

")
Anna Popova, Head of the Federal Service for Supervision of Consumer Protection and Welfare (Rospotrebnadzor)
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » Posts tagged 'preclinical' (Page 7)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

Zifaxaban

Zifaxaban
cas 1378266-98-8
rotation (-)
C20 H16 Cl N3 O4 S
C20H16ClN3O4 S, M = 429.87
Tianjin Institute of Pharmaceutical Research
Deep vein thrombosis; Lung embolism
Factor Xa antagonist
TY-602; zhifeishaban; zifaxaban
Chinese J Struc Chem. 2014, 33 (7), 1091-1095.
(S) -5- chloro -N- ((2- oxo _3_ (4_ (2_ oxo _2H_-1-yl) phenyl) oxazolidin-5 -1,3_ yl) methyl) thiophene-2-carboxamide

The title compound(zifaxaban 2, C20H16ClN3O4 S, Mr = 429.87) was synthesized and its crystal structure was determined by single-crystal X-ray diffraction. Zifaxaban crystallizes in monoclinic, space group P21 with a = 5.7900(12), b = 13.086(3), c = 12.889(3) A, β = 100.86(3)°, V = 959.1(3) A3, Z = 2, Dc = 1.489 g/cm3, F(000) = 444, μ = 0.342 mm-1, the final R = 0.0320 and wR = 0.0640 for 2717 observed reflections(I > 2σ(I)).
The absolute configuration of the stereogenic center in the title compound was confirmed to be S by single-crystal X-ray diffraction. Four existing intermolecular hydrogen bonds help to stabilize the lattice and the molecule in the lattice to adopt an L-shape conformation.
Zifaxaban was slightly more active than rivaroxaban 1 in in vitro assay against human FXa and therefore is promising as a drug candidate.
zifaxaban (first disclosed in CN102464658), useful for treating thromboembolic disorders. Zifaxaban, a factor Xa antagonist, is being developed by Tianjin Institute of Pharmaceutical Research, for treating deep vein thrombosis and pulmonary embolism (preclinical, as of November 2014). In May 2014, an IND was filed in China. In June 2014, the institute was seeking to outlicense this product.
In vivo within the cardiovascular, blood coagulation or blood analysis some have formed out of the process of forming a solid mass with the aggregation, called thrombosis, the formation of a solid mass called a thrombus blocks. Thrombosis is an abnormal flow of blood coagulation status due to platelet activation and coagulation factors are activated in accordance therewith.
The blood coagulation was originally a protective mechanism of the organism, there is a mutual antagonism in blood coagulation system and the anti-clotting system. Under physiological conditions, blood clotting factors continue to be activated to produce thrombin, fibrin formation trace, calm on the vascular endothelium, but these traces of fibrin and constantly being activated fibrinolytic system dissolution, while being activated coagulation factors are constantly mononuclear phagocyte system swallowed. The dynamics of the coagulation system and fibrinolysis system, which ensures the blood coagulation potential can also always ensure that the fluid state of the blood.
Sometimes, however, in certain factors can promote the coagulation process, breaking the above dynamic balance triggered the coagulation process, the blood can form a thrombosis or embolism, such as leading to myocardial infarction, stroke, deep vein thrombosis, pulmonary embolism and other thromboembolic disease.
Thromboembolic disease is cardiovascular disease against the most serious diseases, is the first killer of human health. In China, with the improvement and increased aging of the population’s living standards, the incidence of such diseases, mortality, morbidity is increasing every year.
The existing anti-thromboembolic diseases into anti-platelet drugs, anticoagulants and fibrinolytic drugs. Among them, the anti-clotting drugs are the main contents of antithrombotic therapy, mainly thrombin inhibitors and vitamin K antagonists. Heparin and low molecular weight heparin, represented by the presence of oral thrombin inhibitor invalid, non-selective inhibition and high risk of bleeding and other shortcomings. Although warfarin is representative of vitamin K antagonists can be administered orally, but there are narrow therapeutic index, high risk of bleeding and other shortcomings.
Studies have shown that the coagulation process is usually divided into intrinsic coagulation pathway and the extrinsic coagulation pathway. Coagulation process involves a lot of coagulation factors, coagulation factor activated are each the next inactive clotting factor precursor is converted into the activated form. Endogenous, exogenous pathway final summary, the blood coagulation factor X is converted to Xa.
Therefore, theoretically, the direct inhibition of ¾ factor activity should produce effective anti-clotting effect, without the side effects of thrombin inhibitors with. As direct inhibition) (a factor activity on normal hemostasis reaction / adjustment process produces minimal impact. For example, platelets remain low catalytic activity of thrombin on the ability to respond to, and thus does not affect the formation of platelet thrombi, so bleeding integrated minimize the risk of the levy.
research also proved this point. Recently reported a variety of compounds can selectively inhibit efficient Xa, which play a preventive and / or treatment of thromboembolic disease effect (W003000256A1; CN00818966; US2007259913A1; US2007259913A1). Among them, rivaroxaban (Rivaroxaban) was listed in 2008 for hip or knee replacement surgery prophylaxis and treatment of venous thrombosis, with oral, fixed dose and other advantages.
rivaroxaban drawback is the high price of raw materials, low yield preparation, purification of the product is difficult, high production costs. Patent CN00818966 8 reported rivaroxaban synthetic routes as follows:
4

where the first reaction (Preparation of 4- (4-morpholino-3-yl) nitrobenzene) yield of only 17.6%, and rivaroxaban difficult purification.

………………………………
Patent
http://www.google.com/patents/CN103232446A?cl=en
(S) -5- chloro -N- ((2- oxo-3- (4- (2_ oxo -2H- pyridin-1-yl) phenyl) -1, 3_ oxazolidine -5 – yl) methyl) thiophene-2-carboxamide.
[0011] Meanwhile, patent CN201110337461.4 described formula (I) Preparation of the compound:
[0012]

……………………………………..
Patent
CN102464658
http://www.google.com/patents/CN102464658B?cl=en
Example 1
[0046] (S) -5- chloro -N- ((2- oxo-3- (4_ (2_ Batch oxo _2H_ piperidinyl) phenyl) _1,3_ oxazolidin-5-yl) methyl ) thiophene-2-carboxamide (II)
[0048] A, 1- (4- amino-phenyl) -IH- pyridin _2_ -one (Compound VII) is
[0049] The reaction flask was charged with 104g of pyridine -2 (IH) – one (Compound IX), 200g of iodoaniline (compound VIII), 26gCuI, 151g of potassium carbonate, 18g8- hydroxyquinoline, 500mlDMF, nitrogen, heated to reflux, Insulation reaction was stirred 10h. Filtered hot, the filtrate evaporated under reduced pressure to make the solvent, the residue was added ethyl acetate, IL, 0 ° C incubated with stirring lh, filtered and the solid dried, 2L acetonitrile and purified to give 98g dark red solid. Refined liquor was concentrated to 500ml, the ice bath was stirred lh, filtered to give a dark red solid 19g. Total product were 117g, yield 68.9%.
[0050] 1H-NMR (DMSO-Cl6), δ (ppm):… 5 306 (s, 2H), 6 236 (d, 1H), 6 406 (d, 1H), 6 601 (d,. 2H), 6. 977 (d, 2H), 7. 459 (m, 2H).
[0051] B, (R) -2- (2- hydroxy-3- ((2-oxo–2H- pyridin-1-yl) phenyl) amino) propyl) isoindoline-1,3- -dione (Compound V) is
[0052] The reaction flask was added 40gl_ (4- aminophenyl) -IH- pyridin-2-one (Compound VII), 45g (S) _N_ glycidyl phthalimide (Compound VI), 300ml95% ethanol, heating to reflux, the gradual emergence of solid insulation mixing IOh, cooled to room temperature, filtered, and the filter cake washed with ethanol (150ml X 2), and dried to give an off-white solid 38g.
[0053] The mother liquor was taken, evaporated to dryness under reduced pressure, was added 15g (Q-N_ glycidyl phthalimide (Compound VII), 150ml95% ethanol, heated to reflux, stirred incubated 10h, concentrated under reduced pressure, cooled to room temperature , stirred at room temperature for 2h, washed with ethanol and dried to give an off-white solid 33g.
[0054] A total of an off-white solid 71g, yield of 84.8%, without purification, was used directly in the next step.
[0055] 1H-NMR (DMS0_d6), δ (ppm):… 3 053 (m, 1H), 3 194 (m, 1H), 4 644 (m, 2H), 4 020 (m, 1H). , 5. 168 (d, 1H), 5. 851 (t, 1H), 6. 230 (m, 1H), 6. 404 (d, 1H), 6. 665 (d, 2H), 7. 041 ( d, 2H), 7. 435 (m, 1H), 7. 537 (m, 1H), 7. 855 (m, 4H).
[0056] C, ⑶-2- ((2- oxo-3- (4- (2_ oxo _2H_ pyridyl) phenyl) oxazolidin _5_ -1,3_ yl) methyl ) Preparation of isoindoline-1,3-dione (Compound IV) of the
[0057] The reaction flask was charged 50g Compound V, 27gN, N’- carbonyldiimidazole (⑶I), 4_ catalytic amount of dimethylaminopyridine (DMAP), 150mlN, N- dimethylformamide (DMF), stirred for 90 temperature ° C, the reaction was kept for 8 hours to make the solvent was evaporated under reduced pressure, added to IL of water, stirred and dispersed, filtered, washed with water (150mlX “, washed with ethanol (100ml X 1), dried to give a white solid 48g, yield of 90%.
[0058] 1H-NMR (DMSo-CI6), δ (ppm):…. 3 984 (m, 3H), 4 251 (t, 1H), 4 968 (m, 1H), 6 301 (m, 1H), 6. 459 (d, 1H), 7. 423 (d, 2H), 7. 514 (m, 1H), 7. 615 (m, 3H), 7. 892 (m, 4H).
[0059] D, (S) -5- (aminomethyl) -3- (4- (2_ oxo _2H_-1-yl) phenyl) oxazolidin _2_ -1,3_ one hydrochloride (compound III) Synthesis of
[0060] The reaction flask was charged 50g compound IV, 200ml of ethanol, 60ml aqueous methylamine (40%), heated to reflux, stirred incubated 2h, cooled, evaporated under reduced pressure to make the solvent to give a sticky solid.
[0061] added to 300ml of ethanol, 20ml of hydrochloric acid, heated to reflux, stirred incubated lh, cooled to room temperature, incubated with stirring 2h, filtered, washed with ethanol, and dried to obtain;. 34 5g of white solid, yield 88.7%.
1H-NMR (DMS0_d6), δ (ppm):…. 3 240 (m, 2H), 3 980 (m, 1H), 4 255 (m, 1H), 5 028 (m, 1H) , 6. 321 (m, 1H), 6. 475 (d, 1H), 7. 504 (m, 3H), 7. 634 (m, 3H), 8. 561 (s, 1H).
Ε, (S) -5- chloro -N – ((2- oxo-3- (4- (2-oxo–2Η- pyridin-1-yl) phenyl) oxazolidin _1,3_ 5-yl) methyl) thiophene-2-carboxamide Preparation of thiophene (II) of
The reaction flask was charged 15g Compound III, 200ml of tetrahydrofuran, 40ml of water was added with stirring 6. 2g of sodium carbonate was added dropwise 10g5- chloro-thiophene-2-carbonyl chloride (Compound II-1) in tetrahydrofuran IOOml, 30~35 ° C insulation stirred 5h, point board to control the reaction was complete.
to make the solvent was distilled off under reduced pressure, 50ml of water was added, stirring was filtered, the filter cake washed with water and dried to give 18. 5g of white solid.
200ml of acetic acid and purified room temperature overnight, filtered, and the filter cake washed with ethanol and dried to give a white solid 16g, 80% yield.
Melting point: 204 8 ~205 8 ° C;
1H-NMR (DMSo-CI6), δ (ppm):…. 3 623 (t, 2H), 3 893 (m, 1H), 4 230 (t, 1H), 4 871 (m, 1H), 6. 308 (t, 1H), 6. 468 (d, 1H), 7. 193 (d, 1H), 7. 426 (m, 2H), 7. 500 (m, 1H), 7. 637 (m, 4H), 8. 967 (t, 1H);
MS (ESI): m / z = 430 (M + H);
HPLC: rt (%) = 14. 38 (99. 62);
[a] 20d = -37 6 ° (c 0. 3004, DMS0);
WO-2014183667Acetic acid solvate of oxazolidinone derivative, preparation method for the solvate, and application thereof
WO-2014183665Oxazolidinone derivative crystal form I and preparation method and use thereof
WO-2014183666Oxazolidinone derivate crystal form II, preparation method therefor, and application thereo
SEE ABAN SERIES AT…………http://organicsynthesisinternational.blogspot.in/p/aban-series.html
/////////
Anna Popova, Head of the Federal Service for Supervision of Consumer Protection and Welfare (Rospotrebnadzor)
MOSCOW, August 5 (RIA Novosti) – A Russian vaccine against Ebola hemorrhagic fever is now undergoing preclinical tests, Russian consumer rights watchdog, Rospotrebnadzor, head Anna Popova told journalists.
Russian Ebola Vaccine in Preclinical Trials
RIA Novosti
As of today, it is in a stage of preclinical drug trials. The works are intensified now,” Popova said. She added that there are currently no licensed drugs …
see link
http://en.ria.ru/russia/20140805/191739156/Russian-Ebola-Vaccine-in-Preclinical-Trials.html





(R)-3,3,3-Trifluoro-2-(5-(((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin-1-yl)sulfonyl)thiophen-2-yl)-2-hydroxypropanamide
2-Thiopheneacetamide, 5-[[(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methyl-1-piperazinyl]sulfonyl]-α-hydroxy-α-(trifluoromethyl)-, (αR)-
1257229-37-0
C19 H18 F7 N3 O4 S2
…………………
The glucocorticoid receptor (GR) signaling pathway has been linked to the pathophysiology of diabetes and metabolic syndrome. We developed a series of potent and selective 11ï¢-HSD1 inhibitors. These compounds showed excellent potency against both human and mouse 11ï¢-HSD1 enzymes and displayed good pharmacokinetics and ex vivo inhibition of the target in mice.Compounds HSD-016 and HSD-621 were ultimately selected as clinical development candidates. Both compounds have attractive overall pharmaceutical profiles and demonstrated good oral bioavailability in mouse, rat and dog. When orally dosed in C57/BL6 dietï€-induced-ï€obesity (DIO) mice, HSD-016 and HSDï€621 were efficacious and showed a significant reduction in both fed and fasting glucose and insulin levels. Furthermore, both compoundswere well tolerated in drug safety assessment studies.
| Discovery of HSD-621 as a Potential Agent for the Treatment of Type 2 Diabetes (ACS Medicinal Chemistry Letters) Wednesday November 28th 2012 Author(s): Zhao-Kui Wan, Eva Chenail, Huan-Qiu Li, Manus Ipek, Jason Xiang, Vipin Suri, Seung Hahm, Joel Bard, Kristine Svenson, Xin Xu, Xianbin Tian, Mengmeng Wang, Xiangping Li, Christian E. Johnson, Ariful Qadri, Darrell Panza, Mylene Perreault, Tarek S. Mansour, James F. Tobin, Eddine Saiah, DOI:10.1021/ml300352x GO TO: [Article] http://pubs.acs.org/doi/full/10.1021/ml300352xandhttp://pubs.acs.org/doi/suppl/10.1021/ml300352x/suppl_file/ml300352x_si_001.pdf nmr data as 18b |
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) catalyzes the conversion of inactive glucocorticoid cortisone to its active form, cortisol. The glucocorticoid receptor (GR) signaling pathway has been linked to the pathophysiology of diabetes and metabolic syndrome. Herein, the structure–activity relationship of a series of piperazine sulfonamide-based 11β-HSD1 inhibitors is described. (R)-3,3,3-Trifluoro-2-(5-(((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin-1-yl)sulfonyl)thiophen-2-yl)-2-hydroxypropanamide 18a (HSD-621) was identified as a potent and selective 11β-HSD1 inhibitor and was ultimately selected as a clinical development candidate. HSD-621 has an attractive overall pharmaceutical profile and demonstrates good oral bioavailability in mouse, rat, and dog. When orally dosed in C57/BL6 diet-induced obesity (DIO) mice, HSD-621 was efficacious and showed a significant reduction in both fed and fasting glucose and insulin levels. Furthermore, HSD-621 was well tolerated in drug safety assessment studies.
WO 2010141550
http://www.google.com/patents/WO2010141550A2?cl=en
EXAMPLES The title compounds of Examples 1.1, 1.2, and 1.3 were prepared as shown in
Scheme 1 below. Detailed synthesis procedures are provided below.
Scheme 1

Example 1.1

3,3,3-trifluoro-2-r5-({(2R)-4-r4-fluoro-2-(trifluoromethyl)phenyll-2-methylpiperazin- l-yl}sulfonyl)-2-thienyll-2-hvdroxypropanamide Step IA: A mixture of (R)-2-methyl-piperazine (25.0 g, 250 mmol), 2-bromo 5- fluoro benzotrifluoride (55.1 g, 227 mmol), tris(dibenzylidineacetone)dipalldium (0) (2.08g, 2.27 mmol), rac-2,2′-bis(diphenylphosphino)-l,r-binaphthyl (4.24 g, 6.81 mmol) and sodium tert-butoxide (27.3 g, 280 mmol) was mixed and purged with N2. Anhydrous toluene (500 mL) was added and purged with N2 again. The resulting mixture was heated in an oil bath at 105 0C under N2 for 3.5 hours. After cooling, the reaction mixture was concentrated and then filtered through a pad of Celite, washed with Et2O. The organic layer was concentrated, diluted with Et2O (500 mL), filtered through a pad of Celite again, and washed with IN aq. HCl (2 x 150 mL). The aqueous layer was basified with NaOH at 0 0C (pH = -10) and then was extracted with Et2O (3 x 200 mL). The combined organic layer was dried over MgSO4 and concentrated under vacuum to give (3i?)-l-[4- fluoro-2-(trifluoromethyl)phenyl]-3-methylpiperazine as a brown oil (58.5 g, 98%), which was used without further purification.
Step IB: To a solution of 5-bromothiophene-2-sulfonyl chloride (26.2 g, 100 mmol) and (3R)-l-(4-fluoro-2-(trifluoromethyl)phenyl)-3-methylpiperazine (27.6 g, lOOmmol) in DCM (200 ml) was added Et3N (41.8 ml, 300 mmol) at room temp. The reaction mixture was stirred at room temperature until completion of the reaction (about 6 hours) and then washed with aq. NaHCO3. The basic washes were back extracted with dichloromethane (DCM). The combined organic layers were washed with brine and dried over Na2SO4. The crude product was purified on a SiO2 column using hexanes/DCM as the eluent to give (R)-l-(5-bromothiophen-2-ylsulfonyl)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazine as a white solid (38 g, 78 mmol, 78 % yield).
Step 1C: To a solution of (R)-l-(5-bromothiophen-2-ylsulfonyl)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazine (28.1 g, 57.7 mmol) in anhydrous THF (200 ml) was added Butyllithium (28.8 ml, 57.7 mmol) at -780C. The reaction mixture was Stirred under N2 for 15 min. and then a solution of methyl 3,3,3-trifluoropyruvate (6.07 ml, 57.7 mmol) in THF (20 mL) was added via a cannula. The reaction mixture was stirred at -780C for 2 h. and then quenched with a 10 mL of 10% aq. HCl. The reaction mixture was dried over MgSO4 and CombiFlashed with DCM/hexane (15 – 100%) to provide methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2- methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2-hydroxypropanoate as a sticky, light yellow solid (22 g, 39.0 mmol, 67.6 % yield).
Step ID, Method 1: To a solution of methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fiuoro- 2-(trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanoate (21.5 g, 38.1 mmol) in MeOH (200 ml) was added aq. NH3 (-28-
30%, 50 mL). The reaction mixture was stirred at room temperature o/n and then diluted with ice water (700 mL). The resultant white ppt was collected by filtration, washed with water, and dried in an oven at 60 0C to give the desired product 3,3,3-trifluoro-2-(5-((R)- 4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanamide (15 g, 27.3 mmol, 71.7 % yield). The aqueous layer was extracted with DCM (4 x 100 mL), and the combined organic layers were concentrated. Purification of the concentrate by column chromatography with EA/DCM (0-40%) gave an additional 1.5 g of product.
Method 2: To a solution of methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2-
(trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanoate (200 mg) in MeOH (20 ml) at -780C was bubbled NH3 gas. The resultant mixture was stirred at room temperature overnight, concentrated, and dissolved in fresh DCM. The organic layer was washed with aq. NaHCO3 and dried to give 3,3,3- trifluoro-2-(5 -((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin- 1 – ylsulfonyl)thiophen-2-yl)-2-hydroxypropanamide as a white solid (150 mg). It was found that competing hydrolysis of the ester group to the corresponding acid occurred to a greater extent when using Method 1. Thus, in some instances, it may be preferable to use Method 2 when performing step D.
HRMS: calcd for Ci9Hi8F7N3O4S2 + H+, 550.06997; found (ESI-FTMS,
[M+H]1+), 550.07165. Example 1.2
 desired
desiredαR)-3,3,3-trifluoro-2-r5-ααR)-4-r4-fluoro-2-(trifluoromethyl)phenyll-2- methylpiperazin-l-yl}sulfonyl)thiophen-2-yll-2-hvdroxypropanamide
13.5 grams of 3,3,3-trifluoro-2-(5-((R)-4-(4-fiuoro-2-(trifluoromethyl)phenyl)-2- methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2-hydroxypropanamide (prepared according to a procedure similar to that described in Example 1.1) was separated was separated with a chiral column (Chiralpak ADH) in SFC Analytical Instrument; Mobile Phase was 90% CO2 /10%Methanol at flow rate 5mL/min. Early fraction (Retention 4.4min) was collected to give the title compound (5.7g); late fraction was collected to give the diastereomer described in Example 1.3 (6g, retention time 6. lmin).
HRMS: calcd for Ci9Hi8F7N3O4S2 + H+, 550.06997; found (ESI, [M+H]+), 550.0697. Example 1.3
 undesired
undesiredαS)-3,3,3-trifluoro-2-r5-ααR)-4-r4-fluoro-2-qrifluoromethyl)phenyll-2- methylpiperazin-l-yl}sulfonyl)thiophen-2-yll-2-hvdroxypropanamide The title compound was obtained as the late fraction using the separation method described in Example 1.2.
HRMS: calcd for Ci9Hi8F7N3O4S2 + H+, 550.06997; found (ESI, [M+H]+), 550.0701.
| US8524894 | Jun 4, 2010 | Sep 3, 2013 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2005063247A1 * | Dec 20, 2004 | Jul 14, 2005 | Amgen Sf Llc | Aryl sulfonamide compounds and uses related thereto |
| WO2007092435A2 * | Feb 7, 2007 | Aug 16, 2007 | Wyeth Corp | 11-beta hsd1 inhibitors |
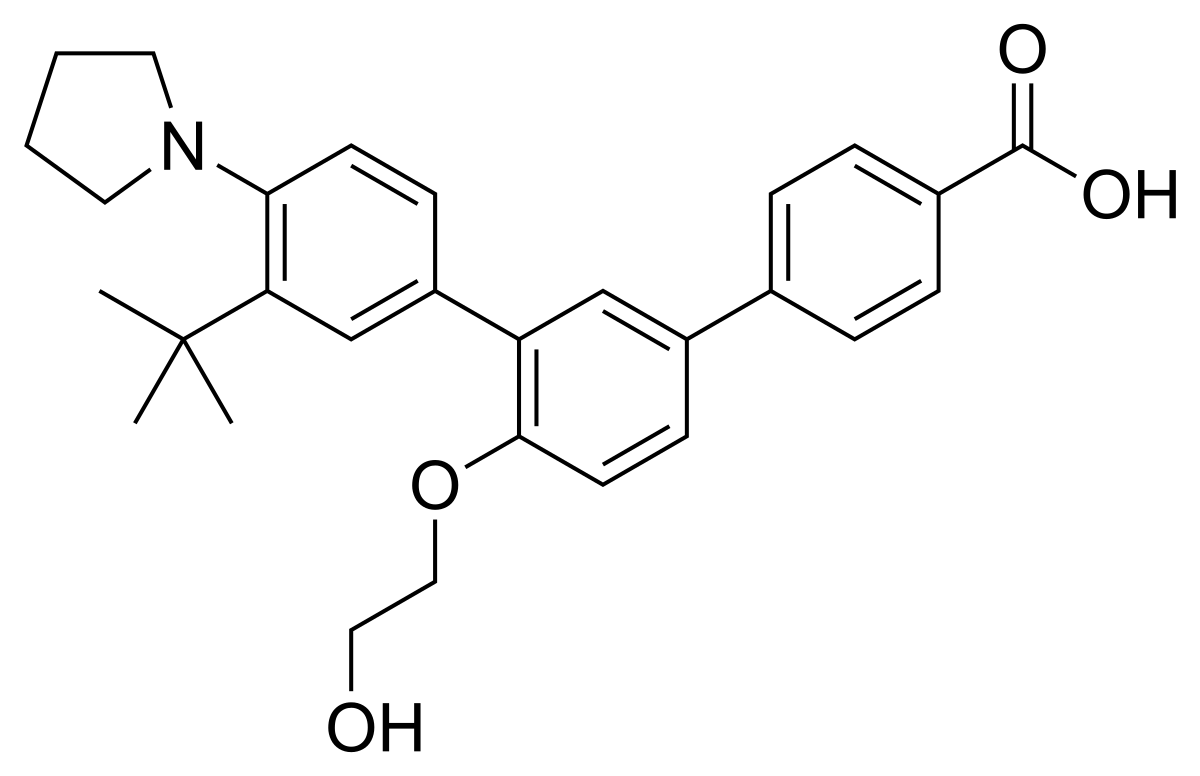
3”-Tert-butyl-4′-(2-hydroxyethoxy)-4”-(pyrrolidin-1-yl)(1,1′:3′,1”)terphenyl-4-carboxylic acid
3′-[3-tert-butyl-4-(pyrrolidin-1-yl)phenyl]-4′-(2-hydroxyethoxy)-[1,1′-biphenyl]-4-carboxylic acid
UNII-0J8RN2W0HK,
Galderma Research & Development
459.5766
C29 H33 N O4
Trifarotene, sold under the brand name Aklief, is a medication for the topical treatment of acne vulgaris in those nine years of age and older.[1] It is a retinoid;[2] more specifically, it is a fourth generation selective retinoic acid receptor (RAR)-γ agonist.[3]
It was approved for medical use in the United States in 2019,[1][4][5] but is not approved in the European Union as of January 2021.[6] Trifarotene was granted orphan drug designation for the treatment of congenital ichthyosis by both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA).[7][8]
State Solid
Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 245C | FDA Label |
| pKa | 5.69 (pKa1) | FDA Label |
USFDA
The drug substance, trifarotene, a terphenyl acid derivative, is a retinoic
acid receptor (RAR) aQonist and is classified as a rotenoid. Trifarotene
intended as a drug for the treatment of acne vulgaris. Since trifarotene
has not been previously approved as an active ingredient in any drug
product in the United States, it is classified as a new molecular entity
(NME).
Trifarotene is produced as a white to off-white to slightly yellow crystalline
powder. It is slightly soluble in acetone, ethanol, and toluene, very slight
soluble in isopropanol, and practically insoluble in water (tiJT4
1
Cb><“JTrifarotene is nonhygroscopic and has pKa1 of 5.69 and pKa2 of 4.55. The chemical name
for trifarotene is 4-{3-[3-tert-butyl-4-(pyrrolidin-1-yl) phenyl]-4-(2-
hydroxyethoxy) phenyl} benzoic acid. It has the chemical formula of
C29H33NQ4, the molecular weiQht of 459.59, …………https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211527Orig1s000ChemR.pdf
Prescription Products
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Aklief | Cream | 50 mcg | Topical | Galderma | 2019-11-28 | Not applicable |  |
|
| Aklief | Cream | 50 ug/1g | Topical | Galderma Laboratories, L.P. | 2019-10-04 | Not applicable |  |
Galderma announced that the U.S. Food and Drug Administration (FDA) granted Orphan Drug Designation status for the company’s trifarotene molecule for the treatment of congenital ichthyosis. Based on this decision, Galderma plans to implement a clinical development plan, reinforcing its commitment to exploring new treatment options for rare diseases, as well as meeting the needs of all patients with skin diseases over the course of their lives.
Galderma治療先天性魚鱗癬的Trifarotene分子取得FDA的孤兒藥資格認定
http://news.msn.com.tw/market3773054.aspx

The company’s molecule trifarotene is a selective agonist of the gamma retinoic acid receptor (RAR-gamma), which is currently in clinical development for use in other more common dermatological conditions. It is the drug’s retinoid functionality and potent keratolytic properties that make it a potentially viable treatment of the lamellar ichthyosis pathology. Galderma has already initiated the program for investigating the treatment of lamellar ichthyosis with trifarotene and is currently working in collaboration with regulatory authorities to implement an innovative and expedient clinical development plan.
Ichthyoses comprise a large group of skin scaling disorders with diverse etiologies. The stereotypic pathophysiology is epidermal hyperplasia and abnormal desquamation, leading to visible accumulation of squames (scales) on the skin’s surface. Congenital ichthyosis is a term used to refer to a specific group of rare inherited forms of ichthyoses that are generally more severe than non-inherited forms of the disease. Lamellar ichthyosis is one such disorder that falls within the congenital ichthyosis category. Lamellar ichthyosis is recognized as a severe disease which persists throughout life. After birth, during the first post-natal weeks, the hyperkeratotic (colloidion) membrane patients are typically born with, is gradually shed and is replaced by scaling and lichenification that involves the entire body, including face, scalp, palms and soles. While usually not life threatening, lamellar ichthyosis can result in disability, partial deafness, poor adaptation to environmental conditions (due to hypohydrosis), severe discomfort (pruritus, fissuring of the skin), and significant psycho-social impact. The estimated prevalence of LI in the US is in the range of 1 per 100,000 to 1 per 200,000 persons.
Synthesis Reference
Thoreau, E. et. al. Structure-based design of Trifarotene (CD5789), a potent and selective RARγ agonist for the treatment of acne. Bioorganic & Medicinal Chemistry Letters, Volume 28, Issue 10. 2018. Pages 1736-1741
https://www.sciencedirect.com/science/article/abs/pii/S0960894X18303482

Reference: Biadatti, Thibaud; Dumais, Laurence; Soulet, Catherine; Talano, Sandrine; Daver, Sebastien. Preparation of [1,1′:3′,1”]terphenyl-4-carboxylic acid and esters a novel ligands modulating retinoic acid receptors (RAR), and use thereof in human medicine and in cosmetics. Assignee Galderma Research & Development, S.N.C., Fr. WO 2006066978. (2016).
PATENT
WO 2006066978
http://www.google.com/patents/WO2006066978A1?cl=en
Example 25 – 3″-ter.-Butyl-4′-(2-hvdroxyethoxy)-4″-pyrrolidin-1-ylM,1′:3′,1″1- terphenyl-4-carboxylic acid

In a manner similar to that of Example 6b, by reacting 500 mg (0.9 mmol) of ethyl 4′-(2- acetoxyethoxy)-3″-terf-butyl-4″-pyrrolidin-1 -yl[1 , 1 ‘;3’, 1 “]terphenyl-4-carboxylate with
300 mg (8 mmol) of sodium hydroxide, 242 mg of 3″-tert-butyl-4′-(2-hydroxyethoxy)-4″- pyrrolidin-1-yl[1l1′;3′,1″]terphenyl-4-carboxylic acid are obtained (yield = 55 %) in the form of a white solid (m.p. = 2230C).
1H NMR (DMSO. 400 MHz): 1.43 (s, 9H); 1.90 (m, 4H); 3.0 (m, 4H); 3.73 (d, J=4.7Hz, 2H); 4.1 (m, 2H); 4.7 (s, 1H); 7.2 (d, 1H, J=8.6Hz); 7.48 (m, 2H); 7.59 (d, J=1.6Hz, 1H); 7.64 (d, J=UHz, 1H); 7.68 (dd, J=2Hz, 7.8Hz, 1H); 7.82 (d, J=8.3Hz, 2H); 7.99 (d, J=8.4Hz, 2H).
PATENT
WO 2013178759
http://www.google.com/patents/WO2013178759A1?cl=en
PATENT
WO 2013178758
http://www.google.com/patents/WO2013178758A1?cl=en
PATENT
WO 2013178760
http://www.google.com/patents/WO2013178760A1?cl=en
The details of skin application are given in the table below.

SYN
New Drug Approvals for 2019: Synthesis and Clinical Applications
New Drug Approvals for 2019: Synthesis and Clinical Applications
Shuo Yuan, Bin Yu, Hong-Min Liu
PII: S0223-5234(20)30639-5
DOI: https://doi.org/10.1016/j.ejmech.2020.112667
Reference: EJMECH 112667
To appear in: European Journal of Medicinal Chemistry
Trifarotene (Aklief). In October 2019, trifarotene, a topical retinoid that
selectively targets retinoic acid receptor gamma (RAR-γ), was approved by the FDA
for the treatment of acne vulgaris [142]. The drug was developed and marketed by
Galderma Pharmaceutical in Switzerland. Trifarotene is considered as the first of the
‘fourth-generation’ retinoids due to its uniquely selective agonism at RAR-γ. The
selective agonism leads to downstream alterations, confering improved efficacy and
reduced side effects [143]. In two phase 3 clinical trials of 2420 patients with
moderate acne on the face and trunk, trifarotene was well tolerated and significantly
reduced inflammatory lesions as early as two weeks on the face and four weeks on the
back, shoulders and chest compared to vehicle (p<0.05) [144].
The synthetic approach of this drug was disclosed by Galderma Research &
Development (Scheme 25) [145]. Bromination of commercially available
2-(tert-butyl)aniline 171 gave 4-bromo-2-(tert-butyl)aniline 172 in quantitative yield,
which then reacted with 1-dibromobutane 173 to give phenylpyrrolidine 174 in 52%
yield. Miyaura reaction of 174 was realized by employing n-BuLi and triisopropyl
borate (TIPB) followed by washed with aqueous HCl, resulting in arylboronic acid
adduct 175 in 66% yield. Treatment of 175 with aromatic bromide 176 in the presence
of Pd(PPh3)4 gave the coupling product 177 in 47% yield, which then underwent
hydrolysis delivering trifarotene (XIX) in 55% yield.
The preparation of coupling partner 176 is depicted in Scheme 26. Esterification of
4-hydroxy-4-biphenylcarboxylic acid 178 gave ethyl benzoate derivative 179 upon
treatment with catalytic H2SO4 in the refluxing EtOH [145]. The resulting ester was
subjected to treatment with tetrabutylammonium bromide (TBAB) in THF, resulting
in bromide 180 in good yields, further NaH-mediated Williamson ether synthesis with
2-bromoethyl acetate 181 gave 176 in 95% yield.

[142] L.J. Scott, Trifarotene: first approval, Drugs 79 (2019) 1905-1909.
[143] E. Thoreau, J.M. Arlabosse, C. Bouix-Peter, S. Chambon, L. Chantalat, S.
Daver, L. Dumais, G. Duvert, A. Feret, G. Ouvry, J. Pascau, C. Raffin, N.
Rodeville, C. Soulet, S. Tabet, S. Talano, T. Portal, Structure-based design of
trifarotene (CD5789), a potent and selective RARγ agonist for the treatment of
acne, Bioorg. Med. Chem. Lett. 28 (2018) 1736-1741.
[144] J. Tan, D. Thiboutot, G. Popp, M. Gooderham, C. Lynde, J.D. Rosso, J. Weiss,
U. Blume-Peytavi, J. Weglovska, S. Johnson, L. Parish, D. Witkowska, N.S.
Colon, A.A. Saenz, F. Ahmad, M. Graeber, L.S. Gold, Randomized phase 3
evaluation of trifarotene 50 µg/g cream treatment of moderate facial and truncal
acne, J. Am. Acad. Dermatol. 80 (2019) 1691-1699.
[145] T. Biadatti, L. Dumais, C. Soulet, S. Talano, S. Daver, Novel ligands that
modulate rar receptors, and use thereof in human medicine and in cosmetics,
2006. WO2006066978.
| WO2006066978A1 * | Dec 21, 2005 | Jun 29, 2006 | Galderma Res & Dev | Novel ligands that modulate rar receptors, and use thereof in human medicine and in cosmetics |
| EP0826366A2 | Aug 1, 1997 | Mar 4, 1998 | Unilever N.V. | Cosmetic compositions containing hydroxy acid or retinoid |
| EP0989846A2 | Sep 22, 1998 | Apr 5, 2000 | E-L Management Corp. | Non-irritating cosmetic and pharmaceutical compositions |
| EP1831149A1 | Dec 21, 2005 | Sep 12, 2007 | Galderma Research & Development | Novel ligands that modulate rar receptors and use thereof in human medicine and in cosmetics |
| FR2915682A1 * | Title not available | |||
| US5851538 | Dec 29, 1995 | Dec 22, 1998 | Advanced Polymer Systems, Inc. | Retinoid formulations in porous microspheres for reduced irritation and enhanced stability |
| WO1999010308A1 * | Aug 21, 1998 | Mar 4, 1999 | Bernardon Jean Michel | Biphenyl derivatives substituted by an aromatic or heteroaromatic radical and pharmaceutical and cosmetic compositions containing same |
| US6150413 * | May 26, 1998 | Nov 21, 2000 | Centre International De Recherches Dermatologiques | Treatment of dermatological, rheumatic, respiratory, cardiovascular, bone and ophthalmological disorders, as well as mammalian skin and hair conditions; 4-(4-(biphenyl-2-yl)but-3-en-1-ynyl)benzoic acid, for example |
 |
|
| Clinical data | |
|---|---|
| Trade names | Aklief |
| Other names | CD5789 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620004 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Topical |
| Drug class | Skin and mucous membrane agents |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.278.901 |
| Chemical and physical data | |
| Formula | C29H33NO4 |
| Molar mass | 459.586 g·mol−1 |
| 3D model (JSmol) | |
| FORM | ROUTE | STRENGTH |
|---|---|---|
| Cream | Topical | 50 mcg |
| Cream | Topical | 50 ug/1g |
| Cream | Topical | 50 MICROGRAMMI/G |
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 4 | Enrolling by Invitation | Treatment | Acne Vulgaris | 1 |
| 3 | Completed | Treatment | Acne Vulgaris | 4 |
| 2 | Completed | Treatment | Acne Vulgaris | 1 |
| 2 | Recruiting | Treatment | Lamellar Ichthyosis | 1 |
| 1 | Completed | Treatment | Malignant Lymphomas | 1 |

read poster
5-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-ylmethyl]-thiophene-3-carboxylic Acid
3-Thiophenecarboxylic acid, 5-[[(5S,9R)-9-(4-cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-yl]methyl]- [ACD/Index Name]
5-{[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-yl]methyl}-3-thiophenecarboxylic acid [ACD/IUPAC Name]
5-{[(5S,9R)-9-(4-Cyanphenyl)-3-(3,5-dichlorphenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-yl]methyl}-3-thiophencarbonsäure [German] [ACD/IUPAC Name]
Acide 5-{[(5S,9R)-9-(4-cyanophényl)-3-(3,5-dichlorophényl)-1-méthyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-yl]méthyl}-3-thiophènecarboxylique [French] [ACD/IUPAC Name]
2IC
BMS-587101
BMS-688521
data
Interaction between leukocyte function-associated antigen-1 (LFA-1), expressed on the surface of cytokine-stimulated cells, and intercellular adhesion molecule (I-CAM), found on the surface of both leukocytes and endothelium, plays a key function in the intercellular immune response, causing T-cell adhesion and subsequent migration through the blood vessel wall to the inflamed area.(1)
Small molecules which inhibit the LFA-1/I-CAM interaction are targeted as potential drugs for the treatment of a variety of autoimmune and inflammatory diseases such as rheumatoid arthritis and psoriasis.(2, 3) The LFA-1 receptor antagonist, BMS-587101, 1,(4, 5) was selected for clinical development, and we required a synthesis that would reliably generate kilogram quantities of API. This paper details the identification and development of a synthesis which enabled the realization of this goal.
BMS-587101 inhibits the interaction between leukocyte function-associated antigen-1 (LFA-1) and the intercellular adhesion molecule (ICAM), thereby offering a potential treatment for various autoimmune and inflammatory diseases, such as rheumatoid arthritis and psoriasis. A four-step multikilogram route to BMS-587101 (22% overall yield ) from the commercial hydantoin B features an efficient dipolar cycloaddition of an azomethine ylide generated by reaction of glycine with hexamethylenetetramine (HMTA).
………….
paper

http://pubs.acs.org/doi/abs/10.1021/op9003168
The process development and the kilogram-scale synthesis of BMS-587101 (1) are described. The synthesis features a [3 + 2] azomethine ylide cycloaddition to efficiently build the spirocyclic core in a diastereoselective fashion followed by a classical resolution which affords the desired enantiomer in >98% enantiomeric excess. The target was prepared in four steps in an overall yield of 22%.
 22.9 kg) was added until a pH of 6.5 was attained. After agitating for 15 min and holding for 30 min, the aqueous layer was discarded, and the organic layer was washed with H2O (470 kg). The solution was then polish filtered, and isopropylacetate (52.2 kg) was used to rinse the polish filter assembly. The solution was concentrated under reduced pressure (240 Torr) to a volume of 718 L at <45 °C. Seeds (500 g) were charged, and the distillation was continued until a volume of 207 L was attained. Heptane (117.8 kg) was charged, the slurry was cooled to 20 °C over 1.5 h and was subsequently wet milled until d90 < 60 μm. The slurry was held for >2 h and filtered. The cake was washed with a 1:1 isopropyl acetate/heptane solution (109.7 kg) isopropyl acetate and dried in vacuum at 35−40 °C to a constant weight. Acid 1 (39.6 kg, 91.5% yield and 99.33 HPLC area % purity) was obtained as a white and sandy crystalline solid.
22.9 kg) was added until a pH of 6.5 was attained. After agitating for 15 min and holding for 30 min, the aqueous layer was discarded, and the organic layer was washed with H2O (470 kg). The solution was then polish filtered, and isopropylacetate (52.2 kg) was used to rinse the polish filter assembly. The solution was concentrated under reduced pressure (240 Torr) to a volume of 718 L at <45 °C. Seeds (500 g) were charged, and the distillation was continued until a volume of 207 L was attained. Heptane (117.8 kg) was charged, the slurry was cooled to 20 °C over 1.5 h and was subsequently wet milled until d90 < 60 μm. The slurry was held for >2 h and filtered. The cake was washed with a 1:1 isopropyl acetate/heptane solution (109.7 kg) isopropyl acetate and dried in vacuum at 35−40 °C to a constant weight. Acid 1 (39.6 kg, 91.5% yield and 99.33 HPLC area % purity) was obtained as a white and sandy crystalline solid.
…………………………
U.S. Patent 7,381,737 B2
http://www.google.com/patents/US7381737
IIIn:

Also provided are crystalline forms of solvates and salts of the substituted spiro-hydantoin compound (IIIn).
5-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid.
EXAMPLES
The following examples illustrate embodiments of the inventive process, and are not intended to limit the scope of the claims. For ease of reference, the following abbreviations are used herein:
ABBREVIATIONS
Preparation 13-(3,5-dichlorophenyl)-1-methylimidazolidine-2,4-dione

Triethylamine (0.78 kg, 7.75 mol) was added in 15-30 minutes with stirring to a thin suspension of sarcosine ethylene hydrochloride (1.00 kg, 6.51 mol) in dichloromethane (6.00 L). After stirring at room temperature for 1.5-2.0 hours, the mixture was filtered to remove the resulting triethylamine hydrochloride salt. The salt cake was washed with dichloromethane (2.00 L). The filtrate was cooled to 0-5° C.
A solution of 3,5-dichlorophenyl isocyanate (1.47 kg, 7.81 mol) in dichloromethane was prepared at 20-25° C. The solution was added to the above cooled filtrate slowly in 30-60 minutes. The temperature was maintained below 10° C. during the addition. After the addition, the mixture was stirred at 20-25° C. for 12-14 hours. The completeness of the reaction was followed by HPLC. Upon reaction completion, TBME (16.00 L) was added in one portion. The resulting suspension was stirred at 20-25° C. for 2-3 hours and was then filtered. The filter cake was washed with TBME (4.50 L) and dried at maximum 40° C. to a constant weight. A suspension of the above filter cake in water (17.0 L, 10 L/kg input) was prepared and stirred at 20-25° C. for at least 16 hours. The suspension was filtered and the filter cake was washed with water (3×1.36 L) and dried at maximum 40° C. to a constant weight to a constant weight. 3-(3,5-dichlorophenyl)-1-methylimidazolidine-2,4-dione (1.52 kg, 90%) was obtained as a white crystalline solid. mp=202-204° C. 1H NMR (DMSO-d6): 7.66 (1H, m), 7.51 (2H, m), 4.10 (2H, s), 3.35 (3H, s). 13C NMR (DMSO-d6): 8 Carbons (169.30, 155.00, 134.98, 134.15, 127.59, 125.30, 51.75, 29.79). Anal. Calcd for C10H8Cl2N2O2: C, 46.35; H, 3.11; N, 10.81; Cl, 27.36. Found: C, 46.43; H, 2.9; N, 10.73; Cl, 27.33.
Preparation 2(E)-4-((1-(3,5-dichlorophenyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl)benzonitrile

A mixture of 3-(3,5-dichlorophenyl)-1-methylimidazolidine-2,4-dione (1.00 kg, 3.86 mol), 4-cyanobenzaldehyde (0.70 kg, 5.79 mol) and pyrrolidone (0.27 kg, 3.86 mmol) was refluxed in EtOH (13.00 L) for 20-24 hours at a temperature of 78° C. The completeness of the reaction was followed by HPLC. Upon reaction completion, the suspension was cooled to 65° C. and THF (4.33 L) was added in 5-10 minutes. The suspension was cooled to 20-25° C. in 3-4 hours and was then filtered. The filter cake was washed with EtOH (4×2.00 L) and dried at maximum 40° C. to a constant weight. (E)-4-((1-(3,5-dichlorophenyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl)benzonitrile (1.24 kg, 86%) was obtained as a fluffy, yellowish crystalline solid. mp=239-241° C. 1H NMR (DMSO-d6): 8.07 (2H, d, J=8.3 Hz), 7.86 (2H, d, J=8.4 Hz), 7.72 (1H, m), 7.59 (2H, m), 6.72 (1H, s), 3.35 (3H, s). 13C NMR (DMSO-d6): 14 Carbons (159.80, 151.48, 137.64, 133.83, 133.70, 131.80, 130.80, 130.68, 127.71, 125.51, 118.83, 114.48, 110.32, 26.72). Anal. Calcd for C18H11Cl2N3O2: C, 58.08; H, 2.97; N, 11.29; Cl, 19.05. Found: C, 58.14; H, 2.72; N, 11.14; Cl, 19.15.
Example 14-[(5S*,9R*)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile hydrochloride salt

A mixture of (E)-4-((1-(3,5-dichlorophenyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl)benzonitrile (1.00 kg, 2.69 mol), glycine (0.50 kg, 6.72 mol) and hexamethylenetetramine (0.28 kg, 2.02 mol) in 1-methyl-2-pyrrolidinone (5.00 L) and toluene (2.50 L) was heated at 140° C. for 7-8 hours. The completeness of the reaction was followed by HPLC. Upon reaction completion, the mixture was cooled to 40-50° C. and filtered. The filtered solid was washed with toluene (0.67 L). To the filtrate was added HCl (1M, 13.33 L, 13.33 mol). The resulting biphasic mixture was heated to 50-60° C. and was stirred for 10-15 minutes. The aqueous phase was separated and the organic phase was washed with HCl (1M, 1.67 L, 1.67 mol) at 60-80° C. The aqueous phases were combined and were stirred at 80° C. for 2 hours. The solution was cooled slowly in 3-4 hours to 20-25° C. with gentle stirring and seeding. Crystallization occurred and the resulting suspension was put aside at 20-25° C. for at least 16 hours with occasional stirring, cooled to 0-5° C. in 2 hours, stirred gently at 0-5° C. for 2 hours and then filtered. The filter cake was washed with ice water (2×2.50 L) and dried at maximum 40° C. to a constant weight. 4-[(5S*,9R*)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile hydrochloride salt (1.09 kg, 90%) was obtained as beige crystalline solid. mp=183-185° C. 1H NMR (DMSO-d6): 7.87(2H, d, J=8.1 Hz), 7.61 (1H, m), 7.40 (2H, d, J=8.1 Hz), 6.68 (2H, m), 4.17 (1H, m), 3.85 (2H, m), 3.76 (2H, m), 3.43 (3H, s), 3.24(2H, s). 13C NMR (DMSO-d6): 14 Carbons (170.84, 152.92, 137.35, 133.94, 132.87, 132.35, 128.01, 124.50, 118.12, 111.30, 71.42, 46.57, 45.11, 25.51). Anal. Calcd for C20H17Cl3N4O2+1.3 H2O: C, 50.51; H, 3.91; N, 11.79; Cl, 22.39. Found: C, 50.56; H, 3.86; N, 11.58; Cl, 21.98; KF, 5.12.
Example 2a4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile semi (+)-DTTA salt

To a suspension of 4-[(5S*,9R*)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile hydrochloric acid salt (1.00 kg, 2.21 mol) in dichloromethane (10.67 L) was added diispopropylethylamine (0.29 kg, 2.21 mol). The mixture was stirred to a clear solution, to which (+)-Di-p-toluoyl-D-tartaric acid (0.21 kg, 0.55 mol) was added. The resulting solution was warmed to 34-36° C. and seeded immediately. It was cooled to 20-25° C. in 1.5-2.0 hours. Crystallization occurred during cooling. TBME (2.75 L) was added in 0.5 hours. The suspension was stirred at 20-25° C. for 16 hours and then filtered. The filter cake was washed with dichloromethane/TBME (2/1, 1.00 L), TBME (1 L) and dried at maximum 35° C. to a constant weight. 4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile semi (+)-DTTA salt (0.47 kg, 35%) was obtained as a white crystalline solid. mp=175-177° C. 1H NMR (DMSO-d6): 7.86 (2H, d, J=8.1 Hz), 7.81 (2H, d, J=8.3 Hz), 7.61 (1H, m), 7.28 (2H, d, J=8.1 Hz), 7.22 (2H, 8.5 Hz), 6.68 (2H, m), 5.71 (1H, s), 3.81(1H, m), 3.50 (4H, m), 3.06 (3H, s), 2.34 (3H, s). 13C NMR (DMSO-d6): 24 Carbons (171.45, 169.40, 165.04, 152.88, 143.61, 138.99, 133.88, 133.08, 132.16, 129.26, 129.20, 128.76, 127.84, 126.99, 124.51, 118.25, 110.78, 72.81, 73.38, 48.15, 47.51, 46.30, 24.90, 21.14). Anal. Calcd for C30H25Cl2N4O6+0.5 H2O: C, 58.40; H, 4.17; N, 9.08; Cl, 11.49. Found C, 58.58; H, 4.06; N, 8.94; Cl, 11.38; KF, 1.59.
Example 2b4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile semi (+)-DTTA salt

A mixture of (E)-4-((1-(3,5-dichlorophenyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl)benzonitrile (10.0 g, 26.9 mmol), glycine (5.06 g, 67.4 mmol), hexamethylenetetramine (2.82 g, 20.1 mmol) in 50 mL N-methylpyrrolidinone and 25 mL of toluene under nitrogen was heated to 138° C. for approximately 12 h. Next, 25 mL toluene and 25 mL H2O were added. The aqueous and nonaqueous layers were split, and the aqueous layer was washed with 25 mL of toluene, and the nonaqueous layers were combined to form a nonaqueous mixture. The nonaqueous mixture was heated to 45-50° C. and ethylene diamine (7.0 mL) was added. The nonaqueous mixture was stirred for 3 hours and then cooled to room temperature. Next, 50 mL H2O was added, followed by the addition of 10 mL brine. The next addition was 25 mL toluene, which was followed by the addition of 125 mL CH2Cl2. The bottom layer of the mixture was removed through a filter. Next, (+)-Di-p-toluoyl-D-tartaric acid (2.59 g, 6.7 mmol) was added and the mixture was stirred for 18 h to form a slurry. Slowly 40 mL of MTBE was added to the slurry. A wash solution containing 7 mL of MTBE and 11 mL of CH2Cl2 was prepared. Filter paper was wetted with 1 mL of the wash solution. The slurry was filtered and then the filtered to form a cake. The filter, the wash reaction flask, and the cake were washed with the remaining 16 mL of the wash solution. Next, the cake was washed with 10 mL MTBE. 4-[(5S, 9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile semi (+)-DTTA salt (4.0 g, 20% yield) was obtained as a white solid (98.7% HPLC AP and 98.3% ee).
Example 2c4-[(5S,9R)-3-3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4,4]non-9-yl]-benzonitrile semi (+)-DTTA salt
A mixture of (E)-4-((1-(3,5)-dichlorophenyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl)benzonitrile (40.0 g, 107.5 mmol), glycine (19.76 g, 263.2 mmol), hexamethylenetetramine (9.07 g, 64.7 mmol) in 200 mL N-methyl-2-pyrrolidinone and 100 mL of toluene was heated under nitrogen to 143° C. for approximately 5.5 h. Next, the mixture was cooled to 50° C. and a solution of 25 mL of ethylenediamine in 200 mL of tetrahydrofuran was added. The mixture was maintained at a temperature of 50° C. for 30 minutes and then was cooled to room temperature. Next, 520 mL of 20 wt % NaCl aqueous solution was added. The aqueous and nonaqueous layers were separated. The nonaqueous layer was transferred to a vacuum distillation apparatus and solvent was distilled off until the temperature of the residue in the flask reached 58° C. at a pressure of 60 torr. Next, 360 mL of methylene chloride was added, followed by the additions of 20 mL of methanol and 2 mL of water. The next addition was (+)-Di-p-toluoyl-D-tartaric acid (10.38 g, 26.9 mmol), followed by 120 mL of methylene chloride and 0.200 g of seeds of 4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4,4]non-9-yl]-benzonitrile semi (+)-DTTA salt. A-slurry was formed and was stirred at room temperature for 24 hours. The slurry was filtered and the cake of crystals was washed with 200 mL of methylene chloride in two portions. The washed cake was then dried at 50° C. under vacuum for 24 hours. A total amount of 20.11 g (yield 31%) of 4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4,4]non-9-yl]-benzonitrile semi (+)-DTTA salt, which was of greater than 99.5% area percent purity, 98.4% potency and 99.2% ee was obtained after drying.
Example 35-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid methyl ester hydrochloride salt

To a suspension of 4-[(5S,9R)-3-(3,5-Dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-9-yl]-benzonitrile semi (+)-DTTA salt (7.50 kg, 12.30 mmol) and methyl 5-formylthiophene-3-carboxylate (2.2 kg, 13.10 mol) was added triethylamine (2.08 kg, 20.60 mol) at 20-25° C. The mixture was stirred to a clear solution, to which acetic acid (1.24 kg, 20.60 mol) was added. The resulting mixture was stirred at 20-25° C. for 1 hour and then cooled to 15° C. Solid sodium triacetoxyborohydride (1.31 kg, 6.17 mol) was added and the reaction mixture was stirred for 0.5 hours. The addition of sodium triacetoxyborohydride was repeated three more times. At the end, a total of 5.22 kg (24.7 mol) sodium triacetoxyborohydride was added in 2 hours. The reaction mixture was stirred at 20-25° C. for 16 hours. The completeness of the reaction was followed by HPLC. Upon reaction completion, TBME (48.1 L) was added to the resulting jelly reaction mixture. The mixture was washed with saturated sodium hydrogen carbonate solution (60.0 L×3). The combined aqueous phase was extracted with TBME (48.1 L). All organic layers were combined, washed with brine (48.1 L) and concentrated in vacuum to a volume of 10.6 L. Isopropanol (192.3 L) was added to the residue and the resulting oil precipitates were dissolved upon warming up to 70-75° C. The solvent volume was reduced to 160.0 L by distillation at 70-75° C. Concentrated HCl (1.5 L) was added at 75° C. in 10 minutes followed by the addition of seed crystals. Crystallization occurred upon cooling to 20-25° C. in 16 hours. The mixture was filtered. The cake was washed with isopropanol (9.6 L×2) and dried at maximum 40° C. to a constant weight. 5-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid methyl ester hydrochloride salt (6.57 kg, 88.0%) was obtained as white crystalline solid. mp=204-207° C. 1H NMR (CDCl3): 14.22 (1H, b), 8.18 (1H, d, J=0.9 Hz), 7.86 (1H, m), 7.67 (2H, d, J=8.1 Hz), 7.24 (1H, m), 7.23 (2H, d, J=8.1 Hz), 6.67 (2H, m), 4.76 (2H, m), 4.46 (1H, m), 4.16 (1H, m), 4.02 (2H, m), 3.86 (3H, s), 3.75 (1H, m), 3.38 (3H, s). 13C NMR (CDCl3): 18 Carbons (171.24, 162.32, 152.98, 136.05, 135.27, 134.03, 132.83, 131.94, 130.46, 128.85, 128.56, 123.92, 117.52, 113.43, 71.13, 52.43, 52.22, 46.73). Anal. Calcd for C27H23Cl3N4O4S: C, 53.52; H, 3.83; N, 9.25; S, 5.29; Cl, 17.55. Found: C, 53.07; H, 3.69; N, 9.08; S, 5.23; Cl, 17.20.
Example 45-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid

To a solution of 5-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid methyl ester hydrochloride salt (20.00 g, 33.00 mmol) and 1,2-propanediol (5.0 g) in tetrahydrofuran (200 mL) and water (100 mL) was added slowly potassium hydroxide solution (0.85M, 116 mL) at 8-12° C. in 0.5 hours. The resulting biphasic mixture was stirred at 8-12° C. for 20-27 hours until the reaction was complete. The reaction mixture was washed with n-heptane (200 mL). The pH was adjusted to 6.5 with addition of water (100 mL) and acetic acid (2.5 mL). Tetrahydrofuran was removed under reduced pressure at internal temperature <40° C. The pH was adjusted to 4.5 with addition of isopropyl acetate (400 mL) and acetic acid (11 mL). After 10 minutes of stirring, the aqueous layer was separated and was extracted with isopropylacetate (200 mL). The organic layers were combined, washed with water (100 mL) and concentrated under reduced pressure to a volume of 190 mL at bath temperature <40° C. Crystallization occurred during concentration. The crystal slurry was stirred at 20-25° C. for 16 hours and was then filtered. The cake was washed with cold isopropylacetate (15 mL×3) and dried in vacuum at 35-40° C. to a constant weight.
5-[(5S,9R)-9-(4-Cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-ylmethyl]-thiophene-3-carboxylic acid (14.35 g, 78.3%) was obtained as white and sandy crystalline solid.
mp=209-230° C. 1H NMR (Acetone-d6): 8.19 (1H, d, J=1.3 Hz), 7.76 (2H, d, J=8.4 Hz), 7.49 (2H, d, J=8.2 Hz), 7.43 (1H, d, J=1.0 Hz), 7.41 (1H, t, J=1.9 Hz), 6.87 (2H, d, J=1.9 Hz), 4.16 (1H, dd, J1=13.9 Hz J2=0.8 Hz), 4.10 (1H, dd, J1=11.7 Hz, J2=6.2 Hz), 3.99 (1H, d, J=14.0 Hz), 3.48(1H, d, J=10.6 Hz), 3.47 (1H, dd, J1=9.6 Hz, J2=6.2 Hz), 3.25 (3H, s), 3.24 (1H, dd, J1=9.6 Hz, J2=11.7 Hz), 3.01 (1H, d, J=11.3 Hz).
13C NMR (Acetone-d6): 22 Carbons (172.69, 163.7, 153.98, 144.55, 142.23, 135.26, 135.09, 134.41, 133.89, 132.96, 130.33, 128.27, 126.98, 125.18, 119.07, 112.44, 74.28, 59.09, 56.45, 54.33, 50.73, 25.75).
Anal. Calcd for C26H20Cl2N4O4S: C, 56.22; H, 3.62; N, 10.08; S, 5.77; Cl, 12.76. Found: C, 56.27; H, 3.20; N, 9.97; S, 5.65; Cl, 12.68.
…………………………..
paper
J. Med. Chem. 2006, 49, 6946
http://pubs.acs.org/doi/abs/10.1021/jm0610806

LFA-1 (leukocyte function-associated antigen-1), is a member of the β2-integrin family and is expressed on all leukocytes. This letter describes the discovery and preliminary SAR of spirocyclic hydantoin based LFA-1 antagonists that culminated in the identification of analog 8 as a clinical candidate. We also report the first example of the efficacy of a small molecule LFA-1 antagonist in combination with CTLA-4Ig in an animal model of transplant rejection.
http://pubs.acs.org/doi/suppl/10.1021/jm0610806/suppl_file/jm0610806si20060913_101747.pdf synthesis as compd 8
says
a white solid: Anal.RP-HPLCtR= 3.09min (method D, purity 99%);
………………….
U.S. Patent 7,199,125 B2
http://www.google.com/patents/US7199125
………………………..
.U.S. Patent 6,710,064 B2
http://www.google.com/patents/US6710064
………….
REFERENCES
For a discussion on the inhibition of LFA-1/ICAM-1as an approach to treating autoimmune diseases see:
For a discussion of therapeutic options for treatment of psoriasis, see:
For other small molecule LFA-1/ICAM-1 antagonists as potential drugs please see:
The Discovery work towards this target compound BMS-587101 is described in:
For additional information related to this compound see:
For the radiolabelled synthesis of BMS-587101 see:
|
10-31-2008
|
CRYSTALLINE FORMS AND PROCESS FOR PREPARING SPIRO-HYDANTOIN COMPOUNDS
|
|
|
6-4-2008
|
Crystalline forms and process for preparing spiro-hydantoin compounds
|
|
|
3-7-2007
|
Pyridyl-substituted spiro-hydantoin compounds and use thereof
|
|
|
7-19-2006
|
Spiro-hydantoin compounds useful as anti-inflammatory agents
|
|
|
6-30-2006
|
Pyridyl-substituted spiro-hydantoin crystalline forms and process
|
|
|
12-21-2005
|
Spiro-hydantoin compounds useful as anti-inflammatory agents
|
| US8710058 * | Dec 4, 2009 | Apr 29, 2014 | Merck Patent Gmbh | Polymorphic forms of 3-(1-{3-[5-(1-methyl-piperidin-4-ylmethoxy)-pyrimidin-2-yl]-benzyl}-6-oxo-1,6-dihydro-pyridazin-3-yl)-benzonitrile hydrochloride salt and processes of manufacturing thereof |
| US20110269767 * | Dec 4, 2009 | Nov 3, 2011 | Merck Patent Gesellschaft Mit Beschrankter Haftung | Novel Polymorphic Forms of 3-(1–6-oxo-1,6-dihydro-pyridazin-3-yl)-benzonitrile Hydrochloride Salt and Processes of Manufacturing Thereof |

AZD 6564
SYNTHESIS SUPP INFO…..http://pubs.acs.org/doi/suppl/10.1021/ml400526d/suppl_file/ml400526d_si_001.pdf
NMR PG 16/32 AS ABOVE
 R1 = NEOPENTYL R2=H
R1 = NEOPENTYL R2=H
5-[(2R,4S)-2-(2,2-Dimethylpropyl)piperidin-4-yl]-1,2-oxazol-3(2H)-one
5-((2R,4S)-2-Neopentylpiperidin-4-yl)isoxazol-3(2H)-one
238.326
C13 H22 N2 O2
Antifibrinolytics
AstraZeneca (Innovator)
SYNTHESIS SUPP INFO…..http://pubs.acs.org/doi/suppl/10.1021/ml400526d/suppl_file/ml400526d_si_001.pdf
NMR PG 16 0F 32
……………………..
Discovery of the fibrinolysis inhibitor AZD6564, acting via interference of a protein – Protein interaction
ACS Med Chem Lett 2014, 5(5): 538
http://pubs.acs.org/doi/abs/10.1021/ml400526d
A class of novel oral fibrinolysis inhibitors has been discovered, which are lysine mimetics containing an isoxazolone as a carboxylic acid isostere. As evidenced by X-ray crystallography the inhibitors bind to the lysine binding site in plasmin thus preventing plasmin from binding to fibrin, hence blocking the protein–protein interaction. Optimization of the series, focusing on potency in human buffer and plasma clotlysis assays, permeability, and GABAa selectivity, led to the discovery of AZD6564 (19) displaying an in vitro human plasma clot lysis IC50 of 0.44 μM, no detectable activity against GABAa, and with DMPK properties leading to a predicted dose of 340 mg twice a day oral dosing in humans.
SUPP INFO…..http://pubs.acs.org/doi/suppl/10.1021/ml400526d/suppl_file/ml400526d_si_001.pdf
Step 9: 5,((2R,4S),2,Neopentylpiperidin,4,yl)isoxazol,3(2H),one

O
L C^O”

METHOD B
O


METHOD C

METHOD D
RIB(OR)2

X = Cl, Br

METHOD E

METHOD F

METHOD G

R1 = 1-methyl-1 H-tetrazol-5-yl and 2-methyl-2H-tetrazol-5-yl
Scheme B. Formation of 5-isoxazol-3-ones
°Y I ‘relative


°Y J ‘relative

………………….
http://www.google.com/patents/EP2417131A1?cl=en
Example 14
5-((2R,4S)-2-Neopentylpiperidin-4-yl)isoxazol-3(2H)-one
Step 1 : Cis-methyl 2-neopentyl-4-(3-oxo-23-dihvdroisoxazol-5-yl)piperidine-l-carboxylate The compound was prepared as described in Example 1, Step 2 starting from cis-methyl 4-(3- ethoxy-3-oxopropanoyl)-2-neopentylpiperidine-l -carboxylate (2.68 g, 8.19 mmol) which resulted in cis-methyl 2-neopentyl-4-(3-oxo-2,3-dihydroisoxazol-5-yl)piperidine-l- carboxylate (1.60 g, 66 %) : IH NMR (400 MHz, cdcl3) δ 0.89 (s, 9H), 1.18 (dd, IH), 1.45 (dd, IH), 1.80 – 1.92 (m, 2H), 1.97 – 2.17 (m, 2H), 2.94 – 3.02 (m, IH), 3.11 – 3.23 (m, IH), 3.71 (s, 3H), 3.88 – 3.99 (m, IH), 4.22 – 4.32 (m, IH), 5.72 (s, IH); m/z (MH+) 297.
Step 2: (2R,4S)-Methyl 2-neopentyl-4-(3-oxo-2,3-dihvdroisoxazol-5-yl)piperidine-l- carboxylate
Following the procedure described in Example 1, Step 3, racemic cis-methyl 2-neopentyl-4- (3-oxo-2,3-dihydroisoxazol-5-yl)piperidine-l -carboxylate (1.60 g, 5.4 mmol) was subjected to chiral separation using Chiralcel IC mobile phase heptane/IP A/FA 60/40/0.1 which resulted in (2R,4S)-methyl 2-neopentyl-4-(3-oxo-2,3-dihydroisoxazol-5-yl)piperidine-l-carboxylate (0.8 g, 2.7 mmol).
Step 3: 5-((2R,4S)-2-Neopentylpiperidin-4-yl)isoxazol-3(2H)-one
5 Starting from (2R,4S)-methyl 2-neopentyl-4-(3-oxo-2,3-dihydroisoxazol-5-yl)piperidine-l- carboxylate (0.8 g, 2.7 mmol) and following the procedure described in Example 1, Step 4 the title compound was obtained (0.44 g, 69 %): 1H NMR (600 MHz, DMSO-d6) δ 0.89 (s, 9H), 1.18 (m, 2H), 1.50 (m, 2H), 1.82-1.90 (m, 2H), 2.70-2.85 (m, 3H), 3.08 (m, IH), 5.71 (s, IH). [α]20 D +43.8 (MeOH/H2O 1:1, c = 1); HRMS calculated for [C13H23N2O2]+: 239.1759; found: 10 239.1753.
—

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D

MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ
VIETNAM
ICELAND
RUSSIA

========================


(4aS,7aR)-1-[5-[(3-Fluorophenyl)ethynyl]pyridin-2-yl]hexahydrocyclopenta[d][1,3]oxazin-2(1H)-one
(4aS,7aR)-l-(5-Phenylethynyl-pyridin-2-yl)-hexahydro-cyclopenta[d] [l,3]oxazin-2-one
336.35
C20 H17 F N2 O2
F. Hoffmann-La Roche Ag, Hoffmann-La Roche Inc.
mgluR5 Positive Allosteric Modulators
Signal Transduction Modulators
http://www.google.com/patents/WO2014056710A1?cl=en
Example 1
(4aS,7aR)-l-(5-Phenylethynyl-pyridin-2-yl)-hexahydro-cyclopenta[d] [l,3]oxazin-2-one

Ste 1 : ((lR,2S)-2-Hydroxymethyl-cyclopentyl)-carbamic acid tert-butyl ester

To a well stirred suspension of 0.94 g (24.7 mmol, 2 equiv.) of L1AIH4 in 30ml of THF at 0°C was added dropwise at 0°C a solution of (lS,2R)-methyl 2-(tert-butoxycarbonylamino)- cyclopentanecarboxylate (CAS: 592503-55-4) (3.0 g, 12.3 mmol) (gas evolution, lightly exo therm). After 15 minutes at 0°C the reaction mixture was allowed to warm up to room temperature and was stirred for 2h. The mixture was cooled to 0°C and water was added dropwise. The precipitated inorganic salts were filtered through Celite and were washed with ethyl acetate. The filtrate was evaporated and the residue was purified by column
chromatography on silica gel eluting with a 0% to 50% ethyl acetate in heptane gradient to yield 1.99 g (75%) of the title compound as a crystalline white solid which was directly used in the next step. Ste 2: (4aS,7aR)-Hexahydro-cyclopenta[d][l,3]oxazin-2-one

To a solution of ((lR,2S)-2-hydroxymethyl-cyclopentyl)-carbamic acid tert-butyl ester (1.6 g, 7.43 mmol) in THF (40 ml) was added potassium tert-butoxide (3.34 g, 29.7 mmol, 4.0 equiv.) at room temperature. After stirring for lh at 60°C the reaction was allowed to warm up to room temperature and after workup with Ethyl acetate/water, drying and concentration in vaccuo, the crude material mixture was adsorbed on silica and chromatographed over a prepacked silica column (50g, 50% to 100% EtOAc in Heptane gradient) to yield 950 mg (91%) of the title compound as a white solid, which was directly used in the next step. -Fluoro-5-phenylethynyl-pyridine
In an 100ml 2-necked round bottomed flask under Argon were dissolved 2-fluoro-5-iodopyridine (5.0 g, 22.4 mmol, 1.0 equiv.) in THF (30 ml). After 5 minutes at room temperature were added bis(triphenylphosphin)palladium(II)chloride (944 mg, 1.35 mmol, 0.06 equiv.), triethylamine (6.81 g, 9.32 ml, 67.3 mmol, 3.0 equiv.), phenyl acetylene (2.75 g, 2.95 ml, 26.9 mmol, 1.2 equiv.) and copper(I)iodide (128 mg, 0.67 mmol, 0.03 equiv.). The brown suspension was cooled with water (exothermic) to room temperature and stirred overnight. Then 200ml of diethylether were added, the mixture was filtered, washed with ether and concentrated in vacuum to yield 5.7g of a brown solid which was adsorbed on silica and was chromatographed in 2 portions over a lOOg prepacked silica column eluting with a 0-10% ethyl acetate in heptane gradient to yield 3.99g (91%) of the title compound as a light brown solid, MS: m/e = 198.1 (M+H+). Step 4: (4aS aR)-l-(5-Phenylethynyl-pyridin-2-yl)-hexahydro-cyclopenta[d][l,3]oxazin-2-one In a 10ml Round bottomed flask were dissolved (4aS,7aR)-hexahydro-cyclopenta[d]- [l,3]oxazin-2-one (80 mg, 0.57 mmol, 1.0 equiv.) and 2-fluoro-5-(phenylethynyl)pyridine (112 mg, 0.57 mmol, 1.0 equiv.) in 2ml of DMF. Sodium hydride (60%> suspension) (29.5 mg, 0.74 mmol, 1.3 equiv.) were added and the brown suspension was stirred at room temperature overnight. The reaction mixture was quenched with water and extracted twice with ethyl acetate. The combined organic phases were dried, filtered and concentrated. The crude material was purified by flash chromatography over a prepacked silica column eluting with 0-50% ethyl acetae in heptane gradient to yield 42.5mg of the title compound as colorless amorphous solid, MS: m/e = 319.1 (M+H+).
Example 2
(4aS,7aR)- 1- [5-(3-Fluorophenylethynyl)-py ridin-2-yl] -hexahydro- cyclopenta[d] [l,3]oxazin-2-one

Step 1 : 2-Fluoro-5-(3-fluoro-phenylethynyl)-pyridine
The title compound was prepared in accordance with the general method of Example 1, step 3 using 3-flurorophenylacetylene instead of phenylacetylene to yield the title compound as a crystalline white solid, MS: m/e = 216.2 (M+H+).
Step 2 : (4aS ,7aR)- 1 – [5 -(3 -Fluorophenylethynyl)-pyridin-2-yl] -hexahydro- cyclopenta[d] [ 1 ,3]oxazin-2-one
The title compound was prepared in accordance with the general method of Example 1, step 4 using (4aS,7aR)-hexahydro-cyclopenta[d]-[l,3]oxazin-2-one (66 mg, 0.47 mmol) (Example 1, step 2) and 2-fluoro-5-((3-fluorophenyl)ethynyl)pyridine (100 mg, 0.47 mmol) to yield 48 mg (31%) of the title compound as a light yellow amorphous solid; MS: m/e = 337.3 (M+H+).

cas no 1416314-55-0
C20 H18 F N5 O3
FYL-67 IS HYDROCHLORIDE
(S)-N-((3-(3-Fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl)phenyl)-2-oxo-oxazolidin-5-yl)methyl)acetamide
N-[[(5S)-3-[3-fluoro-4-[4-(2-pyridinyl)-1H-pyrazol-1-yl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-Acetamide,
(S)-N-((3-(3-fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl) phenyl)-2-oxooxazolidin-5-yl)methyl)acetamide.
| Inventores | Youfu LUO, 罗有福, Zhenling WANG, 王震玲,Yuquan Wei, 魏于全 |
| Requerente | Si Chuan University, 四川大学 |
The discovery and application of antibiotics is one of the greatest achievements of mankind in the 20th century, the field of medicine, called a revolution of the history of the human fight against illness. Since then, the field of medicine into a bacterial disease caused by greatly reducing the golden age. Today, however, due to the widespread use of antibiotics or even abuse, the growing problem of bacterial resistance, humans are gradually approaching the “post-antibiotic era, the efficacy of antibiotics is gradually reduced. Clinical have been found on many new drug-resistant strains of methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant enterococci (VRE), penicillin-resistant Streptococcus pneumoniae (PRSP) has seriously jeopardize the clinical treatment , the number of varieties of drugs less.
The compounds of the oxazolidinone linezolid was in the United States in 2000, mainly used in clinical acquired pneumonia, soft tissue infections, can also be used for the surgical treatment of infectious diseases, bones, lungs, cerebrospinal fluid permeability pharmacokinetic and tissue concentrations. Domestic and foreign the oxazolidinone drug development is a hot field
WO 2012171479
http://www.google.st/patents/WO2012171479A1?cl=en



The object compound (S N-{[3 – (3 – fluoro-4 – (4 – (2 – pyridyl) pyrazol-yl) phenyl) -2 – oxo-oxazol the embankment -5 – yl] methanone yl}
Weigh 150mg of the compound (26f), was dissolved with 10 ml of anhydrous THF was added under nitrogen protection, an ice water bath 154.1 mg t-BuOLi, ice-water bath after stirring for 5 minutes, 149.9 mg Compound 11, followed by ice-water bath was removed, go reaction at room temperature for 36 hours the reaction was stopped, by adding 10 mL of methylene chloride and 10 ml of water and 22μί acetic acid, stirred for 1 minute, the liquid separation, the aqueous phase was extracted with dichloromethane three times, the organic phases were combined, dried and purified by column chromatography to give the product ( 130 white solid 58 mg of yield of 38.2%.
1H-MR (400 MHz, CDC1 3): δ 8.61 (d, J = 4Hz, IH), 8.52 (d, J = 6.8Hz, 2.4H), 8.22 (s, IH), 7.94 (t, J = 8.8 Hz, IH), 7.77-7.69 (m, 2H), 7.55 (d, J = 8Hz, IH), 7.27-7.26 (m, IH), 7.18-7.15 (m, IH), 6.06 (t, J = 6Hz , IH), 4.86-4.80 (m, IH), 4.11 (t, J = 9.2Hz, IH), 3.86-3.82 (m, IH), 3.78-3.62 (m, 2H), 2.04 (s, 3H 😉 .
13 C-MR (DMSO-e): δ 170.51, 154.47, 152.94, 151.26, 149.94, 139.70, 139.15, 137.43 129.96, 125.61, 125.19, 123.42, 122.19, 120.38, 114.52, 106.68, 72.29, 47.70, 41.84, 22.91.
ESI-MSm / z 418.08 (M + Na +).
………………….
Nanoscale (2013), 5(1), 275-283
DOI: 10.1039/C2NR32505E
In this work, a novel oxazolidinone compound FYL-67 was synthesized, and the obtained FYL-67 could form nanoassemblies in aqueous solution by a self-assembly method without using any carrier, organic solvent, or surfactant. The prepared FYL-67 nanoassemblies had a particle size of 264.6 ± 4.3 nm. The FYL-67 nanoassemblies can be lyophilized into a powder form without any cryoprotector or excipient, and the re-dissolved FYL-67 nanoassemblies are stable and homogeneous. The in vitro release profile showed a significant difference between rapid release of free FYL-67 and much slower and sustained release of FYL-67 nanoassemblies. In vitro susceptibility tests were conducted in three strains of methicillin-susceptibleStaphylococcus aureus (MSSA) and three strains of methicillin-resistant Staphylococcus aureus(MRSA), using linezolid as a positive control. FYL-67 nanoassemblies exhibited excellent in vitro activity, with a minimum inhibitory concentration (MIC) value of 0.5 μg mL−1 against MRSA. In the in vitro post-antibiotic effect (PAE) evaluation, FYL-67 nanoassemblies showed a more powerful effect than linezolid. Besides, in vitro cytotoxicity tests indicated that FYL-67 nanoassemblies had a very low cytotoxicity on HEK293 cells and L02 cells. Furthermore, in both MSSA and MRSA systemic infection mouse models, FYL-67 nanoassemblies showed a lower ED50 than linezolid. In a murine model of MRSA systemic infection, FYL-67 nanoassemblies displayed an ED50 of less than 4.0 mg kg−1, which is 2.3-fold better than that oflinezolid. Our findings suggested that the FYL-67 nanoassemblies may be a potential drugcandidate in MRSA therapy.

 |
||
| Fig. 1 Synthetic route of the novel compound FYL-67. (i) 2-(pyridin-2-yl)malonaldehyde, p-TsOH (cat.),ethanol, reflux, 2 h; (ii) Fe, HCl, 95% ethanol, 1 h; (iii) Cbz–Cl, K2CO3, CH2Cl2, 2 h; (iv) (S)-1-acetamido-3-chloropropan-2-yl acetate, LiOt-Bu, THF, r.t.; (v) HCL (g), acetone, ethyl ether | ||
1H-NMR (400 MHz, CDCl3): δ 8.61 (d, J = 4 Hz, 1H), 8.52 (d, J = 6.8 Hz, 2.4H), 8.22 (s, 1H), 7.94 (t, J = 8.8 Hz, 1H), 7.77–7.69 (m, 2H), 7.55 (d, J = 8 Hz, 1H), 7.27–7.26 (m, 1H), 7.18–7.15 (m, 1H), 6.06 (t, J = 6 Hz, 1H), 4.86–4.80 (m, 1H), 4.11 (t, J = 9.2 Hz, 1H), 3.86–3.82 (m, 1H), 3.78–3.62 (m, 2H), 2.04 (s, 3H).
13C-NMR (DMSO-d6): δ 170.51, 154.47, 152.94, 151.26, 149.94, 139.70, 139.15, 137.43, 129.96, 125.61, 125.19, 123.42, 122.19, 120.38, 114.52, 106.68, 72.29, 47.70, 41.84, 22.91.
ESI-MS m/z418.08 (M + Na+).
1H-NMR (400 MHz, DMSO-d6) δ: 9.33 (s, 1H), 8.80 (s, 1H), 8.74 (d, J = 5.6 Hz, 1H), 8.45 (t, J = 7.2 Hz, 1H), 8.38–8.31 (m, 2H), 7.90 (t, J = 8.8 Hz, 1H), 7.81 (dd, J = 2.4 Hz, J = 16.4 Hz, 1H), 7.76 (t,J = 6.0 Hz, 1H); 7.55 (dd, J = 1.6 Hz, J = 8.8 Hz, 1H), 4.83–4.76 (m, 1H), 4.60 (br s, 1H), 4.20 (t, J = 8.8 Hz, 1H), 3.91–3.82 (m, 1H), 3.45 (t, J = 5.2 Hz, 2H), 1.85 (s, 3H);
13C-NMR (DMSO-d6) δ: 170.51, 154.47, 152.94, 151.26, 149.94, 139.70, 139.15, 137.43, 129.96, 125.61, 125.19, 123.42, 122.19, 120.38, 114.52, 106.68, 72.29, 47.70, 41.84, 22.91;
HR-MS(TOF) m/z calcd for C20H18FN5O3 [M + Cl−]: 430.1082, found: 430.1085; for C20H18FN5O3 [M + H+]: 396.1472, found: 396.1472.
PAPER

A concise, environmentally benign, and cost-effective route was developed for the large-scale preparation of 1, a novel oxazolidinone antibacterial candidate. The key intermediate 2-(1-(2-fluoro-4-nitrophenyl)-1H-pyrazol-4-yl)pyridine 7 was prepared with high purity by mild deamination of the regioisomeric mixture 21. The mixture was prepared from a nucleophilic SNAr reaction by selective C–N coupling of the secondary amine functionality of 4-(pyridin-2-yl)-1H-pyrazol-3-amine 14 with 1,2-difluoro-4-nitrobenzene 10 in optimized conditions with the primary amine group remaining intact. The gaseous nitrogen release rate and reaction mixture temperature of the deamination step can be well controlled by altering the feeding manner, thereby providing safety guarantees. The optimized synthetic strategy of 1 with an overall yield of 27.6%, including seven sequential transformations by only five solid–liquid isolations, significantly improved the product separation workup. The strategy bypassed time-consuming and laborious procedures for any intermediate involved as well as for the final API. This study presents a process enabling the rapid delivery of a multikilogram quantity of API with high purity.
\
(S)-N-((3-(3-Fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl)phenyl)-2-oxo-oxazolidin-5-yl)methyl)acetamide (1)
| WO2008143649A2 * | 4 Dez 2007 | 27 Nov 2008 | Das Jagattaran | Novel oxazolidinone compounds as antiinfective agents |
| CN1172484A * | 29 Jan 1996 | 4 Fev 1998 | 法玛西雅厄普约翰美国公司 | Hetero-aromatic ring substituted phenyloxazolidinone antimicrobials |

The compound TIR-199 that holds much promise in the laboratory in fighting renal (kidney) cancer. feb19,2013
RIVERSIDE, Calif. — Chemists at the University of California, Riverside have developed a compound that holds much promise in the laboratory in fighting renal (kidney) cancer.
Named TIR-199, the compound targets the “proteasome,” a cellular complex in kidney cancer cells, similar to the way the drug bortezomib, approved by the Food and Drug Administration, targets and inhibits the proteasome in multiple myeloma cells, a cancer coming from bone marrow.
Michael Pirrung, a distinguished professor of chemistry at UC Riverside, announced the development of TIR-199 in a lecture he gave on Feb. 19 at the 5th International Conference on Drug Discovery and Therapy, held in Dubai, UAE.
Operating like the garbage dump of a cell, the proteasome breaks down proteins. Drugs that block the action of proteasomes are called proteasome inhibitors, and have been shown to have activity against a variety of cancer cell lines, albeit with mixed results. For example, bortezomib, though effective against multiple myeloma, has many side effects because cells other than bone marrow cells are affected.
“The novel feature of our new proteasome inhibitor, TIR-199, is that it is nearly as potent as bortezomib, but is selective in inhibiting the growth of only renal cancer cell lines,” Pirrung said. “It’s what makes TIR-199 attractive.”
The TIR-199 research project at UC Riverside began about four years ago after a multidisciplinary, international team reported on a class of compounds that act on the proteasome. These compounds are the “syringolin” natural products — such as a compound produced naturally by the wheat-infecting bacterium Pseudomonas syringae. TIR-199 is a synthetic relative of syringolin.

Michael Pirrung is a distinguished professor of chemistry at UC Riverside. Photo credit: I. Pittalwala, UC Riverside.

Structure of Syringolin A.
The ring structure of syringolin A is formed by the two nonproteinogenic amino acids 5-methyl-4-amino-2-hexenoic acid and 3,4-dehydrolysine. The α-amino group of the latter is joined by a peptide bond to a valine residue, which is linked to another valine residue via a urea moiety.

