Letermovir, MK 8828, AIC 246
2-[(4S)-8-fluoro-2-[4-(3-methoxyphenyl)piperazin-1-yl]-3-[2-methoxy-5-(trifluoromethyl)phenyl]-4H-quinazolin-4-yl]acetic acid
Letermovir; UNII-1H09Y5WO1F; AIC-246; 2-((4S)-8-Fluoro-2-(4-(3-methoxyphenyl)piperazin-1-yl)-3-(2-methoxy-5-(trifluoromethyl)phenyl)-4H-quinazolin-4-yl)acetic acid; 2-[(4S)-8-fluoro-2-[4-(3-methoxyphenyl)piperazin-1-yl]-3-[2-methoxy-5-(trifluoromethyl)phenyl]-4H-quinazolin-4-yl]acetic acid; Letermovir [INN]
| Molecular Formula: |
C29H28F4N4O4 |
| Molecular Weight: |
572.550633 g/mol |
Letermovir (INN) is an antiviral drug that is being developed for the treatment of cytomegalovirus (CVM) infections. It has been tested in CMV infected patients with allogeneic stem cell transplants and may also be useful for other patients with a compromised immune system such as those with organ transplants or HIV infections.[1]
The drug has been granted fast track status by the US Food and Drug Administration (FDA) and orphan drug status by the European Medicines Agency.[1]
The drug candidate is under development by Merck & Co., Inc as investigative compound MK-8828.[2]
AIC246, also known as letermovir, is a novel anti-CMV compound with IC50 value of 5.1 ± 1.2 nM. It targets the pUL56 (amino acid 230-370) subunit of the viral terminase complex [1].
The subunit pUL56 is a component of the terminase complex which is responsible for packaging unit length DNA into assembling virions.
AIC246 has a novel mode of action targets the enzyme UL56 terminase and keep active to other drug-resistant virus. The anti-HCMV activity of AIC246 was evaluated in vitro by using different HCMV laboratory strains, GCV-resistant viruses. The result showed that the inhibitory potentcy of AIC246 surpasses the current gold standard GCV by more than 400-fold with respect to EC50s (mean, ∼4.5 nM versus ∼2 μM) and by more than 2,000-fold with respect to EC90 values (mean, ∼6.1 nM versus ∼14.5 μM). In the CPE-RA strains, the EC50 values of AIC 246 ranged from 1.8 nM to 6.1 nM [2].
In mouse model with HCMV subcutaneous xenograft, oral administration of AIC246 caused significant a dose-dependent reduction of the HCMV titer. 30 mg/kg/d AIC246 for 9 days induced PFU reduction with maximum efficiency, compared with the gold standard GCV at the ED50 and ED90 level [2].
References:
[1].Verghese PS, Schleiss MR. Letermovir Treatment of Human Cytomegalovirus Infection Anti-infective Agent. Drugs Future. 2013, 38(5):291-298.
[2]. Lischka P1, Hewlett G, Wunberg T, et al.In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246.Antimicrob Agents Chemother. 2010, 54(3):1290-1297.
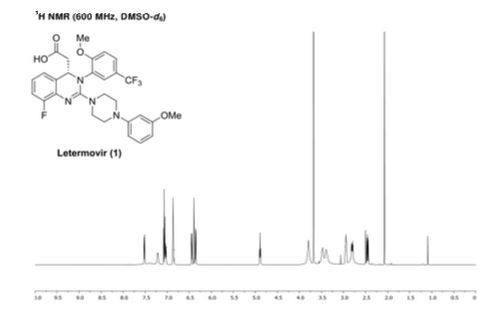
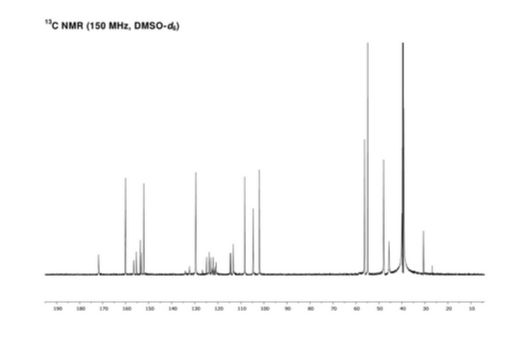
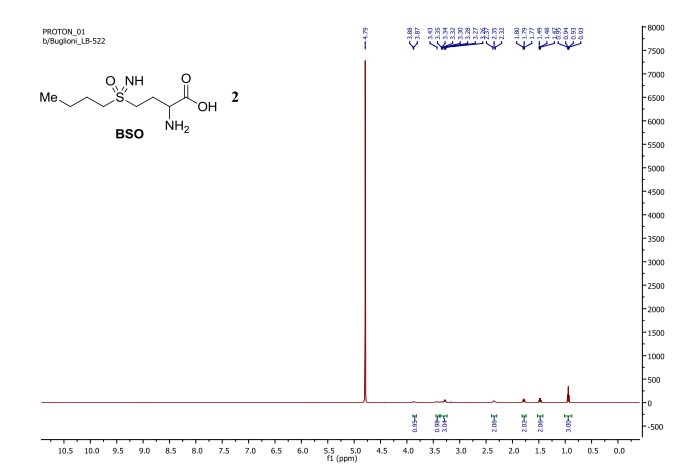
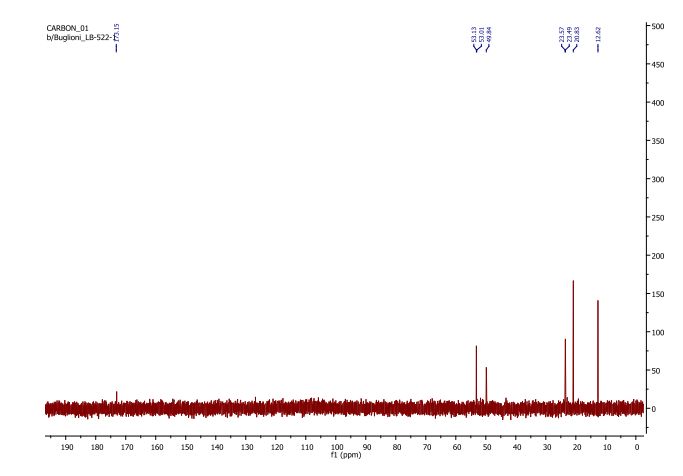
NMR


Human cytomegalovirus (HCMV) remains the leading viral cause of birth defects and life-threatening disease in transplant recipients. All approved antiviral drugs target the viral DNA polymerase and are associated with severe toxicity issues and the emergence of drug resistance. Attempts to discover improved anti-HCMV drugs led to the identification of the small-molecular-weight compound AIC246 (Letermovir). AIC246 exhibits outstanding anti-HCMV activity in vitro and in vivo and currently is undergoing a clinical phase IIb trial. The initial mode-of-action studies suggested that the drug acts late in the HCMV replication cycle via a mechanism distinct from that of polymerase inhibitors. Here, we extend our mode-of-action analyses and report that AIC246 blocks viral replication without inhibiting the synthesis of progeny HCMV DNA or viral proteins. The genotyping of mutant viruses that escaped AIC246 inhibition uncovered distinct point mutations in the UL56 subunit of the viral terminase complex. Marker transfer analyses confirmed that these mutations were sufficient to mediate AIC246 resistance. The mapping of drug resistance to open reading frame UL56 suggests that viral DNA processing and/or packaging is targeted by AIC246. In line with this, we demonstrate that AIC246 affects the formation of proper unit-length genomes from viral DNA concatemers and interferes with virion maturation. However, since AIC246-resistant viruses do not exhibit cross-resistance to previously published terminase inhibitors, our data suggest that AIC246 interferes with HCMV DNA cleavage/packaging via a molecular mechanism that is distinct from that of other compound classes known to target the viral terminase.
PATENT
WO 2006133822

Scheme 2:
Chromatography
on a chiral phase


Scheme 4:

Scheme 5:
Synthesis of {8-fluoro-2- [4- (3-methoxyphenyl) piperazin-l -yl] -3- [2-methoxy-5- (trifluoromethyl) phenyl] -3,4-dihydroquinazolin-4-yl }acetic acid

xample 1
N- (2-bromo-6-fluoφhenyl) -N ‘- [2-methoxy-5- (trifluoromethyl) phenyl] urea

2-methoxy-5-trifluoromethylphenyl isocyanate (274.3 g) are dissolved in acetonitrile (1 L), then 2-bromo-6-fluoroaniline (200 g) was added with acetonitrile (50 mL) flushed. The resulting clear solution is at 38 h reflux (ca. 85 0 stirred C), then under vacuum at 40 0 concentrated C a dogged mush. This is filtered off, with acetonitrile (260 mL, to 0-5 0 C cooled) washed and incubated overnight at 45 0 dried C in the VDO using entraining nitrogen. Thus, a total of 424.3 g of N- (2-bromo-6-fluorophenyl) -N ‘- get [2-methoxy-5- (trifluoromethyl) phenylJ-urea as a solid, corresponding to 99.2% of theory.
1 H NMR (300 MHz, d 6 -DMSO): δ = 8.93 (s, IH), 8.84 (s, IH), 8.52 (d, V = 2.3, 2H), 7, 55 (d, 2 = Vr = 7.7, IH), 7.38 to 7.26 (m, 3H), 7.22 (d, 2 J = 8.5, IH), 4.00 (s, 3H) ppm;
– – MS (API-ES-pos.): M / z = 409 [(M + H) + , 100%];
HPLC (Method 1): R τ = 22.4 and 30.6 min.
example 2
N- (2-bromo-6-fluorophenyl) -N ‘- [2-methoxy-5- (trifluoromethyl) phenyl] urea (Alterhativsynthese)
2-methoxy-5-trifluoromethylphenyl isocyanate (1.19 kg) are at about 35 0 dissolved melted and C in acetonitrile (4.2 L), then 2-bromo-6-fluoroaniline (870 g) was added and with acetonitrile ( 380 mL) rinsed. The resulting clear solution is at 74-88 45 h 0 stirred C, then under vacuum (200 mbar) at 50 0 C to a dogged mush concentrated (amount of distillate 4.4 L). This is at room temperature with diisopropylether (1.5 L), washed aspirated, with diisopropylether (1.15 L) washed and at 45 0 C in the VDO using entraining nitrogen to constant weight (24 h) dried. Thus, a total of 1, 63 kg Η- (2-bromo-6-fluoro-phenyl) -W- – obtained [2-methoxy-5 (trifluoromethyl) phenyl] urea as a solid, corresponding to 87.5% of theory.
HPLC (Method 1): R τ = 22.6 and 30.8 min.
example 3
{8-Fluor-3-[2-methoxy-5-(trifluormethyl)phenyl]-2-oxo-l,2,3,4-tetrahydrochinazolin-4-yl}essigsäuremethylester

N- (2-bromo-6-fluorophenyl) -N- [2-methoxy-5- (trifluoromethyl) phenyl] urea (300 g) under a nitrogen atmosphere in isobutyronitrile (1.2 L) was suspended, then triethylamine
(21O mL), bis (acetonitrile) dichloropalladium (7.5 g), tris- (o-tolyl) phosphine (18.0 g) and
Methyl acrylate (210 mL) were added in this order. The resulting suspension is for 16 hours at reflux (ca. 102 0 stirred C) and then cooled to room temperature. Water (1.2 L) is added and the mixture 1 at room temperature stirred, then aspirated and washed with water / methanol h: washed and acetonitrile (10O mL) (1 1 30O mL). The residue is treated overnight at 45 0 dried C in the VDO using entraining nitrogen. Thus, a total of 208 g as a solid, corresponding to 68.5% of theory.
1 H NMR (300 MHz, d 6 -DMSO): δ = 9.73 (s, IH), 7.72 (d, 2 J = 7.3, IH), 7.71 (s, IH), 7 , 33 (d, 2 J = 9.3, IH), 7.15 (dd, 2 J = 9.6, 2 J = 8.6, IH), 7.01 (d, 2 J = 7.3 , IH), 6.99 to 6.94 (m, IH), 5.16 (t, 2 , J = 5.9, IH), 3.84 (s, 3H), 3.41 (s, 3H) , 2.81 (dd, 2 J = 15.4, 2 J = 5.8, IH), 2.62 (dd, 2 J = 15.4, 2 J = 6.3, IH) ppm;
MS (API-ES-pos.): M / z = 413 [(M + H) + , 100%], 825 [(2M + H) + , 14%];
HPLC (Method 1): R τ = 19.3 min; Pd (ICP): 16,000 ppm.
example 4
{8-Fluor-3-[2-methoxy-5-(trifluormethyl)phenyl]-2-oxo-l,2,3,4-tetrahydrochinazolin-4-yl}essigsäuremethylester (Alternative synthesis)
N- (2-bromo-6-fluorophenyl) -N ‘- [2-methoxy-5- (trifluoromethyl) phenyl] urea (2.5 kg) is suspended under a nitrogen atmosphere in isobutyronitrile (9 L), then triethylamine (1.31 kg), bis (acetonitrile) dichloropalladium (64.9 g), tris (o-tolyl) phosphine (149 g) and methyl acrylate (1.59 kg) were added in this order. The resulting suspension is 22 hours at 90-100 0 stirred C, then cooled to room temperature. Water (9 L) is added and stirred, then aspirated and washed with water / methanol (1: 1, 2.5 L) at room temperature, the mixture for 1 hour and acetonitrile (850 mL). The residue is treated overnight at 45 0 dried C in the VDO using entraining nitrogen to constant weight (21 h). Thus, a total of 1.90 kg as a solid, corresponding to 74.9% of theory.
HPLC (Method 1): R τ = 19.4 min.
example 5
{2-Chlor-8-fluor-3-[2-methoxy-5-(trifluormethyl)phenyl]-3,4-dihydrochinazolin-4-yl}essigsäure-methylester / chlorination

A solution of 2.84 kg {8-fluoro-3- [2-methoxy-5- (trifluoromethyl) phenyl] -2-oxo-l, 2,3,4-tetrahydroquinazolin-4-yl} acetic acid methyl ester in 14.8 l of chlorobenzene is heated to reflux and the solvent is distilled off until water no longer separates. It is to 12O 0 cooled C. Within 10 min phosphorus oxychloride are metered in 3.17 kg, and then is added within a further 10 min 2.10 kg DBU. It is heated to reflux for 9 hours.
For working up the mixture is cooled to 40 0 C., stirred overnight and dosed the reactor contents to 11.4 L of water, previously estimated at 40 0 was tempered C. For dosing an internal temperature of 40-45 to 0 C, are satisfied. The mixture is allowed to cool to room temperature, 11.4 L of dichloromethane, filtered through a Seitz filter plate and the phases are separated. The organic phase is washed with 11.4 L of water, 11.4 L of an aqueous saturated sodium bicarbonate solution and again with 11.4 L of water. The organic phase is concentrated on a rotary evaporator in vacuo and the remaining residue (2.90 kg) is used without further treatment in the next step.
1 H NMR (300 MHz, d 6 -DMSO): δ = 7.93 to 7.82 (m, 2H), 7.38 (d, 2 J = 8.9, IH), 7.17 (m, 2H), 6.97 to 6.91 (m, IH), 5.45 and 5.29 (m and t, 2 , J = 5.4, IH), 3.91 and 3.84 (2s, 3H) , 3.48 (s, 3H), 3.0 to 2.6 (m, 2H) ppm;
MS (CI, NH 3 ): m / z = 431 [(M + H) + , 100%];
HPLC (Method 1): R τ = 23.5 min; typical Pd value (ICP): 170 ppm.
example 6
{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-[2-methoxy-5-(trifluormethyl)phenyl]-3,4-dihydrochinazolin-4-yl}essigsäuremethylester / Amination – –

(52.5 g) is dissolved in 1,4-dioxane (10O mL), then (25.8 g) and DBU (20.4 g) was added at room temperature 3-methoxyphenylpiperazine, whereupon the temperature rises. The mixture is stirred at reflux for 22 h, then cooled to room temperature, with ethyl acetate (500 mL) and water (200 mL) and the phases separated. The organic phase (200 mL) washed with 0.2N hydrochloric acid (three times 100 mL) and water, dried over sodium sulfate and evaporated. Thus, a total of 62.5 g obtained as a solidified foam, which is reacted as the crude product without further purification.
HPLC (Method 1): R τ = 16.6 min.
example 7
{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-[2-methoxy-5-(trifluormethyl)phenyl]-3,4-dihydrochinazolin-4-yl}essigsäuremethylester / Pot chlorination + amination
(50.0 g) is introduced in chlorobenzene (300 mL), then chlorobenzene is partially distilled (5O mL). The mixture is heated to 120 0 cooled C., DBU (36.9 g) is added, then at 120-128 is 0 C phosphorous oxychloride (33.4 mL) over 10 min. metered. The mixture (approximately 130 at reflux for 9 hours 0 C) stirred. Subsequently, at 40 0cooled C, slowly at 40-45 0 C with water (200 mL), cooled to room temperature and diluted with dichloromethane (200 mL), stirred and then the phases separated. The organic phase is washed with water (200 mL), saturated aqueous sodium bicarbonate solution (200 mL) and again water (200 mL), dried over sodium sulfate, concentrated by rotary evaporation and then under high vacuum at 50 0 dried C. The residue (48.1 g) is dissolved in chlorobenzene (20 mL), then with 1,4-dioxane (80 mL) at room temperature and 3-methoxyphenylpiperazine (23.6 g) and DBU (18.7 g) was added, whereupon the temperature rises. The mixture is stirred at reflux for 22 h, then cooled to room temperature, with ethyl acetate (500 mL) and water (200 mL) and the phases separated. The organic phase (200 mL) washed with 0.2N hydrochloric acid (three times 100 mL) and water, dried over sodium sulfate and evaporated. Thus, a total of 55.6 g obtained as a solidified foam, which is reacted as the crude product without further purification.
HPLC (Method 1): R τ = 16.2 min.
example 8
(^)-{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-(2-methoxy-5-trifluormethylphenyl)-3,4-dihydrochinazolin-4-yl} acetate / saponification racemate

(64 g) is dissolved in 1,4-dioxane (45O mL) and IN sodium hydroxide solution (325 mL) and stirred for 2 h at room temperature, then dried in vacuo at 30 0 , a part of the solvent C is distilled off (400 mL). Toluene is added (300 mL) and the phases separated. The aqueous phase is washed with toluene (15O mL twice), then the combined organic phases again with IN sodium hydroxide solution (50 mL) are extracted. The pH of the combined aqueous phases with 2N hydrochloric acid (about 150 mL) to 7.5, then MIBK (15O mL) is added. The phases are separated, the aqueous phase extracted again with MIBK (15O mL), then dried the combined MIBK phases over sodium sulfate and at 45 0 concentrated C. Thus, a total of 64 g as an amorphous solid in quantitative yield.
HPLC (Method 1): R τ = 14.9 min.
Scheme 6:
Separation of enantiomers of {8-fluoro-2- [4- (3-methoxyphenyl) piperazin-l -yl] -3- [2-methoxy-5- (tri-fluoromethyl) phenyl] -3,4-dihydroquinazolin-4-yl } acetate

x (2S, 3S) -2,3-bis [(4-methylbenzoyl) – oxyjbemsteinsäure
x EtOAc
example 9
(2S, 3 £) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid (1: 1 salt) / crystallization

(62.5 g, crude product) is dissolved and filtered in ethyl acetate (495 mL). To the filtrate is (35 25 ‘,) added 2,3-bis [(4-methylbenzoyl) oxy] succinic acid (42.0 g), the mixture for 30 minutes. stirred at room temperature, then with (35 25 “) -2,3-bis [(4-methylbenzoyl) oxy] -succinic acid – (l: l salt) (165 mg) was inoculated and stirred for 3 days at room temperature, then to 0-3 0 cooled C and stirred for a further 3 h, the suspension is suction filtered and washed with cold ethyl acetate (0-10. 0 C, 35 mL ) washed. the crystals are at 40 h 18 0 C in the VDO using entraining nitrogen dried. Thus 37.1 g of the salt are obtained as a solid, corresponding to 30.4% of theory over three stages (chlorination, amination and crystallization) on the racemate, or 60.8% based on the resulting S enantiomer.
– – 1 H NMR (300 MHz, d 6 -DMSO): δ = 7.90 (d, 2 J = 7.8, 4H), 7.56 (d, 2 J = 8.3, IH), 7 , 40 (d, 2 J = 7.8, 4H), 7.28 to 7.05 (m, 4H), 6.91 to 6.86 (m, 2H), 6.45 (d, 2 J = 8.3, IH), 6.39 to 6.36 (m, 2H), 5.82 (s, 2H), 4.94 (m, IH), 4.03 (q, 2 J = 7.1 , 2H), 3.83 (brs, 3H), 3.69 (s, 3H), 3.64 (s, 3H), 3.47 to 3.36 (m, 8H and water, 2H), 2, 98 to 2.81 (m, 5H), 2.58 to 2.52 (m, IH), 2.41 (s, 6H), 1.99 (s, 3H), 1.18 (t, 2 J = 7.2, 3H) ppm;
HPLC (Method 1): R τ = 16.6 and 18.5 min.
example 10
(25,3iS) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid (1: 1 salt) / recrystallization
(2S, 3S) -2,3-bis [(4-methy lbenzoyl) oxy] succinic acid – { (l: l salt) (36.8 g) is suspended in ethyl acetate (37o mL) and (77 by heating to reflux 0 C) dissolved. The mixture is slowly cooled to room temperature. Here there is a spontaneous crystallization. The suspension is stirred at RT for 16 h, then 0-5 0 cooled C and stirred for another 3 h. The suspension is suction filtered and washed with cold ethyl acetate (0-10 0 C, twice 15 ml). The crystals are at 45 h 18 0 C in the VDO using entraining nitrogen dried. Thus 33.6 g of the salt are obtained as a solid, corresponding to 91.3% of theory.
HPLC (Method 1): R τ = 16.9 and 18.8 min .;
HPLC (Method 3): 99.9% ee
example 11
(5)-{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-(2-methoxy-5-trifluormethylphenyl)-3,4-dihydrochinazolin-4-yl}essigsäure

(2IS I , 3S) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid (l: l salt) (10.1 g, containing 14 ppm of Pd) are suspended in ethyl acetate (100 mL) and shaken with saturated aqueous sodium bicarbonate solution (10O mL) shaken until both phases are clear. The phases are separated, the organic phase is evaporated. The residue is dissolved in 1,4-dioxane (100 mL) and IN sodium hydroxide solution (31.2 mL) and stirred for 3 h at room temperature. Subsequently, the pH is adjusted with IN hydrochloric acid (about 17 mL) is set to 7.5, MIBK (8O mL) was added, then the pH is adjusted with IN hydrochloric acid (about 2 mL) adjusted to 7.0. The phases are separated, the organic phase dried over sodium sulfate and concentrated. The residue is dissolved in ethanol and concentrated (40 mL), then again in ethanol (40 mL) and concentrated under high vacuum at 50 0 C dried. The solidified foam is at 45 h 18 0 C in the VDO using entraining nitrogen dried. Thus, a total of 5.05 g as an amorphous solid, corresponding to 85.0% of theory.
1 H NMR (300 MHz, d 6 -DMSO): δ = 7.53 (d, 2 J = 8.4, IH), 7.41 (brs, IH), 7.22 (d, 2 J = 8 , 5, IH), 7.09 to 7.01 (m, 2H), 6.86 (m, 2H), 6.45 (dd, V = 8.2, 3 J = 1.8, IH) 6.39 to 6.34 (m, 2H), 4.87 (t, 2 J = 7.3, IH), 3.79 (brs, 3H), 3.68 (s, 3H), 3.50 -3.38 (m, 4H), 2.96 to 2.75 (m, 5H), 2.45 to 2.40 (m, IH) ppm;
MS (API-ES-neg.): M / z = 571 [(MH), 100%];
HPLC (Method 1): R τ = 15.1 min;
HPLC (Method 2): 99.8% ee; Pd (ICP): <1 ppm.
example 12
(2 / ?, 3Λ) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid (1: 1 salt) / crystallization R-isomer from the mother liquor

The mother liquor from a crystallization of (2IS ‘, 3S) -2,3-bis [(4-methylbenzoyl) oxy] -succinic acid – {8-fluoro-2- [4- (3-methoxyphenyl) piperazin-l -yl] -3- [2-methoxy-5- (trifluoromethyl) phenyl] -3,4-dihydroquinazolin-4-yl} acetic acid methyl ester (l: l-salt) in 279 g scale is washed with saturated aqueous sodium bicarbonate solution (1.5 L ) shaken, the phases are separated and the organic phase is shaken with semi-saturated aqueous sodium bicarbonate solution (1.5 L). The phases are separated, the organic phase dried over sodium sulfate and evaporated. The residue (188.4 g) is dissolved in ethyl acetate (1.57 L), then (2R, 3R) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid (121.7 g) was added and the mixture 10 min. stirred at room temperature. Is then treated with (2R, 3R) -2,3-bis [(4-methyl-benzoyl) oxy] succinic acid – (l: l salt) (0.38 g) was inoculated and stirred for 18 h at room temperature, then to 0-3 0 cooled C and stirred for another 3 h. The suspension is suction filtered and washed with cold ethyl acetate (0-10 0 C, 50O ml). The crystals are at 40 h 18 0 C in the VDO using entraining nitrogen dried. So a total of 160 g of the salt are obtained as a solid.
HPLC (Method 1): R τ = 16.6 and 18.5 min .;
HPLC (Method 3): -99.0% ee
example 13
(i?)-{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-(2-methoxy-5-trifluormethylphenyl)-3,4-dihydrochinazolin-4-yl} acetate / production R-isomer

(2Λ, 3 /?) – 2,3-bis [(4-methylbenzoyl) oxy] succinic acid – {8-fluoro-2- [4- (3-methoxy-phenyl) pipera-tine 1-yl] -3- [ 2-methoxy-5- (trifluormethy l) pheny l] -3, 4-dihydroquinazolin-4-y 1} -acetic acid methyl ester (1: 1 salt) (170 g) are suspended in ethyl acetate (85O mL) and as long as with saturated aqueous sodium bicarbonate (850 mL) shaken until both phases are clear (about 5 min.). The phases are separated, the solvent of the organic phase under normal pressure with 1, 4-dioxane to a final temperature of 99 0 exchanged C (portions distilled total 2.55 L solvent, and 2.55 L of 1,4-dioxane used). The mixture is cooled to room temperature and 18 at room temperature IN sodium hydroxide solution (525 mL) stirred. Subsequently, the pH value with concentrated hydrochloric acid (about 35 mL) is set to 7.5, MIBK (85O mL) was added, then the pH with concentrated hydrochloric acid (ca. 1O mL) adjusted to 7.0. The phases are separated, the organic phase dried over sodium sulfate and concentrated. The residue is dissolved in ethanol and concentrated (350 mL), then again in ethanol (350 mL) at 50 and 0 concentrated C. Thus, a total of 91.6 g as an amorphous solid, corresponding to 91.6% of theory.
HPLC (method 1): R 7 = 14.8 min.
– – Example 14
{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-(2-methoxy-5-trifluormethylphenyl)-3,4-dihydrochinazolin-4-yl} acetate / racemization R-enantiomer
acetic acid (50 g) is dissolved in acetonitrile (500 mL) and treated with sodium methoxide (30% in methanol, 32.4 mL) and then stirred at reflux for 60 h. After cooling to room temperature the mixture is concentrated in vacuo to half, then with hydrochloric acid (20% strength, ca. 20 ml) adjusted to pH 7.5, MIBK (200 mL) was added and hydrochloric acid (20%) on pH 7 adjusted. The phases are separated, the organic phase dried over sodium sulfate and evaporated to the hard foam. The residue is dissolved in ethanol and concentrated (15O mL), then again in ethanol (15O mL) and concentrated. Thus, 54.2 g as an amorphous solid in quantitative yield.
HPLC (Method 1): R τ = 14.9 min .;
HPLC (method 4): 80.8 wt.%.
example 15
{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-[2-methoxy-5-(trifluormethyl)phenyl]-3,4-dihydrochinazolin-4-yl}essigsäuremethylester / Esterification racemate
acetic acid (54 g) (540 g) was dissolved in methanol, then concentrated sulfuric acid (7.85 mL) is added. The mixture is stirred at reflux for 26 h, then cooled and concentrated in vacuo to about one third of the original volume. Water (15O mL) and dichloromethane (15O mL) are added, then the phases are separated. The organic phase is washed with saturated sodium bicarbonate solution (two times 140 mL), dried over sodium sulfate and concentrated to a foamy residue. This is twice in succession in ethanol (150 mL) and concentrated, dried in vacuo using entraining nitrogen then 18 h. Thus, a total of 41.6 g as an amorphous solid, corresponding to 75.2% of theory.
HPLC (Method 1): R τ = 16.8 min .;
HPLC (method 4): 85.3 wt.%;
HPLC (Method 3): -8.5% ee
example 16
(25 1 , 3S) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid – { (1: 1 salt) / crystallization of esterified racemate
(41.0 g) is suspended in ethyl acetate (287 mL), then (2S, 3IS) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid (27.5 g) was added. The mixture is 30 minutes. stirred at room temperature, then with (2 <S ‘, 3S) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid – (1: 1 salt) (0.08 g) was inoculated. The suspension is stirred at RT for 16 h, then 0-5 0 cooled C and stirred for another 3 h, then filtered off with suction and washed with cold ethyl acetate (0-10 0 C, four times 16 ml). The crystals are at 45 h 18 0 C in the VDO using entraining nitrogen dried. So a total of 25.4 g of the salt are obtained as a solid, corresponding to 37.4% of theory.
HPLC (Method 1): R τ = 16.9 and 18.8 min .;
HPLC (method 4): 99.5 wt.%;
HPLC (Method 3): 99.3% ee
example 17
(iS)-{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-(2-methoxy-5-trifluormethylphenyl)-3,4-dihydrochinazolin-4-yl} acetate / saponification crystals
(25,3S) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid – (l rl salt) (25.1 g) is suspended in ethyl acetate (25O mL) and shaken with saturated aqueous sodium bicarbonate solution (250 mL) shaken until both phases are clear. The phases are separated, the organic phase is evaporated. Dissolve the residue in 1, 4-dioxane (25O mL) and IN sodium hydroxide solution (77.4 mL) and stirred for 18 h at room temperature. Subsequently, the pH is adjusted with IN hydrochloric acid (about 50 mL) is set to 7.5, was added MIBK (240 mL), then the pH is adjusted with IN hydrochloric acid (about 15 mL) adjusted to 7.0. The phases are separated, the organic phase dried over sodium sulfate and concentrated. The residue is dissolved in ethanol and concentrated (90 mL), then again in ethanol (90 mL) and concentrated. The solidified foam is at 45 h 180 C in the VDO using entraining nitrogen dried. Thus, a total of 12 g as an amorphous solid, corresponding to 81.2% “ of the theory.
HPLC (Method 1): R τ = 15.1 min;
HPLC (Method 2): 97.5% ee; Pd (ICP): <20 ppm.
Alternative method for the racemization:
example 18
(i)-{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-(2-methoxy-5-trifluormethylphenyl)-3,4-dihydrochinazolin-4-yl} acetic acid / saponification enriched R isomer from the mother liquor after crystallization
The mother liquor from a crystallization of (2 J S ‘, 35) -2,3-bis [(4-methylbenzoyl) oxy] -succinic acid – (l: l-salt) in 207 g scale is shaken with saturated aqueous sodium bicarbonate (500 mL), the phases are separated and the organic phase is shaken with semi-saturated aqueous sodium bicarbonate solution (500 mL). The phases are separated, the organic phase dried over sodium sulfate and evaporated. The residue is dissolved in ethanol (500 mL) and rotary evaporated to a hard foam. This is in 1,4-dioxane (1.6 L) and IN sodium hydroxide solution (1.04 L) and stirred at room temperature for 18 h, then toluene is added (1.5 L) and the phases separated. The aqueous phase is adjusted with hydrochloric acid (20% strength, ca. 155 ml) of pH 14 to pH 8, then is added MIBK (1.25 L) and hydrochloric acid (20% strength, ca. 25 mL) to pH 7 readjusted. The phases are separated, the organic phase dried over sodium sulfate and evaporated to the hard foam. This is at 45 h 18 0 C in the VDO using entraining nitrogen dried. Thus, a total of 150 g obtained as (R / S) mixture as an amorphous solid.
HPLC (Method 2): 14.6% ee
– – Example 19
(i)-{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-(2-methoxy-5-trifluormethylphenyl)-3,4-dihydrochinazolin-4-yl} acetate / racemization
(150 g, R / S mixture with -14.6% ee) is dissolved in acetonitrile (1.5 L) and treated with sodium methoxide (30% in methanol, 97.2 mL) was added, then stirred at reflux for 77 h , After cooling to room temperature the mixture is concentrated in vacuo to half, then with hydrochloric acid (20% strength, ca. 80 mL) made of pH 13 to pH 7.5, was added MIBK (0.6 L) and treated with hydrochloric acid ( 20% strength, ca. 3 mL) adjusted to pH. 7 The phases are separated, the organic phase dried over sodium sulfate and evaporated to the hard foam. The residue is dissolved in ethanol and concentrated (500 mL), then again in ethanol (500 mL) and concentrated, then 18 h at 450 dried C in the VDO using entraining nitrogen. Thus, a total of 148 g as an amorphous solid, corresponding to 98.7% of theory.
HPLC (Method 2): 1.5% ee
example 20
{8-Fluor-2-[4-(3-methoxyphenyl)piperazin-l-yl]-3-[2-methoxy-5-(trifluormethyl)phenyl]-3,4-dihydrochinazolin-4-yl}essigsäuremethylester (Esterification)
(±) – {8-fluoro-2- [4- (3-methoxyphenyl l) piperazin-1 -yl] -3- (2-methoxy-5-trifluormethy lphenyl) -3, 4-dihydroquinazolin-4-yl} acetic acid (148 g) (1480 g) was dissolved in methanol, then concentrated sulfuric acid (21.5 mL) is added. The mixture is stirred at reflux for 6 h, then cooled and concentrated in vacuo to about one third of the original volume. Water (400 mL) and dichloromethane (400 mL) are added, then the phases are separated. The organic phase (diluted twice 375 mL, 300 mL water) with saturated sodium bicarbonate solution, dried over sodium sulfate and concentrated to a foamy residue. This is twice in succession in ethanol (each 400 mL) and concentrated, dried in vacuo using entraining nitrogen then 18 h. Thus, a total of 124 g as an amorphous solid, corresponding to 81.9% of theory.
HPLC (Method 1): R τ = 16.9 min .;
example 21
(25.35) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid – (1: 1 salt) / crystallization of esterified racemate
(2S, 3S) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid – (1: 1 salt) (123 g, 14.4% ee) is suspended in ethyl acetate (861 mL) and filtered, then (2IS ‘, 3IS) -2,3-bis [(4-methylbenzoyl) oxy ] succinic acid (82.5 g). The mixture 30 min. stirred at room temperature, then with (2 £, 3 <S) -2,3-bis [(4-methylbenzoyl) oxy] succinic acid – (1: 1 salt) (0.24 g) was inoculated. The suspension is stirred for 4 days at RT, then concentrated to approximately 600 mL and again with (25 ‘, 3 1 -2,3-bis [(4-methylbenzoyl) oxy] succinic acid S) – (l: l salt) (0.24 g) was inoculated. The suspension is stirred for 1 week at RT, to 0-5 0 cooled C and further stirred for 3 hours, then filtered off with suction and washed with cold ethyl acetate (0-10 0 C, 4 x 40 ml). The crystals are at 45 h 18 0 C in the VDO using entraining nitrogen dried. So a total of 1 1.8 g of salt are obtained as a solid, corresponding to 5.8% of theory.
Scheme 7:

example 22
N- (2-Fluoφhenyl) -N ‘- [2-methoxy-5- (trifluoromethyl) phenyl] urea

2-methoxy-5-trifluoromethylphenyl isocyanate (1057.8 g) is dissolved in acetonitrile (4240 mL), then 2-fluoro aniline (540.8 g) was added with acetonitrile (50 mL) flushed.The resulting clear solution is stirred for 4 h at reflux (about 82 ° C), then seeded at about 78 ° C and about 15 min. touched. The suspension is on 0 0 cooled C, aspirated and the product with acetonitrile (950 mL, to 0-5 0 cooled C) washed. The product is dried overnight at 45 ° C in a vacuum drying oven using entraining nitrogen. Thus, a total of 1380.8 g of N- (2-fluorophenyl) -N ‘- [2-methoxy-5- (trifluoromethyl) phenyl] -harnstqff ‘ obtained as a solid, corresponding to 86.4% of theory.
1 H NMR (500 MHz, d 6 -DMSO): δ = 9.36 (s, IH), 9.04 (s, IH), 8.55 (d, 1.7 Hz, IH), 8.17 ( t, 8.2 Hz, IH), 7.33 (d, 8.5 Hz, IH), 7.20 to 7.26 (m, 2H), 7.14 (t, 7.6 Hz, IH), 7, 02 (m, IH), 3.97 (s, 3H) ppm;
MS (API-ES-pos.): M / z = 329 [(M + H) + , 100%];
HPLC: R τ = 48.7 min.
Instrument: HP 1100 Multiple Wavelength detection; Column: Phenomenex-Prodigy ODS (3) 100A, 150 mm x 3 mm, 3 microns; Eluent A: (1.36 g KH 2 PO 4 +0.7 mL H 3PO 4 ) / L water, eluent B:
acetonitrile; Gradient: 0 min 20% B, 40 min 45% B, 50 min 80% B, 65 min 80% B; Flow: 0.5 mL / min; Temp .: 55 0 C; UV detection: 210 nm.
example 23
Methyl (2E) -3- {3-fluoro-2 – [({[2-methoxy-5 – (trifluormethy l) pheny 1] amino} carbonylation l) amino] pheny 1} acrylate

N- (2-fluorophenyl) -N ‘- [2-methoxy-5- (trifluoromethyl) phenyl] urea (0.225 kg) is dissolved in acetic acid (6.75 L) and (30.3 g) was added with palladium acetate. Then 65% oleum is (247.5 g) is added and then methyl acrylate (90 g). The solution is stirred overnight at room temperature. Then, at about 30 0 C and about 30 mbar acetic acid (3740 g) were distilled off. The suspension is treated with water (2.25 L) and stirred for about 1 hour. The product is drained, washed twice with water (0.5 L) and incubated overnight at 50 0 dried C in a vacuum drying oven using entraining nitrogen. Thus, a total of 210.3 g of methyl (2E) -3- {3-fluoro-be 2 – [({[2-methoxy-5- (trifluoromethyl) phenyl] amino} carbonyl) amino] phenyl} acrylate obtained as a solid, corresponding to 72.2% of theory.
1 H NMR (300 MHz, d 6 -DMSO): δ = 9.16 (s, IH), 8.84 (s, IH), 8.45 (d, 1.7 Hz, IH), 7.73 ( m, 2H), 7.33 (m, 3H), 7.22 (d, 8.6 Hz, IH), 6.70 (d, 16Hz, IH), 3.99 (s, 3H), 3.71 (s, 3H) ppm;
MS (API-ES-pos.): M / z = 429.9 [(M + NH,) + ]; 412.9 [(M + H) + ]
HPLC: R τ = 46.4 min.
Instrument: HP 1100 Multiple Wavelength detection; Column: Phenomenex-Prodigy ODS (3) 100A, 150 mm x 3 mm, 3 microns; Eluent A: (1.36 g KH 2 PO 4 +0.7 mL H 3PO 4 ) / L water, eluent B: acetonitrile; Gradient: 0 min 20% B, 40 min 45% B, 50 min 80% B, 65 min 80% B; Flow: 0.5 mL / min; Temp .: 55 0 C; UV detection: 210 nm.
example 24
{8-FluorO-[2-methoxy-5-(trifluormethyl)phenyl]-2-oxo-l,2,3,4-tetrahydrochinazolin-4-yl}essigsäuremethylester

Methyl (2E) -3- {3-fluoro-2 – [({[2-methoxy-5- (trifluoromethyl) phenyl] amino} carbonyl) amino] phenyl} acrylate (50 g) is dissolved in acetone (1.2 L) was suspended and 3.7 g) was added l, 8-diazabicyclo [5.4.0] undec-7-ene (. The suspension is heated to reflux (ca..56 ° C) and stirred for 4 h. The resulting clear solution is hot through diatomaceous earth (5 g) was filtered. The diatomaceous earth is rinsed with warm acetone (100 ml). Subsequently, acetone (550 g) was distilled off. The resulting suspension is in 3 h at O 0 cooled and stirred C. The product is drained, washed twice with cold acetone (50 ml) and incubated overnight at 45 0 dried C in a vacuum drying oven using entraining nitrogen. Thus, a total of 44.5 g of {8-fluoro-3- [2-methoxy-5- (trifluoromethyl) phenyl] -2-oxo-1, 2, 3, 4-tetrahydrochinazo-lin-4-yl} acetic acid methyl ester as a solid, corresponding to 89% of theory.
1 H NMR (300 MHz, d 6 -DMSO): δ = 9.73 (s, IH), 7.72 (d, 2 J = 7.3, IH), 7.71 (s, IH), 7 , 33 (d, 2 J = 9.3, IH), 7.15 (dd, 2 J = 9.6, 2 J = 8.6, IH), 7.01 (d, 2 J = 7.3 , IH), 6.99 to 6.94 (m, IH), 5.16 (t, 2 J =
5.9, IH), 3.84 (s, 3H), 3.41 (s, 3H), 2.81 (dd, 1 J = 15.4, V = 5.8, IH), 2.62 (dd, 2 Vr = = 15.4, V = 6.3, IH) ppm;
MS (API-ES-pos.): M / z = 413 [(M + H) + , 100%], 825 [(2M + H) + , 14%];
HPLC: R τ = 37.1 min.
Instrument: HP 1100 Multiple Wavelength detection; Column: Phenomenex-Prodigy ODS (3) 100A, 150 mm x 3 mm, 3 microns; Eluent A: (1.36 g KH 2 PO 4 +0.7 mL H 3PO 4 ) / L water, eluent B: acetonitrile; Gradient: 0 min 20% B, 40 min 45% B, 50 min 80% B, 65 min 80% B; Flow: 0.5 mL / min; Temp .: 55 0 C; UV detection: 210 nm.
PATENT
WO 2015088931
Human cytomegalovirus (HCMV) is ubiquitously distributed in the human population. In immunocompetent adults infections are mainly asymptomatic, but in
immunocompromised patients, such as transplant recipients or AIDS patients, life threatening infections occur at a high rate. HCMV is also the leading cause of birth defects among congenitally transmitted viral infections.
Various substituted heterocyclic compounds are inhibitors of the HCMV terminase enzyme. Included in these heterocycles are quinazolines related to Compound A, as defined and described below. These compounds and pharmaceutically acceptable salts thereof are useful in the treatment or prophylaxis of infection by HCMV and in the treatment, prophylaxis, or delay in the onset or progression of HCMV infection. Representative quinazoline compounds that are useful for treating HCMV infection are described, for example, in US Patent Patent No. 7, 196,086. Among the compounds disclosed in US7, 196,086, is (S)-2-(8-fluoro-3-(2-methoxy-5-(trifluoromethyl)phenyl)-2-(4-(3-methoxyphenyl)piperazin-l-yl)-3,4-dihydroquinazolin-4-yl)acetic acid, hereinafter referred to as Compound A. Compound A is a known inhibitor of HCMV terminase. The structure of Compound A is as follows:

Compound A
US Patent Nos. 7,196,086 and 8,084,604 disclose methodology that can be employed to prepare Compound A and related quinazoline-based HCMV terminase inhibitors. These methods are practical routes for the preparation of Compound A and related heterocyclic compounds.
EXAMPLE 6
Preparation of Compound A

To a slurry of compound 7 (20g, 18.9 mmol) in MTBE (40.0 mL) at room temperature was added a solution of sodium phosphate dibasic dihydrate (8.42 g, 47.3 mmol) in water (80 mL) and the resulting slurry was allowed to stir at room temperature for 40 minutes. The reaction mixture was transferred to a separatory funnel and the organic phase was collected and washed with a solution of sodium phosphate dibasic dihydrate (3.37 g, 18.91 mmol) in water (40.0 mL). A solution of KOH (4.99 g, 76 mmol) in water (80 mL) and methanol (10.00 mL) was then added to the organic phase and the resulting mixture was heated to 50 °C and allowed to stir at this temperature for 6 hours. MTBE (20 mL) and water (40 mL) were then added to the
reaction mixture and the resulting solution was transferred to a separatory funnel and the aqueous layer was collected and washed with MTBE (20 mL). Additional MTBE (40 mL) was added to the aqueous layer and the resulting solution was adjusted to pH 4-5 via slow addition of concentrated HCl. The resulting acidified solution was transferred to a separatory funnel and the organic phase was collected, concentrated in vacuo and solvent switched with acetone, maintaining a 30 mL volume. The resulting acetone solution was added dropwise to water and the precipitate formed was filtered to provide compound A as a white solid (10 g, 92%). XH NMR (500 MHz, d6-DMSO): δΗ 12.6 (1H, s), 7.52 (1H, dd, J= 8.6, 1.3 Hz), 7.41 (1H, brs), 7.22 (1H, d, J= 7.2 Hz), 7.08-7.02 (2H, m), 6.87-6.84 (2H, m), 6.44 (1H, dd, J= 8.3, 1.8 Hz), 6.39 (1H, t, J= 2.1 Hz), 6.35 (1H, dd, J= 8.1, 2.0 Hz), 4.89 (1H, t, J= 7.3 Hz), 3.79 (3H, br s), 3.68 (3H, s), 3.47 (2H, br s), 3.39 (2H, br s), 2.96-2.93 (2H, m), 2.82-2.77 (3H, m), 2.44 (1H, dd, J = 14.8, 7.4 Hz).
XAMPLE 1
Preparation of Intermediate Compound 2

N,N-dicyclohexylmethylamine
IPAC, 80°C
To a degassed solution of 2-bromo-6-fluoroaniline (1, 99.5 g, 0.524 mol), methyl acrylate (95.0 mL, 1.05 mol), Chloro[(tri-tert-butylphosphine)-2-(2-aminobiphenyl)] palladium(II) (0.537 g, 1.05 mmol) in isopropyl acetate (796 mL), was added degassed N,N-dicyclohexylmethylamine (135 mL, 0.628 mol). The resulting reaction was heated to 80 °C and allowed to stir at this temperature for 5 hours. The resulting slurry was cooled to 20 °C and filtered. The filtrate was washed with 1 M citric acid to provide a solution that contained compound 2 (99.3 g, 97% assay yield) in isopropyl acrylate, which was used without further purification. ‘H NMR (500 MHz, d-CHCl3): δΗ 7.79 ppm (1H, d, J= 15.9 Hz), 7.17 ppm (1H, d, J= 8.2 Hz), 7.00 ppm (1H, ddd, J= 10.7, 8.2, 1.2 Hz), 6.69 ppm (1H, td, J = 8.2, 5.1 Hz), 6.38 ppm (1H, d, J= 15.9 Hz), 4.06 ppm (2H, br s), 3.81 ppm (3H, s).
EXAMPLE 2
Preparation of Intermediate Compound 3

To a solution of compound 2 (48.8 g, 0.250 mol) in 683 mL of isopropyl acetate was added 244 mL of water, followed by di-sodium hydrogen phosphate (53.2 g, 0.375 mol). To the resulting solution was added phenyl chloroformate (39.2 mL, 0.313 mol) dropwise over 30 minutes. The resulting reaction was heated to 30 °C and allowed to stir at this temperature for 5 hours for 4 hours and then was heated to 60 °C and allowed to stir at this temperature for 5 hours for an additional 2 hours to remove excess phenyl chloroformate. An additional 293 mL of isopropyl acetate was then added and the reaction mixture was allowed to stir at room temperature until the solids completely dissolved into solution. The resulting reaction mixture was transferred to a separatory funnel and the organic phase was washed with 98 mL of water and collected to provide a solution of compound 3 in isopropyl acetate, which was used without further purification. XH NMR (500 MHz, d-acetonitrile): δΗ 7.91 ppm (1H, d, J= 15.9 Hz), 7.85 ppm (1H, br s), 7.63 ppm (1H, d, J= 7.9 Hz), 7.45-7.39 ppm (3H, m), 7.33-7.27 ppm (2H, m), 7.21 ppm (2H, br), 6.60 ppm (1H, d, J= 16.0 Hz).
EXAMPLE 3
Preparation of Intermediate Compound 4

A solution of compound 3 (79.0 g, 0.250 mol), 2-methoxy-5-(trifluoromethyl)aniline (52.7 g, 0.276 mol), and 4-dimethylaminopyridine (0.92 g, 0.0075 mol) in isopropyl acetate (780 mL) was heated to reflux and allowed to stir at this temperature for 5 hours. The resulting slurry was cooled to 20 °C, then allowed to stir at this temperature for for two hours at this temperature, then filtered. The collected filter cake was dried in vacuo to provide compound 5 (95.0 g, 0.230 mol) as a white solid, which was used without further purification. ¾ NMR (500 MHz, d-TFA): δΗ 7.98 ppm (1H, d, J= 16.1 Hz), 7.87 ppm (1H, s), 7.47 ppm (1H, d, J = 7.9 Hz), 7.41 ppm (1H, d, J= 8.5 Hz), 7.35 ppm (1H, q, J= 8.5 Hz), 7.19 ppm (1H, t, J= 8.6 Hz), 6.98 ppm (1H, d, J= 8.6 Hz), 6.56 ppm (1H, d, J= 16.0 Hz), 3.85 ppm (6H, br s).
EXAMPLE 4
Preparation of Intermediate Compound 6

To a stirred suspension of compound 4 (14.0 g, 34.0 mmol) in toluene (140 mL) at room temperature was added 2-picoline (10.1 mL, 102 mmol) followed by PCI5 (8.19 g, 37.3 mmol). The resulting reaction was heated to 40 °C and allowed to stir at this temperature for 4 hours, then was cooled to 0 °C and cautiously (internal temperature kept <15 °C) quenched with KOH (2 M, 102 mL). The resulting solution was allowed to warm to room temperature, allowed to stir for 30 minutes, then was filtered and the filtrate transferred to a separatory funnel. The organic phase was washed sequentially with H3PO4 (1M, 50 mL) and H20 (50 mL) to provide a solution of compound 5 in toluene, which was used without further purification. XH NMR (500 MHz, d6-DMSO): δΗ 7.96 (1H, d, J= 16.2 Hz), 7.74 (1H, d, J= 7.9 Hz), 7.61 (1H, dd, J= 6.7, 1.6 Hz), 7.50 (1H, d, J= 1.9 Hz), 7.43 (1H, t, J= 9.2 Hz), 7.30 (1H, d, J= 8.4 Hz), 7.28 (1H, m), 6.79 (1H, d, J= 16.2 Hz), 3.91 (3H, s), 3.74 (3H, s).
To the solution of compound 5 at room temperature was added an aqueous solution of piperazine hydrochloride (0.40 M, 93.3 mL, 37.3 mmol) followed by Na2HP04 (14.5 g, 102 mmol). The resulting reaction was allowed to stir for 1 hour at room temperature, then transferred to a separatory funnel. The organic phase was washed sequentially with aH2P04 (50 mL) and H20 (50 mL). Salicylic acid (5.16 g, 37.3 mmol) was then added to the organic phase, and the resulting solution was cooled to 0 °C and allowed to stir at this temperature for 1 hour to provide a slurry which was filtered and washed with cold toluene (50 mL). The filter cake was dried under air to provide compound 6 (23.0 g, 31.7 mmol, 93 %) as a white crystalline solid: XH NMR (500 MHz, d6-DMSO): δΗ 12.9 (1H, br s), 7.75 (1H, dd, J= 7.8, 1.8 Hz), 7.72 (1H, d, J= 16.1 Hz), 7.40 (1H, td, J= 7.2, 1.7 Hz), 7.27 (1H, d, J= 7.8 Hz), 7.17 (1H, m), 7.16 (1H, t, J= 8.2 Hz), 7.02 (1H, br s), 6.95 (1H, t, J= 8.6 Hz), 6.88-6.81 (3H, m), 6.78 (1H, br s), 6.60 (1H, dd, J= 8.2, 2.0 Hz), 6.54 (1H, m), 6.48 (1H, d, J= 16.1 Hz), 6.43 (1H, dd, J= 8.0, 2.1 Hz), 3.73 (3H, s), 3.71 (3H, s), 3.69 (4H, br s), 3.68 (3H, s).
Free Base: XH NMR (500 MHz, CD3CN): δΗ 7.91 (1H, d, J= 16.1 Hz), 7.29 (1H, d, J= 8.0 Hz), 7.24 (1H, d, J= 1.4 Hz), 7.20 (1H, t, J= 8.1 Hz), 7.15 (1H, dd, J= 8.6, 1.4 Hz), 6.94 (1H, m), 6.92 (1H, t, J= 8.1 Hz), 6.80 (1H, td, J= 8.1, 5.4 Hz), 6.60 (1H, dd, J= 8.3, 2.2 Hz), 6.54 (1H, t, J= 2.2 Hz), 6.50 (1H, d, J= 16.1 Hz), 6.47 (2H, m), 3.80 (3H, s), 3.79 (3H, s), 3.72 (3H, s), 3.63 (4H, t, J= 5.1 Hz), 3.25 (4H, t, J= 5.0 Hz).
2: 1 NDSA Salt: ‘H NMR (500 MHz, d6-DMSO): δΗ 10.2 (2H, br s), 8.86 (1H, d, J= 8.6 Hz), 7.92 (1H, d, J= 7.0 Hz), 7.47-7.37 (4H, m), 7.27-7.14 (4H, m), 6.96 (1H, d, J= 8.6 Hz), 6.65 (1H, d, J= 8.3 Hz), 6.59 (1H, s), 6.54 (1H, d, J= 15.9 Hz), 6.47 (1H, d, J= 8.3 Hz), 3.91 (4H, m), 3.77 (3H, s), 3.76 (3H, s), 3.74 (3H, s), 3.43 (4H, m). 1,5 -naphthalene disulfonic acid
EXAMPLE 5
Preparation of Intermediate Compound 7

To a suspension of compound 6 (12.5 g, 16.6 mmol) in 125 mL of toluene was added 50 mL of 0.43M aqueous K3P04. The resulting reaction was allowed to stir for 1 hour at room temperature and the reaction mixture was transferred to a separatory funnel. The organic phase was collected, washed once with 30 mL 0.43M aqueous K3P04then cooled to 0 °C and aqueous K3P04 (60 mL, 0.43 M, 25.7 mmol) was added. To the resulting solution was added a room temperature solution of ((lS,2S,4S,5R)-l-(3,5-bis(trifluoromethyl)benzyl)-2-((R)-
hydroxy( 1 -(3 -(trifluoromethyl)benzyl)quinolin- 1 -ium-4-yl)methyl)-5-vinylquinuclidin- 1 -ium bromide) (0.704 g, 0.838 mmol) in 1.45 mL of DMF. The resulting reaction was allowed to stir at 0 °C until the reaction was complete (monitored by HPLC), then the reaction mixture was transferred to a separatory funnel and the organic phase was collected and washed sequentially with 1M glycolic acid (25 mL) and water (25 mL). The organic phase was filtered through solka flok and concentrated in vacuo to a total volume of 60 mL. Ethyl acetate (20 mL) was added to the resulting solution, followed by (S,S)-Di-P-Toluoyl-D-tartaric acid (5.61 g, 14.1 mmol). Penultimate seed (0.2 g) was added the resulting solution was allowed to stir at room
temperature for 12 hours. The solution was then filtered and the collected solid was washed twice with ethyl acetate, then dried in vacuo to provide compound 7 as its DTTA salt ethyl acetate solvate (13.8 g, 78%) . ‘H NMR (500 MHz, d6-DMSO): δΗ 13.95 (2H, br s), 7.90 (4H, d, J= 8.1 Hz), 7.55 (1H, dd, J= 8.6, 1.3 Hz), 7.38 (4H, d, J= 8.1 Hz), 7.26 (1H, d, J= 7.8 Hz), 7.09-7.05 (3H, m), 6.91-6.86 (2H, m), 6.44 (1H, dd, J= 8.2, 1.7 Hz), 6.39 (1H, t, J= 2.0 Hz), 6.36 (1H, dd, J= 8.2, 2.0 Hz), 5.82 (2H, s), 4.94 (1H, t, J= 7.1 Hz), 4.02 (2H, q, J= 7.1 Hz), 3.83 (3H, br s), 3.68 (3H, s), 3.64 (3H, s), 3.47 (2H, br s), 3.37 (2H, br s), 2.95 (2H, br s), 2.87- 2.80 (3H, m), 2.56 (1H, dd, J= 14.3, 7.0 Hz), 2.39 (6H, s), 1.98 (3H, s), 1.17 (3H, t, J= 7.1 Hz).
PAPER
Asymmetric Synthesis of Letermovir Using a Novel Phase-Transfer-Catalyzed Aza-Michael Reaction
Department of Process Chemistry, Merck and Co., Inc., P.O. Box 2000, Rahway, New Jersey 07065, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00076
Publication Date (Web): May 13, 2016
Copyright © 2016 American Chemical Society
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
Abstract
The development of a concise asymmetric synthesis of the antiviral development candidate letermovir is reported, proceeding in >60% yield over a total of seven steps from commercially available materials. Key to the effectiveness of this process is a novel cinchonidine-based PTC-catalyzed aza-Michael reaction to configure the single stereocenter.
http://pubs.acs.org/doi/full/10.1021/acs.oprd.6b00076
(S)-2-(8-Fluoro-3-(2-methoxy-5-(trifluoromethyl)phenyl)-2-(4-(3-methoxyphenyl)piperazin-1-yl)-3,4-dihydroquinazolin-4-yl)acetic Acid (Letermovir, 1)
letermovir (1, 20.2 g, 35.3 mmol, 100 wt %, 94%) as an amorphous white powder. 1H NMR (DMSO-d6, 600 MHz) δH 7.52 (dd, J = 8.7, 1.7 Hz, 1H), 7.40 (brs, 1H), 7.21 (m, 1H), 7.07 (t, J = 8.2 Hz, 1H), 7.04 (m, 1H), 6.87 (m, 2H), 6.44 (dd, J = 8.2, 1.9 Hz, 1H), 6.40 (t, J = 2.3 Hz, 1H), 6.36 (dd, J = 8.0, 2.0 Hz, 1H), 4.89 (t, J = 7.2 Hz, 1H), 3.80 (brs, 3H), 3.68 (s, 3H), 3.39–3.48 (m, 4H), 2.82–2.95 (m, 4H), 2.80 (dd, J = 14.8, 7.4 Hz, 1H), 2.46 (dd, J = 14.9, 7.4 Hz, 1H); 13C NMR (DMSO-d6, 150 MHz) δC 171.8, 160.2, 156.5, 154.6 (d, JCF = 246.3 Hz), 153.2, 152.2, 134.2, 132.3 (d, JCF = 11.2 Hz), 129.6, 124.1 (q, JCF = 271.3 Hz), 123.8 (q, JCF = 3.7 Hz), 122.4, 122.1 (q, JCF = 7.1 Hz), 121.4 (q, JCF = 29.2 Hz), 120.8, 114.5 (d, JCF = 19.5 Hz), 113.3, 108.3, 104.6, 101.9, 59.0, 56.3, 54.8, 47.9, 45.6, 40.0; HR-MS calcd for C29H29F4N4O4+ [M + H]+ 573.2119, found 573.2117 (Δ = 0.2 mmu).
References
Masangkay, Estel Grace (July 29, 2014). “Merck Kicks Off Phase 3 Study Of CMV Drug Letermovir”. Retrieved 8 Oct 2014.
| Patent ID |
Date |
Patent Title |
| US8084604 |
2011-12-27 |
Process for the Preparation of Dihydroquinazolines |
| US2007191387 |
2007-08-16 |
Substituted dihydroquinazolines |
| Patent ID |
Date |
Patent Title |
| US2015133461 |
2015-05-14 |
PHARMACEUTICAL COMPOSITION CONTAINING AN ANTIVIRALLY ACTIVE DIHYDROQUINAZOLINE DERIVATIVE |
| US2015050241 |
2015-02-19 |
METHOD OF TREATING VIRAL INFECTIONS |
| US2015045371 |
2015-02-12 |
Salts of a dihydroquinazoline derivative |
| US2015038514 |
2015-02-05 |
SODIUM AND CALCIUM SALTS OF DIHYDROQUINAZOLINE DERIVATIVE AND USE THEREOF AS ANTIVIRAL AGENTS |
| US2015038728 |
2015-02-05 |
NOVEL ARYLATED CAMPHENES, PROCESSES FOR THEIR PREPARATION AND USES THEREOF |
| US8816075 |
2014-08-26 |
Process for the preparation of dihydroquinazolines |
| US2014193802 |
2014-07-10 |
IDENTIFICATION OF AN ALTERED THERAPEUTIC SUSCEPTIBILITY TO ANTI-HCMV COMPOUNDS AND OF A RESISTANCE AGAINST ANTI-HCMV COMPOUNDS |
| US2014178432 |
2014-06-26 |
PRODUCTION OF DENSE BODIES (DB) FROM HCMV-INFECTED CELLS |
| US8372972 |
2013-02-12 |
Process for the preparation of dihydroquinazolines |
| US8084604 |
2011-12-27 |
Process for the Preparation of Dihydroquinazolines |
/////Letermovir, MK 8828, AIC 246, fast track status, US Food and Drug Administration, orphan drug status , European Medicines Agency
COC1=C(C=C(C=C1)C(F)(F)F)N2[C@H](C3=C(C(=CC=C3)F)N=C2N4CCN(CC4)C5=CC(=CC=C5)OC)CC(=O)O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....