
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

PAL, Palash; (IN).
GINJUPALLI, Sadasiva Rao; (IN).
SHARMA, Uday; (IN).
CHOWDARY, Talluri Bhushaiah; (IN).
MANTRI, Anand Vijaykumar; (IN).
GADE, Bharath Reddy; (IN).
KULKARNI, Gaurav; (IN)
Sugammadex (Org 25969, Bridion) is chemically known as Cyclooctakis-(l-→4)-[6-S-(2-carboxyethyl)-6-thio-a-D-glucopyranosyl]. Sugammadex is an agent for reversal of neuromuscular blockade by the neuromuscular blocking agents (NMBAs) rocuronium, vecuronium, pancuronium in general anesthesia. It is the first selective relaxant binding agent (SRBA). SRBAs are a new class of drugs that selectively encapsulates and binds NMBAs.
The word Sugammadex is derived from Su= Sugar and Gamma cyclodex = Cyclodextrin. Sugammadex is inert chemically and does not bind to any receptor. It acts by rapidly encapsulating steroidal NMBDs to form a stable complex at a 1 : 1 ratio and thus decreasing the free concentration of the drug from the plasma. This creates a concentration gradient favoring the movement of the remaining rocuronium molecules from the neuromuscular junction back into the plasma, where they are encapsulated by free Sugammadex molecules. The latter molecules also enter the tissues and form a complex with rocuronium. Therefore, the neuromuscular blockade of rocuronium is terminated rapidly by the diffusion of rocuronium away from the neuromuscular junction back into the plasma.
NMBDs are quaternary ammonium compounds with at least one charged nitrogen atom. Cyclodextrins have a lipophilic center but a hydrophilic outer core, attributable to negatively charged ions on their surface. These negatively charged ions on the surface of Sugammadex attract the positive charges of the quaternary ammonium relaxant, drawing the drug in to the central core of the cyclodextrin. The binding of the guest molecule into the host cyclodextrin occurs because of vander waal’s forces, hydrophobic and electrostatic interactions. The structure of the cyclodextrin is such that all four hydrophobic rings of the steroidal relaxant fit tightly within the concentric doughnut forming an inclusion complex. This has been confirmed by calorimetry and X-ray crystallography. Such a reaction occurs in the plasma not at the neuromuscular junction and the concentration of free rocuronium in the plasma decrease rapidly after Sugammadex administration.
[0004] US 6670340 disclose process for preparation of Sugammadex sodium. The process as disclosed in example 4 of this patent involves reaction of iodo γ-cyclodextrin intermediate with 3-mercapto propionic acid in presence of sodium hydride and DMF to give 6-per-deoxy-6-per-(3-carboxyethyl)thio-Y-cyclodextrin, sodium salt (Sugammadex sodium). The preparation of iodo intermediate, 6-per-deoxy-6-per-iodo-y-cyclodextrin is as given in example 3 which involves reaction of γ-cyclodextrin with iodine in presence of triphenylphosphine (PPh3) and DMF. In practice, and to develop a process that has to be taken from lab scale to manufacturing scale, purity is one of the most important criteria. Since this process involves use of triphenylphosphine reagent there is formation of triphenylphosphine oxide as a by-product. Removal of triphenylphosphine oxide from the reaction mass is very difficult as it requires repeated washing with the solvent, which leads to inconsistency in yield of final product Sugammadex sodium. Furthermore, the product was dialysed for 36 hours to get pure compound. The dialysis purification is expensive and provides product in lower yield and hence such processes are not feasible and economical at industrial scale.
[0005] Another process for preparing the intermediate compound, 6-perdeoxy-6-per-chloro gamma cyclodextrin as disclosed in WO2012025937 involves use of phosphorous halide in particular, phosphorous pentachloride. WO2012025937 also disclose process for preparation of Sugammadex sodium using this intermediate which involves a) reaction of gamma-cyclodextrin with phosphorous pentachloride and dimethylformamide to obtain 6-perdeoxy-6-per-chloro gamma cyclodextrin and b) reaction of 6-perdeoxy-6-per-chloro gamma cyclodextrin with 3-mercapto propionic acid in presence of alkali metal hydrides and an organic solvent to give Sugammadex sodium. Preparation of chloro gamma cyclodextrine intermediate using phosphorous pentachloride is associated with formation of phosphorous impurities during the reaction, which are difficult to remove and also it involves tedious workup procedure.
[0006] WO2014125501 discloses preparation of 6-perdeoxy-6-per-chloro gamma cyclodextrin using phosphorous pentachloride (see example 1). The process as given in example 1 of this patent application was repeated by the present inventors. The first step provided yellow to brown mass which lacked the powder form and the flow properties. The mass was pasty at times and difficult to filter. Thus the process was unclean and tedious. Overall, no consistent product was obtained. WO2014125501 also disclose preparation of Sugammadex sodium using this intermediate which involves reaction of 6-perdeoxy-6-per-halo-gamma-cyclodextrin with 3-mercapto propionic acid in presence of alkali metal alkoxide such as sodium methoxide and organic solvent, the drawback of this this reaction is that it needs anhydrous conditions for completion of the reaction.
[0007] It has been reported that the generation of impurities and obtaining less pure compounds are major concerns with Sugammadex. Applicant Nippon Organon K.K.in their “Report on the Deliberation Results” submitted to Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare, mentions as follows:
For related substances, specifications for 14 different related substances (Related Substance A, Org 48301, Related Substance B, Related Substance D, Related Substance E, Related Substance F, Related Substance G, Related Substance H, Related Substance I, Related Substance J, Related Substance K, Related Substance L, Related Substance M, Related Substance N), other individual related substances, and total related substances have been set. In the course of regulatory review, the specifications limit for 4 different related substances (Related Substance A, Related Substance D, Related Substance F, Related Substance G) have been changed based on the results of batch analyses. For related substances (degradation products), specifications for Related Substance E, Related Substance I, Related Substance C, Related Substance G, Related Substance D, Related Substance K, other individual degradation products, and total degradation products have been established. In the course of regulatory review, a specification for Impurity A which arises in *** (hidden part) step has been newly set and the specification limits for individual degradation products have been changed based on the results of batch analyses and stability studies.
The cause for change of the colour of the drug product (the light yellow-brown colour darkened) was investigated using liquid chromatography -ultraviolet-visible spectrophotometry (LC-UV/VIS) and liquid chromatography-mass spectrometry (LC-MS), which suggested that trace amounts of varieties of unspecified degradation products (unidentified), instead of a single degradation product, were involved and in addition to *** investigated in formulation development, *** and *** content of the drug substance, *** and *** during the manufacture of the drug product, and *** were considered to affect the color of the drug product. Therefore, *** and *** have been included in the drug substance specification and the relevant manufacturing process steps have been improved.
[0008] In view of the above it is clear that Sugammadex is not only prone to degradation but traces of degradation impurities affect and change the colour to yellowish brown and makes it unacceptable in quality. Therefore, it is crucial to carefully select the process to prepare pure Sugammadex sodium.
[0009] The reported purification techniques for Sugammadex sodium employ column chromatographic and membrane dialysis which are costly and not convenient in large scale operations. Therefore, the reported processes for preparation of Sugammadex sodium as discussed herein are time consuming and not economically and industrially viable.
Thus, there exist a need to provide a process of preparation of Sugammadex sodium which is simple, convenient, with easy work up procedure, economically efficient and the one which provides Sugammadex sodium in good yield and high purity.
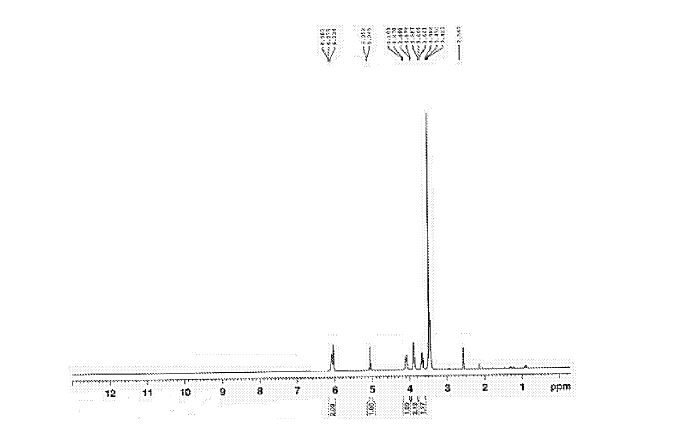
Figure 2 is 1HNMR of 6-perdeoxy-6-per-chloro gamma cyclodextrin
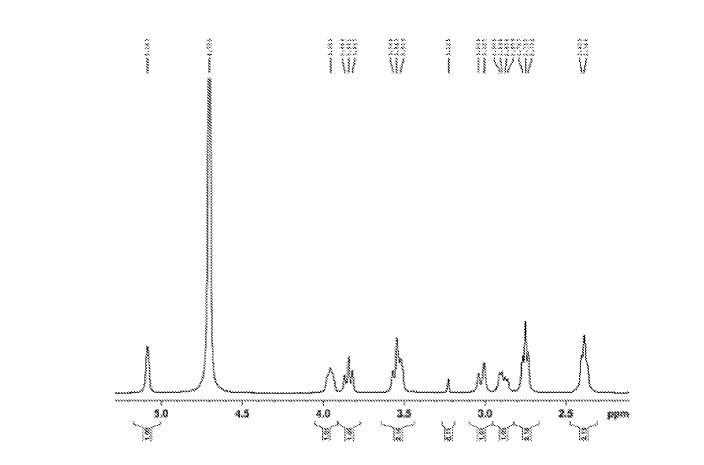
Figure 6 is 1HNMR of Sugammadex prepared according to example 6
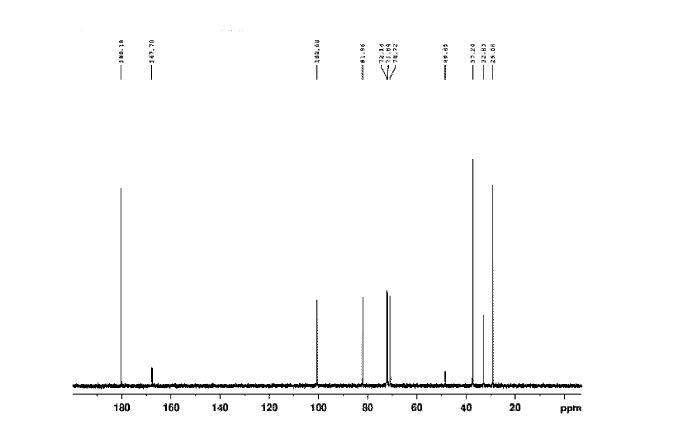
Figure 7 is 13CNMR of Sugammadex prepared according to example 6
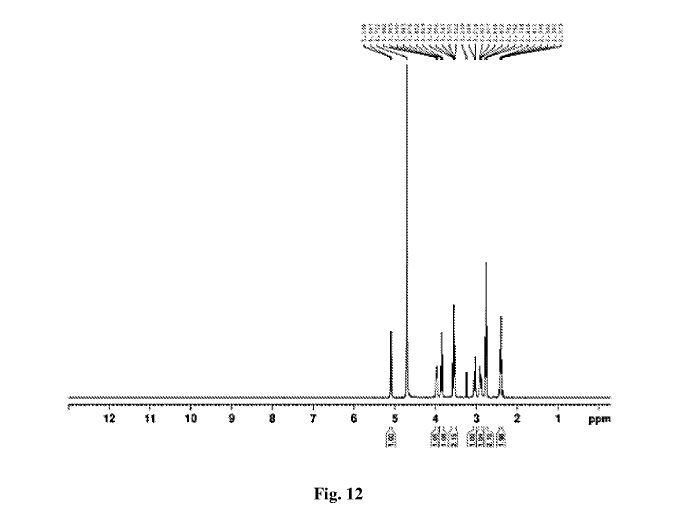
Figure 12 is 1HNMR of Sugammadex prepared according to example 8
SEE PATENT PLEASE
Figure 13 is HPLC profile of Sugammadex prepared according to process of example 1 of WO2014125501.
scheme 1.

scheme 2.

the process for preparation of Sugammadex sodium comprising reaction of 6-perdeoxy-6-per-chloro gamma cyclodextrin (Formula II) with 3-mercaptopropionic acid in presence of alkali metal amide selected from lithium amide, sodium amide (sodamide) or potassium amide to get Sugammadex sodium.

Sugammadex Sodium
scheme 4.

the present invention provides process for preparation of Sugammadex comprising reacting the acid of Sugammadex of formula (IV) with sodium hydroxide to form Sugammadex sodium of formula (I).

Formula IV Formula I
Scheme 6
scheme 7.


scheme 8.


scheme 9.

Examples
Example 1
[0079] Preparation of 6-perdeoxy-6-per-chloro gammacyclodextrin
In a four-neck round bottomed flask (2L) equipped with mechanical stirrer, thermometer pocket in a tub charged anhydrous DMF (250ml) under nitrogen atmosphere. Triphosgene (36.5g, 0.123mol) was added to the flask at 0-15°C and the mixture was stirred for lh. Dry gamma cyclodextrin (20g, 0.015mol) was added to the obtained slurry with stirring for 30 min followed by addition of DMF (50ml). The reaction mixture was heated at 65-70°C 16 h. After the completion of reaction, the reaction mixture was cooled and diisopropyl ether (800ml) was charged to the mixture to precipitate out the material. The solvent mixture of DMF and diisopropyl ether was decanted off from the reaction mixture to obtain gummy brown mass. The reaction mass was treated with saturated sodium bicarbonate solution (800ml) which leads to precipitation of the solid. The precipitated solid was filtered, washed with the water (250x3ml) and dried. This compound was used for the next step without any purification.
Yield: 95%, HPLC Purity: 99%
Example 2
[0080] Preparation of 6-perdeoxy-6-per-chloro gamma-cyclodextrin
In a 5L four-necked flask equipped with stirrer, dropping funnel, nitrogen inlet, and thermometer with pocket, oxalyl chloride (293.8g, 198.5ml, 2315mmol) was added to DMF (1200 ml) and maintained the mixture at 0-5°C under nitrogen followed by stirring at 20-25°C for lhr. A solution of gamma-cyclodextrin (lOOg, 77.16mmol) in DMF (500ml) was added to above mixture at 5-10°C under nitrogen. The mixture was stirred at 65-70°C for 14- 16 hr. After the completion of reaction, the reaction mixture was cooled to 20-25°C and diluted with diisopropyl ether (1.2L). The organic layer was decanted and the viscous residue was treated with 10% NaOH solution at 5- 10°C until PH = 8. The resulting slurry was stirred for one hour at 20-25°C. The slurry was filtered under vacuum and the solid was washed with water (3 x 500ml) and dried under vacuum. The crude material was suspended in methanol (750ml), stirred for 30min, filtered under vacuum and washed with diisopropyl ether (500ml). The solid obtained was dried at 55- 60°C in an oven for 12-16hr to afford the titled compound (95g).
Yield: 85%, Purity: 98%, melting point: 226-228°C
lH NMR (400 MHz, DMSO-d6): δ 6.0 (br s., 16 H), 4.99 (m, 8 H), 4.04 (d, J = 10 Hz, 8 H), 3.87
– 3.78 (m, 16H), 3.64 – 3.56 (m, 8 H), 3.46 – 3.34 (m, 16 H) ppm.
13C NMR (100 MHz, DMSO-d6): δ 101.98, 82.93, 72.30, 72.16, 71.11, 44.92 ppm.
Mass: m/z (M+Na)+ calcd for ![]()
1463.14; found: 1463.06.
Example 3
[0081] Preparation of 6-perdeoxy-6-per-chloro gamma-cyclodextrin
In a clean, dried 50L glass reactor equipped with stirrer, dropping funnel, nitrogen inlet, and thermometer with pocket was charged anhydrous dimethylformamide (15L, moisture content NMT 0.4%) while maintaining the temperature at 0-5°C (using dry ice acetone bath). Oxalyl chloride (2L, 23635mmol, 30eq) was added slowly over a period 4-5hr (while maintaining the temperature below 5°C) and stirring was continued for lhr at the same temperature. A solution of dry gamma-cyclodextrin (1.0kg, 770.94mmol) dissolved in dimethylformamide (5L) was added slowly into the above reaction mixture. The solution was heated at 65-70°C for 16hr. The reaction was monitored by TLC at regular intervals. After the completion of reaction, the reaction mixture was cooled to room temperature and diisopropyl ether (10L) was added to the reaction mixture with stirring. The gummy solid precipitate out. The upper layer solvent was decanted, the gummy brown material was cooled to 0 to 5°C and was neutralized (pH 8.0) with slow addition of aqueous sodium hydroxide solution (20%, 5L) with stirring. The slurry obtained was stirred for lhr at temperature 0 to 5°C. The precipitate was filtered, washed with the water (3 x 2L) and dried under vacuum. The wet cake was suspended into methanol (10L), stirred, filtered, washed with diisopropyl ether (2L) and dried in oven at 60°C for 14-16hr to give the titled compound (980g). Yield: 87.9%, Purity: 98.1% as measured by HPLC.
Example 4
[0082] Preparation of Sugammadex sodium
In a four-neck round bottomed flask (3L) equipped with mechanical stirrer, thermometer pocket in a tub under the nitrogen atmosphere, anhydrous DMF (300ml) and 3-Mercaptopropionic acid (18.3g, 0.172mol) were charged at 0-5°C followed by addition of sodamide (20g, O.38mol). The reaction mixture was stirred at the same temperature for lh. 6-perdeoxy-6-per-chloro gamma cyclodextrin (25g, 0.017mol, as obtained in example 1) was charged slowly. The reaction mixture was heated at 90-95°C for 16h. After completion of reaction, the reaction mixture was cooled to room temperature and methanol (300ml) was added to it. The mixture was stirred and the precipitated material was filtered off. The precipitated material was dissolved in a mixture of methanol (50ml) and water (50ml) and re-precipitated with the excess addition of methanol (450ml). The solid was filtered and dried. Yield: 76%
The dried solid was purified by the preparative HPLC method using formic acid buffer in mixture of acetonitrile and water (80:20%) followed by lyophilization to get acid of Sugammadex which is further converted to Sugammadex sodium using sodium hydroxide.
Example 5
[0083] Preparation of Sugammadex sodium
In a four-neck round bottomed flask (5L) equipped with mechanical stirrer, thermometer pocket in a tub under the nitrogen atmosphere, anhydrous DMF (1500ml) and 3-mercaptopropionic acid (HOg, 1038mmol) were charged at 0-5°C followed by addition of sodamide (81g, 2077mmol). The mixture was stirred at the same temperature for lh. 6-perdeoxy-6-per-chloro gamma cyclodextrin (lOOg, 69.25mmol, as obtained in example 1) was charged slowly. Extra DMF (500ml) was added to the mixture. The temperature of the mixture was raised to 80-85°C and maintained for 16h. After completion of reaction, the reaction mixture was cooled to room temperature and methanol (1500 ml) was added to it. The mixture was stirred and the precipitated material was filtered off. The precipitated material (wet cake) was dissolved in a mixture of methanol (800ml) and water (800ml). Charcoal (50g) was added and the mixture was stirred for 30mins at 50-55°C. The solution was filtered off through a pad of celite. Methanol (2500ml) was added the solution and precipitated solid was filtered and dried furnishing the titled compound (105g). Yield: 69.6%, Purity: 85.3%.
Example 6
[0084] Preparation of Sugammadex sodium
A clean, dried 10L four neck flask equipped with stirrer, dropping funnel, nitrogen inlet, and thermometer with pocket, was charged with a solution of sodium hydroxide (83g, 2077mmol) dissolved in water (100ml) followed by addition of anhydrous DMF (2L) maintained under inert atmosphere using nitrogen. A solution of 3-mercapto propionic acid (HOg, 1037mmol) in DMF (1L) was added slowly under nitrogen maintaining the temperature between 0-5°C. The mixture was stirred for another lhr at this temperature. A mixture of 6-deoxy-6-chloro gamma cyclodextrin (lOOg, 69mmol) in DMF (1L) was added slowly at 5-10°C. The resulting mixture was heated to 75-80°C for 16-20hr. After the completion of reaction, the reaction mixture was cooled to 25-30°C and methanol (1.5L) was added into the reaction mixture, the resulting precipitate was stirred at 20-25°C, filtered, and dried under vacuum. The dried solid was dissolved in water (1L), treated with activated carbon (50 g, 5%) at 50°C, stirred and filtered through celite. The filtrate was stirred at 60°C and excess methanol (2.5L) was added slowly to the filtrate to get the precipitate. The precipitated material was filtered under vacuum as white solid, washed with methanol (500ml) and dried in oven to give pure Sugammadex sodium (90 g).
Yield: 90 g, Purity: 91.2%.
lU NMR (400 MHz, D20): δ 5.09 (m, 8H); 3.98-3.94 (m, 8H); 3.88-3.83 (m, 8H); 3.58-3.52 (m, 16H); 3.07-3.01 (m, 8H); 2.92-2.87 (m, 8H); 2.78-2.74 (m, 16H); 2.34-2.47 (m, 16H) ppm.
13C NMR (100 MHz, D20): δ 180.18, 100.60, 81.96, 72.14, 71.84, 70.72, 37.24, 32.83, 29.06 ppm. Mass: m/z (M-Na7+H6)+ calcd for C72HnoNa048S8: 2023.12; found: 2023.39.
Example 7
Preparation of Sugammadex acid (Compound of formula IV)
In a clean, dried 5L four neck flask equipped with stirrer, dropping funnel, nitrogen inlet, and thermometer with pocket was charged dimethylformamide (1500ml) followed by addition of potassium hydroxide (194.0 g, 3464mmol) and the mixture maintained at 0-5°C. A solution of 3-mercapto propionic acid (186.35g, 153.0ml, 1756mmol) in DMF (500ml) was added to the reactor over a period of 30 minutes under nitrogen while maintaining the temperature between 0-5°C. The
resulting mixture was stirred at this temperature for 60 minutes. A solution of 6-deoxy-6-chloro gamma cyclodextrin (lOOg, 69.22mmol) in DMF (500ml) was added to the flask. The resulting mixture was heated at 110-120°C for 1.5-2hr while monitoring the progress of the reaction through HPLC. After completion of the reaction, the temperature of the reaction mixture was brought to 40-50°C and methanol (1000ml) was added to the mixture. The resulted precipitate was stirred at 20-25°C for lhr, filtered under vacuum and washed with methanol (500ml). The wet solid was dissolved in water (2000ml) with vigorous stirring and the solution was acidified with concentrated hydrochloric acid to give the white solid precipitate. The precipitated solid was filtered and suspended in ethyl acetate (500 ml), stirred for 30 minutes and filtered. The solid was dried to afford the titled compound (75g).
Yield: 55%, Purity: 95.8% as measured by HPLC.
lH NMR (400 MHz, DMSO-d6): δ 5.94 (br. s, 16H), 3.82-3.73 (m, 8H), 3.63-3.54 (m, 8H), 3.43-3.32 (m, 16H), 3.08-3.02 (m, 8H), 2.89-2.81 (m, 8H), 2.78-2.72 (m, 16H), 2.55-2.43 (m, 16H) ppm.
13C NMR (100 MHz, DMSO-d6): δ 173.00, 102.01, 83.94, 72.45, 72.33, 71.36, 34.53, 33.08, 27.87 ppm.
Mass: m/z (M-H2+K) + calcd for C72Hno048S8K: 2039.24; found: 2039.26.
Example 8
Preparation of Sugammadex Sodium
In a clean, dried 3L four neck flask equipped with stirrer, dropping funnel, nitrogen inlet, and thermometer with pocket, the compound (75g) as obtained in example 4 was dissolved in solution of sodium hydroxide (37.5g, 0.937mol) in water (100ml) and methanol (100ml). The pH of resultant mixture was maintained between 8-10. To this mixture methanol (1.5L) was slowly added at room temperature and the mixture was stirred for additional 30 minutes. The precipitated white solid was filtered off under vacuum and thoroughly washed with methanol (500ml). The solid was dried at 50°C under vacuum oven for 24hr to afford Sugammadex sodium (79g).
Yield: 96.9%, Purity: 95.5% measured by HPLC.

