

Ranjit Desai
Inventor of Oxemia (Desidustat), a breakthrough PHD inhibitor approved for Chronic Kidney Diseases (CKD) / Accomplished pharma executive / 4 INDs in 4 years, ZYDUS LIFESCIENCES
DESIDUSTAT
Formal Name
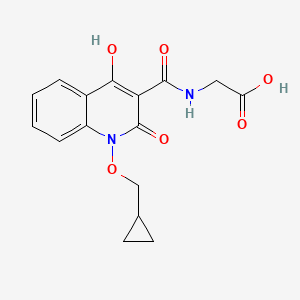
N-[[1-(cyclopropylmethoxy)-1,2-dihydro-4-hydroxy-2-oxo-3-quinolinyl]carbonyl]-glycine
Molecular Formula C16H16N2O6
FormulationA crystalline solid
2-(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamido)acetic acid
desidustat
Glycine, N-((1-(cyclopropylmethoxy)-1,2-dihydro-4-hydroxy-2-oxo-3-quinolinyl)carbonyl)-
N-(1-(Cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine
ZYAN1 compound
BCP29692
EX-A2999
ZB1514
CS-8034
HY-103227
A16921
(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl) glycine in 98% yield, as a solid. MS (ESI-MS): m/z 333.05 (M+H) +. 1H NMR (DMSO-d 6): 0.44-0.38 (m, 2H), 0.62-0.53 (m, 2H), 1.34-1.24 (m, 1H), 4.06-4.04 (d, 2H), 4.14-4.13 (d, 2H), 7.43-7.39 (t, 1H), 7.72-7.70 (d, 1H), 7.89-7.85 (m, 1H), 8.11-8.09 (dd, 1H), 10.27-10.24 (t, 1H), 12.97 (bs, 1H), 16.99 (s, 1H). HPLC Purity: 99.85%
Oxemia (Desidustat) has received approval from the Drug Controller General of India. This was an incredible team effort by Zydans across the organization and I am so proud of what we have accomplished. Oxemia is a breakthrough treatment for Anemia associated with Chronic Kidney Disease in Patients either on Dialysis or Not on Dialysis, and will help improve quality of life for CKD patients. Team #zydus , on to our next effort!

Desidustat (INN, also known as ZYAN1) is a drug for the treatment of anemia of chronic kidney disease. This drug with the brand name Oxemia is discovered and developed by Zydus Life Sciences.[1] The subject expert committee of CDSCO has recommended the grant of permission for manufacturing and marketing of Desidustat 25 mg and 50 mg tablets in India,based on some conditions related to package insert, phase 4 protocols, prescription details, and GCP.[2] Clinical trials on desidustat have been done in India and Australia.[3] In a Phase 2, randomized, double-blind, 6-week, placebo-controlled, dose-ranging, safety and efficacy study, a mean hemoglobin increase of 1.57, 2.22, and 2.92 g/dL in desidustat 100, 150, and 200 mg arms, respectively, was observed.[4] The Phase 3 clinical trials were conducted at additional lower doses as of 2019.[5] Desidustat is developed for the treatment of anemia as an oral tablet, where currently injections of erythropoietin and its analogues are drugs of choice. Desidustat is a HIF prolyl-hydroxylase inhibitor. In preclinical studies, effects of desidustat was assessed in normal and nephrectomized rats, and in chemotherapy-induced anemia. Desidustat demonstrated hematinic potential by combined effects on endogenous erythropoietin release and efficient iron utilization.[6][7] Desidustat can also be useful in treatment of anemia of inflammation since it causes efficient erythropoiesis and hepcidin downregulation.[8] In January 2020, Zydus entered into licensing agreement with China Medical System (CMS) Holdings for development and commercialization of desidustat in Greater China. Under the license agreement, CMS will pay Zydus an initial upfront payment, regulatory milestones, sales milestones and royalties on net sales of the product. CMS will be responsible for development, registration and commercialization of desidustat in Greater China.[9] It has been observed that desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines like IL-6 and oxidative stress [10] A clinical trial to evaluate the efficacy and safety of desidustat tablet for the management of Covid-19 patients is ongoing in Mexico, wherein desidustat has shown to prevent acute respiratory distress syndrome (ARDS) by inhibiting IL-6.[11] Zydus has also received approval from the US FDA to initiate clinical trials of desidustat in chemotherapy Induced anemia (CIA).[12]. Desidustat has met the primary endpoints in the phase 3 clinical trials and Zydus had filed the New Drug Application (NDA) to DCGI in November, 2021.[13]\
CLIP
https://www.businesstoday.in/industry/pharma/story/zydus-receives-dcgi-approval-for-new-drug-oxemia-what-you-need-to-know-324966-2022-03-07
Zydus receives DCGI approval for new drug Oxemia; what you need to know
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, Zydus said in a statement.
- Mar 07, 2022,
- Updated Mar 07, 2022, 10:51 AM IST
Gujarat-based pharma company Zydus Lifesciences on Monday received the Drugs Controller General of India (DCGI) approval for its new drug application for a first-of-its-kind oral treatment for anemia associated with Chronic Kidney Disease (CKD) – Oxemia (Desidustat).
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, the drug firm said in a statement.
Desidustat showed good safety profile, improved iron mobilization and LDL-C reduction in CKD patients in DREAM-D and DREAM-ND Phase III clinical trials, conducted in approximately 1,200 subjects. Desidustat provides CKD patients with an oral convenient therapeutic option for the treatment of anemia. The pharma major did not, however, declare the cost per dose if the drug is available in the market.
“After more than a decade of research and development into the science of HIF-PH inhibitors, results have demonstrated that Oxemia addresses this unmet need and additionally reduces hepcidin, inflammation and enables better iron mobilization. This advancement offers ease of convenience for the patient and will also reduce the disease burden by providing treatment at an affordable cost, thereby improving the quality of life for patients suffering from Chronic Kidney Disease,” Chairman of Zydus Lifesciences Pankaj Patel said.
Chronic Kidney Disease (CKD) is a progressive medical condition characterised by a gradual loss of kidney function and is accompanied by comorbidities like anemia, cardiovascular diseases (hypertension, heart failure and stroke), diabetes mellitus, eventually leading to kidney failure.
PATENT
US277539705
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=C922CC7937C0B6D7F987FE395E8B6F34.wapp2nB?docId=US277539705&_cid=P21-KCEB8C-83913-1
Patent applications WO 2004041818, US 20040167123, US 2004162285, US 20040097492 and US 20040087577 describes the utility of N-arylated hydroxylamines of formula (IV), which are intermediates useful for the synthesis of certain quinolone derivatives (VI) as inhibitors of hepatitis C (HCV) polymerase useful for the treatment of HCV infection. In these references, the compound of formula (IV) was prepared using Scheme 1 which involves partial reduction of nitro group and subsequent O-alkylation using sodium hydride as a base. |
 (MOL) (CDX)
(MOL) (CDX)
The patent application WO 2014102818 describes the use of certain quinolone based compound of formula (I) as prolyl hydroxylase inhibitors for the treatment of anemia. Compound of formula (I) was prepared according to scheme 2 which involved partial reduction of nitro group and subsequent O-alkylation using cesium carbonate as a base. |
 (MOL) (CDX)
(MOL) (CDX)
The drawback of process disclosed in WO 2014102818 (Scheme 2) is that it teaches usage of many hazardous reagents and process requires column chromatographic purification using highly flammable solvent at one of the stage and purification at multi steps during synthesis, which is not feasible for bulk production. |
 (MOL) (CDX)
(MOL) (CDX)
 (MOL) (CDX)
(MOL) (CDX)
The process for the preparation of compound of formula (I-a) comprises the following steps: |
Step 1′a Process for Preparation of ethyl 2-iodobenzoate (XI-a)
In a 5 L fixed glass assembly, Ethanol (1.25 L) charged at room temperature. 2-iodobenzoic acid (250 g, 1.00 mol) was added in one lot at room temperature. Sulphuric acid (197.7 g, 2.01 mol) was added carefully in to reaction mixture at 20 to 35° C. The reaction mixture was heated to 80 to 85° C. Reaction mixture was stirred for 20 hours at 80 to 85° C. After completion of reaction distilled out ethanol at below 60° C. The reaction mixture was cooled down to room temperature. Water (2.5 L) was then added carefully at 20 to 35° C. The reaction mixture was then charged with Ethyl acetate (1.25 L). After complete addition of ethyl acetate, reaction mixture turned to clear solution. At room temperature it was stirred for 5 to 10 minutes and separated aqueous layer. Aqueous layer then again extracted with ethyl acetate (1.25 L) and separated aqueous layer. Combined organic layer then washed with twice 10% sodium bicarbonate solution (2×1.25 L) and twice process water (2×1.25L) and separated aqueous layer. Organic layer then washed with 30% brine solution (2.5 L) and separated aqueous layer. Concentrated ethyl acetate in vacuo to get ethyl 2-iodobenzoate in 95% yield, as an oil, which was used in next the reaction, without any further purification. MS (ESI-MS): m/z 248.75 (M+H). 1H NMR (CDCl 3): 1.41-1.37 (t, 3H), 4.41-4.35 (q, 2H), 7.71-7.09 (m, 1H), 7.39-7.35 (m, 1H), 7.94-7.39 (m, 1H), 7.96-7.96 (d, 1H). HPLC Purity: 99.27% |
Step-2 Process for the Preparation of ethyl 2-((tert-butoxycarbonyl)(cyclopropylmethoxy)aminolbenzoate (XII-a)
In a 5 L fixed glass assembly, toluene (1.5 L) was charged at room temperature. Copper (I) iodide (15.3 g, 0.08 mol) was added in one lot at room temperature. Glycine (39.1 g, 0.520 mol) was added in one lot at room temperature. Reaction mixture was stirred for 20 minutes at room temperature. Ethyl 2-iodobenzoate (221.2 g, 0.801 mol) was added in one lot at room temperature. Tert-butyl (cyclopropylmethoxy)carbamate (150 g, 0.801 mol) was added in one lot at room temperature. Reaction mixture was stirred for 20 minutes at room temperature. Potassium carbonate (885.8 g, 6.408 mol) and ethanol (0.9 L) were added at 25° C. to 35° C. Reaction mixture was stirred for 30 minutes. The reaction mixture was refluxed at 78 to 85° C. for 24 hours. Reaction mixture was cooled to room temperature and stirred for 30 minutes. The reaction mixture was then charged with ethyl acetate (1.5 L). After complete addition of ethyl acetate, reaction mixture turned to thick slurry. At room temperature it was stirred for 30 minutes and the solid inorganic material was filtered off through hyflow supercel bed. Inorganic solid impurity was washed with ethyl acetate (1.5 L), combined ethyl acetate layer was washed with twice water (2×1.5 L) and separated aqueous layer. Organic layer washed with 30% sodium chloride solution (1.5 L) and separated aqueous layer. Ethyl acetate was concentrated in vacuo to get ethyl 2-((tert-butoxycarbonyl)(cyclopropylmethoxy)amino)benzoate in 89% yield, as an oil, which was used in next the reaction, without any further purification. MS (ESI-MS): m/z 357.93 (M+Na). 1H NMR (CDCl 3): 0.26-0.23 (m, 2H), 0.52-0.48 (m, 2H), 1.10-1.08 (m, 1H), 1.38-1.35 (t, 3H), 1.51 (s, 9H), 3.78-3.76 (d, J=7.6 Hz, 2H), 4.35-4.30 (q, J=6.8 Hz, 2H), 7.29-7.25 (m, 1H), 7.49-7.47 (m, 2H), 7.78-7.77 (d, 1H). HPLC Purity: 88.07% |
Step 3 Process for the Preparation of ethyl 2-((cyclopropylmethoxy)amino)benzoate (XIII-a)
In a 10 L fixed glass assembly, dichloromethane (2.4 L) was charged at room temperature. Ethyl 2-((tert-butoxycarbonyl)(cyclopropylmethoxy)amino)benzoate (200 g, 0.596 mol) was charged and cooled externally with ice-salt at 0 to 10° C. Methanolic HCl (688.3 g, 3.458 mol, 18.34% w/w) solution was added slowly drop wise, over a period of 15 minutes, while maintaining internal temperature below 10° C. Reaction mixture was warmed to 20 to 30° C., and stirred at 20 to 30° C. for 3 hours. The reaction mixture was quenched with addition of water (3.442 L). Upon completion of water addition, the reaction mixture turn out to light yellow coloured solution. At room temperature it was stirred for another 15 minutes and separated aqueous layer. Aqueous layer was again extracted with Dichloromethane (0.8 L). Combined dichloromethane layer then washed with 20% sodium chloride solution (1.0 L) and separated aqueous layer. Concentrated dichloromethane vacuo to get Ethyl 2-((cyclopropylmethoxy)amino)benzoate in 92% yield, as an oil. MS (ESI-MS): m/z 235.65 (M+H) +. 1H NMR (CDCl 3): 0.35-0.31 (m, 2H), 0.80-0.59 (m, 2H), 0.91-0.85 (m, 1H), 1.44-1.38 (t, 3H), 3.76-3.74 (d, 2H), 4.36-4.30 (q, 2H), 6.85-6.81 (t, 1H), 7.36-7.33 (d, 1H), 7.92-7.43 (m, 1H), 7.94-7.93 (d, 1H), 9.83 (s, 1H). HPLC Purity: 87.62% |
Step 4 Process for the Preparation of ethyl 24N-(cyclopropylinethoxy)-3-ethoxy-3-oxopropanamido)benzoate (XIV-a)
In a 2 L fixed glass assembly, Acetonitrile (0.6 L) was charged at room temperature. Ethyl 2-((cyclopropylmethoxy)amino)benzoate (120 g, 0.510 mol) was charged at room temperature. Ethyl hydrogen malonate (74.1 g, 0.561 mol) was charged at room temperature. Pyridine (161.4 g, 2.04 mol) was added carefully in to reaction mass at room temperature and cooled externally with ice-salt at 0 to 10° C. Phosphorous oxychloride (86.0 g, 0.561 mol) was added slowly drop wise, over a period of 2 hours, while maintaining internal temperature below 10° C. Reaction mixture was stirred at 0 to 10° C. for 45 minutes. The reaction mixture was quenched with addition of water (1.0 L). Upon completion of water addition, the reaction mixture turns out to dark red coloured solution. Dichloromethane (0.672 L) was charged at room temperature and it was stirred for another 15 minutes and separated aqueous layer. Aqueous layer was again extracted with dichloromethane (0.672 L). Combined dichloromethane layer then washed with water (0.400 L) and 6% sodium chloride solution (0.400 L) and separated aqueous layer. Mixture of acetonitrile and dichloromethane was concentrated in vacuo to get Ethyl 2-(N-(cyclopropylmethoxy)-3-ethoxy-3-oxopropanamido)benzoate in 95% yield, as an oil. MS (ESI-MS): m/z 350.14 (M+H) l. 1H NMR (DMSO-d 6): 0.3-0.2 (m, 2H), 0.6-0.4 (m, 2H), 1.10-1.04 (m, 1H), 1.19-1.15 (t, 3H), 1.29-1.25 (t, 3H), 3.72-3.70 (d, 2H), 3.68 (s, 2H), 4.17-4.12 (q, 2H), 4.25-4.19 (q, 2H), 7.44-7.42 (d, 1H), 7.50-7.46 (t, 1H), 7.68-7.64 (m, 1H), 7.76-7.74 (d, 1H). HPLC Purity: 86.74% |
Step 5: Process for the Preparation of ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2 dihydroquinolline-3-carboxylate (XY-a)
In a 10 L fixed glass assembly under Nitrogen atmosphere, Methanol (0.736 L) was charged at room temperature. Ethyl 2-(N-(cyclopropylmethoxy)-3-ethoxy-3-oxopropanamido)benzoate (160 g, 0.457 mol) was charged at room temperature. Sodium methoxide powder (34.6 g, 0.641 mol) was added portion wise, over a period of 30 minutes, while maintaining internal temperature 10 to 20° C. Reaction mixture was stirred at 10 to 20° C. for 30 minutes. The reaction mixture was quenched with addition of ˜1N aqueous hydrochloric acid solution (0.64 L) to bring pH 2, over a period of 20 minutes, while maintaining an internal temperature 10 to 30° C. Upon completion of aqueous hydrochloric acid solution addition, the reaction mixture turned to light yellow coloured slurry. Diluted the reaction mass with water (3.02 L) and it was stirred for another 1 hour. Solid material was filtered off and washed twice with water (2×0.24 L). Dried the compound in fan dryer at temperature 50 to 55° C. for 6 hours to get crude ethyl 1-(cyclopropylmetboxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylate as a solid. |
Purification
In a 10 L fixed glass assembly, DMF (0.48 L) was charged at room temperature. Crude ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylate (120 g) was charged at room temperature. Upon completion of addition of crude compound, clear reaction mass observed. Reaction mixture stirred for 30 minutes at room temperature. Precipitate the product by addition of water (4.8 L), over a period of 30 minutes, while maintaining an internal temperature 25 to 45° C. Upon completion of addition of water, the reaction mixture turned to light yellow colored slurry. Reaction mixture was stirred at 25 to 45° C. for 30 minutes. Solid material was filtered off and washed with water (0.169 L). Dried the product in fan dryer at temperature 50 to 55° C. for 6 hours to get pure ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylate in 81% yield, as a solid. MS (ESI-MS): m/z 303.90 (M+H) +. 1H NMR (DMSO-d 6): 0.37-0.35 (m, 2H), 0.59-0.55 (m, 2H), 1.25-1.20 (m, 1H), 1.32-1.29 (t, 3H), 3.97-3.95 (d, 2H), 4.36-4.31 (q, 2H), 7.35-7.31 (in, 1H), 7.62-7.60 (dd, 1H), 7.81-7.77 (m, 1H), 8.06-7.04 (dd, 1H). HPLC Purity: 95.52% |
Step 6 Process for the Preparation of ethyl (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycinate (XVI-a)
In a 5 L fixed glass assembly, tetrahydrofuran (0.5 L) was charged at room temperature. Ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylate (100 g, 0.329 mol) was charged at room temperature. Glycine ethyl ester HCl (50.7 g, 0.362 mol) was charged at room temperature. N,N-Diisopropylethyl amine (64 g, 0.494 mol) was added carefully in to reaction mass at room temperature and heated the reaction mass at 65 to 70° C. Reaction mixture was stirred at 65 to 70° C. for 18 hours. The reaction mixture was quenched with addition of water (2.5 L). |
Upon completion of water addition, the reaction mixture turns out to off white to yellow coloured slurry. Concentrated tetrahydrofuran below 55° C. in vacuo and reaction mixture was stirred at 25 to 35° C. for 1 hour. Solid material was filtered off and washed with water (3×0.20 L). Dried the compound in fan dryer at temperature 55 to 60° C. for 8 hours to get crude ethyl (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycinate as a solid. |
Purification
In a 2 L fixed glass assembly, Methanol (1.15 L) was charged at room temperature. Crude ethyl (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycinate (100 g) was charged at room temperature. The reaction mass was heated to 65 to 70° C. Reaction mass was stirred for 1 h at 65 to 70° C. Removed heating and cool the reaction mass to 25 to 35° C. Reaction mass stirred for 1 h at 25 to 35° C. Solid material was filtered off and washed with methanol (0.105 L). The product was dried under fan dryer at temperature 55 to 60° C. for 8 hours to get pure ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylate in 80% yield, as a solid. MS (ESI-MS): m/z 360.85 (M+H) +. 1H NMR (DMSO-d 6): 0.39 (m, 2H), 0.60-0.54 (m, 2H), 1.23-1.19 (t, 3H), 1.31-1.26 (m, 1H), 4.04-4.02 (d, 2H), 4.18-4.12 (q, 2H), 4.20-4.18 (d, 2H), 7.40-7.36 (m, 1H), 7.70-7.68 (d, 1H), 7.87-7.83 (m, 1H), 8.08-8.05 (dd, 1H), 10.27-10.24 (t, 1H). HPLC Purity: 99.84% |
Step 7: Process for the Preparation of (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine (I-a)
In a 5 L fixed glass assembly, methanol (0.525 L) was charged at room temperature. Ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylate (75 g, 0.208 mol) was charged at room temperature. Water (0.30 L) was charged at room temperature. Sodium hydroxide solution (20.8 g, 0.520 mol) in water (0.225 L) was added carefully at 30 to 40° C. Upon completion of addition of sodium hydroxide solution, the reaction mass turned to clear solution. Reaction mixture stirred for 30 minutes at 30 to 40° C. Diluted the reaction by addition of water (2.1 L). Precipitate the solid by addition of hydrochloric acid solution (75 mL) in water (75 mL). Upon completion of addition of hydrochloric acid solution, the reaction mass turned to off white colored thick slurry. Reaction mixture was stirred for 1 h at room temperature. Solid material was filtered off and washed with water (4×0.375 L). The compound was dried under fan dryer at temperature 25 to 35° C. for 6 hours and then dried for 4 hours at 50 to 60° C. to get (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl) glycine in 98% yield, as a solid. MS (ESI-MS): m/z 333.05 (M+H) +. 1H NMR (DMSO-d 6): 0.44-0.38 (m, 2H), 0.62-0.53 (m, 2H), 1.34-1.24 (m, 1H), 4.06-4.04 (d, 2H), 4.14-4.13 (d, 2H), 7.43-7.39 (t, 1H), 7.72-7.70 (d, 1H), 7.89-7.85 (m, 1H), 8.11-8.09 (dd, 1H), 10.27-10.24 (t, 1H), 12.97 (bs, 1H), 16.99 (s, 1H). HPLC Purity: 99.85% |
Polymorphic Data (XRPD):
References[edit]
- ^ “Zydus receives DCGI approval for new drug Oxemia; what you need to know”.
- ^ CDSCO, SEC Committee. “SEC meeting to examine IND proposals, dated 29.12.2021”. CDSCO website Govt of India. CDSCO. Retrieved 19 January 2022.
- ^ Kansagra KA, Parmar D, Jani RH, Srinivas NR, Lickliter J, Patel HV, et al. (January 2018). “Phase I Clinical Study of ZYAN1, A Novel Prolyl-Hydroxylase (PHD) Inhibitor to Evaluate the Safety, Tolerability, and Pharmacokinetics Following Oral Administration in Healthy Volunteers”. Clinical Pharmacokinetics. 57 (1): 87–102. doi:10.1007/s40262-017-0551-3. PMC 5766731. PMID 28508936.
- ^ Parmar DV, Kansagra KA, Patel JC, Joshi SN, Sharma NS, Shelat AD, Patel NB, Nakrani VB, Shaikh FA, Patel HV; on behalf of the ZYAN1 Trial Investigators. Outcomes of Desidustat Treatment in People with Anemia and Chronic Kidney Disease: A Phase 2 Study. Am J Nephrol. 2019 May 21;49(6):470-478. doi: 10.1159/000500232.
- ^ “Zydus Cadila announces phase III clinical trials of Desidustat”. 17 April 2019. Retrieved 20 April 2019 – via The Hindu BusinessLine.
- ^ Jain MR, Joharapurkar AA, Pandya V, Patel V, Joshi J, Kshirsagar S, et al. (February 2016). “Pharmacological Characterization of ZYAN1, a Novel Prolyl Hydroxylase Inhibitor for the Treatment of Anemia”. Drug Research. 66 (2): 107–12. doi:10.1055/s-0035-1554630. PMID 26367279.
- ^ Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR (August 2018). “Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases”. Journal of Medicinal Chemistry. 61 (16): 6964–6982. doi:10.1021/acs.jmedchem.7b01686. PMID 29712435.
- ^ Jain M, Joharapurkar A, Patel V, Kshirsagar S, Sutariya B, Patel M, et al. (January 2019). “Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation”. European Journal of Pharmacology. 843: 113–120. doi:10.1016/j.ejphar.2018.11.023. PMID 30458168. S2CID 53943666.
- ^ Market, Capital (20 January 2020). “Zydus enters into licensing agreement with China Medical System Holdings”. Business Standard India. Retrieved 20 January 2020 – via Business Standard.
- ^ Joharapurkar, Amit; Patel, Vishal; Kshirsagar, Samadhan; Patel, Maulik; Savsani, Hardikkumar; Jain, Mukul (22 January 2021). “Prolyl hydroxylase inhibitor desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines and oxidative stress”. Drug Development Research. 82 (6): 852–860. doi:10.1002/ddr.21792. PMID 33480036. S2CID 231680317.
- ^ “Zydus’ trials of Desidustat shows positive results for Covid-19 management”. The Hindu Business Line. The Hindu. Retrieved 25 January 2021.
- ^ “Zydus receives approval from USFDA to initiate clinical trials of Desidustat in cancer patients receiving chemotherapy”. PipelineReview.com. La Merie Publishing. Retrieved 22 January 2021.
- ^ “Stock Share Price | Get Quote | BSE”.
Desidustat
|
| Clinical data |
| Other names |
ZYAN1 |
| Identifiers |
|
|
| CAS Number |
|
| UNII |
|
| Chemical and physical data |
| Formula |
C16H16N2O6 |
| Molar mass |
332.312 g·mol−1 |
| 3D model (JSmol) |
|
-
C1CC1CON2C3=CC=CC=C3C(=C(C2=O)C(=O)NCC(=O)O)O
|
-
InChI=1S/C16H16N2O6/c19-12(20)7-17-15(22)13-14(21)10-3-1-2-4-11(10)18(16(13)23)24-8-9-5-6-9/h1-4,9,21H,5-8H2,(H,17,22)(H,19,20)
-
Key:IKRKQQLJYBAPQT-UHFFFAOYSA-N
|
| CTID |
Title |
Phase |
Status |
Date |
| NCT04215120 |
Desidustat in the Treatment of Anemia in CKD on Dialysis Patients |
Phase 3 |
Recruiting |
2020-01-02 |
| NCT04012957 |
Desidustat in the Treatment of Anemia in CKD |
Phase 3 |
Recruiting |
2019-12-24 |
////////// DESIDUSTAT, ZYDUS CADILA, COVID 19, CORONA VIRUS, PHASE 3, ZYAN 1, OXEMIA, APPROVALS 2022, INDIA 2022


 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader












