
Home » Posts tagged 'Protein Based Therapies'
Tag Archives: Protein Based Therapies
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Enfortumab vedotin


Enfortumab vedotin
| Formula |
C6642H10284N1742O2063S46
|
|---|---|
| Cas |
1346452-25-2
|
| Mol weight |
149022.148
|
AGS-22M6E, enfortumab vedotin-ejfv
Fda approved 2019/12/18, Padcev
Antineoplastic, Nectin-4 antibody, Tubulin polymerization inhibitor, Urothelial cancer
エンホルツマブベドチン (遺伝子組換え);
protein Based Therapies, Monoclonal antibody, mAb,
UNII DLE8519RWM
Other Names
- AGS 22CE
- AGS 22M6E
- AGS 22ME
- Enfortumab vedotin
- Enfortumab vedotin-ejfv
- Immunoglobulin G1 (human monoclonal AGS-22M6 γ1-chain), disulfide with human monoclonal AGS-22M6 κ-chain, dimer, tetrakis(thioether) with N-[[[4-[[N-[6-(3-mercapto-2,5-dioxo-1-pyrrolidinyl)-1-oxohexyl]-L-valyl-N5-(aminocarbonyl)-L-ornithyl]amino]phenyl]methoxy]carbonyl]-N-methyl-L-valyl-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-hydroxy-1-methyl-2-phenylethyl]amino]-1-methoxy-2-methyl-3-oxopropyl]-1-pyrrolidinyl]-2-methoxy-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-L-valinamide
- Padcev
Protein Sequence
Sequence Length: 1322, 447, 447, 214, 214multichain; modified (modifications unspecified)
Enfortumab vedotin is an antibody-drug conjugate used in the treatment of patients with advanced, treatment-resistant urothelial cancers.3 It is comprised of a fully human monoclonal antibody targeted against Nectin-4 and a microtubule-disrupting chemotherapeutic agent, monomethyl auristatin E (MMAE), joined by a protease-cleavable link.3 It is similar to brentuximab vedotin, another antibody conjugated with MMAE that targets CD-30 instead of Nectin-4.
The clinical development of enfortumab vedotin was the result of a collaboration between Astellas Pharma and Seattle Genetics2 and it was first approved for use in the United States in December 2019 under the brand name PadcevTM.3
The most common side effects for patients taking enfortumab vedotin were fatigue, peripheral neuropathy (nerve damage resulting in tingling or numbness), decreased appetite, rash, alopecia (hair loss), nausea, altered taste, diarrhea, dry eye, pruritis (itching) and dry skin. [4]Enfortumab vedotin[1] (AGS-22M6E) is an antibody-drug conjugate[2] designed for the treatment of cancer expressing Nectin-4.[3]Enfortumab refers to the monoclonal antibody part, and vedotin refers to the payload drug (MMAE) and the linker.
The fully humanized antibody was created by scientists at Agensys (part of Astellas) using Xenomice from Amgen; the linker technology holding the antibody and the toxin together was provided by and licensed from Seattle Genetics.[5]
Results of a phase I clinical trial were reported in 2016.[2]
In December 2019, enfortumab vedotin-ejfv was approved in the United States for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.[4]
Enfortumab vedotin was approved based on the results of a clinical trial that enrolled 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.[4] The overall response rate, reflecting the percentage of patients who had a certain amount of tumor shrinkage, was 44%, with 12% having a complete response and 32% having a partial response.[4] The median duration of response was 7.6 months.[4]
The application for enfortumab vedotin-ejfv was granted accelerated approval, priority review designation, and breakthrough therapydesignation.[4] The U.S. Food and Drug Administration (FDA) granted the approval of Padcev to Astellas Pharma US Inc.[4]
Indication
Enfortumab vedotin is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor, and a platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced, or metastatic setting.3
Associated Conditions
Pharmacodynamics
Enfortumab vedotin is an anti-cancer agent that destroys tumor cells by inhibiting their ability to replicate.3 Patients with moderate to severe hepatic impairment should not use enfortumab vedotin – though it has not been studied in this population, other MMAE-containing antibody-drug conjugates have demonstrated increased rates of adverse effects in patients with moderate-severe hepatic impairment.3 Enfortumab vedotin may also cause significant hyperglycemia leading, in some cases, to diabetic ketoacidosis, and should not be administered to patients with a blood glucose level >250 mg/dl.3
Mechanism of action
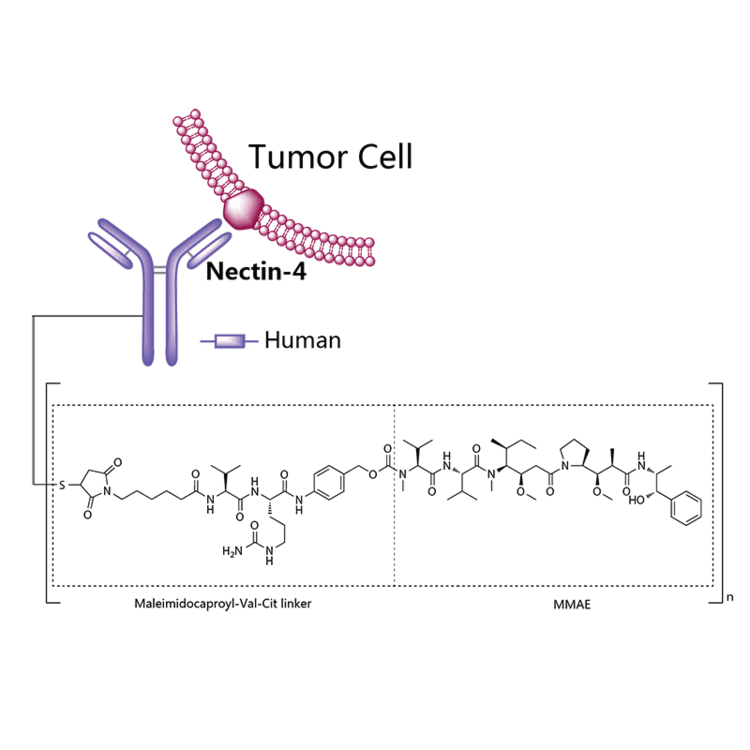
Enfortumab vedotin is an antibody-drug conjugate comprised of multiple components.3 It contains a fully human monoclonal antibody directed against Nectin-4, an extracellular adhesion protein which is highly expressed in urothelial cancers,1 attached to a chemotherapeutic microtubule-disrupting agent, monomethyl auristatin E (MMAE). These two components are joined via a protease-cleavable linker. Enfortumab vedotin binds to cells expressing Nectin-4 and the resulting enfortumab-Nectin-4 complex is internalized into the cell. Once inside the cell, MMAE is released from enfortumab vedotin via proteolytic cleavage and goes on to disrupt the microtubule network within the cell, arresting the cell cycle and ultimately inducing apoptosis.3
PATENT
WO 2016176089
WO 2016138034
WO 2017186928
WO 2017180587
WO 2017200492
US 20170056504
PAPER
Cancer Research (2016), 76(10), 3003-3013.
General References
- Hanna KS: Clinical Overview of Enfortumab Vedotin in the Management of Locally Advanced or Metastatic Urothelial Carcinoma. Drugs. 2019 Dec 10. pii: 10.1007/s40265-019-01241-7. doi: 10.1007/s40265-019-01241-7. [PubMed:31823332]
- McGregor BA, Sonpavde G: Enfortumab Vedotin, a fully human monoclonal antibody against Nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial Carcinoma. Expert Opin Investig Drugs. 2019 Oct;28(10):821-826. doi: 10.1080/13543784.2019.1667332. Epub 2019 Sep 17. [PubMed:31526130]
- FDA Approved Drug Products: Padcev (enfortumab vedotin-ejfv) for IV injection [Link]
References
- ^ World Health Organization (2013). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 109”(PDF). WHO Drug Information. 27 (2).
- ^ Jump up to:a b Seattle Genetics and Agensys, an Affiliate of Astellas, Highlight Promising Enfortumab Vedotin (ASG-22ME) and ASG-15ME Phase 1 Data in Metastatic Urothelial Cancer at 2016 ESMO Congress. Oct 2016
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Enfortumab Vedotin, American Medical Association.
- ^ Jump up to:a b c d e f g “FDA approves new type of therapy to treat advanced urothelial cancer”. U.S. Food and Drug Administration (FDA) (Press release). 18 December 2019. Archived from the original on 19 December 2019. Retrieved 18 December 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Challita-Eid PM, Satpayev D, Yang P, et al. (May 2016). “Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models”. Cancer Research. 76 (10): 3003–13. doi:10.1158/0008-5472.can-15-1313. PMID 27013195.
External links
- “Enfortumab vedotin”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Nectin-4 |
| Clinical data | |
| Trade names | Padcev |
| Other names | AGS-22M6E, AGS-22CE, enfortumab vedotin-ejfv |
| License data | |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChemSID | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C6642H10284N1742O2063S46 |
| Molar mass | 149.0 kg/mol g·mol−1 |
PADCEV™
(enfortumab vedotin-ejfv) for Injection, for Intravenous Use
DESCRIPTION
Enfortumab vedotin-ejfv is a Nectin-4 directed antibody-drug conjugate (ADC) comprised of a fully human anti-Nectin-4 IgG1 kappa monoclonal antibody (AGS-22C3) conjugated to the small molecule microtubule disrupting agent, monomethyl auristatin E (MMAE) via a protease-cleavable maleimidocaproyl valine-citrulline (vc) linker (SGD-1006). Conjugation takes place on cysteine residues that comprise the interchain disulfide bonds of the antibody to yield a product with a drug-to-antibody ratio of approximately 3.8:1. The molecular weight is approximately 152 kDa.
Figure 1: Structural Formula
|
Approximately 4 molecules of MMAE are attached to each antibody molecule. Enfortumab vedotin-ejfv is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells and the small molecule components are produced by chemical synthesis.
PADCEV (enfortumab vedotin-ejfv) for injection is provided as a sterile, preservative-free, white to off-white lyophilized powder in single-dose vials for intravenous use. PADCEV is supplied as a 20 mg per vial and a 30 mg per vial and requires reconstitution with Sterile Water for Injection, USP, (2.3 mL and 3.3 mL, respectively) resulting in a clear to slightly opalescent, colorless to slightly yellow solution with a final concentration of 10 mg/mL [see DOSAGE AND ADMINISTRATION]. After reconstitution, each vial allows the withdrawal of 2 mL (20 mg) and 3 mL (30 mg). Each mL of reconstituted solution contains 10 mg of enfortumab vedotin-ejfv, histidine (1.4 mg), histidine hydrochloride monohydrate (2.31 mg), polysorbate 20 (0.2 mg) and trehalose dihydrate (55 mg) with a pH of 6.0.
///////////////Enfortumab vedotin, AGS-22M6E, エンホルツマブベドチン (遺伝子組換え) , protein Based Therapies, Monoclonal antibody, mAb, FDA 2019
[*]SC1CC(=O)N(CCCCCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(=O)N)C(=O)Nc2ccc(COC(=O)N(C)[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@@H](CC(=O)N3CCC[C@H]3[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c4ccccc4)OC)cc2)C1=O
Burosumab-twza, ブロスマブ
> Burosumab Heavy Chain Sequence QVQLVQSGAEVKKPGASVKVSCKASGYTFTNHYMHWVRQAPGQGLEWMGIINPISGSTSN AQKFQGRVTMTRDTSTSTVYMELSSLRSEDTAVYYCARDIVDAFDFWGQGTMVTVSSAST KGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLY SLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSV FLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTY RVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTK NQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQG NVFSCSVMHEALHNHYTQKSLSLSPGK
> Burosumab Light Chain Sequence AIQLTQSPSSLSASVGDRVTITCRASQGISSALVWYQQKPGKAPKLLIYDASSLESGVPS RFSGSGSGTDFTLTISSLQPEDFATYYCQQFNDYFTFGPGTKVDIKRTVAAPSVFIFPPS DEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTL SKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
ALSO
(Heavy chain)
QVQLVQSGAE VKKPGASVKV SCKASGYTFT NHYMHWVRQA PGQGLEWMGI INPISGSTSN
AQKFQGRVTM TRDTSTSTVY MELSSLRSED TAVYYCARDI VDAFDFWGQG TMVTVSSAST
KGPSVFPLAP SSKSTSGGTA ALGCLVKDYF PEPVTVSWNS GALTSGVHTF PAVLQSSGLY
SLSSVVTVPS SSLGTQTYIC NVNHKPSNTK VDKKVEPKSC DKTHTCPPCP APELLGGPSV
FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY
RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK GQPREPQVYT LPPSRDELTK
NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG
NVFSCSVMHE ALHNHYTQKS LSLSPGK
(Light chain)
AIQLTQSPSS LSASVGDRVT ITCRASQGIS SALVWYQQKP GKAPKLLIYD ASSLESGVPS
RFSGSGSGTD FTLTISSLQP EDFATYYCQQ FNDYFTFGPG TKVDIKRTVA APSVFIFPPS
DEQLKSGTAS VVCLLNNFYP REAKVQWKVD NALQSGNSQE SVTEQDSKDS TYSLSSTLTL
SKADYEKHKV YACEVTHQGL SSPVTKSFNR GEC
(dimer; disulfide bridge:H22-H96, H144-H200, H220-L213, H220-H’226, H229-H’229, H261-H321, H367-H425, H’22-H’96, H’144-H’200, H’220-L’213, H’261-H’321, H’367-H’425, L23-L88, L133-L193, L’23-L’88, L’133-L’193)
Burosumab-twza, KRN 23
ブロスマブ
CAS1610833-03-8
UNII G9WJT6RD29
Protein chemical formulaC6388H9904N1700O2006S46
Protein average weight144100.0 Da
Protein Based Therapies
Monoclonal antibody (mAb)
breakthrough therapy and orphan drug designations
Approval Status:Approved April 2018
Specific Treatments:X-linked hypophosphatemia
Crysvita (burosumab-twza) is a fibroblast growth factor 23 (FGF23) blocking antibody.
This drug is indicated for the treatment of X-linked hypophosphatemia with radiological evidence of bone disease in children of 1 year of age and older and adolescents with growing skeletons [4].

Burosumab (INN, trade name Crysvita) known as KRN23 is a human monoclonal antibody designed for the treatment of X-linked hypophosphatemia.[1][2][3] Burosumab was approved by the FDA for its intended purpose, in patients aged 1 year and older, on 17 April 2018.[4] The FDA approval fell under both the breakthrough therapy and orphan drug designations.[4]
This drug was developed by Ultragenyx and is in a collaborative license agreement with Kyowa Hakko Kirin.[5]
Burosumab (KRN23) is an entirely human monoclonal IgG1 antibody that binds excess fibroblast growth factor 23 (FGF23) and has been successfully tested in clinical trials in children with X-linked hypophosphatemic rickets [1].
The U.S. Food and Drug Administration approved Crysvita (burosumab) in April 2018. This is the first drug approved to treat adults and children ages 1 year and older with X-linked hypophosphatemia (XLH), which is a rare, inherited form of rickets. X-linked hypophosphatemia causes low circulating levels of phosphorus in the blood. It causes impaired bone growth and development in children and adolescents and issues with bone mineralization throughout a patient’s life [3].
XLH is a serious disease which affects about 3,000 children and 12,000 adults in the United States. Most children with XLH suffer from bowed or bent legs, short stature, bone pain and severe dental pain. Some adults with this condition suffer from persistent, unrelenting discomfort and complications, such as joint pain, impaired mobility, tooth abscesses and hearing loss [3]
Crysvita is specifically indicated for the treatment of X-linked hypophosphatemia (XLH) in adult and pediatric patients 1 year of age and older.
Crysvita is supplied as a subcutaneous injection. The recommended starting dose for pediatrics is 0.8 mg/kg of body weight, rounded to the nearest 10 mg, administered every two weeks. The minimum starting dose is 10 mg up to a maximum dose of 90 mg. After initiation of treatment with Crysvita, measure fasting serum phosphorus every 4 weeks for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is above the lower limit of the reference range for age and below 5 mg/dL, continue treatment with the same dose. Follow dose adjustment schedule per the drug label. The recommended dose regimen in adults is 1 mg/kg body weight, rounded to the nearest 10 mg up to a maximum dose of 90 mg, administered every four weeks. After initiation of treatment with Crysvita, assess fasting serum phosphorus on a monthly basis, measured 2 weeks post-dose, for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is within the normal range, continue with the same dose. See drug label for specific dose adjustments.
Mechanism of Action
Crysvita (burosumab-twza) is a fibroblast growth factor 23 (FGF23) blocking antibody. X-linked hypophosphatemia is caused by excess fibroblast growth factor 23 (FGF23) which suppresses renal tubular phosphate reabsorption and the renal production of 1,25 dihydroxy vitamin D. Burosumab-twza binds to and inhibits the biological activity of FGF23 restoring renal phosphate reabsorption and increasing the serum concentration of 1,25 dihydroxy vitamin D.
REFERENCES
1 file:///H:/761068Orig1s000ChemR.pdf
REF
- Kutilek S: Burosumab: A new drug to treat hypophosphatemic rickets. Sudan J Paediatr. 2017;17(2):71-73. doi: 10.24911/SJP.2017.2.11. [PubMed:29545670]
- Kinoshita Y, Fukumoto S: X-linked hypophosphatemia and FGF23-related hypophosphatemic diseases -Prospect for new treatment. Endocr Rev. 2018 Jan 26. pii: 4825438. doi: 10.1210/er.2017-00220. [PubMed:29381780]
- FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia [Link]
- Crysvita Drug Label [Link]
- Burosumab for a rare bone disease [Link]
- DRUG: Burosumab [Link]
- NHS document [Link]
- Burosumab for XLH [Link]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | FGF 23 |
| Clinical data | |
| Trade names | Crysvita |
| Synonyms | KRN23 |
| ATC code | |
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C6388H9904N1700O2006S46 |
| Molar mass | 144.1 kDa |
References
- Jump up^ Statement On A Nonproprietary Name Adopted By The USAN Council – Burosumab, American Medical Association.[permanent dead link]
- Jump up^ World Health Organization (2016). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 115”(PDF). WHO Drug Information. 30 (2): 255.
- Jump up^ “Burosumab (KRN23) for X-Linked Hypophosphatemia (XLH)” (PDF). n.d. Retrieved 2018-04-18.
- ^ Jump up to:a b “FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia” (Press release). FDA. 17 April 2018.
- Jump up^ “Collaboration with Ultragenyx to Develop and Commercialize KRN23 for X-linked Hypophosphatemia” (Press release). Kyowa Kirin. 4 September 2013. Retrieved 2018-04-17.
//////////////Burosumab-twza, Crysvita FDA 2018, BLA 761068, Protein Based Therapies, Monoclonal antibody, mAb, KRN 23, breakthrough therapy, orphan drug designations, Peptide, ブロスマブ

{kind=link}