




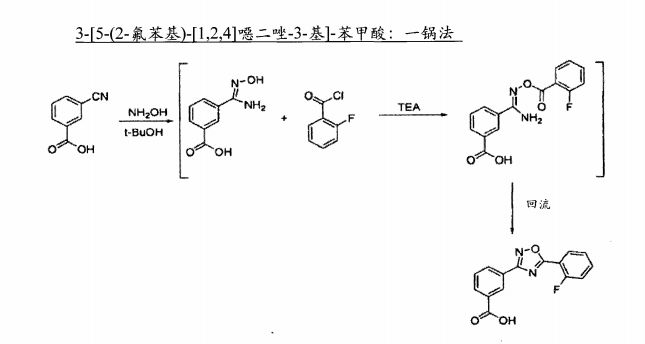
| Novel Tricyclic Compounds [US2011311474] | 2011-12-22 |
Home » Posts tagged 'Orphan Drug' (Page 7)
Tag Archives: Orphan Drug
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
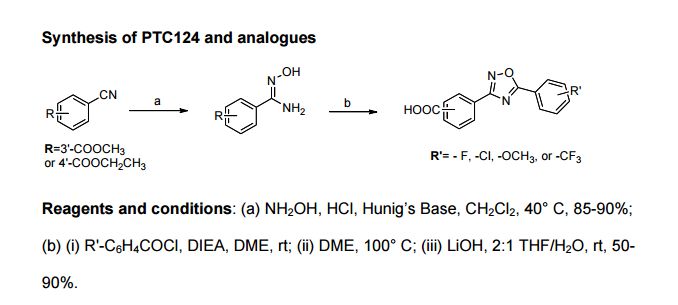
Ataluren (Translarna) drug for Duchenne Muscular Dystrophy

Ataluren (Translarna)
3-(5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl)benzoic acid
3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
CAS 775304-57-9
PTC Therapeutics (Originator)
- Molecular FormulaC15H9FN2O3
- Average mass284.242 Da
- EC-000.2051
NCGC00168759-02PTC-124, PTC124,UNII:K16AME9I3V
- EU 2014-07-31 APPROVED

Ataluren, formerly known as PTC124, is a pharmaceutical drug for the treatment of Duchenne muscular dystrophy and potentially other genetic disorders. It was designed by PTC Therapeutics and is sold under the trade name Translarna in the European Union.
Ataluren was approved by European Medicine Agency (EMA) on July 31, 2014. It was developed and marketed as Translarna® by PTC Therapeutics.

Ataluren was regulator of nonsense mutations indicated for the treatment of Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene, in ambulatory patients aged 5 years and older.
Translarna® is available as granules for oral use, containing 125 mg, 250 mg or 1000 mg of free Ataluren. The recommended dose is 10 mg/kg body weight in the morning, 10 mg/kg body weight at midday, and 20 mg/kg body weight in the evening.
Medical uses
Ataluren has been tested on healthy humans and humans carrying genetic disorders caused by nonsense mutations,[1][2] such as some people with cystic fibrosis and Duchenne muscular dystrophy. It is approved for the use in Duchenne in the European Union.

Mechanism of action
Ataluren makes ribosomes less sensitive to premature stop codons (referred to as “read-through”). This may be beneficial in diseases such as Duchenne muscular dystrophy where the mRNA contains a mutation causing premature stop codons or nonsense codons. Studies have demonstrated that PTC124 treatment increases expression of full-length dystrophin protein in human and mouse primary muscle cells containing the premature stop codon mutation for Duchenne muscular dystrophy and rescues striated muscle function.[3] Studies in mice with the premature stop codon mutation for cystic fibrosis demonstrated increased CFTR protein production and function.[4] The European Medicines Agency review on the approval of ataluren concluded that “the non-clinical data available were considered sufficient to support the proposed mechanism of action and to alleviate earlier concerns on the selectivity of ataluren for premature stop codons.” [5]
In cystic fibrosis, early studies of ataluren show that it improves nasal potential difference.[6] Ataluren appears to be most effective for the stop codon ‘UGA’.[1]

History
Clinical trials
In 2010, PTC Therapeutics released preliminary results of its phase 2b clinical trial for Duchenne muscular dystrophy, with participants not showing a significant improvement in the six minute walk distance after the 48 weeks of the trial.[7] This failure resulted in the termination of a $100 million deal with Genzyme to pursue the drug.
Phase 2 clinical trials were successful for cystic fibrosis in Israel, France and Belgium.[8] Multicountry phase 3 clinical trials are currently in progress for cystic fibrosis in Europe and the USA.[9]
Approval
On 23 May 2014 ataluren received a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA).[10]Translarna was first available in Germany, the first EU country to launch the new medicine.[11]
In August 2014, ataluren received market authorization from the European Commission to treat patients with nonsense mutation Duchenne muscular dystrophy. A confirmatory phase III clinical trial is ongoing.[11] The drug does not yet have approval by the US Food and Drug Administration.
In October 2015, NICE asked for further evidence of benefit to justify the “very high cost”.[12] NICE estimated that for a typical patient, treatment would cost £220,256 per year.
In February 2016, FDA declined to approve or even discuss PTC Therapeutics application for ataluren because it deemed the data presented by the developer “insufficient to warrant a review”.[13]

Ataluren Molecule
PAPER
http://www.pnas.org/content/106/9/3585.full
http://www.pnas.org/content/suppl/2009/02/10/0813345106.DCSupplemental
http://www.pnas.org/content/suppl/2009/02/10/0813345106.DCSupplemental/Appendix_PDF.pdf
Samples were analyzed for purity on an Agilent 1200 series LC/MS equipped with a Luna® C18 reverse phase (3 micron, 3 x 75 mm) column having a flow rate of 0.8-1.0 mL/min. The mobile phase was a mixture of acetonitrile (0.025% TFA) and H2O (0.05% TFA), and temperature was maintained at 50 °C. A gradient of 4% to 100% acetonitrile over 7 minutes was used during analytical analysis. Purity of final compounds was determined to be >95%, using a 5 μL injection with quantitation by AUC at 220 and 254 nM. High resolution mass spectra were obtained with an Agilent 6210 Time-of-Flight LC/MS with a 3.5 um Zorbax SB-C18 column (2.1 x 30 mm) (solvents are Water and ACN with 0.1% Formic Acid). A 3 minute gradient at 1 mL/min from 5% to 100% acetonitrile was used.
3-[5-(2-fluorophenyl)-[1,2,4]-oxadiazol-3-yl]-benzoic acid (1a, PTC124).
1 H NMR (d6-DMSO, 400 MHz) δ 13.15-13.68 (bs, 1H), 8.62 (s, 1H), 8.31 (d, 1H, JHH = 6.8 Hz), 8.24 (t, 1H, JHH = 7.2 Hz), 8.17 (d, 1H, JHH = 7.4 Hz), 7.77-7.82 (m, 1H), 7.73 (t, 1H, JHH = 7.6 Hz), 7.53 (dd, 1H, JHH = 10.8 Hz, JHH = 8.4 Hz), 7.48 (t, 1H, JHH = 6.8 Hz).
13C NMR (d6-DMSO, 400 MHz) δ 172.72 (d, JCF = 4.4 Hz), 167.39, 166.52, 159.95 (d, JCF = 258.0 Hz), 135.80 (d, JCF = 8.8 Hz), 132.28, 131.97, 131.97, 131.04, 130.94, 129.86, 127.76, 125.4 (d, JCF = 3.6 Hz), 117.2 (d, JCF = 20.4 Hz), 111.6 (d, JCF = 11.2 Hz). LC-
MS: rt (min) = 5.713; [M+H]+ 285.1;
HRMS: (CI+, m/z), calcd for C15H10FN2O3 (MH+ ), 285.06814; found, 285.06769.
CLIP
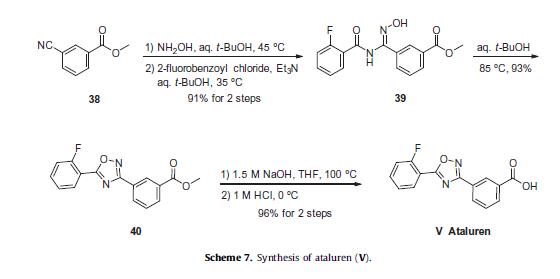
Ataluren (Translarna) Ataluren is a drug marketed under the trade name Translarna which was developed by PTC Therapeutics and approved by the European Union in May 2014 for the treatment of Duchenne’s muscular dystrophy (DMD) and potentially other genetic disorders.50
Ataluren renders ribosomes less sensitive to premature stop or ‘read-through’ codons, which are thought to be beneficial in diseases such as DMD and cystic fibrosis.51 Of the reported synthetic approaches to ataluren,52–55 the most likely process-scale approach consists of the sequence described in Scheme 7, which reportedly has been exemplified on kilogram scale.56
The sequence to construct ataluren, which was described by the authors at PTC Therapeutics, commenced with commercially available methyl 3-cyanobenzoate (38).56 This ester was exposed to hydroxylamine in aqueous tert-butanol and warmed gently until the reaction was deemed complete.
Then this mixture was treated with 2-fluorobenzoyl chloride dropwise and subsequently triethylamine dropwise. To minimize exotherm and undesired side products, careful control of the addition of reagents was achieved through slow dropwise addition of these liquid reagents.
Upon complete consumption of starting materials and formation of amidooxime 39, the aqueous reaction mixture was then heated to 85 C to facilitate 1,2,4-oxadiazole formation, resulting in the tricyclic ester 40 in excellent yield across the three steps.
Finally,saponification of ester 40 through the use of sodium hydroxide followed by acidic quench gave ataluren (V) in 96% over the two-step sequence.57
50. Welch, E. M.; Barton, E. R.; Zhuo, J.; Tomizawa, Y.; Friesen, W. J.; Trifillis, P.;Paushkin, S.; Patel, M.; Trotta, C. R.; Hwang, S.; Wilde, R. G.; Karp, G.; Takasugi,J.; Chen, G.; Jones, S.; Ren, H.; Moon, Y. C.; Corson, D.; Turpoff, A. A.; Campbell,J. A.; Conn, M. M.; Khan, A.; Almstead, N. G.; Hedrick, J.; Mollin, A.; Risher, N.;Weetall, M.; Yeh, S.; Branstrom, A. A.; Colacino, J. M.; Babiak, J.; Ju, W. D.;Hirawat, S.; Northcutt, V. J.; Miller, L. L.; Spatrick, P.; He, F.; Kawana, M.; Feng,H.; Jacobson, A.; Peltz, S. W.; Sweeney, H. L. Nature 2007, 447, 87.
51. Hirawat, S.; Welch, E. M.; Elfring, G. L.; Northcutt, V. J.; Paushkin, S.; Hwang,S.; Leonard, E. M.; Almstead, N. G.; Ju, W.; Peltz, S. W.; Miller, L. L. J. Clin.Pharmacol. 2007, 47, 430.
52Karp, G. M.; Hwang, S.; Chen, G.; Almstead, N. G. US Patent 2004204461A1,2004.
53. Andersen, T. L.; Caneschi, W.; Ayoub, A.; Lindhardt, A. T.; Couri, M. R. C.;Skrydstrup, T. Adv. Synth. Catal. 2014, 356, 3074.
54. Gupta, P. K.; Hussain, M. K.; Asad, M.; Kant, R.; Mahar, R.; Shukla, S. K.; Hajela,K. New J. Chem. 2014, 38, 3062.
55. Lentini, L.; Melfi, R.; Di Leonardo, A.; Spinello, A.; Barone, G.; Pace, A.; PalumboPiccionello, A.; Pibiri, I. Mol. Pharm. 2014, 11, 653.
56. Almstead, N. G.; Hwang, P. S.; Pines, S.; Moon, Y. -C.; Takasugi, J. J. WO Patent2008030570A1, 2008.
57. Almstead, N. G.; Chen, G.; Hirawat, S.; Hwang, S.; Karp, G. M.; Miller, L.; Moon,Y. C.; Ren, H.; Takasugi, J. J.; Welch, E. M.; Wilde, R. G. WO Patent2007117438A2, 2007.
CLIP

Ataluren trial success: trial aborted.
07 September 2011 – Pharma……..http://chem.vander-lingen.nl/info/item/September_2011/id/190/mid/140
Last week the newspaper NRC Handelsblad reported on a court case in which the parents of two young boys sued a pharmaceutical company over access to one of their developmental drugs. The drug in question wasAtaluren, the pharmaceutical companyPTC Therapeutics. The boys suffer from Duchenne muscular dystrophyand had taken part in a clinical trial. Whereas the results of this trial on the whole were inconclusive the boys did seriously benefit from the drug. Hardly any wonder the parents took action when the whole development program was canceled.
And the judge? He threw the case out arguing that doctors do not make the compound themselves and arguing that the compound is not commercially available. Are these arguments valid? and do the boys have options?
It is not that ataluren is a complex molecule. To judge from one of the patents, synthesis is straightforward starting from 2-cyanobenoic acid and 2-fluorobenzoyl chloride, both commercially available. The synthetic steps are methylation of 2-cyanobenoic acid (iodomethane), nitrile hydrolysis with hydroxylamine, esterification with the fluoro acid chloride using DIPEA, high-temperature dehydration to the oxadiazole and finally ester hydrolysis (NaOH).
Except for the fluorine atom in it the compound is unremarkable. If you have to believe the Internet many Chinese companies produce and sell it. Ataluren is also still in the running as a potential treatment for some other diseases. So if need be the compound will be around for some time to come.
CLIP
Ataluren [3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid] is an orally available, small molecule compound that targets nonsense mutation. It is the first drug in its class and appears to allow cellular machinery to read through premature stop codons in mRNA, and thus enables the translation process to produce full-length, functional proteins.
Ataluren is developed and approved for the treatment of nonsense mutation Duchenne muscular dystrophy (nmDMD) by EU in July 2014 [1].

| Ataluren: 2D and 3D Structure |
Nonsense Mutations as Target for DMD
A single nucleotide change in the DNA sequence that introduces a premature stop codon is known as a nonsense mutation, a subset of a major class of premature termination codon (PTC) mutations. Nonsense mutations cause premature termination of translation resulting in the production of truncated polypeptides, which in turn halts the ribosomal translation process at an earlier site than normal, producing a truncated, non-functional protein [1].
Nonsense mutations are implicated in 5-70 % of individual cases of most inherited diseases, including Duchenne muscular dystrophy (DMD) and cystic fibrosis. Ataluren appears to allow cellular machinery to read through premature stop codons in mRNA, enabling the translation process to produce full length, functional proteins.
Ataluren Synthesis

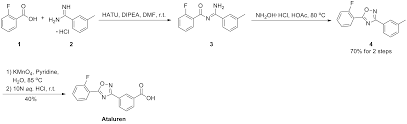
New J Chem 2014, 38, 3062-3070: The text reports one pot synthesis of Ataluren with an overall yield of 40%. It also reports few interesting and potent derivatives too.

WO 2007117438A2: It appears to be the industrial process. The patent also reports various pharmaceutically relevant assay and their results wrt Ataluren.
Identifications:

| 1H NMR (Estimated) for Ataluren |
Experimental: 1H NMR (d6-DMSO, 400 MHz) δ 13.15-13.68 (bs, 1H), 8.62 (s, 1H), 8.31 (d, 1H, JHH= 6.8 Hz), 8.24 (t, 1H, JHH = 7.2 Hz), 8.17 (d, 1H, JHH = 7.4 Hz), 7.77-7.82 (m, 1H), 7.73 (t, 1H, JHH = 7.6 Hz), 7.53 (dd, 1H, JHH = 10.8 Hz, JHH = 8.4 Hz), 7.48 (t, 1H, JHH = 6.8 Hz).

| 13C-NMR (Estimated) for Ataluren |
Experimental: 13C NMR (d6-DMSO, 400 MHz) δ 172.72 (d, JCF = 4.4 Hz), 167.39, 166.52, 159.95 (d, JCF = 258.0 Hz), 135.80 (d, JCF = 8.8 Hz), 132.28, 131.97, 131.97, 131.04, 130.94, 129.86, 127.76, 125.4 (d, JCF = 3.6 Hz), 117.2 (d, JCF = 20.4 Hz), 111.6 (d, JCF = 11.2 Hz)……https://ayurajan.blogspot.in/2016/05/ataluren-treatment-for-duchenne.html
CLIP
It is not that ataluren is a complex molecule. To judge from one of the patents, synthesis is straightforward starting from 2-cyanobenoic acid and 2-fluorobenzoyl chloride, both commercially available. The synthetic steps are methylation of 2-cyanobenoic acid (iodomethane), nitrile hydrolysis with hydroxylamine, esterification with the fluoro acid chloride using DIPEA, high-temperature dehydration to the oxadiazole and finally ester hydrolysis (NaOH).

CLIP
Route 1


Reference:1. WO2004091502A2 / US6992096B2.
2. WO2008045566A1 / US2008114039A1.
3. WO2008030570A1 / US2008139818A1.
Route 2
Reference:1. New. J. Chem. 2014, 38, 3062-3070.
Route 3


Reference:1. Adv. Synth. Catal. 2014, 356, 3074-3082.
CLIP
Carcinogenicity
Carcinogenicity bioassays in transgenic mice (26 weeks) and in rats (24 months):
● For Tg.rasH2 mouse: Ataluren did not increase the incidence of tumors up to the HDs in males (600 mg/kg/day) and in females (300 mg/kg/day). The non-neoplastic findings included endometrial hyperplasia and nephropathy in females.
● For rats: Urinary bladder tumors (benign urothelial cell papilloma [2 rats] and malignant urothelial cell carcinoma [1 rat]) were observed in 3/60 female rats dosed at 300 mg/kg/day. In addition, one case of malignant hibernoma was observed in 1/60 male rats at the dose of 300 mg/kg/day. The non-neoplastic toxicity consisted of a decrease of body weight.
PATENT
Example 1 (prepared by known ataluren)
Method ataluren according to Patent Document 2 is described in Example W02004091502A2 prepared.
Specific methods of preparation:
To a solution of 0.6 l of DMF was 44. 14g3- cyano acid 62.19 g of potassium carbonate was added, followed by stirring at room temperature for 30 minutes. 20 minutes To the suspension was added 28 ml of methyl iodide (450mmol), and the reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was poured into 1.2 l of ice water, stirred for 30 minutes, the precipitate was filtered out thereof. The white cake was dissolved in 70 ml of methanol, and then reprecipitated in cold water. To give 79% yield of 3-cyano-benzoic acid methyl ester.
50 g of 3-cyano-benzoic acid methyl ester was dissolved in 500 ml of ethanol, to which was added 41 ml of 50% aqueous hydroxylamine (620mmol). 100 ° C and the reaction mixture was stirred for 1 hour, the solvent was removed under reduced pressure. So that the oily residue is dissolved in 100 ml of 20/80 ethanol / toluene, concentrated again. To give 61 g 3- (N- hydroxy amidino (carbamimidoyl)) – benzoic acid methyl ester.
60 g of 3- (N- hydroxy amidino (carbamimidoyl)) – benzoic acid methyl ester was dissolved in 200 ml of anhydrous tetrahydrofuran, followed by adding thereto 75 ml of diisopropylethylamine (434 mmol), and then 20 minutes this mixture was added 48.1 ml 2- fluorobenzoyl chloride (403mmol). The reaction mixture was stirred at room temperature for 1 hour. The precipitate was filtered off, the filtrate was concentrated under reduced pressure. The residue was dissolved in 400 ml of ethyl acetate, washed with 400 ml of water and then twice. The solvent was removed under reduced pressure, containing 60% ethyl acetate in hexane to give the desired product, generating 81 g 3- (N-2- amidino-fluorobenzoyl) – benzoate.
at 130 ° C with a Dean-Stark apparatus was dissolved in 500 ml of toluene was heated under reflux in 44 g of 3- (N-2- fluorobenzoyl) -1,2,3,4-_ benzoate 4 hours. 5 ° C and the reaction mixture was stirred for 18 hours. The white precipitate was filtered off, the filtrate was concentrated, recrystallized in toluene. To give 38 g of 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester.
33 g of 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester was dissolved in 400 ml of tetrahydrofuran, to which was added 100 ml of 1. 5M aqueous sodium hydroxide solution. At 100 ° C and the reaction mixture was heated at reflux for 2 hours. The solvent was removed under reduced pressure at 5 ° C the solution was stirred for 2 hours. The organic solvent was removed, washed with 50 mL of water. The aqueous solution was then acidified with hydrochloric acid to pH 1. The white precipitate was filtered off, the filter cake washed with cold water, then dried with a freeze dryer. To give 3.0 g of 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] benzoic acid. 1H-NMR (500MHz, d6-DMS0): 8. 31 (1H), 8 18 (2H), 8 08 (1H), 7 88 (2H), 7 51 (2H)….. Display: ataluren- Sample Preparation Example 1 prepared in Preparation Example 2 and TO2004091502A2 induced.
Each prepared in Example 2 (prepared according to known Form A)
Method [0084] A known polymorph according to Patent Document W02008039431A2 Example 5. 1. 1.1 prepared as described. Specifically: ataluren be prepared 1 100 mg Preparation Example, 60 ° C add 16.2 ml of isopropanol ultrasound clear solution, the solution by 2 square micron filter and the filtrate was kept covered with aluminum foil having a small hole. vial, 60 ° C and evaporated. The solid formed was isolated to give ataluren the A polymorph.
as needles.
its XRPD shown in Figure 1, the display ataluren polymorph A disclosed in Patent Document W02008039431A2 consistent.
SEE
European Journal of Organic Chemistry (2016), 2016(3), 438-442
Russian Chemical Bulletin (2015), 64(1), 142-145.
European Journal of Medicinal Chemistry (2015), 101, 236-244.
Bioorganic & Medicinal Chemistry Letters (2014), 24(11), 2473-2476.
New Journal of Chemistry (2014), 38(7), 3062-3070.
Proceedings of the National Academy of Sciences of the United States of America (2010), 107(11), 4878-4883, S4878/1-S4878/14.
WO 2008039431
WO 2008045566
WO 2008030570
WO 2007117438
WO 2006110483
WO 2007117438
WO 2006110483
US 20040204461
PATENT
novel crystalline forms of 3-[5-(2-fluorophenyl)-
[l,2,4]oxadiazol-3-yl]-benzoic acid, which has the following chemical structure (I):

(I)
In particular, crystalline forms of 3-[5-(2-fluorophenyl)-[l,2,4]oxadiazol-3-yl]- benzoic acid are useful for the treatment, prevention or management of diseases ameliorated by modulation of premature translation termination or nonsense-mediated mRNA decay, as described in U.S. Patent No. 6,992,096 B2, issued January 31, 2006, which is incorporated herein by reference in its entirety. In addition, the present provides a crystalline form of 3-[5-(2-fluorophenyl)-[l,2,4]oxadiazol-3-yl]-benzoic acid which is substantially pure, i.e., its purity greater than about 90%.
Processes for the preparation of 3-[5-(2-fluorophenyl)-[l,2,4]oxadiazol-3-yl]- benzoic acid are described in U.S. Patent No. 6,992,096 B2, issued January 31, 2006, and U.S. patent application no. 1 1/899,813, filed September 9, 2007, both of which are incorporated by reference in their entirety.
PATENT
Example
3- ‘5- (2-fluorophenyl) – “1,2,41 oxadiazol-3-yl benzoate 1- Batch 1
The 3-cyano-benzoic acid methyl ester (105 kg) and t-butanol was added molten drying reactor. Under an inert atmosphere for about 2 hours 48 minutes, 50. /. Aqueous hydroxylamine (43L, 47.4 kg) was added to a clear solution of 3-cyano benzoic acid methyl ester in a molten in t-butanol. The addition of a 50% aqueous solution of hydroxylamine period, the maximum temperature batch of about 43 ° C. 50% aqueous solution of hydroxylamine addition rate of from about 9L / h when the changes start adding to about 30L / hr. To maintain the temperature of the batch by varying the reactor jacket set point. In particular, the set value is about 40.5 ° C, with the addition of a rate increase at the beginning join, change the setting to about 29.6 ° C. After about 40-45t stirred for about 4 hours, the reaction was deemed complete (i.e., less than about 0.5% ester).
The batch was transferred to a drying reactor, additional (chased through) approximately 10L molten tert-butanol. Jacket setpoint from about 33 when the batch was received when dried reactor. C is reduced to about 27 after the completion of the transfer. C. Batch crystallization was observed part, which does not adversely affect stirring. The batch was cooled to about 34.4 ° C, triethylamine (72.6 kg, IOOL) added to the reactor. The jacket temperature set value from about 20.4. C is increased to about 31.0 ° C, in order to maintain the batch temperature in the range of about 30-35t. With molten tert-butanol (IO L) was washed with a linear (line rinse) After the batch was added to the 2-fluorobenzoyl chloride (113.7 kg, 86.0L).Charge is added to the first third of the rate of about 25L / hr. In the meantime, the jacket inlet temperature was lowered to about 15 ° C, the batch temperature is maintained at about 34.6 ° C. In about 5.5 hours after the addition was complete.During the addition, the maximum temperature of the batch was about 38.8 ° C. Near the end of the addition, the addition rate slowed to about 11L / hr was added last 27 liters of 2-fluoro-benzoyl chloride. 30-35. C After stirring for about 2 hours, that the reaction was complete (i.e., less than about 0.5% of methyl 3-amidinophenoxy). Then, after about 1 hour 42 minutes, the batch was heated to reflux temperature (about 82 ° C), and then stirred for about 18 hours. During the stirring, a number of product partially crystallized to form a slurry. The slurry was cooled to about 40. C thus sampled, during which complete crystallization occurs. The batch was then heated to reflux temperature and stirred for about 1 hour 50 minutes.Then, after about two hours, the batch was cooled to about 69 ° C, and after about four hours and 15 minutes, slowly added 630L of pure water, while maintaining the batch temperature at about 66-69 ° C. After about 3 hours 14 minutes, the slurry was cooled to about 22.4 ° C, and transferred to 2x200L ceramic filter, the ceramic filter equipped 25-30n polypropylene mesh filter cloth. In about 55 minutes after the completion of material from the container to the filter transfer. With 50n /. The tert-butanol solution (210L) was washed cake was washed for about 10 minutes so that the cleaning liquid can penetrate into each cake. Then, the cake was dried in a vacuum for about 5-10 minutes. The purified water as a second washing (158L / cake) applied to the filter cake to remove residual t-butanol and triethylammonium chloride salt. Dried in a vacuum for about 5 minutes, the solution was removed. In vacuo and then the cake was dried for about 2 hours, and then sampled using liquid chromatography. The filter cake was measured by liquid chromatography purity of about 99.6%.
The filter cake was dried in vacuo for about 8 hours 25 minutes later, the wet cake (207.4kg) is transferred to an air oven. At about 50-55. C, the oven dried in air for about 52 hours. The product was isolated in a total yield of about 89.9% (174.65kg), in the calculation of cost of materials sampling, you can adjust the overall yield of about 90.7%.
Batch 2
The 3-cyano-benzoic acid methyl ester (105 kg) and t-butanol was added molten drying reactor. Under an inert atmosphere for about 3 hours 29 minutes, 50% aqueous solution of hydroxylamine (47.85 kg) was added to the reactor. During the addition, the temperature is maintained at about 40-45 ° C. At about 40-45. C After stirring for about 3 hours 16 minutes, that the reaction was complete (i.e., less than about 0.5% ester). As for the drying reactor, the batch was transferred to one of the batch in. The batch was cooled
To about 34.4 ° C, and triethylamine (72.6 kg, 100 L). During about 45 minutes was added, while maintaining the batch temperature between about 30-35 ° C. During the addition, the jacket inlet temperature of from about 31.4. C increased to about 32.6. C. After the molten tert-butanol linear washed, was added to the batch 2- fluorobenzoyl chloride (l 13.7 kg, 86.0 L). After about 3 hours, 27 minutes, add the acid chloride. 35. C under stirring for about 8 hours, that the reaction is not complete (i.e., more than about 0.5% residual 3-amidino-benzoyl ester). Then, 1.5% by weight of the original charge of triethylamine and 2-fluorobenzoyl chloride was added to the batch. Linear washed with tert-butanol (IO L) associated with each additional charge. During the addition of the acid chloride, no additional cooling. The batch was maintained at a temperature of about 30-35 ° C, the jacket inlet temperature range was maintained at about 30.3. C to about 33.0 ° C. After stirring for about 2 hours at 30-35t, that reaction was complete (i.e., less than 0.5% of methyl 3-amidinophenoxy).
After about 1 hour and 44 minutes, the batch was heated to reflux temperature (about 83 ° C), and stirred for about 18 hours.The same batch 1, during cooling the sample, the solid was completely crystallized. The batch was then heated to reflux temperature and stirred for about 1 hour and 2 minutes. Then, after about 2 hours and 20 minutes, the batch was cooled to about 69.2 ° C, and after about four hours and 30 minutes, slowly added 630 L of pure water, while the temperature of the batch was maintained at about 65.6-69.2 ° C. After about 3 hours and 30 minutes, the slurry was cooled to about 23.4 ° C, and, as for, the contents were transferred to one of the double batch of the ceramic filter. About 5 hours and 6 minutes, to complete the transfer of the material. With about 50% of t-butanol (2 volumes / cake) was washed filter cake was washed with 10 minutes to allow the cleaning liquid to penetrate into each cake, then dried in vacuo. About 1 hour and 40 minutes, the filter is completed. The purified water was added to a final wash the filter cake. The liquid was removed by drying under vacuum for about 10 minutes. In vacuo and then the cake was dried for about 2 hours and 5 minutes, and then sampled using liquid chromatography. The cake purity liquid chromatography were about 99.5% and 99.6%. After the cake was then dried in vacuo for about 2 hours and 5 minutes, the wet cake (191.5 kg) is transferred to an air oven. At about 50-55. C under dry in an air oven for about 48 hours. The product was isolated in a total yield of about 92.5% (179.7 kg).
Lot 3
The 3-cyano-benzoic acid methyl ester (52.5 kg) and molten tert-butanol (228 kg) added to the reaction vessel. The vessel was sealed, the batch temperature set of about 40-45 ° C, and the stirrer is started. Under an inert atmosphere, after 2 hours 40 minutes, 50% of the shoes amine solution (24 kg) was added to the reactor. During the addition, the temperature is maintained at about 40-45 ° C. In about 42. Under C, then further stirred for about 5 hours to complete the reaction.
The batch was cooled to 30-35 ° C, and after 15 minutes, was added triethylamine (36 kg). After about 2 hours 44 minutes, was added 2-fluorobenzoyl chloride (57 kg). During the addition, batch temperature was maintained at about 30-35 ° C.Under the 32t, the batch was stirred for 2 hours 10 minutes to complete the reaction.
After about 50 minutes, the batch was heated to reflux temperature (about 83-86 ° C), at about 8rc, stirred for about 18 hours. Then, over about two hours, the batch was cooled to about 65-70 ° C, and after about 6 hours 25 minutes, slowly added to purified water (315 L), while the batch temperature was maintained at about 65- 70 ° C. After about 2 hours and 15 minutes, the slurry was cooled to about 22 ° C, and the contents were transferred to a centrifuge filter (2 batches). About 1 hour and 40 minutes, the filter is completed. After about 20 minutes, with about 50% aqueous solution of tert-butyl alcohol (90 kg / cake), dried cake. The purified water (79 kg / cake) as the last added to the filter cake washed. At about 900 rpm drying the cake for about 1 hour and 5 minutes, then filled cylinder. Liquid chromatography wet cake (91.5 kg, LOD = 5% w / w) of a purity of about 99.75% area.
3- ‘5- (2-fluorophenyl) -fl, 2,41 oxadiazol-3-yl l- acid batch 1
3- [5- (2-fluorophenyl) – [l, 2,4] oxadiazol-3-yl] – benzoic acid methyl ester (74.0kg) added to the reaction vessel, the vessel is sealed, evacuated and purification. Jacket set value of about 35. C, start the stirrer in the container. Molten tert-butanol (222 L, 3 volumes) and purified water (355 L, 4.8 vol) was added to the vessel. After the addition was added 25.1% w / w aqueous sodium hydroxide solution (43.5 kg, 1.1 molar equivalents), and with additional purified water (100L, 1.35 mol) was washed linear. During the addition, the batch temperature from about 39.0t reduced to about 38.8 ° C. After about 1 hour and 54 minutes, the batch temperature to about 63-67. C, and then, after about 30 minutes, which was adjusted to about 68-72.C. About 68-72t, stirring the mixture for about 3 hours. Then, after about five hours 11 minutes, the solution was cooled to about 40-45 ° C. Then, after the above process, after about three hours 33 minutes, the solution was then heated to about 68-72. C.
Jacket temperature of the reaction vessel was set to about 60 ° C, the stirrer started, and at about 70 ° C, a slightly positive pressure of nitrogen (1.5 to 5.6 psig), the heat transfer liquid through a micron filter . During the transfer, the product temperature is reduced to about 64.3 ° C, the transfer is completed in about 45 minutes. Was added to the purified water container (61 L, 0.82 vol) and the contents were heated to about 68-72. C.
The batch temperature was adjusted to about 69.4 ° C, and after about four hours and 18 minutes, with 13.9% w / w sulfuric acid (100.7 kg, 1.15 mol equiv.). During the addition, batch temperature was maintained at about 68.0-70.8 ° C. After the addition of the acid, with purified water (50 L, 0.68 vol) line wash at about 68-72 ° C, the stirring was continued for 31 minutes.
After about 4 hours and 10 minutes, the batch in a linear fashion from about 69.2t cooled to about 41.2 ° C. The stirrer Rosenmund filter / dryer was elevated to the highest position and jacket set value is set at about 40 ° C. The slurry was transferred to the two portions of the filter / drier. Applying a constant nitrogen pressure to the first portion (less than about 15 psig). During the transfer, a pressure of about 23.9 to about 28.8 psi, the transfer is complete in about 1 hour and 5 minutes. The second part of the slurry was transferred onto the filter cake, and the composite was stirred briefly to homogenize the batch. Use about 26.1 to about 29.1 psi nitrogen pressure filters the second part, after about three hours, squeeze the cake so that it does not contain liquid. With about 38-42 ° C hot tert-butanol solution (352 kg, 5 volumes) and about 65-70 ° C in 3x hot purified water (370 L, 5 volumes) and the filter 々.
Said filter / dryer jacket temperature was set to about 43 ° C, the product was dried under vacuum for about 26 hours while stirring periodically. Determination of purity of about 99.7%. The product was isolated in a total yield of about 74.4% (52.45 kg).
Batch 2
Was added to the reactor vessel 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester (47 kg, wet cake) and melt-hyun tert-butyl alcohol (111.4 kg). A sealed container, and the batch temperature was set at 30-40t, and start the stirrer. The purified water (51.6 kg) was added to the vessel. After the addition was added 3.47% w / w aqueous sodium hydroxide solution (202.4 kg). After about l hour, the batch temperature to about 67-73. C, then, at about 7 (under TC, stirred for about three hours.
Under a slight positive pressure of nitrogen, with a 1 micron polypropylene bag filter the batch, and then transferred to the new reactor. Was added to the vessel pure water (146 kg), and heating the batch to about 68-72. C.
After about four hours, the 10.7% aqueous hydrochloric acid was added to the batch. During the addition, batch temperature was maintained at about 68-72 ° C. PH was measured by using the batch pH of about 2.2, and then stirring was continued at about 7 (under TC about 1 hour.
After about two hours, the batch in a linear fashion from about 70. C is cooled to about 60 ° C. After about two hours, about 60. C of the batch in a linear fashion from about 6 (TC was cooled to about 40 ° C. In 40t, the batch was stirred for 2 hours, and the slurry was transferred to a centrifuge filter. After about 30 minutes, filtered completion . After about 30 minutes, with about 42Mw / w in t-butanol solution (165kg) cake was washed. The purified water (118kg, 4 (TC) as the last added to the filter cake was washed. The filter cake was dried at about 900rpm about 1 hour, then filled cylinder.
The wet cake was transferred to a paddle dryer (a double cone drier also suitable for this step), the jacket temperature was set to about 70. C. At about 70. C, the product was dried under vacuum for about 48 hours while stirring periodically.Determination of purity of about 99.8%. The product was isolated overall yield of about 74% (68.5 kg).
Lot 3
To the reaction vessel was added 3- [5- (2-fluorophenyl) – [1,2,4] oxadiazol-3-yl] – benzoic acid methyl ester (10 g) and t-butanol fused (128mL ). The batch temperature was set to 30-40 ° C, and the stirrer is started. After about 30 minutes, the aqueous sodium hydroxide solution 4.48% w / w of (32.5 g) was added to the vessel. The batch was maintained at a temperature of about 40-50 ° C. After about l hour, the batch temperature is raised to about 78-82 ° C, and then, at about 78-82t, and then stirred for about one hour. Under positive pressure of nitrogen, a polyethylene bag with a 5 micron filter the batch, and then transferred to a new reaction vessel. The batch was maintained at a temperature of about 78-82 ° C.
It was added to a new vessel 37% aqueous hydrochloric acid (4 mL) and tert-butanol molten (8 mL). The temperature was maintained at about 30-40. Under C, and stirring the mixture for about 30 minutes.
After about four hours, using a metering pump was added to the batch of hydrochloric acid in tert-butanol. After about SO-SO minutes before adding half filled. The stirrer speed is set at about 200rpm. After about 3.5 hours, add the remaining charge. The stirrer speed is set at about 100 rpm. During the addition, batch temperature was maintained at about 78-82 ° C.PH meter with a final batch pH was adjusted to about 1.2, at about 78-82t, then continue stirring for about l hour. After about one hour, the batch in a linear fashion from about 78-82. C is cooled to about 70 ° C. After about four hours, about 7 (TC batches in a linear fashion from 70.C cooled to about 50 ° C, and the stirrer speed was set at about 80 rpm. After about four hours, about 50 ° C Batch linearly cooled from 50 ° C to about 40t, and stirrer speed was set at approximately 60rpm. In 40.C, the batch was stirred for a further 4 hours.
The temperature of the filter is set to about 40-45 ° C. The slurry was transferred to the filter. After about one minute to complete filtration. After about two minutes, with tert-butanol (50 mL, 50.C) washing the filter cake. The pure water (IOO mLx2, 60.C) as the last wash was added to the cake. Under vacuum at about 60-70 ° C the cake was dried for about 12 hours, and then loaded into the container.
Determination of HPLC purity of about 99.9% of the area. The yield of isolated product was about 94% (9.0g).
3- “5- (2-fluorophenyl) -” 1,2,41-oxadiazol-3-yl 1- acid: One-pot
The methyl 3-cyanophenyl Yue (7.35 g) and tert-butanol molten (100 mL) added to the reactor vessel. Sealed containers, the batch temperature was set to 60 ° C, and the stirrer is started. The suspension was stirred for 1 hour and then the batch temperature was set to 40. C. Under an inert atmosphere, after three hours, 50% aqueous solution of hydroxylamine (3.63 g) was added to the reactor. During the addition, batch temperature was maintained at 38-41 ° C. 40. C After stirring for 18 hours, to complete the reaction.
The batch was cooled to 27 ° C, and after two minutes, triethylamine (5.56 g). After 3 hours, was added 2-fluorobenzoyl chloride (7.82 g). During the addition, batch temperature was maintained at 24-27 ° C. 40. C, the batch was stirred for a further 4 hours.
After 30 minutes, the batch was heated to 79 ° C, and at about 79. C was stirred for 16 hours. After 3 hours, the white suspension was added to the water (IOO mL), while the batch temperature was maintained at 70 ° C. After 20 minutes, a 37% aqueous hydrochloric acid were added to the batch. PH was measured by using the batch pH of about 2.2, stirring was continued at about 70t for about 1 hour.
After three hours, the batch in a linear manner from 7 (TC cooled to 30 ° C, and the slurry is transferred to the filter. After 5 minutes, the filtering is done. After five minutes, with tert-butanol (50mL, 40 .C) filter cake was washed. The purified water (IOO mL, 60.C) is added to a final wash the filter cake. In 70.C of the filter cake was dried in a vacuum oven for 18 hours and then removed. Determination of purity approximately 98.68%. The total yield of isolated product of about 76% (10.8g).
PICS

A large-scale, multinational, phase 3 trial of the experimental drug ataluren has opened its first trial site, in Cincinnati, Ohio.
The trial is recruiting boys with Duchenne muscular dystrophy (DMD) or Becker muscular dystrophy (BMD) caused by anonsense mutation — also known as a premature stop codon — in the dystrophin gene. This type of mutation causes cells to stop synthesizing a protein before the process is complete, resulting in a short, nonfunctional protein. Nonsense mutations are believed to cause DMD or BMD in approximately 10 to 15 percent of boys with these disorders.
Ataluren — sometimes referred to as a stop codon read-through drug — has the potential to overcome the effects of a nonsense mutation and allow functional dystrophin — the muscle protein that’s missing in Duchenne MD and deficient in Becker MD — to be produced.
The orally delivered drug is being developed by PTC Therapeutics, a South Plainfield, N.J., biotechnology company, to whichMDA gave a $1.5 million grant in 2005.
PTC124 has been developed by PTC Therapeutics.
References
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL (May 2007). “PTC124 targets genetic disorders caused by nonsense mutations”. Nature 447 (7140): 87–91. Bibcode:2007Natur.447…87W.doi:10.1038/nature05756. PMID 17450125.
- Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL (Apr 2007). “Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers”. Journal of clinical pharmacology 47 (4): 430–444.doi:10.1177/0091270006297140. PMID 17389552.
- Nature. 2007 May 3;447(7140):87-91.
- Proc Natl Acad Sci U S A. 2008 Feb 12;105(6):2064-9.
- Neuromuscul Disord. 2015 Jan;25(1):5-13.
- Wilschanski, M. (2013). “Novel therapeutic approaches for cystic fibrosis”. Discovery Medicine 15 (81): 127–133. PMID 23449115.
- “PTC Therapeutics and Genzyme Corporation announce preliminary results from the phase 2b clinical trial of ataluren for nonsense mutation Duchenne/Becker muscular dystrophy (NASDAQ:PTCT)”. Ptct.client.shareholder.com. Retrieved 2013-11-28.
- Wilschanski, M.; Miller, L. L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatsch, T.; Cohen-Cymberknoh, M.; Miller, N. L.; Reha, A.; Northcutt, V. J.; Hirawat, S.; Donnelly, K.; Elfring, G. L.; Ajayi, T.; Kerem, E. (2011). “Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis”. European Respiratory Journal 38 (1): 59–69. doi:10.1183/09031936.00120910. PMID 21233271.Sermet-Gaudelus, I.; Boeck, K. D.; Casimir, G. J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; Miller, N. L.; Constantine, S.; Reha, A.; Ajayi, T.; Elfring, G. L.; Miller, L. L. (November 2010). “Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis”. American Journal of Respiratory and Critical Care Medicine 182 (10): 1262–1272. doi:10.1164/rccm.201001-0137OC. PMID 20622033.
- “PTC Therapeutics Completes Enrollment of Phase 3 Trial of Ataluren in Patients with Cystic Fibrosis (NASDAQ:PTCT)”. Ptct.client.shareholder.com. 2010-12-21. Retrieved2013-11-28.
- http://www.marketwatch.com/story/ptc-therapeutics-receives-positive-opinion-from-chmp-for-translarna-ataluren-2014-05-23
- “PTC Therapeutics Announces Launch of Translarna™ (ataluren) in Germany”.marketwatch.com. 3 Dec 2014. Retrieved 27 Dec 2014.
- “NICE asks for further evidence for the benefits of a new treatment for Duchenne muscular dystrophy to justify its very high cost”.
- http://uk.reuters.com/article/us-ptc-therapeutics-fda-idUKKCN0VW1FG
External links
References:
1. Ryan, N. J. Ataluren: first global approval. Drugs 2014, 74(14), 1709-14. (FMO only)
2. Gupta, P. K.; et. al. A metal-free tandem approach to prepare structurally diverse N-heterocycles: synthesis of 1,2,4-oxadiazoles and pyrimidinones. New J Chem 2014, 38, 3062-3070 (FMO only)
3. Almstead, N. G.; et. al. Methods for the production of functional protein from dna having a nonsense mutation and the treatment of disorders associated therewith. WO2007117438A2
| WO2004091502A2 | Apr 9, 2004 | Oct 28, 2004 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acid compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8486982 | Jun 22, 2012 | Jul 16, 2013 | Ptc Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acids |
| US8796322 | Jun 19, 2013 | Aug 5, 2014 | Ptc Therapeutics, Inc. | Methods for using 1,2,4-oxadiazole benzoic acid compounds |
| US8975287 | Jun 18, 2014 | Mar 10, 2015 | Ptc Therapeutics, Inc. | Methods for using 1,2,4-Oxadiazole benzoic acid compounds |
| US9205088 | Jan 28, 2015 | Dec 8, 2015 | Ptc Therapeutics, Inc. | Compositions of 1,2,4-oxadiazol benzoic acid compounds and methods for their use |
| US9289398 | Mar 29, 2007 | Mar 22, 2016 | Ptc Therapeutics, Inc. | Methods for the production of functional protein from DNA having a nonsense mutation and the treatment of disorders associated therewith |
| Preparation | CN101535284A | CN101535284B | ||||
| 10 | Crystal | CN101541770A | ||||
| 11 | Crystal | CN104341371A | ||||
| 12 | Crystal | CN102382075A |
| Formula | CN1802360A | CN1802360B | ||||
| 2 | Combination | CN104056278A | ||||
| 3 | Indication | CN101076703A | ||||
| 4 | Indication | CN101076332A | ||||
| 5 | Indication | CN101076337A | ||||
| 6 | Indication | CN101193632A | ||||
| 7 | Formulation | CN103720688A | ||||
| 8 | Indication | CN101505739A |

 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
|
|
| Other names
PTC124
|
|
| Identifiers | |
| 775304-57-9 |
|
| ChEMBL | ChEMBL256997 |
| ChemSpider | 9394889 |
| 7341 | |
| Jmol 3D model | Interactive image |
| KEGG | D09323 |
| PubChem | 11219835 |
| UNII | K16AME9I3V |
| Properties | |
| C15H9FN2O3 | |
| Molar mass | 284.24 g/mol |
| Pharmacology | |
| M09AX03 (WHO) | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
///////ORPHAN DRUG, Ataluren, Translarna, Duchenne Muscular Dystrophy, EU, 775304-57-9, PTC Therapeutics, PTC 124
O=C(O)c1cccc(c1)c2nc(on2)c3ccccc3F
AVORALSTAT
Avoralstat, BCX4161,
CAS 918407-35-9
UNII: UX17773O15
513.5513, C28-H27-N5-O5
2-Pyridinecarboxylic acid, 3-(2-(((4-(aminoiminomethyl)phenyl)amino)carbonyl)-4-ethenyl-5-methoxyphenyl)-6-(((cyclopropylmethyl)amino)carbonyl)-
3-(2-((4-Carbamimidoylphenyl)carbamoyl)-4-ethenyl-5-methoxyphenyl)-6-((cyclopropylmethyl)carbamoyl)pyridine-2-carboxylic acid
Hereditary angioedema (HAE)
Kallikrein inhibitor
BioCryst Pharmaceuticals
![]()
BioCryst is also investigating second-generation plasma kallikrein inhibitors to avoralstat, for treating HAE (in February 2016, this program was listed as being in preclinical development).
Prevent acute attacks in patients with hereditary angioedema (HAE); Treat hereditary angioedema (HAE)
U.S. – Fast Track (Treat hereditary angioedema (HAE));
U.S. – Orphan Drug (Prevent acute attacks in patients with hereditary angioedema (HAE))
26 Feb 2016Clinical trials in Hereditary angioedema (Prevention) in USA (PO, Hard-gelatin capsule) before February 2016
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in France (PO, Soft-gelatin capsule)
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in Germany (PO, Soft-gelatin capsule)

| Conditions | Interventions | Phases | Recruitment | Sponsor/Collaborators |
|---|---|---|---|---|
| Hereditary Angioedema|HAE | Drug: BCX4161|Drug: Placebo | Phase 2|Phase 3 | Recruiting | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161|Drug: Placebo | Phase 2 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
Avoralstat, also known as BCX-4161, is a potent and orally active Kallikrein inhibitor and Bradykinin inhibitor. Avoralstat may be potentially useful for treatment for Hereditary angioedema. Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.

Selective inhibitor of plasma kallikrein that subsequently suppresses bradykinin production
Hereditary angioedema (HAE) is a serious and potentially life-threatening rare genetic illness, caused by mutations in the C1-esterase inhibitor (C1 INH) gene, located on chromosome 11q. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by C1 INH gene mutations which produce low levels of C1 -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective C1 protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; 100(Suppl 2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herin by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that C1 INH is the most important inhibitor of kallikrein in human plasma and that C1 INH deficiency leads to unopposed activation of the kallikrein-bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack.
Approved therapies include purified plasma-derived C1 INH (Cinryze®, Berinert), the recombinant peptide kallikrein inhibitor ecallantide (Kalbitor®), and the bradykinin receptor B2 inhibitor iticabant (Firazyr®). All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
There are many delivery routes for active pharmaceutical ingredients (APIs). Generally, the oral route of administration is favored. Oral administration provides a number of advantages, such as, but not limited to, patient convenience, flexibility of timing of administration, location of administration and non-invasiveness. Oral administration also provides more prolonged drug exposure compared with intermittent intravenous infusion, which may be important for drugs with schedule-dependent efficacy. For example, a drug with a short half-life can achieve a greater exposure time by either continuous infusion or by continuous oral dosing. The use of oral therapy further has the potential to reduce the cost of healthcare resources for inpatient and ambulatory patient care services.
In the pharmaceutical arts, it is known that a number of APIs cannot be administered effectively by the oral route. The main reasons why these compounds cannot be administered by the oral route are: a) rapid enzymatic and metabolic degradation; b) chemical and/or biological instability; c) low solubility in aqueous medium; and/or d) limited permeability in the gastrointestinal tract. For such compounds, non-oral routes of delivery, such as parenteral administration, mainly via intramuscular or subcutaneous injections, may be developed. However, non-oral administration poses a disadvantage for the patient as well as healthcare providers, and for this reason, it is important to develop alternative routes of administration for such compounds, such as oral routes of administration.
While the oral route of administration is the most convenient for the patient and the most economical, designing formulations for administration by the oral route involves many complications. Several methods are available to predict the ease by which an API may be formulated into a formulation suitable for administration by the oral route. Such methods include, but are not limited to, and Lipinski rule (also referred to as the Rule of Five) and the Biopharmaceutical Drug Disposition Classification System (BDDCS).
The BDDCS divides APIs into four classifications, depending on their solubility and permeability. Class I APIs have high solubility and high permeability; Class II APIs have low solubility and high permeability; Class III APIs have high solubility and low permeability; and Class IV APIs have low solubility and low permeability. APIs in higher classes in the BDDCS face greater challenges in formulating into an effective, pharmaceutically acceptable product than those in lower classes. Of the four classes, APIs falling into Class IV are the most difficult to formulate into a formulation for administration by the oral route that is capable of delivering an effective amount of the API as problems of both solubility and permeability must be addressed (note the BDDCS does not inherently address chemical stability). The role of BDDCS in drug development is described generally in L.Z. Benet J Pharm Sci. 2013, 102(1), 34-42.
Lipinski’s rule (described in Lipinski et al. Adv. Drug Deliv. Rev. 46 (1-3): 3-26) states, in general, that in order to develop a successful formulation for administration by the oral route, an API can have no more than one violation of the following criteria:
i) not more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms)
ii) not more than 10 hydrogen bond acceptors (nitrogen or oxygen atoms) iii) a molecular mass less than 500 daltons
iv) an octanol-water partition coefficient log P not greater than 5.
J. Zhang et al. Medicinal Chemistry, 2006, 2, 545-553, describes a number of small molecule amidine compounds which have activity as inhibitors of kallikrein. The molecules described in this document fall into Class IV of the BDDCS as described above. The compounds are poorly soluble in aqueous and physiological fluids, and are poorly permeable as demonstrated by oral dosing in rats and in vitro experiments with Caco-2 cells.
Furthermore, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, one of the compounds described in Zhang et al., is a Class IV API and violates criteria iii) and iv) as set forth in the Lipinski Rule.
Furthermore, the compounds described in Zhang et al., including 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, exhibit poor stability with respect to oxidation in air, to light
(photodegradation) and in aqueous and physiological fluids, as well as to elevated temperatures.
Therefore, the compounds described by Zhang et al. including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, not only exhibit poor solubility and permeability characteristics, but also poor stability characteristics. As a result, such compounds are predicted to be especially difficult to formulate into an effective, orally deliverable
pharmaceutical composition that is capable of delivering an effective amount of the compound to a subject.
Polymorphism, the occurrence of different crystal forms, is a property of some molecules. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties, such as, but not limited to, melting point, thermal behaviors (e.g. measured by thermogravimetric analysis (TGA), or differential scanning calorimetry (DSC), x-ray diffraction pattern, infrared absorption fingerprint, and solid state NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Discovering new polymorphic forms and solvates of a pharmaceutical product can provide alternate forms of the compound that display a number of desirable and advantageous properties, such as, but not limited to, ease of handling, ease of processing, ease of formulation, storage stability, and/or ease of purification. Further, new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof may further provide for improved pharmaceutical products, by providing compounds that are more soluble in a set of pharmaceutical excipients. Still further, the provision of new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof enlarges the repertoire of compounds that a formulation scientist has available for formulation optimization, for example by providing a pharmaceutical product with different properties, such as, but not limited to, improved processing characteristics, improved handling characteristics, improved solubility profiles, improved dissolution profile and/or improved shelf-life. Therefore, there is a need for additional polymorphs of pharmaceutically useful compounds, such as, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid and the compounds disclosed herein.
In one aspect, the present invention provides an oral formulation that is capable of delivering an effective amount of the amidine compounds described by Zhang et al. to a subject. In particular, the present invention provides an oral formulation that is capable of delivering an effective amount of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid to a subject. In one specific aspect, the 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid is present in a particular crystal form designated Form A. In light of the art suggesting the difficulties in formulating such an oral formulation, this result was unexpected.
As described herein, the amidine compounds described in Zhang et al., including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (specifically including particular crystal Form A), may now be conveniently used in oral administration and further used in oral administration for the treatment of a number of diseases and conditions in a subject, such as, but not limited to, HAE as described herein.
Avoralstat & next generation kallikrein inhibitors for HAE
Avoralstat
![]()
May 16 is HAE awareness day
See BioCryst’s video regarding HAE to learn more
Avoralstat is being developed as an oral prophylactic treatment for patients suffering from Hereditary Angioedema (HAE). Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
In May 2014 BioCryst, announced that the OPuS-1 (OralProphylaxiS-1) Phase 2a proof of concept clinical trial met its primary efficacy endpoint, several secondary endpoints and all other objectives established for the trial. OpuS-1 enrolled 24 HAE patients with a history of HAE attack frequency of at least 1 per week. Treatment with avoralstat demonstrated a statistically significant mean attack rate reduction of 0.45 attacks per week versus placebo, p<0.001. The mean attack rate per week was 0.82 on BCX4161 treatment, compared to 1.27 on placebo.
In December 2014, BioCryst initiated enrollment in OPuS-2 (Oral ProphylaxiS-2). OPuS-2 is a blinded, randomized, 12-week, three-arm, parallel cohort design trial evaluating the efficacy and safety of two different dose regimens of avoralstat administered three-times daily, 300 mg and 500 mg, compared with placebo. The primary efficacy endpoint for the trial will be the mean angioedema attack rate, which will be reported for each avoralstat dose group compared to placebo. The trial is being conducted in the U.S., Canada and Europe. On October 8, 2015, announced that it has completed enrollment of approximately 100 HAE patients with a history of moderately frequent to very frequent attacks in OPuS-2. BioCryst expects to report the OPuS-2 trial results in early 2016.
PATENT
WO200234711
http://www.google.com/patents/WO2002034711A1?cl=en
PATENT
PATENT
Examples
Example 1 – Synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl- phenyll-6-(cvclopropylmethyl-carbarnoyl)-pyridine-2-carboxylic acid
The synthesis of the above compound and intermediates is described below. In this section, the following abbreviations are used:

The synthesis of starting material, (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) is described in Scheme 1.
f 0HCY ° ΒΓΥΥ°

Preparation of 6-bromobenzofdl[1,3ldioxole-5-carbaldehvde (1b)

1a 1b
To a mixture of piperonal (1a) (498 g, 3.32 mol) in glacial acetic acid (1000 mL) was added a solution of bromine (200 mL, 3.89 mol) in glacial acetic acid (500 mL) over a period of 30 min and stirred at room temperature for 24h. The reaction mixture was poured into water (2000 mL) and the solid that separated was collected by filtration. The solid was dissolved in boiling ethanol (4000 mL) and cooled to room temperature. The solid obtained on cooling was collected by filtration to furnish 6-bromobenzo[d][1 ,3]dioxole-5-carbaldehyde (lb) (365 g, 48 %) as a white solid, MP 126 °C; HNMR (300 MHz, DMSO-d6): δ 10.06 (s, 1 H), 7.42 (s,1 H), 7.29 (s, 1 H), 6.20 (d, J=12.3, 2H); IR (KBr) 3434, 2866, 1673,1489, 1413, 259, 1112, 1031 , 925 cm“1; Analysis calculated for CeH5BrO3.O 25H C, 41.15; H, 2.37; Found: C, 41.07; H, 2.11.
Preparation of 2-bromo-5-hvdroxy-4-methoxybenzaldehyde (1c)

1c
A solution of potassium tert-butoxide (397 g, 3.36 mol) in DMSO (1.5 L) was heated at 50 °C for 30 min. Methanol (1.5 L) was added to it and continued heating at 50 °C for additional 30 min. To the hot reaction mixture was added 6-bromo-benzo[d][1,3]dioxole-5-carbaldehyde (1 b) (350g, 1.53 mol) and continued heating at 50 °C for 30 min. The reaction mixture was cooled to room temperature and quenched with water (2.3 L) and sodium hydroxide (61.2 g, 1.53 mol). The reaction mixture was washed with ether (2 x 1.5 L), acidified to pH 2 using cone. HCI and extracted with ethyl acetate ( 1 L). The ethyl acetate layers were combined and concentrated under vacuum to dryness. The residue obtained was treated with water (1.5 L) and ethyl acetate (1 L). The solid obtained was collected by filtration to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (97 g, 27.5% as a first crop). The layers from the filtrate were separated and aqueous layer was extracted with ethyl acetate (200 ml_). The ethyl acetate layers were combined dried over MgS04 and concentrated under vacuum to dryness to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (192 g, 54.4%, second crop) as an orange solid, MP 108 °C; ‘HNMR (300MHz, DMSO-cfe): S 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H); IR (KBr) 3477, 2967, 2917,
2837, 2767, 2740, 1657, 1595, 1428, 1270, 1210, 1164, 1022 cm“‘; Analysis calculated for C8H7Br03.H20: C, 38.58; H, 3.64: Found: C, 38.60; H, 3.60.
Preparation of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehvde ( d)

To a solution 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (120 g, 520 mmol) in DMF (1000 mL) was added potassium carbonate (79 g, 572 mmol) and benzyl bromide (68 mL, 572 mmol). The reaction mixture was stirred at room temperature overnight and quenched with water (3000 mL). The solid obtained was collected by filtration, washed with ether and dried under vacuum to furnish 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (113.19 g, 67.9%) as a white solid, MP 144 °C;1HNMR (300 MHz, DMSO-c/6): δ 10.06 (s, 1H), 7.47-7.34 (m, 7H), 5.17 (s, 2H), 3.92 (s, 3H); IR (KBr) 2898, 2851 , 1673, 1592, 1502, 1437, 1402, 1264, 1210, 1158, 1017, 754 cm“1; Analysis calculated for C 5H13Br03: C, 56.10; H, 4.08; Found: C, 55.44; H, 4.08.
Preparation of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e)
15 046578
146

1d 1e
To a solution of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (100 g, 311 mmol) in
ethanol (1500 mL) was added triethyl orthoformate (103 mL, 622 mmol), ammonium nitrate
(7.5 g, 93.3 mmol) and stirred at room temperature overnight. The reaction mixture was
treated with ether (1200 mL) and stirred for 15 min before filtration. The filtrate was
concentrated under vacuum to dryness to give 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (134 g) as a brown syrup; The product was used in the next step
without further purification; 1H N R (300 MHz, DMSO-cf6) δ 7.45 – 7.37 (m, 4H), 7.36 – 7.33
(m, 1 H), 7.17 – 7.14 (m, 1 H), 7.10 (s, 1 H), 5.10 (s, 2H), 3.80 (s, 3H), 3.58 – 3.33 (m, 5H),
1.13 – 1.07 (m, 6H); IR (KBr) 2974, 2879, 1601 , 1503, 1377, 1260, 1163, 1060 cm“1;
Analysis calculated for C19H23Br04: C, 57.73; H, 5.86; Found: C, 57.21 ; H, 5.94.
acid (1fi

To a solution of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (120 g,
300 mmol) in dry ether (1000 mL) at -78 °C was added n-butyllithium (1.6 M solution in
hexanes, 244 mL, 390 mmol) over a period of 30 min and further stirred at -78 °C for 30 min.
A solution of tri-n-butylborate (110 mL, 405 mmol) in dry ether (300 mL) was added to this
solution at -78 °C over a period of 30 min. The reaction mixture was further stirred for 2 h at -78 °C and warmed to 0 °C. The reaction mixture was quenched with 3N HCI (300 mL) at 0
°C and heated at reflux for 1 h. After cooling to room temperature, the solid obtained was
collected by filtration washed with water (250 mL) dried in vaccum to afford (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (30.85 gm, 37.6% as a white solid. The organic
layer from above filtrate was extracted with 1.5 N NaOH (3 x 200 mL). The combined basic
extracts were acidified with cone. HCI (pH about 4). The solid obtained was collected by
filtration, washed with water and dried under vacuum to furnish a second crop of (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (22.3 g, 26%) as a light orange solid
MP 158 °C; 1H NMR (300 MHz, DMSO-cfe) δ 10.08 (s, 1 H), 7.52 (s, 1 H), 7.48 – 7.33 (m, 5H),
7.24 (s, 1H), 5.18 (s, 2H), 3.89 (s, 3H); 1H NMR (300 MHz, DMSO-d6/D20) δ 10.06 (s, 1H),
7.52 (s, 1H), 7.49 – 7.32 (m, 5H), 7.23 (s, 1 H), 5.18 (s, 2H), 3.89 (s, 3H); MS (ES+) 309.1 (M+Na); IR (KBr) 3335, 2937, 1647, 1545, 1388, 1348, 1268, 1146, 1095 cm-1; Analysis calculated for C15H15BO5.0.25H2O: C, 62.00; H, 5.38; Found: C, 61.77; H, 5.19.
Synthesis of methyl-6-(cvclopropylmethylcarbamoyl¾-3-ftrifluoromethylsulfonyloxyVpicolinate
The synthesis of the intermediate methyl 6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethyl sulfonyloxy)picolinate (2h) is described in Scheme 2.

Preparation of 2-bromo-3-hvdroxy-6-methylpyridine (2b)

H3C N Br
2a 2b
To a solution of 3-hydroxy-6-methylpyridine (2a) (3000 g, 27.5 mol) in pyridine (24 L) cooled to 15 °C was added a solution of bromine (4.83 kg, 1.55 L, 30.2 mol) in pyridine (3 L) over a period of 50 min maintaining the internal temperature between 20 to 25 DC. After stirring for 19 h at room temperature the solvent was removed under vacuum and the residue was triturated with water. The solid separated was collected by filtration, washed with water and dried under vacuum to give 2-bromo-3-hydroxy-6-methylpyridine (2b) (3502 g, 67.7 %) as a light brown solid which was used as such without further purification; 1H NMR (300 MHz, DMSO-d6) δ 10.43 (s, 1H), 7.18 (d, J = 8.0 Hz, 1 H), 7.08 (d, J
MS (ES+) 188.35, 186.36 (M+1).
(2c)

2b 2c
A mixture of 2-bromo-3-hydroxy-6-methylpyridine (2b) (3000 g, 15.96 mol), anhydrous potassium carbonate (3308 g, 23.94 mol), and iodomethane (2.491 kg, 1.09 L, 17.556 mol) in 30 L of acetone was heated at 40 °C overnight. The reaction mixture was cooled to room temperature and filtered through Celite. Evaporation of the solvent followed by silica gel chromatography (Hexane: ethyl acetate = 7:3) afforded the desired compound, 2-bromo-3-methoxy-6-methylpyridine (2c) which was used as such for the next step; 1H NMR (300 MHz, DMSO-cfe) δ 7.42 (dd, J = 8.3, 1.5 Hz, 1H), 7.29 – 7.19 (m, 1H), 3.84 (d, J = 1.6 Hz, 3H), 2.37 (d, J = 1.7 Hz, 3H).

2c
2d
To a solution of 2-bromo-3-methoxy-6-methylpyridine (2c) (310 g, 1.53 mol) in 6000 mL of water at 60 °C was added KMnO, (725 g, 4.59 mol) in small portions over a 90 min period with vigorous mechanical stirring. A dark purple solution resulted. This solution was kept at 90 °C for a further 3 h and filtered through Celite while still hot to give a colourless filtrate.
After cooling, the aqueous solution was acidified to pH 1-2 by adding 6 N HCI. The white solid obtained was collected by filtration to give on drying 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (302g, 85%) of product, which was used as such in the next reaction without further purification. An analytical sample was obtained by recrystallization from methanol to give 6-bromo-5-methoxy-2-pyridinecarboxylic acid; 1H NMR (300 MHz, DMSO-tfe) δ 7.40 – 7.28 (m, 1H), 7.17 (d, J = 8.3 Hz, 1 H), 3.83 (d, J = 1.7 Hz, 3H).
Preparation of 6-bromo-N-(cvclopropylmethyl)-5-methoxypicolinamide (2e)

To a solution of 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (12 g, 52 mol) in pyridine (70 mL) was added EDCI (11.5 g, 59 mmol) and cyclopropylmethylamine (3.6 g, 52 mmol). The reaction mixture was stirred at room temperature overnight and then concentrated under vacuum. The reaction mixture was diluted with water (100 mL) and ethyl acetate (100 mL). The organic layer was separated and the water layer was extracted with ethyl acetate (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), brine (500 mL), dried over magnesium sulphate, filtered and concentrated under vacuum to furnish 10.43g of crude product. The crude product was converted into a slurry (silica gel 20 g) and purified by flash column chromatography (silica gel 230 g, eluting with 0-100% ethyl acetate in hexane) to yield compound 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (8.02 g, 54%) as off white solid, mp 67-70 °C; 1HNMR (300 MHz, DMSO-d6) δ 8.51 (t, J = 5.8, 1 H), 8.02 (d, J = 8.4, 1 H), 7.65 (d, J = 8.5, 1 H), 3.96 (s, 3H), 3.14 (t, J = 6.5, 2H), 1.11 -0.99 (m, 1 H), 0.47 – 0.36 (m, 2H), 0.27 – 0.20 (m, 2H); MS (ES+) 307.0, 309.0 (100%
M+Na)
Preparation of methyl 6-(cvclopropylmethylcarbamoyl)-3-methoxypicolinate (2f)

To a solution of 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (7.5 g, 27.6 mol) in methanol (300 mL) in a 2-L stainless steel bomb was added Pd(OAc)2(750 mg), 1 ,1-bis(diphenylphosphino)-ferrocene (750 mg), and triethylamine (3.9 mL, 27.6 mmol). The reaction mixture was vacuum flushed and charged with CO gas to 150 psi. The reaction mixture was and heated with stirring at 150°C overnight and cooled to room temperature. The catalyst was filtered through a pad of celite, and concentrated to dryness to furnish crude product. The crude was purified by flash column chromatography (silica gel 150 g,
eluting with, 0%, 5%, 10%, 20%, 30%, 50% ethyl acetate/hexanes (250 mL each) as eluents to give methyl 6-(cyclopropylmethyl-carbamoyl)-3-methoxypicolinate (2f) (6.29 g, 86.1 %) as a salmon coloured solid, MP 107 °C; 1HNMR (300 MHz, DMSO-cfe) δ 8.28 (t, J = 6.0, 1H), 7.91 (d, J = 8.8, 1H), 7.55 (d, J = 8.8, 1 H), 3.68 (s, 3H), 3.64 (s, 3H), 2.90 (t, J = 6.5, 2H), 0.89 – 0.68 (m, 1 H), 0.26 – 0.09 (m, 2H), 0.08 – 0.00 (m, 2H); MS (ES+) 287.1 (M+Na); IR (KBr) 3316, 2921 , 1730, 1659, 1534, 1472, 1432, 1315, 1272, 1228, 1189, 1099, 1003, 929, 846, 680 cm“1; Analysis calculated for C13H16 204: C, 59.08; H, 6.10; N, 10.60; Found: C, 58.70; H, 5.97; N, 10.23.
Preparation of 6-(cvclopropylmethylcarbamoyl 3-hvdroxypicolinic acid (2q)

2f 2g
Aluminium chloride method:
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-methoxypicolinate (2f) (0.16 mmol) in dichloromethane (840 mL) was added AICI3 (193 g, 1.5 mol). The reaction mixture was heated at reflux for 12 h under nitrogen. After slowly adding ~2L of 1 N HCI, the organic layer was separated. The aqueous layer was re-extracted several times with ethyl acetate/DME. The combined organic layer was washed with brine, dried (MgSO.4), and evaporated in vacuo to furnish crude 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid. To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid was added a solution of acetyl chloride (1 10 mL) in methanol (1.1 L). The reaction mixture was stirred for 12 h at room temperature and then concentrated to dryness in vacuo. After co-evaporating once with methanol, the compound was purified by flash-column chromatography (silica gel, 500 g, eluted with chloroform and 3% methanol in chloroform) to furnish 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g).
Boron tribromide method:
To a stirring solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-ethoxypicolinate (2f) (58.0 g, 208 mmol) was added BBr3 (79 mL, 834 mmol) in CH2CI2 (1.3 L) at 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 18h. The reaction mixture was evaporated to dryness and anhydrous methanol (1 L) was added to the light yellowish solid residue. Insoluble solid was collected by filtration (36 g). Mother liquor was evaporated and co-evaporated with MeOH (2 x 200 mL). The insoluble solid (36 g) was treated with MeOH (500 mL) and acetyl chloride (50 mL) and stirred at room temperature for 18 h (at this point reaction mixture was clear). The mixture was evaporated to dryness and diluted with water and extracted with EtOAc. White solid that separated out from EtOAc layer was collected by filtration, washed with water (2 x 20 mL), dried in vacuo at 50 °C to afford 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (5.36 g, 10 %) as a white solid, MP 92-95 °C. 1HNMR (DMSO-cfe) δ 11.04 (s, 1 H, exchangeable with D20), 8.37 (t, J = 6.0, 1 H, exchangeable with D20), 8.12 (d, J = 8.7 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 3.90 (m, 3 H), 3.15 (m, 2 H), 1.04 ( m, 1 H), 0.41 (m, 2 H), 0.24 (m, 2 H). IR (KBr): 3346, 3205, 1684 cm“1; MS (ES+): 251.1 (M+1); Analysis calculated for C12H14N2O4.0.1 H2O: C, 57.18; H, 5.67; N, 11.14; Found: C, 57.11 ; H, 5.61; N, 11.09.
Preparation of methyl-6-(cvclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy) picolinate (2h

To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (28 mmol) in DMF (200 mL) were added triethylamine (12 mL, 84 mmol) and N-phenyl-bis(trifluoromethanesulfonimide) (12 g, 34 mmol). The reaction mixture was stirred for 1.5 h at room temperature and then poured into ice. After diluting with water and extracting with ethyl acetate, the aqueous phase was re-extracted, and then the combined organic layer was washed with water and concentrated under vacuum to give methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)picolinate (2h), which was used in the next step without purification.
1H NMR (300 MHz, CDCI3) δ 8.50 (d, J = 8.6, 1 H), 8.07 (s, 1 H), 7.88 (d, J = 8.6, 1 H), 4.09 (d, J = 12.6, 3H), 3.48 – 3.24 (m, 2H), 1.18 – 1.01 (m, 1 H), 0.69 – 0.44 (m, 2H), 0.42 – 0.20 (m, 2H). MS (ES*): 405.17, 100%, M+Na.
Synthesis of 3-f2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid:
The synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i) is described as shown in Scheme 3.

3-f4-Benzyloxy-2-formyl-5-methoxy-phenylV6-(cvcloDroDvlmethvl-carbarnovn-pyridine-2-carboxylic acid methyl ester (3a)
5 046578
153

3a
To a solution of methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)
picolinate (2h) (24.3g, 63 mmol) in DME (225 mL) were added water (25 mL), (4- (benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (27.3 g, 95 mmol), NaHC03(15.9 g,
5 189 mmol), and bis(triphenylphosphine)palladium(ll) chloride (0.885 g). The reaction
mixture was stirred at 70°C overnight under nitrogen. After extracting with ethyl acetate, the organic layer was washed with water and brine and dried (MgSO^), and then concentrated
under vacuum. The compound was purified by flash-column chromatography (silica gel, 300 g, eluting with 10%, 20%, 30% and 40% ethyl acetate in hexane) to furnish 3-(4-benzyloxy- 10 2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
methyl ester (3a) (25 g, 83%) as off white solid, MP 48-50°C: 1H NMR (300 MHz, DMSO-cfe) δ 9.61(s, 1 H), 8.40 (d, J= 7.9 Hz, 1H), 8.14 (t, J= 5.0 Hz, 1H), 7.87 (d, J= 8.1 Hz, 1 H), 7.58
(s, 1H), 7.54-7.30 (m, 5H), 6.71 (s, 1 H), 5.24 (s, 2H), 3.93 (s, 3H), 3.70 (s, 3H), 3.45-3.34 (m,
2H), 1.19-1.05 (m, 1 H), 0.64-0.54 (m, 2H), 0.37-0.30 (m, 2H); IR ( Br) 1735, 1678, 1594,
15 1513, 1437, 1283, 1217, 1141, 1092 cm“1; MS (ES+) 497.29 (M+Na); Analysis calculated for
C27H2eN206: C, 68.34; H, 5.52; N, 5.90; Found; C, 68.16; H, 5.62; N, 5.80.
2-(6-(Cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-vn-4-methoxy-5- vinylbenzoic acid (3b)

To a solution of 3-(4-benzyloxy-2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl- carbamoyl)-pyridine-2-carboxylic acid methyl ester (3a) (24g, 50.6 mmol) in acetonitrile (50
mL), 2-methyl-2-propanol (350 mL), and water (125 mL) were added sodium dihydrogen
phosphate (12.5 g) and 2-methyl-2-butene (55 mL, 519 mmol). The reaction mixture was cooled in an ice bath and then sodium chlorite (28 g) was added. After stirring for 1 h, the reaction mixture was extracted with ethyl acetate and washed with water. The aqueous layer was re-extracted and then the combined organic layers were dried (MgS04). The solvent was evaporated in vacuo to furnish 5-(benzyloxy)-2-(6- ((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (29 g) which was used for the next step. MS (ES+): 513.24, (M+Na(; (ES ): 489.26, M-1.
Methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxytoarbonyltohenyl)-6-(cvclopropylmethylcarbamovnpicolinate (3c)

To a mixture of 5-(benzyloxy)-2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxy-carbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (31 g, 63.2 mmol), and triethylamine (17.7 mL, 126.4 mmol) in dichloromethane (300 mL), was added MEM-chloride (9.03 mL, 79 mmol), and stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and dried over MgS04, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 40 g) to furnish methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 89%) as a thick gum; H NMR (300 MHz, CDCI3) δ 8.35 (d, J = 8.0 Hz, 1 H), 8.15 (t, J = 5.7 Hz, 1 H), 7.78 (d, J = 8.0 Hz, 1H), 7.71 (s, 1H), 7.49 (d, J = 6.8 Hz, 2H), 7.36 (ddd, J = 7.5, 14.8, 22.4 Hz, 3H), 6.66 (s, 1 H), 5.37-5.13 (m, 4H), 3.90 (s, 3H), 3.69 (s, 3H), 3.60-3.49 (m, 2H), 3.49 (s, 2H), 3.39 (dd, J = 4.4, 8.4 Hz, 2H), 3.34 (s, 3H), 1.19-1.00 (m, 1H), 0.57 (q, J = 5.8 Hz, 2H), 0.38-0.25 (m, 2H). MS (ES+): 601.24 (M+Na); (ES“): 577.27 (M-1);1H NMR (300 MHz, DMSO-cfe) δ 8.69 (t, 7 = 6.1 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1 H), 7.63 (s, 1H), 7.41 (m, 5H), 6.92 (s, 1 H), 5.20 (m, 4H), 3.83 (s, 3H), 3.57 (s, 3H), 3.44 (m, 2H), 3:33 (m, 2H), 3.21 (m, 5H), 1.14 (m, 1H), 0.44 (m, 2H), 0.27 (m, 2H). IR (KBr):
1732, 1671 cm“1. MS (ES+): 601.1(M+Na); Analysis calculated for C31H 2Oe: C, 64.35; H, 5.92; N, 4.84; Found: C, 64.27; H, 6.04; N, 4.79.
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(4-hvdroxy-5-methoxy-2-(((2-methoxyethoxy¾methoxy)carbonyl)phenyl)picolinate (3d)

3c 3d
To a solution of methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)-carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 56.68 mmol) in ethanol (650 mL) was added 10% Pd/C (4 g) and hydrogenated at 45 psi for 5 h. The catalyst was removed by filtration through Celite and the filtrate was concentrated under vacuum to yield methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)picolinate (3d) (31.87 g, 86%), which was pure enough to be used as such for the next step. An analytical sample of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) was obtained by purification of 350 mg of above crude using flash column chromatography (silica gel, eluting with ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethyl-carbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-phenyl)picolinate (3d) as a clear gum; 1HNMR (300 MHz, DMSO-d6) δ 9.74 (s, 1 H), 8.68 (t, J = 6.1 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1 H), 7.95 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 6.83 (s, 1H), 5.19 (s, 2H), 3.77 (m, 3H), 3.58 (s, 3H), 3.44 (m, 2H), 3.34 (m, 2H), 3.21 (m, 5H), 1.04 (m, 1 H), 0.44 (m, 2H), 0.27 (m, 2H); IR (KBr): 1731 , 1664 cm‘1. MS (ES*): 489.0 (M+1); Analysis calculated for C^e^O,,: C, 59.01; H, 5.78; N, 5.73; Found: C, 58.92; H, 6.15; N, 5.29.
6-(Cvclopropylmethylcarbamovn-3-(5-methoxy-2-(((2-methoxyethoxy^methoxy)-carbonyl)-4- (trifluoromethylsulfonyloxy)phenyl)picolinate (3e)

To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2- methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) (14.3 g, 29.3 mmol) in dichloromethane (150 mL) were added pyridine (12 mL, 146 mmol) and triflic anhydride (7.5 mL g, 44 mmol). After stirring overnight at room temperature under N2. the reaction mixture was poured into ice water and then extracted twice with dichloromethane. After washing the combined organic extracts with water and drying (MgS0 ), the solvent was evaporated in vacuo. The compound was purified by flash chromatography over silica gel column using ethyl acetate: hexane to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)-carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (1 g, 93%); H NMR (300 MHz, CDCy a 8.41 (d, J = 8.0, 1H), 8.17 (s, 1H), 8.03 (s, 1H), 7.79 (d, J = 8.0, 1 H), 6.82 (s, 1H), 5.32 (q, J = 6.1, 2H), 3.97 (s, 3H), 3.74 (s, 3H), 3.67 – 3.57 (m, 2H), 3.55 – 3.45 (m, 2H), 3.41 (dd, J = 8.2, 14.5, 2H), 3.34 (s, 3H), 1.36 – 1.17 (m, 1H), 0.58 (d, J = 7.1 , 2H), 0.33 (d, J = 5.1 , 2H).
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(5-methoxy-2-f((2-methoxyethoxy)- methoxy)carbonvn-4-vinylphenyl)picolinate (3f)

To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (37.4
g, 60.30 mmol) and potassium vinyltrifluoroborate (16.87 g, 120.6 mmol) in DMF (450 mL) and water (45 mL) was bubbled N2 for 5 min. To this mixture was added NaHC03 (20.26 g, 241.2 mmol) and dichloro-bis(triphenylphosphine)palladium (II) (6.34 g, 9.0 mmol). The reaction mixture was stirred at 70 °C for 20 h under N2(reaction progress was checked by 1H N R because product and starting material had same Rf in TLC). The reaction mixture was cooled down to room temperature and diluted with ethyl acetate. The organic layer was separated, washed with water, brine, dried ( gS04) and filtered. The filtrate was concentrated under vacuum to yield crude methyl 6-(cyclopropylmethyl-carbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-4-vinylphenyl)-picolinate (3f). The crude product was purified by flash column chromatography (silica gel, 1 kg, eluting with 0-100% ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate [31) (26.54 g, 88%) as an amber gum; H NMR (300 MHz, DMSO-c¾ δ 8.70 (t, J = 6.1 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1 H), 8.12 (s, 1 H), 8.00 (d, J = 8.0 Hz, 1 H), 6.98 (m, 2H), 5.94 (dd, J = 1.2, 17.8 Hz, 1H), 5.43 (d, J = 12.5 Hz, 1 H), 5.21 (d, J = 6.5 Hz, 2H), 3.88 (s, 3H), 3.64 (s, 3H), 3.48 (d, J = 3.1 Hz, 2H), 3.35 (m, 5H), 3.22 (m, 2H), 1.11 (s, 1H), 0.44 (dt, J = 4.9, 5.5 Hz, 2H), 0.28 (q, J = 4.8 Hz, 2H). IR (KBr); 1732, 1670 cm“1. MS (ES+) 499.1 (M+1).
2-(6-(cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzolc acid (3g)

A mixture of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate (3f) (27.4 mmol) in DME (160 mL) and 6N HCI (40 mL) was stirred at room temperature for 6 h or till TLC showed complete conversion. The solvent was removed under vacuum. The residue obtained was suspended in water, the solid separated out was collected by filtration, washed with water and dried under vacuum to give 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (7.0 g, 63%) as a white
solid MP 40 – 42 °C; H NMR (300 MHz, DMSO-de) δ 8.69 (t, J= 6.0 Hz, 1H, NH), 8.20 (d, J= 7.9 Hz, 1H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1H), 6.97 (dd, J= 18.0, 11.3 Hz, 1H), 6.88 (s, 1H), 5.92 (d, J= 7.9 Hz, 1H), 5.38 (d, J= 11.1 Hz, 1H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); IR (KBr): 3084, 1728, 1650, 1533, 1212, 1143 cm-1; MS (ES+) 433.26 (M+Na); (ES-): 409.28 (M-1); Analysis calculated for θ22Η22Ν2Ο6.0.25Η2Ο; C, 63.68; H, 5.47; N, 6.75; Found C, 63.75; H, 5.56; N, 6.65
Methyl-3-(2-(4-carbamimidoylprienylcarbamoyl)-5-metrioxy-4-vinylphenyl)-6- (cvclopropylmethylcarbamoyl)picolinate (3h)

To a solution of 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (2.35 g, 5.7 mmol) and 4-aminobenzimidamide dihydrochloride (3j) (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 mL) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 in CMA 50) yielding methyl-3-(2-(4-carbamimidoylphenyl-carbamoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (2.2 g, 65%) as a white solid MP 266 °C; 1H NMR (300 MHz, DMSO-c/6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 – 7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for 
C, 58.93; H, 5.63; N,11.85; Found: C, 58.75; H, 5.65; N, 11.92.
46578
159
3-r2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy -vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i)

3h 3i
To a solution of methyl-3-(2-(4-carbamirriidoylphenylcarbarnoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (1 g, 1.9 mmol) in methanol (10 mL) and THF
(10 mL) was added 2 N NaOH (10 mL). The reaction mixture was stirred at room
temperature for 3 h, and concentrated in vacuo to remove methanol and THF. The aqueous layer was acidified with 6N HCI to pH 6-7 and the solid obtained was collected by filtration
washed with water and ether to furnish on drying 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
(3i)(0.775 g, 80%) as the hydrochloride salt as an off white solid.
1H NMR (300 MHz, DMSO-d6) δ 12.67 (s, 1 H), 9.11 (s, 2H), 8.97 (s, 2H), 8.74 (s, 1 H), 7.90
(d, J = 7.8, 1 H), 7.80 (s, 1 H), 7.72 – 7.58 (m, 4H), 6.99 (dd, J = 11.3, 17.7, 1 H), 6.78 (s, 1H),
5.95 (d, J = 17.2, 1H), 5.38 (d, J = 11.9, 1H), 3.82 (s, 3H), 3.18 (s, 2H), 1.06 (s, 1 H), 0.43 (d,
J = 7.9, 2H), 0.25 (d, J = 4.7, 2H); MS (ES+) 514.0 (M+1 ); Analysis calculated for
C2eH27N5O5.HCI.H2O: C, 59.21; H, 5.32; N, 12.33; Found: C, 59.43; H, 5.21; N, 12.06.
Example 1A- Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride in Form
C

The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 to -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1.6 kg) and water (0.57 kg). The mixture was heated to 46 °C. Smopex-234 (21 g) and Acticarbone 2SW (10 g) were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 “C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The
suspension was stirred at 0-3.0 “for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjackel= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 11 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C.
Example-1 B: Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbartiovQpyridine-2-carboxylic acid hydrochloride in Form A
The procedure was carried out in an identical manner to Example 1 A, with the exception that after the final filtration the filter cake was rinsed with 2.87 kg methyl ierf-butyl ether instead of 2.87 kg water, and pulled dry. The product was dried at 40-43 °C and 50 mbar to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) as Form A.
PATENT
Methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (compound 6a) is (I) (pages 85 and 86). Avoralstat hydrochloride (compound of formula XVIII) is (II) (claim 40, page 109). A Markush structures is presented (claim 1, page 99).
The synthesis of (II) via intermediate (I) is described (example 1, pages 80-93).
A synthesis of the compound 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (Compound 3i) is described in Schemes A-C.
O y OHCk n Br^ ^OCH3
B Brr22,, AAccOOHH Y^ V”“ \ \ tt–BBuuOOKK
OHC^^^O ” Br^\^0 MeOH ” OHC
1a 1b 66%

1d 95% 1 e

1f
Scheme A


3h 31
Scheme C
Examples. In this section, the following abbreviations are used:



Example-1 : Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b)

7b
Step (1): Preparation of 6-Bromobenzo 1 ,3]dioxole-5-carbaldehyde (1 b):

1b
A solution of bromine (33.0 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to a solution of piperonal (1a) (29.9 kg, 199.16 mol) in acetic acid (105 L) at room
temperature over a period of 50 min and the reaction mixture was stirred at room temperature for 14.2 h. Additional solution of bromine (33 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to the reaction mixture over a period of 2 h and the reaction mixture was stirred for 22 h. The reaction mixture was quenched by addition of ice water (500 L) with stirring over a period of 6 h and continued stirring for additional 1.25 h. The mixture was allowed to settle and most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (600 L) was added to the solid, stirred, mixture was allowed to settle and then most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (100 L) was added to the decanted mixture, stirred for 15 min and the solid obtained was collected by filtration using a centrifuge. The solid was washed with water (2 x 100 L) and air-dried in a tray drier for 3.75 h to afford the crude product 1 b (52 kg). The crude product (51.2 kg) was stirred in n-hexane (178 L) for 3 h, collected by filtration, washed with n-hexane (25 L) and dried to afford 6-bromobenzo[1 ,3]dioxole-5-carbaldehyde (1b) (40.1 1 kg, 87.9%) as a light brown solid. MP: 109-112°C. 1H NMR (300 MHz, CDCI3) δ 10.21 (s, 1 H), 7.37 (s, 1 H), 7.07 (s, 1 H), 6.10 (s, 2H); HNMR (DMSO-cf6): δ 10.06 (s, 1 H), 7.42 (s, 1 H), 7.29 (s, 1 H), 6.20 (d, J =12.3 Hz, 2H)
The process is also illustrated in Fig. 1.
Average yield of isolated 1 b from step-1 is 78 – 88%.
Step (2): Preparation of 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c)

A solution of potassium terf-butoxide (10.7 kg, 95.36 mol) in DMSO (49 L) was stirred at 50 °C for 30 min. Methanol (49 L) was added slowly over a period of 4.25 h and stirred at 50 °C for 30 min. 6-Bromobenzo[1 ,3]dioxole-5-carbaldehyde (1 b) (9.91 kg, 43.27 mol) was added to the reaction mixture in small portions over a period of 45 min and stirred at 50 °C for 1 h. The reaction mixture was cooled to room temperature and split into two equal portions. Each portion was quenched with water (50.9 L) and basified with 50% aqueous NaOH solution (2.4 L). Each portion was extracted with MTBE (4 x 36 L) to remove impurities. The aqueous layer was acidified with cone. HCI to pH ~ 3 to obtain
product as a yellow solid. The solid was collected by filtration using a centrifuge, washed with water (2 x 35 L) and air-dried to afford 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) (4.37 kg, 40.7%, contains 7 % water); Mp: 100-102°C; 1HNMR (300MHz, DMSO-d6): δ 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H).
The process is also illustrated in Fig. 2.
Average yield of isolated product 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) from step-2 is 40-50%.
Step (3): 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-y benzaldehyde (4a)

2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) [1.3 kg (93%, 7% water content), 5.25 mol] was dissolved in toluene (13 L) in a reaction flask equipped with a Dean Stark apparatus. The solution was heated at reflux with stirring to distil off about 25% of the toluene along with water (90 ml_). The solution was cooled to 90 °C then
bis(pinacolato)diboron (1.5 kg, 5.82 mol), KOAc (772.6 g, 7.87 mol) and Pd(PPh3) (24.3 g, 0.02 mol) were added and the reaction mixture was heated at reflux for 10h. After confirming the completion of reaction by TLC (mobile phase: 100% DCM), the reaction mixture was cooled to room temperature and was kept standing overnight. The reaction mixture was filtered through celite and the celite cake was washed with toluene (4 L). The filtrate of this batch was mixed with the filtrate of another batch (batch size 1.3 kg obtained from an identical reaction). The mixed filtrate was washed with water (17.5 L), brine (17.5 L), dried over Na2S04, filtered and the solution was passed through a pad of silica gel (2 kg, mesh size 230-400). The silica gel pad was washed with toluene. The combined filtrate and washing was concentrated under reduced pressure and the residual crude product was stirred with n-hexane (23 L) for 1 h to obtain a solid product. The solid was collected by filtration, washed with n-hexane (5 L) and dried to afford 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)benzaldehyde (4a) (2.47 kg, 84.6%). H NMR (300 MHz, CDCI3) δ 10.54 (s, 1 H), 7.57 (s, 1 H), 7.33 (s, 1 H), 5.89 (s, 1 H), 4.01 (s, 3H), 1.37 (s, 12H); 1H NMR (300 MHz, DMSO-d6) δ 10.35 (s, 1 H), 9.95 (s, 1 H), 7.33 (s, 1 H), 7.23 (s, 1 H), 3.87 (s, 3H), 1.33 (s, 12H); MS (ES+) 301.1 (M+Na); 579.1 (2M+Na); Analysis calculated for C14H19B05: C, 60.46; H, 6.89; Found: C, 60.60; H, 6.87
The average yield of 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) from step (3) is 78 – 90%.
The process is also illustrated in Fig. 3.
Step (4): Preparation of 3-Bromo-2,6-dimethylpyridine (5b)

2,6-lutidine (5a) (115 kg, 1073.3 mol) was added into pre-chilled oleum (20-23%, 1015 kg, 2276.7 mol) at 0 °C over a period of 4.5 h (temperature r6ached 14 °C during the addition). Bromine (88.18 kg, 1103.6 mol) was then added at 5-10 °C over a period of 1 h. The reaction mixture was slowly heated to 150 °C over a period of 12h. TLC analysis indicated about 40-50% conversion to product and the formation of a dimer by-product (5%). The reaction mixture was cooled to room temperature and then additional bromine (88.18 kg, 1103.6 mol) was added slowly. The reaction mixture was slowly heated to maintain a temperature of 65-75 °C over a period of 15h. TLC analysis indicated a 65-70 % conversion to product and the formation of 5% dimer by product. The reaction mixture was quenched by addition of water (500L) while maintaining the reaction temperature below 20 °C. The mixture was basified with 6.6 M NaOH (3800 L) while maintain the temperature at < 40 °C. EtOAc (220 L) was added and the mixture was stirred for 1 h then allowed to settle over a period of 2 h. The layers were separated and the aqueous layer was treated with NaOH (10 kg) in water (10 L) and extracted with EtOAc (160 L). The organic extracts were combined washed with brine (100 L), dried over Na2S04 (50.0 kg), filtered and the solvent was evaporated under atmospheric pressure. The residue was vacuum distilled and the desired product 3-bromo-2,6-dimethylpyridine (5b) was collected at 58-60 °C, 2 mmHg (98.45 kg, 49.2 %) as a colorless liquid.
The process is also illustrated in Fig. 4.
Step (5): Preparation of 3-Bromopyridine-2,6-dicarboxylic acid (5c)

5b 5c
To a stirred solution of 3-bromo-2,6-dimethylpyridine (5b) (98 kg, 5326 mol) in water (1310 L) was added KMn0 (225 kg, 1423.6 mol) in 5 equal portions in 1 h intervals at 70 °C. After stirring for 1 h at 70 °C, additional KMn04 (225 Kg, 1423.6 mol) was added in 5 equal portion in 1 h intervals at 90 °C. The reaction mixture was stirred for 12 h at 90 °C. The suspension was filtered hot through celite to obtain a clear solution. The solvent was distilled off to remove about 30% of the total volume. The remaining concentrated solution was chilled to 0 °C and made acidic (to pH 3-4) by the addition of cone. HCI (120 L). The white precipitate obtained was collected by filtration and dried at 70 °C to afford 3-bromopyridine-2,6-dicarboxylic acid (5c) as a white solid (109 kg, 84%).
The process is also illustrated in Fig. 5.
Step (6): Preparation of Dimethyl 3-Bromopyridine-2,6-dicarboxylate (5d)

To a stirred solution of 3-bromopyridine-2,6-dicarboxylic acid (5c) (20.0 kg, 81.29 mol) in methanol (100 L) was added cone. H2S04 (4.4 L) over a period of 30 min. The reaction mixture was heated to 65 °C and maintained at that temperature for 5 h (the reaction was monitored by TLC analysis to determine completion of reaction). The reaction mixture was cooled to room temperature basified by careful addition of aqueous NaHC03 solution (prepared from 10 kg NaHC03 in 120 L of water) and further diluted with water (120 L). The white solid obtained was collected by filtration, washed with plenty of water and then oven-dried at 40 °C to obtain dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (9.2 kg, 41.3%) as a white solid; 1HNMR (300 MHz, DMSO-cf6) δ 8.47 (d, J = 8.4, 1 H), 8.08 (dd, J = 4.5, 8.4, 1 H), 3.95 (s, 3H), 3.91 (s, 3H); MS (ES+) 570.6 (2M+Na); Analysis calculated for C9H8BrN04: C, 39.44; H, 2.94; Br, 29.15 N, 5. 1 ;
Found: C, 39.52; H, 2.92; Br, 29.28; N, 5.03.
The process is also illustrated in Fig. 6.
6582
Step (7): Preparation of Methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (

To a stirred solution of dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (27 kg, 98.52 mol) in ierf-butanol (135 L) was added at room temperature cyclopropylmethanamine (7.83 kg, 110.1 mol). The reaction mixture was heated at 65 °C for 17 h. The progress of reaction was monitored by TLC and HPLC (HPLC analysis showed the formation of 74% of the product 5e after 17 h. The reaction mixture was cooled to room temperature and then cone. HCI (2.7 L) was added slowly and the mixture was stirred for 15 min. The reaction mixture was concentrated under reduced pressure to obtain the crude product. The crude product was dissolved in hot /-PrOH (54 L) filtered through a celite pad. The filtrate was cooled with stirring to 10 °C to obtain a white precipitate. The solid obtained was collected by filtration, washed with cold
i-PrOH (13 kg), n-hexane (15 L) and dried to provide pure methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e) (15.7 kg, 50.9%). The filtrate was concentrated under reduced pressure and the crude product can be purified by silica gel column chromatography eluting with tert-butanol in hexanes to furnish additional 10% methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e). HNMR (300 MHz, DMSO-cf6) δ 8.83 (t, J = 5.9, 1 H), 8.47 – 8.41 (m, 1 H), 8.06 (d, J = 8.4, 1 H), 3.96 (s, 3H), 3.16 (t, J = 6.5, 2H), 1.14 – 0.99 (m, 1 H), 0.42 (m, 2H), 0.30 -0.19 (m, 2H); MS (ES+) 337.0 (M+23), 650.8 (2M+23); Analysis calculated for
C12H13BrN203: C, 46.03; H, 4.18; N, 8.95; Br, 25.52; Found: C, 46.15; H, 4.17; N, 8.72; Br, 25.26.
The average isolated yield for step (7) is 50% to 60%.
The process is also illustrated in Fig. 7.
Step (8): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a)
2

6a
THF (37.5 L) was charged to a 100 L reactor followed by ethyl 3-bromo-6- (cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate (5e) (2.5 kg, 7.98 mol) under a nitrogen atmosphere. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) (2.88 kg, 10.36 mol) was added, followed by the addition of PPh3 (53.13 g, 0.20 mol), PdCI2(PPh3)2 (120.4 g, 0.17 mol) and a solution of Na2C03(2.12 kg, 20.00 mol) in demineralized water (10.0 L) under nitrogen atmosphere. The reaction mixture was degassed again two times by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 6.5 h, cooled to room temperature and filtered through a Celite bed. Water (75 L) was added to the filtrate and the product was extracted with ethyl acetate (75 L). The aqueous layer was back extracted with ethyl acetate (2 χ 60 L). The combined ethyl acetate extract was divided into two equal portions and each portion was washed with brine (37 L), dried over Na2S04, filtered and concentrated under reduced pressure to give crude methyl 6- ((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) as a reddish viscous material (-4.5 Kg) which was used as such for the next step without further purification. An analytical sample was prepared by purification of a small sample by flash column chromatography (silica gel, eluting with 0-100% ethyl acetate in hexane) to furnish methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)-picolinate (6a) as an off-white solid; HNMR (300 MHz, DMSO-d6) δ 9.89 (s, 1 H), 9.52 (s, 1 H), 8.79 (t, J = 6.1 Hz, 1 H), 8.23 (d, J = 8.0 Hz, 1 H), 8.09 (d, J = 8.0 Hz, 1 H), 7.34 (s, 1 H), 6.90 (s, 1 H), 3.85 (s, 3H), 3.62 (s, 3H), 3.22 (m, 2H), 1.16 -1.02 (m, 1 H), 0.49 – 0.38 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 791.0 (2M+Na), (ES-) 382.7 (M-1), 767.3 (2M-1); Analysis calculated for C20H20N2O6.0.25 H20: C, 61.77; H, 5.31 ; N, 7.20; Found: C, 61.54; H, 5.13; N, 7.05.
The process is also illustrated in Fig. 8.
46582
Step (9): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b)

6a 6b
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) (2.11 kg, estimated about 3.83 mol from step-8) in dichloromethane (16.0 L) and pyridine (1.4 L, 17.4 mol) cooled to -10°C and maintained at that temperature for 1 h was added a solution of triflic anhydride (980.0 ml_, 5.8 mol) in dichloromethane (6.0 L) drop wise over a period of 3 h at -10 °C. The reaction mixture was stirred at -5°C for 1.3 h, quenched with saturated aqueous NaHCO3(10.4 L) and stirred for 30 mins. The organic layer was separated, washed successively with saturated aqueous NaHC03 (10.4 L), 1 HCI (2 x 16.6 L), water (13.2 L), brine (13.2 L), dried over MgS04, filtered and concentrated under reduced pressure to give the crude product. The crude product was stirred with 15% ethyl acetate in n-hexane (7.0 L) for 1 h. The solid obtained was collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (7.0 L) for 1 h, was collected by filtration and washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (8.0 L) for 1 h, collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was dried to afford methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)-oxy)phenyl)picolinate (6b) as a light brown solid (1.7 kg, 86% yield, for combined steps 8 & 9). Average isolated yield for combined steps 8 and 9 was 70% to 86%; Ή NMR (300 MHz, DMSO-cf6): δ 9.64 (s, 1 H), 8.78 (t, J = 6.1 , 1 H), 8.29 (d, J = 8.0, 1 H), 8.16 (d, J = 8.0, 1 H), 8.03 (s, 1H), 7.39 (s, 1 H), 4.00 (s, 3H), 3.63 (s, 3H), 3.22 (m, 2H), 1.11 (m, 1 H), 0.52 – 0.39 (m, 2H), 0.28 (m, 2H); MS (ES+) 538.9 (M+Na). The process is also illustrated in Fig. 9.
Step (10): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c)

A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4- (((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b) (12 kg, 23.24 mol) in DME (106 L) was charged into reactor under nitrogen. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. Potassium trifluoro(vinyl)borate (3.9 kg, 29.1 1 mol), PdCI2(PPh3)2 (815 g, 1.13 mol), KHC03 (4.65 g, 46.44 mol) and demineralized water (12 L) was then added under a N2 atmosphere. The reaction mixture was degassed by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 5 h. The reaction mixture was cooled to room temperature and then filtered through a Celite bed. Demineralized water (118 L) was added to the filtrate followed by ethyl acetate (124 L). The mixture was stirred for 20 min and then the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 95 L). The combined organic extract was washed with brine (95 L), dried over Na2S04, and filtered. The solvent was evaporated under reduced pressure to give the crude product. The crude product was purified by column chromatography (silica gel, 120 kg, 230-400 mesh size, eluting with ethyl acetate in n-hexane) to obtain methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (6 kg, 72%). 1H NMR (300 MHz, CDCI3): δ (ppm) 9.64 (s, 1 H), 8.35 (d, J = 7.8 Hz, 1 H), 8.06-8.03 (m, 2H), 7.78(d, J = 7.8 Hz, 1 H), 7.02-6.92 (m, 1 H), 6.61 (s, 1 H), 5.86 (d, J = 17.7 Hz, 1 H), 5.38 (d, J = 1 1.4 Hz, 1 H), 3.84 (s, 3H), 3.67 (s, 3H), 3.35-3.29 (m, 2H),1.08-1.03 (m, 1H), 0.55-0.49 (m, 2H), 0.29-0.2 4(m, 2H). 1HNMR (300 MHz, DMSO-d6) 6 9.68 (s, 1 H), 8.77 (t, J = 6.1 , 1 H), 8.35 – 8.21 (m, 1 H), 8.16 – 8.01 (m, 2H), 7.14 -6.87 (m, 2H), 6.01 (dd, J = 1.2, 17.8, 1 H), 5.45 (dd, J = 1.1 , 1 1.3, 1 H), 3.91 (s, 3H), 3.64 (s, 3H), 3.23 (m, 2H), 1.21 – 1.01 (m, 1H), 0.51 – 0.40 (m, 2H), 0.34 – 0.20 (m, 2H). MS
(ES+) 417.0 (M+Na); Analysis calculated for C22H22N205: C, 66.99; H, 5.62; N, 7.10;
Found: C, 66.75; H, 5.52; N, 7.06.
The process is also illustrated in Fig. 10.
Step (1 1): Preparation of 2-(6-((cyclopropylmethyl)carbamoyl)-2- (methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d)

To a stirred solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (1.57 kg, 3.80 mol) in acetonitrile (15.4 L) was added ferf-butyl alcohol (22.2 L), demineralized water (3.2 L) and sodium dihydrogen phosphate monohydrate (323.74 g, 2.346 mol). The reaction mixture was cooled to 0 °C and added 2-methyl-2-butene (5.3 L, 50.0 mol) and stirred at 0 °C for 30 min. A solution of 80% sodium chlorite (1.36 kg, 12.0 mol) in demineralized water (5.2 L) was added to the reaction mixture over a period of 2.5 h at 0 °C [temperature rises to 7 °C during the addition]. The reaction mixture was stirred at 0 °C for 2 h, diluted with water (40 L) and ethyl acetate (24 L). After stirring the mixture, it was allowed to settle and the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 20 L) then acidified with 5.9 % aqueous acetic acid (2 L) and extracted once with ethyl acetate (10 L). The organic extracts were combined washed with water (2 x 20 L), a solution of acetic acid (125 mL) in water (20.0 L), brine (2 χ 20 L), dried over Na2S04, filtered and concentrated under reduced pressure (vapor temperature below 40 °C). The residue obtained was dissolved in acetone (7 L) (residue didn’t dissolve completely). The solution was poured slowly into a reactor containing stirred n-hexane (70.0 L) to precipitate the solid product and the mixture was stirred for 2 h. The solid obtained was collected by filtration, washed with 10% acetone in n-hexane (6.3 L), AJ-hexane (6.3 L), dried to afford 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4- methoxy-5-vinylbenzoic acid (6d) as an off-white solid (1.29 Kg, yield: 79.0%). Average isolated yield for step 1 1 is 74% to 84%. 1H NMR (300 MHz, DMSO-d6): δ (ppm) 12.50 (brs, 1 H), 8.69(t, J= 6.0 Hz, 1 H, NH), 8.20 (d, J= 7.9 Hz, 1 H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1 H), 6.97 (dd, J= 18.0, 1 1.3 Hz, 1 H), 6.88 (s, 1 H), 5.92 (d, J= 7.9 Hz, 1 H), 5.38 (d, J= 1 1.1 Hz, 1 H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); MS (ES+) 433.26, (M+Na); (ES-) 409.28 (M-1). The process is also illustrated in Fig. 1 1.
Step (12): Preparation of Methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate methanesulfonate (7a

Pyridine (3.8 L, 47.17 mol) and EDCI (5.31 kg, 27.66 mol) were sequentially added to a cooled solution (0 °C) of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (9 kg, 21.92 mol) and 4-aminobenzamidine dihydrochloride (5.13 kg, 24.65 mol) in /-PrOH (90 L). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. TLC analysis indicated incomplete reaction. Additional EDCI (1.08 kg, 5.6 mol) was added and the reaction mixture was stirred for 8 h. The reaction was still incomplete as indicated by TLC analysis, additional EDCI (0.54 kg, 2.8 mol) was added and the reaction mixture was stirred for 5 h. TLC analysis indicated there was trace amount of unreacted starting material remaining. The reaction mixture was cooled to 0 °C and a solution of
methanesulfonic acid (MSA) (9.13 kg, 95 mol) in MeOH (38.7 L) was added to the cooled mixture over a period of 4 h. The reaction mixture was allowed to warm to room temperature and stirred for 15 h. The product was collected by filtration, washed with a mixture of /‘-PrOH and MeOH (4:1 , 45 L). The wet cake was slurried in a mixture of /-PrOH and MeOH (2:1 , 135 L) stirred for 1 h and the product was collected by filtration and washed with a mixture of /‘-PrOH and MeOH (4:1 , 46.8 L). The product was dried in
2015/046582
a vacuum oven at 45 °C to afford methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) as a pink-colored solid (12.71 kg, 93%). Average isolated yield for this step: >90%.
1H NMR (300 MHz, DMSO-c/6) δ 10.71 (s, 1 H), 9.16 (s, 2H), 8.80 (s, 2H), 8.68 (t, J = 6.1 Hz, 1 H), 8.22 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.1 Hz, 1 H), 7.93 (s, 1H), 7.84 – 7.72 (m, 4H), 7.12 – 6.97 (m, 2H), 6.04 (dd, J = 17.8, 1.3 Hz, 1 H), 5.45 (d, J = 12.6 Hz, 1H), 3.91 (s, 3H), 3.60 (s, 3H), 3.25 – 3.16 (m, 2H), 2.32 (s, 3H), 1.10 – 1.01 (m, 1 H), 0.48 – 0.37 (m, 2H), 0.30 – 0.22 (m, 2H); MS (ES+) 528.0 (M+1); Analysis calculated for
C29H29N5O5.CH3SO3H.2H2O. C, 54.62; H, 5.65; N, 10.62; S, 4.86; Found: C, 54.95; H, 5.55; N, 10.61 ; S, 4.87.
The process is also illustrated in Fig. 12.
Step (13): Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-rnethoxy-4- vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate

(3i) ,a 3i
A pre-cooled (0-5 °C) aq. NaOH solution [prepared from solid NaOH (4 kg, 100 mol) in water (86 L)] was added to a suspension of methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) (28.7 kg, 46 mol) in acetonitrile (86 L) cooled to 0 to 5 °C over a period of 25 mins. The reaction mixture was stirred at 0 to 5 °C for 2.5 h (TLC analysis showed the reaction was complete). The reaction mixture was filtered through a sparkler filter, washed with a mixture of 1 :1 CH3CN / H20 ( 57.4 L). Acetic acid (3.2 L, 55.9 mol) in water (56 L) was added to the filtrate at room temperature over a period of 25 mins and the resulting mixture was stirred at room temperature for 2.5 h. The solid product obtained was collected by filtration, washed with a 1 :4 mixture of CH3CN / H20 (57.5 L). The solid was dried at 45°C in a vacuum oven to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate (3i) as an off-white solid (12,77 kg, 54.1%). Average yield for this step is 50% to 75%. Mp: >200°C; H NMR (300 MHz, DMSO-d6): δ 13.49 (s, 1 H), 8.94 (bs, 4H), 8.56 (t, 1 H), 7.82 – 7.71 (m, 2H), 7.67 -7.56 (m, 4H), 7.51 (d, J = 7.8, 1 H), 6.98 (dd, J = 11.3, 17.8, 1 H), 6.68 (s, 1 H), 5.92 (d, J = 16.6, 1 H), 5.36 (d, J = 12.4, 1 H), 3.80 (s, 3H), 3.16 (m, 2H), 1.05 (m, 1 H), 0.43 (m, 2H), 0.24 (m, 2H); MS (ES+) 514.1 (M+1), 536.1 (M+Na), (ES-) 512.1 ; Analysis calculated for C28H27N5O5.3H2O: C, 59.25; H, 5.86; N, 12.34; Found C, 59.50; H,
5.75; N, 12.05. If needed this material can be crystallized from a mixture of acetone and water.
The process is also illustrated in Fig. 13.
Step 14: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b

A pre-cooled (5-8 °C) aqueous NaOH solution (prepared from solid NaOH (1.97 kg, 49.25 mol) in demineralized water (41 L) was added to a pre-cooled (0-5 °C) suspension of (3i) (13.8 kg, 26.9 mol) in acetonitrile (41 L). The reaction mixture was stirred at 0-5 °C for 30 min (until the reaction mixture becomes homogeneous). The reaction mixture was filtered through a sparkler filter washed with 50% acetonitrile in demineralized water (4.4 L). The filtrate was charged into a reactor and cooled to 0-5 °C. Aqueous HCI [prepared from cone. HCI (9.3 L) in demineralized water (36 L)] was added slowly with stirring to keep the reaction temperature at or below 15 °C, the resulting mixture was stirred at 10-15 °C for 13 h. The reaction mixture was cooled to 0-5 °C and stirred for 1 h. The solid obtained was collected by filtration and washed with demineralized water (36 L). The solid product was suspended in water (69 L) stirred for 30 mins and collected by filtration washed twice with water (20 L each). The solid product was dried in a vacuum oven at 45°C to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethyl carbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (1 1.21 Kg, 75.77%). Mp: >200°C; 1H NMR (300 MHz, DMSO-ci6): δ 12.98 (br s, 1 H), 10.86 (s, 1 H), 9.24 (s, 3H), 9.04 (s, 2H), 8.22 (d, J = 7.8 Hz, 1 H), 7.96 (d, J = 5.7 Hz, 2H), 7.78 (s, 4H), 7.09-6.99 (m, 2H), 6.07 (d, J = 17.7 Hz, 1 H), 5.45(d, J = 11.4 Hz, 1 H), 3.88 (s, 3H), 3.26-3.24 (m, 2H), 1.09 (m, 1 H), 0.47 (m, 2H), 0.28 (m, 2H).
Average isolated yield for this step varies from 63% to 80%.
The process is also illustrated in Fig. 14.
Example-2: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)

6d 8a
To a solution of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (2.35 g, 5.7 mmol) and 4-aminobenzamidine dihydrochloride (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 ml_) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The
reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and concentrated in vacuum. The residue obtained was purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 and 0-100% chloroform in CMA 50) to furnish methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)-carbamoyl)picolinate hydrochloride (8a) (2.2 g, 65%) as a white solid; MP 266 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 -7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1 H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1 H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for C29H29N505 (H20)1 5 (HCI): C, 58.93; H, 5.63; N, 1 1.85; Found: C, 58.75; H, 5.65; N, 1 1.92.
Step-2: preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)

8a 8b j0 a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml), was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with a solution of sulfuric acid (0.483 ml, 9.00 mmol) in water (5 mL) and stirred for 10 min at room temperature. To this cold water (5 ml) was added and stirred at room temperature until product crystallized out. Cold water (5 mL) was added to the slurry and stir for additional 20 min, additional cold water (5 mL) was added prior to filtration of solid. The solid obtained was collected by filtration washed with water (5 mL and 2.5 mL), dried under vacuum overnight to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b) (1.103 g, 90 % yield) as a white solid; MP 221.7 °C; H NMR (300 MHz, DMSO-d6) δ 12.30 – 10.91 (bs, 1 H, D20 exchangeable), 10.69 (bs, 1 H, D20 exchangeable), 9.24 (t, J = 6.0 Hz, 1 H), 9.16 (s, 2H, D2O exchangeable), 8.78 (s, 2H, D2O exchangeable), 8.24 (d, J = 8.0 Hz, 1 H), 8.04 – 7.91 (m, 2H), 7.84 – 7.67 (m, 4H), 7.13 – 6.94 (m, 2H), 6.03 (dd, J = 17.8, 1 .4 Hz, 1 H), 5.51 – 5.37 (m, 1 H), 3.88 (s, 3H), 3.24 (t, J = 6.4 Hz, 2H), 1.16 – 1.01 (m, 1 H), 0.52 – 0.41 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for: C28H27N605 1.0H2SO4 1.5H20: C, 52.66; H, 5.05; N, 10.97; S, 5.02; Found: C, 52.81 ; H, 4.95; N, 10.94; S, 4.64.
Example-3: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane s

To a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml) was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with methanesulfonic acid (0.584 ml, 9.00 mmol) and stirred for 1 h at room temperature. Cold water (5.00 ml) was added to the reaction mixture and stirred at room temperature until product crystallized out. To the slurry was added water (5 ml) of water stirred for additional 20 min, followed by the addition of water (5 ml) prior to filtration. The solid obtained was collected by filtration washed with water (5 ml and 2.5 ml), dried under vacuum to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane sulfonate salt (8c)
(1 .138 g, 1.867 mmol, 93 % yield) as a white solid; MP 221.2 °C; 1 H NMR (300 MHz,
DMSO-d6) δ 12.89 (s, 1 H, D2O exchangeable), 10.69 (s, 1 H, D2O exchangeable), 9.24
(t, J = 6.0 Hz, 1 H), 9.16 (s, 2H,), 8.85 (s, 2H), 8.24 (d, J = 8.0 Hz, 1 H), 8.06 – 7.91 (m, 2H), 7.86 – 7.70 (m, 4H), 7.15 – 6.96 (m, 2H), 6.03 (dd, J = 17.8, 1.4 Hz, 1 H), 5.52 – 5.35 (m, 1 H), 3.88 (s, 3H), 3.25 (t, J = 6.3 Hz, 2H), 2.34 (s, 3H), 1.17 – 1.01 (m, 1 H), 0.53 -0.43 (m, 2H), 0.32 – 0.23 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for:
CzeH^NsOsCHsSOsH 1.5H20: C, 54.71 ; H, 5.38; N, 11.00; S, 5.04; Found: C, 54.80; H, 5.14; N, 10.94; S, 4.90.
Example-4: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) in Form C (Compound XX)

The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled
46582
to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 – -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1 .6 kg) and 0.57 kg water. The mixture was heated to 46 °C. 21 g of Smopex-234 and 10 g Acticarbone 2SW were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 °C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The suspension was stirred at 0-3.0 °for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjacke,= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 1 17 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C (Compound XX).
/////avoralstat, BCX4161, Fast Track, Treat hereditary angioedema (HAE), Orphan Drug, PRECLINICAL
COc1cc(c(cc1C=C)C(=O)Nc2ccc(cc2)C(=N)N)c3cc(ncc3C(=O)O)C(=O)NCC4CC4
Avoralstat

Avoralstat, BCX4161,
CAS 918407-35-9
UNII: UX17773O15
513.5513, C28-H27-N5-O5
2-Pyridinecarboxylic acid, 3-(2-(((4-(aminoiminomethyl)phenyl)amino)carbonyl)-4-ethenyl-5-methoxyphenyl)-6-(((cyclopropylmethyl)amino)carbonyl)-
3-(2-((4-Carbamimidoylphenyl)carbamoyl)-4-ethenyl-5-methoxyphenyl)-6-((cyclopropylmethyl)carbamoyl)pyridine-2-carboxylic acid
Hereditary angioedema (HAE)
Kallikrein inhibitor
BioCryst Pharmaceuticals
![]()
BioCryst is also investigating second-generation plasma kallikrein inhibitors to avoralstat, for treating HAE (in February 2016, this program was listed as being in preclinical development).
Prevent acute attacks in patients with hereditary angioedema (HAE); Treat hereditary angioedema (HAE)
U.S. – Fast Track (Treat hereditary angioedema (HAE));
U.S. – Orphan Drug (Prevent acute attacks in patients with hereditary angioedema (HAE))
26 Feb 2016Clinical trials in Hereditary angioedema (Prevention) in USA (PO, Hard-gelatin capsule) before February 2016
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in France (PO, Soft-gelatin capsule)
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in Germany (PO, Soft-gelatin capsule)
| Conditions | Interventions | Phases | Recruitment | Sponsor/Collaborators |
|---|---|---|---|---|
| Hereditary Angioedema|HAE | Drug: BCX4161|Drug: Placebo | Phase 2|Phase 3 | Recruiting | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161|Drug: Placebo | Phase 2 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
Avoralstat, also known as BCX-4161, is a potent and orally active Kallikrein inhibitor and Bradykinin inhibitor. Avoralstat may be potentially useful for treatment for Hereditary angioedema. Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
Selective inhibitor of plasma kallikrein that subsequently suppresses bradykinin production
Hereditary angioedema (HAE) is a serious and potentially life-threatening rare genetic illness, caused by mutations in the C1-esterase inhibitor (C1 INH) gene, located on chromosome 11q. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by C1 INH gene mutations which produce low levels of C1 -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective C1 protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; 100(Suppl 2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herin by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that C1 INH is the most important inhibitor of kallikrein in human plasma and that C1 INH deficiency leads to unopposed activation of the kallikrein-bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack.
Approved therapies include purified plasma-derived C1 INH (Cinryze®, Berinert), the recombinant peptide kallikrein inhibitor ecallantide (Kalbitor®), and the bradykinin receptor B2 inhibitor iticabant (Firazyr®). All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
There are many delivery routes for active pharmaceutical ingredients (APIs). Generally, the oral route of administration is favored. Oral administration provides a number of advantages, such as, but not limited to, patient convenience, flexibility of timing of administration, location of administration and non-invasiveness. Oral administration also provides more prolonged drug exposure compared with intermittent intravenous infusion, which may be important for drugs with schedule-dependent efficacy. For example, a drug with a short half-life can achieve a greater exposure time by either continuous infusion or by continuous oral dosing. The use of oral therapy further has the potential to reduce the cost of healthcare resources for inpatient and ambulatory patient care services.
In the pharmaceutical arts, it is known that a number of APIs cannot be administered effectively by the oral route. The main reasons why these compounds cannot be administered by the oral route are: a) rapid enzymatic and metabolic degradation; b) chemical and/or biological instability; c) low solubility in aqueous medium; and/or d) limited permeability in the gastrointestinal tract. For such compounds, non-oral routes of delivery, such as parenteral administration, mainly via intramuscular or subcutaneous injections, may be developed. However, non-oral administration poses a disadvantage for the patient as well as healthcare providers, and for this reason, it is important to develop alternative routes of administration for such compounds, such as oral routes of administration.
While the oral route of administration is the most convenient for the patient and the most economical, designing formulations for administration by the oral route involves many complications. Several methods are available to predict the ease by which an API may be formulated into a formulation suitable for administration by the oral route. Such methods include, but are not limited to, and Lipinski rule (also referred to as the Rule of Five) and the Biopharmaceutical Drug Disposition Classification System (BDDCS).
The BDDCS divides APIs into four classifications, depending on their solubility and permeability. Class I APIs have high solubility and high permeability; Class II APIs have low solubility and high permeability; Class III APIs have high solubility and low permeability; and Class IV APIs have low solubility and low permeability. APIs in higher classes in the BDDCS face greater challenges in formulating into an effective, pharmaceutically acceptable product than those in lower classes. Of the four classes, APIs falling into Class IV are the most difficult to formulate into a formulation for administration by the oral route that is capable of delivering an effective amount of the API as problems of both solubility and permeability must be addressed (note the BDDCS does not inherently address chemical stability). The role of BDDCS in drug development is described generally in L.Z. Benet J Pharm Sci. 2013, 102(1), 34-42.
Lipinski’s rule (described in Lipinski et al. Adv. Drug Deliv. Rev. 46 (1-3): 3-26) states, in general, that in order to develop a successful formulation for administration by the oral route, an API can have no more than one violation of the following criteria:
i) not more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms)
ii) not more than 10 hydrogen bond acceptors (nitrogen or oxygen atoms) iii) a molecular mass less than 500 daltons
iv) an octanol-water partition coefficient log P not greater than 5.
J. Zhang et al. Medicinal Chemistry, 2006, 2, 545-553, describes a number of small molecule amidine compounds which have activity as inhibitors of kallikrein. The molecules described in this document fall into Class IV of the BDDCS as described above. The compounds are poorly soluble in aqueous and physiological fluids, and are poorly permeable as demonstrated by oral dosing in rats and in vitro experiments with Caco-2 cells.
Furthermore, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, one of the compounds described in Zhang et al., is a Class IV API and violates criteria iii) and iv) as set forth in the Lipinski Rule.
Furthermore, the compounds described in Zhang et al., including 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, exhibit poor stability with respect to oxidation in air, to light
(photodegradation) and in aqueous and physiological fluids, as well as to elevated temperatures.
Therefore, the compounds described by Zhang et al. including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, not only exhibit poor solubility and permeability characteristics, but also poor stability characteristics. As a result, such compounds are predicted to be especially difficult to formulate into an effective, orally deliverable
pharmaceutical composition that is capable of delivering an effective amount of the compound to a subject.
Polymorphism, the occurrence of different crystal forms, is a property of some molecules. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties, such as, but not limited to, melting point, thermal behaviors (e.g. measured by thermogravimetric analysis (TGA), or differential scanning calorimetry (DSC), x-ray diffraction pattern, infrared absorption fingerprint, and solid state NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Discovering new polymorphic forms and solvates of a pharmaceutical product can provide alternate forms of the compound that display a number of desirable and advantageous properties, such as, but not limited to, ease of handling, ease of processing, ease of formulation, storage stability, and/or ease of purification. Further, new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof may further provide for improved pharmaceutical products, by providing compounds that are more soluble in a set of pharmaceutical excipients. Still further, the provision of new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof enlarges the repertoire of compounds that a formulation scientist has available for formulation optimization, for example by providing a pharmaceutical product with different properties, such as, but not limited to, improved processing characteristics, improved handling characteristics, improved solubility profiles, improved dissolution profile and/or improved shelf-life. Therefore, there is a need for additional polymorphs of pharmaceutically useful compounds, such as, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid and the compounds disclosed herein.
In one aspect, the present invention provides an oral formulation that is capable of delivering an effective amount of the amidine compounds described by Zhang et al. to a subject. In particular, the present invention provides an oral formulation that is capable of delivering an effective amount of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid to a subject. In one specific aspect, the 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid is present in a particular crystal form designated Form A. In light of the art suggesting the difficulties in formulating such an oral formulation, this result was unexpected.
As described herein, the amidine compounds described in Zhang et al., including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (specifically including particular crystal Form A), may now be conveniently used in oral administration and further used in oral administration for the treatment of a number of diseases and conditions in a subject, such as, but not limited to, HAE as described herein.
Avoralstat & next generation kallikrein inhibitors for HAE
Avoralstat
![]()
May 16 is HAE awareness day
See BioCryst’s video regarding HAE to learn more
Avoralstat is being developed as an oral prophylactic treatment for patients suffering from Hereditary Angioedema (HAE). Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
In May 2014 BioCryst, announced that the OPuS-1 (OralProphylaxiS-1) Phase 2a proof of concept clinical trial met its primary efficacy endpoint, several secondary endpoints and all other objectives established for the trial. OpuS-1 enrolled 24 HAE patients with a history of HAE attack frequency of at least 1 per week. Treatment with avoralstat demonstrated a statistically significant mean attack rate reduction of 0.45 attacks per week versus placebo, p<0.001. The mean attack rate per week was 0.82 on BCX4161 treatment, compared to 1.27 on placebo.
In December 2014, BioCryst initiated enrollment in OPuS-2 (Oral ProphylaxiS-2). OPuS-2 is a blinded, randomized, 12-week, three-arm, parallel cohort design trial evaluating the efficacy and safety of two different dose regimens of avoralstat administered three-times daily, 300 mg and 500 mg, compared with placebo. The primary efficacy endpoint for the trial will be the mean angioedema attack rate, which will be reported for each avoralstat dose group compared to placebo. The trial is being conducted in the U.S., Canada and Europe. On October 8, 2015, announced that it has completed enrollment of approximately 100 HAE patients with a history of moderately frequent to very frequent attacks in OPuS-2. BioCryst expects to report the OPuS-2 trial results in early 2016.
PATENT
WO200234711
http://www.google.com/patents/WO2002034711A1?cl=en
PATENT
PATENT
Examples
Example 1 – Synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl- phenyll-6-(cvclopropylmethyl-carbarnoyl)-pyridine-2-carboxylic acid
The synthesis of the above compound and intermediates is described below. In this section, the following abbreviations are used:
The synthesis of starting material, (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) is described in Scheme 1.
f 0HCY ° ΒΓΥΥ°
Preparation of 6-bromobenzofdl[1,3ldioxole-5-carbaldehvde (1b)
1a 1b
To a mixture of piperonal (1a) (498 g, 3.32 mol) in glacial acetic acid (1000 mL) was added a solution of bromine (200 mL, 3.89 mol) in glacial acetic acid (500 mL) over a period of 30 min and stirred at room temperature for 24h. The reaction mixture was poured into water (2000 mL) and the solid that separated was collected by filtration. The solid was dissolved in boiling ethanol (4000 mL) and cooled to room temperature. The solid obtained on cooling was collected by filtration to furnish 6-bromobenzo[d][1 ,3]dioxole-5-carbaldehyde (lb) (365 g, 48 %) as a white solid, MP 126 °C; HNMR (300 MHz, DMSO-d6): δ 10.06 (s, 1 H), 7.42 (s,1 H), 7.29 (s, 1 H), 6.20 (d, J=12.3, 2H); IR (KBr) 3434, 2866, 1673,1489, 1413, 259, 1112, 1031 , 925 cm“1; Analysis calculated for CeH5BrO3.O 25H C, 41.15; H, 2.37; Found: C, 41.07; H, 2.11.
Preparation of 2-bromo-5-hvdroxy-4-methoxybenzaldehyde (1c)
1c
A solution of potassium tert-butoxide (397 g, 3.36 mol) in DMSO (1.5 L) was heated at 50 °C for 30 min. Methanol (1.5 L) was added to it and continued heating at 50 °C for additional 30 min. To the hot reaction mixture was added 6-bromo-benzo[d][1,3]dioxole-5-carbaldehyde (1 b) (350g, 1.53 mol) and continued heating at 50 °C for 30 min. The reaction mixture was cooled to room temperature and quenched with water (2.3 L) and sodium hydroxide (61.2 g, 1.53 mol). The reaction mixture was washed with ether (2 x 1.5 L), acidified to pH 2 using cone. HCI and extracted with ethyl acetate ( 1 L). The ethyl acetate layers were combined and concentrated under vacuum to dryness. The residue obtained was treated with water (1.5 L) and ethyl acetate (1 L). The solid obtained was collected by filtration to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (97 g, 27.5% as a first crop). The layers from the filtrate were separated and aqueous layer was extracted with ethyl acetate (200 ml_). The ethyl acetate layers were combined dried over MgS04 and concentrated under vacuum to dryness to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (192 g, 54.4%, second crop) as an orange solid, MP 108 °C; ‘HNMR (300MHz, DMSO-cfe): S 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H); IR (KBr) 3477, 2967, 2917,
2837, 2767, 2740, 1657, 1595, 1428, 1270, 1210, 1164, 1022 cm“‘; Analysis calculated for C8H7Br03.H20: C, 38.58; H, 3.64: Found: C, 38.60; H, 3.60.
Preparation of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehvde ( d)
To a solution 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (120 g, 520 mmol) in DMF (1000 mL) was added potassium carbonate (79 g, 572 mmol) and benzyl bromide (68 mL, 572 mmol). The reaction mixture was stirred at room temperature overnight and quenched with water (3000 mL). The solid obtained was collected by filtration, washed with ether and dried under vacuum to furnish 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (113.19 g, 67.9%) as a white solid, MP 144 °C;1HNMR (300 MHz, DMSO-c/6): δ 10.06 (s, 1H), 7.47-7.34 (m, 7H), 5.17 (s, 2H), 3.92 (s, 3H); IR (KBr) 2898, 2851 , 1673, 1592, 1502, 1437, 1402, 1264, 1210, 1158, 1017, 754 cm“1; Analysis calculated for C 5H13Br03: C, 56.10; H, 4.08; Found: C, 55.44; H, 4.08.
Preparation of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e)
15 046578
146
1d 1e
To a solution of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (100 g, 311 mmol) in
ethanol (1500 mL) was added triethyl orthoformate (103 mL, 622 mmol), ammonium nitrate
(7.5 g, 93.3 mmol) and stirred at room temperature overnight. The reaction mixture was
treated with ether (1200 mL) and stirred for 15 min before filtration. The filtrate was
concentrated under vacuum to dryness to give 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (134 g) as a brown syrup; The product was used in the next step
without further purification; 1H N R (300 MHz, DMSO-cf6) δ 7.45 – 7.37 (m, 4H), 7.36 – 7.33
(m, 1 H), 7.17 – 7.14 (m, 1 H), 7.10 (s, 1 H), 5.10 (s, 2H), 3.80 (s, 3H), 3.58 – 3.33 (m, 5H),
1.13 – 1.07 (m, 6H); IR (KBr) 2974, 2879, 1601 , 1503, 1377, 1260, 1163, 1060 cm“1;
Analysis calculated for C19H23Br04: C, 57.73; H, 5.86; Found: C, 57.21 ; H, 5.94.
acid (1fi
To a solution of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (120 g,
300 mmol) in dry ether (1000 mL) at -78 °C was added n-butyllithium (1.6 M solution in
hexanes, 244 mL, 390 mmol) over a period of 30 min and further stirred at -78 °C for 30 min.
A solution of tri-n-butylborate (110 mL, 405 mmol) in dry ether (300 mL) was added to this
solution at -78 °C over a period of 30 min. The reaction mixture was further stirred for 2 h at -78 °C and warmed to 0 °C. The reaction mixture was quenched with 3N HCI (300 mL) at 0
°C and heated at reflux for 1 h. After cooling to room temperature, the solid obtained was
collected by filtration washed with water (250 mL) dried in vaccum to afford (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (30.85 gm, 37.6% as a white solid. The organic
layer from above filtrate was extracted with 1.5 N NaOH (3 x 200 mL). The combined basic
extracts were acidified with cone. HCI (pH about 4). The solid obtained was collected by
filtration, washed with water and dried under vacuum to furnish a second crop of (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (22.3 g, 26%) as a light orange solid
MP 158 °C; 1H NMR (300 MHz, DMSO-cfe) δ 10.08 (s, 1 H), 7.52 (s, 1 H), 7.48 – 7.33 (m, 5H),
7.24 (s, 1H), 5.18 (s, 2H), 3.89 (s, 3H); 1H NMR (300 MHz, DMSO-d6/D20) δ 10.06 (s, 1H),
7.52 (s, 1H), 7.49 – 7.32 (m, 5H), 7.23 (s, 1 H), 5.18 (s, 2H), 3.89 (s, 3H); MS (ES+) 309.1 (M+Na); IR (KBr) 3335, 2937, 1647, 1545, 1388, 1348, 1268, 1146, 1095 cm-1; Analysis calculated for C15H15BO5.0.25H2O: C, 62.00; H, 5.38; Found: C, 61.77; H, 5.19.
Synthesis of methyl-6-(cvclopropylmethylcarbamoyl¾-3-ftrifluoromethylsulfonyloxyVpicolinate
The synthesis of the intermediate methyl 6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethyl sulfonyloxy)picolinate (2h) is described in Scheme 2.
Preparation of 2-bromo-3-hvdroxy-6-methylpyridine (2b)
![]()
H3C N Br
2a 2b
To a solution of 3-hydroxy-6-methylpyridine (2a) (3000 g, 27.5 mol) in pyridine (24 L) cooled to 15 °C was added a solution of bromine (4.83 kg, 1.55 L, 30.2 mol) in pyridine (3 L) over a period of 50 min maintaining the internal temperature between 20 to 25 DC. After stirring for 19 h at room temperature the solvent was removed under vacuum and the residue was triturated with water. The solid separated was collected by filtration, washed with water and dried under vacuum to give 2-bromo-3-hydroxy-6-methylpyridine (2b) (3502 g, 67.7 %) as a light brown solid which was used as such without further purification; 1H NMR (300 MHz, DMSO-d6) δ 10.43 (s, 1H), 7.18 (d, J = 8.0 Hz, 1 H), 7.08 (d, J
MS (ES+) 188.35, 186.36 (M+1).
(2c)
![]()
2b 2c
A mixture of 2-bromo-3-hydroxy-6-methylpyridine (2b) (3000 g, 15.96 mol), anhydrous potassium carbonate (3308 g, 23.94 mol), and iodomethane (2.491 kg, 1.09 L, 17.556 mol) in 30 L of acetone was heated at 40 °C overnight. The reaction mixture was cooled to room temperature and filtered through Celite. Evaporation of the solvent followed by silica gel chromatography (Hexane: ethyl acetate = 7:3) afforded the desired compound, 2-bromo-3-methoxy-6-methylpyridine (2c) which was used as such for the next step; 1H NMR (300 MHz, DMSO-cfe) δ 7.42 (dd, J = 8.3, 1.5 Hz, 1H), 7.29 – 7.19 (m, 1H), 3.84 (d, J = 1.6 Hz, 3H), 2.37 (d, J = 1.7 Hz, 3H).
2c
2d
To a solution of 2-bromo-3-methoxy-6-methylpyridine (2c) (310 g, 1.53 mol) in 6000 mL of water at 60 °C was added KMnO, (725 g, 4.59 mol) in small portions over a 90 min period with vigorous mechanical stirring. A dark purple solution resulted. This solution was kept at 90 °C for a further 3 h and filtered through Celite while still hot to give a colourless filtrate.
After cooling, the aqueous solution was acidified to pH 1-2 by adding 6 N HCI. The white solid obtained was collected by filtration to give on drying 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (302g, 85%) of product, which was used as such in the next reaction without further purification. An analytical sample was obtained by recrystallization from methanol to give 6-bromo-5-methoxy-2-pyridinecarboxylic acid; 1H NMR (300 MHz, DMSO-tfe) δ 7.40 – 7.28 (m, 1H), 7.17 (d, J = 8.3 Hz, 1 H), 3.83 (d, J = 1.7 Hz, 3H).
Preparation of 6-bromo-N-(cvclopropylmethyl)-5-methoxypicolinamide (2e)
To a solution of 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (12 g, 52 mol) in pyridine (70 mL) was added EDCI (11.5 g, 59 mmol) and cyclopropylmethylamine (3.6 g, 52 mmol). The reaction mixture was stirred at room temperature overnight and then concentrated under vacuum. The reaction mixture was diluted with water (100 mL) and ethyl acetate (100 mL). The organic layer was separated and the water layer was extracted with ethyl acetate (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), brine (500 mL), dried over magnesium sulphate, filtered and concentrated under vacuum to furnish 10.43g of crude product. The crude product was converted into a slurry (silica gel 20 g) and purified by flash column chromatography (silica gel 230 g, eluting with 0-100% ethyl acetate in hexane) to yield compound 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (8.02 g, 54%) as off white solid, mp 67-70 °C; 1HNMR (300 MHz, DMSO-d6) δ 8.51 (t, J = 5.8, 1 H), 8.02 (d, J = 8.4, 1 H), 7.65 (d, J = 8.5, 1 H), 3.96 (s, 3H), 3.14 (t, J = 6.5, 2H), 1.11 -0.99 (m, 1 H), 0.47 – 0.36 (m, 2H), 0.27 – 0.20 (m, 2H); MS (ES+) 307.0, 309.0 (100%
M+Na)
Preparation of methyl 6-(cvclopropylmethylcarbamoyl)-3-methoxypicolinate (2f)
To a solution of 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (7.5 g, 27.6 mol) in methanol (300 mL) in a 2-L stainless steel bomb was added Pd(OAc)2(750 mg), 1 ,1-bis(diphenylphosphino)-ferrocene (750 mg), and triethylamine (3.9 mL, 27.6 mmol). The reaction mixture was vacuum flushed and charged with CO gas to 150 psi. The reaction mixture was and heated with stirring at 150°C overnight and cooled to room temperature. The catalyst was filtered through a pad of celite, and concentrated to dryness to furnish crude product. The crude was purified by flash column chromatography (silica gel 150 g,
eluting with, 0%, 5%, 10%, 20%, 30%, 50% ethyl acetate/hexanes (250 mL each) as eluents to give methyl 6-(cyclopropylmethyl-carbamoyl)-3-methoxypicolinate (2f) (6.29 g, 86.1 %) as a salmon coloured solid, MP 107 °C; 1HNMR (300 MHz, DMSO-cfe) δ 8.28 (t, J = 6.0, 1H), 7.91 (d, J = 8.8, 1H), 7.55 (d, J = 8.8, 1 H), 3.68 (s, 3H), 3.64 (s, 3H), 2.90 (t, J = 6.5, 2H), 0.89 – 0.68 (m, 1 H), 0.26 – 0.09 (m, 2H), 0.08 – 0.00 (m, 2H); MS (ES+) 287.1 (M+Na); IR (KBr) 3316, 2921 , 1730, 1659, 1534, 1472, 1432, 1315, 1272, 1228, 1189, 1099, 1003, 929, 846, 680 cm“1; Analysis calculated for C13H16 204: C, 59.08; H, 6.10; N, 10.60; Found: C, 58.70; H, 5.97; N, 10.23.
Preparation of 6-(cvclopropylmethylcarbamoyl 3-hvdroxypicolinic acid (2q)
2f 2g
Aluminium chloride method:
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-methoxypicolinate (2f) (0.16 mmol) in dichloromethane (840 mL) was added AICI3 (193 g, 1.5 mol). The reaction mixture was heated at reflux for 12 h under nitrogen. After slowly adding ~2L of 1 N HCI, the organic layer was separated. The aqueous layer was re-extracted several times with ethyl acetate/DME. The combined organic layer was washed with brine, dried (MgSO.4), and evaporated in vacuo to furnish crude 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid. To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid was added a solution of acetyl chloride (1 10 mL) in methanol (1.1 L). The reaction mixture was stirred for 12 h at room temperature and then concentrated to dryness in vacuo. After co-evaporating once with methanol, the compound was purified by flash-column chromatography (silica gel, 500 g, eluted with chloroform and 3% methanol in chloroform) to furnish 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g).
Boron tribromide method:
To a stirring solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-ethoxypicolinate (2f) (58.0 g, 208 mmol) was added BBr3 (79 mL, 834 mmol) in CH2CI2 (1.3 L) at 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 18h. The reaction mixture was evaporated to dryness and anhydrous methanol (1 L) was added to the light yellowish solid residue. Insoluble solid was collected by filtration (36 g). Mother liquor was evaporated and co-evaporated with MeOH (2 x 200 mL). The insoluble solid (36 g) was treated with MeOH (500 mL) and acetyl chloride (50 mL) and stirred at room temperature for 18 h (at this point reaction mixture was clear). The mixture was evaporated to dryness and diluted with water and extracted with EtOAc. White solid that separated out from EtOAc layer was collected by filtration, washed with water (2 x 20 mL), dried in vacuo at 50 °C to afford 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (5.36 g, 10 %) as a white solid, MP 92-95 °C. 1HNMR (DMSO-cfe) δ 11.04 (s, 1 H, exchangeable with D20), 8.37 (t, J = 6.0, 1 H, exchangeable with D20), 8.12 (d, J = 8.7 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 3.90 (m, 3 H), 3.15 (m, 2 H), 1.04 ( m, 1 H), 0.41 (m, 2 H), 0.24 (m, 2 H). IR (KBr): 3346, 3205, 1684 cm“1; MS (ES+): 251.1 (M+1); Analysis calculated for C12H14N2O4.0.1 H2O: C, 57.18; H, 5.67; N, 11.14; Found: C, 57.11 ; H, 5.61; N, 11.09.
Preparation of methyl-6-(cvclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy) picolinate (2h
To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (28 mmol) in DMF (200 mL) were added triethylamine (12 mL, 84 mmol) and N-phenyl-bis(trifluoromethanesulfonimide) (12 g, 34 mmol). The reaction mixture was stirred for 1.5 h at room temperature and then poured into ice. After diluting with water and extracting with ethyl acetate, the aqueous phase was re-extracted, and then the combined organic layer was washed with water and concentrated under vacuum to give methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)picolinate (2h), which was used in the next step without purification.
1H NMR (300 MHz, CDCI3) δ 8.50 (d, J = 8.6, 1 H), 8.07 (s, 1 H), 7.88 (d, J = 8.6, 1 H), 4.09 (d, J = 12.6, 3H), 3.48 – 3.24 (m, 2H), 1.18 – 1.01 (m, 1 H), 0.69 – 0.44 (m, 2H), 0.42 – 0.20 (m, 2H). MS (ES*): 405.17, 100%, M+Na.
Synthesis of 3-f2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid:
The synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i) is described as shown in Scheme 3.
3-f4-Benzyloxy-2-formyl-5-methoxy-phenylV6-(cvcloDroDvlmethvl-carbarnovn-pyridine-2-carboxylic acid methyl ester (3a)
5 046578
153
3a
To a solution of methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)
picolinate (2h) (24.3g, 63 mmol) in DME (225 mL) were added water (25 mL), (4- (benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (27.3 g, 95 mmol), NaHC03(15.9 g,
5 189 mmol), and bis(triphenylphosphine)palladium(ll) chloride (0.885 g). The reaction
mixture was stirred at 70°C overnight under nitrogen. After extracting with ethyl acetate, the organic layer was washed with water and brine and dried (MgSO^), and then concentrated
under vacuum. The compound was purified by flash-column chromatography (silica gel, 300 g, eluting with 10%, 20%, 30% and 40% ethyl acetate in hexane) to furnish 3-(4-benzyloxy- 10 2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
methyl ester (3a) (25 g, 83%) as off white solid, MP 48-50°C: 1H NMR (300 MHz, DMSO-cfe) δ 9.61(s, 1 H), 8.40 (d, J= 7.9 Hz, 1H), 8.14 (t, J= 5.0 Hz, 1H), 7.87 (d, J= 8.1 Hz, 1 H), 7.58
(s, 1H), 7.54-7.30 (m, 5H), 6.71 (s, 1 H), 5.24 (s, 2H), 3.93 (s, 3H), 3.70 (s, 3H), 3.45-3.34 (m,
2H), 1.19-1.05 (m, 1 H), 0.64-0.54 (m, 2H), 0.37-0.30 (m, 2H); IR ( Br) 1735, 1678, 1594,
15 1513, 1437, 1283, 1217, 1141, 1092 cm“1; MS (ES+) 497.29 (M+Na); Analysis calculated for
C27H2eN206: C, 68.34; H, 5.52; N, 5.90; Found; C, 68.16; H, 5.62; N, 5.80.
2-(6-(Cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-vn-4-methoxy-5- vinylbenzoic acid (3b)
To a solution of 3-(4-benzyloxy-2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl- carbamoyl)-pyridine-2-carboxylic acid methyl ester (3a) (24g, 50.6 mmol) in acetonitrile (50
mL), 2-methyl-2-propanol (350 mL), and water (125 mL) were added sodium dihydrogen
phosphate (12.5 g) and 2-methyl-2-butene (55 mL, 519 mmol). The reaction mixture was cooled in an ice bath and then sodium chlorite (28 g) was added. After stirring for 1 h, the reaction mixture was extracted with ethyl acetate and washed with water. The aqueous layer was re-extracted and then the combined organic layers were dried (MgS04). The solvent was evaporated in vacuo to furnish 5-(benzyloxy)-2-(6- ((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (29 g) which was used for the next step. MS (ES+): 513.24, (M+Na(; (ES ): 489.26, M-1.
Methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxytoarbonyltohenyl)-6-(cvclopropylmethylcarbamovnpicolinate (3c)
To a mixture of 5-(benzyloxy)-2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxy-carbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (31 g, 63.2 mmol), and triethylamine (17.7 mL, 126.4 mmol) in dichloromethane (300 mL), was added MEM-chloride (9.03 mL, 79 mmol), and stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and dried over MgS04, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 40 g) to furnish methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 89%) as a thick gum; H NMR (300 MHz, CDCI3) δ 8.35 (d, J = 8.0 Hz, 1 H), 8.15 (t, J = 5.7 Hz, 1 H), 7.78 (d, J = 8.0 Hz, 1H), 7.71 (s, 1H), 7.49 (d, J = 6.8 Hz, 2H), 7.36 (ddd, J = 7.5, 14.8, 22.4 Hz, 3H), 6.66 (s, 1 H), 5.37-5.13 (m, 4H), 3.90 (s, 3H), 3.69 (s, 3H), 3.60-3.49 (m, 2H), 3.49 (s, 2H), 3.39 (dd, J = 4.4, 8.4 Hz, 2H), 3.34 (s, 3H), 1.19-1.00 (m, 1H), 0.57 (q, J = 5.8 Hz, 2H), 0.38-0.25 (m, 2H). MS (ES+): 601.24 (M+Na); (ES“): 577.27 (M-1);1H NMR (300 MHz, DMSO-cfe) δ 8.69 (t, 7 = 6.1 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1 H), 7.63 (s, 1H), 7.41 (m, 5H), 6.92 (s, 1 H), 5.20 (m, 4H), 3.83 (s, 3H), 3.57 (s, 3H), 3.44 (m, 2H), 3:33 (m, 2H), 3.21 (m, 5H), 1.14 (m, 1H), 0.44 (m, 2H), 0.27 (m, 2H). IR (KBr):
1732, 1671 cm“1. MS (ES+): 601.1(M+Na); Analysis calculated for C31H 2Oe: C, 64.35; H, 5.92; N, 4.84; Found: C, 64.27; H, 6.04; N, 4.79.
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(4-hvdroxy-5-methoxy-2-(((2-methoxyethoxy¾methoxy)carbonyl)phenyl)picolinate (3d)
3c 3d
To a solution of methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)-carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 56.68 mmol) in ethanol (650 mL) was added 10% Pd/C (4 g) and hydrogenated at 45 psi for 5 h. The catalyst was removed by filtration through Celite and the filtrate was concentrated under vacuum to yield methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)picolinate (3d) (31.87 g, 86%), which was pure enough to be used as such for the next step. An analytical sample of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) was obtained by purification of 350 mg of above crude using flash column chromatography (silica gel, eluting with ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethyl-carbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-phenyl)picolinate (3d) as a clear gum; 1HNMR (300 MHz, DMSO-d6) δ 9.74 (s, 1 H), 8.68 (t, J = 6.1 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1 H), 7.95 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 6.83 (s, 1H), 5.19 (s, 2H), 3.77 (m, 3H), 3.58 (s, 3H), 3.44 (m, 2H), 3.34 (m, 2H), 3.21 (m, 5H), 1.04 (m, 1 H), 0.44 (m, 2H), 0.27 (m, 2H); IR (KBr): 1731 , 1664 cm‘1. MS (ES*): 489.0 (M+1); Analysis calculated for C^e^O,,: C, 59.01; H, 5.78; N, 5.73; Found: C, 58.92; H, 6.15; N, 5.29.
6-(Cvclopropylmethylcarbamovn-3-(5-methoxy-2-(((2-methoxyethoxy^methoxy)-carbonyl)-4- (trifluoromethylsulfonyloxy)phenyl)picolinate (3e)
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2- methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) (14.3 g, 29.3 mmol) in dichloromethane (150 mL) were added pyridine (12 mL, 146 mmol) and triflic anhydride (7.5 mL g, 44 mmol). After stirring overnight at room temperature under N2. the reaction mixture was poured into ice water and then extracted twice with dichloromethane. After washing the combined organic extracts with water and drying (MgS0 ), the solvent was evaporated in vacuo. The compound was purified by flash chromatography over silica gel column using ethyl acetate: hexane to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)-carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (1 g, 93%); H NMR (300 MHz, CDCy a 8.41 (d, J = 8.0, 1H), 8.17 (s, 1H), 8.03 (s, 1H), 7.79 (d, J = 8.0, 1 H), 6.82 (s, 1H), 5.32 (q, J = 6.1, 2H), 3.97 (s, 3H), 3.74 (s, 3H), 3.67 – 3.57 (m, 2H), 3.55 – 3.45 (m, 2H), 3.41 (dd, J = 8.2, 14.5, 2H), 3.34 (s, 3H), 1.36 – 1.17 (m, 1H), 0.58 (d, J = 7.1 , 2H), 0.33 (d, J = 5.1 , 2H).
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(5-methoxy-2-f((2-methoxyethoxy)- methoxy)carbonvn-4-vinylphenyl)picolinate (3f)
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (37.4
g, 60.30 mmol) and potassium vinyltrifluoroborate (16.87 g, 120.6 mmol) in DMF (450 mL) and water (45 mL) was bubbled N2 for 5 min. To this mixture was added NaHC03 (20.26 g, 241.2 mmol) and dichloro-bis(triphenylphosphine)palladium (II) (6.34 g, 9.0 mmol). The reaction mixture was stirred at 70 °C for 20 h under N2(reaction progress was checked by 1H N R because product and starting material had same Rf in TLC). The reaction mixture was cooled down to room temperature and diluted with ethyl acetate. The organic layer was separated, washed with water, brine, dried ( gS04) and filtered. The filtrate was concentrated under vacuum to yield crude methyl 6-(cyclopropylmethyl-carbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-4-vinylphenyl)-picolinate (3f). The crude product was purified by flash column chromatography (silica gel, 1 kg, eluting with 0-100% ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate [31) (26.54 g, 88%) as an amber gum; H NMR (300 MHz, DMSO-c¾ δ 8.70 (t, J = 6.1 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1 H), 8.12 (s, 1 H), 8.00 (d, J = 8.0 Hz, 1 H), 6.98 (m, 2H), 5.94 (dd, J = 1.2, 17.8 Hz, 1H), 5.43 (d, J = 12.5 Hz, 1 H), 5.21 (d, J = 6.5 Hz, 2H), 3.88 (s, 3H), 3.64 (s, 3H), 3.48 (d, J = 3.1 Hz, 2H), 3.35 (m, 5H), 3.22 (m, 2H), 1.11 (s, 1H), 0.44 (dt, J = 4.9, 5.5 Hz, 2H), 0.28 (q, J = 4.8 Hz, 2H). IR (KBr); 1732, 1670 cm“1. MS (ES+) 499.1 (M+1).
2-(6-(cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzolc acid (3g)
A mixture of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate (3f) (27.4 mmol) in DME (160 mL) and 6N HCI (40 mL) was stirred at room temperature for 6 h or till TLC showed complete conversion. The solvent was removed under vacuum. The residue obtained was suspended in water, the solid separated out was collected by filtration, washed with water and dried under vacuum to give 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (7.0 g, 63%) as a white
solid MP 40 – 42 °C; H NMR (300 MHz, DMSO-de) δ 8.69 (t, J= 6.0 Hz, 1H, NH), 8.20 (d, J= 7.9 Hz, 1H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1H), 6.97 (dd, J= 18.0, 11.3 Hz, 1H), 6.88 (s, 1H), 5.92 (d, J= 7.9 Hz, 1H), 5.38 (d, J= 11.1 Hz, 1H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); IR (KBr): 3084, 1728, 1650, 1533, 1212, 1143 cm-1; MS (ES+) 433.26 (M+Na); (ES-): 409.28 (M-1); Analysis calculated for θ22Η22Ν2Ο6.0.25Η2Ο; C, 63.68; H, 5.47; N, 6.75; Found C, 63.75; H, 5.56; N, 6.65
Methyl-3-(2-(4-carbamimidoylprienylcarbamoyl)-5-metrioxy-4-vinylphenyl)-6- (cvclopropylmethylcarbamoyl)picolinate (3h)
To a solution of 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (2.35 g, 5.7 mmol) and 4-aminobenzimidamide dihydrochloride (3j) (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 mL) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 in CMA 50) yielding methyl-3-(2-(4-carbamimidoylphenyl-carbamoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (2.2 g, 65%) as a white solid MP 266 °C; 1H NMR (300 MHz, DMSO-c/6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 – 7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for ![]()
C, 58.93; H, 5.63; N,11.85; Found: C, 58.75; H, 5.65; N, 11.92.
46578
159
3-r2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy -vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i)
3h 3i
To a solution of methyl-3-(2-(4-carbamirriidoylphenylcarbarnoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (1 g, 1.9 mmol) in methanol (10 mL) and THF
(10 mL) was added 2 N NaOH (10 mL). The reaction mixture was stirred at room
temperature for 3 h, and concentrated in vacuo to remove methanol and THF. The aqueous layer was acidified with 6N HCI to pH 6-7 and the solid obtained was collected by filtration
washed with water and ether to furnish on drying 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
(3i)(0.775 g, 80%) as the hydrochloride salt as an off white solid.
1H NMR (300 MHz, DMSO-d6) δ 12.67 (s, 1 H), 9.11 (s, 2H), 8.97 (s, 2H), 8.74 (s, 1 H), 7.90
(d, J = 7.8, 1 H), 7.80 (s, 1 H), 7.72 – 7.58 (m, 4H), 6.99 (dd, J = 11.3, 17.7, 1 H), 6.78 (s, 1H),
5.95 (d, J = 17.2, 1H), 5.38 (d, J = 11.9, 1H), 3.82 (s, 3H), 3.18 (s, 2H), 1.06 (s, 1 H), 0.43 (d,
J = 7.9, 2H), 0.25 (d, J = 4.7, 2H); MS (ES+) 514.0 (M+1 ); Analysis calculated for
C2eH27N5O5.HCI.H2O: C, 59.21; H, 5.32; N, 12.33; Found: C, 59.43; H, 5.21; N, 12.06.
Example 1A- Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride in Form
C
The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 to -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1.6 kg) and water (0.57 kg). The mixture was heated to 46 °C. Smopex-234 (21 g) and Acticarbone 2SW (10 g) were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 “C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The
suspension was stirred at 0-3.0 “for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjackel= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 11 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C.
Example-1 B: Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbartiovQpyridine-2-carboxylic acid hydrochloride in Form A
The procedure was carried out in an identical manner to Example 1 A, with the exception that after the final filtration the filter cake was rinsed with 2.87 kg methyl ierf-butyl ether instead of 2.87 kg water, and pulled dry. The product was dried at 40-43 °C and 50 mbar to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) as Form A.
PATENT
Methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (compound 6a) is (I) (pages 85 and 86). Avoralstat hydrochloride (compound of formula XVIII) is (II) (claim 40, page 109). A Markush structures is presented (claim 1, page 99).
The synthesis of (II) via intermediate (I) is described (example 1, pages 80-93).
A synthesis of the compound 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (Compound 3i) is described in Schemes A-C.
O y OHCk n Br^ ^OCH3
B Brr22,, AAccOOHH Y^ V”“ \ \ tt–BBuuOOKK
OHC^^^O ” Br^\^0 MeOH ” OHC
1a 1b 66%
1d 95% 1 e
![]()
1f
Scheme A
3h 31
Scheme C
Examples. In this section, the following abbreviations are used:
Example-1 : Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b)
7b
Step (1): Preparation of 6-Bromobenzo 1 ,3]dioxole-5-carbaldehyde (1 b):
1b
A solution of bromine (33.0 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to a solution of piperonal (1a) (29.9 kg, 199.16 mol) in acetic acid (105 L) at room
temperature over a period of 50 min and the reaction mixture was stirred at room temperature for 14.2 h. Additional solution of bromine (33 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to the reaction mixture over a period of 2 h and the reaction mixture was stirred for 22 h. The reaction mixture was quenched by addition of ice water (500 L) with stirring over a period of 6 h and continued stirring for additional 1.25 h. The mixture was allowed to settle and most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (600 L) was added to the solid, stirred, mixture was allowed to settle and then most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (100 L) was added to the decanted mixture, stirred for 15 min and the solid obtained was collected by filtration using a centrifuge. The solid was washed with water (2 x 100 L) and air-dried in a tray drier for 3.75 h to afford the crude product 1 b (52 kg). The crude product (51.2 kg) was stirred in n-hexane (178 L) for 3 h, collected by filtration, washed with n-hexane (25 L) and dried to afford 6-bromobenzo[1 ,3]dioxole-5-carbaldehyde (1b) (40.1 1 kg, 87.9%) as a light brown solid. MP: 109-112°C. 1H NMR (300 MHz, CDCI3) δ 10.21 (s, 1 H), 7.37 (s, 1 H), 7.07 (s, 1 H), 6.10 (s, 2H); HNMR (DMSO-cf6): δ 10.06 (s, 1 H), 7.42 (s, 1 H), 7.29 (s, 1 H), 6.20 (d, J =12.3 Hz, 2H)
The process is also illustrated in Fig. 1.
Average yield of isolated 1 b from step-1 is 78 – 88%.
Step (2): Preparation of 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c)
A solution of potassium terf-butoxide (10.7 kg, 95.36 mol) in DMSO (49 L) was stirred at 50 °C for 30 min. Methanol (49 L) was added slowly over a period of 4.25 h and stirred at 50 °C for 30 min. 6-Bromobenzo[1 ,3]dioxole-5-carbaldehyde (1 b) (9.91 kg, 43.27 mol) was added to the reaction mixture in small portions over a period of 45 min and stirred at 50 °C for 1 h. The reaction mixture was cooled to room temperature and split into two equal portions. Each portion was quenched with water (50.9 L) and basified with 50% aqueous NaOH solution (2.4 L). Each portion was extracted with MTBE (4 x 36 L) to remove impurities. The aqueous layer was acidified with cone. HCI to pH ~ 3 to obtain
product as a yellow solid. The solid was collected by filtration using a centrifuge, washed with water (2 x 35 L) and air-dried to afford 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) (4.37 kg, 40.7%, contains 7 % water); Mp: 100-102°C; 1HNMR (300MHz, DMSO-d6): δ 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H).
The process is also illustrated in Fig. 2.
Average yield of isolated product 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) from step-2 is 40-50%.
Step (3): 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-y benzaldehyde (4a)
2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) [1.3 kg (93%, 7% water content), 5.25 mol] was dissolved in toluene (13 L) in a reaction flask equipped with a Dean Stark apparatus. The solution was heated at reflux with stirring to distil off about 25% of the toluene along with water (90 ml_). The solution was cooled to 90 °C then
bis(pinacolato)diboron (1.5 kg, 5.82 mol), KOAc (772.6 g, 7.87 mol) and Pd(PPh3) (24.3 g, 0.02 mol) were added and the reaction mixture was heated at reflux for 10h. After confirming the completion of reaction by TLC (mobile phase: 100% DCM), the reaction mixture was cooled to room temperature and was kept standing overnight. The reaction mixture was filtered through celite and the celite cake was washed with toluene (4 L). The filtrate of this batch was mixed with the filtrate of another batch (batch size 1.3 kg obtained from an identical reaction). The mixed filtrate was washed with water (17.5 L), brine (17.5 L), dried over Na2S04, filtered and the solution was passed through a pad of silica gel (2 kg, mesh size 230-400). The silica gel pad was washed with toluene. The combined filtrate and washing was concentrated under reduced pressure and the residual crude product was stirred with n-hexane (23 L) for 1 h to obtain a solid product. The solid was collected by filtration, washed with n-hexane (5 L) and dried to afford 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)benzaldehyde (4a) (2.47 kg, 84.6%). H NMR (300 MHz, CDCI3) δ 10.54 (s, 1 H), 7.57 (s, 1 H), 7.33 (s, 1 H), 5.89 (s, 1 H), 4.01 (s, 3H), 1.37 (s, 12H); 1H NMR (300 MHz, DMSO-d6) δ 10.35 (s, 1 H), 9.95 (s, 1 H), 7.33 (s, 1 H), 7.23 (s, 1 H), 3.87 (s, 3H), 1.33 (s, 12H); MS (ES+) 301.1 (M+Na); 579.1 (2M+Na); Analysis calculated for C14H19B05: C, 60.46; H, 6.89; Found: C, 60.60; H, 6.87
The average yield of 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) from step (3) is 78 – 90%.
The process is also illustrated in Fig. 3.
Step (4): Preparation of 3-Bromo-2,6-dimethylpyridine (5b)
2,6-lutidine (5a) (115 kg, 1073.3 mol) was added into pre-chilled oleum (20-23%, 1015 kg, 2276.7 mol) at 0 °C over a period of 4.5 h (temperature r6ached 14 °C during the addition). Bromine (88.18 kg, 1103.6 mol) was then added at 5-10 °C over a period of 1 h. The reaction mixture was slowly heated to 150 °C over a period of 12h. TLC analysis indicated about 40-50% conversion to product and the formation of a dimer by-product (5%). The reaction mixture was cooled to room temperature and then additional bromine (88.18 kg, 1103.6 mol) was added slowly. The reaction mixture was slowly heated to maintain a temperature of 65-75 °C over a period of 15h. TLC analysis indicated a 65-70 % conversion to product and the formation of 5% dimer by product. The reaction mixture was quenched by addition of water (500L) while maintaining the reaction temperature below 20 °C. The mixture was basified with 6.6 M NaOH (3800 L) while maintain the temperature at < 40 °C. EtOAc (220 L) was added and the mixture was stirred for 1 h then allowed to settle over a period of 2 h. The layers were separated and the aqueous layer was treated with NaOH (10 kg) in water (10 L) and extracted with EtOAc (160 L). The organic extracts were combined washed with brine (100 L), dried over Na2S04 (50.0 kg), filtered and the solvent was evaporated under atmospheric pressure. The residue was vacuum distilled and the desired product 3-bromo-2,6-dimethylpyridine (5b) was collected at 58-60 °C, 2 mmHg (98.45 kg, 49.2 %) as a colorless liquid.
The process is also illustrated in Fig. 4.
Step (5): Preparation of 3-Bromopyridine-2,6-dicarboxylic acid (5c)
![]()
5b 5c
To a stirred solution of 3-bromo-2,6-dimethylpyridine (5b) (98 kg, 5326 mol) in water (1310 L) was added KMn0 (225 kg, 1423.6 mol) in 5 equal portions in 1 h intervals at 70 °C. After stirring for 1 h at 70 °C, additional KMn04 (225 Kg, 1423.6 mol) was added in 5 equal portion in 1 h intervals at 90 °C. The reaction mixture was stirred for 12 h at 90 °C. The suspension was filtered hot through celite to obtain a clear solution. The solvent was distilled off to remove about 30% of the total volume. The remaining concentrated solution was chilled to 0 °C and made acidic (to pH 3-4) by the addition of cone. HCI (120 L). The white precipitate obtained was collected by filtration and dried at 70 °C to afford 3-bromopyridine-2,6-dicarboxylic acid (5c) as a white solid (109 kg, 84%).
The process is also illustrated in Fig. 5.
Step (6): Preparation of Dimethyl 3-Bromopyridine-2,6-dicarboxylate (5d)
To a stirred solution of 3-bromopyridine-2,6-dicarboxylic acid (5c) (20.0 kg, 81.29 mol) in methanol (100 L) was added cone. H2S04 (4.4 L) over a period of 30 min. The reaction mixture was heated to 65 °C and maintained at that temperature for 5 h (the reaction was monitored by TLC analysis to determine completion of reaction). The reaction mixture was cooled to room temperature basified by careful addition of aqueous NaHC03 solution (prepared from 10 kg NaHC03 in 120 L of water) and further diluted with water (120 L). The white solid obtained was collected by filtration, washed with plenty of water and then oven-dried at 40 °C to obtain dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (9.2 kg, 41.3%) as a white solid; 1HNMR (300 MHz, DMSO-cf6) δ 8.47 (d, J = 8.4, 1 H), 8.08 (dd, J = 4.5, 8.4, 1 H), 3.95 (s, 3H), 3.91 (s, 3H); MS (ES+) 570.6 (2M+Na); Analysis calculated for C9H8BrN04: C, 39.44; H, 2.94; Br, 29.15 N, 5. 1 ;
Found: C, 39.52; H, 2.92; Br, 29.28; N, 5.03.
The process is also illustrated in Fig. 6.
6582
Step (7): Preparation of Methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (
To a stirred solution of dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (27 kg, 98.52 mol) in ierf-butanol (135 L) was added at room temperature cyclopropylmethanamine (7.83 kg, 110.1 mol). The reaction mixture was heated at 65 °C for 17 h. The progress of reaction was monitored by TLC and HPLC (HPLC analysis showed the formation of 74% of the product 5e after 17 h. The reaction mixture was cooled to room temperature and then cone. HCI (2.7 L) was added slowly and the mixture was stirred for 15 min. The reaction mixture was concentrated under reduced pressure to obtain the crude product. The crude product was dissolved in hot /-PrOH (54 L) filtered through a celite pad. The filtrate was cooled with stirring to 10 °C to obtain a white precipitate. The solid obtained was collected by filtration, washed with cold
i-PrOH (13 kg), n-hexane (15 L) and dried to provide pure methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e) (15.7 kg, 50.9%). The filtrate was concentrated under reduced pressure and the crude product can be purified by silica gel column chromatography eluting with tert-butanol in hexanes to furnish additional 10% methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e). HNMR (300 MHz, DMSO-cf6) δ 8.83 (t, J = 5.9, 1 H), 8.47 – 8.41 (m, 1 H), 8.06 (d, J = 8.4, 1 H), 3.96 (s, 3H), 3.16 (t, J = 6.5, 2H), 1.14 – 0.99 (m, 1 H), 0.42 (m, 2H), 0.30 -0.19 (m, 2H); MS (ES+) 337.0 (M+23), 650.8 (2M+23); Analysis calculated for
C12H13BrN203: C, 46.03; H, 4.18; N, 8.95; Br, 25.52; Found: C, 46.15; H, 4.17; N, 8.72; Br, 25.26.
The average isolated yield for step (7) is 50% to 60%.
The process is also illustrated in Fig. 7.
Step (8): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a)
2
6a
THF (37.5 L) was charged to a 100 L reactor followed by ethyl 3-bromo-6- (cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate (5e) (2.5 kg, 7.98 mol) under a nitrogen atmosphere. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) (2.88 kg, 10.36 mol) was added, followed by the addition of PPh3 (53.13 g, 0.20 mol), PdCI2(PPh3)2 (120.4 g, 0.17 mol) and a solution of Na2C03(2.12 kg, 20.00 mol) in demineralized water (10.0 L) under nitrogen atmosphere. The reaction mixture was degassed again two times by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 6.5 h, cooled to room temperature and filtered through a Celite bed. Water (75 L) was added to the filtrate and the product was extracted with ethyl acetate (75 L). The aqueous layer was back extracted with ethyl acetate (2 χ 60 L). The combined ethyl acetate extract was divided into two equal portions and each portion was washed with brine (37 L), dried over Na2S04, filtered and concentrated under reduced pressure to give crude methyl 6- ((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) as a reddish viscous material (-4.5 Kg) which was used as such for the next step without further purification. An analytical sample was prepared by purification of a small sample by flash column chromatography (silica gel, eluting with 0-100% ethyl acetate in hexane) to furnish methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)-picolinate (6a) as an off-white solid; HNMR (300 MHz, DMSO-d6) δ 9.89 (s, 1 H), 9.52 (s, 1 H), 8.79 (t, J = 6.1 Hz, 1 H), 8.23 (d, J = 8.0 Hz, 1 H), 8.09 (d, J = 8.0 Hz, 1 H), 7.34 (s, 1 H), 6.90 (s, 1 H), 3.85 (s, 3H), 3.62 (s, 3H), 3.22 (m, 2H), 1.16 -1.02 (m, 1 H), 0.49 – 0.38 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 791.0 (2M+Na), (ES-) 382.7 (M-1), 767.3 (2M-1); Analysis calculated for C20H20N2O6.0.25 H20: C, 61.77; H, 5.31 ; N, 7.20; Found: C, 61.54; H, 5.13; N, 7.05.
The process is also illustrated in Fig. 8.
46582
Step (9): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b)
6a 6b
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) (2.11 kg, estimated about 3.83 mol from step-8) in dichloromethane (16.0 L) and pyridine (1.4 L, 17.4 mol) cooled to -10°C and maintained at that temperature for 1 h was added a solution of triflic anhydride (980.0 ml_, 5.8 mol) in dichloromethane (6.0 L) drop wise over a period of 3 h at -10 °C. The reaction mixture was stirred at -5°C for 1.3 h, quenched with saturated aqueous NaHCO3(10.4 L) and stirred for 30 mins. The organic layer was separated, washed successively with saturated aqueous NaHC03 (10.4 L), 1 HCI (2 x 16.6 L), water (13.2 L), brine (13.2 L), dried over MgS04, filtered and concentrated under reduced pressure to give the crude product. The crude product was stirred with 15% ethyl acetate in n-hexane (7.0 L) for 1 h. The solid obtained was collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (7.0 L) for 1 h, was collected by filtration and washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (8.0 L) for 1 h, collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was dried to afford methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)-oxy)phenyl)picolinate (6b) as a light brown solid (1.7 kg, 86% yield, for combined steps 8 & 9). Average isolated yield for combined steps 8 and 9 was 70% to 86%; Ή NMR (300 MHz, DMSO-cf6): δ 9.64 (s, 1 H), 8.78 (t, J = 6.1 , 1 H), 8.29 (d, J = 8.0, 1 H), 8.16 (d, J = 8.0, 1 H), 8.03 (s, 1H), 7.39 (s, 1 H), 4.00 (s, 3H), 3.63 (s, 3H), 3.22 (m, 2H), 1.11 (m, 1 H), 0.52 – 0.39 (m, 2H), 0.28 (m, 2H); MS (ES+) 538.9 (M+Na). The process is also illustrated in Fig. 9.
Step (10): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c)
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4- (((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b) (12 kg, 23.24 mol) in DME (106 L) was charged into reactor under nitrogen. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. Potassium trifluoro(vinyl)borate (3.9 kg, 29.1 1 mol), PdCI2(PPh3)2 (815 g, 1.13 mol), KHC03 (4.65 g, 46.44 mol) and demineralized water (12 L) was then added under a N2 atmosphere. The reaction mixture was degassed by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 5 h. The reaction mixture was cooled to room temperature and then filtered through a Celite bed. Demineralized water (118 L) was added to the filtrate followed by ethyl acetate (124 L). The mixture was stirred for 20 min and then the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 95 L). The combined organic extract was washed with brine (95 L), dried over Na2S04, and filtered. The solvent was evaporated under reduced pressure to give the crude product. The crude product was purified by column chromatography (silica gel, 120 kg, 230-400 mesh size, eluting with ethyl acetate in n-hexane) to obtain methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (6 kg, 72%). 1H NMR (300 MHz, CDCI3): δ (ppm) 9.64 (s, 1 H), 8.35 (d, J = 7.8 Hz, 1 H), 8.06-8.03 (m, 2H), 7.78(d, J = 7.8 Hz, 1 H), 7.02-6.92 (m, 1 H), 6.61 (s, 1 H), 5.86 (d, J = 17.7 Hz, 1 H), 5.38 (d, J = 1 1.4 Hz, 1 H), 3.84 (s, 3H), 3.67 (s, 3H), 3.35-3.29 (m, 2H),1.08-1.03 (m, 1H), 0.55-0.49 (m, 2H), 0.29-0.2 4(m, 2H). 1HNMR (300 MHz, DMSO-d6) 6 9.68 (s, 1 H), 8.77 (t, J = 6.1 , 1 H), 8.35 – 8.21 (m, 1 H), 8.16 – 8.01 (m, 2H), 7.14 -6.87 (m, 2H), 6.01 (dd, J = 1.2, 17.8, 1 H), 5.45 (dd, J = 1.1 , 1 1.3, 1 H), 3.91 (s, 3H), 3.64 (s, 3H), 3.23 (m, 2H), 1.21 – 1.01 (m, 1H), 0.51 – 0.40 (m, 2H), 0.34 – 0.20 (m, 2H). MS
(ES+) 417.0 (M+Na); Analysis calculated for C22H22N205: C, 66.99; H, 5.62; N, 7.10;
Found: C, 66.75; H, 5.52; N, 7.06.
The process is also illustrated in Fig. 10.
Step (1 1): Preparation of 2-(6-((cyclopropylmethyl)carbamoyl)-2- (methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d)
To a stirred solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (1.57 kg, 3.80 mol) in acetonitrile (15.4 L) was added ferf-butyl alcohol (22.2 L), demineralized water (3.2 L) and sodium dihydrogen phosphate monohydrate (323.74 g, 2.346 mol). The reaction mixture was cooled to 0 °C and added 2-methyl-2-butene (5.3 L, 50.0 mol) and stirred at 0 °C for 30 min. A solution of 80% sodium chlorite (1.36 kg, 12.0 mol) in demineralized water (5.2 L) was added to the reaction mixture over a period of 2.5 h at 0 °C [temperature rises to 7 °C during the addition]. The reaction mixture was stirred at 0 °C for 2 h, diluted with water (40 L) and ethyl acetate (24 L). After stirring the mixture, it was allowed to settle and the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 20 L) then acidified with 5.9 % aqueous acetic acid (2 L) and extracted once with ethyl acetate (10 L). The organic extracts were combined washed with water (2 x 20 L), a solution of acetic acid (125 mL) in water (20.0 L), brine (2 χ 20 L), dried over Na2S04, filtered and concentrated under reduced pressure (vapor temperature below 40 °C). The residue obtained was dissolved in acetone (7 L) (residue didn’t dissolve completely). The solution was poured slowly into a reactor containing stirred n-hexane (70.0 L) to precipitate the solid product and the mixture was stirred for 2 h. The solid obtained was collected by filtration, washed with 10% acetone in n-hexane (6.3 L), AJ-hexane (6.3 L), dried to afford 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4- methoxy-5-vinylbenzoic acid (6d) as an off-white solid (1.29 Kg, yield: 79.0%). Average isolated yield for step 1 1 is 74% to 84%. 1H NMR (300 MHz, DMSO-d6): δ (ppm) 12.50 (brs, 1 H), 8.69(t, J= 6.0 Hz, 1 H, NH), 8.20 (d, J= 7.9 Hz, 1 H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1 H), 6.97 (dd, J= 18.0, 1 1.3 Hz, 1 H), 6.88 (s, 1 H), 5.92 (d, J= 7.9 Hz, 1 H), 5.38 (d, J= 1 1.1 Hz, 1 H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); MS (ES+) 433.26, (M+Na); (ES-) 409.28 (M-1). The process is also illustrated in Fig. 1 1.
Step (12): Preparation of Methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate methanesulfonate (7a
Pyridine (3.8 L, 47.17 mol) and EDCI (5.31 kg, 27.66 mol) were sequentially added to a cooled solution (0 °C) of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (9 kg, 21.92 mol) and 4-aminobenzamidine dihydrochloride (5.13 kg, 24.65 mol) in /-PrOH (90 L). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. TLC analysis indicated incomplete reaction. Additional EDCI (1.08 kg, 5.6 mol) was added and the reaction mixture was stirred for 8 h. The reaction was still incomplete as indicated by TLC analysis, additional EDCI (0.54 kg, 2.8 mol) was added and the reaction mixture was stirred for 5 h. TLC analysis indicated there was trace amount of unreacted starting material remaining. The reaction mixture was cooled to 0 °C and a solution of
methanesulfonic acid (MSA) (9.13 kg, 95 mol) in MeOH (38.7 L) was added to the cooled mixture over a period of 4 h. The reaction mixture was allowed to warm to room temperature and stirred for 15 h. The product was collected by filtration, washed with a mixture of /‘-PrOH and MeOH (4:1 , 45 L). The wet cake was slurried in a mixture of /-PrOH and MeOH (2:1 , 135 L) stirred for 1 h and the product was collected by filtration and washed with a mixture of /‘-PrOH and MeOH (4:1 , 46.8 L). The product was dried in
2015/046582
a vacuum oven at 45 °C to afford methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) as a pink-colored solid (12.71 kg, 93%). Average isolated yield for this step: >90%.
1H NMR (300 MHz, DMSO-c/6) δ 10.71 (s, 1 H), 9.16 (s, 2H), 8.80 (s, 2H), 8.68 (t, J = 6.1 Hz, 1 H), 8.22 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.1 Hz, 1 H), 7.93 (s, 1H), 7.84 – 7.72 (m, 4H), 7.12 – 6.97 (m, 2H), 6.04 (dd, J = 17.8, 1.3 Hz, 1 H), 5.45 (d, J = 12.6 Hz, 1H), 3.91 (s, 3H), 3.60 (s, 3H), 3.25 – 3.16 (m, 2H), 2.32 (s, 3H), 1.10 – 1.01 (m, 1 H), 0.48 – 0.37 (m, 2H), 0.30 – 0.22 (m, 2H); MS (ES+) 528.0 (M+1); Analysis calculated for
C29H29N5O5.CH3SO3H.2H2O. C, 54.62; H, 5.65; N, 10.62; S, 4.86; Found: C, 54.95; H, 5.55; N, 10.61 ; S, 4.87.
The process is also illustrated in Fig. 12.
Step (13): Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-rnethoxy-4- vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate
(3i) ,a 3i
A pre-cooled (0-5 °C) aq. NaOH solution [prepared from solid NaOH (4 kg, 100 mol) in water (86 L)] was added to a suspension of methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) (28.7 kg, 46 mol) in acetonitrile (86 L) cooled to 0 to 5 °C over a period of 25 mins. The reaction mixture was stirred at 0 to 5 °C for 2.5 h (TLC analysis showed the reaction was complete). The reaction mixture was filtered through a sparkler filter, washed with a mixture of 1 :1 CH3CN / H20 ( 57.4 L). Acetic acid (3.2 L, 55.9 mol) in water (56 L) was added to the filtrate at room temperature over a period of 25 mins and the resulting mixture was stirred at room temperature for 2.5 h. The solid product obtained was collected by filtration, washed with a 1 :4 mixture of CH3CN / H20 (57.5 L). The solid was dried at 45°C in a vacuum oven to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate (3i) as an off-white solid (12,77 kg, 54.1%). Average yield for this step is 50% to 75%. Mp: >200°C; H NMR (300 MHz, DMSO-d6): δ 13.49 (s, 1 H), 8.94 (bs, 4H), 8.56 (t, 1 H), 7.82 – 7.71 (m, 2H), 7.67 -7.56 (m, 4H), 7.51 (d, J = 7.8, 1 H), 6.98 (dd, J = 11.3, 17.8, 1 H), 6.68 (s, 1 H), 5.92 (d, J = 16.6, 1 H), 5.36 (d, J = 12.4, 1 H), 3.80 (s, 3H), 3.16 (m, 2H), 1.05 (m, 1 H), 0.43 (m, 2H), 0.24 (m, 2H); MS (ES+) 514.1 (M+1), 536.1 (M+Na), (ES-) 512.1 ; Analysis calculated for C28H27N5O5.3H2O: C, 59.25; H, 5.86; N, 12.34; Found C, 59.50; H,
5.75; N, 12.05. If needed this material can be crystallized from a mixture of acetone and water.
The process is also illustrated in Fig. 13.
Step 14: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b
A pre-cooled (5-8 °C) aqueous NaOH solution (prepared from solid NaOH (1.97 kg, 49.25 mol) in demineralized water (41 L) was added to a pre-cooled (0-5 °C) suspension of (3i) (13.8 kg, 26.9 mol) in acetonitrile (41 L). The reaction mixture was stirred at 0-5 °C for 30 min (until the reaction mixture becomes homogeneous). The reaction mixture was filtered through a sparkler filter washed with 50% acetonitrile in demineralized water (4.4 L). The filtrate was charged into a reactor and cooled to 0-5 °C. Aqueous HCI [prepared from cone. HCI (9.3 L) in demineralized water (36 L)] was added slowly with stirring to keep the reaction temperature at or below 15 °C, the resulting mixture was stirred at 10-15 °C for 13 h. The reaction mixture was cooled to 0-5 °C and stirred for 1 h. The solid obtained was collected by filtration and washed with demineralized water (36 L). The solid product was suspended in water (69 L) stirred for 30 mins and collected by filtration washed twice with water (20 L each). The solid product was dried in a vacuum oven at 45°C to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethyl carbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (1 1.21 Kg, 75.77%). Mp: >200°C; 1H NMR (300 MHz, DMSO-ci6): δ 12.98 (br s, 1 H), 10.86 (s, 1 H), 9.24 (s, 3H), 9.04 (s, 2H), 8.22 (d, J = 7.8 Hz, 1 H), 7.96 (d, J = 5.7 Hz, 2H), 7.78 (s, 4H), 7.09-6.99 (m, 2H), 6.07 (d, J = 17.7 Hz, 1 H), 5.45(d, J = 11.4 Hz, 1 H), 3.88 (s, 3H), 3.26-3.24 (m, 2H), 1.09 (m, 1 H), 0.47 (m, 2H), 0.28 (m, 2H).
Average isolated yield for this step varies from 63% to 80%.
The process is also illustrated in Fig. 14.
Example-2: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)
6d 8a
To a solution of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (2.35 g, 5.7 mmol) and 4-aminobenzamidine dihydrochloride (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 ml_) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The
reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and concentrated in vacuum. The residue obtained was purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 and 0-100% chloroform in CMA 50) to furnish methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)-carbamoyl)picolinate hydrochloride (8a) (2.2 g, 65%) as a white solid; MP 266 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 -7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1 H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1 H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for C29H29N505 (H20)1 5 (HCI): C, 58.93; H, 5.63; N, 1 1.85; Found: C, 58.75; H, 5.65; N, 1 1.92.
Step-2: preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)
8a 8b j0 a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml), was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with a solution of sulfuric acid (0.483 ml, 9.00 mmol) in water (5 mL) and stirred for 10 min at room temperature. To this cold water (5 ml) was added and stirred at room temperature until product crystallized out. Cold water (5 mL) was added to the slurry and stir for additional 20 min, additional cold water (5 mL) was added prior to filtration of solid. The solid obtained was collected by filtration washed with water (5 mL and 2.5 mL), dried under vacuum overnight to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b) (1.103 g, 90 % yield) as a white solid; MP 221.7 °C; H NMR (300 MHz, DMSO-d6) δ 12.30 – 10.91 (bs, 1 H, D20 exchangeable), 10.69 (bs, 1 H, D20 exchangeable), 9.24 (t, J = 6.0 Hz, 1 H), 9.16 (s, 2H, D2O exchangeable), 8.78 (s, 2H, D2O exchangeable), 8.24 (d, J = 8.0 Hz, 1 H), 8.04 – 7.91 (m, 2H), 7.84 – 7.67 (m, 4H), 7.13 – 6.94 (m, 2H), 6.03 (dd, J = 17.8, 1 .4 Hz, 1 H), 5.51 – 5.37 (m, 1 H), 3.88 (s, 3H), 3.24 (t, J = 6.4 Hz, 2H), 1.16 – 1.01 (m, 1 H), 0.52 – 0.41 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for: C28H27N605 1.0H2SO4 1.5H20: C, 52.66; H, 5.05; N, 10.97; S, 5.02; Found: C, 52.81 ; H, 4.95; N, 10.94; S, 4.64.
Example-3: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane s
To a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml) was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with methanesulfonic acid (0.584 ml, 9.00 mmol) and stirred for 1 h at room temperature. Cold water (5.00 ml) was added to the reaction mixture and stirred at room temperature until product crystallized out. To the slurry was added water (5 ml) of water stirred for additional 20 min, followed by the addition of water (5 ml) prior to filtration. The solid obtained was collected by filtration washed with water (5 ml and 2.5 ml), dried under vacuum to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane sulfonate salt (8c)
(1 .138 g, 1.867 mmol, 93 % yield) as a white solid; MP 221.2 °C; 1 H NMR (300 MHz,
DMSO-d6) δ 12.89 (s, 1 H, D2O exchangeable), 10.69 (s, 1 H, D2O exchangeable), 9.24
(t, J = 6.0 Hz, 1 H), 9.16 (s, 2H,), 8.85 (s, 2H), 8.24 (d, J = 8.0 Hz, 1 H), 8.06 – 7.91 (m, 2H), 7.86 – 7.70 (m, 4H), 7.15 – 6.96 (m, 2H), 6.03 (dd, J = 17.8, 1.4 Hz, 1 H), 5.52 – 5.35 (m, 1 H), 3.88 (s, 3H), 3.25 (t, J = 6.3 Hz, 2H), 2.34 (s, 3H), 1.17 – 1.01 (m, 1 H), 0.53 -0.43 (m, 2H), 0.32 – 0.23 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for:
CzeH^NsOsCHsSOsH 1.5H20: C, 54.71 ; H, 5.38; N, 11.00; S, 5.04; Found: C, 54.80; H, 5.14; N, 10.94; S, 4.90.
Example-4: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) in Form C (Compound XX)
The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled
46582
to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 – -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1 .6 kg) and 0.57 kg water. The mixture was heated to 46 °C. 21 g of Smopex-234 and 10 g Acticarbone 2SW were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 °C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The suspension was stirred at 0-3.0 °for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjacke,= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 1 17 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C (Compound XX).
/////avoralstat, BCX4161, Fast Track, Treat hereditary angioedema (HAE), Orphan Drug, PRECLINICAL
COc1cc(c(cc1C=C)C(=O)Nc2ccc(cc2)C(=N)N)c3cc(ncc3C(=O)O)C(=O)NCC4CC4
Pacritinib

Pacritinib
パクリチニブ;
| Formula |
C28H32N4O3
|
|---|---|
| CAS |
937272-79-2
|
| Mol weight |
472.5787
|
UPDATE FDA APPROVED 2/28/2022, Vonjo
To treat intermediate or high-risk primary or secondary myelofibrosis in adults with low platelets
A Jak2 inhibitor potentially for the treatment of acute myeloid Leukemia and myelofibrosis.
UNII-G22N65IL3O
пакритиниб
باكريتينيب
帕瑞替尼
ONX-0803; SB-1518
CAS No. 937272-79-2
472.57868 g/mol, C28H32N4O3
S*Bio Pte Ltd. and concert innovator
11-(2-pyrrolidin-1-ylethoxy)-14,19-dioxa-5,7,26-triazatetracyclo(19.3.1.1(2,6).1(8,12))heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
| Pacritinib (SB1518) is a potent and selective inhibitor of Janus Kinase 2 (JAK2) and Fms-Like Tyrosine Kinase-3 (FLT3) with IC50s of 23 and 22 nM, respectively. | ||||||
UPDATED
Pacritinib, sold under the brand name Vonjo, is an anti-cancer medication used to treat myelofibrosis.[1][2] It is a macrocyclic Janus kinase inhibitor. It mainly inhibits Janus kinase 2 (JAK2) and Fms-like tyrosine kinase 3 (FLT3).
Common side effects include diarrhea, low platelet counts, nausea, anemia, and swelling in legs.[2]
Medical uses
Pacritinib in indicated to treat adults who have a rare form of a bone marrow disorder known as intermediate or high-risk primary or secondary myelofibrosis and who have platelet (blood clotting cells) levels below 50,000/µL.[1][2]
History
The effectiveness and safety of pacritinib were demonstrated in a study that included 63 participants with intermediate or high-risk primary or secondary myelofibrosis and low platelets who received pacritinib 200 mg twice daily or standard treatment.[2] Effectiveness was determined based upon the proportion of participants who had a 35% or greater spleen volume reduction from baseline to week 24.[2] Nine participants (29%) in the pacritinib treatment group had a 35% or greater spleen volume reduction, compared to one participant (3%) in the standard treatment group.[2]
The U.S. Food and Drug Administration (FDA) granted the application for pacritinib priority review, fast track, and orphan drug designations.[2]
Society and culture
Names
Pacritinib is the International nonproprietary name (INN).[3][4]
References
- ^ Jump up to:a b c “Enforcement Reports”. Accessdata.fda.gov. Retrieved 5 March 2022.
- ^ Jump up to:a b c d e f g h “FDA approves drug for adults with rare form of bone marrow disorder”. U.S. Food and Drug Administration. 1 March 2022. Retrieved 3 March 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ World Health Organization (2010). “International nonproprietary names for pharmaceutical substances (INN). proposed INN: list 104” (PDF). WHO Drug Information. 24 (4): 386. hdl:10665/74579.
- ^ World Health Organization (2011). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 66”. WHO Drug Information. 25 (3). hdl:10665/74683.
External links
- “Pacritinib”. Drug Information Portal. U.S. National Library of Medicine.
OLD—
Pacritinib (INN[1]) is a macrocyclic Janus kinase inhibitor that is being developed for the treatment of myelofibrosis. It mainly inhibits Janus kinase 2 (JAK2). The drug is in Phase III clinical trials as of 2013.[2] The drug was discovered in Singapore at the labs of S*BIO Pte Ltd. It is a potent JAK2 inhibitor with activity of IC50 = 23 nM for the JAK2WT variant and 19 nM for JAK2V617F with very good selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively).[3][4] The drug is acquired by Cell Therapeutics, Inc. (CTI) and Baxter international and could effectively address an unmet medical need for patients living with myelofibrosis who face treatment-emergent thrombocytopenia on marketed JAK inhibitors.[5]
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.
Synthesis Reference
A245943 — William AD, Lee AC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Tan E, Chen D, Williams M, Sun ET, Goh KC, Ong WC, Goh SK, Hart S, Jayaraman R, Pasha MK, Ethirajulu K, Wood JM, Dymock BW: Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem. 2011 Jul 14;54(13):4638-58. doi: 10.1021/jm200326p. Epub 2011 Jun 15.
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.

The compound 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) was first described in PCT/SG2006/000352 and shows significant promise as a pharmaceutically active agent for the treatment of a number of medical conditions and clinical development of this compound is underway based on the activity profiles demonstrated by the compound.
-
In the development of a drug suitable for mass production and ultimately commercial use acceptable levels of drug activity against the target of interest is only one of the important variables that must be considered. For example, in the formulation of pharmaceutical compositions it is imperative that the pharmaceutically active substance be in a form that can be reliably reproduced in a commercial manufacturing process and which is robust enough to withstand the conditions to which the pharmaceutically active substance is exposed.
-
In a manufacturing sense it is important that during commercial manufacture the manufacturing process of the pharmaceutically active substance be such that the same material is reproduced when the same manufacturing conditions are used. In addition it is desirable that the pharmaceutically active substance exists in a solid form where minor changes to the manufacturing conditions do not lead to major changes in the solid form of the pharmaceutically active substance produced. For example it is important that the manufacturing process produce material having the same crystalline properties on a reliable basis and also produce material having the same level of hydration.
-
In addition it is important that the pharmaceutically active substance be stable both to degradation, hygroscopicity and subsequent changes to its solid form. This is important to facilitate the incorporation of the pharmaceutically active substance into pharmaceutical formulations. If the pharmaceutically active substance is hygroscopic (“sticky”) in the sense that it absorbs water (either slowly or over time) it is almost impossible to reliably formulate the pharmaceutically active substance into a drug as the amount of substance to be added to provide the same dosage will vary greatly depending upon the degree of hydration. Furthermore variations in hydration or solid form (“polymorphism”) can lead to changes in physico-chemical properties, such as solubility or dissolution rate, which can in turn lead to inconsistent oral absorption in a patient.
-
Accordingly, chemical stability, solid state stability, and “shelf life” of the pharmaceutically active substance are very important factors. In an ideal situation the pharmaceutically active substance and any compositions containing it, should be capable of being effectively stored over appreciable periods of time, without exhibiting a significant change in the physico-chemical characteristics of the active substance such as its activity, moisture content, solubility characteristics, solid form and the like.
-
In relation to 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene initial studies were carried out on the hydrochloride salt and indicated that polymorphism was prevalent with the compound being found to adopt more than one crystalline form depending upon the manufacturing conditions. In addition it was observed that the moisture content and ratio of the polymorphs varied from batch to batch even when the manufacturing conditions remained constant. These batch-to-batch inconsistencies and the exhibited hygroscopicity made the hydrochloride salt less desirable from a commercial viewpoint.
-
Accordingly it would be desirable to develop one or more salts of 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene which overcome or ameliorate one or more of the above identified problems.
PATENT
US 2011263616
http://www.google.com/patents/US20110263616
11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26triaza-tetra-cyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) which have been found to have improved properties. In particular the present invention relates to the maleate salt of this compound. The invention also relates to pharmaceutical compositions containing this salt and methods of use of the salt in the treatment of certain medical conditions.

PATENT
http://www.google.com/patents/US8415338
Representative Procedure for the Synthesis of Compounds Type (XVIIId) [3-(2-Chloro-pyrimidin-4-yl)-phenyl]-methanol (XIIIa2)

Compound (XIIIa2) was obtained using the same procedure described for compound (XIIIa1); LC-MS (ESI positive mode) m/z 221 ([M+H]+).
4-(3-Allyloxymethyl-phenyl)-2-chloro-pyrimidine (XVa2)

Compound (XVa2) was obtained using the same procedure described for compound (XVa1); LC-MS (ESI positive mode) m/z 271 ([M+H]+).
[4-(3-Allyloxymethyl-phenyl)-pyrimidin-2-yl]-[3-allyloxymethyl-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-amine (XVIId1)

Compound (XVIId1) was obtained using the same procedure described for compound (XVIIb1); LC-MS (ESI positive mode) m/z 501.
Macrocycle Example 3 Compound 13

Compound (13) was obtained using the same procedure described for compound (1) HPLC purity at 254 nm: 99%; LC-MS (ESI positive mode) m/z 473 ([M+H]+); 1H NMR (MeOD-d4) δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34-8.31 (m, 1H), 7.98-7.96 (m, 1H), 7.62-7.49 (m, 2H), 7.35 (d, 1H), 7.15-7.10 (m, 1H), 7.07-7.02 (m, 1H), 5.98-5.75 (m, 2H, 2×=CH), 4.67 (s, 2H), 4.67 (s, 2H), 4.39-4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88-3.82 (m, 2H), 3.70 (t, 2H), 2.23-2.21 (m, 2H), 2.10-2.07 (m, 2H).
PAPER
J MC 2011, 54 4638
http://pubs.acs.org/doi/abs/10.1021/jm200326p

Discovery of the activating mutation V617F in Janus Kinase 2 (JAK2V617F), a tyrosine kinase critically involved in receptor signaling, recently ignited interest in JAK2 inhibitor therapy as a treatment for myelofibrosis (MF). Herein, we describe the design and synthesis of a series of small molecule 4-aryl-2-aminopyrimidine macrocycles and their biological evaluation against the JAK family of kinase enzymes and FLT3. The most promising leads were assessed for their in vitro ADME properties culminating in the discovery of 21c, a potent JAK2 (IC50 = 23 and 19 nM for JAK2WT and JAK2V617F, respectively) and FLT3 (IC50 = 22 nM) inhibitor with selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively). Further profiling of 21c in preclinical species and mouse xenograft and allograft models is described. Compound 21c(SB1518) was selected as a development candidate and progressed into clinical trials where it is currently in phase 2 for MF and lymphoma.
 Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
S*BIO Pte. Ltd., 1 Science Park Road, #05-09, The Capricorn, Singapore Science Park II, Singapore 117528
J. Med. Chem., 2011, 54 (13), pp 4638–4658
DOI: 10.1021/jm200326p
Publication Date (Web): May 23, 2011
Copyright © 2011 American Chemical Society
(21c)
The title compound was synthesized from 21a and pyrrolidine (yield, 83%; mixture of trans/cis85:15 by NMR). LC-MS (ESI positive mode) m/z 473 ([M + H]+). HRMS: theoretical C28H32N4O3MW, 472.2474; found, 473.2547. 1H NMR (MeOD-d4): δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34–8.31 (m, 1H, CH), 7.98–7.96 (m, 1H), 7.62–7.49 (m, 2H), 7.35 (d, 1H), 7.15–7.10 (m, 1H), 7.07–7.02 (m, 1H), 5.98–5.75 (m, 2H), 4.67 (s, 2H), 4.67 (s, 2H), 4.39–4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88–3.82 (m, 2H), 3.70 (t, 2H), 2.23–2.21 (m, 2H), 2.10–2.07 (m, 2H); chloride content (titration) 7.7% (1.18 equivs); water content (Karl Fischer) 6.1% (1.85 equivs); Anal. Calcd. for C28H32N4O3·1.18HCl·1.85H2O: C, 61.46; H, 6.46; N, 10.24; Cl, 7.65. Found: C, 61.99; H, 6.91; N, 10.25; Cl, 7.45.
References
1 “International Nonproprietary Names for Pharmaceutical Substances (INN) List 104” (PDF). WHO Drug Information 24 (4): 386. 2010.
2“JAK-Inhibitoren: Neue Wirkstoffe für viele Indikationen”. Pharmazeutische Zeitung (in German) (21). 2013.
3William, A. D.; Lee, A. C. -H.; Blanchard, S. P.; Poulsen, A.; Teo, E. L.; Nagaraj, H.; Tan, E.; Chen, D.; Williams, M.; Sun, E. T.; Goh, K. C.; Ong, W. C.; Goh, S. K.; Hart, S.; Jayaraman, R.; Pasha, M. K.; Ethirajulu, K.; Wood, J. M.; Dymock, B. W. (2011). “Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma”. Journal of Medicinal Chemistry 54 (13): 4638–58. doi:10.1021/jm200326p. PMID 21604762.
4Poulsen, A.; William, A.; Blanchard, S. P.; Lee, A.; Nagaraj, H.; Wang, H.; Teo, E.; Tan, E.; Goh, K. C.; Dymock, B. (2012). “Structure-based design of oxygen-linked macrocyclic kinase inhibitors: Discovery of SB1518 and SB1578, potent inhibitors of Janus kinase 2 (JAK2) and Fms-like tyrosine kinase-3 (FLT3)”. Journal of Computer-Aided Molecular Design 26 (4): 437–50. doi:10.1007/s10822-012-9572-z. PMID 22527961.
5http://www.pmlive.com/pharma_news/baxter_licenses_cancer_drug_from_cti_in_$172m_deal_519143
| US8153632 * | Nov 15, 2006 | Apr 10, 2012 | S*Bio Pte Ltd. | Oxygen linked pyrimidine derivatives |
| US8415338 * | Apr 4, 2012 | Apr 9, 2013 | Cell Therapeutics, Inc. | Oxygen linked pyrimidine derivatives |
| US20110294831 * | Dec 9, 2009 | Dec 1, 2011 | S*Bio Pte Ltd. | 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene citrate salt |
| Patent | Submitted | Granted |
|---|---|---|
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US8153632] | 2009-03-19 | 2012-04-10 |
| ANTIVIRAL JAK INHIBITORS USEFUL IN TREATING OR PREVENTING RETROVIRAL AND OTHER VIRAL INFECTIONS [US2014328793] | 2012-11-30 | 2014-11-06 |
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US2013172338] | 2013-02-20 | 2013-07-04 |
| METHOD OF SELECTING THERAPEUTIC INDICATIONS [US2014170157] | 2012-06-15 | 2014-06-19 |
| CYCLODEXTRIN-BASED POLYMERS FOR THERAPEUTIC DELIVERY [US2014357557] | 2014-05-30 | 2014-12-04 |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE MALEATE SALT [US2011263616] | 2011-10-27 | |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE CITRATE SALT [US2011294831] | 2011-12-01 | |
| BIOMARKERS AND COMBINATION THERAPIES USING ONCOLYTIC VIRUS AND IMMUNOMODULATION [US2014377221] | 2013-01-25 | 2014-12-25 |
| Oxygen linked pyrimidine derivatives [US8415338] | 2012-04-04 | 2013-04-09 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(16E)-11-[2-(1-Pyrrolidinyl)ethoxy]-14,19-dioxa-5,7,26-triazatetracyclo[19.3.1.12,6.18,12]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 46216796 |
| ChemSpider | 28518965 |
| ChEMBL | CHEMBL2035187 |
| Synonyms | SB1518 |
| Chemical data | |
| Formula | C28H32N4O3 |
| Molecular mass | 472.58 g/mol |


S*Bio Pte Ltd
Address: 1 Science Park Rd, Singapore 117528
Phone:+65 6827 5000

S*BIO Pte Ltd. provides research and clinical development services for small molecule drugs for the treatment of cancer in Singapore. The company’s products include JAK2 inhibitors, such as SB1518 for leukemia/myelofibrosis, lymphoma, and polycythemia; and SB1578 for RA/psoriasis. The company also offers SB939, a histone deacetylases for MDS/AML+combo, prostate cancer, sarcoma, pediatric tumor, and myelofibrosis; SB2602, a mTOR inhibitor; SB2343, a mTOR/PI3K inhibitor; and SB1317, a CDK/Flt3 inhibitor. The company was founded in 2000 and is based in Singapore. S*BIO Pte Ltd. operates as a subsidiary of Chiron Corporation Limited.
SEE……..http://apisynthesisint.blogspot.in/2016/01/pacritinib.html

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
|
|
| Clinical data | |
|---|---|
| Trade names | Vonjo |
| Other names | SB1518 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C28H32N4O3 |
| Molar mass | 472.589 g·mol−1 |
| 3D model (JSmol) | |
///////Vonjo, FDA APPTOVESD 2022, APPROVALS 2022, PACRITINIB, パクリチニブ, priority review, fast track, orphan drug, UNII-G22N65IL3O, пакритиниб , باكريتينيب , 帕瑞替尼 , SB 1518
c1cc2cc(c1)-c3ccnc(n3)Nc4ccc(c(c4)COC/C=C/COC2)OCCN5CCCC5
C1CCN(C1)CCOC2=C3COCC=CCOCC4=CC=CC(=C4)C5=NC(=NC=C5)NC(=C3)C=C2
Upadacitinib, ABT-494, упадацитиниб , أوباداسيتينيب , 乌帕替尼 ,
ABT 494
(-)-(3S,4R) cis form
CAS 1310726-60-3 FREE FORM
| MF | C17H19F3N6O |
|---|---|
| MW | 380.36757 g/mol |
Tartrate form
C17 H19 F3 N6 O . C4 H6 O6 . 4 H2 O ………….CAS 1607431-21-9
1-Pyrrolidinecarboxamide, 3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)-, (3S,4R)-, (2R,3R)-2,3-dihydroxybutanedioate, hydrate (1:1:4)
FREE FORM
(3s,,4R)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- l-carboxamide.
(35,,4R)-3-ethyl-4-(3H- imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- l-carboxamide,
(cis,)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide
1-Pyrrolidinecarboxamide, 3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)-, (3S,4R)-
rel-(-)-(3S,4R)-3-Ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide
A Jak1 inhibitor potentially for the treatment of rheumatoid arthritis.
pharmaceutically acceptable salts thereof, stereoisomers thereof, and isomers thereof, is provided in U.S. Patent No. 8,426,411,
Abbott Laboratories ABBOTT ……INNOVATOR
(3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide
1310726-60-3 Cas
1-Pyrrolidinecarboxamide, 3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)-, (3S,4R)-
ABT-494
UNII:4RA0KN46E0
упадацитиниб [Russian] [INN]
أوباداسيتينيب [Arabic] [INN]
乌帕替尼 [Chinese] [INN]
Arthritis, psoriatic PHASE 3 ABBVIE
Upadacitinib (code name ABT-494) is a drug which is currently under investigation for the treatment of rheumatoid arthritis, Crohn’s disease, ulcerative colitis, and psoriatic arthritis. It was developed by the biotech company AbbVie.
Upadacitinib tartrate, a selective Jak1 inhibitor, is in phase III clinical trials at AbbVie (previously Abbott) for the treatment of patients with moderate to severe rheumatoid arthritis or active psoriatic arthritis with inadequate responses to conventional or biologic disease-modifying antirheumatic drugs (DMARDs). Phase III clinical trials are also ongoing for the treatment of moderately to severely active Crohn’s disease, ulcerative colitis, moderate to severe atopic dermatitis, and active ankylosing spondylitis.
In 2015, orphan drug designation was assigned to the compound for the treatment of pediatric juvenile idiopathic arthritis (JIA) categories excluding systemic JIA. In 2017, additional orphan drug designation was assigned in the U.S. for the treatment of pediatric systemic JIA.
In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie.
Upadacitinib tartrate [USAN]
1607431-21-9
Mechanism of action
The Janus kinases (JAKs) are a family of cytoplasmic tyrosine kinases whose function is to transduce cytokine-mediated signals via the JAK-STAT pathway. There are four JAK subtypes, each of which has overlapping receptor responsibilities. Inhibitors of this enzyme family (jakinibs) have shown efficacy in treating certain inflammatory and autoimmune diseases such as rheumatoid arthritis and Crohn’s disease. However, the first generation of these drugs, tofacitinib and ruxolitinib, lacked subtype selectivity, affecting JAK1/JAK3 and JAK1/JAK2 respectively. This has led to dose-limiting side effects in this otherwise promising class of drugs.[2][3] Upadacitinib is a second generation Janus kinase inhibitor that is selective for the JAK1 subtype of this enzyme over the JAK2 (74-fold), JAK3 (58-fold) and TYK2 subtypes.[4]
Clinical trials
Phase I studies
A phase I study revealed that upadacitinib followed a bi-exponential disposition with a terminal half-life of 6–16 hours.[1] There was no significant accumulation over the dose range of 3–36 mg per day. No interaction was found in rheumatoid arthritis patients taking methotrexate. The most common adverse event was headache but its incidence was similar to that when taking placebo (15.6% for upadacitinib vs. 16.7% for placebo). An investigation into absorption and metabolism found that dosing after a high-fat meal had no effect on upadacitinib total drug exposure over time (area under the curve or AUC).[5] Inhibition of CYP3A by ketoconazole increased total AUC, indicating the importance of this metabolic route.
Phase II studies
Two phase IIb studies were initiated to study the efficacy and safety of upadacitinib in patients with rheumatoid arthritis and one phase II study was initiated in patients with Crohn’s disease.
BALANCE I
In the first study, 276 rheumatoid arthritis patients were recruited who had previously experienced inadequate response to anti–tumor necrosis factor (TNF) therapy and were currently on a stable dose of methotrexate.[6] Patients were randomized to receive 3, 6, 12, or 18 mg twice daily or placebo. The primary endpoint was a 20% improvement in symptoms according to the American College of Rheumatology improvement criteria (ACR20). At the completion of the study it was found that response rates were significantly higher in those receiving upadacitinib versus in those receiving placebo alone (36–42% and 22– 26%, respectively). Adverse events included headache, nausea, and infection but no infections were serious.
BALANCE II
In the second phase IIb study, 300 rheumatoid arthritis patients were recruited who have had an inadequate response to methotrexate.[7] Patients were randomized to receive 3, 6, 12, or 18 mg twice daily or placebo. The primary endpoint was a 20% improvement in symptoms according to the American College of Rheumatology improvement criteria (ACR20). At the completion of the study it was found that response rates were significantly higher in those receiving upadacitinib versus in those receiving placebo alone. (62%, 68%, 80%, 64%, and 76% for the 3, 6, 12, 18, and 24 mg doses, respectively) than with placebo (46%). Improvement in symptoms was rapid, with significant changes in disease scores by week 2. Adverse events were mild with infection being the most serious. One case of community-acquired pneumonia occurred at 12 mg.
CELEST
In this 16-week study, 220 patients were recruited with moderately to severely active Crohn’s disease. Participants must have also experienced an inadequate response to or intolerance to Immunotherapy or TNF inhibitors.[8][9] Patients were randomized to therapy with upadacitinib at 3, 6, 12, 24 mg twice daily or 24 mg once daily for 16 weeks or placebo, followed by blinded extension therapy for 36 weeks. The co-primary endpoints were the proportion of patients who achieved clinical remission (soft stool frequency or daily abdominal pain score) at week 16 and endoscopic remission at week 12 or 16. Secondary endpoints included significant clinical response (≥30% reduction in symptoms) at week 16 and endoscopic response (≥25% decrease in symptoms) at week 12 or 16. At 16 weeks 22% of patients taking the 24 mg twice daily dose achieved endoscopic remission with upadacitinib compared to 0% of patients taking placebo. 27% of patients taking the 6 mg twice daily dose achieved clinical remission compared to 11% of patients taking placebo. Adverse events did not appear to be dose-related. A single case of non-melanoma skin cancer was reported in the 24 mg twice daily group.
Phase III studies
Abbvie has planned a total of six phase III trials that will evaluate over 4,000 patients with moderate to severe rheumatoid arthritis.[10] Two phase III trials are planned studying patients with psoriatic arthritis and one in patients with ulcerative colitis.
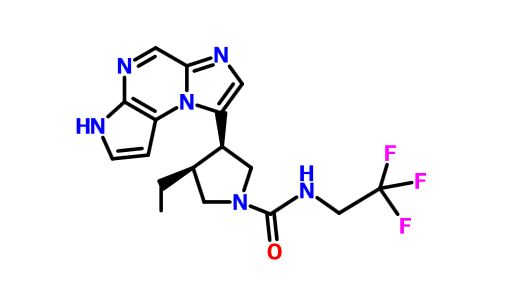
AbbVie, a global biopharmaceutical company, today announced the start of a large Phase 3 clinical trial program to study the use of ABT-494, an investigational, once-daily, oral selective JAK1 inhibitor for the treatment of rheumatoid arthritis (RA). This program will include adult patients with inadequate responses (IR) to conventional or biologic disease-modifying antirheumatic drugs (DMARDs), as well as methotrexate-naive patients.

PATENT
WO2015061665
The synthesis of the compounds of the invention, including (35,,4R)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- l-carboxamide, pharmaceutically acceptable salts thereof, stereoisomers thereof, and isomers thereof, is provided in U.S. Patent No. 8,426,411, the entire content of which is incorporated herein by reference.
For example, (3lS,,4R)-3-ethyl-4-(3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide can be synthesized according to the following scheme:
N-Alkylation using alkyl halide, a-haloketone or oc-haloamide
A round bottom flask is charged with a base such as NaH (60% dispersion in mineral oil), K2CO3, or CS2CO3 (preferably NaH (60% dispersion in mineral oil), 0.9-1.5 equiv., preferably 0.95 equiv.) and an organic solvent (such as N, N-dimethylformamide (DMF), dichloromethane (DCM), 1,4-dioxane, or N-methyl-2-pyrrolidone (NMP), preferably DMF). The mixture is cooled to about -10 °C to ambient temperature (preferably about 0°C) and a solution of an appropriately substituted amine (preferably 1 equiv.) in an organic solvent (such as DMF) is added. Alternatively, the base may be added portionwise to a solution of the amine and an organic solvent at about 0°C to ambient temperature. The reaction mixture is stirred for about 5-90 min (preferably about 15-30 min) at about -10°C to ambient temperature (preferably about 0°C) followed by the addition of an alkyl halide, a-haloketone, or cc-haloamide (1-2 equiv., preferably 1.2 equiv.). Alternatively, a solution of an amine and a base in an organic solvent may be added to a solution of an alkyl halide, α-haloketone, or a-haloamide in an organic solvent at about 0°C. The reaction mixture is stirred at about -10°C to ambient temperature (preferably ambient temperature) for about 0.5-24 h (preferably about 1 h). Optionally, the organic solvent may be removed under reduced pressure.
Optionally, the reaction mixture or residue may be diluted with water, aqueous NH4CI, or aqueous NaHC03. If a precipitate forms the solid may be optionally collected via vacuum filtration to give the target compound. Alternatively, an organic solvent (such as ethyl acetate (EtOAc) or DCM) is added to the aqueous mixture and the layers are separated. The aqueous layer may optionally be extracted further with an organic solvent (such as EtOAc and/or DCM). The combined organic layers are optionally washed with additional aqueous solutions such as brine, dried over anhydrous Na2S04 or MgS04, filtered, and concentrated to dryness under reduced pressure.
The procedure above is illustrated below in the preparation of ie/t-butyl 2-amino-2-oxoethyl(5-tosyl-5H-pyrrolo[3,2-b]pyrazin-2-yl)carbamate from ie/t-butyl (5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-yl)carbamate.
To a solution of iert-butyl 5-tosyl-5H-pyrrolo[3,2-b]pyrazin-2-ylcarbamate (1.00 g, 2.57 mmol, Example #3 Step E) and DMF (13 mL) under nitrogen at about 0 °C was added NaH (60% dispersion in mineral oil, 0.113 g, 2.83 mmol) in one portion. After about 30 min, 2-bromoacetamide (0.391 g, 2.83 mmol) was added in one portion. After about 30 min, the ice bath was removed and the solution was stirred at ambient temperature for about 2 h. Saturated aqueous NH4Cl/water (1: 1, 100 mL) was added. After stirring for about 10 min, the mixture was filtered using water to wash the filter cake. The aqueous phase was extracted with EtOAc (50 mL). The filter cake was dissolved in EtOAc and added to the organic layer. The organic layer was dried over Na2S04, filtered, and concentrated under reduced pressure. The material was purified by silica gel chromatography eluting with a gradient of 20-100% EtOAc/heptane to give tert-butyl 2-amino-2-oxoethyl(5-tosyl-5H-pyrrolo[3,2-b]pyrazin-2-yljcarbamate (0.980 g, 82%): LC/MS (Table 1, Method n) Rt = 0.70 min; MS m/z 446 (M+H)+.
Similar reaction condition can also be used to synthesize benzyl 3-ethyl-4-(2-((5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-yl)amino)acetyl)pyrrolidine-l-carboxylate from iert-butyl (5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-yl)carbamate and benzyl 3-(2-bromoacetyl)-4-ethylpyrrolidine- 1 -carboxylate.
Cyclization of a ketone using a dithiaphosphetane reagent (e.g., synthesizing (3S,4R)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-l-carboxylate from benzyl 3-ethyl-4-(2-((5-tosyl-5H-pyrrolo[2,3-Z>]pyrazin-2-yl)amino)acetyl)pyrrolidine-l-carboxylate)
To a solution of a ketone (preferably 1 equiv.) in an organic solvent such as tetrahydrofuran (THF) or 1,4-dioxane (preferably 1,4-dioxane) is added a thiolating reagent such as Lawesson’s reagent or Belleau’s reagent (2,4-bis(4-phenoxyphenyl)-l,3-dithia-2,4-diphosphetane-2,4-disulfide) (0.5-2.0 equiv., preferably Lawesson’s reagent, 0.5-0.6 equiv.). The reaction is heated at about 30°C to 120°C (preferably about 60-70°C) for about 0.5-10 h (preferably about 1-2 h). Optionally, additional thiolating reagent (0.5-2.0 equiv., preferably 0.5-0.6 equiv.) can be added to the reaction mixture and heating can be continued for about 0.5-10 h (preferably about 1-2 h). The reaction mixture is concentrated under reduced pressure.
Preparation of 8-((ds)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine from (3S,4R)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-l-carboxylate
To a solution of (cis)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-l-carboxylate (0.838 g, 1.541 mmol) is added a solution of HBr (2.50 mL, 15.19 mmol, 33% in acetic acid). The reaction mixture is stirred at ambient temperature for about 1 h. The reaction is diluted with diethyl ether or Et20 (50 mL) and water (20 mL). The layers are stirred for about 3 min and the organic layer is decanted then the procedure is repeated 5 times. The aqueous layer is cooled to about 0°C and is basified with saturated aqueous NaHC03 solution (10 mL) to about pH 7. The aqueous layer is extracted with EtOAc (3 x 50 mL), combined, and dried over anhydrous Na2S04, filtered and concentrated to give a brown solid. The solid is dissolved in DCM (50 mL) and washed with water (3 x 20 mL), dried over anhydrous Na2S04, filtered and concentrated to afford 8-((cis)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine (0.453, 61%) as a brown residue: LC/MS (Table 1, Method a) Rt = 1.73 min; MS m/r. 410 (M+H)+.
Hydrolysis of a sulfonamide (e.g., 8-((3R,4S)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine to 8-((3R,4S)-4-ethylpyrrolidin-3-yl)-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine)
To a flask containing a sulfonamide, for example, a sulfonyl-protected pyrrole, (preferably 1 equiv.) in an organic solvent (such as 1,4-dioxane, methanol (MeOH), or THF/MeOH, preferably 1,4-dioxane) is added an aqueous base (such as aqueous Na2C03 or aqueous NaOH, 1-30 equiv., preferably 2-3 equiv. for aqueous NaOH, preferably 15-20 equiv. for aqueous Na2C03). The mixture is stirred at about 25-100 °C (preferably about 60 °C) for about 1-72 h (preferably about 1-16 h). In cases where the reaction does not proceed to completion as monitored by TLC, LC/MS, or HPLC, additional aqueous base (such as aqueous Na2C03, 10-20 equiv., preferably 10 equiv. or aqueous NaOH, 1-5 equiv., preferably 1-2 equiv.) and/or a cosolvent (such as ethanol (EtOH)) is added. The reaction is continued at about 25-100°C (preferably about 60°C) for about 0.25-3 h (preferably about 1-2 h). In any case where an additional base labile group is present (for example, an ester a
trifluoromethyl, or a cyano group), this group may also be hydrolyzed. The reaction is worked up using one of the following methods. Method 1. The organic solvent is optionally removed under reduced pressure and the aqueous solution is neutralized with the addition of a suitable aqueous acid (such as aqueous HC1). A suitable organic solvent (such as EtOAc or DCM) and water are added, the layers are separated, and the organic solution is dried over anhydrous Na2S04 or MgS04, filtered, and concentrated to dryness under reduced pressure to give the target compound. Method 2. The organic solvent is optionally removed under reduced pressure, a suitable organic solvent (such as EtOAc or DCM) and water are added, the layers are separated, and the organic solution is dried over anhydrous Na2S04 or MgS04, filtered, and concentrated to dryness under reduced pressure to give the target compound. Method 3. The reaction mixture is concentrated under reduced pressure and directly purified by one of the subsequent methods.
Formation of a urea using CDI or thiocarbonyldiimidazole, respectively (e.g., from 8-((3R,45)-4-ethylpyrrolidin-3-yl)-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine to (35,4R)-3-ethyl-4-(3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide)
To a solution or slurry of an amine or amine salt (1-3 equiv., preferably 1-2 equiv.) in an organic solvent such as DCM, THF, or DMF (preferably DMF) at about 20 – 80 °C (preferably about 65 °C) is optionally added an organic base, such as triethylamine (TEA), N,N-diisopropylethylamine (DIEA), pyridine (preferably TEA) (1-10 equiv., preferably 1-5 equiv.) followed by CDI or 1,1 ‘-thiocarbonyldiimidazole (0.5-2 equiv., preferably 1 equiv.). After about 0.5-24 h (preferably about 1-3 h), a second amine or amine salt (1-10 equiv., preferably 1-3 equiv.) is added neat or as a solution or slurry in an organic solvent such as DCM, THF, or DMF (preferably DMF). The reaction is held at about 20 – 80 °C (preferably about 65 °C ) for about 2 – 24 h (preferably about 3 h). If the reaction mixture is heated, it is cooled to ambient temperature. The reaction mixture is partitioned between an organic solvent (such as EtOAc, DCM or 1,4-dioxane) and an aqueous base (such as saturated aqueous NaHC03 or saturated aqueous Na2C03, preferably saturated aqueous NaHC03). Optionally, the reaction mixture is concentrated under reduced pressure and the residue is partitioned as above. In either case, the aqueous layer is then optionally extracted with additional organic solvent such as EtOAc or DCM. The combined organic layers may optionally be washed with brine and concentrated in vacuo or dried over anhydrous Na2S04 or MgS04 and then decanted or filtered prior to concentrating under reduced pressure to give the target compound. Optionally, the reaction mixture is concentrated under reduced pressure and the residue is directly purified.
Chiral preparative HPLC purification
Chiral purification is performed using Varian 218 LC pumps, a Varian CVM 500 with
switching valves and heaters for automatic solvent, column and temperature control and a Varian 701 Fraction collector. Detection methods include a Varian 210 variable wavelength detector, an in-line polarimeter (PDR-chiral advanced laser polarimeter, model ALP2002) used to measure qualitative optical rotation (+/-) and an evaporative light scattering detector (ELSD) (a PS-ELS 2100 (Polymer Laboratories)) using a 100: 1 split flow. ELSD settings are as follows: evaporator: 46 °C, nebulizer: 24 °C and gas flow: 1.1 SLM. The absolute stereochemistry of the purified compounds was assigned arbitrarily and is drawn as such. Compounds of the invention where the absolute stereochemistry has been determined by the use of a commercially available enantiomerically pure starting material, or a stereochemically defined intermediate, or X-ray diffraction are denoted by an asterisk after the example number.
(ci5,)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide isolated using the above method has an Rt min of 1.52, and m/z ESI+ (M+H)+ of 381.
The starting materials and intermediates of the above synthesis scheme may be obtained using the following schemes:
Preparation of starting material of l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid
Step A: ethyl pent-2-ynoate to (Z)-ethyl pent-2-enoate
To a slurry of Lindlar catalyst (0.844 g, 0.396 mmol) in THF (100 mL) and pyridine (10.00 mL) is added ethyl pent-2-ynoate (5.22 mL, 39.6 mmol). The reaction mixture is sparged with hydrogen for about 10 min and an atmosphere of hydrogen is maintained via balloon. After about 15 h the reaction mixture is filtered through a pad of Celite®, diluted with Et20 (30 mL) and washed with saturated aqueous CuS04 (40 mL), followed by water (40 mL). The organic layer is separated, dried over anhydrous MgS04, filtered, and concentrated in vacuo to provide crude (Z)-ethyl pent-2-enoate (5 g, 98%). 1H NMR (DMSO-d6) δ 1.05 (t, 3H), 1.28 (t, 3H), 2.65 (m, 2H), 4.18 (q, 2 H), 5.72 (m, 1H), 6.21 (m, 1H).
Step B: (ds)-ethyl l-benzyl-4-ethylpyrrolidine-3-carboxylate (from (Z)-ethyl pent-2-enoate and N-benzyl-l-methoxy-N-((trimethylsilyl)methyl)methanamine)
To a solution of N-benzyl-l-methoxy-N-((trimethylsilyl)methyl)methanamine (9.98 mL, 39.0 mmol) and (Z)-ethyl pent-2-enoate (5 g, 39.0 mmol) in DCM (50 mL) is added trifluoroacetic acid (TFA) (0.030 mL, 0.390 mmol) at RT. After about 2 days, the reaction mixture is concentrated in vacuo to provide crude (cis)-ethyl 1 -benzyl-4-ethylpyrrolidine-3- carboxylate (9.8 g, 96%) as an oil. LC/MS (Table 1, Method a) Rt = 1.62 min; MS m/z: 262 (M+H)+.
Step C: ethyl l-benzyl-4-ethylpyrrolidine-3-carboxylate to (ds)-ethyl 4-ethylpyrrolidine-3-carboxylate
A Parr shaker is charged with PdOH2 on carbon (2.243 g, 3.19 mmol) and (cis)-et yl l-benzyl-4-ethylpyrrolidine-3-carboxylate (16.7 g, 63.9 mmol) followed by EtOH (100 mL). The reaction mixture is degassed and purged with hydrogen gas and shaken on the parr shaker at 60 psi for about 4 days at ambient temperature. The reaction mixture is degassed and purged with nitrogen. The suspension is filtered through a pad of Celite® washing with EtOH (~ 900 mL). The solvent is removed under reduced pressure to afford (cis)-ethyl 4-ethylpyrrolidine-3 -carboxylate (8.69 g, 79%) as an oil: LC/MS (Table 1, Method a) Rt = 1.11 min; MS m/z: 172 (M+H)+.
Step D: (ds)-ethyl 4-ethylpyrrolidine-3-carboxylate to (ds)-l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid
To a flask charged with (cis)-et yl 4-ethylpyrrolidine-3-carboxylate (8.69g, 50.7 mmol) is added aqueous HCl (6N, 130 mL, 782 mmol). The solution is heated at about 75°C for about 12 h. aqueous HCl (6N, 100 mL, 599 mmol) is added and stirred at about 80 °C for about 20 h. Aqueous HCl (6N, 100 mL, 599 mmol) is added and continued stirring at about 80 °C for about 20 h. The reaction mixture is cooled to ambient temperature and the solvent is removed under reduced pressure. 1,4-Dioxane (275 mL) and water (50 mL) are added followed by portionwise addition of Na2C03 (13.5 g, 127 mmol). Di-ie/t-butyl dicarbonate (13.3 g, 60.9 mmol) is added and the reaction mixture is stirred at ambient temperature for about 16 h. The solid is filtered and washed with EtOAc (250 mL). The aqueous layer is acidified with aqueous HCl (IN) to about pH 3-4. The layers are partitioned and the aqueous layer is extracted with EtOAc (3 x 100 mL). The combined organic layers are dried over anhydrous Na2S04, filtered and removed under reduced pressure. As the organic layer is almost fully concentrated (~ 10 mL remaining), a solid precipitated. Heptane (30 mL) is added and the solid is filtered washing with heptane to afford (cis)-l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid (3.9 g, 32%) as an off white solid as product: LC/MS (Table 1, Method c) Rt = 0.57 min; MS m/z: 242 (M-H)~.
Synthesis of Intermediate benzyl 3-(2-bromoacetyl)-4-ethylpyrrolidine-l-carboxylate
Acidic cleavage of a Boc-protected amine (e.g., l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid to 4-ethylpyrrolidine-3-carboxylic acid
hydrochloride)
To a solution of a Boc-protected amine (preferably 1 equiv.) in an organic solvent (such as DCM, 1,4-dioxane, or MeOH) is added TFA or HC1 (preferably 4 N HC1 in 1,4-dioxane, 2-35 equiv., preferably 2-15 equiv.). The reaction is stirred at about 20-100 °C (preferably ambient temperature to about 60 °C) for about 1-24 h (preferably about 1-6 h). In any case where an additional acid labile group is present (for example, a t-butyl ester), this group may also be cleaved during the reaction. Optionally, additional TFA or HC1
(preferably 4 N HC1 in 1,4-dioxane solution, 2-35 equiv., preferably 2-15 equiv.) may be added to the reaction mixture in cases where the reaction does not proceed to completion as monitored by TLC, LC/MS, or HPLC. Once the reaction has proceeded to an acceptable level, the reaction mixture can be concentrated in vacuo to provide the amine as a salt.
Alternatively, the reaction may be partitioned between an organic solvent (such as EtOAc, DCM or 1,4-dioxane) and an aqueous base (such as saturated aqueous NaHC03 or saturated aqueous Na2C03, preferably saturated aqueous NaHC03). The aqueous layer can be optionally extracted with additional organic solvent such as EtOAc or DCM. The combined organic layers may optionally be washed with brine, dried over anhydrous Na2S04 or MgS04, then decanted or filtered, prior to concentrating under reduced pressure to give the target compound.
Cbz-protection of an amine (e.g., 4-ethylpyrrolidine-3-carboxylic acid hydrochloride to l-((benzyloxy)carbonyl)-4-ethylpyrrolidine-3-carboxylic acid)
A solution of an amine or an amine salt (preferably 1 equiv.) and a base (for example, Na2C03 or NaOH, 1-3 equiv., preferably Na2C03, 1.6 equiv.) in water or aqueous organic solvent (for example, water / 1,4-dioxane or water / acetonitrile (MeCN), preferably water/ 1,4-dioxane) is stirred at ambient temperature for about 1-10 min (preferably 5 min). A solution of benzyl 2,5-dioxopyrrolidin-l-yl carbonate (1-2 equiv., preferably 1.0 equiv.) in an organic solvent such as 1,4-dioxane or MeCN is added to the reaction. The reaction is stirred at ambient temperature for about 8-144 h (preferably about 72 h). Optionally, the reaction mixture is concentrated under reduced pressure. The resulting aqueous solution is diluted with an organic solvent (such as EtOAc or DCM). The organic extracts are optionally washed with water and/or brine, dried over anhydrous Na2S04 or MgS04, filtered or decanted, and concentrated under reduced pressure. Alternatively, the resulting aqueous solution is acidified by adding an acid such as aqueous NH4C1 or HC1 and is then extracted with an organic solvent (such as EtOAc or DCM).
Formation of a bromomethyl ketone from an acid (e.g., l-((benzyloxy)carbonyl)-4-ethylpyrrolidine-3-carboxylic acid to benzyl 3-(2-bromoacetyl)-4-ethylpyrrolidine-l-carboxylate)
To a solution of a carboxylic acid (preferably 1 equiv.) in an organic solvent (DCM or 1,2-dichloroethane (DCE), preferably DCM) is slowly added oxalyl chloride (1.2-3.0 equiv., preferably 2.2 equiv.) followed by dropwise addition of DMF (0.01-0.20 equiv., preferably about 0.15 equiv.). The reaction is stirred at about 0-40 °C (preferably ambient temperature) for about 3-24 h (preferably about 14 h) before it is concentrated under reduced pressure to a constant weight to give the crude acid chloride. A solution of a crude acid chloride
(preferably 1 equiv.) in an organic solvent (such as THF, MeCN, Et20, or THF/MeCN, preferably THF/MeCN) is added to trimethylsilyldiazomethane (2.0 M in Et20) or diazomethane solution in Et20 (prepared from DIAZALD® according to Aldrich protocol or J. Chromatogr. Sci. 1991, 29:8) (2-10 equiv., preferably 3.5 equiv. of
trimethylsilyldiazomethane) at about -20-20 °C (preferably about 0 °C) in a suitable organic solvent such as THF, MeCN, Et20, or THF/MeCN (preferably THF/MeCN). The reaction mixture is stirred for about 0.5-5 h (preferably about 3 h) at about -20-20 °C (preferably about 0 °C) before the dropwise addition of 48% aqueous HBr (5-40 equiv., preferably about 10 equiv.). After about 0-30 min, (preferably about 5 min) the reaction mixture can be concentrated to dryness to give the desired product, neutralized by a dropwise addition of saturated aqueous NaHC03 or is optionally washed with brine after optional addition of an organic solvent (such as EtOAc or DCM, preferably EtOAc). In cases where the reaction mixture is subjected to an aqueous work-up, the organic layer is dried over anhydrous Na2S04 or MgS04 (preferably MgS04), filtered, and concentrated under reduced pressure.
Synthesis of Intermediate tert-butyl (5-tosyl-5H-pyrrolo[2,3-Z>]pyrazin-2-yl)carbamate
Step A: 3,5-dibromopyrazin-2-amine to 5-bromo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine
To a solution of 3,5-dibromopyrazin-2-amine (125 g, 494 mmol), TEA (207.0 mL, 1483 mmol), and copper (I) iodide (0.941 g, 4.94 mmol) in THF (1255 mL) is added
PdCl2(PPh3)2 (3.47 g, 4.94 mmol). The reaction mixture is cooled at about -5-0°C and a solution of (trimethylsilyl)acetylene (65.0 mL, 470 mmol) in THF (157 mL) is added dropwise over about 15 min. The reaction mixture is stirred at about -5-0°C for about 1.5 h and then allowed to warm to room temperature (RT) overnight. The reaction mixture is then filtered through a CELITE® pad and washed with THF until no further product eluted. The filtrate is concentrated under reduced pressure to give a brown-orange solid. The solid is triturated and sonicated with warm petroleum ether (b.p. 30-60°C, 400 mL), cooled to RT, collected, washed with petroleum ether (b.p. 30-60°C; 2 x 60 mL), and dried to give 5-bmmo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine (124 g, 93%, 93% purity) as a brown solid: LC/MS (Table 1, Method b) Rt = 2.51 min; MS m/z: 270, 272 (M+H)+.
Step B: 5-bromo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine to 2-bromo-5-tosyl-5H-pyrrolo[2,3-Z>]pyrazine
To a solution of 5-bromo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine (3.00g, 11.1 mmol) in DMF (60 mL) at about 0 °C is added NaH (60% dispersion in mineral oil, 0.577g, 14.4 mmol) in three portions. After about 15 min, p-toluenesulfonyl chloride (2.75g, 14.4 mmol) is added and the reaction is allowed to warm slowly to ambient temperature. After about 16 h, the reaction mixture is poured onto ice-cold water (120 mL) and the precipitate is collected by vacuum filtration. The crude solid is dissolved in DCM (15 mL) and purified by silica gel chromatography eluting with DCM to give 2-bromo-5-tosyl-5H-pyrrolo[2,3-bjpyrazine (2.16 g, 52%): LC/MS (Table 1, Method c) Rt = 1.58 min; MS m/z: 352, 354 (M+H)+.
Step C: 2-bromo-5-tosyl-5H-pyrrolo[2,3-b]pyrazine to methyl 5-tosyl-5H-pyrrolo[2,3-Z>]pyrazine-2-carboxylate
CO is bubbled into an orange solution of 2-bromo-5-tosyl-5H-pyrrolo[2,3-b]pyrazine (50. Og, 142 mmol) in DMF (2.50 L) within a 5 L round bottom flask for about 2 min.
Bis(triphenylphosphine)-palladium(II) dichloride (9.96g, 14.2 mmol), TEA (59 mL, 423 mmol) and MeOH (173.0 mL, 4259 mmol) are added and the flask is fitted with a balloon of CO. The mixture is heated at about 95°C under an atmosphere of CO (1 atmosphere). After stirring overnight, the reaction mixture is cooled to ambient temperature overnight and poured into ice water (3.2 L). The mixture is stirred for about 10 min and the precipitate is collected by filtration, while washing with water, and dried for 1 h. The crude material is dissolved in DCM, separated from residual water, dried over anhydrous MgS04, filtered, added silica gel, and concentrated under reduced pressure to prepare for chromatography. The crude material is purified by silica gel column chromatography eluting with 0-5% MeOH in DCM to yield methyl 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylate with 5 mol% DCM as an excipient (40.7 g, 86%, 93% purity): LC/MS (Table 1, Method a) Rt = 2.35 min;
MS m/z 332 (M+H)+.
Step D: methyl 5-tosyl-5H-pyrrolo[2,3-Z>]pyrazine-2-carboxylate to 5-tosyl-5H-pyrrolo[2,3-/>]pyrazine-2-carboxylic acid
HC1 (6 N aqueous, 714 mL) is added to a yellow solution of methyl 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylate (17.8g, 53.6 mmol) in 1,4-dioxane (715 mL) within a 2 L round bottom flask, and the mixture is heated at about 60°C for about 16 h. The reaction mixture is cooled to ambient temperature. The organic solvent is removed under reduced pressure and the precipitate is collected, washed with water, and dried to yield 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylic acid (14.4 g, 85%) as a yellow solid: LC/MS (Table 1, Method a) Rt = 1.63 min; MS m/z 316 (Μ-Η)“.
Step E: 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylic acid to tert-butyl 5-tosyl-5H-pyrrolo[2,3-Z>]pyrazin-2-ylcarbamate
In a 500 mL round bottom flask, 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylic acid (14.4 g, 45.3 mmol), diphenylphosphoryl azide (9.78 mL, 45.3 mmol) and TEA (13.9 mL, 100 mmol) in ie/t-butanol (i-BuOH) (200 mL) are added to give an orange suspension. The mixture is heated at about 70°C for about 16 h, cooled to ambient temperature and the insoluble material is removed by filtration. The solvent is removed under reduced pressure and the crude material is purified by silica gel column chromatography eluting with 25-60% EtOAc in heptane to yield tert-butyl 5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-ylcarbamate (9.75 g, 54%) as an off-white solid: LC/MS (Table 1, Method a) Rt = 2.79 min; MS m/z 389 (M+H)+.
PATENT
WO2011068881


PATENT
http://www.google.com/patents/US20110311474
Preparation #F.1.1: 8-((cis)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazine
-

-
To a solution of (cis)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-1-carboxylate (0.838 g, 1.541 mmol, prepared using E from Example #36 Step D with TFA, N, R, S.1 with Example #3 Step E, and T with Lawesson’s reagent) was added a solution of HBr (2.50 mL, 15.19 mmol, 33% in acetic acid). The reaction mixture was stirred at ambient temperature for about 1 h. The reaction was diluted with Et2O (50 mL) and water (20 mL). The layers were stirred for about 3 min and the organic layer was decanted then the procedure was repeated 5 times. The aqueous layer was cooled to about 0° C. was basified with saturated aqueous NaHCO3solution (10 mL) to about pH 7. The aqueous layer was extracted with EtOAc (3×50 mL), combined, and dried over anhydrous Na2SO4, filtered and concd to give a brown solid. The solid was dissolved in DCM (50 mL) and washed with water (3×20 mL), dried over anhydrous Na2SO4, filtered and coned to afford 8-((cis)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazine (0.453, 61%) as a brown residue: LC/MS (Table 1, Method a) Rt=1.73 min; MS m/z: 410 (M+H)+.

SEE…………..1-((cis)-4-ethylpyrrolidin-3-yl)-6-tosyl-6H-pyrrolo[2,3-e][1,2,4]triazolo[4,3-a]pyrazine (0.250 g, 0.609 mmol, Example #36, step F)
PATENTS
WO-2017066775
WO-2015061665
WO-2011068881
References
- ^ Jump up to:a b c Mohamed, Mohamed-Eslam F.; Camp, Heidi S.; Jiang, Ping; Padley, Robert J.; Asatryan, Armen; Othman, Ahmed A. (December 2016). “Pharmacokinetics, Safety and Tolerability of ABT-494, a Novel Selective JAK 1 Inhibitor, in Healthy Volunteers and Subjects with Rheumatoid Arthritis”. Clinical Pharmacokinetics. 55 (12): 1547–1558. doi:10.1007/s40262-016-0419-y. ISSN 1179-1926. PMID 27272171.
- Jump up^ Fleischmann, Roy (May 2012). “Novel small-molecular therapeutics for rheumatoid arthritis”. Current Opinion in Rheumatology. 24 (3): 335–341. doi:10.1097/BOR.0b013e32835190ef. ISSN 1531-6963. PMID 22357358.
- Jump up^ Riese, Richard J.; Krishnaswami, Sriram; Kremer, Joel (August 2010). “Inhibition of JAK kinases in patients with rheumatoid arthritis: scientific rationale and clinical outcomes”. Best Practice & Research. Clinical Rheumatology. 24 (4): 513–526. doi:10.1016/j.berh.2010.02.003. ISSN 1532-1770. PMID 20732649.
- Jump up^ “Characterization of ABT-494, a Second Generation Jak1 Selective Inhibitor”. ACR Meeting Abstracts. Retrieved 2017-05-21.
- Jump up^ Mohamed, Mohamed-Eslam F.; Jungerwirth, Steven; Asatryan, Armen; Jiang, Ping; Othman, Ahmed A. (2017-05-14). “Assessment of effect of CYP3A Inhibition, CYP Induction, OATP1B Inhibition, and High-Fat Meal on Pharmacokinetics of the JAK1 inhibitor Upadacitinib”. British Journal of Clinical Pharmacology. doi:10.1111/bcp.13329. ISSN 1365-2125. PMID 28503781.
- Jump up^ Kremer, Joel M.; Emery, Paul; Camp, Heidi S.; Friedman, Alan; Wang, Li; Othman, Ahmed A.; Khan, Nasser; Pangan, Aileen L.; Jungerwirth, Steven; Keystone, Edward C. (December 2016). “A Phase IIb Study of ABT‐494, a Selective JAK‐1 Inhibitor, in Patients With Rheumatoid Arthritis and an Inadequate Response to Anti–Tumor Necrosis Factor Therapy”. Arthritis & Rheumatology (Hoboken, N.j.). 68 (12): 2867–2877. doi:10.1002/art.39801. ISSN 2326-5191. PMC 5132116
 . PMID 27389975.
. PMID 27389975. - Jump up^ Genovese, Mark C.; Smolen, Josef S.; Weinblatt, Michael E.; Burmester, Gerd R.; Meerwein, Sebastian; Camp, Heidi S.; Wang, Li; Othman, Ahmed A.; Khan, Nasser; Pangan, Aileen L.; Jungerwirth, Steven (December 2016). “Efficacy and Safety of ABT‐494, a Selective JAK‐1 Inhibitor, in a Phase IIb Study in Patients With Rheumatoid Arthritis and an Inadequate Response to Methotrexate”. Arthritis & Rheumatology (Hoboken, N.j.). 68 (12): 2857–2866. doi:10.1002/art.39808. ISSN 2326-5191. PMC 5132065 . PMID 27390150.
- Jump up^ “A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study of ABT-494 for the Induction of Symptomatic and Endoscopic Remission in Subjects With Moderately to Severely Active Crohn’s Disease Who Have Inadequately Responded to or Are Intolerant to Immunomodulators or Anti-TNF Therapy – Full Text View – ClinicalTrials.gov”. Retrieved 2017-05-22.
- Jump up^ “AbbVie Announces Positive Phase 2 Study Results for Upadacitinib (ABT-494), an Investigational JAK1-Selective Inhibitor, in Crohn’s Disease | AbbVie Newsroom”. Retrieved 2017-05-22.
- Jump up^ Phase 3 upadacitinib trials
-
Upadacitinib 
Clinical data Synonyms ABT-494 Routes of
administrationOral Pharmacokinetic data Metabolism Hepatic (CYP3A major, CYP2D6 minor) [1] Biological half-life 6-15 hours[1] Identifiers CAS Number PubChem CID IUPHAR/BPS ChemSpider UNII ChEMBL Chemical and physical data Formula C17H19F3N6O Molar mass 380.38 g·mol−1 3D model (JSmol)

/////Upadacitinib, ABT 494, упадацитиниб , أوباداسيتينيب , 乌帕替尼 , ORPHAN DRUG, PHASE 3
c21cnc4c(n1c(cn2)[C@@H]3[C@@H](CN(C3)C(=O)NCC(F)(F)F)CC)ccn4
OR
CC[C@@H]1CN(C[C@@H]1c4cnc3cnc2nccc2n34)C(=O)NCC(F)(F)F
FDA approves new orphan drug Uptravi (selexipag) to treat pulmonary arterial hypertension

KEEPING WATCHING THIS POSTS FOR SYNTHESIS UPDATES
12/22/2015
On December 21, the U.S. Food and Drug Administration approved Uptravi (selexipag) tablets to treat adults with pulmonary arterial hypertension (PAH), a chronic, progressive, and debilitating rare lung disease that can lead to death or the need for transplantation.
December 22, 2015
On December 21, the U.S. Food and Drug Administration approved Uptravi (selexipag) tablets to treat adults with pulmonary arterial hypertension (PAH), a chronic, progressive, and debilitating rare lung disease that can lead to death or the need for transplantation.
“Uptravi offers an additional treatment option for patients with pulmonary arterial hypertension,” said Ellis Unger, M.D., director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research. “The FDA supports continued efforts to provide new treatment options for rare diseases.”
PAH is high blood pressure that occurs in the arteries that connect the heart to the lungs. It causes the right side of the heart to work harder than normal, which can lead to limitations on exercise ability and shortness of breath, among other more serious complications.
Uptravi belongs to a class of drugs called oral IP prostacyclin receptor agonists. The drug acts by relaxing muscles in the walls of blood vessels to dilate (open) blood vessels and decrease the elevated pressure in the vessels supplying blood to the lungs.
Uptravi’s safety and efficacy were established in a long-term clinical trial of 1,156 participants with PAH. Uptravi was shown to be effective in reducing hospitalization for PAH and reducing the risks of disease progression compared to placebo. Participants were exposed to Uptravi in this trial for a median duration of 1.4 years.
Common side effects observed in those treated with Uptravi in the trial include headache, diarrhea, jaw pain, nausea, muscle pain (myalgia), vomiting, pain in an extremity, and flushing.
Uptravi was granted orphan drug designation. Orphan drug designation provides incentives such as tax credits, user fee waivers, and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Uptravi is marketed by San Francisco-based Actelion Pharmaceuticals US, Inc.
Selexipag, Uptravi
475086-01-2 CAS
(C26H32N4O4S, Mr = 496.6 g/mol)
A prostacyclin receptor (PGI2) agonist used to treat pulmonary arterial hypertension (PAH).
NIPPON SHINYAKU….INNOVATOR
Selexipag (brand name Uptravi) is a drug developed by Actelion for the treatment of pulmonary arterial hypertension (PAH). Selexipag and its active metabolite, ACT-333679 (MRE-269) (the free carboxylic acid), are agonists of the prostacyclin receptor, which leads to vasodilation in the pulmonary circulation.[1]
The US FDA granted it Orphan Drug status[2] (for PAH). It was approved by the U.S. FDA on 22 December 2015.[2]

ACT-333679 or MRE-269, the active metabolite of selexipag




PATENT
US2012/101276
http://www.google.st/patents/US20120101276?hl=pt-PT&cl=en
The present invention relates to a crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide (hereinafter referred to as “compound A”).

BACKGROUND OF THE INVENTION
Compound A has an excellent PGI2 agonistic effect and shows a platelet aggregation inhibitory effect, a vasodilative effect, a bronchodilative effect, a lipid deposition inhibitory effect, a leukocyte activation inhibitory effect, etc. (see, for example, in WO 2002/088084 (“WO ‘084”)).
Specifically, compound A is useful as preventive or therapeutic agents for transient ischemic attack (TIA), diabetic neuropathy, diabetic gangrene, peripheral circulatory disturbance (e.g., chronic arterial occlusion, intermittent claudication, peripheral embolism, vibration syndrome, Raynaud’s disease), connective tissue disease (e.g., systemic lupus erythematosus, scleroderma, mixed connective tissue disease, vasculitic syndrome), reocclusion/restenosis after percutaneous transluminal coronary angioplasty (PTCA), arteriosclerosis, thrombosis (e.g., acute-phase cerebral thrombosis, pulmonary embolism), hypertension, pulmonary hypertension, ischemic disorder (e.g., cerebral infarction, myocardial infarction), angina (e.g., stable angina, unstable angina), glomerulonephritis, diabetic nephropathy, chronic renal failure, allergy, bronchial asthma, ulcer, pressure ulcer (bedsore), restenosis after coronary intervention such as atherectomy and stent implantation, thrombocytopenia by dialysis, the diseases in which fibrosis of organs or tissues is involved [e.g., Renal diseases (e.g., tuburointerstitial nephritis), respiratory diseases (e.g., interstitial pneumonia (pulmonary fibrosis), chronic obstructive pulmonary disease), digestive diseases (e.g., hepatocirrhosis, viral hepatitis, chronic pancreatitis and scirrhous stomachic cancer), cardiovascular diseases (e.g, myocardial fibrosis), bone and articular diseases (e.g, bone marrow fibrosis and rheumatoid arthritis), skin diseases (e.g, cicatrix after operation, scalded cicatrix, keloid, and hypertrophic cicatrix), obstetric diseases (e.g., hysteromyoma), urinary diseases (e.g., prostatic hypertrophy), other diseases (e.g., Alzheimer’s disease, sclerosing peritonitis; type I diabetes and organ adhesion after operation)], erectile dysfunction (e.g., diabetic erectile dysfunction, psychogenic erectile dysfunction, psychotic erectile dysfunction, erectile dysfunction associated with chronic renal failure, erectile dysfunction after intrapelvic operation for removing prostata, and vascular erectile dysfunction associated with aging and arteriosclerosis), inflammatory bowel disease (e.g., ulcerative colitis, Crohn’s disease, intestinal tuberculosis, ischemic colitis and intestinal ulcer associated with Behcet disease), gastritis, gastric ulcer, ischemic ophthalmopathy (e.g., retinal artery occlusion, retinal vein occlusion, ischemic optic neuropathy), sudden hearing loss, avascular necrosis of bone, intestinal damage caused by administration of a non-steroidal anti-inflammatory agent (e.g., diclofenac, meloxicam, oxaprozin, nabumetone, indomethacin, ibuprofen, ketoprofen, naproxen, celecoxib) (there is no particular limitation for the intestinal damage so far as it is damage appearing in duodenum, small intestine and large intestine and examples thereof include mucosal damage such as erosion and ulcer generated in duodenum, small intestine and large intestine), and symptoms associated with lumbar spinal canal stenosis (e.g., paralysis, dullness in sensory perception, pain, numbness, lowering in walking ability, etc. associated with cervical spinal canal stenosis, thoracic spinal canal stenosis, lumbar spinal canal stenosis, diffuse spinal canal stenosis or sacral stenosis) etc. (see, for example, in WO ‘084, WO 2009/157396, WO 2009/107736, WO 2009/154246, WO 2009/157397, and WO 2009/157398).
In addition, compound A is useful as an accelerating agent for angiogenic therapy such as gene therapy or autologous bone marrow transplantation, an accelerating agent for angiogenesis in restoration of peripheral artery or angiogenic therapy, etc. (see, for example, in WO ‘084).
Production of Compound A
Compound A can be produced, for example, according to the method described in WO ‘084, and, it can also be produced according to the production method mentioned below.

Step 1:
6-Iodo-2,3-diphenylpyrazine can be produced from 6-chloro-2,3-diphenylpyrazine by reacting it with sodium iodide. The reaction is carried out in the presence of an acid in an organic solvent (e.g., ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or their mixed solvent). The acid to be used is, for example, acetic acid, sulfuric acid, or their mixed acid. The amount of sodium iodide to be used is generally within a range of from 1 to 10 molar ratio relative to 6-chloro-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the acid to be used, but may be generally within a range of from 60° C. to 90° C. The reaction time varies depending on the kinds of the solvent and the acid to be used and on the reaction temperature, but may be generally within a range of from 9 hours to 15 hours.
Step 2:
5,6-Diphenyl-2-[(4-hydroxybutyl(isopropyl)amino]pyrazine can be produced from 6-iodo-2,3-diphenylpyrazine by reacting it with 4-hydroxybutyl(isopropyl)amine. The reaction is carried out in the presence of a base in an organic solvent (e.g., sulfolane, N-methylpyrrolidone, N,N-dimethylimidazolidinone, dimethyl sulfoxide or their mixed solvent). The base to be used is, for example, sodium hydrogencarbonate, potassium hydrogencarbonate, potassium carbonate, sodium carbonate or their mixed base. The amount of 4-hydroxybutyl(isopropyl)amine to be used may be generally within a range of from 1.5 to 5.0 molar ratio relative to 6-iodo-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from 170° C. to 200° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 5 hours to 9 hours.
Step 3:
Compound A can be produced from 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine by reacting it with N-(2-chloroacetyl)methanesulfonamide. The reaction is carried out in the presence of a base in a solvent (N-methylpyrrolidone, 2-methyl-2-propanol or their mixed solvent). The base to be used is, for example, potassium t-butoxide, sodium t-butoxide or their mixed base. The amount of N-(2-chloroacetyl)methanesulfonamide to be used may be generally within a range of from 2 to 4 molar ratio relative to 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from −20° C. to 20° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 0.5 hours to 2 hours.
The compounds to be used as the starting materials in the above-mentioned production method for compound A are known compounds, or can be produced by known methods.
PATENT
WO 2002088084
and
http://www.google.fm/patents/WO2009157398A1?cl=en
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 21 p. 6692 – 6704
compd 31
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 24 p. 7720 – 7725
 2a isthe drug
2a isthe drug
N-Acylsulfonamide and N-acylsulfonylurea derivatives of the carboxylic acid prostacyclin receptor agonist 1 were synthesized and their potential as prodrug forms of the carboxylic acid was evaluated in vitro and in vivo. These compounds were converted to the active compound 1 by hepatic microsomes from rats, dogs, monkeys, and humans, and some of the compounds were shown to yield sustained plasma concentrations of 1 when they were orally administered to monkeys. These types of analogues, including NS-304 (2a), are potentially useful prodrugs of 1.
http://www.sciencedirect.com/science/article/pii/S0968089607007614
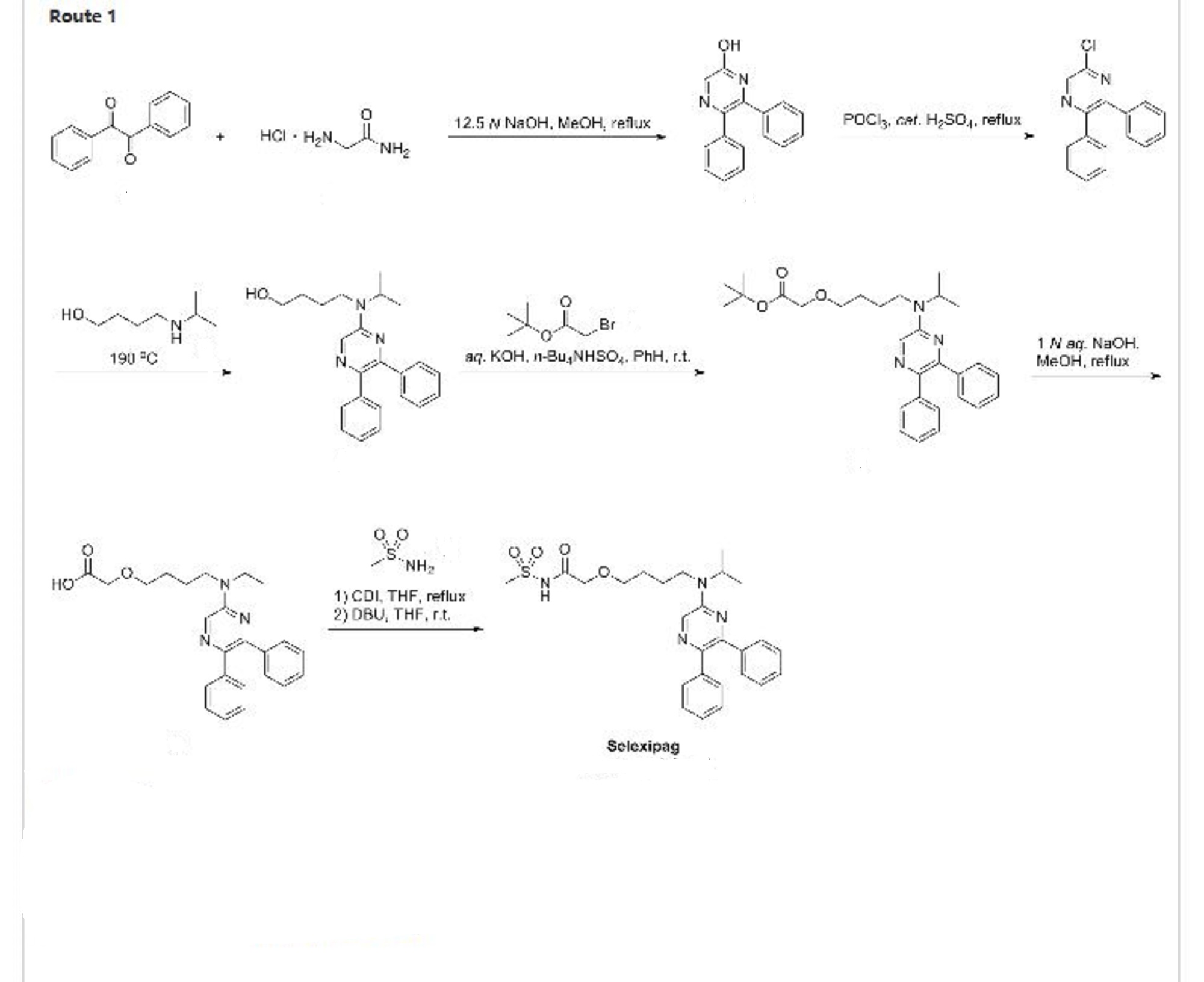
PATENT
Example 1 t- butylamine Form I crystal of the salt
Compound A (40 mg) with 0.5mL dimethoxyethane (hereinafter, referred to as. “DME”) was dissolved in, and t- butylamine (1.1 eq) were added, 25 1 ° C. at 8 it was stirred for hours. Thereafter, the reaction solution was added t- butyl methyl ether (1mL), at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, I-form crystals of t- butylamine salt ( 3 to afford 9.9mg). B Powder X-ray diffraction spectrum of type I crystal obtained t- butylamine salt using the apparatus shown in Figure 1.
Melting point: 152.5 ℃
elemental analysis (C 3 0 H 4 3 N 5 O 4 S + 0.0 3 H 2 as O)
calculated value (%) C: 6 3 .1 8 H: 7 . 6 1 N: 12 .2 8 measured value (%) C: 6 2. 8 5 H: 7 . 6 4 N: 12.52 1 H-NMR (DMSO-D 6 ): delta 8 .15 (s, 1H), 7 .55 – 7 . 8 0 (M, 2H), 7 .10- 7 . .45 (M, 10H), 4 7 . 0-4 8 5 (M, 1H), 3 . 6 6 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 3 (s, 3 H), 1.50-1. 7 5 (M, 4H), 1.2 3 (s, 9H), 1.22 (D, 6 H)
Example 2 I-form crystal of the potassium salt
Compound A tetrahydrofuran with (40mg) 12mL (hereinafter, referred to as. “THF”) was dissolved in, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added, 40 ℃ It was heated and stirred in for 15 minutes. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. After repeated two more times this step, at -20 ° C. 3 and held hours. The resulting precipitated crystals were collected by filtration under reduced pressure, and dried to obtain Form I crystal of the potassium salt. B Powder X-ray diffraction spectrum of type I crystal of the obtained potassium salt using the apparatus shown in Fig. 1 H-NMR (DMSO-D 6 ): delta 8 .14 (s, 1H), 7 .1 8 – 7 . 3 8 . (M, 10H), 4 7 . 2-4 8 4 (M, 1H) , 3 . 6 5 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 2 (s, 3 H), 1.55-1. 7 0 ( M, 4H), 1.2 3 (D, 6 H)
Example 3 II-form crystals of the potassium salt
Compound A with (40mg) was dissolved in THF and 12mL, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added and heated with stirring for 15 min at 40 ℃. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. This operation was repeated two more times, at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, after drying, 40 ℃, relative humidity 7 while 5% of thermo-hygrostat 7 left for days to give crystalline Form II of the potassium salt. B Powder X-ray diffraction spectrum of crystalline Form II of the resulting potassium salt using the apparatus Fig 3 is shown in.
Example 4 III type crystal of the potassium salt
Compound A , in addition to (100mg) acetonitrile (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (1.1 eq) was added and stirred for 200 minutes at 20 ℃. While stirring the mixture 7 after a heated stirring for 1 hour to 0 ° C., and then cooled to 10 ℃ over 10 hours. Further heated while the mixture 6 is heated to 0 ℃, t- butyl methyl ether (0. 3 after adding mL), cooled to 20 ℃ over 10 hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, III type crystal of the potassium salt ( 7 to afford 5mg). The powder X-ray diffraction spectrum of the type III crystal of the obtained potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, of about 7 endothermic peak was observed at around 4 ° C..
Elemental analysis (C 2 6 H 3 1 N 4 O 4 . SK + 0 7 8 H 2 as O)
calculated value (%) C: 5 6 .91 H: 5.9 8 N: 10.21
measured value (%) C: 5 6 . 6 1 H: 5.55 N:. 10 3 6
EXAMPLE 5 IV-type crystal of the potassium salt
Compound A , in addition to (50mg) and ethyl acetate (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (2.2 eq) was added and 2 at 20 ° C. 3 and stirred for hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried to obtain Form IV crystal of the potassium salt (41mg). The powder X-ray diffraction spectrum of crystalline Form IV of the resulting potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, an endothermic peak was observed at around approximately 91 ℃.

Selexipag (C26H32N4O4S, Mr = 496.6 g/mol) ist ein Diphenylpyrazin-Derivat. Es wird in der Leber zum aktiven Metaboliten ACT-333679 (MRE-269) biotransformiert. Selexipag unterscheidet sich strukturell von Prostazyklin und anderen Prostazylin-Rezeptor-Agonisten.

References
- 1 Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
- 2 New Drug Approved for Rare Lung Disorder. PPN. 23 Dec 2015 Has link to GRIPHON study results
- Kuwano et al. NS-304, an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007;322:1181-1188.
- Kuwano et al. A long-acting and highly selective prostacyclin receptor agonist prodrug, NS-304, ameliorates rat pulmonary hypertension with unique relaxant responses of its active form MRE-269 on rat pulmonary artery. J Pharmacol Exp Ther 2008;326:691-699.
- Simonneau G, Lang I, Torbicki A, Hoeper MM, Delcroix M, Karlocai K, Galie N. Selexipag, an oral, selective IP receptor agonist for the treatment of pulmonary arterial hypertension Eur Respir J 2012; 40: 874-880
- Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010;104:9-21.
- Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
| Patent | Submitted | Granted |
|---|---|---|
| Methods of identifying critically ill patients at increased risk of development of organ failure and compounds for the treatment hereof [US8877710] | 2009-12-30 | 2014-11-04 |
| Form-I crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide and method for producing the same [US8791122] | 2010-06-25 | 2014-07-29 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2015057325] | 2013-03-26 | 2015-02-26 |
| INHIBITION OF NEOVASCULARIZATION BY SIMULTANEOUS INHIBITION OF PROSTANOID IP AND EP4 RECEPTORS [US2014275200] | 2014-03-05 | 2014-09-18 |
| INHIBITION OF NEOVASCULARIZATION BY INHIBITION OF PROSTANOID IP RECEPTORS [US2014275238] | 2014-03-05 | 2014-09-18 |
| Fibrosis inhibitor [US8889693] | 2014-04-10 | 20 |
| Patent | Submitted | Granted |
|---|---|---|
| Heterocyclic compound derivatives and medicines [US7205302] | 2004-05-27 | 2007-04-17 |
| METHODS OF IDENTIFYING CRITICALLY ILL PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ORGAN FAILURE AND COMPOUNDS FOR THE TREATMENT HEREOF [US2014322207] | 2014-07-11 | 2014-10-30 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2014329824] | 2014-07-18 | 2014-11-06 |
| Sustained Release Composition of Prostacyclin [US2014303245] | 2012-08-10 | 2014-10-09 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2013261177] | 2011-09-30 | 2013-10-03 |
| METHODS OF TREATMENT OF PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ISCHEMIC EVENTS AND COMPOUNDS HEREOF [US2013040898] | 2011-04-29 | 2013-02-14 |
| Substituted Diphenylpyrazine Derivatives [US2013005742] | 2010-08-06 | 2013-01-03 |
| USE OF FORM-I CRYSTAL OF 2–N-(METHYLSULFONYL)ACETAMIDE [US2014148469] | 2014-01-22 | 2014-05-29 |
| CRYSTALS OF 2- {4- [N- (5,6-DIPHENYLPYRAZIN-2-YL) -N-ISOPROPYLAMINO]BUTYLOXY}-N- (METHYLSULFONYL) ACETAMIDE [US2014155414] | 2014-01-22 | 2014-06-05 |
| PROSTACYCLIN AND ANALOGS THEREOF ADMINISTERED DURING SURGERY FOR PREVENTION AND TREATMENT OF CAPILLARY LEAKAGE [US2014044797] | 2012-03-30 | 2014-02-13 |
 |
|
| Names | |
|---|---|
| IUPAC name
2-{4-[(5,6-diphenylpyrazin-2-yl)(propan-2-yl)amino]butoxy}-N-(methanesulfonyl)acetamide
|
|
| Other names
ACT-293987, NS-304
|
|
| Identifiers | |
| 475086-01-2 |
|
| ChEMBL | ChEMBL238804 |
| ChemSpider | 8089417 |
| 7552 | |
| Jmol interactive 3D | Image |
| KEGG | D09994 |
| PubChem | 9913767 |
| UNII | P7T269PR6S |
| Properties | |
| C26H32N4O4S | |
| Molar mass | 496.6 g·mol−1 |
SEE……….http://apisynthesisint.blogspot.in/2015/12/fda-approves-new-orphan-drug-uptravi.html
//////////
CC(C)N(CCCCOCC(=O)NS(=O)(=O)C)C1=CN=C(C(=N1)C2=CC=CC=C2)C3=CC=CC=C3
Uridine triacetate, ウリジントリアセタート FDA approves first emergency treatment for overdose of certain types of chemotherapy

12/11/2015 12:05 PM EST
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
UPDATED IN SEPT 2016…………..
Uridine triacetate
Uridine, 5-hydroxy-, 2′,3′,5′-triacetate
Orphan drug
FAST TRACK
MF C15H18N2O9, MW 370.314
|
[(2R,3R,4R,5R)-3,4-bis(acetyloxy)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)oxolan-2-yl]methyl acetate
|
Vistogard [Trade name]
Xuriden [Trade name]
(2R,3R,4R,5R)-2-(acetoxymethyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3,4-diyl diacetate
Wellstat (Originator)
PN-401; RG-2133; TAU
MOA:Pyrimidine analog
Indication:Hereditary orotic aciduria; Chemotherapy induced poisoning
To treat patients with hereditary orotic aciduria
| Drug Name(s) | XURIDEN |
| FDA Application No. | (NDA) 208169 |
| Active Ingredient(s) | URIDINE TRIACETATE |
| Company | WELLSTAT THERAP |
| Original Approval or Tentative Approval Date | September 4, 2015 |
FDA APPROVAL SUMMARY
Chemotherapy induced poisoning, VISTOGARD, FDA 2015-12-11
Hereditary orotic aciduria, Xuriden, FIRST APPROVAL, 2015-09-04
|
|||||||
| External Identifiers |
|
|---|
Uridine triacetate is a drug used in the treatment of hereditary orotic aciduria[1] and to treat patients following an overdose ofchemotherapy drugs 5-fluorouracil or capecitabine, or in patients exhibiting early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of 5-fluorouracil or capecitabine administration.[2][3]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics and it is marketed in USA by BTG. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine triacetate is a prodrug of uridine.[4]
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTP incorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [3]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidine metabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
Marketed as the product Xuriden (FDA), uridine triacetate is indicated for the treatment of hereditary orotic aciduria. Marketed as the product Vistogard (FDA), uridine triacetate is indicated for the emergency treatment of adult and pediatric patients in the following situations: following a fluorouracil or capecitabine overdose regardless of the presence of symptoms; or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration.

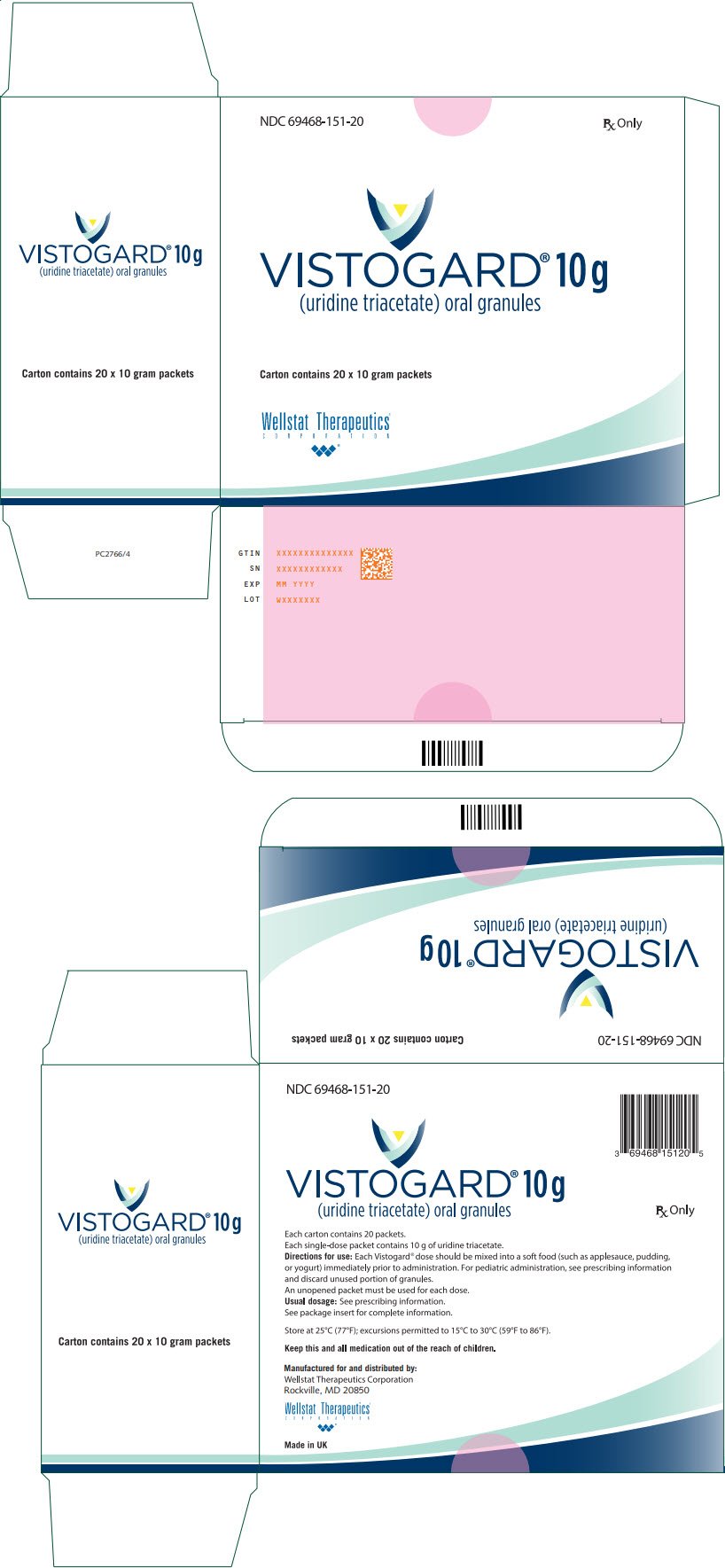
Uridine Triacetate was approved by the U.S. Food and Drug Administration (FDA) on Sep 4, 2015. It was developed by Wellstat Therapeutics, then marketed as Xuriden® by Wellstat Therapeutics in US. Then it was also approved by FDA for overdose of certain types of chemotherapy on Dec 11, 2015 and marketed as Vistogard®.
Uridine Triacetate is a prodrug of the nucleoside uridine used to treat hereditary orotic aciduria. Hereditary orotic aciduria is inherited from a recessive gene. The disease is due to a defective or deficient enzyme, which results in the body being unable to normally synthesize uridine, a necessary component of ribonucleic acid (RNA). Signs and symptoms of the disease include blood abnormalities (anemia, decreased white blood cell count, decreased neutrophil count), urinary tract obstruction due to the formation of orotic acid crystals in the urinary tract, failure to thrive, and developmental delays.
Xuriden® is approved as oral granules that can be mixed with food or in milk or infant formula, and is administered once daily. The starting dosage is 60 mg/kg once daily; the dose may be increased to 120 mg/kg (not to exceed 8 grams) once daily for insufficient efficacy.
Mechanism Of Action
Uridine triacetate is an acetylated form of uridine. Following oral administration, uridine triacetate is deacetylated by nonspecific esterases present throughout the body, yielding uridine in the circulation (Figure 1).
Figure 1: Uridine Triacetate Conversion to Uridine

URIDEN provides uridine in the systemic circulation of patients with hereditary orotic aciduria who cannot synthesize adequate quantities of uridine due to a genetic defect in uridine nucleotide synthesis.
Uridine triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578] such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Uridine triacetate is also used for replacement therapy in the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase (UMPS) deficiency. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria.
Route 1


Reference:1. J. Am. Chem. Soc. 1953, 75, 2017-2019.
2. Angew. Chem. internat. Edit. 1971, 10, 75.
3. US3116282.
PATENT
Production Example 1

5.6 g of uracil and 0.1 g of ammonium sulfate were dissolved in 22.4 ml of 1,1,1,3,3,3-hexamethyldisilazane and reacted at 120° C. for 2.5 hours. After the completion of the reaction, the reaction mixture was distilled to give 11.8 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine. 1H-NMR (400 MHz, in C2D6CO): δ=0.29 (s, 9H), 0.31 (s, 9H), 6.35 (d, J=5.6 Hz, 1H), 8.19 (d, J=5.5Hz, 1H)
Referential Example 11.21 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 1.15 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.8 ml of acetonitrile and cooled to 5° C. Next, 0.94 g of SnCl4 was added dropwise thereinto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted for 3 hours. The reaction mixture was analyzed by HPLC. Thus, β-uridine triacetate was obtained with a reaction yield of 83%.
Example 1

0.93 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 0.92 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.7 ml of acetonitrile and cooled to 4° C. Then 0.49 g of FeCl3 was added thereto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted. The reaction was monitored by HPLC. After the completion of the reaction, the reaction mixture was added dropwise at 4° C. into a cold aqueous solution of sodium hydrogencarbonate which had been preliminarily prepared. After filtering off the catalyst residue, the filtrate was separated and the aqueous layer was extracted with 20 ml portions of ethyl acetate thrice. The organic layers were combined, washed with a saturated aqueous solution of sodium chloride and dried over sodium sulfate. After distilling off the solvent, 1.2 g (purity 80%) of the target compound was obtained as a viscous white solid.
Namely, the target compound could be obtained at a yield comparable to REFERNTIAL EXAMPLE 1 wherein SnCl4 was employed as the catalyst. 1H-NMR (400 MHz, in CDCl3): δ=2.11 (s, 3H), 2.14 (s, 3H), 2.15 (s, 3H), 4.35 (m, 3H), 5.33 (m, 2H), 5.79 (d, J=8.2 Hz, 1H), 6.04 (d, J=4.9 Hz, 1H), 7.39 (d, J=8.2 Hz, 1H)

CLIP
12/11/2015 12:05 PM EST
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
CLIP
With support from Almac, Wellstat delivers for a rare disease.
Proximity of API and finished drug development helps uridine triacetate to market for two indications
By Rick Mullin
“The initial contact was a cold call by Almac in 2010 or 2011,” recalls Mike Bamat, senior vice president of R&D at Wellstat Therapeutics, a small drug company in Gaithersburg, Md. “There were probably a couple of calls. It was one of those things where timing is everything.”
Almac, a Craigavon, Northern Ireland-based pharmaceutical services company, was looking to get in on Wellstat’s development of uridine triacetate, a synthetic pyrimidine analog, as an antidote for fluorouracil and capecitabine toxicity and overdose in cancer patients receiving those chemotherapies. And the calls, which Almac records indicate followed some communication between the companies, happened to come just when Wellstat was looking to change service partners as it moved toward commercial development of the drug.

Uridine triacetate
Discovery: Wellstat Therapeutic’s research on the therapeutic potential of exogenous uridine leads to a determination that uridine triacetate is a safe means of delivering the agent
Applications: Treatment of hereditary orotic aciduria (HOA), an extremely rare disease in which the body does not produce uridine, causing overproduction of orotic acid; emergency treatment of toxic reaction to or overdose of the cancer treatments fluorouracil and capecitabine
Methods of action: Treating HOA, uridine triacetate restores intracellular nucleotide concentrations, normalizing orotic acid production; as a chemotherapy antidote, it increases intracellular levels of uridine to dilute fluorouracil and capecitabine
Years in development: Since 2008 for chemotherapy antidote, and 2013 for HOA
Approved: Xuriden for HOA, Sept. 4, 2015; Vistogard for chemotherapy antidote, Dec. 11, 2015
The job went to Almac, as did work that sprang up as the result of another phone call to Wellstat—this one from the U.S. Food & Drug Administration.
As Bamat explains, uridine triacetate caught FDA’s attention regarding another potential indication—an extremely rare and life-threatening disease called hereditary orotic aciduria, or HOA. A consequence of the body’s inability to produce uridine, a necessary component of ribonucleic acid, HOA can manifest in a range of symptoms including blood abnormalities, developmental delays, and urinary tract obstruction caused by overproduction of orotic acid. There have been 20 reported cases of HOA since the 1950s. Only four cases are currently known in the U.S., Bamat says, and likely fewer than 20 in the world.
Wellstat landed approvals for Xuriden, the HOA treatment, in September of last year and Vistogard, the chemotherapy antidote, in December.
The story of Xuriden centers on a raft of FDA incentives for super-rare diseases that enabled Wellstat to move forward on an expedited application for a drug that will never be made in any great volume. But bringing Xuriden and Vistogard to market may also be viewed as the story of a drug discovery firm becoming a commercial enterprise thanks to its partnership with a service provider.
As Wellstat began late-stage development of the chemotherapy antidote, its research partner at the time, QS Pharma, was acquired by the service firm WIL Research. The look and feel of the partnership changed, according to Bamat.
“We kind of lost the small, easy-to-work-with relationship we had with them,” he says. Wellstat also needed support on development and manufacturing of a finished drug product composed of granules delivered in packets or sachets. The drug is administered orally, usually sprinkled on food such as applesauce or yogurt.
Almac was deemed a good fit because of its experience with developing drugs in granule form for “sachet presentation,” a packaging method more common in Europe than in the U.S. The Northern Ireland firm’s ability to develop and manufacture the active pharmaceutical ingredient (API) and the drug product in one location—at its headquarters—would also prove to be a significant advantage.
The distance between Gaithersburg and Craigavon, however, was a concern, according to Bamat. “We debated it. Especially those of us who knew we would be going there,” he says. “We couldn’t just jump in a car and go. But we looked at a variety of things, including cost and value, and it was all very positive at Almac.”
According to David Downey, vice president of commercial operations at Almac, bringing Wellstat’s work on uridine triacetate to commercial production posed several challenges, the first being to secure supply of uridine starting material, which is extracted from sugar beets by Euticals, an Italian firm. Next was developing a method to control particle size in both the API and the finished product. Almac also had to validate process equipment as it scaled up production.
“Uridine triacetate is Wellstat’s first commercial product,” Downey says. “So we were provided with a process more fit for development than for commercial production.”
The basic formulation of a granule drug product is simple, according to Downey: The API and excipient are mixed in a dry blender. The challenge is developing an analytical regimen to assure the granules are blended uniformly. Meeting the challenge required a high level of coordination between API and drug product process development.
“Wellstat needed a partner that could support them from the API to the drug product,” Downey says. The physical proximity between the Almac facilities in Craigavon conducting API and drug product work was a key advantage, he claims.

Uridine triacetate is formulated into granules presented in packets and sprinkled on food.
Credit: Wellstat Therapeutics
“If you listen to our business development people, you’ll hear them use the term, ‘crossing car parks as opposed to crossing oceans,’ ” Downey says, explaining that many competitors who offer API and finished drug services run these operations thousands of kilometers apart from each other, sometimes on different continents.
Before it signed on with Almac, Wellstat had been working with uridine triacetate for about 10 years. Its focus on developing the antidote drug started in 2008. Branching into the HOA treatment, however, upped the stakes.
Clinical study development for an HOA therapy was expedited via a full house of regulatory incentives from FDA, according to Bamat. “We had orphan drug designation, rare pediatric designation, breakthrough therapy designation, and priority review,” he says. “So they really went all out in helping us develop this.”
Although Wellstat was interested in developing a life saving drug for children, it was concerned about paying for it, given the tiny market. “At that time, the rare pediatric disease priority review voucher program was just on the radar,” Bamat says. “FDA said, ‘Consider this new program. Maybe it’s a way that at some risk you could recoup some of your costs.’ We looked at it and were willing to take the risk.”
It paid off. Wellstat was able to sell its priority review voucher—which entitles a company that brings a rare pediatric drug to market to receive expedited review of a subsequent drug—to AstraZeneca last year for an undisclosed amount. Other vouchers sold in 2015 brought high sums, including $350 million for one that AbbVie bought from United Therapeutics in August.
Bamat says Wellstat is not likely to change focus after its success with uridine triacetate. It continues to investigate new indications for the compound and will likely work with Almac on anything going into commercial development.
He emphasizes the importance of maintaining an effective working relationship with an outsourcing partner. “My main consideration is that these are people we can really work with on a day-to-day, week-to-week basis,” Bamat says. “Will the communication be good? Will they be honest and transparent with us, and will we be the same for them? That was a key factor, and we felt it was a plus with Almac.”
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2-3 hours |
| Biological half-life | 2-2.5 hours |
| Excretion | Renal |
| Identifiers | |
| DrugBank | DB09144 |
| Chemical data | |
| Formula | C15H18Cl0N2O9S0 |
| Molar mass | 370.31 g·mol−1 |
References
- HIGHLIGHTS OF PRESCRIBING INFORMATION OF XURIDEN
- Jump up^ BTG Announces FDA Approval of VISTOGARD® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine
- Jump up^ “FDA Approved Drugs:Uridine Triacetate”. FDA. 2015-12-11. Retrieved 2016-04-29.
- “Uridine triacetate”. DrugBank.
| Patent ID | Date | Patent Title |
|---|---|---|
| US7807654 | 2010-10-05 | Compositions and methods for treatment of mitochondrial diseases |
| US2010222296 | 2010-09-02 | PYRIMIDINES, SUCH AS URIDINE, IN TREATMENTS FOR PATIENTS WITH BIPOLAR DISORDER |
| US7737128 | 2010-06-15 | Pyrimidines, such as uridine, in treatments for patients with bipolar disorder |
| US2010098678 | 2010-04-22 | Methods of Treatment of Mitochondrial Disorders |
| US2010041620 | 2010-02-18 | METHODS FOR IMPROVING FRONTAL BRAIN BIOENERGETIC METABOLISM |
| US2010041621 | 2010-02-18 | METHODS AND COMPOSITIONS FOR IMPROVING COGNITIVE PERFORMANCE |
| US7582619 | 2009-09-01 | Compositions and methods for treatment of mitochondrial diseases |
| US2008226684 | 2008-09-18 | METHOD AND PROCESS FOR THE PRODUCTION OF MULTI-COATED RECOGNITIVE AND RELEASING SYSTEMS |
| US7105498 | 2006-09-12 | Acylated uridine and cytidine and uses thereof |
| US6956028 | 2005-10-18 | Compositions and methods for treatment of mitochondrial diseases |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015307542 | 2015-10-29 | MODIFIED NUCLEIC ACID MOLECULES AND USES THEREOF |
| US2015167017 | 2015-06-18 | ALTERNATIVE NUCLEIC ACID MOLECULES AND USES THEREOF |
| US8821899 | 2014-09-02 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8771713 | 2014-07-08 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8741316 | 2014-06-03 | Highly porous, recognitive polymer systems |
| US2012294869 | 2012-11-22 | Methods for Treating Fatty Liver Disease |
| US2012078529 | 2012-03-29 | DETERMINING THE SEVERITY OF 5-FLUOROURACIL OVERDOSE |
| US8067392 | 2011-11-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7915233 | 2011-03-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7884202 | 2011-02-08 | Nucleobase Having Perfluoroalkyl Group and Process for Producing the Same |
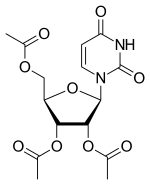
Uridine triacetate
- Molecular FormulaC15H18N2O9
- Average mass370.311 Da
ウリジントリアセタート
[(2R,3R,4R,5R)-3,4-diacetyloxy-5-(2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl acetate
Uridine, 2′,3′,5′-triacetate
uridini triacetas
Vistogard [Trade name]
Xuriden [Trade name]
(2R,3R,4R,5R)-2-(acetoxymethyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3,4-diyl diacetate
[(2R,3R,4R,5R)-3,4-bis(acetyloxy)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)oxolan-2-yl]methyl acetate
FDA APPROVED2015/9/4 . AS Xuriden
Uridine triacetate (INN),[1] formerly known as vistonuridine, is an orally active tri-acetylated prodrug of uridine[2] used:
- in the treatment of hereditary orotic aciduria (brand name Xuriden /ˈzʊərədɛn/ ZOOR-ə-den);[3]
- to treat patients following an overdose of chemotherapy drugs 5-fluorouracil (5-FU) or capecitabine regardless of the presence of symptoms, or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration (brand name Vistogard).[4][5][6]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine Triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. Uridine, a pyrimidine nucleotide, has been used in a variety of diseases including depressive disorders and inherited myopathies. (NCI04)
Pharmacology from NCIt
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTPincorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidinemetabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
from DrugBank
Uridine triacetate is an acetate ester that is uracil in which the three hydroxy hydrogens are replaced by acetate group. A prodrug for uridine, it is used for the treatment of hereditary orotic aciduria and for management of fluorouracil toxicity. It has a role as a prodrug, a neuroprotective agent and an orphan drug. It is a member of uridines and an acetate ester.
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 65” (PDF). World Health Organization. p. 92. Retrieved 12 March 2017.
- ^ “Uridine triacetate — DrugBank Page”. 12 March 2017.
- ^ “Xuriden (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “Vistogard (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “BTG Announces FDA Approval of Vistogard® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine”. BTG International Ltd. 11 December 2015. Retrieved 12 March 2017.
- ^ “Approved Drugs — Uridine Triacetate”. U.S. Food and Drug Administration. Retrieved 12 March 2017.
External links
Patents
- US7776838
- US5968914
- US6258795
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7776838 |
| Expiration | Aug 17, 2027 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
from FDA Orange Book
| FDA Orange Book Patents: 2 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6258795 |
| Expiration | Jul 10, 2019 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
from FDA Orange Book
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2–3 hours |
| Elimination half-life | 2–2.5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.021.710 |
| Chemical and physical data | |
| Formula | C15H18N2O9 |
| Molar mass | 370.31 g·mol−1 |
| 3D model (JSmol) | |
////////////Uridine triacetate, ウリジントリアセタート , FDA 2015, breakthrough therapy designation ,
//////////174105-38-8, Priority review drug , Orphan drug, FDA 2015, Vistogard, uridine triacetate, fast track designations, PN-401, RG-2133, TAU, XURIDEN
|
CC(=O)OC[C@H]1O[C@H]([C@H](OC(C)=O)[C@@H]1OC(C)=O)N1C=CC(=O)NC1=O
|
Umbralisib, TGR-1202, a Phosphoinositide-3 kinase delta inhibitor, Rhizen Pharmaceuticals S.A./TG Therapeutics

TGR 1202, TGR-1202-101, RP 5264, Umbralisib
AK173784;
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one,
2-[(1S)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one
CAS TOSYLATE 1532533-72-4 Umbralisib tosylate
CAS 1532533-67-7, 1514919-95-9
| Molecular Formula: | C31H24F3N5O3 |
|---|---|
| Molecular Weight: | 571.54917 g/mol |
RP-5307
TGR-1202
TGR-1202 PTSA
FU8XW5V3FS (UNII code)
RP-5264 (free base)
A PI3K inhibitor potentially for treatment of chronic lymphocytic leukemia, leukemia,lymphoma,B-cell
TGR‐1202, a next generation PI3K-δ delta inhibitor. TGR-1202 (RP-5264) is a highly specific, orally available, PI3K delta inhibitor, targeting the delta isoform with nanomolar potency and several fold selectivity over the alpha, beta, and gamma isoforms of PI3K.
TG Therapeutics, under license from Rhizen Pharmaceuticals, is developing TGR-1202 (structure shown; formerly RP-5264), a lead from a program of PI3K delta inhibitors, for the potential oral treatment of hematological cancers including Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), B-cell lymphoma and mantle cell lymphoma (MCL)
Incozen Therapeutics Pvt Ltd
TG Therapeutics
TGR-1202 potential to perform as the best PI3K inhibitor in its class and the possible superiority of TG-1101 over Rituxan®.
| Rhizen Pharmaceuticals S.A. | |
| Description | Phosphoinositide 3-kinase (PI3K) delta inhibitor |
Leukemia, chronic lymphocytic PHASE 3, TG Therapeutics
Orphan Drug
Umbralisib is a novel phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor under development at TG Therapeutics in phase III clinical trials, in combination with ublituximab, for the treatment of chronic lymphocytic leukemia (CLL) and for the treatment of diffuse large B-cell lymphoma (DLBCL). The company refers to the combination regimen of ublituximab and TGR-1202 as TG-1303. The drug is also in phase II clinical development for the oral treatment of hematologic malignancies, as a single agent or in combination therapy. Phase I clinical trials are ongoing in patients with select relapsed or refractory solid tumors, such as adenocarcinoma of the pancreas, adenocarcinoma of the colon, rectum, gastric and GE junction cancer, and GI Stromal Tumor (GIST).
In 2016, orphan drug designation was assigned to the compound in the U.S. for the treatment of CLL. In 2017, additional orphan drug designation was granted in the U.S. for the treatment of CLL and DLBCL, in combination with ublituximab.
Originated by Rhizen Pharmaceuticals, the product was jointly developed by Rhizen Pharmaceuticals and TG Therapeutics since 2012. In 2014, exclusive global development and commercialization rights (excluding India) were licensed to TG Therapeutics.
CLINICAL TRIALS……….https://clinicaltrials.gov/search/intervention=TGR-1202
B-cell lymphoma; Chronic lymphocytic leukemia; Hematological neoplasm; Hodgkins disease; Mantle cell lymphoma; Non-Hodgkin lymphoma
Phosphoinositide-3 kinase delta inhibitor
SYNTHESIS


Rhizen Pharmaceuticals Announces Out-licensing Agreement for TGR-1202, a Novel Next Generation PI3K-delta Inhibitor
Rhizen to receive upfront payment of $8.0 million — Rhizen to retain global manufacturing and supply rights — Rhizen to retain development and commercialization for India
Rhizen to retain development and commercialization for India
| Source: Rhizen Pharmaceuticals SA
La Chaux-de-Fonds, Switzerland, Sept. 23, 2014 (GLOBE NEWSWIRE) — Rhizen Pharmaceuticals S.A. today announced an out-licensing agreement for TGR-1202, a novel next generation PI3K-delta inhibitor. TG Therapeutics exercised its option for early conversion to a licensing agreement from a 50:50 joint venture partnership.
In exchange for this licensing agreement, TG Therapeutics will pay Rhizen an upfront payment of $8.0 million ($4.0 million in cash and $4.0 million in TG Therapeutics common stock). In addition to the upfront payment, Rhizen will be eligible to receive regulatory filing, approval and sales based milestones in the aggregate of approximately $240 million, and tiered royalties based on net sales.
Swaroop Vakkalanka, Ph.D. and President of Rhizen stated, “We are extremely happy and take pride in discovering a novel, next generation, once-daily PI3K-delta inhibitor under active development led by TG Therapeutics. We are encouraged by the progress of TRG-1202 to date, and the speed at which TG Therapeutics is developing the asset in various hematological malignancies. We look forward to the day this novel drug reaches cancer patients in need of new and safe therapies.”
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, www.rhizen.com.

TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.
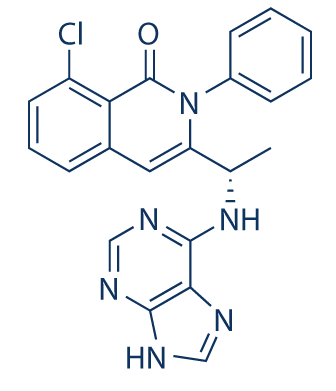
IPI 145
PATENTS
WO 2011055215
http://www.google.com/patents/WO2011055215A2?cl=en

PATENT
WO 2015181728
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015181728
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:

Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)

7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)

1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1

The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8

PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,

(A)
Synthesis of Compound of Formula A
Unless otherwise stated, purification implies column chromatography using silica gel as the stationary phase and a mixture of petroleum ether (boiling at 60-80°C) and ethyl acetate or dichloromethane and methanol of suitable polarity as the mobile phases. The term “RT” refers to ambient temperature (25-28°C).
Intermediate 1 : 2-( l-bromoethyl)-6-fluoro-3-f3-fluorophenyl)-4H-chromen-4-one
Step-1 [l-(5-Fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone]: 3- Fluorophenylacetic acid (7.33 g, 47.56 mmoles) was dissolved in 25 ml dichloromethane. To this mixture, oxalylchloride (7.54 g, 59.46 mmoles) and DMF (3 drops) were added at 0°C and stirred for 30 min. The solvent was evaporated and dissolved in 25 ml dichloromethane. To this mixture, 4-fluoroanisole (5.00 g, 39.64 mmoles) was added and cooled to 0°C. At 0°C A1C13 (7.95 g, 59.46 mmoles) was added and the reaction mixture was warmed to RT and stirred for 12 hours. The reaction mixture was quenched by the addition of 2N HC1, extracted with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as colorless solid (4.5 g, 45% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 11.34 (s, 1H), 7.75 (dd, J=9.4, 3.1 Hz, 1H), 7.42 (m, 2H), 7.12 (m, 3H), 7.05 (dd, J=9.0, 4.5 Hz, 1H), 4.47 (s, 2H).
Step-2 [2-Ethyl-6-fiuoro-3-(3-fluorophenyl)-4H-chromen-4-one]: l-(5-Fluoro-2- hydroxyphenyl)-2-(3-fluorophenyl)ethanone obtained from Step-1 (3.00 g, 12.08 mmoles) was placed in a round bottom flask and to this triethylamine (25 ml) and propionic anhydride (4.92 g, 37.82 mmoles) were added, and the mixture was refluxed for 24 hours. After cooling to RT, the reaction mixture was acidified by the addition of IN HC1 solution, extracted with ethyl acetate, washed with sodium bicarbonate solution, dried with sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as off-yellow solid (1.80 g, 52% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.80 (m, 1H), 7.76 (m, 2H), 7.51 (dd, J=8.0, 6.4 Hz), 7.22 (m, 1H), 7.18 (m, 2H), 2.56 (q, J=7.6 Hz, 2H), 1.20 (t, J=7.6 Hz, 3H).
Step-3: To a solution of 2-Ethyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained from Step-2 (1.80 g, 6.28 mmoles) in carbon tetrachloride (20 ml), N- bromosuccinimide (1.11 g, 6.28 mmoles) was added and heated to 80°C. Azobisisobutyronitrile (10 mg) was added to the reaction mixture at 80°C. After 12 hours, the reaction mixture was cooled to RT, diluted with dichloromethane and washed with water. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the crude title compound as yellow solid (1.25 g, 55% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.91 (dd, J=9.2, 4.3 Hz, 1H), 7.81 (dt, j=8.2, 2.8 Hz, 1H), 7.74 (dd, J=8.3, 3.1 Hz, 1H), 7.57 (m, 1H), 7.32 (dt, J=8.5, 2.4 Hz, 1H), 7.19 (m, 2H), 5.00 (q, J=6.8 Hz, 1H), 1.97 (d, J=6.8 Hz, 3H).
Intermediate 2: 6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To a solution of Intermediate 1 (15.0 g, 40.84 mmol) in DMSO (150 ml), n-butanol (7.5 ml) was added and heated to 120°C for 3 hours. The reaction mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (7.90 g, 64%). 1H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1, 3 Hz, 1H), 7.54 (dd, J = 9.2, 4.2 Hz, 1H), 7.47-7.37 (m, 2H), 7.15-6.98 (m, 3H), 4.74 (quintet, J= 6.8 Hz, 1H), 2.23 (d, J = 7.4 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H).
Intermediate 3 : 2-acetyl-6-fluoro-3-( 3-fluorophenyl)-4H-chromen-4-one

DMSO (5.60 ml, 79.14 mmol) was added to dichloromethane (40 ml), and cooled to – 78°C, followed by oxalyl chloride (3.40 ml, 39.57 mmol). After 10 min., intermediate 2 (6.00 g, 19.78 mmol) in dichloromethane (54 ml) was added dropwise and stirred for 20 min.
Triethylamine (12 ml) was added and stirred for 1 hour. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 4: fS)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To intermediate 3 (2.00 g, 6.66 mmol), R-Alpine borane (0.5 M in THF, 20 ml) was added and heated to 60°C for 20 hours. The reaction mixture quenched with 2N HC1, and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.51 g, 75%).
Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: fR)-l-f6-fluoro-3-f3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4- chlorobenzoate

To a solution of intermediate 4 (1.45 g, 4.78 mmol) in THF (15 ml), 4-chlorobenzoic acid (0.748 g, 4.78 mmol) and triphenylphosphine (1.88 g, 7.17 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (1.4 ml, 7.17 mmol). After 1 hour, the reaction mixture was concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.81 g, 86%) which was used without purification in the next step. Intermediate 6: fR)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

Method A
Intermediate 5 (1.75 g, 3.96 mmol) in methanol (17 ml) was cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HCl solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87% yield). Enantiomeric excess: 93.6%>, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B
Step-1 [(R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one]: To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24 hours. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:
petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%> yield). 1H-NMR (δ ppm, CDCls, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
Step-2: (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained in Step-1 (10.5 g, 26.69 mmol) in dichloromethane (110 ml) was cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6 hours. The reaction mixture was quenched with 2N HCl solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford intermediate 6 a yellow solid (6.1 g, 76% yield). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 7: 4-bromo-2-fluoro-l-isopropoxybenzene

To a solution of 4-bromo-3-fluorophenol (10 g, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8 ml, 62.62 mmol) and triphenylphosphine (20.6 g, 78.52 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (15.4 ml, 78.52 mmol). The mixture was refluxed for 1 hour, concentrated and the residue was purified by column
chromatography with ethyl acetate: petroleum ether to afford the title compound as a colorless liquid (13.1 g, 99% yield), which was used without purification in the next step.
Intermediate 8: 2-f3-fluoro-4-isopropoxyphenyl)-4,4,5.,5-tetramethyl-l,3i2-dioxaborolane

Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15 g, 58.96 mmol) were added to a solution of intermediate 7 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II) CH2CI2 (4.4 g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 9: 3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-dlpyrimidin-4-amine

To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF (110 ml), ethanol (55 ml) and water (55 ml), intermediate 8 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min.
Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
(RS)- 2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 9 (0.080 g, 0.293 mmol) in DMF (2 ml), potassium carbonate (0.081 g, 0.587 mmol) was added and stirred at RT for 10 min. To this mixture intermediate 1 (0.215 g, 0.587 mmol) was added and stirred for 12 hours. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a pale yellow solid (0.045 g). MP: 175-177°C. 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 8.20 (s, 1H), 7.85 (dd, J = 81, 3.0 Hz, 1H), 7.48-7.33 (m, 5H), 7.14 (t, J= 8.3 Hz, 1H), 7.02 (m, 2H), 6.90 (m, 1H), 6.10 (q, J = 7.1 Hz, 1H), 5.42 (s, 2H), 4.64 (quintet, J = 6.0 Hz, 1H), 1.99 (d, J = 7.1 Hz, 3H), 1.42 (d, J= 6.1 Hz, 6H).
fS)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (“S-isomer”)
To a solution of intermediate 9 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 6 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45°C. After 2 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 % yield). MP: 139-142°C. Mass: 571.7 (M+). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64 min.). fR)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-ehromen-4-one
To a solution of intermediate 8 (0.284 g, 0.989 mmol) in THF (5.0 ml), intermediate 4 (0.250 g, 0.824 mmol) and tris(4-methoxy)phenylphosphine (0.435 g, 1.23 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.23 mmol) was added stirred at RT. After 12 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate :
petroleum ether to afford the title compound as an off-white solid (0.105 g, 22 % yield). MP: 145-148°C. Mass: 571.7 (M+). Enantiomeric excess: 95.4% as determined by HPLC on a chiralpak AD-H column, enriched in the late eluting isomer (retention time = 14.83 min.).
PATENT
WO 2014006572
http://www.google.com/patents/WO2014006572A1?cl=en
 B1 IS DESIRED
B1 IS DESIRED
(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
-
To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45° C. After 2 h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:petroleum ether to afford the title compound as an off-white solid (0.049 g, 20%). MP: 139-142° C. Mass: 571.7 (M+).1H-NMR (δ ppm, CDCl3, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J=8.2, 3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J=8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J=8.4 Hz, 1H), 6.11 (q, J=7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J=6.1 Hz, 1H), 2.00 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=10.64 min)
4-Methylbenzenesulfonate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (22.7 g, 39.69 mmol) in isopropanol (600 ml), p-toluenesulphonic acid (8.30 g, 43.66 mmol) was added and refluxed for 1 h. The reaction mixture was concentrated, co-distilled with petroleum ether and dried. To the residue water (300 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (28.2 g, 95%). MP: 138-141° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.80 (d, J=8.2 Hz, 2H), 7.51 (dd, J=9.3, 4.3 Hz, 1H), 7.45 (dd, J=7.5, 3.1 Hz, 1H), 7.42-7.31 (m, 3H), 7.29 (m, 2H), 7.22 (d, J=8.0 Hz, 2H), 7.16 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.5 Hz, 1H), 6.97 (br s, 1H), 6.88 (br s, 1H), 6.11 (q, J=7.2 Hz, 1H), 4.67 (quintet, J=6.0 Hz, 1H), 2.36 (s, 3H), 2.03 (d, J=7.1 Hz, 3H), 1.43 (d, J=6.0 Hz, 6H). Mass: 572.4 (M++1-PTSA). Enantiomeric excess: 93.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.35 min.)

Sulphate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulfate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulphate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g, 26.24 mmol) in isopropanol (600 ml) was cooled to 0° C. To this Sulphuric acid (2.83 g, 28.86 mmol) was added and stirred at room temperature for 24 h. The reaction mass was filtered and washed with petroleum ether and dried under vacuum. To the solid, water (150 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (13.5 g, 76%). MP: 125-127° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.51 (dd, J=9.2, 4.2 Hz, 1H), 7.45-7.31 (m, 3H), 7.29 (m, 1H), 7.15 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.4 Hz, 1H), 6.96 (br s, 1H), 6.88 (br s, 1H), 6.09 (q, J=7.1 Hz, 1H), 4.676 (quintet, J=6.1 Hz, 1H), 2.01 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Mass: 572.2 (M++1-H2SO4). Enantiomeric excess: 89.6% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.08 min.)
-
Various other acid addition salts of compound B1 were prepared as provided in Table 1.
-
TABLE 1 Melting Point Acid Method of preparation (° C.) Hydro- Compound B1 (1 eq.) dissolved in THF, 130-132 chloric excess HCl/Et2O was added, the clear acid solution obtained was evaporated completely. The residue obtained was washed with water. p- Compound B1 (1 eq.) dissolved in 138-141° C. Toluene- isopropyl alcohol (IPA), refluxed for sulfonic 30 min., acid (1.1 eq.) in IPA was added, acid the clear solution obtained was evaporated completely. The residue obtained was washed with water. Benzene- Compound B1 (1 eq.) dissolved in IPA, 170-172 sulphonic refluxed for 30 min., acid(1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Maleic Compound B1 (1 eq.) dissolved in IPA, 107-109 acid refluxed for 30 min., acid (1.1 eq.) in IPA was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Camphor Compound B1 (1 eq.) dissolved in IPA, 120-121 sulfonic refluxed for 30 min., acid (1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Sulphuric Compound B1 (1 eq.) dissolved in IPA, 125-127 acid refluxed for 30 min., acid(1.1 eq.) in IPA was added, the clear solution obtained was evaporated completely. The residue obtained was washed with water.

REFERENCES
WO 2014/006572 and U.S. Patent Publication No. 2014/0011819,
http://www.tgtherapeutics.com/O’ConnorTGR202Single%20AgentEHA&Lugano2015.pdf
-
TGR-1202: Phase I/II started 09/28/2015
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Phase I/II started 09/28/2015
Week in Review, Clinical StatusLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer Molecular target: CD20 Description: Glycoengineered mAb against CD20 … -
The Daily Extra, Company NewsTG Therapeutics Inc. (NASDAQ:TGTX) rose $2.65 (23%) to $14.37 after the company said it received an SPA from FDA for the Phase III UNITY-CLL trial of ublituximab (TG-1101) in combination with TGR-1202 to treat chronic …
-
Targets & Mechanisms: The battle for IRAK 04/23/2015
Nimbus, Aurigene and TG Therapeutics are chasing IRAK4 inhibitors for cancerBC Innovations, Targets & MechanismsNow that Nimbus has put IRAK4 on the map for B cell lymphoma, several companies are closing in with their own inhibitors, and they’re all on track for IND-enabling studies this year. -
TGR-1202: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Ildong Pharmaceutical Co. Ltd. (KSE:000230), Seoul, South Korea Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer … -
TGR-1202: Phase I started 12/15/2014
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Rhizen, TG Therapeutics deal 12/08/2014
Week in Review, DealsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Business: Cancer TG Therapeutics exercised an option under a 2012 deal to license exclusive, worldwide …
| Patent | Submitted | Granted |
|---|---|---|
| NOVEL SELECTIVE PI3K DELTA INHIBITORS [US2014011819] | 2013-07-02 | 2014-01-09 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
////////Umbralisib
CC(C)OC1=C(C=C(C=C1)C2=NN(C3=C2C(=NC=N3)N)C(C)C4=C(C(=O)C5=C(O4)C=CC(=C5)F)C6=CC(=CC=C6)F)F
IPI 504, Retaspamycin, Retaspimycin

IPI 504, Retaspamycin, Retaspimycin
CAS 857402-63-2
Cas 857402-23-4 ( Retaspimycin); 857402-63-2 ( Retaspimycin HCl).
MF C31H45N3O8 BASE
MW: 587.32067 BASE
Infinity Pharmaceuticals Inc, INNOVATOR
[(3R,5S,6R,7S,8E,10S,11S,12Z,14E)-6,20,22-trihydroxy-5,11-dimethoxy-3,7,9,15-tetramethyl-16-oxo-21-(prop-2-enylamino)-17-azabicyclo[16.3.1]docosa-1(22),8,12,14,18,20-hexaen-10-yl] carbamate;hydrochloride
17-Allylamino-17-demethoxygeldanamycin Hydroquinone Hydrochloride
| Retaspimycin hydrochloride; 8,21-didehydro-17-demethoxy-18,21-dideoxo-18,21-dihydroxy-17-(2-propenylamino)-geldanamycin monohydrochloride | |
| Application: | A novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90) |
| Molecular Weight: | 624.17 ……….HCl salt |
| Molecular Formula: | C31H46ClN3O8……….HCl salt |
Introduction
IPI-504 is a novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90).
Orphan drug designation was assigned to the compound by the FDA for the treatment of gastrointestinal stromal cancer (GIST).

Retaspimycin Hydrochloride is the hydrochloride salt of a small-molecule inhibitor of heat shock protein 90 (HSP90) with antiproliferative and antineoplastic activities. Retaspimycinbinds to and inhibits the cytosolic chaperone functions of HSP90, which maintains the stability and functional shape of many oncogenic signaling proteins and may be overexpressed or overactive in tumor cells. Retaspimycin-mediated inhibition of HSP90 promotes the proteasomal degradation of oncogenic signaling proteins in susceptible tumor cell populations, which may result in the induction of apoptosis.
Phase I study of Retaspimycin: A phase 1 study of IPI-504 (retaspimycin hydrochloride) administered intravenously twice weekly for 2 weeks at 22.5, 45, 90, 150, 225, 300 or 400 mg/m(2) followed by 10 days off-treatment was conducted to determine the safety and maximum tolerated dose (MTD) of IPI-504 in patients with relapsed or relapsed/refractory multiple myeloma (MM). Anti-tumor activity and pharmacokinetics were also evaluated. Eighteen patients (mean age 60.5 years; median 9 prior therapies) were enrolled. No dose-limiting toxicities (DLTs) were reported for IPI-504 doses up to 400 mg/m(2).
The most common treatment-related adverse event was grade 1 infusion site pain (four patients). All other treatment-related events were assessed as grade 1 or 2 in severity. The area under the curve (AUC) increased with increasing dose, and the mean half-life was approximately 2-4 h for IPI-504 and its metabolites. Four patients had stable disease, demonstrating modest single-agent activity in relapsed or relapsed/refractory MM. (source: Leuk Lymphoma. 2011 Dec;52(12):2308-15.)

Figure Hsp90 protein partners and clients destabilized by Hsp90 inhibition (Jackson et al., 2004).
In a different approach, Infinity Pharmaceuticals has developed IPI504 (retaspimycin or 17-AAG hydroquinone, Figure 4) (Adams et al., 2005; Sydor et al., 2006), a new GA analogue, in which the quinone moiety was replaced by a dihydroquinone one. Indeed, the preclinical data suggested that the hepatotoxicity of 17-AAG was attributable to the ansamycin benzoquinone moiety, prone to nucleophilic attack.
Furthermore, it was recently reported that the hydroquinone form binds Hsp90 with more efficiency than the corresponding quinone form (Maroney et al., 2006). In biological conditions, the hydroquinone form can interconvert with GA, depending on redox equilibrium existing in cell. It has been recently proposed, that NQ01 (NAD(P)H: quinone oxidoreductase) can produce the active hydroquinone from the quinone form of IPI504 (Chiosis, 2006).
However, Infinity Pharmaceuticals showed that if the overexpression of NQ01 increased the level of hydroquinone and cell sensitivity to IPI504, it has no significant effect on its growth inhibitory activity. These results suggest that NQ01 is not a determinant of IPI504 activity in vivo (Douglas et al., 2009).

Figure 4: GA, 17-AAG, 17-DMAG and IPI504.
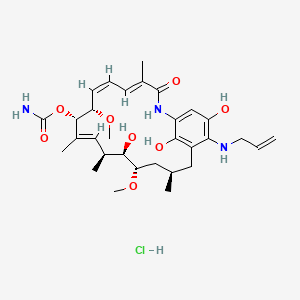
PATENT
http://www.google.com/patents/EP2321645A1?cl=en
Geldanamycin (IUPAC name ([18S-(4E,5Z,8R*.9R*.10E,12R*.13S*,14R*,l6S*)]- 9- [(aminocarbonyl)oxy]- 13- hydroxy- 8,14,19- trimetoxy- 4,10,12,16- tetramethyl- 2- azabicyclo[16.3.1.]docosa- 4,6.10,18,21- pentan- 3.20,22trion) is a benzoquinone ansamycin antibiotic which may be produced by the bacterium Streptorayces hygroscopicus. Geldanamycin binds specifically to HSP90 (Heat Shock Protein 90) and alters its function.
While Hsp90 generally stabilizes folding of proteins and, in particular in tumor cells, folding of overexpressed/mutant proteins such as v-Src. Bcr-Abl and p53. the Hsp90 inhibitor Geldanamycin induces degradation of such proteins.
The respectiv e formula of geldanamycin is given herein below:

E\en though geldanamycin is a potent antitumor agent, the use of geldanamycin also shows some negathe side-effects (e.g. hepatotoxicity) which led to the dev elopment of geldanamycin analogues/derivatives, in particular analogues/deriv atives containing a derivatisation at the 17 position. Without being bound by theory , modification at the 17 position of geldanamycin may lead to decreases hepatotoxicity.
Accordingly geldanamycin analogues/derivatives which are modified at the 17 position, such as 17-AAG (17-N-Allylamino-17-demethoxygeldanamycin), are preferred in context of the present invention. Also preferred herein are geldanamycin derivatives to be used in accordance with the present invention which are water-soluble or which can be dissoh ed in water completely (at least 90 %. more preferably 95 %. 96 %. 97 %, 98 % and most preferably 99 %). 17-AAG ([QS.5S,6RJS$EΛ0R,l \SΛ2E,14E)-2\- (allylamino)-6-hydroxy-5.11-diraethoxy-
3.7.9,15-tetramethyl-16.20.22-trioxo-17-azabicyclo[16.3.1]docosa-8,12.14,18,21-pentaen-10- yl] carbamate) is. as mentioned above a preferred derivative of geldanamycin. 17- AAG is commercially available under the trade name “Tanespimycin“ (also known as KOS-953) for example from Kosan Biosciences Incorporated (Acquired by Bristol-Myers Squibb Company). Tanespimycin is presently studied in phase II clinical trials for multiple myeloma and breast cancer and is usually administered intravenously.
The respective formula of 17- AAG is given herein below:

Preferred geldanamycin-derh ative (HSP90 inhibitor) to be used in context of the present invention are IPI-504 (also known as retaspiimcin or Mcdi-561 : lnfinin Pharmaceuticals (Medlmmunc/ Astra Zeneca)). Clinical trials on the use of IPI-504 (which is usually administered intravenously) in the treatment of non-small cell lung cancer (NSCLC) and breast cancer are performed. Also alvespimycin hy drochloride (Kosan Biosciences Incorporated Acquired By : Bristol-Myers Squibb Company) is a highly potent, water-soluble and orally acti\e derivative of geldanamycin preferably used in context of the present invention.

IPI-504
PATENT
WO 2005063714
http://www.google.co.ug/patents/WO2005063714A1?cl=en
Example 24
Preparation of Air-stable Hydroquinone Derivatives of the Geldanamycin Family of Molecules
,

17-Allylamino-17-Demethoxygeldanamycin (10.0 g, 17.1 mmol) in ethyl acetate
(200 mL) was stirred vigorously with a freshly prepared solution of 10% aqueous sodium hydrosulfite (200 mL) for 2 h at ambient temperature. The color changed from dark purple to bright yellow, indicating a complete reaction. The layers were separated and the organic phase was dried with magnesium sulfate (15 g). The drying agent was rinsed with ethyl acetate (50 mL). The combined filtrate was acidified with 1.5 M hydrogen chloride in ethyl acetate (12 mL) to pH 2 over 20 min. The resulting slurry was stirred for 1.5 h at ambient temperature. The solids were isolated by filtration, rinsed with ethyl acetate (50 mL) and dried at 40 °C, 1 mm Hg, for 16 h to afford 9.9 g (91%) of off-white solid. Crude hydroquinone hydrochloride (2.5 g) was added to a stirred solution of 5% 0.01 N aq. hydrochloric acid in methanol (5 mL). The resulting solution was clarified by filtration then diluted with acetone (70 mL). Solids appeared after 2-3 min. The resulting slurry was stirred for 3 h at ambient temperature, then for 1 h at 0-5 °C. The solids were isolated by filtration, rinsed with acetone (15 mL) and dried
PAPER
J. Med. Chem., 2006, 49 (15), pp 4606–4615
DOI: 10.1021/jm0603116

17-Allylamino-17-demethoxygeldanamycin (17-AAG)1 is a semisynthetic inhibitor of the 90 kDa heat shock protein (Hsp90) currently in clinical trials for the treatment of cancer. However, 17-AAG faces challenging formulation issues due to its poor solubility. Here we report the synthesis and evaluation of a highly soluble hydroquinone hydrochloride derivative of 17-AAG, 1a (IPI-504), and several of the physiological metabolites. These compounds show comparable binding affinity to human Hsp90 and its endoplasmic reticulum (ER) homologue, the 94 kDa glucose regulated protein (Grp94). Furthermore, the compounds inhibit the growth of the human cancer cell lines SKBR3 and SKOV3, which overexpress Hsp90 client protein Her2, and cause down-regulation of Her2 as well as induction of Hsp70 consistent with Hsp90 inhibition. There is a clear correlation between the measured binding affinity of the compounds and their cellular activities. Upon the basis of its potent activity against Hsp90 and a significant improvement in solubility, 1a is currently under evaluation in Phase I clinical trials for cancer.
17-Allylamino-17-demethoxygeldanamycin Hydroquinone Hydrochloride Ia
17-AAG hydroquinone hydrochloride (1a) as an off-white solid (11 g, 18 mmol, 80% yield). HPLC purity: 99.6%;
IR (neat): 3175, 2972, 1728, 1651, 1581, 1546, 1456, 1392, 1316, 1224, 1099, 1036 cm-1;
1H NMR (CDCl3:d6-DMSO, 6:1, 400 MHz):
δ 10.20 (1H, br), 9.62 (2H, br), 8.53 (1H, s), 8.47 (1H, s), 7.74 (1H, s), 6.72 (1H, d, J= 11.6 Hz), 6.28 (1H, dd, J = 11.6, 11.2 Hz), 5.73 (1H, dddd, J = 17.2, 10.0, 3.2, 2.4 Hz), 5.53 (1H, d, J = 10.4 Hz), 5.49 (1H, dd, J = 10.8, 10.0 Hz), 5.32 (2H, s), 5.04 (1H, d, J = 4.8 Hz), 5.02 (1H, d, J = 16.0 Hz), 4.81 (1H, s), 4.07 (1H, d, J = 9.6 Hz), 3.67 (2H, d, J = 6.4 Hz), 3.31 (1H, d,J = 8.8 Hz), 3.07 (3H, s), 3.07−3.04 (1H, m), 2.99 (3H, s), 2.64 (1H, m), 2.52−2.49 (1H, m), 1.76 (3H, s), 1.61−1.39 (3H, m), 0.78 (3H, d, J = 6.4 Hz), 0.64 (3H, d, J = 7.2 Hz);
13C NMR (CDCl3:d6-DMSO, 6:1, 100 MHz): δ 167.3, 155.8, 143.3, 136.3, 135.0, 134.2, 132.9, 132.1, 128.8, 127.6, 125.9, 125.3, 123.7, 123.0, 115.1, 104.5, 80.9, 80.7, 80.1, 72.5, 56.2, 56.2, 52.4, 34.6, 33.2, 31.1, 27.2, 21.6, 12.1, 12.1, 11.7;
HRMS calculated for C31H45N3O8 (M+ + H): 588.3285, Found 588.3273.
POSTER
Synthesis and biological evaluation of IPI-504, an aqueous soluble analog of 17-AAG and potent inhibitor of Hsp90
MEDI 210 |
| IPI-504 is the hydroquinone hydrochloride salt of 17-allylamino-17-demethoxy-geldanamycin (17-AAG), an Hsp90 inhibitor that is currently in clinical trials for the treatment of cancer.
IPI-504 demonstrates high aqueous solubility (>200 mg/mL). Interestingly, in vitro and in vivo IPI-504 interconverts with 17-AAG and exists in a pH and enzyme-mediated redox equilibrium. This occurs due to oxidation of the hydroquinone (IPI-504) to the quinone (17-AAG) at physiological pH and the reduction of 17-AAG by quinone reductases such as NQO1 to IPI-504. Here we report the design and synthesis of the stabilized hydroquinone IPI-504 and its inhibitory effect against Hsp90 and Grp94. Although IPI-504 was originally designed to be a soluble prodrug of 17-AAG, the hydroquinone is more potent than the quinone in the biochemical Hsp90 binding assay. Various hydroquinone analogs have been prepared to investigate the structure activity relationship of hydroquinone binding to Hsp90. Hydroquinone and quinone forms of 17-AAG metabolites show comparable binding affinities for Hsp90 and in cancer cell lines, hydroquinone analogs elicit specific responses consistent with Hsp90 inhibition. The desirable pharmacological properties as well as in vitro and in vivo activity of our lead compound, IPI-504, has led to the initiation of Phase I clinical trials in multiple myeloma. |
| http://oasys2.confex.com/acs/231nm/techprogram/P945016.HTM |
|
Geldanamycin and Beyond: Progress Toward the Development of HSP90 Inhibitors
8:30 AM-12:05 PM, Wednesday, 29 March 2006 Georgia World Congress Center — Georgia Ballroom 1, OralDivision of Medicinal ChemistryThe 231st ACS National Meeting, Atlanta, GA, March 26-30, 2006 |
References
Synthesis and biological evaluation of IPI-504, an aqueous soluble analog of 17-AAG and potent inhibitor of Hsp90
231st Am Chem Soc (ACS) Natl Meet (March 26-30, Atlanta) 2006, Abst MEDI 210
Design, synthesis, and biological evaluation of hydroquinone derivatives of 17-amino-17-demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90
J Med Chem 2006, 49(15): 4606
http://www.biotechduediligence.com/retaspamycin-hcl-ipi-504.html
///////////////////Hsp90, IPI-504, infinity pharma, Retaspamycin, Retaspimycin
Sparsentan, PS433540, RE-021

Sparsentan (PS433540, RE-021)
- C32H40N4O5S
- Average mass592.749
FDA APPROVED 2023/2/17, Filspari
4′-((2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl)-N-(4,5-dimethylisoxazol-3-yl)-2′-(ethoxymethyl)-[1,1′-biphenyl]-2-sulfonamide
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide
Sparsentan
PS433540; RE-021, formerly known as DARA
CAS :254740-64-2
4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(4,5- dimethylisoxazol-3-yl)-2-(ethoxymethyl)biphenyl-2-sulfonamide
Mechanism of Action:acting as both an Endothelin Receptor Antagonist (ERA) and Angiotensin Receptor Blocker (ARB).
Indication: Focal Segmental Glomerulosclerosis (FSGS).Focal Segmental Glomerulosclerosis (FSGS) is a rare and severe nephropathy which affects approximately 50,000 patients in the United States. Most cases of FSGS are pediatric.
Development Stage: Phase II
Developer:Retrophin, Inc
- OriginatorBristol-Myers Squibb
- DeveloperRetrophin
- ClassAntihypertensives; Isoxazoles; Small molecules; Spiro compounds; Sulfonamides
- Mechanism of ActionAngiotensin type 1 receptor antagonists; Endothelin A receptor antagonists
- Orphan Drug Status Yes – Focal segmental glomerulosclerosis
-
- 09 Jan 2015 Sparsentan receives Orphan Drug status for Focal segmental glomerulosclerosis in USA
- 31 Dec 2013 Phase-II/III clinical trials in Focal segmental glomerulosclerosis in USA (PO)
- 07 May 2012I nvestigation in Focal segmental glomerulosclerosis in USA (PO)
Sparsentan is an investigational therapeutic agent which acts as both a selective endothelin receptor antagonist and an angiotensin receptor blocker. Retrophin is conducting the Phase 2 DUET trial of Sparsentan for the treatment of FSGS, a rare and severe nephropathy that is a leading cause of end-stage renal disease. There are currently no therapies approved for the treatment of FSGS in the United States. Ligand licensed worldwide rights of Sparsentan (RE-021) to Retrophin in 2012 .The Food and Drug Administration (FDA) has granted orphan drug designation for Retrophins sparsentan for the treatment of focal segmental glomerulosclerosis (FSGS) in January 2015.
In 2006, the drug candidate was licensed to Pharmacopeia by Bristol-Myers Squibb for worldwide development and commercialization. In 2012, a license was obtained by Retrophin from Ligand. In 2015, Orphan Drug Designation was assigned by the FDA for the treatment of focal segmental glomerulosclerosis.
Sparsentan, also known as RE-021, BMS346567, PS433540 and DARA-a, is a Dual angiotensin II and endothelin A receptor antagonist. Retrophin intends to develop RE-021 for orphan indications of severe kidney diseases including Focal Segmental Glomerulosclerosis (FSGS) as well as conduct proof-of-concept studies in resistant hypertension and diabetic nephropathy. RE-021, with its unique dual blockade of angiotensin and endothelin receptors, is expected to provide meaningful clinical benefits in mitigating proteinuria in indications where there are no approved therapies
Sparsentan, sold under the brand name Filspari, is a medication used for the treatment of primary immunoglobulin A nephropathy.[1] Sparsentan is an endothelin and angiotensin II receptor antagonist.[1][4] It is taken by mouth.[1]
The most common side effects include swelling of the extremities, low blood pressure, dizziness, high blood potassium, anemia, injury to the kidney, and increased liver enzymes in the blood.[5]
It was approved for medical use in the United States in February 2023.[5][6][7] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[8]
PATENT
WO 2000001389
https://www.google.co.in/patents/WO2000001389A1?cl=en


Example 41
4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.4! non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l, l’-biphenyl! -2-sulfonamide

A. 4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl)-N-[(2-trimethylsilylethoxy)methyl]-2′- hydroxym ethyl [1, l’-biphenyl] -2-sulfonamide P14 (243 mg, 0.41 mmol) was used to alkylate 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene hydrochloride according to General Method 4. 41A (100 mg, 35% yield) was isolated as a slightly yellow oil after silica gel chromatography using 1:1 hexanes/ethyl acetate as eluant. B. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methvn -N-0.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l,l’-biphenyn-2- sulfonamide
Deprotection of 41A (100 mg, 0.14 mmol) according to General Method 8 (ethanol) gave the title compound as white solid in 46% yield following silica gel chromatography (96:4 methanol/chloroform eluant):
MS m/e 565 (ESI+ mode); HPLC retention time 3.21 min (Method A);
HPLC purity >98%.
Example 42
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide

A. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3 ,4- dimethyl-5-isoxazolyl)-N-[(2-methoxyethoxy)methyll-2′- hvdroxym ethyl [1 , l’-biphenyl] -2-sulfonamide
Triethylsilane (6 ml) and TFA (6 ml) were added to a solution of 5F (960 mg, 1.5 mmol) in 15 ml dichloromethane at RT. The mixture was stirred at RT for 2 h and was then concentrated. The residue was taken up in ethyl acetate and was washed successively with aqueous sodium bicarbonate, water, and brine. The organic layer was dried over sodium sulfate and concentrated. The residue was chromatographed on silica gel using 100:2 dichloromethane/methanol to afford 42A (740 mg, 77%) as a colorless gum. Rf=0.13, silica gel, 100:5 dichloromethane/methanol. B. 4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-N-r(2-methoxyethoxy)methyll-2′- ethoxymethyl[l.l’-biphenyll-2-sulfonamide A mixture of 42A (100 mg, 0.15 mmol), iodoethane (960 mg, 6.1 mmol) and silver (I) oxide (180 mg, 0.77 mmol) in 0.7 ml DMF was heated at 40 ° C for 16 h.. Additional iodoethane (190 mg, 1.2 mmol) and silver (I) oxide (71 mg, 0.31 mmol) were added and the reaction mixture was heated at 40 ° C for an additional 4 h. The mixture was diluted with 1:4 hexanes/ethylacetate and was then washed with water and brine. The organic layer was dried over sodium sulfate and was then concentrated. The residue was chromatographed on silica gel using 200:3 dichloromethane/methanol as eluant to afford 42B (51mg, 49%) as a colorless gum. Rf=0.35, silica gel, 100:5 dichloromethane/methanol.
C. 4,-[(2-Butyl-4-oxo-1.3-diazaspirof4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl )-2′-ethoxym ethyl [ 1. l’-biphenyll -2-sulfonamide
42B (51 mg) was deprotected according to General Method 7 to afford the title compound in 80% yield following preparative reverse-phase HPLC purification: white solid; m.p. 74-80 ° C (amorphous); IH NMR (CDCL, )δ0.87(tr, J=7Hz, 3H), 0.99(tr, J=7Hz, 3H), 1.32(m, 2H), 1.59(m, 2H), 1.75-2.02(m, 11H), 2.16(s, 3H), 2.35(m, 2H), 3.38 (m, 2H), 4.23(m, 2H), 4.73(s, 2H), 7.11-7.85 (m, 7H); MS m/e 593 (ESI+ mode); HPLC retention time 18.22 min. (Method E); HPLC purity >97%.
PATENT
WO 2001044239
http://www.google.co.in/patents/WO2001044239A2?cl=en
……………………
Dual angiotensin II and endothelin A receptor antagonists: Synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides with improved potency and pharmacokinetics
J Med Chem 2005, 48(1): 171
J. Med. Chem., 2002, 45 (18), pp 3829–3835
DOI: 10.1021/jm020138n
 BMS 248360 A DIFFERENT COMPD
BMS 248360 A DIFFERENT COMPDThe ETA receptor antagonist (2) (N-(3,4-dimethyl-5-isoxazolyl)-4‘-(2-oxazolyl)-[1,1‘-biphenyl]-2-sulfonamide, BMS-193884) shares the same biphenyl core as a large number of AT1 receptor antagonists, including irbesartan (3). Thus, it was hypothesized that merging the structural elements of 2 with those of the biphenyl AT1 antagonists (e.g., irbesartan) would yield a compound with dual activity for both receptors. This strategy led to the design, synthesis, and discovery of (15) (4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide, BMS-248360) as a potent and orally active dual antagonist of both AT1 and ETAreceptors. Compound 15 represents a new approach to treating hypertension.

Scheme 2 a DIFFERENT COMPD
a (a) DIBAL, toluene; (b) NaBH4, MeOH; (c) (Ph)3P, CBr4, THF (51% from 9); (d) compound 7, NaH, DMF; (e) 1 N HCl; (f) compound 4, (Ph3P)4Pd, aqueous Na2CO3, EtOH/toluene; (g) 6 N aqueous HCl/EtOH (60% from 10); (h) 13, sodium triacetoxy borohydride, AcOH, (i) diisopropylcarbodiimide, CH2Cl2 (31% from 12).
PATENT
WO 2010135350
http://www.google.com/patents/WO2010135350A2?cl=en
Compound 1 :

Scheme IV

Scheme V

Formula IV 1
Scheme VII

Formula Vl

A solution of 2-(2,4-dimethylphenyl)benzenesulfonic acid (Compound 12) (0.5 g, 1.9 mmol) in 50 mL of anhydrous acetonitrile was prepared and transferred to a round-bottom flask. After flushing with nitrogen gas, N-bromosuccinimide (0.75 g, 4.2 mmol) was added followed by 50 mg (0.2 mmol) of benzoyl peroxide. The solution was heated at reflux for 3 hours. The solvent was removed in-vacuo and the resulting syrup purified by silica gel chromatography (1 :1 hexanes/EtOAc) to yield Compound 13 as a white solid. 1H NMR (500 MHz, CD3CN) 8.12 (d, J = 7.5 Hz, IH), 7.92 (t, J = 7.5 Hz, IH), 7.78 (d, J= 7.5 Hz, IH), 7.74-7.71 (m, 2H), 7.68-7.65 (m, 2H), 5.12 (s, 2H), 4.70 (s, 2H). Example 4 2-(4-Bromomethyl-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 14)

A solution of 20 mg (0.058 mmol) of (l-bromomethylbenzo[3,4- d])benzo[l,2-f]-2-oxa-l,l-dioxo-l-thiocycloheptane (Compound 13) in ethanol was stirred at elevated temperature until the starting material was consumed to give crude product (compound 14) that was used directly in the next step without isolation or purification.
Example 5
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonic acid (Compound 15)

To the above ethanol solution of crude 2-(4-bromomethyl-2- ethoxymethylphenyl)benzenesulfonic acid (Compound 14) described in Example 4 was added approximately 25 mL of anhydrous DMF. The ethanol was removed from the system under reduced pressure. Approximately 15 mg (0.065 mmol) of 2-butyl-l,3- diazaspiro[4.4]non-l-en-4-one (compound 7 in Scheme IV) was added followed by 300 μL of a IM solution of lithium bis-trimethylsilylamide in THF. The solution was allowed to stir at room temperature for 3 hours. The solvents were removed under reduced pressure and the remaining residue purified by preparative RP-HPLC employing a Cl 8 column and gradient elution (H2O:MeCN) affording the title compound as a white solid; [M+H]+ calcd for C27H34N2O5S 499.21, found, 499.31 ; 1H NMR (500 MHz, CD3CN) 8.04 (t, J= 5.5 Hz, IH), 7.44-7.10 (m, 2H), 7.28 (s, IH), 7.22 (d, J= 8.0 Hz, 2H), 7.08- 7.04 (m, 2H), 4.74 (br s, 2H), 4.32 (d, J= 13.0 Hz IH), 4.13 (d, J= 13.0 Hz IH), 3.40- 3.31 (m, 2H), 2.66 (t, J= 8 Hz, 2H), 2.18-2.13 (m, 5H), 1.96-1.90 (m, 2H obscured by solvent), 1.48 (m, 2H), 1.27 (s, J= 7 Hz, 2H), 1.16 (t, J= 7 Hz, 3H), 0.78 (t, J= 7.5 Hz, 3H).
Example 6
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16)

To a solution of DMF (155 μL, 2 mmol, 2 equiv.) in dichloromethane (5 mL) at 0 0C was added dropwise oxalyl chloride (175 μL, 2 mmol, 2 equiv.) followed by a dichloromethane (5 mL) solution of 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l- en-3-yl)methyl)-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 15) (0.50 g, 1.0 mmol). The resulting mixture was stirred at 0 0C for ~2 hours, diluted with additional dichloromethane (25 mL), washed with saturated sodium bicarbonate solution (10 mL), water (10 mL), and brine (10 mL), dried over sodium sulfate, and then concentrated to give crude sulfonyl chloride (compound 16) that was used without purification.
Example 7
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

[0062] To a solution of 5-amino-3,4-dimethylisoxazole (60 mg, 0.54 mmol) in THF at -60 °C was added dropwise potassium tert-butoxide (1 mL of 1 M solution) followed by a solution of crude 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3- yl)methyl)-2-ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16) (0.28 g, 0.54 mmol) in THF (4 mL). The resulting mixture was stirred at about -60 °C for 1 hour, allowed to warm to room temperature overnight, and then quenched with IN HCl solution to about pH 4. Standard workup of extraction with ethyl acetate, washing with water, drying, and concentration provided the final compounds as a white solid. 1H NMR (400 MHz, CDCl3) 8.03 (dd, J = 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J = 7.5, 2H), 1.37 (sextet, J = 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
Example 8 l-Bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17)

To a solution of ethyl 4-bromo-3-ethoxymethylbenzoate (9.4 g, 33 mmol) in toluene (56 mL) at about -10 0C was added 51 g of a 20% diisobutylaluminum hydride solution in toluene (ca. 70 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy 5% NaOH solution to keep the temperature below about 10 °C. Organic phase of the resulting mixture was separated and the aqueous phase was extracted with toluene. The combined organic phase was concentrated in vacuo to a final volume of ~60 mL toluene solution of l-bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) that was used in next step without purification.
Example 9 l-Bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18)

To a solution of 1 -bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) (8.4 g, 33 mmol) in toluene (60 mL) prepared in Example 8 at about -10 °C was added methanesulfonyl chloride (7.9 g, 68 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy water to keep the temperature at about 0 °C. The organic layer was separated and washed again with icy water to provide a crude product solution of 1 – bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18) that was used without purification.
Example 10
1 -Bromo-4-((2-butyl-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 -yl)methy l)-2- ethoxymethylbenzene bisoxalic acid salt (Compound 19)

To the crude solution of 1 -bromo-2-ethoxymethyl-4- methanesulfonyloxymethylbenzene (Compound 18) (1 1 g, 33 mmol) in toluene (80 mL) prepared in Example 9 was added a 75% solution of methyltributylammonium chloride in water (0.47 mL). The resulting mixture was added to a solution of 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene (compound 7 in Scheme VI) (7.5 g, 32 mmol) in dichloromethane (33 mL) pretreated with a 10 M NaOH solution (23 mL). The reaction mixture was stirred at room temperature for 2 hours until compound 18 was not longer detectable by HPLC analysis and then was quenched with water (40 mL). After stirring about 10 minutes, the organic layer was separated and aqueous layer was extracted with toluene. The combined organic phase was washed with water and concentrated to a small volume. Filtration through a silica gel pad using ethyl acetate as solvent followed by concentration yielded 1 -bromo-4-((2-buty 1-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 – yl)methyl)-2-ethoxymethylbenzene as a crude oil product.
The crude oil was dissolved in ethyl acetate (22 mL) and warmed to around 50 °C. Anhydrous oxalic acid (4.6 g) was added to the warm solution at once and the resulting mixture was stirred until a solution was obtained. The mixture was cooled gradually and the bisoxalic acid salt (compound 19) was crystallized. Filtration and drying provided pure product (compound 19) in 50-60% yield from ethyl 4-bromo-3- ethoxymethylbenzoate in 3 steps. 1H NMR (400 MHz, CDCl3) 12.32 (bs, 4H), 7.58 (d, J = 7.8, IH), 7.36 (s, IH), 7.12 (d, J= 7.8, IH), 4.90 (s, 2H), 4.56 (s, 2H), 3.68 (q, J= 7.5, 2H), 2.87-2.77 (m, 2H), 2.40-1.95 (m, 8H), 1.62-1.53 (m, 2H), 1.38-1.28 (m, 4H), and 1.82 (t, J= 7.5, 3H).
Example 11
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

To a suspension of l-bromo-4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non- l-en-3-yl)methyl)-2-ethoxymethylbenzene bisoxalic acid salt (Compound 19) (5.0 g, 8.3 mmol) in toluene (20 niL) under nitrogen was added water (30 mL) and pH was adjusted to 8-9 by addition of a 2 M NaOH solution at room temperature. The organic phase was separated and mixed with 2-(N-(3,4-dimethyl-5-isoxazolyl)-N- methoxymethylamino)sulfonylphenylboronic acid pinacol ester (Scheme VII, Formula IX, where R8is methoxymethyl and M = boronic acid pinacol ester) (3.6 g, 8.5 mmol), bis(dibenzylideneacetone)palladium(0) (Pd(dba)2) (0.12 g), and a standard phosphine ligand. After a 2 M sodium carbonate solution was added, the reaction mixture was warmed to 70 0C and stirred until the reaction was complete by HPLC analysis. The reaction was cooled to room temperature and quenched with water, and then separated in phases. The organic phase was treated with activated carbon, filtered through a pad of silica gel, and was concentrated to afford a crude mixture.
The crude reaction mixture was dissolved in ethanol (40 mL) after palladium catalyst was removed and was treated with 6 M HCl solution (ca. 40 mL). The mixture was warmed to 75-80 °C and stirred for about 2 hours until the reaction was completed by HPLC analysis. After the mixture was cooled to room temperature, the pH of the mixture was adjusted to 8 by addition of 10 M NaOH solution. The mixture was stirred for 2 more hours and the pH was adjusted to 6 by adding 2 M HCl and the crystal seeds. Filtration of the crystalline solid followed by drying provided N-(3,4-dimethyl-5- isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en-3yl)methyl-2- ethoxymethylphenyl)phenylsulfonamide (Compound 1) as a white solid.1H NMR (400 MHz, CDCIa) 8.03 (dd, J= 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J= 7.5, 2H), 1.37 (sextet, J= 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
| US20040002493 * | Aug 20, 2001 | Jan 1, 2004 | Kousuke Tani | Benzoic acid derivatives and pharmaceutical agents comprising the same as active ingredient |
| US20070054806 * | Sep 6, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Novel sulfonamide-comprising solid formulations |
| US20070054807 * | Sep 8, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Storage-stable formulations of sulfonamides |
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Filspari |
| Other names | RE-021, PS433540 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623018 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEBI | |
| ECHA InfoCard | 100.275.317 |
| Chemical and physical data | |
| 3D model (JSmol) | |
|
show
|
|
|
show
|
|
References
- ^ Jump up to:a b c d e f “Filspari- sparsentan tablet, film coated”. DailyMed. 17 February 2023. Retrieved 6 March 2023.
- ^ Jump up to:a b c d “Filspari EPAR”. European Medicines Agency (EMA). 22 February 2024. Retrieved 24 February 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Filspari Product information”. Union Register of medicinal products. 23 April 2024. Retrieved 7 September 2024.
- ^ Chiu AW, Bredenkamp N (September 2023). “Sparsentan: A First-in-Class Dual Endothelin and Angiotensin II Receptor Antagonist”. The Annals of Pharmacotherapy. 58 (6): 645–656. doi:10.1177/10600280231198925. PMID 37706310. S2CID 261743204.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Drug Trials Snapshots: Filspari”. U.S. Food and Drug Administration (FDA). 17 February 2023. Retrieved 7 September 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Travere Therapeutics Announces FDA Accelerated Approval of Filspari (sparsentan), the First and Only Non-immunosuppressive Therapy for the Reduction of Proteinuria in IgA Nephropathy” (Press release). Travere Therapeutics. 17 February 2023. Retrieved 17 February 2023 – via GlobeNewswire.
- ^ Syed YY (April 2023). “Sparsentan: First Approval”. Drugs. 83 (6): 563–568. doi:10.1007/s40265-023-01864-x. PMC 10232600. PMID 37022667.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ “PHARMACOPEIA LAUNCHES STUDY OF DARA COMPOUND | FDAnews”. http://www.fdanews.com.
- ^ “Ligand Licenses DARA Program to Retrophin”. investor.ligand.com. 21 February 2012.
- ^ https://www.fiercebiotech.com/biotech/retrophin-sheds-shkreli-connection-new-name-travere-therapeutics.
{{cite news}}: Missing or empty|title=(help) - ^ “Ongoing Non-malignant Hematological, Neurological, and Other Disorder Indications Accelerated Approvals”. U.S. Food and Drug Administration (FDA). 21 August 2024. Retrieved 7 September 2024.
- ^ “Travere Therapeutics Announces Full FDA Approval of Filspari (sparsentan), the Only Non-Immunosuppressive Treatment that Significantly Slows Kidney Function Decline in IgA Nephropathy” (Press release). Travere Therapeutics. 5 September 2024. Retrieved 7 September 2024 – via GlobeNewswire.
- ^ “Despite trial scare, Travere’s Filspari gains full FDA nod in kidney disease showdown with Novartis”. fiercepharma.com.
External links
- Clinical trial number NCT03762850 for “A Study of the Effect and Safety of Sparsentan in the Treatment of Patients With IgA Nephropathy (PROTECT)” at ClinicalTrials.gov
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Sparsentan (Filspari). Sparsentan (27), marketed by Travere Therapeutics, is an oral, dual endothelin angiotensin receptor antagonist that received accelerated USFDA approval in February 2023 for reducing proteinuria in adults with primary immunoglobulin A (IgA) nephropathy who are at risk of rapid
disease progression.205206,207 Also known as Berger’s disease, IgAnephropathy is an immune-complex mediated disease characterized by deposits of IgA in the kidneys, resulting in inflammation and damage which can eventually lead to kidney failure. Typical treatment of IgA nephropathy has focused
on supportive care to slow kidney decline, for example, lowering blood pressure, reducing proteinuria, and minimizing lifestyle risk factors; immunosuppressive therapy has also been utilized, though it is controversial and carries risks.208 Sparsentan is the first nonimmunosuppressive treatment for IgA nephropathy and has received first-in-class and orphan drug designations. Accelerated approval was based on reduction of proteinuria (which is a risk factor for disease progression) during interim
analysis in phase III clinical trials. 209 endothelin type A (ETASparsentan blocks ) and angiotensin II type 1 receptors(AT1), interrupting the signaling pathway that contributes to disease progression. 210
The structure of the drug combines 211,212 elements that target both of these receptor types.
213 Thesynthesis of sparsentan (27), as shown in Scheme 50 and Scheme 51, was disclosed by Retrophin Pharmaceuticals (now Travere Therapeutics). Its telescoped sequences and isolation of intermediates as salts suggest that this route may be suitable for large-scale manufacturing.
The synthesis of the spirocyclic imidazolinone intermediate 27.7 is shown in Scheme 50.
Displacement of the benzylic bromide in 27.1 with sodium ethoxide produced ether 27.2. Reduction of the ester with sodium borohydride and zinc chloride yielded alcohol 27.3 which was then converted to mesylate 27.4. Reaction with spirocyclic imidazolinone 27.5 under phase transfer conditions
yielded 27.6 whichwasisolatedasthebisoxalatesalt (27.7).The sequence from 27.1 to 27.7 is telescoped, and no yields were given in the patent.
The construction of the biphenyl framework is shown in Scheme 51. Treatment of aryl bromide 27.8 with n-BuLi and triisopropyl borate followed by reaction with pinacol yielded boronic ester 27.9. Intermediates 27.7 and 27.9 were coupled via a Suzuki reaction to form the biphenyl which was isolated as
the camphorsulfonate salt (27.10). The synthesis was finished with deprotection of the methoxymethyl group under acidic conditions followed by recrystallization from isopropanol and heptane to yield sparsentan (27).
(206) Donadio, J. V.; Grande, J. P. IgA nephropathy. N. Engl. J. Med.2002, 347, 738−748.
(207) Fabiano, R. C. G.; Pinheiro, S. V. B.; Simões e Silva, A. C.Immunoglobulin A nephropathy: a pathophysiology view. Inflammation Res. 2016, 65, 757−770.
(208) Floege, J.; Rauen, T.; Tang, S. C. W. Current treatment of IgAnephropathy. Springer Semin. Immunopathol. 2021, 43, 717−728.
(209) Rovin, B.H.; Barratt, J.; Heerspink, H. J. L.; Alpers, C. E.; Bieler,S.; Chae, D.-W.; Diva, U. A.; Floege, J.; Gesualdo, L.; Inrig, J. K.; et al.Efficacy and safety of sparsentan versus irbesartan in patients with IgA
nephropathy (PROTECT): 2-year results from a randomised, active controlled, phase 3 trial. Lancet 2023, 402, 2077−2090.
(210) Komers, R.; Plotkin, H. Dual inhibition of renin-angiotensin aldosterone system and endothelin-1 in treatment of chronic kidney disease. Am. J. Physiol.: Regul., Integr. Comp. Physiol. 2016, 310, R877−
R884.
(211) Murugesan, N.; Tellew, J. E.; Gu, Z.; Kunst, B. L.; Fadnis, L.;Cornelius, L. A.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; et al. Discovery of N-isoxazolyl biphenylsulfonamides as potent dual
angiotensin II and endothelin A receptor antagonists. J. Med. Chem.2002, 45, 3829−3835.
(212) Murugesan, N.; Gu, Z.; Fadnis, L.; Tellew, J. E.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; Dickinson, K. E.; Valentine,M.T.; et al. Dual angiotensin II and endothelin A receptor antagonists:
synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides withimproved potencyandpharmacokinetics. J. Med. Chem. 2005, 48, 171−179.
(213) Komers, R.; Shih, A. Biphenyl sulfonamide compounds for the treatment of kidney diseases or disorders. WO 2018071784, 2018.

//////////////Sparsentan, PS433540, RE-021, Bristol-Myers Squibb, ORPHAN DRUG, Retrophin, FDA 2023, APPROVALS 2023
O=S(C1=CC=CC=C1C2=CC=C(CN3C(CCCC)=NC4(CCCC4)C3=O)C=C2COCC)(NC5=NOC(C)=C5C)=O,
