
Home » Posts tagged 'preclinical' (Page 6)
Tag Archives: preclinical
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-thio)-methyl)-4-fluorobiphenyl-2-carboxamide
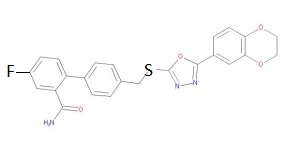
Cas 1820758-44-8
C24 H18 F N3 O4 S
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-thio)-methyl)-4-fluorobiphenyl-2-carboxamide


Glycogen synthase kinase-3 (GSK-3) is a constitutively active, ubiquitous serine/threonine kinase that takes part in a number of physiological processes ranging from glycogen metabolism to apoptosis. GSK-3 is a key mediator of various signaling pathways, such as the Wnt and the insulin/AKT signaling pathways.
Therefore, dysregulation of GSK-3 has been linked to various human diseases, such as cancer, diabetes, and neurodegenerative diseases.Two related isoforms of GSK-3 exist in mammals, GSK-3α and -β, which share a sequence identity within their catalytic domains of 98%.
Beyond the catalytic domains they show significant differences. Although these isoforms are structurally related, they are not functionally equivalent, and one cannot compensate for loss of the other.
The debate on the respective contributions of the isoforms GSK-3α and GSK-3β on the pathogenesis of different diseases is ongoing.
Various studies indicate that the therapies of certain diseases benefit from specific targeting of GSK-3α and GSK-3β. GSK-3α was recently identified as a differentiation target in acute myeloid leukemia (AML). AML is a hematopoietic malignancy defined by uncontrolled proliferation and disrupted myeloid differentiation. AML is the second most common form of leukemia in adults.
The current treatment of AML with conventional chemotherapy is very aggressive yet ineffective for the majority of patients with the disease.Thus, alternative targeted treatment approaches for AML are highly desirable. GSK-3α recently emerged as a potential target in this disease.
PAPER

The challenge for glycogen synthase kinase-3 (GSK-3) inhibitor design lies in achieving high selectivity for one isoform over the other. The therapy of certain diseases, such as acute myeloid leukemia (AML), may require α-isoform specific targeting. The scorpion shaped GSK-3 inhibitors developed by our group achieved the highest GSK-3α selectivity reported so far but suffered from insufficient aqueous solubility. This work presents the solubility-driven optimization of our isoform-selective inhibitors using a scorpion shaped lead. Among 15 novel compounds, compound 27 showed high activity against GSK-3α/β with the highest GSK-3α selectivity reported to date. Compound 27 was profiled for bioavailability and toxicity in a zebrafish embryo phenotype assay. Selective GSK-3α targeting in AML cell lines was achieved with compound 27, resulting in a strong differentiation phenotype and colony formation impairment, confirming the potential of GSK-3α inhibition in AML therapy
Evaluation of Improved Glycogen Synthase Kinase-3α Inhibitors in Models of Acute Myeloid Leukemia
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01200
http://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.5b01200/suppl_file/jm5b01200_si_001.pdf
compound 27 as a colorless solid. HPLC: 96%, tR = 6.93 min.
1H NMR (DMSO-d6, 500 MHz, 300 K): δ (ppm) = 4.32 (td, J = 5.2 Hz, J = 3.7 Hz, 4H), 4.60 (s, 2H), 7.05 (d, J = 8.4 Hz, 1H), 7.25 (dd, J = 9.1 Hz, J = 2.7 Hz, 1H), 7.31 (td, J = 8.6 Hz, J = 2.8 Hz, 1H), 7.38 (m, 3H), 7.41 (d, J = 2.0 Hz, 1H), 7.45 (dd, J = 8.4 Hz, J = 2.1 Hz, 1H), 7.49 (d, J = 8.2 Hz, 2H), 7.73 (s, 1H).
13C NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.6, 64.1, 64.4, 114.3 (d, JC–F = 21 Hz), 115.0, 115.9 (d, JC–F = 21 Hz), 115.9, 118.1, 120.0, 128.6 (2C), 128.8 (2C), 132.0 (d, JC–F = 8 Hz), 134.8, 135.5, 138.9, 139.0 (d, JC–F = 7 Hz), 143.8, 146.7, 160.9 (d, JC–F = 247 Hz), 162.7, 164.9, 169.5.
EI-MS: m/z = 463 (100, [M+]), 464 (26, [M+ + H]), 465 (7, [M+ + 2H].
ABOUT Boris Schmidt
Boris Schmidt
-
Clemens Schöpf-Institute of Chemistry and BiochemistryDarmstadt, Hessen, Germany*Phone: +49 6151 163075. Fax: +49 6151 163278.E-mail: Schmidt_boris@t-online.de.
………………………………………….
ABOUT Theresa Neumann
-
Clemens Schöpf-Institute of Chemistry and BiochemistryDarmstadt, Hessen, Germany
////////FC(C=C1C(N)=O)=CC=C1C(C=C2)=CC=C2CSC3=NN=C(O3)C4=CC5=C(OCCO5)C=C4
New 5-Substituted-N-(piperidin-4-ylmethyl)-1H-indazole-3-carboxamides: Potent Glycogen Synthase Kinase-3 (GSK-3) Inhibitors in Model of Mood Disorders
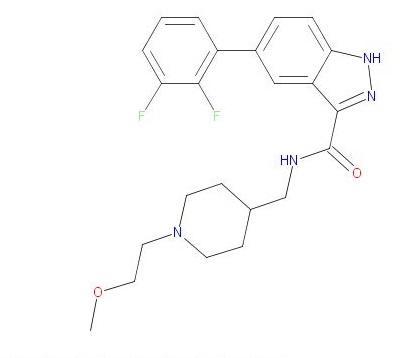
CAS 1452582-16-9, 428.47, C23 H26 F2 N4 O2
1H-Indazole-3-carboxamide, 5-(2,3-difluorophenyl)-N-[[1-(2-methoxyethyl)-4-piperidinyl]methyl]-
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
![]()
1 H-indazole-3-carboxamide compounds acting as glycogen synthase kinase 3 beta (GSK-33) inhibitors and to their use in the treatment of GSK-33-related disorders such as (i) insulin-resistance disorders; (ii) neurodegenerative diseases; (iii) mood disorders; (iv) schizophrenic disorders; (v) cancerous disorders; (vi) inflammation, (vii) substance abuse disorders; (viii) epilepsies; and (ix) neuropathic pain.
Protein kinases constitute a large family of structurally related enzymes, which transfer phosphate groups from high-energy donor molecules (such as adenosine triphosphate, ATP) to specific substrates, usually proteins. After phosphorylation, the substrate undergoes to a functional change, by which kinases can modulate various biological functions.
In general, protein kinases can be divided in several groups, according to the substrate that is phosphorylated. For example, serine/threonine kinase phosphorylates the hydroxyl group on the side chain of serine or threonine aminoacid.
Glycogen synthase kinases 3 (GSK-3) are constitutively active multifunctional enzymes, quite recently discovered, belonging to the serine/threonine kinases group.
Human GSK-3 are encoded by two different and independent genes, which leads to GSK-3a and GSK-33 proteins, with molecular weights of about 51 and 47 kDa, respectively. The two isoforms share nearly identical sequences in their kinase domains, while outside of the kinase domain, their sequences differ substantially (Benedetti et al., Neuroscience Letters, 2004, 368, 123-126). GSK-3a is a multifunctional protein serine kinase and GSK-33 is a serine-threonine kinase.
It has been found that GSK-33 is widely expressed in all tissues, with widespread expression in the adult brain, suggesting a fundamental role in neuronal signaling pathways (Grimes and Jope, Progress in Neurobiology, 2001, 65, 391-426). Interest in glycogen synthase kinases 3 arises from its role in various physiological pathways, such as, for example, metabolism, cell cycle, gene expression, embryonic development oncogenesis and neuroprotection (Geetha et al., British Journal Pharmacology, 2009, 156, 885-898).
GSK-33 was originally identified for its role in the regulation of glycogen synthase for the conversion of glucose to glycogen (Embi et al., Eur J Biochem, 1980, 107, 519-527). GSK-33 showed a high degree of specificity for glycogen synthase.
Type 2 diabetes was the first disease condition implicated with GSK- 3β, due to its negative regulation of several aspects of insulin signaling pathway. In this pathway 3-phosphoinositide-dependent protein kinase 1 (PDK-1 ) activates PKB, which in turn inactivates GSK-33. This inactivation of GSK-33 leads to the dephosphorylation and activation of glycogen synthase, which helps glycogen synthesis (Cohen et al., FEBS Lett, 1997, 410, 3-10). Moreover, selective inhibitors of GSK-33 are expected to enhances insulin signaling in prediabetic insulin- resistant rat skeletal muscle, thus making GSK-33 an attractive target for the treatment of skeletal muscle insulin resistance in the pre-diabetic state (Dokken et al., Am J. Physiol. Endocrinol. Metab., 2005, 288, E1 188-E1 194).
GSK-33 was also found to be a potential drug target in others pathological conditions due to insulin-resistance disorders, such as syndrome X, obesity and polycystic ovary syndrome (Ring DB et al., Diabetes, 2003, 52: 588-595).
It has been found that GSK-33 is involved in the abnormal phosphorylation of pathological tau in Alzheimer’s disease (Hanger et al., Neurosci. Lett, 1992, 147, 58-62; Mazanetz and Fischer, Nat Rev Drug Discov., 2007, 6, 464-479; Hong and Lee, J. Biol. Chem., 1997, 272, 19547- 19553). Moreover, it was proved that early activation of GSK-33, induced by apolipoprotein ApoE4 and β-amyloid, could lead to apoptosis and tau hyperphosphorylation (Cedazo-Minguez et al., Journal of Neurochemistry, 2003, 87, 1 152- 1 164). Among other aspect of Alzheimer’s disease, it was also reported the relevance of activation of GSK-33 at molecular level (Hernandez and Avila, FEBS Letters, 2008, 582, 3848-3854).
Moreover, it was demonstrated that GSK-33 is involved in the genesis and maintenance of neurodegenerative changes associated with Parkinson’s disease (Duka T. et al., The FASEB Journal, 2009; 23, 2820- 2830).
Accordingly to these experimental observations, inhibitors of GSK-33 may find applications in the treatment of the neuropathological consequences and the cognitive and attention deficits associated with tauopathies; Alzheimer’s disease; Parkinson’s disease; Huntington’s disease (the involvement of GSK-33 in such deficits and diseases is disclosed in Meijer L. et al., TRENDS Pharm Sci, 2004; 25, 471 -480); dementia, such as, but not limited to, vascular dementia, post-traumatic dementia, dementia caused by meningitis and the like; acute stroke; traumatic injuries; cerebrovascular accidents; brain and spinal cord trauma; peripheral neuropathies; retinopathies and glaucoma (the involvement of GSK-33 in such conditions is disclosed in WO 2010/109005).
The treatment of spinal neurodegenerative disorders, like amyotrophic lateral sclerosis, multiple sclerosis, spinal muscular atrophy and neurodegeneration due to spinal cord injury has been also suggested in several studies related to GSK-33 inhibition, such as, for example in Caldero J. et al., “Lithium prevents excitotoxic cell death of motoneurons in organotypic slice cultures of spinal cord”, Neuroscience. 2010 Feb 17;165(4):1353-69, Leger B. et al., “Atrogin-1 , MuRF1 , and FoXO, as well as phosphorylated GSK-3beta and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord-injured patients”, Muscle Nerve, 2009 Jul; 40(1 ):69-78, and Galimberti D. et al., “GSK33 genetic variability in patients with Multiple Sclerosis”, Neurosci Lett. 201 1 Jun 1 5;497(1 ):46- 8. Furthermore, GSK-33 has been linked to the mood disorders, such as bipolar disorders, depression, and schizophrenia.
Inhibition of GSK-33 may be an important therapeutic target of mood stabilizers, and regulation of GSK-33 may be involved in the therapeutic effects of other drugs used in psychiatry. Dysregulated GSK-33 in mood disorder, bipolar disorder, depression and schizophrenia could have multiple effects that could impair neural plasticity, such as modulation of neuronal architecture, neurogenesis, gene expression and the ability of neurons to respond to stressful, potentially lethal conditions (Jope and Ron, Curr. Drug Targets, 2006, 7, 1421- 1434).
The role of GSK-33 in mood disorder was highlighted by the study of lithium and valproate (Chen et al., J. Neurochem., 1999, 72, 1327- 1330; Klein and Melton, Proc. Natl. Acad. Sci. USA, 1996, 93, 8455-8459), both of which are GSK-33 inhibitors and are used to treat mood disorders. There are also existing reports from the genetic perspective supporting the role of GSK-33 in the disease physiology of bipolar disorder (Gould, Expert. Opin. Ther. Targets, 2006, 10, 377-392).
It was reported a decrease in AKT1 protein levels and its phosphorylation of GSK-33 at Serine-9 in the peripheral lymphocytes and brains of individuals with schizophrenia. Accordingly, this finding supports the proposal that alterations in AKT1 -GSK-33 signaling contribute to schizophrenia pathogenesis (Emamian et al., Nat Genet, 2004, 36, 131- 137).
Additionally, the role of GSK-33 in cancer is a well-accepted phenomenon.
The potential of small molecules that inhibit GSK-33 has been evidenced for some specific cancer treatments (Jia Luo, Cancer Letters, 2009, 273, 194-200). GSK-33 expression and activation are associated with prostate cancer progression (Rinnab et al., Neoplasia, 2008, 10, 624-633) and the inhibition of GSK3b was also proposed as specific target for pancreatic cancer (Garcea et al., Current Cancer Drug Targets, 2007, 7, 209-215) and ovarian cancer (Qi Cao et al., Cell Research, 2006, 16 671 -677). Acute inhibition of GSK-33 in colon-rectal cancer cells activates p53-dependent apoptosis and antagonizes tumor growth (Ghosh et al., Clin Cancer Res 2005, 1 1 , 4580-4588).
The identification of a functional role for GSK-33 in MLL-associated leukaemia suggests that GSK-33 inhibition may be a promising therapy that is selective for transformed cells that are dependent on HOX overexpression (Birch et al., Cancer Cell, 2010, 1 7, 529-531 ).
GSK-33 is involved in numerous inflammatory signalling pathways, for example, among others GSK-33 inhibition has been shown to induce secretion of the anti-inflammatory cytokine IL-1 0. According to this finding, GSK-33 inhibitors could be useful to regulate suppression of inflammation (G. Klamer et al., Current Medicinal Chemistry, 2010, 17(26), 2873-2281, Wang et al., Cytokine, 2010, 53, 130-140).
GSK-33 inhibition has been also shown to attenuate cocaine-induced behaviors in mice. The administration of cocaine in mice pretreated with a GSK-33 inhibitor demonstrated that pharmacological inhibition of GSK3 reduced both the acute behavioral responses to cocaine and the long- term neuroadaptations produced by repeated cocaine (Cocaine-induced hyperactivity and sensitization are dependent on GSK3, Miller JS et al. Neuropharmacology. 2009 Jun; 56(8):1 1 16-23, Epub 2009 Mar 27).
The role of GSK-33 in the development of several forms of epilepsies has been demonstrated in several studies, which suggest that inhibition of GSK-33 could be a pathway for the treatment of epilepsy (Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy, Lohi H et al., Hum Mol Genet. 2005 Sep 15;14(18):2727-36 and Hyperphosphorylation and aggregation of Tau in laforin-deficient mice, an animal model for Lafora disease, Purl R et al., J Biol Chem. 2009 Aug 21 ;284(34) 22657-63). The relationship between GSK-33 inhibition and treatment of neuropathic pain has been demonstrated in Mazzardo-Martins L. et al., “Glycogen synthase kinase 3-specific inhibitor AR-A014418 decreases neuropathic pain in mice: evidence for the mechanisms of action”, Neuroscience. 2012 Dec 13;226, and Xiaoping Gu et al., “The Role of Akt/GSK33 Signaling Pathway in Neuropathic Pain in Mice”, Poster A525, Anesthesiology 2012 October 13-17, 2012 Washington.
A review on GSK-33, its function, its therapeutic potential and its possible inhibitors is given in “GSK-33: role in therapeutic landscape and development of modulators” (S. Phukan et al., British Journal of Pharmacology (2010), 160, 1- 19).
WO 2004/014864 discloses 1 H-indazole-3-carboxamide compounds as selective cyclin-dependant kinases (CDK) inhibitors. Such compounds are assumed to be useful in the treatment of cancer, through a mechanism mediated by CDK2, and neurodegenerative diseases, in particular Alzheimer’s disease, through a mechanism mediated by CDK5, and as anti-viral and anti-fungine, through a mechanism mediated by CDK7, CDK8 and CDK9.
Cyclin-dependant kinases (CDKs) are serine/threonine kinases, first discovered for their role in regulating the cell cycle. CDKs are also involved in regulating transcription, mRNA processing, and the differentiation of nerve cells. Such kinases activate only after their interaction and binding with regulatory subunits, namely cyclins.
Moreover, 1 H-indazole-3-carboxamide compounds were also described as analgesics in the treatment of chronic and neuropathic pain (see, for example, WO 2004/074275 and WO 2004/101 548) and as 5-HT4 receptor antagonists, useful in the treatment of gastrointestinal disorders, central nervous system disorders and cardiovascular disorders (see, for example, WO 1994/101 74).
Patent
WO 2013124158
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
SEE ENTRY 8

DMSO-de; δ 13.09 (s, 1 H), 8.23-8.42 (m, 2H), 7.72 (dd, J=0.82, 8.69 Hz, 1 H), 7.55 (td, J=1.76, 8.74 Hz, 1 H), 7.24-7.49 (m, 3H), 3.40 (t, J=6.04 Hz, 2H), 3.22 (s, 3H), 3.18 (d, J=6.40 Hz, 2H), 2.84 (d, J=11.53 Hz, 2H), 2.42 (t, J=5.95 Hz, 2H), 1.82- 2.02 (m, 2H), 1.41 -1.71 (m, 3H), 1.06-1.31 (m, 2H)
PAPER

Hit Optimization of 5-Substituted-N-(piperidin-4-ylmethyl)-1H-indazole-3-carboxamides: Potent Glycogen Synthase Kinase-3 (GSK-3) Inhibitors with in Vivo Activity in Model of Mood Disorders
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01208

http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01208
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
Angelini S.p.A., Angelini Research Center,
![]()
/////
COCCN1CCC(CNC(=O)c2n[nH]c3ccc(cc23)c4cccc(F)c4F)CC1
BMS-248360, A NEW SARTAN ON HORIZON

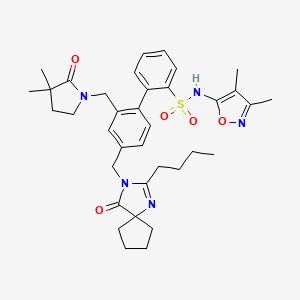
2-[4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2-[(3,3-dimethyl-2-oxopyrrolidin-1-yl)methyl]phenyl]-N-(3,4-dimethyl-1,2-oxazol-5-yl)benzenesulfonamide
4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide,
4′- . (2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non-l-en-3-yl)methyll -N-C3.4- dimethyl-5-isoxazolyl)-2,-[(3.3-dimethyl-2-oxo-l- pyrrolidinvDmethyll [1.1 ‘-biphenyl] -2-sulfonamide
4‘-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N–(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide
BMS-248360
PRECLINICAL …..treating hypertension
Bristol Myers Squibb Co, INNOVATOR
Hypertension remains one of the largest unmet medical needs in the 21st century, especially when one considers that hypertension is the portent of future debilitating cardiovascular disease. While many drugs are available for treating the disease, approximately one-third of the hypertensive population is still not adequately treated. Of the more recent avenues explored for treating hypertension, disruption of the effects of either angiotensin II (AII) or endothelin-1 (ET-1) has shown promise. These endogenous vasoactive peptides are among the most potent vasoconstrictors and cell proliferative factors identified to date. AII is the effector molecule of the renin−angiotensin system (RAS), and a large number of AII receptor (AT1) antagonists, including irbesartan , have been developed for treating hypertension


SYNTHESIS
picked from…….http://www.drugfuture.com/synth/syndata.aspx?ID=324487

EP 1094816; JP 2002519380; US 2002143024; WO 0001389
The intermediate biphenyl aldehyde (XI) is prepared by two related methods. 4-Bromo-3-methylbenzonitrile (I) is oxidized to aldehyde (II) via radical bromination with N-bromosuccinimide/benzoyl peroxide, followed by treatment with trimethylamine N-oxide. Suzuki coupling of aryl bromide (II) with the pinacol boronate (III) affords biphenyl (IV). After protection of the aldehyde moiety of (IV) as the corresponding ethylene ketal (V), its cyano group is reduced to aldehyde (VI) employing DIBAL in THF. Subsequent reduction of (VI) with NaBH4 leads to alcohol (VII), which is further converted into the benzyl bromide (VIII) by means of CBr4/PPh3. Bromide (VIII) is condensed with the spiro imidazolone (IX) in the presence of NaH, to produce (X). Then acidic hydrolysis of the ethylene ketal and SEM groups of (X) gives rise to the intermediate aldehyde (XI)
NEXT

Alternatively, reduction of 4-bromo-3-formylbenzonitrile ethylene ketal (XII) by means of DIBAL leads to aldehyde (XIII), which is further reduced to alcohol (XIV) with NaBH4. After bromination of (XIV) with CBr4/PPh3, the resultant benzyl bromide (XV) is condensed with the spiro imidazolone (IX), yielding (XVI). Then, acidic ketal hydrolysis in (XVI) furnishes aldehyde (XVII). Suzuki coupling between aryl bromide (XVII) and boronic acid (XVIII) gives biphenyl (XIX). The SEM group of (XIX) is then removed under acidic conditions to provide (XI)

Reductive amination of the biphenyl aldehyde (XI) with 4-amino-2,2-dimethylbutanoic acid (XX) in the presence of NaBH(OAc)3 produces aminoacid (XXI). This is finally cyclized to the corresponding lactam by treatment with DIC

Coupling of 2-bromobenzenesulfonyl chloride (I) with 5-amino-3,4-dimethylisoxazole (II) affords sulfonamide (III), which is further protected as the N-methoxyethoxymethyl derivative (IV) employing MEM-chloride in DMF. Lithiation of bromosulfonamide (IV), followed by treatment with trimethyl borate and acidic work up leads to the boronic acid intermediate (V). This is then subjected to Suzuki coupling with 4-bromo-3-methylbenzaldehyde (VI) to yield the biphenyl adduct (VII). After reduction of aldehyde (VII) to the benzylic alcohol (VIII) with NaBH4, reaction with methanesulfonyl chloride and diisopropylethylamine gives rise to the mesylate (IX) (1-3).

Mesylate (IX) is condensed with ethyl 2-propyl-4-ethylimidazole-5-carboxylate (X) yielding (XI). Simultaneous ester group hydrolysis and MEM group deprotection under acidic conditions gives rise to the imidazolecarboxylic acid (XII). This is finally coupled with methylamine via activation with CDI to produce the desired N-methyl carboxamide (1-3).
Reductive amination of the biphenyl aldehyde (XI) with 4-amino-2,2-dimethylbutanoic acid (XX) in the presence of NaBH(OAc)3 produces aminoacid (XXI). This is finally cyclized to the corresponding lactam by treatment with DIC
PAPER
 BMS 248360
BMS 248360The ETA receptor antagonist (2) (N-(3,4-dimethyl-5-isoxazolyl)-4‘-(2-oxazolyl)-[1,1‘-biphenyl]-2-sulfonamide, BMS-193884) shares the same biphenyl core as a large number of AT1 receptor antagonists, including irbesartan (3). Thus, it was hypothesized that merging the structural elements of 2 with those of the biphenyl AT1 antagonists (e.g., irbesartan) would yield a compound with dual activity for both receptors. This strategy led to the design, synthesis, and discovery of (15) (4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide, BMS-248360) as a potent and orally active dual antagonist of both AT1 and ETAreceptors. Compound 15 represents a new approach to treating hypertension.

Scheme 2 a
a (a) DIBAL, toluene; (b) NaBH4, MeOH; (c) (Ph)3P, CBr4, THF (51% from 9); (d) compound 7, NaH, DMF; (e) 1 N HCl; (f) compound 4, (Ph3P)4Pd, aqueous Na2CO3, EtOH/toluene; (g) 6 N aqueous HCl/EtOH (60% from 10); (h) 13, sodium triacetoxy borohydride, AcOH, (i) diisopropylcarbodiimide, CH2Cl2 (31% from 12).
15 as a white solid (40 mg, 31%):
mp 104−110 °C;
1H NMR (CDCl3) δ 0.90 (t, J = 7.0 Hz, 3H), 1.08 (s, 3H), 1.14 (s, 3H), 1.36 (m, 2H), 1.61 (m, 2H), 1.75−2.06 (m, 13H), 2.17 (s, 3H), 2.39 (m, 2H), 4.18 (m, 2H), 4.71 (m, 2H), 7.02−7.93 (m, 7H);
13CNMR (CDCl3 ) δ 7.82, 11.91, 14.79, 23.36, 25.50, 25.61, 27.11, 28.81, 29.88, 35.33, 38.42, 41.48, 44.59, 46.24, 46.47, 109.29, 125.15, 125.76, 129.68, 130.58, 131.76, 133.20, 134.07, 137.15, 138.27, 139.11, 139.57, 155.81, 162.68, 162.91, 181.25, 187.83.
Anal. (C36H45N5O5S) C, H, N, S.
……………………………
PATENT
US 2002143024
http://www.google.com/patents/US20020143024
 Zhang, H.-Y. et al., Tetrahedron, 1994, 50, 11339-11362.
Zhang, H.-Y. et al., Tetrahedron, 1994, 50, 11339-11362.

N-(3,4-Dimethyl-5-iso-xazolyl)-2′-formyl-4′-(hydroxy-methyl)-N-[[2-(tri-methylsilyl)ethoxy]- methyl][1,1′- biphenyl]-2- sulfonamide
Example 3 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
[0414]

Example 3 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
A. 4′-Cyano-2′-(1,3-dioxolan-2-yl)-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
A mixture of 2B (1.28 g, 2.73 mmol), ethylene glycol (1.69 g, 27.3 mmol) and p-toluenesulfonic acid (38 mg) in toluene (30 mL) was heated at 130° C. for 5 h, while a Dean-Stark water separator was used. After cooling, the mixture was diluted with EtOAc. The organic liquid was separated and washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 5:4 hexane/EtOAc to afford 3A (1.1 g, 79%) as a colorless gum: Rf=0.57, silica gel, 1:2 hexane/EtOAc.
B. 2′-(1,3-Dioxolan-2-yl)-4′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
To 3A (1.1 g, 2.14 mmol) in THF (21 mL) at 0° C. was added DIBAL-H (1M in CH2Cl2, 4.28 mL 4.28 mmol) dropwise. The reaction was stirred at RT overnight. MeOH (20 mL) was added and the reaction was stirred for 5 min. The mixture was poured into cold 0.1 N HCl solution (150 mL), shaken for 5 min, and then extracted with 3:1 EtOAc/hexane. The combined organic extracts were washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 3:4 hexane/EtOAc to afford 3B (710 mg, 64%) as a colorless gum: Rf=0.45, silica gel, 2:3 hexane/EtOAc.
C. 2′-(1,3-Dioxolan-2-yl)-4′-hydroxymethyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) [1,1′-biphenyl]-2-sulfonamide
3B (710 mg, 1.4 mmol) was subjected to sodium borohydride reduction according to General Method 11 to afford 3C, which was used for the next reaction step without further purification.
D. 4′-Bromomethyl-2′-(1,3-dioxolan-2-yl)-N-(3,4′-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) [1,1′-biphenyl]-2-sulfonamide
3C was treated with carbon tetrabromide and triphenylphosphine according to General Method 2. The crude residue was chromatographed on silica gel using 3:2 hexane/EtOAc to afford 3D (750 mg, 94%) as a colorless gum: Rf=0.74, silica gel, 1:2 hexane/EtOAc.
E. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-(1,3-dioxolan-2-yl)-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
3D (750 mg, 1.3 mmol) was treated with 2-n-butyl-1,3-diazaspiro[4.4]non-1-en-4-one hydrochloride (387 mg, 1.68 mmol) according to General Method 4. The crude residue was chromatographed on silica gel using 100:1.7 CH2Cl2/MeOH to afford 3E as a gum (830 mg, 93%): Rf=0.40, silica gel, 100:5 CH2Cl2/MeOH.
F. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
3E (830 mg, 1.20 mmol) was subjected to deprotection according to General Method 7. The crude residue was chromatographed on silica gel using 100:1.5 and then 100:4 CH2Cl2 /MeOH to afford the title compound as a gum (480 mg, 72%): Rf=0.16, silica gel, 100:5 CH2Cl2/MeOH.
Example 4 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2′-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl][1,1′-biphenyl]-2-sulfonamide

To 3F (110 mg, 0.20 mmol) in CH2Cl2 (4 mL) was added 4-amino-2,2-dimethylbutanoic acid hydrochloride (98 mg, 0.59 mmol) [Scheinmann, et al., J. Chem. Research (S), 414-415 (1993)] and 3 Å molecular sieves, followed by glacial acetic acid (35 mg, 0.59 mmol) and then sodium acetate (48 mg, 0.59 mmol). The mixture was stirred for 8 minutes, and NaB(AcO)3H (124 mg, 0.59 mmol) was then added. The reaction mixture was stirred at RT for 2 h, diluted with EtOAc and filtered through celite. The filtrate was washed with H2O and brine, dried and concentrated. This material was dissolved in CH2Cl2 (6 mL) and 1,3-diisopropylcarbodiimide (32 mg, 0.25 mmol) was added. The reaction mixture was stirred at RT for 2 h and diluted with CH2Cl2, washed with H2O and brine, dried and concentrated. The residue was purified by preparative HPLC to provide the title compound as a white solid (40 mg, 31%, for two steps): mp 104-110° C. Analysis calculated for C36H45N5O5S.0.8 H2O: Calc’d: C, 64.13; H, 6.97; N, 10.39; S, 4,75. Found: C, 64.18; H, 6.60; N, 10.23; S, 4.50.
Example 5 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide (Alternative Preparation for 3F)
A. 2-[(2′-Bromo-5′-formyl)phenyl)]-1,3-dioxolane
DIBAL-H (1.0 M solution in toluene, 445 mL, 445 mmol, 1.1 eq) was added over 30 minutes to a solution of 2-[(2′-bromo-5′-cyano)phenyl)]-1,3-dioxolane (103 g, 404 mmol, 1.0 eq) [Zhang, H.-Y. et al., Tetrahedron, 50, 11339-11362 (1994)] in toluene (2.0 L) at −78° C. The solution was allowed to warm to 0° C. After 1 hour, a solution of Rochelle’s salt (125 g) in water (200 mL) was added, and the mixture was allowed to warm to room temperature and was stirred vigorously for 16 h. The organic layer was concentrated and the residue partitioned between ethyl acetate (1 L) and 1 N hydrochloric acid (800 mL). The organic layer was washed with saturated aqueous sodium bicarbonate (800 mL), dried over sodium sulfate, and then concentrated to give 70.5 g of crude 5A as a yellow solid, which was used without further purification.
B. 2-[(2′-Bromo-5′-hydroxymethyl)phenyl)]-1,3-dioxolane
Sodium borohydride (3.66 g, 96.7 mmol, 0.5 eq) was added to a solution of crude 5A (49.7 g, approximately 193 mmol, 1.0 eq) in absolute ethanol (1300 mL) at 0° C. After 2 hours, a solution of 10% aqueous sodium dihydrogen phosphate (50 mL) was added and the mixture was stirred and allowed to warm to room temperature. The mixture was concentrated, then partitioned between ethyl acetate (800 mL) and saturated aqueous sodium bicarbonate (500 mL). The organic layer was dried over sodium sulfate and concentrated to give 49.0 g of crude 5B as a yellow oil, which was used without further purification.
C. 2-[(2′-Bromo-5′-bromomethyl)phenyl)]-1,3-dioxolane
Triphenylphosphine (52.7 g, 199 mmol, 1.05 eq) was added in portions over 15 minutes to a solution of crude 5B (49.0 g, approximately 189 mmol, 1.0 eq) and carbon tetrabromide (69.0 g, 208 mmol, 1.1 eq) in THF at 0° C. After 2 hours, saturated aqueous sodium bicarbonate solution (20 mL) was added, and the mixture was allowed to warm to room temperature and was then concentrated. Ether (500 mL) was added, and the resulting mixture was filtered. The filtrate was dried over magnesium sulfate and concentrated. The residue was chromatographed on silica gel (8:1 hexanes/ethyl acetate as eluant) to give 5C as a white solid (31.1 g, 51% yield from 2-[(2′-bromo-5′-cyano)phenyl)]-1,3-dioxolane).
D. 2-(1,3-Dioxolan-2-yl)-4-[(2-n-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]bromobenzene
[0436] Sodium hydride (60% dispersion in mineral oil, 9.65 g, 241 mmol, 2.5 eq) was added in portions over 15 minutes to a mixture of 2-n-butyl-1,3-diazaspiro[4.4]non-1-en-4-one hydrochloride (18.7 g, 96.5 mmol, 1.0 eq) in DMF (400 mL) at 0° C. The mixture was stirred and allowed to warm to room temperature over 15 minutes. To this mixture was added via canula a solution of 5C (31.1 g, 96.5 mmol, 1.0 eq) in DMF (100 mL). After 14 hours, the mixture was concentrated in vacuo and partitioned between ethyl acetate (500 mL) and 10% aqueous sodium dihydrogen phosphate (300 mL). The organic layer was dried over sodium sulfate and concentrated to give crude 5D as an orange oil (42.7 g), which was used without further purification.
E. 4-[(2-n-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2-formyl-bromobenzene
A solution of crude 5D (6.0 g, approximately 13.6 mmol, 1.0 eq) in THF (180 mL) and 1N hydrochloric acid (30 mL) was heated at 65° C. for 1.5 hours. The mixture was cooled and then treated with saturated aqueous sodium carbonate solution (75 mL) and ethyl acetate (200 mL). The organic layer was removed and dried over sodium sulfate, concentrated, and then further dried azeotropically with toluene to give 5E as a crude yellow oil (8.2 g) which contained a small amount of toluene. This material was used without further purification.
F. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
Palladium catalyzed Suzuki coupling of 5E and [2-[[(3,4-dimethyl-5-isoxazolyl)[(2-methoxyethoxy)methyl]amino]sulfonyl]phenyl]boronic acid was performed according to General Method 1 to yield 5F in 60% yield.
G. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
Deprotection of 5F according to General Method 7 provided the title compound (5G=3F) in 73% yield: Rf=0.2 (silica gel using CH2Cl2/MeOH [100:5]).
PATENT
EP 1237888; WO 0144239
Example 3 4′-r(2-Butyl-4-oxo-1.3-diazaspiror4.41non-l-en-3-yl)methvn-2′-formyl-N-
(3, 4-dimethyl-5-isoxazolyl)-[ 1,1 ‘-biphenyl] -2-sulfonamide

A. 4′-Cvano-2>-(1.3-dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [1.1 ‘-biphenyl] -2-sulfonamide
A mixture of 2B (1.28 g, 2.73 mmol), ethylene glycol (1.69 g, 27.3 mmol) and p-toluenesulfonic acid (38 mg) in toluene (30 mL) was heated at 130°C for 5 h, while a Dean-Stark water separator was used. After cooling, the mixture was diluted with EtOAc. The organic liquid was separated and washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 5:4 hexane/EtOAc to afford 3A (1.1 g, 79%) as a colorless gum: R^0.57, silica gel, 1:2 hexane EtOAc.
B. 2,-(1.3-Dioxolan-2-yl)-4′-formyl-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [1 , l’-biphenyl] -2-sulfonamide To 3A (1.1 g, 2.14 mmol) in THF (21 mL) at 0°C was added DIBAL- H (IM in CH2C12, 4.28 mL 4.28 mmol) dropwise. The reaction was stirred at RT overnight. MeOH (20 mL) was added and the reaction was stirred for 5 min. The mixture was poured into cold 0.1 N HCI solution (150 mL), shaken for 5 min, and then extracted with 3:1 EtOAc/hexane. The combined organic extracts were washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 3:4 hexane/EtOAc to afford 3B (710 mg, 64%) as a colorless gum: R^O.45, silica gel, 2:3 hexane/EtOAc. C. 2′-(1.3-Dioxolan-2-yl)-4′-hvdroxymethyl-N-(3.4-dimethyl-5- isoxazolyl)-N-(2-methoxyethoxymethyl) [1.1 ‘-biphenyl] -2- sulfonamide
3B (710 mg, 1.4 mmol) was subjected to sodium borohydride reduction according to General Method 11 to afford 3C, which was used for the next reaction step without further purification.
D. 4l-Bromomethyl-2,-(1.3-dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)- N-(2-methoxyethoxymethyl) [1 , l’-biphenyl] -2-sulfonamide 3C was treated with carbon tetrabromide and triphenylphosphine according to General Method 2. The crude residue was chromatographed on silica gel using 3:2 hexane/EtOAc to afford 3D (750 mg, 94%) as a colorless gum: R^0.74, silica gel, 1:2 hexane/EtOAc.
E. 4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn- 2,-(1.3- dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [ 1. l’-biphenyll -2-sulfonamide 3D (750 mg, 1.3 mmol) was treated with 2-re-butyl-l,3- diazaspiro[4.4]non-l-en-4-one hydrochloride (387 mg, 1.68 mmol) according to General Method 4. The crude residue was chromatographed on silica gel using 100:1.7 CH2CL/MeOH to afford 3E as a gum (830 mg, 93%): R^O.40, silica gel, 100:5 CH2Cl2/MeOH.
F. 4′-r(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2,– formyl-N-(3.4-dimethyl-5-isoxazolyl)-[l.l’-biphenyl1-2-sulfonamide
3E (830 mg, 1.20 mmol) was subjected to deprotection according to General Method 7. The crude residue was chromatographed on silica gel using 100:1.5 and then 100:4 CH2C12 /MeOH to afford the title compound as a gum (480 mg, 72%): R^O.16, silica gel, 100:5 CH.Cl MeOH.
Example 4
4′- . (2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non-l-en-3-yl)methyll -N-C3.4- dimethyl-5-isoxazolyl)-2,-[(3.3-dimethyl-2-oxo-l- pyrrolidinvDmethyll [1.1 ‘-biphenyl] -2-sulfonamide
To 3F (110 mg, 0.20 mmol) in CH2C12 (4 mL) was added 4-amino- 2,2-dimethylbutanoic acid hydrochloride (98 mg, 0.59 mmol) [Scheinmann, et al., J. Chem. Research (S), 414-415 (1993)] and 3A molecular sieves, followed by glacial acetic acid (35 mg, 0.59 mmol) and then sodium acetate (48 mg, 0.59 mmol). The mixture was stirred for 8 minutes, and NaB(AcO)3H (124 mg, 0.59 mmol) was then added. The reaction mixture was stirred at RT for 2 h, diluted with EtOAc and filtered through celite. The filtrate was washed with H2O and brine, dried and concentrated. This material was dissolved in CH2C12 (6 mL) and 1,3-diisopropylcarbodiimide (32 mg, 0.25 mmol) was added. The reaction mixture was stirred at RT for 2 h and diluted with CH2C12, washed with H2O and brine, dried and concentrated. The residue was purified by preparative HPLC to provide the title compound as a white solid (40 mg, 31%, for two steps): mp 104- 110°C. Analysis calculated for C36H45N5O5S • 0.8 H2O: Calc’d: C, 64.13; H, 6.97; N, 10.39; S, 4,75. Found: C, 64.18; H, 6.60; N, 10.23; S, 4.50.
Example 5
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2,-formyl-N-
(3,4-dimethyl-5-isoxazolyl)-[l,l’-biphenyl]-2-sulfonamide (Alternative
Preparation for 3F)
A. 2-[(2′-Bromo-5′-formyl)phenyl)1-1.3-dioxolane
DIBAL-H (1.0 M solution in toluene, 445 mL, 445 mmol, 1.1 eq) was added over 30 minutes to a solution of 2-[(2′-bromo-5′-cyano)phenyl)]-l,3- dioxolane (103 g, 404 mmol, 1.0 eq) [Zhang, H.-Y. et al., Tetrahedron, 50, 11339-11362 (1994)] in toluene (2.0 L) at -78 °C. The solution was allowed to warm to 0 °C. After 1 hour, a solution of Rochelle’s salt (125 g) in water (200 mL) was added, and the mixture was allowed to warm to room temperature and was stirred vigorously for 16 h. The organic layer was concentrated and the residue partitioned between ethyl acetate (1 L) and 1 N hydrochloric acid (800 mL). The organic layer was washed with saturated aqueous sodium bicarbonate (800 mL), dried over sodium sulfate, and then concentrated to give 70.5 g of crude 5A as a yellow solid, which was used without further purification.
B. 2-[(2′-Bromo-5′-hvdroxymethyl)phenyl)l-1.3-dioxolane
Sodium borohydride (3.66 g, 96.7 mmol, 0.5 eq) was added to a solution of crude 5A (49.7 g, approximately 193 mmol, 1.0 eq) in absolute ethanol (1300 mL) at 0 °C. After 2 hours, a solution of 10% aqueous sodium dihydrogen phosphate (50 mL) was added and the mixture was stirred and allowed to warm to room temperature. The mixture was concentrated, then partitioned between ethyl acetate (800 mL) and saturated aqueous sodium bicarbonate (500 mL). The organic layer was dried over sodium sulfate and concentrated to give 49.0 g of crude 5B as a yellow oil, which was used without further purification. C. 2-[(2′-Bromo-5′-bromomethyl)phenyl)]-l,3-dioxolane Triphenylphosphine (52.7 g, 199 mmol, 1.05 eq) was added in portions over 15 minutes to a solution of crude 5B (49.0 g, approximately 189 mmol, 1.0 eq) and carbon tetrabromide (69.0 g, 208 mmol, 1.1 eq) in THF at 0 °C. After 2 hours, saturated aqueous sodium bicarbonate solution (20 mL) was added, and the mixture was allowed to warm to room temperature and was then concentrated. Ether (500 mL) was added, and the resulting mixture was filtered. The filtrate was dried over magnesium sulfate and concentrated. The residue was chromatographed on silica gel (8:1 hexanes/ethyl acetate as eluant) to give 5C as a white solid (31.1 g, 51% yield from 2-[(2′-bromo-5′-cyano)phenyl)]-l,3-dioxolane).
D. 2-( 1 ,3-Dioxolan-2-yl)-4- [ (2-re-butyl-4-oxo- 1 ,3-diazaspiro [4.4] non- 1- en-3-yl)methyl] bromobenzene Sodium hydride (60% dispersion in mineral oil, 9.65 g, 241 mmol,
2.5 eq) was added in portions over 15 minutes to a mixture of 2-rc-butyl- l,3-diazaspiro[4.4]non-l-en-4-one hydrochloride (18.7 g, 96.5 mmol, 1.0 eq) in DMF (400 mL) at 0°C. The mixture was stirred and allowed to warm to room temperature over 15 minutes. To this mixture was added via canula a solution of 5C (31.1 g, 96.5 mmol, 1.0 eq) in DMF (100 mL). After 14 hours, the mixture was concentrated in vacuo and partitioned between ethyl acetate (500 mL) and 10% aqueous sodium dihydrogen phosphate (300 mL). The organic layer was dried over sodium sulfate and concentrated to give crude 5D as an orange oil (42.7 g), which was used without further purification.
E. 4-[(2-n-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2- formyl-bromobenzene
A solution of crude 5D (6.0 g, approximately 13.6 mmol, 1.0 eq) in THF (180 mL) and IN hydrochloric acid (30 mL) was heated at 65°C for 1.5 hours. The mixture was cooled and then treated with saturated aqueous sodium carbonate solution (75 mL) and ethyl acetate (200 mL). The organic layer was removed and dried over sodium sulfate, concentrated, and then further dried azeotropically with toluene to give 5E as a crude yellow oil (8.2 g) which contained a small amount of toluene. This material was used without further purification.
F. 4′-.(2-Butyl-4-oxo-1.3-diazaspiro■4.41non-l-en-3-yl)methyl1-2,– formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) f 1.1 ‘-biphenyl] -2-sulfonamide Palladium catalyzed Suzuki coupling of 5E and [2-[[(3,4-dimethyl-5- isoxazolyl) [(2-methoxyethoxy)methyl] amino] sulfonyl] phenyl]boronic acid was performed according to General Method 1 to yield 5F in 60% yield.
G. 4’-[ 2-Butyl-4-oxo-1.3-diazaspiro[4■41non-l-en-3-yl)methvn-2,– formyl-N-(3 ,4-dimethyl-5-isoxazolyl)- fi .1 ‘-biphenyl] -2-sulfonamide
Deprotection of 5F according to General Method 7 provided the title compound (5G = 3F) in 73% yield: R^0.2 (silica gel using CH2ClJ eOH [100:5]).
| Patent | Submitted | Granted |
|---|---|---|
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6638937] | 2002-10-03 | 2003-10-28 |
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6835741] | 2004-06-03 | 2004-12-28 |
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6852745] | 2004-07-01 | 2005-02-08 |
///////////BMS-248360, Preclinical, SARTAN, BMS, HYPERTENTION
CCCCC1=NC2(CCCC2)C(=O)N1CC3=CC(=C(C=C3)C4=CC=CC=C4S(=O)(=O)NC5=C(C(=NO5)C)C)CN6CCC(C6=O)(C)C
GSK2334470

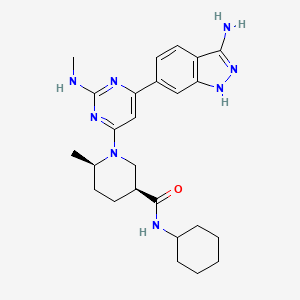
GSK2334470
GSK2334470; 1227911-45-6; GSK-2334470; GSK 2334470;
(3S,6R)-1-[6-(3-Amino-1H-indazol-6-yl)-2-(methylamino)-4-pyrimidinyl]-N-cyclohexyl-6-methyl-3-piperidinecarboxamide
(3S.6/?V1-r6-(3-Amino-1 H-indazol-6-ylV2-(methylaminoV4-pyrimidinyll-Λ/-cvclohexyl-6- methyl-3-piperidinecarboxamide
| Molecular Weight | 462.59 |
| Formula | C25H34N8O |
| CAS Number | 1227911-45-6 |
![]()
Phosphoinositide Dependent Kinase (PDK) 1 Inhibitors
[α]20D = – 32.6 o (c 1.17, MeOH)
[α] D = -27.6 (Concentration = 1.16, Solvent = Methanol)
SOL………DMSO to 100 mM
ethanol to 100 mM
nmr……http://www.chemietek.com/Files/Line2/Chemietek,%20GSK2334470%20(1),%20NMR-DMSO.pdf
http://file.selleckchem.com/downloads/nmr/S708702-GSK2334470-HNMR-Selleck.pdf


GSK2334470 is a potent and selective PDK1 (3-Phosphoinositide dependent protein kinase-1) inhibitor. GSK2334470 blocks the phosphorylation of known PDK1 substrates, but surprisingly find that the potency and kinetics of inhibition vary for different PDK1 targets. GSK2334470 subsequent activation of PDK1 substrates S6K1, SGK and RSK in HEK293, U87 and mouse embryonic fibroblast cell lines.
GSK2334470 inhibited activation of an Akt1 mutant lacking the PH domain (pleckstrin homology domain) more potently than full-length Akt1, suggesting that GSK2334470 is more effective at inhibiting PDK1 substrates that are activated in the cytosol rather than at the plasma membrane. GSK2334470 also suppressed T-loop phosphorylation and activation of RSK2 (p90 ribosomal S6 kinase 2), another PDK1 target activated by the ERK (extracellular-signal-regulated kinase) pathway.
GSK2334470 is a highly specific and potent inhibitor of PDK1 (3-Phosphoinositide dependent protein kinase-1) with IC50 of 10 nM. It does not suppress activity on other 96 kinases, including Aurora, ROCK, p38 MAPK and PI3K. GSK2334470 has been used in cells to ablate T-loop phosphorylation and activate SGK, S6K1 and RSK as well as suppress the activation of Akt.
PATENT
WO 2010059658
http://www.google.com/patents/WO2010059658A1?cl=en
Example 78
(3S.6/?V1-r6-(3-Amino-1 H-indazol-6-ylV2-(methylaminoV4-pyrimidinyll-Λ/-cvclohexyl-6- methyl-3-piperidinecarboxamide
To (3S,6R)-1-[6-(4-cyano-3-fluorophenyl)-2-(methylamino)-4-pyrimidinyl]-Λ/-cyclohexyl-6- methyl-3-piperidinecarboxamide (260 mg, 0.58 mmol) in EtOH (10 ml.) as a suspension at room temperature in a microwave vial was added hydrazine monohydrate (807 uL, 16.7 mmol, 30 equiv) in one portion. The mixture was capped and heated at 100 0C for 48 hours. A duplicate run was performed. The crude reactions from both runs were combined, and concentrated in vacuo. The residue was taken up in 10 ml. of water. The resulting suspension was sonicated briefly, and filtered. The solids collected were dried under vacuum at room temperature over P2O5 for 18 hours, and then at 65 0C under vacuum for another 18 hours to afford the title compound (410 mg) as a cream-colored solid. LC-MS (ES) m/z = 463 [M+H]+. 1H NMR (400 MHz, CD3OD): δ 1.16 – 1.32 (m, 3H),1.29 (d, J = 6.8 Hz, 3H), 1.34 – 1.45 (m, 2H), 1.65 – 1.68 (m, 1 H), 1.76 – 1.81 (m, 5H), 1.85 – 1.92 (m, 2H), 1.97 – 2.05 (m, 1 H), 2.35 – 2.42 (m, 1 H), 2.97 (s, 3H), 3.1 1 – 3.15 (m, 1 H),3.64 – 3.70 (m, 1 H), 4.45 – 4.65 (bs, 1 H), 4.72 – 4.92 (bs, 1 H), 6.45 (s, 1 H), 7.52 (dd, J =8.5, 1.14 Hz, 1 H), 7.75 (d, J = 8.3 Hz, 1 H), 7.85 (s, 1 H).
ntermediate 112
Cis- methyl-6-methyl-3-piperidinecarboxylate

A solution of cis-3-methyl 1-(phenylmethyl)-6-methyl-1 ,3-piperidinedicarboxylate (69 g, 237 mol) in EtOH (50 mL) and EtOAc (300 mL) was added to a slurry of 10% Pd/C (3.7 g) in EtOAc (30 mL) and EtOH (10 mL) EtOH under nitrogen in a Parr Shaker bottle. The mixture was hydrogenated under 65 psi at room temperature for 4 hours. The mixture was filtered through celite, and washed with EtOAc. The filtrate was concentrated in vacuo to give 37 g of the title compound as a liquid. LC-MS (ES) m/z = 158 [M+H]+.
Intermediate 113
Methyl (3S,6f?)-6-methyl-3-piperidinecarboxylate L-(+)-tartaric acid salt 
L-(+)-Tartaric acid salt A suspension of L-(+)-tartaric acid (39 g, 260 mmol, 1.05 equiv) in IPA (200 ml.) and water (13 mL) water was heated in a water bath at 600C until all dissolved. To this hot stirred solution was added neat racemic methyl (3S,6R)-6-methyl-3-piperidinecarboxylate (39 g, 248 mmol), followed by addition of 25 mL of IPA rinse. The resulting mixture was heated to 60 0C, resulting in a clear solution, and then cooled to room temperature, while the hot water bath was removed. This hot solution was seeded with a sample of methyl (3S,6R)-6-methyl-3-piperidinecarboxylate L-(+)-tartaric acid salt that had a chiral purity of 98% ee, and aged at ambient temperature (with the water bath removed) for 20 minutes. The mixture turned into an oily texture with seeds still present. To the mixture was added 5 mL of water, and heated in the warm water bath at 43 0C. The mixture became clear with the seeds still present. The heating was stopped, and the mixture was stirred in the warm water bath. After 20 minutes, the mixture gradually turned into a paste. After another 10 min, the water bath was removed, and the mixture was stirred at ambient temperature for another 1 hour. The resulting paste was filtered. The cake was washed with 50 mL of IPA, giving 62 g of wet solids. This cake was taken up in 150 mL of IPA and 8 mL of water, and stirred as a slurry while being heated in a water bath to 60 0C (internal temp 55 0C) for 5 minutes. The heating was turned off while the mixture was still stirred in the warm water bath. After 30 min, the mixture was filtered. The cake was washed with 100 mL of IPA. Drying under house vacuum at room temperature for 48 hours gave 46.7 g of solids. An analytical sample was derivatised to the corresponding N-Cbz derivative (as in the preparation of intermediate 1 11 ), which was determined by chiral HPLC (methods used to analyze the resolution of intermediate 11 1 above) to have 85% ee. This material was taken up in IPA (420 mL) and water (38 mL) as a suspension. The mixture was heated in a water bath to 65 0C, at which time the mixture became a clear solution. The heating bath was removed. The mixture was seeded and aged at ambient temp for 20 hours. The solids formed were filtered, and washed with 100 mL of IPA. The solids collected were dried under house vacuum at room temperature for 24 h, and then under vacuum at room temperature for another 24 hours to give 28.5 g of the title compound. An analytical sample was converted to the N-Cbz derivative. The ee was determined to be 97.7%. LC-MS (ES) m/z = 158 [M+H]+.
Intermediate 114 4,6-Dichloro-Λ/-methyl-2-pyrimidinamine

Methylamine (2M solution, 113 ml_, 217 mmol, 2.05 equiv) was charged to a 1 L 3-neck flask fitted with a magnetic stirrer and a thermometer. The mixture was chilled in an ice bath. To this stirred solution was added via addition funnel a solution of 4,6-dichloro-2-(methylsulfonyl)pyrimidine (25 g, 1 10 mmol) in EtOAc (250 ml.) portionwise over a 25 minutes period. The temp was between 5-10 0C. After completion of addition, the ice bath was removed, and the mixture was stirred for 1 hour at ambient temperature. LCMS showed conversion complete. The suspension was filtered, and washed with EtOAc. The filtrate was concentrated in vacuo. The residue was partitioned between water (100 ml.) and EtOAc (450 ml_). The organic was washed with brine, dried over MgSO4, filtered and concentrated in vacuo to give white solids, which were triturated in 150 ml. of CH2CI2. These solids were collected by filtration and washing with cold CH2CI2 (50 ml_). Drying under house vacuum at room temperature for 20 hours, and then high vacuum at room temperature for 3 hours gave 9.31 g of the title compound as a solid. LC-MS (ES) m/z = 179 [M+H]+.
Intermediate 121 (3S,6/?)-1-r6-Chloro-2-(methylamino)-4-pyrimidinyll-Λ/-cvclohexyl-6-methyl-3-piperidinecarboxamide

To a suspension of (3S,6/?)-1-[6-chloro-2-(methylamino)-4-pyrimidinyl]-6-methyl-3-piperidinecarboxylic acid (3.05 g, 10.71 mmol) in CH2CI2 (50 ml.) at room temperature was added Hunig’s base (2.70 ml_, 15.43 mmol, 1.3 equiv) and cyclohexylamine (1.60 ml_, 14.2 mmol, 1.2 equiv), and the resulting mixture was chilled in an ice bath. To this stirred solution was added HATU (4.96 g, 13.1 mmol, 1.1 equiv) in one portion, and the resulting suspension was stirred in the ice bath for 30 minutes. LCMS showed conversion complete. The mixture was diluted with CH2CI2 (50 ml.) and filtered through celite. The filtrate was washed water (2 X 25 ml.) and then brine. The organic was dried over Na2SO4, filtered, and concentrated in vacuo. Silica gel column chromatography using gradient elution of 1 % EtOAc in CHCI3 to 50% EtOAc in CHCI3 afforded the title compound (4.26 g) as a foam. LC-MS (ES) m/z = 366 [M+H]+.
PAPER
Journal of Medicinal Chemistry (2011), 54(6), 1871-1895.
http://pubs.acs.org/doi/full/10.1021/jm101527u

Phosphoinositide-dependent protein kinase-1(PDK1) is a master regulator of the AGC family of kinases and an integral component of the PI3K/AKT/mTOR pathway. As this pathway is among the most commonly deregulated across all cancers, a selective inhibitor of PDK1 might have utility as an anticancer agent. Herein we describe our lead optimization of compound 1toward highly potent and selective PDK1 inhibitors via a structure-based design strategy. The most potent and selective inhibitors demonstrated submicromolar activity as measured by inhibition of phosphorylation of PDK1 substrates as well as antiproliferative activity against a subset of AML cell lines. In addition, reduction of phosphorylation of PDK1 substrates was demonstrated in vivo in mice bearing OCl-AML2 xenografts. These observations demonstrate the utility of these molecules as tools to further delineate the biology of PDK1 and the potential pharmacological uses of a PDK1 inhibitor.
REFERENCES
Najafov, et al., Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem.J. (2011), 433 (2) 357.
For a PDK1 inhibitor, the substrate matters.
Knight ZA. Biochem J. 2011 Jan 15;433(2):e1-2. PMID: 21175429.
Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1.
Najafov A, et al. Biochem J. 2011 Jan 15;433(2):357-69. PMID: 21087210.

Jeffrey Axten
Director, Medicinal Chemistry, Virtual Proof of Concept DPU at GlaxoSmithKline
/////
GSK1904529A, GSK 4529

GSK1904529A, GSK 4529
GSK1904529A is a selective inhibitor of IGF1R with IC50 of 27 nM.
| 851.96 | |
| Formula | C44H47F2N9O5S |
| CAS Number | 1089283-49-7 |
N-(2,6-difluorophenyl)-5-[3-[2-[5-ethyl-2-methoxy-4-[4-(4-methylsulfonylpiperazin-1-yl)piperidin-1-yl]anilino]pyrimidin-4-yl]imidazo[1,2-a]pyridin-2-yl]-2-methoxybenzamide,
N-(2,6-Difluorophenyl)-5-[3-[2-[[5-ethyl-2-(methyloxy)-4-[4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl]phenyl]amino]-4-pyrimidinyl]imidazo[1,2-a]pyridin-2-yl]-2-(methyloxy)benzamide
NMR……http://www.abmole.com/download/gsk1904529a-hnmr.pdf

GSK1904529A, selectively inhibits IGF-IR and IR with IC50s of 27 and 25 nmol/L, respectively. It is a promising candidate for therapeutic use in solid and hematologic cancers. IC50s for GSK1904529A in tumor cell lines ranged from 35 nmol/L to >30 umol/L. The tumor histologic types showing the greatest sensitivity to this compound were Ewing’s sarcoma and multiple myeloma, where IC50s in three of five Ewing’s sarcoma cell lines were <100 nmol/L and IC50s in five of eight multiple myeloma cell lines were <200 nmol/L.
GSK1904529A is a small-molecule inhibitor of the insulin-like growth factor-I receptor (IGF-IR) with IC50 value of 27 nM 1.
GSK1904529A is a reversible and ATP-competitive inhibitor with Ki value of 1.6 nM. In NIH-3T3/LISN cells, GSK1904529A potently inhibited phosphorylation of IGF-IR with IC50 value of 22 nM. It also demonstrated to be a selective inhibitor since it showed poor inhibitory activity against 45 other serine/threonine and tyrosine kinases. When treated with whole-cell extracts, GSK1904529A significantly inhibited the ligand-induced phosphorylation of IGF-IR and decreased phosphorylation of downstream signaling including AKT, IRS-1 and ERK at concentrations > 0.01μM. GSK1904529A suppressed cell proliferation in a variety of tumor cells. The IC50 values for NCI-H929, TC-71, SK-N-MC, COLO 205, MCF7 and PREC are 81, 35, 43, 124, 137 and 68 nM, respectively. In COLO 205, MCF-7, and NCI-H929 cells, GSK1904529A treatment resulted in cell accumulation in G1 and decrease in S and G2-M phases. Moreover, in NIH-3T3/LISN xenograft model, once daily administration of GSK1904529A at 30 mg/kg inhibited 56% of tumor growt

…………..
Intermediates
 ,
,  ,
,  ,
, 
 ,
,

 ,
, ![]() ,
, ![]()
 ,
, 
 u can construct your synthesis
u can construct your synthesis
http://www.google.com/patents/US20080300242
Intermediate Example 2 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
Step A: Methyl 3-formyl-4-hydroxybenzoate
Methyl 4-hydroxybenzoate (3.00 g, 19.7 mmol) and magnesium chloride (2.81 g, 29.5 mmol) were stirred in 100 mL of acetonitrile. TEA (10.3 mL, 73.9 mmol) was added via syringe. Paraformaldehyde (12.0 g, 133 mmol) was added in a single portion and the reaction was heated to reflux. The reaction was stirred at reflux for 24 hours and cooled to rt. The reaction was quenched by the addition of approximately 100 mL of 1N HCl and poured into EtOAc. The layers were separated, and the organic layer was washed with brine. The combined aqueous layers were extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 2.06 g (58%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 11.54 (s, 1H), 10.27 (s, 1H), 8.21 (d, J=2.4 Hz, 1H), 8.03 (dd, J=8.8, 2.4 Hz, 1H), 7.07 (d, J=8.8 Hz, 1H), 3.79 (s, 3H).
Step B: methyl 3-formyl-4-(methyloxy)benzoate
Methyl 3-formyl-4-hydroxybenzoate (2.06 g, 11.4 mmol) and K2CO3 (2.36 g, 17.1 mmol) were stirred in 50 mL of DMF. Methyl iodide (1.42 mL, 22.8 mmol) was added via syringe, and the reaction was stirred for 6 hours at rt. The reaction was poured into H2O and diethyl ether, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted with diethyl ether. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo to afford 2.24 g of crude desired product. 1H NMR (400 MHz, DMSO-d6): δ 10.33 (s, 1H), 8.23 (d, J=2.2 Hz, 1H), 8.20 (dd, J=8.8, 2.2 Hz, 1H), 7.36 (d, J=8.8 Hz, 1H), 3.99 (s, 3H), 3.83 (s, 3H).
Step C: 2-(methyloxy)-5-[(methyloxy)carbonyl]benzoic acid
Crude methyl 3-formyl-4-(methyloxy)benzoate from the previous step was dissolved in 40 mL of dioxane with stirring. Sulfamic acid (5.87 g, 60.5 mmol) in 20 mL of H2O was added to the stirring solution. Sodium chlorite (1.68 g, 80% by weight, 18.6 mmol) in 20 mL of H2O was added dropwise via addition funnel. The reaction was stirred for 40 min and poured into EtOAc and H2O. The layers were separated, and the organic layer was washed with brine. The combined aqueous layers were extracted with EtOAc, and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The solid was transferred to an Erlenmeyer flask with the aid of 30-40 mL of DCM. Approximately 50 mL of hexanes was added. Air was blown over the solution to allow most of the DCM to evaporate. Diethyl ether was added (20-30 mL), and the suspension was filtered. The solid was washed with hexanes, collected, and dried to afford 1.96 g (82% over 2 steps) of the desired compound. 1H NMR (400 MHz, DMSO-d6): δ 12.92 (brs, 1H), 8.22 (d, J=2.2 Hz, 1H), 8.07 (dd, J=8.8, 2.2 Hz, 1H), 7.24 (d, J=8.8 Hz, 1H), 3.88 (s, 3H), 3.82 (s, 3H).
Step D: methyl 3-{[(2,6-difluorophenyl)amino]carbonyl}-4-(methyloxy)benzoate
2-(Methyloxy)-5-[(methyloxy)carbonyl]benzoic acid (1.96 g, 9.33 mmol) was suspended in 60 mL of DCM with stirring. DMF (0.036 mL, 0.46 mmol) was added via syringe. Oxalyl chloride (7.0 mL, 2.0M in dichloromethane, 14 mmol) was added dropwise via addition funnel. The addition funnel was rinsed with 10 mL of DCM. The reaction was stirred for 2 hours and concentrated in vacuo. The resultant solid was further dried under high vacuum pressure. The solid was dissolved in 60 mL of DCM with stirring. Pyridine (3.8 mL, 47 mmol), (4-dimethylamino)pyridine (0.0570 g, 0.467 mmol), and 2,6-difluoroaniline (3.0 mL, 28 mmol) were added to the solution. The reaction was stirred for 18 hours and poured into 1N HCl. The layers were separated, and the aqueous layer was washed once with DCM and once with diethyl ether. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 1.56 g (52%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 9.81 (s, 1H), 8.31 (d, J=2.0 Hz, 1H), 8.10 (dd, J=8.8, 2.0 Hz, 1H), 7.38 (m, 1H), 7.31 (d, J=88 Hz, 1H), 7.22-7.13 (m, 2H), 3.97 (s, 3H), 3.82 (s, 3H).
Step E: 5-[(2-Chloro-4-pyrimidinyl)acetyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide and 5-[(E)-2-(2-chloro-4-pyrimidinyl)-1-hydroxyethenyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
Methyl 3-{[(2,6-difluorophenyl)amino]carbonyl}-4-(methyloxy)benzoate (1.56 g, 4.86 mmol) was dissolved in 50 mL of THF with stirring and cooled to 0° C. Lithium bis(trimethylsilyl)amide (14.6 mL, 1.0M in THF, 14.6 mmol) was added slowly via syringe. 2-Chloro-4-methylpyrimidine (0.750 g, 5.83 mmol) was dissolved in 10 mL of THF and added dropwise via addition funnel. The addition funnel was rinsed with 10 mL of THF. The reaction was stirred at 0° C. for 1 hour and quenched with saturated ammonium chloride solution. The mixture was poured into H2O and EtOAc, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 1.26 g (62%) of the desired product. The proton NMR is a mixture of the keto and enol tautomers (˜2:1). 1H NMR (400 MHz, DMSO-d6): δ 13.58 (s, 1H, enol), 9.83 (s, 1H, keto), 9.82 (s, 1H, enol), 8.72 (m, 1H, keto), 8.54 (m, 1H, enol), 8.34 (s, 1H, keto), 8.22 (m, 1H, both), 8.06 (m, 1H, enol), 7.56 (m, 1H, keto), 7.42-7.31 (m, 2H, both+1H, enol), 7.22-7.14 (m, 2H, both), 6.55 (s, 1H, enol), 4.66 (s, 2H, keto), 4.00 (s, 3H, keto), 3.97 (s, 3H, enol).
Step F: 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
A tautomeric mixture of 5-[(2-Chloro-4-pyrimidinyl)acetyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide and 5-[(E)-2-(2-chloro-4-pyrimidinyl)-1-hydroxyethenyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (1.26 g, 3.02 mmol) was dissolved in 60 mL of DCM with stirring. NBS (0.538 g, 3.02 mmol) was added in a single portion. The reaction was stirred for 20 minutes and concentrated in vacuo. The residue was dissolved in 60 mL of dioxane with stirring, and 2-aminopyridine (0.853 g, 9.06 mmol) was added in a single portion. The reaction was heated at 60° C. with an oil bath for 24 hours and cooled to rt. The reaction was stirred at rt for an additional 40 hours. The reaction was poured into half-saturated NaHCO3 solution and EtOAc, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted twice with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. Impure fractions were concentrated and further purified by flash chromatography. The combined clean fractions (by TLC) from both runs were combined and concentrated in vacuo to afford 1.07 g (72%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 9.80 (s, 1H), 9.40 (d, J=7.0 Hz, 1H), 8.57 (d, J=5.1 Hz, 1H), 8.10 (d, J=1.5 Hz, 1H), 7.84-7.77 (m, 2H), 7.57 (m, 1H), 7.39 (m, 1H), 7.33-7.26 (m, 2H), 7.24-7.14 (m, 3H), 3.99 (s, 3H).
Step A: 1,1-dimethylethyl 4-(methylsulfonyl)-1-piperazinecarboxylate
To 1,1-dimethylethyl 1-piperazinecarboxylate (568 g, 3.05 mol) in DCM (4 L) was added TEA (617 g, 6.10 mol). After stirring for 10 min at 0° C., methanesulfonyl chloride (384 g, 3.35 mol) was added via addition funnel. The mixture was stirred at rt overnight. The mixture was poured into H2O (1 L) and extracted with DCM (1 L). The organic layer was separated, washed with H2O (1 L), dried (Na2SO4), and rotovapped down to provide the title compound of step A (720 g, 2.72 mol, 90%) which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 1.44 (s, 9H), 2.76 (s, 3H), 3.11-3.17 (m, 4H), 3.50-3.53 (m, 4H).
Step B: 1-(methylsulfonyl)piperazine hydrochloride
To 1,1-dimethylethyl 4-(methylsulfonyl)-1-piperazinecarboxylate (360 g, 1.36 mol) in MeOH (1 L) was added HCl (6 M in MeOH, 2 L) dropwise. The mixture was stirred at rt for 1 h. About 1 L of MeOH was rotovapped off. The resultant precipitate was filtered, washed with MeOH, and dried on high vacuum to provide the title compound of Step B (A combination of 2 batches, 570 g) which was used without further purification. 1H NMR (400 MHz, D2O) δ 2.95 (s, 3H), 3.27-3.29 (m, 4H), 3.42-3.46 (m, 4H).
Step C: 1-(methylsulfonyl)-4-(4-piperidinyl)piperazine dihydrochloride
To 1-(methylsulfonyl)piperazine hydrochloride (150 g, 632 mmol) in DCE (3.5 L) was added TEA (192 g, 1.90 mol). The mixture was stirred at rt for 1 h and then acetic acid (94.8 g, 1.58 mol) and 1,1-dimethylethyl 4-oxo-1-piperidinecarboxylate (251 g, 1.26 mol) was added. After stirring another h, the reaction was cooled with an ice water bath and NaBH(OAc)3 (294 g, 1.39 mol) was added in four portions. The mixture was stirred overnight at rt. The reaction mixture was neutralized with saturated Na2CO3 to pH 8-9. The organic phase was washed with brine and H2O, dried (Na2SO4), and rotovapped down to provide the crude Boc-protected amine (A combination of 3 batches, 720 g). This amount was split into 2 batches and used without further purification. To 1,1-dimethylethyl 4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinecarboxylate (360 g, 1.04 mol) in MeOH (1 L) was added HCl (6 M in MeOH, 2 L). The mixture was stirred at rt for 30 min. About 1 L of MeOH was rotovapped off. The resultant precipitate was filtered, washed with MeOH, and dried on high vacuum to provide the title compound of Step C (A combination of 2 batches, 600 g, 1.87 mol, 89% over 2 steps). 1H NMR (400 MHz, D2O) δ 1.87-1.91 (m, 2H), 2.33-2.36 (m, 2H), 2.97 (s, 3H), 2.99-3.05 (m, 2H), 3.45-3.59 (m, 11H).
Step A: 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine
A mixture of 1-ethyl-2-fluoro-4-(methyloxy)-5-nitrobenzene (Example 187, step C) (0.93 g, 4.67 mmol), 1-(methylsulfonyl)-4-(4-piperidinyl)piperazine (Example 204, step C) (1.16 g, 4.67 mmol) and K2CO3 (0.774 g, 5.60 mmol) in DMSO (20 mL) was heated at 90° C. for 48 h. The reaction had not progressed sufficiently so the reaction was then heated at 120° C. for an additional 4 h. The reaction was cooled to rt, poured into H2O and extracted with DCM. Some saturated brine solution was added and the resultant was exhaustively extracted with DCM. The combined organics were washed with H2O then dried over MgSO4. The resultant solution was concentrated onto silica and purified by flash chromatography to afford 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine (1.12 g, 56%). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.73-7.80 (m, 1H), 6.75 (s, 1H), 3.91 (s, 3H), 3.23-3.30 (m, 1H), 3.05-3.19 (m, 3H), 2.87 (s, 2H), 2.70-2.84 (m, 2H), 2.53-2.67 (m, 5H), 1.77-1.94 (m, 2H), 1.48-1.67 (m, 2H), 1.19 (t, J=7.42 Hz, 3H).
Step B: 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline
A mixture of 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine (1.12 g, 2.63 mmol) and sulfided platinum on carbon (0.410 g, 0.105 mmol) in EtOAc (40 mL) was sealed in a round bottom flask with a rubber septum. The reaction mixture was purged with N2 gas and then a balloon of H2 gas was connected and the vessel was flushed with the H2 gas. The reaction was stirred at rt for 2 d. TLC analysis showed the complete consumption of the starting nitro compound so the reaction mixture was filtered through celite to remove the catalyst. The filtrate was concentrated onto silica gel and purified by flash chromatography to afford 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (0.479 g, 46%).
1H NMR (400 MHz, DMSO-d6) δ ppm 6.60 (s, 1H), 6.46 (s, 1H), 4.35 (br. s., 2H), 3.71 (s, 3H), 3.03-3.16 (m, 4H), 2.81-2.93 (m, 5H), 2.56-2.68 (m, 6H), 2.29-2.42 (m, 1H), 1.72-1.89 (m, 2H), 1.44-1.62 (m, 2H), 1.09 (t, J=7.51 Hz, 3H).
Example 237 N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide
A mixture of 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (Intermediate Example 2) (0.60 g, 1.22 mmol), 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (Example 206, Step B) (0.48 g, 1.22 mmol) and HCl (4N,1,4-Dioxane, 0.61 mL, 2.44 mmol) in trifluoroethanol (15 mL) was heated at 170° C. for 40 min in the microwave. The reaction mixture was concentrated onto silica gel and purified by flash column chromatography. Recrystallization from DCM and EtOH afforded the title compound N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide (0.61 g, 56%).
1H NMR (400 MHz, DMSO-d6)
δ ppm 9.80 (s, 1H), 9.36 (br. s., 1H), 8.50 (s, 1H), 8.26 (d, J=5.22 Hz, 1H), 8.12 (d, J=2.11 Hz, 1H), 7.80 (dd, J=8.80, 2.02 Hz, 1H), 7.71 (d, J=9.07 Hz, 1H), 7.53 (s, 1H), 7.36-7.50 (m, 2H), 7.30 (d, J=8.80 Hz, 1H), 7.14-7.25 (m, 2H), 6.91-7.00 (m, 1H), 6.83 (s, 1H), 6.58 (d, J=5.22 Hz, 1H), 4.00 (s, 3H), 3.80 (s, 3H), 3.08-3.15 (m, 4H), 3.00-3.07 (m, 2H), 2.88 (s, 3H), 2.67-2.76 (m, 2H), 2.61-2.66 (m, 4H), 2.56 (q, J=7.51 Hz, 2H), 2.38-2.46 (m, 1H), 1.80-1.91 (m, 2H), 1.50-1.68 (m, 2H), 1.11 (t, J=7.51 Hz, 3H).
MS (M+H, ES+) 852.
Separately, the Title Compound was Prepared in the Following Manner:
A mixture of 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (Intermediate Example 2) (23.0 g, 46.8 mmol), 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (Example 206, Step B) (18.6 g, 46.8 mmol) and HCl (4N,1,4-Dioxane, 23.4 mL, 93.6 mmol) in trifluoroethanol (200 mL) was heated in a sealed vessel at 85° C. for 48 h. After cooling to rt, the reaction mixture was treated with an excess of 7N NH3 in MeOH and then subjected to filtration. The filtrate was concentrated onto silica gel and purified by flash chromatography. The chromatographed product was dissolved in DCM and treated with an excess of diethyl ether. The resultant bright yellow precipitate was collected by filtration and then recrystallized from DCM and EtOH to afford the title compound N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide (28.2 g, 67%).
……………..
Discovery and optimization of imidazo[1,2-a]pyridine inhibitors of insulin-like growth factor-1 receptor (IGF-1R)
Bioorg Med Chem Lett 2009, 19(3): 1004……http://www.sciencedirect.com/science/article/pii/S0960894X08014376


Scheme 1.
Reagents and conditions: (a) (ClCO)2, DMF, CH2Cl2; (b) 2,6-difluoroaniline, pyridine, CH2Cl2 (84%, 2 steps); (c) LiN(SiMe3)2, THF (83%); (d) NBS, CH2Cl2, then 2-aminopyridine, dioxane, 60 °C (77%); (e) HCl or p-TSA·H2O, trifluoroethanol or isopropanol, 80–100 °C or 140–180 °C (μw) (50–90%).
References
Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin-like growth factor-I receptor tyrosine kinase.
Sabbatini et al. Clin Cancer Res. 2009 May 1;15(9):3058-67. PMID: 19383820.
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
/////////////GSK1904529A, IGF1R, GSK 4529, preclinical
What is SBM-TFC-039 an SGLT Inhibitor from Sirona Biochem !!
A new “flozin” seems to me appearing on the horizon in form of SBM-TFC-039 an SGLT Inhibitor from Sirona Biochem, picked up a list from WO 2012160218, from TFChem…….see link , Sirona Biochem Announces SGLT2 Inhibitor and Skin Lightening Patent Granted, 29 Jun 2015, Patent entitled “Family of aryl, heteroaryl, o-aryl and o-heteroaryl carbasugars”
This led me to search, “Family of aryl, heteroaryl, o-aryl and o-heteroaryl carbasugars”
WO 2012160218 A1, IN 2013-DN10635, CN 103649033Tf化学公司
| Applicant | Tfchem |

List above as in http://www.google.com/patents/WO2012160218A1?cl=en
FROM THE ABOVE LIST, SBM-TFC-039 MAY BE PREDICTED/OR AS SHOWN BELOW
COMPD 16 as in/WO2012160218
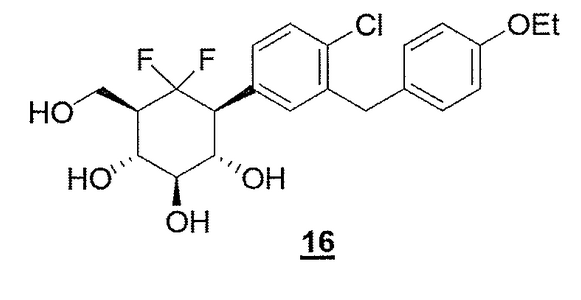
COMPD 16, PREDICTED/LIKELY SBM-TFC-039 has CAS 1413373-30-4, name D-myo-Inositol, 1-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1,2,3-trideoxy-2,2-difluoro-3-(hydroxymethyl)-
Just scrolling through the patent gave me more insight
MORE EVIDENCE….http://www.google.com/patents/WO2012160218A1?cl=en, this patent descibes compd 16 as follows
Compound 16 according to the invention has been compared to Dapaglifozin to underline the improvement of the duration of action, i.e. the longer duration of glucosuria, of the compound when the intracyclic oxygen atom of the glucose moiety is replaced by a CF2 moiety.

This assay has been carried out at a dose of 3 mg/ kg.
The results obtained are presented on Figure 5. It appears thus that 16 (3 mg/kg) triggered glucosuria that lasted beyond 24 hours compared to Dapagliflozin.
• Compound 16 according to the invention has been compared to the compound 9 of WO 2009/1076550 to underline the improvement of the duration of action of the compound when a mimic of glucose bearing a CH-OH moiety instead of the intracyclic oxygen atom is replaced by a mimic of glucose bearing a CF2 in place of the CH-OH moiet .

%7D)
| Company | Sirona Biochem Corp. |
| Description | Sodium-glucose cotransporter 2 (SGLT2) inhibitor |
| Molecular Target | Sodium-glucose cotransporter 2 (SGLT2) |
| Mechanism of Action | Sodium-glucose cotransporter 2 (SGLT2) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Preclinical |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
| Regulatory Designation | |
| Partner | Shanghai Fosun Pharmaceutical Group Co. Ltd. |
SBM-TFC-039
PATENT
WO 2012160218
http://www.google.com/patents/WO2012160218A1?cl=en
Examples within this first subclass include but are not limited to:

Synthesis of compound 8
C35H34O5 M = 534.64 g.mol“
Mass: (ESI ): 535.00 (M + H); 552.00 (M + H20); 785.87; 1086.67 (2M + H20)

A.

Procedure A:
To a solution of 4 (10.5g, 15.89mmol, leq) in toluene (400mL) were added 18-crown-6 (168mg, 0.64mmol, 0.04eq) and potassium carbonate (6.69g, 48.5mmol, 3.05eq.). The mixture was stirred overnight at room temperature, and then the remising insoluble material was filtered off and washed with toluene. The filtrate and the washings were combined, washed with 2N hydrochloric acid aqueous solution followed by saturated sodium hydrogencarbonate aqueous solution, dried over sodium sulphate, filtered and concentrated under reduced pressure. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford cyclohexenone 8 (4.07g; 48% yield) as yellowish oil.
Procedure B:
A solution of 7 (3.27g, 5.92mmol, leq) in pyridine (14mL) was cooled to 0°C before POCl3 (2.75mL, 29.6mmol, 5eq) was added dropwise. The mixture was stirred at this temperature for 10 min before the cooling bath was removed. The reaction mixture was stirred overnight at room temperature before being re-cooled to 0°C. POCI3 (2.75mL, 29.6mmol, 5eq) was added once again trying to complete the reaction. The mixture was stirred for an additional 20h at room temperature before being diluted with Et20 (20mL) and poured onto crushed ice. 1M HC1 aqueous solution (lOOmL) was added, and the mixture was extracted with Et20 (200mL & l OOmL). The combined organic extracts were washed with brine (lOOmL), dried over sodium sulphate, filtered and concentrated before being purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 80:20) to afford compound 8 (1.46g, 46% yield) as an orange oil. Synthesis of compound 9
C15H12BrC102 M = 339.61 g.moF1
Mass: (GC-MS): 338-340

The synthesis of this product is described in J. Med. Chem. 2008, 51, 1 145—1149.Synthesis of compound 10
C15H14B1CIO M = 325.63 g.mof1

10 The synthesis of this product is described in J. Med. Chem. 2008, 51, 1145-1 149.
Synthesis of compound 11
C50H49CIO6 M = 781.37 g.moF1
Mass: ESI+): 798.20 (M + H20)

Under inert atmosphere, Mg powder (265mg, 10.9mmol, 2.4eq) was charged into a three necked flask, followed by addition of a portion of 1/3 of a solution of the 4- bromo-l-chloro-2-(4-ethylbenzyl)benzene (2.95g, 9.1mmol; 2eq) in dry THF (25mL) and 1 ,2-dibromoethane (10 mol % of Mg; 85mg; 0.45mmol). The mixture was heated to reflux. After the reaction was initiated (exothermic and consuming of Mg), the remaining solution of 2-(4-ethylbenzyl)-4-bromo-l-chlorobenzene in dry TFIF was added dropwise. The mixture was then allowed to react for another one hour under gentle reflux until most of the Mg was consumed.
The above Grignard reagent was added dropwise into the solution of cyclohexenone 8 (2.42g, 4.53mmol, leq) in dry THF (25mL) under inert atmosphere at room temperature (about 25°C), then allowed to react for 3h. A saturated aqueous solution of ammonium chloride was added into the mixture to quench the reaction. The mixture was extracted with Et20, washed with brine, dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 100:0 to 80:20) to afford the target compound 11 as a yellow oil (3.01g, 86%).
Synthesis of compound 12
C5oH49C105 M = 765.37 g.mol“1
+): 782.13 (M + H20)

Triethylsilane (0.210mL, 1.30mmol, 3eq) and boron-trifluoride etherate (48% BF3, O. l lOmL, 0.866mmol, 2eq) were successively added into a solution of alcohol 1 1 (338mg, 0.433mmol, leq) in dichloromethane (5mL) under inert atmosphere at -20°C. After stirring for 2.5h, a saturated aqueous solution of sodium chloride was added to quench the reaction. The mixture was extracted with CH2C12 (10mLx3) and the organic layer was washed with brine, dried over Na2S04, filtrated and concentrated. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 9.8:0.2 to 8:2) to afford the target compound 12 as a white powder (278 mg, 0.363mmol, 84%).
Synthesis of compound 13
C5oH5tC106 M = 783.39g.moF1
Mass: (ESI+): 800 (M + H20); 1581 (2M + H20)

Under inert atmosphere, borane-dimethyl sulfide complex (2M in THF, 16.7mL, 33mmol, 10.5eq) was added to a solution of 12 (2.41g; 3.15mmol, leq) in dry THF (lOOmL) cooled to 0°C. The reaction mixture was then refluxed for lh,cooled to 0°C and treated carefully with sodium hydroxide (3M in H20, 10.5mL, 31.5mmol, lOeq), followed by hydrogen peroxide (30% in H20, 3.2mL, 31.5mmol, l Oeq) at room temperature (above 30°C). The mixture was allowed to react overnight at room temperature (~25°C) before a saturated aqueous solution of ammonium chloride was added to quench the reaction. The mixture was extracted with ethyl acetate and the organic layer was washed with brine, dried over Na2S04, filtered, and concentrated. The residue was purified by silica gel chromatography (cyclohexane/ethyl acetate 97:3 to 73:27) to afford the desired compound 13 (1.05g; 43%) as a yellowish oil.
Synthesis of compound 14
C50H49CIO6 M = 781.37g.mol“1
Mass: (ESI+): 798 (M + H20); 1471; 1579 (2M + H20)

13 14
Dess-Martin periodinane (81mg; 1.91mmol; 1.5eq) was added portion wise to a solution of alcohol 13 (l .Og; 1.28mmol, leq) in anhydrous dichloromethane (20mL) at 0°C. The reaction was then stirred overnight at room temperature before being quenched with IN aqueous solution of sodium hydroxide. The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 82: 18), to afford the target ketone 14 (783mg, 79% yield) as a colorless oil. Synthesis of compound 15
C5oH49ClF206 M = 803.37g.moF1
19 F NMR (CDCU, 282.5MHz): -100.3 (d, J=254Hz, IF, CFF); -1 13.3 (td, Jl=254Hz, J2=29Hz, IF, CFF).
Mass: (ESI+): 820.00 (M+H20)

14 15
A solution of ketone 14 (421mg, 0.539mmol, leq) in DAST (2mL, 16.3mmol, 30eq.) was stirred under inert atmosphere at 70°C for 12h. The mixture was then cooled to room temperature and dichloromethane was added. The solution was poured on a mixture of water, ice and solid NaHC03. Agitation was maintained for 30min while reaching room temperature. The aqueous layer was extracted with dichloromethane and the organic phase was dried over Na2S04, filtered and concentrated. The crude product was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford the desired compound 15 as a yellowish oil ( 182mg, 42% yield).
Synthesis of compound 16
C22H25CIF2O5 M = 442.88g.mor1
19 F NMR (MeOD, 282.5MHz): -96.7 (d, J=254Hz, IF, CFF); 12.2 (td,
Jl=254Hz, J2=28Hz, IF, CFF).
Mass: (ESI+): 465.3 (M+Na)

o-Dichlorobenzene (0.320mL, 2.82mol, lOeq) followed by Pd/C 10% (0.342g, 0.32mol, l .leq) were added to a solution of 15 (228mg, 0.28mmol, leq) in a mixture of THF and MeOH (2: 1, v/v, 160mL). The reaction was placed under hydrogen atmosphere and stirred at room temperature for 2h. The reaction mixture was filtered and concentrated before being purified on silica gel chromatography (dichloromethane/methanol 100: 1 to 90: 10) to afford compound 16 (105mg, 83% yield).
Sirona Biochem’s SGLT Inhibitor Performs Better Than Johnson and Johnson’s SGLT Inhibitor, According to Study
Vancouver, British Columbia – December 7, 2012 – Sirona Biochem Corp. (TSX-V: SBM), announced its sodium glucose transporter (SGLT) inhibitor for Type 2 diabetes reduced blood glucose more effectively than Johnson and Johnson’s canagliflozin, an advanced SGLT inhibitor being considered for market approval in Europe and the U.S. Studies compared Sirona Biochem’s SGLT Inhibitor, SBM-TFC-039, with canagliflozin and were conducted on Zucker Diabetic Fatty (ZDF) rats.
In the study, SBM-TFC-039 significantly and rapidly reduced blood glucose levels at a dose of 1.0 mg/kg. Six (6) hours after administration, SBM-TFC-039 reduced blood glucose by 44% compared to canagliflozin at 26%. SBM-TFC-039 also had a longer duration of effect than canagliflozin. At 36 and 48 hours after treatment, SBM-TFC-039, at a dose of 1.0 mg/kg, was still effective at reducing blood glucose, whereas canagliflozin lost its effect after 36 hours. Studies were conducted at the Institut Universitaire de Cardiologie et de Pneumologie de Québec (IUCPQ) by Principal Investigator Dr. Denis Richard, Research Chair on Obesity and Professor, Faculty of Medicine, Department of Anatomy & Physiology at Laval University.
“SGLT Inhibitors are a ground-breaking new treatment for Type 2 diabetes and these results demonstrate that SBM-TFC-039 will be a significant competitor for other SGLT Inhibitors,” said Neil Belenkie, Chief Executive Officer of Sirona Biochem. “The first SGLT Inhibitor,Forxiga™, was approved last month by the European Commission. We believe there is tremendous market potential worldwide for SGLT Inhibitors in the treatment of diabetes.”
SBM-TFC-039 is a sodium glucose transporter (SGLT) inhibitor. SGLT inhibitors are a new class of drug candidates for the treatment of diabetes. In the kidneys, SGLT inhibitors reduce the reabsorption of glucose into the bloodstream by eliminating excess glucose into the urine.
About Sirona Biochem Corp.
Sirona Biochem is a biotechnology company developing diabetes therapeutics, skin depigmenting and anti-aging agents for cosmetic use, biological ingredients and cancer vaccine antigens. The company utilizes a proprietary chemistry technique to improve pharmaceutical properties of carbohydrate-based molecules. For more information visit www.sironabiochem.com.
![]()
Laboratory – France
TFChem
Voie de l’innovation
Pharma Parc II
Chaussée du Vexin
27100 Val de Reuil
France
Phone:+33(0)2.32.09.01.16
Fax:+33(0)2.32.25.07.64



……………………………………………………………………………….
Shanghai Fosun Pharmaceutical Group Co. Ltd.

![]()
//////
AZD 3264 an IKK2 Inhibitor from Astra Zeneca

AZD 3264
MW 441.50
CAS 1609281-86-8
Inhibition of IkB-kinase IKK2 has been identified as one of the novel pathways to treat inflammatory conditions such as asthma, chronic pulmonary obstructive disorder (COPD) and rheumatoid arthritis
……………………..
PATENT
WO 2003010158
https://www.google.com/patents/WO2003010158A1?cl=en

The synthesis began with the aromatic nucleophilic substitution reaction of 2-fluorobromobenzene (2) with (S)-N-Boc-3-pyrrolidinol 3 to give the bromo intermediate 4, which was borylated via halogen metal exchange using n-hexLi in THF followed by treatment with triisopropyl borate and acidic work-up to give the boronic acid intermediate 5. Suzuki coupling of the boronic acid 5 with bromothiophene 6(2)afforded the intermediate 7. Intermediate 7 was subjected to regioselective bromination using bromine in acetic acid. This reaction was nonregioselective and yielded 17% of the required isomer 8. The bromo compound 8 was coupled with isoxazole boronate ester 9 by another Suzuki reaction to get the title compound. The overall yield of the synthesis was <6%.


………………………..
PAPER
http://pubs.acs.org/doi/full/10.1021/op500105n

An efficient and scalable synthesis of AZD3264 is described in which the differential reactivities of various halogen atoms have been employed. The process involves five linear chemical steps with three isolated stages starting from commercially available fragments.
What is SMU-B?

cas 1253286-89-3
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 5-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B
or is it
1253286-90-6
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 6-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B

A series of novel aminopyridyl/pyrazinyl-substituted spiro[indoline-3,4′-piperidine]-2-ones were designed, synthesized, and tested in various in vitro/in vivo pharmacological and antitumor assays. 6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B) was identified as a potent, highly selective, well-tolerated, and orally efficacious c-Met/ALK dual inhibitor, which showed pharmacodynamics effect by inhibiting c-Met phosphorylation in vivo and significant tumor growth inhibitions (>50%) in GTL-16 human gastric carcinoma xenograft models.
see..http://pubs.acs.org/doi/abs/10.1021/ml400203d
cas 1253286-90-6
6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B)
SEE

1_4,3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-nitro-approved P set

obtained in Step 1-3 (IS) -I- (2,6- dichloro-3-fluorophenyl) ethanol (2. 09g, IOmmol) was dissolved in dry THF (80 ml). Then, at room temperature under a nitrogen atmosphere, a solution of 3-hydroxy-2-nitro-pyridine (1.54g, llmmol) and triphenylphosphine (3. 409g, 13mmol), and so is completely dissolved, cooled to 0 ° C, was added Diisopropyl azodicarboxylate (DIAD, 2.63g, 13mmol), After the addition, the mixture was stirred at 0 ° C for 16 hours, the solvent was removed by rotary evaporation and the oily residue was purified by silica gel column chromatography (petroleum ether / ethyl acetate : 4/1) to give the desired product as a white solid (3. 046g, yield: 92%) o 1H-NMR (CDClySOOMHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), 6 . 10 (q, J = 6. 4Hz, 1H), 7. 09 (dd, J = 7. 6,8. 8Hz, 1H), 7. 21 (dd, J = 8. 4, I. 2Hz, 1H ), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H),
7. 37 (dd, J = 4. 8,8. OHz, 1H), 8. 04 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 330 94 [M + H, 35C1,35Cl], 332. 92 [M + H, 35Cl, 37Cl].
1_5,3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere based grant given P

to take steps 1-4 to get the 3 – [(lR) _l- (2,6_ dichloro-3-fluorophenyl) ethoxy] -2_ nitro than Li Jie (2. 649g, 8mmol) was dissolved in ethanol (15mL) was added iron powder (3. 575g, 64mmol) were mixed under nitrogen with vigorous stirring at 90 ° C oil bath, was added via syringe 0.8mL IM HCl (aq), after 10 minutes, was added 0. 8mL IMHCl (aq). Stirring was continued for 30 minutes, TLC showed the reaction. Cooled to room temperature, filtered through Celite, the filter residue washed with ethanol (3X IOmL). The combined organic phase was removed by rotary evaporation of the solvent gave the desired product as a light brown solid (2. 41g, yield: 100%) o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H ), 5. 03 (s, br, 2H), 6. 01 (q, J = 6. 8Hz, 1H), 6. 47 (dd, J = 4. 8,7. 6Hz, 1H), 6. 70 (d, J = 8. OHz, 1H), 7. 05 (t, J = 8. 8Hz, 1H), 7. 28 (dd, J = 4. 0,8. 0Hz, 1H), 7. 57 ( d, J = 5.2Hz, lH). Mass spectrum m / z:. 301 00 [M + H, 35Cl, 35Cl], 302. 77 [M + H, 35Cl, 37Cl].
l-6,5_ desert _3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere base than Li Jie

The steps 1-5 obtained 3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl atmosphere than Li Jie (1.506g, 5mmol) dissolved in acetonitrile (20mL) in. Then, at 0 ° C and the degree of stirring in added portionwise N- bromosuccinimide (0.908g, 5. Lmmol), After the addition, stirring was continued for 30 minutes. The solvent was removed by rotary evaporation, the crude product was obtained as a white solid was the desired product (1.045g, yield: 55%) was purified by column chromatography on silica gel. 1H-NMR (⑶Cl3,500MHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H), 4 85 (s, br, 2H), 6 98 (q, J = 6. 8Hz.. , 1H), 6. 82 (d, J =
2. 0Hz, 1H), 7. 08 (t, J = 8. 4Hz, 1H), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H), 7. 65 (d, J = 2 . OHz, 1H). Mass spectrum m / z:… 378 84 [M + H, 35Cl, 35Cl, 79Br], 380 82 [M + H, 35Cl, 35Cl, 81Br or 35Cl, 37Cl, 79Br], 382 80 [M + H, 35Cl , 37Cl, 81Bror 37Cl, 37Cl, 79Br].
Step 2, I ‘- methyl-5- (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one

2-1,5- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] _2_ one

[0300] 5-bromo – indol-2-one (I. 272g, 6mmol) was suspended THF (15mL) at, and cooled to -78 ° C, added dropwise with stirring IM NaN (SiMe3) THF solution of 2 (30mL, 30mmol). After the addition was stirred at _78 ° C 30 min, then 2-chloro -N- (2- chloro-ethyl) -N- methyl-ethylamine hydrochloride solid (I. 155g, 6mmol). After the addition stirring was continued for 30 minutes, then warmed to room temperature and stirred for two days. TLC showed the reaction was completed, to the pink suspension was carefully added aqueous 4M hydrochloric acid (IOmL), and then adjusted with concentrated aqueous ammonia to pH ^ 9, and extracted with DCM (3 X 80mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (I. 38g, yield: 78%) was purified. 1H-NMR (CD3ODjOOMHz):. 8 (ppm) I. 86-1 92 (m, 2H), I 94-1 98 (m, 2H), 2 44 (s, 3H), 2 62-…. 2. 68 (m, 2H), 2. 86-2. 91 (m, 2H), 6. 76 (d, J = 7. 6Hz, 1H), 7. 33 (dd, J = I. 2,7 . 6Hz, 1H), 7. 44 (d, J = I. 6Hz, 1H), 7. 81 (s, br, 1H). Mass spectrum m / z:. 294 99 [M + H, 79Br], 296 82 [M + H, 81Br]..
2-2, V – methyl-5- (4,4,5,5-tetramethyl–1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one

Under nitrogen, obtained in Step 2-1 to 5-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] _2_ one (147. 6mg, 0. 5mmol) , the United pinacols drop acid unitary purpose (140mg, 0. 55mmol) and acetic acid Bell (147mg, I. 5mmol) in DMSO (0. 2ml) was added in PdCl2 (dppf) • CH2Cl2 (20. 4mg, 0. 025mmol ), to the resulting solution was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 16 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (2mL), extracted with DCM (3X5mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the desired product (170mg, yield: 100%) o MS m / z:. 342 07 [M + H], 343. 08 [M + H, 100%], 344. 11 [M + H].
Step 3,5_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ batch P fixed base] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2_ one
The steps 1-6 5_ desert obtained _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl batch atmosphere pyridine (75. 8mg , 0. 2mmol), I’- step 2_2 obtained methyl 5- (4,4,5,5-tetramethyl-l, 3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4′-piperidin] -2-one (82mg, 0. 24mmol) and potassium carbonate (82. 9mg, 0. 6mmol) was dissolved in DME / water mixture solution (4 / 1,2. Oml ). Then, under nitrogen, was added Pd (PPh3) 4 (II. 6mg, 0. Olmmol), to the resulting mixture was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 18 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (5mL), extracted (3 X IOmL) with DCM. The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (88. 6mg, yield: 86%) was purified. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I 93-2 02 (m, 4H), 2 44 (s, 3H),…
2. 66-2. 72 (m, 2H), 2. 89-2. 93 (m, 2H), 4. 87 (s, br, 2H), 6. ll (q, J = 6. 4Hz, 1H ), 6. 88 (d, J =
8. OHz, 1H), 6. 94 (d, J = I. 2Hz, 1H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 19 (dd, J = I. 2,8 . OHz, 1H),
7. 31 (m, 1H), 7. 36 (s, 1H), 7. 66 (s, br, 1H), 7. 80 (d, J = 2. OHz, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
Example 2: 6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ than Li Jie base] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one

Step I, I ‘- methyl-6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one
1-1,6- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] -2_ one

As described in Example I steps 2-1 of the method from the commercially available 6-bromo – indol-2-one was prepared, Yield: 82%. Analysis of the data obtained the desired product are = 1H-Nmr (Cd3OdJOOmHz): 8 (ppm) 1.90-1.98 (m, 4H),
2. 44 (s, 3H), 2. 64-2. 68 (m, 2H), 2. 86-2. 92 (m, 2H), 7. 05 (d, J = 2. 0Hz, 1H), 7. 16-7. 21 (m, 2H), 7. 91 (s, br, 1H). Mass spectrum m / z: 295 00 [M + H, 79Br], 296 78 [M + H, 81Br]… [0312] 1-2, 1 ‘- methyl-6- (4,4,5,5-tetramethyl-_1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one

In the step 1-1 of the obtained 6-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] -2_ ketone and commercially available linking pinacol boronic ester material, the method of Example I was prepared in accordance with steps 2-2, Yield: 95%. Analysis of the data obtained of the target product are as follows: Mass spectrum m / z:. 342 06 [M + H], 343 04 [M + H, 100%], 344. 12 [M + H]..
Step 2,6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3 ratio Li Jie base] -I ‘- methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one
Example I steps 1-6 to obtain 5-bromo -3 – [(IR) -I- (2,6- dichloro-3-fluorophenyl) ethoxy] -2-amino- pyridine, I obtained in Example 1-2 of the present embodiment in step ‘- methyl-6- (4,4,5,5-tetramethyl-l, 3,2-dioxolane-2-yl borane) spiro [indoline-_3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 82%. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I. 91-1 95 (m, 2H), I 97-2 03 (m, 2H… ), 2. 45 (s, 3H), 2. 65-2. 72 (m, 2H), 2. 89-2. 95 (m, 2H), 5. 12 (s, hr, 2H),
6. 12 (q, J = 6. 4Hz, 1H), 6. 94-7. 00 (m, 3H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 31 (m, 1H ), 7. 35 (d, J = 7. 2Hz, 1H), 7. 90 (d, J = 2. 0Hz, 1H), 9. 28 (s, br, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
5- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’–methyl-spiro [indole: 3 [0317] Example morpholine-3,4 ‘- piperidin] -2-one
H2N N


Step I, 5_ desert _3_ (2,6_ two gas -3- integrity oxy) _2_ atmosphere based grant given P
1-1,2,6_ two gas acid gas _3_
Cl OF
Sodium hydroxide (13g, 325mmol) in water (IlOmL) was cooled to _5 ° C was added dropwise under vigorous stirring of liquid bromine (12. 5g, 78. 2mmol), added after the addition of pre-cooled to 10 ° C dioxane (75mL). The above mixture under vigorous stirring was added dropwise a pre-cooled to 5 ° C of I- (2,6- dichloro-3-fluorophenyl) ethanone (5g, 21. 2mmol) in dioxane (330mL) and water (90mL) was added. After the addition, at room temperature for 2 hours Lan Xiang, Xiang Lan then 90 C for 30 minutes. TLC was not shown with the S starting material disappeared, and was acidified with concentrated hydrochloric acid to PH~9. The resulting mixture was rotary evaporated to dryness, added water (20mL), and extracted with diethyl ether (2X80mL), the organic phases were combined, dried (Na2SO4), and concentrated to give an oily product solidified after cooling to a transparent, slightly yellow solid (3. 4g, Yield: 67%). 1H-Nmr (Cdci3AOOmHz):. 8 (ppm) 7. 21 (. Dd, J = 8. 0,8 8Hz, 1H), 7 35 (. Dd, J = 4. 4,9 2Hz, 1H), 9 . 79 (s, br, 1H). Mass spectrum m / z (ES “:. 207 11 [M_H, 35Clj35Cl], 209 10 [MH, 35Cl, 37Cl]..
1-2,2,6–dichloro-3-fluoro-benzyl alcohol
^ Coh
F
[0325] To be filled with 2,6-dichloro-3-fluoro benzoic acid (3g, 14. 35mmol) added dropwise to the flask IM BH3. THF (43mL, 43mmol), added after the mixture was stirred under reflux for 24 hours. TLC showed the reaction was complete, methanol (50mL) to destroy excess borane, and the solvent was distilled off under reduced pressure and the resulting trimethyl borate, the process is repeated twice more to give a viscous product 2. I g, yield: 75% . 1H-Nmr (Cdci3JOOmHz): 8 (ppm) 2. 09 (t, J = 6. 4Hz, 1H), 4. 97 (d, J = 6. 4Hz, 2H), 7 09 (t, J = 8. . 8Hz, 1H), 7. 32 (dd, J = 4. 8,9. 1Hz, 1H). Mass spectrum m / z (ES-):.. 193 08 [M_H, 35Cl, 35Cl], 195 12 [MH, 35Cl, 37Cl].
1-3,3_ (2,6-gas _3_ integrity oxy) _2_ nitro grant given P

Following the procedure of steps 1-4 of Example I, was prepared from 2,6-dichloro-3-fluoro-benzyl alcohol and 3-hydroxy-2-nitropyridine prepared in yield (in this example embodiment steps 1_2) : 90%. 1H-Nmr (Cdci3AOOmHz): 8 (ppm) 5. 45 (s, 2H), 7 20, 7 37 (dd, J = 4. 8. (Dd, J = 8. 0,9 2Hz, 1H.). , 9. 2Hz, 1H), 7. 59 (dd, J = 4. 4,8. 4Hz, 1H),
7. 74 (dd, J = L 2,8. 4Hz, 1H), 8. 17 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 316 89 [M + H, 35Cl, 35Cl], 318. 89 [M + H, 35Cl, 37Cl].
1_4,3_ (2,6-gas _3_ integrity oxy) _2_ atmosphere based grant given P

The method according to Example I step 1_5 from 3- (2,6-gas -3- integrity oxy) _2_ nitro Jie ratio 唳 preparation (in this case, steps 1-3), that Yield: 95% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 65 (s, br, 2H), 5 31 (s, 2H), 6 66 (dd, J = 5. 2,8.. . 0Hz, 1H), 7. 14 (dd, J = I. 2,8. 0Hz, 1H), 7. 18 (dd, J =
8. 4,9. 2Hz, 1H), 7. 37 (dd, J = 4. 8,8. 8Hz, 1H), 7. 73 (dd, J = I. 6,5. 6Hz, 1H). Mass spectrum m / z:. 286 95 [M + H, 35Cl, 35Cl], 288 85 [M + H, 35Cl, 37Cl]..
1-5,5_ desert -3- (2,6-gas -3_ integrity oxy) ~ 2 ~ atmosphere based grant given P

Following the procedure of Example I step 1_6 embodiment, starting from 3- (2,6-gas _3_ integrity yloxy) _2_ atmosphere group given the preparation of the batch P (in the example of the present embodiment in step 1-4), Yield: 60% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 68 (s, br, 2H), 5 28 (s, 2H), 7 21 (dd, J = 8. 0,8.. . 8Hz, lH), 7. 24 (dd, J = 2. OHz, 1H), 7. 39 (dd, J = 4. 8,
9. 2Hz, 1H), 7. 78 (d, J = 2. OHz, 1H). Mass spectrum m / z:. 364 83 [M + H, 35Cl, 36Cl, 79Br], 366 77 [M + H], 368 69 [M + H]…
Step 2,5_ [6_ atmosphere base _5_ [(2,6_ two gas -3- gas) methoxy] -3_ than Li Jie base] -I-methyl-spiro [indoline _ 3,4 ‘- piperidin] -2-one
The present embodiment 5_ desert steps 1_5 obtained _3_ (2,6_ two gas _3_ integrity yloxy) pyridine ~ 2 ~ atmosphere, Examples 2-2 obtained in step I I ‘- methyl-5- (4,4,5,5-tetramethyl-borane _1,3,2- dioxolane-2-yl) spiro [indoline-_3,4’ – piperidine ] -2-one, prepared as in Example I Step 3. Yield: 85 V0o 1H-Nmr (Cdci3JOOmHz):.. 8 (ppm) I. 92-2 02 (m, 4H), 2. 43 (s, 3H), 2. 65-2 71 (m, 2H) , 2. 90-2. 91 (m, 2H), 4. 92 (s, br, 2H), 5. 52 (s, 2H), 6. 89 (d, J = 8. 4Hz, 1H), 6 . 90 (d, J = L 2Hz, 1H), 7. 06 (t, J = 8. OHz, 1H), 7. 21 (dd, J = L 2,8. OHz, 1H), 7. 31 ( m, 1H),
7. 37 (s, 1H), 7. 79 (s, br, 1H), 7. 80 (d, J = 2.0Hz, lH). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
6- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’- methyl-spiro [indole: 4 [0337] Example morpholine _3,4 ‘- piperidin] -2-one



H2N N
Following the procedure in Example I step of Example 3, the procedure of Example 3 to give 5-bromo-1-5 _3_ (2,6-dichloro-3-fluoro-benzyloxy) -2-amino-pyridine and Step 2 in Example I to give the embodiment 1-2 ‘- methyl-6- (4,4,5,5-tetramethyl-1,3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4 ‘- piperidine] _2_ ester -one, yield:. 78 V0o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 96-2 00 (m, 2H), 2. 01 -2. 12 (m, 2H), 2. 46 (s, 3H), 2. 66-2. 73 (m, 2H), 2. 90-2. 96 (m, 2H), 5. 30 (s , hr, 2H), 6. 94-7. 01 (m, 3H), 7. 07 (t, J =
8. 4Hz, 1H), 7. 30 (m, 1H), 7. 34 (d, J = 7. 2Hz, 1H), 7. 89 (d, J = 2. OHz, 1H), 8. 56 ( s, br, 1H). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
Example 5: 5_ [5_ atmosphere base -6- [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one
J0A = o
. | J: too
[0342] Step 1,5_ desert _2_ atmosphere base _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Jie than exposure
Cl 6, / ISL / Br
xy
H2N N
In at 0 ° C, NaH (80mg of NaH in mineral oil, 2mmol) force the mouth (1R) -1_ (2,6- dichloro-3-fluorophenyl) ethanol (418mg, 2mmol. See example Example I Step 1_3) in anhydrous THF (6mL) and stirred for half an hour, a solution of 2-amino-3,5-dibromo-pyrazine (506mg, 2mmol) in THF (6mL) was added. The resulting mixture was warmed to room temperature, heated under reflux for 20 hours. TLC showed the reaction was substantially complete. After cooling to room temperature, water was added (IOmL), the mixture was extracted three times with ethyl acetate (3x20mL), the organic phases were combined, dried, concentrated, and the residue to give 594mg product was purified by column chromatography (l-3Me0H inhexanes), yield: 78%. 1H-NMR (O) Cl3, 500MHz):. 8 (ppm) I. 83 (d, J = 7. 2Hz, 3H), 5. 12 (s, br, 2H), 6 73 (q, J = 6 . 8Hz, 1H), 7. 05 (t, J = 8. OHz, 1H), 7. 28 (dd, J = 4. 8,
8. 8Hz, 1H), 7. 58 (s, 1H). Mass spectrum m / z:. 379 83 [M + H, 35Cl, 35Cl, 79Br], 381. 81 [M + H, 35Cl, 35Cl, 81Br], 383 79 [M + H, 35Cl, 37Cl, 81Br]..
Step 2,5_ [5_ atmosphere base _6_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2-one
5_ bromide present embodiment obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, implemented I’- methyl step 2-2 obtained in Example I-5 (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron
2-yl) spiro [indoline-3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 54%. 1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 85-1 88 (m, 2H), I 97-2 04 (m, 2H…. ), 2. 46 (s, 3H), 2. 76-2. 82 (m, 2H), 2. 97-3. 02 (m, 2H), 6. 74 (q, J = 6. 4Hz, 1H ), 6. 85 (d, J = 8. OHz, 1H), 7. 15 (t, J = 8. 4Hz, 1H), 7. 41 (dd, J = 4. 8,9. 2Hz, lH) , 7. 54 (dd, J = I. 6,
8. OHz, 1H), 7. 69 (d, J = I. 8Hz, 1H), 7. 81 (dt, J = 2. 0,8. 0Hz, 1H), 7. 87 (s, 1H). Mass spectrum m / z:. 515 92 [M + H, 35Cl, 35Cl], 517. 90 [M + H, 35Cl, 37Cl].
Example 6: 6- [5-amino -6 – [(lR) -l_ (2,6- dichloro _3_ fluorophenyl) ethoxy] pyrazin-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one

The embodiment of Example 5, 5_ bromo obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, Example I’- methyl-2 obtained in steps 1-2 6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indole morpholine -3,4’_ piperidin] -2-one, prepared as in Example I Step 3. Yield: 67% 0
1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 88-1 96 (m, 4H), 2 48 (s… , 3H), 2. 76-2. 82 (m, 2H), 2. 98-3. 05 (m, 2H), 6. 75 (q, J = 6. 4Hz, 1H), 7. 16 (t , J = 8. 8Hz, 1H), 7. 31 (d, J = 2. OHz, 1H), 7. 36-7. 43 (m, 3H), 7. 88 (s, 1H).
Mass spectrum m / z:. 515 99 [M + H, 35Clj35Cl], 517 90 [M + H, 35Cl, 37Cl]..
SEE
Bioorganic & Medicinal Chemistry Letters (2014), 24(16), 3673-3682.
School of Pharmaceutical Sciences, Southern Medical University,



 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
amcrasto@gmail.com





 Stockholm, Sweden
Stockholm, Sweden Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide

N-[2-(7-Benzyl-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl)ethyl]acetamide
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide
Acetamide, N-[2-[7-(cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl]-
339.47, C22 H29 N O2
cas 1287785-08-3
Melatonin MT2 Agonists
Takeda……..innovator
Treatment of Sleep Disorders,
-
Melatonin (N-acetyl-5-methoxytryptamine), which is a hormone synthesized and secreted principally in the pineal gland, increases in dark circumstances and decreases in light circumstances. Melatonin exerts suppressively on pigment cells and the female gonads, and acts as a synchronous factor of biological clock while taking part in transmittance of photoperiodic code. Therefore, melatonin is expected to be used for the therapy of diseases related with melatonin activity, such as reproduction and endocrinic disorders, sleep-awake rhythm disorders, jet-lag syndrome and various disorders related to aging, etc.
-
Recently, it has been reported that the production of melatonin melatonin could reset the body’s aging clock (see Ann. N. Y. Acad. Sci., Vol. 719, pp. 456-460 (1994)). As previously reported, however, melatonin is easily metabolized by metabolic enzymes in vivo (see Clinical Examinations, Vol. 38, No. 11, pp. 282-284 (1994)). Therefore, it cannot be said that melatonin is suitable as a pharmaceutical substance.
-
Various melatonin agonists and antagonists such as those mentioned below are known.
- (1) EP-A-578620 discloses compounds of:
-
-

- (2) EP-A-420064 discloses a compound of:

- (3) EP-A-447285 discloses a compound of:

- (4) EP-A-662471 discloses a compound of:

- (5) EP-A-591057 discloses a compound of:

- (6) EP-A-527687 discloses compounds of:

X=S, 0, Y=CH
X=0, NH, Y=N - (7) EP-A-506539 discloses compounds of:

-
-
Tricyclic or more poly-cyclic compounds with a cyclic ether moiety, such as those mentioned below, are known.
- (1) Compounds of:

are disclosed in Tetrahedron Lett., Vol. 36, p. 7019 (1995).
- (2) Compounds of:


are disclosed in J. Med. Chem., Vol. 35, p. 3625 (1992).
- (3) Compounds of:

are disclosed in Tetrahedron, Vol. 48, p. 1039 (1992).
- (4) Compounds of:

are disclosed in Tetrahedron Lett., Vol. 32, p. 3345 (1991).
- (5) A compound of:

is disclosed in Bioorg. Chem., Vol. 18, p. 291 (1990).
- (6) A compound of:

is disclosed in J. Electroanal. Chem. Interfacial Electrochem., Vol. 278, p. 249 (1990).
see
- (1) Compounds of:














http://www.google.co.in/patents/EP0885210B1?cl=en
Highly Potent MT2-Selective Agonists
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide (15)
MSA 100 a serotonin receptor antagonist.

(2E)-N-[2-[2-[(2S)-1-Methyl-2-piperidinyl]ethyl]phenyl]-3-phenyl-2-propenamide (1)
(S)-2′[2-1-(methyl-2-piperidyl) ethyl] cinnamanilide
(2E)-Λ/-[2-[2-[(2S)-1-Methyl-2-piperidinyl]ethyl]phenyl]-3-phenyl-2- propenamide
CAS 951155-17-2
C23H28 N2O, 348.48
It was reported that the (S)-enantiomer of 2′-[2-(1-methyl-2-piperidyl)ethyl]cinnamanilide (MSA100, 1) is an active 5-HT (5-hydroxytryptamine or serotonin) receptor antagonist; however, its (R)-isomer is totally or substantially devoid of the same activity.(1)
-
1.(a) Amer, M. S., U.S. Patent 5,780, 487, 1998.(b) Prashad, M.; Liu, Y.; Hu, B.; Girgis, M. J.; Schaefer, F., WO2007/111705, 2007.(c) Prashad, M.; Liu, Y.; Hu, B.; Girgis, M. J.; Schaefer, F., WO2007/111706, 2007.
…………………………………………………………………..
http://www.google.com/patents/WO2007111706A2?cl=en





Example 4:
Synthesis of (2E)-/V-[2-[2-[(2S)~1 -Methyl-2-piperidinyl]ethyI]phenyl]-3-phenyl-2- propenamide
(a) Free Base Generation:
A 250-mL, 4-necked, round-bottomed flask, equipped with a mechanical stirrer, digital thermometer and nitrogen inlet-outlet, heating cooling bath, and addition funnel, is charged with 6.28 g (S)-2-[2-(1-methyl-2-piperidinyl)ethyl]-benzenamine (1R,3S)-(+)-camphoric acid salt (1 :1) and 60 mL of isopropyl acetate. Stir the mixture at 20-25 0C under nitrogen and add a solution of 1.60 g of sodium hydroxide in 20 mL of water over a period of 5 min while maintaining an internal temperature at 20-25 0C. Stir the suspension efficiently until all the solid dissolves (5 min). Separate the organic layer and save. Extract the aqueous layer with 20 mL of isopropyl acetate. Combine the organic layers and wash it with 20 mL of water. Separate the organic layer and concentrate it under vacuum (20-100 mbar) at an internal temperature at 20-40 0C (external temperature 30-60 0C) to obtain ~65 mL of a solution of (S)-2-[2-(1-methyl-2-piperidinyl)ethyl]-benzenamine (containing 3.28 g of free base) in isopropyl acetate. Save this solution for the next step and store it under nitrogen.
(b) Reaction:
A 250-mL, 4-necked, round-bottomed flask, equipped with a mechanical stirrer, digital thermometer, nitrogen inlet-outlet, heating mantle, condenser, and addition funnel is charged with -65 mL of a solution of (S)-2-[2-(1-methyl-2-piperidinyl)ethyl]-benzenamine (containing 3.28 g of free base) in isopropyl acetate and 6.22 g of potassium carbonate. Stir the reaction mixture under nitrogen at an internal temperature at 23 ± 3 0C to afford a suspension. Add 3.75 g of cinnamoyl chloride over a period of 5 min while maintaining an internal temperature at 23 ± 3 0C to obtain a slurry. Heat the reaction mixture to an internal temperature at 85 ± 5 0C (external temperature 90-100 0C) over a period of 30-60 min. Stir the reaction mixture at this temperature for an additional 2 h. Cool the reaction mixture to 23 ± 3 0C over a period of 1 h. Add 50 mL of water. Stir the reaction mixture at 23 ± 3 0C for 30-60 min to obtain a bi-phasic solution. Separate the organic layer. Add 80 mL of 0.5 N HCI solution over a period of 10 min while maintaining an internal temperature at 23 ± 3 0C to afford a bi-phasic solution. Separate the aqueous layer. Add 60 mL of isopropyl acetate. Stir the reaction mixture and add a solution of 2.00 g of sodium hydroxide in 25 mL of water over a period of 10 min while maintaining an internal temperature at 23 ± 3 0C to afford a bi- phasic solution. Separate the organic layer and save. Extract the aqueous layer with 60 mL of isopropyl acetate. Combine the organic layers and wash it with 40 mL of water. Separate the organic layer and concentrate it under vacuum (20-100 mbar) at an internal temperature at 20-40 0C (external temperature 30-60 0C) to obtain 22mL (19.3 g) of a solution of (iii) in isopropyl acetate. Stir and heat the reaction mixture to an internal temperature at 85 ± 5 0C (external temperature 90-100 0C) over a period of 30-60 min. Add 96 mL of hepatane over a period of 10 min while maintaining an internal temperature at 85 ± 5 0C. Stir and Cool the reaction mixture to 23 ± 3 0C over a period of 1 h. Stir the resulting slurry at 23 ± 3 0C for an additional 2 h. Collect the solid by filtration over a polypropylene filter paper in a Buchner funnel with suction. Wash the solid with a total of 28 mL of a mixture of isopropyl acetate and heptane (1/6) in two equal portions of 14 mL each. Dry the solid at 45-50 0C under vacuum (13-40 mbar) with nitrogen bleeding to obtain a constant weight (LOD < 1%, 4 h) of 4.06 g of (2£)-A/-[2-[2-[(2S)-1-methyl-2-piperidinyl]ethyl]phenyl]-3-phenyl-2-propenamide as an off white solid.
Theoretical Yield: 5.23 g
Yield: 77.6%
Purity: 99.8% (HPLC area %).
Enantiomeric purity: (R)-(iii) was not detected by Chiral HPLC. Example 5:
Alternative synthesis of (2£)-/V-[2-[2-[(2S)-1-MethyI-2-piperidinyl]ethyl]phenyI]-3- phenyl-2-propenamide
(a) Free base generation:
In a 500 ml round bottomed flask equipped with a mechanical stirrer the resolved camphoric acid salt (IV) (20 g) in isopropyl acetate (120 g) is added at an internal temperature of 20 to 25 0C (external temperature 20 0C). Then, at an internal temperature of 25 to 30 0C (external temperature 20 0C) a solution of sodium hydroxide (38.24 g) in water (60 g) is added to the reaction mixture over a period of 5 minutes. The reaction mixture (suspension) is then stirred for a further 30 minutes. The resulting orange emulsion is then allowed to separate into a two-phase mixture and the water phase is removed. The organic phase is then subjected to a rotary evaporator and the isopropyl acetate is distilled at an internal temperature of 60 0C and under reduced pressure (250 mbar). Approximately 90 g of isopropyl acetate is distilled. Prior to distilling, the organic phase is a clear, bright orange colour and of a volume of approximately 160 ml ( 13Og),
(b) Reaction:
In a 1.5 I flask equipped with a mechanical stirrer and at an internal temperature of 35 0C (external temperature 38 0C) and under inert conditions (nitrogen) 2-butanone (160 g) and isopropyl acetate (20 g) is added to the reaction mixture of part (a). Then, at an internal temperature of 35 0C (external temperature of 38 0C) a solution of cinnamoyl chloride (8.9 g) in 2-butanone (20 g) is added drop wise. Then, the reaction mixture is treated with more 2- butanone (2 x 5 g). The resulting suspension is then stirred for 20 minutes at an internal temperature of 350C. The pH of the mixture is between 6 and 8.
(c) Resolution:
The suspension of step (b) is then cooled to an internal temperature of 25 0C (external temperature 20 0C) and at the same time a mixture of water (200 g) and isopropyl acetate (60 g) is added. The reaction mixture is then stirred for a further 15 minutes at an internal temperature of 25 0C (external temperature 20 0C). The resulting two-phase reaction mixture is then separated and the water phase removed. The resulting yellow upper layer is then treated with 2.5 mol/l hydrochloric acid (200 g). The resulting two-phase mixture is then separated and the water phase is transferred into a 750 ml flask equipped with a mechanical stirrer. The organic phase is then washed with 2.5 mol/l hydrochloric acid (200 g) and the resulting two-phase mixture is separated and the water phase is added to the first water phase. The combined water phases are then treated with acetic acid (300 g) and sodium hydroxide (150 g) is added. The reaction mixture is then stirred at an internal temperature of 25 to 30 0C (external temperature 20 0C) for 15 minutes. The resulting two-phase reaction mixture is then separated.
(d) Crystallization
The organic phase from the above reaction step (c) is reduced in volume on a rorary evaporator at an external temperature of 60 0C and at 250 mbar. Then, the reduced-volume reaction mixture is treated with isopropanol (60 g) and the resulting reaction mixture is reduced in volume on a rotary evaporator at an external temperature of 60 0C and under a vacuum of 150 mbar. Then, at an internal temperature of 50 to 55 0C (external temperature 60 0C) the reaction mixture is treated with water (20 g) and the resulting suspension is further treated with the product (iv) (10 mg) in isopropanol (0.01 g). The reaction mixture is then stirred for a further 15 minutes at an internal temperature of 50 to 55 0C (external temperature 60 0C). Then, further water is added over a period of 15 to 30 minutes and the reaction mixture is maintained at an internal temperature of 50 to 55 0C (external temperature 60 0C). Then, the resulting suspension is cooled to an internal temperature of 22 to 22 0C (external temperature 20 0C. Then, the suspension is stirred for a further 30 minutes at an internal temperature of 22 to 22 0C (external temperature 20 0C) and the resulting solid is collected by filtration and washed with a mixture of water and isopropyl acetate (2 x 20 g), where the water: isopropyl acetate ratio is of 5:1 g/g. The resulting solid may then be dried under a vacuum at a temperature of 55 0C.
Yield: 14.8 g (89.3% of theory). mp: 127.3 to 130.2 0C
Example 6: Recrystallisation of (2E)-Λ/-[2-[2-[(2S)-1-Methyl-2-piperidinyl]ethyl]phenyl]-3-phenyl-2- propenamide
(a) In a 200 ml round bottomed flask equipped with a magnetic stirrer, containing the product (iv) (15 g) is added isopropanol (25 g) and heptane (heptane fraction from petroleum having a boiling point of 65 to 100 0C) (25 g) is added. Then, the reaction mixture is heated to an internal temperature of 75 0C (external temperature 95 0C) and refluxed for approximately 30 minutes, whilst stirring. Then, the reaction mixture is filtered over a glass fibre filter at an internal temperature of 70 to 75 0C (external temperature 85 0C) in to a 350 ml flask equipped with a magnetic stirrer. Then, a mixture of isopropanol (5 g) and heptane (5 g) is added and the reaction mixture is heated to an internal temperature of 70 0C (external temperature 95 0C). Then, further heptane is added drop wise to the reaction mixture at an internal temperature of 65 to 75 0C (external temperature 75 0C).
(b) Crystallization
The solution from step (a) is then cooled to an internal temperature of 40 0C (external temperature 40 0C) over a period of 15 minutes. The, at an internal temperature of 40 0C, the solution is treated with a suspension of the recrystallized product (v) (11 mg) in heptane is added and the reaction mixture is stirred for 30 minutes at an internal temperature of 40 0C (external temperature 40 to 45 0C). Then, the reaction mixture is treated with some further heptane (15 g) at an internal temperature of 40 0C. The resulting suspension is then cooled to an internal temperature of -10 0C (external temperature -10 to -15 0C) over a period of 30 minutes and then further stirred for a further hour. The reaction mixture is then filtered at an internal temperature of -10 0C (external temperature -10 to -15 0C) and the resulting solid may be washed in a mixture of isopropanol and heptane, where the isopropanohheptane ratio is 1 :1.5. The solid may be washed twice (2 x 11.25 g). The solid may then be dried in a vacuum at a temperature of 60 0C.
Yield: 17.8 g (89% of theory) mp: 127.4 to 132.0 0C.
………………………………………………………………..

An efficient process was developed for the manufacture of MSA100, a serotonin receptor antagonist, via a five-step synthetic route furnishing a high quality of active pharmaceutical ingredient. Highlights of this synthesis include: (1) replacing carcinogenic methyl iodide with methyl p-toluenesulfonate as the methylating reagent; (2) a hydrogenation protocol with optimized temperature, pressure, and mass-transfer conditions that avoided one side product and reduced the other one effectively; (3) chemical resolution employing D-camphoric acid in a mixed-solvent system; (4) amidation under anhydrous conditions for controlling a Michael adduct impurity; and (5) plausible mechanisms for the formation of side products.
Example I PREPARATION AND CONFIRMATION OF S-MPEC


racemic-APEMEP-HI -5/1-


– 6 –
1. 2 – nitrobenzaldehyde
2. 2 – picoline
3. 2 – (o-nitrostyryl) pyridine (NSP)
4. 2 – “(o-nitrostyryl)- 1 -methylpyridinium iodide 5. RS-2- (o-aminophenethyl)-l-methylpiperidine. HI
6. S-[2-(o-aminophenethyl)- 1 -methylpiperidine-dibenzoyl-L-tartrate] (S-APEMP. DBLT OR .L-DBT)
7. S-2′- [2-(l-methyl-2-piperidyl) ethyl] cinnamanilide (S-MPEC) 7a. Cinnamoyl chloride S-MPEC CHEMICAL PROCESS
(A) 2-(O-Nitrostyryl) – 1 -Methylpyridinium Iodide fNSMP-P
To a 50 L round bottomed flask was added 2-nitrobenzaldehyde (3,500 g. 23.2 moles), 2-picoline (3.2L., 32.8 moles) and acetic anhydride. The mixture was stirred efficiently under an inert atmosphere (nitrogen or another inert gas) and heated to reflux for 27 hrs. The mixture was cooled to under 100 C, for safe handling, and quenched in a suitable vessel equipped with external cooling and efficient stirring on 10.5 Kg. of ice. The pH was adjusted to 11 with 45% aqueous sodium hydroxide at a rate to keep the temperature below 50°C. After cooling to 20-30°C, the granular solid was collected by filtration, washed well with water. Yield 6572 g. of crude 2-(o-nitrostyryl) pyridine (NSP).
This solid was transferred to a 50L, round bottomed flask, dissolved in acetone (14L.) and iodomethane (2.94L., 47.7 moles) (quaternizing methylating agent) was added. (Other such (alkylating) agents may be used, generally having the formula CH3X, X being an anion such as sulfate, methyl sulfate, halide (Cl, Br, I), etc.). The mixture was heated to reflux under an inert atmosphere (nitrogen or another inert gas) for 18 hrs. After cooling to 20°C. the precipitate was collected by filtration and washed with acetone or a 1 :1 mixture of acetone:ethyl acetate (3×3.5L.). Drying to constant weight at 50-60°C. yielded 6,839 g. (80%) of NSMP.I. (B) RS- 2-(o-Aminophenethyl)-l-Methylpiperidine. Hvdroiodide
ΓRS-APEMP.HΠ
In a 5 gallon reactor, a solution of NSNP.I (935 g., 2.5 moles) in – 7 – methanol (14L.) was reduced in a hydrogen atmosphere (Psi. 55) in the presence of Pt/C (5 or 10%, 98g.). After removal of the catalyst and evaporation of the filtrate in the usual manner, the residue was dissolved in hot methanol (2.8L.). Ethyl acetate (2.8L) was added to the hot mixture to induce crystallization, yield 516.3 g. (59%) of RS-APEMP.HI.
(C) S-[2-(o-Aminophenethv0- 1 -Methylpiperidine Dibenzoyl-L-Tartrate] (S- APEMP.DBLT)
A solution of RS-APEMP.HI (516g., 1.5 mole) ethyl acetate (5.5g.) (or other low boiling water immiscible solvent such as benzene, toluene etc.) was extracted with 5% aqueous sodium hydroxide to liberate the free base (organic phase), washing the organic phase with water, drying over a suitable drying agent (such as anhydr. sodium sulfate, magnesium sulfate, potassium carbonate etc.) After separating the solvent from the drying agent the solution was evaporated in vacuo and the residual RS-APEMP free base was dissolved in methanol (l .O.L.) and a solution of dibenzoyl-L-tartaric acid (540 g., 1.5 moles) in methanol (2.3 L.) was added. The mixture was held overnight at room temperature. The crystalline precipitate was collected and recrystallized from methanol (3.4 L.), yield 246g. of S-APEMP.DBLT. (28.6%, wt; 57.2% of the S-APEMP). (O) S-2′-r2-π-Methyl-2-Piperidvnethyll Cinnamanilide f S-MPEC) A solution of S-APEMP.DBLT (287 g, 0.5 mole) in ethyl acetate
(3.2 L.) (or other low boiling water immiscible solvent) was extracted with 7.5% aqueous sodium bicarbonate (3.2 L.) to liberate the S-APEMP. After a water wash and drying over a suitable drying agent the solvent was removed in vacuo. The oily residue, S-APEMP, was dissolved in ethyl acetate (1.0 L.) and anhydrous potassium carbonate (412 g, 3.0 moles) (or other suitable acid acceptor such as triethyl amine, pyridine etc.) was added. Cinnamoyl chloride (143 g., 0.7 mole) in 700 ml. of ethyl acetate was added slowly. After the initial reaction, the mixture was refluxed for 14 hrs. After cooling to room temperature the mixture was extracted with water (1.7 L.) and dried over a suitable drying agent. After removing the drying agent the solvent was removed in vacuo and the residue was dissolved in hot ethyl acetate (280 ml.) and allowed to slowly cool to room temperature; filtration yielded S-MPEC, (136 g., 79% yield). Analysis: Calcd. For C, H, N : C, 79.27; H, 8.10;N, 8.04. Found: C, 79.27; H, 8.06; N, 8.07. HPLC(chiral)purity: 99.5%, [oc ]D25, -46° (c=0.01,EtOH); Melting point: 128°C.
TABLE 2 CERTIFICATE OF ANALYSIS Compound Name: (-)-2′-[2-(l-Methyl-2 piperidyl)ethyl]cinnamanilide(/-MPEC,S- MPEC

……………………………….
synthesis of 2′[2-1-(methyl -2-piperidyl) ethyl] cinnamanilide (Y1), which is a compound of formula (Y) where Ra is hydrogen and R1 is methyl:

Processes for the preparation of compounds of formula (Y) are described in U.S. Pat. No. 3,931,195 which comprises the step of alkylating compounds of formula (i) (below) with an alkyl halide, such as methyl iodide for methylation. The same methylation step is described in EP0973741 for the synthesis of compound (Y1).

Thus, the processes described in the prior art involve the use of a highly toxic reagent (e.g. methyl iodide) and provides a yield of about 50%

