PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Mazisotine (also known as LY3556050 or CNTX-0290) is an experimental, non-opioid chemical compound designed to treat chronic and neuropathic pain. It functions as a selective somatostatin receptor 4 (SSTR4) agonist, meaning it activates specific peripheral nerve pathways to block pain signals without activating the central nervous system’s opioid receptors.
While it showed early promise in animal models, Eli Lilly and Company removed mazisotine from its clinical development pipeline after disappointing results in Phase II clinical trials. It currently remains in use strictly as a chemical tool for laboratory pain research.
Key Facts and Clinical History
Mechanism of Action: It binds to and activates SSTR4. This triggers a cellular response that suppresses pain and inflammation in peripheral sensory neurons.
Intended Indications: It was being evaluated to treat diabetic peripheral neuropathic pain, osteoarthritis pain, and chronic low back pain.
Development Partners: The compound was originally licensed by Eli Lilly from Centrexion Therapeutics in 2019 for an upfront fee of $47.5 million.
Discontinuation: In mid-2025, Eli Lilly officially dropped the drug. Phase II clinical trials revealed that its efficacy did not meet the high success thresholds required to continue human testing.
Side Effects: In clinical studies, reported treatment-emergent adverse effects were generally mild to moderate. They included constipation, nausea, dizziness, fatigue, and headaches.
Mazisotine (LY3556050, CNTX-0290) is a chemical compound which acts as an agonist at somatostatin receptor 4. It has analgesic effects and has been researched for the treatment of pain associated with arthritis and neuropathic pain. It was not pursued for human medical use following disappointing results in Phase II clinical trials, but continues to be used in research into the role of SST4 receptors in pain perception.[1][2]
A Study of LY3556050 in Adult Participants With Diabetic Peripheral Neuropathic PainCTID: NCT06074562Phase: Phase 2Status: CompletedDate: 2025-07-03
Chronic Pain Master Protocol (CPMP): A Study of LY3556050 in Participants With OsteoarthritisCTID: NCT04627038Phase: Phase 2Status: CompletedDate: 2023-11-02
Chronic Pain Master Protocol (CPMP): A Study of LY3556050 in Participants With Diabetic Peripheral Neuropathic PainCTID: NCT04707157Phase: Phase 2Status: TerminatedDate: 2023-11-02
Chronic Pain Master Protocol (CPMP): A Study of LY3556050 in Participants With Chronic Low Back PainCTID: NCT04874636Phase: Phase 2Status: CompletedDate: 2023-11-02
A Study of Effect of LY3556050 on Metformin in Healthy ParticipantsCTID: NCT05615467Phase: Phase 1Status: CompletedDate: 2023-01-18
A Study of Single and Repeated Doses of LY3556050 in Healthy ParticipantsCTID: NCT05341102Phase: Phase 1Status: CompletedDate: 2022-04-22
A Study of LY3556050 in Healthy ParticipantsCTID: NCT04156750Phase: Phase 1Status: CompletedDate: 2020-08-19
[0082] Methyl 4-bromobut-2-enoate (36.29g, 202.7mmol) was dissolved in MeCN (500 mL) at 15-25 ºC. tert-Butyl tosylcarbamate (50.00 g, 184.3 mmol) was added at 15-25 ºC. K2CO3 (30.57 g, 221.2mmol) and KI (3.06 g, 202.7 mmol) were added to the solution at 15-25 ºC, and warmed under nitrogen at 30 ºC for 20 hrs. The solution was cooled to 20 ºC and the mixture filtered. The filtered residue was washed with MeCN (100 mL) to give methyl (E)-4-((N-(tert-butoxycarbonyl)-4-methylphenyl)sulfonamido)but-2-enoate. TFA (101.03 g, 886.06 mmol) was added to the methyl (E)-4-((N-(tert-butoxycarbonyl)-4-methylphenyl)sulfonamido)but-2-enoate in MeCN solution (483.72 g, 143 mmol) and heated to 55-60 ºC for 16 hrs. The reaction solution was concentrated in vacuo to ~50 mL and solvent exchanged with toluene (2 x 250 mL). Toluene (500 mL) was added followed by EtOAc (50 mL) at 15-25 °C, and heated to 60 ºC for 1 hr., then cooled to 0 ºC for 12 hrs. The solution was filtered, and the wet cake was rinsed with n-heptane (50 mL). The cake was dried in vacuum at 50 ºC to give the title compound (37.85 g, 74.4%) as a white solid.1H NMR (CDCl3) δ 7.68 (d, J = 8.0Hz, 2H) 7.25 (d, J = 8.0Hz, 2H) 6.71 (dt, J = 15.6, 5.2Hz, 1H) 5.88 (dt, J = 15.6, 1.6Hz, 1H) 4.55 (t, J = 6.4Hz, 1H) 3.71 – 3.67 (m, 2H) 3.65 (s, 3H) 2.37 (s, 3H); HRMS (ESI+) Calculated for [C12H15NO4S+H] +: 270.0795, Found: 270.0788 (M+H).
0084] Methyl (E)-4-((4- 2-enoate (51.90 g) was dissolved in 2-MeTHF (600mL) at 0 ºC . Added (2-bromoethyl)diphenylsulfonium triflate (36.50 g), KF (6.47 g), KOH (18.75 g) to the solution at 0 ºC. Warmed the solution to 15 ºC for 22 hrs. then 30 ºC for 3 hrs. Added water (100 mL) and MeOH (100 mL) into the solution. Added LiOH.H2O (4.77 g) and stirred at 30 ºC for 16 hrs. Cooled to 15-25oC and added n-heptane (100 mL). Stirred at 15-25oC for 10 min. Separated and collected the aqueous phase and washed the aqueous phase with n-heptane/2-MeTHF (50 mL/200 mL × 2). Concentrated the aqueous phase in vacuo to ~50 mL and added 3M aq. HCl
dropwise to adjust pH to 1~2. Stirred the mixture at 20-30 ºC for 2 hrs. Filtered the solution and rinsed through with EtOH/H2O (15 mL1:4). Dried the wet cake at 45 ºC for 8-10 hrs. to give the title compound as white solid (20.37 g, 65%) 1H NMR (CDCl3) δ 7.67 (d, J = 8.2 Hz, 2 H) 7.34 (d, J = 8.2 Hz, 2 H) 3.63 (d, J = 9.4 Hz, 2 H) 3.12 (d, J = 9.4 Hz, 2 H) 2.46 – 2.40 (m, 4 H) 2.07 – 2.01 (m, 2 H); HRMS (ESI+) Calcd. for [C13H15NO4S+H] +: 282.0795, Found: 282.0795 (M+H).
0086] 2-Amino-2-methylpropan- mmol) was dissolved in toluene (250 mL) at 15-25 ºC. Added potassium tert-butoxide (60.59 g, 534.6 mmol) to the solution and warmed to 30-40 ºC. Added 2-fluoro-3-methylpyridine (50.00 g, 450.0 mmol) to the solution and warmed to 80 ºC. Stirred the mixture for 18 hrs. at 80 ºC. Cooled the mixture to 15-25 ºC, added water (100 mL) and stirred for 30 min. Separated the aqueous layer and washed the organics with 10% aq. NaCl (300 mL x 3). Added toluene (250 mL) to the organics and concentrated in vacuo to give the title compound as a liquid (107.50 g, 98%). 1H NMR (CDCl3) δ 7.92 – 7.82 (m, 1H) 7.32 – 7.22 (m, 1H) 6.74 – 6.66 (m, 1H) 3.97 (s, 3H) 2.14 (s, 3H) 1.15 (s, 6H).
[0088] Dissolved 6-carboxylic acid (32.33 g, 88.2% purity) in toluene (320 mL) at 15~25 ºC. Added DMF (741.0 mg) and (COCl)2 (19.20 g) to the solution and heated to 45-55 ºC for 3~4 hrs. Concentrated the mixture in vacuo and exchanged with THF (100 mL × 2). Added THF (320 mL) and cooled to 0-10 ºC. Added 2-methyl-1-((3-methylpyridin-2-yl)oxy)propan-2-amine (23.60 g), TEA (30.80 g), DMAP (620.0 mg) at 0-10 ºC then warmed to 15-25 ºC for 2~4 hrs.
Concentrated the mixture in vacuo and solvent exchanged with EtOH (140 mL × 2). Concentrated in vacuo to 140 mL and heated to 50 ºC until the solid was dissolved. Added water (210 mL) dropwise into the solution at 40 ºC. Cooled the solution to 10 ºC for 14 hrs. Filtered and rinsed with EtOH/H2O (75 mL, 1:1.5). Dried the wet cake at 45 ºC for 20 hrs. to give the title compound as a solid (41.87 g, 87.4%).1H NMR (CDCl3) δ 7.94 – 7.87 (m, 1 H) 7.60 (d, J=8.0 Hz, 2 H) 7.37 (d, J=6.6 Hz, 1 H) 7.26 (d, J=8.0 Hz, 2 H) 6.78 (dd, J=6.6, 5.0 Hz, 1 H) 6.48 (s, 1 H) 4.25 (s, 2 H) 3.52 (d, J=9.4 Hz, 2 H) 2.95 (d, J=9.4 Hz, 2 H) 2.39 – 2.33 (m, 3 H) 2.17 (s, 3 H) 1.84 (s, 2 H) 1.39 (s, 6 H); HRMS (ESI+) Calcd. for [C23H29N3O4S+H] +: 444.1952, Found: 444.2089 (M+H).
[0090] Dissolved 2-yl)oxy)propan-2-yl)-3-tosyl-3-azabicyclo[3.1.0]hexane-6-carboxamide (5.00 g) in MTBE (50 mL) under nitrogen. Cooled to -70-60 ºC and added dropwise 1M Ph2PK in THF (56 mL, 56 mmol) into the solution. Stirred the mixture for 6-8 hrs. at -70-60 ºC. Added 2M aq. HCl (50 mL) to the solution allowing the temperature to rise to 15-25 ºC. Separated and collected the aqueous phase and washed with 2-MeTHF (50 mL × 3). Added 2-MeTHF (50 mL) and adjusted the pH to 8~9 with K2CO3 powder. Separated and extracted the aqueous phase with 2-MeTHF (50 mL). Combined the organics and concentrated in vacuo to give the title compound as a yellow-brown solid (3.05 g, 89.4%) 1H NMR (CDCl3) δ 8.01 – 7.92 (m, 1 H) 7.45 – 7.36 (m, 1 H) 6.81 (dd, J=7.0, 5.0 Hz, 1 H) 6.39 (s, 1 H) 4.31 (s, 2 H) 3.09 – 2.89 (m, 4 H) 2.21 (s, 3 H) 1.94 – 1.86 (m, 2 H) 1.69 (s, 1 H) 1.47 (s, 6 H) 1.12 (t, J=2.8 Hz, 1 H); HRMS (ESI+) Calcd. for [C16H23N3O2+H] +: 290.1863, Found:
Stevens R, Corradini L, Doods H (April 2019). “Preclinical Evaluation of Human Somatostatin Receptor 4 (hSSTR4) Agonist CNTX-0290 for Mixed Pain Conditions”. The Journal of Pain. 20 (4): S73. doi:10.1016/j.jpain.2019.02.092.
Difluprednate is a topical corticosteroid used for the symptomatic treatment of inflammation and pain associated with ocular surgery.
Difluprednate is a corticosteroid, It is chemically a butyrate ester of 6(alpha),9(alpha)-difluoro prednisolone acetate. Accordingly, difluprednate is sometimes abbreviated DFBA, for difluoroprednisolone butyrate acetate.
Difluprednate is a topical corticosteroid indicated for the treatment of infammation and pain associated with ocular surgery. It is a butyrate ester of 6(α), 9(α)-difluoro prednisolone acetate. Difluprednate is abbreviated DFBA, or difluoroprednisolone butyrate acetate. It is indicated for treatment of endogenous anterior uveiti.
Approval
On June 24, 2008, the US Food and Drug Administration (FDA) approved difluprednate for the treatment of post-operative ocular inflammation and pain.[1] It is marketed by Alcon under the tradename Durezol.
WO/2022/118271DIFLUPREDNATE FOR REDUCING THE ADVERSE EFFECTS OF OCULAR INFLAMMATION
SYN 1
Synthetic Reference
Process for preparation of Difluprednate from sterol fermentation product; Ding, Kai; Xu, Feifei; Assignee Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Peop. Rep. China; East China University of Science and Technology; 2014; Patent Information; Aug 06, 2014; CN; 103965277; A
SYN 2
Synthetic Reference
Preparation method of Difluprednate; Tian, Yuan; Zhou, Shengan; Guo, Bin; Xu, Zhiguo; Assignee Guangzhou Renheng Pharmaceutical Technology Co., Ltd., Peop. Rep. China 2017; Patent Information; May 10, 2017; CN; 106632561; A
SYN3
Synthetic Reference
Shailesh, Singh; Bharat, Suthar; Jain, Ashish; Gaikwad, Vinod; Kulkarni, Kuldip. Process for preparing difluprednate. Assignee Ajanta Pharma Ltd., India. IN 2013MU02535. (2015).
SYN4
Synthetic Reference
Sun, Hongbin; Chen, Bo. Method for preparation of Difluprednate. Assignee China Pharmaceutical University, Peop. Rep. China. CN 103509075. (2014).
Embodiment 1:4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound)
10g hydrocortisone-21 acetic ester (formula II compound) is joined in 250mL eggplant type bottle, add 50mL N, dinethylformamide and 8.8mL pyridine, slowly heat up and make material dissolution complete, slowly cooling afterwards, slowly be added dropwise to 4.4mL methylsulfonyl chloride, add rear solution to be yellow completely.Be warming up to 85 ℃ of stirrings, the reaction solution thick one-tenth that can slowly become sticky is faint yellow, adds slightly some DMFs and makes reaction solution dilution, can normally stir, and keeps this thermotonus one hour, and reaction solution slowly becomes grey black during this period.TLC follows the tracks of (sherwood oil: ethyl acetate=1: 1) show that reaction finishes.Stop heating, treat that the backward reaction solution of slow cooling adds 200mL methyl alcohol, stir 1min, reaction flask is placed in to crystallization under ice-water bath.Suction filtration after 1h, makes water and methanol wash filter cake, crude product productive rate 100%.With methyl alcohol-methylene dichloride mixed solvent system recrystallization, obtain sterling, M.P.231-235 ℃, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.55(1H,s,11-H),5.07(1H,d,J=5Hz,21-H),4.84(1H,d,J=5Hz,21-H),2.15(3H,s,H-21-OAc),1.31(3H,s,19-CH 3),0.65(3H,s,18-CH 3),0.66-2.90(m,17H,backbone).
By 9.4g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound) and 10g4-Dimethylamino pyridine add in 1000mL eggplant-shape bottle, add again 50mL diethylene glycol dimethyl ether and 260mL methylene dichloride, heated and stirred makes dissolution of solid, slowly adds 32mL butyryl oxide slightly after cooling, is warming up to 80 ℃ of return stirrings.After 23h, TLC follows the tracks of, and raw material primitive reaction is complete, stops heating and stirs.Vacuum concentration is removed methylene dichloride.After being down to room temperature, add frozen water in reaction flask, white solid standing to be separated out.Suction filtration, saturated sodium bicarbonate aqueous solution washing leaching cake, dries under infrared lamp, obtain 4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-ketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound) sterling 10.65g, M.P220-224 ℃, productive rate 95.9%. 1H-NMR(500MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.54(1H,m,11-H),4.87(1H,d,J=4.8Hz,O=C-CH 2-O,21-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.75(2H,m,2-H),0.70(3H,s,18-CH 3),0.95(3H,t,J=4.4Hz),1.34(3H,s,18-CH 3),1.66(2H,m,-CH 2CH 3),2.17(3H,s,O=C-CH 3),2.32(2H,t,J=4.3Hz,O=C-CH 2),? 13C-NMR(75MHz,CDCl 3):δ(ppm)199.1,198.9,173.4,170.4,169.1,144.1,124.1,118.5,94.5,66.9,48.2,46.3,40.9,37.5,36.4,34.2,33.8,32.7,32.2,32.1,30.6,26.2,24.5,20.5,18.3,13.7,13.6;ESI-MS?m/z:457.2[M+H +],479.2[M+Na +];HRMS?for?C 27H 36O 6+Na +?calcd?479.2410,found479.2402.
10g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters add in 250mL eggplant type bottle, then add 80mL methylvinyl acetate, slowly drip while stirring the 1mL vitriol oil.Be warming up to 80 ℃ of stirring reactions, solution is thin out yellow clarification slowly.(sherwood oil: ethyl acetate=3: 1), raw material reaction is complete produces new point to TLC after 30min.Stop heating, wait to be cooled to 50 ℃, add 1mL triethylamine, be stirred to and be down to room temperature.Add water in reaction solution, ethyl acetate aqueous layer extracted three times, saturated common salt water washing organic phase twice, anhydrous sodium sulfate drying.After 30min, steam organic solvent and obtain brown color oily matter.Column chromatography is purified and is obtained 3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.74(1H,s,4-H),5.53(1H,s,11-H),5.45(1H,s,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.17(3H,s,19-CH 3),0.96(3H,t,J=7.5Hz),0.70(3H,s,18-CH 3).
14g4, 9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3, 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI) adds in dry three-necked bottle, add while stirring 400mL acetum, under room temperature, slowly pass into anhydrous hydrogen chloride gas (98% vitriol oil is added dropwise in 37% concentrated hydrochloric acid solution and makes) until saturated, be stirred to raw material and be dissolved into yellow solution completely, continue to stir 2h, TLC monitoring reacts completely, stop stirring, in reaction solution, add the aqueous solution, after separating out solid, suction filtration, saturated sodium bicarbonate aqueous solution washing, dry, be weighed as 13g, productive rate is 93%. 1H?NMR(300MHz,CDCl 3):δ(ppm)6.10(s,1H),5.61(s,1H),5.41-5.16(m,1H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.82(dd,J=28.3,15.7Hz,3H),2.50(s,2H),2.32(t,J=7.4Hz,2H),2.17(s,3H),1.96(s,5H),1.66(d,J=7.4Hz,2H),1.46(s,2H),1.33(s,3H),0.96(s,3H),0.71(s,3H).
13g 6 α-fluoro-4; 9; (11)-diene-pregnant steroid-3,20-22 ketone-17-butyric ester-20-acetic ester is dissolved in and fills 300mL1, in the eggplant type bottle of 4 dioxane; add while stirring 40mL 0.46mol/L high chloro acid solution; under room temperature, stir after several minutes, add 14g N-succinimide in reaction system, under nitrogen protection, stir; raw material dissolves gradually, and it is faint yellow that reaction solution is.(the sherwood oil: ethyl acetate=12: 5) monitoring, raw material primitive reaction is complete, adds 10%Na of TLC after 2h 2sO 3unnecessary N-succinimide is fallen in aqueous solution cancellation, and checks (it is blue that test paper no longer becomes) with starch-kalium iodide test paper.Add water in reaction flask, ethyl acetate extraction three times, twice of saturated common salt water washing organic phase, anhydrous sodium sulfate drying organic phase, after 30min, be spin-dried for organic phase, obtain faint yellow oily matter, column chromatography purification (sherwood oil: ethyl acetate=12: 1) obtain white solid 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3, the about 14g of 20-diketone-17-butyric ester-20-acetic ester, productive rate is 89%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.93(1H,d,J=4.5,4-H),5.06(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.84(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.02(3H,s,19-CH 3),4.72(1H,s,11-H);ESI-MS?m/z:593.3,595.3[M+Na +].
100mg 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in the Plastic Bottle of tetrafluoroethylene, adds 2mL methylene dichloride to dissolve, and stirs at-20 ℃.1mL Olah reagent with under 1mL methylene dichloride low temperature, mix after, be slowly added dropwise in reaction system, maintain low temperature and stir 2 hours, TLC monitoring reaction finishes.Reaction flask shifts out low-temp reaction groove, is slowly added dropwise to the 1mol/L NaOH aqueous solution by excessive HF cancellation, is adjusted to pH7~8.Add chloroform in reaction system, extraction, organic layer is used respectively aqueous hydrochloric acid and the saturated common salt water washing of 3mol/L, anhydrous sodium sulfate drying, after standing 30min, steams except organic solvent, column chromatography is further purified and is obtained white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)?6.11(1H,d,J=4.5Hz,4-H),5.27(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),4.40(1H,d,J=4.5Hz,11-H),1.02(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.52(3H,s,19-CH 3);ESI-MS?m/z:533.3[M+Na +]
40mg 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester is dissolved in 3mL dioxane, adds 28mgDDQ, and 100 ℃ of return stirrings heat up.TLC monitoring reaction (sherwood oil: ethyl acetate=12: 8) after 13h, generate the larger product of polarity, steam except organic solvent dioxane, obtain brown color oily matter, add a small amount of methylene dichloride lysate, suction filtration, elimination solid residue, filtrate is washed with sodium bicarbonate aqueous solution after adding a small amount of methylene dichloride again, steams except organic phase rear pillar Chromatographic purification, obtain white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, be title molecule difluprednate, productive rate 70%. 1h-NMR (300MHz, CDCl 3): δ (ppm) 7.20 (1H, d, J=4.5Hz, 1-H), 6.43 (1H, s, 4-H), 6.38 (1H, d, J=6Hz, 2-H), 5.36 (1H, m, 6-H), 4.64-4.91 (2H, ABq, J=16.6Hz, 21-H), 4.43 (1H, d, J=4.5Hz, 11-H), 2.27 (2H, m ,-CH 2-CH 3), 2.17 (3H, s, O=C-CH 3), 1.55 (3H, s, 19-CH 3), 1.02 (3H, s, 18-CH 3), 0.93 (3H, t, J=4.5Hz, 0=C-CH 2cH 2cH 3); ESI-MS m/z:509.3[M+H +]; HRMS for C 27h 35o 7f 2+ H +calcd 509.2351, found 509.2356.M.P.188-190 ℃ (literature value M.P.190-194 ℃); [α] d22=+30.1 ° of (literature values [α] d22=+31.7 °).
Claims (6)
Hide Dependent
1. a method of preparing difluprednate, as following reaction formula:
Specifically comprise the following steps:
(1) by hydrocortisone-21-acetic ester (formula II compound):
Carry out dehydration reaction, generate formula III compound:
(2) formula III compound is carried out to butyric acid esterification, obtains formula IV compound:
(3) formula IV compound is carried out to the reaction of enolization esterifying reagent, obtains formula V compound:
(4) formula V compound is reacted with fluoro reagent and obtains formula VI and formula VII compound:
(5) by formula VI compound, through configuration reversal, reaction obtains formula VII compound;
(6) formula VII compound is reacted with N-bromo-succinimide and water, obtains formula VIII compound:
(7) formula VIII compound epoxidation under alkaline condition is obtained to formula IX compound:
(8) formula IX compound is reacted with fluorination reagent and obtains formula X compound:
(9) dehydrogenation of formula X compound oxidation is obtained to formula I compound (difluprednate).
2. method as claimed in claim 1, is characterized in that, in step (2), formula III compound is obtained to formula IV compound through fourth esterification, and the fourth esterifying reagent adopting is butyryl oxide or butyryl chloride; The alkaline catalysts adopting is pyridine, triethylamine or DMAP; The solvent adopting is methylene dichloride, diethylene glycol dimethyl ether, 1, the mixture of the optional solvents in 2-ethylene dichloride, dioxane, trichloromethane, DMF, methyl-sulphoxide, N,N-dimethylacetamide or above-mentioned solvent.
3. method as claimed in claim 1, is characterized in that, in step (3), formula IV compound is obtained to formula V compound through enolization esterification, and the enolization esterifying reagent adopting is diacetyl oxide, Acetyl Chloride 98Min., methylvinyl acetate or vinyl-acetic ester; The catalyzer adopting is the vitriol oil or tosic acid; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, methylvinyl acetate, vinyl-acetic ester or above-mentioned solvent.
4. method as claimed in claim 1, is characterized in that, in step (4), formula V compound is obtained to formula VI compound and formula VII compound through fluoridizing, and the fluoro reagent adopting is Selectfluor or Accufluor; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, acetonitrile or above-mentioned solvent.
5. method as claimed in claim 1, it is characterized in that, in step (8), formula IX compound is obtained to formula X compound through fluoridizing open loop, the fluorination reagent adopting is aqueous hydrogen fluoride solution, hydrogen fluoride pyridine solution (Olah reagent) or hydrogen fluoride triethylamine solution; The solvent adopting is methylene dichloride, chloroform, 1, the mixture of the optional solvents in 2-ethylene dichloride, tetrahydrofuran (THF), toluene or above-mentioned solvent; Range of reaction temperature is-50~50 ℃.
6. a key intermediate compound for synthetic difluprednate, shown in IV compound:
Difluprednate ophthalmic emulsion 0.05% is also being studied in other ocular inflammatory diseases, including a phase 3 study evaluating difluprednate for the treatment of anterior uveitis[2][3]
Arrys Therapeutics (under license from AskAt ) and affiliate Ikena Oncology (formerly known as Kyn Therapeutics ) are developing ARY-007 , an oral formulation of grapiprant, for treating cancers; in December 2019, preliminary data were expected in 2020
Grapiprant (trade name Galliprant) is a small molecule drug that belongs in the piprant class. This analgesic and anti-inflammatory drug is primarily used as a pain relief for mild to moderate inflammation related to osteoarthritis in dogs. Grapiprant has been approved by the FDA’s Center for Veterinary Medicine and was categorized as a non-cyclooxygenase inhibiting non-steroidal anti-inflammatory drug (NSAID) in March 2016.[1]
Preclinical studies also indicate that grapiprant is not only efficacious as a acute pain but also in chronic pain relief and inflammation drug. The effect of the drug is directly proportional to the dosage and its effects were comparable to human medication such as rofecoxib and piroxicam.[2]
Grapiprant, a prostanoid EP4 receptor antagonist, is in phase II clinical trials at AskAt for the treatment of chronic pain. Phase I/II clinical trials are ongoing at Arrys Therapeutics in combination with pembrolizumab for the treatment of patients with microsatellite stable colorectal cancer and in patients with advanced or metastatic PD-1/L1 refractory non-small cell lung cancer (NSCLC).
Grapiprant is also used in humans, and was researched to be used as a pain control and inflammation associated with osteoarthritis. The effect of grapiprant could be explained through the function of prostaglandin E2, in which acts as a pro-inflammatory mediator of redness of the skin, edema and pain which are the typical signs of inflammation. The effect of PGE2 stems from its action through the four prostaglandin receptor subgroups EP1, EP2, EP3 and EP4, in which the prostaglandin EP4 receptor acts as the main intermediary of the prostaglandin-E2-driven inflammation. Grapiprant is widely accepted in veterinary medicine due to its specific and targeted approach to pain management in dogs. The serum concentration of grapiprant is increased when used in conjunction with other drugs such as acetaminophen, albendazole, and alitretinoin.
Common side effects are intestinal related effects such as mild diarrhea, appetite loss, and vomiting.[3] Additionally, it is found that it might lead to reduced tear production due to it being a sulfa-based medication and also reduced albumin levels.
Grapiprant, a prostanoid EP4 receptor antagonist, is in phase II clinical trials at AskAt for the treatment of chronic pain. Phase I/II clinical trials are ongoing at Arrys Therapeutics in combination with pembrolizumab for the treatment of patients with microsatellite stable colorectal cancer and in patients with advanced or metastatic PD-1/L1 refractory non-small cell lung cancer (NSCLC).
Medical uses
Grapiprant is used once a day as an oral pain relief for dogs with inflammation-related osteoarthritis. It is a non-steroidal anti-inflammatory (NSAID) that functions as a targeted action to treat osteoarthritis pain and inflammation in dogs.
Mechanism of action
Grapiprant acts as a specific antagonist that binds and blocks the prostaglandin EP4 receptor, one out of the four prostaglandin E2 (PGE2) receptor subgroups. The EP4 receptor then mediates the prostaglandin-E2-elicited response to pain, and hence grapiprant was proven to be effective in the decrease of pain in several inflammatory pain models of rats. It was also proven to be effective in reducing osteoarthritis-related pain in humans, which serves as a proof for its mechanism of action. The approximate calculation for canine efficacy dose is between the range of 1.3 and 1.7 mg/kg, in conjunction with a methylcellulose suspending agent. Based on the calculations from the comparisons of binding affinity of grapiprant to the EP4 receptors of dogs, rats, and humans, the study of plasma and serum protein binding determinations, the effective doses determined in inflammation pain models of rats, and human-related clinical studies, it is evaluated that Grapiprant should be administered just once a day. The approved dose of the commercial Grapiprant tablet by the FDA for the pain relief and inflammation associated with osteoarthritis to dogs is reported to be 2 mg/kg a day.[4]
Absorption
Studies in animals such as horses have shown the presence of Grapiprant in serum 72 hours with a concentration >0.005 ng/ml after the initial administration of a dose of 2 mg/kg. Grapiprant is expeditiously absorbed and the reported serum concentration was reported to be 31.9 ng/ml in an amount of time of 1.5 hours. The actual body exposure to grapiprant after administration of one dose was shown to be 2000 ng.hr/ml. The degree and rate at which grapiprant is absorbed into the body, presents a mean bioavailability of 39%. A significant reduction in the bioavailability, concentration time and maximal concentration were reported to have occurred after food intake.[1] And thus, grapiprant is usually not administered with food as it will not be as efficient.[5]
Distribution
The volume of distribution in cat studies was reported to be 918 ml/kg.[1]
Route of elimination
Following an oral administration, the majority of the dose was metabolized within the first 72 hours. Equine studies have shown that grapiprant is present in urine 96 hours after the first administration of a dose of 2 mg/kg and has a concentration >0.005 ng/ml. From the excreted dose conducted in horses, it is found that 55%, 15% and 19% of the orally-administered dose was excreted in bile, urine, and faeces respectively.[1]
Toxicity
Safety studies conducted on grapiprant have demonstrated that it generally possesses an exceptional safety profile and a wide safety margin in veterinary studies.[6] In animal studies, a research on 2.5-12 times overdose was conducted for grapiprant and the study resulted in soft-blobs and mucous-filled faeces, occasional bloody stools and emesis.
PATENT
WO-2020014465
Novel crystalline forms of grapiprant and their salts eg HCl (designated as Form A), useful for inhibiting prostaglandin EP4 receptor activity and treating cancers.
Prostaglandins are mediators of pain, fever and other symptoms associated with inflammation. Prostaglandin E2 (PGE2) is the predominant eicosanoid detected in inflammation conditions. In addition, it is also involved in various physiological and/or pathological conditions such as hyperalgesia, uterine contraction, digestive peristalsis, awakeness, suppression of gastric acid secretion, blood pressure, platelet function, bone metabolism, angiogenesis or the like.
[0003] Four PGE2 receptor subtypes (EP1, EP2, EP3 and EP4) displaying different pharmacological properties exist. The EP4 subtype, a Gs-coupled receptor, stimulates cAMP production as well as PI3K and GSK3P signaling, and is distributed in a wide variety of tissue suggesting a major role in PGE2-mediated biological events. Various EP4 inhibitors have been described previously, for example, in WO 2002/032900, WO 2005/021508, EiS 6,710,054, and US 7,238,714, the contents of which are incorporated herein by reference in their entireties.
[0004] Accordingly, there is a need for treating, preventing, and/or reducing severity of a proliferative disorder associated with prostaglandin EP4 receptor activity. The present invention addresses such a need.
It has now been found that compounds of the present invention, and compositions thereof, are useful for treating, preventing, and/or reducing severity of a proliferative disorder associated with prostaglandin EP4 receptor activity. In general, salt forms and co-crystal forms, and pharmaceutically acceptable compositions thereof, are useful for treating or lessening the severity of proliferative disorders associated with prostaglandin EP4 receptor activity, as described in detail herein. Such compounds are represented by the chemical structure below, denoted as compound A (also known as grapiprant):
A
or a pharmaceutically acceptable salt thereof.
United States Patent 7,960,407, filed March 1, 2006 and issued June 14, 2011 (“the ‘407 patent,” the entirety of which is hereby incorporated herein by reference), describes certain EP4 inhibitor compounds. Such compounds include compound A:
or a pharmaceutically acceptable salt thereof.
[0037] Compound A, N-[({2-[4-(2-Ethyl-4,6-dimethyl-lH-imidazo[4,5-c]pyridin-l-yl) phenyl]ethyl}amino)carbonyl]-4-methylbenzenesulfonamide, is described in detail in the ‘407
patent, including its synthetic route. The ‘407 patent also discloses a variety of physical forms of compound A.
[0038] It would be desirable to provide a solid form of compound A (e.g., as a co-crystal thereof or salt thereof) that imparts characteristics such as improved aqueous solubility, stability and ease of formulation. Accordingly, the present invention provides both co-crystal forms and salt forms of compound A:
A.
PATENT
WO 2002032900
PATENT
WO 2002032422
Family members of the product case ( WO0232422 ) of grapiprant have protection in most of the EU states until October 2021 and expire in the US in October 15, 2021.
PATENT
WO 2003086371
PATENT
WO2020014445 covering combinations of grapiprant and an immuno-oncology agent.
^Nagahisa, A.; Okumura, T. (2017). “Pharmacology of grapiprant, a novel EP4 antagonist: receptor binding, efficacy in a rodent postoperative pain model, and a dose estimation for controlling pain in dogs”. Journal of Veterinary Pharmacology and Therapeutics. 40 (3): 285–292. doi:10.1111/jvp.12349. ISSN1365-2885. PMID27597397.
Anal. Calcd For C28H35FN6O5S: C, 57.32; H, 6.01; N, 14.32. Found: C, 57.34; H, 6.13; N, 14.29.
Activated by a wide range of stimuli such as capsaicin, acid, or heat, the transient receptor potential vanilloid-1 (TRPV1) has been identified as a potential treatment for chronic pain.TRPV1 is a highly characterized member of the TRP cation channel family believed to be involved in a number of important biological roles and plays a role in the transmission of pain.TRPV1 activation inhibits the transition of pain signals from the periphery to the central nervous system (CNS), leading to the possible development of analgesic and anti-inflammatory agents. TRPV1 antagonists have also been evaluated in multiple clinical trials where hyperthermic effects seen preclinically are also observed in humans
Heat a solution of 2,5-dibromo-3-methyl-pyridine (Chontech Inc., Waterford, Conn.) (2.0 g, 7.97 mmol), (R)-2-methyl-piperazine (ChemPacific Corp., Baltimore, Md.; 3.2 g, 31.9 mmol) in DMA at 130° C. for 16 h. Partition the reaction mixture between water and EtOAc. Wash the EtOAc layer with water (1×) and brine (1×), dry (Na2SO4) and concentrate under reduced pressure to give 1-(5-bromo-3-methyl-pyridin-2-yl)-3-(R)-methyl-piperazine as a solid.
2. 2,4-dichloro-6-(4-fluorophenyl)pyrimidine
Dissolve 4-fluorobromobenzene (8.75 g, 0.05 moles) in anhydrous ether (80 mL) under nitrogen atmosphere and cool to −78° C. Add dropwise 1.6 M n-BuLi (34 mL, 0.055 moles) and stir at −78° C. for 45 min. Dissolve 2,4-dichloropyrimidine (7.45 g, 0.05 moles) in Et2O (100 mL) and add dropwise to the reaction mixture. Warm the reaction mixture to −30° C. and stir at this temperature for 30 min followed by 0° C. for 30 min. Quench the reaction mixture with AcOH (3.15 mL, 0.055 moles) and water (0.5 mL, 0.027 moles) dissolved in THF (5.0 mL). Add dropwise a THF (40 mL) solution of DDQ (11.9 g, 0.053 moles) to the reaction mixture. Bring the reaction mixture to room temperature and stir at room temperature for 30 min. Cool the reaction mixture to 0° C., add 3.0 N aq. NaOH (35 mL) and stir for 30 min. Decant the organic layer from the reaction mixture and wash the brown solid with Et2O (3×100 mL). Combine the organic layers, wash several times with saturated NaCl solution and dry with MgSO4. Filter and evaporate under vacuum to afford a brown colored solid. Purify by flash column chromatography using 5% EtOAc/hexane to afford the title product as a white solid.
Heat a mixture of 2,4-dichloro-6-(4-fluoro-phenyl)-pyrimidine (6.0 g, 24.7 mmol), 1-(5-bromo-3-methyl-pyridin-2-yl)-3-(R)-methyl-piperazine (7.0 g, 25.9 mmol) and K2CO3 (6.8 g, 49.4 mmol) in DMA at 60° C. for 16 h. Partition the mixture between EtOAc and water, dry (Na2SO4) the organic layer and concentrate under reduced pressure. Purify with flash silica gel column eluting with 15% EtOAc/hexanes. Concentrate under reduced pressure to give the title compound.
Heat a mixture of 4-[4-(5-bromo-3-methyl-pyridin-2-yl)-2-(R)-methyl-piperazin-1-yl]-2-chloro-6-(4-fluoro-phenyl)-pyrimidine (7.7 g, 16.2 mmol), (R)-2-methylpyrrolidine hydrobromide [prepared essentially as described by Nijhuis et. al. (1989) J. Org. Chem. 54(1):209] (3.5 g, 21.1 mmol) and K2CO3 (5.1 g, 37.3 mmol) in DMA at 110° C. for 16 h. Partition the mixture between EtOAc and water, dry (Na2SO4) the organic layer and concentrate under reduced pressure. Purify with flash silica gel column eluting with 10% EtOAc/hexanes. Concentrate under reduced pressure to give the title compound.
To a mixture of 4-[4-(5-bromo-3-methyl-pyridin-2-yl)-2-(R)-methyl-piperazin-1-yl]-6-(4-fluoro-phenyl)-2-(2-methyl-pyrrolidin-1-yl)-pyrimidine (700 mg, 1.33 mmol) and Zn(CN)2 (94 mg, 0.799 mmol) in DMF, add Pd(PPh3)4 (77 mg, 0.067 mmol). Purge the reaction mixture for 10 min with dry N2. Heat the stirring reaction mixture overnight at 80° C., cool to room temperature and partition between water and EtOAc. Dry the solution (Na2SO4), concentrate under reduced pressure. Purify the residue by flash column eluting with EtOAc-Hexanes (1:1) to afford the title compound as a white solid.
A highly efficient, regioselective five-step synthesis of the TRPV1 antagonist 1 is described. The coupling of piperazine 7 with dichloropyrimidine 8 proceeded via a regioselective Pd-mediated amination affording product 11 in excellent yield. Conversion of the penultimate product 14 afforded 1 through formation of a magnesium ate complex and trapping with CO2.
Branford, CT — January 16, 2004 — Neurogen Corporation (Nasdaq: NRGN) today announced that it has consummated its previously announced alliance with Merck & Co ., Inc. (NYSE: MRK) to discover and develop next-generation drugs for the treatment of pain. The deal received clearance from the Federal Trade Commission under the Hart-Scott-Rodino Act and the companies have now commenced the collaboration. The alliance, announced December 1, 2003, enables Merck , through a subsidiary, and Neurogen to pool drug candidates targeting the vanilloid receptor (VR1 ), a key integrator of pain signals in the nervous system, and combine their ongoing VR1 programs to form a global research and development collaboration.
With consummation of the deal, Neurogen has received $30 million from Merck , including a $15 million up-front license fee payment and a $15 million equity investment in Neurogen common stock. Under the agreement, Merck has purchased 1,783,252 shares of newly issued Neurogen common stock at $8.41 per share, the average market price per share for the 25 trading days preceding regulatory clearance. Merck ‘s new shareholder position represents approximately 9% of Neurogen ‘s 19,873,464 total shares outstanding.
Neurogen Corporation targets new small molecule drugs to improve the lives of patients suffering from disorders with significant unmet medical need. Neurogen has generated a portfolio of compelling new drug candidates through its Accelerated Intelligent Drug Discovery (AIDD(TM)) system, its expertise in cellular functional assays, and its depth in medicinal chemistry. Neurogen conducts its research and development independently and, when advantageous, collaborates with world-class pharmaceutical companies to obtain additional resources and to access complementary expertise.
The process development of Mavatrep (1), a potent transient receptor potential vanilloid-1 (TRPV1) antagonist, is described. The two key synthetic transformations are the synthesis of (E)-6-bromo-2-(4-(trifluoromethyl)styryl)1H-benzo[d]imidazole (4) and the Suzuki coupling of 4 with 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol (5). Compound 1a was prepared in four chemical steps in 63% overall yield.
Example 10 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol(Cpd 18)
Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
[0278]
A solution of 4-trifluoromethylbenzaldehyde (7.7 mL, 57.7 mmol), malonic acid (12.0 g, 115.4 mmol), 0.567 μL piperidine (5.75 mmol) in 30 mL of pyridine was stirred at 70° C. for 18 h. The reaction solution was cooled to room temperature. Water (300 mL) was added and the resulting mixture was acidified to pH 4 (litmus) using concentrated hydrochloric acid to give a precipitate. The solid was filtered, and washed with water until the filtrate was neutral. The solid product was dried in vacuo to give the title Compound 10a as a white powder (11.2 g, 90%). 1HNMR (400 MHz, DMSO-d6) δ (ppm): 12.60 (bs, 1H), 7.92 (d, 2H, J=8.2 Hz), 7.77 (d, 2H, J=8.2 Hz), 7.66 (d, 1H, J=16.0 Hz), 6.70 (d, 1H, J=16.0 Hz).
[0000]
Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
[0279]
A solution of Compound 10a (20.6 g, 95.4 mmol) in anhydrous methylene chloride (200 mL) was treated with oxalyl chloride (16.6 mL, 190 mmol) and “3 drops” of anhydrous dimethylformamide. The resulting solution was stirred at room temperature under an argon atmosphere for 18 h. The solvent was concentrated to give 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b as a solid, which was used without further purification in the next step.
[0280]
To a solution of 4-bromo-benzene-1,2-diamine (16.1 g, 86.7 mmol) in acetic acid (100 mL) was added dropwise a solution of Compound 10b (assumed 95.4 mmol) in acetic acid (100 mL). The reaction mixture was stirred at 100° C. for 18 h. The reaction mixture was cooled to room temperature, and a mixture of ethyl acetate and hexanes 3:7 (500 mL) was added. The mixture was triturated at room temperature for 3 h to give a precipitate. The solid was filtered, and dried in vacuo to give the title Compound 10c (23.2 g, 73%). 1H NMR (400 MHz, DMSO-d6/CDCl3) δ (ppm): 8.45 (d, 1H, J=16.7 Hz), 7.84-7.90 (m, 1H), 7.74 (d, 2H, J=8.3
To a solution of methyl 2-bromobenzoate (20.76 g, 96 mmol) in 120 mL of anhydrous ether under Argon at 0° C. was slowly added methylmagnesium bromide (77 mL, 3.26 M) at a rate that the internal temperature of the mixture was below 20° C. A white suspension resulted, and the mixture was stirred at room temperature for 2 h. The mixture was cooled in an ice-water bath. To the reaction mixture was very slowly added hydrochloric acid (400 mL, 0.5 M). The pH of the final mixture was adjusted to less than about 6 with few drops of 2M hydrochloric acid. The layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined and dried over magnesium sulfate. The organic fraction was filtered, and the filtrate was concentrated to yield the title compound as a pale yellow liquid, which was distilled under vacuum to afford the title Compound 10d as a colorless liquid (16.9 g, 82%, b.p. about 65-70° C./0.3 mmHg). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.67 (dd, 1H, J=1.7, 7.9 Hz), 7.58 (dd, 1H, J=1.3, 7.9 Hz), 7.30 (ddd, 1H, J=1.4, 7.4, 7.9 Hz), 7.10 (ddd, 1H, J=1.7, 7.4, 7.8 Hz), 2.77 (br s, 1H), 1.76 (s, 6H).
[0000]
Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
[0283]
To a solution of n-BuLi (166 mL, 2.6 M, 432 mmol) in 200 mL of THF at −78° C. under argon was slowly added a solution of Compound 10d (42.2 g, 196 mmol) in 60 mL of THF at a rate that the internal temperature remained below −70° C. The mixture was stirred at −75° C. for 2 h. To the reaction mixture was then added triisopropylborate (59 mL, 255 mmol) in three portions. The mixture was allowed to warm slowly to room temperature overnight. The mixture was then cooled to 0° C., and was carefully quenched with dilute hydrochloric acid (250 mL, 2N). The mixture was then stirred at room temperature for 1 h. The pH of the mixture was checked and adjusted to acidic using additional 2N HCl if prophetic. The two layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined, and dried with magnesium sulfate and filtered. The filtrate was concentrated under reduced pressure to yield a pale yellow oil. The residue was then diluted with ethyl acetate (400 mL) and, washed with 1N sodium hydroxide solution (150 mL×3). The basic aqueous layers were combined and acidified with 2N HCl. The clear solution turned cloudy when the acid was added. The mixture was extracted with ether (150 mL×3). The organic layers were combined and dried with magnesium sulfate. The solution was filtered, and the filtrate was concentrated under reduced pressure to yield the title Compound 10e as a colorless oil (26.2 g, 82%) which was used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.00 (s, 1H), 7.66 (dm, 1H, J=7.3 Hz), 7.45 (dt, 1H, J=1.1, 7.7 Hz), 7.40 (dm, 1H, J=7.6 Hz), 7.31 (dt, 1H, J=1.2, 7.1 Hz), 1.44 (s, 6H).
[0000]
Step E. (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
[0284]
To a mixture of Compound 10e (11.7 g, 71 mmol), Compound 10c (19.9 g, 54 mmol), sodium carbonate (46 g, 435 mmol) and PdCl2(dppf).CH2Cl2 (8.9 g, 11 mmol) in a 1 L round bottom flask equipped with water condenser was added 400 mL of anhydrous DME and 200 mL of water. The mixture was evacuated and filled with Argon three times. The mixture was heated to 100° C. for 20 h. The mixture was then cooled to room temperature. The biphasic system was transferred to a 1 L separatory funnel and the two layers were separated. The organic layer was washed with brine (2×300 mL). The aqueous layers were combined and extracted with ethyl acetate once (about 300 mL). The organic layers were combined, dried with sodium sulfate, and filtered. The volume of the filtrate was reduced to about 170 mL under reduced pressure. The mixture was then filtered through a pad of silica gel and the pad was washed with ethyl acetate until the filtrate did not contain any product. After concentration, a light pink/beige solid was obtained. The solid was triturated with 50 mL ethyl acetate, and the mixture was heated to 85° C. for 5 min. The mixture was slowly cooled to r.t., then cooled at 0° C. for 0.5 h. The mixture was filtered, and the solid was washed with cold ethyl acetate twice, and dried under vacuum at 40° C. to yield the title Compound 18 as a light beige solid (7.58 g, 33%). RP-HPLC 95% pure.
Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3.
m.p. (uncorr.) 250-251° C.
Example 10.1 Scale Up Preparation of (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol (Cpd 18) Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
[0286]
A 2-L 4-neck round bottom flask equipped with an air condenser/argon inlet, mechanical stirrer, thermocouple and a stopper was charged with 4-(trifluoromethyl)benzaldehyde (250 g, 196.2 mL, 1.44 mol), malonic acid (302.6 g, 2.87 mol), and pyridine (750 mL). An exotherm developed (about 38-40° C.), which was maintained for 30 min. Piperidine (14.202 mL, 143.58 mmol) was then added to the reaction and a second exotherm developed (Tmax about 42° C. after about 10 min.). The reaction was stirred for 30 min and then heated to 60° C. for 18 h (overnight). The reaction appeared to be complete by TLC, and was cooled to about 40° C., diluted into water (2 L; done to prevent reaction freezing), cooled to room temperature, and further diluted with water (4 L, 6 L total). The slurry was acidified to pH=2.0-3.0 with concentrated hydrochloric acid (about 675-700 mL). The material was stirred for 30 min., and a white solid was collected by filtration. The filter cake was washed with water until the filtrate was neutral (pH about 5.5-6, 2.5 L), air-dried in a Buchner funnel for 2 h, and then further dried in a vacuum oven at 60° C. overnight to provide 300.5 g (96%) of the title Compound 10a as a white solid.
Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
[0287]
To a 5-L 4-neck round bottom flask equipped with a magnetic stirrer, argon inlet-argon outlet to a carbonate scrub, two stoppers, and a room temperature water bath was charged with 4-(trifluoromethyl)cinnamic acid (315 g, 1.46 mol) and dichloromethane (3.15 L) to give a slurry. To the slurry was added oxalyl chloride (151.71 mL, 1.75 mol) and DMF (1.13 mL, 14.57 mmol). Upon addition of DMF, gas evolution commenced, and the reaction was continued for about 3 h during which time a solution developed. When the reaction was complete (LC-MS), it was concentrated to dryness to give 342.4 g of 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b (>100%) as a yellow oily solid.
[0288]
A 5-L 4-neck round bottom flask equipped with mechanical stirrer, thermocouple, air condenser with argon inlet, and a stopper was charged with 4-bromo-benzene-1,2-diamine (244 g, 1.27 mol) and acetic acid (2.13 L). To this solution was added a solution of Compound 10b (327 g, 1.39 mol) in toluene (237 mL). After this addition, the temperature spiked to 45° C. in about 30 seconds and then subsided. The reaction was then heated to 90° C. for 16 h (overnight). The reaction was cooled to 40° C., and poured into a mixed solution of EtOAc and heptane (about 1:3, 5.75 L) and a precipitate occurred. The resulting slurry was stirred for 3 h, and the solid was collected by filtration, washed with EtOAc:heptane (1:3, 3 L), and then dried in a vacuum oven (60° C.) to give 324.3 g (65%) of the title Compound 10c as a partial acetate salt.
Step C. 2-(2-bromo-phenyl)-propan-2-ol
[0289]
A 12-Liter 4-neck flask equipped with a thermocouple, condenser, septum, addition funnel and overhead mechanical stirrer under argon was charged with methyl-2-bromobenzoate (226.5 g, 1.05 mol) and THF (1.6 L, 19.66 mol). The mixture was cooled to a temperature between 2 and 5° C. with stirring and held for 30 min. To the solution was slowly added methyl magnesium bromide in diethyl ether (3M, 1.05 L; 3.15 mol) via the addition funnel at a rate to maintain the reaction temperature below 15° C. An exotherm was observed during the addition, the reaction temperature warmed from 3 to 15° C. The addition of 1.05 L Grignard was complete in 4 h (approximate feed rate was 4.17 mL/min). The reaction mixture appeared to be off-white/yellow slurry. The reaction was allowed to warm to room temperature and stirred overnight (15 h). The reaction was sampled by HPLC/TLC and showed no starting material present. The ice bath was again applied to the reaction flask and a 0.5 M HCl solution (4.5 L; 2.25 mol) was slowly added over a period of 2 h. The temperature increased dramatically from 0 to 15° C. After the quench was complete, the reaction was stirred at room temperature for 30 min. Additional 2 N HCl (500 mL; 1.00 mol) was slowly added to maintain a pH less than 6. MTBE (1 L) was added to help with the phase split. The reaction was stirred at room temperature for 1 to 2 h to dissolve the solid material into the aqueous phase (most likely Mg(OH)2 which is very basic). The pH must be checked and adjusted with additional acid when necessary. The phases were separated and the aqueous layer was washed with an additional 1 L MTBE (2×500 mL). The organic phases were combined, washed with NaHCO3 solution (2×300 mL), dried over MgSO4, filtered and the filtrate was concentrated under vacuum to yield the title Compound 10d (220.83 g, 97.48% yield) as a clear yellow oil.
Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
[0290]
A 12-Liter 4-neck round bottom flask equipped with a thermocouple, condenser, addition funnel and overhead mechanical stirrer under dry Argon was charged with anhydrous THF, (3 L) and chilled to −70 to −78° C. via a dry ice/acetone bath. n-Butyl lithium (2.5N in hexanes, 860 mL, 2.15 mol) was slowly added via addition funnel. An exotherm was observed as the temperature rose from −78 to −70° C. To the addition funnel was added a solution of Compound 10d (220 g, 979.97 mmol) in anhydrous THF (1 L). The 2-(2-bromophenyl)propan-2-ol solution was slowly added to the n-BuLi solution. The addition took 90 min in order to maintain a reaction temperature below −70° C. After the addition was complete, the reaction mixture was stirred at −70 to −75° C. for 30 min. The triethylborate (230 mL, 1.35 mol) was quickly added in 3 portions at −70° C. An exotherm was observed, the batch temperature rose from −70 to −64° C. The reaction was stirred at −70° C. and slowly warmed to room temperature over night. After the reaction was cooled to 0-5° C., the reaction was slowly quenched with 2 M HCl (1 L, 2.00 mol) added via the addition funnel while maintaining the batch temperature 0-5° C. The reaction mixture was stirred for 1 h. The aqueous phase pH was 9-10. The pH was then adjusted to acidic (4-5) with 2 M HCl (200 mL). The two phases were separated and the aqueous layer was extracted with MTBE (2×500 mL). The combined organic phases were dried with anhydrous magnesium sulfate. The solution was filtered and concentrated to yield a yellow oil. The yellow oil was diluted with MTBE (1.5 L) and washed with 1M NaOH (3×500 mL). The product containing basic aqueous phases were combined and acidified with 2 M HCl (800 mL) (the clear solution turns turbid with the addition of acid). After stirring the turbid solution for 15 min (pH=4-5) (Note 1), it was extracted with MTBE (2×500 mL). The organic phases were combined and dried over MgSO4. The solution was filtered and the filtrate was concentrated to yield the title Compound 10e as a clear yellow oil (121.78 grams, 77% yield).
Step E. (E)-2-(2-{2-[2-(4-Trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
A 5-L 4-neck flask equipped with a thermocouple controller, condenser, overhead mechanical stirrer, Firestone Valve® and a nitrogen inlet/outlet was charged with dimethoxyethane (2 L), DI water (1 L) and sodium carbonate (230.9 g, 2.18 mol). The solution was degassed and purged with N2 three times. Compound 10e (71.7 g, 0.35 mol) and Compound 10c (100.0 g, 0.27 mol) were added to the degassed solution. The solution was degassed and purged with N2 three times. PdCl2(dppf) (44.48 g, 54.4 mmol) was added to the solution, and the solution was degassed and purged with N2 three times. The resulting two-phase suspension was heated to reflux for 18 h, and then cooled to room temperature. The reaction mixture was transferred to a 12-L separatory funnel, and the layers were separated. The organic layer was washed with brine (1 L). The two aqueous layers were combined and extracted with EtOAc (1 L). The combined organic layers were dried (Na2SO4), filtered, and the filtrate was concentrated to an oil. Two separate 100 g coupling reactions were combined and purified by chromatography in 10 successive chromatography runs on an ISCO preparative chromatography system (10×1.5 Kg SiO2, 5 column volumes of EtOAc, 250 mL/min flow rate). The combined fractions were transferred to two 22 L 4-neck round bottom flasks, and Silicycle Si-thiol functionalized silica gel (2 g) was added to each solution. The solutions were warmed to 40° C. and aged for 1 h. The solutions were filtered thru a medium glass funnel and washed with EtOAc (4 L) and combined. The filtrate was evaporated to a semi solid, which was transferred to a 2 L round bottom flask, to which EtOAc (0.4 L) was added. The resulting white precipitate slurry was cooled to −5° C. and stirred for 1 h. The slurry was filtered and washed twice with cold EtOAc (100 mL). The solids were dried in a vacuum oven at 40° C. for 40 h to afford 84.0 g (36.5% yield, 98.8 area % purity) of the title Compound 18 as a white solid. Anal. Calcd for C25H21N2OF3.0.04% H2O.0.15 mol MeOH: C, 70.48; H, 5.14: N, 6.42; F, 13.06 Found: C, 70.54; H, 4.83: N, 6.18; F, 13.33
Example 10.2 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol monosodium salt (Cpd 18)
A 5-L 4-neck flask equipped with a thermocouple controller, an overhead mechanical stirrer, and a nitrogen inlet/outlet was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (125.0 g, 0.510 mol) and MeOH (1.25 L). A solution of sodium methoxide in methanol (0.5 M, 592 mL, 0.3 mol) was added. The reaction was heated to 65° C. for 30 min and all solids dissolved. The solution was cooled and evaporated to dryness. The foam was collected by scraping it out of the flask. The solids were placed in vacuum oven for 24 h at 40° C. to afford 139 g (about 100% isolated yield) of the title Compound 18 monosodium salt as a yellowish solid.1H NMR (400 MHz, DMSO-d6) δ 7.80-7.84 (m, 3H), 7.74 (d, 2H, J=8.59 Hz), 7.65 (d, 1H, J=16.4 Hz), 7.40-7.44 (m, 2H), 7.25-7.37 (m, 2H), 7.16-7.20 (m, 1H), 7.01-7.05 (m, 1H), 6.84-6.87 (m, 1H), 1.23 (s, 6H). Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3. m.p. (uncorr.) 258-259° C.
Example 10.3 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol hydrochloride salt (Cpd 18)
A 250-mL separatory funnel was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (1.0 g, 2.4 mmol) and EtOAc (20 mL). Aqueous HCl (1M, 20 mL) was added to the white slurry, and the separatory funnel was shaken. The solid product quickly dissolved, and a white precipitate started to form. The organic layer was transferred to a 100 mL round bottom flask equipped with a magnetic stir bar, and was stirred for 2 h. The thick slurry was filtered, rinsed with EtOAc (2×5 mL), and put into a vacuum oven at 40° C. for 36 h to afford 0.95 g (87.5%) of the title Compound 18 hydrochloride salt.
The immune cell enters the nerve. Credit: Dr. Marzia Malcangio, King’s College London
Scientists have identified new pain relief targets that could be used to provide relief from chemotherapy-induced pain. BBSRC-funded researchers at King’s College London made the discovery when researching how pain occurs in nerves in the periphery of the body.
Dr Marzia Malcangio said: “We have been investigating and identifying mechanisms underlying pain generation and our findings could help chemotherapy patients who suffer pain related side effects.”
One potential side effect of some chemotherapy drugs (such as vincristine) is damage to nerves. This is particularly prominent in hands and feet as the drugs affect nerves in the periphery of the body. This causes pain which doctors treat with painkillers. However, some people find that the pain persists.
Dr Malcangio’s team investigated why the chemotherapy drugs were causing pain in hope to solve the problem. The used mice in…
The process development of Mavatrep (1), a potent transient receptor potential vanilloid-1 (TRPV1) antagonist, is described. The two key synthetic transformations are the synthesis of (E)-6-bromo-2-(4-(trifluoromethyl)styryl)1H-benzo[d]imidazole (4) and the Suzuki coupling of 4 with 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol (5). Compound 1a was prepared in four chemical steps in 63% overall yield.
Example 10 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol(Cpd 18)
Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
[0278]
A solution of 4-trifluoromethylbenzaldehyde (7.7 mL, 57.7 mmol), malonic acid (12.0 g, 115.4 mmol), 0.567 μL piperidine (5.75 mmol) in 30 mL of pyridine was stirred at 70° C. for 18 h. The reaction solution was cooled to room temperature. Water (300 mL) was added and the resulting mixture was acidified to pH 4 (litmus) using concentrated hydrochloric acid to give a precipitate. The solid was filtered, and washed with water until the filtrate was neutral. The solid product was dried in vacuo to give the title Compound 10a as a white powder (11.2 g, 90%). 1HNMR (400 MHz, DMSO-d6) δ (ppm): 12.60 (bs, 1H), 7.92 (d, 2H, J=8.2 Hz), 7.77 (d, 2H, J=8.2 Hz), 7.66 (d, 1H, J=16.0 Hz), 6.70 (d, 1H, J=16.0 Hz).
[0000]
Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
[0279]
A solution of Compound 10a (20.6 g, 95.4 mmol) in anhydrous methylene chloride (200 mL) was treated with oxalyl chloride (16.6 mL, 190 mmol) and “3 drops” of anhydrous dimethylformamide. The resulting solution was stirred at room temperature under an argon atmosphere for 18 h. The solvent was concentrated to give 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b as a solid, which was used without further purification in the next step.
[0280]
To a solution of 4-bromo-benzene-1,2-diamine (16.1 g, 86.7 mmol) in acetic acid (100 mL) was added dropwise a solution of Compound 10b (assumed 95.4 mmol) in acetic acid (100 mL). The reaction mixture was stirred at 100° C. for 18 h. The reaction mixture was cooled to room temperature, and a mixture of ethyl acetate and hexanes 3:7 (500 mL) was added. The mixture was triturated at room temperature for 3 h to give a precipitate. The solid was filtered, and dried in vacuo to give the title Compound 10c (23.2 g, 73%). 1H NMR (400 MHz, DMSO-d6/CDCl3) δ (ppm): 8.45 (d, 1H, J=16.7 Hz), 7.84-7.90 (m, 1H), 7.74 (d, 2H, J=8.3
To a solution of methyl 2-bromobenzoate (20.76 g, 96 mmol) in 120 mL of anhydrous ether under Argon at 0° C. was slowly added methylmagnesium bromide (77 mL, 3.26 M) at a rate that the internal temperature of the mixture was below 20° C. A white suspension resulted, and the mixture was stirred at room temperature for 2 h. The mixture was cooled in an ice-water bath. To the reaction mixture was very slowly added hydrochloric acid (400 mL, 0.5 M). The pH of the final mixture was adjusted to less than about 6 with few drops of 2M hydrochloric acid. The layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined and dried over magnesium sulfate. The organic fraction was filtered, and the filtrate was concentrated to yield the title compound as a pale yellow liquid, which was distilled under vacuum to afford the title Compound 10d as a colorless liquid (16.9 g, 82%, b.p. about 65-70° C./0.3 mmHg). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.67 (dd, 1H, J=1.7, 7.9 Hz), 7.58 (dd, 1H, J=1.3, 7.9 Hz), 7.30 (ddd, 1H, J=1.4, 7.4, 7.9 Hz), 7.10 (ddd, 1H, J=1.7, 7.4, 7.8 Hz), 2.77 (br s, 1H), 1.76 (s, 6H).
[0000]
Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
[0283]
To a solution of n-BuLi (166 mL, 2.6 M, 432 mmol) in 200 mL of THF at −78° C. under argon was slowly added a solution of Compound 10d (42.2 g, 196 mmol) in 60 mL of THF at a rate that the internal temperature remained below −70° C. The mixture was stirred at −75° C. for 2 h. To the reaction mixture was then added triisopropylborate (59 mL, 255 mmol) in three portions. The mixture was allowed to warm slowly to room temperature overnight. The mixture was then cooled to 0° C., and was carefully quenched with dilute hydrochloric acid (250 mL, 2N). The mixture was then stirred at room temperature for 1 h. The pH of the mixture was checked and adjusted to acidic using additional 2N HCl if prophetic. The two layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined, and dried with magnesium sulfate and filtered. The filtrate was concentrated under reduced pressure to yield a pale yellow oil. The residue was then diluted with ethyl acetate (400 mL) and, washed with 1N sodium hydroxide solution (150 mL×3). The basic aqueous layers were combined and acidified with 2N HCl. The clear solution turned cloudy when the acid was added. The mixture was extracted with ether (150 mL×3). The organic layers were combined and dried with magnesium sulfate. The solution was filtered, and the filtrate was concentrated under reduced pressure to yield the title Compound 10e as a colorless oil (26.2 g, 82%) which was used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.00 (s, 1H), 7.66 (dm, 1H, J=7.3 Hz), 7.45 (dt, 1H, J=1.1, 7.7 Hz), 7.40 (dm, 1H, J=7.6 Hz), 7.31 (dt, 1H, J=1.2, 7.1 Hz), 1.44 (s, 6H).
[0000]
Step E. (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
[0284]
To a mixture of Compound 10e (11.7 g, 71 mmol), Compound 10c (19.9 g, 54 mmol), sodium carbonate (46 g, 435 mmol) and PdCl2(dppf).CH2Cl2 (8.9 g, 11 mmol) in a 1 L round bottom flask equipped with water condenser was added 400 mL of anhydrous DME and 200 mL of water. The mixture was evacuated and filled with Argon three times. The mixture was heated to 100° C. for 20 h. The mixture was then cooled to room temperature. The biphasic system was transferred to a 1 L separatory funnel and the two layers were separated. The organic layer was washed with brine (2×300 mL). The aqueous layers were combined and extracted with ethyl acetate once (about 300 mL). The organic layers were combined, dried with sodium sulfate, and filtered. The volume of the filtrate was reduced to about 170 mL under reduced pressure. The mixture was then filtered through a pad of silica gel and the pad was washed with ethyl acetate until the filtrate did not contain any product. After concentration, a light pink/beige solid was obtained. The solid was triturated with 50 mL ethyl acetate, and the mixture was heated to 85° C. for 5 min. The mixture was slowly cooled to r.t., then cooled at 0° C. for 0.5 h. The mixture was filtered, and the solid was washed with cold ethyl acetate twice, and dried under vacuum at 40° C. to yield the title Compound 18 as a light beige solid (7.58 g, 33%). RP-HPLC 95% pure.
Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3.
m.p. (uncorr.) 250-251° C.
Example 10.1 Scale Up Preparation of (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol (Cpd 18) Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
[0286]
A 2-L 4-neck round bottom flask equipped with an air condenser/argon inlet, mechanical stirrer, thermocouple and a stopper was charged with 4-(trifluoromethyl)benzaldehyde (250 g, 196.2 mL, 1.44 mol), malonic acid (302.6 g, 2.87 mol), and pyridine (750 mL). An exotherm developed (about 38-40° C.), which was maintained for 30 min. Piperidine (14.202 mL, 143.58 mmol) was then added to the reaction and a second exotherm developed (Tmax about 42° C. after about 10 min.). The reaction was stirred for 30 min and then heated to 60° C. for 18 h (overnight). The reaction appeared to be complete by TLC, and was cooled to about 40° C., diluted into water (2 L; done to prevent reaction freezing), cooled to room temperature, and further diluted with water (4 L, 6 L total). The slurry was acidified to pH=2.0-3.0 with concentrated hydrochloric acid (about 675-700 mL). The material was stirred for 30 min., and a white solid was collected by filtration. The filter cake was washed with water until the filtrate was neutral (pH about 5.5-6, 2.5 L), air-dried in a Buchner funnel for 2 h, and then further dried in a vacuum oven at 60° C. overnight to provide 300.5 g (96%) of the title Compound 10a as a white solid.
Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
[0287]
To a 5-L 4-neck round bottom flask equipped with a magnetic stirrer, argon inlet-argon outlet to a carbonate scrub, two stoppers, and a room temperature water bath was charged with 4-(trifluoromethyl)cinnamic acid (315 g, 1.46 mol) and dichloromethane (3.15 L) to give a slurry. To the slurry was added oxalyl chloride (151.71 mL, 1.75 mol) and DMF (1.13 mL, 14.57 mmol). Upon addition of DMF, gas evolution commenced, and the reaction was continued for about 3 h during which time a solution developed. When the reaction was complete (LC-MS), it was concentrated to dryness to give 342.4 g of 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b (>100%) as a yellow oily solid.
[0288]
A 5-L 4-neck round bottom flask equipped with mechanical stirrer, thermocouple, air condenser with argon inlet, and a stopper was charged with 4-bromo-benzene-1,2-diamine (244 g, 1.27 mol) and acetic acid (2.13 L). To this solution was added a solution of Compound 10b (327 g, 1.39 mol) in toluene (237 mL). After this addition, the temperature spiked to 45° C. in about 30 seconds and then subsided. The reaction was then heated to 90° C. for 16 h (overnight). The reaction was cooled to 40° C., and poured into a mixed solution of EtOAc and heptane (about 1:3, 5.75 L) and a precipitate occurred. The resulting slurry was stirred for 3 h, and the solid was collected by filtration, washed with EtOAc:heptane (1:3, 3 L), and then dried in a vacuum oven (60° C.) to give 324.3 g (65%) of the title Compound 10c as a partial acetate salt.
Step C. 2-(2-bromo-phenyl)-propan-2-ol
[0289]
A 12-Liter 4-neck flask equipped with a thermocouple, condenser, septum, addition funnel and overhead mechanical stirrer under argon was charged with methyl-2-bromobenzoate (226.5 g, 1.05 mol) and THF (1.6 L, 19.66 mol). The mixture was cooled to a temperature between 2 and 5° C. with stirring and held for 30 min. To the solution was slowly added methyl magnesium bromide in diethyl ether (3M, 1.05 L; 3.15 mol) via the addition funnel at a rate to maintain the reaction temperature below 15° C. An exotherm was observed during the addition, the reaction temperature warmed from 3 to 15° C. The addition of 1.05 L Grignard was complete in 4 h (approximate feed rate was 4.17 mL/min). The reaction mixture appeared to be off-white/yellow slurry. The reaction was allowed to warm to room temperature and stirred overnight (15 h). The reaction was sampled by HPLC/TLC and showed no starting material present. The ice bath was again applied to the reaction flask and a 0.5 M HCl solution (4.5 L; 2.25 mol) was slowly added over a period of 2 h. The temperature increased dramatically from 0 to 15° C. After the quench was complete, the reaction was stirred at room temperature for 30 min. Additional 2 N HCl (500 mL; 1.00 mol) was slowly added to maintain a pH less than 6. MTBE (1 L) was added to help with the phase split. The reaction was stirred at room temperature for 1 to 2 h to dissolve the solid material into the aqueous phase (most likely Mg(OH)2 which is very basic). The pH must be checked and adjusted with additional acid when necessary. The phases were separated and the aqueous layer was washed with an additional 1 L MTBE (2×500 mL). The organic phases were combined, washed with NaHCO3 solution (2×300 mL), dried over MgSO4, filtered and the filtrate was concentrated under vacuum to yield the title Compound 10d (220.83 g, 97.48% yield) as a clear yellow oil.
Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
[0290]
A 12-Liter 4-neck round bottom flask equipped with a thermocouple, condenser, addition funnel and overhead mechanical stirrer under dry Argon was charged with anhydrous THF, (3 L) and chilled to −70 to −78° C. via a dry ice/acetone bath. n-Butyl lithium (2.5N in hexanes, 860 mL, 2.15 mol) was slowly added via addition funnel. An exotherm was observed as the temperature rose from −78 to −70° C. To the addition funnel was added a solution of Compound 10d (220 g, 979.97 mmol) in anhydrous THF (1 L). The 2-(2-bromophenyl)propan-2-ol solution was slowly added to the n-BuLi solution. The addition took 90 min in order to maintain a reaction temperature below −70° C. After the addition was complete, the reaction mixture was stirred at −70 to −75° C. for 30 min. The triethylborate (230 mL, 1.35 mol) was quickly added in 3 portions at −70° C. An exotherm was observed, the batch temperature rose from −70 to −64° C. The reaction was stirred at −70° C. and slowly warmed to room temperature over night. After the reaction was cooled to 0-5° C., the reaction was slowly quenched with 2 M HCl (1 L, 2.00 mol) added via the addition funnel while maintaining the batch temperature 0-5° C. The reaction mixture was stirred for 1 h. The aqueous phase pH was 9-10. The pH was then adjusted to acidic (4-5) with 2 M HCl (200 mL). The two phases were separated and the aqueous layer was extracted with MTBE (2×500 mL). The combined organic phases were dried with anhydrous magnesium sulfate. The solution was filtered and concentrated to yield a yellow oil. The yellow oil was diluted with MTBE (1.5 L) and washed with 1M NaOH (3×500 mL). The product containing basic aqueous phases were combined and acidified with 2 M HCl (800 mL) (the clear solution turns turbid with the addition of acid). After stirring the turbid solution for 15 min (pH=4-5) (Note 1), it was extracted with MTBE (2×500 mL). The organic phases were combined and dried over MgSO4. The solution was filtered and the filtrate was concentrated to yield the title Compound 10e as a clear yellow oil (121.78 grams, 77% yield).
Step E. (E)-2-(2-{2-[2-(4-Trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
A 5-L 4-neck flask equipped with a thermocouple controller, condenser, overhead mechanical stirrer, Firestone Valve® and a nitrogen inlet/outlet was charged with dimethoxyethane (2 L), DI water (1 L) and sodium carbonate (230.9 g, 2.18 mol). The solution was degassed and purged with N2 three times. Compound 10e (71.7 g, 0.35 mol) and Compound 10c (100.0 g, 0.27 mol) were added to the degassed solution. The solution was degassed and purged with N2 three times. PdCl2(dppf) (44.48 g, 54.4 mmol) was added to the solution, and the solution was degassed and purged with N2 three times. The resulting two-phase suspension was heated to reflux for 18 h, and then cooled to room temperature. The reaction mixture was transferred to a 12-L separatory funnel, and the layers were separated. The organic layer was washed with brine (1 L). The two aqueous layers were combined and extracted with EtOAc (1 L). The combined organic layers were dried (Na2SO4), filtered, and the filtrate was concentrated to an oil. Two separate 100 g coupling reactions were combined and purified by chromatography in 10 successive chromatography runs on an ISCO preparative chromatography system (10×1.5 Kg SiO2, 5 column volumes of EtOAc, 250 mL/min flow rate). The combined fractions were transferred to two 22 L 4-neck round bottom flasks, and Silicycle Si-thiol functionalized silica gel (2 g) was added to each solution. The solutions were warmed to 40° C. and aged for 1 h. The solutions were filtered thru a medium glass funnel and washed with EtOAc (4 L) and combined. The filtrate was evaporated to a semi solid, which was transferred to a 2 L round bottom flask, to which EtOAc (0.4 L) was added. The resulting white precipitate slurry was cooled to −5° C. and stirred for 1 h. The slurry was filtered and washed twice with cold EtOAc (100 mL). The solids were dried in a vacuum oven at 40° C. for 40 h to afford 84.0 g (36.5% yield, 98.8 area % purity) of the title Compound 18 as a white solid. Anal. Calcd for C25H21N2OF3.0.04% H2O.0.15 mol MeOH: C, 70.48; H, 5.14: N, 6.42; F, 13.06 Found: C, 70.54; H, 4.83: N, 6.18; F, 13.33
Example 10.2 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol monosodium salt (Cpd 18)
A 5-L 4-neck flask equipped with a thermocouple controller, an overhead mechanical stirrer, and a nitrogen inlet/outlet was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (125.0 g, 0.510 mol) and MeOH (1.25 L). A solution of sodium methoxide in methanol (0.5 M, 592 mL, 0.3 mol) was added. The reaction was heated to 65° C. for 30 min and all solids dissolved. The solution was cooled and evaporated to dryness. The foam was collected by scraping it out of the flask. The solids were placed in vacuum oven for 24 h at 40° C. to afford 139 g (about 100% isolated yield) of the title Compound 18 monosodium salt as a yellowish solid.1H NMR (400 MHz, DMSO-d6) δ 7.80-7.84 (m, 3H), 7.74 (d, 2H, J=8.59 Hz), 7.65 (d, 1H, J=16.4 Hz), 7.40-7.44 (m, 2H), 7.25-7.37 (m, 2H), 7.16-7.20 (m, 1H), 7.01-7.05 (m, 1H), 6.84-6.87 (m, 1H), 1.23 (s, 6H). Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3. m.p. (uncorr.) 258-259° C.
Example 10.3 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol hydrochloride salt (Cpd 18)
A 250-mL separatory funnel was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (1.0 g, 2.4 mmol) and EtOAc (20 mL). Aqueous HCl (1M, 20 mL) was added to the white slurry, and the separatory funnel was shaken. The solid product quickly dissolved, and a white precipitate started to form. The organic layer was transferred to a 100 mL round bottom flask equipped with a magnetic stir bar, and was stirred for 2 h. The thick slurry was filtered, rinsed with EtOAc (2×5 mL), and put into a vacuum oven at 40° C. for 36 h to afford 0.95 g (87.5%) of the title Compound 18 hydrochloride salt.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


































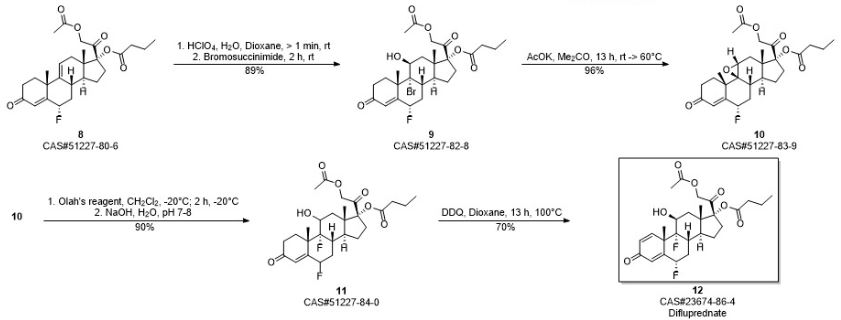





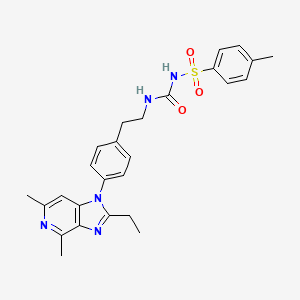













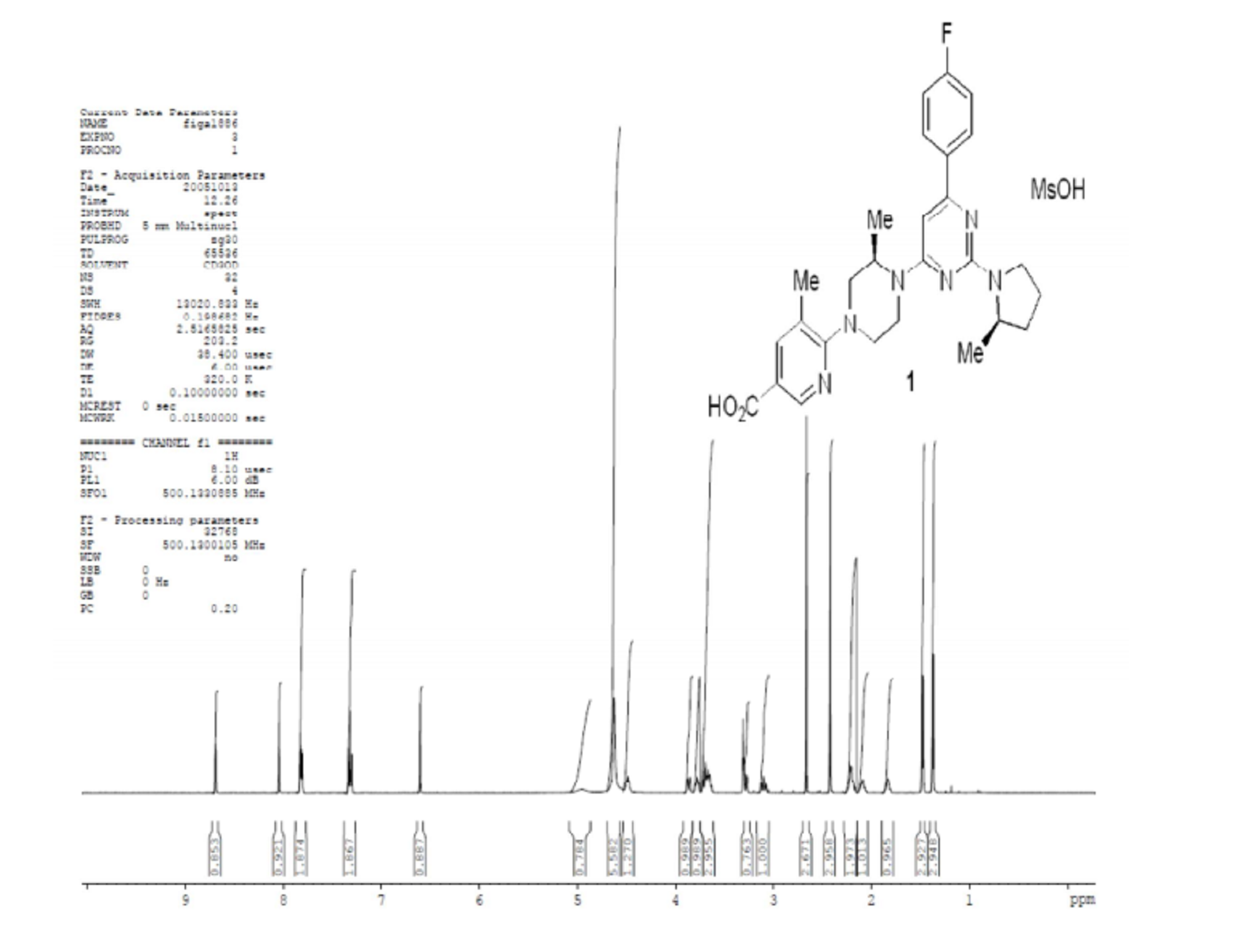
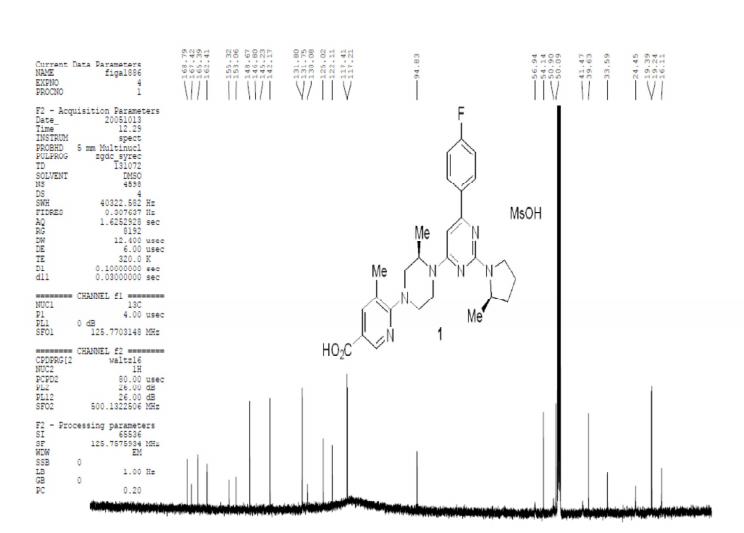

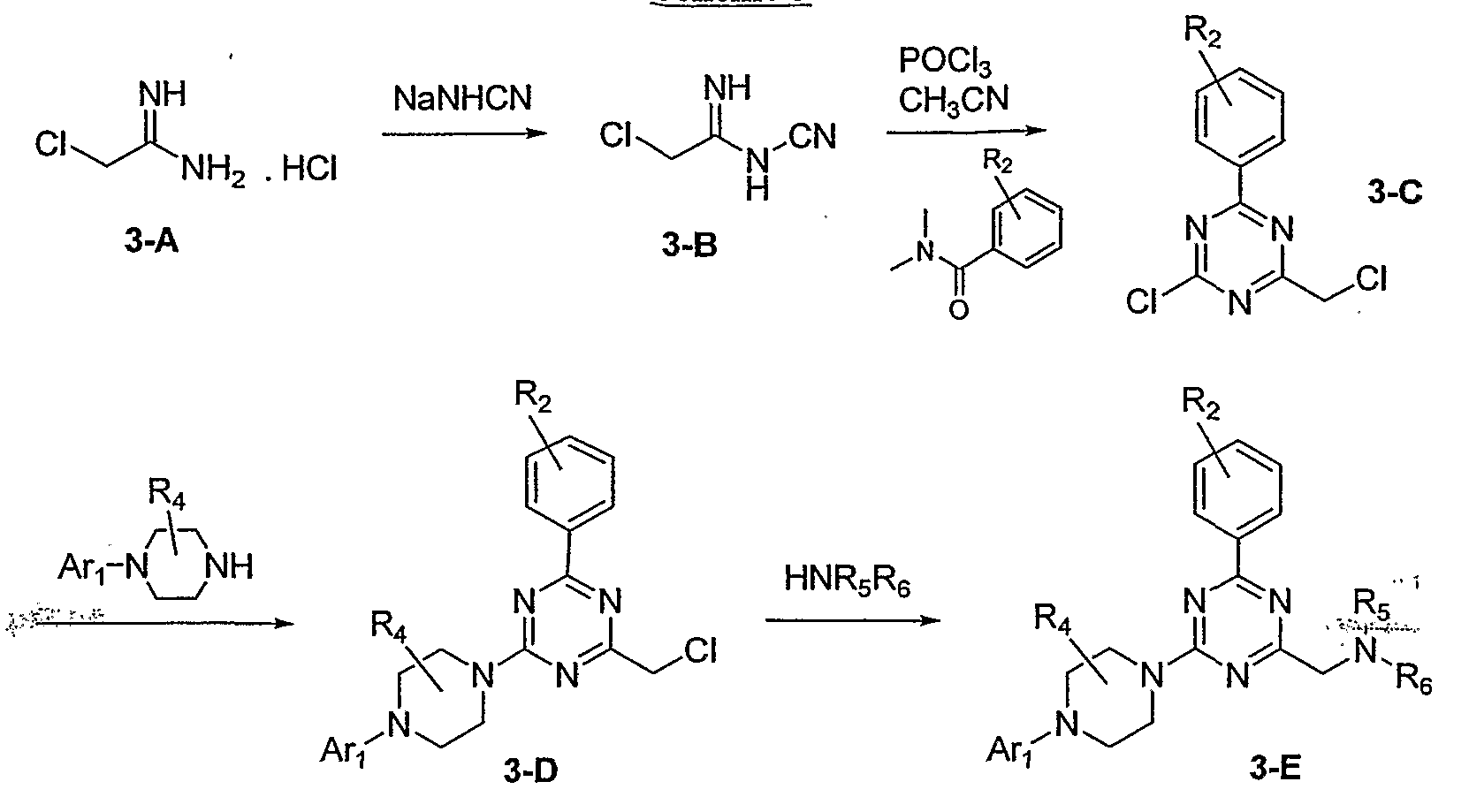

























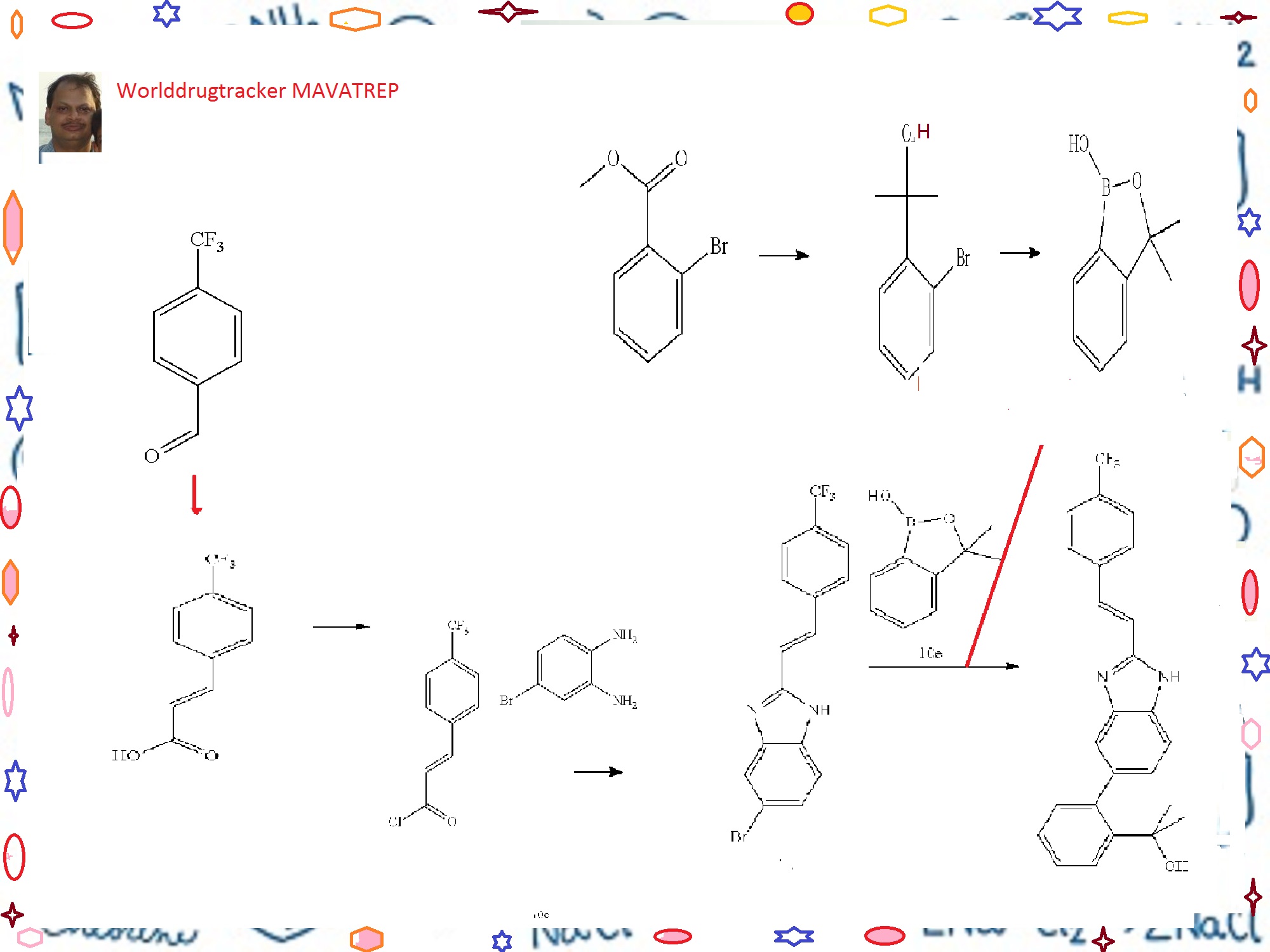



{kind=link}
{kind=link}
{kind=link}