

| Patent | Submitted | Granted |
|---|---|---|
| Compound useful for the treatment of degenerative and inflammatory diseases [US8088764] | 2010-12-30 | 2012-01-03 |
| NOVEL COMPOUNDS USEFUL FOR THE TREATMENT OF DEGENERATIVE AND INFLAMMATORY DISEASES [US2011190260] | 2011-08-04 |
Home » Posts tagged 'phase 2' (Page 13)
Tag Archives: phase 2
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
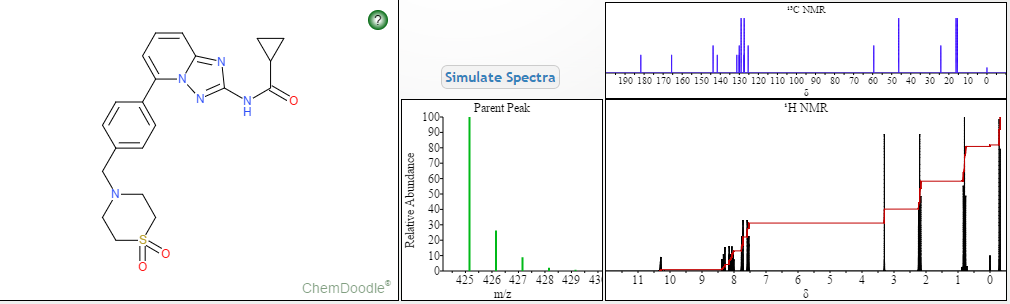
Filgotinib

Filgotinib
EU APPROVED 2020/9/24, JYSELECA
JAPAN APPROVED2020/9/25
- C21H23N5O3S
- MW425.504
- Elemental Analysis: C, 59.28; H, 5.45; N, 16.46; O, 11.28; S, 7.54
1206161-97-8
Cyclopropanecarboxamide, N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]-
G146034
GLPG0634
N-(5-(4-((1,1-dioxidothiomorpholino)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide
Galapagos Nv INNOVATOR
PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulcerative colitis
Filgotinib is an orally available inhibitor of JAK1/JAK2 and TYK2 in phase III clinical development at Galapagos and Gilead for the treatment of rheumatoid arthritis, moderate or severe Crohn’s disease and ulcerative colitis
IL-6 antagonist; Jak1 tyrosine kinase inhibitor; Tyk2 tyrosine kinase inhibitor; Jak3 tyrosine kinase inhibitor; Jak2 tyrosine kinase inhibitor
Autoimmune disease; Cancer; Colitis; Crohns disease; Inflammatory disease; Neoplasm; Rheumatoid arthritis; Transplant rejection
In 2017, orphan drug designation was assigned to the compound in the U.S. for the treatment of pediatric Crohn’s disease and pediatric ulcerative colitis.
GlaxoSmithKline had been developing filgotinib preclinically for the treatment of rheumatoid arthritis pursuant to a license; however, in 2010, the compound was re-acquired by Galapagos. In 2012, the product was licensed to Abbott for development and marketing. In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie. The license agreement between Galapagos and Abbott was terminated in September 2015, Galapagos regaining all rights to the product. The same year, Galapagos and Gilead entered into a global partnership and Gilead obtained the global rights of codevelopment and commercialization for the treatment of inflammatory diseases
Filgotinib (GLPG0634), by the Belgian biotech company Galápagos NV, is a drug which is currently under investigation for the treatment of rheumatoid arthritis and Crohn’s disease.
Filgotinib (GLPG0634) is an orally-available, selective inhibitor of JAK1 (Janus kinase 1) for the treatment of rheumatoid arthritis and potentially other inflammatory diseases. Filgotinib (GLPG0634) dose-dependently inhibited Th1 and Th2 differentiation and to a lesser extent the differentiation of Th17 cells in vitro. GLPG0634 was well exposed in rodents upon oral dosing, and exposure levels correlated with repression of Mx2 expression in leukocytes. The JAK1 selective inhibitor GLPG0634 (Filgotinib) is a promising novel therapeutic with potential for oral treatment of rheumatoid arthritis and possibly other immune-inflammatory diseases. Filgotinib (GLPG0634) is currently in a Phase 2 study in Crohn’s disease.

Mechanism of action
Filgotinib is a Janus kinase inhibitor with selectivity for subtype JAK1 of this enzyme. It is considered a promising agent as it inhibits JAK1 selectively. Less selective JAK inhibitors (e.g. tofacitinib) are already being marketed. They show long-term efficacy in the treatment of various inflammatory diseases. However, their lack of selectivity leads to dose-limiting side effects.[1] It is thought that inhibition of all JAK isoenzymes is beneficial in rheumatoid arthritis. However, pan-JAK inhibition might also lead to unwanted side effects that might not outweigh its benefits. This is the rationale for the development of newer and more selective inhibitors like filgotinib.
The signal transmission of large numbers of proinflammatory cytokines is dependent on JAK1. Inhibition of JAK2 may also contribute to the efficacy against RA. Nonetheless it is thought that JAK2 inhibition might lead to anemia and thrombopenia by interference witherythropoietin and thrombopoietin and granulocyte-macrophage colony-stimulating factor. Therefore one might prefer to choose a more selective JAK1 inhibitor as a primary therapeutic option. Filgotinib exerts a 30-fold selectivity for JAK1 compared to JAK2.[2] It is however still to be seen to what extent JAK2 inhibition should be avoided.
Novel crystalline forms of filgotinib salts, particularly hydrochloride salt, useful for treating JAK-mediated diseases eg inflammatory diseases, autoimmune diseases, proliferative diseases, allergy and transplant rejection. Galapagos and licensee AbbVie are developing filgotinib, a selective JAK-1 inhibitor, for treating rheumatoid arthritis (RA) and Crohn’s disease (CD). In August 2015, the drug was reported to be in phase 2 clinical development for treating RA and CD. The drug is also being investigated for the treatment of colitis and was discovered as part of the company’s arthritis alliance with GSK; however in August 2010 Galapagos reacquired the full rights. See WO2013189771, claiming use of filgotinib analog for treating inflammatory diseases. Also see WO2010010190 (co-assigned with GSK and Abbott) and WO2010149769 (assigned to Galapagos) claiming filgotinib, generically and specifically, respectively.
Clinical trials and approval
The efficacy of filgotinib is currently studied in a phase2b program (DARWIN trial 1, 2) with involvement of 886 rheumatoid arthritis patients and 180 Crohn’s disease patients.
Phase 1 study
It was shown in phase 1 studies that the pharmacokinetics of filgotinib metabolism is independent of hepatic CYP450 enzymatic degradation. The drug metabolism is however mediated by carboxylesterases. There is no interference reported with the metabolism of methotrexate nor with any of the investigated transport proteins.[3]
Phase 2 study: Proof of concept (2011)
In november 2011 Galápagos released the results of their phase 2 study (identification: NCT01384422, Eudract: 2010-022953-40) in which 36 patients were treated who showed a suboptimal clinical response to methotrexate treatment. Three groups of twelve patients were treated either with 200 mg filgotinib in a single dose, 200 mg divided in two doses or placebo. The primary end-point was the ACR20 score, which monitors improvements in the symptomatology of the patient. After the scheduled 4 weeks of treatment, 83% of the respondents showed an improved ACR20-score. Half of the treated patients showed a complete (or near complete) remission of the disease. There were no reports ofanemia nor changes in lipidemia. The company stated in their press release that filgotinib is the first selective JAK1 inhibitor that shows clinical efficacy. As a result of this study, the company stated that “GLPG0634 shows one of the highest initial response rates ever reported for rheumatoid arthritis treatments”.[4]
DARWIN 1 trial
The DARWIN 1 trial is a 24 week double blind placebo-controlled trial with 599 rheumatoid arthritis patients enrolled. All participants have moderate to severe RA and showed an insufficient response to standard methotrexate treatment. The trial compares three dosages of filgotinib as a once or twice per day regimen. During the trial all participants remain on their methotrexate treatment. According to the company, the results of this trial are expected in July 2015.[5]
DARWIN 2 trial
The DARWIN 2 trial is a double blind placebo-controlled trial with 280 rheumatoid arthritis patients enrolled who show an insufficient response to standard methotrexate treatment. This trial, in contrast to the previous DARWIN 1 trial, methotrexate is discontinued. Therefore, this trial investigates filgotinib as a monotherapy.[6] The recruitment of DARWIN trial 2b ended in november 2014.[7] Preliminary results are expected in the second quarter of 2015 and a full completion of the study is expected in the third quarter of 2015.
DARWIN 3 trial
Patients who complete DARWIN 1 and 2 will be eligible for DARWIN 3.
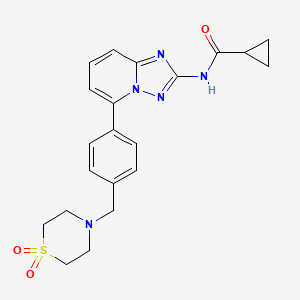
COSY PREDICT
Time line
- june 2011: results of first phase 2 trial
- november 2014: initiation of DARWIN 1 and 2 trials
- april 2015: expected date of DARWIN 1 trial results
- june 2015: expected date of DARWIN 2 trial results
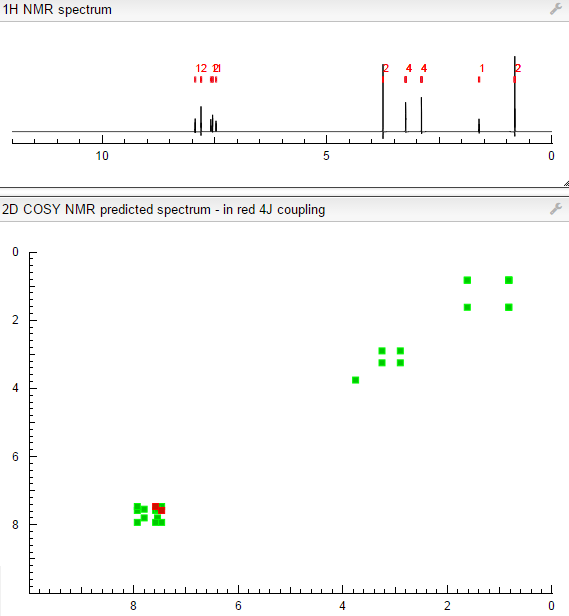
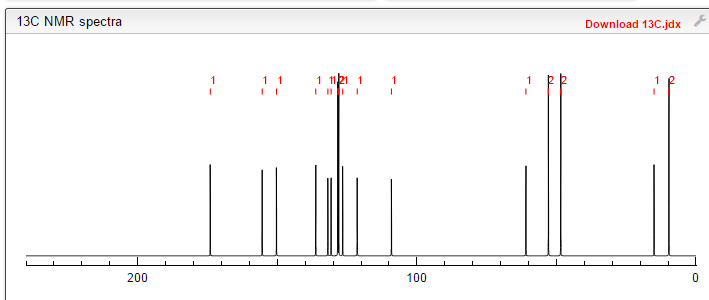
NMR FROM NET….ABMOLE, DMSOD6
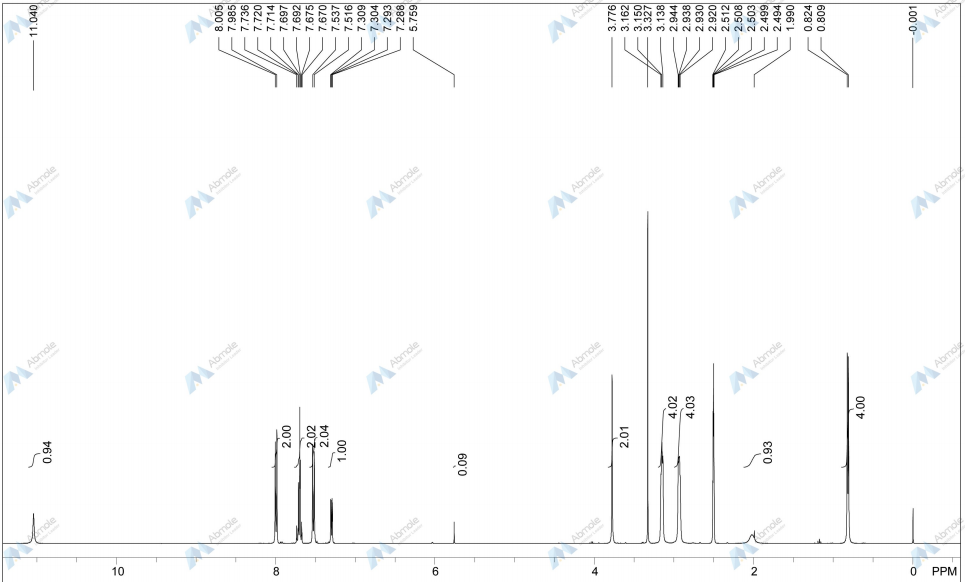
NMR MEDKOO DMSOD6

CHEMIETEK
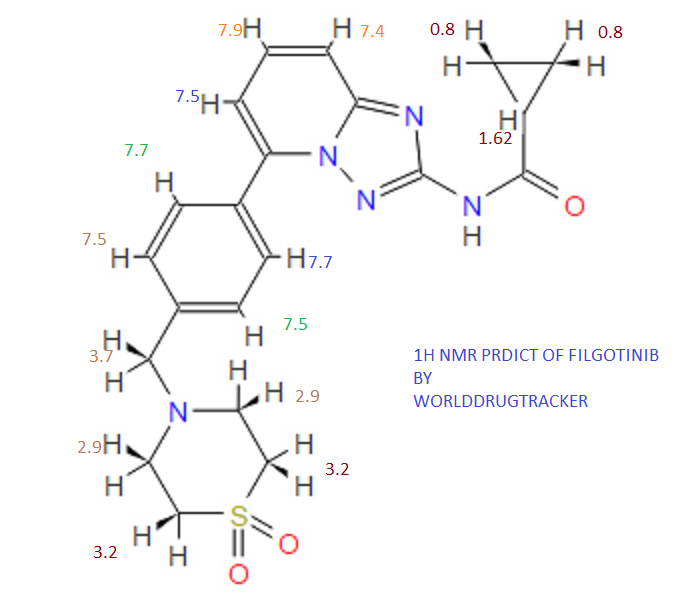
1H NMR PREDICT


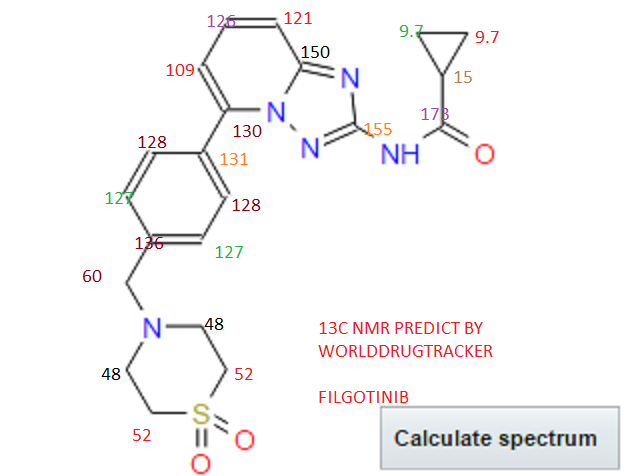
13C NMR PREDICT
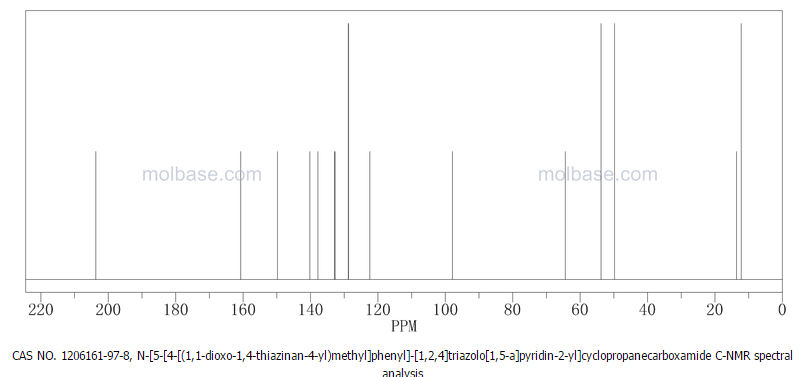

……………………
MORE PREDICTS
1H NMR PREDICT
13C NMR PREDICT
PRODUCT PATENT
http://www.google.com/patents/WO2010149769A1?cl=en
| Applicants: | GALAPAGOS NV [BE/BE]; Generaal De Wittelaan L11/A3 B-2800 Mechelen (BE) (For All Designated States Except US). MENET, Christel Jeanne Marie [FR/BE]; (BE) (For US Only). SMITS, Koen Kurt [BE/BE]; (BE) (For US Only) |
| Inventors: | MENET, Christel Jeanne Marie; (BE). SMITS, Koen Kurt; (BE) |
| International Filing Date: | 25.06.2010 |
ESTIMATED EXP 2030
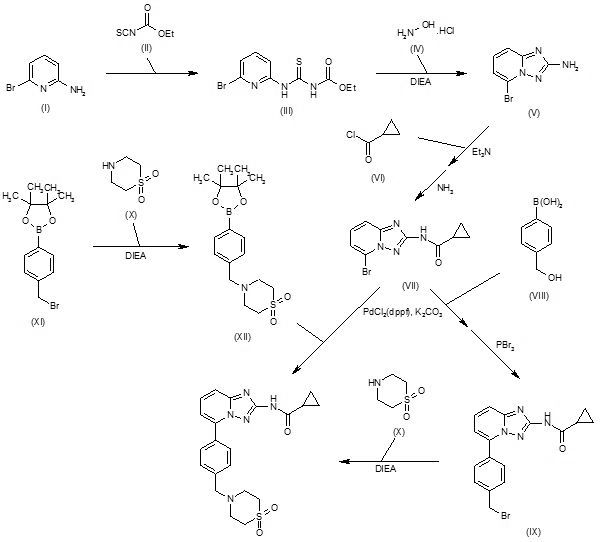
Condensation of 2-amino-6-bromopyridine (I) with ethoxycarbonyl isothiocyanate (II) in CH2Cl2 gives 1-(6-bromopyridin-2-yl)-3-carboethoxythiourea (III), which upon cyclization with hydroxylamine hydrochloride (IV) in the presence of DIEA in EtOH/MeOH yields 2-amino-5-bromo[1,2,4]triazolo[1,5-a]pyridine (V). N-Acylation of amine (V) with cyclopropanecarbonyl chloride (VI) using Et3N in acetonitrile, and subsequent treatment with methanolic ammonia furnishes the carboxamide (VII) (1-3), which upon Suzuki coupling with 4-(hydroxymethyl)phenylboronic acid (VIII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C, followed by bromination with PBr3 in CHCl3 affords intermediate (IX). Condensation of benzyl bromide derivative (IX) with thiomorpholine-1,1-dioxide (X) using DIEA in CH2Cl2/MeOH yields filgotinib (1,2). Alternatively, condensation of (4-bromomethylphenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (XI) with thiomorpholine 1,1-dioxide (X) in the presence of DIEA in CH2Cl2/MeOH gives intermediate (XII), which undergoes Suzuki coupling with aryl bromide (VII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C to afford the target filgotinib
The present invention is based on the discovery that the compound of the invention is able to act as an inhibitor of JAK and that it is useful for the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6. In a specific aspect the compound is an inhibitor of JAKl and JAK2. The present invention also provides methods for the production of this compound, a pharmaceutical composition comprising this compound and methods for treating inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 by administering the compound of the invention.
Accordingly, in a first aspect of the invention, a compound of the invention is provided having a formula (I):

[0017] The compound of the invention is a novel inhibitor of JAK that appears to exhibit a dramatically improved in vivo potency as compared to structurally similar compounds. In a particular embodiment the compound of the invention is an inhibitor of JAKl and JAK2. In particular it appears to exhibit this increase in potency at lower in vivo exposure levels compared to structurally similar compounds. The use of a compound with these improvements is expected to result in a lower dosage requirement (and therefore an improved dosing schedule).
General Synthetic Method Scheme 1

1. RCOCI, Et3N 2. NH3 / MeOH CH3CN, 20 0C 2O 0C

wherein Ar represents phenyl-Ll-heterocycloalkyl, where Ll is a bond, -CH2– or -CO- and the heterocycloalkyl group is optionally substituted.
General
1.1.1 l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

(2)
[00117] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5 0C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20 0C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
7.7.2 5-Bromo-[l, 2, 4]triazolo[l, 5-a]pyridin-2-ylamine (3)

[00118] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH
(1 :1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 0C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition Of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-t/β) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (IH, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 :1, M+H+, 100%).
7.7.3 General procedure for mono-acylation to afford intermediate (4):

[00119] To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN
(150 mL) at 5 0C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2x50mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A
Preparation of compounds of the invention via Suzuki coupling (5):
[00120] An appropriate boronic acid (2eq.) is added to a solution of bromo intermediate (4) in
1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters
XBridge Prep Cl 8 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using
MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

Bl. 4 4-[2-(Cyclopropanecarbonyl-amino)-[ 1 , 2, 4]triazolo[l, 5-a] pyridin-5-yl] -benzoyl chloride

[00121] 2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)- [l,2,4]triazolo[l,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide formation (General Method)

[00122] An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 00C. The acid chloride (Bl, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method C

Wherein R3a or R3b together with the nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reductive alkylation (general method)
[00123] An appropriate amine (2 eq.), cyclopropanecarboxylic acid (for example cyclopropanecarboxylic acid [5-(4-formyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridine-2-yl]-amide) prepared by method A (1 eq.) and Ti(OPr)4 are mixed and stirred at room temperature for 3 hrs. The mixture is diluted in ethanol and Na(CN)BH3 (leq.) is added. The resulting solution is stirred at room temperature for 16 hrs. The mixture is diluted in water and filtered. The filtrate is washed with ethanol. The combined solvent phases are evaporated under vacuum. The final compound is isolated by preparative HPLC.
Method D 
wherein R1 and R2 together with the Nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reaction ofalkylation

[00124] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and Et3N (2 eq) (or AgCO3) are dissolved in DCM/MeOH (4:1 v:v) under N2 and an amine (2 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSθ4. Organic layers are filtered and evaporated. The final compound is isolated by flash chromatography.
Suzuki coupling

[00125] The obtained boronic acid (2eq.) is added to a solution of cyclopropanecarboxylic acid
(5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide (4) in 1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (This reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Synthesis of the compound of the invention and comparative examples
Compound l(the compound of the invention)
Step 1:

[00126] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and DIPEA
(2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgS O4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
Step 2: Suzuki coupling

[00127] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide
(l.leq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00128] Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 500C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative route to Compound l(the compound of the invention):
Step 1:

[00129] 4-(Hydroxymethyl)phenylboronic acid (l.leq.) was added to a s o luti o n o f cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

[00130] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)- [l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSθ4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

[00131] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin- 2-yl]-amide (leq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO^ Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
PATENT
WO 2010010190
WO 2013173506
WO 2013189771
WO 2015117980
WO 2015117981
POLYMORPH
CN 105061420
https://encrypted.google.com/patents/CN105061420A?cl=en
JAK inhibitor N-(5-(4-(1,1-dioxothiomorpholinyl)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide, and methods for preparing the four crystal forms, wherein the four crystal forms respectively are a crystal form H1, a crystal form H2, a crystal form H3 and a crystal form H4,
POLYMORPH
E CRYSTAL
CN 105111206
D CRYSTAL
CN 105111207
H CRYSYAL
CN 105198876
G
CN 105198877
F CN 105198878
C CN 105198880
POLYMORPH
WO 2016105453
POLYMORPH
Preparation of solid forms of filgotinib free base
WO 2017012773
The present invention relates to cryst. filgotinib free base (I), a method of its prepn. and a pharmaceutical compn. comprising the same. Thus, reacting the compd. II with thiomorpholine dioxide afforded 92% I (purity of 98.6%) which was then converted into its hydrochloride salt. Pharmaceutical compn. comprising compd. I was disclosed.
POLYMORPH
CN 105669669
The present invention provides a crystal form A, B, D, G and M of N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide hydrochloride.
PAPER
Future Medicinal Chemistry (2015), 7(2), 203-235. | Language: English, Database: CAPLUSA review. The discovery of the JAK-STAT pathway was a landmark in cell biol. The identification of these pathways has changed the landscape of treatment of rheumatoid arthritis and other autoimmune diseases. The two first (unselective) JAK inhibitors have recently been approved by the US FDA for the treatment of myelofibrosis and rheumatoid arthritis and many other JAK inhibitors are currently in clin. development or at the discovery stage. Research groups have demonstrated the different roles of JAK member and the therapeutic potential of targeting them selectively. ………..
https://www.future-science.com/doi/10.4155/fmc.14.149
PAPER
Journal of Pharmaceutical Sciences (Philadelphia, PA, United States) (2018), 107(6), 1624-1632.
PATENT
US2010/331319 A1, ; Page/Page column 13-14
http://www.google.com/patents/US20100331319
Synthetic Preparation of the Compound of the Invention and Comparative Examples
The compound of the invention and the comparative examples can be produced according to the following scheme.

wherein Ar represents phenyl-L1-heterocycloalkyl, where L1 is a bond, —CH2— or —CO— and the heterocycloalkyl group is optionally substituted.
General 1.1.1 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5° C. is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20° C.) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (1H, br s, NH), 8.81 (1H, d, J=7.8 Hz, H-3), 8.15 (1H, br s, NH), 7.60 (1H, t, J=8.0 Hz, H-4), 7.32 (1H, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.2 5-Bromo-[1,2,4]triazolo[1,5-a]pyridin-2-ylamine (3)

To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1:1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20° C.) for 1 h. 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1:1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-d6) δ 7.43-7.34 (2H, m, 2×aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1:1, M+H+, 100%).
1.1.3 General Procedure for Mono-Acylation to Afford Intermediate (4)

To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN (150 mL) at 5° C. is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp. (for 1-16 h) to hydrolyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2×50 mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A Preparation of Compounds of the Invention Via Suzuki Coupling (5):
An appropriate boronic acid (2 eq.) is added to a solution of bromo intermediate (4) in 1,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140° C. for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 90° C. for 16 h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSO4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

B1. 4 4-[2-(Cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoyl chloride

2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide Formation (General Method)

An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 0° C. The acid chloride (B1, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
…
Synthesis of the Compound of the Invention and Comparative Examples Compound 1 (the Compound of the Invention) Step 1:

2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
STEP 2: Suzuki coupling

4-[4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-1,1-dioxide (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 50° C., the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative Route to Compound 1 (the Compound of the Invention): Step 1:

4-(Hydroxymethyl)phenylboronic acid (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSO4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
…………………….
PATENT
Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders
Also claims a method for preparing filgotinib hydrochloride trihydrate. The present filing forms a pair with this week’s filing, WO2015117980, claiming a tablet composition comprising filgotinib hydrochloride.
The compound cyclopropanecarboxylic acid {5-[4-(l,l-dioxo-thiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5-a]pyridin-2-yl -amide (Compound 1), which has the chemical structure:

is disclosed in our earlier application WO 2010/149769 (Menet C. J., 2010) as being an inhibitor of JAK and as being useful in the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, allergy, transplant rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 or interferons. Hereafter this compound is named Compound 1. The data presented in WO 2010/149769 demonstrate that despite similar in vitro activities, Compound 1 has unexpectedly high in vivo potency compared with structurally similar compounds.
Example 1. Preparation of Compound 1
1.1. Route 1
1.1.1. 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide

[00205] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) is added portionwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated without further purification.
1.1.2. Cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide

1.1.2.1. Step i): l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea
[00206] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5°C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction
mixture is then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3 x 600 niL) and air-dried to afford the desired product. The thiourea may be used as such for the next step without any purification. lH (400 MHz, CDC13) δ 12.03 (1H, br s), 8.81 (1H, d), 8.15 (1H, br s), 7.60 (1H, t), 7.32 (1H, dd), 4.31 (2H, q), 1.35 (3H, t).
1.1.2.2. Step ii): 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine
[00207] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) is added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids are washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2.3. Step Hi): Cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide
[00208] To a solution of the 2-amino-triazolopyridine obtained in the previous step (7.10 g, 33.3 mmol) in dry MeCN (150 mL) at 5°C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et20 (50 mL). The solids are collected by filtration, washed with H20 (2x50mL), acetone (50 mL) and Et20 (50 mL), then dried in vacuo to give the desired compound.
1.1.3. Compound 1

[00209] 4-[4-(4,4,5,5-Tetramethyl-[l ,3,2]dioxaborolan-2-yl)-benzyl] hiomoφholine , l -dioxide (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo.
[00210] The final compound is obtained after purification by flash chromatography.
[00211] Alternatively, after completion of the reaction, a palladium scavenger such as 1 ,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cool down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HC1 is added, and after stirring at room temperature, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at room temperature, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H20, treated with a palladium scavenger (e.g. SMOPEX 234) at 50°C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the desired compound as a free base.
1.2. Route 2
1.2.1. Step 1: cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2, 4]triazolo[l, 5- a] pyridin-2-yl] -amide

[00212] 4-(Hydroxymethyl)phenylboronic acid (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water
(4:1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo. The resulting mixture is used without further purification.
1.2.2. Step 2: Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5- a Jpyridin-2-ylJ -amide

[00213] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl] -amide (1.0 eq) in chloroform is slowly added phosphorus tribromide (1.0 eq.). The reaction mixture is stirred at room temperature for 20 h, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer is dried over anhydrous MgSO i, filtered and concentrated to dryness. The resulting white residue is triturated in dichloromethane/diethyl ether 2:1 to afford the desired product.
1.2.3. Step 3:

[00214] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (l eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5: 1 v:v) under N2 and thiomorpho line 1,1-dioxide (1.1 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is dissolved in DCM, washed with water and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated by column chromatography using EtOAc to afford the desired product.
…………………
PATENT
http://www.google.co.in/patents/WO2013189771A1?cl=en
Example 1. Synthesis of the compounds
1.1. Route 1
1.1.1. Synthesis of 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine (Intermediate 3)

led to 5 °C was added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture was then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gave a solid which was collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea was used as such in the next step without any purification.
[00157] lH (400 MHz, CDC13) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.1.2. 5-Bromo-f 1,2, 4]triazolo[ 1 ,5-a] pyridin-2-ylamine (3)
[00158] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) was added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture was stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) was then added and the mixture slowly heated to reflux (Note: bleach scrubber was required to quench H2S evolved). After 3 h at reflux, the mixture was allowed to cool and filtered to collect the precipitated solid. Further product was collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids were washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound was used as such in the next step without any purification.
[00159] lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2. Synthesis of 4-[ 4-(4, 4, 5, 5-Tetramethyl-f 1, 3,2] ‘ dioxaborolan-2-yl) -benzyl] ‘- thiomor holine- 1, 1 -dioxide (Intermediate 4)

[00160] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portion wise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
1.1.3. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- a ridin-2-ylamine (Formula I)

[00161] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide (l .leq.) was added to a solution of 5-bromo-[l,2,4]triazolo[l,5-a]pyrid in-2-ylamine (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90°C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhydrous MgSC>4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00162] lH (400 MHz, CDC13) δ 7.94-7.92 (d, 2H), 7.52-7.48 (m, 3H), 7.37-7.34 (m, 1H), 7.02-7.00 (m, 1H), 6.00 (d, 2H), 3.76 (d, 2H), 3.15-3.13 (m, 4H), 2.93-2.91 (m, 4H).
[00163] m/z 358.2 (M+H+, 100%). 1.2. Route 2
1.2.1. Cyclopropanecarboxylic acid {5-[4-(l, l-dioxo-thiomorpholin-4-ylmethyl)-phenylJ- [l,2,4]triazolo[l,5-a]pyridin-2-yl}-amide (Formula II)
[00164] The compound according to Formula II may be synthesized according to the procedure described in WO 2010/149769.
1.2.2. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- aJpyridin-2-ylamine (Formula I)
[00165] The compound according to Formula I can also be produced by hydrolysis of the compound accor ing to Formula II:

[00166] Hydrochloric acid 30% aq (12.06 kg; 3.9 rel. volumes) was added to a slurry of the compound according to Formula II (3.45 kg; 1.0 equiv.) in demineralized water (10.0 kg; 3.0 rel. volumes). Subsequently, a line rinse was performed with demineralized water (3.4 kg; 1.0 rel. volumes). The reaction mixture was heated to 80±5°C for 14.5 h. After completion of the reaction (conversion > 99%>), the reaction mixture was cooled to 20±5°C. The reaction mixture was diluted with demineralized water (6.8 kg; 2.0 rel. volumes) and sodium hydroxide 33%> aq (9.52 kg; 3.7 rel volumes) was dosed at such a rate that the temperature of the reactor contents remained below 35°C. An additional amount of sodium hydroxide 33%> aq (2.55 kg; 1.0 rel. volumes) was needed to get the pH > 10. The product was filtered off, washed twice with demineralized water (1.5 rel. volumes) and dried under vacuum for 1 h, thus yielding the crude compound according to Formula I.
[00167] The crude compound according to Formula I (5.70 kg) was re-slurried in demineralized water (23.0 kg; 8.5 rel. volumes). Hydrochloric acid 30%> aq (1.65 kg; 0.7 rel. volumes) and demineralized water (4.3 kg; 1.6 rel. volumes) were added and the reaction mixture was stirred at 20±5°C for 45 min. As the compound according to Formula I was not dissolved completely, the reaction mixture was stirred at 45±5°C for 1 h. The reaction mixture was filtered and the residue was washed with demineralized water (2.0 kg 0.75 rel. volumes). Sodium hydroxide 33%> aq (1.12 kg; 0.6 rel volumes) was added to the filtrate. An additional amount of sodium hydroxide 33%> aq (1.01 kg) was needed to get the pH > 10. The resulting reaction mixture was stirred at 20±5°C for about 3 h. The product was filtered off, washed twice with demineralized water (4.1 kg; 1.5 rel. volumes), and twice with methyl tert-butyl ether (MTBE; 3.0 kg; 1.5 rel. volumes) and dried under vacuum for 15.5 h on the filter. The product was further dried in a vacuum oven at 40±5°C for 202 h, thus affording the desired compound according to Formula I.
Update
Scheme 1: General S nthesis of Compounds of Formula I or A

Formula A
Scheme 7.

(16) (17) (18)
(18a): R3a=R3b=R2a=R (18b): R3a=R3b=D; R2a 18c): R3a=R3b=H; R2a

References
- Namour, Florence; Diderichsen, Paul Matthias; Cox, Eugène; Vayssière, Béatrice; Van der Aa, Annegret; Tasset, Chantal; Van’t Klooster, Gerben (2015-02-14). “Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection”. Clin Pharmacokinet. Epub ahead of print.doi:10.1007/s40262-015-0240-z.
- Van Rompaey, L; Galien, R; Van der Aar, E; Clement-Lacroix, P; Van der Aar, E; Nelles, L; Smets, B; Lepescheux, L; Cristophe, T; Conrath, K; Vandeghinste, N; Vayssiere, B; De Vos, S; Fletcher, S; Brys, R; Van’t Klooster, G; Feyen, J; Menet, C (2013-10-01). “Preclinical characterization of GLPG0634, a selective inhibitor of JAK1 for the treatment of inflammatory diseases”. J Immunol. 191(7). doi:10.4049/jimmunol.1201348.
- http://acrabstracts.org/abstracts/phase-1-and-phase-2-data-confirm-that-glpg0634-a-selective-jak1-inhibitor-has-a-low-potential-for-drug-drug-interactions/
- “Galapagos’ GLPG0634 shows excellent efficacy and safety in rheumatoid arthritis Phase II study” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos reports that the last patient in DARWIN 1 has completed 12 weeks of treatment” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos completes recruitment for Darwin 1 study with GLPG0634 (filgotinib) in RA”. EuroInvestor. Retrieved 2015-02-26.
- NASDAQ OMX Corporate Solutions. “Galapagos completes recruitment for Darwin 2 monotherapy study with GLPG0634 (filgotinib) in RA”. Yahoo Finance. Retrieved 2015-02-26.
| US8551980 | Nov 17, 2010 | Oct 8, 2013 | Bayer Intellectual Property Gmbh | Substituted triazolopyridines |
| US8796457 | Jun 25, 2010 | Aug 5, 2014 | Galapagos Nv | Compound useful for the treatment of degenerative and inflammatory diseases |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-[4-[(1,1-dioxo-1,4-thiazinan-4-yl)methyl]phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Biological half-life | 6 hours[1] |
| Identifiers | |
| CAS Registry Number | 1206161-97-8 |
| ATC code | L01XE18 |
| IUPHAR/BPS | 7913 |
| ChemSpider | 28189566 |
| UNII | 3XVL385Q0M |
| ChEMBL | CHEMBL3301607 |
| Chemical data | |
| Formula | C21H23N5O3S |
| Molecular mass | 425.50402 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
/////////Galapagos, GLPG0634, Filgotinib, PHASE 2, orphan drug designation, PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulceraticolitis
ve SMILES code: O=C(C1CC1)NC2=NN3C(C4=CC=C(CN5CCS(CC5)(=O)=O)C=C4)=CC=CC3=N2
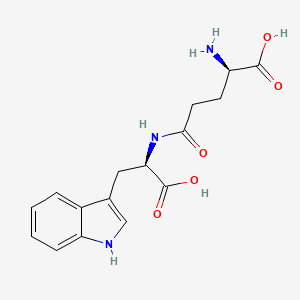
Orilotimod
Orilotimod
(2R)-2-amino-5-{[(1R)-1-carboxy-2-(1H-indol-3-yl)ethyl]amino}-5-oxopentanoic acid
Apo805,UNII-Q66Z43C5XM; Thymodepressin; Orilotimod [USAN]; AC1OIBUF;
- C16H19N3O5
- MW 333.339
Apotex Technologies Inc. INNOVATOR
Orilotimod potassium,
-
APO805K1
D-Tryptophan, D-gamma-glutamyl-, potassium salt (1:1), CAS 960155-19-5
The drug, orilotimod, was originally developed and launched by Immunotech Developments; however, ApoPharma (a subsidiary of Apotex) is developing orilotimod, presumably a topical formulation, for the treatment of psoriasis. In August 2015, the ApoPharma’s drug was reported to be in phase 2 clinical development.
Thymodepressin is the free diacid having Chemical Abstracts Service (CAS) Registry Number@ of 186087-26-3. U.S. Pat. No. 5,736,519 discloses H-D-iGlu-D-Trp-OH and a process for its preparation wherein it is purified by ion exchange chromatography. It is an immunosuppressant and selectively inhibits proliferation of hemopoietic precursor cells and stimulates granulocyte and lymphocyte apoptosis (Sapuntsova, S. G., et al. (May 2002), Bulletin of Experimental Biology and Medicine, 133(5), 488-490).
Thymodepressin is currently being sold in Russia as the disodium salt of D-isoglutamyl-D-tryptophan in liquid formulation for injection and intranasal administration for the treatment of psoriasis and atopic dermatitis. The solid form of the disodium salt of D-isoglutamyl-D-tryptophan is an amorphous powder which is hygroscopic and very difficult to handle. The disodium salt of D-isoglutamyl-D-tryptophan has the molecular formula C16H17N3Na2O5 and is reported in Kashirin, D. M., et al. (2000), Pharmaceutical Chemistry Journal, 34(11), 619-622.
PAPENT
BEAWARE EXAMPLE WITH AN ESTER GP
http://www.google.im/patents/WO2012129671A1?cl=en
Preparation of H-D-Glu( -Trp-OH)-0-Et hydrochloride salt (Apo836.HCI)

A. Preparation of Boc-D-Glu(D-Trp-0-Bzl)-0-Et
Proceeding in a similar manner as described under Example 3A, Boc-D- Glu(D-Trp-0-Bzl)-0-Et was prepared in 87% yield.1H NMR ( DMSO-D6l 400 MHz) δ ppm: 10.87, (s, 1 H), 8.35 (d, J = 7.2 Hz, 1 H), 7.48 (d, J = 7.8 Hz, 1 H), 7.35 (d, J = 7.9 Hz, 1 H), 7.29-7.33 (m, 3H), 7.23 (d, J = 7.7 Hz, 1H), 7.09-7.22 (m, 3H), 7.08 (t, J = 7.6 Hz, 1H), 6.98 (t, J = 7,7 Hz, 1 H), 4.98 – 5.06 (m, 2H), 4.55 (apparent q, J = 7.3 Hz, 1 H), 4.04 – 4.11 (m, 2H), 3.90 – 3.95 (m, 1 H), 3.04 – 3.19 (m, 2H), 2.18 – 2.23 (m, 2H), 1.84 – 1.89 (m, 1 H), 1.70 – 1.77 (m, 1 H), 1.38 (s, 9H), 1.16 (t, J = 7.1 Hz, 3H); MS-ESI (m/z): 552 [ +1]+.
B. Preparation of Boc-D-Glu(D-Trp-OH)-0-Et
Proceeding in a similar manner as described under Example 3B, Boc-D-
Glu(D-Trp-OH)-0-Et was prepared in quantitative yield. 1H NMR ( DMSO-D6, 400 MHz) δ ppm: 12.62 (br. 1H), 10.82, (s, 1 H), 8.10 (d, J = 7.7 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1 H), 7.33 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 7.5 Hz, 1 H), 7.12 (s, 1 H), 7.06 (t, J = 7.3 Hz, 1 H), 6.98 (t, J = 7.5 Hz, 1 H)„ 4.45 (apparent q, J = 7.7 Hz, 1 H), 4.03 – 4.11 (m, 2H), 3.87 – 3.92 (m, 1 H), 3.13 – 3.18 (m, 1H), 2.96 – 3.03 (m,
1 H), 2.13 – 2.20 (m, 2H), 1.82 – 1.88 (m, 1H), 1.69-1.75 (m, 1 H), 1.38 (s, 9H>, 1.17 (t, J = 7.1 Hz, 3H); MS-ESI (m/z): 462 [M+1]+.
C. Preparation of H-D-Glu(D-Trp-OH)-0-Et.HCI (Apo836 HCI)
To an ice-cooled solution of Boc-D-Glu(D-Trp-OH)-0-Et (4.55 g, 9.8 mmol) obtained in Section B above in dichloromethane (100 mL) was bubbled HCI gas for 15 min. The reaction mixture was concentrated under vacuum by rotary evaporation to give H-D-Glu(D-Trp-OH)-0-Et hydrochloride (Apo836.HCI, 4.0 g) as a foamy solid. 1 H NMR ( DMSO-D6, 400 MHz) δ ppm: 12.68 (br. s, 1 H), 10.90, (s, 1H), 8.66 (br, s, 3H), 8.33 (d, J = 7.8 Hz, 1 H), 7.52 (d, J = 7.8 Hz, 1 H), 7.33 (d, J = 8.0 Hz, 1 H), 7.12 (d, J = 1.5 Hz, 1H), 7.06 (t, J = 7.2 Hz, 1 H), 6.98 (t, J = 7.2 Hz, 1 H), 4.47 (apparent q, J = 4.8 Hz, 1 H), 4.13 – 4.19 (m, 2H), 3.90 (br, 1 H), 3.16 – 3.20 (m, 1H), 2.98 – 3.04 (m, 1 H), 2.29 – 2.33 (m, 2H), 1.94 – 1.98
(m, 2H), 1.20 (t, J = 7.1 Hz, 3H); MS-ESI (m/z): 362 [M+1]+ (free base).
……………………..
file:///H:/ORILOTIMODUS20150225341A1.pdf
Novel crystalline and amorphous salts of thymodepressin (orilotimod), particularly potassium salt, useful for treating psoriasis and atopic dermatitis. Also claims salt exchange method for preparing thymodepressin salts.
hymodepressin is the free diacid having Chemical Abstracts Service (CAS) Registry Number@ of 186087-26-3. U.S. Pat. No. 5,736,519 discloses H-D-iGlu-D-Trp-OH and a process for its preparation wherein it is purified by ion exchange chromatography. It is an immunosuppressant and selectively inhibits proliferation of hemopoietic precursor cells and stimulates granulocyte and lymphocyte apoptosis (Sapuntsova, S. G., et al. (May 2002), Bulletin of Experimental Biology and Medicine, 133(5), 488-490).
Thymodepressin is currently being sold in Russia as the disodium salt of D-isoglutamyl-D-tryptophan in liquid formulation for injection and intranasal administration for the treatment of psoriasis and atopic dermatitis. The solid form of the disodium salt of D-isoglutamyl-D-tryptophan is an amorphous powder which is hygroscopic and very difficult to handle. The disodium salt of D-isoglutamyl-D-tryptophan has the molecular formula C16H17N3Na2O5 and which is reported in Kashirin, D. M., et al. (2000), Pharmaceutical Chemistry Journal, 34(11), 619-622.
Through investigations in our laboratory, we have determined that the freeze-dried disodium salt of D-isoglutamyl-D-tryptophan is extremely hygroscopic turning into a gel in a matter of minutes in air and cannot easily be handled.
A powdery or amorphous form of a compound, intended for pharmaceutical use may give rise to manufacturing problems due to bulk density issues, hygroscopicity and variable water content that cannot be corrected by vacuum drying. D-isoglutamyl-D-tryptophan is a dipeptide and the drying of an amorphous form at elevated temperature, for example, 80-100° C. under vacuum is not recommended. Thus, there are serious difficulties experienced during the purification of the disodium salt of D-isoglutamyl-D-tryptophan and obtaining the pure disodium salt on a manufacturing scale. Further, there is no published procedure for its preparation.
The monosodium salt of D-isoglutamyl-D-tryptophan is identified by the CAS Registry System and is listed in the CAS REGISTRYSM File with a CAS Registry Number@ of 863988-88-9. However, there are no references citing the substance and thus no publication of its identity, its physical and/or chemical properties, its characterization or a procedure for its preparation. Freeze-dried powders of mono sodium and disodium salts of peptide drugs may not have controllable powder bulk density ranges for formulation. They may require significant investment in freeze-dried dispersion technology.
EXAMPLES
Example 1
Preparation of potassium salt of D-isoglutamyl-D-tryptophan (1:1) from D-isoglutamyl-D-tryptophan and potassium hydroxide
In a 100-mL round bottom flask equipped with a magnetic stir bar was placed 5 mL of potassium hydroxide solution (0.5 N). The solution was cooled to 0° C. in an ice-water bath, and solid H-D-iGlu-D-Trp-OH (1.00 g, 3 mmol) was added. The mixture was stirred while the pH of the solution was adjusted to ca. 6.0 by adding a few drops of potassium hydroxide solution (0.5 N). The solution was filtered to remove any solid particulates. The filtrate was evaporated to dryness at a bath temperature of about 30° C. to afford a solid. After drying under vacuum at room temperature for overnight, the salt was obtained in quantitative yield, with a HPLC purity (peak area percent) of 98.3%. HPLC method; Column: XTerra MS C18; 5 μm, 4.6×250 mm; Mobile phase: A=the aqueous phase: 4 mM Tris, 2 mM EDTA, pH 7.4; B=the organic phase: CH3CN; gradient: B %: 0 min. 5%, 15 min. 55%, 30 min. 55%, 32 min. 5%, 35 min. 5%; Flow rate: 1 mL/min; injection volume: 5 μL; λ: 222, 254, 282, 450 nm; retention time of the product: 6.41 min. The XRPD pattern of this crystalline material is shown in FIG. 1A; the water content by Karl-Fischer test is 0.7%; UV (water, c=23.8 ρM, λmax nm): 221 (ε 33270), 280 (ε 5417); MS (m/z): 372.0 [M]+, 334.2 [C16H20N3O5]+, 187.9 (100%). The FT-IR (KBr) spectrum is shown in FIG. 1B.
Example 2
A. Preparation of mono potassium salt of D-isoglutamyl-D-tryptophan (1:1) from the mono ammonium salt of D-isoglutamyl-D-tryptophan (1:1)
A solution of H-D-iGlu-D-Trp-OH, mono ammonium salt (1:1), (1.66 g, 4.05 mmol) and potassium hydroxide (253 mg, 4.50 mmol) in water (20 mL) was stirred at room temperature for 15 min. The pH of the solution was about 9. The reaction mixture was evaporated under reduced pressure to a volume of about 1 mL. After cooling to room temperature, isopropanol was added until a solid precipitated out. The resulting suspension was stirred at room temperature for 15 min, then filtered. The solid was washed with isopropanol (2×20 mL) and ethyl acetate (20 mL), then dried under vacuum in an oven at 42° C. overnight. An off white solid was obtained (1.49 g, 99% yield). The water content by Karl-Fischer test is 2.5%. Analytical data (XRPD pattern, FT-IR and MS spectra) are similar to those described in Example 1.
B. Preparation of amorphous form of potassium salt of D-isoglutamyl-D-tryptophan (1:1) from the mono ammonium salt of D-isoglutamyl-D-tryptophan (1:1)
A solution of H-D-iGlu-D-Trp-OH, mono ammonium salt (1:1), (517 mg, 1.40 mmol) and potassium hydroxide (82 mg, 1.46 mmol) in water (10 mL) was stirred at room temperature for 30 minutes. The resulting mixture was freeze-dried overnight. An off white solid was obtained in quantitative yield. The XRPD pattern spectrum confirmed that this material is amorphous.
1H NMR (D2O) δ: 7.69 (d, J=7.9 Hz, 1H), 7.48 (d, J=8.2 Hz, 1H), 7.23 (t, J=7.6 Hz, 1H), 7.22 (s, 1H), 7.16 (t, J=7.4 Hz, 1H), 4.59 (dd, J=8.7, 4.8 Hz, 1H), 3.51 (dd, J=6.8, 5.8 Hz, 1H), 3.38 (dd, J=14.8, 4.8 Hz, 1H), 3.11 (dd, J=14.8, 8.8 Hz, 1H), 2.20-2.49 (m, 2H) and 1.85-1.94 (m, 2H);
13C NMR (D2O) δ: 181.4, 177.0, 176.6, 138.8, 129.9, 126.9, 124.5, 121.9, 121.4, 114.5, 113.2, 58.6, 57.0, 34.6 (CH2), 30.2 (CH2) and 29.3 (CH2);
the water content by Karl-Fischer test is 5.4%;
the FT-IR (KBr) spectrum is shown in FIG. 1C;
MS (m/z): 371.7 [M]+, 334.2 [C16H20N3O5]+, 187.9 (100%);
HPLC purity (peak area percent): 99.8%, Retention time: 5.04 min; HPLC conditions: Column Waters Symmetry C18, 3.9×150 mm, 5 μm; Mobile phase: 0.035% HClO4, pH 2/CH3CN, 85/15, isocratic, Flow rate: 1 mL/min; λ: 220, 254, 280 nm.
| Patent | Submitted | Granted |
|---|---|---|
| GAMMA-GLUTAMYL AND BETA-ASPARTYL CONTAINING IMMUNOMODULATOR COMPOUNDS AND METHODS THEREWITH [EP1042286] | 2000-10-11 | 2010-08-25 |
| CRYSTALLINE D-ISOGLUTAMYL-D-TRYPTOPHAN AND THE MONO AMMONIUM SALT OF D-ISOGLUTAMYL-D-TRYPTOPHAN [US8119606] | 2010-01-21 | 2012-02-21 |
| Pharmaceutically Acceptable Salts of Thymodepressin and Processes for their Manufacture [US8138221] | 2010-03-04 | 2012-03-20 |
| CRYSTALLINE FORMS OF THE MONO-SODIUM SALT OF D-ISOGLUTAMYL-D-TRYPTOPHAN [US8207217] | 2010-02-04 | 2012-06-26 |
////////Orilotimod, PHASE 2, thymodepressin, APO 805K1
C1=CC=C2C(=C1)C(=CN2)CC(C(=O)O)NC(=O)CCC(C(=O)O)N
Odalasvir
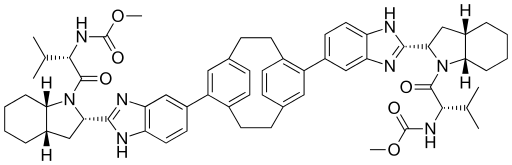
ACH-3102 , Odalasvir
Odalasvir
ACH-0143102; ACH-3102
CAS : 1415119-52-6
Dimethyl N, N ‘- (tricyclo [8.2.2.24,7] hexadeca-1 (12), 4,6, 10,13,15-hexaene-5,11-diylbis {1H-benzimidazole-5,2-diyl [(2S, 3aS, 7aS) -octahydro-1H-indole-2,1-diyl] [(1S) -1 – (1-methylethyl) -2-oxoethylene]}) biscarbamate
Carbamic acid, N,N’-(tricyclo(8.2.2.24,7)hexadeca-4,6,10,12,13,15-hexaene-5,11-diylbis(1H-benzimidazole-6,2-diyl((2S,3aS,7aS)-octahydro-1H-indole-2,1-diyl)((1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl)))bis-, C,C’-dimethyl ester
Dimethyl N,N’-(1,4(1,4)-dibenzenacyclohexaphane-12,42-diylbis(1hbenzimidazole-5,2-diyl((2S,3aS,7aS)-octahydro-1H-indole-2,1-diyl)((2S)-3-methyl-1-oxobutan-1,2-diyl)))biscarbamate
Mechanism of Action: HCV NS5A Protein inhibitor
Indication: Hepatitis C
Developer: Achillion Pharmaceuticals, Inc.

-
C60-H72-N8-O6
- 1001.2788
Odalasvir[1] is an investigational new drug in development for the treatment hepatitis C.
Achillion Pharmaceuticals Inc’s Odalasvir (ACH-3102) is an investigational new drug in development for the treatment hepatitis C. Achillion’s ongoing study tests its NS5A inhibitor, ACH-3102, with Sovaldi in previously untreated genotype 1 hepatitis C patients over six and eight weeks of therapy. The main goal is to achieve a cure, or sustained virological response, 12 weeks after the completion of therapy.
Odalasvir is a hepatitis C virus (HCV NS5A) inhibitor in phase II clinical studies at Achillion for the treatment of hepatitis C.
In 2012, fast track designation was assigned to the compound in the U.S. for the treatment of chronic hepatitis C.
WILL BE UPDATED………….
WO 2012166716
http://www.google.com/patents/US20120302538

General Considerations
All nonaqueous reactions were performed under an atmosphere of dry argon gas using oven-dried glassware and anhydrous solvents. The progress of reactions and the purity of target compounds were determined using one of the following two HPLC methods: (1) Waters AQUITY HPLC BEH C18 1.7 μm 2.1×50 mm column with an isocratic elution of 0.24 min at 90:10 water:acetonitrile containing 0.05% formic acid followed by a 4.26-min linear gradient elution from 90:10 to 10:90 at a flow rate of 1.0 mL/min with UV (PDA), ELS, and MS (SQ in APCI mode) detection (method 1); and (2) Waters AQUITY HPLC BEH C18 1.7 μm 2.1×50 mm column with an isocratic elution of 0.31 min at 95:5 water:acetonitrile containing 0.05% formic acid followed by a 17.47-min linear gradient elution from 95:5 to 5:95 at a flow rate of 0.4 mL/min with UV (PDA), ELS, and MS (SQ in APCI mode) detection (method 2).
Target compounds were purified via preparative reverse-phase HPLC using a YMC Pack Pro C18 5 μm 150×20 mm column with an isocratic elution of 0.35 min at 95:5 water:acetonitrile containing 0.1% trifluoroacetic acid followed by a 23.3-min linear gradient elution from 95:5 to 5:95 at a flow rate of 18.9 mL/min with UV and mass-based fraction collection.

Example 1
Synthesis of Compound 10
Compound 10 was prepared via bromination of [2.2]paracyclophane as outlined previously (Reich, H. J.; Cram, D. J. J. Am. Chem. Soc. 1969, 91, 3527-3533; Reich, H. J.; Cram, D. J. J. Am. Chem. Soc. 1969, 91, 3534-3543). Compounds 1, 2, 6, 8, and 10 can be obtained from commercial sources. Compounds 3-7 and 9 were prepared using general synthetic methods known in the art.
Example 2Synthesis of Compound 11
A deoxygenated (argon) mixture of 9 (284.2 mg), 10 (52.3 mg), K3PO4 (248.1 mg), and PdCl2dppf.CH2Cl2 (7.4 mg) in dioxane/water (5.5 mL/0.55 mL) was irradiated in a microwave for 2 h at 80° C. The resulting mixture was evaporated under reduced pressure and the remaining solid was extracted with DCM. This crude material was purified by PTLC (20 cm×20 cm×2000 μm glass plates; eluted with 45:50:5 v/v/v DCM:EtOAc:MeOH, Rf 0.28) to give 75.3 mg of 11. The purity of 11 was determined via analytical reverse-phase HPLC using a 3.5-min gradient elution of increasing concentrations of ACN in water (10-90%) containing 0.05% formic acid with a flow rate of 1.0 mL/min on a Waters AQUITY HPLC BEH C18 1.7 μm 2.1×50 mm column with UV (PDA), ELS, and MS (SQ in APCI mode) detection. HPLC: tR 1.57 min (98% purity). MS m/z calculated for C56H64N8O6 ([M]+), 945. found, 946 ([M+1]+).
SEE ALSO
US 2012302538
http://www.google.com/patents/US20120302538
……………
see
US 20150023913
http://www.google.com/patents/US20150023913
…………..
see
WO 2015005901
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Dimethyl N,N′-(1,4(1,4)-Dibenzenacyclohexaphane-12,42-diylbis(1hbenzimidazole-5,2-diyl((2S,3aS,7aS)-octahydro-1H-indole-2,1-diyl)((2S)-3-methyl-1-oxobutan-1,2-diyl)))biscarbamate
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 1415119-52-6 |
| ATC code | None |
| Chemical data | |
| Formula | C60H72N8O6 |
| Molecular mass | 1001.28 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin

Join me on Facebook 
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
//////////
GOSOGLIPTIN
GOSOGLIPTIN
CAS 869490-23-3 FREE BASE
DIHYDROCHLORIDE..869490-47-1
GOSOGLIPTIN; UNII-GI718UO477; PF-00734200; PF-734200;
(3,3-difluoropyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-ylpiperazin-1-yl)pyrrolidin-2-yl]methanone
| Molecular Formula: | C17H24F2N6O |
|---|---|
| Molecular Weight: | 366.408866 g/mol |
| Company | Pfizer Inc. |
| Description | Dipeptidyl peptidase-4 (DPP-4) inhibitor |
| Molecular Target | Dipeptidyl peptidase-4 (DPP-4) (CD26) |
| Mechanism of Action | Dipeptidyl peptidase-4 (DPP-4) inhibitor |
| Latest Stage of Development | Phase II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
Type 2 diabetes mellitus is a chronic disorder characterized by hyperglycemia coupled with a gradual decline in insulin sensitivity and insulin secretion. The incretin hormone glucagon-like peptide-1 (GLP-1), which is released post-prandially from the L-cells of the intestine, stimulates the release of insulin from pancreatic β-cells. However, GLP-1 is rapidly degraded in vivo by peptidases, including dipeptidyl peptidase IV (DPP-4), which is a widely distributed serine protease that specifically cleaves N-terminal dipeptides from polypeptides with proline or alanine at the penultimate position.
In vivo administration of DPP-4 inhibitors to human subjects results in higher circulating concentrations of endogenous GLP-1 and subsequent decrease in plasma glucose. Long term treatment with a DPP-4 inhibitor leads to a reduction in circulating HbA1c (glycosylated hemoglobin). DPP-4 inhibition also offers the potential to improve the insulin producing function of the pancreas through either β-cell preservation or regeneration. Therefore, DPP-4 inhibition has emerged as a promising new treatment of Type 2 diabetes
PF-734200 is a potent, selective, orally active dipeptidyl peptidase IV inhibitor. It had been in phase II clinical development at Pfizer for the treatment of type 2 diabetes; however, in 2010 the company discontinued these trials. In 2012, the product was licensed to SatRx, a spin-off of the ChemRar High Tech Center, by Pfizer on an exclusive worldwide basis (with the exception of China) for the development and commercialization as monotherapy or in combination with other therapies for the treatment of type 2 diabetes. SatRx is conducting phase II clinical trials for the treatment of type 2 diabetes.
……………………….
PAPER
New synthetic route to a dipeptidyl peptidase-4 inhibitor
Org Process Res Dev 2012, 16(3): 409
http://pubs.acs.org/doi/abs/10.1021/op200309z

A new synthetic route to a dipeptidyl peptidase-4 (DPP4) inhibitor was developed and demonstrated on a multigram scale. This approach takes advantage of the cheap and readily available Boc-trans-4-hydroxy-l-proline methyl ester as starting material which was derivatized through an SN2 reaction. Several leaving groups were studied, and the nosylate group showed superiority over other derivatives. Formation of an amide using the most costly starting material, 3,3-difluoropyrrolidine, was performed late in the synthesis to minimize its economical impact on the overall cost of the API.
(3,3-Difluoropyrrolidin-1-yl)-(2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)methanone.FREE BASE
Mp 149 °C (decomp).
[α]d = −31.1 (T = 24 °C, c = 1, CHCl3). Specific rotation of product 4 prepared using the initial route: [α]d = −31.5 (T = 24 °C, c = 1, CHCl3).
1H NMR (400 MHz; CDCl3) δ 8.30 (d, J = 4 Hz, 2H), 6.48 (t, J = 4 Hz, 1H), 3.95–3.6 (m, 9H), 3.25–2.85 (m, 4H), 2.6–2.25 (m, 7H), 1.75–1.6 (m, 1H).
13C NMR (100 MHz; CDCl3) δ 172.28; 161.55; 157.70; 127.22 (t, 1J C–F = 248 Hz), 126.22 (t, 1J C–F = 246 Hz), 109.95; 66.54; 58.87; 57.99; 52.71 (t, 2 J C–F = 32 Hz); 52.00; 50.41; 43.03; 34.46, 34.37, 34.25; 19F NMR (377 MHz, CDCl3) δ −102.1 (m, 2F).
IR (neat): 2951w, 2864w, 2799w, 2759w, 1630s, 1585vs, 1547m, 1449m, 1172m, 1254m, 1129m, 982w, 923m, 796m, 638w.
HRMS (ES, N2) Calcd for C17H24F2N6O: 367.20524, found: 367.20592.
……………………….
PAPER
(3,3-difluoro-pyrrolidin-1-yl)-((2S,4S)-(4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl)-methanone: A potent, selective, orally active dipeptidyl peptidase IV inhibitor
Bioorg Med Chem Lett 2009, 19(7): 1991
http://www.sciencedirect.com/science/article/pii/S0960894X09001966?np=y
- Pfizer Global Research & Development, Groton/New London Laboratories, Pfizer Inc, Groton, CT 06340, United States
A series of 4-substituted proline amides was evaluated as inhibitors of dipeptidyl pepdidase IV for the treatment of type 2 diabetes. (3,3-Difluoro-pyrrolidin-1-yl)-[(2S,4S)-(4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl]-methanone (5) emerged as a potent (IC50 = 13 nM) and selective compound, with high oral bioavailability in preclinical species.

………………….
PATENT
WO 2005116014
http://www.google.co.in/patents/WO2005116014A1?cl=en
Example 113 (3.3-Difluoropyrrolidin-1-yl)-((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)-methanone

Step 1 – (S)-2-(3.3-Difluoro-pyrrolidine-1-carbonyl)-4-oxo-pyrrolidine-1 -carboxylic acid tert-butyl ester
(S)-4-Oxo-pyrrolidine-1 ,2-dicarboxylic acid 1-tert-butyl ester (6.6 kg, 1.0 equivalent) was charged to a reactor, followed by addition of dichloromethane (15 volumes). The reaction mixture was cooled to 0°C. Triethylamine (4.82 liters, 1.2 equiv) was added over 30 minutes. The mixture turned from suspension to a clear solution at the end of triethylamine addition. The mixture was held at 0°C to 5°C for 10 minutes. Pivaloyl chloride (3.65 kg, 1.05 equivalents) was added slowly while keeping the reaction temperature at 0°C to 5°C. The reaction mixture turned back to aslurry. The reaction mixture was sampled for completion by HPLC (using diethylamine to derivatize) after held for 1 hour at 0°C to 5°C.
3,3-Difluoro- pyrrolidine hydrochloride (4.13 kg, 1.0 equivalent) was charged to the above mixture over 10 minutes at – 10°C to 0°C. Triethylamine (4.0 liters, 1.0 equiv) was introduced slowly over 70 minutes at -10°C to 0°C. Upon completion of triethylamine addition, the mixture was stirred for 1h at 0 to 5°C. The reaction was complete by HPLC assay (-1% starting material). The reaction was quenched with water (10 volumes) at 0°C to 5 °C. The mixture was heated to 20°C to 25 °C. The layers were separated, and the organic layer was washed with 0.5 M HCI (5 volumes). The organic layer was again washed with combined 5% NaHC03 (2 volumes) and half saturated brine solution (1.64 M, 3 volumes). The organic solution was concentrated atmospherically to a low stirrable volume (approximately 20 liters). Ethyl acetate (12.6 volumes, 82.8 liters) was added, the solution was concentrated atmospherically to -6 volumes. The mixture was held at 60°C to 65 °C for 2 hours and cooled to room temperature over 3 hours. The mixture was held at 20°C to 25 °C for 8 hours. Heptane (8 volumes) was added, and the mixture was granulated for a minimum of 2 hours. The solid was filtered, rinsed with 2:1 heptane/ethyl acetate (1 volume), and dried in a tray dryer at 25°C to 35°C for a minimum of 12 h. Yield: 7.26 kg, 79%. HPLC purity: 99.7%. The mother liquor (86 liters) was concentrated to 12 liters under partial vacuum at 65°C to 70°C. The mixture was cooled to 60°C to 65 °C. Ethyl acetate (4.0 liters) was added slowly over 15 minutes. The mixture was cooled to 20°C to 25 °C over 2 hours and was held at that temperature for at least 2 hours. The solid was filtered and rinsed with heptane/ethyl acetate (3:1 v/v, 1.7 liters). Drying in a tray dryer for 12 hours at 35°C to 45 °C yielded 435 grams of product. HPLC purity: 96.4%.
Step 2 – (2S.4S)-2-(3.3-Dif luoro-pyrrolidine-1 -carbonyl)-4-(4-pyrimidin-2-yl-piperazin-1 -yl)-pyrrolidine-1 – carboxylic acid tert-butyl ester A reactor was charged with THF (20 volumes), 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was held at 20°C to 25°C until all material was dissolved over 30 minutes. Acetic acid (0.792 kg, 1.05 equivalents) as added. The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60°C to 70°C until a steady temperature of 66.9°C was observed in the overheads indicating complete removal of water from the system. More THF was added as necessary. At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. The reaction mixture was cooled to -3°C to 7°C and sampled for complete formation of imine by HPLC (using sodium triacetoxyborohydride to reduce imine). Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at -5°C to 15°C. The reaction mixture was heated to 20°C to 25°C and held for 12 hours. HPLC results confirmed the reaction was complete by 99.8%. Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30°C to 60°C. Ethyl acetate (10 volumes) was added to the suspension after it cooled to 20°C to 25CC. The organic phase was separated and the aqueous phase was checked by HPLC. It contained less than 2% of the product. The organic phase was washed with water (5 volumes), saturated brine solution (5 volumes) and concentrated to a small volume (2 volumes) under partial vacuum at 45°C to 50°C. To the slurry was added heptane (10 volumes) at 45°C to 50°C over 30 minutes. The mixture was cooled to 20°C to 25°C and granulated for 2 hours. Solid was collected by filtration, rinsed with heptane (2 volumes). Drying in a tray dryer for 12 hours at 35°C to 45°C yield 5.35 kg (91.3%) of the product. Step 3 – (3.3-Dif luoro-pyrrolidin-1 -yl)-f(2S.4S)-4-(4-pyrimidin-2-yl-piperazin-1 -yl)-pyrrolidin-2-yll- methanone Water (19 liters, 2 volumes) was charged to a reactor followed by the product from Step 2 (9.57 kg,
1.0 equivalent). To the slurry was added concentrated HCI (37 wt% in water, 19.1 liters, 2 volumes) slowly at 20°C to 30°C over 4 hours. The slurry went into solution after 12 liters of HCI was added. After the addition completion, the reaction was complete by HPLC assay. The reaction mixture was cooled to 5°C to 15°C. To the mixture was added 50% NaOH aqueous solution slowly with agitation to pH 10 to pH 11. The pH was monitored with a pH meter closely during the neutralization. The total volume of 50% NaOH added was 12.45 liters. The mixture was warmed to 20°C to 25°C and extracted with ethyl acetate twice (115 liters, 12 volumes and 57 liters, 6 volumes, respectively). The sample from aqueous layer after second extraction was analyzed by HPLC and showed only 1% of the product in that aqueous solution.
The organic layers were combined and treated with magnesium sulfate (5 kg) for 1 hour. The mixture was filtered. The filter cake was rinsed with ethyl acetate (10 liters). The filtrate was charged back to the reactor via a 0.2 micron in-line filter for speck free operation. (The following operations were performed under speck free conditions.) The solution was concentrated to 20 liters (2 volumes) under partial vacuum at 50°C to 60°C. The mixture was cooled to 20°C to 25°C over 30 minutes. Upon cooling to room temperature, crystallization occurred. The mixture was held for 30 minutes. Hexanes (20 liters, 2 volumes) was added slowly over 1 hour. The mixture was granulated for 2 hours. The solid product was collected by filtration and rinsed with hexanes/ethyl acetate (10 liters, 1 :1 v/v). The filter was blown dry with nitrogen for a minimum of 2 hours. The product was dried in a tray dryer at 44°C for 12 hours.
Yield: 5.7 kg, 75.9%.
m.p. 156°C. MS m/z 367 (MH+).
FREE BASE
1H NMR (400 MHz, D20): δ 8.15 (d, 2H, J = 5.0 Hz, CH of pyrimidine), 6.55 (t, 1 H, J = 4.8 Hz, CH of pyrimidine), 3.87-3.81 (dd, 1 H, H2b of proline, rotomeric), 3.78-3.50 (m, 4H, N-CH2 of pyrrolidide), 3.55-3.40 (m, 4H, N-CH2 of piperazine), 2.97 (dd, 1 H, J = 10.2, 6.6 Hz, H5a of proline), 2.85-2.75 (m, 1 H, H4b of proline), 2.69 (dd, 1 H, J = 10.0, 9.1 Hz, H5b of proline), 2.55-2.20 (m, 7H, overlapping N-CH2 of piperazine, CH2 of pyrrolidide and H3b of proline), 1.47-1.38 (m, 1 H, H3a of proline).
Alternatively, the dihydrochloride salt of the titled compound was prepared according to the method of Example 1.
………………
US 2005/0256310
http://www.google.com/patents/US20050256310

This approach begins with N–t-Boc-4-oxo-l-proline (1) that undergoes a mixed anhydride activation with pivaloyl chloride at 0 °C, followed by amidation with 3,3-difluoropyrrolidine to yield the intermediate 2. Reductive amination with 1-(2-pyrimidyl)piperazine using sodium triacetoxyborohydride in THF/AcOH provided the desired stereoisomer 3 in high yield and selectivity, the undesired diastereomer being completely removed by crystallization. Deprotection of 3 with 6 N HCl, followed by neutralization with 50% NaOH and extraction provided PF-734200 (4) in good yield.
EXAMPLE 113 (3,3-Difluoropyrrolidin-1-yl)-((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)-methanone

Step 1—(S)-2-(3,3-Difluoro-pyrrolidine-1-carbonyl)-4-oxo-pyrrolidine-1-carboxylic acid tert-butyl
(S)-4-Oxo-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester (6.6 kg, 1.0 equivalent) was charged to a reactor, followed by addition of dichloromethane (15 volumes). The reaction mixture was cooled to 0° C. Triethylamine (4.82 liters, 1.2 equiv) was added over 30 minutes. The mixture turned from suspension to a clear solution at the end of triethylamine addition. The mixture was held at 0° C. to 5° C. for 10 minutes. Pivaloyl chloride (3.65 kg, 1.05 equivalents) was added slowly while keeping the reaction temperature at 0° C. to 5° C. The reaction mixture turned back to a slurry. The reaction mixture was sampled for completion by HPLC (using diethylamine to derivatize) after held for 1 hour at 0° C. to 5° C. 3,3-Difluoro-pyrrolidine hydrochloride (4.13 kg, 1.0 equivalent) was charged to the above mixture over 10 minutes at −10° C. to 0° C. Triethylamine (4.0 liters, 1.0 equiv) was introduced slowly over 70 minutes at −10° C. to 0° C. Upon completion of triethylamine addition, the mixture was stirred for 1 h at 0 to 5° C. The reaction was complete by HPLC assay (˜1% starting material). The reaction was quenched with water (10 volumes) at 0° C. to 5 ° C. The mixture was heated to 20° C. to 25 ° C. The layers were separated, organic layer was washed with 0.5 M HCl (5 volumes). The organic layer was again washed with combined 5% NaHCO3 (2 volumes) and half saturated brine solution (1.64 M, 3 volumes). The organic solution was concentrated atmospherically to a low stirrable volume (approximately 20 liters). Ethyl acetate (12.6 volumes, 82.8 liters) was added, the solution was concentrated atmospherically to ˜6 volumes. The mixture was held at 60° C. to 65° C. for 2 hours and cooled to room temperature over 3 hours. The mixture was held at 20° C. to 25 ° C. for 8 hours. Heptane (8 volumes) was added, and the mixture was granulated for a minimum of 2 hours. The solid was filtered, rinsed with 2:1 heptane/ethyl acetate (1 volume), and dried in a tray dryer at 25° C. to 35° C. for a minimum of 12 h. Yield: 7.26 kg, 79%. HPLC purity: 99.7%. The mother liquor (86 liters) was concentrated to 12 liters under partial vacuum at 65° C. to 70° C. The mixture was cooled to 60° C. to 65° C. Ethyl acetate (4.0 liters) was added slowly over 15 minutes. The mixture was cooled to 20° C. to 25° C. over 2 hours and was held at that temperature for at least 2 hours. The solid was filtered and rinsed with heptane/ethyl acetate (3:1 v/v, 1.7 liters). Drying in a tray dryer for 12 hours at 35° C. to 45° C. yielded 435 grams of product. HPLC purity: 96.4%.
Step 2—(2S,4S)-2-(3,3-Difluoro-pyrrolidine-1-carbonyl)-4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester
A reactor was charged with THF (20 volumes), 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was held at 20° C. to 25° C. until all material was dissolved over 30 minutes. Acetic acid (0.792 kg, 1.05 equivalents) as added. The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60° C. to 70° C. until a steady temperature of 66.9° C. was observed in the overheads indicating complete removal of water from the system. More THF was added as necessary. At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. The reaction mixture was cooled to −3° C. to 7° C. and sampled for complete formation of imine by HPLC (using sodium triacetoxyborohydride to reduce imine). Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at −5° C. to 15° C. The reaction mixture was heated to 20° C. to 25° C. and held for 12 hours. HPLC results confirmed the reaction was complete by 99.8%. Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30° C. to 60° C. Ethyl acetate (10 volumes) was added to the suspension after it cooled to 20° C. to 25° C. The organic phase was separated and the aqueous phase was checked by HPLC. It contained less than 2% of the product. The organic phase was washed with water (5 volumes), saturated brine solution (5 volumes) and concentrated to a small volume (2 volumes) under partial vacuum at 45° C. to 50° C. To the slurry was added heptane (10 volumes) at 45° C. to 50° C. over 30 minutes. The mixture was cooled to 20° C. to 25° C. and granulated for 2 hours. Solid was collected by filtration, rinsed with heptane (2 volumes). Drying in a tray dryer for 12 hours at 35° C. to 45° C. yield 5.35 kg (91.3%) of the product.
Step 3—(3,3-Difluoro-pyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl]-methanone
Water (19 liters, 2 volumes) was charged to a reactor followed by the product from Step 2 (9.57 kg, 1.0 equivalent). To the slurry was added concentrated HCl (37 wt % in water, 19.1 liters, 2 volumes) slowly at 20° C. to 30° C. over 4 hours. The slurry went into solution after 12 liters of HCl was added. After the addition completion, the reaction was complete by HPLC assay. The reaction mixture was cooled to 5° C. to 15° C. To the mixture was added 50% NaOH aqueous solution slowly with agitation to pH 10 to pH 11. The pH was monitored with a pH meter closely during the neutralization. The total volume of 50% NaOH added was 12.45 liters. The mixture was warmed to 20° C. to 25° C. and extracted with ethyl acetate twice (115 liters, 12 volumes and 57 liters, 6 volumes, respectively). The sample from aqueous layer after second extraction was analyzed by HPLC and showed only 1% of the product in that aqueous solution. The organic layers were combined and treated with magnesium sulfate (5 kg) for 1 hour. The mixture was filtered. The filter cake was rinsed with ethyl acetate (10 liters). The filtrate was charged back to the reactor via a 0.2 micron in-line filter for speck free operation. (The following operations were performed under speck free conditions.) The solution was concentrated to 20 liters (2 volumes) under partial vacuum at 50° C. to 60° C. The mixture was cooled to 20° C. to 25° C. over 30 minutes. Upon cooling to room temperature, crystallization occurred. The mixture was held for 30 minutes. Hexanes (20 liters, 2 volumes) was added slowly over 1 hour. The mixture was granulated for 2 hours. The solid product was collected by filtration and rinsed with hexanes/ethyl acetate (10 liters, 1:1 v/v). The filter was blown dry with nitrogen for a minimum of 2 hours. The product was dried in a tray dryer at 44° C. for 12 hours.
Yield: 5.7 kg, 75.9%. m.p. 156° C. MS m/z 367 (MH+).
1H NMR (400 MHz, D2O): δ 8.15 (d, 2H, J=5.0 Hz, CH of pyrimidine), 6.55 (t, 1H, J=4.8 Hz, CH of pyrimidine), 3.87-3.81 (dd, 1H, H2b of proline, rotomeric), 3.78-3.50 (m, 4H, N—CH2 of pyrrolidide), 3.55-3.40 (m, 4H, N—CH2 of piperazine), 2.97 (dd, 1H, J=10.2, 6.6 Hz, H5a of proline), 2.85-2.75 (m, 1H, H4b of proline), 2.69 (dd, 1H, J=10.0, 9.1 Hz, H5b of proline), 2.55-2.20 (m, 7H, overlapping N—CH2 of piperazine, CH2 of pyrrolidide and H3b of proline), 1.47-1.38 (m, 1H, H3a of proline).
Alternatively, the dihydrochloride salt of the titled compound was prepared according to the method of Example 1.
……………..
PAPER

Scheme 1.
Reagents and conditions: (a) 3,3-difluoropyrrolidine hydrochloride, EDC, HOBt, TEA, DCM, rt; (b) NaBH4, MeOH, (c) (1) trifluoromethane-sulphonyl chloride, DIPEA, DCM; (2) 2-(1-piperazinyl)pyrimidine, DCM, −10 °C; (d) 4 N HCl in dioxane, rt; (e) 2-(1-piperazinyl)pyrimidine, NaBH(OAc)3, AcOH, DCE; (f) R1R2NH hydrochloride, EDC, HOBt TEA, DCM, 0–rt; (g) N-heterocyclic piperazine, NaBH(OAc)3, AcOH, DCE.
……………………….
if image is not clear see at………..http://www.allfordrugs.com/2015/07/03/gosogliptin/
| Patent | Submitted | Granted |
|---|---|---|
| Therapeutic compounds [US7291618] | 2005-11-17 | 2007-11-06 |
| (2S,4S)-4-(piperazin-1-yl)pyrrolidine-2-methanone derivatives [US7465732] | 2007-05-03 | 2008-12-16 |
| THERAPEUTIC COMPOUNDS [US2007161664] | 2007-07-12 | |
| Therapeutic compounds [US2006079498] | 2006-04-13 |
//////////
see gliptins at…………http://drugsynthesisint.blogspot.in/p/gliptin-series.html
BEZ 235 (NVP-BEZ235), Dactolisib

BEZ235 (NVP-BEZ235)Dactolisib
4-[2,3-dihydro-3-methyl-2-oxo-8-(3-quinolinyl)-1H-imidazo[4, 5-c]quinolin-1-yl]-α,α-dimethyl-benzeneacetonitrile
2-methyl-2-{4-[3-methyl-2-oxo-8-(quinolin-3-yl)-1H,2H,3H-imidazo[4,5-c]quinolin-1-yl]phenyl}propanenitrile
2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]-propionitrile
Chemical Formula: C30H23N5O
CAS Number: 915019-65-7
Molecular Weight: 469.54
PHASE 2, NOVARTIS
CANCER, BLADDER
NVP-BEZ235 is a dual inhibitor of phosphatidylinositol 3-kinase (P13K)and the downstream mammalian target of rapamycin (mTOR) by binding to the ATP-binding cleft of these enzymes. It specifically blocks the dysfunctional activation of the P13K pathway and induce G(1) arrest. NPV-BEZ235 has been shown to inhibit VEGF induced cell proliferation and survival in vitro and VEGF induced angiogenesis in vivo. It has also been shown to inhibit the growth of human cancer in animal models.
BEZ-235 is an orally active phosphatidylinositol 3-kinase (PI3K) inhibitor in early clinical trials at Novartis for the treatment of advanced breast cancer, renal cell carcinoma, solid tumors and castration-resistant prostate cancer. Phase I clinical trials were also under way at the company for the treatment of glioma, however, no developments in this indication has been reported. Phase II clinical trials are ongoing at Johann Wolfgang Goethe Universität for the treatment of relapsed or refractory acute leukemia.
PI3Ks perform various functions, promoting cell growth, proliferation, differentiation, motility, survival and intracellular trafficking. Mutations leading to increased activity of PI3Ks, including faulty production or action of PI3K antagonists, have been found in many cancers.

……………………………..
WO 2006122806
http://www.google.com/patents/WO2006122806A2?cl=en
![]()
…………………………..
WO 2008064093
2-methyl-2-[4-(3-methyl- 2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile of formula I (compound I),

Example 1
2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]-propionitrile

In a suitable lab glass reactor are placed 45.0 g of starting 2[4-(8-bromo-3-methyl-2-oxo-2,3- dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]2-methyl-propionitrile together with 2.25 g of bistriphenylphosphine’palladium dichloride in 445 ml N,N-dimethylformamide. This mixture is heated to 95 0C and then a solution of 22.2 g of 3-quinoline boronic acid in a mixture of 225 ml DMF, 300 ml H2O and 60 g of KHCO3 is added. This mixture is heated for 2 h at 95 0C. Then 1080 ml H2O are added. The product 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl- 2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile precipitates. The mixture is cooled within 1.5 h to 0 – 5 °C. After stirring at that temperature for 2 h the crude product is filtered and washed with 300 ml H2O. This product is dried in vacuo at 60 0C for 18 h, to yield crude product.
40 g of this crude product is dissolved in 200 ml formic acid at 60 0C. 8 g of active charcoal and Smopex 234 are added. The mixture is stirred at 60 0C for 1 h, the charcoal is filtered, the residue washed with 80 ml formic acid and then 175 ml formic acid are distilled off in vacuo. Then 320 ml methanol are added and the mixture is heated at reflux for 3 h. The purified product precipitates from the reaction mixture. The mixture is cooled to 0 – 5 0C within 1 h, then stirred 2 h at that temperature is finally filtered and washed with 80 ml cold methanol. This recrystallisation procedure is repeated again. Finally the twice recrystallised material is dried in vacuo at 60 0C to yield purified 2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin- 3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile.
Example 1a 5-Bromo-2-(2-nitro-vinylamino)-benzoic acid

A suspension of 25 g (16 mmol) of 2-amino-5-bromo-benzoic acid (Fluka, Buchs, Switzerland) in H2O-HCI (37%) (10:1) is stirred for 8 h and then filtered (solution A). 8.17 g (255 mmol) of nitromethane (Fluka, Buchs, Switzerland) are added over 10 min to an ice- bath cooled mixture of 35 g of ice and 15.3 g (382 mmol) of NaOH. After stirring for 1 h at 0 0C and 1 h at rt, the solution is added at 0 0C to 28 g of ice and 42 ml of HCI (37%) (solution B). Solutions A and B are combined and the reaction mixture is stirred for 18 h at rt. The yellow precipitate is filtered off, washed with H2O and dried in vacuo at 400C to give the title compound. ES-MS: 287, 289 (M + H)+, Br pattern; 1H NMR (DMSO-d6): δ 13.7-14.6/br s (1 H), 12.94/d (1 H), 8.07/d (1 H), 8.03/dd (1 H), 7.83/dd (1 H), 7.71/d (1 H), 6.76/d (1 H).
Example 1b 6-Bromo-3-nitro-quinolin-4-ol

29 g (101 mmol) of 5-bromo-2-(2-nitro-vinylamino)-benzoic acid (Example 1a) and 11.9 g (121 mmol) of potassium acetate in 129 ml (152 mmol) of acetic anhydride are stirred for 1.5 h at 120 0C. The precipitate is filtered off and washed with acetic acid until the filtrate is colorless, then is washed with H2O and dried in vacuo to give the title compound. ES-MS: 269, 271 (M + H)+, Br pattern; analytical HPLC: W= 2.70 min (Grad 1).
Example 1c 6-Bromo-4-chloro-3-nitro-quinoline

20 g (74.3 mmol) of 6-bromo-3-nitro-quinolin-4-ol (Example 1b) in 150 ml (1.63 mol) of POCI3 are stirred for 45 min at 120 °C. The mixture is cooled to rt and poured slowly into ice- water. The precipitate is filtered off, washed with ice-cold water, and dissolved in CH2CI2. The organic phase is washed with cold brine, and the aqueous phase is discarded. After drying over MgSO4, the organic solvent is evaporated to dryness to provide the title compound. 1H NMR (CDCI3): J9.20/S (1H), 8.54/d (1H), 8.04/d (1H), 7.96/dd (1H); analytical HPLC: W= 4.32 min (Grad 1).
Example 1d 2-Methyl-2-(4-nitro-phenyl)-propionitrile
O .

To 15 g (92.5 mmol) of (4-nitro-phenyl)-acetonitrile (Fluka, Buchs, Switzerland), 1.64 mg (5.09 mmol) of tetrabutylammonium bromide (Fluka, Buchs, Switzerland) and 43.3 g (305 mmol) of iodomethane in 125 mL of CH2CI2 are added 1O g (250 mmol) of NaOH in 125 ml of water. The reaction mixture is stirred for 20 h at RT. After this time, the organic layer is separated, dried over MgSO4, and evaporated to dryness. The residue is dissolved in diethylether and treated with black charcoal for 30 min, filtered over Celite and evaporated in vacuo to give the title compound as a pale yellow solid. Analytical HPLC: tret= 3.60 minutes (Grad 1).Example 1e (2-(4-Amino-phenyl)-2-methyl-propionitrile

16 g (84.1 mmol) of 2-methyl-2-(4-nitro-phenyl)-propionitrile (Example 1d) and 4.16 g of Raney-Ni are shacked in 160 ml of THF-MeOH (1:1) under 1.1 bar of H2 for 12 h at rt. After completion of the reaction, the catalyst is filtered-off and the filtrate is evaporated to dryness. The residue is purified by flash chromatography on silica gel (hexane-EtOAc 3:1 to 1:2) to provide the title compound as an oil. ES-MS: 161 (M + H)+; analytical HPLC: tret= 2.13 minutes (Grad 1).
Example 1f 2-[4-(6-Bromo-3-nitro-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile

18 g (62.6 mmol) of 6-bromo-4-chloro-3-nitro-quinoline (Example 1c) and 11 g (68.9 mmol) of (2-(4-amino-phenyl)-2-methyl-propionitrile (Example 1e) are dissolved in 350 ml of acetic acid and stirred for 2 h. After this time, water is added and the yellow precipitate is filtered off and washed with H2O. The solid is dissolved in EtOAc-THF (1 :1), washed with sat. aqueous NaHCO3 and dried over MgSO4. The organic phase is evaporated to dryness to give the title compound as a yellow solid. ES-MS: 411 , 413 (M + H)+, Br pattern; analytical HPLC: tret= 3.69 min (Grad 1).
Example 1q 2-[4-(3-Amino-6-bromo-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile

24 g (58.4 mmol) of 2-[4-(6-bromo-3-nitro-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile (Example 1e) is shacked in 300 ml of MeOH-THF (1:1) under 1.1 bar of H2 in the presence of 8.35 g of Raney-Ni for 1 h. After completion of the reaction, the catalyst is filtered off and the filtrate is evaporated to dryness to give the title compound as a yellow foam. ES-MS: 381 , 383 (M + H)+, Br pattern; analytical HPLC: W= 3.21 min (Grad 1).
Example 1h
2-[4-(8-Bromo-2-oxo-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-2-methyl- propionitrile

A solution of 5 g (13.1 mmol) of 2-[4-(3-amino-6-bromo-quinolin-4-ylamino)-phenyl]-2- methyl-propionitrile (Example 1g) and 1.59 g (15.7 mmol) of triethylamine in 120 ml CH2CI2 is added over 40 min to a solution of 2.85 g (14.4 mmol) of trichloromethyl chloroformate (Fluka, Buchs, Switzerland) in 80 ml of CH2CI2 at 00C with an ice-bath. The reaction mixture is stirred for 20 min at this temperature then is quenched with sat. aqueous NaHCO3, stirred for 5 min and extracted with CH2CI2. The organic layer is dried over Na2SO4, filtered and evaporated in vacuo to give crude title compound as a brownish solid. ES-MS: 407, 409 (M + H)+, Br pattern; analytical HPLC: tret= 3.05 min (Grad 1). Example 1i
2-[4-(8-Bromo-3-methyl-2-oxo-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-2- methyl-propionitrile

To a solution of 3.45 g (8.47 mmol) of 2-[4-(8-bromo-2-oxo-2,3-dihydro-imidazo[4,5- c]quinolin-1-yl)-phenyl]-2-methyl-propionitrile (Example 1h), 1.8 g (12.7 mmol) of iodomethane (Fluka, Buchs, Switzerland) and 273 mg (0.847 mmol) of tetrabutylammonium bromide (Fluka, Buchs, Switzerland) in 170 ml of CH2CI2 is added a solution of 508 mg (12.7 mmol) of NaOH (Fluka, Buchs, Switzerland) in 85 ml of H2O. The reaction mixture is stirred for 2 days and 900 mg (6.35 mmol) of iodomethane and 254 mg (6.35 mmol) of NaOH in 5 ml of H2O are added. The reaction mixture is stirred for 1 day at rt . After this time, the reaction is quenched with H2O and extracted with CH2CI2 (2*). The organic layer is washed with brine, dried over Na2SO4, filtered and evaporated in vacuo to give the title compound as a beige solid. ES-MS: 421 , 423 (M + H)+, Br pattern; analytical HPLC: tret= 3.15 min (Grad 1).
Example 2
2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]propionitrile p-toluenesulfonate salt
26.5 g of 2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1- yl)-phenyl]propionitrile are placed together with 55 ml formic acid into a glass reactor. This mixture is heated to 60 0C to get a clear solution. This solution is clearfiltered and washed with 36 ml formic acid. Then formic acid is distilled off until the volume of the residual solution is 55 ml. Then a solution of 11.3 g of p-toluenesulfonic acid in 228 ml acetone is added at 50 0C, followed by further addition of 822 ml acetone within 30 minutes. The salt precipitates from the reaction mixture. The mixture is cooled to 0 0C within 2 h, stirred at that temperature for 3 h, is then filtered and washed with 84 ml acetone. The product‘ is dried at 60 0C in vacuo for 18 h to yield 29.8 g (82.4 %) of the 2-Methyl-2-[4-(3-methyl-2-oxo-8- quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile p-toluenesulfonate salt (crystalline form A). The crystalline forms of the present invention are synthesized in accordance with the following examples which are illustrative without limiting the scope of the present invention.
Example 3:
Preparation of form A of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro- imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile
Form A of compound I can be manufactured in the following way: 241 g of free base are dissolved 2.4 I acetic acid at 50 0C. The solution is clearfiltered, washed with 250 ml acetic acid and then at 50 0C 7.2 I of water are added. The free base starts precipitating. The mixture is cooled within 1 h to 25 0C, is then filtered and washed with 10 I H2O. The free base is then dried in vacuo at 50 0C over night to yield 204 g of free base.
References
| WO2005054237A1 | 19 Nov 2004 | 16 Jun 2005 | Hans-Georg Capraro | 1h-imidazoquinoline derivatives as protein kinase inhibitors |
| WO2006122806A2 | 18 May 2006 | 23 Nov 2006 | Novartis Ag | 1,3-dihydro-imidazo [4,5-c] quinolin-2-ones as lipid kinase inhibitors |
| CL11872006A | Title not available |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2009118324A1 * | 24 Mar 2009 | 1 Oct 2009 | Novartis Ag | 5imidazoquinolines and pyrimidine derivatives as potent modulators of vegf-driven angiogenic processes |
| WO2013049300A1 * | 27 Sep 2012 | 4 Apr 2013 | Dana-Farber Cancer Institute, Inc. | Method of treating mucoepidermoid carcinoma |
| WO2013152717A1 | 9 Apr 2013 | 17 Oct 2013 | Shanghai Yunyi Healthcare Management Co., Ltd. | Fused pyrimidine compound, and preparation method, intermediate, composition, and uses thereof |
| EP2474323A2 * | 24 Mar 2009 | 11 Jul 2012 | Novartis AG | Imidazoquinolines and pyrimidine derivatives as potent modulators of vegf-driven angiogenic processes |
| US8476294 | 2 Jun 2010 | 2 Jul 2013 | Novartis Ag | 1H-imidazo[4,5-c]quinolinone derivatives |
VX 787, PIMODIVIR, for Avian influenza

(2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
(2S,3S)-3-((5-Fluoro-2-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic Acid
399.394
C20 H19 F2 N5 O2
JNJ-872
VRT-0928787
VX-787
VRT-0928787
VX-787
vx 787
| Vertex Pharmaceuticals |
Janssen Pharmaceuticals, under license from Vertex Pharmaceuticals, is developing VX-787 and its back-up compound VX-353, an influenza A viral replication inhibitor, for treating influenza A virus infection, including pandemic and avian influenza strains. In May 2015, VX-787 was in phase II clinical trial.

Useful for treating influenza virus infection. For concurrent filing see WO2015073476 (claiming the polymorphic forms of VX-787) and WO2015073491 (claiming the composition comprising the hydrochloride salt of VX-787).
Polymorphic forms of hydrochloride (A,F and D) and tosylate salts (form A) of VX-787 are claimed. , useful for treating influenza virus infection. For concurrent filing see WO2015073481 (claiming the processes for the synthesis of VX-787 ) and WO2015073491 (claiming the composition comprising the hydrochloride salt of VX-787).
WO2010148197
http://www.google.com/patents/WO2010148197A1?cl=en
(1070) (2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
(1070) (2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
Compound 1070 was made in a similar fashion as described above for compounds 946 and 947.
………………….
WO 2013019828
http://www.google.com/patents/WO2013019828A1?cl=en
WO 2012083122
http://www.google.co.in/patents/WO2012083122A1?cl=en
Synthetic Scheme 1

(a) CHC13; (b) NaOMe, MeOH; (c) DPPA, Et3N, BnOH; (d) H2, Pd/C;
Synthetic Scheme 2

(a) Et3N, CH3CN; (b) cone. H2S04; (c) 9M H2S04; (d) Ag2C03, HOAc, DMSO, 100 °C; (e) X- phos, Pd2(dba)3, K3PO4, 2-methyl THF, H20, 120 °C (f) LiOH, THF, MeOH, 70 °C
Synthetic Scheme 3

(a) Et3N, THF; (b) chiral SFC separation; (c) 5-fluoro- l -(p-tolylsulfonyl)-3-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-
………………………
See new patents
WO2015073491
……………………………..
Discovery of a Novel, First-in-Class, Orally Bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2
J. Med. Chem., 2014, 57 (15), pp 6668–6678
DOI: 10.1021/jm5007275
http://pubs.acs.org/doi/abs/10.1021/jm5007275
Vertex Pharmaceuticals Inc

1H NMR (300 MHz, DMSO-d6) δ 12.71 (br s, 1H), 8.58 (s, 1H), 8.47 (dd, J = 9.6, 2.8 Hz, 1H), 8.41 (d, J = 4.8 Hz, 1H), 8.39–8.34 (m, 1H), 4.89–4.76 (m, 1H), 2.94 (d, J = 6.9 Hz, 1H), 2.05 (br s, 1H), 1.96 (br s, 1H), 1.68 (complex m, 7H);
13C NMR (300 MHz, DMSO-d6) δ 174.96, 157.00, 155.07, 153.34, 152.97, 145.61, 142.67, 140.65, 134.24, 133.00, 118.02, 114.71, 51.62, 46.73, 28.44, 28.00, 24.90, 23.78, 20.88, 18.98;
LCMS gradient 10–90%, 0.1% formic acid, 5 min, C18/ACN, tR = 2.24 min, (M + H) 400.14;
HRMS (ESI) of C20H20F2N5O2 [M + H] calcd, 400.157 95; found, 400.157 56.
Article
June 18, 2014
Vertex Licenses VX-787 to Janssen Pharmaceuticals for the Treatment of Influenza
Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) today announced that it has entered into a licensing agreement with Janssen Pharmaceuticals, Inc. for the worldwide development and commercialization of VX-787, a novel medicine discovered by Vertex for the treatment of influenza. As part of the agreement, Vertex will receive an up-front payment of $30 million from Janssen and has the potential to receive additional development and commercial milestone payments as well as royalties on future product sales. Vertex completed a Phase 2a study of VX-787 in 2013 that showed statistically significant improvements in viral and clinical measurements of influenza infection. VX-787 is designed to directly inhibit replication of the influenza virus.
“With a deep history in developing new medicines for viral infections and diseases, Janssen is well-positioned to advance the global development of VX-787 for the treatment of influenza,” said Jeffrey Leiden, M.D., Ph.D., Chairman, President and Chief Executive Officer of Vertex. “This collaboration provides important support for the continued development of VX-787 in influenza and contributes to our financial strength to enable continued investment in our key development programs for cystic fibrosis and in research aimed at discovering new medicines.”
About the Collaboration
Under the terms of the collaboration, Janssen will have full global development and commercialization rights to VX-787. Vertex will receive a $30 million up-front payment from Janssen and could receive additional development and commercial milestone payments as well as royalties on future product sales. The collaboration, and the related $30 million up-front payment, is subject to the expiration of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act.
About VX-787
VX-787 is an investigational medicine that is designed to directly inhibit replication of influenza A, including recent H1 (pandemic) and H5 (avian) influenza strains, based on in-vitro data. VX-787’s mechanism represents a new class of potential medicines for the treatment of influenza, distinct from neuraminidase inhibitors, the current standard of care for the treatment of influenza. VX-787 is intended to provide a rapid onset of action and an expanded treatment window.
In a Phase 2a influenza challenge study, statistically significant improvements in viral and clinical measurements of influenza infection were observed after treatment with VX-787. The study met its primary endpoint and showed a statistically significant decrease in the amount of virus in nasal secretions (viral shedding) over the seven-day study period. In addition, at the highest dosing regimen evaluated in the study, there was a statistically significant reduction in the severity and duration of influenza-like symptoms. In this study, VX-787 was generally well-tolerated, with no adverse events leading to discontinuation. Those who took part in the study volunteered to be experimentally exposed to an attenuated form of live H3N2 influenza A virus. H3N2 is a common type of influenza virus and was the most common type observed in the 2012/2013 influenza season in the United States.
VX-787 was discovered by Vertex scientists.
About Influenza
Often called “the flu,” seasonal influenza is caused by influenza viruses, which infect the respiratory tract.1 The flu can result in seasonal epidemics2 and can produce severe disease and high mortality in certain populations, such as the elderly.3 Each year, on average 5 to 20 percent of the U.S. population gets the flu4 resulting in more than 200,000 flu-related hospitalizations and 36,000 deaths.5 The overall national economic burden of influenza-attributable illness for adults is $83.3 billion.5 Direct medical costs for influenza in adults totaled $8.7 billion including $4.5 billion for adult hospitalizations resulting from influenza-attributable illness.5 The treatment of the flu consists of antiviral medications that have been shown in clinical studies to shorten the disease and reduce the severity of symptoms if taken within two days of infection.6 There is a significant need for new medicines targeting flu that provide a wider treatment window, greater efficacy and faster onset of action.
About Vertex
Vertex is a global biotechnology company that aims to discover, develop and commercialize innovative medicines so people with serious diseases can lead better lives. In addition to our clinical development programs focused on cystic fibrosis, Vertex has more than a dozen ongoing research programs aimed at other serious and life-threatening diseases.
Founded in 1989 in Cambridge, Mass., Vertex today has research and development sites and commercial offices in the United States, Europe, Canada and Australia. For four years in a row, Science magazine has named Vertex one of its Top Employers in the life sciences. For additional information and the latest updates from the company, please visit www.vrtx.com.
Vertex’s press releases are available at www.vrtx.com.
| WO2002024705A1 | 13 Sep 2001 | 28 Mar 2002 | Charles Jackson Barnett | Stereoselective process for preparing cyclohexyl amine derivatives |
| WO2003015798A1 | 13 Aug 2002 | 27 Feb 2003 | Toyama Chemical Co Ltd | Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue |
| WO2005095400A1 | 30 Mar 2005 | 13 Oct 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2006069258A1 * | 20 Dec 2005 | 29 Jun 2006 | Amgen Inc | Substituted heterocyclic compounds and methods of use |
| WO2007084557A2 | 17 Jan 2007 | 26 Jul 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2008079346A1 | 21 Dec 2007 | 3 Jul 2008 | Vertex Pharma | 5-cyan0-4- (pyrrolo [2, 3b] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| WO2009073300A1 | 31 Oct 2008 | 11 Jun 2009 | Vertex Pharma | [1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2010011756A1 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridine kinase inhibitors |
| WO2010011768A1 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010011772A2 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010148197A1 * | 17 Jun 2010 | 23 Dec 2010 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| WO2011008915A1 * | 15 Jul 2010 | 20 Jan 2011 | Abbott Laboratories | Pyrrolopyridine inhibitors of kinases |
| US20100038988 | 12 Aug 2008 | 18 Feb 2010 | Gannon Ramy | Stator and Method of Making the Same |
| WO2003015798A1 | Aug 13, 2002 | Feb 27, 2003 | Toyama Chemical Co Ltd | Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue |
| WO2005095400A1 | Mar 30, 2005 | Oct 13, 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 | Jan 17, 2007 | Jul 26, 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2009073300A1 | Oct 31, 2008 | Jun 11, 2009 | Vertex Pharma | [1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2010011756A1 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridine kinase inhibitors |
| WO2010011768A1 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010011772A2 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010148197A1 * | Jun 17, 2010 | Dec 23, 2010 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US20100038988 | Aug 12, 2008 | Feb 18, 2010 | Gannon Ramy | Stator and Method of Making the Same |
……
.

Vertex Pharmaceuticals’ Boston Campus, United States of America
Lynette Hopkinson VP Commercial Regulatory Affairs, Global Regulatory Affairs Vertex Pharmaceuticals Incorporated, United States

swati Patel, a lead analyst, shared a toast with Mir Hussain, a systems engineer, at Vertex Pharmaceuticals during the Friday beer hour, which features beer and chips for employees.
On Fridays around 5 o’clock, after a hard week of work, Frank Holland likes to unwind with a beer. And he doesn’t have to leave work to get one.
Holland is a research scientist at Vertex Pharmaceuticals, which every Friday rings in “beer hour,” offering free adult beverages and munchies to its 1,300 Boston employees.
For Holland, the weekly ritual is a chance to escape the bubble of his chemistry lab and bump into colleagues from other departments — as well as Vertex’s top executives, who regularly attend. For those who prefer grapes to hops, there is also wine.
“Some of the other companies I worked at, you really had to go out of your way to meet people,” said Holland, 32. “At Vertex all you have to do is show up in the cafeteria on a Friday afternoon.”
Sure, free beer is common at hip tech offices; some even have their own bars. But Vertex, best known for its treatment for cystic fibrosis, was doing this way before it was cool. The beer-hour tradition goes back to the company’s founding days, in 1989. Back then, it was just two dozen people in a small office in Cambridge. Someone went to a corner store, bought a case of beer and some chips, and beer hour was born.

Virginia Carden Carnahan
Vice President, New Product Planning and Strategy, Vertex Pharmaceuticals

A scientist works in the lab at Boston-based Vertex Pharmaceuticals.
Vertex Pharmaceuticals Headquarters Lobby
…………
ZSTK 474

4-[4-[2-(difluoromethyl)benzimidazol-1-yl]-6-morpholin-4-yl-1,3,5-triazin-2-yl]morpholine
ZSTK474; 475110-96-4; 4,4′-(6-(2-(Difluoromethyl)-1H-benzo[d]imidazol-1-yl)-1,3,5-triazine-2,4-diyl)dimorpholine; ZSTK-474; ZSTK 474; TCMDC-137004;
2-(2-Difluoromethylbenzimidazol-1-yl)-4,6-bis(morpholino)-1,3,5-triazine
2-(2-difluoromethylbenzimidazol-1-yl)-4,6-dimorpholino-1,3,5-triazine
Zenyaku Kogyo (Innovator)
phase2………Treatment of Solid Tumors Therapy
ZSTK474 is a cell permeable and reversible P13K inhibitor with an IC₅₀ at 6nm. It was identified as part of a screening library, selected for its ability to block tumor cell growth. ZSTK474 has shown strong antitumor activities against human cancer xenographs when administered orally to mice without a significant toxic effect.
Phosphatidylinositol 3-kinase (PI3K) has been implicated in a variety of diseases including cancer. A number of PI3K inhibitors have recently been developed for use in cancer therapy. ZSTK474 is a highly promising antitumor agent targeting PI3K. We previously reported that ZSTK474 showed potent inhibition against four class I PI3K isoforms but not against 140 protein kinases.
However, whether ZSTK474 inhibits DNA-dependent protein kinase (DNA-PK), which is structurally similar to PI3K, remains unknown. To investigate the inhibition of DNA-PK, we developed a new DNA-PK assay method using Kinase-Glo. The inhibition activity of ZSTK474 against DNA-PK was determined, and shown to be far weaker compared with that observed against PI3K. The inhibition selectivity of ZSTK474 for PI3K over DNA-PK was significantly higher than other PI3K inhibitors, namely NVP-BEZ235, PI-103 and LY294002.
Other Names: ZSTK-474
Chemical Formula: C19H21F2N7O2
CAS Number: 475110-96-4
Molecular Weight: 417.41
WO 2002088112
http://www.google.co.in/patents/EP1389617A1?cl=en
The condensation of 2,4-dichloro-6-(4-morpholinyl)-1,3,5-triazine

with 2-(difluoromethyl)-1H-benzimidazole by means of K2CO3 in DMF gives

2-chloro-4-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-6-(4-morpholinyl)-1,3,5-triazine ,

which is then condensed with morpholine by means of K2CO3 in DMF to afford the target trisubstituted triazine.
aReagents and conditions: (i) K2CO3, DMF, room temp; (ii) morpholine, DMF or THF, room temp; (iii) NaH or K2CO3, DMF or DMSO, 120 °C.

- 2-(2-difluoromethylbenzimidazol-1-yl)-4,6-dimorpholino-1,3,5-triazine(compound 19)
Melting point: 211-214°C
NMR(CDCl3) δ : 3.79(8H, t, J=4Hz), 3.88(8H, t, J=4Hz), 7.3-7.4(2H, m), 7.56(1H, t, J=53Hz), 7.88(1H, d, J=7Hz), 8.32(1H, d, J=7Hz)
MS m/z: 417(M+
……………………
J. Med. Chem., 2011, 54 (20), pp 7105–7126
DOI: 10.1021/jm200688y
1 (0.35 g, 84% yield): mp (EtOH) 217–219 °C (lit. 211–214 °C);
1H NMR (CDCl3) δ 8.33 (dd, J = 7.3, 1.4 Hz, 1H), 7.89 (dd, J = 7.2, 1.5 Hz, 1H), 7.56 (t, JHF= 53.6 Hz, 1H), 7.46–7.37 (m, 2H), 3.91–3.86 (m, 8H), 3.81–3.76 (m, 8H).
Kawashima, S.; Matsuno, T.; Yaguchi, S.; Sasahara, H.; Watanabe, T.Preparation of Heterocyclic Compounds as Antitumor Agents. PCT Int. Appl. WO 02088112, 2002;
Chem. Abstr. 2002, 137, 370113.
………………………………….
2-(difluoromethyl)-1H-benzimidazole
A mixture of o-phenylenediamine (5.41 g, 50 mmol) and difluoroacetic acid (9.6 g, 100 mmol) in 4 M HCl (20 mL) was heated under reflux for 1 h and diluted with hot water (50 mL). The solution was treated with charcoal and filtered through Celite before being neutralized with aqueous NH3. The resulting white precipitate was collected, washed with water, and dried to give 2-(difluoromethyl)-1H-benzimidazole (6.07 g, 72% yield): mp 156–158 °C; 1H NMR (DMSO-d6) δ 13.28 (br, 1H), 7.76–7.68 (m, 1H), 7.61–7.54 (m, 1H), 7.36–7.26 (m, 2H), 7.26 (t,JHF= 53.3 Hz, 1H).
Ge, F.; Wang, Z.; Wan, W.; Lu, W.; Hao, J.One-pot synthesis of 2-trifluoromethyl and 2-difluoromethyl substituted benzo-1,3-diazoles Tetrahedron Lett. 2007, 48, 3251–3254
TRIAZINE, PYRIMIDINE AND PYRIDINE ANALOGS AND THEIR USE AS THERAPEUTIC AGENTS AND DIAGNOSTIC PROBES [US2011275762]2011-11-10
| Patent | Submitted | Granted |
|---|---|---|
| Heterocyclic compound and antitumor agent containing the same as active ingredient [US7071189] | 2004-06-17 | 2006-07-04 |
| Treatment of prostate cancer, melanoma or hepatic cancer [US2007244110] | 2007-10-18 | |
| Heterocyclic compound and antitumor agent containing the same as effective ingredient [US7307077] | 2006-11-02 | 2007-12-11 |
| IMMUNOSUPPRESSIVE AGENT AND ANTI-TUMOR AGENT COMPRISING HETEROCYCLIC COMPOUND AS ACTIVE INGREDIENT [US7750001] | 2008-05-15 | 2010-07-06 |
| PYRIMIDINYL AND 1,3,5-TRIAZINYL BENZIMIDAZOLES AND THEIR USE IN CANCER THERAPY [US2011009405] | 2011-01-13 | |
| SUBSTITUTED PYRIMIDINES AND TRIAZINES AND THEIR USE IN CANCER THERAPY [US2011053907] | 2011-03-03 | |
| IMMUNOSUPPRESSIVE AGENT AND ANTI-TUMOR AGENT COMPRISING HETEROCYCLIC COMPOUND AS ACTIVE INGREDIENT [US2010267700] | 2010-10-21 | |
| AMORPHOUS BODY COMPOSED OF HETEROCYCLIC COMPOUND, SOLID DISPERSION AND PHARMACEUTICAL PREPARATION EACH COMPRISING THE SAME, AND PROCESS FOR PRODUCTION OF THE SAME [US8227463] | 2010-09-30 | 2012-07-24 |
| PYRAZOLO[1,5-a]PYRIDINES AND THEIR USE IN CANCER THERAPY [US2010226881] | 2010-09-09 | |
| PYRIMIDINYL AND 1,3,5-TRIAZINYL BENZIMIDAZOLE SULFONAMIDES AND THEIR USE IN CANCER THERAPY [US2010249099] | 2010-09-30 |
…………..
Zenyaku Kogyo


Sector: Health Care
Industry: Biotech & Pharma
Sub-Industry: Specialty Pharma
Zenyaku Kogyo Co. Ltd. produces pharmaceuticals. The Company manufactures and sells over-the-counter drugs, health foods, and prescription medicines, as well as skin care products.
Address:
5-6-15 Otsuka
Bunkyo, 112-8650
Japan
Phone: 81-3-3946-1111
Fax: 81-3-3946-1130
Web url: www.zenyaku.co.jp
Otsuka
Bunkyo




……
Motesanib (AMG-706)

Motesanib (AMG-706)
Amgen Inc.

Motesanib (AMG 706) is an experimental drug candidate originally developed by Amgen[1] but is now being investigated by theTakeda Pharmaceutical Company. It is an orally administered small molecule belonging to angiokinase inhibitor class which acts as an antagonist of VEGF receptors, platelet-derived growth factor receptors, and stem cell factor receptors.[2] It is used as thephosphatesalt motesanib diphosphate.
Motesanib, also known as AMG-706, is an orally administered multikinase inhibitor that selectively targets VEGF receptors, platelet-derived growth factor receptors, and Kit receptors.
Clinical trials
Motesanib was originally investigated for effectiveness against advanced nonsquamous non-small-cell lung cancer (NSCLC), withPhase II trials indicating an effectiveness comparable to bevacizumab when they were both used in combination withpaclitaxel/carboplatin.[3] However a later and more detailed Phase III trial failed to show any benefit for the treatment of NSCLC.[2][4]A second Phase III trial was started in 2012,[5] which focused on patients from Asian backgrounds (performed on the bases ofsubgroup analysis)[6] however this also failed to meet its primary endpoint.[7]
The drug has undergone a Phase II evaluation as first-line therapy for breast cancer[2] however this study found no evidence to support further investigation.[8] Phase II testing against persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas was also unsuccessful.[9]
There have also been 2 separate Phase II clinical trials for thyroid cancer which have both shown promising results.[10][11][12]
Developed at Amgen, the compound is also being evaluated as both monotherapy and in combination with other agents in the treatment of breast, colorectal, lung, thyroid and ovarian cancers. Clinical trials for the treatment of bladder cancer have been terminated.
The National Cancer Institute had been evaluating the potential of the drug in patients with low-grade neuroendocrine tumors; however, no recent development has been reported for this research. The FDA awarded fast track status to motesanib in 2004. In 2008, the compound was licensed to Takeda in Japan.

AMG-706 is synthesized as follows: 1-Acetyl-3,3-dimethyl-6-nitroindoline (I) is reduced by catalytic hydrogenation over Pd/C, giving the aminoindoline (II), which is then coupled with 2-chloronicotinoyl chloride (III) in the presence of DIEA to yield the corresponding nicotinamide (IV). Subsequent condensation of (IV) with neat 4-(aminomethyl)pyridine (V) at 120 °C affords the 2-aminonicotinamide derivative (VI). The N-acetyl group of (VI) is finally removed by acidic hydrolysis to furnish the title compound (1,2).
,………………………………………
US 2003125339
http://www.google.com/patents/US20030125339
………………………………………………….
US 2003225106
https://www.google.com/patents/US20030225106
EXAMPLE 133
[2295]

N-(3,3-Dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
Step A—Preparation of 1-acetyl-6-amino-3,3-dimethylindoline
1-Acetyl-3,3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL), the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc:CH2Cl2 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O—204.27.
Step B—Preparation of N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from 1-acetyl-6-amino-3,3-dimethylindoline (Step A) by the method described in Example 82.
Step C—Preparation of N-(3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N5O—373.45.
…………………….
http://www.google.com/patents/WO2012063085A3?cl=en
Example 133

N- (3, 3-Dimethy1indolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) }carboxamide Step A – Preparation of l-acetyl-6-amino-3 , 3- dimethylindoline l-Acetyl-3 , 3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL) , the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc :CH2C12 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O-204.27.
Step B – Preparation of N-(l-acetyl- 3 , 3-dimethylindolin-6- yl) (2-[ (4-pyridylmethyl) amino] (3-pyridyl) } carboxamide The titled compound was prepared from l-acetyl-6- amino-3 , 3-dimethylindoline (Step A) by the method described in Example 82.
Step C – Preparation of N- (3 , 3-dimethylindolin-6-yl) {2- [ (4- pyridylmethyl) amino] (3-pyridyl) }carboxamide
The titled compound was prepared from N-(l-acetyl- 3 , 3-dimethylindolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) } carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N50-373.45.
References
- Stafford, edited by Rongshi Li, Jeffrey A. (2009). “Chapter 5. Discovery of Motesanib”. Kinase inhibitor drugs. Hoboken, N.J.: Wiley. pp. 113–130. ISBN 978-0-470-27829-1.
- “Amgen and Takeda’s NSCLC Drug Fails in Phase III Study”. 30 Mar 2011.
- Blumenschein Jr, G. R.; Kabbinavar, F.; Menon, H.; Mok, T. S. K.; Stephenson, J.; Beck, J. T.; Lakshmaiah, K.; Reckamp, K.; Hei, Y.- J.; Kracht, K.; Sun, Y.- N.; Sikorski, R.; Schwartzberg, L. (14 February 2011). “A phase II, multicenter, open-label randomized study of motesanib or bevacizumab in combination with paclitaxel and carboplatin for advanced nonsquamous non-small-cell lung cancer”. Annals of Oncology 22 (9): 2057–2067. doi:10.1093/annonc/mdq731.
- Jump up^ Scagliotti, G. V.; Vynnychenko, I.; Park, K.; Ichinose, Y.; Kubota, K.; Blackhall, F.; Pirker, R.; Galiulin, R.; Ciuleanu, T.-E.; Sydorenko, O.; Dediu, M.; Papai-Szekely, Z.; Banaclocha, N. M.; McCoy, S.; Yao, B.; Hei, Y.-j.; Galimi, F.; Spigel, D. R. (2 July 2012). “International, Randomized, Placebo-Controlled, Double-Blind Phase III Study of Motesanib Plus Carboplatin/Paclitaxel in Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer: MONET1”. Journal of Clinical Oncology 30 (23): 2829–2836. doi:10.1200/JCO.2011.41.4987. PMID 22753922.
- “Takeda Initiates Phase 3 Trial of Motesanib in Japan and Additional Asian Countries”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Kubota, K.; Ichinose, Y.; Scagliotti, G.; Spigel, D.; Kim, J. H.; Shinkai, T.; Takeda, K.; Kim, S.- W.; Hsia, T.- C.; Li, R. K.; Tiangco, B. J.; Yau, S.; Lim, W.- T.; Yao, B.; Hei, Y.- J.; Park, K. (13 January 2014). “Phase III study (MONET1) of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous nonsmall-cell lung cancer (NSCLC): Asian subgroup analysis”.Annals of Oncology 25 (2): 529–536. doi:10.1093/annonc/mdt552.
- Jump up^ “Takeda Announces Phase 3 MONET-A Study Evaluating Motesanib (AMG 706) in Patients with Advanced Non-Squamous Non-Small Cell Lung Cancer Does Not Meet Primary Endpoint”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Martin, Miguel; Roche, Henri; Pinter, Tamas; Crown, John; Kennedy, M John; Provencher, Louise; Priou, Frank; Eiermann, Wolfgang; Adrover, Encarna; Lang, Istvan; Ramos, Manuel; Latreille, Jean; Jagiełło-Gruszfeld, Agnieszka; Pienkowski, Tadeusz; Alba, Emilio; Snyder, Raymond; Almel, Sachin; Rolski, Janusz; Munoz, Montserrat; Moroose, Rebecca; Hurvitz, Sara; Baños, Ana; Adewoye, Henry; Hei, Yong-Jiang; Lindsay, Mary-Ann; Rupin, Matthieu; Cabaribere, David; Lemmerick, Yasmin; Mackey, John R (April 2011). “Motesanib, or open-label bevacizumab, in combination with paclitaxel, as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: a phase 2, randomised, double-blind, placebo-controlled study”. The Lancet Oncology 12 (4): 369–376. doi:10.1016/S1470-2045(11)70037-7. PMID 21429799.
- Schilder, R.J.; Sill, M.W.; Lankes, H.A.; Gold, M.A.; Mannel, R.S.; Modesitt, S.C.; Hanjani, P.; Bonebrake, A.J.; Sood, A.K.; Godwin, A.K.; Hu, W.; Alpaugh, R.K. (April 2013). “A phase II evaluation of motesanib (AMG 706) in the treatment of persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas: A Gynecologic Oncology Group study”. Gynecologic Oncology 129 (1): 86–91. doi:10.1016/j.ygyno.2013.01.006. PMID 23321064.
- Motesanib Diphosphate Provides Anticancer Activity Among Patients with Progressive Thyroid Cancer, CancerConnect.com
- Jump up^ Schlumberger, M. J.; Elisei, R.; Bastholt, L.; Wirth, L. J.; Martins, R. G.; Locati, L. D.; Jarzab, B.; Pacini, F.; Daumerie, C.; Droz, J.-P.; Eschenberg, M. J.; Sun, Y.-N.; Juan, T.; Stepan, D. E.; Sherman, S. I. (29 June 2009). “Phase II Study of Safety and Efficacy of Motesanib in Patients With Progressive or Symptomatic, Advanced or Metastatic Medullary Thyroid Cancer”.Journal of Clinical Oncology 27 (23): 3794–3801. doi:10.1200/JCO.2008.18.7815. PMID 19564535.
- Sherman, Steven I.; Wirth, Lori J.; Droz, Jean-Pierre; Hofmann, Michael; Bastholt, Lars; Martins, Renato G.; Licitra, Lisa; Eschenberg, Michael J.; Sun, Yu-Nien; Juan, Todd; Stepan, Daniel E.; Schlumberger, Martin J. (3 July 2008). “Motesanib Diphosphate in Progressive Differentiated Thyroid Cancer”. New England Journal of Medicine 359 (1): 31–42.doi:10.1056/NEJMoa075853. PMID 18596272.
External links

Motesanib Diphosphate (AMG-706)
857876-30-3 diphosphate
453562-69-1 (free base)
N-(2,3-Dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-3-pyridinecarboxamide diphosphate
3-Pyridinecarboxamide, N-(2,3-dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-, phosphate (1:2)
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-(pyridin-4-ylmethylamino)pyridine-3-carboxamide diphosphate
| 569.4 | |
| Formula | C22H23N5O.2H3PO4 |
|---|
 |
|
| Names | |
|---|---|
| IUPAC name
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-[(pyridin-4-ylmethyl)amino]pyridine-3-carboxamide
|
|
| Other names
AMG 706
|
|
| Identifiers | |
| 453562-69-1 |
|
| ChEMBL | ChEMBL572881 |
| ChemSpider | 9842625 |
| Jmol-3D images | Image |
| PubChem | 11667893 |
| Properties | |
| C22H23N5O | |
| Molar mass | 373.45 g·mol−1 |
Stats today
6.8 lakh views on this blog
…………………..
TAKEDA, JAPAN
![]()

TOKYO HO

Takeda Pharmaceutical CEO Yasuchika Hasegawa
 Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.
Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.

Christophe Weber (L), the new president of Takeda Pharmaceutical Co., and CEO Yasuchika Hasegawa pose

 Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
 OSAKA
OSAKA

Dotonbori, Osaka, Japan
 OSAKA
OSAKA

Allisartan isoproxil

Allisartan isoproxil
CAS: 947331-05-7
553.01, C27 H29 Cl N6 O5
An angiotensin II receptor antagonist used to treat mild to moderate essential hypertension.
Approved china, cfda July 1 2012
![]()
Shanghai Allist Pharmaceutical, Inc.
Allist Shanghai Pharmaceutical Co., Ltd.

2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carboxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester,
2-Butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)biphenyl-4-ylmethyl]-1H-imidazole-5-carboxylic acid isopropoxycarbonyloxymethyl ester
2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester
Allisartan is an orally-available angiotensin AT1 antagonist in phase II clinical trials at Shanghai Allist Pharmaceutical for the treatment of mild to moderate essential hypertension.
Shanghai Allist Pharmaceutical PHASE 2 for Hypertension

The prior art discloses Arleigh medoxomil illiquid, low bulk density, electrostatic phenomena evident. Chinese patent discloses a CN200710094131.0 Alicante medoxomil polymorph and method of preparation. Allie medoxomil based crystal prepared by the method has high stability characteristics, but relatively small bulk density of the crystal clear after the electrostatic phenomenon and poor liquidity dried, crushed and used for easy dispensing generate dust, operating the site clean and labor protection inconvenience, on the other hand also for accurate weighing and packaging products inconvenience.
CN200710094021.4 and CN201110289695.6 disclose the preparation of Alicante medoxomil, the inventor repeated, the proceeds of crystal and Chinese patent CN200710094131.0 consistent disclosed.

Allisartan isoproxil
Angiotensin II AT-1 receptor antagonist
Essential hypertension
Amorphous form of allisartan isoproxil is claimed in WO 2015062498. Useful for treating hypertension. Shenzhen Salubris Pharmaceuticals, in collaboration with Allist, has developed and launched allisartan isoproxil. In October 2012, Shenzhen Salubris signed a strategic cooperation framework agreement with Allist Pharmaceutical for the production and marketing of allisartan isoproxil. Family members of the product case of allisartanWO2007095789, expire in the EU and in the US in 2026. For a prior filing see WO2009049495 (assigned to Allist Pharmaceuticals), claiming the crystalline form of allisartan and its method of preparation.
The compound of formula (I) is an Ang II receptor antagonist. Its chemical name is 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carb-oxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester. Chinese Patent CN101024643A describes the structure, and its use as antihypertensive drugs.

As regards to the solid physical properties of the compound of formula (I), the patent document of CN101024643A discloses that it is a white solid, and its melting point is 134.5-136° C. However, CN101024643A dose not disclose the crystalline structure of the compound of formula (I).

 CHINA
CHINA
NEW PATENT
WO-2015062498
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015062498
2-butyl-4-chloro -1- [2 ‘- (1H- tetrazol-5-yl) -1,1’-biphenyl- – methyl] – imidazole-5-carboxylic acid, 1 – [(isopropoxy) – oxy] -, methyl ester, is a novel angiotensin Ⅱ receptor antagonist. China Patent CN200680000397.8 disclosed structural formula Alicante medoxomil compound. Allie medoxomil toxicity, blood pressure better than the same type of products (such as losartan), which by generating active metabolite (EXP3174) in vivo metabolism, and thus play its antihypertensive effect.
The prior art discloses Arleigh medoxomil illiquid, low bulk density, electrostatic phenomena evident. Chinese patent discloses a CN200710094131.0 Alicante medoxomil polymorph and method of preparation. Allie medoxomil based crystal prepared by the method has high stability characteristics, but relatively small bulk density of the crystal clear after the electrostatic phenomenon and poor liquidity dried, crushed and used for easy dispensing generate dust, operating the site clean and labor protection inconvenience, on the other hand also for accurate weighing and packaging products inconvenience.
CN200710094021.4 and CN201110289695.6 disclose the preparation of Alicante medoxomil, the inventor repeated, the proceeds of crystal and Chinese patent CN200710094131.0 consistent disclosed.
……………………..
PATENT
http://www.google.com/patents/CN103965171A?cl=en
Hypertension is a major disease threat to human health, looking for efficiency, low toxicity anti-hypertensive drugs can help relieve social pressures and family responsibilities, with good social and economic benefits.
Angiotensin II (Ang II) is the renin – angiotensin – aldosterone system (RAAS) main vasoconstrictor hormone, which plays an important role in the pathobiology of many chronic diseases, particularly its the role of blood pressure regulation is particularly prominent, and therefore Ang II receptor is believed to be a good target for the development of anti-hypertensive drugs.
EP0253310 discloses a series of imidazole derivatives, DuPont declared and obtained by the study of losartan potassium-listed in 1994, was the first non-peptide Ang II receptor antagonist anti-hypertensive drugs. Thereafter, he listed a series of losartan antihypertensive drugs: candesartan cilexetil, valsartan, irbesartan, telmisartan and olmesartan medoxomil, etc. (EP0253310, W02005049587, GB2419592, EP1719766, US5196444) .
The losartan potassium in the body, the active metabolite EXP3174 has a stronger antihypertensive effect than losartan potassium, but EXP3174 polar molecular structure, is difficult to form passive absorption by diffusion through the cell membrane. US5298915 discloses five carboxyl ester group transformation EXP3174 is a series of derivatives, focusing on the compound HN-65021, and discloses hypotensive test results HN-65021 administered by the oral route, its hypotensive activity with chlorine Similar losartan potassium (BritishJouurnal ofClinical Pharmacology, 40,1995,591).
CN200680000397.8 _5_ discloses a class of imidazole carboxylic acid derivatives, namely Alicante medoxomil compound 8 has a good blood pressure lowering effect, the structure of formula I, the preparation method disclosed in this patent document follows the route A, losartan potassium by oxidation, the protecting group into an ester, deprotected to give a compound of formula I, the route step oxidation process of hydroxyl to carboxyl groups, will be reduced to very fine granular potassium permanganate, manganese dioxide, filtration This manganese mud time-consuming, inefficient, polluting; the second step conversion was about 70%, and post-processing cumbersome; byproducts and produced the first two steps more. This makes the high cost of the entire route, not suitable for the production of amplification.

CN200710094021.4 discloses another method for preparing the compounds of formula I, the following route B, the starting material by nucleophilic substitution, oxidation, an ester, a tetrazole ring to obtain a compound of formula I, the first step of the method nucleophilic substitution easy to generate an imidazole ring -3 para isomer impurities difficult to remove; the last step into the ring to use sodium azide, operating dangerous.

CN201210020174.5 disclosed a series of anti-hypertensive compound and preparation method, the following line C, the temperature control in the first step of its preparation O ~ 5 ° C, a mixed solution of acetone and water, with a 5% aqueous solution of sodium hypochlorite oxidation, yield 70%, the second step use of potassium permanganate, manganese dioxide will produce the same, and a yield of only 40%, the first two steps total yield of 28%, is very low, and the post-treatment methods are by column separation, the first two steps are used are organic and inorganic mixed solvent is not conducive to recovery, not suitable for scale-up.






Example 8 2-Butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil crude)

To a 20L reactor 9800ml of methanol, stirring was started, the rotational speed is added at 200r / min 1225.3g solid compound of formula II, and heated to reflux. The reaction 8-10h evacuation HPLC detection, the formula II compound residue <1.0% seen as a response endpoint. After reaching the end of the reaction the heating was stopped, continued stirring speed of 180r / min. About 3_4h fell 20_25 ° C, colorless transparent crystalline solid precipitated. The reaction mixture was cooled to continue to 15-20 ° C, to maintain 15-20 ° C with stirring 3h, the reaction mixture was filtered to give a pale yellow clear filtrate. The filtrate was concentrated under reduced pressure to move 20L flask, vacuum degree of 0.075MPa, 40_45 ° C methanol distilled off under until no distillate. 800ml of absolute ethanol was added, a vacuum degree of 0.075MPa, 40-45 ° C under distillation until no distillate.
900ml of absolute ethanol was added, heated to reflux. N-heptane was added slowly 1100ml, reflux 15min, to -10 ° c / h speed cooled to 15 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration lh, was Allie medoxomil crude (800.lg, yield 93.8%).Purification was used directly in the next step without drying.
Example 9 2-butyl-4-chloro-_1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole-5-carboxylic acid, 1 – [(isopropylamino oxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

850ml of absolute ethanol was added to the 3L reaction vessel was charged with crude Alicante medoxomil (800.lg, 1.45mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1300ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration, the purified Alicante medoxomil (780.9g, 97.6% yield).
Example 10 2-butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

950ml of absolute ethanol was added to the 5L reaction vessel was charged with crude Alicante medoxomil (549.9g, 1.72mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1200ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = cake was washed with a mixture of 1/3, and dried under reduced pressure after filtration to obtain a purified Alicante medoxomil (540.0g, 98.2% yield).
……………….
PATENT
Example 122-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester (compound 8)

To a 100 ml of one-necked flask, 0.523 g of material, 0.124 g of potassium carbonate, 5 ml of N,N-dimethylacetamide were added in turn. The solution was stirred at room temperature for 20 minutes. Then 0.562 g of 1-chloromethyl isopropyl carbonate was added and the mixture was reacted at 45-50° C. for 16 hours. After the reaction was completed, the mixture solution was filtered, and 30 ml of water was added into the filtrate. The resulting mixture was extracted with 30 ml of ethyl acetate twice. The organic phase was dried and concentrated to give 1.724 g of oil, which was directly used in the next reaction without purification.
10 ml of dioxane and 5 ml of 4 mol/L HCl were added, and the resulting mixture was reacted at room temperature for 16 hours. The reaction was stopped and the solution was adjusted to pH 6-7 using aqueous sodium bicarbonate solution. The solution went turbid, and was extracted with ethyl acetate. The organic phase was washed with saturated brine, dried, concentrated to give 0.436 g of 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester.
In addition, the following reaction condition can be used to deprotect the protecting group. To 1.7 g of oily product, 5 ml absolute methanol was added and the mixture was heated slowly to reflux and stirred for 8 hours. When the insoluble solid disappeared totally, the mixture was discontinued to heating and cooled to 5° C. The white solid precipitated, and was separated by filtration, and the filter cake was washed with a small quantity of methanol. The combined filtrate was concentrated to dryness to give 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester with the yield of 70%.
1H-NMR (CDCl3) δ H (ppm): 0.89 (t, 3H, J=14.6), 1.24 (d, 6H, J=6.3), 0.37 (m, 2H, J=22.1), 1.69 (m, 2H, J=30.5), 2.64 (t, 2H, J=15.5), 4.81 (m, 1H, J=12.4), 5.54 (s, 2H), 5.86 (s, 2H), 6.95-7.64 (8H), 8.08 (d, 1H, J=7.42)
ESI(+) m/z: 552.7
Mp: 134.5-136° C.

USE CTRLand + Key, or click on picture
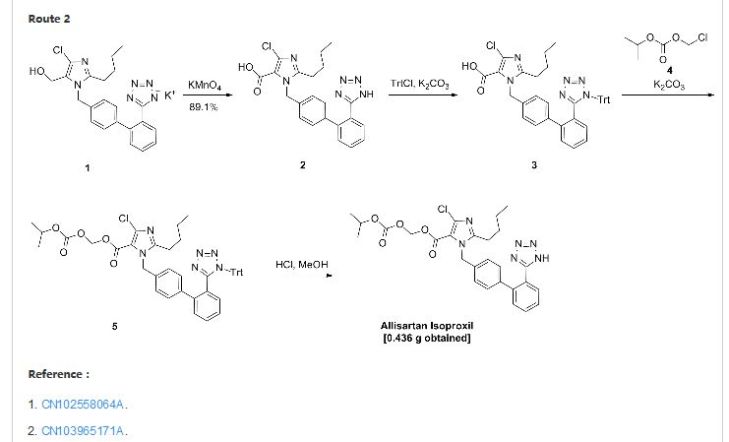
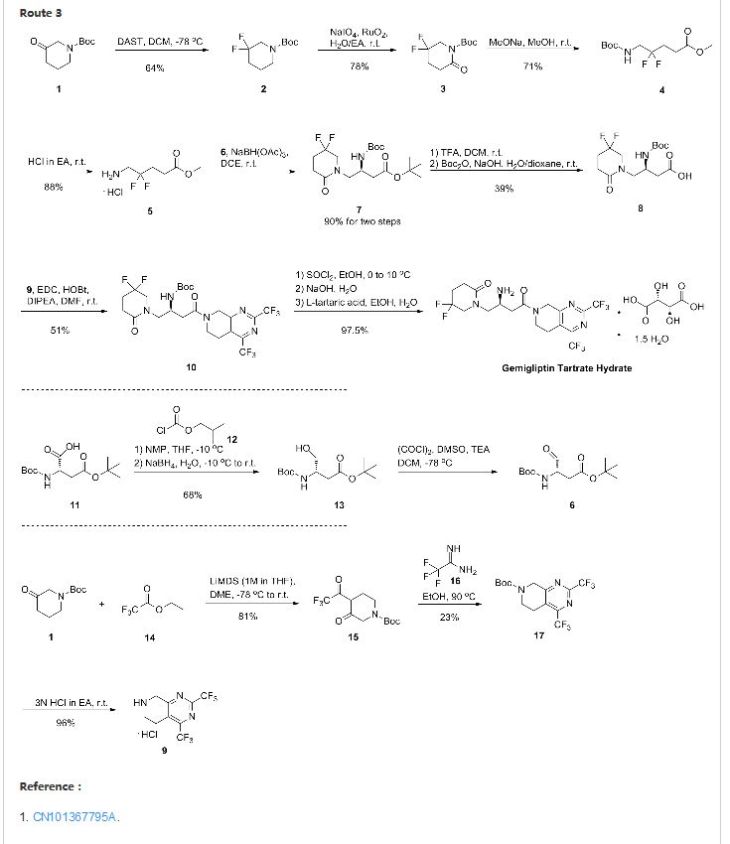



| WO2005011646A2 * | 20 Jul 2004 | 10 Feb 2005 | Nicoletta Almirante | Nitrooxy derivatives of losartan, valsatan, candesartan, telmisartan, eprosartan and olmesartan as angiotensin-ii receptor blockers for the treatment of cardiovascular diseases |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8455526 * | 6 Jun 2008 | 4 Jun 2013 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| US20100168193 * | 6 Jun 2008 | 1 Jul 2010 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| USRE44873 | 31 Jul 2006 | 29 Apr 2014 | Salubris Asset Management Co., Ltd. | Imidazole-5-carboxylic acid derivatives, the preparation method therefor and the uses thereof |
| CN101024643A | 20 Feb 2006 | 29 Aug 2007 | 上海艾力斯医药科技有限公司 | Imidazo-5-carboxylic-acid derivatives, its preparing method and use |
| US5298519 * | 24 Sep 1992 | 29 Mar 1994 | Chemish Pharmazeutische Forschungsgesellschaft M.B.H. | Acylals of imidazole-5-carboxylic acid derivatives, and their use as angiotensin (II) inhibitors |
……………….
update……………..
Example 1
Weigh 25g 2- butyl-4-chloro-1- [2 ‘- (1-trityl–1H- tetrazol-5-yl) -1,1’-biphenyl – methyl] – imidazole 5-carboxylic acid, 1 – [(isopropoxy) – carbonyloxy] -, methyl ester, was added to a 500ml three-necked flask, methanol was added 200ml, refluxed for 9h, methanol was distilled off under reduced pressure to give crude Alicante medoxomil .
To the residue (i.e., medoxomil crude Alicante) were added 33ml of isopropanol and 66ml of n-heptane, heated to 76 ℃ stirred for 2h. After cooling to 60 ℃ stirring for 1h, and then the system was slowly cooled to 0 ℃, stirring was continued for 3h. Filtered, the filter cake was washed with n-heptane. At 40 ℃ 8 hours and dried in vacuo to give 15.3g Alicante medoxomil (purity 99.3%) as a XRD spectrum as shown in Figure, the main peak of the diffraction peaks as shown in the following table, the DSC spectrum shown in figure II . Compared with the published crystal, the crystal obtained by the absence of significant electrostatic phenomena.
Shanghai , CHINA







RG-1577, EVT 302, Sembragiline, RO-4602522
RG-1577, EVT 302, Sembragiline, RO-4602522
CAS 676479-06-4, MW 342.36
- C19 H19 F N2 O3
- Acetamide, N-[(3S)-1-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-3-pyrrolidinyl]-
UNII-K3W9672PNJ
RG-1577, a selective and reversible monoamine oxidase B inhibitor, for treating AD (phase 2 clinical, as of May 2015).
Family members of the product case for RG-1577 (WO2004026825) hold protection in EU until 2023 and expire in US in 2024 with US154 extension. Follows on from WO2006097197, claiming a process for preparing RG-1577.
Alzheimer‘s Disease is a brain disease that slowly destroys memory and thinking skills, up to loss of the ability to carry out the simplest tasks. It is the most common cause of dementia among older people. Mild Alzheimer‘s Disease manifests itself in memory loss and small changes in other cognitive abilities, e.g getting lost, trouble handling money and managing daily tasks, having some mood and personality changes, etc.
In the stage of Moderate Alzheimer‘s Disease, the control of language, reasoning, sensory processing, and conscious thought are impacted. Memory loss and con usion grow worse, e.g patients have problems recognizing family and friends and become unable to learn new things, etc. hallucinations, delusions, and paranoia may occur. .Severe Alzheimer‘s Disease is the final stage. Patients cannot communicate anymore and are completely dependent.
N-[(3S)-l-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide has previously been described in the art. 1 WO 2006/097197 2 and WO 2006/0972703 relate to methods for preparing enantiomerically pure 4-pyrrolidinophenylbenzyl ether derivatives.
![]()
The processes of the prior art hamper from several drawbacks (e.g. long reaction sequence, low overall yield also due to loss of half of the product in the classical resolution step, the need for a chromatographic purification to remove by-products formed in the Mitsunobu reaction) and are therefore less suitable for the preparation of N-[(3S)-l-[4-[(3-fluorophenyl) methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide on large scale.
Most Recent Events
- 01 Aug 2014Roche completes a phase I trial in volunteers in USA (NCT02104648)
- 14 May 2014Roche completes enrolment in the MAyflOwer RoAD trial for Alzheimer’s disease (combination therapy, adjunctive treatment) in Australia, Canada, Czech Republic, France, Germany, Italy, Poland, South Korea, Spain, Sweden the United Kingdom and the USA (NCT01677754)
- 01 Apr 2014Roche initiates enrolment in a phase I trial in healthy volunteers in USA (NCT02104648)
http://www.evotec.com/uploads/media_library/10/2012-09_Evotec_Company_presentation_September_e.pdf

……………………..
WO2004026825
http://www.google.com/patents/WO2004026825A1?cl=en
………………….
WO2006097197
http://www.google.com/patents/WO2006097197A1?cl=en
……………………………………………..
PATENT
WO 2015063001
Novel, crystalline polymorphic forms A and B of a pyrrolidone derivative ie RG-1577, useful for treating Alzheimer’s disease (AD). Roche and its Japanese subsidiary Chugai, under license from Evotec, which previously licensed the drug from Roche, are developing RG 1577
formula 1 via the following routes

In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route A

1
In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route B

In a certain embodiment, present invention relates to a crystalline polymorph of a compound of formula 1.

synthesize a compound of formula 1 from a compound of formula 7

compound of formula 6 to a compound of formula 7

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 6 via the intermediate 6a to a compound of formula 7

further comprising reacting a compound of formula 3 with a compound of formula 5 to a compound of formula 6

comprising reacting a compound of formula 2 to a compound of formula 3

2 3
In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 10 to a compound of formula 6

eacting a compound of formula 9 with a compound of formula 5 to a compound of formula 10

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 8 to a compound of formula 9

(lS’)-N-[l-[4-(3-fluoro-benzyloxy)-phenyl]-5-oxo-pyrrolidin-3-yl-]acetamide (1)
To a suspension of chloride (7) (37.9 g, 100 mmol) in 2-methyltetrahydrofurane (600 ml) was added under vigorous stirring at 0°C 1.65 M potassium ie/t-butoxide in THF (75.5 ml, 125 mmol, ACROS) over 2.5 h. After additional stirring at 0°C for 1 h, the cold suspension was hydrolyzed with 0.1 M HCl (600 ml) and the reaction mixture was stirred at 30°C for 0.5 h. The organic layer was washed with water (300 ml), dried (Na2S04) and filtered. Removal of the solvent by rotary evaporation (50°C/>10 mbar) afforded 32.1 g crystalline residue, which was dissolved in 2-butanone (400 ml) at ca. 95°C and hot filtered. Crystallization, which was induced by seeding and cooling to room temperature and 0°C (4 h) afforded 25.4 g (74.2%) of the titled compound (1) as an off-white, crystalline powder,
Mp. 162-164°C (polymorph B).
Ee >99.8%, [cc]D20 = – 17.8 (DMF; c = 1).
1H NMR (400 MHz, DMSO- 6) δ ppm 1.82 (s, 3H), 2.34 (dd, J1=n. l, J2=3.9, 1H), 2.84 (dd, J/=17.1, J2=8.2, 1H), 3.55 (dd, J/=10.2, J2=3.2, 1H), 4.07 (dd, J/=10.2, J2=6.7, 1H), 4.32-4.41 (m, 1H), 5.13 (s, 2H), 7.02 & 7.55 (d, J=9.1, each 1H), 7.11-7.19 (m, 1H), 7.24-7.31 (m, 1H), 7.40-7.47 (m, 1H), 8.40 (d, J=6.4, 1H).
ESI-MS (m/z) 343 [M+H]+, 365 [M+Na]\. Anal.Calcd for Ci9H19FN203 (342.37): Calcd. C, 66.66; H, 5.59; N, 8.18; F, 5.02; O, 14.02. Found C, 66.76; H, 5.48; N, 8.13; F, 5.03; O, 13.99.
Crystallized (1) form previous step (9.5 g, 0.028 mol) was dissolved in 2-butanone (290 mL) upon heating. The hot solution was filtered over charcoal. The solution was concentrated by removal of 2-butanone (200 mL) by distillation prior to seeded cooling crystallization. Filtration, washing with chilled 2-butanone and drying at 50°C/25 mbar/16h afforded 9.18 g (93.9% corrected yield) of the title compound (1) as a crystalline powder of polymorphic form B with an assay of 100.4 %(w/w) and a purity of 99.97 %(area) (by HPLC).
Alternatively, to a stirred suspension of hydroxyamide (6) (30.0 g, 0.083 mol) in toluene (500 ml) was added at 50°C within 45 minutes thionyl chloride (10.40 g, 0.087 mol) and the resulting mixture was stirred for 3h at 50°C. The mixture was then heated up to 92°C and subsequently stirred at this temperature for 15 h. The Suspension was then cooled to 50°C and toluene was removed by distillation under reduced pressure. The distillation residue was cooled to ambient temperature and treated with N-methylpyrrolidone (210 ml) to obtain an almost clear solution. This solution was then cooled to -10°C and subsequently treated at this temperature within 2h with a solution of potassium iert-butoxide (12.40 g, 0.111 mol) in THF (60 g). The resulting mixture was stirred for another 60 minutes at -10°C, then warmed up to room temperature within 60 minutes and subsequently stirred at room temperature for 6 h. The reaction mixture was quenched with water (150 g) and the pH was adjusted with acetic acid (approx. 1.8 g) to pH 7-8. The mixture was then heated to 30-45°C and THF and toluene were distilled off under reduced pressure (<200 mbar) to obtain a clear NMP/water mixture (400 ml). This mixture was heated to 45°C and 260 mg of seed crystals were added. Water (320 ml) was then added within 3 h whereby the product crystallized. The resulting suspension was cooled to room temperature within 3 h and subsequently stirred at this temperature for 2 h. Filtration and washing of the filter cake with a mixture of water (100 ml) and N-methylpyrrolidone (20 ml) and subsequently only with water (150 ml) afforded after drying (70°C/10 mbar/20 h) 26.2 g (92%) of the title compound (1) as a crystalline powder with an assay of 99.6 %(w/w) and a purity of 99.7 %(area) (by HPLC).

HPLC
Purity (HPLC): Column: XSelect Phenyl Hexyl x2, 150 x 4.6mm, 3.5um. Starting
Pressure: 226 bar; temp.: 50°C. Inj. vol.: 2.0 μΐ^ + wash. Flow: 1.0 ml/min. Det: 204 nm. A: Water + 5% ACN, 77-2% in 7 min., hold for 1 min.; B: 0.1% HCOOH, 18% isocratic; C: MeOH, 5-80% in 7 min., hold for 1 min. Sample prep.: 2 mg/ml ACN. Retention times: β-acid 5.93 min., diacid 6.18 min., cc-acid 6.89 min., diester 6.96 min.
ee determination(HPLC): Column: Chiralpak IA-3 100 x 4.6mm, 3um; 91 bar, 2ml/min; temp.: 30°C. Inj. vol.: 10.0 μL· Det.: 206 nm. A: n-heptane, 80%; B: EtOH, 20%. Sample prep.: 4 mg/ml EtOH. Retention times: D-enantiomer 2.21 min., L-enantiomer 2.71 min
………………….
US 20050065204
EXAMPLE 11
Preparation of (S)-1-(4-Hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic Acid
8.00 g Polyethyleneglycol 6000 was dissolved in 150 mL (100 mM) magnesium acetate buffer pH 6.0 under stirring, and the solution added to a stirred suspension of 10.00 g (42.51 mmol) (RS)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid methyl ester (99.7%) in 40 mL methylcyclohexane. The mixture was heated to 28° C. and the pH readjusted to 6.0 with 2 M NaOH. The reaction was started by adding 33.2 mg Candida cylindraceae cholesterase (16.88 kU/g), and the pH was maintained at 6.0 by the controlled addition of 1.0 M NaOH solution under stirring. After a total consumption of 20.35 mL (20.35 mmol) 1.0 M sodium hydroxide solution (after 17.1 h; 47.9% conversion) the reaction mixture was passed through a sintered glass filter. The filtrate spontaneously separated into an aqueous and an organic phase.The aqueous phase was washed with 2×200 mL ethyl acetate to remove uncleaved ester. The aqueous phase was set to pH 4.0 with 25% sulfuric acid and concentrated in vacuo to a volume of ca. 80 mL (bath 60° C.). The solution was cooled to 1° C. (formation of white precipitate/crystals) and the pH set to 1.5 with 25% sulfuric acid. The precipitate/crystals were stirred overnight at 1° C., filtered off on a sintered glass filter (washed with a minimum amount of water) and dried overnight on high vacuum (RT, 6×10−2 mbar) to give 4.32 g (19.53 mmol; 45.9%) (S)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid. Analysis: HPLC (area A226nm): 99.3%, 0.7% ester. 98.9%ee. The product contains 5.3% water (according to Karl Fischer determination) and 2.1% (w/w) PEG (according to NMR).
| Company | Evotec AG |
| Description | Small molecule monoamine oxidase B (MAO-B) inhibitor |
| Molecular Target | Monoamine oxidase B (MAO-B) |
| Mechanism of Action | Monoamine oxidase B (MAO-B) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase II |
| Standard Indication | Alzheimer’s disease (AD) |
| Indication Details | Treat Alzheimer’s disease (AD) |
| Regulatory Designation | |
| Partner |
//////////
Chūō, japan




A Chūō Line (Rapid) E233 series (right) and A Chūō-Sōbu Line E231 series (June 2007)
 Chuo Dori street on a weekend afternoon
Chuo Dori street on a weekend afternoon

