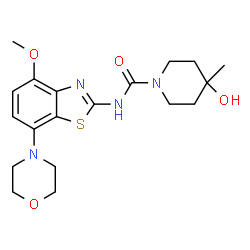
Tozadenant
RO-449351
SYN-115
- Molecular Formula C19H26N4O4S
- Average mass 406.499 Da
A2 (3); A2a-(3); RO4494351; RO4494351-000; RO4494351-002; SYN-115
Phase III clinical trials at Biotie Therapies for the treatment of Parkinson’s disease as an adjunctive therapy with levodopa
1-Piperidinecarboxamide, 4-hydroxy-N-[4-methoxy-7-(4-morpholinyl)-2-benzothiazolyl]-4-methyl-
4-Hydroxy-N-[4-methoxy-7-(4-morpholinyl)-1,3-benzothiazol-2-yl]-4-methyl-1-piperidinecarboxamide
4-Hydroxy-N-[4-methoxy-7-(4-morpholinyl)-2-benzothiazolyl]-4-methyl-1-piperidinecarboxamide
4-Hydroxy-4-methyl-piperidine-1-carboxylic acid(4-methoxy-7-morpholin-4-yl-benzothiazol-2-yl)-amide
CAS 870070-55-6
- Originator Roche
- Developer Acorda Therapeutics
- Class Amides; Antiparkinsonians; Benzothiazoles; Carboxylic acids; Morpholines; Piperidines; Small molecules
- Mechanism of Action Adenosine A2A receptor antagonists
Highest Development Phases
- Phase III Parkinson’s disease
- Phase I Liver disorders
Most Recent Events
- 30 Jun 2017 Biotie Therapies plans a phase I trial in Healthy volunteers in Canada (NCT03200080)
- 30 Jun 2017 Phase-I clinical trials in Liver disorders (In volunteers) in USA (PO) (NCT03212313)
- 27 Apr 2017 Acorda Therapeutics initiates enrolment in a phase III trial for Parkinson’s disease in Germany (EudraCT2016-003961-25)(NCT03051607)
Biotie Therapies Holding , under license from Roche , is developing tozadenant (phase 3, as of August 2017) for the treatment of Parkinson’s disease.
SYN-115, a potent and selective adenosine A2A receptor antagonist, is in phase III clinical trials at Biotie Therapeutics for the treatment of Parkinson’s disease, as an adjunjunctive therapy with levodopa. Phase 0 trials were are underway at the National Institute on Drug Abuse (NIDA) for the treatment of cocaine dependency, but no recent development has been reported.
The A2A receptor modulates the production of dopamine, glutamine and serotonin in several brain regions. In preclinical studies, antagonism of the A2A receptor resulted in increases in dopamine levels, which gave rise to the reversal of motor deficits.
Originally developed at Roche, SYN-115 was acquired by Synosia in 2007, in addition to four other drug candidates with potential for the treatment of central nervous system (CNS) disorders. Under the terms of the agreement, Synosia was responsible for clinical development and in some cases commercialization, while Roche retained the right to opt-in to two preselected programs.
In 2010, the compound was licensed to UCB by Synosia Therapeutics for development and commercialization worldwide.
In February 2011, Synosia (previously Synosis Therapeutics) was acquired by Biotie Therapeutics, and in 2014, Biotie regained global rights from UCB.



Representative examples of A2AAdoR antagonists.
Tozadenant, also known as 4-hydroxy-N-(4-methoxy-7-(4-morpholinyl)benzo[d]thiazol-2-yl)-4-methylpiperidine-l-carboxamide or SYN115, is an adenosine A2A receptor antagonist. The A2A receptor modulates the production of
dopamine, glutamine and serotonin in several brain regions. In preclinical studies, antagonism of the A2A receptor resulted in increases in dopamine levels, which gave rise to the reversal of motor deficits.
Tozadenant is currently phase III clinical trials for the treatment of Parkinson’s disease as an adjunctive therapy with levodopa. It has also been explored for the treatment of cocaine dependency.
(F. Hoffmann-La Roche AG)


Claus Riemer

Sonia Poli

PAPER
Fredriksson, Kai; Lottmann, Philip; Hinz, Sonja; Onila, Iounut; Shymanets, Aliaksei; Harteneck, Christian; Müller, Christa E.; Griesinger, Christian; Exner, Thomas E. – Angewandte Chemie – International Edition, 2017, vol. 56, 21, pg. 5750 – 5754, Angew. Chem., 2017, vol. 129, pg. 5844 – 5848,5
PAPER
Mancel, Valérie; Mathy, François-Xavier; Boulanger, Pierre; English, Stephen; Croft, Marie; Kenney, Christopher; Knott, Tarra; Stockis, Armel; Bani, Massimo – Xenobiotica, 2017, vol. 47, 8, pg. 705 – 718
Paper
Design, Synthesis of Novel, Potent, Selective, Orally Bioavailable Adenosine A2A Receptor Antagonists and Their Biological Evaluation
Sujay Basu*  , Dinesh A. Barawkar, Sachin Thorat, Yogesh D. Shejul, Meena Patel, Minakshi Naykodi, Vaibhav Jain, Yogesh Salve, Vandna Prasad, Sumit Chaudhary, Indraneel Ghosh, Ganesh Bhat, Azfar Quraishi, Harish Patil, Shariq Ansari, Suraj Menon, Vishal Unadkat, Rhishikesh Thakare, Madhav S. Seervi, Ashwinkumar V. Meru, Siddhartha De, Ravi K. Bhamidipati, Sreekanth R. Rouduri, Venkata P. Palle, Anita Chug, and Kasim A. Mookhtiar
, Dinesh A. Barawkar, Sachin Thorat, Yogesh D. Shejul, Meena Patel, Minakshi Naykodi, Vaibhav Jain, Yogesh Salve, Vandna Prasad, Sumit Chaudhary, Indraneel Ghosh, Ganesh Bhat, Azfar Quraishi, Harish Patil, Shariq Ansari, Suraj Menon, Vishal Unadkat, Rhishikesh Thakare, Madhav S. Seervi, Ashwinkumar V. Meru, Siddhartha De, Ravi K. Bhamidipati, Sreekanth R. Rouduri, Venkata P. Palle, Anita Chug, and Kasim A. Mookhtiar
Drug Discovery Facility, Advinus Therapeutics Ltd., Quantum Towers, Plot-9, Phase-I, Rajiv Gandhi Infotech Park, Hinjawadi, Pune 411 057, India
J. Med. Chem., 2017, 60 (2), pp 681–694
DOI: 10.1021/acs.jmedchem.6b01584
Patent
https://www.google.com/patents/US20050261289
-
Adenosine modulates a wide range of physiological functions by interacting with specific cell surface receptors. The potential of adenosine receptors as drug targets was first reviewed in 1982. Adenosine is related both structurally and metabolically to the bioactive nucleotides adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP) and cyclic adenosine monophosphate (cAMP); to the biochemical methylating agent S-adenosyl-L-methione (SAM); and structurally to the coenzymes NAD, FAD and coenzyme A; and to RNA. Together adenosine and these related compounds are important in the regulation of many aspects of cellular metabolism and in the modulation of different central nervous system activities.
-
[0003]
The adenosine receptors have been classified as A1, A2A, A2B and A3receptors, belonging to the family of G protein-coupled receptors. Activation of aderosine receptors by adenosine initiates signal transduction mechanisms. These mechanisms are dependent on the receptor associated G protein. Each of the adenosine receptor subtypes has been classically characterized by the adenylate cyclase effector system, which utilises cAMP as a second messenger. The A1and A3 receptors, coupled with Gi proteins inhibit adenylate cyclase, leading to a decrease in cellular cAMP levels, while A2A and A2Breceptors couple to Gs proteins and activate adenylate cyclase, leading to an increase in cellular cAMP levels. It is known that the A1receptor system activates phospholipase C and modulates both potassium and calcium ion channels. The A3 subtype, in addition to its association with adenylate cyclase, also stimulates phospholipase C and activates calcium ion channels.
-
[0004]
The A1 receptor (326-328 amino acids) was cloned from various species (canine, human, rat, dog, chick, bovine, guinea-pig) with 90-95% sequence identify among the mammalian species. The A2Areceptor (409-412 amino acids) was cloned from canine, rat, human, guinea pig and mouse. The A2B receptor (332 amino acids) was cloned from human and mouse and shows 45% homology with the human A1 and A2A receptors. The A3 receptor (317-320 amino acids) was cloned from human, rat, dog, rabbit and sheep.
-
[0005]
The A1 and A2A receptor subtypes are proposed to play complementary roles in adenosine’s regulation of the energy supply. Adenosine, which is a metabolic product of ATP, diffuses from the cell and acts locally to activate adenosine receptors to decrease the oxygen demand (A1) or increase the oxygen supply (A2A) and so reinstate the balance of energy supply: demand within the tissue. The actions of both subtypes is to increase the amount of available oxygen to tissue and to protect cells against damage caused by a short term imbalance of oxygen. One of the important functions of endogenous adenosine is preventing damage during traumas such as hypoxia, ischemia, hypotension and seizure activity.
-
[0006]
Furthermore, it is known that the binding of the adenosine receptor agonist to mast cells expressing the rat A3 receptor resulted in increased inositol triphosphate and intracellular calcium concentrations, which potentiated antigen induced secretion of inflammatory mediators. Therefore, the A3 receptor plays a role in mediating asthmatic attacks and other allergic responses.
-
[0007]
Adenosine is a neurotransmitter able to modulate many aspects of physiological brain function. Endogenous adenosine, a central link between energy metabolism and neuronal activity, varies according to behavioral state and (patho)physiological conditions. Under conditions of increased demand and decreased availability of energy (such as hypoxia, hypoglycemia, and/or excessive neuronal activity), adenosine provides a powerful protective feedback mechanism. Interacting with adenosine receptors represents a promising target for therapeutic intervention in a number of neurological and psychiatric diseases such as epilepsy, sleep, movement disorders (Parkinson or Huntington’s disease), Alzheimer’s disease, depression, schizophrenia, or addiction. An increase in neurotransmitter release follows traumas such as hypoxia, ischemia and seizures. These neurotransmitters are ultimately responsible for neural degeneration and neural death, which causes brain damage or death of the individual. The adenosine A1agonists mimic the central inhibitory effects of adenosine and may therefore be useful as neuroprotective agents. Adenosine has been proposed as an endogenous anticonvulsant agent, inhibiting glutamate release from excitatory neurons and inhibiting neuronal firing. Adenosine agonists therefore may be used as antiepileptic agents. Furthermore, adenosine antagonists have proven to be effective as cognition enhancers. Selective A2A antagonists have therapeutic potential in the treatment of various forms of dementia, for example in Alzheimer’s disease, and of neurodegenerative disorders, e.g. stroke. Adenosine A2A receptor antagonists modulate the activity of striatal GABAergic neurons and regulate smooth and well-coordinated movements, thus offering a potential therapy for Parkinsonian symptoms. Adenosine is also implicated in a number of physiological processes involved in sedation, hypnosis, schizophrenia, anxiety, pain, respiration, depression, and drug addiction (amphetamine, cocaine, opioids, ethanol, nicotine, and cannabinoids). Drugs acting at adenosine receptors therefore have therapeutic potential as sedatives, muscle relaxants, antipsychotics, anxiolytics, analgesics, respiratory stimulants, antidepressants, and to treat drug abuse. They may also be used in the treatment of ADHD (attention deficit hyper-activity disorder).
-
[0008]
An important role for adenosine in the cardiovascular system is as a cardioprotective agent. Levels of endogenous adenosine increase in response to ischemia and hypoxia, and protect cardiac tissue during and after trauma (preconditioning). By acting at the A1 receptor, adenosine A1 agonists may protect against the injury caused by myocardial ischemia and reperfusion. The modulating influence of A2Areceptors on adrenergic function may have implications for a variety of disorders such as coronary artery disease and heart failure. A2Aantagonists may be of therapeutic benefit in situations in which an enhanced anti-adrenergic response is desirable, such as during acute myocardial ischemia. Selective antagonists at A2A Areceptors may also enhance the effectiveness of adenosine in terminating supraventricula arrhytmias.
-
[0009]
Adenosine modulates many aspects of renal function, including renin release, glomerular filtration rate and renal blood flow. Compounds which antagonize the renal affects of adenosine have potential as renal protective agents. Furthermore, adenosine A3 and/or A2Bantagonists may be useful in the treatment of asthma and other allergic responses or and in the treatment of diabetes mellitus and obesity.
-
[0010]
Numerous documents describe the current knowledge on adenosine receptors, for example the following publications:
-
- Bioorganic & Medicinal Chemistry, 6, (1998), 619-641,
- Bioorganic & Medicinal Chemistry, 6, (1998), 707-719,
- J. Med. Chem., (1998), 41, 2835-2845,
- J. Med. Chem., (1998), 41, 3186-3201,
- J. Med. Chem., (1998), 41, 2126-2133,
- J. Med. Chem., (1999), 42, 706-721,
- J. Med. Chem., (1996), 39, 1164-1171,
- Arch. Pharm. Med. Chem., 332, 39-41, (1999),
- Am. J. Physiol., 276, H1113-1116, (1999) or
- Naunyn Schmied, Arch. Pharmacol. 362,375-381, (2000)
EXAMPLE 14-Hydroxy-4-methyl-piperidine-1-carboxylic acid(4-methoxy-7-morpholin-4-yl-benzothiazol-2-yl)-amide (I)
-
[0065]
To a solution of (4-methoxy-7-morpholin-4-yl-benzothiazol-2-yl)-carbamic acid phenyl ester (3.2 g, 8.3 mmol) and N-ethyl-diisopropyl-amine (4.4 ml, 25 mmol) in trichloromethane (50 ml) is added a solution of 4-hydroxy-4-methyl-piperidine in trichloromethane (3 ml) and tetrahydrofurane (3 ml) and the resulting mixture heated to reflux for 1 h. The reaction mixture is then cooled to ambient temperature and extracted with saturated aqueous sodium carbonate (15 ml) and water (2×5 ml). Final drying with magnesium sulphate and evaporation of the solvent and recrystallization from ethanol afforded the title compound as white crystals (78% yield), mp 236° C. MS: m/e=407(M+H+).



PATENT
WO-2017136375
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017136375&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
Novel deuterated forms of tozadenant are claimed. Also claimed are compositions comprising them and method of modulating the activity of adenosine A2A receptor (ADORA2A), useful for treating Parkinson’s diseases. Represents new area of patenting to be seen from CoNCERT Pharmaceuticals on tozadenant. ISR draws attention towards WO2016204939 , claiming controlled-release tozadenant formulations.
This invention relates to deuterated forms of morpholinobenzo[d]thiazol-2-yl)-4-methylpiperidine-1-carboxamide compounds, and pharmaceutically acceptable salts thereof. This invention also provides compositions comprising a compound of this invention and the use of such compositions in methods of treating diseases and conditions that are beneficially treated by administering an adenosine A2A receptor antagonist.
Many current medicines suffer from poor absorption, distribution, metabolism and/or excretion (ADME) properties that prevent their wider use or limit their use in certain indications. Poor ADME properties are also a major reason for the failure of drug candidates in clinical trials. While formulation technologies and prodrug strategies can be employed in some cases to improve certain ADME properties, these approaches often fail to address the underlying ADME problems that exist for many drugs and drug candidates. One such problem is rapid metabolism that causes a number of drugs, which otherwise would be highly effective in treating a disease, to be cleared too rapidly from the body. A possible solution to rapid drug clearance is frequent or high dosing to attain a sufficiently high plasma level of drug. This, however, introduces a number of potential treatment problems such as poor patient compliance with the dosing regimen, side effects that become more acute with higher doses, and increased cost of treatment. A rapidly metabolized drug may also expose patients to undesirable toxic or reactive metabolites.
[3] Another ADME limitation that affects many medicines is the formation of toxic or biologically reactive metabolites. As a result, some patients receiving the drug may experience toxicities, or the safe dosing of such drugs may be limited such that patients receive a suboptimal amount of the active agent. In certain cases, modifying dosing intervals or formulation approaches can help to reduce clinical adverse effects, but often the formation of such undesirable metabolites is intrinsic to the metabolism of the compound.
[4] In some select cases, a metabolic inhibitor will be co- administered with a drug that is cleared too rapidly. Such is the case with the protease inhibitor class of drugs that are used to treat HIV infection. The FDA recommends that these drugs be co-dosed with ritonavir, an inhibitor of cytochrome P450 enzyme 3A4 (CYP3A4), the enzyme typically responsible for their metabolism (see Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60). Ritonavir, however, causes adverse effects and adds to the pill burden for HIV patients who must already take a combination of different drugs. Similarly, the
CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose of reducing rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar affect.
Quinidine, however, has unwanted side effects that greatly limit its use in potential combination therapy (see Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at http://www.accessdata.fda.gov).
[5] In general, combining drugs with cytochrome P450 inhibitors is not a satisfactory strategy for decreasing drug clearance. The inhibition of a CYP enzyme’s activity can affect the metabolism and clearance of other drugs metabolized by that same enzyme. CYP inhibition can cause other drugs to accumulate in the body to toxic levels.
[6] A potentially attractive strategy for improving a drug’s metabolic properties is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the formation of undesirable metabolites by replacing one or more hydrogen atoms with deuterium atoms. Deuterium is a safe, stable, non-radioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the ADME properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
[7] Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, MI et al, J Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res 1985, 14: 1-40 (“Foster”); Kushner, DJ et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, MB et al, Curr Opin Drug Discov Devel, 2006, 9: 101-09 (“Fisher”)). The results have been variable and unpredictable. For some compounds deuteration caused decreased metabolic clearance in vivo. For others, there was no change in metabolism. Still others demonstrated increased metabolic clearance. The variability in deuterium effects has also led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting adverse metabolism (see Foster at p. 35 and Fisher at p. 101).
[8] The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem. 1991, 34, 2871-76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
///////////////TOZADENANT, phase III, clinical trials, Parkinson’s disease , adjunctive therapy, levodopa, RO-449351, SYN-115
CC1(CCN(CC1)C(=O)NC2=NC3=C(C=CC(=C3S2)N4CCOCC4)OC)O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....













