
Home » Posts tagged 'phase 2' (Page 16)
Tag Archives: phase 2
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Lifitegrast, SAR 1118

Lifitegrast, SAR 1118
SAR-1118-023
CAS 1025967-78-5
Xiidra (lifitegrast ophthalmic solution)
L-Phenylalanine, N-[[2-(6-benzofuranylcarbonyl)-5,7-dichloro-1,2,3,4-tetrahydro-6-isoquinolinyl]carbonyl]-3-(methylsulfonyl)-
INNOVATOR
PLEASE, ALL THESE ARE NOT MINE, FROM THE NET

 PLEASE NOT MINE FROM THE NET
PLEASE NOT MINE FROM THE NET




SYN CONSTRUCTED FROM WO 2014018748,
3(2H)-Benzofuranone, 6-hydroxy- cas 6272-26-0
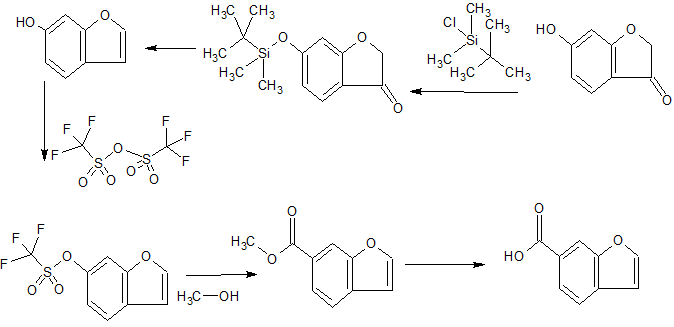
6-Benzofurancarboxylic acid cas 77095-51-3
NEXT…….
L-Phenylalanine, 3-bromo- cas 82311-69-1

L-Phenylalanine, 3-(methylsulfonyl)-, phenylmethyl ester, hydrochloride (1:1) cas 1194550-59-8
NEXT………..


WO 2014018748,
SAR 1118 ophthalmic solution from SARcode Bioscience (Brisbane, Calif.) is a first-in-class molecule that inhibits T-cell inflammation by blocking the binding of two key cellular surface proteins (LFA-1 and ICAM-1) that mediate the chronic inflammatory cascade, so it may be able to reduce the inflammation associated with dry-eye disease.
A growing body of evidence points to a role for inflammation mediated by lymphocyte function-associated antigen-1 (LFA-1) and its ligand intercellular adhesion molecule-1 in the pathogenesis of diabetic macular oedema. This phase 1b clinical trial assessed the safety, tolerability, and pharmacokinetics of topically administered SAR 1118, a novel LFA-1 antagonist, in human subjects
Topical SAR 1118 was safe and well tolerated, and dose-dependent levels of drug were detected in aqueous. However, vitreous levels were below the threshold of detection with the concentrations tested. Further investigation of this medication for posterior segment applications would require intravitreal delivery or chemical modification of the drug.
In a Phase 2 dry eye trial, subjects receiving SAR 1118 demonstrated a reduction in corneal staining, increased tear production, and improved visual-related function as compared to placebo. These data were part of the scientific program of the Association for Research in Vision and Ophthalmology (ARVO) Annual Meeting held in Fort Lauderdale, Florida. SAR 1118 is a first-in-class topically administered small molecule integrin antagonist that inhibits T-cell mediated inflammation, a key component of dry eye disease.
In the randomized, placebo-controlled, multi-center trial, which included 230 subjects with dry eye disease, SAR 1118 demonstrated dose-dependent significant improvements (p<0.05) in inferior corneal staining over 12 weeks. As early as two weeks, a statistically significant(p<0.05) increase in tear production and improvement in visual-related functions (ability to read, drive at night, use a computer, and watch television) were observed, demonstrating early onset of action. Visual-related function was assessed using the Ocular Surface Disease Index (OSDI), a validated instrument designed to measure the severity of dry eye disease and the impact on vision-related function and quality of life. SAR 1118 was safe and well-tolerated with no serious ocular adverse events reported. Most ocular adverse events were transient and related to initial instillation.
“We are encouraged by the clinical effects of SAR 1118 in improving both signs and symptoms of dry eye, which supports the broad anti-inflammatory mechanism of this novel molecule,” commented Charles Semba, MD, Chief Medical Officer of SARcode Corporation. “We are excited to begin Phase 3 development in the later part of 2011, and have discussed appropriate and acceptable endpoints with the regulatory bodies to facilitate a smooth path towards approval.”
“It is well known that dry eye disease can have a deleterious effect on visual function, daily activities, workplace productivity, and quality of life. The potential to impact a patient’s quality of life in as early as 2 weeks could be a major advance in the current dry eye treatment model,” said Quinton Oswald, Chief Executive Officer of SARcode Corporation. “We hope to achieve similar results in our Phase 3 program.”
About Dry Eye Syndrome
Dry eye syndrome is a prevalent and often chronic condition estimated to affect approximately 20 million people in the US. It is among the most common diseases treated by ophthalmologists throughout the world, and has been shown to have a significant impact upon quality of life. Dry eye varies in severity and etiology, and symptoms most commonly manifest as discomfort, visual disturbances, and tear film instability due to decreased quality or quantity of tears. A major contributing factor towards the development of dry eye is inflammation caused by T-cell infiltration, proliferation and inflammatory cytokine production that can lead to reduction in tear film quality and ocular surface damage.
About SAR 1118 – SAR 1118 is a potent novel small molecule lymphocyte function-associated antigen-1 (LFA-1; CD11a/CD18; alphaLbeta2) antagonist under investigation for a broad range of ocular inflammatory conditions including dry eye and diabetic macular edema. LFA-1 is member of the integrin family of adhesion receptors found on the surface of all leukocytes and represents a therapeutic target central to a number of inflammatory stimuli. SAR 1118 has demonstrated potency in blocking LFA-1 binding to its cognate ligand, intercellular adhesion molecule-1 (ICAM-1; CD54), thereby inhibiting cell adhesion, cytokine production, and cellular proliferation in in vitro models.
About SARcode Corporation – SARcode Corporation, founded in 2006, is a venture-backed ophthalmic biopharmaceutical company based in Brisbane, CA. SARcode’s lead development program is a novel class of lymphocyte function-associated antigen-1 (LFA-1) antagonists for the treatment T-cell mediated inflammatory diseases. Institutional investors include Alta Partners and Clarus Venture Partners. For more information, visit http://www.sarcode.com
WO 2006125119

Tom Gadek
| Inventors | Thomas Gadek, John Burnier |
| Applicant | Sarcode Corporation |
http://www.google.com.mx/patents/WO2005044817A1?cl=en
EXAMPLE 14 [0305] This example describes the synthesis of

[0306] which was prepared according to Scheme 9 and the procedure below.
[0307] SCHEME 9

[0308] a) To a solution of 3-carboxylbenzenesulfonyl chloride (3.54 g, 16 mmol) in ethyl acetate (50 mL) at 0 °C was added concentrated ammonia (2.5 mL). The reaction was neutralized with HCl in dioance (20 mL), diluted with ethyl acetate (100 mL), dried with anhydrous sodium sulfate and filtered. Concentration of the filtrate yielded the title compound, which was used without purification. [0309] b) Crude compound 14.1 was dissolved in THF (50 mL), to it was added borane (1.0 M in THF, 50 L) over 20 minute period. After the reaction was stirred at room temperature for 15 hours, the reaction was diluted with brine (20 mL) and water (10 mL), extracted with ethyl acetate (100 mL). The organic extract was dried over anhydrous sodium sulfate and filtered. Concentration of the filtrate yielded the title compound, which was used without further purification. [0310] c) To crude compound 14.2 solution in DCM (100 mL) was added activated 4A molecular sieve powder (8 g), pyridinium dichromate (7.55 g, 20 mmol). After the reaction was stirred at room temperature for 2 hours, the reaction mixture was filtered through silica gel (50 g), rinsed with ethyl acetate. The residue after concentration of the filtrate was purified by silca gel column with 30-50% ethyl acetate in hexane to give compound 14.3 (477mg, 16%, 3 steps). ESI-MS (m/z): (M+H4″) 186. [0311] d) Compound 14.4 was made according to Example 8e except that compound 14.3 was used instead of compound 8.7. MS (ESI4) m/z: 260 (M+H4″). [0312] e) Compound 14 was made according to Example 3g except that compound 14.4 was used instead of compound 3.4. 1H NMR (400 MHz, CD3OD) δ 7.89 (s, 1 H), 7.80 (s, 1 H), 7.75 (m, 2 H), 7.64 (s, 1 H), 7.57(d, 1 H), 7.34 (d, 2 H), 6.93 9s, 1 H), 5.00 (m, 1 H), 3.99 (m, 1 H), 3.73 (m, 1 H), 3.40 (dd, 1 H), 3.12 (dd, 1 H), 2.89 (m, 2 H) ppm; ESI-MS (m/z) 616 (M+H4″). [0313] EXAMPLE 15 [0314] This example describes the synthesis of

which was prepared according to Scheme 10 and the procedure below. [0315] SCHEME 10 rr–λ I BuLi, THF m-CPBA

s ) 2. DMF CH2CI2
15.1 15.2

[0316] a) To a solution of 0.2 mol of furan in 200 mL of dry THF was added 0.2 mol of «-BuLi (1.6 M in hexanes) at -78 °C, the resulting solution was stirred at room temperature for 4 hours. Subsequently, the mixture was cooled to -78 °C and treated with 0.21 mol of dimethyl disulfide, and the mixture was stirred at room temperature overnight, followed by adding 10 mL of saturated aqueous NH C1. The mixture was concentrated at room temperature, and the residue was diluted with 200 mL of saturated aqueous NH4C1 and extracted with ether. The extract was then washed with brine and dried with anhydrous Na2SO . The solvent was removed, and the residue was distilled to collect, the fraction at 135-140 °C/760 mmHg to give compound 15.1 in 55% yield. 1H NMR (400 MHz, CD3C1): δ 7.50 (s, IH), 6.45 (m, IH), 6.39 (s, IH), 2.42 (s, 3H) ppm. [0317] b) To a solution of 0.1 mol of compound 15.1 in 100 mL of dry THF was added 0.1 mol of n- uLi (1.6 M in hexanes) at -78 °C, the resulting solution was stirred at room temperature for 4 hours. Subsequently, the mixture was cooled to -78 °C and treated with 0.12 mol of dry DMF, and the mixture was stirred at room temperature overnight. The reaction was quenched by adding 10 mL of saturated aqueous NH4C1, and the mixture was concentrated. The residue was diluted with 100 mL of brine and extracted with EtOAC. The extract was washed with brine and dried with anhydrous Na2SO4. The solvent was removed and the residue was purified to give the title compound in 65% yield. 1H NMR (400 MHz, CD3C1): δ 9.52 (s, IH), 7.24 (d, J= 3.4 Hz, IH), 6.42 (d, J= 3.4Hz, IH), 2.60 (s, 3H) ppm; ESI-MS (m/z) (M+H4) 143.1. [0318] c) A mixture of 50 mmol of compound 15.2 and 120 mmol of -CPBA in 100 mL of CH2C12 was stirred at room temperature overnight. The mixture was diluted with 150 mL of CH2C12, and the mixture was washed with saturated aqueous NaHCO3 for several times. The solution was then dried with anhydrous Na2SO4 and concentrated. The residue was purified to give compound 15.3 in 70% yield. 1H NMR (400 MHz, CD3C1): δ 9.83 (s, IH), 7.33 (m, 2H), 3.27 (s, 3H) ppm; ESI-MS (m/z): (M+H4″) 175.0.
[0319] d) Compound 15.4 was made according to Example 8e except that compound 15.3 was used instead of 8.7. ESI-MS (m/z): (M+H4″) 248.1. [0320] e) Compound 15 was made according to Example except that compound 15.4 was used instead of 3.4. 1H NMR (400 MHz, CD3OD): δ 7.92 (s, IH), 7.76 (m, IH), 7.67 (s, IH), 7.34 (m, IH), 7.13 (s, IH), 6.69 (s, IH), 6.49 (s, IH), 5.11 (m, IH), 4.73 and 4.88 (m, 2H), 3.76 and 4.02 (m, 2H), 3.46 (m, IH), 3.30 (m, IH), 3.17 (s, 3H), 2.94 (m, 2H) ppm; ESI-MS (m/z): (M+H4) 605.05. [0321]
…………………………………….
US 20110092707
http://www.google.com/patents/US20110092707
Formula I:

has been found to be an effective inhibitor of Lymphocyte Function-Associated Antigen-1 (LFA-1) interactions with the family of Intercellular Adhesion Molecules (ICAM), and has desirable pharmacokinetic properties, including rapid systemic clearance. Improved forms, including crystalline forms, and their uses in treatment of disorders mediated by the interaction of LFA-1 and ICAM are described herein. Novel polymorphs of the compound of Formula I which may afford improved purity, stability, bioavailability and other like characteristics for use in pharmaceutical formulations and methods of use thereof are useful in treating disease.
Methods of Manufacture of the Compound of Formula I
In one embodiment, the compound of Formula I was synthesized as in the following Schemes 1-7. Alternate steps were used in the process as described below. The variants of this overall route yield superior yields, cost of goods and superior chiral purity compared to previously described methods. The final product of this synthesis yields crystalline Form A directly.

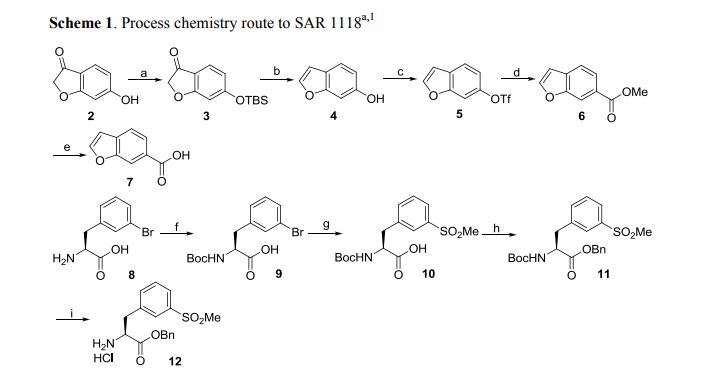
A first alternative protecting strategy produces compound 5, a trityl protected species as shown in Scheme 1. The synthesis begins by reductively aminating 3, 5, dichlorobenzaldehyde, compound 1, with 1-chloro-2-aminoethane and sodium cyanoborohydride in 35% yield. Cyclization of compound 2 using aluminum chloride catalysis and ammonium chloride at 185° C. provided compound 3 in 91% yield. Protection of the free amine of compound 3 as the trityl protected species afforded compound 4 in 89% yield. A carboxylic acid functionality was introduced by treatment of compound 4 with n-butyllithium (nBuLi) and Tetramethylethylenediamine (TMEDA), with subsequent introduction of carbon dioxide, to produce compound 5 in 75% yield.

Bromophenylalanine was used as the starting material for the right hand portion of the final molecule as shown in Scheme 2. t-Butylcarbamate (Boc) protection of the amino group was accomplished, using sodium bicarbonate (3 equivalents), t-butyl dicarbonate (Boc2O, 1.1 equivalent) in dioxane and water, to obtain compound 7 in 98% yield. A methyl sulfone functionality was introduced by treating the bromo compound 7 with copper iodide (0.4 equivalents), cesium carbonate (0.5 equivalents), L-proline (0.8 equivalents), and the sodium salt of methanesulfinic acid (3.9 equivalents) in dimethylsulfoxide (DMSO) at 95-100° C. for a total of 9 hours, with two further additions of copper iodide (0.2 equivalents) and L-proline (0.4 equivalents) during that period. Compound 8 was isolated in 96% yield. The carboxylic acid of compound 8 was converted to the benzyl ester, compound 9, in 99% yield, using benzyl alcohol (1.1 equivalent), dimethylaminopyridine (DMAP, 0.1 equivalent) and N-(3-dimethylaminopropyl)-N-ethylcarbodiimide (EDC, 1.0 equivalent). The amino group of compound 9 is deprotected by adding a 4N solution of HCl in dioxane to compound 9 at 0° C. in methylene chloride. The HCl salt of the free amino species, compound 10 was isolated in 94% yield.

Compound 5 was treated with triethylamine (TEA, 5 equivalents) and 2-(7-Aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU, 1.25 equivalents) for 10 minutes in dimethylformamide (DMF), and then compound 10 was added to the solution. After stirring at room temperature for 18 hours, the product, compound 11 was isolated in 70% yield. Removal of the trityl protecting group was accomplished by treating compound 1, with HCl in dioxane (4N, excess) at room temperature for 2 hours, diethyl ether added, and the solid product, compound 12, was isolated by filtration in 95% yield.

The benzofuranyl carbonyl moiety of the compound of Formula I was prepared using two alternative schemes, Scheme 4 and Scheme 4″. In one embodiment, the benzofuranyl carbonyl moiety was prepared by protecting the hydroxyl group of compound 13 by reacting with tert-butyldimethylsilyl chloride (1.0 equivalents) and triethylamine (TEA, 1.1 equivalents) in acetone, to give compound 14 in 79% yield. A solution of compound 14 in methanol was then treated with sodium borohydride (1.0 equivalent) at room temperature overnight. The reaction was quenched with an addition of acetone, stirred at room temperature for a further 2.5 hours, aqueous HCl (4N) was added with the temperature controlled to below 28C, tetrahydrofuran (THF) was added, and the solution stirred overnight under argon and in the absence of light. The product, compound 15, was isolated quantitatively by extraction into methylene chloride, concentrated at low heat, and used without further purification. The triflate ester, compound 16, was produced in 69% yield from compound 15 by reacting it with N-phenyl-bis(trifluoromethanesulfonimide) (1.0 equivalent) in methylene chloride for 72 hours. Compound 16 in a mixture of DMF, methanol, and triethylamine, was added to a prepared solution of palladium acetate, diphenyl, DMF and methanol in an autoclave. Carbon monoxide was charged into the autoclave to a pressure of 8 bar, and the reaction mixture was heated at 70° C. for 6 hours. After workup, compound 17 was isolated in 91% yield. Lithium hydroxide (4 equivalents) in methanol and water was used to hydrolyze the ester and permit the isolation of compound 18 in 97% yield.

In one embodiment, the benzofuranyl carbonyl moiety of the compound of Formula I was prepared according to Scheme 4″. By way of an Arbuzov reaction, diethyl 2-(1,3-dioxolan-2-yl)ethylphosphonate, compound 1″, was prepared from 2-(2-bromoethyl)-1,3-dioxolane by the addition of triethyl phosphate. After removal of ethyl bromide through distillation at 210° C. the crude reaction mixture was cooled and then by way of vacuum distillation, compound 1″ was collected as a colorless oil in 94% yield.
In the next step, n-butyllithium (2.15 equivalents) in hexane was cooled to −70° C. and diisopropylamine (2.25 equivalents) was added while keeping the temperature below −60° C. Compound 1″ (1 equivalent) dissolved in tetrahydrofuran (THF) was added over 30 min at −70° C. After 10 min, diethyl carbonate (1.05 equivalents) dissolved in THF was added over 30 min keeping the reaction temperature below −60° C. After stirring for one hour at −60° C., the reaction was allowed to warm to 15° C. and furan-2-carbaldehyde (1.3 equivalents) dissolved in THF was added. After stirring for 20 hrs at room temperature, the reaction was rotary evaporated to dryness to yield ethyl 2-(1,3-dioxolan2-yl)methyl-3-(furan-2-yl)acrylate, compound 5″. Crude compound 5″ was used directly in the next reaction.
The crude compound 5″ (1 equivalent) was dissolved in ethanol and added to a mixture of water and phosphoric acid (85%, 15 equivalents) over 30 min while keeping the temperature below 50° C. After stirring for 20 hrs at room temperature, another 200 ml of phosphoric acid (85%) was added and the mixture was heated to 50° C. for an additional two hrs. After removal of ethanol by rotary evaporation, the material was extracted with toluene, washed with water, dried with sodium sulfate, treated with charcoal, filtered and dried down to an oil. This oil was distilled to afford ethyl benzofuran-6-carboxylate, compound 6″, (bp 111-114.5° C.) which crystallized on standing. Compound 6″ was recovered at 57% yield based on compound 1″.
Compound 6″ (875 mmol) was dissolved in methanol and tetrahydrofuran (THF). Sodium hydroxide (4 M, 3 equivalents) was added and the reaction was stirred overnight. After concentration via rotary evaporation, the aqueous solution was extracted with methyl tert-butyl ether (MTBE), acidified to pH 2 with the addition of hydrochloric acid (HCl) and cooled resulting in fine crystals of benzofuran-6-carboxylic acid, i.e., compound 18. Compound 18 was isolated, washed with water and dried to a final yield of 97% yield.

The benzofuran carboxylic acid 18 was treated with oxalyl chloride (1.2 equivalents) and a catalytic amount of DMF, stirring for 5.5 hours until a clear solution was obtained. The solvent was removed under reduced pressure and the acid chloride of compound 18 was stored under argon until use, on the next day. The acid chloride, in methylene chloride was added slowly to a methylene chloride solution of the compound of Formula I and diisopropylethylamine (DIPEA) which was cooled to 0-5° C. The reaction was not permitted to rise above 5° C., and after completion of addition, was stirred at 5° C. for a further 0.5 hour. Upon aqueous workup and extraction with methylene chloride, the product, compound 19, was isolated in quantitative yield.
Taking the compound of Formula I directly as the crude reaction product after transfer hydrogenolysis, and reconcentrating down from a solution in methylene chloride, the amorphous form of the compound of Formula I was obtained in 97% purity.

An alternative protection strategy in this synthetic approach is illustrated in Scheme 6.
…………………….
WO 2014018748
http://www.google.com/patents/WO2014018748A1?cl=en
[0040] Methods of Manufacture of the Compound of Formula I

[0041] In one embodiment, the compound of Formula I is synthesized as in the following Schemes 1-7. The final product of this synthesis yields the compound of Formula I as an amorphous solid or as a crystalline form such as Forms A-E, or a pharmaceutically acceptable salt, either directly or indirectly. Variants of this overall route may provide superior yields, cost of goods, and/or superior chiral purity.
[0042] Protecting groups for amino and carboxy groups are known in the art. For example, see Greene, Protective Groups in Organic Synthesis, Wiley Interscience, 1981, and subsequent editions.
[0043] In various embodiments in the subsequent schemes, HATU is used as a reagent in amide- bond forming reactions. Alternatively, HATU is not used. In various embodiments, at least one amide-bond forming reaction is performed with thionyl chloride as a reagent in place of HATU. In various embodiments, all amide-bond forming reactions are performed with thionyl chloride as a reagent to form acid chlorides.
[0044] Scheme 1

[0045] A first alternative protecting strategy produces compound 5′, a protected species as shown in Scheme 1. The synthesis begins by reductively aminating 3, 5, dichlorobenzaldehyde, compound . Cyclization of compound 2′ provides compound 3′. Protection of the free amine of compound 3′ as a protected species provides compound 4′. A carboxylic acid functionality is introduced by treatment of compound 4′ with introduction of carbon dioxide, to produce compound 5′. In various embodiments, the protecting group of compound 4′ is a benzofuranyl carbonyl moiety derived from compound 18′.
[0046] In various embodiments, upon scaleup to multikilogram and larger scale reactions, treatment of compound 4′ with strong base (such as n-butyllithium (nBuLi) to generate a lithio species, or lithium diisopropyl amide (LDA) to generate the lithio species) is performed in flow mode rather than batchwise reaction due to instability of lithio species except at cold temperatures. Flow rates and residence times may be adjusted to maximize yield.
[0047] Scheme IB


5′ 4″”
[0048] In various embodiments, 6-hydroxy-l, 2,3, 4-tetrahydro-isoquino line (Compound 3″) is used as a starting material for Compound 5′. The starting material is chlorinated (x2) for example, with N-chlorosuccinimide. In various embodiments, the chlorination is performed in the presence of a sulfonic acid. In various embodiments, the sulfonic acid is selected from p- toluenesulfonic acid and methanesulfonic acid. Following protection of the amino group, the hydroxy group is functionalized, for example, as the triflate ester, which is carbonylated to yield the amino-protected methyl ester. Hydrolysis of the methyl ester yields the amino protected carboxylic acid.
[0049] Scheme 2

[0050] In various embodiments, bromophenyl alanine is used as the starting material for a portion of the final molecule as shown in Scheme 2. The starting material is protected with an amino protecting group to allow for introduction of a methyl sulfone functionality in compound 8′. Protecting groups are rearranged by introduction of an orthogonal protecting group for the carboxylic moiety, followed by deprotection of the amino group to provide compound 10′. In various embodiments, expensive or exotic bases are replaced with carbonate base such as potassium carbonate or calcium carbonate as a reagent.
[0051] Scheme 2A

10
[0052] In various embodiments, 3-methylsulfonylbenzaldehyde is converted into the 3- methylsulfonylphenylalanine derivative and functionalized to yield compound 10 as shown above.
[0053] Scheme 3

12′
[0054] Compounds 5′ and 10′ are joined through amide bond formation followed by deprotection of the remaining amino group in the presence of the carboxylic protecting group to yield compound 12′ or a salt thereof, such as the HCL salt.
[0055] Scheme 3

[0056] As an alternative to Scheme 3, compound 10″ is coupled with compound 5′ to yield the bromo compound 12″”, with subsequent introduction of a methyl sulfone functionality in place of bromine at a later step to produce compound 19′. Alternatively, instead of a bromine, compound 10″ includes X, where X is any halide (CI, I, Br, F) or a leaving group such as OTs, OTf, or the like.
[0057] Scheme 4

[0058] The benzofuranyl carbonyl moiety of the compound of Formula I can be prepared using various alternative schemes. In one embodiment, the benzofuranyl carbonyl moiety is prepared by protecting the hydroxyl group of compound 13′, reducing the carbonyl of compound 13′ to yield the benzofuranyl moiety, followed by carboxylation to yield compound 18′.
[0059] Scheme 4A
[0060] In one embodiment, compound 18′ is prepared from 6-hydroxybenzofuran via the triflate ester and the 6-carboxy methyl ester as intermediates, as shown in Example 4A.
[0061] Schem

[0062] The benzofuran carboxylic acid 18′ is coupled with compound 12′ (or a salt thereof) by amide bond formation to yield protected compound 19′, as shown in Scheme 5. Amide bond formation is known in the art
[0063] Schem

[0064] As an alternative to Schemes 3-5, compounds 18′ and 5″ may be coupled through amide bond formation followed by deprotection of the remaining carboxylic group to form compound 12″. Amide bond formation between compound 12″ and 10′ yields compound 19′ with a protected carboxylic group.
[0065] Scheme 5B

[0066] As an alternative to Schemes 1-5, compounds 12″ and 10″ may be coupled through amide bond formation followed by introduction of a methyl sulfone functionality in place of the bromine in converting compound 19″ to compound 19′ (similar to Scheme 2). Alternatively, instead of a bromine, compound 10″ includes X, where X is any halide (CI, I, Br, F) or a leaving group such as OTs, OTf, or the like. Compound 12″ can also be made using the following scheme:

[0067] Scheme 6

[0068] Final deprotection of compound 19′ to yield the compound of Formula I or a salt thereof is accomplished in a variety of ways. In various embodiments, the resulting compound of Formula I is provided in higher optical purity and/or higher overall purity and/or higher overall yield.
EXAMPLES
[00111] Example 1

Scheme El
[00112] Reductively aminating 3,5-dichlorobenzaldehyde, compound 1, with l-chloro-2- aminoethane and sodium cyanoborohydride provided 35% yield of compound 2. Cyclization of compound 2 using aluminum chloride catalysis and ammoniun chloride at 185°C provided compound 3 in 91% yield. Protection of the free amine of compound 3 as the trityl protected species afforded compound 4 in 89%> yield. A carboxylic acid functionality was introduced by treatment of compound 4 with n-butyllithium (nBuLi) and tetramethylethylenediamine (TMEDA), with subsequent introduction of carbon dioxide, to produce trityl protected compound 5 in 75% yield.
[00113] Example 1 A

2″

Scheme El A
[00114] To a glass reactor was charged 3,5-dichlorobenzaldehyde. Absolute ethanol was added to the batch slowly (this addition is mildly exothermic) and agitation started. 2,2- Diethoxyethyl amine (1.03 equiv) was slowly added to the batch, keeping the batch temperature at 20-78 °C. The batch was then heated to 76-78 °C for 2 h. GC-MS analysis indicated reaction completion (starting material < 1%). The batch was cooled to ambient temperature for work-up. The batch was concentrated in vacuo to a residue and azeotroped with heptanes (x2). The residue was cooled and held at 0-5 °C for 12 h to form a suspension. The solids were collected by filtration and the cake was washed with cold (0-5 °C) heptanes, and dried under hot nitrogen (45-50 °C) to afford Compound 2′ as a white solid (94% yield).
[00115] To a glass reactor was charged concentrated 95-98%) sulfuric acid (25.9 equiv).
The batch was heated to 120-125 °C and a solution of Compound 2′ in CH2CI2 was added slowly over 1 h, keeping the batch temperature between 120-125 °C. The batch was then stirred at 120— 125 °C for 6 h. The batch was cooled to < 50 °C. To a glass reactor was charged DI water and the batch temperature was adjusted to 0-5 °C. The reaction mixture was slowly transferred, keeping the batch temperature between 0-50 °C. DI water was used to aid the transfer. To the batch was added Dicalite 4200. The batch was filtered through a pad of Dicalite 4200. To the filtrate was added 50% aqueous sodium hydroxide solution slowly over 3 h, keeping the batch temperature between 0-50 °C to adjust the pH to 12. The resulting suspension was stirred at 45- 50 °C for 2 h and the solids were collected by filtration. The filter cake was slurried in DI water at 30-35 °C for 1 h. The batch was filtered. The cake was washed with heptanes and dried in vacuum oven at 45-50 °C for 22 h to give crude compound 2″ as a tan solid (75% yield), which was further purified by recrystallization.
[00116] To a reactor was added platinum dioxide (0.012 equiv), Compound 2″, and
MeOH (10 vol) and the suspension was stirred at room temperature under argon for 10 minutes. The reaction mixture was inerted with argon three times and then stirred under 125 psi of hydrogen at room temperature for 25 hours. HPLC analysis indicated complete reaction with less than 1% of the starting material remaining. After standing, the supernatant was decanted from the solids (catalyst) by vacuum. To the solids was added methanol and the slurry was mixed under nitrogen. The solids were allowed to settle on the bottom over several hours. The supernatant was decanted from the solids by vacuum. The combined supernatants were filtered through Celite under a blanket of nitrogen and the filter pad was washed with MeOH (x2). The combined filtrate and washes were concentrated to dryness. The residue was slurried in MTBE. The mixture was treated with 3 M HC1 while maintaining the temperature <40 °C resulting in the formation of a heavy precipitate. The mixture was stirred at 35-40 °C for 60 to 90 minutes. The batch was cooled to 0-5 °C, stirred for 60 to 90 minutes and then filtered. The filter cake was washed with cold DI water (x2) followed by a displacement wash with MTBE (x2). The filter cake was dried under reduced pressure to afford Compound 3 Hydrochloride Salt (86% yield). The hydrogenation catalyst can be recovered and re-used.
[00117] Compound 3 and trityl chloride were added to the reaction flask. DCM (10 vol) was added to the reactor and agitation was started to form slurry. The reaction mixture was cooled to 10-15 °C. N,N-Diisopropylethylamine (2.5 equiv) was slowly added to the reaction mixture, maintaining the temperature at 15-25 °C during the addition. Once addition was complete, the batch was stirred at 15 to 25 °C for a minimum of 60 minutes. The reaction was assayed by HPLC by diluting a sample with acetonitrile and then injecting it on the HPLC. The first assay after 30 minutes indicated that the reaction was complete with <1% of starting material observed by HPLC analysis. The reaction mixture was diluted with DI water (5 vol). The reaction mixture was stirred for 5 minutes after which it was transferred into a separation funnel and the phases were allowed to separate. The DCM layer was washed with DI water (5 vol) by stirring for 5 minutes and then allowing the phases to separate. The DCM layer was washed with brine (5 vol) by stirring for 5 minutes and then allowing the phases to separate. The DCM layer was dried over magnesium sulfate, filtered and the filter cake was washed with DCM (x2). The combined filtrate and washes were concentrated to a residue that was azeotroped with EtOAc (x2). The residue was suspended in EtOAc and stirred for 1 hour in a 40 °C water bath. The resulting slurry was cooled to 0-5 °C for 1 hour and then filtered. The filter cake was washed twice with EtOAc and then dried under reduced pressure to afford Compound 4.
[00118] Exam le IB

21 4″
[00119] To 1, 2,3, 4-tetrahydro-6-hydroxy-isoqino line in acetonitrile was added p- toluenesulfonic acid and N-chlorosuccinimide. The suspension was cooled to ambient temperature, and the product isolated by filtration for a yield of approximately 61% with purity greater than 95%. The isolated TsOH salt was recrystallized until purity was greater than 99.7%. To one equivalent of the TsOH salt suspended in methanol was added 2M sodium carbonate (0.55 eq.) and 1.2 eq. of Boc anhydride. The suspension was stirred at room temperature overnight. The reaction was monitored by HPLC. Upon completion, the mixture was cooled to below 10 °C, water was added, and the Boc-protected dichloro compound was isolated by filtraton. The product was washed and dried at 40 °C for a yield of 95% and purity of >97%. The Boc-protected dichloro compound was suspended in dichloromethane (10 volumes) and pyridine (5 volumes) was added. The mixture was cooled to below 2 °C, and triflic anhydride (1.25 eq) was added. The mixture was stirred at 0-2 °C for 10 minutes, and then poured into 10 volumes of 6%) aqueous sodium hydrogen carbonate solution. After washing with dichloromethane, the organic phases were combined and dried over magnesium sulphate. Following purification, the product (Compound 4′) was obtained in 90% yield and >98% purity. Compound 4′ was dissolved in dimethylformamide and methanol at room temperature. Diisopropylamine (4 eq) was added. Under CO atmosphere, l,3-bis(diphenylphosphino)propane (0.1 eq) and palladium acetate (0.1 eq) was added. The reaction was heated to refiux, and monitored by HPLC. Upon near completion, the mixture was cooled to ambient temperature. Workup with water, ethyl aceate, and brine yielded Compound 4″, which was used without further purification. Compound 4″ was dissolved in methanol and 2.4 M sodium hydroxide (10 volumes each) and refiuxed. The mixture was cooled to ambient temperature, and toluene was added. Following aqueous workup, the pH was adjusted to 2.3 with 3M hydrochloric acid, and crude product was isolated by filtration in 53% yield with greater than 80% purity.
[00120] Exam le 2

Scheme E2
[00121] t-Butylcarbamate (Boc) protection of the amino group of bromophenyl alanine was accomplished, using sodium bicarbonate (3 equivalents), t-butyl dicarbonate (Boc20, 1.1 equivalent) in dioxane and water, to obtain compound 7 in 98% yield. A methyl sulfone functionality was introduced by treating the bromo compound 7 with copper iodide (0.4 equivalents), cesium carbonate (0.5 equivalents), L-proline (0.8 equivalents), and the sodium salt of methanesulfinic acid (3.9 equivalents) in dimethylsulfoxide (DMSO) at 95-100°C for a total of 9 hours, with two further additions of copper iodide (0.2 equivalents) and L-proline (0.4 equivalents) during that period. Compound 8 was isolated in 96%> yield. The carboxylic acid of compound 8 was converted to the benzyl ester, compound 9, in 99% yield, using benzyl alcohol (1.1 equivalent), dimethylaminopyridine (DMAP, 0.1 equivalent) and N-(3- dimethylaminopropyl)-N-ethylcarbodiimide (EDC, 1.0 equivalent). The amino group of compound 9 is deprotected by adding a 4N solution of HC1 in dioxane to compound 9 at 0°C in methylene chloride. The HCl salt of the free amino species, compound 10 was isolated in 94% yield.
[00122] Example 2 A
[00123] Example 2 was repeated with potassium carbonate in place of cesium carbonate.
[00124] Example 2B
[00125] Boc-protected bromophenylalanine (Compound 7) (100g) was dissolved in
DMSO (400 mL) with stirring and degassing with argon. Sodium methane sulfmate (98g), copper iodide (28.7g), potassium carbonate (40 g) and L-proline (26.75g) were added at 28-30 °C. Reaction was heated to about 87 °C for about 17-19 hours. Reaction was cooled and quenched with crushed ice, stirred for 30-40 minutes, and the pH was adjusted from about 12 to about 3-4 with citric acid (350 g). Quenched reaction mixture was filtered, extracted with dichloromethane x3, washed with ammonium chloride solution, washed with sodium bisulphite solution, and washed with brine. Crude product in dichloromethane was concentrated in vacuo until moisture content was below about 0.5%, and used in next step without further isolation. Crude compound 8 in dichloromethane was charged with benzyl alcohol and DMPA with stirring under nitrogen. Reaction cooled to 0-5 °C. EDC-HCL (1.03 equiv) added with stirring for 30 minutes. Upon completion of reaction by TLC and HPLC, the reaction was quenched with sodium bicarbonate solution, the organic layer was separated, and the aqueous layer was extracted with dichloromethane. The organic layer was washed with citric acid solution, and combined organic layers were washed with brine solution. Dichloromethane was removed at 45- 50 °C, and the concentrate was used for next step without further isolation. The amino group of compound 9 was deprotected by adding a 4N solution of HCl in dioxane to compound 9 at 10- 15°C in methylene chloride. The HCl salt of the free amino species, compound 10 was isolated by filtration from diethyl ether. Isolation of compound 10 was performed through recrystallization using a dimethylformamide/dichloromethane solvent system.
[00126] Example 3

Scheme E3
[00127] Compound 5 was treated with triethylamine (TEA, 5 equivalents) and 2-(7-Aza- lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU, 1.25 equivalents) for 10 minutes in dimethylformamide (DMF), and then compound 10 was added to the solution. After stirring at room temperature for 18 hours, the product, compound 11 was isolated in 70% yield. Removal of the trityl protecting group was accomplished by treating compound 11, with HC1 in dioxane (4 N, excess) at room temperature for 2 hours, diethyl ether added, and the solid product, compound 12, was isolated by filtration in 95% yield. The compound 12 exists in both amorphous and crystalline form and can be isolated in either form.
[00128] Example 3 A
[00129] Compound 5 was dissolved in isopropyl acetate and cooled to 20 to 25 °C.
Thionyl chloride was added, with cooling to 10 to 15 °C, and N-methylmorpholine was added slowly. The reaction was monitored by HPLC. Compound 10, water, and isopropyl acetate were stirred at 15 to 20°C until a solution was achieved. N-methylmorpholine was added followed by addition of the Compound 5 reaction mixture (acid chloride of Compound 5). The reaction was monitored by HPLC. Upon completion, the biphasic layers were allowed to settle, and the aqueous layer was removed. The upper organic layer was extracted with water, and the remaining organic layer was distilled under vacuum. Dioxane and IpAc were added with further distillation. Once dry, 4N anhydrous HC1 in dioxane was added. The mixture was stirred at 20 to 25°C for 12 hours, and checked for complete deprotection by HPLC. Once complete, the thick slurry was filtered, washed with IP Ac and dried under vacuum at 45 to 55°C. Yield of Compound 12 was 88%.
[00130] Example 4
[00131] The benzofuranyl carbonyl moiety of the compound of Formula I was prepared using various schemes, (Schemes E4, E4A, and E4B).

15
Phenyl-bis-triflate

18 ‘
Scheme E4
[00132] The benzofuranyl carbonyl moiety was prepared by protecting the hydroxyl group of compound 13 by reacting with tert-butyldimethylsilyl chloride (1.0 equivalents) and triethylamine (TEA, 1.1 equivalents) in acetone, to give compound 14 in 79% yield. A solution of compound 14 in methanol was then treated with sodium borohydride (1.0 equivalent) at room temperature overnight. The reaction was quenched with an addition of acetone, stirred at room temperature for a further 2.5 hours, aqueous HCl (4N) was added with the temperature controlled to below 28 °C, tetrahydrofuran (THF) was added, and the solution stirred overnight under argon and in the absence of light. The product, compound 15, was isolated quantitatively by extraction into methylene chloride, concentrated at low heat, and used without further purification. The triflate ester, compound 16, was produced in 69% yield from compound 15 by reacting it with N- phenyl-bis(trifluoromethanesulfonimide) (1.0 equivalent) in methylene chloride for 72 hours. Compound 16 in a mixture of DMF, methanol, and triethylamine, was added to a prepared solution of palladium acetate, l,3-Bis(diphenylphosphino)propane (dppp), DMF and methanol in an autoclave. Carbon monoxide was charged into the autoclave to a pressure of 8 bar, and the reaction mixture was heated at 70 °C for 6 hours. After workup, compound 17 was isolated in 91% yield. Lithium hydroxide (4 equivalents) in methanol and water was used to hydro lyze the ester and permit the isolation of compound 18′ in 97% yield.
[00133] Example 4A
[00134] Example 4 was repeated with triflic anhydride and sodium hydroxide as reagents for the ester hydrolysis.
[00135] Compound 15 (6-Hydroxybenzofuran) was stirred in dichloromethane and diisopropylethylamine. Triflic anhydride (1.2 eq.) was added, keeping the temperature below 20C. The reaction was monitored by HPLC. The reaction was quenched with methanol, solvent was removed with vacuum, and the crude residue of Compound 16 was used without further purification. Compound 16 as crude residue was dissolved in 4 volumes of dimethylformamide and 2 volumes methanol. To the solution was added 0.02 eq. of palladium acetate, 0.02 eq. of dppp, and CO under pressure. The reaction was monitored by HPLC. Following workup, Compound 17 was isolated as a crude oily residue without further purification. The residue of compound 17 was dissolved in methanol (5 volumes) and 1 volume of sodium hydroxide (27.65%) was added. The mixture was heated to 40C until full conversion of HPLC. The mixture was cooled to ambient temperature and 3 volumes of water were added. The pH was adjusted to about 2 with 3M hydrochloric acid. The suspension was filtered, washed with water, and dried to give Compound 18’ in about 75% overall yield with purity >99.5%.
[00136] Example 4B

Scheme E4B [00137] Diethyl 2-(l,3-dioxolan-2-yl)ethylphosphonate, compound 1″, was prepared from
2-(2-bromoethyl)-l,3-dioxolane by the addition of triethyl phosphate. After removal of ethyl bromide through distillation at 210°C the crude reaction mixture was cooled and then by way of vacuum distillation, compound 1″ was collected as a colorless oil in 94% yield.
[00138] In the next step, n-butyllithium (2.15 equivalents) in hexane was cooled to -70 °C and diisopropylamine (2.25 equivalents) was added while keeping the temperature below -60 °C. Compound 1″ (1 equivalent) dissolved in tetrahydrofuran (THF) was added over 30 min at -70 °C. After 10 min, diethyl carbonate (1.05 equivalents) dissolved in THF was added over 30 min keeping the reaction temperature below -60 °C. After stirring for one hour at -60 °C, the reaction was allowed to warm to 15 °C and furan-2-carbaldehyde (1.3 equivalents) dissolved in THF was added. After stirring for 20 hrs at room temperature, the reaction was rotary evaporated to dryness to yield ethyl 2-((l,3-dioxolan2-yl)methyl-3-(furan-2-yl)acrylate, which was used directly in the next reaction.
[00139] The crude compound (1 equivalent) was dissolved in ethanol and added to a mixture of water and phosphoric acid (85%>, 15 equivalents) over 30 min while keeping the temperature below 50°C. After stirring for 20 hrs at room temperature, another 200 ml of phosphoric acid (85%>) was added and the mixture was heated to 50 °C for an additional two hrs.
After removal of ethanol by rotary evaporation, the material was extacted with toluene, washed with water, dried with sodium sulfate, treated with charcoal, filtered and dried down to an oil. This oil was distilled to afford ethyl benzofuran-6-carboxylate, compound 6″, (bp 111-114.5°C) which crystallized on standing. Compound 6″ was recovered at 57%> yield based on compound
1″.
[00140] Compound 6″ (875 mmol) was dissolved in methanol and tetrahydrofuran (THF).
Sodium hydroxide (4 M, 3 equivalents) was added and the reaction was stirred overnight. After concentration via rotary evaporation, the aqueous solution was extracted with methyl tert-butyl ether (MTBE), acidified to pH 2 with the addition of hydrochloric acid (HC1) and cooled resulting in fine crystals of benzofuran-6-carboxylic acid, i.e., compound 18′. Compound 18′ was isolated, washed with water and dried to a final yield of 97%> yield.
[00141] Example 5

10% Pd/C, HCOOH/NEt3
MeOH/THF 5:1

Form A of Formula I
Scheme E5
[00142] The benzofuran carboxylic acid 18′ was treated with oxalyl chloride (1.2 equivalents) and a catalytic amount of DMF, stirring for 5.5 hours until a clear solution was obtained. The solvent was removed under reduced pressure and the acid chloride of compound 18′ was stored under argon until use, on the next day. The acid chloride, in methylene chloride was added slowly to a methylene chloride solution of the compound of Formula 12 and diisopropylethylamine (DIPEA) which was cooled to 0-5 °C. The reaction was not permitted to rise above 5°C, and after completion of addition, was stirred at 5°C for a further 0.5 hour. Upon aqueous workup and extraction with methylene chloride, the product, compound 19, was isolated in quantitative yield.
[00143] The benzyl ester of compound 19 was removed by transfer hydrogenolysis using
10% palladium on carbon, using formic acid and triethylamine in a 5: 1 mixture of methanol:THF, to produce the compound of Formula I in 95% yield.
[00144] A final step of slurrying in methyl ethylketone (MEK) produced Form A of the compound of Formula I. The product was washed with water to remove residual MEK. Alternatively, the product of the hydrogenolysis step was slurried in acetonitrile to yield Form A of the compound of Formula I.
[00145] Taking the compound of Formula I directly as the crude reaction product after transfer hydrogenolysis, and reconcentrating down from a solution in methylene chloride, the amorphous form of the compound of Formula I was obtained in 97% purity.
[00146] Example 6
[00147] An alternative protection strategy was performed in Scheme E6.

Scheme E6
[00148] Boc-protection was used for the ring nitrogen in the intermediates 21 and 22.
Compound 5 was deprotected with HC1 in dioxane to produce compound 23 in better than 97%> yield. Boc-protection was introduced, using di-tert-butyl dicarbonate (1.1 equivalent), and compound 21 was obtained in better than 95% yield. Compound 10 was coupled with compound 21 to obtain compound 22, using HATU and triethylamine in DMF. The product, compound 22, was obtained in quantitative yield, and greater than 90% purity. Deprotection with HC1 yielded the compound of Formula 12 in 97.4% yield.
[00149] Transfer hydrogeno lysis of compound 19 produced the compound of Formula I with optical purity of 98.5% (S) enantiomer compared to 79-94.5% (S) enantiomer optical purity obtained by hydrolysis of the corresponding methyl ester.
PATENT
http://www.google.co.in/patents/WO2009139817A2?cl=en
The compound of Formula I:

Formula I has been found to be an effective inhibitor of Lymphocyte Function- Associated Antigen -1 (LFA-I) interactions with the family of Intercellular Adhesion Molecules (ICAM), and has desirable pharmacokinetic properties, including rapid systemic clearance. Improved forms, including crystalline forms, and their uses in treatment of disorders mediated by the interaction of LFA-I and ICAM are described herein. Novel polymorphs of the compound of Formula I which may afford improved purity, stability, bioavailability and other like characteristics for use in pharmaceutical formulations and methods of use thereof are useful in treating disease.
Sch
Scheme 1

[00174] The first alternative protecting strategy produces compound 5, a trityl protected species as shown in Scheme 1. The synthesis begins by reductively aminating 3, 5, dichlorobenzaldehyde, compound 1, with 1-chloro- 2-aminoethane and sodium cyanoborohydride in 35% yield. Cyclization of compound 2 using aluminum chloride catalysis and ammoniun chloride at 185°C provided compound 3 in 91% yield. Protection of the free amine of compound 3 as the trityl protected species afforded compound 4 in 89% yield. A carboxylic acid functionality was introduced by treatment of compound 4 with n-butyllithium and Tetramethylethylenediamine (TMEDA), with subsequent introduction of carbon dioxide, to produce compound 5 in 75% yield. Scheme 2
l

[00175] Bromophenylalanine was used as the starting material for the right hand portion of the final molecule as shown in Scheme 2. t-Butylcarbamate (Boc) protection of the amino group was accomplished, using sodium bicarbonate (3 equivalents), f-butyl dicarbonate (BoC2O, 1.1 equivalent) in dioxane and water, to obtain compound
7 in 98% yield. A methyl sulfone functionality was introduced by treating the bromo compound 7 with copper iodide (0.4 equivalents), cesium carbonate (0.5 equivalents), L-proline ( 0.8 equivalents), and the sodium salt of methanesulfinic acid (3.9 equivalents) in dimethylsulfoxide (DMSO) at 95-1000C for a total of 9 hours , with two further additions of copper iodide (0.2 equivalents) ) and L-proline ( 0.4 equivalents) during that period. Compound
8 was isolated in 96% yield. The carboxylic acid of compound 8 was converted to the benzyl ester, compound 9, in 99% yield, using benzyl alcohol (1.1 equivalent), dimethylaminopyridine (DMAP, 0.1 equivalent) andN-(3- dimethylaminopropyl)-N-ethylcarbodiimide (EDC, 1.0 equivalent). The amino group of compound 9 is deprotected by adding a 4N solution of HCl in dioxane to compound 9 at O0C in methylene chloride. The HCl salt of the free amino species, compound 10 was isolated in 94% yield.
Scheme 3

DMF

[00176] Compound 5 was treated with triethylamine (TEA, 5 equivalents) and 2-(7-Aza-lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium hexafluorophosphate (HATU, 1.25 equivalents) for 10 minutes in dimethylformamide (DMF), and then compound 10 was added to the solution. After stirring at room temperature for 18 hours, the product, compound 11 was isolated in 70% yield. Removal of the trityl protecting group was accomplished by treating compound 1, with HCL in dioxane (4N, excess) at room temperature for 2 hours, diethyl ether added, and the solid product, compound 12, was isolated by filtration in 95% yield.
scheme 4


The benzofuranyl carbonyl moiety of the compound of Formula I was prepared by protecting the hydroxyl group of compound 13 by reacting with tert- butyldimethylsilyl chloride (1.0 equivalents) and TEA (1.1 equivalents) in acetone, to give compound 14 in 79% yield. A solution of compound 14 in methanol was then treated with sodium borohydride (1.0 equivalent) at room temperature overnight. The reaction was quenched with an addition of acetone, stirred at room temperature for a further 2.5 hours, aqueous HCl (4N) was added with the temperature controlled to below 28C, tetrahydrofuran (THF) was added, and the solution stirred overnight under argon and in the absence of light. The product, compound 15, was isolated quantitatively by extraction into methylene chloride, concentrated at low heat, and used without further purification. The triflate ester, compound 16, was produced in 69% yield from compound 15 by reacting it with N- phenyl-bis(trifluoromethanesulfonimide) (1.0 equivalent) in methylene chloride for 72 hours. Compound 16 in a mixture of DMF, methanol, and triethylamine, was added to a prepared solution of palladium acetate, diphenyl, DMF and methanol in an autoclave. Carbon monoxide was charged into the autoclave to a pressure of 8 bar, and the reaction mixture was heated at 700C for 6 hours. After workup, compound 17 was isolated in 91% yield. Lithium hydroxide (4 equivalents) in methanol and water was used to hydrolyze the ester and permit the isolation of compound 18 in 97% yield.
Scheme 5

10% Pd/C, HCOOH/NEt3 MeOH/THF 5:l

Form A of Formula I
The benzofuran carboxylic acid 18 was treated with oxalyl chloride (1.2 equivalents) and a catalytic amount of DMF, stirring for 5.5 hours until a clear solution was obtained. The solvent was removed under reduced pressure and the acid chloride of compound 18 was stored under argon until use, on the next day. The acid chloride, in methylene chloride was added slowly to a methylene chloride solution of the compound of Formula I and diisopropylethylamine (DIPEA) which was cooled to 0-50C. The reaction was not permitted to rise above 5°C, and after completion of addition, was stirred at 5°C for a further 0.5 hour. Upon aqueous workup and extraction with methylene chloride, the product, compound 19, was isolated in quantitative yield. The benzyl ester of compound 19 was removed by transfer hydrogenolysis using 10% palladium on carbon, using formic acid and triethylamine in a 5:1 mixture of methanol:THF, to produce the compound of Formula I in 95% yield. A final step of slurrying in methyl ethylketone (MEK) produced Form A of the compound of Formula I. The product was washed with water to remove residual MEK. Alternatively, the product of the hydrogenolysis step was slurried in acetonitrile yielded Form A of the compound of Formula I.
[00179] Taking the compound of Formula I directly as the crude reaction product after transfer hydrogenolysis, and reconcentrating down from a solution in methylene chloride, the amorphous form of the compound of Formula I was obtained in 97% purity.
Scheme 6
DMF

12 22
In this alternative approach, Boc-protection was used for the ring nitrogen in the intermediates 20, 21, and 22. Compound 5 was deprotected with HCl in dioxane to produce compound 20 in better than 97% yield. Boc- protection was introduced, using di-tert-butyl dicarbonate (1.1 equivalent), and compound 21 was obtained in better than 95% yield. Compound 10 was coupled with compound 21 to obtain compound 22, using HATU and triethylamine in DMF. The product, compound 22, was obtained in quantitative yield, and greater than 90% purity. Deprotection with HCl yields the compound of Formula I in 97.4% yield and the synthesis rejoins the process described in Scheme 5.
clip
Lifitegrast clinical efficacy for treatment of signs and symptoms of dry eye disease across three randomized controlled trials
Current Medical Research and Opinion (2016), 32, (10), 1759-1765. Publisher: (Taylor & Francis Ltd., )
PAPER
LFA-1/ICAM-1 interaction is essential in support of inflammatory and specific T-cell regulated immune responses by mediating cell adhesion, leukocyte extravasation, migration, antigen presentation, formation of immunological synapse, and augmentation of T-cell receptor signaling. The increase of ICAM-1 expression levels in conjunctival epithelial cells and acinar cells was observed in animal models and patients diagnosed with dry eye. Therefore, it has been hypothesized that small molecule LFA-1/ICAM-1 antagonists could be an effective topical treatment for dry eye. In this letter, we describe the discovery of a potent tetrahydroisoquinoline (THIQ)-derived LFA-1/ICAM-1 antagonist (SAR 1118) and its development as an ophthalmic solution for treating dry eye.
http://pubs.acs.org/doi/suppl/10.1021/ml2002482/suppl_file/ml2002482_si_001.pdf

| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US8927574 | No | 2010-11-12 | 2030-11-12 |  |
| US7928122 | No | 2004-11-05 | 2024-11-05 | |
| US9085553 | No | 2013-07-25 | 2033-07-25 | |
| US9353088 | No | 2010-10-21 | 2030-10-21 | |
| US9216174 | No | 2004-11-05 | 2024-11-05 | |
| US7314938 | No | 2005-03-10 | 2025-03-10 | |
| US8084047 | No | 2006-05-17 | 2026-05-17 | |
| US8367701 | No | 2009-04-15 | 2029-04-15 | |
| US8168655 | No | 2009-05-09 | 2029-05-09 | |
| US8592450 | No | 2006-05-17 | 2026-05-17 | |
Proprietary Name: XIIDRA
Dosage Form; Route of Administration: SOLUTION/DROPS; OPHTHALMIC
Strength: 5%
Reference Listed Drug: Yes
Reference Standard: Yes
TE Code:
Application Number: N208073
Product Number: 001
Approval Date: Jul 11, 2016
Applicant Holder Full Name: SHIRE DEVELOPMENT LLC
Marketing Status: Prescription
Patent and Exclusivity Information
Patent and Exclusivity for: N208073
“ALL FOR DRUGS, New drug approvals, Drug approvals international, ” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
///////
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8084047 * | Jul 23, 2009 | Dec 27, 2011 | Sarcode Bioscience Inc. | Compositions and methods for treatment of eye disorders |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8367701 | Nov 4, 2011 | Feb 5, 2013 | Sarcode Bioscience Inc. | Crystalline pharmaceutical and methods of preparation and use thereof |
| US8592450 | Feb 16, 2012 | Nov 26, 2013 | Sarcode Bioscience Inc. | Compositions and methods for treatment of eye disorders |
| US8758776 | Jan 21, 2011 | Jun 24, 2014 | Sarcode Bioscience Inc. | Compositions and methods for treatment |
| US8771715 | Jan 21, 2011 | Jul 8, 2014 | Sarcode Bioscience Inc. | Compositions and methods for treatment |
| WO2012121659A1 * | Mar 8, 2012 | Sep 13, 2012 | Kat2Biz Ab C/O Interpares Konsult Ab | Reduction of c-0 bonds by catalytic transfer hydrogenolysis |
| WO2014018748A1 * | Jul 25, 2013 | Jan 30, 2014 | Sarcode Bioscience Inc. | Lfa-1 inhibitor and polymorph thereof |
Sage Therapeutics receives fast track designation for status epilepticus therapy

Sage Therapeutics (Originator)
For Epilepsy, status epilepticus
SGE-102; SAGE-547; allopregnanolone; allosteric GABA A receptor modulators (CNS disorders),
Sage Therapeutics receives fast track designation for status epilepticus therapy
Ligand Pharmaceuticals announced that its partner Sage Therapeutics has received fast track designation from the US Food and Drug Administration (FDA) for the Captisol-enabled SAGE-547 to treat status epilepticus.
read at
| Chemical Name: (3α)-Allopregnanolone | ||
| Synonyms: (+)-3α-Hydroxy-5α-pregnan-20-one; (3α,5α)-3-Hydroxypregnan-20-one; 3α,5α-THP; 3α,5α-Tetrahydroprogesterone; 3α-Hydroxy-5α-dihydroprogesterone; 3α-Hydroxy-5α-pregnan-20-one; 3α-Hydroxy-5α-pregnane-20-one; 5α-Pregnan-3α-ol-20-one; 5α-Pregnane-3α-ol-20-one; Allopregnan-3α-ol-20-one; Allopregnanolone; Allotetrahydroprogesterone; | ||
| CAS Number: 516-54-1 | ||
|
||
| Mol. Formula: C21H34O2 | ||
| Appearance: White Solid | ||
| Melting Point: 174-176°C | ||
| Mol. Weight: 318.49 |
SAGE-547 is a GABA(A) receptor modulator in phase I/II clinical trials at Sage Therapeutics as adjunctive therapy for the treatment of adults with super-refractory status epilepticus (SRSE).
In 2014, orphan drug designation was assigned in the U.S for the treatment of status epilepticus. In July 2014, fast track designation was received in the U.S. for the treatment of adults with super-refractory status epilepticus (SRSE).
July 22, 2014
SAGE Therapeutics, a biopharmaceutical company developing novel medicines to treat life-threatening, rare central nervous system (CNS) disorders, announced today that the U.S. Food and Drug Administration (FDA) has granted fast track designation to the SAGE-547 development program. SAGE-547 is an allosteric modulator of GABAA receptors in development for the treatment of adult patients with refractory status epilepticus who have not responded to standard regimens (super-refractory status epilepticus, or SRSE). SAGE is currently evaluating SAGE-547 in a Phase 1/2 clinical trial for the treatment of SRSE. Preliminary data indicate that the first four patients enrolled in the clinical trial met the key efficacy endpoint, in that each was successfully weaned off his or her anesthetic agent while SAGE-547 was being administered. There have also been no reported drug-related serious adverse events in these four patients to date.
“The fast track designation for SAGE-547 recognizes the significant unmet need that exists in the treatment of super-refractory status epilepticus,” said Jeff Jonas, MD, chief executive officer of SAGE Therapeutics. “The receipt of orphan drug designation earlier this year for status epilepticus and the fast track designation are both significant regulatory milestones for SAGE-547, and we will continue to work closely with the FDA to advance our lead compound and the additional programs in our pipeline for the treatment of life-threatening CNS disorders.”
Fast track designation is granted by the FDA to facilitate the development and expedite the review of drug candidates that are intended to treat serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs.
About SAGE-547
SAGE-547 is an allosteric modulator of both synaptic and extra-synaptic GABAA receptors. GABAA receptors are widely regarded as validated drug targets for a variety of CNS disorders, with decades of research and multiple approved drugs targeting these receptor systems. SAGE-547 is an intravenous agent in Phase 1/2 clinical development as an adjunctive therapy, a therapy combined with current therapeutic approaches, for the treatment of SRSE.
About Status Epilepticus (SE)
SE is a life-threatening seizure condition that occurs in approximately 150,000 people each year in the U.S., of which 30,000 SE patients die.1 We estimate that there are 35,000 patients with SE in the U.S. that are hospitalized in the intensive care unit (ICU) each year. An SE patient is first treated with benzodiazepines, and if no response, is then treated with other, second-line, anti-seizure drugs. If the seizure persists after the second-line therapy, the patient is diagnosed as having refractory SE (RSE), admitted to the ICU and placed into a medically induced coma. Currently, there are no therapies that have been specifically approved for RSE; however, physicians typically use anesthetic agents to induce the coma and stop the seizure immediately. After a period of 24 hours, an attempt is made to wean the patient from the anesthetic agents to evaluate whether or not the seizure condition has resolved. Unfortunately, not all patients respond to weaning attempts, in which case the patient must be maintained in the medically induced coma. At this point, the patient is diagnosed as having SRSE. Currently, there are no therapies specifically approved for SRSE.
About SAGE Therapeutics
SAGE Therapeutics (NASDAQ: SAGE) is a biopharmaceutical company committed to developing and commercializing novel medicines to treat life-threatening, rare CNS disorders. SAGE’s lead program, SAGE-547, is in clinical development for super-refractory status epilepticus and is the first of several compounds the company is developing in its portfolio of potential seizure medicines. SAGE’s proprietary chemistry platform has generated multiple new compounds that target GABAA and NMDA receptors, which are broadly accepted as impacting many psychiatric and neurological disorders. SAGE Therapeutics is a public company launched in 2010 by an experienced team of R&D leaders, CNS experts and investors. For more information, please visitwww.sagerx.com.
| Allopregnanolone | |
|---|---|
 |
|
| Identifiers | |
| PubChem | 262961 |
| ChemSpider | 17216124 |
| ChEMBL | CHEMBL38856 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C21H34O2 |
| Molar mass | 318.49 g/mol |
Allopregnanolone (3α-hydroxy-5α-pregnan-20-one or 3α,5α-tetrahydroprogesterone), generally abbreviated as ALLO or as 3α,5α-THP, is an endogenous inhibitory pregnane neurosteroid.[1] It is synthesized from progesterone, and is a potent positive allosteric modulator of the GABAA receptor.[1] Allopregnanolone has effects similar to those of other potentiators of the GABAA receptor such as the benzodiazepines, including anxiolytic, sedative, and anticonvulsant activity.[1]
The 21-hydroxylated derivative of this compound, tetrahydrodeoxycorticosterone (THDOC), is an endogenous inhibitory neurosteroid with similar properties to those of allopregnanolone, and the 3β-methyl analogue of allopregnanolone, ganaxolone, is under development to treat epilepsy and other conditions.[1]
Biosynthesis
The biosynthesis of allopregnanolone starts with the conversion of progesterone into 5α-dihydroprogesterone by 5α-reductase type I. After that, 3α-hydroxysteroid dehydrogenase converts this intermediate into allopregnanolone.[1]
Depression, anxiety, and sexual dysfunction are frequently-seen side effects of 5α-reductase inhibitors such as finasteride, and are thought to be caused, in part, by interfering with the normal production of allopregnanolone.[2]
Mechanism
Allopregnanolone acts as a potent positive allosteric modulator of the GABAA receptor.[1] While allopregnanolone, like other inhibitory neurosteroids such as THDOC, positively modulates all GABAA receptor isoforms, those isoforms containing δ subunits exhibit the greatest potentiation.[1] Allopregnanolone has also been found to act as a positive allosteric modulator of the GABAA-ρ receptor, though the implications of this action are unclear.[3][4] In addition to its actions on GABA receptors, allopregnanolone, like progesterone, is known to be a negative allosteric modulator of nACh receptors,[5] and also appears to act as a negative allosteric modulator of the 5-HT3 receptor.[6] Along with the other inhibitory neurosteroids, allopregnanolone appears to have little or no action at other ligand-gated ion channels, including the NMDA, AMPA, kainate, and glycine receptors.[7]
Unlike progesterone, allopregnanolone is inactive at the nuclear progesterone receptor (nPR).[7] However, allopregnanolone can be intracellularly oxidized into 5α-dihydroprogesterone, which is an agonist of the nPR, and thus/in accordance, allopregnanolone does appear to have indirect nPR-mediated progestogenic effects.[8] In addition, allopregnanolone has recently been found to be an agonist of the newly-discovered membrane progesterone receptors (mPR), including mPRδ, mPRα, and mPRβ, with its activity at these receptors about a magnitude more potent than at the GABAA receptor.[9][10] The action of allopregnanolone at these receptors may be related, in part, to its neuroprotective and antigonadotropic properties.[9][11] Also like progesterone, recent evidence has shown that allopregnanolone is an activator of the pregnane X receptor.[7][12]
Similarly to many other GABAA receptor positive allosteric modulators, allopregnanolone has been found to act as an inhibitor of L-type voltage-gated calcium channels (L-VGCCs),[13] including α1 subtypes Cav1.2 and Cav1.3.[14] However, the threshold concentration of allopregnanolone to inhibit L-VGCCs was determined to be 3 μM (3,000 nM), which is far greater than the concentration of 5 nM that has been estimated to be naturally produced in the human brain.[14] Thus, inhibition of L-VGCCs is unlikely of any actual significance in the effects of endogenous allopregnanolone.[14] Also, allopregnanolone, along with several other neurosteroids, has been found to activate the G protein-coupled bile acid receptor (GPBAR1, or TGR5).[15] However, it is only able to do so at micromolar concentrations, which, similarly to the case of the L-VGCCs, are far greater than the low nanomolar concentrations of allopregnanolone estimated to be present in the brain.[15]
Function
Allopregnanolone possesses a wide variety of effects, including, in no particular order, antidepressant, anxiolytic, stress-reducing, rewarding,[16] prosocial,[17] antiaggressive,[18] prosexual,[17] sedative, pro-sleep,[19] cognitive and memory-impairing, analgesic,[20] anesthetic, anticonvulsant, neuroprotective, and neurogenic effects.[1]
Fluctuations in the levels of allopregnanolone and the other neurosteroids seem to play an important role in the pathophysiology of mood, anxiety, premenstrual syndrome, catamenial epilepsy, and various other neuropsychiatric conditions.[21][22][23]
Increased levels of allopregnanolone can produce paradoxical effects, including negative mood, anxiety, irritability, and aggression.[24][25][26] This appears to be because allopregnanolone possesses biphasic, U-shaped actions at the GABAA receptor – moderate level increases (in the range of 1.5–2 nM/L total allopregnanolone, which are approximately equivalent to luteal phase levels) inhibit the activity of the receptor, while lower and higher concentration increases stimulate it.[24][25] This seems to be a common effect of many GABAA receptor positive allosteric modulators.[26][21] In accordance, acute administration of low doses of micronized progesterone (which reliably elevates allopregnanolone levels), have been found to have negative effects on mood, while higher doses have a neutral effect.[27]
Therapeutic applications
Allopregnanolone and the other endogenous inhibitory neurosteroids have very short half-lives, and for this reason, have not been pursued for clinical use themselves. Instead, synthetic analogs with improved pharmacokinetic profiles, such as ganaxolone, have been synthesized and are being investigated. However, exogenous progesterone, such as oral micronized progesterone (OMP), reliably elevates allopregnanolone levels in the body with good dose-to-serum level correlations.[28] Due to this, it has been suggested that OMP could be described as a prodrug of sorts for allopregnanolone.[28] As a result, there has been some interest in using OMP to treat catamenial epilepsy,[29] as well as other menstrual cycle-related and neurosteroid-associated conditions.
……………………………………….
http://www.google.com/patents/WO2006037016A2?cl=en
Materials and Methods
[0181] The materials and methods used for the follwing experiments have been described in Griffin L.D., et al, Nature Medicine 10: 704-711 (2004). This reference is hereby incorporated by reference in its entirety.
Example 1: Allopregnanolone Treatment of Niemann Pick type-C Mice Substantially Reduces Accumulation of the Gangliosides GMl, GM2, and GM3 in the Brain [0182] Mice were given a single injection of allopregnanolone, prepared in 20% βcyclodextrin in phosphate buffered saline, at a concentration of 25 mg/kg. The injection was on day 7 of life (P7, postnatal day 7). Concentrations of gangliosides GMl, GM2, GM3, were measured as well as other lipids such as ceramides and cerebrosides.
…………………………………………….
WO-2014031792 OR EQ
http://www.google.com/patents/US20140057885?cl=en
…………………………………….
WO-2013112605
http://www.google.com/patents/WO2013112605A2?cl=en
References
- Reddy DS (2010). “Neurosteroids: endogenous role in the human brain and therapeutic potentials”. Prog. Brain Res. 186: 113–37. doi:10.1016/B978-0-444-53630-3.00008-7. PMC 3139029. PMID 21094889.
- Römer B, Gass P (December 2010). “Finasteride-induced depression: new insights into possible pathomechanisms”. J Cosmet Dermatol 9 (4): 331–2. doi:10.1111/j.1473-2165.2010.00533.x. PMID 21122055.
- Morris KD, Moorefield CN, Amin J (October 1999). “Differential modulation of the gamma-aminobutyric acid type C receptor by neuroactive steroids”. Mol. Pharmacol. 56 (4): 752–9. PMID 10496958.
- Li W, Jin X, Covey DF, Steinbach JH (October 2007). “Neuroactive steroids and human recombinant rho1 GABAC receptors”. J. Pharmacol. Exp. Ther. 323 (1): 236–47. doi:10.1124/jpet.107.127365. PMID 17636008.
- Bullock AE, Clark AL, Grady SR, et al. (June 1997). “Neurosteroids modulate nicotinic receptor function in mouse striatal and thalamic synaptosomes”. J. Neurochem. 68 (6): 2412–23. PMID 9166735.
- Wetzel CH, Hermann B, Behl C, et al. (September 1998). “Functional antagonism of gonadal steroids at the 5-hydroxytryptamine type 3 receptor”. Mol. Endocrinol. 12 (9): 1441–51. doi:10.1210/mend.12.9.0163. PMID 9731711.
- Mellon SH (October 2007). “Neurosteroid regulation of central nervous system development”. Pharmacol. Ther. 116 (1): 107–24. doi:10.1016/j.pharmthera.2007.04.011. PMC 2386997. PMID 17651807.
- Rupprecht R, Reul JM, Trapp T, et al. (September 1993). “Progesterone receptor-mediated effects of neuroactive steroids”. Neuron 11 (3): 523–30. PMID 8398145.
- Thomas P, Pang Y (2012). “Membrane progesterone receptors: evidence for neuroprotective, neurosteroid signaling and neuroendocrine functions in neuronal cells”. Neuroendocrinology 96 (2): 162–71. doi:10.1159/000339822. PMC 3489003. PMID 22687885.
- Pang Y, Dong J, Thomas P (January 2013). “Characterization, neurosteroid binding and brain distribution of human membrane progesterone receptors δ and {epsilon} (mPRδ and mPR{epsilon}) and mPRδ involvement in neurosteroid inhibition of apoptosis”. Endocrinology 154 (1): 283–95. doi:10.1210/en.2012-1772. PMC 3529379. PMID 23161870.
- Sleiter N, Pang Y, Park C, et al. (August 2009). “Progesterone receptor A (PRA) and PRB-independent effects of progesterone on gonadotropin-releasing hormone release”. Endocrinology 150 (8): 3833–44. doi:10.1210/en.2008-0774. PMC 2717864. PMID 19423765.
- Lamba V, Yasuda K, Lamba JK, et al. (September 2004). “PXR (NR1I2): splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators”. Toxicol. Appl. Pharmacol. 199 (3): 251–65. doi:10.1016/j.taap.2003.12.027. PMID 15364541.
- Hu AQ, Wang ZM, Lan DM, et al. (July 2007). “Inhibition of evoked glutamate release by neurosteroid allopregnanolone via inhibition of L-type calcium channels in rat medial prefrontal cortex”. Neuropsychopharmacology 32 (7): 1477–89. doi:10.1038/sj.npp.1301261. PMID 17151597.
- Earl DE, Tietz EI (April 2011). “Inhibition of recombinant L-type voltage-gated calcium channels by positive allosteric modulators of GABAA receptors”. J. Pharmacol. Exp. Ther. 337 (1): 301–11. doi:10.1124/jpet.110.178244. PMC 3063747. PMID 21262851.
- Keitel V, Görg B, Bidmon HJ, et al. (November 2010). “The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain”. Glia 58 (15): 1794–805. doi:10.1002/glia.21049. PMID 20665558.
- Rougé-Pont F, Mayo W, Marinelli M, Gingras M, Le Moal M, Piazza PV (July 2002). “The neurosteroid allopregnanolone increases dopamine release and dopaminergic response to morphine in the rat nucleus accumbens”. Eur. J. Neurosci. 16 (1): 169–73. PMID 12153544.
- Frye CA (December 2009). “Neurosteroids’ effects and mechanisms for social, cognitive, emotional, and physical functions”. Psychoneuroendocrinology. 34 Suppl 1: S143–61. doi:10.1016/j.psyneuen.2009.07.005. PMC 2898141. PMID 19656632.
- Pinna G, Costa E, Guidotti A (February 2005). “Changes in brain testosterone and allopregnanolone biosynthesis elicit aggressive behavior”. Proc. Natl. Acad. Sci. U.S.A. 102 (6): 2135–40. doi:10.1073/pnas.0409643102. PMC 548579. PMID 15677716.
- Terán-Pérez G, Arana-Lechuga Y, Esqueda-León E, Santana-Miranda R, Rojas-Zamorano JÁ, Velázquez Moctezuma J (October 2012). “Steroid hormones and sleep regulation”. Mini Rev Med Chem 12 (11): 1040–8. PMID 23092405.
- Patte-Mensah C, Meyer L, Taleb O, Mensah-Nyagan AG (February 2014). “Potential role of allopregnanolone for a safe and effective therapy of neuropathic pain”. Prog. Neurobiol. 113: 70–8. doi:10.1016/j.pneurobio.2013.07.004. PMID 23948490.
- Bäckström T, Andersson A, Andreé L, et al. (December 2003). “Pathogenesis in menstrual cycle-linked CNS disorders”. Ann. N. Y. Acad. Sci. 1007: 42–53. PMID 14993039.
- Guille C, Spencer S, Cavus I, Epperson CN (July 2008). “The role of sex steroids in catamenial epilepsy and premenstrual dysphoric disorder: implications for diagnosis and treatment”. Epilepsy Behav 13 (1): 12–24. doi:10.1016/j.yebeh.2008.02.004. PMID 18346939.
- Finocchi C, Ferrari M (May 2011). “Female reproductive steroids and neuronal excitability”. Neurol. Sci. 32 Suppl 1: S31–5. doi:10.1007/s10072-011-0532-5. PMID 21533709.
- Bäckström T, Haage D, Löfgren M, et al. (September 2011). “Paradoxical effects of GABA-A modulators may explain sex steroid induced negative mood symptoms in some persons”. Neuroscience 191: 46–54. doi:10.1016/j.neuroscience.2011.03.061. PMID 21600269.
- Andréen L, Nyberg S, Turkmen S, van Wingen G, Fernández G, Bäckström T (September 2009). “Sex steroid induced negative mood may be explained by the paradoxical effect mediated by GABAA modulators”. Psychoneuroendocrinology 34 (8): 1121–32. doi:10.1016/j.psyneuen.2009.02.003. PMID 19272715.
- Bäckström T, Bixo M, Johansson M, et al. (February 2014). “Allopregnanolone and mood disorders”. Prog. Neurobiol. 113: 88–94. doi:10.1016/j.pneurobio.2013.07.005. PMID 23978486.
- Andréen L, Sundström-Poromaa I, Bixo M, Nyberg S, Bäckström T (August 2006). “Allopregnanolone concentration and mood–a bimodal association in postmenopausal women treated with oral progesterone”. Psychopharmacology (Berl.) 187 (2): 209–21. doi:10.1007/s00213-006-0417-0. PMID 16724185.
- Andréen L, Spigset O, Andersson A, Nyberg S, Bäckström T (June 2006). “Pharmacokinetics of progesterone and its metabolites allopregnanolone and pregnanolone after oral administration of low-dose progesterone”. Maturitas 54 (3): 238–44. doi:10.1016/j.maturitas.2005.11.005. PMID 16406399.
- Orrin Devinsky; Steven Schachter; Steven Pacia (1 January 2005). Complementary and Alternative Therapies for Epilepsy. Demos Medical Publishing. pp. 378–. ISBN 978-1-934559-08-6.
Additional reading
- Herd, MB; Belelli, D; Lambert, JJ (2007). Neurosteroid modulation of synaptic and extrasynaptic GABA(A) receptors. Pharmacol. Ther. 116(1):20-34. doi:10.1016/j.pharmthera.2007.03.007.
BMS-582949 in phase 2 for Treatment of Antipsoriatics , Rheumatoid arthritis
BMS 582949, PS-540446
UNII-CR743OME9E
CAS 623152-17-0
4-[5-(N-Cyclopropylcarbamoyl)-2-methylphenylamino]-5-methyl-N-propylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide
4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide
Bristol-Myers Squibb Company
M.Wt: 406.48
Cas : 623152-17-0 Formula: C22H26N6O2
BMS-582949 had been in phase II clinical trials at Bristol-Myers Squibb for the oral treatment of moderate to severe psoriasis and for the treatment of rheumatoid arthritis (RA) in combination with methotrexate and for the treatment of inflammation in atherosclerotic plaque. However, no recent development has been reported for this research.
…………………..
http://www.google.com/patents/WO2012031057A1?cl=en
The present invention generally relates to a method of treating resistant rheumatic disease, such as refractory rheumatoid arthritis, with a therapeutically effective amount of a dual action p38 inhibitor that is safe and well-tolerated. A dual action p38 kinase inhibitor is a compound that inhibits both activation of p38 kinase and p38 kinase activity in cells.
A large number of cytokines participate in the inflammatory response, including IL- 1 , IL-6, IL-8 and TNF-a. Overproduction of cytokines such as IL-1 and TNF-a are implicated in a wide variety of diseases, including inflammatory bowel disease, rheumatoid arthritis, psoriasis, multiple sclerosis, endotoxin shock, osteoporosis, Alzheimer’s disease, and congestive heart failure, among others. See e.g., Henry et al., Drugs Fut. , 24: 1345- 1354 ( 1999); Salituro et al., Curr. Med. Ckem., 6:807-823 (1999)]. Important mediators of proinflammatory cytokines such as TNFct and IL-1 β,. as well as cellular responses to such cytokines production, are the mitogen-activated protein (MAP) kinases, and in particular, p38 kinase. See e.g., Schieven, G.L., “The biology of p38 kinase: a central role in inflammation”, Current Topics in Medicinal Chemistry, 5 :921 – 928 (2005). Accordingly, modulation of p38 kinase may be useful in the treatment of inflammatory disease including rheumatic diseases such as rheumatoid arthritis (RA).
Compounds that reportedly inhibit p38 kinase and cytokines such as IL-1 and TNF-a for use in treating inflammatory diseases are disclosed in U.S. Patent Nos.
6,277,989 and 6, 130,235 to Scios, Inc; U.S. Patent. Nos. 6, 147,080 and 5,945,41 8 to Vertex Pharmaceuticals Inc; U.S. Patent Nos. 6,251 ,914, 5,977, 103 and 5,658,903 to Smith-Kline Beecham Corp.; U.S. Patent Nos. 5,932,576 and 6,087,496 to G.D. Searle & Co.; WO 00/56738 and WO 01 /27089 to Astra Zeneca; WO 01/34605 to Johnson & Johnson; WO 00/12497 (quinazoHne derivatives as p38 kinase inhibitors); WO 00/56738 (pyridine and pyrimidine derivatives for the same purpose); WO 00/12497 (discusses the relationship between p38 kinase inhibitors); and WO 00/12074 (piperazine and piperidine compounds useful as p38 inhibitors). Other compounds that inhibit p38 kinase are pyrrolotriazine aniline compounds, information on these compounds is disclosed in U.S. Patent Nos. 6,670,357; 6,867,300; 7,034, 151 ; 7, 160,883; 7,21 1,666; 7,253, 167; and U.S. Publication Nos. 2003/023283 1 (published Dec. 18, 2003); 2004/0229877 (published Nov. 1 8, 2004); 2005/0043306 (published Feb. 24, 2005; 2006/0003967 (published Jan. 5, 2006); 2006/0030708 (published Feb. 9, 2006); 2006/0041 124 (published Feb. 23, 2006); 2006/0229449 (published Oct. 12, 2006); 2006/0235020 (published Oct. 19, 2006); and 2007/0213300 (published Sept 13, 2007).
In particular, WO 2003/090912 (U.S. Patent Nos. 7, 160,883, 7,388,009, p38 inhibitor, BMS-582949 (Example 7,

including processes of making and uses thereof.
……………………
http://www.google.com/patents/WO2003090912A9?cl=en
Examples 4-22

Compounds having the formula (Id), above, wherein R4 has the values listed in the following Table, were prepared following the same procedure described for Example 3, using the appropriate amine in place of ra-butylamine.



…………………………
WO 2006020904
http://www.google.com.br/patents/WO2006020904A1?cl=en
EXAMPLE IA St


Part a.
A solution of Example 1 (0.86 g, 2.20 mmol, 1.0 eq.) in THF (4.0 mL) and 1 N aqueous NaOH (9.0 mL, 4.1 eq.) was stirred at 6O0C overnight. After cooling to RT, the reaction mixture was concentrated in vacuo but not to dryness. To the solution at O0C was added 1 N aqueous hydrochloric acid until it was acidic and the precipitate was collected and dried to afford crude Example IA acid (0.51 g, 64.0 % yield). HPLC Ret. t. = 2.400 min.; LC/MS (M+H) + = 366.06+. The filtrate was then extracted with EtOAc (3x) and the organic layers were combined, dried over sodium sulfate, and concentrated in vacuo to give Example IA acid (0.035 g, 4.4 % yield). Part b.

A solution of Part a. acid (0.026 g, 0.071 mmol, 1.0 eq.), EDC (0.021 g, 0.11 mmol, 1.5 eq.), HOBt (0.015 g, 0.11 mmol, 1.5 eq), ^-propylamine (0.015 mL, 0.15 mmol, 2.1 eq.) and DIPEA (0.040 mL, 0.23 mmol, 3.2 eq.) in DMF (0.20 mL) was shaken at RT overnight. Water (1 mL) was added and the precipitate collected by filtration, washed with water, and dried to give Example IA amide (0.021 g, 70% yield); HPLC Ret. t. = 2.883 min.; LC/MS (M+H)+ = 421.18 +.
EJiAMPLE 2 Direct Aminolysis Procedure

n-Buli/THF
Ester Compound I or Hexyllithium/THF
-^
,NH9

1. Aminolysis with hexyllithium
To a dried 100 ml flask was added THF (10 ml) under nitrogen, which was then cooled to -100C. Hexyllithium (2.3 M in hexane, 6.5 ml, 15.0 mmol) was added slowly (exothermic, temperature was up to 5°C), followed by dropwise addition of propylamine (1.01 g, 1.4 ml, 17.1 mmol) at such a rate to maintain the temperature below 5°C. The resulting mixture was stirred at O0C for 20 minutes. A suspension of ester compound I (1.0 g, 2.5 mmol) in THF (12 ml) was added over a 10 minute period (exothermic, T<5°C). After being stirred at 00C for 20 minutes, the mixture was allowed to warm to room temperature and stirred for 5 hours. Ester compound I was <0.1 AP at this point by HPLC analysis. The mixture was cooled to -50C. Acetic acid (2 ml) was added slowly to maintain the temperature <10°C. The resulting thick slurry was stirred at room temperature for 20 minutes, and then solvents were exchanged with DMF (15 ml) on a rotavapor. To the resulting yellow slurry, water (15 ml) was added slowly to keep T<25°C. During the addition of water, the slurry became a clear solution, and a new slurry was formed. The slurry was stirred at room temperature for overnight. In the morning the slurry was filtered and the solid was washed with DMF/water (1:1, 5 ml), water (5 ml) and acetone (5 ml). The cake was dried under vacuum at 55°C for 24 hours to afford 0.90 g of amide product II (yield: 87.2%) as a white solid. HPLC: 99.70 AP.
2. Aminolysis with n-butyllithium
To a dried 100 ml of flask was added THF (10 ml) under nitrogen and then cooled to -100C. n-Butyllithium (2.5 M in hexane, 6.0 ml, 15.0 mmol) was added slowly, followed by dropwise addition of propylamine (0.98 g, 16.5 mmol) at such a rate to keep the temperature below 00C. The resulting mixture was stirred at O0C for 20 minutes. A suspension of ester compound I (1.0 g, 2.5 mmol) in THF (12 ml) was added over a 10 minute period (T<5°C). After being stirred at O0C for 30 minutes, the mixture was allowed to warm to room temperature and stirred for overnight (~22h, Note 1). Compound I was not detected at this point by HPLC analysis. The mixture was cooled to -7°C. Acetic acid (2 ml) was added dropwise to maintain the temperature <10°C. The resulting thick slurry was stirred at 50C for 2 hours and at room temperature for 20 minutes, followed by evaporation on a rotavapor to give a wet yellow solid. To this solid was added acetone (10 ml) and water (20 ml). The slurry was stirred at room temperature for one and half hours. Filtration gave a white solid. This solid was washed with 35% acetone in water (10 ml), water (5 ml) and acetone (5 ml). The cake was dried under vacuum at 55°C for the weekend to afford 0.94g of amide product II (yield: 91.0%) as a white solid. HPLC: 99.76 AP. Note 1: Compound I was -0.056 AP at 2.5 hours.
……………………
WO 2003090912
http://www.google.com/patents/WO2003090912A1?cl=en
……………………..
Discovery of 4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (BMS-582949), a clinical p38a MAP kinase inhibitor for the treatment of inflammatory diseases
J Med Chem 2010, 53(18): 6629
http://pubs.acs.org/doi/abs/10.1021/jm100540x
The discovery and characterization of 7k (BMS-582949), a highly selective p38α MAP kinase inhibitor that is currently in phase II clinical trials for the treatment of rheumatoid arthritis, is described. A key to the discovery was the rational substitution of N-cyclopropyl for N-methoxy in 1a, a previously reported clinical candidate p38α inhibitor. Unlike alkyl and other cycloalkyls, the sp2 character of the cyclopropyl group can confer improved H-bonding characteristics to the directly substituted amide NH. Inhibitor 7k is slightly less active than 1a in the p38α enzymatic assay but displays a superior pharmacokinetic profile and, as such, was more effective in both the acute murine model of inflammation and pseudoestablished rat AA model. The binding mode of 7k with p38α was confirmed by X-ray crystallographic analysis.

EXAMPLE 3

Direct Aminolysis
Ester Compound I

Amide Product II
Method A:
A solution of n-propylamine (6.5 eq) in THF (20 ml/g of ester compound I) was cooled to — 5°C and was slowly treated with 2.5 M solution of n-butyllithium (6.1 eq). The mixture was stirred for 10 minutes. At the end of the period, a slurry of ester compound I (1 eq) in THF (14 ml/g of ester compound I) was cannulated into the performed Li-NHPr solution. The reaction mixture was warmed to 25°C and stirred till all of ester compound I was consumed (~ 3 hours). After the reaction was judged to be completed by HPLC, the reaction mixture was cooled to ~0°C and was slowly treated with acetic acid (5 ml/g of ester compound I). The slurry was then warmed to -2O0C and was stirred for 1 hour. At the end of the period, the solvent was distilled under vacuum to the minimum volume and the concentrated slurry was diluted with a solution of acetone (10 ml/g of ester compound I) and water (20 ml/g of ester compound I). The slurry was stirred for 1 hour and was cooled to ~5°C. The slurry was filtered and the cake was washed with acetone (5 ml/g of ester compound I). The cake was dried to give the amide product II (typically in 85% yield and 99 AP).
Method B:
A solution of n-propylamine (20 eq) in 2,2,2-trifmoroethanol (10 ml/g of ester compound I) was slowly treated with 2.5 M solution of n-butyllithium (1.5 eq). The mixture was stirred for 5 minutes. At the end of the period, the starting material, ester compound I, was added and the reaction mixture was warmed to 900C. The reaction mixture was held at 900C for 24 hours and was allowed to cool to ~20°C. The reaction mixture was then analyzed by HPLC. Typically, analysis indicated there was only 1.57 AP of starting material left.
Method C:
A solution of n-propylamine (2 eq) in methylene chloride (10 ml/g of ester compound I) at 200C was slowly treated with 2.0 M solution of trimethylaluminum (4 eq) in hexanes. The mixture was stirred for 15 minutes. At the end of the period, the starting material, ester compound 1 (1 eq), was added and the reaction mixture was warmed to 600C. The reaction mixture was held at 600C for 24 hours and was allowed to cool to ~20°C. The reaction mixture was then slowly quenched with aqueous HCl solution and analyzed by HPLC. Typically, analysis indicated there was 96.8AP of amide compound II product with 0.03 AP of the dipropylamide impurity.
…………………………………….
| WO2003090912A1 * | 15 abr. 2003 | 6 nov. 2003 | Squibb Bristol Myers Co | Pyrrolo-triazine aniline compounds useful as kinase inhibitors |
Liu C, Lin J, Everlof G, Gesenberg C, Zhang H, Marathe PH, Malley M, Galella MA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K.
Bioorg Med Chem Lett. 2013 May 15;23(10):3028-33. doi: 10.1016/j.bmcl.2013.03.022. Epub 2013 Mar 15.
Freebern WJ, Bigwarfe TJ, Price KD, Haggerty HG.
J Immunotoxicol. 2013 Jan-Mar;10(1):106-17. doi: 10.3109/1547691X.2012.736427. Epub 2012 Nov 23.
Liu C, Lin J, Wrobleski ST, Lin S, Hynes J, Wu H, Dyckman AJ, Li T, Wityak J, Gillooly KM, Pitt S, Shen DR, Zhang RF, McIntyre KW, Salter-Cid L, Shuster DJ, Zhang H, Marathe PH, Doweyko AM, Sack JS, Kiefer SE, Kish KF, Newitt JA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K.
J Med Chem. 2010 Sep 23;53(18):6629-39. doi: 10.1021/jm100540x.
BMS-582949: crystalline form of a p38alpha inhibitor? WO2008079857.
Norman P.
Expert Opin Ther Pat. 2009 Aug;19(8):1165-8. doi: 10.1517/13543770902816160.
| WO2000012074A2 | Aug 27, 1999 | Mar 9, 2000 | Sarvajit Chakravarty | Use of piperidines and/or piperazines as inhibitors of p38-alpha kinase | |
| WO2000012497A2 | Aug 27, 1999 | Mar 9, 2000 | Sarvajit Chakravarty | Quinazoline derivatives as medicaments | |
| WO2000056738A1 | Mar 17, 2000 | Sep 28, 2000 | Astrazeneca Ab | Pyridine and pyrimidine derivatives and their use as inhibitors of cytokine mediated disease | |
| WO2001027089A1 | Oct 10, 2000 | Apr 19, 2001 | Astrazeneca Ab | Pyrimidine derivatives | |
| WO2001034605A1 | Oct 27, 2000 | May 17, 2001 | Ortho Mcneil Pharm Inc | SUBSTITUTED 2-ARYL-3-(HETEROARYL)-IMIDAZO[1,2-a]PYRIMIDINES, AND RELATED PHARMACEUTICAL COMPOSITIONS AND METHODS | |
| WO2003090912A1 | Apr 15, 2003 | Nov 6, 2003 | Squibb Bristol Myers Co | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US4200750 | Dec 8, 1977 | Apr 29, 1980 | Westwood Pharmaceuticals Inc. | 4-Substituted imidazo [1,2-a]quinoxalines | |
| US5658903 | Jun 3, 1996 | Aug 19, 1997 | Smithkline Beecham Corporation | Cytokine inhibitors | |
| US5932576 | May 22, 1998 | Aug 3, 1999 | G. D. Searle & Company | 3(5)-heteroaryl substituted pyrazoles as p38 kinase inhibitors | |
| US5945418 | Mar 20, 1997 | Aug 31, 1999 | Vertex Pharmaceuticals Incorporated | Administering to the mammal to inhibit a mammalian protein kinase p38 which causes cell proliferation, cell death and response to extracellular stimuli | |
| US5977103 | Jan 10, 1997 | Nov 2, 1999 | Smithkline Beecham Corporation | Substituted imidazole compounds | |
| US6087496 | Apr 1, 1999 | Jul 11, 2000 | G. D. Searle & Co. | Enzyme inhibitors | |
| US6130235 | Aug 3, 1998 | Oct 10, 2000 | Scios Inc. | Piperidine moieties coupled to indole, benzimidazole or benzotriazole. | |
| US6147080 | Jun 10, 1997 | Nov 14, 2000 | Vertex Pharmaceuticals Incorporated | Inhibitors of p38 | |
| US6251914 | Jul 1, 1998 | Jun 26, 2001 | Smithkline Beecham Corporation | Treating cytokine mediated diseases | |
| US6277989 | Mar 14, 2000 | Aug 21, 2001 | Scios, Inc. | Quinazoline derivatives as medicaments | |
| US6670357 | Nov 7, 2001 | Dec 30, 2003 | Bristol-Myers Squibb Company | Antiinflammatory agents | |
| US6867300 | Nov 6, 2002 | Mar 15, 2005 | Bristol-Myers Squibb Company | Methods for the preparation of pyrrolotriazine compounds useful as kinase inhibitors | |
| US7034151 | Feb 5, 2004 | Apr 25, 2006 | Bristol-Myers Squibb Company | 1,4-dihydro-4-oxo-pyrrolo[2,1-f][1,2,4]triazine-6-carboxylates; novel approach to the formation of the bicyclic heterocyclic ring system | |
| US7041501 | Oct 31, 2002 | May 9, 2006 | Bristol-Myers Squibb Company | Methods of screening for toxicity of test compounds | |
| US7160883 | Apr 22, 2003 | Jan 9, 2007 | Bristol-Myers-Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US7211666 | Dec 22, 2004 | May 1, 2007 | Bristol-Myers Squibb Company | N-Cyclopropyl-4-[[5-[(methoxyamino)carbonyl]-2-methylphenyl]amino]-5-methylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide; aminating with chloramine to produce a pyrrole with a Nitrogen nitrogen bond; reacting with formamide, cyclizing to form the pyrrolotriazine core; kinase inhibitors | |
| US7253167 | Jun 29, 2005 | Aug 7, 2007 | Bristol-Myers Squibb Company | Tricyclic-heteroaryl compounds useful as kinase inhibitors | |
| US7388009 | Oct 3, 2003 | Jun 17, 2008 | Bristol-Myers Squibb Company | Heterocyclic drugs as enzyme inhibitors for Kinase enzymes or prodrugs | |
| US7462616 | Oct 24, 2006 | Dec 9, 2008 | Bristol-Myers Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US7759343 | Oct 28, 2008 | Jul 20, 2010 | Bristol-Myers Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US61379001 | Title not available | ||||
| US20030232831 | Apr 22, 2003 | Dec 18, 2003 | Alaric Dyckman | Aryl ketone pyrrolo-triazine compounds useful as kinase inhibitors | |
| US20040229877 | Oct 29, 2003 | Nov 18, 2004 | Katerina Leftheris | Administering pyrrolotriazine carboxamide and benzamide compounds for therapy of p38 kinase-associated conditions | |
| US20050043306 | Oct 3, 2003 | Feb 24, 2005 | Katerina Leftheris | Heterocyclic drugs as enzyme inhibitors for Kinase enzymes or prodrugs | |
| US20060003967 | Jun 28, 2005 | Jan 5, 2006 | Zhongping Shi | Method for preparing pyrrolotriazine compounds | |
| US20060030708 | Aug 5, 2005 | Feb 9, 2006 | Lobben Paul C | Methods for the preparation of pyrrolotriazine compounds | |
| US20060041124 | Oct 14, 2005 | Feb 23, 2006 | Bang-Chi Chen | Process for preparing pyrrolotriazine kinase inhibitors | |
| US20060229449 | Apr 3, 2006 | Oct 12, 2006 | Apurba Bhattacharya | Reacting with chloramine in presence of aqueous base, phase transfer catalyst; anti-cancer agents, kinase inhibitors | |
| US20060235020 | Apr 4, 2006 | Oct 19, 2006 | Soojin Kim | Process for preparing salts of 4-[[5-[(cyclopropylamino)carbonyl]-2-methylphenyl]amino]-5-methyl-N-propylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide and novel stable forms produced therein | |
| US20070213300 | Mar 6, 2007 | Sep 13, 2007 | Bristol-Myers Squibb Company | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
Japan First to Approve Alectinib アレクチニブ 塩酸塩 (AF 802) for ALK+ NSCLC
Alectinib (AF802, CH5424802, RG7853, RO5424802)
CAS 1256580-46-7 FREE
1256589-74-8 (Alectinib Hydrochloride)
9-Ethyl-6,11-dihydro-6,6-dimethyl-8-[4-(4-morpholinyl)-1-piperidinyl]-11-oxo-5H-benzo[b]carbazole-3-carbonitrile
| Formula: | C30H34N4O2 |
| M.Wt: | 482.62 |
Mechanism of Action:ALK inhibitor
Indication:Non-small cell lung cancer (NSCLC)
Current Status:Phase II (US,EU,UK), NDA(Japan)
Company:中外製薬株式会社 (Chugai), Roche
Japan First to Approve Alectinib for ALK+ NSCLC
Roche announced that the Japanese Ministry of Health, Labor and Welfare (MHLW) has approved alectinib for the treatment of people living with non-small cell lung cancer (NSCLC) that is anaplastic lymphoma kinase fusion gene-positive (ALK+). The approval was based on results from a Japanese Phase 1/2 clinical study (AF-001JP) for people whose tumors were advanced, recurrent or could not be removed completely through surgery (unresectable).

| Company | Chugai Pharmaceutical Co. Ltd. |
| Description | Anaplastic lymphoma kinase (ALK) inhibitor |
| Molecular Target | Anaplastic lymphoma kinase (ALK) |
| Mechanism of Action | Anaplastic lymphoma kinase (Ki-1) (ALK) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Registration |
| Standard Indication | Non-small cell lung cancer (NSCLC) |
| Indication Details | Treat advanced ALK-positive non-small cell lung cancer (NSCLC); Treat non-small cell lung cancer (NSCLC); Treat unresectable progressive or recurrent ALK-positive non-small cell lung cancer (NSCLC) |
| Regulatory Designation |
U.S. – Breakthrough Therapy (Treat advanced ALK-positive non-small cell lung cancer (NSCLC)); |
| Partner |

Alectinib (also known as CH5424802,RO5424802), a second generation oral inhibitor of anaplastic lymphoma kinase (ALK), is being developed by Chugai and Roche for the treatment of patients with ALK-positive non-small cell lung cancer (NSCLC) that has progressed on Xalkori (Crizotinib).
Alectinib was discovered by Chugai Pharmaceutical Co. Ltd. Chugai became a subsidiary of Roche in 2002 and the Swiss group currently owns 59.9 percent of the company.
On October 8, 2013, Chugai Pharmaceutical announced that it has filed a new drug application to Japan’s Ministry of Health, Labour and Welfare (MHLW) for alectinib hydrochloride for the treatment of ALK fusion gene positive non-small cell lung cancer (NSCLC).
IT is a potent and selective ALK inhibitor with IC50 of 1.9 nM.Alterations in the anaplastic lymphoma kinase (ALK) gene have been implicated in human cancers. Among these findings, the fusion gene comprising EML4 and ALK has been identified in non-small cell lung cancer (NSCLC) and fusion of ALK to NPM1 has been observed in anaplastic large cell lymphoma (ALCL). The possibility of targeting ALK in human cancer was advanced with the launch of crizotinib for NSCLC in the U.S. in 2011. The development of resistance to crizotinib in tumors, however, has led to the need for second-generation ALK inhibitors. One of these, alectinib hydrochloride, has been found to be an orally active, potent and highly selective ALK inhibitor with activity in ALK-driven tumor models. Alectinib has shown preclinical activity against cancers with ALK gene alterations, including NSCLC cells expressing the EML4-ALK fusion and ALCL cells expressing the NPM-ALK fusion. Alectinib was well tolerated and active in a phase I/II study conducted in Japan in patients with ALK-rearranged advanced NSCLC and in patients with ALK-positive NSCLC who had progressed on crizotinib. Alectinib has been submitted for approval in Japan for the treatment of ALK fusion gene-positive NSCLC and is in phase I/II development for ALK-rearranged NSCLC in the U.S.

……………..

………………….
WO2012023597
http://www.google.fm/patents/WO2012023597A1?cl=en
(Preparation 30)
Compound F6-20
9 – ethyl-6, 6 – dimethyl-8 – (4 – morpholin-4 – yl – piperidin-1 – yl) -11 – oxo-6 ,11 – dihydro-5H-benzo [b] carbazol-3 – carbonitrile

Under the same conditions as the synthesis of the compound B3-13-1, and the title compound was synthesized from compound F5-49.
1 H-NMR (400MHz, DMSO-D 6) δ: 12.70 (1H, s), 8.32 (1H, d, J = 7.9 Hz), 8.04 (1H, s), 8.00 (1H, s), 7.61 (1H , d, J = 8.5 Hz), 7.34 (1H, s), 3.64-3.57 (4H, m), 3.27-3.18 (2H, m), 2.82-2.66 (4H, m), 2.39-2.28 (1H, m ), 1.96-1.87 (2H, m), 1.76 (6H, s), 1.69-1.53 (2H, m), 1.29 (3H, t, J = 7.3 Hz)
LCMS: m / z 483 [M + H] +
HPLC retention time: 1.98 minutes (analysis conditions U)
Hydrochloride 9 of compound F6-20 – ethyl-6, 6 – dimethyl-8 – (4 – morpholin-4 – yl – piperidin-1 – yl) -11 – oxo-6 ,11 – dihydro-5H-benzo [b I was dissolved at 60 ℃ in a mixture of 10 volumes of methyl ethyl ketone, 3 volumes of water and acetic acid volume 4-carbonitrile -] carbazol-3. I was dropped hydrochloric acid (2N) 1 volume of solution. After stirring for 30 minutes at 60 ℃, and the precipitated solid was filtered and added dropwise to 25 volume ethanol, 9 – Dry ethyl -6,6 – dimethyl-8 – (4 – morpholin-4 – yl – piperidin-1 – yl) I got a one-carbonitrile hydrochloride – 11 – oxo-6 ,11 – dihydro-5H-benzo [b] carbazol-3. Ethyl-6, 6 – 9 – obtained dimethyl-8 – (4 – morpholin-4 – yl – piperidin-1 – yl) -11 – oxo-6 ,11 – dihydro-5H-benzo [b] carbazol-3 – I was pulverized with a jet mill carbonitrile monohydrochloride.
1 H-NMR (400MHz, DMSO-D 6) δ: 12.78 (1H, s), 10.57 (1H, br.s), 8.30 (1H, J = 8.4 Hz), 8.05 (1H, s), 7.99 (1H , s), 7.59 (1H, d, J = 7.9 Hz), 7.36 (1H, s) ,4.02-3 .99 (2H, m) ,3.84-3 .78 (2H, m) ,3.51-3 .48 (2H, m), 3.15-3.13 (1H, s) ,2.83-2 .73 (2H, s) ,2.71-2 .67 (2H, s) ,2.23-2 .20 (2H, m) ,1.94-1 .83 (2H, m), 1.75 (6H, s ), 1.27 (3H, t, J = 7.5 Hz)
FABMS: m / z 483 [M + H] +
I was dissolved at 90 ℃ to 33 volume dimethylacetamide F6-20 F6-20 mesylate. Was added to 168 volumes mesylate solution (2 N) 1.2 volume, ethyl acetate solution was stirred for 4 hours. The filtered crystals were precipitated, and dried to obtain a F6-20 one mesylate. I was milled in a jet mill F6-20 one mesylate salt was obtained.
……………………
Journal of Medicinal Chemistry, 54(18), 6286-6294; 2011
http://pubs.acs.org/doi/abs/10.1021/jm200652u
| WO2002043704A1 * | 30 Nov 2001 | 6 Jun 2002 | Yasuki Kato | Composition improved in solubility or oral absorbability |
| WO2008051547A1 * | 23 Oct 2007 | 2 May 2008 | Cephalon Inc | Fused bicyclic derivatives of 2,4-diaminopyrimidine as alk and c-met inhibitors |
| WO2009073620A2 * | 1 Dec 2008 | 11 Jun 2009 | Newlink Genetics | Ido inhibitors |
| WO2010143664A1 * | 9 Jun 2010 | 16 Dec 2010 | Chugai Seiyaku Kabushiki Kaisha | Tetracyclic compound |
| JP2008280352A | Title not available | |||
| JP2009100783A | Title not available | |||
| JPH0892090A * | Title not available |
|
References |
1: Ignatius Ou SH, Azada M, Hsiang DJ, Herman JM, Kain TS, Siwak-Tapp C, Casey C, He J, Ali SM, Klempner SJ, Miller VA. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol. 2014 Apr;9(4):549-53. doi: 10.1097/JTO.0000000000000094. PubMed PMID: 24736079.
2: Gouji T, Takashi S, Mitsuhiro T, Yukito I. Crizotinib can overcome acquired resistance to CH5424802: is amplification of the MET gene a key factor? J Thorac Oncol. 2014 Mar;9(3):e27-8. doi: 10.1097/JTO.0000000000000113. PubMed PMID: 24518097.
3: Latif M, Saeed A, Kim SH. Journey of the ALK-inhibitor CH5424802 to phase II clinical trial. Arch Pharm Res. 2013 Sep;36(9):1051-4. doi: 10.1007/s12272-013-0157-8. Epub 2013 May 23. Review. PubMed PMID: 23700294.
4: Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, Hida T, Yamamoto N, Yoshioka H, Harada M, Ohe Y, Nogami N, Takeuchi K, Shimada T, Tanaka T, Tamura T. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013 Jun;14(7):590-8. doi: 10.1016/S1470-2045(13)70142-6. Epub 2013 Apr 30. PubMed PMID: 23639470.
5: Kinoshita K, Asoh K, Furuichi N, Ito T, Kawada H, Hara S, Ohwada J, Miyagi T, Kobayashi T, Takanashi K, Tsukaguchi T, Sakamoto H, Tsukuda T, Oikawa N. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem. 2012 Feb 1;20(3):1271-80. doi: 10.1016/j.bmc.2011.12.021. Epub 2011 Dec 22. PubMed PMID: 22225917.
6: Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011 May 17;19(5):679-90. doi: 10.1016/j.ccr.2011.04.004. PubMed PMID: 21575866.
Gadgeel S, Ou SH, Chiappori A, et al: A phase I dose escalation study of a new ALK inhibitor, CH542480202, in ALK+ non-small cell lung cancer patients who have failed crizotinib. Abstract O16.06. Presented at the 15th World Conference on Lung Cancer, Sydney, Australia, October 29, 2013.
Ou SH, Gadgeel S, Chiappori AA, et al: Consistent therapeutic efficacy of CH5424802/RO5424802 in brain metastases among crizotinib-refractory ALK-positive non-small cell lung cancer patients in an ongoing phase I/II study. Abstract O16.07. Presented at the 15th World Conference on Lung Cancer, Sydney, Australia, October 29, 2013.
Kinoshita, Kazuhiro et al,Preparation of tetracyclic compounds such as 11-oxo-5,6-dihydrobenzo[b]carbazole-3-carbonitrile derivatives as anaplastic lymphoma kinase (ALK) inhibitors,Jpn. Kokai Tokkyo Koho, 2012126711, 05 Jul 2012
Furumoto, Kentaro et al, Composition containing tetracyclic compound and dissolution aid (4環性化合物を含む組成物), PCT Int. Appl., WO2012023597, 23 Feb 2012, Also published as CA2808210A1, CN103052386A, EP2606886A1, EP2606886A4, US20130143877
Kinoshita, Kazutomo et al,Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802), Bioorganic & Medicinal Chemistry, 20(3), 1271-1280; 2012
Kinoshita, Kazutomo et al,9-Substituted 6,6-Dimethyl-11-oxo-6,11-dihydro-5H-benzo[b]carbazoles as Highly Selective and Potent Anaplastic Lymphoma Kinase Inhibitors, Journal of Medicinal Chemistry, 54(18), 6286-6294; 2011
Kinoshita, Kazuhiro et al, Preparation of tetracyclic compounds such as 11-oxo-5,6-dihydrobenzo[b]carbazole-3-carbonitrile derivatives as anaplastic lymphoma kinase (ALK) inhibitors,Jpn. Tokkyo Koho, 4588121, 24 Nov 2010

RedHill Biopharma Ltd. Acquires Phase 2 Oncology Drug Upamostat MESUPRON From Wilex AG

Upamostat
CAS: 590368-25-5
Chemical Formula: C32H47N5O6S
Exact Mass: 629.32470
Synonym: WX 671; WX-671; WX671. Upamostat; Brand name: Mesupron.
IUPAC/Chemical name:
(S)-ethyl 4-(3-(3-(N-hydroxycarbamimidoyl)phenyl)-2-(2,4,6-triisopropylphenylsulfonamido)propanoyl)piperazine-1-carboxylate

RedHill Biopharma Ltd. , an Israeli biopharmaceutical company focused on late clinical-stage drugs for inflammatory and gastrointestinal diseases, including cancer, and WILEX AG , a biopharmaceutical company focused on oncology, based in Munich, Germany, today announced that they have signed an exclusive license agreement for the oncology drug … (more)
Upamostat, also known as Mesupron, WX-671, is an orally bioavailable, 3-amidinophenylalanine-derived, second generation serine protease inhibitor prodrug targeting the human urokinase plasminogen activator (uPA) system with potential antineoplastic and antimetastatic activities. After oral administration, serine protease inhibitor WX-671 is converted to the active Nα-(2,4,6-triisopropylphenylsulfonyl)-3-amidino-(L)-phenyla lanine-4-ethoxycarbonylpiperazide (WX-UK1), which inhibits several serine proteases, particularly uPA; inhibition of uPA may result in the inhibition of tumor growth and metastasis. uPA is a serine protease involved in degradation of the extracellular matrix and tumor cell migration and proliferation.

|
Information about this agent |
WX-671 (Mesupron) is an orally available prodrug of WX-UK1, a serine protease inhibitor that inhibits uPA as well as other serine proteases. WX-UK1 (Setyono-Han et al., Thromb Haemost 2005) and WX-671 have shown to efficiently reduce primary tumor growth and metastasis formation in a variety of animal models. The proteolytic factor uPA and its inhibitor PAI-1 belong to those biological factors which have provided the highest level of evidence (LOE1) in terms of their prognostic and predictive significance. WX-671 is currently the only drug in Phase II aiming at this target.Results: All 95 patients were accrued between Jun 2007 and Aug 2008. Efficacy is assessed by a central reader at regular intervals based on digital CT images. By end of 2009, 2 patients were still on treatment without signs of progression, 64 patients had died. Preliminary analysis of overall survival showed an increase in overall survival from 10.2 mo (gemcitabine alone) to 13.5 mo for the combination of gemcitabine and WX-671. 1-year survival increased from 37% with gemcitabine to 53% when combined with 400 mg WX- 671. Conclusions: The combination of daily oral WX-671 in combination with weekly i.v. gemcitabine was well tolerated. see asco.com’s website.

|
References |
1. Analysis of highly potent amidine containing inhibitors of serine proteases and their N-hydroxylated prodrugs (amidoximes) By Kotthaus, Joscha; Steinmetzer, Torsten; van de Locht, Andreas; Clement, Bernd From Journal of Enzyme Inhibition and Medicinal Chemistry (2011), 26(1), 115-122.
2. Combined treatment of cancer by urokinase inhibition and a cytostatic anti-cancer agent for enhancing the anti-metastatic effect By Schmalix, Wolfgang; Schneider, Anneliese; Setyono-Han, Buddy; Foekens, Johannes From U.S. Pat. Appl. Publ. (2008), US 20080226624 A1 20080918.
3. Peptides and small molecules targeting the plasminogen activation system: towards prophylactic anti-metastasis drugs for breast cancer By Tyndall, Joel D. A.; Kelso, Michael J.; Clingan, Phillip; Ranson, Marie From Recent Patents on Anti-Cancer Drug Discovery (2008), 3(1), 1-13.
4. Synthesis of hydroxyamidine and hydroxyguanidine amino acid or oligopeptide derivatives for use as urokinase plasminogen activator inhibitors for the treatment of cancer and its metastasis By Sperl, Stefan; Buergle, Markus; Schmalix, Wolfgang; Wosikowski, Katja; Clement, Bernd From U.S. Pat. Appl. Publ. (2006), US 20060142305 A1 20060629.
5. Crystalline modifications of N-α-(2,4,6-triisopropylphenylsulfonyl)-3-hydroxyamidino-(l)-phenylalanine-4-ethoxycarbonylpiperazide and/or its salts By Grunenberg, Alfons; Lenz, Jana From PCT Int. Appl. (2006), WO 2006056448 A1 20060601.
6. Synthesis of hydroxyamidine and hydroxyguanidine amino acid or oligopeptide derivatives for use as urokinase plasminogen activator inhibitors for the treatment of cancer and its metastasis By Sperl, Stefan; Burgle, Markus; Schmalix, Wolfgang; Wosikowski, Katja; Clement, Bernd From PCT Int. Appl. (2004), WO 2004103984 A1 20041202.
7. Preparation of 3-amidinophenylalanine derivatives from 3-cyanophenylalanines via reduction and hydrogenation under mild conditions By Ziegler, Hugo; Wikstroem, Peter From PCT Int. Appl. (2003), WO 2003072559 A1 20030904.
1. Buddy et al, Suppression of Rat Brest Cancer Metastasis and Reduction of Primary Tumor Growth by the Small Synthetic Urokinase Inhibitor WX-UK1. Thromb Haemost. 2005, 93:779-786.
2. Ertongur S, Lang S, Mack B, Wosikowski K, Muehlenweg B, Gires O. Inhibition of the invasion capacity of carcinoma cells by WX-UK1, a novel synthetic inhibitor of the urokinase-type plasminogen activator system. Int J Cancer. 2004, 110(6):815-24.
3. Setyono-Han B, Stürzebecher J, Schmalix WA, Muehlenweg B, Sieuwerts AM, Timmermans M, Magdolen V, Schmitt M, Klijn JG, Foekens JA. Suppression of rat breast cancer metastasis and reduction of primary tumour growth by the small synthetic urokinase inhibitor WX-UK1. Thromb Haemost. 2005, 93(4):779-86.
FDA approves Alcobra’s protocol for Phase IIb study of metadoxine drug candidate

Metadoxine
Israel-based Alcobra has received approval from the US Food and Drug Administration (FDA) for its protocol for planned Phase IIb clinical study of Metadoxine Extended Release (MDX) drug candidate for the treatment of Fragile X Syndrome.

The multi-center, randomized, placebo-controlled, Phase IIb study, will be conducted primarily in the US and patient enrollment is expected to begin in the near future.
The study is supported by data collected from multiple earlier pre-clinical studies which demonstrated significant improvement in behavioral and cognitive outcomes based on evaluations of memory, learning, and social interaction.
Metadoxine, or Pyridoxol L-2-pyrrolidone-5-carboxylate, whose structure formula is reported hereinbelow

is known for its effectiveness in acute and chronic alcoholism and for the prevention of alcohol related pathology
 |
|
| Systematic (IUPAC) name | |
|---|---|
| L-Proline, 5-oxo-, compd. with 5-hydroxy-6-methylpyridine-3,4-dimethanol (1:1) | |
| Clinical data | |
| Legal status | PHASE 2 |
| Routes | Oral, IV |
| Identifiers | |
| CAS number | 0074536-44-0 |
| ATC code | N07BB |
| Chemical data | |
| Formula | C13H18N2O6 |
| Mol. mass | 298 g/mol |
………..
Metadoxine, also known as pyridoxine-pyrrolidone carboxylate, is a drug used to treat chronic and acute alcohol abuse.[1] Metadoxine improved the clinical signs of acute alcohol intoxication and accelerated alcohol clearance from the blood [2]It is presently in human clinical trials as an attention-deficit/hyperactivity disorder predominantly inattentive treatment.[3]
Pyridoxine is one form of vitamin B6 and a precursor to the metabolically active pyridoxal phosphate. Pyridoxal phosphate is a coenzyme to many enzymes: see vitamin B6 metabolic functions.
Pyrrolidone carboxylate is involved in amino acid metabolism through the glutathione pathway.[4] Glutathione is an important antioxidant and combats redox imbalance. It also supports de novo ATP synthesis.[5]
Alcohol-induced liver diseases are a common disorder in modern communities and societies. For example, in Europe there are more than 45 million individuals showing signs of alcohol-related damage such as liver disease and myopathies. Chronic alcohol consumption increases hepatic accumulation of triglycerides and leads to hepatic steatosis, which is the earliest and most common response to severe alcohol intoxication.
Thus, severe alcohol intoxication is a serious disease that should be treated with medication in order to reduce the damage to the human body of the alcohol intoxicated individual. For example, alcohol intoxication can be treated with metadoxine (pyridoxine L-2-pyrrolidone-5-carboxylate). Metadoxine is a salt of the corresponding anion of L-2-pyrrolidone-5-carboxylic acid (L-2-pyroglutamic acid) (1) and the protonated derivative of pyridoxine (vitamin B6) (2), having the following structures:

(1) (2)
WO 2008/066353 discloses the use of Metadoxine in the treatment of alcohol intoxication either alone or in combination with other active agents. WO 2008/066353 mentions that metadoxine does not inhibit the expression and activation of an alcohol-induced cytochrome P450 2El, which is the key enzyme involved in alcohol-induced toxicity. Thus, the use of metadoxine may be limited.
Several studies have shown that in order to effectively treat alcohol intoxication, there is a need for a relatively high daily dose (ca. 900 mg) administered intravenously (see, e.g., Lu et al. Chin. Med. J. 2007, 120 (2), 155-168 and Shpilenya et al. Alcohol Clin. Exp. Res. 2002, 26 (3), 340-346). These studies disclose side effects associated with the use of metadoxine, including nausea and vomiting.
Thus, there exists a need in the art for effective and safe drugs for treating alcohol intoxication and other associated diseases.
History
Metadoxine is predominantly used in developing nations for acute alcohol intoxication. Alternate names include: Abrixone (Eurodrug, Mexico), Alcotel (Il Yang, South Korea), Ganxin (Qidu Pharmaceutical, China), Metadoxil (Baldacci, Georgia; Baldacci, Italy; Baldacci, Lithuania; CSC, Russian Federation; Eurodrug, Colombia; Eurodrug, Hungary; Eurodrug, Thailand; Micro HC, India), Viboliv (Dr. Reddy’s, India), and Xin Li De (Zhenyuan Pharm, China).[6]
Fatty liver refers to a pathogenic condition where fat comprises more than 5% of the total weight of the liver. Liver diseases including the fatty liver, hepatitis, fibrosis and cirrhosis are known to be the most serious disease next to cancer causing death in people with ages 40 to 50, in the advanced countries. In advanced countries, nearly about 30% of the population is with fatty liver, and about 20% of people with fatty liver progresses to cirrhosis. About half of the cirrhosis patients die of liver diseases within 10 years after the diagnosis. Fatty liver and steatohepatitis are frequently found in people who intake excessive alcohols and who have obesity, diabetes, hyperlipemia, etc. Among them, alcoholic steatohepatitis (ASH), which is caused by excessive alcohol intake, is at high risk of progressing to hepatitis, cirrhosis and hepatoma, along with non-alcoholic steatohepatitis (NASH).
When taken in, alcohol is carried to the liver and oxidized to acetaldehyde by such enzymes as alcohol dehydrogenase, catalase, etc. The acetaldehyde is metabolized and converted into acetate and is used as energy source. Repeated alcohol intake induces the increase of NADH and NADP+ during the metabolism and acetaldehyde which as the metabolite product of alcohol depletes GSH, thereby changing intracellular oxidation-reduction homeostasis and inducing oxidative stress. Oxidative stress may cause mitochondrial dysfunction, lipid peroxidation and protein modification, thereby leading to death of hepatocytes, inflammation, activation of astrocytes, and the like. In addition, the increase of NADH promotes lipid synthesis, thereby inducing fatty liver.
At present, there are few therapeutically effective drugs for treating fatty liver. Exercise and controlled diet are recommended, but these are not so effective in treating fatty liver. The development of an effective treatment drug is in desperate need. As it is known that fatty liver is related with insulin resistance which is found in diabetes and obesity, the therapeutic effect of some anti-diabetic drugs, e.g., metformin, on fatty liver has been reported. But, the drug has the problem that it may induce adverse reactions such as hepatotoxicity or lactic acidosis. Betaine, glucuronate, methionine, choline and lipotrophic agents are often used as alternative supplementary drug therapy, but they are not fully proven on medical or pharmaceutical basis. Accordingly, development of a fatty liver treatment having superior effect and safety with no adverse reactions is in need.
Metadoxine (pyridoxol 1-2-pyrrolidone-5-carboxylate) is a complex compound of pyridoxine and pyrrolidone carboxylate represented by the formula (1) below:

Metadoxine is a drug used to treat alcoholic liver disease. It is used to treat liver fibrosis and fatty liver through increasing alcohol metabolism and turnover, reducing toxicity of free radicals and restoring the level of ATP and glutathione (Arosio, et al., Pharmacol. Toxicol. 73: 301-304, 1993; Calabrese, et al., Int. J. Tissue React. 17: 101-108, 1995; Calabrese, et al., Drugs Exp. Clin. Res. 24: 85-91, 1998; Caballeria, et al., J. Hepatol. 28: 54-60, 1998; and Muriel, et al., Liver Int. 23: 262-268, 2003).
However, metadoxine is unable to inhibit the expression and activation of alcohol-induced cytochrome P4502E1 (CYP2E1), which is a key enzyme involved in alcohol-induced toxicity, and thus unable to control the augmentation of inflammation mediated by CYP2E1. Therefore, the treatment of alcohol-induced fatty liver using metadoxine is very limited. Further, the expression of CYP2E1 is related with insulin resistance, thus metadoxine cannot not overcome insulin resistance.
Garlic oil is a liquid including about 1% of allicin along with reduced allicin and other sulfur-containing substances. Upon binding to vitamin B1, allicin is turned into allithiamin, which is chemically stable, acts swiftly, and is easily absorbed by the digestive organs. The substance inhibits carcinogenesis induced by chemicals in white rats (Brady, et al., Cancer Res. 48: 5937-5940, 1988; and Reddy, et al., Cancer Res. 53: 3493-3498, 1993), induces phase II enzyme (Hayes, et al., Carcinogenesis 8: 1155-1157, 1987; and Sparnins, et al., Carcinogenesis 9: 131-134, 1988), and inactivates CYP2E1 (Brady, et al., Chem. Res. Toxicol. 4:642-647, 1991). In addition, garlic oil is reported to have antithrombotic, anti-atherosclerotic, antimutagenic, anticancer and antibacterial activities (Agarwal, Med. Res. Rev. 16: 111-124, 1996; and Augusti, Indian J. Exp. Biol. 34: 634-660, 1996).
Pharmacology
Treatment for acute alcohol abuse
In an animal model, metadoxine treatment increased the clearance of alcohol and acetaldehyde, reduced the damaging effect of free radicals, and enabled cells to restore cellular ATP and glutathione levels. [7][8] It increases the urinary elimination of ketones, which are formed when the oxidation rate of acetaldehyde into acetate is exceeded on massive alcohol intoxication.[8][4]
As a medical treatment, it is typically given intravenously.
Treatment for AD/HD-PI
Metadoxine is a selective antagonist to the 5-HT2B receptor, a member of the serotonin receptor family.[3] Electrophysiological studies also showed that Metadoxine caused a dose-dependent, reversible reduction in glutamatergic excitatory transmission and enhancement of GABAergic inhibitory transmission, changes that may be associated with cognitive regulation.[3] It is given orally in an extended release pill, which differs from the instant release alcohol treatment.
Treatment for liver disease
Metadoxine may block the differentiation step of preadipocytes by inhibiting CREB phosphorylation and binding to the cAMP response element, thereby repressing CCAAT/enhancer-binding protein b during hormone-induced adipogenesis.[7]
Treatment for Fragile X Syndrome
Metadoxine treatment led to significant improvement in blood and brain biological markers (AKT and ERK), which may have a role in learning and memory.[3] The study also demonstrated a reduction in the amount of immature neurons and abnormally increased protein levels.[3]
…………………..
PATENT
http://www.google.com/patents/WO2010013242A1?cl=en
Scheme 1

[0054] In another aspect, the invention provides methods of synthetically preparing, e.g., carboxylated lactam ring of formula (II) (e.g. wherein n=2 for a reactant of formula (IVb) in Scheme 2), carboxylated lactam ring of (III) (e.g. wherein n=3 for a reactant of formula (IVb) in Scheme 2) and carboxylated lactam ring of formula (IV) (e.g. wherein n=4 for a reactant of formula (IVb) in Scheme 2), as depicted in Scheme 2.
Scheme 2
pyridoxine

Compound IVb, n=2,3,4

[0055] In another aspect, the invention provides methods of preparing a salt adduct including a positively charged pyridoxine moiety, or a derivative thereof, and a carboxylated 5- to 7-membered lactam ring, including the steps of:
(a) suspending an optionally substituted amino dioic acid in water and heating for a sufficient period of time to allow completing lactamization reaction;
(b) optionally decolorizing the reaction mixture to eliminate impurities;
(c) isolating the lactam carboxylate;
(d) optionally purifying the obtained lactam carboxylate by crystallization;
(e) admixing the obtained lactam carboxylate and a pyridoxine base or a derivative thereof in a solvent mixture optionally under heating; and
(f) isolating the product.
In certain embodiments, a solvent mixture of step (e) includes a mixture of an alcohol such as methanol, ethanol, isopropanol and the like, and water. [0059] According to yet another embodiment, there is provided methods of preparing N-substituted L-pyroglutamic acid and the carboxylate thereof, such as, for example, N-methyl-L-pyroglutamic acid (1-methyl-L-pyroglutamic acid), starting from L-pyroglutamic acid ethyl ester, as depicted in Scheme 3 below. Scheme 3


1-methyl-L-pyroglutamic acid
[0060] The invention further provides methods of preparing a salt adduct of the invention, wherein said positively charged moiety is a substituted pyridoxine, as depicted in Scheme 4 below. The starting reagent is 2-methyl-3-hydroxy-4- methoxymethyl-5-hydroxymethyl-pyridine hydrochloride (Compound (V)). The preparation of the corresponding salt is described in Example 1. Scheme 4
HCI NH 3 / MeOH 2 L-pyroglutamic acid


Compound V Compound Vl

Salt lid
[0061] The invention further provides methods of preparing a salt adduct of the invention, wherein said positively charged moiety is a substituted pyridoxine, as depicted in Scheme 5 below. The starting reagent in scheme 5 is 2-methyl-3-hydroxy- 4-methoxymethyl-5-hydroxymethyl-pyridine hydrochloride (Compound V). The preparation of the corresponding salt is described in Example 2. Scheme 5

Compound V
HCI

Compound VIII IX Compound L-pyroglutamic acid

, SaIt IIe
| WO2010150261A1 * | June 24, 2010 | Dec 29, 2010 | Alcobra Ltd. | A method for the treatment, alleviation of symptoms of, relieving, improving and preventing a cognitive disease, disorder or condition |
| WO2011061743A1 * | Nov 18, 2010 | May 26, 2011 | Alcobra Ltd. | Metadoxine and derivatives thereof for use in the treatment of inflammation and immune-related disorders |
| US8476304 | Jul 3, 2012 | Jul 2, 2013 | Alcobra Ltd. | Method for decreasing symptoms of alcohol consumption |
| US8710067 | Jul 3, 2012 | Apr 29, 2014 | Alcobra Ltd. | Method for the treatment, alleviation of symptoms of, relieving, improving and preventing a cognitive disease, disorder or condition |
| WO2008066353A1 * | Nov 30, 2007 | June 5, 2008 | Jae Hoon Choi | Pharmaceutical composition comprising metadoxine and garlic oil for preventing and treating alcohol-induced fatty liver and steatohepatitis |
| WO2009004629A2 * | Jul 3, 2008 | Jan 8, 2009 | Alcobra Ltd | A method for decreasing symptoms of alcohol consumption |
| FR2172906A1 * | Title not available | |||
| US4313952 * | Dec 8, 1980 | Feb 2, 1982 | Maximum Baldacci | Method of treating acute alcoholic intoxication with pyridoxine P.C.A. |
References
- Addolorato, G; Ancona C, Capristo E, Gasbarrini G (2003). “Metadoxine in the treatment of acute and chronic alcoholism: a review”. International Journal of Immunopathology and Pharmacology.
- Martinez, Diaz; Villamil Salcedo; Cruz Fuentes (2001). “Efficacy of Metadoxine in the Management of Acute Alcohol Intoxication”. Journal of International Medical Research.
- “Metadoxine extended release (MDX) for adult ADHD” (in English). Alcobra Ltd. 2014. Retrieved 2014-05-07.
- Shpilenya, Leonid S.; Alexander P. Muzychenko; Giovanni Gasbarrini; Giovanni Addolorato (2002). “Metadoxine in Acute Alcohol Intoxication: A Double-Blind, Randomized, Placebo-Controlled Study”. Alcoholism:Clinical and Experimental Research.
- Shull, Kenneth H.; Robert Kisilevsky (1996). “Effects of Metadoxine on cellular status of glutathione and on enzymetric defence system following acute ethanol intoxication in rats”. Drugs Exp Clin Res.
- “Metadoxine – Drugs.com” (in English). Drugs.com. 2014. Retrieved 2014-05-08.
- Yang, YM; HE Kim; SH Ki; SG Kim (2009). “Metadoxine, an ion-pair of pyridoxine and L-2-pyrrolidone-5-carboxylate, blocks adipocyte differentiation in association with inhibition of the PKA-CREB pathway.”. Archives of Biochemistry and Biophysics.
- Calabrese, V; A Calderone; N Ragusa; V Rizza (1971). “Effects of l-2-pyrrolidone-5-carboxylate on hepatic adenosine triphosphate levels in the ethionine-treated rat”. Biochemical Pharmacology.
XenoPort begins phase II trial of XP-23829 in patients with psoriasis
![]()
XP 23829 from Xenoport is an interesting molecule and as on 27 July 2014, I did not find conclusive evidence
See some structures below
 Not sure about the structure of XP 23829
Not sure about the structure of XP 23829
OR
 Best fit
Best fit
OR
 Not sure?
Not sure?(N,N-dimethylcarbamoyl)methyl methyl(2E)but-2-ene-1,4-dioate.
OR

I AM NOT SURE ABOUT THIS ONE ALSO????????
As Football worldcup2014 goes on in Brazil

A thought for it is due…………
……………………………………………………
Best fit is probably is as shown below, and there are reasons
(N,N- Diethylcarbamoyl)methyl methyl (2E)but-2-ene-l,4-dioate
Introduction
(N,N-Diethylcarbamoyl)methyl methyl (2E)-but-2-ene-1,4-dioate


C11 H17 N O5, mw 243.13
M.p.: 53-56 °C.
1 H NMR (CDCI3, 400 MHz): δ 6.99-6.90 (m, 2H), 4.83 (s, 2H), 3.80 (s, 3H), 3.39 (q, J = 1.1 Hz, 2H), 3.26 (q, J = 7.2 Hz, 2H), 1 .24 (t, J = 7.2 Hz, 3H), 1 .14 (t, J = 7.2 Hz, 3H). MS (ESI): m/z 244.13 (M+H)+.
Cas…….1208229-58-6
XP-23829 PROBABLE
For the treatment of moderate-to-severe chronic plaque-type psoriasis.
XP-H-093
US8148414 Basic patent
Xenoport, Inc. Innovator
XenoPort has initiated a phase II trial of XP-23829, a proprietary investigational next-generation fumaric acid product candidate (ClinicalTrials.gov Identifier NCT02173301). The multicenter, randomized, double-blind, placebo-controlled study is designed to assess the efficacy and safety of XP-23829 as a potential treatment of patients with moderate to severe chronic plaque-type psoriasis. XenoPort expects to enroll approximately 200 subjects in this trial, which is being conducted in the U.S. The study will include a screening and washout phase of up to 4 weeks, a 12-week treatment phase and a 4-week post-treatment phase. Eligible study subjects will be randomized to placebo or one of three treatment arms of XP-23829: 400 or 800 mg once daily or 400 mg twice daily. The primary endpoint will examine the percent change in Psoriasis Area and Severity Index (PASI) score from baseline at the end of week 12. Secondary endpoints will include the proportion of subjects who achieve a reduction of 75% or greater from baseline in PASI (PASI75) score and subjects who achieve a Static Physicians Global Assessment score of “clear” or “almost clear.” Topline results are expected in the third quarter of 2015 (XenoPort News Release).
XP23829 — A Prodrug of Monomethyl Fumarate
Our third product candidate, XP23829, is in Phase 1 clinical development. Provided we are able to demonstrate the safety and desired pharmacokinetic, or PK, profile of XP23829 in our Phase 1 trials, we believe that XP23829 could be a potential treatment of patients with RRMS, psoriasis and/or certain other disorders where the mechanism of action of XP23829 may be relevant. For example, we are exploring the potential of XP23829 to protect against neurodegeneration in experimental preclinical models of Parkinson’s disease through a grant from The Michael J. Fox Foundation. We hold a composition-of-matter patent and a formulation patent in the United States on XP23829 and hold patents or pending patent applications directed to the XP23829 methods of synthesis and use in the United States. We have also filed applications directed to the XP23829 composition of matter and methods of synthesis and use in other jurisdictions.
Prodrug Background
XP23829 is a fumaric acid ester compound and a patented prodrug of MMF. Fumaric acid ester compounds have shown immuno-modulatory and neuroprotective effects in cell-based systems and preclinical models of disease. A product containing a combination of fumaric acid ester compounds, known as Fumaderm, is approved in Germany for the treatment of psoriasis. Tecfidera (a formulation of DMF, also known as BG-12) from Biogen Idec Inc. is another fumaric acid ester prodrug that converts to MMF in the body. Phase 3 clinical trials of Tecfidera as a potential treatment for RRMS showed statistically significant benefits of Tecfidera versus placebo. Tecfidera is currently under U.S. regulatory review as a potential treatment for RRMS.
Our Prodrug
XP23829 is a novel prodrug of MMF that we believe may provide improved tolerability and efficacy compared to DMF. In preclinical studies that compared molar equivalent doses of XP23829 to DMF, XP23829 provided higher blood levels of the biologically active molecule MMF and a similar or greater degree of efficacy in MS and psoriasis animal models. Toxicology studies conducted in two species showed that XP23829 caused less stomach irritation when compared to DMF.
Phase 1 Clinical Trial in Healthy Volunteers
In October 2012, we reported favorable preliminary results from our first Phase 1 clinical trial in healthy adults designed to assess the pharmacokinetics, safety and tolerability of single doses of four different formulations of XP23829. The trial was a randomized, double-blind, two-period crossover, food effect comparison clinical trial of XP23829. Sixty subjects were assigned to five cohorts of 12, with each cohort receiving one of four different formulations of XP23829 or placebo. The trial demonstrated that administration of XP23829 resulted in the expected levels of MMF in the blood. As anticipated, the four formulations produced

April 4, 2012
http://investor.xenoport.com/releasedetail.cfm?ReleaseID=708145
XenoPort Awarded U.S. Patent Directed to Composition and Formulations of XP23829, a Novel Fumarate Analog for the Potential Treatment of Relapsing-Remitting Multiple Sclerosis and Psoriasis
SANTA CLARA, Calif.–(BUSINESS WIRE)–Apr. 4, 2012– XenoPort, Inc. (Nasdaq: XNPT) announced today that it was awarded U.S. Patent 8,148,414 for “Prodrugs of Methyl Hydrogen Fumarate, Pharmaceutical Compositions Thereof, and Methods of Use.” The term of the patent extends until 2029, subject to potential Hatch-Waxman patent term extensions.
The patent is directed to the XP23829 compound, analogs thereof and formulations thereof. A related U.S. patent application directed to therapeutic uses of XP23829 is now pending.
XP23829 is a prodrug of methyl hydrogen fumarate, also known as monomethyl fumarate (MMF). In cell- and animal-based models, MMF has been shown to exhibit immuno-modulatory properties and inhibit damage from oxidative stress.
In XenoPort’s preclinical animal studies that compared molar equivalent doses of XP23829 to dimethyl fumarate (DMF), another prodrug of MMF, XP23829 demonstrated a greater degree of efficacy in animal models of both multiple sclerosis (MS) and psoriasis. Toxicology studies conducted in two species showed that XP23829 caused less stomach irritation compared to DMF.
XenoPort intends to file an Investigational New Drug Application (IND) for XP23829 for the treatment of relapsing remitting MS with the U.S. Food and Drug Administration (FDA) in the second quarter of 2012 and expects to initiate human clinical trials later this year.
XenoPort owns all rights to XP23829.
About XenoPort
XenoPort is a biopharmaceutical company focused on developing and commercializing a portfolio of internally discovered product candidates for the potential treatment of neurological disorders. Horizant® (gabapentin enacarbil) Extended-Release Tablets is XenoPort’s first FDA-approved product. GlaxoSmithKline holds commercialization rights and certain development rights for Horizant in the United States. Regnite® (gabapentin enacarbil) is approved for the treatment of moderate-to-severe primary restless legs syndrome in Japan. Astellas Pharma Inc. holds all development and commercialization rights for Regnite in Japan and five Asian countries. XenoPort holds all other world-wide rights and has co-promotion and certain development rights to gabapentin enacarbil in the United States. XenoPort’s pipeline of product candidates includes potential treatments for patients with postherpetic neuralgia, spasticity and Parkinson’s disease.
To learn more about XenoPort, please visit the company Website at http://www.XenoPort.com.
More info about this drug
SEE a patent
WO 2010022177
……………………………………….
WO 2013181451
http://www.google.com/patents/WO2013181451A1?cl=en
Scheme 5:

ONE OUT OF THESE
Example 6: (/V,/V-Diethylcarbamoyl)methyl methyl (2£)but-2-ene-1 ,4-dioate

[0138] Following general procedure A, methyl hydrogen fumarate (MHF) (0.39 g, 3.00 mmol) dissolved in NMP was reacted at about 55 °C with 2-chloro-/V,/V-diethylacetamide (0.44 g, 3.00 mmol) in the presence of CsHC03 (0.69 g, 3.60 mmol) to afford 0.37 g (51 % yield) of the title compound after purification by silica gel column chromatography (Biotage) using a mixture of ethyl acetate (EtOAc) and hexanes (1 :1 ) as eluent. M.p.: 53-56 °C. 1 H NMR (CDCI3, 400 MHz): δ 6.99-6.90 (m, 2H), 4.83 (s, 2H), 3.80 (s, 3H), 3.39 (q, J = 1.1 Hz, 2H), 3.26 (q, J = 7.2 Hz, 2H), 1 .24 (t, J = 7.2 Hz, 3H), 1 .14 (t, J = 7.2 Hz, 3H). MS (ESI): m/z 244.13 (M+H)+.
Example 7: Methyl 2-morpholin-4-yl-2-oxoethyl (2 £)but-2-ene-1 ,4-dioate
[0139] Following general procedure A, methyl hydrogen fumarate (MHF) (0.50 g, 3.84 mmol) dissolved in NMP was reacted at about 55 °C with 4-(chloroacetyl) morpholine (0.75 g, 4.61 mmol) in the presence of CsHC03 (0.89 g, 4.61 mmol) to afford 0.34 g (35% yield) of the title compound as a white solid after purification by mass-guided preparative HPLC and lyophilization. M.p.: 124 to 126°C; 1 H NMR (CDCI3, 400 MHz): δ 6.97-6.91 (m, 2H), 4.84 (s, 2H), 3.82 (s, 3H), 3.72-3.70 (m, 4H), 3.64-3.62 (m, 2H), 3.46-3.41 (m, 2H). MS (ESI): m/z 258.04 (M+H)+. Example 8: A/,A/-Dimethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4-dioate

[0140] Following general procedure A, methyl hydrogen fumarate (MHF) (0.50 g, 3.84 mmol) dissolved in NMP was reacted at about 55 °C with /V,/V-dimethyl chloroacetamide (0.56 g, 4.61 mmol) in the presence of CsHC03 (0.89 g, 4.61 mmol). The crude material was precipitated out from a mixture of ethyl acetate (EtOAc) and hexanes (Hxn) (1 :1 ) to provide a white solid. This solid was further dissolved in dichloromethane (DCM) and the organic layer washed with water. After removal of the solvents 0.55 g (67% yield) of the title compound was obtained as a white solid. 1 H NMR (CDCI3, 400 MHz): δ 6.98- 6.90 (m, 2H), 4.84 (s, 2H), 3.80 (s, 3H), 2.99-2.97 (2s, 6H). MS (ESI): m/z 216 (M+H)+.
Example 9: Methyl (2-morpholino-4-ylethyl) fumarate

[0141] Following general Procedure A, methyl hydrogen fumarate (MHF) dissolved in NMP is reacted at about 55 °C with 4-(chloroethyl) morpholine (0.75 g, 4.61 mmol) in the presence of CsHC03 to afford the title compound after purification by mass-guided preparative HPLC and lyophilization. Example 10: Methyl (3-mor holino-4-ylpropyl) fumarate

[0142] Following the procedure of Methyl (2-morpholino-4-ylethyl) fumarate, and replacing 4-(chloroethyl) morpholine with 4-(chloropropyl) morpholine provides the title compound.
Example 11 : Methyl (4-morpholino-4-ylbutyl) fumarate

[0143] Following the procedure of Methyl (2-morpholino-4-ylethyl) fumarate, and replacing 4-(chloroethyl) morpholine with 4-(chlorobutyl) morpholine provides the title compound. Example 12: Methyl 5-morpholino-4-ylpentyl) fumarate

[0144] Following the procedure of Methyl (2-morpholino-4-ylethyl) fumarate, and replacing 4-(chloroethyl) morpholine with 4-(chloropentyl) morpholine provides the title compound. Example 13: (A/-cyclopropyl-W-ethylcarbamoyl)methyl methyl 2(E)but-2-ene-1 ,4-dioate

[0145] Following the general procedure A, methyl hydrogen fumarate (MHF) (38.7 g, 0.297 mol) suspended in toluene (100 mL) was reacted at about 80 °C with 2-chloro-/V-cyclopropyl- N-ethylacetamide (48 g, 0.297 mol) in the presence of W,/V-diisopropylethylamine (DIEA; 42.3 g, 57 mL, 0.327 mol) to afford 50 g (63.3%) of the title compound after recrystallization using methyl ferf-butyl ether. The crystalline compound had a melting point of 92.1 °C. 1 H NMR (CDCI3, 400 MHz): δ 7.01 -6.92 (m, 2H), 4.99 (s, 2H), 3.81 (s, 3H), 3.44 (q, J = 7.2 Hz, 2H), 2.69-2.66 (m, 1 H), 1 .14 (t, J = 7.2 Hz, 3H), 0.94-0.91 (m, 2H), 0.83-0.81 (m, 2H). MS (ESI): m/z 256.2 (M+H)+.
Example 14: (/V-cyclopropyl-/V-methylcarbamoyl)methyl methyl 2(E)but-2-ene-1 , 4- dioate

[0146] Following general procedure A, methyl hydrogen fumarate (MHF) (38.7 g, 0.40 mol) suspended in toluene (100 mL) was reacted at about 80 °C with 2-chloro-/V-cyclopropyl-/V- methylacetamide (60 g, 0.40 mol) in the presence of Ν,Ν-diisopropylethylamine (DIEA; 57.8 g, 78 mL, 0.44 mol) to afford 50 g (50.86%) of the title compound after recrystallization using methyl fe/t-butyl ether. The crystalline compound had a melting point of 93.6 °C. 1 H NMR (CDCI3, 400 MHz): δ 7.01 -6.91 (m, 2H), 5.01 (s, 2H), 3.82 (s, 3H), 2.94 (s, 3H), 2.73-2.68 (m, 1 H), 0.94-0.86 (m, 2H), 0.83-0.78 (m, 2H). MS (ESI): m/z 242.2 (M+H)+.
Example 15: Methyl 2-oxo-2-pyrrolidinylethyl 2(E)but-2-ene-1 ,4-dioate

[0147] Following general procedure A, methyl hydrogen fumarate (MHF) (20.78 g, 0.159 mol) suspended in toluene (60 mL) was reacted at about 80 °C with 2-chloro-1 -pyrrolidin-1 -yl- ethanone (23.5 g, 0.159 mol) in the presence of N,N-diisopropylethylamine (DIEA; 22.69 g, 31 .5 mL, 0.175 mol) to afford 24 g (62.3%) of the title compound after recrystallization using methyl fe/t-butyl ether. The crystalline compound had a melting point of 102.1 °C. 1 H NMR (CDCI3, 400 MHz): δ 7.00-6.92 (m, 2H), 4.75 (s, 2H), 3.81 (s, 3H), 3.53-3.49 (t, J = 6.8 Hz, 2H), 3.42-3.39 (t, J = 6.8 Hz, 2H), 2.20-1 .97 (m, 2H), 1 .91 -1 .82 (m, 2H). MS (ESI): m/z 242 (M+H)+.
………………………………
Patent
http://www.google.co.in/patents/US8148414
(I):

Following general procedure A, methyl hydrogen fumarate (MHF) (0.39 g, 3.00 mmol) dissolved in NMP was reacted at ca. 55° C. with 2-chloro-N,N-diethylacetamide (0.44 g, 3.00 mmol) in the presence of CsHCO3 (0.69 g, 3.60 mmol) to afford 0.37 g (51% yield) of the title compound (1) after purification by silica gel column chromatography (Biotage) using a mixture of ethyl acetate (EtOAc) and hexanes (1:1) as eluent. M.p.: 53-56° C. 1H NMR (CDCl3, 400 MHz): δ 6.99-6.90 (m, 2H), 4.83 (s, 2H), 3.80 (s, 3H), 3.39 (q, J=7.2 Hz, 2H), 3.26 (q, J=7.2 Hz, 2H), 1.24 (t, J=7.2 Hz, 3H), 1.14 (t, J=7.2 Hz, 3H). MS (ESI): m/z 244.13 (M+H)+.

Methyl 2-oxo-2-piperazinylethyl(2E)but-2-ene-1,4-dioate hydrochloride (14) (0.20 g, 0.68 mmol) was reacted with acetyl chloride (AcCl) (0.60 mL, 0.66 g, 0.84 mmol) and diisopropylethylamine (0.70 mL, 0.52 g, 4.0 mmol) in dichloromethane (DCM). Following aqueous work-up, the crude product was purified by silica gel flash chromatography to afford 0.12 g (54% yield) of the title compound (16) as a white solid. 1H NMR (CDCl3, 400 MHz): δ 6.98-6.93 (m, 2H), 4.86 (s, 2H), 3.83 (s, 3H), 3.66 3.63 (m, 4H), 3.50-3.40 (m, 4H), 2.14 (s, 3H). MS (ESI): m/z 299.12 (M+H)+.
Example 9N,N-Dimethylcarbamoyl)methyl methyl(2E)but-2-ene-1,4-dioate (9)
Following general procedure A, methyl hydrogen fumarate (MHF) (0.50 g, 3.84 mmol) dissolved in NMP was reacted at ca. 55° C. with N,N-dimethyl chloroacetamide (0.56 g, 4.61 mmol) in the presence of CsHCO3 (0.89 g, 4.61 mmol). The crude material was precipitated out from a mixture of ethyl acetate (EtOAc) and hexanes (Hxn) (1:1) to provide a white solid. This solid was further dissolved in dichloromethane (DCM) and the organic layer washed with water. After removal of the solvents 0.55 g (67% yield) of the title compound (9) was obtained as a white solid. 1H NMR (CDCl3, 400 MHz): δ 6.98-6.90 (m, 2H), 4.84 (s, 2H), 3.80 (s, 3H), 2.99-2.97 (2s, 6H). MS (ESI): m/z 216 (M+H)+.
………………………..
http://www.google.com/patents/WO2014031844A1?cl=en
Compound (1).
Table 1 : Flushing Incidence as a Function of MMF Cmax


*Formulation 2 is the dosage form described in Example 10; Formulation 3 is the dosage form described in Example 3 ; Formulation 4 is the dosage form described in Example 5 ;
** maximum average Concentration; ***average Cmax; Poster (see above); Compound (1) referred to in the above table is an MMF prodrug of Formula (II); (N,N- Diethylcarbamoyl)methyl methyl (2£)but-2-ene-l,4-dioate having the following chemical structure:
Compound (1).
The maximum slope values ( dose and ng) for different dosage treatments are given in Table 2. The Figures 15-16 show plots of maximum MMF slope vs flushing incidence. The curves in the figures were fitted using a Hill Emax model. Table 2

Compound, Flushing
Table 3: Composition of Enteric Coated Sustained Release Tablet (15% HPMC in Core)

Quantity Quantity
Component Manufacturer Role
(mg tablet) (%w/w)
Vertellus (Greensboro,
Triethyl Citrate Plasticizer 1.25 0.42
NC)
Emerson Resources Anti- tacking
PlasAC YL™ T20 2.41 0.80
(Norristown, PA) agent
Total Enteric
27.87 9.30 Coating
Total Tablet 334.69 111.68
[00191] The tablets were made according to the following steps. The core tablets were prepared using a wet granulation process. The granulation was performed in two batches at 456 g per batch. Compound (1) and hydroxypropyl cellulose were passed through a conical mill with a 610 micron round holed screen. Compound (1) and hydroxypropyl cellulose were then combined in a Key KG- 5 granulator bowl and mixed with water addition for approximately 7 minutes. The wet granules were dried in a Glatt GPCG-1 fluid bed dryer at 40 °C. The two portions of dried granules were sized by passing through a conical mill with an approximately 1300 micron grater type screen. The milled granules were blended with the hypromellose 2208, silicon dioxide, and lactose monohydrate for 10 minutes in an 8 quart (7.6 1) V-blender. This blend was passed through an 850 micron mesh screen. The magnesium stearate was passed through a 600 micron mesh screen and blended with the additional core materials in the V-blender for 5 minutes. Core tablets (299.69 mg) were compressed using a GlobePharma Minipress II rotary tablet press with 8.6 mm round concave tooling. The core tablets had a final mean hardness of approximately 12 kp. For the coating, an aqueous suspension was prepared by mixing with an impeller 63.8 g Opadry 03019184 with 770.7 g of purified water. The water contained in the suspension is removed during the film coating process and therefore not included in the final formulation in Table 3. The tablets were coated with the aqueous suspension in an O’ Hara Technologies Labcoat M coater with a 12″ (30.5 cm) diameter perforated pan until the desired weight gain of barrier coat was achieved. The coating process occurred at an inlet temperature of approximately 52 °C and an outlet temperature of 36 °C. After coating, the tablets were dried for 2 hours at 40 °C. An aqueous suspension was prepared by mixing with an impeller 405.1 g methacrylic acid copolymer dispersion, 6.3 g triethyl citrate, 60.6 g PlasACRYL™ T20 with 228.1 g water. The water contained in the methacrylic acid copolymer dispersion and the
PlasACRYL™ T20 is removed during the film coating process and therefore not included in the final formulation in Table 3. The tablets were coated with the aqueous suspension in the O’ Hara Technologies Labcoat M coater until the desired weight gain of enteric film was achieved. The coating process occurred at an inlet temperature of approximately 40 °C and an outlet temperature of 30 °C. After coating, the tablets were dried for 2 hours at 40 °C.
Example 2
In Vitro Dissolution Profile of Example 1 Dosage Form
[00192] A two-stage dissolution method was used to determine the in vitro dissolution profile of dosage forms prepared according to Example 1. The 2-stage dissolution test was used to better approximate the pH conditions experienced by a dosage form after swallowing by a patient, i.e., low pH of the stomach followed by near neutral pH of the intestines. The dosage forms were first placed into a dissolution vessel (USP, Type I, basket) containing 750 mL of 0.1 N hydrochloric acid (pH 1.2). After 2 hours, 250 mL of 200 mM tribasic sodium phosphate was added to the vessel resulting in a pH adjustment from 1.2 to 6.8. The dissolution medium was kept at 37 °C and was agitated at 100 rpm.
[00193] For the Example 1 dosage forms, samples of the dissolution medium were withdrawn after 1 and 2 hours in the low pH stage, and at 0.5, 2, 4, 7, 10, and 14 hours following buffer addition. The released amount of the MMF prodrug in the samples was determined by reverse phase HPLC using a C18 column and a 7 minute gradient method according to Table 4 where Mobile Phase A is water/0.1 ]¾Ρθ4 and Mobile Phase B is water/acetonitrile/H3PC>4 (10/90/0.1 by volume) with UV detection at 210 nm.
Table 4: HPLC Gradient Conditions

[00194] As shown in FIG. 1, for dosage forms prepared according to Example 1, drug release is delayed for approximately 2 hours, followed by sustained release reaching >90 at 12 hours.
Example 3
Preparation of Delayed Sustained Release Dosage Form (Enteric Coated, 15% HPMC in Core, without Barrier Layer) [00195] Delayed sustained release tablets containing compound (1) were made having the ingredients shown in Table 5:
Table 5: Composition of Enteric Coated Sustained Release Tablet (15% HPMC in Core, without Barrier Layer)

[00196] The tablets were made according to the following steps. The core tablets were prepared using a wet granulation process. The granulation was performed in two batches at 463.9 g per batch. Compound (1) and hydroxypropyl cellulose were passed through a conical mill with a 610 micron round holed screen. Compound (1) and hydroxypropyl cellulose were then combined in a Key KG- 5 granulator bowl and mixed with water addition for approximately 10 minutes. The wet granules were dried in a Glatt GPCG-1 fluid bed dryer at 40 °C. The two portions of dried granules were blended with silicon dioxide and sized by passing through a conical mill with an approximately 1300 micron grater type screen. The milled granules were blended with the hypromellose 2208 and lactose monohydrate for 10 minutes in an 8 quart (7.6 1) V-blender. This blend was passed through an 850 micron mesh screen. The magnesium stearate was passed through a 600 micron mesh screen and blended with the additional core materials in the V-blender for 5 minutes. Core tablets (299.68 mg) were compressed using a GlobePharma Minipress II rotary tablet press with 11/32″ round concave tooling. The core tablets had a final mean hardness of approximately 11 kp. For the coating, an aqueous suspension was prepared by mixing with an impeller 578.7 g methacrylic acid copolymer dispersion, 9.0 g triethyl citrate, 86.5 g PlasACRYL™ T20 with 325.8 g water. The water contained in the methacrylic acid copolymer dispersion and the
PlasACRYL™ T20 is removed during the film coating process and therefore not included in the final formulation in Table 4. The tablets were coated with the aqueous suspension in the O’ Hara Technologies Labcoat M coater until the desired weight gain of enteric film was achieved. The coating process occurred at an inlet temperature of approximately 41 °C and an outlet temperature of 31 °C. After coating, the tablets were dried for 2 hours at 40 °C.
…………………………
WO 2014071371
http://www.google.com/patents/WO2014071371A1?cl=en
(N,N-Diethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4-dioate has the following chemical structure:

This compound was synthesized in Example 1 of Gangakhedkar et al., U.S. Patent No. 8,148,414. The compound is a prodrug of methyl hydrogen fumarate (MHF) and has a disclosed melting point of between 53 °C and 56 °C.
Cocrystals are crystals that contain two or more non-identical molecules that form a crystalline structure. The intermolecular interactions between the non-identical molecules in the resulting crystal structures can result in physical and chemical properties that differ from the properties of the individual components. Such properties can include, for example, melting point, solubility, chemical stability, mechanical properties and others. Examples of cocrystals may be found in the Cambridge Structural Database and in Etter, et al.,
“The use of cocrystallization as a method of studying hydrogen bond preferences of 2-aminopyridine” J. Chem. Soc, Chem. Commun. (1990), 589-591 ; Etter, et al., “Graph-set analysis of hydrogen-bond patterns in organic crystals” Acta Crystallogr., Sect. B, Struct. Sci. (1990), B46: 256-262; and Etter, et al., “Hydrogen bond directed cocrystallization and molecular recognition properties of diarylureas” J. Am. Chem. Soc. (1990), 1 12: 8415-8426. Additional information relating to cocrystals can be found in: Carl Henrik Gorbotz and Hans-Petter Hersleth,
“On the inclusion of solvent molecules in the crystal structures of organic compounds”; Acta Cryst. (2000), B56: 625-534; and Senthil Kumar, et al., “Molecular Complexes of Some Mono- and Dicarboxylic Acids with trans-1 ,4,-Dithiane-1 ,4-dioxide” American Chemical Society, Crystal Growth & Design (2002) , 2(4) : 313-318.
(N,N-Diethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4-dioate is a prodrug of methyl hydrogen fumarate. Once administered, the compound is metabolized in vivo into an active metabolite, namely, methyl hydrogen fumarate (MHF) which is also referred to herein as monomethyl fumarate (MMF). The in vivo metabolism of (N,N-Diethylcarbamoyl)methyl

(N,N-Diethylcarbamoyl)methyl methyl Methyl hydrogen fumarate N ^ diethyl glycolamide
(2E)but-2-ene-1 ,4-dioate
Table 1

As can be seen from the data in Table 1 , the six cocrystals disclosed herein each exhibit a higher melting point than crystalline (N,N-Diethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4- dioate.

…………………………….
Steady state pharmacokinetics of formulations of XP23829, a novel prodrug of monomethyl fumarate (MMF), in healthy subjects
66th Annu Meet Am Acad Neurol (AAN) (April 26-May 3, Philadelphia) 2014, Abst P1.188
………………………………….
Lymphocyte and eosinophil responses in healthy subjects dosed with Tecfidera and XP23829, a novel fumaric acid ester (FAE)
66th Annu Meet Am Acad Neurol (AAN) (April 26-May 3, Philadelphia) 2014, Abst P1.201
………………………..
A comparison of XP23829 with DMF, the active ingredient of BG-12
4th Cooperative Meet Consorti Mult Scler Cent (CMSC) Am Comm Treat Res Mult Scler (ACTRIMS) (May 30-June 2, San Diego) 2012, Abst SC03
http://annualmeeting.mscare.org/index.php?option=com_content&view=article&id=174&Itemid=101
…………………………..
Favorable metabolism and pharmacokinetics of formulations of XP23829, a novel fumaric acid ester, in healthy subjects
65th Annu Meet Am Acad Neurol (AAN) (March 16-23, San Diego) 2013, Abst P05.189
…………………………………..
Comparison of the efficacy and tolerability of a novel methyl hydrogenfumarate prodrug with dimethyl fumarate in rodent EAE and GI irritation models
Neurology 2011, 76(9): Abst P05.040
| WO2013119791A1 * | Feb 7, 2013 | Aug 15, 2013 | Xenoport, Inc. | Morpholinoalkyl fumarate compounds, pharmaceutical compositions, and methods of use |
| US20120034303 * | Jan 8, 2010 | Feb 9, 2012 | Forward Pharma A/S | Pharmaceutical formulation comprising one or more fumaric acid esters in an erosion matrix |
| US20120095003 * | Oct 14, 2011 | Apr 19, 2012 | Xenoport, Inc. | Methods of using prodrugs of methyl hydrogen fumarate and pharmaceutical compositions thereof |
| US20120157523 * | Oct 14, 2011 | Jun 21, 2012 | Xenoport, Inc. | Prodrugs of methyl hydrogen fumarate, pharmaceutical compositions thereof, and methods of use |
| K Gogas ET AL: “Comparison of the efficacy and tolerability of a novel methylhydrogenfumarate prodrug with dimethylfumarate in rodent experimental autoimmune encephalomyelitis and GI irritation models“, 26th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) & 15th Annual Conference of Rehabilitation in MS (RIMS), 15 October 2010 (2010-10-15), XP055076728, Retrieved from the Internet: URL:http://registration.akm.ch/einsicht.php?XNABSTRACT_ID=115706&XNSPRACHE_ID=2&XNKONGRESS_ID=126&XNMASKEN_ID=900 [retrieved on 2013-08-27] |
![]()
| WO2013119791A1 * | Feb 7, 2013 | Aug 15, 2013 | Xenoport, Inc. | Morpholinoalkyl fumarate compounds, pharmaceutical compositions, and methods of use |
| US20100048651 * | Aug 19, 2009 | Feb 25, 2010 | Xenoport, Inc. | Prodrugs of methyl hydrogen fumarate, pharmaceutical compositions thereof, and methods of use |
| US8669281 | 20 Sep 2013 | 11 Mar 2014 | Alkermes Pharma Ireland Limited | Prodrugs of fumarates and their use in treating various diseases |
| WO2014031894A1 | 22 Aug 2013 | 27 Feb 2014 | Xenoport, Inc. | Oral dosage forms of methyl hydrogen fumarate and prodrugs thereof |
| WO2014071371A1 | 5 Nov 2013 | 8 May 2014 | Xenoport, Inc. | Cocrystals of (n,n-diethylcarbamoyl)methyl methyl (2e)but-2-ene-1,4-dioate |
Inovio Kicks Off Study of Cervical Cancer Immunotherapy INO 3112

Inovio Kicks Off Study of Cervical Cancer Immunotherapy
Inovio Pharmaceuticals Inc. announced it has initiated a Phase 1/2a clinical trial to evaluate safety, immunogenicity, clinical responses and disease-free survival of its DNA immunotherapy product, INO-3112, in treating human papillomavirus (HPV)-associated cervical cancer. Read more…
Inovio Pharmaceuticals Inc. announced it has initiated a Phase 1/2a clinical trial to evaluate safety, immunogenicity, clinical responses and disease-free survival of its DNA immunotherapy product, INO-3112, in treating human papillomavirus (HPV)-associated cervical cancer. INO-3112 is a combination of Inovio’s lead active immunotherapy product, VGX-3100, and its proprietary immune activator expressing interleukin-12 (IL-12). VGX-3100 is currently being evaluated in a randomized Phase 2 efficacy trial for the treatment of high grade cervical dysplasia (pre-cancer).
DARA BioSciences receives FDA orphan drug designation for KRN5500 (SPK 241) …..Antitumor agent

KRN5500
Antitumor agent
151276-95-8 cas
IUPAC/Chemical name:
(2E,4E)-N-(2-(((2R,3R,4R,5R,6S)-6-((7H-purin-6-yl)amino)-2-((S)-1,2-dihydroxyethyl)-4,5-dihydroxytetrahydro-2H-pyran-3-yl)amino)-2-oxoethyl)tetradeca-2,4-dienamide
C28H43N7O7
Exact Mass: 589.32240
L-glycero-beta-L-manno-Heptopyranosylamine, 4-deoxy-4-((((1-oxo-2,4-tetradecadienyl)amino)acetyl)amino)-N-1H-purin-6-yl-, (E,E)-
L-glycero-beta-L-manno-Heptopyranosylamine, 4-deoxy-4-(((((2E,4E)-1-oxo-2,4-tetradecadienyl)amino)acetyl)amino)-N-1H-purin-6-yl-

-
- (1) Melting point: 182-183 °C,
- (2) Specific rotation [a]0 2S = 0 (c = 0.1, in methanol),
- (3) Elementary analysis:

- (4) FD mass spectrum (m/z): 590 (M + H) , C28 H4 3 N707
- (5) Infrared spectrum (KBr disc): 3250 cm-1, 1650 cm-1, 1620 cm-1,
- (6) Proton nuclear magnetic resonance spectrum (500 MHz, in CD30D) δH: 0.89 (3H, t, J = 7.3 Hz), 1.20-1.50 (14H, m), 2.18 (2H, dt, J = 7.3, 7.3 Hz), 3.6-3.8 (5H, m), 3.95 (1 H, d, J = 16.3 Hz), 3.98 (1H, d, J = 16.3 Hz), 4.00 (1H, dd, J = <1, 2.9 Hz), 4.15 (1H, dd, J = 10.8, 10.8 Hz), 5.66 (1 H, brs), 5.98 (1 H, d, J = 15.7 Hz), 6.12 (1 H, dt, J = 7.3, 15.7 Hz), 6.22 (1 H, dd, J = 10.0, 15.7 Hz), 7.17 (1 H, dd, J = 10.0, 15.7 Hz), 8.15 (1 H, s), 8.30 (1 H, s).
- EP 0525479; JP 1993186494; US 5461036; US 5631238

DARA BioSciences receives FDA orphan drug designation for KRN5500
DARA BioSciences has received orphan drug designation from the US Food and Drug Administration’s (FDA) Office of Orphan Products Development for KRN5500, for treating multiple myeloma
Multiple myeloma is a hematologic cancer or cancer of the blood.
KRN5500 is a non-opioid, non-narcotic compound that is currently being tested in Phase I clinical trial.
Earlier this year, KRN5500 received orphan status to be developed for the parenteral treatment of painful, chronic, chemotherapy-induced peripheral neuropathy (CCIPN) that is refractory to conventional analgesics in patients with cancer.
“We believe this myeloma-specific orphan designation enhances both the viability and the future market opportunity for this valuable pipeline product.”
DARA BioSciences MD, CEO and chief medical officer David J Drutz said: “It is noteworthy in this regard that up to 20% of myeloma patients have intrinsic peripheral neuropathy, an incidence that increases to the range of 75% in patients treated with neurotoxic drugs such as thalidomide or bortezomib.
KRN5500 is a semisynthetic derivative of the nucleoside-like antineoplastic antibiotic spicamycin, originally isolated from the bacterium Streptomyces alanosinicus. KRN 5500 inhibits protein synthesis by interfering with endoplasmic reticulum and Golgi apparatus functions. This agent also induces cell differentiation and caspase-dependent apoptosis.
KRN5500 is available as a solution for intravenous (IV) administration. KRN5500 was discovered in an effort to identify new agents that induced differentiation of myeloid leukemia cells.
Safety and efficacy data from Phase I trials have been leveraged to support DARA Therapeutics’ active IND and ongoing Phase 2a clinical trial. The objective of this Phase 2a feasibility study is to determine the potential of KRN5500 (a spicamycin analogue) to be a breakthrough medicine for the treatment of neuropathic pain in cancer patients.
Four clinical trials have been conducted in cancer patients, including one in Japan and 3 in the United States. Three of these studies are complete; the fourth was closed to patient accrual and treatment in December 2004.
A total of 91 patients with solid tumors have been treated with single IV KRN5500 doses of up to 21 mg/m2 and weekly doses of up to 42 mg/m2. While KRN5500 has not shown anti-cancer efficacy in any trial, its use in pain elimination is encouraging. (source: http://www.darabiosciences.com/krn5500.htm).

Chemical structures of KRN5500 and its known metabolites.
………………..
http://www.google.com/patents/EP0525479A1?cl=en
spk 241
- 6-[4′-N-(N’-trans,trans-2,4-tridecadienylglycyl)spicamynyl-amino]purine,
- (20) SPK241:


Example 52: Preparation of SPK241
-
[0214]To trans-2-dodecenal (4.5 g) dissolved in methylene chloride (80 ml) was added (carbomethoxymethylene)triphenylphosphorane (8.3 g), and the mixture was stirred for 2 hours. The reaction mixture was subjected to chromatography on a silica gel column with eluent systems of n-hexane- ethyl acetate (from 100:1 to 20:1) to give the methyl ester of trans,trans-2,4-tetradecadienoic acid (5.4 g). Potassium hydroxide (6.5 g) was dissolved in a mixed solvent of ethanol-water (1:1) (100 ml). The methyl ester of trans,trans-2,4-tetradecadienoic acid (5.4 g) was added to the mixture, and the resulting mixture was stirred at 60 °C for 40 minutes. After the reaction mixture was cooled, it was adjusted to the weak acidic range of pH with citric acid and extracted with ethyl acetate. The ethyl acetate layer was dried over anhydrous sodium sulfate and concentrated to give trans,trans-2,4-tetradecadienoic acid (4.4 g). Hereafter, the title compound can be synthesized by the two methods described below.
-
[0215]In the first method, trans,trans-2,4-tetradecadienoic acid (4.3 g) is first dissolved in N,N-dimethylformamide (DMF, 50 ml). Para-nitrophenol (2.67 g) and N,N’-dicyclohexylcarbodiimide (3.9 g) were added to trans,trans-2,4-tetradecadienoic acid solution, and the mixture was stirred for 12 hours. After precipitates produced were removed by filtration and the solvent (DMF) was removed by distillation, the residue was subjected to chromatography on a silica gel column with eluent systems of n-hexane-ethyl acetate (from 200:1 to 50:1) to give the active ester of trans,trans-2,4-tetradecadienoic acid (5.1 g). To the active ester (500 mg) dissolved in DMF (30 ml) were added 6-(4′-N-glycyl-spicamynyl-amino)purine hydrochloride (556 mg) and triethylamine (1.2 ml). The mixture was stirred for 12 hours. After the solvent was removed by distillation, the residue was subjected to chromatography on a silica gel column with eluent systems of chloroform-methanol (from 7:1 to 5:1) to give SPK241 in the yield of 398 mg.
-
[0216]In the second method, trans,trans-2,4-tetradecadienoic acid (99.6 g) was dissolved in thionyl chloride (87 ml), and the mixture was stirred at room temperature. The excessive thionyl chloride was removed by distillation to give trans,trans-2,4-tetradecadienoic acid chloride (102.0 g). To glycine (66.8 g) dissolved in an aqueous 2N sodium hydroxide solution (540 ml) were added at the same time trans,trans-2,4-tetradecadienoic acid chloride (102.0 g) and 2N sodium hydroxide (270 ml) with 1/10 portions at a 3 minute interval. After the addition was completed, the mixture was warmed to room temperature, stirred for 15 minutes and acidified with concentrated hydrochloric acid (140 ml) under ice-cooling. Precipitates thus produced were collected by filtration and desiccated to give trans,trans-2,4-tetradecadienoyl glycine (75.0 g). To the solution of trans,trans-2,4-tetradecadienoyl glycine (4.7 g) and 6-(4′-N-glycyl-spicamynyl-amino)-purine (5.1 g) in N,N-dimethylformamide (DMF, 60 ml) was added N-hydroxysuccinimide (2.1 g), and the mixture was ice-cooled. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (3.4 g) dissolved in DMF (100 ml) was added dropwise to the mixture. After the addition was completed, the mixture was heated to room temperature and stirred for 12 hours. Water (500 ml) was added to the reaction mixture, and precipitates produced were collected by filtration and desiccated. Sodium methoxide (3.1 g) was added to a suspension of the precipitates in methanol (100 ml), and the mixture was stirred at room temperature, then ice-cooled and acidified by adding dropwise thereto a 10% methanolic hydrochloric acid solution. Precipitates produced were filtered, dried and subjected to chromatography on a silica gel column with eluent systems of chloroform-methanol (from 7:1 to 5:1) to give SPK241 in the yield of 5.00 g.
Physicochemical properties of SPK241
-
[0217]
- (1) Melting point: 182-183 °C,
- (2) Specific rotation [a]0 2S = 0 (c = 0.1, in methanol),
- (3) Elementary analysis:
- (4) FD mass spectrum (m/z): 590 (M + H) , C28 H4 3 N707
- (5) Infrared spectrum (KBr disc): 3250 cm-1, 1650 cm-1, 1620 cm-1,
- (6) Proton nuclear magnetic resonance spectrum (500 MHz, in CD30D) δH: 0.89 (3H, t, J = 7.3 Hz), 1.20-1.50 (14H, m), 2.18 (2H, dt, J = 7.3, 7.3 Hz), 3.6-3.8 (5H, m), 3.95 (1 H, d, J = 16.3 Hz), 3.98 (1H, d, J = 16.3 Hz), 4.00 (1H, dd, J = <1, 2.9 Hz), 4.15 (1H, dd, J = 10.8, 10.8 Hz), 5.66 (1 H, brs), 5.98 (1 H, d, J = 15.7 Hz), 6.12 (1 H, dt, J = 7.3, 15.7 Hz), 6.22 (1 H, dd, J = 10.0, 15.7 Hz), 7.17 (1 H, dd, J = 10.0, 15.7 Hz), 8.15 (1 H, s), 8.30 (1 H, s).

| DE3407979A1 * | Mar 3, 1984 | Sep 6, 1984 | Kirin Brewery | Spicamycin sowie verfahren zu seiner herstellung |
| JPS59161396A | Title not available | |||
| US3155647 | Jul 24, 1963 | Nov 3, 1964 | Olin Mathieson | Septaciding and derivatives thereof |
| WO1990015811A1 | Jun 14, 1990 | Dec 27, 1990 | Kirin Brewery | Spicamycin x and its use |
| EP1328236A2 * | Sep 20, 2001 | Jul 23, 2003 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
| EP2305264A1 * | Sep 20, 2001 | Apr 6, 2011 | The General Hospital Corporation | Spicamycin derivatives for use in decreasing or preventing pain |
| EP2349285A2 * | Oct 9, 2009 | Aug 3, 2011 | Dara Biosciences, Inc. | Methods for treating or preventing pain using spicamycin derivatives |
| EP2597082A1 | Nov 24, 2011 | May 29, 2013 | Symrise AG | Compounds for masking an unpleasant taste |
| US5905069 * | Jan 26, 1998 | May 18, 1999 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin or derivatives thereof |
| US7196071 | Sep 20, 2001 | Mar 27, 2007 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
| US7375094 | Mar 15, 2007 | May 20, 2008 | The General Hospital Corporation | Produced via Streptomyces; antitumor agents; time-release agents; for opiod-resistant pain; drug screening |
| US7632825 | Apr 30, 2008 | Dec 15, 2009 | Bayer Pharmaceuticals Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
|
References 1: Mizumura Y. [Spicamycin derivative]. Nippon Rinsho. 2006 Feb;64(2):322-8. Review. Japanese. PubMed PMID: 16454188. 2: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2004 Apr;26(3):211-44. PubMed PMID: 15148527. 3: Borsook D, Edwards AD. Antineuropathic effects of the antibiotic derivative spicamycin KRN5500. Pain Med. 2004 Mar;5(1):104-8. PubMed PMID: 14996243. 4: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2003 Dec;25(10):831-55. PubMed PMID: 14735233. 5: Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2003 Nov;25(9):747-71. PubMed PMID: 14685303. 6: Supko JG, Eder JP Jr, Ryan DP, Seiden MV, Lynch TJ, Amrein PC, Kufe DW, Clark JW. Phase I clinical trial and pharmacokinetic study of the spicamycin analog KRN5500 administered as a 1-hour intravenous infusion for five consecutive days to patients with refractory solid tumors. Clin Cancer Res. 2003 Nov 1;9(14):5178-86. PubMed PMID: 14613997. 7: Yamamoto N, Tamura T, Kamiya Y, Ono H, Kondoh H, Shirao K, Matsumura Y, Tanigawara Y, Shimada Y. Phase I and pharmacokinetic study of KRN5500, a spicamycin derivative, for patients with advanced solid tumors. Jpn J Clin Oncol. 2003 Jun;33(6):302-8. PubMed PMID: 12913085. 8: Kobierski LA, Abdi S, DiLorenzo L, Feroz N, Borsook D. A single intravenous injection of KRN5500 (antibiotic spicamycin) produces long-term decreases in multiple sensory hypersensitivities in neuropathic pain. Anesth Analg. 2003 Jul;97(1):174-82, table of contents. PubMed PMID: 12818962. 9: Gadgeel SM, Boinpally RR, Heilbrun LK, Wozniak A, Jain V, Redman B, Zalupski M, Wiegand R, Parchment R, LoRusso PM. A phase I clinical trial of spicamycin derivative KRN5500 (NSC 650426) using a phase I accelerated titration “2B” design. Invest New Drugs. 2003 Feb;21(1):63-74. PubMed PMID: 12795531. 10: Byrd JC, Lucas DM, Mone AP, Kitner JB, Drabick JJ, Grever MR. KRN5500: a novel therapeutic agent with in vitro activity against human B-cell chronic lymphocytic leukemia cells mediates cytotoxicity via the intrinsic pathway of apoptosis. Blood. 2003 Jun 1;101(11):4547-50. Epub 2003 Feb 20. PubMed PMID: 12595316. |
BMS-791325, Beclabuvir In Phase 2 for Hepatitis C (HCV)
BMS-791325, Beclabuvir
IN PHASE 2 for Hepatitis C (HCV)
An NS5B inhibitor.

BMS-791325 preferably is


958002-33-0
958002-36-3 (as hydrochloride)
C36 H45 N5 O5 S, 659.838
Cycloprop(d)indolo(2,1-a)(2)benzazepine-9-carboxamide, 12-cyclohexyl-N-((dimethylamino)sulfonyl)-4b,5,5a,6-tetrahydro-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-, (4bS,5aR)-
(4bS,5aR)-12-Cyclohexyl-N-(dimethylsulfamoyl)-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-4b,5,5a,6-tetrahydrocyclopropa(d)indolo(2,1-a)(2)benzazepine-9-carboxamide
(4bS,5aR)-12-Cyclohexyl-N-(dimethylsulfamoyl)-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-4b,5,5a,6-tetrahydrocyclopropa(d)indolo(2,1-a)(2)benzazepine-9-carboxamide
(1aR,12bS)-8-Cyclohexyl-N-(dimethylsulfamoyl)-11-methoxy-1a-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8-yl)carbonyl]-1,1a,2,12b-tetrahydrocyclopropa[d]indolo[2,1-a][2]benzazepine-5-carboxamide
Cycloprop [d] indolo [2, 1 -a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (laR,12bS)-
Bristol-Myers Squibb (Originator)
RNA-Directed RNA Polymerase (NS5B) Inhibitors
UNII-MYW1X5CO9S
BMS-791325 is in phase II clinical studies at Bristol-Myers Squibb for the treatment of chronic hepatitis C. In 2013, the company received breakthrough therapy designation in the U.S. for the treatment of chronic hepatitis C in combination with daclatasvir and asunaprevir.
| Patent | WO 2007136982 |
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
http://www.google.com/patents/WO2007136982A1?cl=en
Scheme 1.

N-protected piperazines can also be coupled to the intermediate indolobenzazepine acids and the resultant piperazine carboxamides can be deprotected using methods known in the art and derivatized using a variety of synthetic protocols, some illustrative examples of which are shown below (See Scheme 2).
Scheme 2.

An intermediate useful for the synthesis of some compounds of the invention involves the preparation of the tert-butyl ester indolobenzazepine shown in Scheme 3. Scheme 3.

t-Butylation either:

This methodology involves base catalyzed hydrolysis of the indole methyl ester shown, followed by its reaction with either thionyl chloride and potassium tertiary butoxide, or alkylation with silver carbonate and tertiary butyl bromides. The resultant compound can be transformed using chemistry analogous to that outlined previously to provide the mixed ester indolobenzazepines shown above.
Scheme 4.

Some examples exist as stereoisomeric mixtures. The invention encompasses all stereoisomers of the compounds. Methods of fractionating stereoisomeric mixtures are well known in the art, and include but are not limited to; preparative chiral supercritical fluid chromatography (SFC) and chiral high performance liquid chromatography (HPLC). An example using this approach is shown in scheme 5. Scheme 5.

An additional method to achieve such separations involves the preparation of mixtures of diastereomers which can be separated using a variety of methods known in the art. One example of this approach is shown below (Scheme 6).
Scheme 6.

Diastereomers separated by reverse phase HPLC
Some diastereomeric amides can be separated using reverse phase HPLC. After hydroysis, the resultant optically active acids can be coupled with bridged piperazine derivatives (Scheme 6). For example, O-(lH-benzotriazol-l-yl)-N,N, N’,N’-tetramethyluronium tetrafluoroborate and diisopropyl ethyl amine in DMSO can be used to give the alkyl bridged piperazine carboxamides. Other standard acid amine coupling methods can also be used to give optically active carboxamides.

Schemes 7-9 illustrate other methods of making intermediates and compounds.

Scheme 8.

Scheme 9.


Biological Methods
The compounds demonstrated activity against HCV NS5B as determined in the following HCV RdRp assays.
DESCRIPTION OF SPECIFIC EMBODIMENTS
Unless otherwise specified, analytical LCMS data on the following intermediates and examples were acquired using the following columns and conditions. Stop time: Gradient time + 1 minute; Starting cone: 0% B unless otherwise noted; Eluent A: 5% CH3CN / 95% H2O with 10 mM NH4OAc (for columns A, D and E); 10 % MeOH / 90 % H2O with 0.1% TFA (for columns B and C); Eluent B: 95% CH3CN / 5% H2O with 10 mM NH4OAc (for columns A, D and E); 90 % MeOH / 10 % H2O with 0.1% TFA (for columns B and C); Column A:
Phenomenex lOμ 4.6 x 50 mm C18; Column B: Phenomenex C18 lOμ 3.0 x 50 mm; Column C: Phenomenex 4.6 x 50 mm C18 lOμ; Column D: Phenomenex Lina C18 5μ 3.0 x 50 mm; Column E: Phenomenex 5μ 4.6 x 50 mm Cl 8.
Intermediate 1

lH-Indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-, methyl ester. Freshly recrystallized pyridinium tribromide (recrystallization from hot AcOH (5 mL per 1 g), rinsed with cold AcOH and dried under high vacuum over KOH) was added in portions (over 10 min.) to a stirring solution of methyl 3-cyclohexyl-lH-indole-6- carboxylate (60 g, 233 mmol) (prepared using procedures describe in WO2004/065367) in CHC1/THF (1: 1, 1.25 L) at 2o C. The reaction solution was stirred at 0-5 °C for 2.5h, and washed with sat. aq. NaHSO3 (1 L), 1 N HCl (1 L) and brine (1 L). The organic layer was dried (MgSO4) and concentrated. The resulting red oil was diluted with Et2θ and concentrated. The resulting pink solid was dissolved into Et2θ (200 mL) treated with hexanes (300 mL) and partially concentrated. The solids were collected by filtration and rinsed with hexanes. The mother liquor was concentrated to dryness and the procedure repeated. The solids were combined to yield lH-indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-, methyl ester (64 g, 190 mmol, 82%) as a fluffy pink solid, which was used without further purification. IHNMR (300 MHz, CDCl3) δ 8.47 (br s, IH), 8.03 (d, J = 1.4 Hz, IH), 7.74 (dd, J = 1.4, 8.8 Hz, IH), 7.69 (d, J = 8.8 Hz, IH), 3.92 (s, 3H), 2.82 (tt, J = 3.7, 11.7 Hz, IH), 1.98 – 1.72 (m, 7H), 1.50 – 1.27 (m, 3H). 13CNMR (75 MHz, CDC13) δ 168.2, 135.6, 130.2, 123.1, 120.8, 120.3, 118.7, 112.8, 110.7, 52.1, 37.0, 32.2(2), 27.0(2), 26.1. LCMS: m/e 334 (M-H)“, ret time 3.34 min, column A, 4 minute gradient.
Intermediate 2

lH-Indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-. A solution of methyl 2- bromo-S-cyclohexyl-lH-indole-ό-carboxylate (20 g, 60 mmol) and LiOH (3.8 g, 160 mmol) in MeOΗ/TΗF/Η2O ( 1 : 1 : 1 , 300 mL) was heated at 90 °C for 2h. The reaction mixture was cooled in an ice/H2O bath, neutralized with IM HCl (-160 mL) diluted with H2O (250 mL) and stirred for Ih at rt. The precipitates were collected by filtration rinse with H2O and dried to yield lH-indole-6-carboxylic acid, 2-bromo-3- cyclohexyl- (quant.) which was used without further purification.
An alternative procedure that can by used to provide lH-indole-6-carboxylic acid, 2-bromo-3-cyclohexyl- is described below: A solution of methyl 2-bromo-3-cyclohexyl-lH-indole-6-carboxylate (117 g, 349 mmol) and LiOKH2O (26.4 g, 629 mmol) in MeOH/THF/H2O (1: 1: 1, 1.8 L) was heated at reflux for 3h. The reaction mixture was cooled in an ice/H2O bath to ~2 °C, neutralized with IM HCl (-650 mL) (added at such a rate that temperature did not exceed 5 °C), diluted with H2O (1 L) and stirred while warming to ambient temperature. The precipitates were collected by filtration rinsed with H2O and dried to yield the mono THF solvate of lH-indole-6-carboxylic acid, 2-bromo-3- cyclohexyl- (135.5 g, 345 mmol, 99%) as a yellow solid, which was used without further purification. IHNMR (300 MHz, CDCl3) δ 11.01 (br s, IH), 8.77 (s, IH), 8.07 (d, J = 1.5 Hz, IH), 7.82 (dd, J = 1.5, 8.8 Hz, IH), 7.72 (d, J = 8.8 Hz, IH), 3.84 – 3.74 (m, 4H), 2.89 (m, IH), 1.98 – 1.72 (m, HH), 1.50 – 1.24 (m, 3H). 13CNMR (75 MHz, CDC13) δ 172.7, 135.5, 130.7, 122.3, 120.9(2), 118.8, 113.3, 111.1, 67.9(2), 37.0, 32.2(2), 27.0(2), 26.1, 25.5(2). LCMS: m/e 320 (M-H)“, ret time 2.21 min, column A, 4 minute gradient.
Intermediate 3

lH-Indole-6-carboxamide, 2-bromo-3-cyclohexyl-N-
[(dimethylamino)sulfonyl]-. l,l’-Carbonyldiimidazole (1.17 g, 7.2 mmol) was added to a stirred solution of 2-bromo-3-cyclohexyl-lH-indole-6-carboxylic acid (2.03 g, 6.3 mmol) in THF (6 mL) at 22 °C. The evolution of CO2 was instantaneous and when it slowed the solution was heated at 50°C for 1 hr and then cooled to 220C. N,N-Dimethylsulfamide (0.94 g, 7.56 mmol) was added followed by the dropwise addition of a solution of DBU (1.34 g ,8.8 mmol) in THF (4 mL). Stirring was continued for 24 hr. The mixture was partitioned between ethyl acetate and dilute HCl. The ethyl acetate layer was washed with water followed by brine and dried over Na2SO4. The extract was concentrated to dryness to leave the title product as a pale yellow friable foam, (2.0 g, 74 %, >90 % purity , estimated from NMR). 1H NMR (300 MHz, DMSO-D6) δ ppm 1.28 – 1.49 (m, 3 H) 1.59 – 2.04 (m, 7 H) 2.74 – 2.82 (m, 1 H) 2.88 (s, 6 H) 7.57 (dd, J=8.42, 1.46 Hz, 1 H) 7.74 (d, J=8.78 Hz, 1 H) 7.91 (s, 1 H) 11.71 (s, 1 H) 12.08 (s, 1 H).
An alternative method for the preparation of lH-indole-6-carboxamide, 2- bromo-3-cyclohexyl-N-[(dimethylamino)sulfonyl]- is described below.
To a 1 L four necked round bottom flask equipped with a mechanical stirrer, a temperature controller, a N2 inlet , and a condenser, under N2, was added 2-bromo-3- cyclohexyl-lH-indole-6-carboxylic acid (102.0 g, 0.259 mol) and dry TΗF (300 mL). After stirring for 10 min, CDI (50.3 g, 0.31 mol) was added portion wise. The reaction mixture was then heated to 50 oC for 2 h. After cooling to 30 oC, N,N- dimethylaminosulfonamide (41.7 g, 0.336 mol) was added in one portion followed by addition of DBU (54.1 mL, 0.362 mol) drop wise over a period of 1 h. The reaction mixture was then stirred at rt for 20 h. The solvent was removed in vacuo and the residue was partitioned between EtOAc and 1 Ν HCl (1 : 1, 2 L). The organic layer was separated and the aqueous layer was extracted with EtOAc (500 mL). The combined organic layers were washed with brine (1.5 L) and dried over MgSO4. The solution was filtered and concentrated in vacuo to give the crude product (111.0 g). The crude product was suspended in EtOAc (400 mL) at 60 oC. To the suspension was added heptane (2 L) slowly. The resulting suspension was stirred and cooled to 0 oC. It was then filtered. The filter cake was rinsed with small amount of heptane and house vacuum air dried for 2 days. The product was collected as a white solid (92.0 g, 83%). 1H ΝMR (MeOD, 300 MHz) δ 7.89 (s, H), 7.77 (d, J= 8.4 Hz, IH), 7.55 (dd, J= 8.4 and 1.8 Hz, IH), 3.01 (s, 6H), 2.73-2.95 (m, IH), 1.81-2.05 (m, 8H), 1.39-1.50 (m, 2H); m/z 429 (M +H)+. Intermediate 4

lH-Indole-6-carboxamide, 3-cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2- formyl-4-methoxyphenyl)-. A mixture of the 2-Bromo-3-cyclohexyl- N- [(dimethylamino)sulfonyl]-lH-indole-6-carboxamide (4.28g, 0.01 mol), 4-methoxy- 2-formylphenyl boronic acid (2.1%, 0.015 mol), 2-dicyclohexylphosphino-2′,6′- dimethoxy-biphenyl (41 mg, 0.0001 mol), palladium acetate (11.2 mg), and finely ground potassium carbonate (4.24g, 0.02 mol) in toluene (30 mL) was stirred under reflux and under nitrogen for 30 min, at which time LC/MS analysis showed the reaction to be complete. The reaction mixture was then diluted with ethyl acetate and water, and then acidified with an excess of dilute HCl. The ethyl acetate layer was then collected and washed with dilute HCl, water and brine. The organic solution was then dried (magnesium sulfate), filtered and concentrated to give a gum. The gum was diluted with hexanes (250 ml) and ethyl acetate (25 mL), and the mixture was stirred for 20 hr at 22° C during which time the product was transformed into a bright yellow granular solid (4.8 g) which was used directly without further purification.
An alternative procedure for the preparation of lH-indole-6-carboxamide, 3- cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2-formyl-4-methoxyphenyl)- is provided below:
To a slurried solution of 2-bromo-3-cyclohexyl-N-[(dimethylamino)sulfonyl]- indole-6-carboxamide (54.0 g, 126 mmol), 4-methoxy-2-formylphenylboronic acid (29.5 g, 164 mmol) and LiCl (13.3 g, 315 mmol) in EtOH/toluene (1 : 1, 1 L) was added a solution of Na2CO3 (40.1 g, 379 mmol) in water (380 mL). The reaction mixture was stirred 10 min. and then Pd(PPh3)4 (11.3 g, 10.0 mmol) was added. The reaction solution was flushed with nitrogen and heated at 70 °C (internal monitoring) overnight and then cooled to rt. The reaction was diluted with EtOAc (1 L) and EtOH (100 mL), washed carefully with IN aqueous HCl (1 L) and brine (500 mL), dried (MgSO4), filtered and concentrated. The residual solids were stirred with Et20 (600 mL) for Ih and collected by filtration to yield lH-indole-6-carboxamide, 3- cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2-formyl-4-methoxyphenyl)- (52.8g, 109 mmol, 87%) as a yellow powder which was used without further purification. IHNMR (300 MHz, d6-DMSO) δ 11.66 (s, IH), 8.17 (s, IH), 7.75 (d, J = 8.4 Hz, IH), 7.74 (d, J = 8.4 Hz, IH), 7.59 (dd, J = 1.4, 8.4 Hz, IH), 7.23 – 7.16 (m, 2H), 7.08 (dd, J = 2.6, 8.4 Hz, IH), 6.54 (d, J = 8.8 Hz, IH), 3.86 (s, 3H), 3.22 – 3.08 (m, IH), 2.91 (s, 6H), 2.00 – 1.74 (m, 7H), 1.60 – 1.38 (m, 3H). 13CNMR (75 MHz, CDC13) δ 165.7, 158.8, 147.2, 139.1, 134.3, 132.0, 123.4, 122.0, 119.2, 118.2, 114.8, 112.3, 110.4, 109.8, 79.6, 45.9, 37.2(2), 34.7, 32.0(2), 25.9(2), 24.9. LCMS: m/e 482 (M- H)“, ret time 2.56 min, column A, 4 minute gradient.
Intermediate 5

6H-Isoindolo[2,l-a]indole-3-carboxamide, 11-cyclohexyl-N-
[(dimethylamino)sulfonyl]-6-ethoxy-8-methoxy-. To a 5 L four necked round bottom flask equipped with a temperature controller, a condenser, a N2 inlet and a mechanical stirrer, was charged toluene (900 mL), EtOH (900 mL), 2-bromo-3- cyclohexyl-N^NjN-dimethylsulfamoyiyiH-indole-ό-carboxamide (90 g, 0.21 mol), 2-formyl-4-methoxyphenylboronic acid (49.2 g, 0.273 mol) and LiCl (22.1 g, 0.525 mol). The resulting solution was bubbled with Ν2 for 15 mins. A solution of Na2CO3 (66.8 g, 0.63 mol) in Η2O (675 mL) was added and the reaction mixture was bubbled with N2 for another (10 mins). Pd(PPh3)4 (7.0 g, 6.3 mmol) was added and the reaction mixture was heated to 70 °C for 20 h. After cooling to 35 °C, a solution of 1 N HCl (1.5 L) was added slowly. The resulting mixture was transferred to a 6 L separatory funnel and extracted with EtOAc (2 X 1.5 L). The combined organic extracts were washed with brine (2 L), dried over MgSO4, filtered and concentrated in vacuo to give a yellow solid, which was triturated with 20% EtOAc in hexane (450 mL, 50 °C to 0 °C) to give 3-cyclohexyl-N-(N,N-dimethylsulfamoyl)-2-(2-formyl-4- methoxyphenyl)-lH-indole-6-carboxamide(65.9 g) as a yellow solid. HPLC purity, 98%.
The mother liquid from the trituration was concentrated in vacuo. The residue was refluxed with EtOH (50 mL) for 3 h. The solution was then cooled to 0 °C. The precipitates were filtered and washed with cooled TBME (5 °C) (20 mL). The filter cake was house vacuum air dried to give a further quantity of the title compound as a white solid (16.0 g). HPLC purity, 99%. 1H NMR (CDC13, 300 MHz) δ 8.75 (s, IH), 7.96 (s, IH), 7.73 (d, J= 8.4 Hz, IH), 7.67 (d, J= 8.4 Hz, IH), 7.45 (dd, J= 8.4 and 1.4 Hz, IH), 7.09 (d, J= 2.2 Hz, IH), 6.98 (dd, J= 8.4 and 2.2 Hz, IH), 6.50 (s, IH), 3.86 (s, 3H), 3.05 (s, 6H), 2.92-3.13 (m, 3H), 1.85-1.93 (m, 7 H), 1.40-1.42 (m, 3H), 1.05 (t, J= 7.1 Hz, 3H). m/z 512 (M + H)+.
Intermediate 6

lH-indole-6-carboxamide, 3-cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2- formyl-4-methoxyphenyl)-. 1 l-cyclohexyl-N-(N,N-dimethylsulfamoyl)-6-ethoxy-8- methoxy-6H-isoindolo[2,l-a]indole-3-carboxamide was dissolved in THF (75 mL). To the solution was added a solution of 2 N HCl (300 mL). The mixture was vigorously stirred under N2 at rt for 16 h. The resulting suspension was filtered and washed with cooled TBME (2 X 30 mL). the filer cake was vacuum air dried overnight to give the title compound as a yellow solid. HPLC purity, 99% 1H NMR (DMSO-d6, 300 MHz) δ 11.65 (s, IH), 8.16 (s, IH), 7.76 (d, J= 5.9 Hz, IH), 7.73 (d, J= 5.9 Hz, IH), 7.58 (dd, J= 8.5 and 1.5 Hz, IH), 7.17-7.20 (m, 2H), 7.08 (dd, J = 8.5 and 1.4 Hz, IH), 6.55 (d, J= 8.6 Hz, IH), 3.86 (s, 3H), 3.14-3.18 (m, IH), 2.91 (s, 6H), 1.75-1.99 (m, 7H), 1.48-1.60 (m, 3H); m/z 484 (M + H)+.
Intermediate 7

7H-Indolo[2, 1-a] ‘ [2] benzazepine-6-carboxylic acid, 13-cyclohexyl-10- [[[(dimethylamino)sulfonyl] amino] carbonyl]-3-methoxy-, methyl ester. A mixture of the 3-cyclohexyl-N-(N,N-dimethylsulfamoyl)-2-(2-formyl-4-methoxyphenyl)-lH- indole-6-carboxamide (4.8g, 0.01 mol), methyl 2-(dimethoxyphosphoryl)acrylate (9.7 g, 0.02 mol) and cesium carbonate (7.1g, 0.02 mol) in DMF (28mL) was stirred for 20 hr at an oil bath temperature of 55 ° C. The mixture was poured into ice-water and acidified with dilute HCl to precipitate the crude product. The solid was collected, dried and flash chromatographed on Siθ2 (11Og) using an ethyl acetate and methylene chloride (1: 10) solution containing 2% acetic acid. Homogeneous fractions were combined and evaporated to afford the title compound as a pale yellow solid (3.9g, 71 % yield). MS: 552 (M=H+).
An alternate procedure for the preparation of 7H-indolo[2,l- a] [2]benzazepine-6-carboxylic acid, 13-cyclohexyl-10- [[[(dimethylamino)sulfonyl]amino]carbonyl]-3-methoxy-, methyl ester is provided below. A solution of l l-cyclohexyl-N-[(dimethylamino)sulfonyl]-6-hydroxy-8- methoxy-6H-isoindolo[2,l-a]indole-3-carboxamide (cyclic hemiaminal) (63.0 g, 130 mmol), methyl 2-(dimethoxyphosphoryl)acrylate (60 g, 261 mmol), cesium carbonate (106 g, 326 mmol) in DMF (400 mL) was heated at 60 °C (bath temp) for 4.5h. Additional methyl 2-(dimethoxyphosphoryl)acrylate (15 g, 65 mmol) and cesium carbonate (21.2 g, 65 mmol) were added and the reaction was heated at 60 °C overnight then and cooled to rt. The stirring reaction mixture was diluted with H2O (1 L), slowly neutralized with IN aqueous HCl (800 mL), stirred 3h, and then the precipitates were collected by filtration. The solids were triturated with Et20 (800 mL) and dried to yield methyl 7H-indolo[2,l-a][2]benzazepine-6-carboxylic acid, 13- cyclohexyl-10-[[[(dimethylamino)sulfonyl]amino]carbonyl]-3-methoxy-, methyl ester (70.2 g, 127 mmol, 98%) as a yellow solid which was used without further purification. IHNMR (300 MHz, CDC13) δ 8.67 (s, IH), 8.09 (s, IH), 7.86 (d, J = 8.4 Hz, IH), 7.80 (s, IH), 7.50 (d, J = 8.4 Hz, IH), 7.42 (d, J = 8.8 Hz, IH), 7.08 (dd, J = 2.6, 8.8 Hz, IH), 6.98 (d, J = 2.6 Hz, IH), 5.75 – 5.51 (m, IH), 4.29 – 4.01 (m, IH), 3.89 (s, 3H), 3.82 (s, 3H), 3.05 (s, 6H), 2.87 – 2.73 (m, IH), 2.11 – 1.12 (m, 10H). LCMS: m/e 550 (M-H)-, ret time 3.21 min, column A, 4 minute gradient.
Example 1

Cycloprop[d]indolo[2,l-a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (+/-)-. TBTU (43.7 mg, 0.136mmol) and DIPEA (0.095 mL, 0.544 mmol) were added to a solution of (+/-) cycloprop[d]indolo[2,l-a][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (50 mg, 0.0906 mmol) in DMSO (2.0 mL). The reaction mixture was stirred at rt for 15 min. 3-Methyl-3,8-diaza-bicyclo[3.2. l]octane dihydrochloride {J & W PharmLab, LLC Morrisville, PA 19067-3620}. (27.1 mg, 0. 136 mmol) was then added and the reaction mixture was stirred at rt for 3 hr. It was then concentrated and the residue was purified by preparative reverse phase HPLC to give the final product as a yellow solid, (32 mg, 46% yield). MS m/z 660(MH+), Retention time: 2.445 min IH NMR (300 MHz, MeOD) δ ppm 0.20 (m, 0.23 H) 1.11 – 2.25 (m, 15.77 H) 2.58 (m, 0.23 H) 2.69 (m, 0.77 H) 2.75 – 3.11 (m, 10 H) 3.28 – 3.75 (m, 5 H) 3.91 (s, 2.31 H) 3.92 (s, 0.69 H) 4.15 – 4.37 (m, 1 H) 4.68 (m ,br, 1 H) 4.94 – 5.00 (m, 0.23 H) 5.16 (d, J=15.00 Hz, 0.77 H) 7.00 – 7.09 (m, 1 H) 7.18 (d, J=2.56 Hz, 0.23 H) 7.21 (d, J=2.56 Hz, 0.77 H) 7.33 (d, J=8.41 Hz, 0.77 H) 7.35 (d, J=8.42 Hz, 0.23 H) 7.57 (dd, J=8.42, 1.46 Hz, 0.77 H) 7.62 (dd, J=8.78, 1.46 Hz, 0.23 H) 7.91 (d, J=8.42 Hz, 0.77 H) 7.93 (d, J=8.42 Hz, 0.23 H) 8.00 (s, 0.77 H) 8.07 (s, 0.23 H).
Example 4

Cycloprop[d]indolo[2,l-a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonylj ‘- 1 , Ia, 2, 12b-tetrahydro-ll-methoxy-la-[(8-methyl-3, 8- diazabicyclo[3.2.1]oct-3-yl)carbonyl]-, (+/-)-. To a solution of (+/-) cycloprop[d]indolo[2,l-a][2]benzazepine-5-carboxamide, 8-cyclohexyl-la-(3,8- diazabicyclo[3.2.1]oct-3-ylcarbonyl)-N-[(dimethylamino)sulfonyl]-l,la,2,12b- tetrahydro-11-methoxy- (54 mg, 0.071 mmol) in methanol (3 mL), paraformaldehyde (6.4 mg, 0.213 mmol), ZnCl2 (29 mg, 0.213 mmol) and
Na(CN)BH3 (13.4 mg, 0.213 mmol) were added. The resultant mixture was heated at 60°C for 2hr, and then cooled to rt. The solid present was removed by filtration, and the filtrate was concentrated under vacuum and the residue purified by preparative reverse phase HPLC to give the title compound as a light yellow colored solid, (37 mg, 67% yield). MS ml 660(MH+), Retention time: 2.495 min. IH NMR (500 MHz, MeOD) δ ppm 0.21 (m, 0.3 H) 1.13 (m, 0.3 H) 1.18 – 2.22 (m, 15.4 H) 2.58 (m, 0.3 H) 2.68 (m, 0.7 H) 2.76 – 3.11 (m, 11 H) 3.32 – 3.37 (m, 1 H) 3.63 (d, J=15.56 Hz, 0.7 H) 3.82 – 4.32 (m, 7.3 H) 4.88 – 4.92 (m, 0.3 H) 5.08 (d, J=15.56 Hz, 0.7 H) 7.00 – 7.08 (m, 1 H) 7.18 (d, J=2.14 Hz, 0.3 H) 7.21 (d, J=2.14 Hz, 0.7 H) 7.32 (d, J=8.55 Hz, 0.7 H) 7.35 (d, J=8.55 Hz, 0.3H) 7.57 (d, J=7.93 Hz, 0.7 H) 7.62 (dd, J=8.39, 1.37 Hz, 0.3 H) 7.91 (d, J=8.55 Hz, 0.7 H) 7.93 – 7.99 (m, 1 H) 8.09 (s, 0.3 H).
Example 6

Cycloprop [d] indolo [2, 1 -a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (laR,12bS)-. To a solution of (-) cycloprop[d]indolo[2,l-a][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (204 mg, 0.37 mmol) in DMSO (8.0 mL), TBTU (178 mg, 0.555 mmol) and DIPEA (0.39 mL, 2.22 mmol) were added. The reaction mixture was stirred at rt for 15 min. Then 3- methyl-3,8-diaza-bicyclo[3.2.1]octane dihydrochloride (111 mg, 0. 555 mmol) was added and the reaction mixture was stirred at rt for 2 hr. It was then concentrated and the residue was purified by preparative reverse phase HPLC to give a yellow solid as final TFA salt. (265 mg, 92% yield). Average Specific Rotation: -53.56° Solvent, MeOH.; Wavelength 589 nm; 50 cm cell. MS m/z 660(MH+), Retention time: 3.035 min. 1H NMR (300 MHz, MeOD) δ ppm 0.20 (m, 0.23 H) 1.11 – 2.25 (m, 15.77 H) 2.58 (m, 0.23 H) 2.69 (m, 0.77 H) 2.75 – 3.11 (m, 10 H) 3.28 – 3.75 (m, 5 H) 3.91 (s, 2.31 H) 3.92 (s, 0.69 H) 4.15 – 4.37 (m, 1 H) 4.68 (m ,br, 1 H) 4.94 – 5.00 (m, 0.23 H) 5.16 (d, J=15.00 Hz, 0.77 H) 7.00 – 7.09 (m, 1 H) 7.18 (d, J=2.56 Hz, 0.23 H) 7.21 (d, J=2.56 Hz, 0.77 H) 7.33 (d, J=8.41 Hz, 0.77 H) 7.35 (d, J=8.42 Hz, 0.23 H) 7.57 (dd, J=8.42, 1.46 Hz, 0.77 H) 7.62 (dd, J=8.78, 1.46 Hz, 0.23 H) 7.91 (d, J=8.42 Hz, 0.77 H) 7.93 (d, J=8.42 Hz, 0.23 H) 8.00 (s, 0.77 H) 8.07 (s, 0.23 H). An alternate procedure for the synthesis of cycloprop[d]indolo[2,l- a][2]benzazepine-5-carboxamide, 8-cyclohexyl-N-[(dimethylamino)sulfonyl]- l,la,2,12b-tetrahydro-l l-methoxy-la-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8- yl)carbonyl]-, (laR,12bS)-rel-(-)-is provided below. To a mixture of (-) cycloprop[<i]indolo[2,l-α][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (25.2 g, 45.68 mmol) and 3-methyl-3,8-diazabicyclo-[3.2.1]octane dihydrochloride (10.0 g, 50.22 mmol) in anhydrous MeCN (300 mL) was added DIPEA (23.62 g, 182.72 mmol) under N2. After 15 min, TBTU (16.12 g, 50.22 mmol) was added. The reaction solution was stirred for 30 min under N2. The ΗPLC indicated the disappearance of starting material. The solvent in the solution was evaporated to give a foam. This was dissolved in EtOAc (2.5 L), washed with H2O (1.5 L), H2O/brine (8:2) (1.5 L), brine (1.5 L), dried over Na2SO4 and evaporated to give 28.8 g of crude product. This solid was pooled with 45.4 g of material obtained from five separated reactions to afford a total of 74.2 g of crude product. This was passed through a pad of silica gel (E. Merck 230-400 mesh, 1 kg), eluting with MeOH/CH2Cl2 (2.5:97.5). After evaporation, it gave a foam, which was treated with EtOAc and hexane to turn into a solid. After drying at 50 °C under vacuum for 7 h, the GC analysis indicated it has 1.4% each of EtOAc and hexane. After further drying at 61-64 °C, the GC analysis indicated it still has 1.0% of hexane and 1.4% of EtOAc. The product was dissolved in Et2O and slowly evaporated in vacuum three times, dried at 60 °C under vacuum for 3 h to give 68.3 g. This was washed with H2O (900 mL) and redried at 68 °C under vacuum for 7 h to give 67.1 g (77% yield) of the compound of example 6. The GC analysis indicated it has 0.97% Of Et2O. HPLC conditions column: Cadenza CD-C18 3 x 250 mm; UV: 257 and 220 nm; 25 °C; flow rate: 0.5 mL/min; gradient time: 38 min, 0 – 80% B (0 – 35 min) and 80% B (35 – 38 min); solvent A: 25 nM CH3COONH4 at pH 4.7 in water, solvent B: MeCN. HPLC purity 99.7% (Rt 26.54 min); Chiral HPLC conditions column: Regis (S5S) Whelk-Ol 250 x 4.6 mm; UV 258nm; 35 °C; flow rate 2.0 mL/min; mobile phase C02/Me0H; gradient time 20 min, 30% MeOH (0 – 1 min), 30 – 48% MeOH (1 – 19 min), 48% MeOH (19 – 20 min). Chiral HPLC purity > 99.8% (Rt 16.60 min); LC/MS (ES+) 660.36 (M+H, 100); HRMS: calcd. 660.3220, found 660.3197; [α]D 25 C – 79.66 ° (c 1.06, MeOH); Anal. Calcd for C36H45N5O5S-O-O H2O»0.09 Et2O: C, 64.53; H, 7.00; N, 10.35; S, 4.74; H2O, 1.51; Et2O, 0.97. Found: C, 64.50; H, 7.12; N, 10.41; S, 5.14; H2O, 1.52; Et2O, 0.97. The absolute stereochemistry of cycloprop[d]indolo[2,l- a][2]benzazepine-5-carboxamide, 8-cyclohexyl-N-[(dimethylamino)sulfonyl]- l,la,2,12b-tetrahydro-l l-methoxy-la-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8- yl)carbonyl]-, (laR,12bS)-rel-(-)- is as drawn above, and was determined from an x- ray crystal structure obtained on the (R)-camphorsulfonic acid salt.
Additionally, the following salts were prepared: hydrochloride, phosphate, acetate, sulfate, camsylate, sodium, calcium, and magnesium. The hydrochloride salt had the following characteristics. DSC: small, broad endotherm from 25°C to 75°C, and potential melt/degradation endotherm with peak at temperatures ranging between 253 °C and 258 °C; TGA: Early weight loss from 25°C to 75°C ranging between 0.003% and 1.5%, and degradation weight loss starting at approximately 200°C.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
| WO2006020082A1 * | Jul 15, 2005 | Feb 23, 2006 | Squibb Bristol Myers Co | Inhibitors of hcv replication |
| WO2006046030A2 * | Oct 25, 2005 | May 4, 2006 | Angeletti P Ist Richerche Bio | Tetracyclic indole derivatives as antiviral agents |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008111978A1 | Mar 13, 2007 | Sep 18, 2008 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2008112473A1 * | Mar 5, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112841A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112848A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112851A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv inhibitors |
| WO2008112863A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2009067108A1 * | Nov 20, 2007 | May 28, 2009 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2009067392A1 * | Nov 17, 2008 | May 28, 2009 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine derivatives for the treatment of hepatitis c |
| WO2009067481A1 * | Nov 19, 2008 | May 28, 2009 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2010080874A1 | Jan 7, 2010 | Jul 15, 2010 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| WO2013059265A1 * | Oct 17, 2012 | Apr 25, 2013 | Bristol-Myers Squibb Company | A compound for the treatment of hepatitis c |
| WO2014014885A1 * | Jul 16, 2013 | Jan 23, 2014 | Bristol-Myers Squibb Company | Novel methods and intermediates for the preparation of (4bs,5ar)-12-cyclohexyl-n-(n,n-dimethylsulfamoyl)-3-methoxy-5a-((1 r,5s) -3-methyl-3,8-diazabicyclo[3.2.1]octane-8-carbonyl)-4b,5,5a,6-tetrahydrobenzo [3,4]cyclopropa[5,6]azepino[1,2-a]indole-9-carboxamide |
| CN101679442B | Mar 13, 2008 | Feb 20, 2013 | 百时美施贵宝公司 | Compounds for the treatment of hepatitis c |
| EP2518073A1 * | Nov 19, 2008 | Oct 31, 2012 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
The First Kilogram Synthesis of Beclabuvir, an HCV NS5B Polymerase Inhibitor
 , Kenneth J. Fraunhoffer, Zhinong Gao, Chao Hang, Yi Hsiao, Wenhao Hu, Kishta Katipally, Adam Littke, Aghogho Pedro, Yuping Qiu, Maria Sandoval, Richard Schild, Michelle Soltani, Anthony Tedesco, Dale Vanyo, Purushotham Vemishetti, and Robert E. Waltermire
, Kenneth J. Fraunhoffer, Zhinong Gao, Chao Hang, Yi Hsiao, Wenhao Hu, Kishta Katipally, Adam Littke, Aghogho Pedro, Yuping Qiu, Maria Sandoval, Richard Schild, Michelle Soltani, Anthony Tedesco, Dale Vanyo, Purushotham Vemishetti, and Robert E. Waltermire
The process development and kilogram-scale synthesis of beclabuvir (BMS-791325, 1) is described. The convergent synthesis features the use of asymmetric catalysis to generate a chiral cyclopropane fragment and coupling with an indole fragment via an alkylation. Subsequent palladium-catalyzed intramolecular direct arylation efficiently builds the central seven-membered ring. The target was prepared in 12 linear steps with five isolations in an overall yield of 8%.
Preparation of (4bS,5aR)-12-Cyclohexyl-N-(N,N-dimethylsulfamoyl)-3-methoxy-5a-((1R,5S)-3-methyl-3,8-diazabicyclo[3.2.1]octane-8-carbonyl)-4b,5,5a,6-tetrahydrobenzo[3,4]cyclopropa[5,6]azepino[1,2-a]indole-9-carboxamide Hydrochloride (1·HCl)
BMS-791325·HCl (1·HCl) was isolated in 89.5% yield.
1H NMR (600 MHz, 10:1 v/v CD3CN/D2O): major rotamer: 7.91 (br s, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.55 (br d, J = 8.5 Hz, 1H), 7.29 (d, J = 8.5 Hz, 1H), 7.20 (d, J = 2.5 Hz, 1H), 7.00 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 5.03 (br d, J = 12.7 Hz, 1H), 4.58 (br d, J = 4.9 Hz, 2H), 3.87 (s, 3H), 3.56 (d, J = 15.5 Hz, 1H), 3.40 (br s, 3H), 3.32–3.28 (m, 4H), 2.96 (s, 6H), 2.92 (tt, J= 12.2, 3.6 Hz, 1H), 2.59 (br t, J = 7.0 Hz, 1H), 2.05–1.90 (m, 2H), 1.79–1.71 (m, 4H), 1.55 (br d, J= 12.2 Hz, 2H), 1.46–1.36 (m, 4H), 1.26 (t, J = 5.3 Hz, 2H), 1.23–1.15 (m, 2H);
minor rotamer: 8.05 (br s, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.58 (dd, J = 8.5, 1.4 Hz, 1H), 7.34 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 2.6 Hz, 1H), 6.98 (d, 1H, overlap with major rotamer), 4.91 (d, J = 15.0 Hz, 1H), 4.58 (br d, J = 4.9 Hz, 2H), 4.11 (d, J = 15.0 Hz, 1H), 3.89 (s, 3H), 3.46 (br d, J = 12.5 Hz, 2H), 3.17 (br d, J = 12.5 Hz, 2H), 2.97 (s, 6H), 2.85 (br s, 3H), 2.76 (tt, J = 12.1, 3.5 Hz, 1H), 2.49 (br s, 1H), 2.05–1.90 (m, 2H), 1.79–1.71 (m, 4H), 1.46–1.36 (m, 6H), 1.23–1.15 (m, 2H), 1.10 (m, 1H), 0.03 (t, J = 6.1 Hz, 1H).
13C NMR (125 MHz, 10:1 v/v CD3CN/D2O): major rotamer: 170.1, 167.7, 161.0, 140.4, 139.3, 135.9, 133.6, 131.1, 124.9, 123.0, 121.7, 120.8, 119.0, 118.6, 114.3, 110.7, 59.2, 56.2, 53.1, 48.3, 44.5, 38.9, 37.6, 34.8, 33.77, 33.72, 27.92, 27.77, 26.82, 26.5, 23.6, 18.5;
minor rotamer: 168.3, 168.0, 161.3, 138.4, 137.5, 135.8, 134.2, 130.0, 125.4, 121.9, 120.0, 119.64, 119.58, 117.9, 113.3, 111.3, 59.6, 56.3, 53.1, 44.6, 42.2, 38.9, 38.3, 37.4, 33.8, 33.6, 28.3, 27.74, 26.79, 26.5, 24.84, 11.9.
HRMS (ESI) calcd for C36H45N5O5S (free base) [M + H]+660.3214, found m/z 660.3220.
////////BMS-791325, Beclabuvir, Phase 2, Hepatitis C, HCV,
