(R = R1 = X = Y = H,Z = Cl, 230-2° (HCONMe2) The following III were prepd.: n, R, m, R1, R2, m.p.; 3, Me, 1, H, A(R = R1 = X = Y = H, Z= Cl), 190-1° and 213-15°; 2, H, 2, H, A(R = X = Y = H, R1 = Me, Z =Cl), 198° (PrOH); 3, Me, 2, H, A(R = R1 = X = Y = H,Z = Cl), 160-2°; 1, Me, 1, H, A(R = R1 = X = Y = H,Z = Cl), 178°; 1, Me, 1, Me, A(R1 = X = Z = H,R = Me, Y =AcNH), 330° (decompn.) (EtOH); 2, H, 2, H, A(R1 = X = Y = H,R = 4-ClC6H4,Z = Cl), 320-1° (HCONMe2); 2, H, 2, H, A(R = Y = Z = H, R1 = Me, X = Cl) 96° (iso-PrOH); 1, Me, 1, Me, A(R = R1 = X = Z = H, Y = Cl), 220° and 246-8°; 1, Me, 1, Me, A(R1 = X = Z = H, R = Me, Y = NH2), 305° (EtOH-H2O); 1, Me, 1, Me, A(R1 = X = Z = H, R = Me, Y = MeO, 244° (EtOH) Also prepd. were (m.p. given): 1,4-bis[2-(7-chloro-4-quinolylamino)propyl]hexahydro-1,4-diazepine, 169°; 1-[5-(7-chloro-4-quinolylamino)-2-pentyl]-4-[2-(7-chloro-4-quinolylamino)propyl] piperazine, 210-12°(HCONMe2); 1,4-bis[3-(7-chloro- 4-quinolylamino)propyl] hexahydro-1,4-diazepine, 186° (HCONMe2). The following were prepd. (m.p. and optical rotation given):L(+)-1,4-bis[2-(7-chloro-4-quinolylamino)propyl]piperazine, 250-1°, [α]23.5D 382° ± 1° (c 4, 50:50 MeOH-H2O); D(-)-1,4- bis[2-(7-chloro-4-quinolylamino)propyl] piperazine, 250-1°, [α]25D -382.5° ± 1° (c 4, 50:50 MeOH-H2O); DL-1,4-bis[2-(7-chloro-4-quinolylamino)propyl]piperazine (IV), 266-8°, -; meso-1,4-bis [2-(7-chloro-4-quinolylamino)propyl] piperazine (V), 270-1° (HCONMe2), -; equimol. mixt. of IV and V, 250-2°, -; 1,4-bis[2-(6-chloro-4-quinolylamino)propyl]piperazine-form A (VI-form A), 227° -; VI-form B, 110° and 245°, -. Also prepd. are the following intermediates of the general formula VII (R = H) (X, Y, Z, and m.p. given): OH, H, SO2NMe2, ∼288°; Cl, H,SO2NMe2, 170°; HO(CH2)3CHMeNH, H, H, 158° (EtOH); AcO(CH2)3CHMeNAc, H, H, -; HO(CH2)3CHMeNAc, H, H, -; MeSO3(CH2)3CHMeNAc, H, H, -; N-(5-piperazino-2-pentyl)acetamido, H, H, -; HO(CH2)3CHMeNH, MeO, H, -; AcO(CH2)3CHMeNAc, MeO, H, -; HO(CH2)3CHMeNAc, MeO, H, -; MeSO3(CH2)3CHMeNAc, MeO, H, -; N-(5-piperazino-2-pentyl)acetamido, MeO, H, -; Me(HOCH2)CH, H, Cl, 210°; Me(ClCH2)CH, H, Cl, 148-50°; Me(HOCH2)CH, Cl, H, 192°; Me(ClCH2)CH, Cl, H, 142°; Me(HOCH2)CH, H, MeO, 170°; Me(ClCH2)CH, H, MeO, 160°. Also prepd. were (m.p. given): VII (R = CO2Et, X = OH, Y = H, Z = SO2NMe2), ∼335°; VII (R = CO2H, X = OH, Y = H, Z = SO2HMe2), 310° (decompn.); 1,4-bis(2-oxopropyl)hexahydro-1,4-diazepine, -; 1,4-bis(2-oximinopropyl)hexahydro-1,4-diazepine, 180-1°; 1,4-bis(2-aminopropyl)hexahydro-1,4-diazepine, -; 1,4-bis(2-cyanoethyl)-hexahydro-1,4-diazepine, -. The following were prepd. (m.p. and optical rotation given): L(+)-4-(3-hydroxy-2-propylamino)-7-chloroquinoline, 223-4°, [α]24D 28.5° ± 2° (c 1, EtOH); L(+)-4-(3-chloro-2-propylamino)-7-chloroquinoline, 146-7°, [α]24D 103 ± 1° (c 2, EtOH); L(+)-4-(3-piperazino-2-propylamino)-7-chloroquinoline, 128-30°, [α]23D 139 ± 1° (c 2, EtOH); D(-)-4-(3-hydroxy-2-propylamino)-7-chloroquinoline, 223-4°, [α]25D – 31 ± 2° (c 1, EtOH); D(-)-4-(3-chloro-2-propylamino)-7-chloroquinoline, 147-8°, [α]24D -101 ± 1° (c 2, EtOH); D(-)-4-(3-piperazino-2-propylamino)-7-chloroquinoline, 131-2°, [α]23D -137 ± 1° (c 2, EtOH)
PATENT
FR CAM42 19631007.
Piperazines (I) are antiinflammatory and anthelmintic agents. A mixt. of 8.25 g. MeCH(NH2)CH2OH, 19.8 g. 4,6-dichloroquinoline, and 55 g. PhOH is heated to give 16.0 g. 6-chloro-4-[(3-hydroxy-2-propyl)-amino]quinoline (II), m. 192°. II (14.0 g.) is treated with a soln. of 10.6 g. SOCl2 in 40 ml. CHCl3 to give 12.5 g. 6-chloro-4-[(3-chloro-2-propyl)amino]quinoline (III), m. 142°. A mixt. of 13.2 g. 1-[2-(7-chloro-4-quinolylamino)propyl]piperazine, 11.0 g. III, 6.4 g. NaI, 2.3 g. anhyd. Et3N, and 200 ml. AcEt is refluxed 18 hrs., the solvent is distd. in vacuo, and the residue is taken up in 100 ml. MeOH. The mixt. is made alk. with 110 ml. NaOH (d. 1.33), poured into 1000 ml. H2O, and the ppt. that forms is filtered off, washed with H2O, and recrystd. in HCONMe2 to give 11.0 g. 1-[2-(7-chloro-4-quinolylamino)propyl]-4-[2-(6-chloro-4-quinolylamino)propyl]piperazine, m. 208-10°. Similarly prepd. are the following I (R, m, R1, n, R2, R3, R4, and m.p. given): H, 2, H, 2, H, MeO, H, 245°; H, 2, H, 2, H, H, SO2NMe2, 271°; H, 2, H, 2, H, H, CF3, 293°; Me, 3, Me, 3, H, MeO, H, 180° and 190°; Me, 3, H, 1, H, H, Cl, 190-1° and 213-15°; H, 2, H, 2, H, Cl, H, 264°; Me, 1, Me, 1, H, H, CF3, 240°; H, 2, H, 2, H, H, MeO, 200°; Me, 3, H, 2, H, H, Cl, 160-2°; Me, 1, H, 1, H, H, Cl, 178°; Me, 1, Me, 1, Me, AcNH, H, 330°; H, 2, H, 2, p-ClC6H4, H, Cl, 320-1°; Me, 1, Me, 1, H, Cl, H, 227° (form A); Me, 1, Me, 1, H, Cl, H, 110° and 245° (form B); H, 3, H, 3, H, H, Cl, 239-41°; Me, 1, Me, 1, Me, NH2, H, 305°; Me, 1, Me, 1, Me, MeO, H, 244°; Me, 3, Me, 3, Me, 3, H, H, MeO, 218°; H, 3, H, 3, H, H, Cl, 240-2°. Also prepd. are (m.p. given): 1,4-bis[2-(7-chloro-4-quinolylamino)propyl]hexahydrodiazepine, 169°; 2,5-dimethyl-1,4-bis[2-(7-chloro-4-quinolylamino)propyl)piperazine, 264°; 1-[5-(7-chloro-4-quinolylamino [-2-pentyl]-4-[2-(7-chloro -4-quinolylamino)propyl]piperazine, 210-12°; 2,5-dimethyl-1,4-bis[3-(7-methoxy-4-quinolylamino)propyl]piperazine, 216°; 1,4-bis[3-(3-methyl-7-chloro-4-quinolylamino)propyl] piperazine, 198°; 1,4-bis[3-(7-chloro-4-quinolylamino)propyl]hexahydrodiazepine, 186°; 1-[2(7-chloro-4-quinolylamino)propyl]-4-[2-(7-methoxy-4-quinolylamino)propyl]piperazine, 206-8°; 1,4-bis[3-(3-methyl-5-chloro-4- quinolylamino)propyl]piperazine, 96°; 1 – [2 -[2 -(p – chlorophenyl)- 7- chloro- 4- quinolylamino]propyl] -4 – [2 – (7 – chloro – 4-quinolylamino)propyl]piperazine, 230-2°; L(+) 1,4-bis[2-(7-chloro-4-quinolylamino)propyl]piperazine, 250-1°, [α]23.5D + 382° ± 1° (c 4, 50/50 MeOH-H2O); L(+)-7-chloro-4-(3-hydroxy-2-propylamino)quinoline, 223-4°, [α]24D 28.5° ± 2° (c 1, EtOH); L(+)-7-chloro-4-(3-chloro-2-propylamino)quinoline, 146-7°, [α]24D 103° + 1° (c 2, EtOH); L(+)-7-chloro-4-(3-piperazino-2-propylamino)quinoline 128-30°, [α]23D 139° ± 1° (c 2, EtOH); D(–)-1,4-bis[2-(7-chloro-4-quinolylamino)propyl]piperazine, 250-1°, [α]25D -382° ± 1° (c 4, 50:50 MeOH-H2O); meso- 1,4 – bis [2 – (7 – chloro – 4 – quinolylamino)propyl] piperazine, 270-1°.
Patent Information
BE 612207

| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2016045487-A1 | Compositions and methods for treating neuropathy | 2013-03-27 | |
| WO-2014160811-A1 | Compositions and methods for treating neuropathy | 2013-03-27 | |
| AU-2014234258-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | |
| AU-2014234258-B2 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | 2019-02-14 |
| CA-2907628-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-2976069-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | |
| EP-2976069-B1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | 2020-05-06 |
| US-2014322296-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | |
| US-2016045447-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | |
| US-9668979-B2 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | 2017-06-06 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2014147242-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | |
| AU-2009215107-A1 | Treatments for neuropathy | 2008-02-12 | |
| AU-2009215107-B2 | Treatments for neuropathy | 2008-02-12 | 2013-05-09 |
| AU-2013203934-A1 | Treatments for neuropathy | 2008-02-12 | |
| CA-2714676-A1 | Treatments for neuropathy | 2008-02-12 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| CA-2714676-C | Treatments for neuropathy | 2008-02-12 | 2015-04-14 |
| EP-2240177-A2 | Treatments for neuropathy | 2008-02-12 | |
| US-2009203735-A1 | Treatments for neuropathy | 2008-02-12 | |
| US-2011086878-A1 | Treatments for Neuropathy | 2008-02-12 | |
| US-2016058749-A1 | Treatments for neuropathy | 2008-02-12 |
////////////////Dichlorquinazine, BRN 0867697, Dichlorquinazine, EINECS 234-130-6, NSC 129790, RP 12278, UNII-HT3GAD2SCM, WR 3863
CC(C)(NC1=C2C=CC(=CC2=NC=C1)Cl)N3CCN(CC3)C(C)(C)NC4=C5C=CC(=CC5=NC=C4)Cl
WRONG
CC(CN1CCN(CC(C)Nc2ccnc3cc(Cl)ccc23)CC1)Nc4ccnc5cc(Cl)ccc45.CS(=O)(=O)O
AND
Clc1ccc2c(c1)nccc2NC(C)CN1CCN(CC(C)Nc2ccnc3cc(Cl)ccc32)CC1
CORRECT

NEW DRUG APPROVALS
ONETIME
$10.00
RP 12146


RP 12146, EX-A7910
cas 2732869-64-4
4-[[5-[(3R)-3-hydroxy-3-methyl-2-oxoindol-1-yl]pyridin-3-yl]methyl]-2H-phthalazin-1-one
| M.Wt | 398.422 | |
| Formula | C23H18N4O3 | |
RP12146 (RP12146) is a novel, selective, and potent small molecule inhibitor of PARP1/2 with IC50 of 0.6/0.5 nM, with several fold selectivity over other isoforms.
SCHEME
Ref
WO2021220120
Rhizen Pharmaceuticals AG
WO2022215034
Rhizen Pharmaceuticals AG; Incozen Therapeutics Pvt. Ltd.
RP-12146 is an oral poly (ADP-ribose) polymerase (PARP) inhibitor in phase I clinical development at Rhizen Pharmaceuticals for the treatment of adult patients with locally advanced or metastatic solid tumors.
Solid TumorExtensive-stage Small-cell Lung CancerLocally Advanced Breast CancerMetastatic Breast CancerPlatinum-sensitive Ovarian CancerPlatinum-Sensitive Fallopian Tube CarcinomaPlatinum-Sensitive Peritoneal Cancer
Poly(ADP-ribose) polymerase (PARP) defines a family of 17 enzymes that cleaves NAD+ to nicotinamide and ADP-ribose to form long and branched (ADP-ribose) polymers on glutamic acid residues of a number of target proteins, including PARP itself. The addition of negatively charged polymers profoundly alters the properties and functions of the acceptor proteins. Poly(ADP-ribosyl)ation is involved in the regulation of many cellular processes, such as DNA repair, gene transcription, cell cycle progression, cell death, chromatin functions and genomic stability. These functions have been mainly attributed to PARP-1 that is regarded as the best characterized member of the PARP family. However, the identification of novel genes encoding PARPs, together with the characterization of their structure and subcellular localization, have disclosed different roles for poly(ADP-ribosyl)ation in cells, including telomere replication and cellular transport.
Recently, poly(ADP-ribose) binding sites have been identified in many DNA damage checkpoint proteins, such as tumor suppressor p53, cyclin-dependent kinase inhibitor p21Cip1/waf1, DNA damage recognition factors (i.e., the nucleotide excision repair xeroderma pigmentosum group A complementing protein and the mismatch repair protein MSH6), base excision repair (BER) proteins (i.e. DNA ligase III, X-ray repair cross-complementing 1, and XRCC1), DNA-dependent protein kinase (DNA-PK), cell death and survival regulators (i.e.,
NF-kB, inducible nitric oxide synthase, and telomerase). These findings suggest that the different components of the PARP family might be involved in the DNA damage signal network, thus regulating protein-protein and protein-DNA interactions and, consequently, different types of cellular responses to genotoxic stress. In addition to its involvement in BER and single strand breaks (SSB) repair, PARP-1 appears to aid in the non-homologous end-joining (NHEJ) and homologous recombination (HR) pathways of double strand breaks (DSB) repair. See Lucio Tentori et al., Pharmacological Research, Vol. 45, No. 2, 2002, page 73-85.
PARP inhibition might be a useful therapeutic strategy not only for the treatment of BRCA mutations but also for the treatment of a wider range of tumors bearing a variety of deficiencies in the HR pathway. Further, the existing clinical data (e.g., Csaba Szabo et al., British Journal of Pharmacology (2018) 175: 192-222) also indicate that stroke, traumatic brain injury, circulatory shock and acute myocardial infarction are some of the indications where PARP activation has been demonstrated to contribute to tissue necrosis and inflammatory responses.
As of now, four PARP inhibitors, namely olaparib, talazoparib, niraparib, and rucaparib have been approved for human use by regulatory authorities around the world.
Patent literature related to PARP inhibitors includes International Publication Nos. WO 2000/42040, WO 2001/016136, WO 2002/036576, WO 2002/090334, WO2003/093261, WO 2003/106430, WO 2004/080976, WO 2004/087713, WO 2005/012305, WO 2005/012524, WO 2005/012305, WO 2005/012524, WO 2005/053662, W02006/033003, W02006/033007, WO 2006/033006, WO 2006/021801, WO 2006/067472, WO 2007/144637, WO 2007/144639, WO 2007/144652, WO 2008/047082, WO 2008/114114, WO 2009/050469, WO 2011/098971, WO 2015/108986, WO 2016/028689, WO 2016/165650, WO 2017/153958, WO 2017/191562, WO 2017/123156, WO 2017/140283, WO 2018/197463, WO 2018/038680 and WO 2018/108152, each of which is incorporated herein by reference in its entirety for all purposes.
There still remains an unmet need for new PARP inhibitors for the treatment of various diseases and disorders associated with cell proliferation, such as cancer.
PATENT
Illustration 1
CLIP
https://cancerres.aacrjournals.org/content/81/13_Supplement/1233
Abstract 1233: Preclinical profile of RP12146, a novel, selective, and potent small molecule inhibitor of PARP1/2
Srikant Viswanadha, Satyanarayana Eleswarapu, Kondababu Rasamsetti, Debnath Bhuniya, Gayatriswaroop Merikapudi, Sridhar Veeraraghavan and Swaroop VakkalankaProceedings: AACR Annual Meeting 2021; April 10-15, 2021 and May 17-21, 2021; Philadelphia, PA
Abstract
Background: Poly (ADP-ribose) polymerase (PARP) activity involves synthesis of Poly-ADP ribose (PAR) polymers that recruit host DNA repair proteins leading to correction of DNA damage and maintenance of cell viability. Upon combining with DNA damaging cytotoxic agents, PARP inhibitors have been reported to demonstrate chemo- and radio-potentiation albeit with incidences of myelosuppression. A need therefore exists for the development selective PARP1/2 inhibitors with a high therapeutic window to fully exploit their potential as a single agent or in combination with established therapy across various tumor types. Additionally, with the emerging concept of ‘synthetic lethality’, the applicability PARP inhibitors can be expanded to cancers beyond the well-defined BRCA defects. Herein, we describe the preclinical profile of RP12146, a novel and selective small molecule inhibitor of PARP1 and PARP2.
Methods: Enzymatic potency was evaluated using a PARP Chemiluminescent Activity Assay Kit (BPS biosciences). Cell growth was determined following incubation with RP12146 in BRCA1 mutant and wild-type cell lines across indications. Apoptosis was evaluated following incubation of cell lines with compound for 120 h, subsequent staining with Annexin-V-PE and 7-AAD, and analysis by flow cytometry. For cell cycle, cells were incubated with compound for 72 h, and stained with Propidium Iodide prior to analysis by flow cytometry. Expression of downstream PAR, PARP-trapping, phospho-γH2AX and cleaved PARP expression were determined in UWB1.289 (BRCA1 null) cells by Western blotting. Anti-tumor potential of RP12146 was tested in OVCAR-3 Xenograft model. Pharmacokinetic properties of the molecule were also evaluated. Results: RP12146 demonstrated equipotent inhibition of PARP1 (0.6 nM) and PARP2 (0.5 nM) with several fold selectivity over the other members of the PARP family. Compound caused a dose-dependent growth inhibition of both BRCA mutant and non-mutant cancer cell lines with GI50 in the range of 0.04 µM to 9.6 µM. Incubation of UWB1.289 cells with RP12146 caused a G2/M arrest with a corresponding dose-dependent increase in the percent of apoptotic cells. Expression of PAR was inhibited by 86% at 10 nM with a 2.3-fold increase in PARP-trapping observed at 100 nM in presence of RP12146. A four-fold increase in phospho-γH2AX and > 2-fold increase in cleaved PARP expression was observed at 3 µM of the compound. RP12146 exhibited anti-tumor potential with TGI of 28% as a single agent in OVCAR-3 xenograft model. Efficay was superior compared to Olaparib tested at an equivalent dose. Pharmacokinetic studies in rodents indicated high bioavailability with favorable plasma concentrations relevant for efficacy
Conclusions: Data demonstrate the therapeutic potential of RP12146 in BRCA mutant tumors. Testing in patients is planned in H1 2021.
Citation Format: Srikant Viswanadha, Satyanarayana Eleswarapu, Kondababu Rasamsetti, Debnath Bhuniya, Gayatriswaroop Merikapudi, Sridhar Veeraraghavan, Swaroop Vakkalanka. Preclinical profile of RP12146, a novel, selective, and potent small molecule inhibitor of PARP1/2 [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021 Apr 10-15 and May 17-21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract nr 1233.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
CLIP
Rhizen Pharmaceuticals AG Announces First Patient Dosing in a Phase I/Ib Study of Its Novel PARP Inhibitor (RP12146) in Patients With Advanced Solid Tumors
RHIZEN’S PARP INHIBITOR EFFORTS ARE PART OF A LARGER DDR PLATFORM THAT ALSO INCLUDES AN EARLY STAGE POLθ-DIRECTED PROGRAM; PLATFORM ENABLES PROPRIETARY IN-HOUSE COMBINATIONS
- Rhizen Pharma commences dosing in a phase I/Ib trial to evaluate its novel PARP inhibitor (RP12146) in patients with advanced cancers.
- Rhizen indicated that RP12146 has comparable preclinical activity vis-à-vis approved PARP inhibitors and shows improved preclinical safety that it expects will translate in the clinic.
- The two-part multi-center phase I/Ib study is being conducted in Europe and is designed to initially determine safety, tolerability and MTD/RP2D of RP12146 and to subsequently assess its anti-tumor activity in expansion cohorts with HRR mutation-enriched ES-SCLC, ovarian and breast cancer patients.
- RP12146 is part of a larger DDR platform at Rhizen that includes a preclinical-stage Polθ inhibitor program; the DDR platform enables novel, proprietary, in-house combinations
November 01, 2021 07:24 AM Eastern Daylight Time
BASEL, Switzerland–(BUSINESS WIRE)–Rhizen Pharmaceuticals AG (Rhizen), a Switzerland-based privately held, clinical-stage oncology & inflammation-focused biopharmaceutical company, announced today that it has commenced dosing in a multi-center, phase I/Ib trial to evaluate its novel poly (ADP-ribose) polymerase (PARP) inhibitor (RP12146) in patients with advanced solid tumors. This two-part multi-center phase I/Ib study is being conducted in Europe and has been designed to initially determine safety, tolerability, maximum tolerated dose (MTD), and/or recommended phase II dose (RP2D) of RP12146 and to subsequently assess its anti-tumor activity in expansion cohorts with HRR mutation-enriched ES-SCLC, ovarian and breast cancer patients.
“Our PARP program is foundational for our DDR platform efforts and will be the backbone for several novel proprietary combinations that we hope to bring into development going forward.”
Rhizen indicated that RP12146 has shown preclinical activity and efficacy comparable to the approved PARP inhibitor Olaparib, and shows improved safety as seen in the preclinical IND-enabling toxicology studies; an advantage that Rhizen hopes will translate in the clinical studies. Rhizen also announced that its PARP program is part of a larger DNA Damage Response (DDR) platform effort, which includes a preclinical-stage polymerase theta (Polθ) inhibitor program. Rhizen expects the platform to enable novel proprietary combinations of its PARP and Polθ assets given the mechanistic synergy and opportunity across PARP resistant/refractory settings.
“PARP inhibitors are a great success story in the DNA damage response area, but they are not without safety concerns that have limited realization of their full potential. Although our novel PARP inhibitor is competing in a crowded space, we expect its superior preclinical safety to translate into the clinic which will differentiate our program and allow us to extend its application beyond the current landscape of approved indications and combinations”, said Swaroop Vakkalanka, Founder & CEO of Rhizen Pharma. Swaroop also added that “Our PARP program is foundational for our DDR platform efforts and will be the backbone for several novel proprietary combinations that we hope to bring into development going forward.”
About Rhizen Pharmaceuticals AG.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel oncology & inflammation therapeutics. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways.
Rhizen has proven expertise in the PI3K modulator space with the discovery of our first PI3Kδ & CK1ε asset Umbralisib, that has been successfully developed & commercialized in MZL & FL by our licensing partner TG Therapeutics (TGTX) in USA. Beyond this, Rhizen has a deep oncology & inflammation pipeline spanning discovery to phase II clinical development stages.
Rhizen is headquartered in Basel, Switzerland.
REF
Safety, Pharmacokinetics and Anti-tumor Activity of RP12146, a PARP Inhibitor, in Patients With Locally Advanced or Metastatic Solid Tumors….https://clinicaltrials.gov/ct2/show/NCT05002868
//////////RP 12146, oral poly (ADP-ribose) polymerase (PARP) inhibitor, phase I, clinical development, INCOZEN, Rhizen Pharmaceuticals, adult patients, locally advanced, metastatic solid tumors, PARP, CANCER, EX-A7910, EX A7910

NEW DRUG APPROVALS
ONE TIME
$10.00
LERIGLITAZONE
LERIGLITAZONE
C19H20N2O4S,
| MW 372.4 |
Hydroxypioglitazone, CAS 146062-44-4
MIN 102, Hydroxy Pioglitazone (M-IV)лериглитазон [Russian] [INN]ليريغليتازون [Arabic] [INN]乐立格列酮 [Chinese] [INN]
5-[[4-[2-[5-(1-hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione
Hydroxypioglitazone is a member of the class of thiazolidenediones that is the hydroxy derivative of pioglitazone. It has a role as a human xenobiotic metabolite. It is a member of thiazolidinediones, a member of pyridines and an aromatic ether. It derives from a pioglitazone.
- OriginatorIDIBELL
- DeveloperMinoryx Therapeutics
- ClassNeuroprotectants; Phenyl ethers; Pyridines; Small molecules; Thiazolidinediones
- Mechanism of ActionPeroxisome proliferator-activated receptor gamma agonists
- Orphan Drug StatusYes – Adrenoleucodystrophy; Friedreich’s ataxia
- Phase II/IIIAdrenoleucodystrophy
- Phase IIFriedreich’s ataxia
- PreclinicalCNS disorders
- 23 Sep 2020Leriglitazone receives Rare Pediatric Disease designation from the US FDA for X-linked adrenoleukodystrophy before September 2020
- 23 Sep 2020Minoryx Therapeutics licenses leriglitazone to Sperogenix Therapeutics in China, Hong Kong and Macau for X-linked adrenoleukodystrophy (X-ALD)
- 14 Sep 2020Minoryx Therapeutics completes the phase II FRAMES trial in Friedreich’s ataxia (In adolescents, In adults) in Spain, Germany, France and Belgium (PO) (NCT03917225)
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Leriglitazone (Hydroxypioglitazone), a metabolite of pioglitazone. Leriglitazone (Hydroxypioglitazone) PioOH is a PPARγ agonist, stabilizes the PPARγ activation function-2 (AF-2) co-activator binding surface and enhances co-activator binding, affording slightly better transcriptional efficacy. Leriglitazone (Hydroxypioglitazone) binds to the PPARγ C-terminal ligand-binding domain (LBD) with Ki of 1.2 μM,induces transcriptional efficacy of the PPARγ (LBD) with EC50 of 680 nM.
Leriglitazone is under investigation in clinical trial NCT03917225 (A Clinical Study to Evaluate the Effect of MIN-102 on the Progression of Friedreich’s Ataxia in Male and Female Patients).
Treatment of X-Linked Adrenoleukodystrophy
PATENT
WO 9218501
WO 9322445
PAPER
Chemical & Pharmaceutical Bulletin (1995), 43(12), 2168-72
https://www.jstage.jst.go.jp/article/cpb1958/43/12/43_12_2168/_article
The metabolites of (±)-5-[p-[2-(5-ethyl-2-pyridyl)ethoxy]benzyl]-2, 4-thiazolidinedione (1, pioglitazone), which is a representative insulin-sensitizing agent, were synthesized to confirm their structures and for studies of their pharmacological properties. Of the metabolites identified, a compound hydroxylated at the 2-position of the ethoxy chain (3) and compounds oxygenated at the ethyl side chain attached to the pyridine ring (4, 5) were found to be active, although the potency was slightly lower than that of the parent compound.




PAPER
Journal of Medicinal Chemistry (1996), 39(26), 5053-5063.
https://pubs.acs.org/doi/10.1021/jm9605694
Pioglitazone (5-(4-(2-(5-ethyl-2-pyridyl)ethoxy)benzyl)-2,4-thiazolidinedione, 2) is a prototypical antidiabetic thiazolidinedione that had been evaluated for possible clinical development. Metabolites 6−9 have been identified after dosing of rats and dogs. Ketone 10 has not yet been identified as a metabolite but has been added to the list as a putative metabolite by analogy to alcohol 6 and ketone 7. We have developed improved syntheses of pioglitazone (2) metabolites 6−9 and the putative metabolite ketone 10. These entities have been compared in the KKAy mouse model of human type-II diabetes to pioglitazone (2). Ketone 10 has proven to be the most potent of these thiazolidinediones in this in vivo assay. When 6−10 were compared in vitro in the 3T3-L1 cell line to 2, for their ability to augment insulin-stimulated lipogenesis, 10 was again the most potent compound with 6, 7, and 9 roughly equivalent to 2. These data suggest that metabolites 6, 7, and 9 are likely to contribute to the pharmacological activity of pioglitazone (2), as had been previously reported for ciglitazone (1).
PATENT
WO 2015150476
Compound 5-[4-[2-(5-(1 -hydroxyethyl)-2-pyridinyl)ethoxy]benzyl]-2,4-thiazolidinedione of formula (1 ) can be prepared according to Scheme 1 (see e.g. J.Med.Chem. 1996, 39(26),5053).
Scheme 1
Scheme 2
Yet another method to prepare mixtures (c) – comprising compound (2) and (4) – and (d) – comprising compounds (3) and (5) – (scheme 3), includes the resolution of the racemic mixture VIII using the already described methods (chiral HPLC separation, enzymatic resolution, chiral resolution, etc) followed by double bond reduction in each of the enantiomers Villa and Vlllb.
Scheme 4
Compounds of formula (2), (3), (4) and (5) may be obtained from mixtures (c) and (d) (Scheme 45) by chiral HPLC separation. Alternatively, the desired enantiomerically pure compounds can be prepared by chiral synthetic procedures known to those skilled in the art (for example: asymmetric hydrogenolysis of the corresponding single isomer of compound VI).
HPLC Method
Column: Symmetry Shield RP-18, 5 μηη (4.6 x 250 mm); wavelength: 210 nm; flow: 1 mL/min; run time: 28 min; mobile phase-gradient: (t/%B): 0/10, 8/10, 12/60, 16/80, 20/80, 24/10, 28/10 [A: Water (potassium dihydrogen o-phosphate (pH~3)), B: Acetonitrile]
A mixture of compounds (2) and (4) (mixture (c)) and a mixture of compounds (3) and (5) (mixture (d)) were prepared according to Scheme 7.
Example 6: Preparation of diastereomeric mixtures D-1 and D-2 of M-IV:
Scheme 1 :
Ent-1 (VIII) Ent-2 (VIII)
Step 3 Step 3
MIV D-1 MIV D-2
Step 1 : Synthesis of compound VIII: HCI (48 ml, 2N) was added to a solution of compound VI (10 g, 0.024 mol) in methanol (200 ml) and the mixture was heated to reflux. After 4 h of reflux, the reaction mixture was cooled to r.t. and concentrated under reduced pressure to afford a yellow solid. The solid was suspended in water (70 ml) and neutralized using a saturated NaHC03 solution. The resulting pale yellow precipitate was collected by filtration and vacuum dried to afford compound VIII (7.5 g; 84% yield).
ES-MS [M+1]+: 371.0.
Step 2: Chiral prep. HPLC
Compound VIII (1 .0 g) was dissolved in a mixture containing equal volumes of acetonitrile, methanol and dichloromethane; injected (150 μΙ injections) in chiral prep-HPLC column (Chiralpak-IA 250 x 20 mm, 5 micron) and separated [Mobile phase- n-Hexane/0.05% Et3N in EtOH (50:50); flow Rate: 18ml/min; run time: 60 min]. The fractions containing the enantiomers Villa and Vlllb were separately concentrated under reduced pressure to minimum volume and the respective residues were diluted with EtOAc (100 ml), followed by water (50 ml). The resultant organic phases were
dried over anhydrous Na2S04 and concentrated to afford compounds Villa and Vlllb as off-white solids. Enantiomers Villa and Vlllb were isolated but the absolute configuration of each enantiomer has not been determined.
Compound Ent-1 (VIII): 250 mg (Yield: 50%); tR (Chiral HPLC) = 14.8 min; ES-MS [M+1]+: 371 .0; 1H NMR (400 MHz, DMSO-d6): δ 12.5 (br S, 1 H), 8.47 (s, 1 H), 7.71 (s, 1 H), 7.67 (dd, J = 8.0, 2.0 Hz, 1 H), 7.53 (d , J = 9.2 Hz, 2H), 7.31 (d, J = 7.6 Hz, 1 H), 7.08 (d, J = 8.8 Hz, 2H), 5.25 (d, J = 4.0 Hz, 1 H), 4.74-4.76 (m, 1 H), 4.43 (dd, J = 6.8, 6.4 Hz, 2H), 3.18 (t, J = 6.4 Hz, 2H), 1.34 (d, J = 6.4 Hz, 3H).
Compound Ent-2 (VIII): 237 mg (Yield: 47%); tR (Chiral HPLC) = 16.7 min; ES-MS [M+1]+: 371 .0; 1H NMR (400 MHz, DMSO-d6): δ 12.5 (br S, 1 H), 8.47 (s, 1 H), 7.71 (s, 1 H), 7.67 (dd, J = 8.0, 2.0 Hz, 1 H), 7.53 (d , J = 8.8 Hz, 2H), 7.31 (d, J = 8.0 Hz, 1 H), 7.08 (d, J = 9.2 Hz, 2H), 5.23 (d, J = 3.6Hz, 1 H), 4.75 (m, 1 H), 4.43 (dd, J = 6.8, 6.4 Hz, 2H), 3.18 (dd, J = 6.8, 6.4 Hz, 2H), 1 .34 (d, J = 6.4 Hz, 3H).
Synthesis of diastereomeric mixtures of M-IV
Synthesis of D-1 MIV
Step 3: A solution of NaBH4 (77 mg, 2.02 mmol) in 0.1 N NaOH (2 ml) was added slowly to a stirred solution of compound Ent-1 (VIII) (250 mg, 0.675 mmol), dimethylglyoxime (32 mg, 0.27 mmol) and CoCI2.6H20 (16 mg, 0.067 mmol) in a mixture of water (10 ml), THF (10 ml) and 1 M NaOH (0.5ml) solution at 10 °C, and the reaction mixture was stirred at r.t. for 1 h. After color of the reaction medium faded, additional quantity of NaBH4 (26 mg, 0.675 mmol) and CoCI2.6H20 (16 mg, 0.067 mmol) were added and stirring was continued at r.t. [additional quantities of CoC|2 and NaBH4 were added at 12 h intervals till the starting material was consumed, as monitored by LCMS]. After 90-96 h, the reaction mixture was neutralized with AcOH (pH~7); diluted with water (10 ml) and extracted in EtOAc (3 χ 50 ml). The combined organic extract was dried over anhydrous Na2S04 and concentrated to afford crude compound which was purified by flash column chromatography (Si02; 4% methanol in CH2CI2) to afford diastereomeric mixture of MIV D-1 (125 mg) as off-white solid.
Synthesis of D-2 MIV
Step 3: A solution of NaBH4 (72 mg, 1 .921 mmol) in 0.1 N NaOH (2 ml) was added slowly to a stirred solution of compound Ent-2 (VIII) (237 mg, 0.64 mmol), dimethylglyoxime (30 mg, 0.256 mmol) and CoCI2.6H20 (15 mg, 0.064 mmol) in a mixture of water (10 ml), THF (10 ml), and 1 M NaOH (0.5ml) solution at 10 °C, and the
reaction mixture was stirred at r.t. for 1 h. After color of the reaction medium faded, additional quantity of NaBH4 (24 mg, 0.64 mmol) and CoCI2.6H20 (15 mg, 0.064 mmol) were added and stirring was continued at r.t. [additional quantities of CoCI2.6H20 and NaBH4 were added at 12 h intervals till the starting material was consumed, as monitored by LCMS]. After 96 h, the reaction mixture was neutralized with AcOH (pH~7); diluted with water (10 ml) and extracted in EtOAc (3 χ 50 ml). The combined organic extract was dried over anhydrous Na2S04 and concentrated to afford crude compound, which was purified by flash column chromatography (Si02; 4% methanol in CH2CI2) to afford diastereomeric mixture of MIV D-2 (100 mg) as off-white solid.
MIV D-1 : yield: 125 mg (50%); tR (Chiral HPLC) = 17.8, 14.7 min; ES-MS [M+1]+: 373.0, 1H NMR (400 MHz, DMSO-d6): δ 12.00 (br s, NH), 8.46 (d, J = 2.0 Hz, 1 H), 7.67 (dd, J = 8.0, 2.4 Hz, 1 H), 7.30 (d, J = 8.0 Hz, 1 H), 7.13 (d, J = 8.8Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 5.27 (d, J = 4.0 Hz, 1 H), 4.88-4.85 (m, 1 H), 4.76-4.74 (m, 1 H), 4.30 (t, J = 6.8 Hz, 2H), 3.30 (m, 1 H), 3.14 (dd, J = 6.8, 6.4 Hz, 2H), 3.08-3.02 (m, 1 H), 1 .34 (d, J = 6.4 Hz, 3H).
MIV D-2: yield: 100 mg (42%); tR (Chiral HPLC) = 19.4, 16.5 min; ES-MS [M+1]+: 373.0; 1H NMR (400 MHz, DMSO-d6): δ 12.01 (br s, -NH), (d, J = 2.0 Hz, 1 H), 7.67 (dd, J = 8.0, 2.0 Hz, 1 H), 7.31 (d, J = 8.0 Hz, 1 H), 7.13 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.27 (d, J = 4.0 Hz, 1 H), 4.88-4.85 (m, 1 H), 4.76-4.74 (m, 1 H), 4.30 (dd, J = 6.8, 6.4 Hz, 2H), 3.30 (m, 1 H), 3.14 (dd, J = 6.8, 6.4 Hz, 2H), 3.08-3.02 (m, 1 H), 1.34 (d, J = 6.8 Hz, 3H).
Diastereomeric mixtures D-1 and D-2 of MIV correspond to mixtures (c) and (d) described above, but the specific diastereomers present in each diastereomeric mixture have not been assigned.
Example 7: in vitro ADME and toxicological characterization
Protocol: The assays performed include cytochrome P450 inhibition with the different isoforms, microsomal and hepatocyte stability, neurotoxicity in neural cells and hERG safety assays using a patch clamp electrophysiology measurement (FDA Draft Guidance for Industry. Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labelling Recommendations 2012, The European Medicines Agency (EMA) Guideline on the Investigation of Drug Interactions Adopted in 2012, Schroeder K et al. 2003 J Biomol Screen 8 (1 ); 50-64, Barter ZE et al. 2007
Curr Drug Metab 8 (1 ); 33-45, LeCluyse EL and Alexandre E 2010 Methods Mol Biol 640; 57-82). The results indicate a safe and favourable ADME profile for the compounds of the invention.
Example 8: The brain plasma ratios of Pioglitazone, MIV, Mill and Mil following oral dosing of a single administration of Pioglitazone at 4.5 mg/kg in male C57BL/6 mice.
The brain-plasma ratio was calculated based on levels of Pioglitazone, MIV, Mill and Mllin plasma and brain quantified at C max (maximal concentration) following oral dosing of a single administration of Pioglitazone at 4.5 mg/kg in male C57BL/6 mice. The percentage brain plasma ratio was 9, 13, 7 and 1 %, respectively, for Pioglitazone, Mil and Mill as shown in the Figure 4. Thus, active metabolites Mill and Mil crossed the BBB at much lower extent than Pioglitazone as it was predicted based on the physicochemical properties of the compounds (see Tablel ). In contrast, unexpectedly metabolite MIV crossed the BBB in a higher percentage than the parent compound Piolgitazone
The calculations of the both indexes (ClogP and QPIogBB) for Pioglitazone and its metabolites Mil and Mill are shown in Table 1 . For both indexes the 2 metabolites are lower than for pioglitazone, suggesting for Mil, and Mill a less favored penetration and distribution within CNS.
TABLE 1
PATENT
WO 2018116281
https://patents.google.com/patent/WO2018116281A1/enPioglitazone is a “dirty” drug which is converted to many metabolites in vivo. The metabolic pathway of pioglitazone after oral administration has been studied in several animal species and in humans and the metabolites have been described in the literature (see e.g. Sohda et al, Chem. Pharm. Bull., 1995, 43(12), 2168-2172) and Maeshiba et al, Arzneim.-Forsch/Drug Res, 1997, 47 (I), 29-35). At least six metabolites have been identified, named M-I to M-VI. Amongst these metabolites, M-II, M-III and M-IV show some pharmacological activity but are less active than Pioglitazone in diabetic preclinical models.
[0005] 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione has the following structure:

[0006] Tanis et al. (J. Med. Chem. 39(26 ):5053-5063 (1996)) describe the synthesis of 5-[[4-[2-[5-( 1 -hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione as follows:Scheme 1

[0007] Tanis et al. describe that the intermediate 14 was obtained in a 27% yield by reacting compound 13 in an aqueous 37% formaldehyde at 170°C for 6 hours. In this process, 5-[[4- [2-[5-( 1 -hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione (compound 6 in Scheme 1) was obtained in a 2.47% overall yield.[0008] WO 2015/150476 Al describes the use of 5-[[4-[2-[5-(l-hydroxyethyl)-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione, and its pharmaceutically acceptable salts, in the treatment of central nervous system (CNS) disorders. WO 2015/150476 Al describes that 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione was prepared according to the process of Tanis et al. (supra) where the intermediate corresponding to compound 14 of Tanis et al. was prepared similarly at 160°C for 5 hours providing a 17% yield. The overall yield of 5-[[4-[2-[5-(l-hydroxyethyl)-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione was about 1.5%.[0009] Due to the low yield of the intermediate 2-[5-(l-methoxymethoxy-ethyl)pyridine-2- yl]ethanol, the process step for preparing this intermediate is critical for the overall yield of the product, 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione. In addition, the prior art process to obtain compound 14 is difficult to scale because the reaction is carried out in a pressure vessel at a very high temperature and it is a very dirty reaction.[0010] Accordingly, the processes described in the art afford the product 5-[[4-[2-[5-(l- hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione only in a very low overall yield and, therefore, they are not suitable for large scale synthesis. In addition, the prior art process employs CH3OCH2CI, a known carcinogen, for protecting the hydroxyl group in the key intermediate. There is a need for an improved process for synthesizing 5- [[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione, and its pharmaceutically acceptable salts.Formula I illustrated by Scheme 2:Scheme 2 r
B


deprotectionoptional saltformation

I (HCI salt)[0255] In another embodiment, the disclosure provides a process for preparing the compound of Formula I illustrated by Scheme 3 : Scheme 3C
Br. e

step ‘< step b step c

step step g

[0256] In another embodiment in Scheme 3, step c, the order of mixing of the reagents can be as follows: 1. n-BuLi, 2. ethylene oxide, and 3. Cul. This order of mixing is described in Example 2.[0257] In the step a, 2,5-dibromopyridine (1) is reacted with i-PrMgCl in THF and then further with acetaldehyde to obtain compound 2. The reaction mixture is preferably filtered over Celite® after the reaction to remove most of the salts. In one embodiment, the addition of acetaldehyde is conducted at a temperature between -15°C and -10°C to control the exothermic reaction. [0258] In the step b, compound 2 is reacted with TBDMS-C1 in the presence of imidazole having DMF as a solvent. The crude product 3 is advantageously purified by a short plug filtration.[0259] In the step c, the hydroxyl protected compound 3 is reacted with ethylene oxide in the presence of n-BuLi and Cu(I)iodide while maintaining the reaction temperature, i.e., the reaction mixture temperature, below -20°C. In one embodiment, the reaction temperature is maintained below -55°C while adding n-BuLi and Cu(I)iodide into the reaction mixture. In another embodiment, the temperature of the reaction mixture is maintained below -55°C while adding n-BuLi, followed by ethylene oxide and then Cu(I)iodide into the reaction mixture. In another embodiment, the temperature of the reaction mixture is maintained below -55°C while adding n-BuLi into the reaction mixture, followed by ethylene oxide. In this embodiment, Cu(I)iodide is added then into the reaction mixture while the reaction mixture temperature is maintained below -20°C, and preferably below -55 °C. The reaction mixture is then allowed to slowly warm to room temperature after the addition of the reagents and stirred at room temperature, e.g., 20-25°C, overnight. This process is described in detail in Example 2. After the reaction, the complexed copper is advantageously removed by washing with 10% ammonia. The crude compound 4 can be purified by column chromatography to give >99% pure product with a yield of about 52%.[0260] The following examples are illustrative, but not limiting, of the methods of the present invention. Suitable modifications and adaptations of the variety of conditions and parameters normally encountered in clinical therapy and which are obvious to those skilled in the art in view of this disclosure are within the spirit and scope of the invention.ExamplesCOMPARATIVE EXAMPLE 1Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]- 2,4-thiazolidinedione (9a) according to the process described in WO 2015/150476 Al Scheme 4

8a 9a[0261] (a) Synthesis of l-(6-methyl-pyridin-3-yl)-ethanol (3a)[0262] LiHMDS (1.0 M in tetrahydrofuran, 463 ml, 0.463 mol) was added drop wise to a cooled solution of methyl 6-methylnicotinate (la) (20 g, 0.132 mol) and ethyl acetate (82 g, 0.927 mol) in dimethylformamide at -50°C; gradually raised the temperature to room temperature and stirred at the same temperature. After 1 h, the reaction mixture was cooled to 0°C; slowly diluted with 20% sulphuric acid and heated to reflux. After 4 h, the reaction mixture was cooled to room temperature, and further to 0°C and basified with potassium carbonate. The reaction medium was diluted with water and extracted in ethyl acetate (3×50 mL). Combined organic extract was dried over sodium sulphate and concentrated to afford crude l-(6-methylpyridin-3-yl)ethan-l-one (2a) (20.0 g) which was taken to the next step without any purification. ES-MS [M+l]+: 136.1.Sodium borohydride (2.3 g, 0.06 mol) was added in small portions over 30 min, to a solution of compound 2a (16.4 g, 0.121 mol) in ethanol (160 mL) at 0°C and the reaction mixture was stirred at same temperature. After 1 h, the reaction mixture was diluted with sodium bicarbonate solution (sat) (2×200 mL) and extracted with dichloromethane (2×500 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford a pale yellow oil, which was purified by flash column chromatography (5% methanol/dichloromethane) to afford compound 3a (17.0 g; 93% yield over 2 steps) as a pale yellow oil. ES-MS [M+l]+: 138.1. 1H NMR (400 MHz, CDC13): δ 8.35 (d, J = 2.0 Hz, 1H), 7.63 (dd, J = 8.0, 2.4 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H), 4.89 (q, J = 6.5 Hz, 1H), 3.30 (br s, 1H), 2.50 (s, 3H), 1.48 (d, J = 6.5 Hz, 3H).[0263] (b) Synthesis of 5-(l-methoxymethoxy-ethyl)-2-methyl-pyridine (4a):Compound 3a (15 g, 0.109 mol) was added, drop wise, to a cooled suspension of sodium hydride (6.56 g, 0.164 mol) in tetrahydrofurane (150 mL) and stirred at 0°C. After 30 min, chloromethyl methyl ether (13.2 g, 0.164 mol) was added drop wise while stirring and keeping the internal temperature around 0°C. After addition is over, the reaction mixture was stirred at the same temperature for 1 h. The reaction was quenched with ice cold water (80 mL) and extracted with ethyl acetate (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford an orange color oil, which was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 4a (10.0 g; 51% yield) as a pale yellow oil. ES-MS [M+l]+: 182.2. 1H NMR (400 MHz, CDC13): δ 8.45 (d, J = 2.0 Hz, 1H), 7.56 (dd, J = 8.0, 2.0 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 4.75 (q, J = 6.4 Hz, 1H), 4.57 (ABq, 2H), 3.36 (s, 3H), 2.53 (s, 3H), 1.48 (d, J = 6.6 Hz, 3H).[0264] (c) Synthesis of 2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethanol (5a):A mixture of compound 4a (7.0 g, 0.0386 mol) and 37% formaldehyde solution (5.8 g, 0.077 mol) was heated to 160°C in a sealed glass tube for 5 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure to afford a crude compound which was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 5 (1.2 g; 17% yield) as pale yellow oil. ES-MS [M+l]+: 212.1. 1H NMR (400 MHz, CDC13): δ 8.42 (d, J = 2.0 Hz, 1H), 7.65 (dd, J = 8.0, 2.4 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 4.72 (q, J = 6.6 Hz, 1H), 4.65 (t, J = 5.6 Hz, 1H), 4.52 (ABq, 2H), 3.73 (m, 2H), 3.24 (s, 3H), 2.86 (t, J = 7.2 Hz, 2H), 1.49 (d, J = 6.4 Hz, 3H).[0265] The total yield for compound 5a from compound la was 8% molar.[0266] (d) Synthesis of 4-{2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethoxy}- benzaldehyde (6a): Methanesulphonylchloride (1.19 g, 0.01 mol) was added, drop wise, to a cooled suspension of compound 5a (1.7 g, 0.008 mol) and triethylamine (1.79 ml, 0.013 mol) in dichloromefhane (20 mL) at 0°C and stirred at same temperature for 1 h. The reaction mixture was diluted with water (50 mL) and extracted with dichloromethane (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford 2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethyl methanesulfonate (2.04 g; 88% yield) as a yellow oil, which was taken to next step without purification. ES-MS [M+l]+: 290.[0267] 2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethyl methanesulfonate was added (2.3 g, 0.008 mol) to a stirred suspension of 4-hydroxybenzaldehyde (1.65 g, 0.0137 mol) and potassium carbonate (1.86 g, 0.0137 mol) in mixture of toluene (25 mL) and ethanol (25 mL); stirred at 85°C for 5 h. After consumption of the starting materials, the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate (2×100 mL). The combined organic extract was washed with water; dried over anhydrous sodium sulphate and concentrated to afford a crude dark yellow liquid. The crude was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 6a (1.5 g; 60% yield) as pale yellow liquid. ES-MS [M+l]+: 316.1.[0268] (e) Synthesis of 5-(4-{2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethoxy}- benzylidene)-thiazolidine-2,4-dione (7a):Piperidine (80 mg, 0.95 mmol) was added to a solution of compound 6a (0.6 g, 1.9 mmol) and thiazolidine-2,4-dione (0.22 g, 1.9 mmol) in ethanol (15 mL) and the mixture was heated to reflux overnight. After 15 h, the reaction mixture was cooled to room temperature and concentrated under reduced pressure to afford crude mixture, which was purified by flash column chromatography (2% methanol/dichloromethane) to afford compound 7 (500 mg; 64% yield) as a yellow solid. ES-MS [M+l]+: 415.1. 1H NMR (400 MHz, DMSO-d6): δ 12.25 (br s, 1H), 8.47 (d, J = 2.0 Hz, 1H), 7.70 (dd, J = 8.0, 2.0 Hz, 1H), 7.54 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 8.8 Hz, 2H), 4.73 (m, 1H), 4.60-4.40 (m, 4H), 4.22 (t, J = 6.2 Hz, 1H), 3.24 (s, 3H), 3.20 (t, J = 6.8 Hz, 2H), 1.41 (d, J = 6.0 Hz, 3H).[0269] (f) Synthesis of 5-(4-{2-[5-(l-hydroxy-ethyl)-pyridin-2-yl]-ethoxy}-benzyl)- thiazolidine-2,4-dione (9a): [0270] A solution of sodium borohydride (115 mg, 3.017 mmol) in 0.2N sodium hydroxide(1.2 mL) was added slowly to a stirred solution of compound 7 (0.5 g, 1.207 mmol), dimethylglyoxime (42 mg, 0.36 mmol) and C0CI2.6H2O (23 mg, 0.096 mmol) in a mixture of water (6 mL): tetrahydrofurane (6 mL) and 1M sodium hydroxide (1 mL) solution at 10°C and after addition, the reaction mixture was stirred at room temperature. After 1 h, the reaction color lightened and additional quantities of sodium borohydride (46 mg, 1.207 mmol) and C0CI2.6H2O (22 mg, 0.096 mmol) were added and stirring was continued at room temperature. After 12 h, the reaction was neutralized with acetic acid (pH~7); diluted with water (10 mL) and extracted in ethyl acetate (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford crude compound 8a, 5-(4- (2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethoxy)benzyl)thiazolidine-2,4-dione, (0.4 g) as pale yellow semi solid, which was taken to next step without purification. ES-MS [M+l]+: 417.5.[0271] 2N HC1 (2 mL) was added to a solution of compound 8a (0.4 g, 0.96 mmol) in methanol (20 ml) and the mixture was heated to reflux. After 4 h, the reaction mixture was cooled to room temperature and then concentrated under reduced pressure to afford a residue which was dissolved in water and the solution was neutralized using sodium bicarbonate solution (sat). The resulting white precipitate was collected by filtration to afford compound 9a (250 mg; 56% yield over 2 steps) as an off-white solid. ES-MS [M+l]+: 373.4. 1H NMR (400 MHz, DMSO-de): δ 12.00 (br s, -NH), 8.46 (d, J = 2.0 Hz, 1H), 7.66 (dd, J = 8.0, 2.4 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.13 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 5.25 (d, J = 4.4 Hz, 1H), 4.86 (m, 1H), 4.75 (m, 1H), 4.30 (t, J = 6.8 Hz, 2H), 3.30 (m, 1H), 3.14 (t, J = 6.4 Hz, 2H), 3.04 (m, 1H), 1.34 (d, J = 6.4 Hz, 3H).[0272] The overall yield of compound 9a was 1.5% molar.EXAMPLE 2Synthesis of 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol[0273] The synthesis of 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol was conducted according to the Scheme 5 using the reagents and solvents listed in Table 1 below: Scheme 5TBDMS-CI OTBDMS 1 . n-BuLi, <-55°C OTBDMSImidazole
DMF

[0274] The 1H-NMR spectra were recorded with Agilent MercuryPlus 300 NMR spectrometer.[0275] LC-MS data were obtained on an Agilent 1290 series with UV detector and HP 6130MSD mass detector using as column Waters XB ridge BEH XP (2.1 x 50 mm; 2.5 μιτι) and as eluent Ammonium acetate (10 mM); Water/ Methanol/ Acetonitrile.[0276] (a) l-(6-bromopyridin-3-yl)ethan-l-ol (2)[0277] A 20 L vessel was placed under nitrogen atmosphere and charged with tetrahydrofuran (5.5 L) and 2,5-dibromopyridine (1) (2000 g, 8.44 mol, 1.0 eq) (OxChem Corporation). The mixture was cooled to -10°C and isopropyl magnesium chloride (20% in THF, 6.02 L, 11.82 mol, 1.4 eq) (Rockwood Lithium) was added slowly over 1 h, keeping the reaction temperature below 5°C. After addition, the cooling bath was removed and the temperature was kept below 30°C (some additional cooling was needed to achieve this) and the reaction mixture was stirred overnight. After 16 h, a sample was taken; quenched with saturated aqueous ammonium chloride and extracted with methyl tert-buty\ ether (TBME). The TBME was evaporated under vacuum. 1H-NMR in deuterated chloroform showed complete conversion.[0278] The reaction mixture was cooled to -15°C and a solution of acetaldehyde (472 g,10.72 mol, 1.27 eq) (Acros) in tetrahydrofuran (200 mL) was added dropwise, while keeping temperature below -10°C. After the addition was complete, the cooling bath was removed and the temperature was allowed to rise to maximum of 5-8°C. After 1.5 h, a sample was taken and the reaction was quenched with aqueous ammonium chloride as described above. 1H-NMR showed the reaction was complete.[0279] Two batches were combined for work up.[0280] The reaction mixture was quenched by pouring the mixture into a solution of aqueous ammonium chloride (1 kg in 5 L water) and stirred for 15 min, filtered over Celite and rinsed thoroughly with toluene. The filtrate was transferred to a separation funnel and the obtained two layers system was separated. The aqueous layer was extracted with toluene (2 L). The combined organic layers were dried over sodium sulfate and filtered. Evaporation of the filtrate to dryness under vacuum yielded 3.49 kg (99%) of the desired crude material. XH NMR (300 MHz, CDC13): δ 8.30 (d, J = 2.5 Hz, 1H), 7.59 (dd, J = 8.0, 2.5 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 4,91 (q, J = 6.5 Hz, 1H), 1.49 (d, J = 6.5 Hz, 3H).[0281] (b) 2-bromo-5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridine (3)[0282] A 50 L reactor under nitrogen atmosphere was charged with compound 2 (10.0 kg, around 49.5 mol) and DMF (16 L). The mixture was cooled to 10°C and imidazole (6.74 kg, 99 mol, 2.0 eq) (Apollo Scientific Ltd.) was added portion wise within 30 min. The mixture was cooled to 0°C and TBDMS-Cl (7.46 kg, 49.5 mol, 1.0 eq) (Fluorochem) was added portion wise within 5 h, keeping the temperature below 3°C. The mixture reaction temperature was allowed to reach room temperature and stirred overnig ht. H NMR of a sample showed complete conversion.[0283] The reaction mixture was transferred to a 100 L extraction-vessel and the product was extracted with heptane (2×7.5 L, 10 L). The combined heptane-layers were washed with water (2×6 L, 3 L) to remove small amounts of DMF, dried over sodium sulfate and evaporated under vacuum to give crude compound 3 (15.5 kg, 49.0 mol) in a 99.0% yield. This crude product was purified by a short plug filtration, using 10 kg silica/heptane and eluted with heptane (approx. 50 L). The product-fractions were combined and evaporated under vacuum to give 12.0 kg of purified compound 3 (38 mol) as a brown oil in a 76.8% molar yield. (Average yield for 3 experiments was 78%). HPLC-MS: Rt= 2.6 min, M+l=316.1 and 318.1; 1H NMR (300 MHz, CDC13): δ 8.55 (d, J = 2.2 Hz, 1H), 7.54 (dd, J = 8.2, 2.2 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 4,86 (q, J = 6.5 Hz, 1H), 1.40 (d, J = 6.5 Hz, 3H), 0.88 (s, 9H), 0.02 (d, J = 26 Hz, 2x3H).[0284] (c) 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol (4)[0285] The ethylene oxide solution in diethylether was prepared in advance. Diethylether(1.2 L) in a 3 L three-necked flask was cooled at -65 °C and ethylene oxide (462.3 g, 10.5 mol, 1.06 eq) (Linde) was added and stirred at -70°C. Alternatively, the ethylene oxide solution can be made at about -20°C and then added gradually to the reaction mixture having a temperature at about -60°C. [0286] To a solution of 2-bromo-5-(l-((ieri-butyldimethylsilyl)oxy)ethyl)pyridine (3) (3.13 kg, 9.90 mol, 1.0 eq) in diethylether (7.5 L) cooled at -59°C, n-butyllithium (4 L, 10.0 mol, 2.5M in hexanes, 1.01 eq) (Aldrich Chemistry) was added while keeping temperature between -58°C and -62°C. After addition, the mixture was stirred for 1 h while keeping temperature between -60°C and -68°C. The upfront prepared ethylene oxide solution was added at once to the reaction mixture, while temperature was around -62°C. Subsequently, copper(I) iodide (962.3 g, 5.05 mol, 0.51 eq) (Acros Organics) was added in portions of 120 g, every 10 min, keeping the temperature between -61°C and -63°C. Stirring was continued for 1 h after addition keeping temperature between -61°C and -63°C. The cooling bath was removed and allowing the temperature to rise to about 15°C and further to 25 °C with a water bath overnight.[0287] Workup: The reaction-mixture was poured into a solution of 1 kg ammonium- chloride in 5 L water and stirred for 30 min, then the layers were separated. The organic layer was washed with aqueous ammonium hydroxide (10%, 2.5 L, 4x) to remove Cu-complex (blue color disappeared). The combined organic layers were dried over sodium sulfate and evaporated to give 3.12 kg (max. 9.90 mol) crude compound 4 as a brown oil. The crude compound was purified over 20 kg silica (heptane/EtOAc) by eluting with 80 L heptane/EtOAc, 20 L EtOAc, 25 L EtOAc/MeOH 95/5, 25 L EtOAc/MeOH 9/1 and 10 L EtOAc/MeOH 8/2, to give 1.47 kg of purified compound 4 (5.22 mol) as a brown oil (with tendency to solidify) in a 52.7% average molar yield (HPLC-purity of 99.5%). (Average yield over 12 experiments 52%). HPLC-MS: Rt= 2.3 min, M+l=282.1; 1H NMR (300 MHz, CDC13): δ 8.42 (d, J = 2.1 Hz, 1H), 7.61 (dd, J = 8.3, 2.1 Hz, 1H), 7.11 (d, J = 8.3 Hz, 1H), 4,88 (q, J = 7.0 Hz, 1H), 4.01 (t, J=6.0 Hz, 2 H), 3.00 (t, J=6.0 Hz, 2 H), 1.41 (d, J =7.0 Hz, 3H), 0.90 (s, 9H), 0.02 (d, J = 26 Hz, 2x3H).[0288] Another 2.5% of the product was isolated by re -purifying impure product fraction.The total yield of compound 4 from compound 1 was 39.6% molar.EXAMPLE 3Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]- 2,4-thiazolidinedione hydrochloride (9) 2. Sodium bisulfiteethanol/water mixture
3. Addition 10% aqueous sodium hydroxide solution

until pH 12

from step e dimethylglyoxime7step g step f

step h[0289] The 1H-NMR spectra were recorded with a 400 MHz Avance Bruker NMR spectrometer. LC-MS data were obtained on a Agilent Technologies 6130 Quadrapole LC/MS using as column Agilent XDB-C18 and as eluent 0.1% formic acid (aq) and 0.05% formic acid in acetonitrile.[0290] Steps d and e: Synthesis of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl] ethoxy] -benzaldehyde (6)[0291] To a well stirred solution of 5-[[[(l,l-dimethylethyl)dimethylsilyl]-oxy]ethyl]-2- pyridineethanol (4) (obtained as described in Example 2) (1.91 kg) in toluene (8.6 L) at 5°C were added sodium hydroxide (30% aqueous, 2.79 L) and tetrabutylammonium bromide (7.2 g). p-Toluenesulfonyl chloride (1.62 kg) was next added in portions during 5 min. After the addition, the reaction mixture was allowed to reach room temperature in 0.5 h and stirred at this temperature for 18 h. Water (7.3 L) was then added and the mixture was mixed well. Once the solids were dissolved, the layers were allowed to settle and the organic layer was separated. This organic phase was washed with water (5.7 L, 2x), followed by washing with a solution of sodium chloride (57 g) in water (5.7 L). The solvents were concentrated at reduced pressure to an amount of 2.5 kg of a brown oil (compound 5).[0292] To this well stirred brown oil were added subsequently ethanol (7.8 L), water (0.86L), 4-hydroxybenzaldehyde (0.88 kg) and potassium carbonate (1.17 kg) and then the mixture was heated at 75 °C for 18 h. Then, the solvent was evaporated while adding toluene (7.7 L) during 6 h and then the reaction mixture was allowed to cool. At 30°C, water (7.6 L) was added, stirred until all solids were dissolved and the mixture was cooled to room temperature. The layers were allowed to settle and separated. The organic layer was washed with water (7.6 L). The first aqueous extract was extracted with toluene (2.8 L) and this organic extract was used to also extract the aqueous washing. The organic extracts were combined and concentrated under vacuum to give 3.49 kg of a black oil (crude title compound 6).[0293] 1.73 kg of this black oil was dissolved in ethanol (0.74 L) and added to a well stirred solution of sodium bisulfite (1.36 kg) in a mixture of water (3.27 L) and ethanol (0.74 L). The container of the black oil was rinsed with ethanol (0.37 L) twice and these two rinses were also added to the bisulfite reaction mixture. After 75 min, heptane (5.3 L) was added, well mixed for 5 min, and the layers were allowed to settle and separated. To the organic layer was added a solution of sodium bisulfite (0.55 kg) in water (2.65 L), and ethanol (1.06 L). After stirring for 30 min, the layers were allowed to settle and separated. The two bisulfide aqueous extracts were combined and flasks rinsed with water (2.12 L). Next, toluene (4.5 L) and heptane (4.5 L) were added, the mixture was well stirred and the pH was adjusted to 12 using sodium hydroxide (10% aq) (temperature became 32°C). After stirring for an additional 5 min, the layers were allowed to settle and separated at 30°C. The aqueous layer was extracted with a mixture of toluene (1.5 L) and heptane (3.0 L). The layers were separated and the organic layers were combined. The combined organic layers were washed with water (5 L, 2x) and concentrated under vacuum to give the purified title compound 6. This procedure was repeated with another 1.73 kg of the black oil (crude title compound 6) to give in total 2.77 kg of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl]ethoxy]-benzaldehyde (6) as brown oil which contained 24% m/m of toluene according to 1H NMR (yield = 80%, calculated from compound 4 and corrected for residual toluene). [0294] 1H NMR (CDC13) δ: 0.00 (s, 3H), 0.09 (s, 3H), 0.91 (s, 9H), 1.44 (d, = 6 Hz, 3H),3.30 (t, = 7 Hz, 2H), 4.47 (t, = 7 Hz, 2H), 4.92 (q, = 6 Hz, 1H), 6.99 – 7.30 (m, 3H), 7.62- 7.67 (m, 1H), 7.80 – 7.85 (m, 2H), 8.5- 8.54 (m, 1H) and 9.88 (s, 1H).[0295] LC-MS; rt 7.5 min: ES: M+ 387, 386.[0296] Step f: Synthesis of (5Z)-5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methylene]-2,4-thiazolidinedione (7)[0297] A solution of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]- ethoxy]-benzaldehyde (6) (2.75 kg, containing 24% m/m of toluene) and piperidine (6.0 g) in methanol (3.16 L) was concentrated at 40°C under reduced pressure. The residue was dissolved in methanol (10.4 L) and 2,4-thiazolidinedione (759 g) and piperidine (230 g) were added. The mixture was heated at 47°C. After 25 h, the reaction mixture was allowed to cool to room temperature. The mixture was kept at pH 5-6 by adjusting it with acetic acid, if necessary. After a night at room temperature, water (1.56 L) was added and the suspension was stirred at room temperature for additional 2 h. The solids were isolated by filtration, washed with methanol (1 L, 2x) and dried under vacuum to give crude compound 7 (1.65 kg). The crude compound was mixed with methanol (10 L) and dichloromethane (8.6 L) and heated at 32°C until all solids dissolved. Then, the solvents were removed by distillation until the temperature of the mixture reached 34°C at a pressure of 333 mbar. Then, it was allowed to cool to room temperature overnight and stirred at 2°C for additional 2 h. The solids were isolated by filtration, washed with methanol (0.5 L, 2x) and dried under vacuum to give title compound 7 (1.50 kg) (yield = 61%).[0298] 1H NMR (CDCI3) δ 0.00 (s, 3H), 0.08 (s, 3H), 0.90 (s, 9H), 1.43 (d, = 6 Hz, 3H),3.32 (t, = 7 Hz, 2H), 4.48 (t, = 7 Hz, 2H), 4.92 (q, = 6 Hz, 1H), 6.95 – 7.00 (m, 2H), 7.24 – 7,28 (m, 1H), 7.38 – 7.42 (m, 2H), 7.67 (s, 1H), 7.69 – 7.73 (m, 1H) and 8.48 (d, = 3 Hz, 1H).[0299] LC-MS; rt 7.5 min: ES: M+ 487, 486, 485.[0300] Step g: Synthesis of 5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione (8)[0301] To a stirred suspension of (5Z)-5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]- ethyl]-2-pyridinyl]ethoxy]phenyl]methylene]-2,4-thiazolidinedione (7) (10 g) in THF (10 mL) and sodium hydroxide (IN aq, 21 mL) was added of a solution of cobalt chloride (26 mg) and of dimethylglyoxime (930 mg) in THF (2.3 mL) and water (1.0 mL). Then the suspension was put under a nitrogen atmosphere by applying the sequence of vacuum and flushing with nitrogen (4x). Thereafter, the suspension was heated to 30°C. Then, a stock solution of sodium borohydride was prepared by dissolving sodium borohydride (2.7 g) in a mixture of water (15.8 mL) and a solution of sodium hydroxide (1 N aq, 3.5 mL), which was put under a nitrogen atmosphere by applying a sequence of vacuum and flushing with nitrogen (3x). This was added to the suspension of compound 7 at a rate of 4.5 mL/h. Simultaneously, nitrogen gas-saturated acetic acid was added to the suspension at a rate of 0.7 mL/h to maintain a pH of 10.0-10.5. After 1 h 30 min the rate of addition of the sodium borohydride solution and acetic acid were both reduced by half. Next, 3 h 45 min after start of addition, the addition of sodium borohydride and acetic acid were stopped. The mixture was allowed to cool down to room temperature and acetone (2.5 mL) was added over a period of 1 minute. After stirring the reaction mixture for 15 min acetic acid was added until the pH was 5.5-6.0 (about 3 mL required). Next, a mixture of ethyl acetate/toluene (1/3 v/v, 30 mL) was added, well mixed and layers were allowed to settle. The aqueous layer was separated and washed with ethyl acetate/toluene (1/3 v/v, 10 mL). Both organic extracts were pooled and water (40 mL) was added, well mixed and layers were allowed to settle. The pH of the aqueous layer was adjusted to 5.5-6 using saturated sodium hydrogen carbonate solution (aq) and again mixed with the organic layer. Layers were allowed to settle and the organic layer was separated and concentrated under vacuum to give 11.09 g of yellow oil (crude mixture containing title compound 8 and its borane complex). Several batches were combined for work up.33.1 g of the crude mixture containing title compound 8 and its borane complex (not corrected for residual solvents) was dissolved in toluene (30 mL) and filtered. The filtrate was submitted to column chromatography (silica gel, gradient of toluene to toluene/ethyl acetate 1/1) to give 30.0 g of mixture of 5-[[4-[2-[5-[[[(l,l- dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione (8) and its borane complex as a slightly yellow oil (yield = 100% from compound 4, not corrected for residual solvents). [0303] 1H NMR (CDC13) δ: -0.03 – 0.10 (m, 6H), 0.87 – 0.93 (m, 9H), 1.42 (d, / = 6 Hz, 3H),3.05-3.71 (m, 4H), 4.30 – 4.51 (m, 3H), 4.87 – 4.94 (m, 1H), 6.82 – 6.88 (m, 2H), 7.10-7.92 (m, 5H), 8.49 (d, / = 3 Hz, 0.6H) and 8.72 (brs, 0.4H).[0304] LC-MS; rt 6.8 min: ES: M+ 489, 488, 487, M“ 487, 486, 485; rt 8.1 min: ES M“ 501,500, 499, 498, 485.[0305] Step h: Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]- methyl]-2,4-thiazolidinedione hydrochloride (9)[0306] To a stirred solution of the mixture of (5-[[4-[2-[5-[[[(l,l-dimethylethyl)- dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione and its borane complex (8) (5.17 g) in methanol (25.2 mL) at 22°C was added hydrochloric acid (30%, 2.75 mL) in about 5 min to give a temperature rise to 28°C. This solution was heated to 40 °C. Three hours after addition, the 11 g of volatiles were removed under reduced pressure. Then, acetonitrile (40.3 mL) was added and the mixture was heated at reflux for 0.5 h. Next, the suspension was allowed to cool down to room temperature and stirred for 1 h at room temperature. Solids were isolated by filtration, washed with a mixture of acetonitrile/water (20/1 v/v, 10 mL) and with acetonitrile (10 mL) and dried under vacuum at 40 °C to give 4.00 g of white solids (crude 9) (yield = 77%, not corrected for residual solvents).[0307] Purification of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione hydrochloride (9):[0308] The crude mixture of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]- methyl]-2,4-thiazolidinedione hydrochloride (3.95 g, crude 9) was dissolved in methanol/water (7/2 v/v, 80 mL) by heating it to 49°C. To this solution was added washed norit (obtained by heating a suspension of norit (6 g) in methanol/water (7/2 v/v, 90 mL) at 45°C for 1 h, then isolating the norit by filtration and washing it twice with methanol/water (7/2 v/v, 30 mL) and drying it under vacuum at 40°C). Equipment was rinsed with methanol/water (7/2 v/v, 18 mL). After 0.5 h of stirring at 46°C, the warm suspension was filtered to remove the norit and filter was washed twice with methanol/water (7/2 v/v, 18 mL). The filtrate was concentrated under vacuum at a bath temperature of 60°C to a mass of 11.8 g (1 v of compound and 2 v of water). To the suspension was added butanone (19.7 mL, 5 v) and the mixture was heated at a bath temperature of 95°C. Under distillation at a constant volume, butanone (95 mL) was added. Next, heating was stopped and the suspension was allowed to reach room temperature in about 0.5 h. Subsequently it was stirred for 0.75 h at room temperature. The solids were isolated by filtration, washed with a mixture of butanone/water (95/5 v/v, 18 mL) and butanone (18 mL) and dried under vacuum at 40°C to give 3.57 g of compound 9 as white solids (yield = 91%).[0309] 1H NMR (DMSO-de): δ 12.00 (br s, -NH), 8.71 (d, = 2.0 Hz, 1H), 8.45 (dd, = 8.3,1.7 Hz, 1H), 7.98 (d, = 8.3 Hz, 1H), 7.15 (d, = 8.7 Hz, 2H), 6.88 (d, = 8.7 Hz, 2H), 5.57 (s, OH), 4.95 (q, = 6.5 Hz, 1H), 4.86 (dd, = 8.9, 4.4 Hz, 1H), 4.40 (t, = 6.3 Hz, 2H), 3.49 (t, = 6.2 Hz, 2H), 3.29 (dd, = 14.2, 4.4 Hz, 1H), 3.06 (dd, = 14.2, 9.0 Hz, 1H), 1.41 (d, = 6.5 Hz, 3H).[0310] LC-MS; rt 3.5 min: ES: M+ 374, 373, M“ 372, 371.EXAMPLE 4Conditions tested in the preparation of compound 5 in the Step d[0311] The conditions described in Table 2 below were tested in the step d in the preparation of compound 5 from compound 4 providing a good yield of compound 5:Table 2Entry Reaction Conditions Amount of p-Ts-Cl / Eq1 Toluene/water/Bu4NBr/NaOH 1.052 1.083 1.074 1.07+0.035 1.076 Et3N / DCM 1.187 1.408 Pyridine / DCM 1.40 EXAMPLE 5Conditions tested in the preparation of compound 6 in the Step e[0312] The conditions described in Table 3 below were tested in the step e in the preparation of compound 6 from compound 5 providing a good yield of compound 6:Table 3

PATENT
Compound 1 is administered to the subject. The structure of 5-[[4-[2-[5-(l -hydroxy ethyljpyri din-2 – yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione is:
[0047] The present disclosure encompasses the use of stereoisomers of 5-[[4-[2-[5-(l- hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione. 5-[[4-[2-[5- (l-hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione has two asymmetric centers and thus four stereoisomers are possible as follows:
//////////LERIGLITAZONE, MIN 102 , лериглитазон , ليريغليتازون , 乐立格列酮 , Hydroxy Pioglitazone, M-IV, PHASE 2
CC(C1=CN=C(C=C1)CCOC2=CC=C(C=C2)CC3C(=O)NC(=O)S3)O

NEW DRUG APPROVALS
ONE TIME
$10.00
DOMPERIDONE
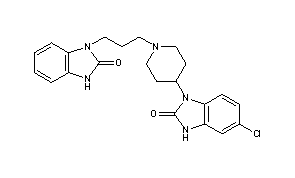
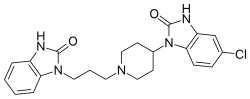
DOMPERIDONE
- Molecular FormulaC22H24ClN5O2
- Average mass425.911 Da
1H-Benzimidazol-2-ol, 5-chloro-1-[1-[3-(2-hydroxy-1H-benzimidazol-1-yl)propyl]-4-piperidinyl]-
260-968-7[EINECS]
2H-Benzimidazol-2-one, 5-chloro-1-[1-[3-(2,3-dihydro-2-oxo-1H-benzimidazol-1-yl)propyl]-4-piperidinyl]-1,3-dihydro-
4-(5-Chloro-2-oxo-1-benzimidazolinyl)-1-[3-(2-oxobenzimidazolinyl)propyl]piperidine
57808-66-9[RN]домперидон
دومبيريدون
多潘立酮
CAS Registry Number: 57808-66-9
CAS Name: 5-Chloro-1-[1-[3-(2,3-dihydro-2-oxo-1H-benzimidazol-1-yl)propyl]-4-piperidinyl]-1,3-dihydro-2H-benzimidazol-2-one
Additional Names: 5-chloro-1-[1-[3-(2-oxo-1-benzimidazolinyl)propyl]-4-piperidyl]-2-benzimidazolinone
Manufacturers’ Codes: R-33812
Trademarks: Euciton (Roux-Ocefa); Evoxin (Sterling Winthrop); Gastronorm (Janssen); Mod (Irbi); Motilium (Janssen); Nauzelin (Janssen); Peridon (Italchimici); Peridys (Robapharm)
Molecular Formula: C22H24ClN5O2
Molecular Weight: 425.91
Percent Composition: C 62.04%, H 5.68%, Cl 8.32%, N 16.44%, O 7.51%
Literature References: A novel in vitro dopamine antagonist with antinauseant properties.Prepn: J. Vandenberk et al.,DE2632870; eidem,US4066772 (1977, 1978 both to Janssen). Pharmacology: C. Ennis et al.,J. Pharm. Pharmacol.31, Suppl., 14P (1979). Gastrokinetic properties: J. M. Van Neuten et al.,Life Sci.23, 453 (1978). 3H-domperidone studies: M. P. Martres et al.,ibid. 1781; M. Baudry et al.,Arch. Pharmacol.308, 231 (1979). Clinical studies: A. J. Reyntjens et al.,Arzneim.-Forsch.28, 1194 (1978); D. B. Wilson, J. W. Dundee, Anaesthesia34, 765 (1979). Review of pharmacology, pharmacokinetics and therapeutic efficacy: R. N. Brogden et al.,Drugs24, 360-400 (1982).
Properties: Crystals from DMF/water, mp 242.5°.
Melting point: mp 242.5°
Therap-Cat: Antiemetic.
Keywords: Antiemetic; Dopamine Receptor Antagonist.
Domperidone, sold under the brand name Motilium among others, is a medication used as an antiemetic, gastric prokinetic agent, and galactagogue.[1][6][7] It may be taken by mouth or rectally, and is available as a tablet, orally disintegrating tablets,[8] suspension, and suppositories.[9] The drug is used to relieve nausea and vomiting; to increase the transit of food through the stomach (by increasing gastrointestinal peristalsis); and to promote lactation (breast milk production) by release of prolactin.[1][7]
It is a peripherally selective dopamine D2 receptor antagonist and was developed by Janssen Pharmaceutica.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
syn
Prepn: J. Vandenberk et al., DE 2632870; eidem, US 4066772 (1977, 1978 both to Janssen).

syn
Ruben Vardanyan, in Piperidine-Based Drug Discovery, 2017
Domperidone (5565)
Domperidone (7.1.6) (Motilium), a peripherally selective D2-like receptor antagonist, regulates the motility of the gastric and small intestinal smooth muscles and has been shown to have some effects on the motor function of the esophagus. It effectively prevents bile reflux but does not affect gastric secretion. As a result of the blockade of dopamine receptors in the chemoreceptor trigger zone it also has an antiemetic activity. Domperiodone provided relief of such symptoms as anorexia, nausea, vomiting, abdominal pain, early satiety, bloating, and distension in patients with symptoms of diabetic gastropathy. It also provided short-term relief of symptoms in patients with dyspepsia or gastroesophageal reflux, prevented nausea and vomiting associated with emetogenic chemotherapy, and prevented the gastrointestinal and emetic adverse effects of antiparkinsonian drugs. Because domperidone does not readily cross the blood brain barrier and does not inhibit dopamine receptors in the brain, reports of adverse effects on the CNS, such as dystonic reactions, are rare [52–61]. Domperidone is widely used in many countries and can now be officially prescribed to patients in the United States. There are very few treatment options currently available for patients with gastrointestinal motility disorders, especially for patients with gastroparesis. Domperidone has been successfully used in the United States and in many countries as a second-line treatment option for the treatment of gastroparesis.
Synthesis of domperidone (7.1.6) started with arylation of ethyl 4-aminopiperidine-1-carboxylate (7.1.28) with 1,4-dichloro-2-nitrobenzene (7.1.29) on heating at 150°C in cyclohexanol in the presence of sodium carbonate and potassium iodide (in a later disclosure in toluene in presence of sodium carbonate [62]) to give compound (7.1.30), which on reflux in 48% hydrobromic acid solution yielded N-(4-chloro-2-nitrophenyl)piperidin-4-amine (7.1.31). The obtained product was alkylated with 1-(3-chloropropyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (7.1.32) on reflux in MBIK in the presence of sodium carbonate and potassium iodide to give compound (7.1.33). The ring closure could be effected by heating o-phenylene diamine (7.1.33) with an appropriate cyclizing agent, such as phosgene, urea, potassium isocyanate [63], and the like. In this patent potassium isocyanate dissolved in water was carefully added to a solution of compound (7.1.34) in 10 N hydrochloric acid solution (exothermic reaction) to give desired domperidone (7.1.6) [64,65] (Scheme 7.4).

Medical uses
Nausea and vomiting
There is some evidence that domperidone has antiemetic activity.[10] It is recommended by the Canadian Headache Society for treatment of nausea associated with acute migraine.[11]
Gastroparesis
Gastroparesis is a medical condition characterised by delayed emptying of the stomach when there is no mechanical gastric outlet obstruction. Its cause is most commonly idiopathic, a diabetic complication or a result of abdominal surgery. The condition causes nausea, vomiting, fullness after eating, early satiety (feeling full before the meal is finished), abdominal pain and bloating.
Domperidone may be useful in diabetic and idiopathic gastroparesis.[12][13]
However, increased rate of gastric emptying induced by drugs like domperidone does not always correlate (equate) well with relief of symptoms.[14]
Parkinson’s disease
Parkinson’s disease is a chronic neurological condition where a decrease in dopamine in the brain leads to rigidity (stiffness of movement), tremor and other symptoms and signs. Poor gastrointestinal function, nausea and vomiting is a major problem for people with Parkinson’s disease because most medications used to treat Parkinson’s disease are given by mouth. These medications, such as levodopa, can cause nausea as a side effect. Furthermore, anti-nausea drugs, such as metoclopramide, which do cross the blood–brain barrier may worsen the extra-pyramidal symptoms of Parkinson’s disease.
Domperidone can be used to relieve gastrointestinal symptoms in Parkinson’s disease; it blocks peripheral D2 receptors but does not cross the blood–brain barrier in normal doses (the barrier between the blood circulation of the brain and the rest of the body) so has no effect on the extrapyramidal symptoms of the disease.[15]
Functional dyspepsia
Domperidone may be used in functional dyspepsia in both adults and children.[16][17]
Lactation
The hormone prolactin stimulates lactation (production of breast milk). Dopamine, released by the hypothalamus stops the release of prolactin from the pituitary gland. Domperidone, by acting as an anti-dopaminergic agent, results in increased prolactin secretion, and thus promotes lactation (that is, it is a galactogogue). Domperidone moderately increases the volume of expressed breast milk in mothers of preterm babies where breast milk expression was inadequate, and appears to be safe for short-term use for this purpose.[18][19][20] In the United States, domperidone is not approved for this or any other use.[21][22]
A study called the EMPOWER trial was designed to assess the effectiveness and safety of domperidone in assisting mothers of preterm babies to supply breast milk for their infants.[23] The study randomized 90 mothers of preterm babies to receive either domperidone 10 mg orally three times daily for 28 days (Group A) or placebo 10 mg orally three times daily for 14 days followed by domperidone 10 mg orally three times daily for 14 days (Group B). Mean milk volumes at the beginning of the intervention were similar between the 2 groups. After the first 14 days, 78% of mothers receiving domperidone (Group A) achieved a 50% increase in milk volume, while 58% of mothers receiving placebo (Group B) achieved a 50% increase in milk volume.[24]
To induce lactation, domperidone is used at a dosage of 10 to 20 mg 3 or 4 times per day by mouth.[25] Effects may be seen within 24 hours or may not be seen for 3 or 4 days.[25] The maximum effect occurs after 2 or 3 weeks of treatment, and the treatment period generally lasts for 3 to 8 weeks.[25] A 2012 review shows that no studies support prophylactic use of a galactagogue medication at any stage of pregnancy, including domperidone.[26]
Reflux in children
Domperidone has been found effective in the treatment of reflux in children.[27] However some specialists consider its risks prohibitory of the treatment of infantile reflux.[28]
Contraindications
- QT-prolonging drugs like amiodarone[29]
Side effects
Side effects associated with domperidone include dry mouth, abdominal cramps, diarrhea, nausea, rash, itching, hives, and hyperprolactinemia (the symptoms of which may include breast enlargement, galactorrhea, breast pain/tenderness, gynecomastia, hypogonadism, and menstrual irregularities).[25] Due to blockade of D2 receptors in the central nervous system, D2 receptor antagonists like metoclopramide can also produce a variety of additional side effects including drowsiness, akathisia, restlessness, insomnia, lassitude, fatigue, extrapyramidal symptoms, dystonia, Parkinsonian symptoms, tardive dyskinesia, and depression.[1][7] However, this is not the case with domperidone, because, unlike other D2 receptor antagonists, it minimally crosses the blood-brain-barrier, and for this reason, is rarely associated with such side effects.[1][7]
Excess prolactin levels
Due to D2 receptor blockade, domperidone causes hyperprolactinemia.[30] Hyperprolactinemia can suppress the secretion of gonadotropin-releasing hormone (GnRH) from the hypothalamus, in turn suppressing the secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) and resulting in hypogonadism (low sex hormone (e.g., testosterone, estradiol) levels).[31] As such, male patients may experience low libido, erectile dysfunction, and impaired spermatogenesis.[31] Also in accordance with hyperprolactinemia, 10–15% of female patients have been reported to experience mammoplasia (breast enlargement), mastodynia (breast pain/tenderness), galactorrhea (inappropriate or excessive milk production/secretion), and amenorrhea (cessation of menstrual cycles) with domperidone treatment.[30] Gynecomastia has been reported in males treated with domperidone,[32] and galactorrhea could occur in males as well.[31]
Rare reactions
Cardiac reactions
Domperidone use is associated with an increased risk of sudden cardiac death (by 70%)[33] most likely through its prolonging effect of the cardiac QT interval and ventricular arrhythmias.[34][35] The cause is thought to be blockade of hERG voltage-gated potassium channels.[36][37] The risks are dose-dependent, and appear to be greatest with high/very high doses via intravenous administration and in the elderly, as well as with drugs that interact with domperidone and increase its circulating concentrations (namely CYP3A4 inhibitors).[38][39] Conflicting reports exist, however.[40] In neonates and infants, QT prolongation is controversial and uncertain.[41][42]
UK drug regulatory authorities (MHRA) have issued the following restriction on domperidone in 2014 due to increased risk of adverse cardiac effects:
Domperidone (Motilium) is associated with a small increased risk of serious cardiac side effects. Its use is now restricted to the relief of nausea and vomiting and the dosage and duration of use have been reduced. It should no longer be used for the treatment of bloating and heartburn. Domperidone is now contraindicated in those with underlying cardiac conditions and other risk factors. Patients with these conditions and patients receiving long-term treatment with domperidone should be reassessed at a routine appointment, in light of the new advice.
However, a 2015 Australian review concluded the following:[39]
Based on the results of the two TQT (the regulatory agency gold standard for assessment of QT prolongation) domperidone does not appear to be strongly associated with QT prolongation at oral doses of 20 mg QID in healthy volunteers. Further, there are limited case reports supporting an association with cardiac dysfunction, and the frequently cited case-control studies have significant flaws. While there remains an ill-defined risk at higher systemic concentrations, especially in patients with a higher baseline risk of QT prolongation, our review does not support the view that domperidone presents intolerable risk.
Possible central toxicity in infants
In Britain a legal case involved the death of two children of a mother whose three children had all had hypernatraemia. She was charged with poisoning the children with salt. One of the children, who was born at 28 weeks gestation with respiratory complications and had a fundoplication for gastroesophageal reflux and failure to thrive was prescribed domperidone. An advocate for the mother suggested the child may have suffered neuroleptic malignant syndrome as a side effect of domperidone due to the drug crossing the child’s immature blood-brain-barrier.[43]
Interactions
In healthy volunteers, ketoconazole increased the Cmax and AUC concentrations of domperidone by 3- to 10-fold.[44] This was accompanied by a QT interval prolongation of about 10–20 milliseconds when domperidone 10 mg four times daily and ketoconazole 200 mg twice daily were administered, whereas domperidone by itself at the dosage assessed produced no such effect.[44] As such, domperidone with ketoconazole or other CYP3A4 inhibitors is a potentially dangerous combination.[44]
Pharmacology
Pharmacodynamics
Domperidone is a peripherally selective dopamine D2 and D3 receptor antagonist.[7] It has no clinically significant interaction with the D1 receptor, unlike metoclopramide.[7] The medication provides relief from nausea by blocking D receptors.[10] It blocks dopamine receptors in the anterior pituitary gland increasing release of prolactin which in turn increases lactation.[45][46] Domperidone may be more useful in some patients and cause harm in others by way of the genetics of the person, such as polymorphisms in the drug transporter gene ABCB1 (which encodes P-glycoprotein), the voltage-gated potassium channel KCNH2 gene (hERG/Kv11.1), and the α1D—adrenoceptor ADRA1D gene.[47]
Effects on prolactin levels
A single 20 mg oral dose of domperidone has been found to increase mean serum prolactin levels (measured 90 minutes post-administration) in non-lactating women from 8.1 ng/mL to 110.9 ng/mL (a 13.7-fold increase).[7][48][49][50] This was similar to the increase in prolactin levels produced by a single 20 mg oral dose of metoclopramide (7.4 ng/mL to 124.1 ng/mL; 16.7-fold increase).[49][50] After two weeks of chronic administration (30 mg/day in both cases), the increase in prolactin levels produced by domperidone was reduced (53.2 ng/mL; 6.6-fold above baseline), but the increase in prolactin levels produced by metoclopramide, conversely, was heightened (179.6 ng/mL; 24.3-fold above baseline).[7][50] This indicates that acute and chronic administration of both domperidone and metoclopramide is effective in increasing prolactin levels, but that there are differential effects on the secretion of prolactin with chronic treatment.[49][50] The mechanism of the difference is unknown.[50] The increase in prolactin levels observed with the two drugs was, as expected, much greater in women than in men.[49][50] This appears to be due to the higher estrogen levels in women, as estrogen stimulates prolactin secretion.[51]
For comparison, normal prolactin levels in women are less than 20 ng/mL, prolactin levels peak at 100 to 300 ng/mL at parturition in pregnant women, and in lactating women, prolactin levels have been found to be 90 ng/mL at 10 days postpartum and 44 ng/mL at 180 days postpartum.[52][53]
Pharmacokinetics
With oral administration, domperidone is extensively metabolized in the liver (almost exclusively by CYP3A4/5, though minor contributions by CYP1A2, CYP2D6, and CYP2C8 have also been reported)[54] and in the intestines.[5] Due to the marked first-pass effect via this route, the oral bioavailability of domperidone is low (13–17%);[1] conversely, its bioavailability is high via intramuscular injection (90%).[1] The terminal half-life of domperidone is 7.5 hours in healthy individuals, but can be prolonged to 20 hours in people with severe renal dysfunction.[1] All of the metabolites of domperidone are inactive as D2 receptor ligands.[1][5] The drug is a substrate for the P-glycoprotein (ABCB1) transporter, and animal studies suggest that this is the reason for the low central nervous system penetration of domperidone.[55]
Chemistry
Domperidone is a benzimidazole derivative and is structurally related to butyrophenone neuroleptics like haloperidol.[56][57]
History
- 1974 – Domperidone synthesized at Janssen Pharmaceutica[58] following the research on antipsychotic drugs.[59] Janssen pharmacologists discovered that some of antipsychotic drugs had a significant effect on dopamine receptors in the central chemoreceptor trigger zone that regulated vomiting and started searching for a dopamine antagonist that would not pass the blood–brain barrier, thereby being free of the extrapyramidal side effects that were associated with drugs of this type.[59] This led to the discovery of domperidone as a strong anti-emetic with minimal central effects.[59][60]
- 1978 – On 3 January 1978 Domperidone was patented in the United States under patent US4066772 A. The application has been filed on 17 May 1976. Jan Vandenberk, Ludo E. J. Kennis, Marcel J. M. C. Van der Aa and others has been cited as the inventors.
- 1979 – Domperidone marketed under trade name “Motilium” in Switzerland and (Western) Germany.[61]
- 1999 – Domperidone was introduced in the forms of orally disintegrating tablets (based on Zydis technology).[62]
- Janssen Pharmaceutical has brought domperidone before the United States Federal Drug Administration (FDA) several times, including in the 1990s.
- 2014 – In April 2014 Co-ordination Group for Mutual Recognition and Decentralised Procedures – Human (CMDh) published official press-release suggesting to restrict the use of domperidone-containing medicines. It also approved earlier published suggestions by Pharmacovigilance Risk Assessment Committee (PRAC) to use domperidone only for curing nausea and vomiting and reduce maximum daily dosage to 10 mg.[9]
Society and culture
Generic names
Domperidone is the generic name of the drug and its INN, USAN, BAN, and JAN.[63][6][64]
Regulatory approval
It was reported in 2007 that domperidone is available in 58 countries, including Canada,[65] but the uses or indications of domperidone vary between nations. In Italy it is used in the treatment of gastroesophageal reflux disease and in Canada, the drug is indicated in upper gastrointestinal motility disorders and to prevent gastrointestinal symptoms associated with the use of dopamine agonist antiparkinsonian agents.[66] In the United Kingdom, domperidone is only indicated for the treatment of nausea and vomiting and the treatment duration is usually limited to 1 week.
In the United States, domperidone is not currently a legally marketed human drug and it is not approved for sale in the U.S. On 7 June 2004, FDA issued a public warning that distributing any domperidone-containing products is illegal.[67]
It is available over-the-counter to treat gastroesophageal reflux and functional dyspepsia in many countries, such as Ireland, the Netherlands, Italy, South Africa, Mexico, Chile, and China.[68]
Domperidone is not generally approved for use in the United States. There is an exception for use in people with treatment-refractory gastrointestinal symptoms under an FDA Investigational New Drug application.[1]
Formulations
| showFormulations |
|---|
Research
Domperidone has been studied as a potential hormonal contraceptive to prevent pregnancy in women.[72]
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s Reddymasu, Savio C.; Soykan, Irfan; McCallum, Richard W. (2007). “Domperidone: Review of Pharmacology and Clinical Applications in Gastroenterology”. The American Journal of Gastroenterology. 102 (9): 2036–2045. ISSN 0002-9270. PMID 17488253.
- ^ “БРЮЛІУМ ЛІНГВАТАБС” [BRULIUM LINGUATABS]. Нормативно-директивні документи МОЗ України (in Ukrainian). 18 March 2014. Retrieved 29 May 2015.
- ^ “Domperidone”. Archived from the original on 22 May 2013. Retrieved 30 June 2013.
- ^ Jump up to:a b Suzanne Rose (October 2004). Gastrointestinal and Hepatobiliary Pathophysiology. Hayes Barton Press. pp. 523–. ISBN 978-1-59377-181-2.
- ^ Jump up to:a b c d Simard, C.; Michaud, V.; Gibbs, B.; Massé, R.; Lessard, É; Turgeon, J. (2008). “Identification of the cytochrome P450 enzymes involved in the metabolism of domperidone”. Xenobiotica. 34 (11–12): 1013–1023. doi:10.1080/00498250400015301. ISSN 0049-8254. PMID 15801545. S2CID 27426219.
- ^ Jump up to:a b Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 366–. ISBN 978-3-88763-075-1.
- ^ Jump up to:a b c d e f g h Barone JA (1999). “Domperidone: a peripherally acting dopamine2-receptor antagonist”. The Annals of Pharmacotherapy. 33 (4): 429–40. doi:10.1345/aph.18003. PMID 10332535. S2CID 39279569.
- ^ “MOTILIUM INSTANTS PL 13249/0028” (PDF). Medicines and Healthcare products Regulatory Agency. 23 February 2010. Archived from the original (PDF) on 31 October 2014. Retrieved 2014-10-31.
- ^ Jump up to:a b “CMDh confirms recommendations on restricting use of domperidone-containing medicines: European Commission to take final legal decision”. European Medicines Agency. 25 April 2014. Archived from the original on 30 January 2016. Retrieved 2014-10-31.
- ^ Jump up to:a b Reddymasu SC, Soykan I, McCallum RW. (2007). “Domperidone: review of pharmacology and clinical applications in gastroenterology”. Am J Gastroenterol. 102 (9): 2036–45. PMID 17488253.
- ^ Worthington I, Pringsheim T, Gawel MJ, Gladstone J, Cooper P, Dilli E, Aube M, Leroux E, Becker WJ (September 2013). “Canadian Headache Society Guideline: acute drug therapy for migraine headache”. The Canadian Journal of Neurological Sciences. 40(5 Suppl 3): S1–S80. doi:10.1017/S0317167100118943. PMID 23968886.
- ^ Stevens JE, Jones KL, Rayner CK, Horowitz M (June 2013). “Pathophysiology and pharmacotherapy of gastroparesis: current and future perspectives”. Expert Opinion on Pharmacotherapy. 14(9): 1171–86. doi:10.1517/14656566.2013.795948. PMID 23663133. S2CID 23526883.
- ^ Silvers D, Kipnes M, Broadstone V, Patterson D, Quigley EM, McCallum R, Leidy NK, Farup C, Liu Y, Joslyn A (1998). “Domperidone in the management of symptoms of diabetic gastroparesis: efficacy, tolerability, and quality-of-life outcomes in a multicenter controlled trial. DOM-USA-5 Study Group”. Clinical Therapeutics. 20 (3): 438–53. doi:10.1016/S0149-2918(98)80054-4. PMID 9663360.
- ^ Janssen P, Harris MS, Jones M, Masaoka T, Farré R, Törnblom H, Van Oudenhove L, Simrén M, Tack J (September 2013). “The relation between symptom improvement and gastric emptying in the treatment of diabetic and idiopathic gastroparesis”. The American Journal of Gastroenterology. 108 (9): 1382–91. doi:10.1038/ajg.2013.118. PMID 24005344. S2CID 32835351.
- ^ Ferrier J (2014). “Domperidone as an unintended antipsychotic”. Can Pharm J. 147 (2): 76–7. doi:10.1177/1715163514521969. PMC 3962062. PMID 24660005.
- ^ Xiao M, Qiu X, Yue D, Cai Y, Mo Q (2013). “Influence of hippophae rhamnoides on two appetite factors, gastric emptying and metabolic parameters, in children with functional dyspepsia”(PDF). Hellenic Journal of Nuclear Medicine. 16 (1): 38–43. PMID 23529392.
- ^ Huang X, Lv B, Zhang S, Fan YH, Meng LN (December 2012). “Itopride therapy for functional dyspepsia: a meta-analysis”. World Journal of Gastroenterology. 18 (48): 7371–7. doi:10.3748/wjg.v18.i48.7371. PMC 3544044. PMID 23326147.
- ^ Grzeskowiak LE, Smithers LG, Amir LH, Grivell RM (October 2018). “Domperidone for increasing breast milk volume in mothers expressing breast milk for their preterm infants: a systematic review and meta-analysis”. BJOG. 125 (11): 1371–1378. doi:10.1111/1471-0528.15177. hdl:2440/114203. PMID 29469929.
- ^ Grzeskowiak LE, Lim SW, Thomas AE, Ritchie U, Gordon AL (February 2013). “Audit of domperidone use as a galactogogue at an Australian tertiary teaching hospital”. Journal of Human Lactation. 29 (1): 32–7. doi:10.1177/0890334412459804. hdl:2440/94368. PMID 23015150. S2CID 26535783.
- ^ Donovan TJ, Buchanan K (2012). “Medications for increasing milk supply in mothers expressing breastmilk for their preterm hospitalised infants”. The Cochrane Database of Systematic Reviews. 3 (3): CD005544. doi:10.1002/14651858.CD005544.pub2. PMID 22419310.
- ^ da Silva OP, Knoppert DC (September 2004). “Domperidone for lactating women”. CMAJ. 171 (7): 725–6. doi:10.1503/cmaj.1041054. PMC 517853. PMID 15451832.
- ^ “FDA warns against women using unapproved drug, domperidone to increase milk production.” U.S. Food and Drug Administration 7 June 2004.
- ^ Asztalos EV, Campbell-Yeo M, daSilva OP, Kiss A, Knoppert DC, Ito S (2012). “Enhancing breast milk production with Domperidone in mothers of preterm neonates (EMPOWER trial)”. BMC Pregnancy and Childbirth. 12: 87. doi:10.1186/1471-2393-12-87. PMC 3532128. PMID 22935052.
- ^ Asztalos EV, Campbell-Yeo M, da Silva OP, Ito S, Kiss A, Knoppert D, et al. (EMPOWER Study Collaborative Group) (2017). “Enhancing human milk production with Domperidone in mothers of preterm infants”. Journal of Human Lactation. 33 (1): 181–187. doi:10.1177/0890334416680176. PMID 28107101. S2CID 39041713.
- ^ Jump up to:a b c d Henderson, Amanda (2003). “Domperidone: Discovering New Choices for Lactating Mothers”. AWHONN Lifelines. 7 (1): 54–60. doi:10.1177/1091592303251726. ISSN 1091-5923. PMID 12674062.
- ^ Donovan, Timothy J; Buchanan, Kerry (14 March 2012). “Medications for increasing milk supply in mothers expressing breastmilk for their preterm hospitalised infants”. Cochrane Database of Systematic Reviews (3): CD005544. doi:10.1002/14651858.cd005544.pub2. PMID 22419310.
- ^ Kapoor, A.K.; Raju, S.M. (2013). “7.2 Gastrointestinal Drugs”. Illustrated Medical Pharmacology. JP Medical Ltd. p. 677. ISBN 978-9350906552. Retrieved 31 October 2014. (Google Books)
- ^ Rebecca Smith (1 August 2014). “Fear that reflux treatment for babies will be denied under new Nice guidance”. The Daily Telegraph. Archived from the original on 31 October 2014. Retrieved 2014-10-31.
- ^ Jump up to:a b Swannick G. (ed.) “MIMS Australia.” December 2013
- ^ Jump up to:a b Elliott Proctor Joslin; C. Ronald Kahn (2005). Joslin’s Diabetes Mellitus: Edited by C. Ronald Kahn … [et Al.]. Lippincott Williams & Wilkins. pp. 1084–. ISBN 978-0-7817-2796-9.
- ^ Jump up to:a b c Edmund S. Sabanegh, Jr. (20 October 2010). Male Infertility: Problems and Solutions. Springer Science & Business Media. pp. 83–. ISBN 978-1-60761-193-6.
- ^ Gerald G. Briggs; Roger K. Freeman; Sumner J. Yaffe (28 March 2012). Drugs in Pregnancy and Lactation: A Reference Guide to Fetal and Neonatal Risk. Lippincott Williams & Wilkins. pp. 442–. ISBN 978-1-4511-5359-0.
- ^ Leelakanok N, Holcombe A, Schweizer ML (2015). “Domperidone and Risk of Ventricular Arrhythmia and Cardiac Death: A Systematic Review and Meta-analysis”. Clin Drug Investig. 36 (2): 97–107. doi:10.1007/s40261-015-0360-0. PMID 26649742. S2CID 25601738.
- ^ van Noord C, Dieleman JP, van Herpen G, Verhamme K, Sturkenboom MC (November 2010). “Domperidone and ventricular arrhythmia or sudden cardiac death: a population-based case-control study in the Netherlands”. Drug Safety. 33 (11): 1003–14. doi:10.2165/11536840-000000000-00000. PMID 20925438. S2CID 21177240.
- ^ Johannes CB, Varas-Lorenzo C, McQuay LJ, Midkiff KD, Fife D (September 2010). “Risk of serious ventricular arrhythmia and sudden cardiac death in a cohort of users of domperidone: a nested case-control study”. Pharmacoepidemiology and Drug Safety. 19(9): 881–8. doi:10.1002/pds.2016. PMID 20652862. S2CID 20323199.
- ^ Rossi M, Giorgi G (2010). “Domperidone and long QT syndrome”. Curr Drug Saf. 5 (3): 257–62. doi:10.2174/157488610791698334. PMID 20394569.
- ^ Doggrell SA, Hancox JC (2014). “Cardiac safety concerns for domperidone, an antiemetic and prokinetic, and galactogogue medicine” (PDF). Expert Opin Drug Saf. 13 (1): 131–8. doi:10.1517/14740338.2014.851193. PMID 24147629. S2CID 30668496.
- ^ Marzi, Marta; Weitz, Darío; Avila, Aylén; Molina, Gabriel; Caraballo, Lucía; Piskulic, Laura (2015). “Efectos adversos cardíacos de la domperidona en pacientes adultos: revisión sistemática”. Revista Médica de Chile. 143 (1): 14–21. doi:10.4067/S0034-98872015000100002. ISSN 0034-9887. PMID 25860264.
- ^ Jump up to:a b Buffery PJ, Strother RM (2015). “Domperidone safety: a mini-review of the science of QT prolongation and clinical implications of recent global regulatory recommendations”. N. Z. Med. J. 128(1416): 66–74. PMID 26117678.
- ^ Ortiz, Arleen; Cooper, Chad J.; Alvarez, Alicia; Gomez, Yvette; Sarosiek, Irene; McCallum, Richard W. (2015). “Cardiovascular Safety Profile and Clinical Experience With High-Dose Domperidone Therapy for Nausea and Vomiting”. The American Journal of the Medical Sciences. 349 (5): 421–424. doi:10.1097/MAJ.0000000000000439. ISSN 0002-9629. PMC 4418779. PMID 25828198.
- ^ Djeddi D, Kongolo G, Lefaix C, Mounard J, Léké A (November 2008). “Effect of domperidone on QT interval in neonates”. The Journal of Pediatrics. 153 (5): 663–6. doi:10.1016/j.jpeds.2008.05.013. PMID 18589449.
- ^ Günlemez A, Babaoğlu A, Arisoy AE, Türker G, Gökalp AS (January 2010). “Effect of domperidone on the QTc interval in premature infants”. Journal of Perinatology. 30 (1): 50–3. doi:10.1038/jp.2009.96. PMC 2834362. PMID 19626027.
- ^ Coulthard MG, Haycock GB (January 2003). “Distinguishing between salt poisoning and hypernatraemic dehydration in children”. BMJ (Clinical Research Ed.). 326 (7381): 157–60. doi:10.1136/bmj.326.7381.157. PMC 1128889. PMID 12531853.
- ^ Jump up to:a b c Jeffrey K. Aronson (27 November 2009). Meyler’s Side Effects of Antimicrobial Drugs. Elsevier. pp. 2244–. ISBN 978-0-08-093293-4.
- ^ Saeb-Parsy K. “Instant pharmacology.” John Wiley & Sons, 1999 ISBN 0471976393, 9780471976394 p216.
- ^ Sakamoto Y, Kato S, Sekino Y, Sakai E, Uchiyama T, Iida H, Hosono K, Endo H, Fujita K, Koide T, Takahashi H, Yoneda M, Tokoro C, Goto A, Abe Y, Kobayashi N, Kubota K, Maeda S, Nakajima A, Inamori M (2011). “Effects of domperidone on gastric emptying: a crossover study using a continuous real-time 13C breath test (BreathID system)”. Hepato-gastroenterology. 58 (106): 637–41. PMID 21661445.
- ^ Parkman HP, Jacobs MR, Mishra A, Hurdle JA, Sachdeva P, Gaughan JP, Krynetskiy E (January 2011). “Domperidone treatment for gastroparesis: demographic and pharmacogenetic characterization of clinical efficacy and side-effects”. Digestive Diseases and Sciences. 56 (1): 115–24. doi:10.1007/s10620-010-1472-2. PMID 21063774. S2CID 39632855.
- ^ Gabay MP (2002). “Galactogogues: medications that induce lactation”. J Hum Lact. 18 (3): 274–9. doi:10.1177/089033440201800311. PMID 12192964. S2CID 29261467.
- ^ Jump up to:a b c d Hofmeyr GJ, Van Iddekinge B, Blott JA (1985). “Domperidone: secretion in breast milk and effect on puerperal prolactin levels”. Br J Obstet Gynaecol. 92 (2): 141–4. doi:10.1111/j.1471-0528.1985.tb01065.x. PMID 3882143. S2CID 25489895.
- ^ Jump up to:a b c d e f Brouwers JR, Assies J, Wiersinga WM, Huizing G, Tytgat GN (1980). “Plasma prolactin levels after acute and subchronic oral administration of domperidone and of metoclopramide: a cross-over study in healthy volunteers”. Clin. Endocrinol. 12 (5): 435–40. doi:10.1111/j.1365-2265.1980.tb02733.x. PMID 7428183. S2CID 27266775.
- ^ Fujino T, Kato H, Yamashita S, Aramaki S, Morioka H, Koresawa M, Miyauchi F, Toyoshima H, Torigoe T (1980). “Effects of domperidone on serum prolactin levels in human beings”. Endocrinol. Jpn. 27 (4): 521–5. doi:10.1507/endocrj1954.27.521. PMID 7460861.
- ^ Jan Riordan (January 2005). Breastfeeding and Human Lactation. Jones & Bartlett Learning. pp. 76–. ISBN 978-0-7637-4585-1.
- ^ Kenneth L. Becker (2001). Principles and Practice of Endocrinology and Metabolism. Lippincott Williams & Wilkins. pp. 147–. ISBN 978-0-7817-1750-2.
- ^ Youssef AS, Parkman HP, Nagar S (2015). “Drug-drug interactions in pharmacologic management of gastroparesis”. Neurogastroenterol. Motil. 27 (11): 1528–41. doi:10.1111/nmo.12614. PMID 26059917. S2CID 34728070.
- ^ Stan K. Bardal; Jason E. Waechter; Douglas S. Martin (2011). Applied Pharmacology. Elsevier Health Sciences. pp. 184–. ISBN 978-1-4377-0310-8.
- ^ Hospital Formulary. HFM Publishing Corporation. 1991. p. 171.
Domperidone, a benzimidazole derivative, is structurally related to the butyrophenone tranquilizers (eg, haloperidol (Haldol, Halperon]).
- ^ Giovanni Biggio; Erminio Costa; P. F. Spano (22 October 2013). Receptors as Supramolecular Entities: Proceedings of the Biannual Capo Boi Conference, Cagliari, Italy, 7-10 June 1981. Elsevier Science. pp. 3–. ISBN 978-1-4831-5550-0.
- ^ Wan EW, Davey K, Page-Sharp M, Hartmann PE, Simmer K, Ilett KF (27 May 2008). “Dose-effect study of domperidone as a galactagogue in preterm mothers with insufficient milk supply, and its transfer into milk”. British Journal of Clinical Pharmacology. 66(2): 283–289. doi:10.1111/j.1365-2125.2008.03207.x. PMC 2492930. PMID 18507654.
- ^ Jump up to:a b c Sneader, Walter (2005). “Plant Product Analogues and Compounds Derived from Them”. Drug discovery : a history. Chichester: John Wiley & Sons Ltd. p. 125. ISBN 978-0-471-89979-2.
- ^ Corsini, Giovanni Umberto (2010). “Apomorphine: from experimental tool to therpeutic aid” (PDF). In Ban, Thomas A; Healy, David & Shorter, Edward (eds.). The Triumph of Psychopharacology and the Story of CINP. CINP. p. 54. ISBN 978-9634081814. Archived from the original (PDF) on 1 November 2014.
- ^ “Domperidone”. Pharmaceutical Manufacturing Encyclopedia, 3rd Edition (Vol. 1-4). William Andrew Publishing. 2013. p. 138. ISBN 9780815518563. Retrieved 12 December 2014.
- ^ Rathbone, Michael J.; Hadgraft, Jonathan; Roberts, Michael S. (2002). “The Zydis Oral Fast-Dissolving Dosage Form”. Modified-Release Drug Delivery Technology. CRC Press. p. 200. ISBN 9780824708696. Retrieved 31 October 2014.
- ^ Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 466–. ISBN 978-1-4757-2085-3.
- ^ “Domperidone”.
- ^ Reddymasu SC, Soykan I, McCallum RW (2007). “Domperidone: review of pharmacology and clinical applications in gastroenterology”. Am. J. Gastroenterol. 102 (9): 2036–45. PMID 17488253.
- ^ “Domperidone – heart rate and rhythm disorders.” Canadian adverse reactions newsletter. Government of Canada. January 2007 17(1)
- ^ “How to Obtain”. Food and Drug Administration. 10 February 2015. Retrieved 24 February 2016.
- ^ Fais, Paolo; Vermiglio, Elisa; Laposata, Chiara; Lockwood, Robert; Gottardo, Rossella; De Leo, Domenico (2015). “A case of sudden cardiac death following Domperidone self-medication”. Forensic Science International. 254: e1–e3. doi:10.1016/j.forsciint.2015.06.004. ISSN 0379-0738. PMID 26119456.
- ^ “De Standaard: “Motilium from now on only with prescription””. standaard.be. 7 May 2013. Retrieved 3 October 2013.
- ^ “ipcalabs.com”. ipcalabs.com. Retrieved 30 June 2013.
- ^ “torrentpharma.com”. torrentpharma.com. Retrieved 30 June2013.
- ^ Hofmeyr, G. J.; Van Iddekinge, B.; Van Der Walt, L. A. (2009). “Effect of domperidone-induced hyperprolactinaemia on the menstrual cycle; a placebo-controlled study”. Journal of Obstetrics and Gynaecology. 5 (4): 263–264. doi:10.3109/01443618509067772. ISSN 0144-3615.
External links
//////////////DOMPERIDONE, Antiemetic, Dopamine Receptor Antagonist, домперидон , دومبيريدون , 多潘立酮

NEW DRUG APPROVALS
ONE TIME
$10.00
MEBEVERINE
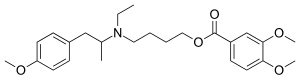
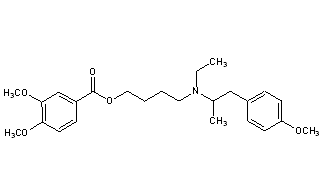
MEBEVERINE
- Molecular FormulaC25H35NO5
- Average mass429.549 Da
3,4-Dimethoxybenzoic Acid 4-[Ethyl[2-(4-methoxyphenyl)-1-methylethyl]amino]butyl Ester
3625-06-7[RN]
222-830-4[EINECS]
3,4-Diméthoxybenzoate de 4-{éthyl[1-(4-méthoxyphényl)-2-propanyl]amino}butyle
мебеверин
ميبيفيرين
美贝维林
- EINECS:222-830-4
- LD50:24 mg/kg (M, i.v.); 995 mg/kg (M, p.o.)
Derivatives
hydrochloride
- Formula:C25H35NO5 • HCl
- MW:466.02 g/mol
- CAS-RN:2753-45-9
- EINECS:220-400-0
- LD50:17.7 mg/kg (R, i.v.); 1540 mg/kg (R, p.o.)
Mebeverine
CAS Registry Number: 3625-06-7
CAS Name: 3,4-Dimethoxybenzoic acid 4-[ethyl[2-(4-methoxyphenyl)-1-methylethyl]amino]butyl ester
Additional Names: veratric acid 4-[ethyl(p-methoxy-a-methylphenethyl)amino]butyl ester;3,4-dimethoxybenzoic acid 4-[ethyl(p-methoxy-a-methylphenethyl)amino]butyl ester;4-[ethyl(p-methoxy-a-methylphenethyl)amino]butyl 3,4-dimethoxybenzoate;4-[N-[2-(p-methoxyphenyl)-1-methylethyl]-N-ethylamino]butyl 3,4-dimethoxybenzoate
Molecular Formula: C25H35NO5
Molecular Weight: 429.55
Percent Composition: C 69.90%, H 8.21%, N 3.26%, O 18.62%
Literature References: Smooth muscle relaxant. Prepn: BE609490C.A.59, 517b (1963) and T. Kralt et al.,DE1126889; eidem,US3265577 (1962, 1962, 1966 to N. V. Philips). Pharmacology: G. Bertaccini et al.,Farmaco Ed. Sci.30, 823 (1975).
Derivative Type: Hydrochloride
CAS Registry Number: 2753-45-9
Trademarks: Colofac (Duphar); Duspatalin (Duphar); Duspatal (Duphar)
Molecular Formula: C25H35NO5.HCl
Molecular Weight: 466.01
Percent Composition: C 64.43%, H 7.79%, N 3.01%, O 17.17%, Cl 7.61%
Properties: Crystals from ethyl methyl ketone, mp 105-107° (Ger. patent); also reported as mp 129-131° (Belg. patent).
Melting point: mp 105-107° (Ger. patent); mp 129-131° (Belg. patent)
Therap-Cat: Antispasmodic.
Keywords: Antispasmodic.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PAT
Indian Pat. Appl., 201841023171

PAPER
Pharma Chemica, 2(2), 366-378; 2010
In a quest of novel antispasmodic agents with antimicrobial properties, the present study describes design and synthesis of novel analogs for veratric acid ester 4-[ethyl-{2-(4- methoxyphenyl)-1-methylethyl} amino] butan-1-ol, an antispasmodic drug which is expected to be a potent antimicrobial agent may be due to the presence of two benzene rings and a secondary or tertiary nitrogen in the basic structural framework of the molecule. The reaction between substituted 2-ethylamino-1-(4’-methoxyphenyl) propane and various haloaryl benzoates derivatives obtained from reaction between different homologs of benzoic acid and dibromoalkanes in a two step process to give corresponding structurally diverse analogs of lead compound has been achieved. The structures of these novel analogs were confirmed by different structure elucidation techniques. All the compounds have been screened for their anti-spasmodic activity and the study extended further to evaluate their sedative, antibacterial and antifungal potency. The novel analogs of lead compound exhibited pronounced antispasmodic activities and also gave encouraging results of antimicrobial and sedative activity as anticipated.
General method of preparation of veratric acid ester 4-[ethyl-{2-(4-methoxyphenyl)-1- methylethyl} amino] butan-1-ol hydrochloride (5) and its analogs (5a-5p) A mixture of compound (3) (149 g, 0.47 mol) and Compound (4) (183 g, 0.95 mol) in ethyl methyl ketone (MEK) was refluxed for a period of 30 h at 75-80oC. The progress of the reaction was monitored by TLC to ensure formation of product and complete conversion of starting. On reaction completion solvent was distilled off and water (750 ml) was added to the reaction mass followed by toluene (300 ml). The resulting solution was cooled to 30oC and stirred for 30 minutes before layer separation. The organic layer was washed further with water (2×100 ml) and dried over sodium sulphate. To the organic layer IPA-HCl (72 g, 20 %) was added till pH is acidic (2-2.5).The product precipitated as solid hydrochloride salt was isolated by filtration and recrystallized from methanol. Yield: 181 g, 82% m.p., 105-107°C.
-C-H stretching (2959-2840), -C=O stretching (1717), -C=C stretching (1605, 1514, 1459), asymmetrical -C-O-C and –C-O stretching (1265-1130), symmetrical -C-O-C stretching (1023)
Chemical Shift ð, 1.25-1.22 ppm (t, 3H, -CH3), 1.57-1.51 (m, 3H, -CH3), 1.90-1.81 (d, 2H, -CH2), 2.16-2.11(m, 2H, – CH2), 2.54-2.25 (m, 1H, -CH), 3.11-3.05 (m, 4H, -CH2), 3.58-3.54 (t, 2H, -CH2), 3.77 (s, 3H, -OCH3), 3.91 (s, 6H, – OCH3), 4.37-4.33 (d, 2H, -CH2), 6.87-6.79 (m, 3H, Ar-H), 7.14-7.12 (d, 2H, Ar-H), 7.52-7.50 (d, 1H, Ar-H), 7.68-7.65 (m, 1H, Ar-H)
Antispasmodic drugs relieve cramps or spasms of the stomach, intestines, and bladder. Antispasmodics are classes (group) of drugs that can help to control some symptoms that arise from the gut, in particular, gut spasm. There are two main types namely “Antimuscarinics” and “Smooth muscle relaxants”. Antispasmodics are commonly used in “Irritable bowel syndrome” (IBS) to help relieve some of the symptoms of IBS such as spasm (colic), bloating and abdominal (stomach) pain and to reduce the motility (movement) of the intestines (gut) [1].
After understanding further the medicinal importance of antispasmodics and their ever increasing demand worldwide, we pursue to undertake the detailed synthetic and pharmacological study of antispasmodics to identify novel candidates as potential drug substances. Our parallel interest also lies on identifying novel antimicrobials since over the years; antibiotics are known to be the major protective agents against bacterial infections. However, the usage of antibiotics and antibacterial chemotherapeutics is becoming more and more restricted in the present age, despite the fact that there exist a large number of antibiotics. This is largely attributed to the emergence of drug-resistant bacteria, which render even some of the most broad spectrum antibiotics ineffective. In addition, most antibiotics have side effects. Thus, it becomes essential to investigate newer drugs with less resistance. Different studies on search of newer antimicrobials and antibacterial have revealed that moderate to remarkable antimicrobial or antibacterial action is present in several compounds, belonging to various pharmacological categories, such as antihistamines [2-4], tranquilizers [5], antihypertensive [6], anti-psychotics [7-11] anti-spasmodic [12] and anti-inflammatory agents [13]. Such compounds, having antibacterial properties in addition to their predesignated pharmacological actions, are termed as non-antibiotics [12]. Many of these compounds possess two or three benzene rings and nitrogen in the secondary or tertiary state in their molecular structure which is expected to be one of the bases for exhibiting antimicrobial potency [14]. Based on this rationale and to pursue our interest to identify newer antispasmodic agents with sedative and antimicrobial properties
[1] M. H. Pittler, E. Ernst, Am. J. Gastroenterol., 1998, 93 (7), 1131–5. [2] S. G. Dastidar, P. K. Saha, B. Sanyamat, A. N. Chakrabarty, J. Appl. Bacteriol., 1976, 41, 209- 214. [3] D. Chattopadyay, S. G. Dastidar, A. Chakrabarty, Arzneimittelforschang, 1988, 38, 869-872. [4] A. Chakrabarty, D. P. Acharya, D. K. Neogi, S. G. Dastidar, Indian J. Med. Res., 1989, 89, 233-237. [5] S. K. Dash, S. G. Dastidar, A. Chakrabarty, Indian J. Exp. Biol., 1977, 15, 324-326. [6] S. G. Dastidar, U. Mondal, S. Niyogi, A. Chakrabarty, Indian J. Med. Res., 1986, 84, 142- 147. [7] J. Molnar, Y. Mandi, J. Kiral, Acta Microbiol Acad Sci Hung., 1976, 23, 45-54. [8] J. E. Kristiansen, Acta Pathol. Microbial Immunol. Scand., 1992, 100 (Suppl. 30), 7-14 [9] S. G. Dastidar, A. Chaudhury, S. Annadurai, M. Mookerjee, A. Chakrabarty, J. Chemother., 1995, 7, 201-206. [10] V. Radhakrishnan, K. Ganguly, M. Ganguly, S. G. Dastidar, A. Chakrabarty, Indian J. Exp. Biol., 1999, 37, 671-675. [11] P. Bourlioux, J. M. Moreaux, W. J. Su, H. Boureau, Acta Pathol. Microbial. Immunol Scand., 1992, 100 (Suppl. 30), 40-43. [12] S. G. Dastidar, A. Chakrabarty, J. Molnar, N. Motohashi, National Institute of Science Communication (NISCOM), New Delhi, 1998, pp. 15. [13] S. Annadurai, S. Basu, S. Ray, S. G. Dastidar, A. C
Mebeverine is a drug used to alleviate some of the symptoms of irritable bowel syndrome. It works by relaxing the muscles in and around the gut.[1]
Medical use
Mebeverine is used to alleviate some of the symptoms of irritable bowel syndrome (IBS) and related conditions; specifically stomach pain and cramps, persistent diarrhoea, and flatulence.[2]
Data from controlled clinical trials have not found a difference from placebo or statistically significant results in the global improvement of IBS.[3][4]
It has not been tested in pregnant women nor in pregnant animals so pregnant women should not take it; it is expressed at low levels in breast milk, while no adverse effects have been reported in infants, breastfeeding women should not take this drug.[1]
Adverse effects
Adverse effects include hypersensitivity reactions and allergic reactions, immune system disorders, skin disorders including hives, oedema and widespread rashes.[2]
Additionally, the following adverse effects have been reported: heartburn, indigestion, tiredness, diarrhoea, constipation, loss of appetite, general malaise, dizziness, insomnia, headache, and decreased pulse rate.[1]
It does not have systemic anticholinergic side effects.[2]
Mebeverine can, on highly rare occasions, cause drug-induced acute angle closure glaucoma.[5]
Mechanism of action
Mebeverine is an anticholinergic but its mechanism of action is not known; it appears to work directly on smooth muscle within the gastrointestinal tract and may have an anaesthetic effect, may affect calcium channels, and may affect muscarinic receptors.[2]
It is metabolized mostly by esterases, and almost completely. The metabolites are excreted in urine.[2]
Mebeverine exists in two enantiomeric forms. The commercially available product is a racemic mixture of them. A study in rats indicates that the two have different pharmacokinetic profiles.[6]
History
It is a second generation papaverine analog, and was first synthesized around the same time as verapamil.[7]
It was first registered in 1965.[8]
Availability
Mebeverine is a generic drug and is available internationally under many brand names.[9]
SYN
Anon., Belgian Patent 609,490 (1962); T. Kralt,
H. O. Moes, A. Lindner and W. J. Asma, German Patent 1,126,889 (1962); Chem. Abstr., 59: 517b
(1963).
SYN

SYN
https://www.sciencedirect.com/science/article/abs/pii/S0731708502000237
References
- ^ Jump up to:a b c “Colofac data sheet” (PDF). New Zealand Medicines and Medical Devices Safety Authority. 14 June 2017. Retrieved 21 July2017.
- ^ Jump up to:a b c d e “Colofac Tablets 135mg – Summary of Product Characteristics (SPC)”. UK Electronic Medicines Compendium. 26 August 2016. Retrieved 21 July 2017.
- ^ Annaházi A, Róka R, Rosztóczy A, Wittmann T (May 2014). “Role of antispasmodics in the treatment of irritable bowel syndrome”. World Journal of Gastroenterology. 20 (20): 6031–43. doi:10.3748/wjg.v20.i20.6031. PMC 4033443. PMID 24876726.
- ^ Darvish-Damavandi M, Nikfar S, Abdollahi M (February 2010). “A systematic review of efficacy and tolerability of mebeverine in irritable bowel syndrome”. World Journal of Gastroenterology. 16(5): 547–53. doi:10.3748/wjg.v16.i5.547. PMC 2816265. PMID 20128021.
- ^ Lachkar Y, Bouassida W (March 2007). “Drug-induced acute angle closure glaucoma”. Current Opinion in Ophthalmology. 18 (2): 129–33. doi:10.1097/ICU.0b013e32808738d5. PMID 17301614. S2CID 30903966.
- ^ Hatami M, Farhadi K, Tukmechi A (August 2012). “Fiber-based liquid-phase micro-extraction of mebeverine enantiomers followed by chiral high-performance liquid chromatography analysis and its application to pharmacokinetics study in rat plasma”. Chirality. 24(8): 634–9. doi:10.1002/chir.22057. PMID 22700279.
- ^ Sneader W (2005). Drug Discovery: A History. John Wiley & Sons. p. 132. ISBN 9780471899792.
- ^ “Mebeverine”. druginfosys. Retrieved 1 February 2015.
- ^ “Mebeverine”. International. drugs.com. Retrieved 1 February2015.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | By mouth |
| ATC code | A03AA04 (WHO) |
| Legal status | |
| Legal status | UK: P (Pharmacy medicines)US: Not approvedIn general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 3625-06-7 HCl: 2753-45-9 |
| PubChem CID | 4031 |
| ChemSpider | 3891 |
| UNII | 7F80CC3NNVHCl: 15VZ5AL4JN |
| KEGG | D04868 |
| ChEMBL | ChEMBL282121 |
| CompTox Dashboard (EPA) | DTXSID6023238 |
| ECHA InfoCard | 100.020.756 |
| Chemical and physical data | |
| Formula | C25H35NO5 |
| Molar mass | 429.557 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| showSMILES | |
| showInChI | |
| (verify) |
//////////mebeverine, мебеверин ,ميبيفيرين ,美贝维林 , Antispasmodic

NEW DRUG APPROVALS
ONE TIME
$10.00
Sotradecol, Sodium tetradecyl sulfate

Sotradecol
Sodium tetradecyl sulfate
cas 139-88-8, Na FORM
free form 300-52-7
- Molecular FormulaC14H29NaO4S
- Average mass316.432 Da
139-88-8 [RN], cas 1191-50-0,
- CAS No.68043-79-8
4-Undecanol, 7-ethyl-2-methyl-, hydrogen sulfate, sodium salt (1:1)
7-Ethyl-2-methyl-4-undecyl sulfate sodium salt
UNII:Q1SUG5KBD6
натрия тетрадецилсульфат [Russian] [INN]
تتراديسيل سولفات صوديوم [Arabic] [INN]
十四烷硫酸钠 [Chinese] [INN]
CAS Registry Number: 139-88-8
CAS Name: 7-Ethyl-2-methyl-4-undecanol hydrogen sulfate sodium salt
Additional Names: 7-ethyl-2-methyl-4-hendecanol sulfate sodium salt; sodium 2-methyl-7-ethyl-4-undecyl sulfate; sodium 7-ethyl-2-methylundecyl-4-sulfate
Trademarks: Sotradecol (Elkins-Sinn); Tergitol 4; Trombavar; Trombovar
Molecular Formula: C14H29NaSO4, Molecular Weight: 316.43
Percent Composition: C 53.14%, H 9.24%, Na 7.27%, S 10.13%, O 20.22%
Properties: White, waxy solid. Sol in water, alcohol, ether. The pH of a 5% soln is from 6.5 to 9.0. Surface tension (dynes/cm) of aq soln at 25°: 56.5 dynes/cm (0.05% w/w); 52 (0.10%); 47 (0.20%); 40 (0.50%); 35 (1.0%). LD50 orally in rats: 4.95 g/kg, H. F. Smyth, C. P. Carpenter, J. Ind. Hyg. Toxicol.30, 63 (1948).
Toxicity data: LD50 orally in rats: 4.95 g/kg, H. F. Smyth, C. P. Carpenter, J. Ind. Hyg. Toxicol.30, 63 (1948)
Use: Wetting agent.
Therap-Cat: Sclerosing agent., Keywords: Sclerosing Agent.
Synonyms of Sodium Tetradecyl Sulfate [INN]
- 4-Ethyl-1-isobutyloktylsiran sodny
- EINECS 205-380-3
- Natrii tetracylis sulfas
- Natrii tetradecylis sulfas
- Natrii tetradecylis sulfas [Latin]
- Natrii tetradecylsulfas
- NSC 755887
- Obliterol
- Sodium sotradecol
- Sodium tetradecyl sulfate
- Sotradecol
- Tergitol
- Tergitol 4
- Tergitol anionic 4
- Tergitol penetrant 4
- Tetradecilsulfato sodico
- Tetradecilsulfato sodico [Spanish]
- Tetradecyl sulfate de sodium
- Trombovar
- UNII-Q1SUG5KBD6
- Varicol
An anionic surface-active agent used for its wetting properties in industry and used in medicine as an irritant and sclerosing agent for hemorrhoids and varicose veins.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Sodium tetradecyl sulfate is an anionic surfactant which occurs as a white, waxy solid. The structural formula is as follows:
 |
C14H28NaS04 7-Ethyl -2-methyl -4-hendecanol sulfate sodium salt MW 316.44Sotradecol® (sodium tetradecyl sulfate injection) is a sterile nonpyrogenic solution for intravenous use as a sclerosing agent.
1% (10 mg/mL): Each mL contains sodium tetradecyl sulfate 10 mg, benzyl alcohol 0.02 mL and dibasic sodium phosphate, anhydrous 4.0 mg in Water for Injection. pH 7.9; monobasic sodium phosphate and/or sodium hydroxide added, if needed, for pH adjustment.
3% (30 mg/mL): Each mL contains sodium tetradecyl sulfate 30 mg, benzyl alcohol 0.02 mL and dibasic sodium phosphate, anhydrous 9.0 mg in Water for Injection. pH 7.9; monobasic sodium phosphate and/or sodium hydroxide added, if needed, for pH adjustment.
Sodium tetradecyl sulfate (STS) is a commonly used synonym for 7-ethyl-2-methyl-4-undecanyl sulfate sodium salt[1] which is anionicsurfactant that is the active component of the sclerosant drug Sotradecol. It is commonly used in the treatment of varicose and spider veins of the leg, during the procedure of sclerotherapy.[2] Being a detergent, its action is on the lipid molecules in the cells of the vein wall, causing inflammatory destruction of the internal lining of the vein and thrombus formation eventually leading to sclerosis of the vein. It is used in concentrations ranging from 0.1% to 3% for this purpose. It is occasionally used for the treatment of stabilisation of joints that regularly dislocate, particularly in patients with Ehlers-Danlos syndrome.[3] In the UK, Ireland, Italy, Australia, New Zealand and South Africa, it is sold under the trade-name Fibro-Vein in concentrations of 0.2%, 0.5%, 1.0%, and 3%.[4]
Synthesis
It may be prepared by the aldol condensation of methyl isobutyl ketone and 2-ethylhexanal (which is itself formed by the aldol self-concensation of butyraldehyde), followed by sulfonation of the resulting alcohol.
SYN

RSC Advances, 10(22), 12788-12799; 2020
https://pubs.rsc.org/en/content/articlelanding/2020/RA/D0RA00386G

PAT
CN 106278961
CN 106278958
U.S.S.R., 1051067,
NMR
| Compound name: | Sodium Tetradecyl Sulfate |
|---|---|
| Spectrum type: | 1H NMR Spectrum (1D, 400 MHz, DMSO-d6, experimental) |

References
- ^ “SOTRADECOL® (Sodium tetradecyl sulfate)” (PDF). Retrieved 29 August 2014.
- ^ Jenkinson HA, Wilmas KM, Silapunt S (November 2017). “Sodium Tetradecyl Sulfate: A Review of Clinical Uses”. Dermatologic Surgery. 43 (11): 1313–1320. doi:10.1097/DSS.0000000000001143. PMID 28430735.
- ^ Burling F (2019). “Comparison of tetradecyl sulfate versus polidocanol injections for stabilisation of joints that regularly dislocate in an Ehlers-Danlos population”. BMJ Open Sport & Exercise Medicine. 5 (1): e000481. doi:10.1136/bmjsem-2018-000481. PMC 6350757. PMID 30792884.
- ^ Fibro-Vein history and details
| Clinical data | |
|---|---|
| Other names | 7-Ethyl-2-methyl-4-hendecanol sulfate sodium salt |
| AHFS/Drugs.com | Consumer Drug Information |
| Routes of administration | Intravenous injection |
| ATC code | C05BB04 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 139-88-8 |
| PubChemCID | 14492 |
| ChemSpider | 8440 |
| UNII | Q1SUG5KBD6 |
| ChEMBL | ChEMBL1200354 |
| CompTox Dashboard (EPA) | DTXSID3041530 |
| ECHA InfoCard | 100.004.892 |
| Chemical and physical data | |
| Formula | C14H29NaO4S |
| Molar mass | 316.43 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////Sodium tetradecyl sulfate, sotradecol, Sclerosing Agent, varicose veins
CCCCCCCCCCCCCCOS(=O)(=O)[O-].[Na+]

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
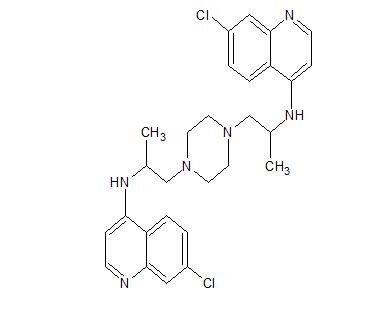{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
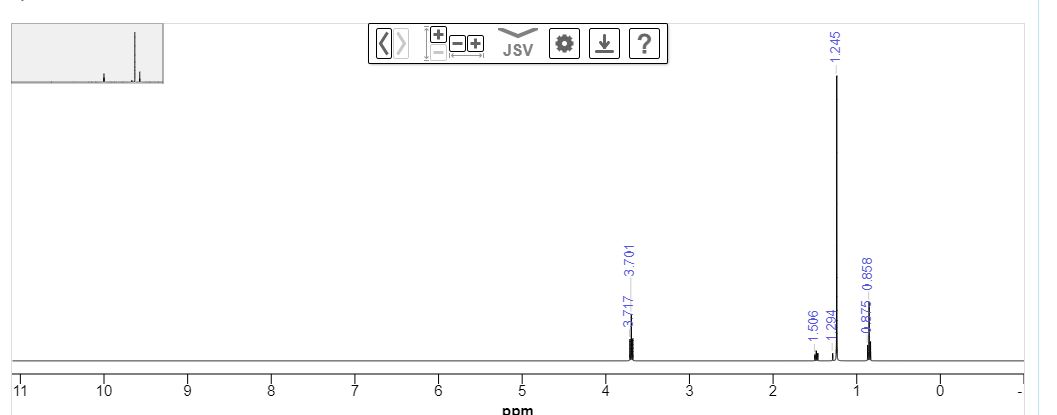{kind=link}
{kind=link}