
Home » Posts tagged 'protease inhibitor'
Tag Archives: protease inhibitor
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Imocitrelvir
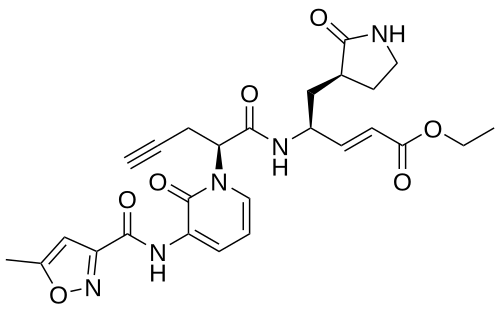
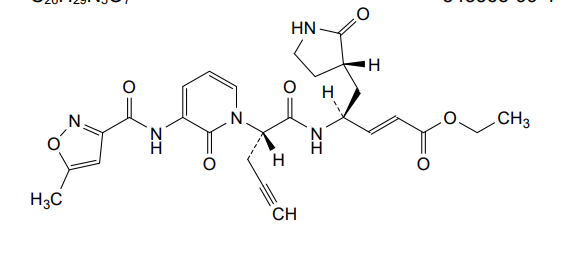
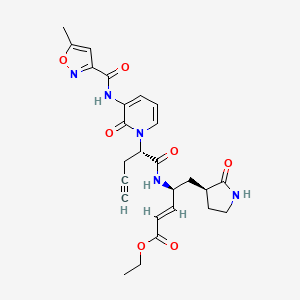
Imocitrelvir
CAS 343565-99-1
MFC26H29N5O7 MW523.5 g/mol
ethyl (2E,4S)-4-{(2S)-2-[3-(5-methyl-1,2-oxazole-3-carboxamido)-2-oxopyridin-1(2H)-yl]pent-4-ynamido}-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate
ethyl (E,4S)-4-[[(2S)-2-[3-[(5-methyl-1,2-oxazole-3-carbonyl)amino]-2-oxo-1-pyridinyl]pent-4-ynoyl]amino]-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate
protease inhibitor, antiviral, AG-7404, V-7404, AG 7404, V 7404, VQ1AN3OO42
Imocitrelvir is an investigational new drug that is being evaluated for the treatment of viral infections. It is a 3C protease inhibitor in picornaviruses. Originally developed by Pfizer for treating human rhinovirus infections,[1] this small molecule has shown promise against a broader range of viruses, including polioviruses.[2][3]
SYN
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2003-09-17
PMID: 14521419
DOI: 10.1021/jm030166l
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016044656&_cid=P21-MHBDH2-20719-1
PAT
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001040189&_cid=P21-MHBDI9-21481-1
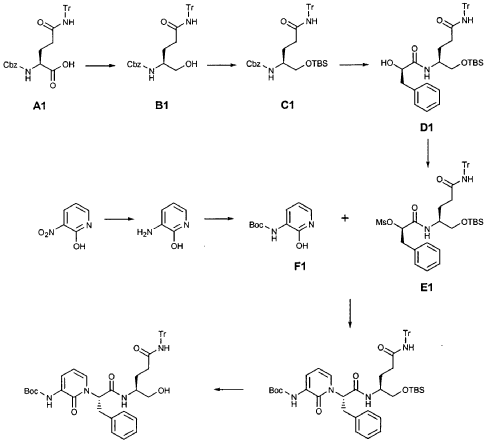
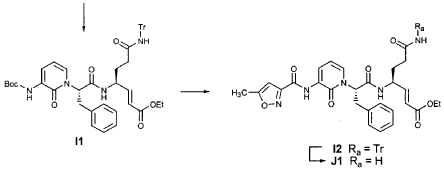
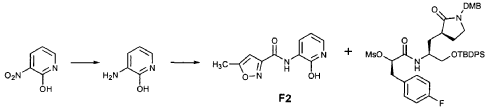
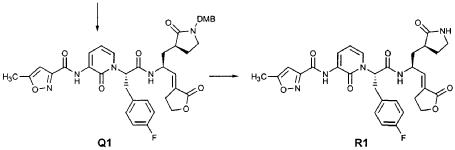
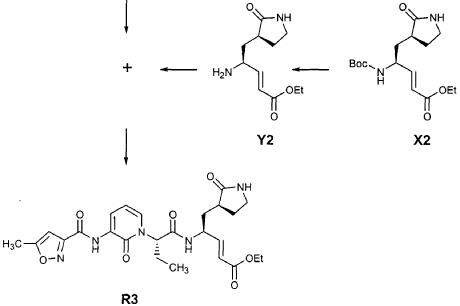
EXAMPLE 21
Preparation of Compound 22: tra«5-(4S,3″”S)-4-(2′-{3″-[(5′”-Methylisoxazole-3′”-carbonyl)amino]-2″-oxo-2″H-pyridin- 1 “-yl} acetylamino)-5-(2″”-oxopyrrilidin-3″”-yl)pent-2-enoic Acid Ethyl Ester
Preparation of Intermediate {3-[(5′-Methylisoxazole-3′-carbonyl)amino]-2-oxo-2H-pyridin-l-yl} acetic Acid tert-Butyl Ester
To a solution of 5-methylisoxazole-3-carboxylic acid (2′-hydroxy-4′-methylpyridin-3′-yl)amide (F2, Example 19) (0.520 g, 2.37 mmol, 1 equiv) in TΗF (20 mL) at 0 °C was added NaΗ (0.095 g, 2.37 mmol, 1.0 equiv). The resulting mixture was stirred at 0 °C for 20 min, and then t-butyl bromoacetate (0.385 mL, 2.61 mmol, 1.1 equiv) was added. The reaction mixture was stirred and warmed to room temperature for 30 min, then was partitioned between 0.5 N ΗC1 (100 mL) and EtOAc (2 x 100 mL). The combined organic layers were dried over Na2SO and were concentrated. Purification of the residue by flash column chromatography (30% EtOAc in hexanes) provided the title intermediate (0.628 g, 79%) as a white solid: IR (cm-1) 3343, 1743, 1651, 1581, 1156; Η NMR (CDC13) δ 1.52 (s, 9H), 2.53 (s, 3H), 4.65 (s, 2H), 6.32 (t, 1H, 7= 7.2), 6.51 (s, IH), 7.01 (dd, 1H, 7= 6.9, 1.8), 8.50 (dd, 1H, 7= 7.5, 1.8), 9.63 (s, br. IH); Anal. C16H19N3O5: C, H, N.
Preparation of Compound 22
The preceding intermediate was transformed into Compound 22 by a process that was analogous to that described in Example 25 for the transformation of V3 to product R3: mp = 102-106 °C; IR (cm”1) 3336, 1684, 1534, 1457; JH NMR (CDCI3) δ 1.27 (t, 3H, 7= 7.2), 1.67-1.75 (m, IH), 1.98-2.09 (m, IH), 2.37-2.49 (m, IH), 2.53 (s, 3H), 2.55-2.61 (m, IH), 3.34-3.46 (m, 2H), 3.51-3.52 (m, IH), 4.17 (q, 2H, 7= 7.2), 4.61-4.78 (m, 3H), 5.98 (dd, IH, 7 = 15.6, 1.5), 6.20 (s, br. IH), 6.35 (t, 1H, 7= 7.8), 6.51 (s, IH), 6.85 (dd, IH, 7= 15.6, 5.1), 7.17 (d, IH, 7= 7.2), 8.33 (d, IH, 7= 7.2), 8.49 (d, IH, 7= 7.5), 9.57 (s, br. IH); Anal.
C23H27N5O7: C, H, N.
EXAMPLE 24
Preparation of Compound 25: trans-(2’S,3″”‘S,4S)-4-(3,-(4″-Fluorophenyl)-2′-{3″‘-[(5″”-methylisoxazole-3″”-carbonyl)amino]-2′”-oxo-2′”H-pyridin- “-yl}propionylamino)-5-(2″ oxopyrrolidin-3′””-yl)pent-2-enoic Acid Ethyl Ester
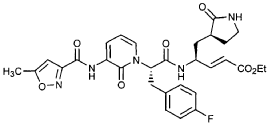
The title compound was prepared from F2 (Example 19) in a manner analogous to that described for the conversion of U2 to 13 in Example 23 utilizing intermediate Y2 (Example 25) where appropriate: IR (cm-1) 3331, 1690, 1590, 1531, 1455; !H NMR (CDCI3) δ 1.30 (t, 3H, 7= 7.0), 1.45-1.55 (m, IH), 1.64-1.75 (m, IH), 2.03-2.31 (m, 3H), 2.49 (s, 3H), 3.10 (dd, IH, 7= 13.7, 7.9), 3.20-3.46 (m, 3H), 4.20 (q, 2H, 7= 7.0), 4.36-4.47 (m, IH), 5.67 (dd, IH, 7 = 15.7, 1.4), 5.85-5.92 (m, IH), 6.29 (t, 1H, 7= 7.2), 6.45 (s, IH), 6.70 (dd, IH, 7= 15.7, 5.7), 6.86 (s, IH), 6.90-6.97 (m, 2H), 7.10-7.16 (m, 2H), 7.60 (dd, IH, 7= 7.2, 1.6), 8.37 (dd, IH, 7 = 7.2, 1.6), 8.51 (d, IH, 7= 6.6), 9.47 (s, IH).
EXAMPLE 25
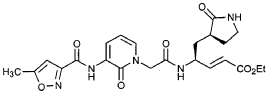
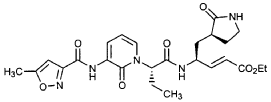
Preparation of Compound 26: tr_.«5-(2’S,3″”S,4S)-4-(2′-{3″-[(5″‘-Methyl-isoxazole-3′”-carbonyl)amino]-2″-oxo-2″H-pyridin-l”-yl}butyrylamino)-5-(2″”-oxopyrrolidin-3″”-yl)pent-2-enoic Acid Ethyl Ester (R3)
Preparation of Intermediate (2R)-2-Trifluoromethanesulfonyl-oxybutyric acid tert-butyl ester (U3)
Commercially available T3 (0.575 g, 3.59 mmol, 1 equiv) was dissolved in CH2CI2 (25 mL) and cooled in an ice bath. 2,6-Lutidine (0.836 mL, 7.18 mmol, 2 equiv) and trifluoromethanesulfonic anhydride (1.15 mL, 6.84 mmol, 1.9 equiv) were added and the reaction mixture was stirred 30 min. It was then diluted with MTBE (400 mL), washed with a mixture of brine and 1 N HCl (2:1, 100 mL) and brine (100 mL), dried over Na2SO4 and evaporated to provide the title intermediate which was used without further purification.
Preparation of Intermediate (2S)-2- {3′-[(5″-Methylisoxazole-3″-carbonyl)amino]-2′-oxo-2’H-pyri din- l’-yl} butyric Acid tert-Butyl Ester (V3)
Intermediate F2 from above (0.200 g, 0.912 mmol, 1.1 equiv) was suspended in TΗF (6 mL). Sodium hydride (60% dispersion in mineral oil, 0.0332 g, 0.830 mmol, 1 equiv) was added in one portion. After stirring 30 min, a solution of intermediate U3 (0.830 mmol, 1 equiv, based on T3) in TΗF (7 mL) was added dropwise. The resulting mixture was stirred 2 hours, then diluted with EtOAc (200 mL) and washed with brine (2 x 50 mL). The organic phase was dried over MgSO4 and evaporated. The residue was purified by flash column chromatography (25% EtOAc in hexanes) to provide the title intermediate (0.178 g, 59%) as an oil: R/= 0.30 (25% EtOAc in hexanes); IR (cm”1) 3331, 1731, 1690, 1649, 1602, 1531 ; *Η NMR (CDCI3) δ 0.93 (t, 3H, 7= 7.3), 1.45 (s, 9H), 1.83-2.01 (m, IH), 2.17-2.31 (m, IH), 2.50 (s, 3H), 5.44-5.51 (m, IH), 6.32 (t, IH, 7= 7.2), 6.48 (s, IH), 7.10 (dd, IH, 7= 7.2, 1.8), 8.45 (dd, 1H, 7= 7.2, 1.8), 9.64 (s, IH); Anal. C18H23N3O5: C, H, N.
Preparation of Intermediate (2S)-2- {3′-[(5″-Methylisoxazole-3″-carbonyl)amino]-2′-oxo-2’H-pyridin-l’-yl}butyric Acid (W3)
Intermediate V3 from above (0.143 g, 0.397 mmol, 1 equiv) was stirred for 1 h in a solution of TFA (2 mL) in CΗ2CI2 (3 mL). The volatiles were evaporated. The residue was suspended in toluene (10 mL) and concentrated to dryness, providing the title intermediate which was used without further purification.
Preparation of Intermediate trα«5-(3’S,4S)-4-Amino-5-(2′-oxopyrrolidin-3′-yl)pent-2-enoic Acid Ethyl Ester (Y2)
Intermediate X2, prepared according to the method disclosed in the co-pending application, U.S. Provisional Patent Application No. 60/150,358, filed August 24, 1999(0.130 g, 0.398 mmol, 1 equiv), was stirred for 30 min in a solution of TFA (2 mL) in CH2CI2 (3 mL). The volatiles were evaporated. The residue was suspended in toluene (10 mL) and concentrated to dryness, providing the title intermediate which was used without further purification.
Preparation of Product R3 (Compound 26)
Intermediates W3 and Y2 (as prepared above) were combined in CH2CI2 (7 mL) and cooled in an ice bath. HOBt (0.064 g, 0.47 mmol, 1.2 equiv), iP^NEt (0.484 mL, 2.78 mmol, 7 equiv) and EDC (0.084 g, 0.44 mmol, 1.1 equiv) were added sequentially. The reaction mixture was allowed to warm to 23 °C overnight, then diluted with EtOAc (500 mL) and washed with 5% KHSO4 , half saturated NaHCO3, and brine (100 mL each). The organic phase was dried over MgSO4 and evaporated. The residue was purified by flash column chromatography (gradient elution, 2→3% CH3OH in CH2CI2) to provide the title intermediate (0.119 g, 58%) as a white foam: IR (cm”1) 3331, 1684, 1649, 1590, 1531; JH NMR (CDCI3) δ 0.92 (t, 3H, J = 7.3), 1.29 (t, 3H, J = 7.1), 1.47-1.58 (m, IH), 1.62-1.77 (m, IH), 1.85-2.00 (m, IH), 2.08-2.33 (m, 4H), 2.49 (s, 3H), 3.25-3.42 (m, 2H), 4.19 (q, 2H, J = 7.1), 4.39-4.50 (m, IH), 5.73 (dd, IH, J = 8.8, 6.8), 5.97 (dd, IH, J = 15.7, 1.4), 6.34 (t, IH, J = 7.2), 6.46 (s, IH), 6.86 (dd, IH, J = 15.7, 5.9), 7.18 (s, IH), 7.59 (dd, IH, J = 7.2, 1.8), 8.42 (dd, IH, J = 7.2, 1.8), 8.58-8.62 (m, IH), 9.56 (s, 1); Anal. C25H31N5O7O.5OH2O: C, H, N.
PAT
- Treatment of infection by human enterovirus d68Publication Number: US-2020016243-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus d68Publication Number: WO-2016044656-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus d68Publication Number: US-2021052708-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus D68Publication Number: US-11191817-B2Priority Date: 2014-09-17Grant Date: 2021-12-07
- Therapeutic compounds and methodsPublication Number: US-2025051283-A1
- Protease Inhibitors for Treatment or Prevention of Coronavirus DiseasePublication Number: US-2023192660-A1Priority Date: 2020-05-08
- Composition and combined medication method for treating enterovirus infectionPublication Number: US-2019030027-A1Priority Date: 2016-01-29
- Composition and combined medication method for treating enterovirus infectionPublication Number: US-10864210-B2Priority Date: 2016-01-29Grant Date: 2020-12-15
- Treatment of infection by human enterovirus D68Publication Number: US-10328128-B2Priority Date: 2014-09-17Grant Date: 2019-06-25
- Treatment of infection by human enterovirus d68Publication Number: US-2017290893-A1Priority Date: 2014-09-17
- Nucleotide and nucleoside therapeutic compositions, combinations and related uses thereofPublication Number: CN-117881402-APriority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: EP-4333859-A1Priority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations, and related usesPublication Number: JP-2024517807-APriority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: WO-2022235874-A1Priority Date: 2021-05-05
- Protease inhibitors for treatment or prevention of coronavirus diseasePublication Number: EP-4146267-A1Priority Date: 2020-05-08
- 4′-substituted nucleosides and nucleotides as antiviral agentsPublication Number: WO-2024227159-A2Priority Date: 2023-04-28
- Therapeutic compoundsPublication Number: WO-2024206284-A2Priority Date: 2023-03-27
- Antibody molecules binding to sars-cov-2Publication Number: WO-2024168061-A2Priority Date: 2023-02-07
- Predictive model for variants associated with drug resistance and theranostic applications thereofPublication Number: WO-2023172635-A1Priority Date: 2022-03-08
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: CA-3216679-A1Priority Date: 2021-05-05
LIT
- Structure and inhibition of SARS-CoV-1 and SARS-CoV-2 main proteases by oral antiviral compound AG7404Publication Name: Antiviral ResearchPublication Date: 2022-12PMCID: PMC9632241PMID: 36336176DOI: 10.1016/j.antiviral.2022.105458
- Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug DesignPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-09-30PMID: 34591488DOI: 10.1021/acs.jmedchem.1c01215
- In Vitro Antiviral Activity of New Oxazoline Derivatives as Potent Poliovirus InhibitorsPublication Name: Journal of Medicinal ChemistryPublication Date: 2018-12-04PMCID: PMC9169555PMID: 30512950DOI: 10.1021/acs.jmedchem.8b01482
- A Novel Series of Highly Potent Small Molecule Inhibitors of Rhinovirus ReplicationPublication Name: Journal of Medicinal ChemistryPublication Date: 2017-06-15PMID: 28581749DOI: 10.1021/acs.jmedchem.7b00175
- Anti-poliovirus activity of protease inhibitor AG-7404, and assessment of in vitro activity in combination with antiviral capsid inhibitor compoundsPublication Name: Antiviral ResearchPublication Date: 2013-05PMID: 23499651DOI: 10.1016/j.antiviral.2013.03.003



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
| Clinical data | |
|---|---|
| Other names | AG-7404, V-7404 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 343565-99-1 |
| PubChem CID | 5280053 |
| IUPHAR/BPS | 13223 |
| UNII | VQ1AN3OO42 |
| ChEMBL | ChEMBL141157 |
| Chemical and physical data | |
| Formula | C26H29N5O7 |
| Molar mass | 523.546 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Imocitrelvir”. PatSnap.
- Xie H, Rhoden EE, Liu HM, Ogunsemowo F, Mainou BA, Burke RM, et al. (November 2024). “Antiviral Development for the Polio Endgame: Current Progress and Future Directions”. Pathogens. 13 (11). Basel, Switzerland: 969. doi:10.3390/pathogens13110969. PMC 11597170. PMID 39599522.
- Bandyopadhyay AS, Burke RM, Hawes KM (June 2024). “Polio Eradication: Status, Struggles and Strategies”. The Pediatric Infectious Disease Journal. 43 (6): e207-211. doi:10.1097/INF.0000000000004330. PMID 38564755.
////////Imocitrelvir, protease inhibitor, antiviral, AG-7404, V-7404, AG 7404, V 7404, VQ1AN3OO42
AMPRENAVIR For the treatment of HIV-1 infection in combination with other antiretroviral agents.

Amprenavir
KVX-478, 141W94, VX-478,
(3S)-Tetrahydro-3-furanyl ((1S,2R)-3-(((4-aminophenyl)sulfonyl)(2-methylpropyl)amino)-2-hydroxy-1-(phenylmethyl)propyl)carbamate
(3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl] carbamate
CAS NO. 161814-49-9, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate
| 161814-49-9 | |
| Weight | 505.224656557 |
|---|---|
| Chemical Formula | C25H35N3O6S |
| Amprenavir is a protease inhibitor used to treat HIV infection. |
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available.
| Amprenavir is a protease inhibitor with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Protease inhibitors block the part of HIV called protease. HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Amprenavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. Protease inhibitors are almost always used in combination with at least two other anti-HIV drugs. |

| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| United States | 5585397 | 1993-12-17 | 2013-12-17 |
| United States | 6730679 | 1997-11-11 | 2017-11-11 |
Background
Research aimed at development of renin inhibitors as potential antihypertensive agents had led to the discovery of compounds that blocked the action of this peptide cleaving enzyme. The amino acid sequence cleaved by renin was found to be fortuitously the same as that required to produce the HIV peptide coat. Structure–activity studies on renin inhibitors proved to be of great value for developing HIV protease inhibitors. Incorporation of an amino alcohol moiety proved crucial to inhibitory activity for many of these agents. This unit is closely related to the one found in the statine, an unusual amino acid that forms part of the pepstatin, a fermentation product that inhibits protease enzymes.
Synthesis
R.D. Tung, M.A. Murcko, G.R. Bhisetti, U.S. Patent 5,558,397 (1996). The scheme shown here is partly based on that used to prepare darunavir and fosamprenavir due to difficulty in deciphering the patent.
AGENERASE (amprenavir) is an inhibitor of the human immunodeficiency virus (HIV) protease. The chemical name of amprenavir is (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulfonamido)-1-benzyl-2-hydroxypropyl]carbamate. Amprenavir is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H35N3O6S and a molecular weight of 505.64. It has the following structural formula:
 |
Amprenavir is a white to cream-colored solid with a solubility of approximately 0.04 mg/mL in water at 25°C.
AGENERASE Capsules (amprenavir capsules) are available for oral administration. Each 50- mg capsule contains the inactive ingredients d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), polyethylene glycol 400 (PEG 400) 246.7 mg, and propylene glycol 19 mg. The capsule shell contains the inactive ingredients d-sorbitol and sorbitans solution, gelatin, glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink. Each 50- mg AGENERASE Capsule contains 36.3 IU vitamin E in the form of TPGS. The total amount of vitamin E in the recommended daily adult dose of AGENERASE is 1,744 IU.
………………………………
paper
DOI: 10.1039/B404071F
http://pubs.rsc.org/en/content/articlelanding/2004/ob/b404071f#!divAbstract

Efficient and industrially applicable synthetic processes for precursors of HIV protease inhibitors (Amprenavir, Fosamprenavir) are described. These involve a novel and economical method for the preparation of a key intermediate, (3S)-hydroxytetrahydrofuran, from L-malic acid. Three new approaches to the assembly of Amprenavir are also discussed. Of these, a synthetic route in which an (S)-tetrahydrofuranyloxy carbonyl is attached to L-phenylalanine appears to be the most promising manufacturing process, in that it offers satisfactory stereoselectivity in fewer steps.

Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
……………

The reaction of N,N-dibenzyl-L-alaninal (I) with nitromethane, catalyzed by the chiral ammonium salt (II) and KF in THF gives the chiral nitroalcohol (III), which is reduced with NiCl2 and NaBH4 to yield the aminoalcohol (IV). The condensation of (IV) with isobutyraldehyde (V) affords the Schiff base (VI), which is reduced with NaBH4 to provide the secondary amine (VII). The reaction of (VII) with 4-nitrobenzenesulfonyl chloride (VIII) and TEA in dichloromethane furnishes the sulfonamide (IX), which is deprotected by hydrogenation with H2 over Pd/C in methanol, giving the diamino compound (X). Finally, this compound is condensed with 3(S)-tetrahydrofuryl (N-oxysuccinimidyl) carbonate (XI) by means of TEA in dichloromethane to afford the target carbamate.
Angew Chem. Int Ed Engl1999,38,(13-14):1931
……………………………………………………….

The reaction of the chiral epoxide (I) with isobutylamine (II) in refluxing ethanol gives the secondary amine (III), which is protected with benzyl chloroformate (IV) and TEA, yielding the dicarbamate (V). Selective deprotection of (V) with dry HCl in ethyl acetate affords the primary amine (VI), which is treated with 3(S)-tetrahydrofuryl N-succinimidinyl carbonate (VII) (prepared by condensation of tetrahydrofuran-3(S)-ol (VIII) with phosgene and N-hydroxysuccinimide (IX)) and DIEA in acetonitrile to provide the corresponding carbamate (X). The deprotection of (X) by hydrogenation with H2 over Pd/C in ethanol gives the secondary amine (XI), which is condensed with 4-nitrophenylsulfonyl chloride (XII) by means of NaHCO3 in dichloromethane/water to yield the sulfonamide (XIII). Finally, the nitro group of (XIII) is reduced with H2 over Pd/C in ethyl acetate to afford the target compound.
https://www.google.com/patents/WO1999048885A1?cl=ensynthesis of (3S)-tetrahydro-3-furyl N-[(1S,2R)-3(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl]carbamate, hereinafter referred to as the compound of formula (I), and to novel intermediates thereto.The compound of formula (I) has the following structure

and was first described in PCT patent publication number WO94/05639 at Example 168. Currently there is considerable interest in the compound of formula (I) as a new chemotherapeutic compound in the treatment of human immunodeficiency virus (HIV) infection and the associated conditions such as acquired immune deficiency syndrome (AIDS) and AIDS dementia.
There exists at the present time a need to produce large quantities of the compound of formula (I) for clinical investigation into the efficacy and safety of the compound as a chemotherapeutic agent in the treatment of HIV infections.
An ideal route for the synthesis of the compound should produce the compound of formula (I) in high yields at a reasonable speed and at low cost with minimum waste materials and in a manner that is of minimum impact to the environment in terms of disposing of waste-materials and energy consumption.
We have found a new process for the synthesis of the compound of formula (I) with many advantages over previously known routes of synthesis. Such advantages include lower cost, less waste and more efficient use of materials. The new process enables advantageous preparation of the compound of formula (I) on a manufacturing scale.
The route of synthesis of the compound of formula (I) described in the specification of WO94/05639 is specifically described therein in examples 39A, 51A, 51B, 51C, 51D, 167 and 168. The overall yield from these examples is 33.2% of theory.
Generally the route described in WO94/05639 involves protecting the amino alcohol of formula (A) (Ex.39)

wherein P is a protecting group to form a compound of formula (B);

wherein P and P′ are each independently a protecting group;
deprotecting the compound of formula (B) to form a compound of formula (C) (Ex 51A);

wherein P′ is a protecting group;
forming a hydrochloride salt of compound (C) (Ex 51B) then reacting with N-imidazolyl-(S)-tetrahydrofuryl carbamate to form the compound of formula (D) (Ex 51C);

wherein P′ is a protecting group;
deprotecting the compound of formula (D) (Ex 51D) wherein P′ is a protecting group to form the compound of formula (D) wherein P′ is H (Ex 51E); and coupling the resultant secondary amine on the compound of formula (D) to a p-nitrophenylsulphonyl group to form a compound of formula (E) (Ex 167);

the resultant compound of formula (E) is then reduced to form the compound of formula (I) (Ex 168).
In summary, the process disclosed in WO94/05639 for producing the compound of formula (I) from the compound of formula (A) comprises 6 distinct stages:
1) protecting,
2) deprotecting,
3) reacting the resultant compound with an activated tetrahydrofuranol group,
4) deprotecting,
5) coupling with a p-nitrophenylsulfonyl group, and
6) reducing the resultant compound to form a compound of formula (I).
Applicants have now found a process by which the compound of formula (I) may be prepared on a manufacturing scale from the same starting intermediate, the compound of formula (A), in only 4 distinct stages instead of 6. In addition to the associated benefits of fewer stages, such as savings in time and cost, the improved process reduces the number of waste products formed. Furthermore, product may be obtained in a higher yield, of approximately 50% of theory
 .
.
EXAMPLESExample 1
(1S,2R)-tert-butyl N-[1-benzyl-2-hydroxy-3-(isobutylamino)propyl]carbamate (127.77 g, 379.7 mmol) was heated in toluene (888 ml) to 80° C. and triethylamine (42.6 g, 417.8 mmol) added. The mixture was heated to 90° C. and a solution of p-nitrobenzene sulphonyl chloride (94.3 g, 425.4 mmol) in toluene (250 ml) was added over 30 minutes then stirred for a further 2 hours. The resultant solution of the nosylated intermediate {(1S,2R)-tert-butyl N-[1-benzyl-2-hydroxy-3-(N-isobutyl- 4-nitrobenzenesulphonamido)propyl]carbamate } was then cooled to 80° C. The solution was maintained at approximately 80° C., and concentrated hydrochloric acid (31.4 ml, 376.8 mmol) was added over 20 minutes. The mixture was heated to reflux (approx 86° C.) and maintained at this temperature for an hour then a further quantity of concentrated hydrochloric acid (26.4 ml, 316.8 mmol) was added. Solvent (water and toluene mixture) was removed from the reaction mixture by azeotropic distillation (total volume of solvent removed approx 600 ml), and the resultant suspension was cooled to 70-75° C. Denatured ethanol (600 ml) was added, and the solution was cooled to 20° C. The mixture was further cooled to approximately −10° C. and the precipitate formed was isolated by filtration, washed with denatured ethanol (50 ml) and dried at approximately 50° C., under vacuum, for approximately 12 hours, to give (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride (160 g; 73% of theory yield corrected for assay). NMR: 1H NMR (300Mhz, dmso-d6): 8.37(2H, d, J=9 Hz), 8.16(NH3 +s), 8.06(2H, d, J=9 Hz), 7.31(5H, m), 5.65(1H, d, J=5 Hz), 3.95(1H, m), 3.39(2H, m), 2.95(5H, m), 1.90(1H, m), 0.77(6H, dd, J=21 Hz and 6 Hz).
1,1′-carbonyidiimidazole (27.66 kg, 170.58 mol) was added to ethyl acetate (314.3 kg) with stirring to give 3-(S)-tetrahydrofuryl imidazole-1-carboxylate. (S)-3-hydroxytetrahydrofuran (157 kg, 178.19 mol) was added over 30 minutes, washed in with ethyl acetate (9.95 kg), then the mixture was stirred for a further hour. (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride (65.08 kg, 142.10 mol) was added and the mixture heated to reflux for approximately 22 hours. The solution was cooled slightly, and denatured ethanol (98 l) was added. The solution was stirred at 60° C. for 10 minutes then cooled and the product allowed to crystallise. The mixture was cooled to <10° C. and stirred for 2 hours. The product was isolated by filtration, washed with denatured ethanol (33 l) and dried at approximately 50° C., under vacuum to give (3S)-tetrahydro-3-furyl N-[(1S,2R)-1-benzyl-2-hydroxy-3-(N-isobutyl-4-nitrobenzene sulphonamido)propyl]carbamate in a yield of 82% of theory.
NMR: 1H NMR (500 Mhz, dmso-d6): 8.38(2H, d, J=9Hz), 8.06(2H, d, J=9 Hz), 7.20(6H, m), 5.02(1H, d, J=5 Hz), 4.94(1H, m), 4.35(EtOH, broad s), 3.71(EtOH, q), 3.65(1H, m), 3.60(1H, m), 3.51(2H, broad m), 3.40(2H, m), 3.15(1H, dd, J=8 Hz and 14 Hz), 3.07(1H, dd, J=8 Hz and 15 Hz), 2.94(2H, m), 2.48(1H, m), 2.06(1H, m), 1.97(1H, m), 1.78(1H, m), 1.05(EtOH, t), 0.83(6H, dd, J=7 Hz and 16 Hz).
Product from the above stage (80.0 g, 149.4 mmol) was hydrogenated in isopropanol (880 ml) with 5% palladium on carbon (16 g, of a wet paste) and hydrogen pressure (approx 0.5 to 1.5 bar) at 25-50° C. for approximately 5 hours. The mixture was cooled and the catalyst removed by filtration. The solution was distilled to a volume of approximately 320 ml and water (80 ml) was added. This solution was divided into two for the crystallisation step.
To half of the above solution, decolourising charcoal (2 g) was added, the mixture stirred at approximately 32° C. for 4 hours, then filtered. The filtercake was washed with isopropanol (20 ml) then further water (40 ml) was added to the filtrate. The solution was seeded to induce crystallisation and stirred for 5 hours. Water (130 ml) was added slowly over 1 hour then the mixture was stirred for 4 hours. The resultant slurry was cooled to approximately 20° C. and the product was isolated by filtration and washed with a 1:4 mixture of isopropano/water (120 ml). The product was dried at approximately 50° C., under vacuum, for approximately 12 hours to give (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl] carbamate (30.3 g; 80% of theory yield).
NMR: 1H NMR (300 Mhz, dmso-d6): 7.39(2H, d, J=9 Hz), 7.18(6H, m), 6.60(2H, d, J=9 Hz), 6.00(2H, s), 4.99(1H, d, J=6 Hz), 4.93(1H, ddt), 3.64(5H, m), 3.34(1H, m), 3.28(1H, dd, J=14 Hz and 3 Hz), 3.01(1H, m, J=14 Hz and 3 Hz), 2.91(1H, m), 2.66(2H, m), 2.50(1H, m), 2.05(1H, m), 1.94(1H, m), 1.78(1H, m), 0.81(6H, dd, J=16 Hz and 7 Hz). m/z: 506.2(M+H+)
Example 11Synthesis of Amprenavir (1)To a solution of carbamate nitro derivative 15 (0.05 g, 0.09 mmol) in 2 mL of EtOAc was added SnCl2.2H2O (0.1 g, 0.5 mmol) at 70° C. The reaction mixture was heated for 1 h until starting material disappeared and the solution cooled to room temperature. It was then poured into saturated aq. NaHCO3 solution and extracted with EtOAc. The organic extract was dried over anhyd. Na2SO4 and concentrated under reduced pressure. It was purified over chromatography using petroleum ether:EtOAc (3:2) to give amprenavir 1 (0.04 g, 90%).IR: (CHCl3, cm−1): υmax 757, 1090, 1149, 1316, 1504, 1597, 1633, 1705, 2960, 3371; 1H NMR (200 MHz, CDC3): δ 0.86 (d, J=5.7 Hz, 3H), 0.90 (d, J=6.6 Hz, 3H), 1.78-2.21 (m, 3H), 235-3.11 (m, 6H), 3.58-4.11 (m, 7H), 4.25 (s, 2H), 5.01 (br s, 1H), 5.07 (br s, 1H), 6.65 (d, J=8.4 Hz, 2H), 7.20-7.28 (m, 5H), 7.51 (d, J=8.4 Hz, 2H); 13C NMR (50 MHz, CDC3): δ 19.9, 20.2, 27.3, 32.8, 35.4, 35.7, 53.8, 55.0, 58.6, 66.8, 72.6, 73.2, 75.3, 114.0, 125.9, 126.5, 1280.4, 129.5, 137.7, 150.9, 155.9;
Anal. Calcd for C25H35N3O6S: C, 59.39; H, 6.98; N, 8.31; S, 6.34. Found: C, 59.36; H, 6.81; N, 8.25; S, 6.29%.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-23/000/115/669/161814-49-9-1h.png)
![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-23/000/115/669/161814-49-9-13c.png)
COSY PREDICTION
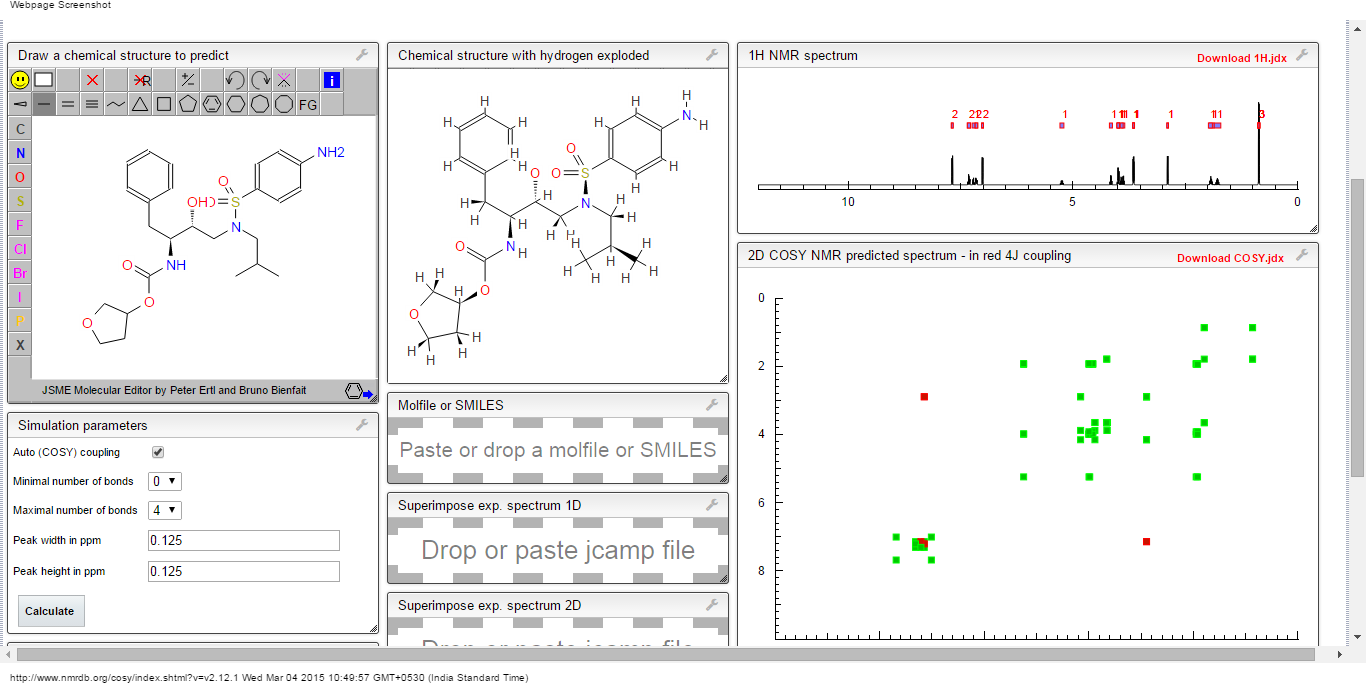
See also
- Fosamprenavir, a prodrug of amprenavir
External links
- Amprenavir bound to proteins in the PDB
- Shen, C. H.; Wang, Y. F.; Kovalevsky, A. Y.; Harrison, R. W.; Weber, I. T. (2010). “Amprenavir complexes with HIV-1 protease and its drug-resistant mutants altering hydrophobic clusters”. FEBS Journal 277 (18): 3699–3714. doi:10.1111/j.1742-4658.2010.07771.x. PMC 2975871. PMID 20695887.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3S)-oxolan-3-yl N-[(2S,3R)-3-hydroxy-4-[N-(2-methylpropyl)(4-aminobenzene)sulfonamido]-1-phenylbutan-2-yl]carbamate | |
| Clinical data | |
| Trade names | Agenerase |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699051 |
| Licence data | EMA:Link, US FDA:link |
|
|
| Legal status |
?
|
| Routes | oral |
| Pharmacokinetic data | |
| Protein binding | 90% |
| Metabolism | hepatic |
| Half-life | 7.1-10.6 hours |
| Excretion | <3% renal |
| Identifiers | |
| CAS number | 161814-49-9 |
| ATC code | J05AE05 |
| PubChem | CID 65016 |
| DrugBank | DB00701 |
| ChemSpider | 58532 |
| UNII | 5S0W860XNR |
| KEGG | D00894 |
| ChEBI | CHEBI:40050 |
| ChEMBL | CHEMBL116 |
| NIAID ChemDB | 006080 |
| Chemical data | |
| Formula | C25H35N3O6S |
| Molecular mass | 505.628 g/mol |
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor…….

AMPRENAVIR
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available.
| Systematic (IUPAC) name | |
|---|---|
| (3S)-oxolan-3-yl N-[(2S,3R)-3-hydroxy-4-[N-(2-methylpropyl)(4-aminobenzene)sulfonamido]-1-phenylbutan-2-yl]carbamate | |
| Clinical data | |
| Trade names | Agenerase |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699051 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. | C (US) |
| Routes | oral |
| Pharmacokinetic data | |
| Protein binding | 90% |
| Metabolism | hepatic |
| Half-life | 7.1-10.6 hours |
| Excretion | <3% renal |
| Identifiers | |
| CAS number | 161814-49-9 |
| ATC code | J05AE05 |
| PubChem | CID 65016 |
| DrugBank | DB00701 |
| ChemSpider | 58532 |
| UNII | 5S0W860XNR |
| KEGG | D00894 |
| ChEBI | CHEBI:40050 |
| ChEMBL | CHEMBL116 |
| NIAID ChemDB | 006080 |
| Chemical data | |
| Formula | C25H35N3O6S |
| Mol. mass | 505.628 g/mol |
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available
………………….
New approaches to the industrial synthesis of HIV protease inhibitors
http://pubs.rsc.org/en/content/articlelanding/2004/ob/b404071f/unauth#!divAbstract
Efficient and industrially applicable synthetic processes for precursors of HIV protease inhibitors (Amprenavir, Fosamprenavir) are described. These involve a novel and economical method for the preparation of a key intermediate, (3S)-hydroxytetrahydrofuran, from L-malic acid. Three new approaches to the assembly of Amprenavir are also discussed. Of these, a synthetic route in which an (S)-tetrahydrofuranyloxy carbonyl is attached to L-phenylalanine appears to be the most promising manufacturing process, in that it offers satisfactory stereoselectivity in fewer steps.
AGENERASE (amprenavir) is an inhibitor of the human immunodeficiency virus (HIV) protease. The chemical name of amprenavir is (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulfonamido)-1-benzyl-2-hydroxypropyl]carbamate. Amprenavir is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H35N3O6S and a molecular weight of 505.64. It has the following structural formula:
|
Amprenavir is a white to cream-colored solid with a solubility of approximately 0.04 mg/mL in water at 25°C.
AGENERASE Capsules (amprenavir capsules) are
available for oral administration. Each 50- mg capsule contains the inactive ingredients d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), polyethylene glycol 400 (PEG 400) 246.7 mg, and propylene glycol 19 mg. The capsule shell contains the inactive ingredients d-sorbitol and sorbitans solution, gelatin, glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink. Each 50- mg AGENERASE Capsule contains 36.3 IU vitamin E in the form of TPGS. The total amount of vitamin E in the recommended daily adult dose of AGENERASE is 1,744 IU.
See also
- Fosamprenavir, a prodrug of amprenavir
External links
- Amprenavir bound to proteins in the PDB

{kind=link}