
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
EU GMP Annex 1 Revision 2016 – what does the pharmaceutical industry expect?
DRUG REGULATORY AFFAIRS INTERNATIONAL

Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim
Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim talked in his keynote speech at the Pharma Congress 2016 about the revision of Annex 1 of the EU GMP Guide. Read here what the pharmaceutical industry expects form the new Annex 1.
Europe’s biggest Pharma Congress of its kind took place in Düsseldorf on 12 and 13 April. With more than 1000 participants, 90 exhibitors and 10 GMP conferences this Congress 2016 has been the biggest since the first one 18 years ago. 50 lectures, almost exclusively case studies from pharmacuetical companies such as Pfizer, Novartis, Boehringer Ingelheim and many more were discussed. Special attention was paid to the keynotes at the beginning of each congress day.
 Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim talked in his keynote…
Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim talked in his keynote…
View original post 876 more words
CFI-402257
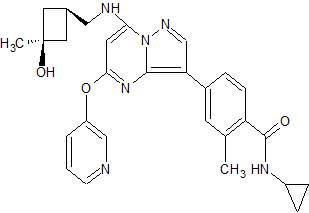
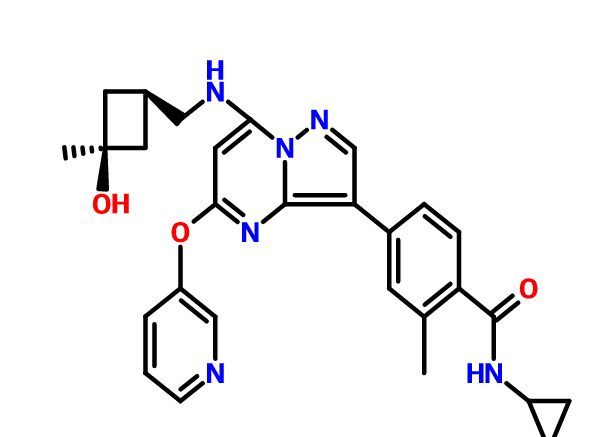

CFI-402257
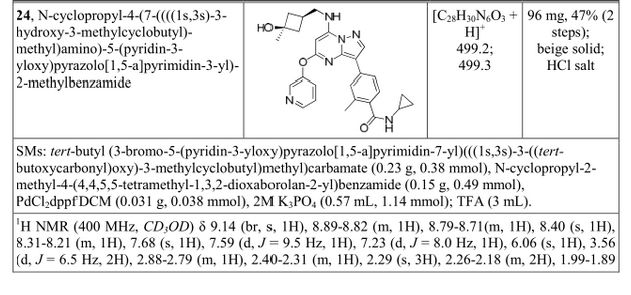
N-cyclopropyl-4-(7-((((1s,3s)-3-hydroxy-3-methylcyclobutyl)methyl)amino)-5-(pyridin-2-yloxy)pyrazolo[1,5-a]pyridin-3-yl)-2-methylbenzamide
N-cyclopropyl-4-(7-( (((Is, 3s)-3-hydroxy-3-methylcyclobutyl)methyl)amino)-5- (pyridin-3-yloxy)pyrazolol 1 , 5-a ]pyrimidin-3-yl)-2-methylbenzamide
CAS 1610759-22-2 (free base); 1610677-37-6 (HCl)
MF: C29H31N5O3
MW: 497.2427
CFI-402257 is a highly potent and selective TTK (threonine tyrosine kinase) Inhibitor ((TTK Ki = 0.1 nM) with potential anticancer activity. TTK is an essential chromosomal regulator and is overexpressed in aneuploid tumors. High TTK levels correlate with a high tumor grade11 and poor patient outcomes. TTK inhibition are associated with a disabled mitotic checkpoint, resulting in chromosome segregation errors, aneuploidy, and cell death.
Synthesis
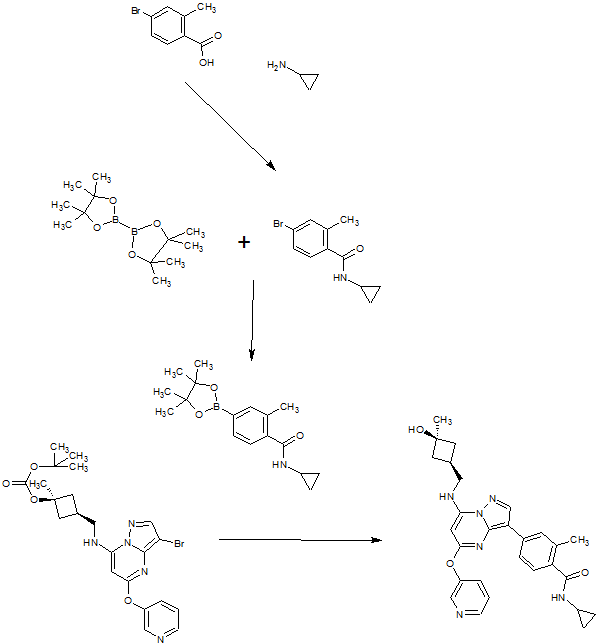
SYN OF INTERMEDIATE
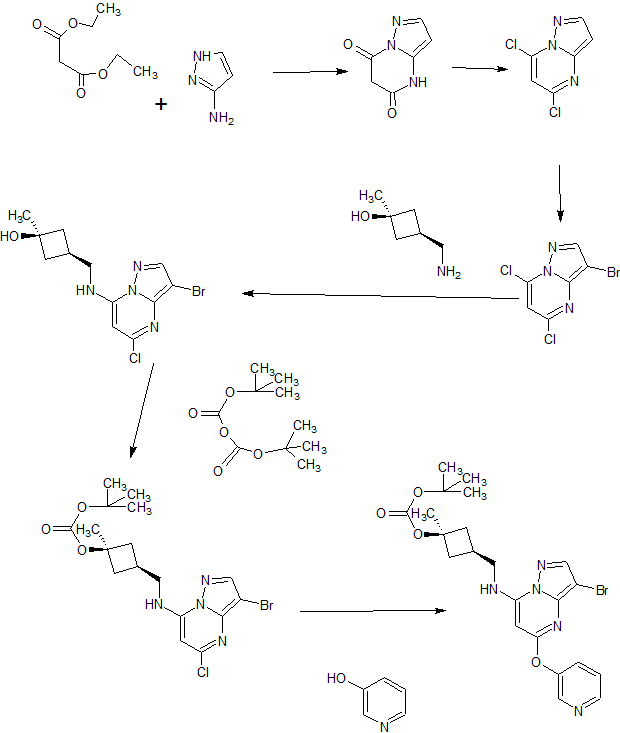
SYNTHESIS COLOUR INDICATED
SYN OF INTERMEDIATES
IF YOU HAVE ENJOYED IT ………EMAIL ME amcrasto@gmail.com, +919323115463, India

INDIA FLAG

DR ANTHONY CRASTO , WORLDDRUGTRACKER, HELPING MILLIONS, MAKING INDIA AND INDIANS PROUD
Protein kinases have been the subject of extensive study in the search for new therapeutic agents in various diseases, for example, cancer. Protein kinases are known to mediate intracellular signal transduction by effecting a phosphoryl transfer from a nucleoside triphosphate to a protein acceptor that is involved in a signaling pathway. There are a number of kinases and pathways through which extracellular and other stimuli cause a variety of cellular responses to occur inside the cell.
Human TTK protein kinase (TTK), also known as tyrosine threonine kinase, dual specificity protein kinase TTK, Monopolar Spindle 1 (Mpsl) and Phosphotyrosine -Picked Threonine Kinase (PYT), is a conserved multispecific kinase that is capable of phosphorylating serine, threonine and tyrosine residues when expressed in E. coli (Mills et al., J. Biol. Chem. 22(5): 16000-16006 (1992)). TTK mRNA is not expressed in the majority of physiologically normal tissues in human (Id). TTK mRNA is expressed in some rapidly proliferating tissues, such as testis and thymus, as well as in some tumors (for example, TTK mRNA was not expressed in renal cell carcinoma, was expressed in 50% of breast cancer samples, was expressed in testicular tumors and ovarian cancer samples) (Id). TTK is expressed in some cancer cell lines and tumors relative to normal counterparts (Id.; see also WO 02/068444 Al).
Therefore, agents which inhibit a protein kinase, in particular TTK, have the potential to treat cancer. There is a need for additional agents which can act as protein kinase inhibitors, in particular TTK inhibitors.
In addition, cancer recurrence, drug resistance or metastasis is one of the major challenges in cancer therapies. Cancer patients who responded favorably to the initial anticancer therapy often develop drug resistance and secondary tumors that lead to the relapse of the disease. Recent research evidences suggest that the capability of a tumor to grow and propagate is dependent on a small subset of cells within the tumor. These cells are termed tumor-initiating cells (TICs) or cancer stem cells. It is thought that the TICs are responsible for drug resistance, cancer relapse and metastasis. Compounds that can inhibit the growth and survival of these tumor-initiating cells can be used to treat cancer, metastasis or prevent recurrence of cancer. Therefore, a need exists for new compounds that can inhibit the growth and survival of tumor- imitating cells.
PATENT
WO 2015070349
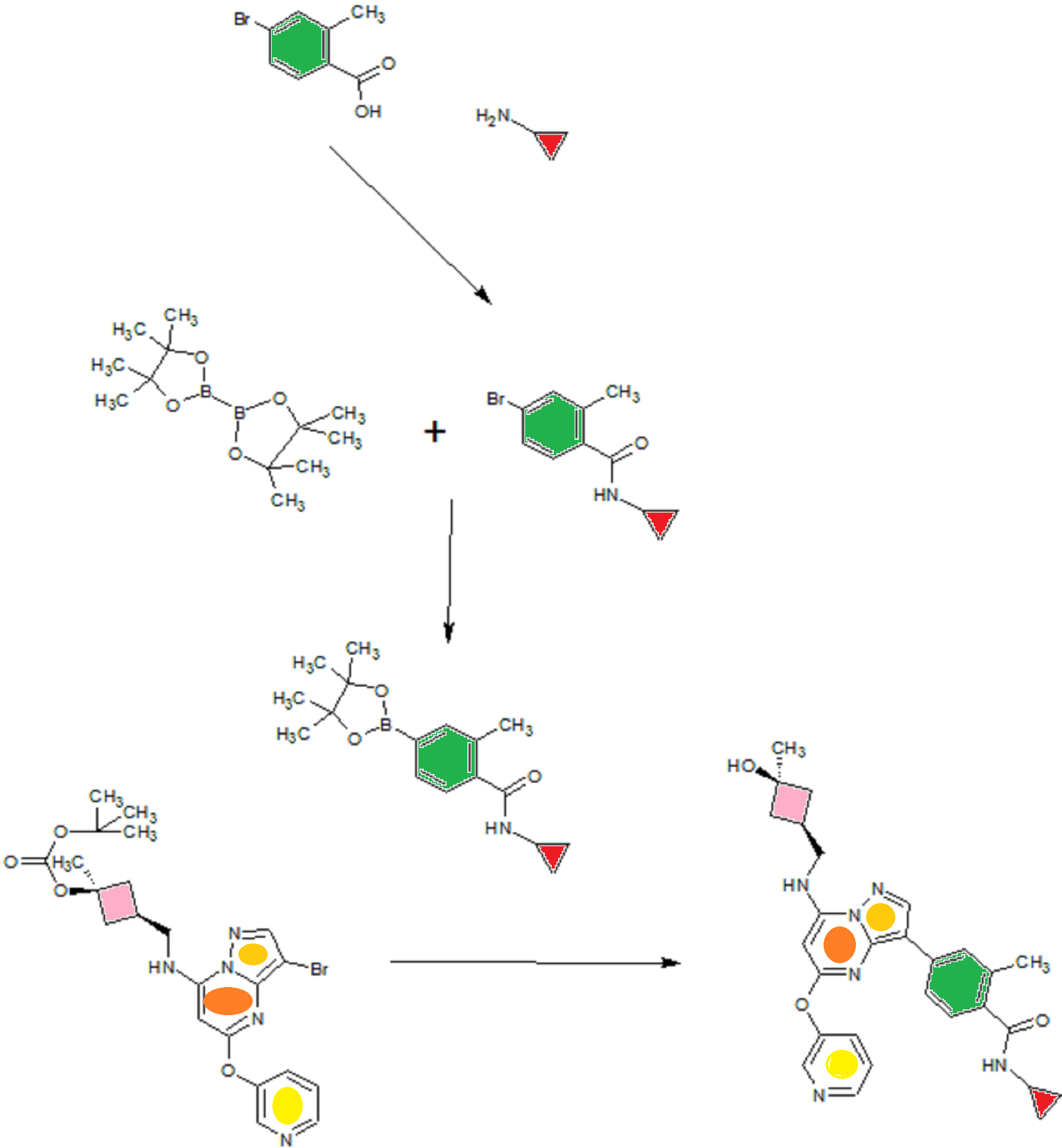
A4: N-cyclopropyl-4-(7-( (((Is, 3s)-3-hydroxy-3-methylcyclobutyl)methyl)amino)-5- (pyridin-3-yloxy)pyrazolol 1 , 5-a ]pyrimidin-3-yl)-2-methylbenzamide hydrochloride and its free base
A). Through Boc deprotection: A mixture of tert-butyl (3- bromo-5-(pyridin-3-yloxy)pyrazolo[l,5-a]pyrimidin-7- yl)(((ls,3s)-3-((tert-butoxycarbonyl)oxy)-3- methylcyclobutyl)methyl)carbamate (0.23 g, 0.38 mmol), N- cyclopropyl-2-methyl-4-(4,4,5,5-tetramethyl-l,3,2- 
dioxaborolan-2-yl)benzamide (0.15 g, 0.49 mmol), PdC dppfDCM (0.15 g, 0.49 mmol), and 2M K3P04 (0.57 mL, 1.14 mmol) in THF (4 mL) was charged with Ar and heated in the microwave at 130 °C for 3 h. Water and EtOAc were added to separate the phases and the aqueous phase was extracted with EtOAc. The combined organic extracts were dried over NaSC>4, filtered and concentrated. The crude product was purified by flash chromatography (gradient: EtOAc/hex 20-60%) to give a yellow oil.
The above intermediate was dissolved in DCM (10 mL) and treated with TFA (3 mL) at rt for 3 h. After reaction completion, solvent was removed in vacuo and the crude product was dissolved in MeOH (5 mL). The mixture was filtered and purified by prep-HPLC. The compound was passed through a PoraPak cartridge and triturated with Et20 to give the title compound as a free base (white solid). The free base was dissolved in MeOH (5 mL), and HC1 (1 M Et20, 2 equiv) was then added slowly. Solvent was removed in vacuo to give the title compound as a beige solid in HC1 salt (96 mg, 47% over 2 steps). ¾ NMR (400 MHz, CD3OD) δ ppm 9.14 (br. s, 1H), 8.89-8.82 (m, 1H), 8.79-8.71 (m, 1H), 8.40 (s, 1H), 8.31-8.21 (m, 1H), 7.68 (s, 1H), 7.59 (d, J = 9.5 Hz, 1H), 7.23 (d, J= 8.0 Hz, 1H), 6.06 (s, 1H), 3.56 (d, J= 6.5 Hz, 2H), 2.88-2.79 (m, 1H),
2.40-2.31 (m, 1H), 2.29 (s, 3H), 2.26-2.18 (m, 2H), 1.99-1.89 (m, 2H), 1.37 (s, 3H),
0.85-0.76 (m, 2H), 0.63-0.53 (m, 2H); MS ESI [M + H]+ 499.3, calcd for [C^HsoNeOs +
H]+ 499.2. HPLC purity: 99.5% at 254 nm.
B). Through PMB deprotection: A mixture of N- cyclopropyl-4-(7-((((ls,3s)-3-hydroxy-3- methylcyclobutyl)methyl)(4-methoxybenzyl)amino)-5- (pyridin-3-yloxy)pyrazolo[l,5-a]pyrimidin-3-yl)-2- methylbenzamide (9.6 g, 15.5 mmol), TFA (50 mL) in DCE
(70 mL) was heated in an oil bath at 50 °C for 4 h. After reaction completion, solvent was removed in vacuo and the crude product was dissolved in a mixture of MeOH/DCM (100 mL/25 mL). 2M Na2CC (150 mL) was then added and the resulting mixture was stirred at rt for 30 min. The reaction mixture was diluted with DCM and the phases were separated. The aqueous phase was extracted with DCM and the combined organic extracts were washed with water, dried over MgSC , filtered and concentrated. The crude product was triturated and sonicated in a mixture of DCM/Et20 (10 mL/70 mL) to give the title compound as a off white solid in free base (5.9 g, 77%). Ti NMR (400 MHz, CD3OD) δ ppm 8.58-8.53 (m, 1H), 8.50-8.46 (m, 1H), 8.36 (s, 1H), 7.86-7.80 (m, 1H), 7.76-7.72 (m, 1H), 7.61-7.55 (m, 2H), 7.18 (d, J = 8.0 Hz, 1H), 5.92 (s, 1H), 3.52 (d, J = 6.8 Hz, 2H), 2.86-2.77 (m, 1H), 2.38-2.28 (m, 1H), 2.25 (s, 3H), 2.24-2.18 (m, 2H), 1.99-1.88 (m, 2H), 1.37 (s, 3H), 0.84-0.75 (m, 2H), 0.64-0.54 (m, 2H); MS ESI [M + H]+ 499.2, calcd for [CzsHsoNgOs + H]+ 499.2. HPLC purity: 96.1% at 235 nm.
PATENT
WO 2014075168
PAPER
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00485

This work describes a scaffold hopping exercise that begins with known imidazo[1,2-a]pyrazines, briefly explores pyrazolo[1,5-a][1,3,5]triazines, and ultimately yields pyrazolo[1,5-a]pyrimidines as a novel class of potent TTK inhibitors. An X-ray structure of a representative compound is consistent with 11/2 type inhibition and provides structural insight to aid subsequent optimization of in vitro activity and physicochemical and pharmacokinetic properties. Incorporation of polar moieties in the hydrophobic and solvent accessible regions modulates physicochemical properties while maintaining potency. Compounds with enhanced oral exposure were identified for xenograft studies. The work culminates in the identification of a potent (TTK Ki = 0.1 nM), highly selective, orally bioavailable anticancer agent (CFI-402257) for IND enabling studies.
Discovery of Pyrazolo[1,5-a]pyrimidine TTK Inhibitors: CFI-402257 is a Potent, Selective, Bioavailable Anticancer Agent
REFERENCES
Discovery of Pyrazolo[1,5-a]pyrimidine TTK Inhibitors: CFI-402257 is a Potent, Selective, Bioavailable Anticancer Agent
Yong Liu, Radoslaw Laufer, Narendra Kumar Patel, Grace Ng, Peter B. Sampson, Sze-Wan Li, Yunhui Lang, Miklos Feher, Richard Brokx, Irina Beletskaya, Richard Hodgson, Olga Plotnikova, Donald E. Awrey, Wei Qiu, Nickolay Y. Chirgadze, Jacqueline M. Mason, Xin Wei, Dan Chi-Chia Lin, Yi Che, Reza Kiarash, Graham C. Fletcher, Tak W. Mak, Mark R. Bray, and Henry W. Pauls
Publication Date (Web): May 6, 2016 (Letter)
DOI: 10.1021/acsmedchemlett.5b00485
////TTK inhibitors, CFI-402257, pyrazolo[1,5-a]pyrimidines, 11/2 type inhibitors, 1610759-22-2, 1610677-37-6
C[C@]1(O)C[C@H](C1)CNc2cc(nc3c(cnn23)c5ccc(C(=O)NC4CC4)c(C)c5)Oc6cccnc6
Antimycobacterial Agents

Styryl Hydrazine Thiazole Hybrids
Will be updated………kindly email amcrasto@gmail.com
DATA
ABOUT Dehydrozingerone

Dehydrozingerone; Feruloylmethane; 1080-12-2; 4-(4-Hydroxy-3-methoxyphenyl)-3-buten-2-one; 4-(4-hydroxy-3-methoxyphenyl)but-3-en-2-one; Vanillalacetone;
http://pubs.acs.org/doi/abs/10.1021/np300465f

Dehydrozingerone (1) is a pungent constituent present in the rhizomes of ginger (Zingiber officinale) and belongs structurally to the vanillyl ketone class. It is a representative of half the chemical structure of curcumin (2), which is an antioxidative yellow pigment obtained from the rhizomes of turmeric (Curcuma longa). Numerous studies have suggested that 2 is a promising phytochemical for the inhibition of malignant tumors, including colon cancer. On the other hand, there have been few studies on the potential antineoplastic properties of 1, and its mode of action based on a molecular mechanism is little known. Therefore, the antiproliferative effects of1 were evaluated against HT-29 human colon cancer cells, and it was found that 1 dose-dependently inhibited growth at the G2/M phase with up-regulation of p21. Dehydrozingerone additionally led to the accumulation of intracellular ROS, although most radical scavengers could not clearly repress the cell-cycle arrest at the G2/M phase. Furthermore, two synthetic isomers of1 (iso-dehydrozingerone, 3, and ortho-dehydrozingerone, 4) were also examined. On comparing of their activities, accumulation of intracellular ROS was found to be interrelated with growth-inhibitory effects. These results suggest that analogues of 1 may be potential chemotherapeutic agents for colon cancer
PAPER

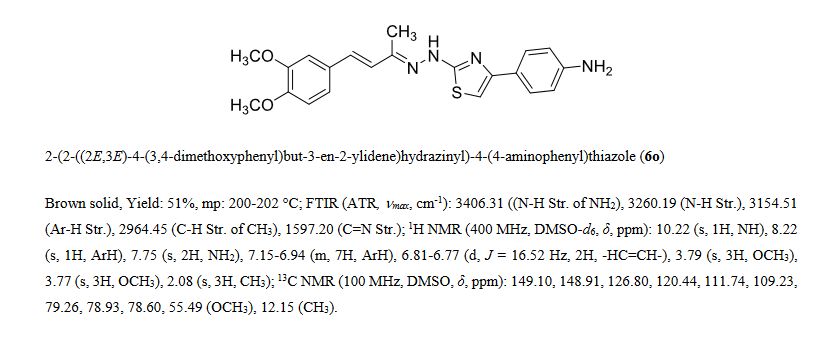
Series of styryl hydrazine thiazole hybrids inspired from dehydrozingerone (DZG) scaffold were designed and synthesized by molecular hybridization approach. In vitro antimycobacterial activity of synthesized compounds was evaluated against Mycobacterium tuberculosis H37Rv strain. Among the series, compound 6o exhibited significant activity (MIC = 1.5 μM; IC50 = 0.48 μM) along with bactericidal (MBC = 12 μM) and intracellular antimycobacterial activities (IC50 = <0.098 μM). Furthermore, 6o displayed prominent antimycobacterial activity under hypoxic (MIC = 46 μM) and normal oxygen (MIC = 0.28 μM) conditions along with antimycobacterial efficiency against isoniazid (MIC = 3.2 μM for INH-R1; 1.5 μM for INH-R2) and rifampicin (MIC = 2.2 μM for RIF-R1; 6.3 μM for RIF-R2) resistant strains of Mtb. Presence of electron donating groups on the phenyl ring of thiazole moiety had positive correlation for biological activity, suggesting the importance of molecular hybridization approach for the development of newer DZG clubbed hydrazine thiazole hybrids as potential antimycobacterial agents.
Dehydrozingerone Inspired Styryl Hydrazine Thiazole Hybrids as Promising Class of Antimycobacterial Agents
IF YOU HAVE ENJOYED IT ………EMAIL ME amcrasto@gmail.com, +919323115463, India

INDIA FLAG

DR ANTHONY CRASTO , WORLDDRUGTRACKER, HELPING MILLIONS, MAKING INDIA AND INDIANS PROUD
///////Antimycobacterial activity, bactericidal, dehydrozingerone, NIAID, thiazole, PRECLINCAL
c1(ccc(c(c1)OC)OC)/C=C/C(C)=N/Nc2nc(cs2)c3ccc(cc3)N
FDA approved a switchover from batch to the new technology for production of HIV drug Prezista, Darunavir on a line at its plant in Gurabo, Puerto Rico

Above is an Illustration example,
FDA urges companies to get on board with continuous manufacturing
The FDA gave Johnson & Johnson’s ($JNJ) Janssen drug unit the thumbs up last week for the continuous manufacturing process that it has been working on for 5 years. The agency approved a switchover from batch to the new technology for production of HIV drug Prezista on a line at its plant in Gurabo, Puerto Rico……http://www.fiercepharma.com/manufacturing/fda-urges-companies-to-get-on-board-continuous-manufacturing
 |
|
 |
SEE……http://www.en-cphi.cn/news/show-29367.html
Just after opening a refurbished manufacturing facility in Cape Town, South Africa earlier this year, pharma giant Johnson & Johnson ($JNJ) recently opened the doors to its Global Public Health Africa Operations office there.
The company has invested $21 million (300 million rand) in the facilities. The global public health facility will focus on HIV, tuberculosis and maternal, newborn and child health, South Africa – The Good News reported.
“This (investment) tells us that South Africa has the capability to provide a facility for world-class manufacturing,” Rob Davies, minister of the Department of Trade and Industry told the publication.
Johnson & Johnson, which has operated in South Africa for more than 86 years, planned to close the Cape Town manufacturing plant by the end of 2008 but was persuaded to keep the facility open for local manufacturing to serve sub-Saharan business. By 2015, the plant was cited by J&J as the most-improved in cost competitiveness from 30 company plants worldwide.
Earlier this month, the FDA gave J&J’s Janssen drug unit the go-ahead to proceed with the continuous manufacturing process it’s been working on for 5 years. The agency approved a switchover from batch to the new technology for production of HIV drug Prezista, Darunavir on a line at its plant in Gurabo, Puerto Rico.

AN EXAMPLE NOT RELATED TO DARUNAVIR
References
International Symposium on Continuous Manufacturing of Pharmaceuticals
Implementation, Technology & Regulatory
![]()
![]()
May 20-21, 2014 (Link to 2016 Meeting Website)
Continuous Bioprocessing
https://iscmp.mit.edu/white-papers/white-paper-4
READ
Achieving Continuous Manufacturing: Technologies and Approaches for Synthesis, Work-Up and Isolation of Drug Substance
https://iscmp.mit.edu/white-papers/white-paper-1
//////
//////FDA, HIV drug, Prezista, Darunavir, Gurabo, Puerto Rico
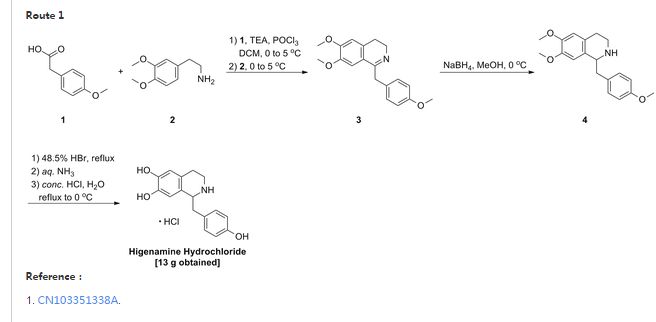
Higenamine Hydrochloride


Higenamine Hydrochloride
- 6,7-Isoquinolinediol, 1,2,3,4-tetrahydro-1-[(4-hydroxyphenyl)methyl]-, hydrochloride (9CI)
- 6,7-Isoquinolinediol, 1,2,3,4-tetrahydro-1-[(4-hydroxyphenyl)methyl]-, hydrochloride, (±)-
- (±)-Demethylcoclaurine hydrochloride
NDA Filed in china
A β-adrenoceptor partial agonist potentially for the treatment of coronary heart disease.
![]()

CAS No.11041-94-4 (Higenamine hydrochloride)

CAS 5843-65-2(free)
Higenamine (norcoclaurine) is a chemical compound found in a variety of plants including Nandina domestica (fruit), Aconitum carmichaelii (root), Asarum heterotropioides, Galium divaricatum (stem and vine), Annona squamosa, and Nelumbo nucifera (lotus seeds).
Legality
Higenamine, also known as norcoclaurine HCl, is legal to use within food supplements in the UK, EU, the USA and Canada. but banned use in The NCAA. Its main is within food supplements developed for weight management, also known as ‘fat burners’. However, products containing (or claiming to contain) pharmacological relevant quantities still require registration as a medicine. The regulatory boundaries for higenamine are unclear as modern formulations have not been clinically evaluated. Traditional formulations with higenamine have been used for thousands of years within Chinese medicine and come from a variety of sources including fruit and orchids. There are no studies comparing the safety of modern formulations (based on synthetic higenamine) with traditional formulations. Nevertheless, it will not be added to the EU ‘novel foods’ catalogue, which details all food supplements that require a safety assessment certificate before use.[1]

Pharmacology
Since higenamine is present in plants which have a history of use in traditional medicine, the pharmacology of this compound has attracted scientific interest. A variety of effects have been observed in in vitro studies and in animal models, but its effects in humans are unknown.
The results of a 2009 study exposed the compound as a β2 adrenergic receptor agonist.[2]
In animal models, higenamine has been demonstrated to be a β2 adrenoreceptor agonist.[2][3][4][5][6] Adrenergic receptors, or adrenoceptors, belong to the class of G protein–coupled receptors, and are the most prominent receptors in the adipose membrane, besides also being expressed in skeletal muscle tissue. These adipose membrane receptors are classified as either α or β adrenoceptors. Although these adrenoceptors share the same messenger, cyclic adenosine monophosphate (cAMP), the specific transduction pathway depends on the receptor type (α or β). Higenamine partly exerts its actions by the activation of an enzyme,adenylate cyclase, responsible for boosting the cellular concentrations of the adrenergic second messenger, cAMP.[7]
In a rodent model, it was found that higenamine produced cardiotonic, vascular relaxation, and bronchodilator effects.[8][9] In particular, higenamine, via a beta-adrenoceptor mechanism, induced relaxation in rat corpus cavernosum, leading to improved vasodilation and erectile function.
Related to improved vasodilatory signals, higenamine has been shown in animal models to possess antiplatelet and antithrombotic activity via a cAMP-dependent pathway, suggesting higenamine may contribute to enhanced vasodilation and arterial integrity.[2][7][9][10]
Toxicity
Regarding toxicity, researchers have suggested that the levels of higenamine reported in food consumption (estimated 47.5 mg in a 9-ounce serving of Lotus) would be comparable to the amount used in food supplements.[citation needed] Higenamine is a beta-adrenergic agonist which has effects on the function of trachea and heart muscles.[11][12]During a study of acute toxicity, mice were orally administered the compound at a dose of 2 g per kg of bodyweight. No mice died during the study.[13] higenamine is one of the main chemicals in a plant called aconite. Aconite has been shown to cause serious heart-related side effects including arrhythmias and even death. in some sources of HIGENAMINE from certain plants that have Aconite
PAPER
Chemical & Pharmaceutical Bulletin (1978), 26(7), 2284-5
https://www.jstage.jst.go.jp/article/cpb1958/26/7/26_7_2284/_pdf
PATENT
CN 103554022
http://google.com/patents/CN103554022B?cl=en
Example 1:
[0024] to the S-necked flask 200mL of anhydrous ammonia clever four furans, lOg instrument crumbs, olive mix was added 0. 5g ship, continue to embrace the mix was added 10 minutes after which 2 drops of 1,2-B burning desert, Continue mixing until the reaction mixture embrace color disappeared, the reaction was cooled to square ° C, and slowly mixed solution thereto 31. 6g4- methoxy Desert Festival and 50mL tetraammine clever furans dropped, about 60min addition was complete, the reaction fluid continues to cool to -65 ° C, to which was slowly dropping 20 percent, 7-dimethoxy-3,4-diamine different wow beep and a mixed solution of ammonia lOOmL four clever furans, the addition was complete continue to maintain – 65 ° C for 2 hours after the embrace slowly warmed 0 ° C, maintaining the internal temperature of 100 ° C 〇 blood slowly added to the reaction mixture, the addition was completed adding 200 blood continues to embrace mixed with ethyl acetate after 0.5 hours, allowed to stand liquid separation, organic phase was separated, dried over anhydrous sulfate steel, concentrated to afford 6, 7-dimethoxy -l- (4- methoxy section yl) -1,2, 3, 4-isopropyl tetraammine wow toot 24. 9g, a yield of 76.1%.
[00 Qiao] to the reaction flask prepared above 6, 7-dimethoxy -l- (4- methoxybenzyl) -1,2, 3, 4 tetraammine different wow beep 24. 9g , 47% aqueous ammonia desert 200 blood acid heated to 130 ° C reflux of cooled to room temperature, precipitation of large amount of solid, filtered higenamine ammonia salt desert, the solid was added 1. of water and continue to add 50 Blood mixed with ammonia football ground, filtered higenamine to higenamine was added lL4mol / L aqueous hydrochloric acid, 80 ° C heat to embrace mixed, cooled to 25 ° C filtration and drying to obtain a final product hydrochloric acid higenamine 11. 7g, a yield of 73.3%.
References
- http://ec.europa.eu/food/food/biotechnology/novelfood/novel_food_catalogue_en.htm
- Tsukiyama, M; Ueki, T; Yasuda, Y; Kikuchi, H; Akaishi, T; Okumura, H; Abe, K (2009). “Beta2-adrenoceptor-mediated tracheal relaxation induced by higenamine from Nandina domestica Thunberg”. Planta Medica 75 (13): 1393–9. doi:10.1055/s-0029-1185743. PMID 19468973.
- Kashiwada, Y; Aoshima, A; Ikeshiro, Y; Chen, YP; Furukawa, H; Itoigawa, M; Fujioka, T; Mihashi, K; et al. (2005). “Anti-HIV benzylisoquinoline alkaloids and flavonoids from the leaves of Nelumbo nucifera, and structure-activity correlations with related alkaloids”.Bioorganic & Medicinal Chemistry 13 (2): 443–8. doi:10.1016/j.bmc.2004.10.020.PMID 15598565.
- Kimura, I; Chui, LH; Fujitani, K; Kikuchi, T; Kimura, M (1989). “Inotropic effects of (+/-)-higenamine and its chemically related components, (+)-R-coclaurine and (+)-S-reticuline, contained in the traditional sino-Japanese medicines “bushi” and “shin-i” in isolated guinea pig papillary muscle”. Japanese journal of pharmacology 50 (1): 75–8.doi:10.1254/jjp.50.75. PMID 2724702.
- Kang, YJ; Lee, YS; Lee, GW; Lee, DH; Ryu, JC; Yun-Choi, HS; Chang, KC (1999). “Inhibition of activation of nuclear factor kappaB is responsible for inhibition of inducible nitric oxide synthase expression by higenamine, an active component of aconite root”. The Journal of Pharmacology and Experimental Therapeutics 291 (1): 314–20.PMID 10490919.
- Yun-Choi, HS; Pyo, MK; Park, KM; Chang, KC; Lee, DH (2001). “Anti-thrombotic effects of higenamine”. Planta Medica 67 (7): 619–22. doi:10.1055/s-2001-17361.PMID 11582538.
- Kam, SC; Do, JM; Choi, JH; Jeon, BT; Roh, GS; Chang, KC; Hyun, JS (2012). “The relaxation effect and mechanism of action of higenamine in the rat corpus cavernosum”.International Journal of Impotence Research 24 (2): 77–83. doi:10.1038/ijir.2011.48.PMID 21956762.
- Bai, G; Yang, Y; Shi, Q; Liu, Z; Zhang, Q; Zhu, YY (2008). “Identification of higenamine in Radix Aconiti Lateralis Preparata as a beta2-adrenergic receptor agonist1”. Acta pharmacologica Sinica 29 (10): 1187–94. doi:10.1111/j.1745-7254.2008.00859.x.PMID 18817623.
- Pyo, MK; Lee, DH; Kim, DH; Lee, JH; Moon, JC; Chang, KC; Yun-Choi, HS (2008). “Enantioselective synthesis of (R)-(+)- and (S)-(-)-higenamine and their analogues with effects on platelet aggregation and experimental animal model of disseminated intravascular coagulation”. Bioorganic & Medicinal Chemistry Letters 18 (14): 4110–4.doi:10.1016/j.bmcl.2008.05.094. PMID 18556200.
- Liu, W; Sato, Y; Hosoda, Y; Hirasawa, K; Hanai, H (2000). “Effects of higenamine on regulation of ion transport in guinea pig distal colon”. Japanese journal of pharmacology 84(3): 244–51. doi:10.1254/jjp.84.244. PMID 11138724.
- Wong, KK; Lo, CF; Chen, CM (1997). “Endothelium-dependent higenamine-induced aortic relaxation in isolated rat aorta”. Planta Medica 63 (2): 130–2. doi:10.1055/s-2006-957628. PMID 9140225.
- Ueki, T; Akaishi, T; Okumura, H; Morioka, T; Abe, K (2011). “Biphasic tracheal relaxation induced by higenamine and nantenine from Nandina domestica Thunberg”. Journal of pharmacological sciences 115 (2): 254–7. doi:10.1254/jphs.10251sc. PMID 21282929.
- Lo, CF; Chen, CM (1997). “Acute toxicity of higenamine in mice”. Planta Medica 63 (1): 95–6. doi:10.1055/s-2006-957619. PMID 9063102.
banned in ncaa https://www.ncaa.org/sites/default/files/2015-16%20NCAA%20Banned%20Drugs.pdf
| CN1539823A * | Oct 27, 2003 | Oct 27, 2004 | 中国医学科学院药物研究所 | Method for preparing new demethyl conclaurine and medinal salt |
| CN1764647A * | Mar 23, 2004 | Apr 26, 2006 | 埃科特莱茵药品有限公司 | Tetrahydroisoquinolyl acetamide derivatives for use as orexin receptor antagonists |
| CN103351338A * | Jun 17, 2013 | Oct 16, 2013 | 张家港威胜生物医药有限公司 | Simple preparation process of higenamine hydrochloride |
| US20060030586 * | Sep 27, 2004 | Feb 9, 2006 | Education Center Of Traditional Chinese Medicine Co. | Method and health food for preventing and/or alleviating psychiatric disorder, and/or for effectuating sedation |
| WO2011038169A2 * | Sep 24, 2010 | Mar 31, 2011 | Mallinckrodt Inc. | One-pot preparation of hexahydroisoquinolines from amides |
 |
|
| Names | |
|---|---|
| IUPAC name
1-[(4-Hydroxyphenyl)methyl]-1,2,3,4-tetrahydroisoquinoline-6,7-diol
|
|
| Other names
norcoclaurine, demethylcoclaurine
|
|
| Identifiers | |
| 5843-65-2 106032-53-5 (R) 22672-77-1 (S) |
|
| ChEBI | CHEBI:18418 |
| ChEMBL | ChEMBL19344 |
| ChemSpider | 102800 |
| Jmol 3D model | Interactive image |
| KEGG | C06346 |
| MeSH | higenamine |
| PubChem | 114840 |
| Properties | |
| C16H17NO3 | |
| Molar mass | 271.32 g·mol−1 |
/////
DSM 265 a promising Antimalarial

DSM265
DSM-265; PfSPZ
2-(1,1-difluoroethyl)-5-methyl-N-(4-(pentafluoro-l6-sulfanyl)phenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine
2-(l,l-difluoroethyl)-5-methyl-N-[4-(pentafluoro- 6– sulfanyl)phenyl] [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
(OC-6-21)-[4-[[2-(1,1-Difluoroethyl)-5-methyl[1,2,4]triazolo[1,5-a]pyrimidin-7-yl]amino]phenyl]pentafluorosulfur
1282041-94-4
Chemical Formula: C14H12F7N5S
Exact Mass: 415.0702
Board Of Regents, University Of Texas System, Monash University, Medicines For Malaria Venture
DSM265 is a long-duration, potent and selective dihydroorotate dehydrogenase (DHODH)) inhibitor. DSM265 is potential useful for the prevention and treatment of malaria. DSM265 is the first DHODH inhibitor to reach clinical development for treatment of malaria. DSM265 is highly selective toward DHODH of the malaria parasite Plasmodium, efficacious against both blood and liver stages of P. falciparum, and active against drug-resistant parasite isolates. DSM265 has advantages over current treatment options that are dosed daily or are inactive against the parasite liver stage.
- OriginatorMonash University; University of Texas Southwestern Medical Center; University of Washington
- Developer Center for Infectious Disease Research; Fred Hutchinson Cancer Research Center; Medicines for Malaria Venture; Takeda; United States Department of Defense
- Class Antimalarials; Pyrimidines; Small molecules; Triazoles
- Mechanism of Action Dihydroorotate dehydrogenase inhibitors
- Phase II Malaria
- Phase I Malaria
Most Recent Events
- 25 Apr 2016 Medicines for Malaria Venture and AbbVie plan a phase I bioavailability trial in Healthy volunteers in USA (PO, Granule) (NCT02750384)
- 01 Mar 2016 Phase-I clinical trials in Malaria prevention (In volunteers) in USA (PO) (NCT02562872)
- 01 Jan 2016 Phase-II clinical trials in Malaria in Peru (PO) (NCT02123290)
Malaria is one of the most significant causes of childhood mortality, but disease control efforts are threatened by resistance of the Plasmodium parasite to current therapies. Continued progress in combating malaria requires development of new, easy to administer drug combinations with broad-ranging activity against all manifestations of the disease. DSM265, a triazolopyrimidine-based inhibitor of the pyrimidine biosynthetic enzyme dihydroorotate dehydrogenase (DHODH), is the first DHODH inhibitor to reach clinical development for treatment of malaria. We describe studies profiling the biological activity, pharmacological and pharmacokinetic properties, and safety of DSM265, which supported its advancement to human trials. DSM265 is highly selective toward DHODH of the malaria parasite Plasmodium, efficacious against both blood and liver stages of P. falciparum, and active against drug-resistant parasite isolates. Favorable pharmacokinetic properties of DSM265 are predicted to provide therapeutic concentrations for more than 8 days after a single oral dose in the range of 200 to 400 mg. DSM265 was well tolerated in repeat-dose and cardiovascular safety studies in mice and dogs, was not mutagenic, and was inactive against panels of human enzymes/receptors. The excellent safety profile, blood- and liver-stage activity, and predicted long half-life in humans position DSM265 as a new potential drug combination partner for either single-dose treatment or once-weekly chemoprevention. DSM265 has advantages over current treatment options that are dosed daily or are inactive against the parasite liver stage.


A new single-dose malaria drug is offering promise as both a cure to malaria and also a way to prevent the disease according to researchers at UT Southwestern Medical Center. The new drug, which is known as DSM265, kills the drug-resistant malaria parasites in the blood and liver by targeting the ability of the parasites to replicate.

Malaria is a very infectious disease that is transmitted by mosquitoes, and it kills about 600,000 people worldwide every year. Most of the people who are killed by malaria are under 5-years-old, and it’s more common in sub-Saharan Africa. Almost 200 million cases of malaria are reported every year, with about 3 billion people in 97 countries at risk for the disease. Lead author Dr. Margaret Phillips, who is a professor of Pharmacology at UT Southwestern said that this could be the first single-dose cure for malaria, and would be used in partnership with another drug. This drug could also be developed into a once-a-week preventive vaccination as well, and the results of the study were just published in Science Translational Medicine. Not only was UT Southwestern involved in the research study, but Monash Institute of Pharmaceutical Sciences in Australia, the University of Washington, and the not-for-profit Medicines for Malaria Venture was also involved.



SYNTHESIS
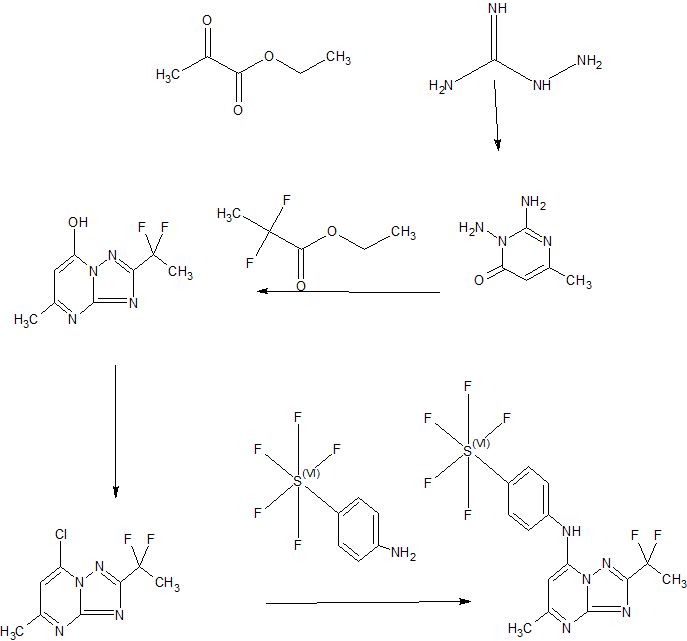

Plasmodium falciparum causes approximately 1 million deaths annually. However, increasing resistance imposes a continuous threat to existing drug therapies. We previously reported a number of potent and selective triazolopyrimidine-based inhibitors of P. falciparum dihydroorotate dehydrogenase that inhibit parasite in vitro growth with similar activity. Lead optimization of this series led to the recent identification of a preclinical candidate, showing good activity against P. falciparum in mice. As part of a backup program around this scaffold, we explored heteroatom rearrangement and substitution in the triazolopyrimidine ring and have identified several other ring configurations that are active as PfDHODH inhibitors. The imidazo[1,2-a]pyrimidines were shown to bind somewhat more potently than the triazolopyrimidines depending on the nature of the amino aniline substitution. DSM151, the best candidate in this series, binds with 4-fold better affinity (PfDHODH IC50 = 0.077 μM) than the equivalent triazolopyrimidine and suppresses parasites in vivo in the Plasmodium berghei model.
Scheme 3

Example 44: Synthesis of 2-(l,l-difluoroethyl)-5-methyl-N-[4-(pentafluoro- 6– sulfanyl)phenyl] [ 1 ,2,4]triazolo[ 1 ,5-a]pyrimidin-7-amine.
A suspension of Intermediate 3 (5.84 g, 25.09 mmol) and 4-aminophenylsulfur pentafluoride (MANCHESTER, 5.5 g, 25.09 mmol) in ethanol (150 mL) was heated at 50 °C for 1 h. Heating resulted in the precipitation of a solid. The reaction mixture was concentrated under vacuum, redissolved in DCM (300 mL) and washed with aq. Na2C03 (2 x 350 mL). The organic layer was dried over Na2S04 and filtered. Then 8 g of silica gel were added and the mixture was concentrated under vacuum to dryness. The residue was purified (silica gel column, eluting with Hexane/EtOAc mixtures from 100:0 to 50:50%) to afford the title compound as a white solid.

1H NMR (400 MHz, DMSO-d6) δ ppm: 10.60 (bs, 1H), 7.97 (d, 2H), 7.67 (d, 2H), 6.79 (s, 1H), 2.47 (s, 3H), 2.13 (t, 3H); [ES+ MS] m/z 416 (MH)+.
PAPER
Journal of Medicinal Chemistry (2011), 54(15), 5540-5561
http://pubs.acs.org/doi/abs/10.1021/jm200592f

Drug therapy is the mainstay of antimalarial therapy, yet current drugs are threatened by the development of resistance. In an effort to identify new potential antimalarials, we have undertaken a lead optimization program around our previously identified triazolopyrimidine-based series of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitors. The X-ray structure of PfDHODH was used to inform the medicinal chemistry program allowing the identification of a potent and selective inhibitor (DSM265) that acts through DHODH inhibition to kill both sensitive and drug resistant strains of the parasite. This compound has similar potency to chloroquine in the humanized SCID mouse P. falciparum model, can be synthesized by a simple route, and rodent pharmacokinetic studies demonstrated it has excellent oral bioavailability, a long half-life and low clearance. These studies have identified the first candidate in the triazolopyrimidine series to meet previously established progression criteria for efficacy and ADME properties, justifying further development of this compound toward clinical candidate statu
PAPER

Malaria persists as one of the most devastating global infectious diseases. The pyrimidine biosynthetic enzyme dihydroorotate dehydrogenase (DHODH) has been identified as a new malaria drug target, and a triazolopyrimidine-based DHODH inhibitor 1 (DSM265) is in clinical development. We sought to identify compounds with higher potency against PlasmodiumDHODH while showing greater selectivity toward animal DHODHs. Herein we describe a series of novel triazolopyrimidines wherein the p-SF5-aniline was replaced with substituted 1,2,3,4-tetrahydro-2-naphthyl or 2-indanyl amines. These compounds showed strong species selectivity, and several highly potent tetrahydro-2-naphthyl derivatives were identified. Compounds with halogen substitutions displayed sustained plasma levels after oral dosing in rodents leading to efficacy in the P. falciparum SCID mouse malaria model. These data suggest that tetrahydro-2-naphthyl derivatives have the potential to be efficacious for the treatment of malaria, but due to higher metabolic clearance than 1, they most likely would need to be part of a multidose regimen
Tetrahydro-2-naphthyl and 2-Indanyl Triazolopyrimidines TargetingPlasmodium falciparum Dihydroorotate Dehydrogenase Display Potent and Selective Antimalarial Activity
REFERENCES
1: Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, Njoroge JW, El
Mazouni F, Lao Y, Kokkonda S, Tomchick DR, Deng X, Laird T, Bhatia SN, March S,
Ng CL, Fidock DA, Wittlin S, Lafuente-Monasterio M, Benito FJ, Alonso LM,
Martinez MS, Jimenez-Diaz MB, Bazaga SF, Angulo-Barturen I, Haselden JN, Louttit
J, Cui Y, Sridhar A, Zeeman AM, Kocken C, Sauerwein R, Dechering K, Avery VM,
Duffy S, Delves M, Sinden R, Ruecker A, Wickham KS, Rochford R, Gahagen J, Iyer
L, Riccio E, Mirsalis J, Bathhurst I, Rueckle T, Ding X, Campo B, Leroy D, Rogers
MJ, Rathod PK, Burrows JN, Charman SA. A long-duration dihydroorotate
dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci
Transl Med. 2015 Jul 15;7(296):296ra111. doi: 10.1126/scitranslmed.aaa6645.
PubMed PMID: 26180101; PubMed Central PMCID: PMC4539048.
2: Held J, Jeyaraj S, Kreidenweiss A. Antimalarial compounds in Phase II clinical
development. Expert Opin Investig Drugs. 2015 Mar;24(3):363-82. doi:
10.1517/13543784.2015.1000483. Epub 2015 Jan 7. Review. PubMed PMID: 25563531.
3: Gamo FJ. Antimalarial drug resistance: new treatments options for Plasmodium.
Drug Discov Today Technol. 2014 Mar;11:81-88. doi: 10.1016/j.ddtec.2014.03.002.
Review. PubMed PMID: 24847657.
4: Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El
Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen
I, Ferrer SB, Jiménez-Díaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst I,
Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA.
Structure-guided lead optimization of triazolopyrimidine-ring substituents
identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors
with clinical candidate potential. J Med Chem. 2011 Aug 11;54(15):5540-61. doi:
10.1021/jm200592f. Epub 2011 Jul 14. PubMed PMID: 21696174; PubMed Central PMCID:
PMC3156099.


/////DSM-265, PfSPZ, DSM-265, DSM 265, 1282041-94-4, (OC-6-21)-
FS(F)(F)(F)(C1=CC=C(NC2=CC(C)=NC3=NC(C(F)(F)C)=NN23)C=C1)F
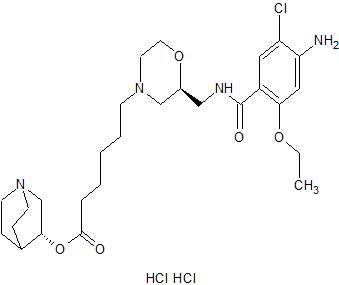
Quisapride Hydrochloride
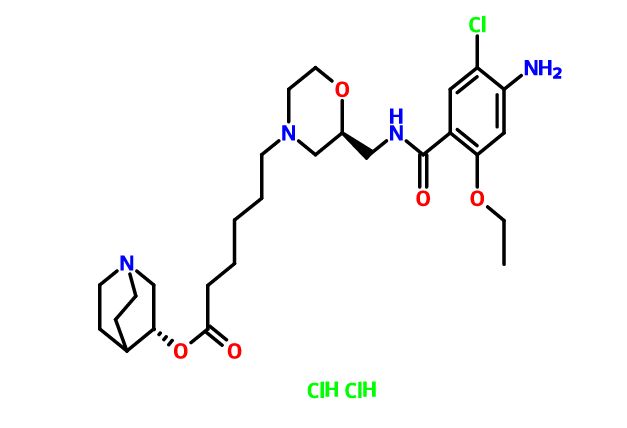
Quisapride Hydrochloride
(R) – quinuclidine-3-5 – ((S) -2 – (( 4 – amino-5-chloro-2-ethoxy benzoylamino) methyl) morpholino) hexanoate
IND Filed china
A 5-HT4 agonist potentially for the treatment of gastrointestinal motility disorders.
![]()
SHR-116 958, SHR 116958
CAS 1132682-83-7 (Free)
| Shanghai Hengrui Pharmaceutical Co., Ltd. |

CAS 1274633-87-2 (dihcl)
- (3R)-1-Azabicyclo[2.2.2]oct-3-yl (2S)-2-[[(4-amino-5-chloro-2-ethoxybenzoyl)amino]methyl]-4-morpholinehexanoate hydrochloride (1:2)
- SHR 116958
-
C27 H41 Cl N4 O5 . 2 Cl H,4-Morpholinehexanoic acid, 2-[[(4-amino-5-chloro-2-ethoxybenzoyl)amino]methyl]-, (3R)-1-azabicyclo[2.2.2]oct-3-yl ester, hydrochloride (1:2), (2S)-
5-HT is a neurotransmitter Chong, widely distributed in the central nervous system and peripheral tissues, 5-HT receptor subtypes at least seven, and a wide variety of physiological functions of 5-HT receptor with different interactions related. Thus, the 5-HT receptor subtypes research is very necessary.
The study found that the HT-5 4 receptor agonists useful for treating a variety of diseases, such as gastroesophageal reflux disease, gastrointestinal disease, gastric motility disorder, non-ulcer dyspepsia, functional dyspepsia, irritable bowel syndrome, constipation, dyspepsia, esophagitis, gastroesophageal disease, nausea, postoperative intestinal infarction, central nervous system disorders, Alzheimer’s disease, cognitive disorder, emesis, migraine, neurological disease, pain, cardiovascular disease, heart failure , arrhythmias, intestinal pseudo-obstruction, gastroparesis, diabetes and apnea syndrome.
The HT-5 4 receptor agonists into benzamides, benzimidazole class and indole alkylamines three kinds, which benzamides derivatives act on the neurotransmitter serotonin in the central nervous system by modulation, It showed significant pharmacological effect. The role of serotonin and benzamides derivatives and pharmacologically related to many diseases. Therefore, more and more people will focus on the human body produce serotonin, a storage position and the position of serotonin receptors, and to explore the relationship between these positions with a variety of diseases.
Commonly used in clinical cisapride (cisapride) and Mosapride (Tony network satisfied) is one of the novel benzamides drugs.

These drugs mainly through the intestinal muscle between the excited 5-HT neurofilament preganglionic and postganglionic neurons 4 receptor to promote the release of acetylcholine and enhancing cholinergic role in strengthening the peristalsis and contraction of gastrointestinal smooth muscle. In large doses, it can antagonize the HT-53 receptors play a central antiemetic effect, when typical doses, through the promotion of gastrointestinal motility and antiemetic effect. These drugs can increase the lower esophageal smooth muscle tension and promote esophageal peristalsis, improving the antrum and duodenum coordinated motion, and promote gastric emptying, but also promote the intestinal movement and enhanced features, increase the role of the proximal colon emptying, It is seen as the whole digestive tract smooth muscle prokinetic effect of the whole gastrointestinal drugs.
Mainly used for reflux esophagitis, functional dyspepsia, gastroparesis, postoperative gastrointestinal paralysis, functional constipation and intestinal pseudo-obstruction patients. Since there is slight antagonism cisapride the HT-5 3 and anti-D2 receptor, can cause cardiac adverse reactions, prolonged QT occurs, and therefore, patients with severe heart disease, ECG QT prolonged, low potassium, and low blood magnesium prohibited drug. Liver and kidney dysfunction, lactating women and children is not recommended. Due to increase between drug diazepam, ethanol, acenocoumarol, cimetidine and ranitidine the absorption of anticholinergic drugs may also antagonize the effect of this product to promote peristalsis of the stomach, should be aware of when using these, such as when diarrhea should reduce, anticoagulant therapy should pay attention to monitoring the clotting time. Mosapride selective gastrointestinal tract the HT-5 4 receptor agonists, there is no antagonism of D2 receptors, does not cause QT prolonged, reduce adverse reactions, mainly fatigue, dizziness, loose stools, mild abdominal pain , the efficacy of cisapride equivalent clinical effect broader Puka cisapride (prucalopride, Pru) of benzimidazole drugs, with high selectivity and specificity of the HT-5 4 receptor, increasing cholinergic neurotransmitters quality release, stimulate peristalsis reflex, enhance colon contraction, and accelerate gastric emptying, gastrointestinal motility to promote good effect, can effectively relieve the patient’s symptoms of constipation, constipation and for treatment of various gastrointestinal surgery peristalsis slow and weak, and intestinal pseudo-obstruction.
WO2005068461 discloses as the HT-5 4 receptor agonists benzamides compounds, particularly discloses compounds represented by the formula:

ATI-7505
ATI-7505 is stereoisomeric esterified. Cisapride analogs, safe and effective treatment of various gastrointestinal disorders, including gastroparesis, gastroesophageal reflux disease and related disorders. The drug can also be used to treat a variety of central nervous system disorders. ATI-7505 for the treatment or prevention of gastroesophageal reflux disease, also taking cisapride significantly reduced side effects. These side effects include diarrhea, abdominal cramps and blood pressure and heart rate rise.
Further, the compounds and compositions of this patent disclosure also useful in treating emesis and other diseases. Such as indigestion, gastroesophageal reflux, constipation, postoperative ileus, and intestinal pseudo-obstruction. In the course of treatment, but also taking cisapride reduce the side effects.
ΑΉ-7505 as the HT-5 4 receptor ligands may be mediated by receptors to treat the disease. These receptors are located in several parts of the central nervous system, modulate the receptor can be used to affect the CNS desired modulation.
ATI-7505 contained in the ester moiety does not detract from the ability of the compounds to provide treatment, but to make the compound easier to serum and / or cytosolic esterases degraded, so you can avoid the drug cytochrome P450 detoxification system, and this system with cisapride cause side effects related, thus reducing side effects.
The HT-Good 5 4 receptor agonists and should the HT-5 4 receptor binding powerful, while the other hardly shows affinity for the receptor, and show functional activity as agonists. They should be well absorbed from the gastrointestinal tract, metabolically stable and possess desirable pharmacokinetic properties. When targeting the receptor in the central nervous system, they should cross the blood-free, selectively targeting peripheral nervous system receptors, they should not pass through the blood-brain barrier. They should be non-toxic, and there is little proof of side effects. Furthermore, the ideal drug candidate will be a stable, non-hygroscopic and easily formulated in the form of physical presence.
Based on the HT-5 4 receptor agonists current developments, the present invention relates to a series of efficacy better, safer, less side effects of the benzamide derivatives.

Synthesis
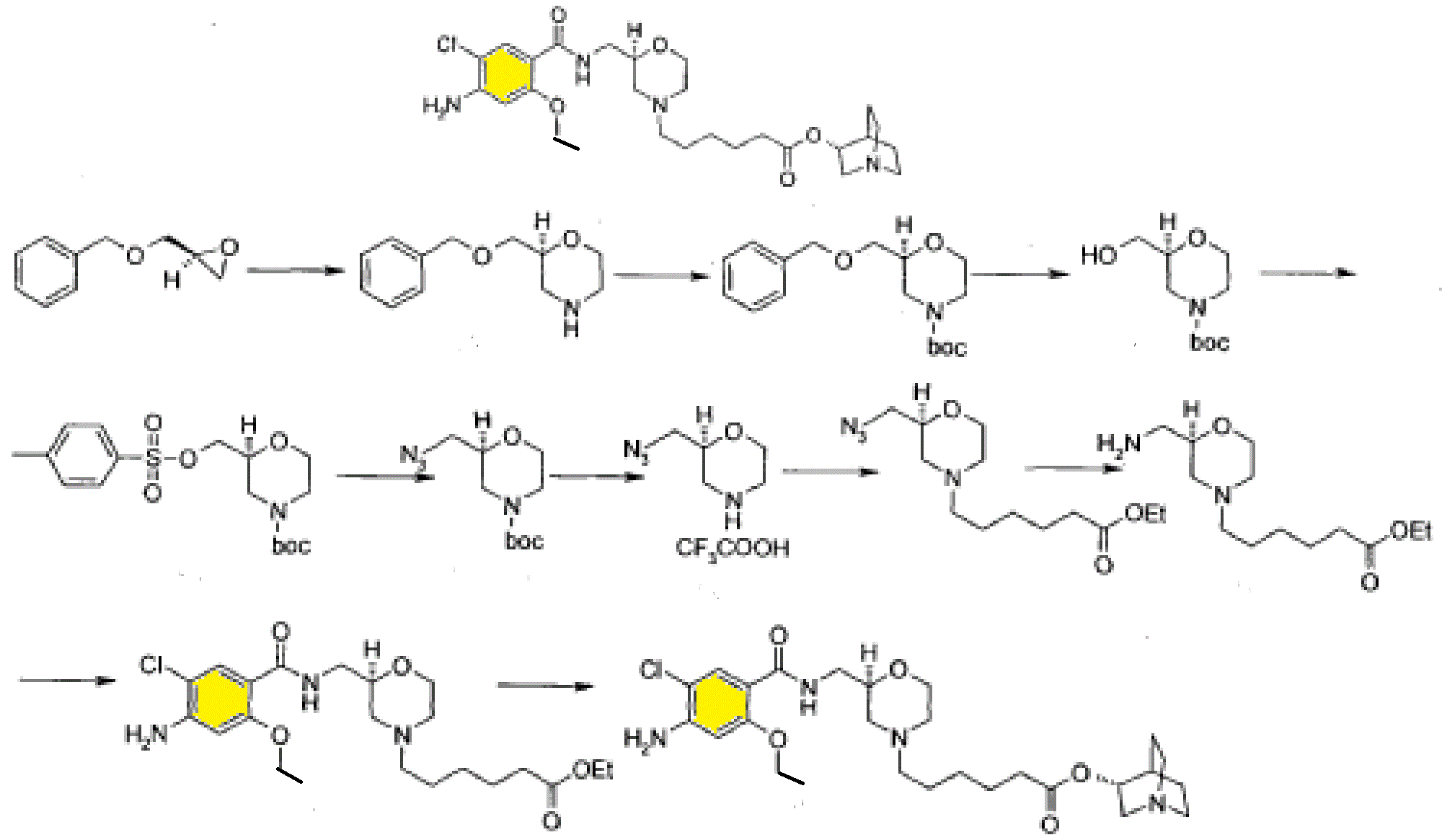
PATENT
Example 3
(R) – quinuclidine-3-5 – ((S) -2 – (( 4 – amino-5-chloro-2-ethoxy benzoylamino) methyl) morpholino) hexanoate



REFERENCES
China Pharmaceuticals: Asia Insight: China Has R&D
Nov 6, 2012 – levofolinate, sevoflurane inhalation, ambroxol hydrochloride, ioversol, etc ….. dextromethorphan hydrochloride 复方沙芬那敏. 3.2 …… quisapride.
//////SHR-116 958, SHR 116958, Quisapride Hydrochloride, preclinical
Cl.Cl.Clc1cc(c(OCC)cc1N)C(=O)NC[C@H]4CN(CCCCCC(=O)O[C@H]3CN2CCC3CC2)CCO4
PDE4 inhibitor , Sumitomo Dainippon Pharma Company

2-[2-Methyl-1-(tetrahydro-2H-pyran-4-yl)-1H-benzimidazol-5-yl]-1,3-benzoxazole Hemifumarate
Sumitomo Dainippon Pharma Company,
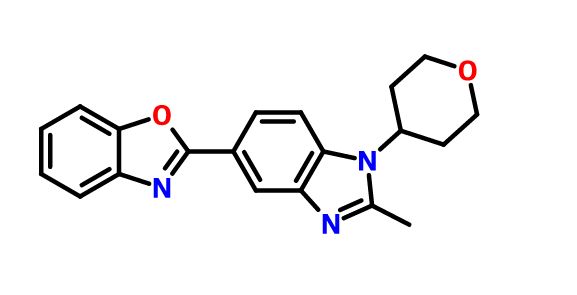
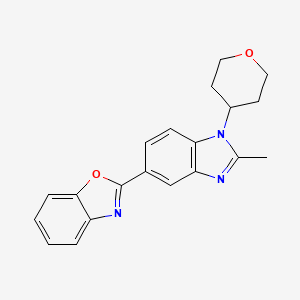
CAS FREE FORM 1256966-65-0
Benzoxazole, 2-[2-methyl-1-(tetrahydro-2H-pyran-4-yl)-1H-benzimidazol-5-yl]-
1H NMR (400 MHz, DMSO-d6)
13C NMR (100 MHz, DMSO-d6)


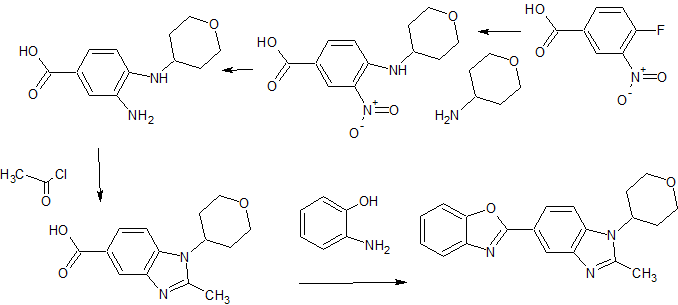
5- (benzoxazol-2-yl) -2-methyl -1-(tetrahydropyran-4-yl) benzimidazole eggplant flask (100 mL), 2- methyl-1- (tetrahydropyran – 4-yl) reference benzimidazole-5-carboxylic acid (example 4-3) (0.64 g, 2.46 mmol ), 2- amino-phenol (0.32 g, 2.95 mmol), and polyphosphoric acid (about 18 g) put, heated to 160 ℃, and the mixture was stirred for 17 hours. After cooling, ice was added, and the mixture was about pH 9 the liquid with concentrated aqueous ammonia (28%). Extraction with chloroform (about 50 mL X 3 times), dried over anhydrous magnesium sulfate, the crude product obtained by distilling off the solvent (0.08 g) PTLC (CHCl 3 by weight deploy purified), the title compound ( 0.002 g, 0.2% yield) was obtained as a yellow-brown semi-solid. 1H-NMR (CDCl 3 ) Deruta (Ppm): 1.88-1.92 (M, 2 H), 2.58-2.68 (M, 2 H), 2.70 (S, 3 H), 3.57-3.64 (M , 2 H), 4.21-4.25 (m , 2 H), 4.43-4.49 (m, 1 H), 7.29 (d, 1H, J = 9.2 Hz), 7.33-7.35 (m, 2 H ), 7.59-7.62 (m, 1 H ), 7.76-7.78 (m, 1 H), 8.18 (dd, 1 H, J = 8.6, 1.6 Hz), 8.57 (d, 1 H, J = 1.4 Hz).

PAPER

A short and practical synthetic route of a PDE4 inhibitor (1) was established by using Pd–Cu-catalyzed C–H/C–Br coupling of benzoxazole with a heteroaryl bromide. The combination of Pd(OAc)2-Cu(OTf)2-PPh3 was found to be effective for this key step. Furthermore, telescoping methods were adopted to improve the yield and manufacturing time, and a two-step synthesis of1 was accomplished in 71% overall yield.
Direct Synthesis of a PDE4 Inhibitor by Using Pd–Cu-Catalyzed C–H/C–Br Coupling of Benzoxazole with a Heteroaryl Bromide
///////////PDE4 inhibitor , Sumitomo Dainippon Pharma Company
Cc1nc3cc(ccc3n1C2CCOCC2)c4nc5ccccc5o4
ICH M7
DRUG REGULATORY AFFAIRS INTERNATIONAL

ICH M7


View original post 1,776 more words
EMA publishes Q A on data required for sterilized primary packaging materials used in aseptic manufacturing processes
DRUG REGULATORY AFFAIRS INTERNATIONAL

The European Medicines Agency, EMA, recently published questions and answers on what data is required for sterilisation processes of primary packaging materials subsequently used in an aseptic manufacturing process. Read more about “What data is required for sterilisation processes of primary packaging materials subsequently used in an aseptic manufacturing process?“.
The European Medicines Agency, EMA, recently published questions and answers on quality of packaging materials (H+V April 2016):
“3. What data is required for sterilisation processes of primary packaging materials subsequently used in an aseptic manufacturing process?
Terminal sterilisation of the primary packaging, used subsequently during aseptic processing of the finished product, is a critical process and the sterility of the primary container is a critical quality attribute to ensure the sterility of the finished product. Both need to be assured for compliance with relevant Pharmacopoeial requirements for the finished product and product approval.
The site where sterilisation…
View original post 556 more words
