Graphical abstract

WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Men suffering from sexual dysfunction can be successful at reversing their problem, by focusing on lifestyle factors and not just relying on medication, according to new research at the University of Adelaide.
In a new paper published in the Journal of Sexual Medicine, researchers highlight the incidence of erectile dysfunction and lack of sexual desire among Australian men aged 35-80 years.
Over a five-year period, 31% of the 810 men involved in the study developed some form of erectile dysfunction.
“Sexual relations are not only an important part of people’s wellbeing. From a clinical point of view, the inability of some men to perform sexually can also be linked to a range of other health problems, many of which can be debilitating or potentially fatal,” says Professor Gary Wittert, Head of the Discipline of Medicine at the University of Adelaide and Director of the University’s Freemasons Foundation Centre for…
View original post 255 more words

DS-8587
Daiichi Sankyo (Japan)
7-[3a(R)-Amino-6a(S)-fluoroperhydrocyclopenta[c]pyrrol-2-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropyl]-8-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride dihydrate
7-[(1S,6S)-1-amino-4-oxa-8-azabicyclo[4.3.0]nonan-8-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropan-1-yl]-1,4-dihydro-8-methoxy-4-oxoquinoline-3-carboxylic acid
| C21 H22 F3 N3 O3 . Cl H . 2 H2 O | |
| Mw | 493.904 |
DS-8587, a new broad-spectrum antibacterial agent, is in phase I clinical trials at Daiichi Sankyo for the treatment of bacterial infection.
DS-8587, from Daiichi Sankyo, is a fluoroquinolone with improved activity against both Gram-negative and Gram-positive bacteria. The compound is especially effective against Acinetobacter baumannii but also has improved activity against streptococci, staphylococci, enterococci, E. coli, and anaerobes . The compound is currently under Phase I of clinical development .
DS-8587, a new generation of fluoroquinolone, against Acinetobacter baumannii. The MICs against clinical isolates and inhibitory activity against target enzymes of DS-8587 was superior to ciprofloxacin and levofloxacin. Furthermore, the antibacterial activity of DS-8587 was less affected by adeA/adeB/adeC or abeM efflux pumps and frequency of single-step mutations with DS-8587 was lower as compared to those with ciprofloxacin. DS-8587 might be an effective agent against A. baumanniiinfection.
WO 2008082009 or
http://www.google.com/patents/EP2540715A1?cl=en
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

[Reference Example 72]
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-4-fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

[Reference Example 73]
(3S)-4-Fluoro-3-(3-hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

[Reference Example 74]
(3S)-3-(3-benzenesulfonyloxy-1-propyl)-4-Fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

[Reference Example 75]
(1S,5R)-5-Fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octan-1-ylcarboxylic acid tert-butyl ester

[Reference Example 76]
(1R,5R)-1-(tert-Butoxycarbonylamino)-5-fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane

[Reference Example 77]
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane

(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-azabicyclo[3.3.0]octane

7-[(1R,5S)-1-Amino-5-fluoro-3-azabicyclo[3.3.0]octan-3-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid

WO 2012018105
http://www.google.st/patents/WO2012018105A1?cl=en
The following structural formula

compound represented by, 7 – [(1R, 5S) -1 – amino-5 – fluoro-3 – azabicyclo [3.3.0] octan-3 – yl] -6 – fluoro -1 – [(1R, . the 2S) -2 – fluoro-cyclopropane-1 – yl] -1,4 – dihydro-8 – methyl-4 – oxo-3-quinoline -) is referred to as carboxylic acid (hereinafter referred to as Compound A multi-agent containing quinolone resistance including resistant Gram-positive cocci resistant pneumococcus, etc., widely against gram-negative bacteria from Gram-positive bacteria, and, in addition to show strong antibacterial activity, convulsions, which is known in the art as a side effect of the antimicrobial agent of the present system potential cardiotoxicity light and toxicity-inducing activity (photosensitivity), has been reported recently in clinical further (QT prolongation), blood sugar abnormalities, and to express the side effects of delayed-type drug 疹等 is excellent safety low, Then, it is excellent oral absorbability and organ migration properties become apparent, is expected as an antimicrobial agent superior (Patent Document 1).
WO 2008/082009 pamphlet
The compound A, was synthesized according to the method described in Patent Document 1.
Preparation 7 1 hydrochloride dihydrate Ratings (1) Compound A crystalline acid addition salt preparation of acid addition salts of (Example 1) Compound A, and Compound A – [(1R, 5S) – 1 – amino-5 – fluoro-3 – azabicyclo [3.3.0] octan-3 – yl] -6 – fluoro -1 – [(1R, 2S) -2 – fluoro-cyclo-1 – yl] -1 , 4 – dihydro-8 – methyl-4 – oxo-3-quinoline – was added 1mol / L hydrochloric acid (74μL) carboxylic acid (Compound A) (31.3mg,, 0.074mmol) in, and dried under reduced pressure at room temperature.10% aqueous 2 the residue – was added to (100μL) propanol was dissolved by heating at 60 ℃, and allowed to stand day out on the room temperature. Collected by filtration the precipitated crystals, and the 1st air dried, 19.9mg (yield: 54%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 2H 2 O
Theoretical value: C; 51.07, H; 5.51, N; 8.51, F; 11.54, Cl; 7.18
Measured value: C; 50.93, H; 5.40, N; 8.49, F; 11.30, Cl; 7.47
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 5.3,7.9,10.6,13.3,21.1,23.0,25.1,27.6 (°)
2 5% aqueous (1001.6mg, 0.746mmol) in the preparation of Compound A one hydrochloride monohydrate (2) Compound A – was added propanol (30mL), was dissolved by heating at 60 ℃. After stirring day out on the room temperature and stirred for 6 hours at 10 ℃. Collected by filtration the precipitated crystals, and the 1st dried air, 839.3 mg (yield: 87%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 1H 2 O
Theoretical value: C; 53.00, H; 5.30, N; 8.83, F; 11.98, Cl; 7.45
Measured value: C; 53.25, H; 5.43, N; 8.51, F; 11.58, Cl; 7.18
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 11.3,14.0,20.1,21.4,22.8,24.0,26.0,26.6 (°)
ref
| EP0343524A1 | May 19, 1989 | Nov 29, 1989 | Shionogi Seiyaku Kabushiki Kaisha | Pyridonecarboxylic acids and antibacterial agents |
| JPH0395176A | Title not available | |||
| JPH02231475A | Title not available | |||
| JPH08225567A | Title not available | |||
| JPS6345261A | Title not available | |||
| JPS6456673A | Title not available | |||
| JPS61282382A | Title not available | |||
| US5017708 * | Sep 8, 1989 | May 21, 1991 | Shionogi & Co., Ltd. | Azabicycloalkanes |
| WO1994014794A1 | Dec 28, 1993 | Jul 7, 1994 | Hideki Ao | 8-methoxyquinolonecarboxylic acid derivative |
| WO1995021163A1 | Feb 2, 1995 | Aug 10, 1995 | Katsumi Chiba | Pyridonecarboxylic acid derivative substituted by bicyclic amino group, ester thereof, salt thereof, and bicyclic amine as intermediate therefor |
| WO1996023782A1 | Feb 1, 1996 | Aug 8, 1996 | Daiichi Seiyaku Co | Heterocyclic compounds |

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.

1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid
1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
Ferrer Internacional (Spain), phase 3 Gram-positive
Ferrer Internacional has completed one Phase III clinical trial to evaluate the topical formulation of ozenoxacin in the treatment of impetigo [
|
poster……http://landing.quotientbioresearch.com/blog/bid/50380/Ozenoxacin-Activity-against-Atypical-Bacteria
Ozenoxacin is active against a great number of pathogens, such as Propionibacterium acnes, Staphylococcus aureus, methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA) including ciprofloxacin-resistant strains, methicillin-susceptible Staphylococcus epidermidis (MSSE), methicillin-resistant Staphylococcus epidermidis (MRSE), Streptococcus pyogenes, Group G Streptococci, penicillin-resistant Streptococcus pneumoniae, Beta-lactamase positive Haemophilus influenzae, non-typeable strains of Haemophilus influenzae, Beta-lactamase positive Moraxella catarrhalis, Neisseria meningitides, Legionella pneumophila, Mycoplasma pneumoniae, Legionella pneumophila, Mycobacterium tuberculosis, Streptococcus agalactiae group B, Neisseria gonorrhoeae, Chlamydia trachomatis, Mycoplasma hominis, Ureaplasma urealyticum Helicobacter pylori, Bacteroides fragilis, Clostridium perfringens, Escherichia coli, quinolone-resistant Escherichia coli, Salmonella spp., Shigella spp., ciprofloxacin-susceptible Pseudomonas aeruginosa, Clostridium difficile, and Listeria monocytogenes.
Ozenoxacin is a novel non-fluorinated quinolone antibacterial agent. It is currently in late stage phase 3 trials for the topical treatment of impetigo. The bacterial action of ozenoxacin is through the dual inhibition of DNA gyrase and topoisomerase IV. Excellent in vitro and in vivo antibacterial activity has been demonstrated in pre-clinical and clinical studies against a broad range of bacterial organisms. This includes organisms with emerging resistance to quinolones. Phase I and II clinical trials have also shown that ozenoxacin is a safe and effective antibacterial agent. No evidence of adverse effects as linked to topically formulated halogenated quinolones has been shown.
Ozenoxacin (I) was firstly disclosed in US6335447, and equivalent patents. Its chemical name is 1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid. Its chemical formula is: H

Ozenoxacin (I)
Topical application of antimicrobial agents is a useful tool for therapy of skin and skin structures infections, sexually transmitted diseases and genital tract infections and some systemic infections susceptible to topical treatment. Topical antimicrobial therapy has several potential advantages compared with systemic therapy.
Firstly, it can avoid an unnecessary exposure of the gut flora which may exert selection for resistance. Secondly, it is expected that the high local drug concentration in topical application and the negligible systemic absorption should overwhelm many mutational resistances. Thirdly, topical applications are less likely than systemic therapy to cause side effects. Accordingly, some topical compositions comprising ozenoxacin have been reported in the art.
JP2002356426A discloses ointments and gels for skin. An ointment comprising ozenoxacin 1%, N-methyl-2-pyrrolidone 8%, propylene glycol 14.9%, oleic acid 0.9%, diisopropanolamine 2.3%, polyethylene glycol 400 20.2%, polyethylene glycol 4000 50.2%, and water 3.2% is reported in Example 2.
JP2003226643A discloses aqueous solutions comprising ozenoxacin, cyclodextrin, and a viscous agent.
EP1731138A1 discloses fine particle dispersion liquid comprising ozenoxacin to be used in the manufacture of pharmaceutical compositions.
WO2007015453A1 discloses lotions comprising ozenoxacin.
JP2007119456A discloses aqueous suspensions containing nanoparticles and solution granules of ozenoxacin to be used in the manufacture of pharmaceutical compositions. Ophthalmic solutions are mentioned preferably. A combined use of ozenoxacin, magnesium ions, and hydroxypropyl-β-cyclodextrin specially for ophthalmic use is disclosed in Yamakawa, T. et al., Journal of Controlled Release (2003), 86(1 ), 101-103.
Semisolid topical compositions are useful alternatives to liquid compositions, because of their better manipulation and consequent patient preferences. However, in spite of the great diversity of components present in the semisolid compositions disclosed in the art, no quantitative stability studies are available for them.
Thus, there is a need of proved stable semisolid topical compositions comprising ozenoxacin as active ingredient, wherein microbiological and therapeutic activities are warranted because of demonstrated durable and prolonged pharmaceutical stability.
Synthesis
US6335447
http://www.google.co.in/patents/US6335447
EXAMPLE 5
To a solution of 0.80 g of 7-[6-({[(benzyloxy)-carbonyl] (methyl)amino}methyl)-5-methyl-3-pyrdyl]-1-cyclo-propyl-8-methyl-4-oxo-1,4-dihydro-3-quinoline-carboxylic acid in 16 ml of acetic acid was added 0.20 g of 5% (w/w) palladium-carbon and the mixture was stirred at ambient temperature and atmospheric pressure for 2 hours under a hydrogen atmosphere. The reaction mixture was filtered and the solvent was evaporated under reduced pressure. The obtained residue was dissolved in a mixed solvent consisting of 3.8 ml of ethanol and 3.8 ml of water. After adding 3.8 ml of an aqueous 1 mol/l sodium hydroxide solution thereto and adjusting the solution to pH 5.5with 1 mol/l hydrochloric acid, 10 ml of chloroform was added thereto. An organic layer was separated and dried over anhydrous magnesium sulfate and the solvent was evaporated under reduced pressure. Addition of diethyl ether to the obtained residue and filtration of crystals afforded 0.25 g of colorless crystals of 1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
IR (KBr) cm−1: 3322, 1721; NMR(d1-TFA) δ: 1.2-1.9 (4H, m), 2.94 (3H, s), 3.05 (3H, s), 3.29 (3H, s), 4.6-5.0 (1H, m), 5.12 (2H, s), 7.91 (1H, d, J=8.5 Hz), 8.6-9.0 (2H, m), 9.0-9.3 (1H, brs), 9.75 (1H, s). Melting point: 199° C.

FINAFLOXACIN
(S-cyano-1-cyclopropyl-ό-fluoro-T-^aS, 7aS)-hexahydropyrrolo [3,4- b]-1,4-oxazin-6(2H)-yl]-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid)
7-[(4aS,7aS)-3,4,4a,5,7,7a-hexahydro-2H-pyrrolo[3,4-b][1,4]oxazin-6-yl]-8-cyano-1-cyclopropyl-6-fluoro-4-oxoquinoline-3-carboxylic acid |
BAY-35-3377
BY-377
CAS Registry Number: 209342-40-5
HYD SALT
(-)-(4aS,7aS)-8-Cyano-1-cyclopropyl-6-fluoro-4-oxo-7-(perhydropyrrolo[3,4-b]-1,4-oxazin-6-yl)-1,4-dihydroquinoline-3-carboxylic acid hydrochloride
209342-41-6,
| C20 H19 F N4 O4 . Cl H | |
| MW | 434.849 |
Synonyms: Finafloxacin, UNII-D26OSN9Q4R,
MerLion Pharmaceuticals (Singapore)…POSTER…….http://www.merlionpharma.com/sites/default/files/file/PPS/F1-2036_Wohlert.pdf
H. pylori, Broad-Spectrum
Finafloxacin is a novel fluoroquinolone being developed by MerLion Pharmaceuticals. Under neutral pH conditions (pH 7.2–7.4), the compound has shown in vitro activity equivalent to that of ciprofloxacin. However, under slightly acidic pH5.8 the compound shows enhanced potency.
Other marketed fluoroquinolones, such as ciprofloxacin, levofloxacin and moxifloxacin, exhibit reduced activity at slightly acidic pH 5.0–6.5. This feature of finafloxacin makes the compound suitable for use in the treatment of infections in acidic foci of infections such as urinary tract infections
Finafloxacin hydrochloride, a novel highly potent antibiotic, is in phase III clinical trials at Alcon for the treatment of ear infections. MerLion Pharmaceuticals is evaluating the product in phase II clinical trials at for the treatment of Helicobacter pylori infection and for the treatment of lower uncomplicated urinary tract infections in females.
A quinolone, finafloxacin holds potential for the treatment of Helicobacter pylori infection and urinary tract infection. Unlike existing antibiotics, finafloxacin demonstrates a unique acid activated activity whereby it becomes increasingly active under acidic conditions.
In 2009, a codevelopment agreement was signed between Chaperone Technologies and MerLion Pharmaceuticals. In 2011, finafloxacin hydrochloride was licensed to Alcon by MerLion Pharmaceuticals in North America for the treatment of ear infections.
MerLion Pharmaceuticals has announced that the FDA has granted a Qualified Infectious Disease Product Designation and Fast Track Status for finafloxacin. The company is currently recruiting patients for the Phase II clinical trial of the compound for the treatment of complicated urinary tract infections (cUTI) and/or acute pyelonephritis compared to ciprofloxacin
Finafloxacin and derivatives thereof can be synthesized according to the methods described in U.S. Patent No. 6,133,260 to Matzke et al., the contents of which are herein incorporated by reference in their entirety. The compositions of the invention are particularly directed toward treating mammalian and human subjects having or at risk of having a microbial tissue infection. Microbial tissue infections that may be treated or prevented in accord with the method of the present invention are referred to in J. P. Sanford et al., “The Sanford Guide to Antimicrobial Therapy 2007” 37 Edition (Antimicrobial Therapy, Inc.). Particular microbial tissue infections that may be treatable by embodiments of the present invention include those infections caused by bacteria, protozoa, fungi, yeast, spores, and parasites.
SYNTHESIS
WO1998026779A1
http://www.google.sc/patents/WO1998026779A1 COPY PASTE ON BROWSER
8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-di-azabicyclo [4.3.0] non-8-yl)-l, 4-dihydro-4-oxo-3-quinolinecarboxylic acid.
The compounds, which are suitable for use in the invention are known already to some extent in EP-A-0350733, EP-A-0550903 as well as from DE-A-4329600 or can be prepared according to the processes described in .
If, for example 9,10-difluoro-3 ,8-dimethyl-7-oxo-2 ,3-dihydro-7H-pyrido [l ,2,3-d, e] [l, 3,4] benzoxadiazine-6 -carboxylic acid and 2-oxa-5 ,8-diazabicyclo [4.3.0] nonane, the reaction can be represented by the following equation:

The 7-halo-quinolonecarboxylic acid derivatives used for preparing the compounds of Fomel (I) of the invention are known or can be prepared by known methods. Thus, the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid, or of the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro- l been ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester described in EP-A-0 276 700th The corresponding 7-fluoro derivatives can be, for example, via the following reaction sequence to build:

An alternative process for preparing the intermediate compound 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride as the starting material for the preparation of 7-chloro-
8-cyano-1-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is used (EP-A-0276700) and in the 3-cyano-2 ,4,5-trifluoro- benzoyl can be converted, is based on 5-fluoro-l ,3-xylene, 5-fluoro-l ,3-xylene, in the presence of a catalyst under ionic conditions in the nucleus disubstituted to 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene, and this is subsequently chlorinated chlorinated under free radical conditions in the side chains of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methylbenzene. This is the 2,4-dichloro-5-fluoro-3-dichloromethyl-benzoic acid to give 2,4-dichloro-5-fluoro-3-formyl-benzoic acid, and then hydrolyzed to 2,4-dichloro-5-fluoro-3 N-hydroxyiminomethyl acid implemented. By treatment with thionyl chloride, 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride is obtained, which can still be ,4,5-trifluoro-ben-zoylfluorid converted by a chlorine / fluorine exchange on-3-cyano-2 .



The amines used for the preparation of compounds of formula (I) according to the invention are known from EP-A-0550903, EP-A-0551653 as well as from DE-A-4 309 964th
An alternative to the synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-dihydro-drobromid or the free base 1 S, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0 ] nonane and the corresponding IR, 6R enantiomer provides the following path represents:
Starting material for this synthesis is the cis-l ,4-dihydroxy-2-butene, which is converted to the bis-mesylate with mesylation tosylamide for 1-tosylpyrrolidine. This is converted into the epoxide m-chloroperbenzoic. The ring opening of the epoxide by heating in isopropanol with ethanolamine to trans-3-hydroxy-4 – (2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl)-pyrrolidine in 80% yield. Tetrahydrofuran is then in pyridine / reacted with tosyl chloride, with cooling to Tris-tosylate, which as a crude product in a mixture with some tetra-tosyl derivative with basichen reaction conditions to give the racemic trans-5 ,8-bis-tosyl-2-oxa-5, 6 – diazabicyclo [4.3.0] nonane is cylisiert. At this stage occurs with high selectivity of a chromatographic resolution kieselgelgebundenem poly (N-methacryloyl-L-leucine-d menthylamide) as the stationary phase. The desired enantiomer, (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane, is of a purity of
> 99% ee. Cleavage of the p-tosyl protecting groups is carried out with HBr-acetic acid to the lS, 6S-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide, the one with a base such as sodium or potassium hydroxide or with the aid of ion exchanger can be converted into the free base. The analogous sequence may be used for the preparation of lR, 6R-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide.
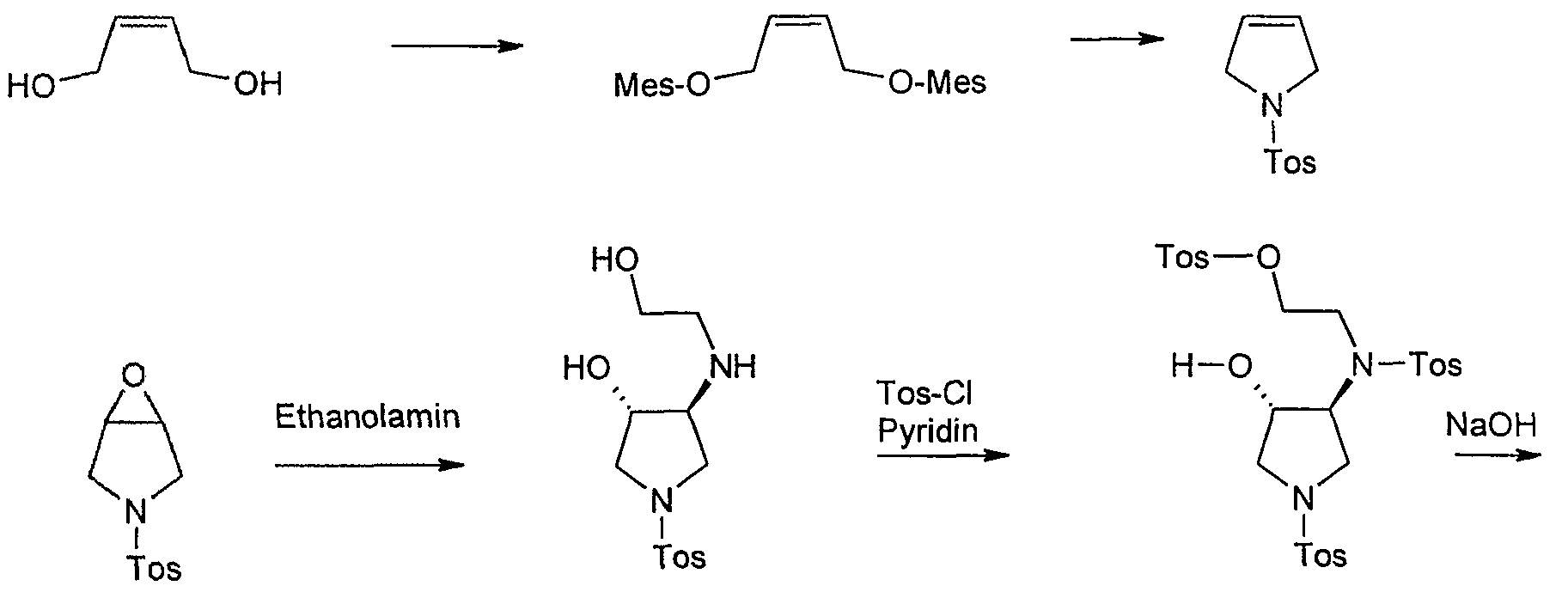

HBr / AcOH

Synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Examples of compounds of the invention are mentioned in addition to the compounds listed in the preparation examples, the compounds listed in Table 1 below, which can be used both in racemic form as well as enantiomerically pure or diastereomerically pure compounds. Table 1:


Example 1 Z
8-cyano-1-cyclopropyl-6 ,7-difluoro-1 ,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester

a 3-bromo-2 ,4,5-trifluoro-benzoate
To a mixture of 1460 ml of methanol and 340 g of triethylamine, 772 g of 3-bromo-2 ,4,5-trifluoro-benzoyl fluoride was added dropwise under ice cooling. There is one
Stirred for an hour at room temperature. The Reaktionsgemsich is concentrated, the residue dissolved in water and methylene chloride, and the aqueous phase was extracted with methylene chloride. After drying the organic phase over sodium sulfate, concentrated, and the residue was distilled in vacuum. This gives 752.4 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester of boiling point 122 ° C/20 mbar.
b. 3-Cyano-2 ,4,5-trifluoro-benzoic acid methyl ester:
269 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester and 108 g of copper cyanide are heated to reflux in 400 ml of dimethylformamide for 5 hours. , All volatile components of the reaction mixture are then distilled off in vacuo. The distillate was then fractionated on a column. This gives 133 g of 3-cyano-2 ,4,5-trifluoro-benzoate of boiling point 88-89 ° C / 0.01 mbar.
c. 3-Cyano-2 ,4,5-trifluoro-benzoic acid
A solution of 156 g of 3-cyano-2 ,4,5-trifluoro-benzoate in 960 ml of glacial acetic acid, 140 ml of water and 69 ml concentrated sulfuric acid is heated for 8 hours under reflux. Then the acetic acid is distilled off under vacuum and the residue treated with water. Of failed-ne solid is filtered off, washed with water and dried. Obtained
118.6 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid as a white solid, mp 187-190 ° C.
d 3-cyano-2 ,4,5-trifluoro-benzoyl chloride:
111 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid and 84 g of oxalyl chloride are stirred in 930 ml of dry methylene chloride with the addition of a few drops of dimethylformamide for 5 hours at room temperature. The methylene chloride is evaporated and the residue distilled in vacuo. This gives 117.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl chloride as a yellow oil.
e 2 – (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester:
To a solution of 36.5 g of 3-dimethylamino-acrylate and 26.5 g of triethylamine in 140 ml toluene, a solution of 55 g 3-cyano-2, 4,5 – trifluoro-benzoyl chloride are added dropwise in 50 ml of toluene so that the temperature 50-55 ° C remains. Then stirred for 2 hours at 50 ° C.
The reaction mixture is concentrated in vacuo and used without further
Processing used in the next step. f 2 – (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-cyclopropylamino-acrylic acid ethyl ester:
To the reaction product of step e 30 g of glacial acetic acid are added dropwise at 20 ° C. A solution of 15.75 g of cyclopropyl amine in 30 ml of toluene is added dropwise. The mixture is stirred at 30 ° C for 1 hour. Are then added 200 ml of water, stirred 15 minutes, the organic phase is separated off and shakes it again with 100 ml of water. The organic phase is dried over sodium sulfate and concentrated in vacuo. The crude product thus obtained is a set-without further purification in the next step.
g 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester:
The reaction product from stage f and 27.6 g of potassium carbonate are stirred in 80 ml dimethylformamide for 16 hours at room temperature. The reaction mixture is then poured into 750 ml ice water, the solid filtered off with suction and washed with 80 ml cold methanol. After drying, 47 g of 8 – cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinoline carboxylic acid ethyl ester, mp 209-211 ° C.
Example 2 Z
2,4-dichloro-5-fluoro-l ,3-dimethylbenzene

a solvent-free
In 124 g of 3,5-dimethyl-fluorobenzene 1 g of anhydrous iron (III) chloride are pre-loaded and launched with the speed of chlorine (about 4 h), with which the reaction. This is initially slightly exothermic (temperature increase from 24 to 32 ° C) and is maintained by cooling below 30 ° C. After addition of 120 g of chlorine, the mixture is determined. According to GC analysis are 33.4% monochloro compound, formed 58.4% desired product and 5%> overchlorinated connections. The hydrogen chloride is removed and the reaction mixture is then distilled in a column in a water jet vacuum:
In the run 49 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene obtained at 72-74 ° C/22 mbar. After 5 g of an intermediate fraction proceed at 105 ° C/22 mbar 75 g of 2,4 – dichloro-5-fluoro-l ,3-dimethylbenzene via, Melting range: 64 – 65 ° C.
b in 1,2-dichloroethane
1 kg of 3,5-dimethyl-fluorobenzene and 15 g of anhydrous iron (III) chloride are placed in 1 1 1 ,2-dichloroethane and chlorine is introduced in the same extent as the reaction proceeds (about 4 h). The reaction is initially exothermic (temperature rise from 24 to 32 ° C) and is kept below 30 ° C by cooling. After the introduction of 1200 g of chlorine are according to GC analysis 4% monochloro compound, 81.1% and 13.3% desired product overchlorinated connections emerged. After distilling off the solvent and the hydrogen chloride is distilled in a column in a water jet vacuum:
In the run 40 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene receive. After some intermediate run going at 127-128 ° C/50 mbar 1115 g of 2,4-dichloro-5-fTuor-l ,3-dimethyl-ethylbenzene over.
Example 3 Z
2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloromethylbenzene

In a photochlorination using chlorine inlet and outlet for the hydrogen chloride to a scrubber and a light source in the vicinity of the chlorine inlet tube, 1890 g of 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene pre-loaded and at 140 to 150 ° C. Chlorine metered. Within 30 hours 3850 g of chlorine are introduced. The content of the desired product according to GC analysis is 71.1% and the proportion of connections minderchlorierten 27.7%. The DestiUaton a 60 cm column with Wilson spirals provides a flow of 1142 g, which can be reused in the chlorination. The main fraction at 160-168 ° C / 0.2 mbar gives 2200 g of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methyl benzene having a melting range of 74-76 ° C. After one recrystallization
Sample from methanol, the melting point 81-82 ° C.
Example Z 4
2,4-dichloro-5-fluoro-3-formyl-benzoic acid

In a 2500 ml stirred apparatus with gas discharge are presented 95% sulfuric acid at 70 ° C. and under stirring, 500 g of molten added dropwise 2,4-dichloro-5-fluoro-3-dichloromethyl-1 trichloromethylbenzene. It is after a short while hydrochloric development. Is metered during a 2 h and stirred until the evolution of gas after. After cooling to 20 ° C., the mixture is discharged ice to 4 kg and the precipitated solid is filtered off with suction. The product is after-washed with water and dried.
Yield: 310 g, melting range: 172-174 ° C
Example Z 5
2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl-benzoic acid

In a stirred reactor 80 g of hydroxylamine hydrochloride in 500 ml of ethanol are charged and added dropwise 200 ml of 45% strength sodium hydroxide solution and then with 40 – 200 g of 2,4-dichloro-5-fluoro-3-formyl-benzoic acid added 45.degree.The reaction is slightly exothermic and it is stirred for 5 h at 60 ° C. After cooling to
Room temperature is provided by the dropwise addition of hydrochloric acid to pH <3, the product taken up in tert-butyl methyl ether, the organic phase separated and the solvent distilled off. The residue obtained 185 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid, melting range: 190 – 194 ° C.
Example No. 6
2,4-dichloro-3-cyano-5-benzoyl-fιuor

In a stirred vessel with metering and gas outlet via a reflux condenser to a scrubber 600 ml of thionyl chloride are introduced and registered at 20 ° C. 210 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid in the proportion as hydrochloric developed and sulfur dioxide. After the addition the mixture is heated until the gas evolution under reflux. Mixture is then distilled, and boiling in the range of 142-145 ° C/10 mbar, 149 g of 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride (98.1% purity by GC) Melting range: 73-75 ° C.
Example No. 7
3-Cyano-2 ,4,5-trifluoro-benzoyl

50 g of potassium fluoride are suspended in 120 ml of tetramethylene sulfone and at 15 mbar for drying distilled (ca. 20 mL).Then, 50.4 g of 2,4 – dichloro-3-cyano-5-fluoro-benzoyl chloride was added and stirred at an internal temperature with exclusion of moisture for 12 hours at 180 ° C. Are removed by vacuum distillation to 32.9 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride in the boiling range of 98 –
Obtain 100 ° C/12 mbar.
Example No. 8
3-Cyano-2 ,4,5-trifluoro-benzoyl chloride

76.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride together with 1 g of anhydrous
Aluminum chloride introduced at 60-65 ° C and then added dropwise 25 g of silicon tetrachloride gas in the course of development. After the evolution of gas at 65 ° C is distilled in a vacuum. Boiling range 120-122 ° C/14 mbar, 73.2 g of 3 – cyano-2 ,4,5-trifluoro-benzoyl chloride over.
Example No. 9
1 – (toluene-4-sulfonyl-pyrroline

In a 20 1 HC4-HWS boilers are 2.016 kg (17.6 mol)
Submitted methanesulfonyl chloride in dichloromethane and 12 1 at -10 ° C internal temperature under strong cooling (-34 ° C) solution of 705 g (8.0 mol) of 2-butene-l ,4-diol in 1.944 kg (2.68 1 , 19.2 mol) of triethylamine was added dropwise over 30 minutes. A yellow suspension stirred for 1 hour at -10 ° C and then treated with 4 1 of water, the temperature rises to 0 ° C.The suspension is warmed to room temperature, stirred for 10 minutes at room temperature and then fed in a 30 1 separating funnel. The phases are stirred separately (good phase separation) and the aqueous phase extracted with 2 1 of dichloromethane. The combined dichloromethane phases are presented in a pre-cooled 20 1 HC4 vessel and kept at 0 ° C.
In another 20-1 HC4 boiler distillation 1.37 kg (8.0 mol) toluenesulfonamide be submitted in 6 1 toluene. It is mixed with 3.2 kg of 45% sodium hydroxide solution, 0.8 1 of water and 130.5 g Tetrabutylammomiimhydrogensulfat, heated to 40 ° C maximum temperature inside and creates a vacuum. Then, the previously obtained
Dichloromethane (15.2 1) was added dropwise over 1.5 hours while the dichloromethane was removed by distillation at 450 mbar (bath temperature: 60 ° C). During the distillation is foaming. In the end, a solution is available at an internal temperature of 33-40 ° C. After the addition of dichloromethane is distilled off, until barely distillate is (duration: about 85 minutes; internal temperature 40 ° C at 60 ° C bath temperature at the end). The vessel contents will be warm transferred to a separating funnel and rinsed the tank with water and 5 1 2 1 toluene at 50 ° C. Before phase separation, the solids are extracted in the intermediate phase and washed with 0.5 1 of toluene. The organic phase is extracted with 2.4 1 of water, separated and evaporated to dryness on a rotary evaporator. The solid residue (1758 g) is suspended in 50 ° C bath temperature in 1.6 1 of methanol, the suspension is transferred into a 10 1-flanged flask and the flask rinsed with diisopropyl 2,4 1. The mixture is heated to reflux temperature (59 ° C) and stirred for 30 minutes under reflux. The suspension is cooled to 0 ° C., stirred at 0 ° C for 1 hour and extracted with 0.8 1 of a cold mixture of ether Methanol/Diisopropyl-: washed (1 1.5). The crystals are dried under a nitrogen atmosphere at 50 ° C/400 mbar.
Yield: 1456 g (81.5% of theory)
Example Z 10
3 – (toluene-4-sulfonylV6-oxa-3-aza-bicvclo [3.1.0] hexane
o “|” h “CH3
334.5 g (1.5 mol) of l-(toluene-4-sulphonyl)-pyrroline are dissolved in 1.5 1 of dichloromethane at room temperature and over 15 minutes with a suspension of 408 g (approx. 1.65 to 1, 77 mol) of 70-75% m-chloroperbenzoic acid in 900 ml of dichloromethane (cools added in manufacturing from). The mixture is heated under reflux for 16 hr (test for
Peroxide with KI / starch paper shows yet to peroxide), the suspension was cooled to 5 ° C, sucks the precipitated m-chlorobenzoic acid and washed with 300 ml of dichloromethane (peroxide with Precipitation: negative; precipitate was discarded). The filtrate is to destroy excess peroxide with 300 ml of 10% sodium sulfite solution, washed twice (test for peroxide runs now negative), extracted with 300 ml of saturated sodium bicarbonate solution, washed with water, dried with sodium sulfate and about a quarter of the volume evaporated. Again on test peroxide: negative. The mixture is concentrated and the solid residue is stirred with ice cooling, 400 ml of isopropanol, the precipitate filtered off and dried at 70 ° C in vacuum.
Yield: 295 g (82.3%),
Mp: 136-139 ° C,
TLC (dichloromethane methanol 98:2): 1 HK (Jodkammer)
Example CLOSED
trans-3-Hydroxy-4-(2-hydroxy-ethylamino-l-(‘toluene-4-sulfonyl’) pyrrolidine

643.7 g (2.65 mol) 3 – (Toluoι-4-sulfonyl)-6-oxa-3-aza-bicyclo [3.1.0] hexane to 318.5 ml with ethanolamine in 4 1 of isopropanol at reflux for 16 hours cooked. After TLC monitoring, further 35.1 ml (total 5.86 mol) of ethanolamine added to the mixture and boiled again until the next morning. The mixture is filtered hot with suction and the filtrate concentrated on a rotary evaporator to 3.5 ltr. After seeding and stirring at room temperature for 3.5 1 diisopropyl ether are added, and stirred at 0 ° C for 6 hours. The precipitated crystals are filtered off, with 250 ml of a mixture of isopropanol / diisopropyl ether (1: 1) and washed 2 times with 300 ml of diisopropyl ether and dried overnight under high vacuum.
Yield: 663.7 g (83% of theory), content: 96.1% (area% by HPLC). Example Z 12
trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l-(toluene-4-sulfonyl)-pyrrolidin-3-yl] – ftoluol-4-sulfonyl)-amino]-ethyl ester)

552 g (1.837 mol) of trans-3-hydroxy-4-(2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl) – pyrrolidine are dissolved under argon in 1.65 1 tetrahydrofuran and 0.8 1 of pyridine dissolved and at -10 ° C in portions 700 g (3.675 mol) p-toluenesulfonyl chloride are added thereto. The mixture is then stirred at this temperature for 16 hours. The work is done by adding 4.3 18.5 1% aqueous hydrochloric acid, extraction twice with dichloromethane (3 1, 2 1), washing the combined organic phases with saturated Natriurnhydrogencarbonatlösung (3 1, 2 1), drying over sodium sulfate, extracting and distilling off the solvent in vacuo. The residue is dried overnight at the oil pump and crude in the next reaction. There were 1093 g as a hard foam (content [area% by HPLC]: 80% Tris-tosyl-product and 13% tetra-tosyl-product, yield see next step). Example Z 13
rac. trans-5 ,8-bis-tosyl-2-oxa-5 .6-diazabicyclor4 .3.01 nonane

1092 g of crude trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l-(toluene-4-sulfonyl) – pyrrolidin-3-yl] – (toluene-4-sulfonyl)-amino]-ethyl} were dissolved in tetrahydrofuran and 9.4 1 at 0-3 ° C with 1.4 1 of a 1.43 molar solution of sodium hydroxide in
Methanol reacted. After half an hour at this temperature, 2.1 1 of water and 430 ml of diluted (2:1) was added to the mixture and acetic acid with previously isolated crystals of trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l – (toluene-4-sulfo-phenyl)-pyrrolidin-3-yl] – (toluene-4-sulfonyl)-amino] ethyl}-seeded. The suspension is stirred overnight at 0 to -4 ° C. The next morning, the crystals are filtered off, washed twice with 400 ml of cold mixture of tetrahydrofuran / water (4:1) and dried at 3 mbar at 50 ° C overnight.
Yield: 503 g of white crystals (62.7%> of theory over 2 steps), content: 99.7% (area% by HPLC). Example Z 14
Preparative chromatographic resolution of racemic rac. trans-5.8-bis-tosyl-2-oxa-5.6-diazabicyclor4.3.0] nonane
The chromatography of the racemate at room temperature in a column (inner diameter 75 mm), which with 870 g of a chiral stationary phase (kie-selgelgebundenes poly (N-methacryloyl-L-leucine-d menthylamide) based on the mer captomodifizierten silica Polygosil 100 , 10 microns; see EP-A 0 379 917) is filled (bed height: 38 cm). Detection is carried out using a UV detector at 254 nm
For the sample application using a solution of a concentration of 100 g of rac. trans-5 ,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 3000 ml of tetrahydrofuran. A Trenncyclus is carried out under the following conditions: with the aid of a pump is required for 2 min at a flow of 50 ml / min, a part of the sample solution and the same time at a flow rate of 50 ml / min, pure n-heptane to the column.
Thereafter eluted at a flow rate of 100 ml / min 18 minutes with a mixture of n-Heptan/Tetrahydrofuran (3/2 vol / vol). This is followed for 3 minutes at a flow of 100 ml / min elution with pure tetrahydrofuran. Thereafter, further eluted with n-Heptan/Tetrahydro-furan (3/2 vol / vol). This cycle is repeated several times.
The first eluted enantiomer is the (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo-[4.3.0] nonane, which is isolated by concentration. The eluate of the more retarding enantiomers is largely evaporated in vacuo, and the precipitated crystals are filtered off with suction and dried. From the separation of 179 g of racemate in this
As 86.1 g (96.2% of theory) of the enantiomer (lS, 6S) -5,8-bis-tosyl-2-oxa-5, 6 – diazabicyclo [4.3.0] nonane having a purity of> 99 % ee. Example Z 15
(LR, 6R-2-oxa-5.6-diazabicvclo [4.3.0] nonane dihydrobromide

38.3 g (87 mmol) of (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 500 ml of 33 -% HBr / glacial acetic acid 10 g added anisole and heated for 4 hours at 60 ° C (bath). After standing overnight, the suspension is cooled, the precipitate filtered, with
100 ml of abs. Ethanol and dried at 70 ° C under high vacuum.
Yield: 23.5 g (93%) of white solid product, mp 309-310 ° C (dec.), DC (dichloromethane/methanol/17% aq ammonia 30:8:1.): 1 HK
[Α] D: + 0.6 ° (c = 0.53, H 2 O) (fluctuating).
Example Z 16
(LS.6S-2-oxa-5.6-diazabicvclor4.3.01nonan-Dihvdrobromid

Z is analogous to Example 15 from (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] no-nan (1S, 6S)-2-oxa-5, 6-diazabicyclo [4.3.0] nonane dihydrobromide receive. Example Z 17
(1 R.6R-2-oxa-5.8-diazabicvclo [4.3.Olnonan

1 Method: 5,8 g (20 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydro-drobromid are suspended in 100 ml of isopropanol at room temperature with 2.4 g ( 42.9 mmol) and powdered potassium hydroxide while leaving about 1 hour in an ultrasonic bath. The suspension is cooled in an ice bath, filtered, washed with isopropanol and the undissolved salt, the filtrate was concentrated and distilled in a Kugelrohr oven at 150-230 ° C oven temperature and 0.7 mbar. Obtained 2.25 g (87.9% of theory) of a viscous oil which crystallizes. [Α] D -21.3 ° (c = 0.92, CHC1 3) Accordingly, this reaction can be carried out in ethanol.
2 Method: A homosexual genie catalyzed mixture of (lR, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide and 620 mg (11 mmol) of powdered potassium hydroxide is dry in a Kugelrohr apparatus at 0.2 mbar and increasing oven temperature to 250 ° C distilled. Obtained 490 mg (76.6% of theory) of (lR, 6R) -2 – oxa-5 ,8-diazabicyclo [4.3.0] nonane as a viscous oil which slowly crystallized.
3 Method: 100 g of moist, pretreated cation exchanger (Dowex 50WX, H + – form, 100-200 mesh, capacity: 5.1 meq / g of dry or 1.7 meq / mL) are charged into a column with about 200 ml 1 N HC1 activated and washed neutral with water 3 1. A solution of 2.9 g (10 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Dihydrobromide in 15 ml of water is added to the ion exchanger, and then washed with 2 1 water, and eluted with approximately 1 1 1 N ammonia solution. The eluate is evaporated. concentrated. Yield: 1.3 g of a viscous oil (quantitative), DC (dichloromethane/methanol/17% NH 3 30:8:1): 1 HK, GC: 99.6% (area).
Example Z 18
(LS.6SV2-oxa-5.8-diazabicvclor4.3.01nonan

Z is analogous to Example 17 from (lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-di-hydrobromide the free base (lS, 6S)-2-oxa-5 ,8-diazabicyclo [ 4.3.0] nonane made.
Example Z 19
2 – (2,4-dichloro-3-cyano-5-fluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester

To a solution of 626 g (4.372 mol) of 3-dimethylamino-acrylate and 591 g (4.572 mol) of ethyl-diisopropyl-amine (Hunigs base) in 1060 ml of dichloromethane, a solution of 1075 g starting at room temperature 2,4-dichloro -3-cyano-5-fluoro-benzoyl chloride (94% pure, corresponding to 1010.5 g = 4.00 mol) was dropped in 850 ml of dichloromethane. The temperature rises to 50-55 ° C (dropwise addition about 90 minutes). Then stirred for 2 hours at 50 ° C and the reaction mixture was used without further purification in the next step.
Example Z 20
2 – (2,4-dichloro-3-Cyano-5-fluoro-benzoyl-3-cvclopropylamino-acrylate

To the reaction mixture from the above step 306 g (5.1 mol) of glacial acetic acid are added dropwise under cooling at about 15 ° C. Then, with further cooling at 10-15 ° C. 267.3 g (4.68 mol) of cyclopropyl amine is added dropwise. Immediately after which the reaction mixture is mixed with 1300 ml of water under ice-cooling and 15 minutes stirred well. The dichloromethane layer was separated and used in the next step.
Example 21 Z
7-chloro-8-cyano-1-cyclopropyl-6-fluoro-1.4-dihydro-4-oxo-3-chinolincarbonsäureethyl ester

To a heated to 60-70 ° C suspension of 353 g (2.554 mol) of potassium carbonate in 850 ml of N-methylpyrrolidone, the dichloromethane phase is dropped from the precursor (about 90 minutes). During the addition of the dichloromethane at the same time
Reaction mixture was distilled off. Then the reaction mixture for 5 Vz hours at 60-70 ° C is well stirred. The mixture is cooled to about 50 ° C. and distilled under a vacuum of about 250 mbar residual dichloromethane from. At room temperature is added dropwise 107 ml 30% hydrochloric acid under ice cooling, then to obtain a pH of 5-6 is set. Then, 2,200 ml of water are added under ice cooling. The reaction mixture is thoroughly stirred for 15 minutes, the solid was then filtered off and washed on the filter twice with 1000 ml of water and extracted three times with 1000 ml of ethanol and then dried in a vacuum oven at 60 ° C.
Yield: 1200 g (89.6% of theory).
This product can be purified, if desired by, the solid is stirred in 2000 ml of ethanol for 30 minutes at reflux. You filtered hot with suction, washed with 500 ml of ethanol and dried at 60 ° C in vacuum. Melting point: 180-182 ° C.
Η-NMR (400 MHz, CDC1 3): d = 1.2 to 1.27 (m, 2H), 1.41 (t, 3H), 1.5-1.56 (m, 2H), 4, 1 to 4.8 (m, 1H), 4.40 (q, 2H), 8.44 (d, J = 8.2 Hz, H), 8.64 (s, 1H) ppm.
Example Z 22
7-chloro-8-cyano-1-cvclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid

33.8 g (0.1 mol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylate dissolved in a mixture of 100 ml of acetic acid, 20 ml water and 10 ml concentrated sulfuric acid was heated for 3 hours under reflux. After cooling, the mixture is poured onto 100 ml of ice water, the precipitate filtered off, washed with water and ethanol and dried at 60 ° C in vacuum.
Yield: 29.6 g (96% of theory),
Mp 216-21 C. (with decomposition)
Example 1

A 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS.6S-2-oxa-5.8-diazabicvclo [4.3.0] non-8-yl – 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid
1.00 g (3.26 mmol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are heated with 501 mg (3.91 mmol) of ( lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane and 0.9 ml of triethylamine in 30 ml of acetonitrile was stirred at 40-45 ° C under argon for 25 hours. All volatile components in vacuo. removed and the residue recrystallized from ethanol. Yield: 1.22 g (94%)
Melting point: 294 ° C. (with decomposition)
B) 8-Cyano-l-cyclopropyl-6-fluoro-7-(‘(lS.6S-2-oxa-5 ,8-diazabicvclo [4.3.01nonan-8-YLV 1.4-dihydro-4-oxo-3- quinoline carboxylic acid Hvdrochlorid
200 mg (0.63 mmol) of 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester to be 97 mg (0.75 mmol) of (lS, 6S)-2-oxa-5, 8 – diazabicyclo [4.3.0] nonane and 0.17 ml of triethylamine in 3 ml of acetonitrile was stirred at 40-45 ° C for 2 hours under argon. All volatile components in vacuo. removed, the residue treated with water, insolubles filtered off and the filtrate was extracted with dichloromethane. The organic phase is dried over sodium sulfate and then concentrated under reduced pressure. a. The resulting residue is dissolved in 6 ml of tetrahydrofuran and 2 ml of water and 30 mg (0.72 mmol) of lithium hydroxide monohydrate was added. After 16 hours of stirring at room temperature, acidified with dilute hydrochloric acid and the resulting precipitate was filtered off with suction and dried. Yield: 155 mg (57%) Melting point:> 300 ° C
C) 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS, 6S-2-oxa-5.8-diazabicvclo [4.3.01non-8 yiyi.4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride
1 g (2.5 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] non-8-yl )-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is suspended in 20 ml of water was added to the suspension, 10 ml hydrochloric acid and stirred for In at room temperature for 3 hours. The resulting precipitate is filtered off, washed with ethanol and dried at 80 ° C under high vacuum.
Yield: 987 mg (90.6% of theory), Melting point: 314-316 ° C. (with decomposition).
D) 8-Cyano-l-cvclopropyl-6-fluoro-7-(iS, 6S)-2-oxa-5.8-diazabicyclo [4.3.0] non-8-YLV 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid hydrochloride
86.4 g (217 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5, 8 – diazabicyclo [4.3.0] non-8-yl) – l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are dissolved at room temperature in 963 ml of water and 239 ml of 1 N aqueous sodium hydroxide solution. After filtration and washing with 200 ml of water is added to 477 ml in aqueous hydrochloric acid and the precipitated crystals placed at 95 ° C to 100 ° C in solution. The solution is cooled overnight, the precipitated crystals are filtered off with suction and washed three times with 500 ml of water and dried in vacuum.
Yield 90 g (94.7% of theory), content:> 99% (area% by HPLC) 99.6% ee. [] D 23: -112 ° (c = 0.29, N NaOH).
……………….
Tetrahedron Lett 2009, 50(21): 2525
A novel approach to Finafloxacin hydrochloride (BAY35-3377)Pages 2525-2528 |
||
Finafloxacin hydrochloride, an important clinical compound was synthesized by a novel synthetic approach. An active intermediate ethyl 7-chloro-8-cyano-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate 19 was prepared by a new route. The chiral (S,S′)-N-Boc 10 was derived from protected pyrrolidine and the absolute stereochemistry was established by X-ray analysis.
http://www.sciencedirect.com/science/article/pii/S0040403909005875
……………….



| WO2011003091A1 * | 2 Jul 2010 | 6 Jan 2011 | Alcon Research, Ltd. | Compositions comprising finafloxacin and methods for treating ophthalmic, otic, or nasal infections |
| US7723524 | 29 Sep 2004 | 25 May 2010 | Daiichi Pharmaceutical Co., Ltd. | 8-cyanoquinolonecarboxylic acid derivative |
| US8536167 | 2 Jul 2010 | 17 Sep 2013 | Alcon Research, Ltd. | Methods for treating ophthalmic, otic, or nasal infections |
| DE4329600A1 * | 2 Sep 1993 | 9 Mar 1995 | Bayer Ag | Pyrido [1,2,3-d,e] [1,3,4] benzoxadiazinderivate |
| EP0276700A1 * | 15 Jan 1988 | 3 Aug 1988 | Bayer Ag | 8-Cyano-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids, process for their preparation, and antibacterial agents containing them |
| EP0350733A2 * | 30 Jun 1989 | 17 Jan 1990 | Bayer Ag | 7-(1-Pyrrolidinyl)-3-quinolone- and -naphthyridone-carboxylic-acid derivatives, method for their preparation and for substituted mono- and bi-cyclic pyrrolidine intermediates, and their antibacterial and feed additive compositions |
| EP0550903A1 * | 28 Dec 1992 | 14 Jul 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| EP0603887A2 * | 23 Dec 1993 | 29 Jun 1994 | Daiichi Pharmaceutical Co., Ltd. | Bicyclic amine derivatives |
| EP0676199A1 * | 23 Mar 1995 | 11 Oct 1995 | Pfizer Inc. | Use of trovafloxacin or derivatives thereof for the manufacture of a medicament for the treatment of H. pylori infections |
| GB2289674A * | Title not available |


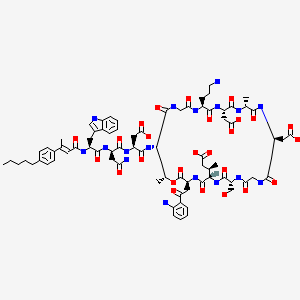
Surotomycin
Click to access surotomycin.pdf
N-[(2E)-3-(4-Pentylphenyl)-2-butenoyl]-D-tryptophyl-D-asparaginyl-N-[(3S,6S,9R,15S,18R,21S,24S,30S,31R)-3-[2-(2-aminophenyl)-2-oxoethyl]-24-(3-aminopropyl)-15,21-bis(carboxymethyl)-6-[(2R)-1-carboxy-2 -propanyl]-9-(hydroxymethyl)-18,31-dimethyl-2,5,8,11,14,17,20,23,26,29-decaoxo-1-oxa-4,7,10,13,16,19,22,25,28-nonaazacyclohentriacontan-30-yl]-L-α-asparagine
MOLECULAR FORMULA C77H101N17O26
MOLECULAR WEIGHT 1680.7
SPONSOR Cubist Pharmaceuticals, Inc.
CODE DESIGNATION CB-183,315
CB-315, CB-183315, CB-183,315
CAS REGISTRY NUMBER 1233389-51-9
U.S. – Fast Track (Treat Clostridium difficile-associated diarrhea (CDAD));
U.S. – Qualified Infectious Disease Program (Treat Clostridium difficile-associated diarrhea (CDAD))
| Company | Cubist Pharmaceuticals Inc. |
| Description | Oral antibacterial lipopeptide |
| Therapeutic Modality | Macrocycle |
| Latest Stage of Development | Phase III |
| Standard Indication | Diarrhea (infectious) |
| Indication Details | Treat Clostridium difficile-associated diarrhea (CDAD) |
EMEA……..
| Name | |||
|---|---|---|---|
| P/0096/2013: EMA decision of 29 April 2013 on the agreement of apaediatric investigation plan and on the granting of a deferral for surotomycin (EMEA-001226-PIP01-11) |
Surotomycin is an investigational oral antibiotic. This antibiotic is under investigation for the treatment of life-threatening Diarrhea, commonly caused by the bacteria Clostridium difficile.[1]
CB-183315 is an investigational antibacterial drug candidate in phase III clinical trials at Cubist for the treatment of Clostridium difficile-associated diarrhea. It is a potent, oral, cidal lipopeptide. In 2012, Qualified Infectious Disease Product Designation was assigned in the U.S. for the treatment of clostridium difficile-associated diarrhea (CDAD).

 Surotomycin Overview
Surotomycin Overview  Surotomycin Fact Sheet
Surotomycin Fact SheetSurotomycin is an antibacterial lipopeptide discovered by Cubist scientists in our research laboratories in Lexington, Massachusetts. Surotomycin is both bactericidal against Clostridium difficile and more potent than vancomycin in vitro. Surotomycin stays at the site of infection in the bowel, with minimal systemic absorption and it does not interfere with normal bowel flora. Based on its features and its preclinical safety profile, Cubist filed an Investigational New Drug (IND) Application for surotomycin in December 2008.
Following safety and pharmacokinetic studies in healthy human volunteers, Cubist began a Phase 2 study in April 2010 to assess the safety and efficacy of surotomycin in patients with CDAD, in particular to assess its ability to reduce relapse rates. In this trial of 209 patients, two different doses of surotomycin were studied and compared with oral vancomycin. The higher dose demonstrated a high clinical cure rate as evidenced by resolution of diarrhea, comparable to oral vancomycin. The most interesting results in this study, however, relate to recurrence rates. The percent of patients who had an initial response to treatment but who subsequently had a recurrence or relapse was 36 percent in the oral vancomycin arm and was 17 percent in the surotomycin 250mg treatment group — about a 50% reduction in relapse rate, which was statistically significant. In this trial, 32% of patients were infected with the hypervirulent NAP-1 strain of C. difficile. The clinical response rate in the subset of patients infected with the NAP-1 strain was comparable across the surotomycin and oral vancomycin groups. Though not statistically significant, there was a modest reduction in the relapse rates in the subset of surotomycin patients infected with NAP-1 strains.
The ability to reduce relapses is important to both patients and health care providers. In the Phase 2 study we assessed the impact of surotomycin and oral vancomycin on normal bowel flora. Treatment with surotomycin had a very minimal impact on levels of Bacteroides, a key normal bowel bacterial species, compared to oral vancomycin which resulted in a marked depletion of stool levels of these bacteria during treatment. Why does this matter? The reason is — bowel flora like Bacteroides are critical in providing a competitive environment in the bowel that prevents C. difficile overgrowth. We believe that it is this difference in impact on normal bowel flora that helps explain the differences seen in recurrence rates following treatment with Surotomycin versus oral vancomycin.
Surotomycin’s Phase 3 program includes two identical global, randomized, double-blind, active-controlled, multi-center trials. The primary objective is to demonstrate non-inferiority of surotomycin versus the comparator, oral vancomycin, in clinical response at the end of treatment in adult subjects with CDAD, using a non-inferiority margin of 10%. We also have designed this trial to allow us to demonstrate that sustained clinical response to surotomycin at the end of the study is superior to oral vancomycin. Also, we will fully evaluate the safety of surotomycin in the study subjects.
In late 2012 Cubist received from the FDA a Qualified Infectious Disease Product (QIDP) designation for surotomycin. Additionally, in early 2013 Cubist was granted Fast track status for surotomycin. The QIDP designation and subsequent granting of Fast Track status was made possible by the GAIN Act, Title VIII (Sections 801 through 806) of the Food and Drug Administration Safety and Innovation Act. The GAIN Act provides pharmaceutical and biotechnology companies with incentives to develop new antibacterial and antifungal drugs for the treatment of life-threatening infectious diseases caused by drug resistant pathogens. Qualifying pathogens are defined by the GAIN Act to include multi-drug resistant Gram-negative bacteria, including Pseudomonas, Acinetobacter, Klebsiella, and Escherichia coli species; resistant Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus; multi-drug resistant tuberculosis; and Clostridium difficile.
CDAD is a disease caused by an overgrowth of, and subsequent toxin production by, C. difficile, a resident anaerobic spore-forming Gram-positive bacterium of the lower gastrointestinal tract. This overgrowth is caused by the use of antibiotics for the treatment of common community and hospital acquired infections (HAIs). Although they treat the underlying infection, many antibiotics disrupt the natural gut flora and allow C. difficile to proliferate. C. difficile produces enterotoxin and cytotoxin, which can lead to severe diarrhea, sepsis and even death. While most types of HAIs are declining, the infection caused by C. difficile remains at historically high levels. According to the latest data from the Centers for Disease Control, C. difficile continues to be the leading cause of death associated with gastroenteritis in the US. For CDAD alone, there was more than a five-fold increase in deaths between 1999 and 2007. C. difficile causes diarrhea linked to 14,000 American deaths each year. About 25% of C. difficile infections first show symptoms in hospital patients; 75% first show in nursing home patients or in people recently cared for in doctors’ offices and clinics. C. difficile infections cost at least $1 billion in extra health care costs annually.
SUROTOMYCIN
CB-183,315 is a cyclic lipopeptide antibiotic currently in Phase III clinical trials for the treatment of Clostridium difficile-associated disease (CDAD). As disclosed in International Patent Application WO 2010/075215, herein incorporated by reference in its entirety, CB-183,315 has antibacterial activity against a broad spectrum of bacteria, including drug-resistant bacteria and C. difficile. Further, the CB-183,315 exhibits bacteriacidal activity.
CB-183,315 (Figure 1) can be made by the deacylation of BOC-protected daptomycin, followed by acylation and deprotection as described in International Patent Application WO 2010/075215.
During the preparation and storage of CB-183,315, the CB-183,315 molecule can convert to structurally similar compounds as shown in Figures 2-4, leading to the formation of anhydro-CB-183,315 (Figure 3) and beta-isomer of CB-183,315 (“B- isomer CB183,315” in Figure 2). Accordingly, one measure of the chemical stability of CB- 183 ,315 is the amount of CB- 183 ,315 (Figure 1 ) present in the CB- 183 ,315 composition relative to the amount of structurally similar compounds including anhydro-CB-183,315 (Figure 3) and beta-isomer of CB-1 83,315 (Figure 2). The amount of CB-183,315 relative to the amount of these structurally similar compounds can be measured by high performance liquid chromatography (FIPLC) after reconstitution in an aqueous diluent (e.g., as described in Example 10). In particular, the purity of CB-183,315 and amounts of structurally similar compounds (e.g., Figures 2, 3 and 4) can be determined from peak areas obtained from HPLC (e.g., according to Example 10 herein), and measuring the rate of change in the amounts of CB-183,315 over time can provide a measure of CB-183,315 chemical stability in a solid form.
There is a need for solid CB-183,315 compositions with improved chemical stability in the solid form (i.e., higher total percent CB-183,315 purity over time), providing advantages of longer shelf life, increased tolerance for more varied storage conditions (e.g., higher temperature or humidity) and increased chemical stability.
……………..
WO2010075215A1
http://www.google.com/patents/WO2010075215A1?cl=en ………… copy paste link
Example 1
Preparation of N-{1 -[(E)-3-(4-pentylphenyl)but-2-enoyl]}-L-tryptophyl-D- asparaginyl-L-α-aspartyl-L-threonylglycyl-L-ornithyl-L-α-aspartyl-D-alanyl-L-α- aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2-diamino-γ- oxobenzenebutanoic acid (13→4)-lactone (49).


1003 1004

Step 1 : Preparation of (E)-ethyl 3-(4-pentylphenyl) but-2-enoate (1002).
A mixture of commercially available 1-(4-pentylphenyl)ethanone (5 g, 26.3 mmol) and (ethoxycarbonylmethylene)-triphenylphosphorane (18.3 g, 52.5 mmol) was stirred at 150 0C for 48 hours under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature and diluted with ethyl acetate (50 ml_) and petroleum ether (200 ml_). The suspension was filtered through a fritted funnel. The concentrated filtrate was purified by flash column chromatography with silica gel (petroleum ether : ethyl acetate = 80:1 ) to give the title compound (1.6 g) having the following physical data: 1H NMR (300 MHz, δ, CDCI3) 0.90 (br, 3H), 1.36 (br, 7), 1.63 (br, 2H), 2.58 (s, 3H), 2.63 (br, 2H), 4.22 (q, 2H), 6.15 (s, 1 H), 7.20 (d, 2H), 7.41 (d, 2H).
Step 2: Preparation of (E)-3-(4-pentylphenyl) but-2-enoic acid (1003).
A solution of compound 1002 (1.5 g, 5.77 mmol) in ethanol (50 ml_) and 3N potassium hydroxide (25 ml_) was stirred at 45 0C for 3 hours. The reaction mixture was concentrated and the resulting residue was diluted with water (50 ml_). The aqueous solution was acidified to pH 2 with 1 N hydrochloric acid and extracted with EtOAc (2 * 30 ml_). The combined organic layers were dried with anhydrous sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography (silica gel, petroleum ether : ethyl acetate = 10:1) to afford the title compound (0.95 g) having the following physical data: 1 H NMR (300 MHz, δ, CDCI3) 0.90 (br, 3H), 1.33 (br, 4H), 1.62 (br, 2H), 2.60 (br, 5H), 6.18 (s, 1 H), 7.18 (d, 2H), 7.42 (d, 2H).
Step 3: Preparation of (E)-3-(4-pentylphenyl)but-2-enoyl chloride (1004).
Oxalyl chloride (3.2 mL, 36.60 mmol) and DMF (50 μl_) were added drop wise to a solution of compound 1003 (5.0 g, 21.52 mmol) in dichloromethane (100 mL) at 0 0C. The reaction solution was warmed up to room temperature and stirred for 4 hours. The reaction mixture was concentrated in vacuum and the residue was dried under hi-vacuum for 3 hours. The crude product was used in the next step without further purification.
Step 4: Preparation of N-{1 -[(E)-3-(4-pentylphenyl)but-2-enoyl]}-L-tryptophyl-D- asparaginyl-L-α-aspartyl-L-threonylglycyl-L-[(N-tert-butoxycarbonyl)-ornithyl]-L-α- aspartyl-D-alanyl-L-α-aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2- diamino-γ-oxobenzenebutanoic acid (13-→4)-lactone (1005).
Deacylated BOC-protected daptomycin (3.5Og, 2.23 mmol) and sodium bicarbonate (1.13 g, 61.0 mmol) were dissolved in THF (130 mL) and water (50 mL). The deacylated BOC-protected daptomycin sodium bicarbonate solution was cooled to 0 0C. and a solution of compound 1004 (1.96 g, 7.82 mmol) in THF (20 mL) was then introduced. The reaction mixture was warmed to room temperature and stirred for 4 hours. The mixture was concentrated in vacuum to remove THF. The remaining aqueous solution was loaded on a C18 flash chromatography column (35mηnχ 300mm, Bondesil HF C18 resin purchased from Varian). The column was first washed with water to remove salt and then with methanol to wash out product. Crude compound 1005 (3.46 g) was afforded as a white solid after removal of methanol. MS m/z 1780.8 (M + H)+.
Steps 5-6: Preparation of N-{1-[(E)-3-(4-pentylphenyl)but-2-enoyl]}-L-tryptophyl- D-asparaginyl-L-α-aspartyl-L-threonylglycyl-L-ornithyl-L-α-aspartyl-D-alanyl-L-α- aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2-diamino-γ- oxobenzenebutanoic acid (13→4)-lactone (49).
TFA (10 ml_) was added to a solution of compound 1005 (3.46 g) in DCM (50 mL) at room temperature. The reaction mixture was stirred vigorously for 45 minutes and added slowly to vigorously stirring diethyl ether (100 mL). The resulting yellow precipitation was collected by filtration. The crude product was purified by Preparative HPLC to afford the TFA salt of compound 6 (0.75 g). MP carbonate resin (purchased from Biotage) was added to the solution of compound 6 TFA salt (0.70 g, 0.39 mmol) in anhydrous methanol (30.0 mL). The mixture was stirred at room temperature for 4 hours. The resins were removed by filtration and rinsed with methanol. The methanol solution was concentrated under vacuum to give product as off-white solid (408 mg). MS m/z 1680.7 (M + H)+.
Example 1 b
Alternative preparation of N-{1-[(E)-3-(4-pentylphenyl)but-2-enoyl]}- L-tryptophyl-D-asparaginyl-L-α-aspartyl-L-threonylglycyl-L-ornithyl-L-α-aspartyl-D- alanyl-L-α-aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2-diamino-γ- oxobenzenebutanoic acid (13→4)-lactone (49).
daptomycin,

1003

A solution of (E)-3-(4-pentylphenyl)but-2-enoic acid (1 100 g, 4.73 mol), Λ/-Ethyl-Λ/’-(3-dimethylaminopropyl)carbodiimide hydrochloride (907 g, 4.73 mol), HOBT (640 g, 4.73 mol) and 4-(dimethylamino)pyridine (22 g, 0.18 mol) in DMF (11 L) was stirred at room temperature for 4 hours at which point the activation of the (E)-3-(4-pentylphenyl)but-2-enoic acid was deemed complete by HPLC.
This reaction mixture was added to a suspension of Deacylated BOC- protected daptomycin (2600 g, 1.66 mol), sodium bicarbonate (804 g, 9.57 mol) in water (11.25 L) and 1 ,4-dioxane (33.75 L). The mixture was stirred at room temperature for 2.5 hours at which time HPLC indicated complete consumption of Deacylated BOC-protected daptomycin. The reaction mixture was diluted with water (22.5 L) and cooled with an ice bath. Concentrated hydrochloric acid (5.25 L) was added while maintaining the internal temperature below 30 0C. After the addition, the solution was stirred at room temperature for 5 days at which time HPLC indicated complete consumption of the Boc protected intermediate.
The reaction mixture was washed with methyl terf-butyl ether (90 L then approximately 60 L then approximately 45 L then approximately 45 L) to remove 1 ,4-dioxane. The remaining solution (approximately 44 L) was adjusted to pH 2.69 with 2N sodium hydroxide (11.3 L) and water (53.4 L). This material was processed by Tangential Flow Filtration (TTF) with a 1 K membrane until the total volume was reduced to 54 L.Water (120 L) was added in two portions and the solution was concentrated to 52 L by continued TTF. The aqueous solution (30 L of 52 L) was purified by chromatography using the following protocol: The aqueous solution was brought to three times of its volume (30 L→90l_) with 20% IPA in aqueous ammonium acetate solution (50 mM). The diluted solution was applied to a 38 L HP20SS resin column at 1.5 L/min. The column was eluted with IPA solution in aqueous 50 mM ammonium acetate (25%→30%→35%, 60 L each concentration).
Fractions (approximately 11 L) were collected and analyzed by HPLC. The fractions with HPLC purity less than 80% were combined and purified again using the same method. The key fractions from both chromatographic separations (with HPLC purity >80%) were combined and acidified with concentrated HCI to pH 2-3. The resulting solution was desalted on an ion exchange column (HP20SS resin, 16 L) which was eluted with WFI (until conductivity = 4.8 μS) followed by IPA in WFI (36 L 10%→ 40 L 60%). The yellow band which was eluted with 60% IPA (approximately 19L) was collected, adjusted to pH 2-3 with concentrated HCI and lyophilized to yield 636.5 g of Compound 49 (HPLC purity of 87.0%). MS m/z 1680.7 (M + H)+.
……………………………..
see formulation
| WO2012162567A1 | May 24, 2012 | Nov 29, 2012 | Cubist Pharmaceuticals, Inc. | Cb-183,315 compositions and related methods |
| WO2012162567A1 | May 24, 2012 | Nov 29, 2012 | Cubist Pharmaceuticals, Inc. | Cb-183,315 compositions and related methods |
| WO2001097851A2 * | Jun 18, 2001 | Dec 27, 2001 | Cubist Pharm Inc | Compositions and methods to improve the oral absorption of antimicrobial agents |
| WO2010075215A1 | Dec 18, 2009 | Jul 1, 2010 | Cubist Pharmaceuticals, Inc. | Novel antibacterial agents for the treatment of gram positive infections |
| WO2011063419A2 * | Nov 23, 2010 | May 26, 2011 | Cubist Pharmaceuticals Inc. | Lipopeptide compositions and related methods |

 artemisinin
artemisininOther common name(s): absinthium, absinth wormwood
Scientific/medical name(s): Artemisia absinthium
Wormwood is a shrubby perennial plant whose upper shoots, flowers, and leaves are used in herbal remedies and as a bitter flavoring for alcoholic drinks. It is native to Europe, northern Africa, and western Asia, and now also grows in North America.
Available scientific evidence does not support claims that wormwood is effective in treating cancer, the side effects of cancer treatment, or any other conditions. The plant contains a volatile oil with a high level of thujone (see Thuja). There are reports that taking large doses of wormwood internally can cause serious problems with the liver and kidneys. It can also cause nausea, vomiting, stomach pain, headache, dizziness, seizures, numbness of the legs and arms, delirium, and paralysis.
Wormwood, or Artemisia absinthium, should not be confused with sweet wormwood, or Artemisia annua. Although wormwood is related to sweet wormwood, they are used in different ways. Extracts of sweet wormwood have been used in traditional herbal medicine, and an active ingredient, artemisinin, is now used in conventional medical treatment of malaria.
Wormwood is promoted as a sedative and anti-inflammatory. There are also claims that it can treat loss of appetite, stomach disorders, and liver and gallbladder complaints. In folk medicine it is used for a wide range of stomach disorders, fever, and irregular menstruation. It is also used to fight intestinal worms. Externally, it is applied to poorly healing wounds, ulcers, skin blotches, and insect bites. It is used in Moxibustion treatments for cancer (seeMoxibustion). Available scientific evidence does not support these claims.
Wormwood is taken in small doses for a short period of time, usually a maximum of 4 weeks. It is available as a capsule and as a liquid that can be added to water to make a tincture. The whole herb is sometimes brewed as a tea. Wormwood oil, washes, or poultices can also be used on the skin. Although pure wormwood is not available, “thujone-free” wormwood extract has been approved by the US Food and Drug Administration (FDA) for use in foods and as a flavoring in alcoholic drinks such as vermouth.
Artemisia absinthium was used by Hippocrates, and the earliest references to wormwood in Western civilization can be found in the Bible. Extract of wormwood was also used in ancient Egypt. The herb is mentioned often in first-century Greek and Roman writings and reportedly was placed in the sandals of Roman soldiers to help soothe their sore feet. It was taken as a treatment for tapeworms as far back as the Middle Ages.
In 1797, Henri Pernod developed absinthe, an alcoholic drink containing distilled spirits of wormwood, fennel, anise and sometimes other herbs. Absinthe became very popular in Europe and the United States in the nineteenth century. It was eventually banned in several countries in the early twentieth century due to its purported ill effects and addictive qualities. More recent analysis has suggested that, when properly prepared and distilled, the thujone content in these drinks was very low. It appears more likely that the addictiveness and other ill effects of absinthe were due to its alcohol content, which is around 60% to 85%. Varying additives or impurities from different distillers may have also produced some of these effects. Even though absinthe is illegal in some countries, various types can be found in some European countries. However, their thujone content is strictly limited. Wormwood is also an ingredient in vermouth and other drinks.
Available scientific studies do not support the use of wormwood for the treatment of cancer or the side effects of conventional cancer treatment. There is not enough evidence available to support its use for other conditions. Wormwood oil has been tested in laboratory studies and appears to inhibit the growth of some fungi. However, human tests have not been completed.
Some derivatives of Artemisia annua, or sweet wormwood, a relative of wormwood, have been shown to be effective in the treatment of malaria. In fact, the World Health Organization approved artemisinin for use against malaria in Africa in 2004. These extracts also show some promise in laboratory studies as cancer treatment drugs. Further studies are required to find out whether the anti-cancer results apply to people. It is important to remember that extracted compounds are not the same as the whole herb, and study results are not likely to show the same effects.
Wormwood should be avoided, especially by women who are pregnant or breast-feeding, by people who have had seizures, and by those with ulcers or stomach irritation. Thujone, a component of wormwood, is known to cause muscle spasms, seizures, and hallucinations if taken internally. In high doses it is known to damage the liver and the kidneys.
Because of its thujone content, large doses of wormwood taken internally can lead to vomiting, stomach and intestinal cramps, headaches, dizziness, nervous system problems, and seizures. Wormwood can also lead to liver failure. The New England Journal of Medicine reported that a man who ordered essential oil of wormwood over the Internet, thinking he had purchased absinthe, suffered liver failure shortly after drinking the oil. Wormwood may also make seizures more likely and may interfere with the anti-convulsant effects of medicines such as phenobarbital.
The plant is a relative of ragweed and daisies. Those with allergies to these types of plants may also be allergic to wormwood. Contact with wormwood can cause rash in some people.
Relying on this type of treatment alone and avoiding or delaying conventional medical care for cancer may have serious health consequences.


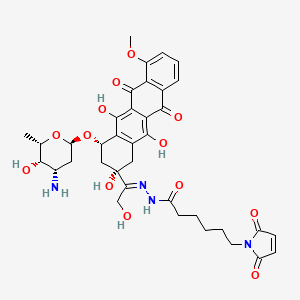
Aldoxorubicin
Click to access aldoxorubicin.pdf
 in phase 3
in phase 3
(E)-N’-(1-((2S,4S)-4-(((2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-2-yl)-2-hydroxyethylidene)-6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanehydrazide hydrochloride
1H-Pyrrole-1-hexanoic acid, 2,5-dihydro-2,5-dioxo-, (2E)-2-[1-[(2S,4S)-4-[(3-amino-
2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-1,2,3,4,6,11-hexahydro-2,5,12-trihydroxy-
7-methoxy-6,11-dioxo-2-naphthacenyl]-2-hydroxyethylidene]hydrazide
N’-[(1E)-1-{(2S,4S)-4-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-2,5,12-
trihydroxy-7-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-2-yl}-2-
hydroxyethylidene]-6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanohydrazide
MOLECULAR FORMULA C37H42N4O13
MOLECULAR WEIGHT 750.7
SPONSOR CytRx Corp.
CODE DESIGNATION INNO-206
CAS REGISTRY NUMBER 1361644-26-9
CAS: 151038-96-9 (INNO-206); 480998-12-7 (INNO-206 HCl salt), 1361644-26-9

hydrochloride
CAS: 151038-96-9
Chemical Formula: C37H42N4O13
Exact Mass: 750.27484
Molecular Weight: 750.75
|
Certificate of Analysis: |
|
|
QC data: |
|
|
Safety Data Sheet (MSDS): |

|
In vitro protocol: |
Clin Cancer Res. 2012 Jul 15;18(14):3856-67 |
|
In vivo protocol: |
Clin Cancer Res. 2012 Jul 15;18(14):3856-67. Invest New Drugs. 2010 Feb;28(1):14-9. Invest New Drugs. 2012 Aug;30(4):1743-9. Int J Cancer. 2007 Feb 15;120(4):927-34. |
|
Clinical study: |
Expert Opin Investig Drugs. 2007 Jun;16(6):855-66. |

Aldoxorubicin (INNO-206): Aldoxorubicin, also known as INNO-206, is the 6-maleimidocaproyl hydrazone derivative prodrug of the anthracycline antibiotic doxorubicin (DOXO-EMCH) with antineoplastic activity. Following intravenous administration, doxorubicin prodrug INNO-206 binds selectively to the cysteine-34 position of albumin via its maleimide moiety. Doxorubicin is released from the albumin carrier after cleavage of the acid-sensitive hydrazone linker within the acidic environment of tumors and, once located intracellularly, intercalates DNA, inhibits DNA synthesis, and induces apoptosis. Albumin tends to accumulate in solid tumors as a result of high metabolic turnover, rapid angiogenesis, hyervasculature, and impaired lymphatic drainage. Because of passive accumulation within tumors, this agent may improve the therapeutic effects of doxorubicin while minimizing systemic toxicity.
“Aldoxorubicin has demonstrated effectiveness against a range of tumors in both human and animal studies, thus we are optimistic in regard to a potential treatment for Kaposi’s sarcoma. The current standard-of-care for severe dermatological and systemic KS is liposomal doxorubicin (Doxil®). However, many patients exhibit minimal to no clinical response to this agent, and that drug has significant toxicity and manufacturing issues,” said CytRx President and CEO Steven A. Kriegsman. “In addition to obtaining valuable information related to Kaposi’s sarcoma, this trial represents another opportunity to validate the value and viability of our linker technology platform.” The company expects to announce Phase-2 study results in the second quarter of 2015.
Kaposi’s sarcoma is an orphan indication, meaning that only a small portion of the population has been diagnosed with the disease (fewer than 200,000 individuals in the country), and in turn, little research and drug development is being conducted to treat and cure it. The FDA’s Orphan Drug Act may grant orphan drug designation to a drug such as aldoxorubicin that treats a rare disease like Kaposi’s sarcoma, offering market exclusivity for seven years, fast-track status in some cases, tax credits, and grant monies to accelerate research
INNO-206 is an anthracycline in early clinical trials at CytRx Oncology for the treatment of breast cancer, HIV-related Kaposi’s sarcoma, glioblastoma multiforme, stomach cancer and pancreatic cancer. In 2014, a pivotal global phase 3 clinical trial was initiated as second-line treatment in patients with metastatic, locally advanced or unresectable soft tissue sarcomas. The drug candidate was originally developed at Bristol-Myers Squibb, and was subsequently licensed to KTB Tumorforschungs. In August 2006, Innovive Pharmaceuticals (acquired by CytRx in 2008) licensed the patent rights from KTB for the worldwide development and commercialization of the drug candidate. No recent development has been reported for research that had been ongoing for the treatment of small cell lung cancer (SCLC).
INNO-206 is a doxorubicin prodrug. Specifically, it is the 6-maleimidocaproyl hydrazone of doxorubicin. After administration, the drug candidate rapidly binds endogenous circulating albumin through the acid sensitive EMCH linker. Circulating albumin preferentially accumulates in tumors, bypassing uptake by other non-specific sites including the heart, bone marrow and the gastrointestinal tract. Once inside the acidic environment of the tumor cell, the EMCH linker is cleaved and free doxorubicin is released at the tumor site. Like other anthracyclines, doxorubicin inhibits DNA and RNA synthesis by intercalating between base pairs of the DNA/RNA strand, thus preventing the replication of rapidly-growing cancer cells. It also creates iron-mediated free oxygen radicals that damage the DNA and cell membranes. In 2011, orphan drug designation was assigned in the U.S. for the treatment of pancreatic cancer and for the treatment of soft tissue sarcoma.
CytRx Corporation (NASDAQ:CYTR) has announced it has initiated a pivotal global Phase 3 clinical trial to evaluate the efficacy and safety of aldoxorubicin as a second-line treatment for patients with soft tissue sarcoma (STS) under a Special Protocol Assessment with the FDA. Aldoxorubicin combines the chemotherapeutic agent doxorubicin with a novel linker-molecule that binds specifically to albumin in the blood to allow for delivery of higher amounts of doxorubicin (3.5 to 4 times) without several of the major treatment-limiting toxicities seen with administration of doxorubicin alone.
According to a news from Medicalnewstoday.com; CytRx holds the exclusive worldwide rights to INNO-206. The Company has previously announced plans to initiate Phase 2 proof-of-concept clinical trials in patients with pancreatic cancer, gastric cancer and soft tissue sarcomas, upon the completion of optimizing the formulation of INNO-206. Based on the multiple myeloma interim results, the Company is exploring the possibility of rapidly including multiple myeloma in its INNO-206 clinical development plans.
According to CytRx’s website, In preclinical models, INNO-206 was superior to doxorubicin with regard to ability to increase dosing, antitumor efficacy and safety. A Phase I study of INNO-206 that demonstrated safety and objective clinical responses in a variety of tumor types was completed in the beginning of 2006 and presented at the March 2006 Krebskongress meeting in Berlin. In this study, doses were administered at up to 4 times the standard dosing of doxorubicin without an increase in observed side effects over historically seen levels. Objective clinical responses were seen in patients with sarcoma, breast, and lung cancers.
INNO-206 – Mechanism of action:
According to CytRx’s website, the proposed mechanism of action is as the follow steps: (1) after administration, INNO-206 rapidly binds endogenous circulating albumin through the EMCH linker. (2) circulating albumin preferentially accumulates in tumors, bypassing uptake by other non-specific sites including heart, bone marrow and gastrointestinal tract; (3) once albumin-bound INNO-206 reaches the tumor, the acidic environment of the tumor causes cleavage of the acid sensitive linker; (4) free doxorubicin is released at the site of the tumor.
INNO-206 – status of clinical trials:
CytRx has announced that, in December 2011, CytRx initiated its international Phase 2b clinical trial to evaluate the preliminary efficacy and safety of INNO-206 as a first-line therapy in patients with soft tissue sarcoma who are ineligible for surgery. The Phase 2b clinical trial will provide the first direct clinical trial comparison of INNO-206 with native doxorubicin, which is dose-limited due to toxicity, as a first-line therapy. (source:http://cytrx.com/inno_206, accessed date: 02/01/2012).
Results of Phase I study:
In a phase I study a starting dose of 20 mg/m2 doxorubicin equivalents was chosen and 41 patients with advanced cancer disease were treated at dose levels of 20–340 mg/m2 doxorubicin equivalents . Treatment with INNO-206 was well tolerated up to 200 mg/m2 without manifestation of drug-related side effects which is a ~3-fold increase over the standard dose for doxorubicin (60 mg/kg). Myelosuppression and mucositis were the predominant adverse effects at dose levels of 260 mg/m2 and became dose-limiting at 340 mg/m2. 30 of 41 patients were assessable for analysis of response. Partial responses were observed in 3 patients (10%, small cell lung cancer, liposacoma and breast carcinoma). 15 patients (50%) showed a stable disease at different dose levels and 12 patients (40%) had evidence of tumor progression. (source: Invest New Drugs (2010) 28:14–19)
|
References |
1: Kratz F, Azab S, Zeisig R, Fichtner I, Warnecke A. Evaluation of combination therapy schedules of doxorubicin and an acid-sensitive albumin-binding prodrug of doxorubicin in the MIA PaCa-2 pancreatic xenograft model. Int J Pharm. 2013 Jan 30;441(1-2):499-506. doi: 10.1016/j.ijpharm.2012.11.003. Epub 2012 Nov 10. PubMed PMID: 23149257.
2: Walker L, Perkins E, Kratz F, Raucher D. Cell penetrating peptides fused to a thermally targeted biopolymer drug carrier improve the delivery and antitumor efficacy of an acid-sensitive doxorubicin derivative. Int J Pharm. 2012 Oct 15;436(1-2):825-32. doi: 10.1016/j.ijpharm.2012.07.043. Epub 2012 Jul 28. PubMed PMID: 22850291; PubMed Central PMCID: PMC3465682.
3: Kratz F, Warnecke A. Finding the optimal balance: challenges of improving conventional cancer chemotherapy using suitable combinations with nano-sized drug delivery systems. J Control Release. 2012 Dec 10;164(2):221-35. doi: 10.1016/j.jconrel.2012.05.045. Epub 2012 Jun 13. PubMed PMID: 22705248.
4: Sanchez E, Li M, Wang C, Nichols CM, Li J, Chen H, Berenson JR. Anti-myeloma effects of the novel anthracycline derivative INNO-206. Clin Cancer Res. 2012 Jul 15;18(14):3856-67. doi: 10.1158/1078-0432.CCR-11-3130. Epub 2012 May 22. PubMed PMID: 22619306.
5: Kratz F, Elsadek B. Clinical impact of serum proteins on drug delivery. J Control Release. 2012 Jul 20;161(2):429-45. doi: 10.1016/j.jconrel.2011.11.028. Epub 2011 Dec 1. Review. PubMed PMID: 22155554.
6: Elsadek B, Kratz F. Impact of albumin on drug delivery–new applications on the horizon. J Control Release. 2012 Jan 10;157(1):4-28. doi: 10.1016/j.jconrel.2011.09.069. Epub 2011 Sep 16. Review. PubMed PMID: 21959118.
7: Kratz F, Fichtner I, Graeser R. Combination therapy with the albumin-binding prodrug of doxorubicin (INNO-206) and doxorubicin achieves complete remissions and improves tolerability in an ovarian A2780 xenograft model. Invest New Drugs. 2012 Aug;30(4):1743-9. doi: 10.1007/s10637-011-9686-5. Epub 2011 May 18. PubMed PMID: 21590366.
8: Boga C, Fiume L, Baglioni M, Bertucci C, Farina C, Kratz F, Manerba M, Naldi M, Di Stefano G. Characterisation of the conjugate of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin with lactosaminated human albumin by 13C NMR spectroscopy. Eur J Pharm Sci. 2009 Oct 8;38(3):262-9. doi: 10.1016/j.ejps.2009.08.001. Epub 2009 Aug 18. PubMed PMID: 19695327.
9: Graeser R, Esser N, Unger H, Fichtner I, Zhu A, Unger C, Kratz F. INNO-206, the (6-maleimidocaproyl hydrazone derivative of doxorubicin), shows superior antitumor efficacy compared to doxorubicin in different tumor xenograft models and in an orthotopic pancreas carcinoma model. Invest New Drugs. 2010 Feb;28(1):14-9. doi: 10.1007/s10637-008-9208-2. Epub 2009 Jan 8. PubMed PMID: 19148580.
10: Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008 Dec 18;132(3):171-83. doi: 10.1016/j.jconrel.2008.05.010. Epub 2008 May 17. Review. PubMed PMID: 18582981.
11: Unger C, Häring B, Medinger M, Drevs J, Steinbild S, Kratz F, Mross K. Phase I and pharmacokinetic study of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin. Clin Cancer Res. 2007 Aug 15;13(16):4858-66. PubMed PMID: 17699865.
12: Lebrecht D, Walker UA. Role of mtDNA lesions in anthracycline cardiotoxicity. Cardiovasc Toxicol. 2007;7(2):108-13. Review. PubMed PMID: 17652814.
13: Kratz F. DOXO-EMCH (INNO-206): the first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin Investig Drugs. 2007 Jun;16(6):855-66. Review. PubMed PMID: 17501697.
14: Kratz F, Ehling G, Kauffmann HM, Unger C. Acute and repeat-dose toxicity studies of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin (DOXO-EMCH), an albumin-binding prodrug of the anticancer agent doxorubicin. Hum Exp Toxicol. 2007 Jan;26(1):19-35. PubMed PMID: 17334177.
15: Lebrecht D, Geist A, Ketelsen UP, Haberstroh J, Setzer B, Kratz F, Walker UA. The 6-maleimidocaproyl hydrazone derivative of doxorubicin (DOXO-EMCH) is superior to free doxorubicin with respect to cardiotoxicity and mitochondrial damage. Int J Cancer. 2007 Feb 15;120(4):927-34. PubMed PMID: 17131338.
16: Di Stefano G, Lanza M, Kratz F, Merina L, Fiume L. A novel method for coupling doxorubicin to lactosaminated human albumin by an acid sensitive hydrazone bond: synthesis, characterization and preliminary biological properties of the conjugate. Eur J Pharm Sci. 2004 Dec;23(4-5):393-7. PubMed PMID: 15567293.
| EP0169111A1 * | Jun 18, 1985 | Jan 22, 1986 | Sanofi | Cytotoxic conjugates useful in therapy, and process for obtaining them |
| EP0269188A2 * | Jun 18, 1985 | Jun 1, 1988 | Elf Sanofi | Cytotoxic conjugates useful in therapy, and process for obtaining them |
| EP0306943A2 * | Sep 8, 1988 | Mar 15, 1989 | Neorx Corporation | Immunconjugates joined by thioether bonds having reduced toxicity and improved selectivity |
| EP0328147A2 * | Feb 10, 1989 | Aug 16, 1989 | Bristol-Myers Squibb Company | Anthracycline immunoconjugates having a novel linker and methods for their production |
| EP0398305A2 * | May 16, 1990 | Nov 22, 1990 | Bristol-Myers Squibb Company | Anthracycline conjugates having a novel linker and methods for their production |
| EP0457250A2 * | May 13, 1991 | Nov 21, 1991 | Bristol-Myers Squibb Company | Novel bifunctional linking compounds, conjugates and methods for their production |

Microscopic view of Islet of Langerhans in the pancreas. Beta cells in the Islets are responsible for producing insulin
Sanford-Burnham and UC San Diego School of Medicine scientists have shown that by encapsulating immature pancreatic cells derived from human embryonic stem cells (hESC), and implanting them under the skin in animal models of diabetes, sufficient insulin is produced to maintain glucose levels without unwanted potential trade-offs of the technology. The research suggests that encapsulated hESC-derived insulin-producing cells hold great promise as an effective and safe cell-replacement therapy for insulin-dependent diabetes. “Our study critically evaluates some of the potential pitfalls of using stem cells to treat insulin-dependent diabetes,” said Pamela Itkin-Ansari, Ph.D., adjunct assistant professor in the Development, Aging, and Regenerative Program at Sanford-Burnham, with a joint appointment at UC San Diego. – See more at: http://beaker.sanfordburnham.org/2014/03/replacing-insulin-though-stem-cell-derived-pancreatic-cells-under-the-skin/#sthash.GDnpkm3h.dpuf
See more at: http://beaker.sanfordburnham.org/2014/03/replacing-insulin-though-stem-cell-derived-pancreatic-cells-under-the-skin/


 Isoliquiritigenin
IsoliquiritigeninLiquorice or licorice (/ˈlɪk(ə)rɪʃ/ lik-(ə-)rish or /ˈlɪk(ə)rɪs/ lik-(ə-)ris)[2] is the root of Glycyrrhiza glabra from which a somewhat sweet flavor can be extracted. The liquorice plant is a legume that is native to southern Europe and parts of Asia. It is not botanically related to anise, star anise, or fennel, which are sources of similar flavouring compounds. The word ‘liquorice’/’licorice’ is derived (via the Old French licoresse), from the Greek γλυκύρριζα (glukurrhiza), meaning “sweet root”,[3] from γλυκύς (glukus), “sweet”[4] + ῥίζα (rhiza), “root”,[5][6] the name provided by Dioscorides.[7]
It is a herbaceous perennial, growing to 1 m in height, with pinnate leaves about 7–15 cm (3–6 in) long, with 9–17 leaflets. The flowers are 0.8–1.2 cm (⅓–½ in) long, purple to pale whitish blue, produced in a loose inflorescence. The fruit is an oblong pod, 2–3 cm (1 in) long, containing several seeds.[8]The roots are stoloniferous.[9]
The scent of liquorice root comes from a complex and variable combination of compounds, of which anethole is the most minor component (0-3% of total volatiles). Much of the sweetness in liquorice comes from glycyrrhizin, which has a sweet taste, 30–50 times the sweetness of sugar. The sweetness is very different from sugar, being less instant and lasting longer.
The isoflavene glabrene and the isoflavane glabridin, found in the roots of liquorice, are xenoestrogens.[10][11]

A, phase I metabolites of ILG formed during incubation with rat liver microsomes and NADPH. Based on accurate mass measurements, HPLC retention times, MS/MS analyses, and comparison with data reported by Guo et al. (18), the structures of metabolites M1, M2, M3, M4, M5, M6, and M7 were assigned as liquiritigenin, 7,8,4′-trihydroxychalcone, sulfuretin, 7,3′,4′-trihydroxychalcone, davidigenin, trans-6,4′-dihydroxyaurone, and cis-6,4′-dihydroxyaurone, respectively. B, structures of ILG glucuronide conjugates formed by rat liver microsomes in the presence of UDPGA.
Liquorice grows best in deep valleys, well-drained soils, with full sun, and is harvested in the autumn, two to three years after planting.[8] Countries producing liquorice include Iran, Afghanistan, the People’s Republic of China, Pakistan, Iraq, Azerbaijan, Uzbekistan, Turkmenistan and Turkey.[12]
The world’s leading manufacturer of liquorice products is M&F Worldwide, which manufactures more than 70% of the worldwide liquorice flavors sold to end-users.[13]

Most liquorice is used as a flavoring agent for tobacco. For example, M&F Worldwide reported in 2011 that approximately 63% of its liquorice product sales are to the worldwide tobacco industry for use as tobacco flavor enhancing and moistening agents in the manufacture of American blend cigarettes, moist snuff, chewing tobacco and pipe tobacco.[12] American blend cigarettes made up a larger portion of worldwide tobacco consumption in earlier years,[14] and the percentage of liquorice products used by the tobacco industry was higher in the past. M&F Worldwide sold approximately 73% of its liquorice products to the tobacco industry in 2005,[15] and a consultant to M&F Worldwide’s predecessor company stated in 1975 that it was believed that well over 90% of the total production of liquorice extract and its derivatives found its way into tobacco products.[16]
Liquorice provides tobacco products with a natural sweetness and a distinctive flavor that blends readily with the natural and imitation flavoring components employed in the tobacco industry, represses harshness, and is not detectable as liquorice by the consumer.[16] Tobacco flavorings such as liquorice also make it easier to inhale the smoke by creating bronchodilators, which open up the lungs.[17] Chewing tobacco requires substantially higher levels of liquorice extract as emphasis on the sweet flavor appears highly desirable.[16]
Liquorice flavour is found in a wide variety of liquorice candies or sweets. In most of these candies the taste is reinforced by aniseed oil, and the actual content of liquorice is very low. Liquorice confections are primarily purchased by consumers in the European Union.[12]
In the Netherlands, where liquorice candy (“drop”) is one of the most popular forms of sweet, only a few of the many forms that are sold contain aniseed, although mixing it with mint, menthol or with laurel is quite popular. Mixing it with ammonium chloride (‘salmiak’) is also popular. The most popular liquorice, known in the Netherlands as zoute drop (salty liquorice) actually contains very little salt, i.e. sodium;[18] the salty taste is probably due to ammonium chloride, and the blood pressure raising effect is due to glycyrrhizin, see below. Strong, salty candies are popular in Scandinavia.
Pontefract in Yorkshire was the first place where liquorice mixed with sugar began to be used as a sweet in the same way it is in the modern day.[19] Pontefract cakes were originally made there. In County Durham, Yorkshire and Lancashire it is colloquially known as Spanish, supposedly because Spanish monks grew liquorice root at Rievaulx Abbey near Thirsk.[20]

Liquorice root

Various liquorice products.

Different flavoured liquorice sticks
Liquorice is popular in Italy (particularly in the South) and Spain in its natural form. The root of the plant is simply dug up, washed and chewed as a mouth freshener. Throughout Italy unsweetened liquorice is consumed in the form of small black pieces made only from 100% pure liquorice extract; the taste is bitter and intense. In Calabria a popular liqueur is made from pure liquorice extract. Liquorice is also very popular in Syria where it is sold as a drink. Dried liquorice root can be chewed as a sweet. Black liquorice contains approximately 100 calories per ounce (15 kJ/g).[21]

Foliage

Glycyrrhiza glabra from Koehler’sMedicinal-Plants
The compound glycyrrhizin (or glycyrrhizic acid), found in liquorice, has been proposed as being useful for liver protection in tuberculosis therapy, however evidence does not support this use which may in fact be harmful.[22] Glycyrrhizin has also demonstrated antiviral, antimicrobial, anti-inflammatory, hepatoprotective and blood-pressure increasing effects in vitro and in vivo, as is supported by the finding that intravenous glycyrrhizin (as if it is given orally very little of the original drug makes it into circulation) slows the progression of viral and autoimmune hepatitis.[23][24][25][26] Liquorice has also demonstrated promising activity in one clinical trial, when applied topically, against atopic dermatitis.[27] Additionally liquorice has also proven itself effective in treating hyperlipidaemia (a high amount of fats in the blood).[28] Liquorice has also demonstrated efficacy in treating inflammation-induced skin hyperpigmentation.[29][30] Liquorice may also be useful in preventing neurodegenerative disorders and cavities.[31][32][33] Anti-ulcer, laxative, anti-diabetic, anti-inflammatory, immunomodulatory, antitumour and expectorant properties of liquorice have also been noted.[34][35][36]
In traditional Chinese medicine, liquorice (मुलेठी, 甘草, شیرین بیان) is commonly used in herbal formulae to “harmonize” the other ingredients in the formula and to carry the formula to the twelve “regular meridians”.[37]
Liquorice may be useful in conventional and naturopathic medicine for both mouth ulcers[38] and peptic ulcers.[39]
Its major dose-limiting toxicities are corticosteroid, in nature, due to the inhibitory effect its chief active constituents, glycyrrhizin and enoxolone have oncortisol degradation and include: oedema, hypokalaemia, weight gain or loss and hypertension.[40][41]
Sliver of liquorice root
.jpg)
Various liquorice root slivers
.JPG)
Liquorice root with bark

Inflorescence ofGlycyrrhiza glabra
Regular consumption of fruits, vegetables and Herbs & Spices are linked with some anti-cancer benefits.

Ginger (Zingiber officinale) is also coming under increasing scrutiny and is known to be an source of several bioactive phenolics (gingerols, paradols, shogaols and gingerones).
On these pages recently we have seen how Ginger has been studied and can display anti-inflammatory & antioxidant activities.
In this study, Karna et al investigated ginger extract and its growth-inhibitory and death-inductory effects in a spectrum of prostate cancer cells.
Comprehensive studies have confirmed that ginger extracts disturbed cell-cycle progression, impaired reproductive capacity, modulated cell-cycle and apoptosis regulatory molecules and induced apoptosis in human prostate cancer cells.
Tumour tissue from an animal model showed reduced proliferation index and widespread apoptosis compared with controls and did not exert any detectable toxicity in normal, rapidly dividing tissues such as gut and bone marrow.
The authors add that to the best of their knowledge…
View original post 23 more words