
Home » Posts tagged 'penciclovir sodium'
Tag Archives: penciclovir sodium
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Penciclovir

Penciclovir
- Molecular FormulaC10H15N5O3
- Average mass253.258 Da
Cas 39809-25-1
97845-62-0 (Na salt)
Launched – 1996 PERRIGO, Herpes labialis
BRL-39123; penciclovir; BRL 39123A; penciclovir sodium; Denavir; Vectavir; Euraxvir; Fenivir
Penciclovir [USAN:INN:BAN]
- BRL 39123
- BRL-39123
- CCRIS 9213
- Denavir
- HSDB 8123
- Penciclovir
- Penciclovirum
- Penciclovirum [INN-Latin]
- UNII-359HUE8FJC
Penciclovir was approved for medical use in 1996.[2]
Developed and launched by SmithKline Beecham (SB; now GlaxoSmithKline) and now marketed in the US by Prestium Pharma and ex-US by Novartis, penciclovir (Vectavir; Fenivir; Denavir; Euraxvir) is a 1% topical cream indicated for the treatment of recurrent herpes labialis (cold sores) in adults and children 12 years of age and older
APPROVALS
THE US
In September 1996, the compound was approved by the US FDA for cold sore treatment , and was launched in the US in 1997.
EUROPE
In December 1995, SB filed for European approvals of the drug . In 1997, the drug was approved in Belgium Iceland Denmark Norway Ireland . In January 2003, the drug was launched in Sweden . In May 2007, the drug was launched in Portugal .
JAPAN
In December 1995, SB filed for Japanese approval of the drug .
CHINA
In September 1999, the compound was approved in China
FDA
Click to access 020629s016lbl.pdf
Chemically, penciclovir is known as 9-[4-hydroxy-3-(hydroxymethyl)butyl] guanine. Its molecular formula is C10H15N5O3; its molecular weight is 253.26. It is a synthetic acyclic guanine derivative
Penciclovir is a white to pale yellow solid. At 20°C it has a solubility of 0.2 mg/mL in methanol, 1.3 mg/mL in propylene glycol, and 1.7 mg/mL in water. In aqueous buffer (pH 2) the solubility is 10.0 mg/mL. Penciclovir is not hygroscopic. Its partition coefficient in n-octanol/water at pH 7.5 is 0.024 (logP = -1.62).
Medical use
In herpes labialis, the duration of healing, pain and detectable virus is reduced by up to one day,[3] compared with the total duration of 2–3 weeks of disease presentation.
Mechanism of action
Penciclovir is inactive in its initial form. Within a virally infected cell a viral thymidine kinase adds a phosphate group to the penciclovir molecule; this is the rate-limiting step in the activation of penciclovir. Cellular (human) kinases then add two more phosphate groups, producing the active penciclovir triphosphate. This activated form inhibits viral DNA polymerase, thus impairing the ability of the virus to replicate within the cell.
The selectivity of penciclovir may be attributed to two factors. First, cellular thymidine kinases phosphorylate the parent form significantly less rapidly than does the viral thymidine kinase, so the active triphosphate is present at much higher concentrations in virally infected cells than in uninfected cells. Second, the activated drug binds to viral DNA polymerase with a much higher affinity than to human DNA polymerases. As a result, penciclovir exhibits negligible cytotoxicity to healthy cells.
The structure and mode of action of penciclovir are very similar to that of other nucleoside analogues, such as the more widely used aciclovir. A difference between aciclovir and penciclovir is that the active triphosphate form of penciclovir persists within the cell for a much longer time than the activated form of aciclovir, so the concentration within the cell of penciclovir will be higher given equivalent cellular doses.
SYN
Choudary, B.M.; Geen, G.R.; Grinter, T.J.; MacBeath, F.S.; Parratt, M.J.
Influence of remote structure upon regioselectivity in the N-alkylation of 2-amino-6-chloropurine: Application to the synthesis of penciclovir
Nucleosides Nucleotides 1994, 13(4): 979
PATENT
US 6573378
PATENT
CN 102070636
PAPER
https://www.tandfonline.com/doi/abs/10.1081/SCC-120026312?journalCode=lsyc20Selective and Practical Synthesis of Penciclovir


9-[4-Hydroxy-3-(hydroxymethyl)butyl]guanine
(Penciclovir)[3a,b] (1)
………………………as a colorless crystalline solid: m.p. 268.4–269.2C [Lit.[3a,b]
275–277C]. UV (H2O) max 252 and 273 (sh) nm [Lit[3a,b] 253 and 270
(sh) nm].
1HNMR (DMSO-d6) 1.42 (pseudo septet, 1H, J¼7 Hz, H-30),
1.69 (pseudo q, 2H, J¼7 Hz, H-20), 3.37 (ddd, 2H, J¼11, 7 and 7 Hz)
and 3.41 (ddd, 2H, J¼11, 7 and 7 Hz) (CH2OH), 3.98 (t, 2H, J¼7 Hz,
H-10), 4.42 (t, 2H, J¼7 Hz, OH), 6.42 (br s, 2H, NH2), 7.67 (s, 1H, H-8),
10.50 (br s, 1H, H-1).
The 1HNMR spectrum is in good agreement with
the literature data.[3a,b]
3 (a) Harnden, M.R.; Jarvest, R.L.Tetrahedron Lett. 1985, 26, 4265–4268;
(b) Harnden, M.R.;Jarvest, R.L.; Bacon, T.H.; Boyd, M.R. J. Med. Chem. 1987, 30,1636–1642;
PAPER
Improved industrial syntheses of penciclovir and famciclovir using N2-acetyl-7-benzylguanine and a cyclic side chain precursor.




| Journal of Zhejiang University SCIENCE A 2013 Vol.14 No.10 P.760-766 |
Accelerated effect on Mitsunobu reaction via bis-N-tert-butoxycarbonylation protection of 2-amino-6-chloropurine and its application in a novel synthesis of penciclovir
| Author(s): Li-yan Dai, Qiu-long Shi, Jing Zhang, Xiao-zhong Wang, Ying-qi Chen | ||
| Affiliation(s): . Department of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China | ||
| Corresponding email(s): dailiyan@zju.edu.cn | ||
| Key Words: 2-amino-6-chloropurine, Mitsunobu reaction, bis-Boc protection, Penciclovir (PCV) | ||
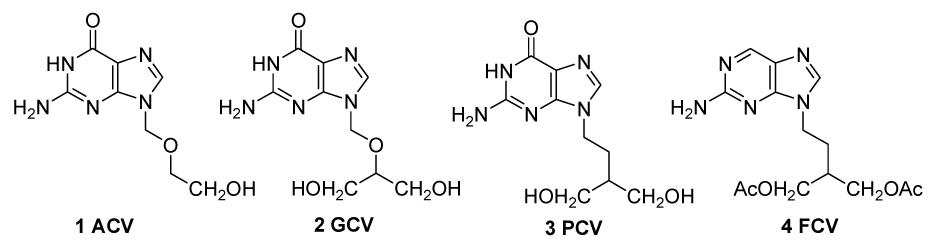
Purine derivatives
To synthesize 3 and 4, 2-amino-6-chloropurine (ACP) is commonly used as a starting material, coupling with alkyl halide side chains (Geen et al., 1990; Geen et al., 1992; Kim et al., 1998; Brand et al., 1999; Toyokuni et al., 2003). However, considering its isomerization at N7 and N9 positions under acidic or alkaline conditions, the most challengeable issue is the selectivity of a N-alkylation at the N7 or N9 position of ACP. Normally, alkylation takes place at the N9 position as well as at the N7 position of the purine moiety, and the N9/N7 ratio is usually less than 6:1 (Kim et al., 1998). Accordingly, to improve this ratio, several approaches have been reported, mainly involving changing the structure of the side chains (Geen et al., 1992) and modification of the ACP (Brand et al., 1999). For example, as reported by Zheng et al. (2004) (Fig. 2), a side chain 6 was synthesized and separated readily at 0 °C. After coupling 6 with 2-amino-6-chloropurine 7, the ratio of the product 9-isomer purine (8a) and the 7-isomer purine (8b) could reach about 10:1. However, the reaction temperature must be strictly controlled as 6 decomposes easily even at room temperature and then an extra careful column chromatography separation procedure would be required to obtain pure 8a. Thus, finding a more practical and efficient method, which could avoid the formation of N7-alkylated compound and shorten the synthetic steps to obtain ACP, becomes attractive.
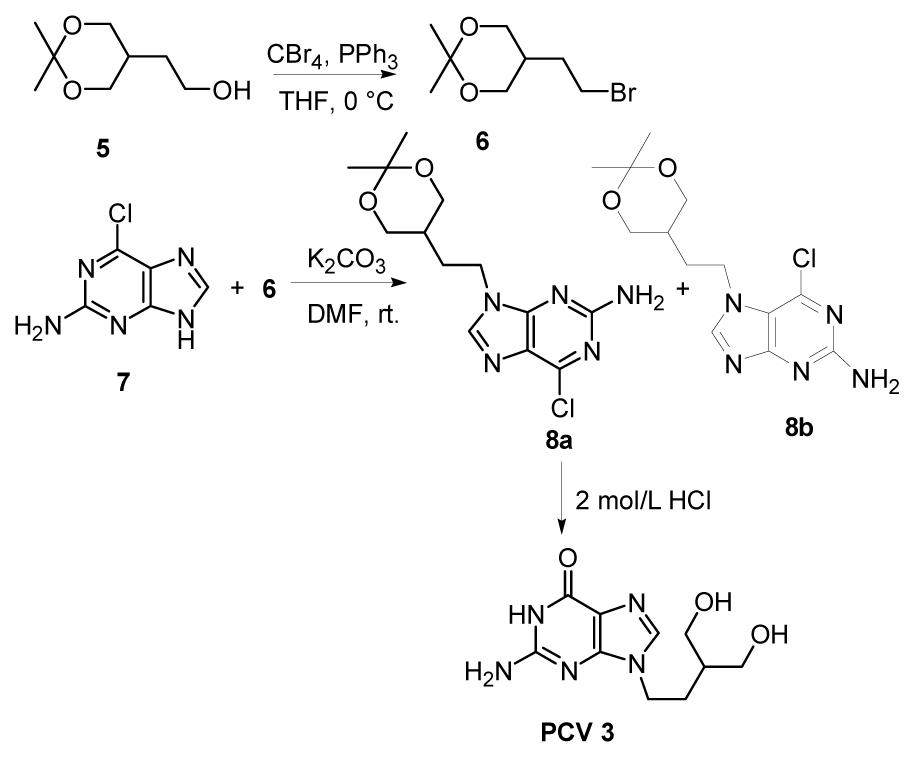
Synthesis of penciclovir (PCV) with conventional method
The Mitsunobu reaction might be an alternative (potential) approach (Mitsunobu, 1981; Swamy et al., 2009). This reaction has become a very popular chemical transformation due to its mildness, occurring under essentially neutral conditions, and its stereospecificity, proceeding with complete Walden inversion of stereochemistry (Mitsunobu, 1981). Moreover, it permits C-O, C-S, C-N, or C-C bonds formed by the condensation of an acidic component with a primary or a secondary alcohol. Actually, some literature has already reported successful Mitsunobu coupling of ACP and adenine with allylic and benzylic alcohol, showing a good N9 selectivity (Yang et al., 2005; Kitade et al., 2006; Yin et al., 2006). However, a poor to modest yield (20%–50%) and a limited substrate scope were observed. In order to improve these yields, Lu et al. (2007) developed a modified Mitsunobu method to couple purine with alcohols in a higher temperature (70 °C), along with two rounds of the Mitsunobu reaction; yet its long reaction procedure and poor atom economy weaken its potential. The poor solubility of ACP or its derivatives in THF, the preferred solvent for Mitsunobu reactions, is likely the primary reason for these defects being observed.
A possible process to improve the solubility of ACP is to make use of the tert-butoxycarbonyl group (Boc), which can serve as the protection of the exocylic amino groups functionality and increase the lipophilicity of the base portion of the purine. Another advantage of the Boc protection group is that its acidolytic removal is less sensitive to steric factors and can also be removed under neutral conditions (Hwu et al., 1996; Siro et al., 1998). In contrast, a few studies have recently been reported that apply the Boc group in the protection of nucleobase (Sikchi and Hultin, 2006; Porcheddu et al., 2008). As described by Porcheddu et al. (2008), solubility of nucleobases, including guanine, was increased in some organic solvents after protected by Boc groups. In addition, some results in our previous study (Yang et al., 2011) demonstrated a very good improvement in coupling purine derivatives under Mitsunobu conditions. Thus, it could be safer to presume that protecting amino groups of ACP with Boc would be an ideal way for its application in the synthesis of PCV 3 and offer similar results as shown under Mitsunobu conditions.
In this study, we firstly synthesized a bis-Boc protected ACP, namely, bis-Boc-2-amino-6-chloropurine 9 (Fig. 3) and investigated its solubility in several different Mitsunobu solvents, then coupling bis-Boc-2-amino-6-chloropurine 9 with a large scope of alcohols confirmed its good reactivity for a Mitsunobu reaction and successfully developed a new and efficient method for the preparation of PCV using Mitsunobu coupling reaction as the key step.

Synthesis of bis-Boc-6-chloropurine 9
a: 2-amino-6-chloropurine, 4,4-dimethylaminopyridine (DMAP), THF and Boc2O, 25 °C, N2; b: MeOH, NaHCO3, 55 °C
To a 250 ml N2-flushed flask with dry THF (100 ml), equipped with a magnetic stir bar, 2-amino-6-chloropurine (2.0 g, 11.8 mmol) and DMAP (0.14 g, 1.18 mmol) were added. Boc2O (10.3 g, 47.2 mmol) was added to the stirred suspension under an N2atmosphere, then the reaction mixture was stirred for 6 h at room temperature (TLC analysis indicated the disappearance of 2-mino-6-chloropurine). The excess amount of THF was removed, and the crude product was dissolved in AcOEt (400 ml), washed with HCl aqueous (2 mol/L, 1×30 ml) and brine (2×50 ml), dried with Na2SO4 and concentrated in vacuo to give a white solid (5.2 g, 94.5%). mp 51–52 °C; 1H NMR (500 MHz, CDCl3): δ=1.47 (s, 18H, C(CH3)3), 1.69 (s, 9H, C(CH3)3), 8.58 (s, 1H, CH); 13C NMR (125 MHz, CDCl3) δ=153.8, 152.0, 151.8, 150.6, 145.5, 144.7, 130.8, 88.0, 83.9, 28.0.
2. Bis-Boc-2-amino-6-chloropurine (9)
A solution of the white solid obtained above (14 g, 30 mmol) in MeOH (400 ml) was added to saturated NaHCO3 aqueous (200 ml), then the turbid solution was stirred at 55 °C for 2 h, at which point clean conversion to bis-Boc protected adenine was observed by TLC. After evaporation of MeOH, the residue mixture was cooled, added 5 mol/L hydrochloric acid to get pH=7 (approximate). A large amount of white solid formed, the reaction mixture was filtrated and then dried under a vacuum to give a white solid 9 (10.5 g, 95.5%). mp 101.3–103.3 °C; 1H NMR (500 MHz, CDCl3): δ=1.50 (s, 18H, C(CH3)3), 8.41 (s, 1H, CH); 13C NMR (125 MHz, CDCl3) δ=153.5, 151.9, 151.6, 151.3, 145.6, 128.5, 82.7, 28.5.
2.3. 5-(2-hydroxyethyl)-2,2-dimethyl-1,3-dioxane (5)
2-hydroxymethyl-1,4-butanediol 11 (8.10 g, 67.4 mmol) and 2,2-dimethoxypropane (13 ml, 105.7 mmol) were dissolved in dry THF (20 ml). The mixture was stirred and p-toluenesulfonic acid monohydrate (0.64 g, 3.4 mmol) was added, the clear solution was stirred at room temperature for 12 h, triethylamine (10 ml) was added to quench the reaction, and the solution was stirred for 30 min. Then solvents were removed to leave a colorless liquid, the residue was subject to column chromatography on silica gel eluted with 2:1 EtOAc/hexane to give a colorless liquid 5 (6.2 g, 61.5%), R f=0.46 (2:1 EtOAc/hexane). 1H NMR (500 MHz, CDCl3): δ=3.99 (dd, 2H, Heq. J 1=11.80 Hz, J 2=4.45 Hz, CH2); 3.80 (t, 2H, J=6.71 Hz, CH2), 3.34 (dd, 2H, Hax. J 1=11.80 Hz, J 2=8.11 Hz, CH2), 1.90–1.98 (m, 2H, CH and OH), 1.62 (q, 2H, J=6.85 Hz, CH2 ); 13C NMR (125 MHz, CDCl3): δ=100.5, 69.8, 60.4, 31.9, 30.3, 21.2.
2.4. Bis-Boc-2-amino-6-chloro-9-[2-(2,2-dimethyl-1,3-dioxan-5-yl)ethyl] purine (12)
Bis-Boc-2-amino-6-chloropurine 9 (1.0 equivalent) was added to a solution of the side chain 5 (1.1 equivalent) and phosphine reagent (1.1 equivalent) in anhydrous THF under N2 atmosphere at 0 °C, the resulting solution was treated with di-p-nitrobenzyl azocarboxylate (DNAD) (1.1 equivalent) dropwise and the reaction mixture was continued at room temperature for 8 h, then the solvent was evaporated and the residue dissolved in cyclohexane. The triphenylphosphane oxide precipitated and was filtered off and then the filtrate evaporated under reduced pressure. The product was purified by a column chromatography on silica gel to obtain the pure products as a white solid. mp>280 °C (dec); 1H NMR (500 MHz, CDCl3): δ=8.36 (s, 1H, CH), 4.02 (t, 2H, J=7.23 Hz, CH2), 3.79 (dd, 2H, Heq. J 1=11.57 Hz, J 2=4.46 Hz, CH2), 3.56 (dd, 2H, Hax. J 1=11.57 Hz, J 2=8.77 Hz, CH2), 1.67 (q, 2H, J=7.22 Hz, CH2), 1.53–1.61 (m, 1H, CH), 1.47 (s, 18H, C(CH3)3), 1.39 (s, 3H, CH3), 1.36 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ=154.3, 151.7, 151.5, 151.1, 128.0, 104.8, 81.7, 71.5, 50.8, 33.7, 28.6, 26.2, 25.7.
2.5. 2-amino-6-chloro-9-[2-(2,2-dimethyl-1,3-dioxan-5-yl) ethyl]purine (8a)
A mixture of compound 12 (2.56 g, 5.0 mmol), 2,6-dimethyl pyridine (1.18 ml, 10 mmol) and dry DCM (20 ml) was stirred at 0 °C, then TBTMS-OTf was added dropwise; after the addition, the reaction mixture was stirred at room temperature until TLC showed that compound 12 had completely disappeared. Then 30 ml saturated ammonium chloride solution was added, separated the organic layer, extracted with DCM (2×20 ml), combined and washed by saturated NaCl (2×40 ml), dried with anhydrous sodium sulfate and evaporated to give a white solid (1.21 g, 78%). mp 125–126 °C; 1H NMR (500 MHz, CDCl3): δ=8.07 (s, 1H, CH) , 6.99 (s, 2H, NH2), 4.12 (t, 2H, J=7.31 Hz, CH2), 3.82 (dd, 2H, 4′-Heq, J 1=11.79 Hz, J 2=4.50 Hz, CH2), 3.53 (dd, 2H, 4′-Hax, J 1=11.79 Hz, J 2=8.80 Hz, CH2), 1.74 (q, 2H, J=7.30 Hz, CH2), 1.53–1.65 (m, 1H, CH), 1.36 (s, 3H, CH3), 1.31 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ=159.94, 150.31, 150.26, 141.84, 132.11, 100.52, 68.14, 52.90, 31.32, 26.84, 26.05.
2.6. 9-[4-hydroxy-3-(hydroxymethyl)butyl] guanine (PCV 3)
Compound 12 (5.12 g, 10 mmol) was dissolved in THF (20 ml) hydrochloric acid (2 mol/L, 20 ml). The mixture was stirred for 2 h at 70 °C, and then slowly warmed to reflux for 2 h. After evaporation of the THF under reduced vacuum, 10% aqueous NaOH solution was added to neutralize the residual liquid, and a large amount of off-white solid formed, filtered, washed with acetone and then water, and dried under vacuum to give an off-white solid 3 (2.07 g, 82%). mp 274.6–276.9 °C.
Table 1
Mole fraction solubility x of bis-Boc-2-amino-6-chloropurine 9 in different Mitsunobu solvents
| T (K) (±0.05 K) | Solubility x a (%)
|
|||
| THFb | DCMb | Methylbenzeneb | Acetonitrileb | |
| 273.15 | 0.1141 | 0.0493 | 0.0213 | 0.0150 |
| 278.15 | 0.1191 | 0.0552 | 0.0253 | 0.0178 |
| 283.15 | 0.1251 | 0.0613 | 0.0303 | 0.0210 |
| 288.15 | 0.1299 | 0.0664 | 0.0349 | 0.0244 |
| 293.15 | 0.1352 | 0.0734 | 0.0405 | 0.0288 |
| 298.15 | 0.1399 | 0.0809 | 0.0470 | 0.0347 |
| 303.15 | 0.1463 | 0.0894 | 0.0544 | 0.0417 |
| 308.15 | 0.1523 | 0.0983 | 0.0634 | 0.0501 |
| 313.15 | 0.1581 | 0.1081 | 0.0734 | 0.0617 |
- a: the solubility of bis-Boc-2-amino-6-chloropurine 9 was measured by our previous method with temperature ranging from 273.15 K to 313.15 K (Wang et al., 2008) at atmospheric pressure. The laser monitoring observation technique was used to determine the disappearance of the solid phase in a solid and liquid mixture
b: all the solvents were further purified by distillation in dry agent (Na/benzophenone) and the sample bis-boc-2-amino-6-chloropurine 9 was dried in vacuum for over 2 d
As shown in Table 1, THF, which is the most common solvent in Mitsunobu reaction, has great solubility for bis-Boc-2-amino-6-chloropurine 9. Afterwards, the best solvent THF was taken for coupling 9 with a number of alcohols under normal Mitsunobu conditions to investigate its reactivity. The results were illustrated in Table 2. We clearly learned that bis-Boc-2-amino-6-chloropurine 9, as an excellent nucleophilic precursor, was able to react with a large number of alcohols, including primary alcohol, secondary alcohol, allyl alcohol, benzyl alcohol, etc., with high N9 selectivity and yields. Moreover, tert-Butyl alcohol still could not react with a protected purine as in the previous study (Yang et al., 2011), owing to its steric hindrance in tertiary carbon.
Table 2
Investigation of the reactivity of bis-Boc-2-amino-6-chloropurine 9 with different alcohols
| Entry | Alcohol | Product | Isolated yield (%) |
| 1 | 10a | 90.2 | |
| 2 | 10b | 86.6 | |
| 3 | 10c | 83.3 | |
| 4 | 10d | 84.8 | |
| 5 | 10e | 86.4 | |
| 6 | 10f | 81.2 | |
| 7 | 10g | 81.5 | |
| 8 | 10h | 80.7 | |
| 9 | 10i | 0 |
- a): a mixture of 9 (1.0 equivalent), alcohol (1.1 equivalent) and phosphine reagent (1.1 equivalent) in anhydrous THF stirring under N2 atmosphere at 0 °C, then treated with azo-reagent DNAD (1.1 equivalent) warmed to room temperature; b): the mixture of the products from procedure a, THF (20 ml) and aqueous hydrochloric acid (2 mol/L, 20 ml) was refluxed for 2 h at 70 °C
- According to the research results above, it is more reasonable and assuring to prepare PCV via a Mitsunobu reaction. This novel method for the preparation of PCV is indicated in Fig. 4. First, the side chain of 5-(2-hydroxyethyl)-2,2-dimethyl-1,3 -dioxane 5 was achieved through the commercially available starting material 2-hydroxymethyl-1,4-butanediol 11 reacting with 2,2-dimethoxypropane catalyzed by p-toluenesulfonic acid. The free –OH group of compound 5 is not necessary to be converted to the other leaving group such as chlorine, tosylate or methanesulphonate, which is always taken as a necessary step in the previous method or many other previous studies for the preparation of PVC till now (Harnden and Jarvest, 1985; Harnden et al., 1987; Zheng et al., 2004), making the synthesis of the side chain part of our method much more convenient and practical.

Synthesis of penciclovir (PCV) with new method
a: 2,2-dimethoxypropane, p-toluenesulfonic acid, THF; b: 1.1 equivalent of the side chain 5, 1.1 equivalent of PPh3, and 1.1 equivalent of azodicarboxylate reagent at rt. in THF; c: TBDMS-OTf, DCM; d: aqueous hydrochloric acid (2 mol/L), THF; e: aqueous hydrochloric acid (2 mol/L)
Our next objective was the synthesis of PCV. As was expected, bis-Boc-2-amino-6-chloropurine 9 combined with the side chain 5(1.1 equivalent) under normal Mitsunobu conditions successfully obtained the desired N9-alkylated compound 12 in 92% yield without the undesired N7 alkylation by-product being formed. Importantly, the reaction conditions were significantly milder than those reported in recent studies (Geen et al., 1990; 1992; Kim et al., 1998; Brand et al., 1999; Toyokuni et al., 2003), requiring only 1.1 equivalent of each of the alcohol, PPh3 and DNAD, and proceeding to completion within 60 min at room temperature. This is mainly due to the enhanced solubility of the compound 9 as mentioned above. By process c in Fig. 4, compound 8a was obtained under neutral conditions. It is 1H and 13C NMR spectra further indicated that no 7-isomer purine (8b) was formed. Subsequently, we could obtain PCV 3 in an acid condition as procedure e; or directly starting from 12, where hydrolytic dechlorination and deprotection step(s) were accomplished in one pot under mild acid conditions (2mol/L, hydrochloric acid in THF at room temperature) to afford the target PCV 3 in 80%–85% yield (process d). The overall yield of PCV from 11 was 44.5% higher than that in previous study (16%) (Zheng et al., 2004).
References
 [1] Ashton, W.T., Karkas, J.D., Field, A.K., Tolman, R.L., 1982. Activation by thymidine kinase and potent antiherpetic activity of 2’-nor-2’-deoxyguanosine (2’NDG). Biochemical and Biophysical Research Communications, 108(4):1716-1721.[2] Brand, B., Reese, C.B., Song, Q., Visintin, C., 1999. Convenient syntheses of 9-[4-hydroxy-3-(hydroxymethyl)butyl] guanine (penciclovir) and 9-[4-acetoxy-3-(acetoxymethyl) butyl]-2-amino-9H-purine (famciclovir). Tetrahedron, 55(16):5239-5252.[3] De Clercq, E., 1991. Broad-spectrum anti-DNA virus and anti-retrovirus activity of phosphonylmethoxyalkylpurines and pyrimidines. Biochemical Pharmacology, 42(5):963-972.[4] Dey, S., Garner, P., 2000. Synthesis of tert-butoxycarbonyl (Boc)-protected purines. The Journal of Organic Chemistry, 65(22):7697-7699.[5] Geen, G.R., Grinter, T.J., Kincey, P.M., Jarvest, R.L., 1990. The effect of the C-6 substituent on the regioselectivity of N-alkylation of 2-aminopurines. Tetrahedron, 46(19):6903-6914.[6] Geen, G.R., Kincey, P.M., Choudary, B.M., 1992. Regiospecific Michael additions with 2-aminopurines. Tetrahedron Letters, 33(32):4609-4612.[7] Harnden, M.R., Jarvest, R.L., 1985. An improved synthesis of the antiviral acyclonucleoside 9-(4-hydroxy-3-hydroxymethylbut-1-yl) guanine. Tetrahedron Letters, 26(35):4265-4268.[8] Harnden, M.R., Jarvest, R.L., Bacon, T.H., Boyd, M.R., 1987. Synthesis and antiviral activity of 9-[4-hydroxy-3-(hydroxymethyl) but-1-yl] purines. Journal of Medicinal Chemistry, 30(9):1636-1642.[9] Hwu, J.R., Jain, M.L., Tsay, S.C., Hakimelahi, G.H., 1996. Ceric ammonium nitrate in the deprotection of tert-butoxycarbonyl group. Tetrahedron Letters, 37(12):2035-2038.[10] Kim, D.K., Lee, N., Kim, Y.W., Chang, K.Y., Kim, J.S., Im, G.J., Choi, W.S., Jung, I.H., Kim, T.S., Hwang, Y.Y., 1998. Synthesis and evaluation of 2-amino-9-(3-hydroxymethyl-4-alkoxycarbonylo-xybut-1-yl) purines as potential prodrugs of penciclovir. Journal of Medicinal Chemistry, 41(18):3435-3441.[11] Kitade, Y., Ando, T., Yamaguchi, T., Hori, A., Nakanishi, M., Ueno, Y., 2006. 4’-fluorinated carbocyclic nucleosides: synthesis and inhibitory activity against S-adenosyl-l-homocysteine hydrolase. Bioorganic & Medicinal Chemistry, 14(16):5578-5583.[12] Korba, B.E., Boyd, M.R., 1996. Penciclovir is a selective inhibitor of hepatitis B virus replication in cultured human hepatoblastoma cells. Antimicrobial Agents and Chemotherapy, 40(13):1282-1284.[13] Lu, W., Sengupta, S., Petersen, J.L., Akhmedov, N.G., Shi, X., 2007. Mitsunobu coupling of nucleobases and alcohols: an efficient, practical synthesis for novel nonsugar carbon nucleosides. Journal of Organic Chemistry, 72(13):5012-5015.[14] Martin, J.C., Dvorak, C.A., Smee, D.F., Matthews, T.R., Verheyden, J.P.H., 1983. 9-(1,3-dihydroxy-2-propoxymethyl) guanine: a new potent and selective antiherpes agent. Journal of Medicinal Chemistry, 26(5):759-761.[15] Mitsunobu, O., 1981. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis, 1981(1):1-28.[16] Ogilvie, K.K., Cheriyan, U.O., Radatus, B.K., Smith, K.O., Galloway, K.S., Kennell, W.L., 1982. Biologically active acyclonucleoside analogues. II. The synthesis of 9-[[2-hydroxy-1-(hydroxymethyl)ethoxy]methyl] guanine (BIOLF-62). Canadian Journal of Chemistry, 60(24):3005-3010.[17] Porcheddu, A., Giacomelli, G., Piredda, I., Carta, M., Nieddu, G., 2008. A Practical and efficient approach to PNA monomers compatible with Fmoc-mediated solid-phase synthesis protocols. European Journal of Organic Chemistry, 2008(34):5786-5797.[18] Schaeffer, H.J., Beauchamp, L., Miranda, P.D., Elion, G.B., Bauer, D.J., Collins, P., 1978. 9-(2-hydroxyethoxymethyl) guanine activity against viruses of the herpes group. Nature, 272(5654):583-585.[19] Shaw, T., Amor, P., Civitico, G., Boyd, M., Locarnini, S., 1994. In vitro antiviral activity of penciclovir, a novel purine nucleoside, against duck hepatitis B virus. Antimicrobial Agents and Chemotherapy, 38(4):719-723.[20] Smith, K.O., Galloway, K.S., Kennell, W.L., Ogilvie, K.K., Radatus, B.K., 1982. A new nucleoside analog, 9-[[2-hydroxy-1-(hydroxymethyl)ethoxyl]methyl] guanine, highly active in vitro against herpes simplex virus types 1 and 2. Antimicrobial Agents and Chemotherapy, 22(1):55-61.[21] Tippie, M.A., Martin, J.C., Smee, D.F., Matthews, T.R., Verheyden, J.P.M., 1984. Antiherpes simplex virus activity of 9-[4-hydroxy-3-(hydroxymethyl)-1-butyl] guanine. Nucleosides and Nucleotides, 3(5):525-535.[22] Toyokuni, T., Walsh, J.C., Namavari, M., Shinde, S.S., Moore, J.R., Barrio, J.R., Satyamurthy, N., 2003. Selective and practical synthesis of penciclovir. Synthetic Communications, 33(22):3897-3905.[23] Sikchi, S.A., Hultin, P.G., 2006. Solventless protocol for efficient Bis-N-Boc protection of adenosine, cytidine, and guanosine derivatives. Journal of Organic Chemistry, 71(16):5888-5891.[24] Siro, J.G., Martin, J., Garcia-Navio, J.L., Remuinan, M.J., Vaquero, J.J., 1998. Easy microwave assisted deprotection of N-Boc derivatives. Synlett, 1998(2):147-148.[25] Swamy, K.C.K., Kumar, N.N.B., Balaraman, E., Kumar, K.V.P.P., 2009. Mitsunobu and related reactions: advances and applications. Chemical Reviews, 109(6):2551-2651.[26] Wang, L., Dai, L.Y., Lei, M., Chen, Y., 2008. Solubility of hexamethylenetetramine in a pure water, methanol, acetic acid, and ethanol+water mixture from (299.38 to 340.35) K. Journal of Chemical & Engineering Data, 53(12):2907-2909.[27] Yang, J., Dai, L., Wang, X., Chen, Y., 2011. Di-p-nitrobenzyl azodicarboxylate (DNAD): an alternative azo-reagent for the Mitsunobu reaction. Tetrahedron, 67(7):1456-1462.[28] Yang, M.M., Schneller, S.W., Korba, B., 2005. 5’-homoneplanocin a inhibits hepatitis B and hepatitis C. Journal of Medicinal Chemistry, 48(15):5043-5046.[29] Yin, X.Q., Li, W.K., Schneller, S.W., 2006. An efficient Mitsunobu coupling to adenine-derived carbocyclic nucleosides. Tetrahedron Letters, 47(52):9187-9189.[30] Zheng, Q.H., Wang, J.Q., Liu, X., Fei, X.S., Mock, B.H., Glick-Wilson, B.E., Sullivan, M.L., Raikwar, S.P., Gardner, T.A., Kao, C.H., 2004. An improved total synthesis of PET HSV-tk gene reporter probe 9-(4-[18F] fluoro-3-hydroxymethylbutyl) guanine ([18F]FHBG). Synthetic Communications, 34(4):689-704.
[1] Ashton, W.T., Karkas, J.D., Field, A.K., Tolman, R.L., 1982. Activation by thymidine kinase and potent antiherpetic activity of 2’-nor-2’-deoxyguanosine (2’NDG). Biochemical and Biophysical Research Communications, 108(4):1716-1721.[2] Brand, B., Reese, C.B., Song, Q., Visintin, C., 1999. Convenient syntheses of 9-[4-hydroxy-3-(hydroxymethyl)butyl] guanine (penciclovir) and 9-[4-acetoxy-3-(acetoxymethyl) butyl]-2-amino-9H-purine (famciclovir). Tetrahedron, 55(16):5239-5252.[3] De Clercq, E., 1991. Broad-spectrum anti-DNA virus and anti-retrovirus activity of phosphonylmethoxyalkylpurines and pyrimidines. Biochemical Pharmacology, 42(5):963-972.[4] Dey, S., Garner, P., 2000. Synthesis of tert-butoxycarbonyl (Boc)-protected purines. The Journal of Organic Chemistry, 65(22):7697-7699.[5] Geen, G.R., Grinter, T.J., Kincey, P.M., Jarvest, R.L., 1990. The effect of the C-6 substituent on the regioselectivity of N-alkylation of 2-aminopurines. Tetrahedron, 46(19):6903-6914.[6] Geen, G.R., Kincey, P.M., Choudary, B.M., 1992. Regiospecific Michael additions with 2-aminopurines. Tetrahedron Letters, 33(32):4609-4612.[7] Harnden, M.R., Jarvest, R.L., 1985. An improved synthesis of the antiviral acyclonucleoside 9-(4-hydroxy-3-hydroxymethylbut-1-yl) guanine. Tetrahedron Letters, 26(35):4265-4268.[8] Harnden, M.R., Jarvest, R.L., Bacon, T.H., Boyd, M.R., 1987. Synthesis and antiviral activity of 9-[4-hydroxy-3-(hydroxymethyl) but-1-yl] purines. Journal of Medicinal Chemistry, 30(9):1636-1642.[9] Hwu, J.R., Jain, M.L., Tsay, S.C., Hakimelahi, G.H., 1996. Ceric ammonium nitrate in the deprotection of tert-butoxycarbonyl group. Tetrahedron Letters, 37(12):2035-2038.[10] Kim, D.K., Lee, N., Kim, Y.W., Chang, K.Y., Kim, J.S., Im, G.J., Choi, W.S., Jung, I.H., Kim, T.S., Hwang, Y.Y., 1998. Synthesis and evaluation of 2-amino-9-(3-hydroxymethyl-4-alkoxycarbonylo-xybut-1-yl) purines as potential prodrugs of penciclovir. Journal of Medicinal Chemistry, 41(18):3435-3441.[11] Kitade, Y., Ando, T., Yamaguchi, T., Hori, A., Nakanishi, M., Ueno, Y., 2006. 4’-fluorinated carbocyclic nucleosides: synthesis and inhibitory activity against S-adenosyl-l-homocysteine hydrolase. Bioorganic & Medicinal Chemistry, 14(16):5578-5583.[12] Korba, B.E., Boyd, M.R., 1996. Penciclovir is a selective inhibitor of hepatitis B virus replication in cultured human hepatoblastoma cells. Antimicrobial Agents and Chemotherapy, 40(13):1282-1284.[13] Lu, W., Sengupta, S., Petersen, J.L., Akhmedov, N.G., Shi, X., 2007. Mitsunobu coupling of nucleobases and alcohols: an efficient, practical synthesis for novel nonsugar carbon nucleosides. Journal of Organic Chemistry, 72(13):5012-5015.[14] Martin, J.C., Dvorak, C.A., Smee, D.F., Matthews, T.R., Verheyden, J.P.H., 1983. 9-(1,3-dihydroxy-2-propoxymethyl) guanine: a new potent and selective antiherpes agent. Journal of Medicinal Chemistry, 26(5):759-761.[15] Mitsunobu, O., 1981. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis, 1981(1):1-28.[16] Ogilvie, K.K., Cheriyan, U.O., Radatus, B.K., Smith, K.O., Galloway, K.S., Kennell, W.L., 1982. Biologically active acyclonucleoside analogues. II. The synthesis of 9-[[2-hydroxy-1-(hydroxymethyl)ethoxy]methyl] guanine (BIOLF-62). Canadian Journal of Chemistry, 60(24):3005-3010.[17] Porcheddu, A., Giacomelli, G., Piredda, I., Carta, M., Nieddu, G., 2008. A Practical and efficient approach to PNA monomers compatible with Fmoc-mediated solid-phase synthesis protocols. European Journal of Organic Chemistry, 2008(34):5786-5797.[18] Schaeffer, H.J., Beauchamp, L., Miranda, P.D., Elion, G.B., Bauer, D.J., Collins, P., 1978. 9-(2-hydroxyethoxymethyl) guanine activity against viruses of the herpes group. Nature, 272(5654):583-585.[19] Shaw, T., Amor, P., Civitico, G., Boyd, M., Locarnini, S., 1994. In vitro antiviral activity of penciclovir, a novel purine nucleoside, against duck hepatitis B virus. Antimicrobial Agents and Chemotherapy, 38(4):719-723.[20] Smith, K.O., Galloway, K.S., Kennell, W.L., Ogilvie, K.K., Radatus, B.K., 1982. A new nucleoside analog, 9-[[2-hydroxy-1-(hydroxymethyl)ethoxyl]methyl] guanine, highly active in vitro against herpes simplex virus types 1 and 2. Antimicrobial Agents and Chemotherapy, 22(1):55-61.[21] Tippie, M.A., Martin, J.C., Smee, D.F., Matthews, T.R., Verheyden, J.P.M., 1984. Antiherpes simplex virus activity of 9-[4-hydroxy-3-(hydroxymethyl)-1-butyl] guanine. Nucleosides and Nucleotides, 3(5):525-535.[22] Toyokuni, T., Walsh, J.C., Namavari, M., Shinde, S.S., Moore, J.R., Barrio, J.R., Satyamurthy, N., 2003. Selective and practical synthesis of penciclovir. Synthetic Communications, 33(22):3897-3905.[23] Sikchi, S.A., Hultin, P.G., 2006. Solventless protocol for efficient Bis-N-Boc protection of adenosine, cytidine, and guanosine derivatives. Journal of Organic Chemistry, 71(16):5888-5891.[24] Siro, J.G., Martin, J., Garcia-Navio, J.L., Remuinan, M.J., Vaquero, J.J., 1998. Easy microwave assisted deprotection of N-Boc derivatives. Synlett, 1998(2):147-148.[25] Swamy, K.C.K., Kumar, N.N.B., Balaraman, E., Kumar, K.V.P.P., 2009. Mitsunobu and related reactions: advances and applications. Chemical Reviews, 109(6):2551-2651.[26] Wang, L., Dai, L.Y., Lei, M., Chen, Y., 2008. Solubility of hexamethylenetetramine in a pure water, methanol, acetic acid, and ethanol+water mixture from (299.38 to 340.35) K. Journal of Chemical & Engineering Data, 53(12):2907-2909.[27] Yang, J., Dai, L., Wang, X., Chen, Y., 2011. Di-p-nitrobenzyl azodicarboxylate (DNAD): an alternative azo-reagent for the Mitsunobu reaction. Tetrahedron, 67(7):1456-1462.[28] Yang, M.M., Schneller, S.W., Korba, B., 2005. 5’-homoneplanocin a inhibits hepatitis B and hepatitis C. Journal of Medicinal Chemistry, 48(15):5043-5046.[29] Yin, X.Q., Li, W.K., Schneller, S.W., 2006. An efficient Mitsunobu coupling to adenine-derived carbocyclic nucleosides. Tetrahedron Letters, 47(52):9187-9189.[30] Zheng, Q.H., Wang, J.Q., Liu, X., Fei, X.S., Mock, B.H., Glick-Wilson, B.E., Sullivan, M.L., Raikwar, S.P., Gardner, T.A., Kao, C.H., 2004. An improved total synthesis of PET HSV-tk gene reporter probe 9-(4-[18F] fluoro-3-hydroxymethylbutyl) guanine ([18F]FHBG). Synthetic Communications, 34(4):689-704.
SYN
EP 0141927; ES 8602791; ES 8603887; ES 8603888; JP 1994293764; US 5075445
This compound has been obtained by two similar ways: 1) The reaction of 6-chloropurine-2-amine (I) with 6,6-dimethyl-5,7-dioxaspiro[2.5]octane-4,8-dione (II) by means of K2CO3 in DMF gives the expected condensation product (III), which is methanolized with HCl/methanol yielding 2-[2-(2-amino-6-methoxypurin-9-yl)ethyl]malonic acid dimethyl ester (IV). The reduction of (IV) with NaBH4 in tert-butanol/methanol affords the corresponding diol (V), which is finally converted into pecnciclovir by hydrolysis with 2N NaOH. 2) The reaction of purine (I) with 3-bromopropane-1,1,1-tricarboxylic acid triethyl ester (VI) by means ofK2CO3 in DMF gives the expected condensation product (VII), which is partially decarboxylated with sodium methoxide in methanol yielding 2-[2-(2-amino-6-chloropurin-9-yl)ethyl]malonic acid diethyl ester (VIII). The reduction of (VIII) with NaBH4 in tert-butanol/methanol followed by acetylation with acetic anhydride affords the corresponding diol diacetate (IX), which is finally converted into penciclovir by hydrlysis with 2N HCl.
References
- Jump up^ “Penciclovir”. Merriam-Webster Dictionary. Retrieved 2016-01-22.
- Jump up^ Long, Sarah S.; Pickering, Larry K.; Prober, Charles G. (2012). Principles and Practice of Pediatric Infectious Disease. Elsevier Health Sciences. p. 1502. ISBN 1437727026.
- Jump up^ Farmaceutiska Specialiteter i Sverige – the Swedish official drug catalog. [http://www.fass.se Fass.se –> Vectavir. Retrieved on August 12, 2009. Translated from “Tiden för läkning, smärta och påvisbart virus förkortas med upp till ett dygn.”
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ˌpɛnˈsaɪkloʊˌvɪər/[1] |
| Trade names | Denavir |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a697027 |
| Pregnancy category |
|
| Routes of administration |
Topical |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 1.5% (oral), negligible (topical) |
| Protein binding | <20% |
| Metabolism | Viral thymidine kinase |
| Elimination half-life | 2.2–2.3 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.189.687 |
| Chemical and physical data | |
| Formula | C10H15N5O3 |
| Molar mass | 253.258 g/mol |
| 3D model (JSmol) | |
/////////////Penciclovir, BRL-39123, BRL 39123A, penciclovir sodium, Denavir, Vectavir, Euraxvir, Fenivir,
C1=NC2=C(N1CCC(CO)CO)NC(=NC2=O)N
