
Home » Posts tagged 'Viltolarsen'
Tag Archives: Viltolarsen
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Viltolarsen
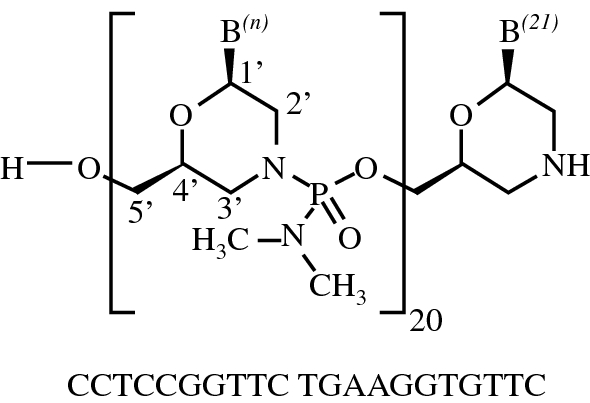
Viltolarsen
维托拉生
ビルトラルセン
| Formula | C244H381N113O88P20 |
|---|---|
| CAS | 2055732-84-6 |
| Mol weight | 6924.8155 |
APPROVED FDA 2020/8/12, Viltepso
APPROVED JAPAN PMDA 2020/3/25, VILTEPSO
- NCNP-01
- NS-065
- NS-065/NCNP-01
- WHO 10771
- WHO-10771
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Viltepso | Injection, solution | 250 mg/1 | Intravenous | Ns Pharma, Inc. | 2020-08-13 | Not applicable |  |
SYNWatanabe N, Nagata T, Satou Y, Masuda S, Saito T, Kitagawa H, Komaki H, Takagaki K, Takeda S: NS-065/NCNP-01: An Antisense Oligonucleotide for Potential Treatment of Exon 53 Skipping in Duchenne Muscular Dystrophy. Mol Ther Nucleic Acids. 2018 Dec 7;13:442-449. doi: 10.1016/j.omtn.2018.09.017.
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US9079934 | No | 2011-08-31 | 2031-08-31 | |
Viltolarsen
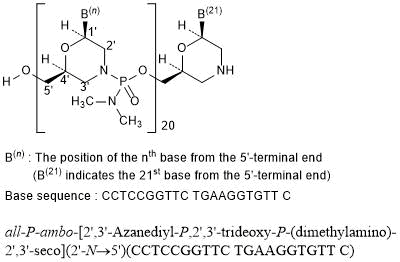
all-P-ambo-[2′,3′-Azanediyl-P,2′,3′-trideoxy-P-(dimethylamino)-2′,3′-seco](2′-N→5′)(CCTCCGGTTC TGAAGGTGTT C)
C244H381N113O88P20 : 6924.82
[2055732-84-6]
Viltolarsen, sold under the brand name Viltepso, is a medication used for the treatment of Duchenne muscular dystrophy (DMD).[3][4][2] Viltolarsen is an antisense oligonucleotide.[3][2]
The most common side effects include upper respiratory tract infection, injection site reaction, cough, and pyrexia (fever).[3][4][2]
Viltolarsen was approved for medical use in the United States in August 2020.[3][4] After golodirsen was approved in December 2019, viltolarsen is the second approved targeted treatment for people with this type of mutation in the United States.[3][5] Approximately 8% of people with DMD have a mutation that is amenable to exon 53 skipping.[3]
Medical uses
Viltolarsen is indicated for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping.[3][2]
DMD is a rare genetic disorder characterized by progressive muscle deterioration and weakness.[3] It is the most common type of muscular dystrophy.[3] DMD is caused by mutations in the DMD gene that results in an absence of dystrophin, a protein that helps keep muscle cells intact.[3] The first symptoms are usually seen between three and five years of age and worsen over time.[3] DMD occurs in approximately one out of every 3,600 male infants worldwide; in rare cases, it can affect females.[3]
Adverse effects
The most common side effects include upper respiratory tract infection, injection site reaction, cough, and pyrexia (fever).[3][4][2]
Although kidney toxicity was not observed in the clinical studies, the clinical experience is limited, and kidney toxicity, including potentially fatal glomerulonephritis, has been observed after administration of some antisense oligonucleotides.[3]
History
Viltolarsen was evaluated in two clinical studies with a total of 32 participants, all of whom were male and had genetically confirmed DMD.[3] The increase in dystrophin production was established in one of those two studies, a study that included sixteen DMD participants, with eight participants receiving viltolarsen at the recommended dose.[3] In the study, dystrophin levels increased, on average, from 0.6% of normal at baseline to 5.9% of normal at week 25.[3] Trial 1 provided data for evaluation of the benefits of viltolarsen.[4] The combined populations from both trials provided data for evaluation of the side effects of viltolarsen.[4] Trial 1 was conducted at six sites in the United States and Canada and Trial 2 was conducted at five sites in Japan.[4] All participants in both trials were on a stable dose of corticosteroids for at least three months before entering the trials.[4]
The U.S. Food and Drug Administration (FDA) concluded that the applicant’s data demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit in people with DMD who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping.[3] A clinical benefit of the drug has not been established.[3] In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of available therapies.[3]
The application for viltolarsen was granted priority review designation and the FDA granted the approval to NS Pharma, Inc.[3]
References
- ^ https://www.drugs.com/pregnancy/viltolarsen.html
- ^ Jump up to:a b c d e f “Viltepso- viltolarsen injection, solution”. DailyMed. 12 August 2020. Retrieved 18 August 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u “FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation”. U.S. Food and Drug Administration (FDA) (Press release). 12 August 2020. Retrieved 12 August 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g h “Drug Trials Snapshots: Viltepso”. U.S. Food and Drug Administration. 12 August 2020. Retrieved 18 August 2020. This article incorporates text from this source, which is in the public domain.
- ^ Anwar S, Yokota T (August 2020). “Golodirsen for Duchenne muscular dystrophy”. Drugs of Today. 56 (8): 491–504. doi:10.1358/dot.2020.56.8.3159186. PMID 33025945.
Further reading
- Dhillon S (July 2020). “Viltolarsen: First Approval”. Drugs. 80 (10): 1027–1031. doi:10.1007/s40265-020-01339-3. PMID 32519222. S2CID 219542850.
- Dzierlega K, Yokota T (June 2020). “Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy”. Gene Ther. 27 (9): 407–416. doi:10.1038/s41434-020-0156-6. PMID 32483212. S2CID 219157034.
- Hwang J, Yokota T (October 2019). “Recent advancements in exon-skipping therapies using antisense oligonucleotides and genome editing for the treatment of various muscular dystrophies”. Expert Rev Mol Med. 21: e5. doi:10.1017/erm.2019.5. PMID 31576784.
- Roshmi RR, Yokota T (October 2019). “Viltolarsen for the treatment of Duchenne muscular dystrophy”. Drugs Today. 55 (10): 627–639. doi:10.1358/dot.2019.55.10.3045038. PMID 31720560.
External links
- “Viltolarsen”. Drug Information Portal. U.S. National Library of Medicine (NLM).
- Clinical trial number NCT02740972 for “Safety and Dose Finding Study of NS-065/NCNP-01 in Boys With Duchenne Muscular Dystrophy (DMD)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Viltepso |
| Other names | NS-065/NCNP-01 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Viltolarsen |
| Pregnancy category | US: N (Not classified yet)[1] |
| Routes of administration | Intravenous |
| Drug class | Antisense oligonucleotide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2]In general: ℞ (Prescription only) |
| Identifiers | |
| CAS Number | 2055732-84-6 |
| DrugBank | DB15005 |
| ChemSpider | 71115970 |
| UNII | SXA7YP6EKX |
| KEGG | D11528 |
| ChEMBL | ChEMBL4298062 |
| Chemical and physical data | |
| Formula | C244H381N113O88P20 |
| Molar mass | 6924.910 g·mol−1 |
//////////Viltolarsen, Viltepso, 维托拉生 , FDA 2020, EU 2020, APPROVALS 2020, NCNP-01, NS-065, NS-065/NCNP-01, WHO 10771, WHO-10771, ビルトラルセン
