
Home » Posts tagged 'REGULATORY'
Tag Archives: REGULATORY
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ozonization of Pharmaceutical Water and the Biocidal Products Regulation

With the new biocidal products regulation from 2013 in-situ generated ozone now also falls into the scope of this directive. Ozone generation systems with a biocide application (such as disinfection of pharma water) thus require an approval after the transitional period expires in the September 2017. The ozone registration group is active for this purpose. Read more about the Ozonization of Pharmaceutical Water and the Biocidal Products Regulation.
With the new biocidal products regulation from 2013 in-situ generated ozone now also falls into the scope of this regulation. Ozone generation systems with a biocide application (such as disinfection of pharma water) thus require an approval after the transitional period expires in the September 2017. We already reported about the impact of the new Biocidal Products Regulation – please see the GMP News “Pharmaceutical Water: Uncertainty caused by the New Biocidal Products Regulation” from 21 May 2014.
Admission will take place in two stages. In the first step, ozone is certified as an active ingredient and registered in the list of active substances authorised in the EU. In the second step, the ozone generation system is approved as a biocidal product. The major manufacturers of ozone generation systems have joined forces for this in the ozone registration group (ORG). It aims at relieving users of ozone systems from the registration procedure. That means the documents should be provided to the users. The access to the marketing authorisation dossier is supposed to be assured through a Letter of Access (LoA). One of the open questions seems to be resolved now: the question whether an authorisation document will be required for each ozone precurser (i.e. water, oxygen or air). As this seems to be unnecessary, only one authorisation document is currently being processed.
The question with regard to how reasonable it is to include ozone from pharmaceutical water systems in the biocidal products regulation cannot be clarified at this point. The same is true with regard to the question on who is supposed to control pharmaceutical companies and whether their ozone comes from approved ozone systems.
You can find more information on the page Ozone registration group.


EMA publishes New Process Validation Guideline
![]()
EMA publishes New Process Validation Guideline
After the publication of the Annex 15 draft at the beginning of February 2014, the EMA made a move towards the revision of its process validation guideline. The final document was published on 27 February 2014. For a long time now, the EMA had already announced this revision in a concept paper. What’s new? click here
After the publication of the Annex 15 draft at the beginning of February 2014, the EMA made a move towards the revision of its process validation guideline. The final document was published on 27 February 2014. For a long time now, the EMA had already announced this revision in a concept paper. The objective of the revision was to integrate modern GMP aspects:
- Integration of the ICH Q8, Q9 and Q10 Guidelines
- Incorporation of Process Analytical Technology (PAT), Quality by Design (QbD) and Real-Time Release Testing (RTRT).
- Extension with regard to an “enhanced approach” and integration of “continuous process verification”
- Integration of the Annexes to the current Note for Guidance
- Harmonisation with the current FDA Guidance on Process Validation
The deadline for comments on the draft for the revision of the process validation guideline ended in October 2012 already. Now, elements in accordance with the Annex 15 have also flowed into the final document. In the following, you will read a short evaluation of the document with regard to the original draft from March 2012, the (still) applicable Note for Guidance on Process Validation and FDA’s Guidance on Process Validation. The GMP relevant aspects of the documents will also be addressed.
The original 7-page long Note for Guidance on Process Validation has more than doubled and now contains 15 pages. Even the original revision draft had only 11 pages. The change in the title to “Guideline on process validation for finished products- information and data to be provided in regulatory submissions” is noticeable. The title itself gives indication about the content of the document, namely marketing authorisation matters.
Like in the draft, the document is composed of 8 numerated chapters, a summary, definitions, references, an Annex I (Process validation scheme) and an Annex II (Standard/non-standard processes) which is a new part compared to the draft. A sub section on “Design space verification” has been newly added to the chapter on process validation.
There haven’t been big changes to the draft document released in 2012. Only the chapter “Design space verification” is brand new, all other parts have been mostly updated. The chapter on ongoing process validation has been removed. Compared to the draft, indications about standard/ non-standard processes are now available in the Annex II – like in the currently applicable Note for Guidance.
What are the changes to the currently applicable Note for Guidance on Process Validation?
Compared to the current Note for Guidance, the revision remains in its final version pretty difficult to read and rather general. This is a marketing authorisation document, which is clearly addressed in the title and only applies to finished dosage forms of chemical medicinal products for human and veterinary use but not for old ones, which are already authorised and on the market. The introduction of a validation life cycle and the integration of continued process verification (CPV) are completely new although this approach is already acquainted from ICH Q8. The “traditional approach” remains accepted. Like in the Annex 15 draft the hybrid approach remains here in the final document “nebulous”. The idea to integrate modern elements from ICH Q8, Q10 (and Q11) into the document is clearly noticeable. Yet, far less concrete references are made to ICH Q9.
A stronger overlap of the FDA Guidance would have been desirable. FDA’s Guidance also deals with APIs and biologicals, and the process validation life cycle runs like a thread through the whole FDA document. FDA’s Guidance also contains GMP aspects. The FDA Guidance explicitly addresses old products which should be integrated to stage 3 of the life cycle. Yet, there is another big difference. The revised document doesn’t highlight statistical methods like the FDA Guidance.
Before the finalisation, a comparison with the Annex 15 has been made which is a nice thing. This explains the long period between the publication of the draft (March 2012) and that of the finalisation (February 2014).
What is significant for the GMP world? On the one hand almost nothing, on the other hand quite a lot: one may wonder why? Direct references to the Annex 15 can be found with regard to the “ongoing process verification” and “concurrent validation”, which is almost nothing looking at the whole document. Moreover, validation in general is required to be executed according to the GMP regarding “continuous process verification” and “change control”; these are the essential parts of the document, and (almost) the complete document should therefore be seen from a GMP perspective.
The new EMA guideline on process validation will apply by the end of August 2014.

Japanese Pharmacopoeia and Japanese GMP Regulations available online
Japanese Pharmacopoeia and Japanese GMP Regulations available online
On Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) website, you can download documents on GMP as well as on marketing authorisations for medicinal products. An English version of the Japanese Pharmacopoeia (JP) is also available. You will find the direct links in the News.
On Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) website, you can find in the section “Regulations and Procedures” under the heading “GMP” requirements regarding the inspection of manufacturers of medicinal products and APIs who want to introduce their products into Japan.
Now, a document was supplemented in January 2014 which describes which documents have to be submitted to the Japanese Agency within a pre-approval inspection and/ or a periodical post-approval inspection.
Go to the PMDA webpage to get more information.
There, you can also access the current Japanese Pharmacopoeia Sixteenth Edition in English.
Source: PMDA, Japan
Indian Regulators promote two levels of GMP
GMP deviations and even data falsification have been identified in a number of companies in India. How is it possible that interpretation of FDA and EU authorities on one side and the Indian authority on the other side come to a completely different picture? Read more in our GMP News
GMP deviations and even data falsification have been identified in a number of companies in India. The FDA has issued numerous Warning Letters, the EU has published GMP Non Compliance Reports in its EudraGMDP database and EDQM has withdrawn various CEPs because of GMP inspection findings.
In an article published by Regulatory Focus on 28 January 2014 the question has been raised whether Indian companies have a chronic data falsification problem. The article lists 7 companies in India which have received a Warning Letter in the past months – all of them because of GMP deviations and because of “actually or potentially tampering with their data”. In addition to the 7 companies the Ranbaxy case is a story of its own. Not only one facility was found to manipulate data but several sites of the company are involved. For this reason the US FDA has issued a consent decree of permanent injunction against Ranbaxy. All manufactured products in the facilities concerned are now subject to an FDA import alert. In a press release the FDA states: “Because this company continued to violate current good manufacturing practice regulations and falsify information on drug applications, the FDA took these actions in an effort to protect consumers.” Dara Corrigan, FDA associate commissioner for regulatory affairs goes on: “The FDA continues to be committed to protecting consumers from potentially unsafe products that may be offered on the market.” On January 23, 2014 the FDA added an additional facility of Ranbaxy to the existing consent decree.
So far, the Indian Authority did not initiate the same measures like US and European counterparts. This also questions the supervision system in India. If inspections have been performed by Indian Inspectors at the concerned facilities why did they fail to make the same findings? The Drug Controller General of India, Mr. G.N. Singh, gave an interesting interpretation: According to an interview published by live mint & Wall Street Journal he said: “…it must be stated that every country has different measures and we cannot judge Ranbaxy by standards set up by the American drug regulator“. When Mr Singh was asked about the problems identified at three Ranbaxy plants he stated: “Some of those were found to be true and my office had told Ranbaxy to take corrective measures. Similar procedures will be followed in this case as well. But I do not think this is a situation which will warrant withdrawal of drugs from the domestic market. Our biggest objective is to maintain good quality of medicines and we are doing that. There are no drugs in the Indian market that are not up to the standards stated under the Drugs and Cosmetics Act.” In a final statement in the interview he also mentioned that he is “not worried about issues of quality.” In another interview with the Business Standard Press Mr Singh made an alarming statement for all customers of medicinal products and APIs in Europe and the US. “If I follow US standards, I will have to shut almost all drug facilities“. If this is the truth EU and US customers are in big trouble because products not complying to EU/US GMP standard (e.g. ICH Q7 GMP for APIs) would need to be taken from the market immediately.
This all looks like it will not fit together. How is it possible that interpretation of FDA and EU authorities on one side and the Indian authority on the other side come to a completely different picture? It can only mean that dual standards exist. This would result in two quality levels, an international and a domestic quality level. Such a policy possibly causes questions by Indian patients who have to accept a different and probably lower quality standard.
It does not look like the Indian Regulators will re-think the GMP inspection approach and the quality standard in their country. Instead of acting in his own country the Drug Controller General of India announced inspections in the US and the EU.
But what are the international implications of this strategy? European Regulators need to react as they require from the Indian Authority to issue Written Confirmations of GMP compliance. Without a Written Confirmation APIs can not enter EU market. Currently more than 200 Written Confirmations have been issued by Indian Authority. If the inspections which have been performed as a prerequisite for issuing a Written confirmation were not based on the international standard ICH Q7 (GMP for APIs) the Written Confirmations are no longer valid documents. This issue might be raised by an EU court if a substandard API in a medicinal product will cause a health risk to patients in Europe.
FDA publishes new Guidance on Validation of Analytical Methods
The FDA has published a new Guidance on the validation of analytical methods which shall replace the 14 years old existing Guideline on the topic. More details about the contents of this highly topical document can be found here.
A new FDA Guidance for Industry entitled “Analytical Procedures and Methods Validation for Drugs and Biologics” was published a few days ago. This Guideline replaces the Guidance for Industry “Analytical Procedures and Methods Validation” from 2000 (this document has never been finalised and has had a draft status 14 years long) and – when finalised – should also replace the “Guidelines for Submitting Samples and Analytical Data for Methods Validation” which came into force in 1987.
Unlike the previous Guideline from 2000, the new document explicitly mentions biologics in its title. The objective of the Guideline is to inform applicants about what data are expected by the FDA in marketing authorisation dossiers. The provisions of the Guideline apply to new drug applications (NDAs), abbreviated new drug applications (ANDAs), biologics license applications (BLAs), and variation applications regarding these types of application, as well as to Type II Drug Master Files. The Guideline can’t be directly used for investigational new drug applications (INDs) as the scope of data with regard to analytical procedures and methods validation varies with the development phase. Nevertheless, IND applicants should orientate themselves to the provisions of the new Guideline.
When comparing it with the former and now invalid “Methods Validation” Guidance, it is apparent that the Draft Guidance has been kept much shorter. There are no detailed descriptions available: for example the table about recommended validation parameters for different analytical tests has been deleted without substitution. Yet, new chapters have been added, like chapter “VIII. Life cycle management of analytical procedures” and its following chapter on the verification of analytical methods in FDA’s own laboratories (“IX: FDA methods verification”).
The document contains plenty of cross-references to corresponding 21 CFR paragraphs and provides – in the last chapter “X. References” – an extensive list of essential FDA Guidelines which also have to be considered in this context, as well as references to corresponding USP chapters, and technical literature on statistical topics. The fact that many aspects of methods validation are addressed in those referenced Guidelines explains the reason why the new Guidance has become shorter.
The document can be commented within 90 days.
read all at

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
http://amcrasto.bravesites.com/
http://anthonycrasto.jimdo.com/
Congratulations! Your presentation titled “Anthony Crasto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
join my process development group on google
organic-process-development group
you can post articles and will be administered by me on the google group which is very popular across the world
https://sites.google.com/site/amcrasto/
LinkedIn group
blogs are
- Green Chemistry International,
- Eurekamoments in Organic Chemistry ,
- WIX CHEMISTRY BLOG ,
- Drug regulatory affairs international
- ORGANIC CHEMISTRY SELECT
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ORGANIC CHEMISTRY INTERNATIONAL
MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ
MY CHINA AND JAPAN BLOGS
http://blog.sina.com.cn/u/5030268874
http://ameblo.jp/medchem-amcrasto/
VIETNAM
ICELAND

FDA Approves Xgeva,denosumab to Treat Giant Cell Tumor of the Bone
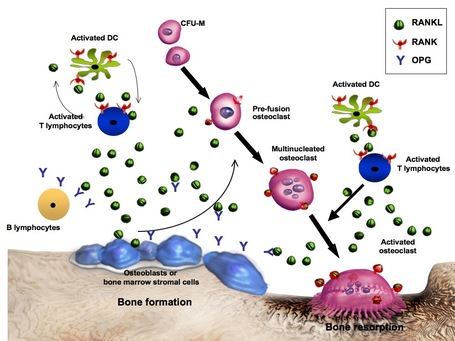
June 13, 2013 — The U.S. Food and Drug Administration today expanded the approved use of Xgeva (denosumab) to treat adults and some adolescents with giant cell tumor of the bone (GCTB), a rare and usually non-cancerous tumor.
GCTB generally occurs in adults between the ages of 20 and 40 years. In most cases, GCTB does not spread to other parts of the body but destroys normal bone as it grows, causing pain, limited range of motion and bone fractures. Rarely, GCTB can transform into a cancerous tumor and spread to the lungs.
Xgeva is a monoclonal antibody that binds to RANKL, a protein essential for maintenance of healthy bone. RANKL is also present in GCTB. Xgeva is intended for patients whose GCTB cannot be surgically removed (unresectable) or when surgery is likely to result in severe morbidity, such as loss of limbs or joint removal. It should only be used in adolescents whose bones have matured
http://www.drugs.com/newdrugs/fda-approves-xgeva-giant-cell-tumor-bone-3815.html

Denosumab is a fully human monoclonal antibody for the treatment of osteoporosis, treatment-induced bone loss, bone metastases, rheumatoid arthritis, multiple myeloma, and giant cell tumor of bone. It was developed by the biotechnology companyAmgen.
Denosumab is designed to inhibit RANKL (RANK ligand), a protein that acts as the primary signal for bone removal. In many bone loss conditions, RANKL overwhelms the body’s natural defenses against bone destruction.
In June 2010, denosumab was approved by the U.S. Food and Drug Administration (FDA) for use in postmenopausal women with risk of osteoporosis under the trade nameProlia, and in November 2010, as Xgeva, for the prevention of skeleton-related events in patients with bone metastases from solid tumors.Denosumab is the first RANKL inhibitor to be approved by the FDA. In the summer of 2011 clinical trials were investigating denosumab in giant cell tumors, multiple myeloma with bone metastases, and hypercalcemia of malignancy, and further investigating its dosing and safety.
A positive genotoxicity result can throw the fate of a promising drug candidate—in which a firm has invested significant time and money—into doubt

A positive genotoxicity result can throw the fate of a promising drug candidate-in which a firm has invested significant time and money-into doubt. The statistical improbability and challenges of bringing a drug to market become paramount.
READ ALL AT
BY WORLD DRUG TRACKER
Automating Lead Optimization
 This diagram illustrates the methods used to determine solubility as a compound advances toward further clinical study, and the increasing reach of automation and informatics systems. Initially, screens are run in silico on a library after hits are determined through a high-throughput screen; then various kinetic solubility assays are used to determine the compound’s potency at various concentrations. Two rounds of kinetic solubility assays determine gross and broad solubility (mmol/L) and finite solubility (less than 20 µmol/L) before the compound is advanced into thermodynamic solubility assays. Figure modified from Petereit A, Saal C. What is the Solubility of My Compound? Assessing Solubility for Pharmaceutical Research and Development Compounds. Am Pharm Rev. 2011; 14
This diagram illustrates the methods used to determine solubility as a compound advances toward further clinical study, and the increasing reach of automation and informatics systems. Initially, screens are run in silico on a library after hits are determined through a high-throughput screen; then various kinetic solubility assays are used to determine the compound’s potency at various concentrations. Two rounds of kinetic solubility assays determine gross and broad solubility (mmol/L) and finite solubility (less than 20 µmol/L) before the compound is advanced into thermodynamic solubility assays. Figure modified from Petereit A, Saal C. What is the Solubility of My Compound? Assessing Solubility for Pharmaceutical Research and Development Compounds. Am Pharm Rev. 2011; 14
The drug discovery business is changing rapidly. More pharmaceutical companies are working with smaller biotech firms to create early-stage compounds, and thus need quicker and standardized solutions to early-stage development problems.
READ ALL AT
by
WORLD DRUGTRACKER
