









如果您有关于yaopha.com的问题或建议请与我们联系,具体方式如下:
E-mail:contactus@yaopha.com
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » Posts tagged 'Orphan Drug' (Page 10)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

MIDOSTAURIN
READ …COMPLETE SYNTHESIS AT

An antineoplastic agent and p110delta inhibitor
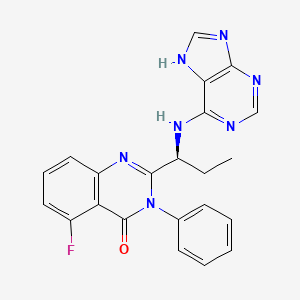
(S)-2-(1-(9H-purin-6-ylamino)propyl)-5-fluoro-3-phenylquinazolin-4(3H)-one
Icos (Originator)
M.Wt: 415.43
Formula: C22H18FN7O
CAS No.: 870281-82-6
CAL-101 Solubility: DMSO ≥80mg/mL Water <1.2mg/mL Ethanol ≥33mg/mL
5-Fluoro-3-phenyl-2-[(1S)-1-(7H-purin-6-ylamino)propyl]-4(3H)-quinazolinone
idelalisib
Idelalisib (codenamed GS-1101 or CAL-101) is a drug under investigation for the treatment of chronic lymphocytic leukaemia. It is in Phase III clinical trials testing drug combinations with rituximab and/or bendamustine as of 2013. The substance acts as aphosphoinositide 3-kinase inhibitor; more specifically, it blocks P110δ, the delta isoform of the enzyme phosphoinositide 3-kinase.[1][2]
GDC-0032 is a potent, next-generation beta isoform-sparing PI3K inhibitor targeting PI3Kα/δ/γ with IC 50 of 0.29 nM/0.12 nM/0.97nM,> 10 fold over Selective PI3K [beta].
GS-1101 is a novel, orally available small molecule inhibitor of phosphatidylinositol 3-kinase delta (PI3Kdelta) develop by Gilead and is waiting for registration in U.S. for the treatment of patients with indolent non-Hodgkin’s lymphoma that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy and for the treatment of chronic lymphocytic leukemia. The compound is also in phase III clinical evaluation for the treatment of elderly patients with previously untreated small lymphocytic lymphoma (SLL) and acute myeloid leukemia. Clinical trials had been under way for the treatment of inflammation and allergic rhinitis; however, no recent development has been reported. Preclinical studies have shown that GS-1101 has desirable pharmaceutical properties. The compound was originally developed by Calistoga Pharmaceuticals, acquired by Gilead on April 1, 2011.
clinical trials, click link
http://clinicaltrials.gov/search/intervention=CAL-101%20OR%20GS-1101%20OR%20Idelalisib
FOSTER CITY, Calif.–(BUSINESS WIRE)–Jan. 13, 2014– Gilead Sciences, Inc. (Nasdaq: GILD) announced today that the U.S. Food and Drug Administration (FDA) has accepted for review the company’s New Drug Application (NDA) for idelalisib, a targeted, oral inhibitor of PI3K delta, for the treatment of refractory indolent non-Hodgkin’s lymphoma (iNHL). FDA has granted a standard review for the iNHL NDA and has set a target review date under the Prescription Drug User Fee Act (PDUFA) of September 11, 2014.
The NDA for iNHL, submitted on September 11, 2013, was supported by a single arm Phase 2 study (Study 101-09) evaluating idelalisib in patients with iNHL that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy. Following Gilead’s NDA submission for iNHL, FDA granted idelalisib a Breakthrough Therapy designation for relapsed chronic lymphocytic leukemia (CLL). The FDA grants Breakthrough Therapy designation to drug candidates that may offer major advances in treatment over existing options. Gilead submitted an NDA for idelalisib for the treatment of CLL on December 6, 2013.
Idelalisib is an investigational, highly selective oral inhibitor of phosphoinositide 3-kinase (PI3K) delta. PI3K delta signaling is critical for the activation, proliferation, survival and trafficking of B lymphocytes and is hyperactive in many B-cell malignancies. Idelalisib is being developed both as a single agent and in combination with approved and investigational therapies.
Gilead’s clinical development program for idelalisib in iNHL includes Study 101-09 in highly refractory patients and two Phase 3 studies of idelalisib in previously treated patients. The development program in CLL includes three Phase 3 studies of idelalisib in previously treated patients. Combination therapy with idelalisib and GS-9973, Gilead’s novel spleen tyrosine kinase (Syk) inhibitor, also is being evaluated in a Phase 2 trial of patients with relapsed or refractory CLL, iNHL and other lymphoid malignancies.
Additional information about clinical studies of idelalisib and Gilead’s other investigational cancer agents can be found at http://www.clinicaltrials.gov. Idelalisib and GS-9973 are investigational products and their safety and efficacy have not been established.
Indolent non-Hodgkin’s lymphoma refers to a group of largely incurable slow-growing lymphomas that run a relapsing course after therapy and can lead ultimately to life-threatening complications such as serious infections and marrow failure. Most iNHL patients are diagnosed at an advanced stage of disease, and median survival from time of initial diagnosis for patients with the most common form of iNHL, follicular lymphoma, is 8 to 10 years. The outlook for refractory iNHL patients is significantly poorer.
Gilead Sciences is a biopharmaceutical company that discovers, develops and commercializes innovative therapeutics in areas of unmet medical need. The company’s mission is to advance the care of patients suffering from life-threatening diseases worldwide. Headquartered in Foster City, California, Gilead has operations in North and South America, Europe and Asia Pacific.
The delta form of PI3K is expressed primarily in blood-cell lineages, including cells that cause or mediate hematologic malignancies, inflammation, autoimmune diseases and allergies. By specifically inhibiting only PI3K delta, a therapeutic effect is exerted without inhibiting PI3K signalling that is critical to the normal function of healthy cells. Extensive studies have shown that inhibition of other PI3K forms can cause significant toxicities, particularly with respect to glucose metabolism, which is essential for normal cell activity.
In 2011, orphan drug designation was assigned to GS-1101 in the U.S. for the treatment of CLL. In 2013, several orphan drug designations were assigned to the compound in the E.U. and U.S.: for the treatment of follicular lymphoma, for the treatment of mucosa-associated lymphoid tissue lymphoma (MALT), for the treatment of nodal marginal zone lymphoma, for the treatment of splenic marginal zone lymphoma, and for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma. Orphan drug designation was also assigned in the U.S. for the treatment of lymphoplasmacytic lymphoma with or without Walenstom’s macroglobulinemia and, in the E.U., for the treatment of Waldenstrom’s macroglobulinemia (lymphoplasmacytic lymphoma).
Later in 2013, some of these orphan drug designations were withdrawn in the E.U.; for the treatment of chronic lymphocytic leukemia / small lymphocytic lymphoma, for the treatment of extranodal marginal-zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma), for the treatment of of nodal marginal-zone lymphoma and for the treatment of splenic marginal-zone lymphoma. In 2013, the FDA granted a breakthrough therapy designation for the treatment of chronic lymphocytic leukemia.
| US8207153 | 6-27-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2012015964 | 1-20-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2011306622 | 12-16-2011 | METHODS OF TREATING HEMATOLOGICAL DISORDERS WITH QUINAZOLINONE COMPOUNDS IN SELECTED SUBJECTS |
| US7932260 | 4-27-2011 | Quinazolinones as Inhibitors of Human Phosphatidylinositol 3-Kinase Delta |
| US2011044942 | 2-25-2011 | METHODS OF TREATMENT FOR SOLID TUMORS |
| US2010256167 | 10-8-2010 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2010202963 | 8-13-2010 | THERAPIES FOR HEMATOLOGIC MALIGNANCIES |
| WO2005113556A1 * | 12 May 2005 | 1 Dec 2005 | Icos Corp | Quinazolinones as inhibitors of human phosphatidylinositol 3-kinase delta |
| WO2005117889A1 * | 12 Nov 2004 | 15 Dec 2005 | Didier Bouscary | Methods for treating and/or preventing aberrant proliferation of hematopoietic |
| WO2005120511A1 * | 4 Jun 2005 | 22 Dec 2005 | Joel S Hayflick | Methods for treating mast cell disorders |
| WO2006089106A2 * | 16 Feb 2006 | 24 Aug 2006 | Icos Corp | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| US20060106038 * | 25 May 2005 | 18 May 2006 | Icos Corporation | Methods for treating and/or preventing aberrant proliferation of hematopoietic cells |
The synthesis of a compound in accordance with formula I is first exemplified using steps A-E below, which provide a synthetic procedure for compound 107, the structure of which is shown below.

(107) is idelalisib
……………….
Synthesis of 2-fluoro-6-nitro-N-phenyl-benzamide (108)
Step A: A solution of 2-fluoro-6- nitrobenzoic acid (100 g, 0.54 mol) and dimethylformamide (5 mL) in dichloromethane (600 mL) was treated dropwise with oxalyl chloride (2 M in dichloromethane, 410 mL, 0.8 mol, 1.5 eq) over 30 min. After stirring 2 h at room temperature, the reaction was concentrated to an orange syrup with some solids present. The syrup was dissolved in dry dioxane (80 mL) and slowly added to a suspension of aniline (49 mL, 0.54 mol, 1 eq) and sodium bicarbonate (90 g, 1.08 mol, 2 eq) in a mixture of dioxane (250 mL) and water (250 mL) at 6 0C. The temperature reached 27°C at the end of the addition. After 30 min, the reaction mixture was treated with water (1.2 L). The precipitate was collected by vacuum filtration, washed with water (300 mL) , air dried in the funnel, and dried in vacuo at 50°C for 24 h to afford an off-white solid product (139 g, 99%). 1H NMR (300 MHz, DMSO-d6) δ 10.82 (s, IH), 8.12 (d, J = 7.7 Hz, IH), 7.91-7.77 (m, 2H), 7.64 (d, J = 7.7 Hz, 2H), 7.38 (t, J = 7.9 Hz, 2H), 7.15 > (t, J = 7.4 Hz, IH), ESI-MS m/z 261 (MH+). The reaction described above and compound 108 are shown below.

………………………..
Synthesis of(S) – [1- (2-fluoro-6-nitro-benzoyl) -phenyl-aminocarbonyl] – propyl-carbamic acid tert-butyl ester (109)
Step B: A suspension of compound 108 (0.5 mol) and dimethylformamide (5 mL) in thionyl chloride (256 mL, 2.5 mol, 5 eq) was stirred at 85°C for 5 hours. The reaction mixture was concentrated in vacuo to a brown syrup. The syrup was dissolved in dichloromethane (200 mL) and was slowly added to a solution of N-BOC-L-2-aminobutyric acid (112 g, 0.55 mol, 1.1 eq) and triethylamine (77 mL, 0.55 mol, 1.1 eq) in dichloromethane (600 mL) at 10 0C. After stirring at room temperature for 3 h, salts were removed by filtration, and the solution was washed with 100 mL of water, saturated sodium bicarbonate, water, 5% citric acid, and saturated sodium chloride. The organic phase was dried with magnesium sulfate and concentrated to a red syrup. The syrup was dissolved in dichloromethane (450 mL) and purified by flash chromatography on a silica gel plug (15 x 22 cm, 4 L dry silica) eluted with hexanes/ethyl acetate (10%, 8 L; 15%, 8 L; 20%, 8 L; 25%, 4 L) to yield the compound 109 as an off-white solid (147 g, 66%). 1H NMR (300 MHz, DMSO-d6) δ 8.13 (d, J = 8.0 Hz, IH), 7.84 (t, J = 8.6 Hz, IH), 7.78- 7.67 (m, IH), 7.65-7.49 (m, 3H), 7.40-7.28 ( m, 2H), 7.19 (d, J = 7.5 Hz, IH), 4.05 (broad s, IH), 1.75- 1.30 (m, 2H), 1.34 (s, 9H), 0.93 (broad s, 3H). ESI- MS m/z 446.3 (MH+) . The reaction described above and compound 109 are shown below.

Synthesis of(S) – [1- (5-fluoro-4-oxo-3-phenyl-3 , 4-dihydro-quinazolin-2- yl) -propyl] -carbamic acid tert-butyl ester (110)
Step C: A solution of compound 109 (125 mmol, 1 eq) in acetic acid (500 mL) was treated with zinc dust (48.4 g, 740 mmol, 6 eq) added in 3 portions, and the reaction mixture was allowed to cool to below 35°C between additions. After stirring for 2 h at ambient temperature, solids were filtered off by vacuum filtration and washed with acetic acid (50 mL) . The filtrate was concentrated in vacuo, dissolved in EtOAc (400 mL) , washed with water (300 mL) , and the water layer was extracted with EtOAc (300 mL) . The combined organic layers were washed with water (200 mL) , sat’d sodium bicarbonate (2 x 200 mL) , sat’d NaCl (100 mL) , dried with MgSO4, and concentrated to a syrup. The syrup was dissolved in toluene (200 mL) and purified by flash chromatography on a silica gel plug (13 x 15 cm, 2 L dry silica) eluted with hexanes/ethyl acetate (10%, 4 L; 15%, 4 L; 17.5%, 8 L; 25%, 4 L) to yield compound 110 as an off-white foamy solid (33.6 g, 69%). 1H NMR (300 MHz, DMSO-d6) δ 7.83 (td, J = 8.2, 5.7 Hz, IH), 7.64-7.48 (m, 5H), 7.39 (broad d, J = 7.6 Hz, IH), 7.30 (dd, J = 8.3 Hz, IH), 7.23 (d, J = 7.6 Hz, IH), 4.02-3.90 (m, IH), 1.76-1.66 (m, IH), 1.62-1.46 (m, IH), 1.33 (s, 9H), 0.63 (t, J= 7.3 Hz, 3H). ESI-MS m/z 398.3 (MH+). The reaction described above and compound 110 are shown below.

…………..
Syn of (S) -2- (1-amino-propyl) -5-fluoro-3-phenyl-3H-quinazolin-4- one (111)
Step D: A solution of compound 110 (85 mmol) in dichloromethane (60 mL) was treated with trifluoroacetic acid (60 mL) . The reaction mixture was stirred for 1 h, concentrated in vacuo, and partitioned between dichloromethane (150 mL) and 10% K2CO3 (sufficient amount to keep the pH greated than 10) . The aqueous layer was extracted with additional dichloromethane (100 raL) , and the combined organic layers were washed with water (50 mli) and brine (50 mL) . After drying with Mg SO4, the solution was concentrated to provide compound 111 as an off-white solid (22 g, 88%) . 1H NMR (300 MHz,
CDCl3) δ 7.73-7.65 (m, IH), 7.62-7.49 (m, 4H), 7.32- 7.22 (m, 2H), 7.13-7.06 (m, IH), 3.42 (dd, J= 7.5, 5.2 Hz, IH), 1.87-1.70 (m, IH), 1.58-1.43 (m, IH), 0.80 (t, J = 7.4 Hz, 3H) . ESI-MS m/z 298.2 (MH+) . The reaction described above and compound 111 are shown below.

………………
syn of (S) -5-fluoro-3-phenyl-2- [1- (9H-purin-6-ylamino) -propyl] – 3H-quinazolin-4-one (107)
Step E: A suspension of compound 111(65.6 mmol, 1 eq) , 6-bromopurine (14.6 g, 73.4 mmol, 1.1 eq) , and DIEA (24.3 mL, 140 mmol, 2 eq) in tert- butanol (40 mL) was stirred for 24 h at 800C. The reaction mixture was concentrated in vacuo and treated with water to yield a solid crude product that was collected by vacuum filtration, washed with water, and air dried. Half of the obtained solid crude product was dissolved in MeOH (600 mL) , concentrated onto silica gel (300 mL dry) , and purified by flash chromatography (7.5 x 36 cm, eluted with 10 L of 4% MeOH/CH2Cl2) to yield a solid product. The solid product was then dissolved in EtOH (250 mL) and concentrated in vacuo to compound 107 idelalisib as a light yellow solid (7.2 g, 50%).
1H NMR (300 MHz, 80 0C, DMSO-d5) δ 12.66 (broad s, IH), 8.11 (s, IH), 8.02 (broad s, IH), 7.81-7.73 (m, IH),7.60-7.42 (m, 6H), 7.25-7.15 (m, 2H), 4.97 (broad s, IH), 2.02-1.73 (m, 2H), 0.79 (t, J= 7.3 Hz, 3H).
ESI-MS m/z 416.2 (MH+).
C, H, N elemental analysis (C22Hi8N7OF-EtOH- 0.4 H2O).
Chiral purity 99.8:0.2 (S:R) using chiral HPLC (4.6 x 250 mm Chiralpak ODH column, 20 °C, 85:15 hexanes : EtOH, 1 rnL/min, sample loaded at a concentration of 1 mg/mL in EtOH) . The reaction described above and compound 107 idelalisib are shown below.

| WO2001030768A1 * | 26 Oct 2000 | 3 May 2001 | Gustave Bergnes | Methods and compositions utilizing quinazolinones |
| WO2001081346A2 * | 24 Apr 2001 | 1 Nov 2001 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2003035075A1 * | 27 Aug 2002 | 1 May 2003 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2005016348A1 * | 13 Aug 2004 | 24 Feb 2005 | Jason Douangpanya | Method of inhibiting immune responses stimulated by an endogenous factor |
| WO2005016349A1 * | 13 Aug 2004 | 24 Feb 2005 | Thomas G Diacovo | Methods of inhibiting leukocyte accumulation |
| WO2005067901A2 * | 7 Jan 2005 | 28 Jul 2005 | Carrie A Northcott | Methods for treating and preventing hypertension and hypertension-related disorders |

TOSEDOSTAT
An aminopeptidase inhibitor with antineoplastic activity.
| CAS No. | 238750-77-1 |
| Chemical Name: | Tosedostat |
| Synonyms: | BB-76163;Chr-2797;tosedostat;CHR2797 (Tosedostat);Tosedostat (CHR2797);α-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]-benzeneaceticacidcyclopentlyester;alpha-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]benzeneacetic acid cyclopentyl ester;Benzeneacetic acid, alpha-(((2R)-2-((1S)-1-hydroxy-2-(hydroxyamino)-2-oxoethyl)-4-methyl-1-oxopentyl)amino)-, cyclopentyl ester, (alphas)- |
| Molecular Formula: | C21H30N2O6 |
| Formula Weight: | 406.47 |
CHR-2797 is an oral, once-daily experimental cancer therapy in phase II clinical development at Chroma Therapeutics for the oral treatment of refractory acute myeloid leukemia in elderly patients. It is also in early clinical development for the treatment of refractory solid tumors alone or in combination with chemotherapy.
No recent development has been reported for phase I/II studies evaluating CHR-2797 as monotherapy in hematologic/blood cancer. A phase I/II clinical trial of the compound in combination with erlotinib for non-small cell lung cancer was terminated in 2010 due to very poor recruitment of patients to the study.
Cell Therapeutics is also conducting phase II clinical trials of the compound for the treatment of myelodysplasia and acute myeloid leukemia.
CHR- 2797 is an inhibitor of aminopeptidases and has demonstrated strong preclinical efficacy as monotherapy in addition to demonstrating strong synergy with a number of leading cancer therapies in a range of cancer cells. It was originally licensed from Vernalis, where it was being evaluated for its potential in treating multiple sclerosis; however development in this indication has been discontinued.
In 2008, orphan drug designation was assigned to CHR-2797 in the U.S. for the treatment of acute myeloid leukemia. In 2011, the compound was licensed to Cell Therapeutics by Chroma Therapeutics in Central America, North America and South America for exclusive marketing and codevelopment for the oral treatment of blood-related cancers and other cancers.
In corporate news, biopharmaceutical company Cell Therapeutics, Inc. (CTIC) was up more than 6% and near 52 week highs after saying Thursday that the U.S. FDA has removed the partial clinical hold on tosedostat and all studies underway have been allowed to continue. Tosedostat is under development for the treatment of blood-related cancers. It is currently being studied in Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia and high-risk myelodysplastic syndromes.

Tosedostat is a proprietary orally bioavailable inhibitor of the M1 family of aminopeptidases with potential antineoplastic activity.
Tosedostat is converted intracellularly into a poorly membrane-permeable active metabolite (CHR-79888) which inhibits the M1 family of aminopeptidases, particularly puromycin-sensitive aminopeptidase (PuSA), and leukotriene A4 (LTA4) hydrolase; inhibition of these aminopeptidases in tumor cells may result in amino acid deprivation, inhibition of protein synthesis due to a decrease in the intracellular free amino acid pool, an increase in the level of the proapoptotic protein Noxa, and cell death.
Noxa is a member of the BH3 (Bcl-2 homology 3)-only subgroup of the proapoptotic Bcl-2 (B-cell CLL/lymphoma 2) protein family
Cell Therapeutics announced that it has received notification from the U.S. Food and Drug Administration (FDA) that the partial clinical hold on tosedostat (IND 075503) has been removed and all studies underway may continue. Tosedostat is a first-in-class selective inhibitor of aminopeptidases, which are required by tumor cells to provide amino acids necessary for growth and tumor cell survival, and is under development for the treatment of blood-related cancers.
Tosedostat is currently being studied in the United States and European Union in investigator-sponsored and cooperative group-sponsored Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia (AML) and high-risk myelodysplastic syndromes (MDS).
“We are pleased that the FDA has responded favorably to the tosedostat clinical trial data provided and removed the partial clinical hold to allow further development of tosedostat in ongoing and future studies,” said John Pagel, MD, PhD, Associate Member, Clinical Research Division, Fred Hutchinson Cancer Research Center; Associate Professor, Medical Oncology Division, University of Washington School of Medicine; and Principal Investigator in the tosedostat first-line AML/MDS trial.
Recently, WO 93/20047 disclosed a class of hydroxamic acid based MMP inhibitors which also are active in inhibiting TNF production.
As mentioned above, MMP inhibitors have been proposed with hydroxamic acid or carboxylic acid zinc binding groups. The following patent publications disclose hydroxamic acid-based MMP inhibitors:
US 4599361 (Searle) EP-A-0236872 (Roche) EP-A-0274453 (Bellon) WO 90/05716 (British Bio-technology) WO 90/05719 (British Bio-technology) WO 91/02716 (British Bio-technology) EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) EP-A-0497192 (Roche) WO 92/13831 (British Bio-technology) WO 92/17460 (SmithKline Beecham) WO 92/22523 – (Research Corporation Technologies) WO 93/09090 (Yamanouchi) WO 93/09097 (Sankyo) WO 93/20047 (British Bio-technology) WO 93/24449 (Celltech) WO 93/24475 (Celltech) EP-A-0574758 (Roche) The following patent publications disclose carboxylic acid-based MMP inhibitors:
EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) WO 93/24449 (Celltech) WO 93/24475 (Celltech)
|
|
|
TOSEDOSTAT
| WO1996033166A1 * | 17 Apr 1996 | 24 Oct 1996 | Du Pont Merck Pharma | Hydroxamic and carboxylic acids as metalloprotease inhibitors |
| WO1998011063A1 * | 8 Sep 1997 | 19 Mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| GB2268934A * | Title not available |
| US5652262 * | 14 mar 1994 | 29 lug 1997 | British Biotech Pharmaceutical, Ltd. | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| US5821262 * | 4 ott 1994 | 13 ott 1998 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as inhibitors of cytokine production |
| US5861436 * | 29 apr 1997 | 19 gen 1999 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| EP0423943A2 | 19 set 1990 | 24 apr 1991 | Beecham Group p.l.c. | Use of collagenase inhibitors in the treatment of demyelinating diseases, in particular multiple sclerosis |
| JPH03157372A | Titolo non disponibile | |||
| WO1997049674A1 | 20 giu 1997 | 31 dic 1997 | Francesca Abrate | Matrix metalloproteinase inhibitors |
| WO1998011063A1 | 8 set 1997 | 19 mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| WO1999040910A1 | 27 gen 1999 | 19 ago 1999 | Andrew Paul Ayscough | Anti-inflammatory agents |
| WO1999044602A1 | 5 mar 1999 | 10 set 1999 | British Biotech Pharm | Inflammatory cell inhibitors |
| WO1999046241A1 | 12 mar 1998 | 16 set 1999 | British Biotech Pharm | Cytostatic agents |
| WO2000044373A1 * | Jan 27, 2000 | Aug 3, 2000 | Raymond Paul Beckett | Antibacterial hydroxamic acid derivatives |
| US6545051 | Jan 27, 2000 | Apr 8, 2003 | British Biotech Pharmaceuticals, Ltd. | Antibacterial hydroxamic acid derivatives |
Drugs Fut 2009, 34(2): 115
PLoS One (2013), 8(2), e57641.
WO 1999046241
WO 1995019956
WO 1998011063
US 6462023
US 20100260674
WO 2000044373
WO 9940910
NMR
http://file.selleckchem.com/downloads/nmr/S152202-CHR-2797-NMR-Selleck.pdf
Anti-Metastatic and Anti-Invasive Agents Compounds which have the property of inhibiting the action of the metalioproteinase enzymes involved in connective tissue breakdown and remodelling, such as fibroblast collagenase (Type 1 ), PMN-collagenase, 72 kDa-gelatinase, 92 kDa- gelatinase, stromelysin, stromelysin-2 and PUMP-1 (known as “matrix metalloproteinases”, and herein referred to as MMPs) have been proposed and are being tested in the clinic for the treatment of solid tumours. Cancer cells are particularly adept at utilising the MMPs to achieve rapid remodelling of the extracellular matrix, thereby providing space for tumour expansion and permitting metastasis. MMP inhibitors should minimise these processes and thus slow or prevent cancer progression.
In view of the rapid emergence of multidrug-resistant bacteria, the development of antibacterial agents with novel modes of action that are effective against the growing number of resistant bacteria, particularly the vancomycin resistant enterococci and β-lactam antibiotic-resistant bacteria, such as methicillin-resistant Staphylocccus aureus, is of utmost importance.
The natural antibiotic actinonin (see for example J. C. S Perkin I, 1975, 819) is a hydroxamic acid derivative of Structure (A):

In ddition to actinonin, various structural analogues of actinonin have also been shown to have antibacterial activity (see for example Broughton et al. (Devlin et al. Journal of the Chemical Society. Perkin Transactions 1 (9):830-841, 1975; Broughton et al. Journal of the Chemical Society. Perkin Transactions 1 (9):857-860, 1975).
The matlystatin group of compounds, share a number of structural similarities with actinonin. Both are peptidic molecules with functional hydroxamic acid metal binding groups (Ogita et al., J. Antibiotics. 45(11):1723-1732; Tanzawa et al., J. Antibiotics. 45(11):1733-1737; Haruyama et al., J. Antibiotics. 47(12):1473-1480; Tamaki et al., J. Antibiotics. 47(12):1481-1492).
………………………………………………………….
EXAMPLE 44 2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl-pentanoylamine]-2-phenyl-ethanoic acid cyclopentyl ester

The above compound was prepared using procedures similar to those described in example 8 using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1H, s), 5.2-5.14 (1H, m), 4.02 (1H, d, J=6.9 Hz), 2.94-2.85 (1H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1H, m) and 0.86 (6H, dd, J=6.5, 11 5 Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1, 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; 8 (MeOD), 7.33-7.19 (5H, m), 5.3 (1H, s), 5.11-5.06 (1H, m), 3.81 (1H, d, J=7.3 Hz), 2.83-2.74 (lH, m), 1.83-1.45 (10H, bm), 1.12-1.03 (lH, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3 Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2
Example 1
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

HO Ξ CONHOH
Prepared using procedures similar to those described in Preparative Example A using phenylglycine cyclopentyl ester.
Diastereoisomer A
Η-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d,
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, 14 dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2,
58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
Η-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d, J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.
tosedostat
http://www.google.it/patents/US6545051

42
| WO98/11063 | WO99/46241 ex 1b | WO 98/11063 analogy ex 8 |

43
| WO98/11063 | WO99/46241 ex 1a | WO 98/11063 analogy ex 8 |
……………………………………………………………………
entry 65 in http://www.google.com/patents/WO2000044373A1
……………………………………………………………………………………………………….
http://www.google.com/patents/WO1999044602A1
Example 43
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester
TC

HO Ξ CONHOH
Prepared using procedures similar to those described in example 8 of WO 98/11063, using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d, 34
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d,
J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and
0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3,
129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.
……………………………..
3R-isobutyl-4S-methoxy-dihydrofuran-2,5-dione (WO 97/02239)
…………………………………………………………………………..
2(S)-Amino(phenyl)ethanoic acid cyclopentyl ester

…………………………………………………………………..
2(R)-[2,2-Dimethyl-5-oxo-1,3-dioxolan-4(S)-yl]-4-methylpentanoic acid pentafluorophenyl ester

…………………………………………………………..
intermediates
238750-91-9
α-amino-, cyclopentyl ester Benzeneacetic acid,
……………….
cas 240489-34-3
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

…………………..
will be updated very soon… keep watching


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family


TEMSIROLIMUS
Proline CCI-779
Torisel, NCGC00167518-01
LAUNCHED 2007
PFIZER
Inhibits mTOR protein
For the treatment of renal cell carcinoma (RCC). Also investigated for use/treatment in breast cancer, lymphoma (unspecified), rheumatoid arthritis, and multiple myeloma.
An ester analog of rapamycin. Temsirolimus binds to and inhibits the mammalian target of rapamycin (mTOR), resulting in decreased expression of mRNAs necessary for cell cycle progression and arresting cells in the G1 phase of the cell cycle. mTOR is a serine/threonine kinase which plays a role in the PI3K/AKT pathway that is upregulated in some tumors
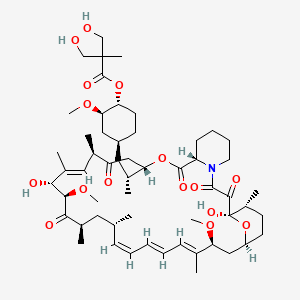
(1R,2R,4S)-4-{(2R)-2-[(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-1,5,11,28,29-pentaoxo-1,4,5,6,9,10,11,12,13,14,21,22,23,24,25,26,27,28,29,31,32,33,34,34a-tetracosahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontin-3-yl]propyl}-2-methoxycyclohexyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate
cas 162635-04-3
Temsirolimus is an intravenous drug for the treatment of renal cell carcinoma (RCC), developed by Wyeth Pharmaceuticals and approved by the FDA in late May 2007, and was also approved by the European Medicines Agency (EMEA) on November 2007. It is a derivative of sirolimus and is sold as Torisel.
Molecular Formula: C56H87NO16
Molecular Weight: 1030.28708
Temsirolimus (CCI-779) is an intravenous drug for the treatment of renal cell carcinoma (RCC), developed by WyethPharmaceuticals and approved by the U.S. Food and Drug Administration (FDA) in late May 2007, and was also approved by the European Medicines Agency (EMEA) on November 2007. It is a derivative of sirolimus and is sold as Torisel.
Temsirolimus is a specific inhibitor of mTOR and interferes with the synthesis of proteins that regulate proliferation, growth, and survival of tumor cells. Treatment with temsirolimus leads to cell cycle arrest in the G1 phase, and also inhibits tumor angiogenesis by reducing synthesis of VEGF.
The product had been under development by Wyeth Pharmaceutical for the treatment of pancreas cancer and metastatic breast cancer, multiple sclerosis (MS) and rheumatoid arthritis (RA); however, no recent development for these indications has been reported. Pfizer had been developing the compound for the treatment of sarcoma.
Temsirolimus holds orphan drug designation in both the U.S. and the E.U. for the treatment of renal cell carcinoma. Orphan drug designation was received in the U.S. in 2006 for the treatment of mantle-cell lymphoma.
mTOR (mammalian target of rapamycin) is a kinase enzyme inside the cell that collects and interprets the numerous and varied growth and survival signals received by tumor cells. When the kinase activity of mTOR is activated, its downstream effectors, the synthesis of cell cycle proteins such as cyclin D and hypoxia-inducible factor-1a (HIF-1a) are increased. HIF-1a then stimulates VEGF. Whether or not mTOR kinase is activated, determines whether the tumor cell produces key proteins needed for proliferation, growth, survival, and angiogenesis.
mTOR is activated in tumor cells by various mechanisms including growth factor surface receptor tyrosine kinases, oncogenes, and loss of tumor suppressor genes. These activating factors are known to be important for malignant transformation and progression.mTOR is particularly important in the biology of renal cancer (RCC) owing to its function in regulating HIF-1a levels. Mutation or loss of the von Hippel Lindau tumor-suppressor gene is common in RCC and is manifested by reduced degradation of HIF-1a. In RCC tumors, activated mTOR further exacerbates accumulation of HIF-1a by increasing synthesis of this transcription factor and its angiogenic target gene products.
Rapamycin 42-ester with 3-hydroxy-2-(hydroxymethyl)-2-methylpropionic acid (CCl-779) is an ester of rapamycin which has demonstrated significant inhibitory effects on tumor growth in both in vitro and in vivo models.
CCl-779 may delay the time to progression of tumors or time to tumor recurrence which is more typical of cytostatic rather than cytotoxic agents. CCl-779 is considered to have a mechanism of action that is similar to that of sirolimus. CCl-779 binds to and forms a complex with the cytoplasmic protein FKBP, which inhibits an enzyme, mTOR (mammalian target of rapamycin, also known as FKBP12-rapamycin associated protein [FRAP]). Inhibition of mTOR’s kinase activity inhibits a variety of signal transduction pathways, including cytokine-stimulated cell proliferation, translation of mRNAs for several key proteins that regulate the G1 phase of the cell cycle, and IL-2-induced transcription, leading to inhibition of progression of the cell cycle from G1 to S. The mechanism of action of CCl-779 that results in the G1-S phase block is novel for an anticancer drug.
The preparation and use of hydroxyesters of rapamycin, including CCl-779, are disclosed in U.S. Pat. No. 5,362,718. A regiospecific synthesis of CCl-779 is described in U.S. Pat. No. 6,277,983.
CCl-779 can be synthesized by the non-regioselective acylation of rapamycin, as described in U.S. Pat. No. 5,362,718. The synthesis, however, is complicated by mixtures of the desired 42-ester, with 31-esterified rapamycin, as well as 31, 42-diesterified rapamycin and unreacted rapamycin.
CCl-779 can also be prepared by the acylation of the 31-silyl ether of rapamycin with a ketal of bis-(hydroxymethyl)propionic acid, followed by removal of the 31-silyl ether and ketal protecting group from the bis-(hydroxymethyl) propionic acid, as described in U.S. Pat. No. 6,277,983. However, the crude 42-monoester produced from this regioselective synthesis requires further purification by column chromatography to remove residual amounts of diester by-products and unreacted rapamycin starting material.
Temsirolimus (CCI-779), an mTOR kinase Inhibitor of formula (I) is an antineoplastic agent indicated for the treatment of advanced renal cell carcinoma.Temsirolimus is a Rapamycin 42 ester with [3-hydroxy-2-(hydroxymethyl)-2-methylpropanoic acid and was first disclosed by Skotnicki et al in US Patent No. 5,362,718.

Several processes for the preparation of Temsirolimus have been reported in the literature such as those described in US 5,362,718; US 6,277,983 and US 7, 153,957.
US Patent No 5,362,718 discloses a process for the preparation of different rapamycin 42 esters including Temsirolimus as per the scheme given below (Scheme-I).

Scheme-I: Synthesis of Temsirolimus as disclosed in US Patent No. 5,362,718
The process is non-regioselective and hence results in 31-estehfied rapamycin, 31 , 42 diesterified rapamycin and unreacted rapamycin along with the desired rapamycin-42 ester.
US Patent No. 6,277,983 reports a process for the preparation of Temsirolimus by using 31 , 42 bis silyl intermediates as per the scheme shown below (Scheme-ll).

Scheme-ll: Synthesis of Temsirolimus as disclosed in US Patent No. 6,277,983 US Patent No. 7, 153,957 reports a process for the preparation of Temsirolimusby using boronate intermediate as per the scheme shown below (Scheme-Ill).

Scheme-Ill: Synthesis of Temsirolimus as disclosed in US Patent No. 7, 153,957
 Temsirolimus synthesis by Sirolimus (sirolimus, also known as rapamycin Rapamycin) esterification from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites.Sirolimus 31 and 42 have two alcohol, but 42 slightly smaller steric hindrance. Protected with trimethylsilyl 31 and 42 of the secondary alcohol to give intermediate 1 , 42 for selective removal of sulfuric acid trimethylsilyl obtain 2 , 2 with an acid chloride 3 and a carboxylic acid4 formed by esterification of acid anhydride reaction of 5 under acidic conditions after removal of the 31-bit trimethylsilyl get 6 , 6 with an alcohol 7 boronate protection is removed Temsirolimus. This synthetic route as 31 and 42 to protect the hydroxyl group appear more cumbersome. Later, the development of an enzyme-catalyzed synthesis route (OL2005, 3945). Lipase PS “Amano” (Burkholderia cepacia) of the catalyst, sirolimus and ester 8 reaction of compound 9 .Good selectivity for the enzyme, so that the esterification reaction occurs only in 42, and slightly larger steric hindrance is no response 31. 9 with sulfuric acid for removal of protection is acetonide Temsirolimus.
Temsirolimus synthesis by Sirolimus (sirolimus, also known as rapamycin Rapamycin) esterification from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites.Sirolimus 31 and 42 have two alcohol, but 42 slightly smaller steric hindrance. Protected with trimethylsilyl 31 and 42 of the secondary alcohol to give intermediate 1 , 42 for selective removal of sulfuric acid trimethylsilyl obtain 2 , 2 with an acid chloride 3 and a carboxylic acid4 formed by esterification of acid anhydride reaction of 5 under acidic conditions after removal of the 31-bit trimethylsilyl get 6 , 6 with an alcohol 7 boronate protection is removed Temsirolimus. This synthetic route as 31 and 42 to protect the hydroxyl group appear more cumbersome. Later, the development of an enzyme-catalyzed synthesis route (OL2005, 3945). Lipase PS “Amano” (Burkholderia cepacia) of the catalyst, sirolimus and ester 8 reaction of compound 9 .Good selectivity for the enzyme, so that the esterification reaction occurs only in 42, and slightly larger steric hindrance is no response 31. 9 with sulfuric acid for removal of protection is acetonide Temsirolimus.
………………………………………………..
SYNTHESIS
https://www.google.co.in/patents/EP0763039A1
Example 11
Rapamycin 42-ester with 2.2-bis-(hydroxymethyl)propionic acid
A solution of the product of Example 10 (2.8 g, 2.65 mmol) in 50 mL THF and
25 mL IN HCl was stirred at room temperature for 4 h. The mixture was diluted with water and extracted three times with EtOAc. The combined organic phases were washed with saturated NaHCO3 solution, saturated NaCl solution, dried over MgSO4, filtered and evaporated to a yellow oily solid. Purification by flash chromatography (3X with EtOAc) afforded the title compound (1.6 g, 59 %).
(-)FAB-MS mlz 1029.6 (M-), 590.4 (southern fragment), 437.3 (northern fragment). !H NMR (400 MHz, d-6 DMSO) δ 4.5 (m, 1 H, C(42)H), 3.45 (s, 4 H), 1.04 (s, 3 H).
*3C NMR (100.6 MHz, d-6 DMSO) δ 174.2, 63.7, 63.6, 49.9, 16.8.
Example 10 Rapamycin 42-ester with 2.2.5-trimethyl.1.3_dioxane-5-carboxyric acid
To a solution of the 2,2-bis(hydroxymethyl)propionic acid isopropylidene ketal (1.041 g, 5.98 mmol) (prepared according to the procedure of Bruice, J. Am. Chem. Soc. 89: 3568 (1967)) and triethylamine (0.83 mL, 5.98 mmol) in 20 mL anhydrous THF at 0 °C under nitrogen was added 2, 4, 6-trichlorobenzoyl chloride (0.93 mL, 5.98 mmol) and the resultant white suspension was stirred 5 h at room temperature. The precipitate was removed by vacuum filtration, rinsing the flask and filter cake with an additional 10 mL dry THF. The filtrate was concentrated by rotary evaporation to a white solid. The residue was dissolved in 20 mL dry benzene, then rapamycin (5.47 g, 5.98 mmol) and DMAP (0.731 g, 5.98 mmol) were added. After stirring overnight at room temperature, the mixture was diluted with EtOAc, washed with H2O and saturated NaCl (aq), dried over MgSO4, filtered and evaporated to a yellow oil. Flash chromatography (5X with 60% EtOAc-hexane) afforded the title compound (2.2 g, 34 %) as a white solid.
(-)FAB-MS mlz 1069.5 (M-), 590.3 (southern fragment), 477.2 (northern fragment). –■H NMR (400 MHz, d-6 DMSO) δ 4.57 (m, 1 H, C(42)H), 4.02 (d, 2 H), 3.60 (d, 2 H), 1.34 (s, 3 H), 1.24 (s, 3 H), 1.06 (s, 3 H). 1 C NMR (100.6 MHz, d-6 DMSO) δ 173.2, 99.0, 65.0, 22.2, 18.1.
…………………………………………..
SYNTHESIS
https://www.google.co.in/patents/US7153957
This scheme



Preparation of 5-Methyl-2-phenyl-1,3,2-dioxaborinane-5-carboxylic acid, [A]
To a suspension of 2,2-bis(hydroxymethyl)propionic acid (131 g, 0.98 mole) in tetrahydrofuran (500 ml) was added a solution of phenylboronic acid (122 g, 1.0 mole) in tetrahydrofuran (500 ml). The mixture was stirred for 3 h and toluene (1.0 L) was added. Water was removed by azeotropic distillation with toluene. Heptanes (500 ml) was added to the precipitated product, heated to reflux and cooled. The mixture was filtered and washed with heptanes (2×300 ml). The solids were dried under vacuum at 70–75° C. until constant weight to give 94% yield. 1H NMR: δ (DMSO-d6) 7.65 (d, 2H, Ar), 7.40 (m, 3H, Ar), 4.35 (d, 2H, CH2), 3.92 (d, 2H, CH2), 1.17 (s, 3H, CH3)
Preparation of Rapamycin 42-ester with 5-methyl-2-phenyl-1,3,2-dioxaborinane-5-carboxylic acid, [B]
As described in U.S. Pat. No. 6,277,983 (2001) a 3 L flask was charged with rapamycin (100 g, 0.104 mole) and dissolved in ethyl acetate (1.50 L). The solution was cooled to 5–10° C. Imidazole (30 g, 0.44 moles, 4.23 eq.) was added and dissolved. Under nitrogen protection, trimethylsilyl chloride (44 g, 0.405 mole, 4.0 eq.) was added over 30–40 min while maintaining the temperature at 0–5° C. during the addition. The mixture was held for a minimum of 0.5 h. The reaction was monitored by TLC (30:70 acetone:heptane eluent). The reaction was complete when all of the rapamycin was consumed.
Two to three drops of the reaction mixture were removed and retained as a 31,42-bis(trimethylsilyl) rapamycin reference standard. 0.5 N Sulfuric acid (300 mL) was added to the 3 L flask over 0.5 h maintaining the temperature 0–5° C. The mixture was stirred vigorously and held for 5 h. The reaction was monitored by thin layer chromatography (TLC) (30:70 acetone:heptane eluent). The reaction was complete when essentially no 31,42-bis-(trimethylsilyl) rapamycin was present. The layers were separated and the lower aqueous layer was back extracted with ethyl acetate (500 mL). The combined organic layers were washed with saturated brine (500 mL) and saturated sodium bicarbonate (2×200 mL) until pH 8 was obtained. The organic layer was washed with water (2×500 mL) and brine (500 ml) until pH 6 to 7 was obtained. The solution was dried over magnesium sulfate (100 g) for 30 min, filtered into a 2 L flask and concentrated to a volume of 135 ml. Ethyl acetate (500 ml) was added and concentrated to a volume of 135 ml. The water chase was repeated once more with ethyl acetate (500 ml). Methylene chloride (300 ml) was added and the solution held until needed in the next step.
A 3 L flask equipped with mechanical stirrer was charged with compound [A] (75 g, 0.341 mole) in methylene chloride (400 mL). Diisopropylethylamine (66.1 g, 0.51 mole) was added dropwise over 20 mins and rinsed with methylene chloride (25 mL). 2,4,6-Trichlorobenzoyl chloride (80 g, 0.328 mole) was added and rinsed with methylene chloride (25 mL). The mixture was held at 0–5° C. for 4 h, and cooled to −10±5° C.
The solution of 31-trimethylsilyl rapamycin was added to the 3 L flask containing the mixed anhydride, and rinsed with methylene chloride (25 mL). A solution of dimethylamino pyridine (48.5 g, 0.397 mole) in methylene chloride (150 mL) was prepared, added over 1.5 h, maintaining the temperature <−8° C., and rinsed with methylene chloride (25 mL). The mixture was held for 12 h at −11 to −5° C. The reaction mixture was quenched with 1 N sulfuric acid (600 ml) keeping the temperature <10° C. The mixture was stirred and held for 30 mins. The pH of the upper aqueous layer was ≦2. The layers were separated, and the lower organic layers washed with brine (450 ml), saturated sodium bicarbonate (500 mL) until pH ≧8. The organic layer was washed with water (450 ml) until pH 6–7 was obtained. The solution was concentrated, acetone (250 ml) added and concentrated. This was repeated with another portion of acetone (250 ml) and concentrated.
The solution was diluted with acetone. 0.5 N Sulfuric acid (500 ml) was added dropwise over 30 mins keeping the pot temperature 0–5° C. The mixture was held for a minimum of 5 h, during which time, the product precipitated out of solution. Aqueous sodium bicarbonate (30 g in 375 ml water) was added dropwise over 30 minutes keeping the pot temperature 0 to 5° C.; the mixture was held for a minimum of 30 minutes. Acetic acid (25 ml) was added until pH was 5–6 keeping the pot temperature <10° C. The mixture was warmed to room temperature and held for 16 h. The solid product was filtered and washed with water (2×100 ml) followed by 1:1 acetone:water (2×100 ml). The cake was purified in acetone (375 ml) to give 65 g (58% overall from rapamycin) of product [B]. LC/MS: using an electrospray interface in the positive ion mode afforded the molecular ion [M+Na]=1138.5 atomic mass units (amu).
Preparation of Rapamycin 42-ester with 2,2-bis(hydroxymethyl)-propionic acid, [C]
Compound [B] (200 g, 0.179 mole), was dissolved in tetrahydrofuran (600 ml), 2-methyl-2,4-pentanediol (42.3 g, 0.358 mole, 2.0 eq.) was added and the mixture stirred for a minimum of 3 h. The reaction mixture was concentrated to a foam. Diethyl ether (1.0 L) was added and the mixture stirred for 2 h. Heptanes (1.0 L) was added dropwise over 1 h and the mixture stirred for 2 h. The mixture was filtered and the solid product washed with heptanes (500 ml). The solids were re-dissolved in acetone (400 ml), re-treated with 2-methyl-2,4-pentanediol (21.1 g, 0.179 mole, 1 eq.) in acetone (200 ml), clarified through a 0.2 micron cartridge filter, and rinsed with acetone (200 ml). The solution was concentrated to a foam, diethyl ether (1.0 L), pre-filtered through a 0.2 micron cartridge filter, was added and the mixture stirred for 2 h. The mixture was co-precipitated by adding pre-filtered heptanes (1.0 L). The precipitated solids were filtered and washed with ether:heptane (2×500 ml). The solids were dried (55 to 60° C., 10 mm Hg, minimum 24 h) to give 159 g (86%) of product [C]. LC/MS: using APCl in the positive ion mode afforded the molecular ion [M+NH4]=1047.0 amu. The 1H NMR of the product (CCl-779) was identical to the product described in example 11 of U.S. Pat. No. 5,362,718 (1994).
…………………………………
Synthesis
http://www.google.com/patents/WO2005100366A1
Example 1 – Synthesis of Proline CCI-779

This example describes a method for the synthesis of the proline analog of CCI- 779, which is illustrated in the scheme provided above.
A.
Preparation of 31, 42-Bis (trimethylsilyl) proline rapamycin (Compound B)
A 3 -neck 50 mL flask was charged with proline rapamycin (compound A in the scheme) (1.47 g, 1.63 mmol), imidazole (0.45 g, 6.6 mmol, 4 eq.) and ethyl acetate (22.5 mL). The magnetically stirred mixture became cloudy. The mixture was cooled to 0-5°C. Under nitrogen protection, trimethylsilyl chloride (0.62 g, 5.7 mmol, 3.5 eq.) was added over 0.5 h via syringe while maintaining the temperature at 0-5°C during the addition. The syringe was rinsed with 2.5 ml ethyl acetate and the mixture held for 0.75 hours (0.75 h), whereupon a white precipitate was formed. The reaction was monitored by thin layer chromatography (TLC) (30:70 acetone :heptane eluent). The TLC sample was prepared by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 10 drops ethyl acetate. The mixture was shaken and allowed to settle. The upper organic layer was spotted against the starting material (proline rapamycin). The reaction was complete when no more starting material was present.
B.
Preparation of 31 -trimethylsilyl proline rapamycin, Compound E
When the above reaction was complete, 2-3 drops of the reaction mixture was removed and retained for the following step as the 31,42-bis(trimethylsilyl) proline rapamycin reference standard. To the 50 ml flask was added 0.5 N sulfuric acid (4.5 mL) over 0.5 h maintaining the temperature at 0-5 °C. The mixture became less cloudy. The mixture was held for 2.5 h and was monitored by thin layer chromatography (TLC, 30:70 acetone:heptane eluent). The TLC sample was prepared by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 10 drops ethyl acetate. The reaction aliquot was shaken and allowed to settle. The upper organic layer was spotted against the 31 ,42-bis(trimethylsilyl) proline rapamycin reference. The reaction was complete when essentially no 31,42-bis(trimethylsilyl) proline rapamycin was present. Ethyl acetate (5 mL) was added and the layers separated. The lower aqueous layer is extracted with ethyl acetate (7.5 mL). The combined organic layers were washed with brine (7.5 mL), by washing with saturated sodium bicarbonate (6 mL) followed by washing water (3 x 7.5 mL), in that order. The pH of the last water wash was 6-7. The organic layer was washed again with brine (7.5 mL) and dried over sodium sulfate (4 g) for 20 min. The mixture was filtered into a 250 mL flask and concentrated to dryness.
The solid was dried at room temperature under high vacuum (10 mmHg or less) for 20 h.
Weight = 1.51 g of an off-white foam.
C.
Preparation of Intermediate, Compound F:
A 3 -neck 100 mL flask equipped with mechanical stirrer was charged with
2,2,5-trimethyl[l,3-dioxane]-5-carboxylic acid, Compound C (0.63 g, 3.6 mmol) in methylene chloride (7.5 mL). Dusopropylethylamine (0.77 g, 5.9 mmol) was added, followed by a rinse with methylene chloride (1 mL). 2,4,6-Trichlorobenzoyl chloride (0.85 g, 3.5 mmol) was added, followed by a rinse with methylene chloride (1.5 mL).
The mixture was held at room temperature for 4.5 h. The solution was cooled to -12 ±
2°C. 31 -Trimethylsilyl proline rapamycin, compound E, (1.51 g) in methylene chloride (8 mL) was dissolved and added to the 100 mL flask. Methylene chloride (2 mL) was added as a rinse. A solution of dimethylamino pyridine (DMAP) (0.77 g, 6.8 mmol) in methylene chloride (3 mL) was prepared and added to the 100 mL flask over
2.5 h maintaining the temperature -12 ± 2 °C. Methylene chloride (1 mL) was added as a rinse. The mixture was held for 16 h and was monitored by HPLC by quenching 3-4 drops of reaction mixture into 0.25 mL water and 0.2 mL ethyl acetate. The HPLC sample was prepared by withdrawing 2 drops of the upper organic layer, blowdrying the sample under nitrogen in an HPLC vial and redissolving using the mobile phase.
HPLC column : CSC Hypersil ODS / BDS 5 μm.
Mobile phase : 68.5 % dioxane:water + 0.01M KH2P04
Wavelength : λ = 280 nm Flow rate : 1 mL / min
Time : 60 min
Retention times : Compound E ~14.0-14.5 min Compound F -33.4-33.8 min
The reaction was complete when < 0.5% of starting material was present. The reaction mixture was quenched with water (6 mL). Methylene chloride (10 mL) was added and the layers separated. The aqueous layer was extracted with methylene chloride (10 mL). The combined organic layers were washed with 0.5 N sulfuric acid (12 mL), brine (10 mL), saturated sodium bicarbonate (6 mL), and water (3 x 10 mL) in that order. The pH of the last water wash was 6-7. The clear yellow solution was concentrated to a foam. The solid was dried at room temperature under high vacuum (10 mmHg or less) for 24 h. Weight = 1.88 g of a yellow foam.
D.
Preparation of crude proline CCI-779
A 1-neck 50 mL flask equipped with mechanical stirrer was charged with Compound F in THF (18.8 mL, 10 vols) and then cooled to 0 – 5 °C (or about -2.5°C). 2 N sulfuric acid (9.4 mL, 5 vols) was added over 2.5 h. After complete addition, the mixture was warmed to 2.5 °C and then held for 45 h. The reaction was monitored by HPLC by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 0.25 mL ethyl acetate. The HPLC sample was prepared by withdrawing 5 drops of the upper organic layer, blow drying the sample under nitrogen in an HPLC vial and redissolving using the mobile phase.
HPLC column : CSC Hypersil ODS / BDS 5 μm.
Mobile phase : 68.5 % dioxane:water + 0.01M KH2P04 Wavelength : λ= 280 nm Flow rate : 1 mL / min Time : 60 min Retention times Compound F ~33.4-33.8 min Desilylated Compound F ~10.5-11.5 min (intermediate) Proline CCI-779 -5.0-5.5 min The desilylated intermediate of compound F was formed first. The reaction was complete when < 0.5% of the silylated analog remained. Ethyl acetate (27 mL) and brine (7.5 mL) was added and the layers separated. The aqueous layer was extracted with ethyl acetate (10 mL). The combined organic layers were washed with brine (10 mL), saturated sodium bicarbonate (7.5 mL), and water (3 x 7.5 mL) in that order. The pH of the last water wash was 6-7. The mixture was dried over sodium sulfate (5 g) for 30 min, filtered into a 250 L flask and concentrated to dryness. Weight = 1.58 g of a yellow foam.
E.
Chromatographic purification of crude proline CCI-779
A silica gel column (31.6 g, 60 A, 200-400 mesh) (22 cm length x 2.5 cm diameter) was prepared and conditioned with 15:85 acetone:HPLC grade hexane (1 L). The yellow crude proline CCI-779 (1.58 g) in acetone (1.58 mL) was prepared and chromatographed. The column was eluted with the remaining 15:85 acetone :hexane mixture followed by 25:75 acetone:hexane (4 L). The positive fractions were combined and concentrated to dryness. The resulting foam was dried at 35 °C, high vacuum (i.e., 10 mmHg or less) for 24 h. Weight = 1.12 g of a light yellow foam.
F.
Ether treatment of proline CCI-779
A 1 -neck 50 mL flask was charged with proline CCI-779 ( 1.12 g) and dissolved in ether (1.5 mL). The mixture was held for 2 h. The ether was stripped to give a foam. The foam was dried at 35 °C, under high vacuum (10 mmHg or less) for 12 h then at room temperature overnight (12 h). Weight = 1.09 g.
*H NMR (500 and 600 MHz, DMSO-d6) δ 5.45 (H-l), 6.12 (H-2), 6.27 (H-3), 6.41 (H-4), 6.20 (H-5), 3.66 (H-7), 1.14 and 1.86 (H-8), 4.02 (H-9), 1.19 and 1.81 (H-10), 1.52 (H-11), 2.03 (H-12), 3.23 and 3.54 (H-18), 1.76 (H-19), 2.20 and 1.89 (H-21), 4.22 (H-22), 4.87 (H-25), 2.28 and 2.70 (H-26), 3.22 (H-28), 5.11 (H-29), 4.04 (H-31), 4.17 (H-32), 2.25 (H-34), 0.985 and 1.38 (H-35), 2.22 (H-36), 1.76 (H-37), 0.961 and 1.11 (H-38), 1.31 (H-39), 0.726 and 1.90 (H- 40), 3.14 (H-41), 4.46 (H-42), 1.22 and 1.81 (H-43), 0.888 and 1.60 (H-44), 1.60 (H-45), 3.05 (H-46, OCH3), 0.697 (H-47), 6.48 (H-48), 0.821 (H-49), 1.76 (H-50), approx. 5.1- 5.3 (H-51), 3.17 (H-52, OCH3), 0.755 (H-53), 0.966 (H-54), 0.805 (H-55), 3.29 (H-56, OCH3), 3.46 (H-59), 1.01 (H-60), approx. 4.3-4.7 (0-61)
13C NMR (75 MHz, DMSO- d6) δ 139.12 (C-1), 130.53 (C-2), 132.49 (C-3), 127.08 (C-4), 127.21 (C-5), 137.12 (C-6), 81.93 (C-7), 40.40 (C-8), 65.83 (C-9), 29.45 (C-10), 25.87 (C-l l), 34.21 (C-12), 99.25 (C-13), 198.17 (C-15), 165.55 (C-16), 47.01 (C-18), 24.04 (C-19), 28.93 (C-21), 58.50 (C-22), 170.44 (C-23), 73.24 (C-25), 39.96 (C-26), 207.67 (C-27), 44.51 (C-28), 123.92 (C-29), 136.56 (C-30), 75.84 (C-31), 84.86 (C-32), 209.49 (C-33), 40.76 (C-34), 39.20 (C-35), 35.05 (C-36), 32.73 (C-37), 38.42 (C-38), 32.06 (C-39), 36.01 (C-40), 80.12 (C- 41), 75.92 (C-42), 29.25 (C-43), 30.24 (C-44), 10.27 (C-45), 55.48 (C-46, OCH3), 15.46 (C-47), 15.59 (C-49), 14.41 (C-50), 56.56 (C-52, OCH3), 12.67 (C-53), 21.50 (C-54), 14.89 (C-55), 57.27 (C-56, OCH3), 174.22 (C-57), 49.90 (C-58), 63.59 and 63.98 (C-59), 16.82 (C-60). MS [M+NH ] 1033.5, [ESI(+), M+Na+] 1038.7.
Example 3 – Synthesis of CCI-779:

A. Synthesis of CCI-779 via intermediate A Method 1 : A mixture of rapamycin (6 g), vinyl ester I (2 g), lipase PS-C “Amano” II (6 g) in anhydrous TBME (36 mL) was heated at 45 °C under Ar2 for 2 days. The mixture was cooled to room temperature and enzyme was removed by filtration, the filtrate was concentrated, the oily residue was added to heptane while stirring. The batch was then cooled to -15 °C for 2 h, collect the solid on the Buchner funnel and washed with cold heptane, A was obtained as off-white solid, crude yield : 98%.MS (El): 1070 Above crude A (6g), dissolved in n-PrOH (24 mL) cooled to 0 °C with an ice-water bath, to this solution was added aqueous H2S04 (12 mL, 1.2N). The mixture was stirred for 24 h at 0°C and was then added to cold phosphate buffer (300 ml, pH=7.8), collect the solid on a Buchner funnel and washed with DI water and dry under vacuum, silica gel column purification eluting with hexane-acetone furnished CCI-779 as a white solid (5.2 g, 90%). MS (El): 1030 Method 2: A mixture of rapamycin (30.0 g, 32.8 mmol), vinyl ester I (10.0 g, 50 mmol), lipase PS-C “Amano” II (30 g) and molecular sieves (5 A) (10.0 g) in anhydrous TBME (150 mL) was heated at 42-43 °C under Ar2 for 48 hours. THF (100 mL) was added to dissolve the precipitation and the mixture was cooled to room temperature. Enzyme was removed by filtration and washed with THF (200 mL), the filtrate was concentrated to about 60 mL and diluted with THF (320 mL). The solution was then cooled to 0-5 °C, H2S04 (180 mL, 2N) was added dropwise over lh. The mixture was stirred for 48 h at 0-5 °C or until the disappearance of A as monitored by TLC. The mixture was diluted with brine (300 mL) and extracted with EtOAc (three times). The combined organic layer was washed with H20, 5% NaHC03, then brine and dried
(MgS04). Evaporation of solvent gave a light yellowish semi solid which was purified by flash chromatography (hexane/acetone, 2:1) to give CCI-779 as a white solid (30.77 g, 91% for two steps). B. Synthesis of CCI-779 via intermediate B: A mixture of rapamycin (3 g), vinyl ester II (1.2 g), lipase PS-C “Amano” II (5 g) in anhydrous TBME (45 mL) was heated at 45 °C under Ar2 for 60 h. The mixture was cooled to room temperature and enzyme was removed by filtration, the filtrate was concentrated, MeOH (20 mL) was added to the residue and concentrated to dryness. Silica gel column purification of crude eluting with hexane-acetone furnished CCI-779 as a white solid (2.3 g), and recovered rapamycin (0.81 g). The yield is 93% based on the recovered rapamycin.
proline analog of CCI-779 (proline-rapamycin42-ester with 2,2-bis(hydroxymethyl)propionic acid or proline-CCI-779) and methods of synthesizing same. Proline-CCI-779 is an active drug substance useful in oncology and other associated indications (immunosuppression, anti-inflammatory, anti-proliferation and anti-tumor). In one aspect, the synthesis of proline-CCI-779 is accomplished through bis- silylation of proline rapamycin, mono-de-protecting 31 ,42-bis-trimethylsilyl proline rapamycin, and acylating the mono-silyl proline rapamycin followed by hydrolysis. In another aspect, the invention provides a two-step enzymatic process involving a regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to give CCI-779.
Example 4 – Synthesis of Proline-CCI-779 The enzymatic procedure of the invention can also be applied to the synthesis of proline CCI-779 from proline-rapamycin under essentially the same conditions as described in Example 2, procedure A for the synthesis of CCI-779 from rapamycin.

proline-rapamycin proline-CCI-779
………………….
more info added for readers
synthesis of CCI-779 or Proline CCI-779 (Temsirolimus) which is useful as an antineoplastic agent having the structure

It is stated to be effective in multiple applications, including inhibition of tumor growth, the treatment for multiple sclerosis and rheumatoid arthritis.
2. The Prior Arts

U.S. Pat. No. 7,202,256 disclosed methods for the synthesis of CCI-779 (Temsirolimus), providing two-step enzymatic process involving regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to obtain the CCI-779 (as shown in scheme 1). A number of drawbacks of the synthesis route depicted in scheme 1 are high-priced PdCl2 and poisonous trimethylboroxine.


A selective synthesis of 42-monoacylated product was previously conducted by reacting rapamycin 31,42-bis-silyl ether, and then the 42-sily ether protection group is selectively removed to provide rapamycin-OH-31-sily ether (U.S. Pat. No. 5,563,145). In addition, a regioselective process for the preparation of CCI-779 is also described in U.S. Pat. No. 6,277,983 (Scheme2). First, rapamycin (compound 4b) is treated with excess chlorotrimethylsilane to form rapamycin31,42-bis-trimethylsilyl ether (compound 5), and then 42-trimethylsilyl ether protection group is selectively removed in mild acid to provide rapamycin 42-OH-31-trimethylsilyl ether (compound 6). This free 42-OH was then acylated with 2,4,6-trichlorobenzyl mixed anhydride of 2,2,5-trimethyl[1,3-dioxane]-5-carboxylic acid (compound 7) at −15° C. for 16 h to give rapamycin 31-trimethylsilyl ether 42-ester (compound 8). Following treatment with mild acid for a certain period, CCI-779 can be isolated. 2,4,6-trichlorobenzyl chloride is irritant, moisture sensitive and costly.

Further, as below-depicted in Scheme 3, U.S. Pat. No. 7,153,957 disclose another method for the CCI-779. It can be prepared by the acylation of 31-silyl ether of rapamycin with the anhydride derived from the 2-phenylboronate acid to give rapamycin 31-silyl ether, 42-boronate. Thereafter, it is hydrolyzed under mild acid condition to form rapamycin 42-ester boronate. After being treated with a suitable diol, CCI-779 was obtained (Scheme 3). Mixed anhydride is not satisfactory for commercial scale synthesis because it can be kept stable only for 48 hr at −5˜0° C., not durable for longer time.
synthesis ofTemsirolimus in a more economic way.

| United States | 5362718 | APPROVED 1994-04-18 | EXPIRY 2014-04-18 |
| Canada | 2429020 | 2009-05-26 | 2021-11-13 |
| Canada | 2187024 | 2004-08-10 | 2015-04-14 |
|
6-13-2012
|
N-HYDROXYAMIDE DERIVATIVES AND USE THEREOF
|
|
|
11-18-2011
|
N-HYDROXYAMIDE DERIVATIVES AND USE THEREOF
|
|
|
8-17-2011
|
N-Hydroxyamide Derivatives and Use Thereof
|
|
|
7-6-2011
|
Sulfonyl Amino Cyclic Derivatives and Use Thereof
|
|
|
11-24-2010
|
Benzothiazole Formulations and Use Thereof
|
|
|
11-19-2010
|
Indazole Compounds for Treating Inflammatory Disorders, Demyelinating Disorders and Cancers
|
|
|
9-31-2010
|
Process for preparation of temsirolimus
|
|
|
4-23-2010
|
COMBINATION OF BENZIMIDAZOLE ANTI-CANCER AGENT AND A SECOND ANTI-CANCER AGENT
|
|
|
10-21-2009
|
Processes for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants
|
|
|
6-12-2009
|
Administration of an Inhibitor of HDAC and an mTOR Inhibitor
|
|
6-8-2007
|
Methods for preparing crystalline rapamycin and for measuring crystallinity of rapamycin compounds using differential scanning calorimetry
|
|
|
4-11-2007
|
Proline CCI-779, production of and uses therefor, and two-step enzymatic synthesis of proline CCI-779 and CCI-779
|
|
|
1-5-2007
|
Methods for treating neurofibromatosis 1
|
|
|
7-12-2006
|
CCI-779 Isomer C
|
| US5362718 | 18 Apr 1994 | 8 Nov 1994 | American Home Products Corporation | Rapamycin hydroxyesters |
| US6197967 | 13 Dec 1999 | 6 Mar 2001 | Clariant Gmbh | Process for the preparation of paraoxadiazolyphenylboronic acids |
| US6277983 | 27 Sep 2000 | 21 Aug 2001 | American Home Products Corporation | Regioselective synthesis of rapamycin derivatives |
| WO1995028406A1 | 14 Apr 1995 | 26 Oct 1995 | American Home Prod | Rapamycin hydroxyesters, process for their preparation and pharmaceutical compositions containing them |
| US7553843 | 6 Dec 2006 | 30 Jun 2009 | Wyeth | Process for the preparation of purified crystalline CCI-779 |
| US7605258 | 16 Oct 2007 | 20 Oct 2009 | Wyeth | Processes for the synthesis of individual isomers of mono-peg CCI-779 |
| US7622578 | 6 Dec 2006 | 24 Nov 2009 | Wyeth | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| US7625726 | 29 Sep 2008 | 1 Dec 2009 | Wyeth | Process for preparing rapamycin 42-esters and FK-506 32-esters with dicarboxylic acid, precursors for rapamycin conjugates and antibodies |
| US7875612 | 24 Apr 2002 | 25 Jan 2011 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| US7910594 | 13 May 2003 | 22 Mar 2011 | Endocyte, Inc. | Vitamin-mitomycin conjugates |
| US8026276 | 25 Jul 2003 | 27 Sep 2011 | Wyeth Llc | Parenteral CCI-779 formulations containing cosolvents, an antioxidant, and a surfactant |
| US8044200 | 14 Mar 2006 | 25 Oct 2011 | Endocyte, Inc. | Synthesis and purification of pteroic acid and conjugates thereof |
| US8105568 | 10 Jul 2009 | 31 Jan 2012 | Endocyte, Inc. | Vitamin receptor binding drug delivery conjugates |
| US8288557 | 22 Jul 2005 | 16 Oct 2012 | Endocyte, Inc. | Bivalent linkers and conjugates thereof |
| US8299116 | 10 Aug 2011 | 30 Oct 2012 | Wyeth Llc | CCI-779 concentrate formulations |
| US8455539 | 15 Oct 2012 | 4 Jun 2013 | Wyeth Llc | CCI-779 concentrate formulations |
| US8465724 | 18 Aug 2006 | 18 Jun 2013 | Endocyte, Inc. | Multi-drug ligand conjugates |
| US8470822 | 7 May 2010 | 25 Jun 2013 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| US8524893 | 28 Jan 2011 | 3 Sep 2013 | Fresenius Kabi Oncology Limited | Process for the preparation of temsirolimus and its intermediates |
| WO2011092564A2 | 20 Jan 2011 | 4 Aug 2011 | Fresenius Kabi Oncology Ltd | Process for the preparation of temsirolimus and its intermediates |

Tasimelteon
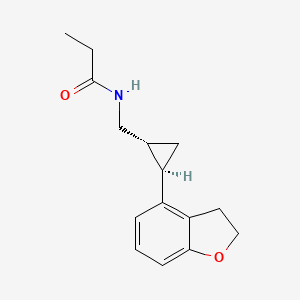
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide, 609799-22-6 cas
As expected, advisors to the US Food and Drug Administration have recommended approval of Vanda Pharmaceuticals’ tasimelteon, to be sold as Hetlioz, for the treatment of non-24-hour disorder in the totally blind.http://www.pharmatimes.com/Article/13-11-14/FDA_panel_backs_Vanda_body_clock_drug_for_blind.aspx
Tasimelteon (BMS-214,778) is a drug which is under development for the treatment of insomnia and other sleep disorders.[1] It is a selective agonistfor the melatonin receptors MT1 and MT2 in the suprachiasmatic nucleus of the brain, similar to older drugs such as ramelteon.[2] It has been through Phase III trials successfully and was shown to improve both onset and maintenance of sleep, with few side effects.[3]
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]

A drug being developed to treat transient insomnia in circadian rhythm sleep disorders (eg jet-lag. The drug appears to be effective in the dose range of 20 to 100mg with an advance in the melatonin rhythm of 2-3 hours with the higher dose
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the
suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of Cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands. This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder
(N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non- 24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping.
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary.
In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime. Tasimelteon
Tasimelteon is a circadian regulator which binds specifically to two high affinity melatonin receptors, Mella (MT1R) and Mellb (MT2R). These receptors are found in high density in the suprachiasmatic nucleus of the brain (SCN), which is responsible for synchronizing our sleep/wake cycle. Tasimelteon has been shown to improve sleep parameters in prior clinical studies, which simulated a desynchronization of the circadian clock. Tasimelteon has so far been studied in hundreds of individuals and has shown a good tolerability profile.
Tasimelteon has the chemical name: tr ns-N-[[2-(2,3-dihydrobenzofuran- 4-yl)cycloprop-lyl] methyl] propanamide, has the structure of Formula I:

Formula I
and is disclosed in US 5856529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78°C (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG- 400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MTIR. It’s affinity (¾) for MTIR is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
Metabolites of tasimelteon include, for example, those described in “Preclinical Pharmacokinetics and Metabolism of BMS-214778, a Novel
Melatonin Receptor Agonist” by Vachharajani et al., J. Pharmaceutical Sci., 92(4):760-772, which is hereby incorporated herein by reference. The active metabolites of tasimelteon can also be used in the method of this invention, as can pharmaceutically acceptable salts of tasimelteon or of its active metabolites. For example, in addition to metabolites of Formula II and III, above, metabolites of tasimelteon also include the monohydroxylated analogs M13 of Formula IV, M12 of Formula V, and M14 of Formula VI.
Formula IV

Formula V
MO

Formula VI
Thus, it is apparent that this invention contemplates entrainment of patients suffering free running circadian rhythm to a 24 hour circadian rhythm by administration of a circadian rhythm regulator (i.e., circadian rhythm modifier) capable of phase advancing and/or entraining circadian rhythms, such as a melatonin agonist like tasimelteon or an active metabolite oftasimelteon or a pharmaceutically acceptable salt thereof. Other MT1R and MT2R agonists, i.e., melatonin agonists, can have similar effects on the master body clock. So, for example, this invention further contemplates the use of melatonin agonists such as but not limited to melatonin, N-[l-(2,3-dihydrobenzofuran-4- yl)pyrrolidin-3-yl]-N-ethylurea and structurally related compounds as disclosed in US 6,211,225, LY-156735 ((R)-N-(2-(6-chloro-5-methoxy-lH-indol- 3yl) propyl) acetamide) (disclosed in U.S. Patent No. 4,997,845), agomelatine (N- [2-(7-methoxy-l-naphthyl)ethyl]acetamide) (disclosed in U.S. Patent No.
5,225,442), ramelteon ((S)-N-[2-(l,6,7,8-tetrahydro-2H-indeno- [5,4-b] furan-8- yl)ethyl]propionamide), 2-phenylmelatonin, 8-M-PDOT, 2-iodomelatonin, and 6- chloromelatonin.
Additional melatonin agonists include, without limitation, those listed in U.S. Patent Application Publication No. 20050164987, which is incorporated herein by reference, specifically: TAK-375 (see Kato, K. et al. Int. J.
Neuropsychopharmacol. 2000, 3 (Suppl. 1): Abst P.03.130; see also abstracts P.03.125 and P.03.127), CGP 52608 (l-(3-allyl-4-oxothiazolidine-2-ylidene)-4- met- hylthiosemicarbazone) (See Missbach et al., J. Biol. Chem. 1996, 271, 13515-22), GR196429 (N-[2-[2,3,7,8-tetrahydro-lH-fur-o(2,3-g)indol-l- yl] ethyl] acetamide) (see Beresford et al., J. Pharmacol. Exp. Ther. 1998, 285, 1239-1245), S20242 (N-[2-(7-methoxy napth-l-yl) ethyl] propionamide) (see Depres-Brummer et al., Eur. J. Pharmacol. 1998, 347, 57-66), S-23478 (see Neuropharmacology July 2000), S24268 (see Naunyn Schmiedebergs Arch. June 2003), S25150 (see Naunyn Schmiedebergs Arch. June 2003), GW-290569, luzindole (2-benzyl-N-acetyltryptamine) (see U.S. Patent No. 5,093,352), GR135531 (5-methoxycarbonylamino-N-acetyltrypt- amine) (see U.S. Patent Application Publication No. 20010047016), Melatonin Research Compound A, Melatonin Agonist A (see IMSWorld R&D Focus August 2002), Melatonin
Analogue B (see Pharmaprojects August 1998), Melatonin Agonist C (see Chem. Pharm. Bull. (Tokyo) January 2002), Melatonin Agonist D (see J. Pineal Research November 2000), Melatonin Agonist E (see Chem. Pharm. Bull. (Tokyo) Febrary 2002), Melatonin Agonist F (see Reprod. Nutr. Dev. May 1999), Melatonin Agonist G (see J. Med. Chem. October 1993), Melatonin Agonist H (see Famaco March 2000), Melatonin Agonist I (see J. Med. Chem. March 2000), Melatonin Analog J (see Bioorg. Med. Chem. Lett. March 2003), Melatonin Analog K (see MedAd News September 2001), Melatonin Analog L, AH-001 (2-acetamido-8- methoxytetralin) (see U.S. Patent No. 5,151,446), GG-012 (4-methoxy-2- (methylene propylamide)indan) (see Drijfhout et al., Eur. J. Pharmacol. 1999, 382, 157-66), Enol-3-IPA, ML-23 (N-2,4-dinitrophenyl-5-methoxy-tryptamine ) (see U.S. Patent No. 4,880,826), SL-18.1616, IP-100-9 (US 5580878), Sleep Inducing Peptide A, AH-017 (see U.S. Patent No. 5,151,446), AH-002 (8-methoxy- 2-propionamido-tetralin) (see U.S. Patent No. 5,151,446), and IP-101.
Metabolites, prodrugs, stereoisomers, polymorphs, hydrates, solvates, and salts of the above compounds that are directly or indirectly active can, of course, also be used in the practice of this invention.
Melatonin agonists with a MT1R and MT2R binding profile similar to that of tasimelteon, which has 2 to 4 time greater specificity for MT2R, are preferred.
Tasimelteon can be synthesized by procedures known in the art. The preparation of a 4-vinyl-2,3-dihydrobenzofuran cyclopropyl intermediate can be carried out as described in US7754902, which is incorporated herein by reference as though fully set forth.
Pro-drugs, e.g., esters, and pharmaceutically acceptable salts can be prepared by exercise of routine skill in the art.
In patients suffering a Non-24, the melatonin and Cortisol circadian rhythms and the natural day/night cycle become desynchronized. For example, in patients suffering from a free-running circadian rhythm, melatonin and Cortisol acrophases occur more than 24 hours, e.g., >24.1 hours, prior to each previous day’s melatonin and Cortisol acrophase, respectively, resulting in desynchronization for days, weeks, or even months, depending upon the length of a patient’s circadian rhythm, before the melatonin, Cortisol, and day /night cycles are again temporarily synchronized.
Chronic misalignment of Cortisol has been associated with metabolic, cardiac, cognitive, neurologic, neoplastic, and hormonal disorders. Such disorders include, e.g., obesity, depression, neurological impairments.


WASHINGTON, June 5, 2013 /PRNewswire/ — Vanda Pharmaceuticals Inc. (Vanda) presented additional entrainment and patient-level clinical data at SLEEP 2013, the 27th Annual Meeting of Associated Professional Sleep Societies in Baltimore, from its SET (Safety and Efficacy of Tasimelteon) and RESET (Randomized-withdrawal study of the Efficacy and Safety of Tasimelteon to treat Non-24-Hour Disorder) Phase III studies of tasimelteon, a circadian regulator for the treatment of Non-24-Hour Disorder (Non-24) in totally blind individuals. Non-24 is a serious, rare and chronic circadian rhythm disorder that affects a majority of totally blind individuals who lack light perception and cannot entrain (synchronize) their master body clock to the 24-hour day. Currently there is no approved FDA treatment for Non-24.
In the SET study, tasimelteon achieved the primary endpoints of entrainment (synchronizing) of the melatonin (aMT6s) rhythm as compared to placebo and clinical response as measured by entrainment plus a score of greater than or equal to 3 on the Non-24 Clinical Response Scale (N24CRS). Tasimelteon also demonstrated significant improvement versus placebo across a number of sleep and wake parameters including measures of total sleep time, nap duration, and timing of sleep, as well as in the Clinical Global Impression of Change (CGI-C), an overall global functioning scale. In treated patients, daytime naps decreased by 46 minutes per day in the worst 25% of days in a cycle and nighttime sleep increased by 57 minutes per day during the worst 25% of nights in a cycle.
The RESET study demonstrated that continued treatment with 20mg of tasimelteon was required to maintain entrainment of melatonin and cortisol circadian rhythms in individuals with Non-24. Patients treated with tasimelteon maintained their clinical benefits while patients who received placebo showed significant deterioration in measures of nighttime sleep, daytime naps and timing of sleep. Furthermore, discontinuation of tasimelteon resulted in a rapid relapse of circadian entrainment and a return to misaligned circadian rhythms, reinforcing the importance of chronic therapy.
Study investigator, Steven W. Lockley, Ph.D., Associate Professor of Medicine, Division of Sleep Medicine, Brigham and Women’s Hospital, Harvard Medical School, commented, “the results clearly demonstrate that tasimelteon can entrain the circadian clock, and that continued treatment is necessary to maintain entrainment.”
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
UPDATED ON JAN 2014
TASIMELTION, an orphan drug for non24
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide
(1R-trans)-N-[[2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]methyl]pro- pananamide VEC162
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
N-(((1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl)methyl)propanamide
Bristol-Myers Squibb Company
PRODUCT PATENT
U.S. Pat. No. 5,856,529
| CAS number | 609799-22-6 |
|---|
| Formula | C15H19NO2 |
|---|---|
| Mol. mass | 245.3 g/mol |
January 31, 2014 — The U.S. Food and Drug Administration today approved Hetlioz (tasimelteon), a melatonin receptor agonist, to treat non-24- hour sleep-wake disorder (“non-24”) in totally blind individuals. Non-24 is a chronic circadian rhythm (body clock) disorder in the blind that causes problems with the timing of sleep. This is the first FDA approval of a treatment for the disorder.
Non-24 occurs in persons who are completely blind. Light does not enter their eyes and they cannot synchronize their body clock to the 24-hour light-dark cycle.
VEC-162, BMS-214778, 609799-22-6, Hetlioz, Tasimelteon (USAN/INN), Tasimelteon [USAN:INN], UNII-SHS4PU80D9,
Tasimelteon
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]
SEQUENCE
Discovered by Bristol-Myers Squibb (BMS) and co-developed with Vanda Pharmaceuticals, tasimelteon is a hypnotic family benzofuran. In Phase III development, it has an orphan drug status.
JAN2014.. APPROVED FDA
In mid-November 2013 the FDA announced their recommendation for the approval of Tasimelteon for the treatment of non-24-disorder.Tasimelteon effectively resets the circadian rhythm, helping to restore normal sleep patterns.http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM374388.pdf
January 2010: FDA granted orphan drug tasimelteon to disturbed sleep / wake in blind without light perception.
February 2008: Vanda has completed enrollment in its Phase III trial in chronic primary insomnia.
June 2007: Results of a Phase III trial for transient insomnia tasimelteon presented by Vanda at the 21st annual meeting of the Associated Professional Sleep Societies. These results demonstrated improvements in objective and subjective measures of sleep and its maintenance.
2004 Vanda gets a license tasimelteon (or BMS-214778 and VEC-162) from Bristol-Myers Squibb.
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
Previously, BMS-214778, identified as an agonist of melatonin receptors, has been the subject of pre-clinical studies for the treatment of sleep disorders resulting from a disturbance of circadian rhythms.The first Pharmacokinetic studies were performed in rats and monkeys.
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands.
This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non-24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping. TASIMELTION
TASIMELTION
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24 h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24 h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary. In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime.
INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:

and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
………………………..
SYNTHESIS
(1R-trans)-N-[[2 – (2,3-dihydro-4 benzofuranyl) cyclopropyl] methyl] propanamide PATENT: BRISTOL-MYERS SQUIBB PRIORITY DATE: 1996 HYPNOTIC

PREPARATION OF XV
XXIV D-camphorsulfonic acid IS REACTED WITH THIONYL CHLORIDE TO GIVE
…………XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
TREATED WITH
XXVI ammonium hydroxide
TO GIVE
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
TREATED WITH AMBERLYST15
….XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
TREATED WITH LAH, ie double bond is reduced to get
…..XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide

Intermediate
I 3-hydroxybenzoic acid methyl ester
II 3-bromo-1-propene
III 3 – (2-propenyloxy) benzoic acid methyl ester
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
XXIII propanoyl chloride
XXIV D-camphorsulfonic acid
XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
XXVI ammonium hydroxide
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Bibliography
– Patents: Benzofuran and dihydrobenzofuran melatonergic agents: US5856529 (1999)
Priority: US19960032689P, 10 Dec. 1996 (Bristol-Myers Squibb Company, U.S.)
– Preparation III (quinazolines): US2004044015 (2004) Priority: EP20000402845, 13 Oct. 2000
– Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
– Preparation XXVIII: Towson et al. Organic Syntheses, Coll. Vol. 8, p.104 (1993) Vol. 69, p.158 (1990)
– Preparation XV: Weismiller et al. Organic Syntheses, Coll. Vol. 8, p.110 (1993) Vol. 69, p.154 (1990).
– G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
– Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
…………………….
SYNTHESIS CONSTRUCTION AS IN PATENT
GENERAL SCHEMES
Reaction Scheme 1

The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2

The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3

Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4

The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5


Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6

Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7

Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
EXPERIMENTAL PROCEDURES
SEE ORIGINAL PATENT FOR CORECTIONS
Preparation 1
Benzofuran-4-carboxaldehyde
Step 1 : N-Methoxy-N-methyl-benzofuran-4-carboxamide
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Preparation 16
(±)-(trans)-2-(2,3-Dihyd robenzofuran-4-yl)cyclopropane- carboxaldehyde
Step 1 : (±Htrans)-N-Methoxy-N-methyl-2-(2.3-dihydrobenzofuran-4- yhcyclopropanecarboxamide
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
Step 2: (±)-(trans)- 2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
Preparation 18
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde
Step 1 : (-Htrans)-N-[3-(2.3-Dihvdrobenzofuran-4-yl)-propenoyll-2.10- camphorsultam
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
Step 2: (-)-N-[[(trans)-2-(2,3-Dihydrobenzofuran-4-yl)-cyclopropylj- carbonylj-2, 10-camphorsultam
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
Step 3: (-)-(trans)-2-(2,3-Dihydrobenzofuran-4-yl)cyclopropane- methanol
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
FINAL PRODUCT TASIMELTEON
Example 2
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58.
Curr Med Chem. 2012;19(21):3532-49. Review.
7 Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist.
Vachharajani NN, Yeleswaram K, Boulton DW.J Pharm Sci. 2003 Apr;92(4):760-72.
TASIMELTION
PATENTS
| US2010261786 | 10-15-2010 | PREDICTION OF SLEEP PARAMETER AND RESPONSE TO SLEEP-INDUCING COMPOUND BASED ON PER3 VNTR GENOTYPE |
| US2009209638 | 8-21-2009 | TREATMENT FOR DEPRESSIVE DISORDERS |
| US6060506 | 5-10-2000 | Benzopyran derivatives as melatonergic agents |
| US5981571 | 11-10-1999 | Benzodioxa alkylene ethers as melatonergic agents |
| WO9825606 | 6-19-1998 | BENZODIOXOLE, BENZOFURAN, DIHYDROBENZOFURAN, AND BENZODIOXANE MELATONERGIC AGENTS |
| WO2007137244A1 * | May 22, 2007 | Nov 29, 2007 | Gunther Birznieks | Melatonin agonist treatment |
| US4880826 | Jun 25, 1987 | Nov 14, 1989 | Nava Zisapel | Melatonin antagonist |
| US4997845 | May 10, 1990 | Mar 5, 1991 | Eli Lilly And Company | β-alkylmelatonins as ovulation inhibitors |
| US5093352 | May 16, 1990 | Mar 3, 1992 | Whitby Research, Inc. | Antidepressant agents |
| US5151446 | Mar 28, 1991 | Sep 29, 1992 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5225442 | Jan 3, 1992 | Jul 6, 1993 | Adir Et Compagnie | Compounds having a naphthalene structure |
| US5580878 | Jun 7, 1995 | Dec 3, 1996 | Interneuron Pharmaceuticals, Inc. | Substituted tryptamines phenalkylamines and related compounds |
| US5856529 | Dec 9, 1997 | Jan 5, 1999 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US6211225 | Jun 6, 2000 | Apr 3, 2001 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7754902 | May 18, 2006 | Jul 13, 2010 | Vanda Pharmaceuticals, Inc. | Ruthenium(II) catalysts for use in stereoselective cyclopropanations |
| US20010047016 | Apr 12, 2001 | Nov 29, 2001 | Gregory Oxenkrug | Method for treating depression |
| US20050164987 | Dec 22, 2004 | Jul 28, 2005 | Barberich Timothy J. | Melatonin combination therapy for improving sleep quality |
| US20090105333 | May 22, 2007 | Apr 23, 2009 | Gunther Birznieks | Melatonin agonist treatment |
extra info
|
 |

back to home for more updates

DR ANTHONY MELVIN CRASTO Ph.D

Macimorelin
CAS 381231-18-1
Chemical Formula: C26H30N6O3
Exact Mass: 474.23794
Molecular Weight: 474.55480
Elemental Analysis: C, 65.80; H, 6.37; N, 17.71; O, 10.11
945212-59-9 (Macimorelin acetate)
AEZS-130
ARD-07
D-87875
EP-01572
EP-1572
JMV-1843
USAN (ab-26)
MACIMORELIN ACETATE
THERAPEUTIC CLAIM
Diagnostic agent for adult growth hormone deficiency (AGHD)
CHEMICAL NAMES
1. D-Tryptophanamide, 2-methylalanyl-N-[(1R)-1-(formylamino)-2-(1H-indol-3-yl)ethyl]-, acetate (1:1)
2. N2-(2-amino-2-methylpropanoyl-N1-[(1R)-1-formamido-2-(1H-indol-3-yl)ethyl]- D-tryptophanamide acetate
MOLECULAR FORMULA
C26H30N6O3.C2H4O2
MOLECULAR WEIGHT
534.6
SPONSOR
Aeterna Zentaris GmbH
CODE DESIGNATIONS
D-87575, EP 1572, ARD 07
CAS REGISTRY NUMBER
945212-59-9
Macimorelin (also known as AEZS-130, EP-1572) is a novel synthetic small molecule, acting as a ghrelin agonist, that is orally active and stimulates the secretion of growth hormone (GH). Based on results of Phase 1 studies, AEZS-130 has potential applications for the treatment of cachexia, a condition frequently associated with severe chronic diseases such as cancer, chronic obstructive pulmonary disease and AIDS. In addition to the therapeutic application, a Phase 3 trial with AEZS-130 as a diagnostic test for growth hormone deficiencies in adults has been completed.
http://www.ama-assn.org/resources/doc/usan/macimorelin-acetate.pdf
QUEBEC, Nov. 5, 2013 /PRNewswire/ – Aeterna Zentaris Inc. (the “Company”) today announced that it has submitted a New Drug Application (“NDA”) to the U.S. Food and Drug Administration (“FDA”) for its ghrelin agonist, macimorelin acetate (AEZS-130). Phase 3 data have demonstrated that the compound has the potential to become the first orally-approved product that induces growth hormone release to evaluate adult growth hormone deficiency (“AGHD”), with accuracy comparable to available intravenous and intramuscular testing procedures. read at
http://www.drugs.com/nda/macimorelin_acetate_131105.html
http://www.ama-assn.org/resources/doc/usan/macimorelin-acetate.pdf
macimorelin (JMV 1843), a ghrelin-mimetic growth hormone secretagogue in Phase III for adult growth hormone deficiency (AGHD)

Macimorelin, a growth hormone modulator, is currently awaiting registration in the U.S. by AEterna Zentaris as an oral diagnostic test of adult growth hormone deficit disorder. The company is also developing the compound in phase II clinical trials for the treatment of cancer related cachexia. The compound was being codeveloped by AEterna Zentaris and Ardana Bioscience; however, the trials underway at Ardana were suspended in 2008 based on a company strategic decision. AEterna Zentaris owns the worldwide rights of the compound. In 2007, orphan drug designation was assigned by the FDA for the treatment of growth hormone deficit in adults.
New active series of growth hormone secretagogues
J Med Chem 2003, 46(7): 1191
WO 2001096300
WO 2007093820
…………………………
J Med Chem 2003, 46(7): 1191
http://pubs.acs.org/doi/full/10.1021/jm020985q


Synthetic Pathway for JMV 1843 and Analoguesa
a Reagents and conditions: (a) IBCF, NMM, DME, 0 °C; (b) NH4OH; (c) H2, Pd/C, EtOH, HCl; (d) BOP, NMM, DMF, Boc-(d)-Trp-OH; (e) Boc2O, DMAP cat., anhydrous CH3CN; (f) BTIB, pyridine, DMF/H2O; (g) 2,4,5-trichlorophenylformate, DIEA, DMF; (h) TFA/anisole/thioanisole (8:1:1), 0 °C; (i) BOP, NMM, DMF, Boc-Aib-OH; (j) TFA/anisole/thioanisole (8:1:1), 0 °C; (k) RP preparative HPLC.
TFA, H-Aib-(d)-Trp-(d)-gTrp-CHO (7). 6 (1 g, 1.7 mmol) was dissolved in a mixture of trifluoroacetic acid (8 mL), anisole (1 mL), and thioanisole (1 mL) for 30 min at 0 °C. The solvents were removed in vacuo, the residue was stirred in ether, and the precipitated TFA, H-Aib-(d)-Trp-(d)-gTrp-CHO was filtered. 7 was purified by preparative HPLC and obtained in 52% yield. 1H NMR (400 MHz, DMSO-d6) + correlation 1H−1H: δ 1.21 (s, 3H, CH3 (Aib)), 1.43 (s, 3H, CH3 (Aib)), 2.97 (m, 2H, (CH2)β), 3.1 (m, 2H, (CH2)β‘), 4.62 (m, 1H, (CH)αA and (CH)αB), 5.32 (q, 0.4H, (CH)α‘B), 5.71 (q, 0.6H, (CH)α‘A), 7.3 (m, 4H, H5 and H6 (2 indoles)), 7.06−7.2 (4d, 2H, H2A and H2B (2 indoles)), 7.3 (m, 2H, H4 or H7 (2 indoles)), 7.6−7.8 (4d, 2H, H4A and H4B or H7A and H7B), 7.97 (s, 3H, NH2 (Aib) and CHO (formyl)), 8.2 (d, 0.4H, NH1B (diamino)), 8.3 (m,1H, NHA and NHB), 8.5 (d, 0.6H, NH1A (diamino)), 8.69 (d, 0.6H, NH2A (diamino)), 8.96 (d, 0.4H, NH2B (diamino)), 10.8 (s, 0.6H, N1H1A (indole)), 10.82 (s, 0.4H, N1H1B (indole)), 10.86 (s, 0.6H, N1H2A (indole)), 10.91 (s, 0,4H, N1H2B (indole)). MS (ES), m/z: 475 [M + H]+, 949 [2M + H]+. HPLC tR: 16.26 min (conditions A).
…………………………..
http://www.google.com/patents/US8192719
The inventors have now found that the oral administration of growth hormone secretagogues (GHSs) EP 1572 and EP 1573 can be used effectively and reliably to diagnose GHD.
EP 1572 (Formula I) or EP 1573 (Formula II) are GHSs (see WO 01/96300, Example 1 and Example 58 which are EP 1572 and EP 1573, respectively) that may be given orally.

EP 1572 and EP 1573 can also be defined as H-Aib-D-Trp-D-gTrp-CHO and H-Aib-D-Trp-D-gTrp-C(O)NHCH2CH3. Wherein, His hydrogen, Aib is aminoisobutyl, D is the dextro isomer, Trp is tryptophan and gTrp is a group of Formula III:

…………………………….
http://www.google.com/patents/US6861409
H-Aib-D-Trp-D-gTrp-CHO: 
Example 1 H-Aib-D-Trp-D-gTrp-CHO
Total synthesis (percentages represent yields obtained in the synthesis as described below):

Z-D-Tr-NH2
Z-D-Trp-OH (8.9 g; 26 mmol; 1 eq.) was dissolved in DME (25 ml) and placed in an ice water bath to 0° C. NMM (3.5 ml; 1.2 eq.), IBCF (4.1 ml; 1.2 eq.) and ammonia solution 28% (8.9 ml; 5 eq.) were added successively. The mixture was diluted with water (100 ml), and the product Z-D-Trp-NH2 precipitated. It was filtered and dried in vacuo to afford 8.58 g of a white solid.
Yield=98%.
C19H19N3O3, 337 g.mol−1.
Rf=0.46 {Chloroform/Methanol/Acetic Acid (180/10/5)}.
1H NMR (250 MHZ, DMSO-d6): δ 2.9 (dd, 1H, Hβ, Jββ′=14.5 Hz; Jβα=9.8 Hz); 3.1 (dd, 1H, Hβ′, Jβ′β=14.5 Hz; Jβ′α=4.3 Hz); 4.2 (sextuplet, 1H, Hα); 4.95 (s, 2H, CH2 (Z); 6.9-7.4 (m, 11H); 7.5 (s, 1H, H2); 7.65 (d, 1H, J=7.7 Hz); 10.8 (s, 1H, N1H).
Mass Spectrometry (Electrospray), m/z 338 [M+H]+, 360 [M+Na]+, 675 [2M+H]+, 697 [2M+Na]+.
Boc-D-Trp-D-Trp-NH2
Z-D-Trp-NH2 (3 g; 8.9 mmol; 1 eq.) was dissolved in DMF (100 ml). HCl 36% (845 μl; 1.1 eq.), water (2 ml) and palladium on activated charcoal (95 mg, 0.1 eq.) were added to the stirred mixture. The solution was bubbled under hydrogen for 24 hr. When the reaction went to completion, the palladium was filtered on celite. The solvent was removed in vacuo to afford HCl, H-D-Trp-NH2 as a colorless oil.
In 10 ml of DMF, HCl, H-D-Trp-NH2 (8.9 mmol; 1 eq.), Boc-D-Trp-OH (2.98 g; 9.8 mmol; 1.1 eq.), NMM (2.26 ml; 2.1 eq.) and BOP (4.33 g; 1.1 eq.) were added successively. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (100 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo to afford 4.35 g of Boc-D-Trp-D-Trp-NH2 as a white solid.
Yield=85%.
C27H31N5O4, 489 g.mol−1.
Rf=0.48 {Chloroform/Methanol/Acetic Acid (85/10/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.28 (s, 9H, Boc); 2.75-3.36 (m, 4H, 2 (CH2)β; 4.14 (m, 1H, CHα); 4.52 (m, 1H, CHα′); 6.83-7.84 (m, 14H, 2 indoles (10H), NH2, NH (urethane) and NH (amide)); 10.82 (d, 1H, J=2 Hz, N1H); 10.85 (d, 1H, J=2 Hz, N1H).
Mass Spectrometry (Electrospray), m/z 490 [M+H]+, 512 [M+Na]+, 979 [2M+H]+.
Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2
Boc-D-Trp-D-Trp-NH2 (3 g; 6.13 mmol; 1 eq.) was dissolved in acetonitrile (25 ml).
To this solution, di-tert-butyl-dicarbonate (3.4 g; 2.5 eq.) and 4-dimethylaminopyridine (150 mg; 0.2 eq.) were successively added. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate/hexane {5/5} to afford 2.53 g of Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2 as a white solid.
Yield=60%.
C37H47N5O8, 689 g.mol−1.
Rf=0.23 {ethyl acetate/hexane (5/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.25 (s, 9H, Boc); 1.58 (s, 9H, Boc); 1.61 (s, 9H, Boc); 2.75-3.4 (m, 4H, 2 (CH2)β); 4.2 (m, 1H, CHα′); 4.6 (m, 1H, CHα); 7.06-8 (m, 14H, 2 indoles (10H), NH (urethane), NH and NH2 (amides)).
Mass Spectrometry (Electrospray), m/z 690 [M+H]+, 712 [M+Na]+, 1379 [2M+H]+, 1401 [2M+Na]+.
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-H
Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2 (3 g; 4.3 mmol; 1 eq.) was dissolved in the mixture DMF/water (18 ml/7 ml). Then, pyridine (772 μl; 2.2 eq.) and Bis(Trifluoroacetoxy)IodoBenzene (2.1 g; 1.1 eq.) were added. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and aqueous saturated sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. Boc-D-NiBoc)Trp-D-g(NiBoc)Trp-H was used immediately for the next reaction of formylation.
Rf=0.14 {ethyl acetate/hexane (7/3)}.
C36H47N5O7, 661 g.mol−1.
1H NMR (200 MHZ, DMSO-d6): δ 1.29 (s, 9H, Boc); 1.61 (s, 18H, 2 Boc); 2.13 (s, 2H, NH2 (amine)); 3.1-2.8 (m, 4H, 2 (CH2)β); 4.2 (m, 1H, CHα′); 4.85 (m, 1H, CHα); 6.9-8 (m, 12H, 2 indoles (10H), NH (urethane), NH (amide)).
Mass Spectrometry (Electrospray), m/z 662 [M+H]+, 684 [M+Na]+.
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-H (4.3 mmol; 1 eq.) was dissolved in DMF (20 ml). Then, N,N-diisopropylethylamine (815 μl; 1.1 eq.) and 2,4,5-trichlorophenylformate (1.08 g; 1.1 eq.) were added. After 30 minutes, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate/hexane {5/5} to afford 2.07 g of Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO as a white solid.
Yield=70%.
C37H47N5O8, 689 g.mol−1.
Rf=0.27 {ethyl acetate/hexane (5/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.28 (s, 9H, Boc); 1.6 (s, 9H, Boc); 1.61 (s, 9H, Boc); 2.75-3.1 (m, 4H, 2 (CH2)β); 4.25 (m, 1H, (CH)αA&B); 5.39 (m, 0.4H, (CH)α′B); 5.72 (m, 0.6H, (CH)α′A); 6.95-8.55 (m, 14H, 2 indoles (10H), NH (urethane), 2 NH (amides), CHO (formyl)).
Mass Spectrometry (Electrospray), m/z 690 [M+H]+, 712 [M+Na]+, 1379 [2M+H]+.
Boc-Aib-D-Trp-D-gTrp-CHO
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO (1.98 g; 2.9 mmol; 1 eq.) was dissolved in a -mixture of trifluoroacetic acid (16 ml), anisole (2 ml) and thioanisole (2 ml) for 30 minutes at 0° C. The solvents were removed in vacuo, the residue was stirred with ether and the precipitated TFA, H-D-Trp-D-gTrp-CHO was filtered.
TFA, H-D-Trp-D-gTrp-CHO (2.9 mmol; 1 eq.), Boc-Aib-OH (700 mg; 1 eq.), NMM (2.4 ml; 4.2 eq.) and BOP (1.53 g; 1.2 eq.) were successively added in 10 ml of DMF. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate to afford 1.16 g of Boc-Aib-D-Trp-D-gTrp-CHO as a white solid.
Yield=70%.
C31H38N6O5, 574 g.mol−1.
Rf=0.26 {Chloroform/Methanol/Acetic Acid (180/10/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.21 (s, 6H, 2 CH3(Aib)); 1.31 (s, 9H, Boc); 2.98-3.12 (m, 4H, 2 (CH2)β); 4.47 (m, 1H, (CH)αA&B); 5.2 (m, 0.4H, (CH)α′B); 5.7 (m, 0.6H, (CH)α′A); 6.95-8.37 (m, 15H, 2 indoles (10H), 3 NH (amides), 1 NH (urethane) CHO (formyl)); 10.89 (m, 2H, 2 N1H (indoles)).
Mass Spectrometry (Electrospray), ml/z 575 [M+H]+, 597 [M+Na]+, 1149 [2M+H]+, 1171 [2M+Na]+.
H-Aib-D-Trp-D-gTrT-CHO
Boc-Aib-D-Trp-D-gTrp-CHO (1 g; 1.7 nmmol) was dissolved in a mixture of trifluoroacetic acid (8 ml), anisole (1 ml) and thioanisole (1 ml) for 30 minutes at 0° C. The solvents were removed in vacuo, the residue was stirred with ether and the precipitated TFA, H-Aib-D-Trp-D-gTrp-CHO was filtered.
The product TFA, H-Aib-D-Trp-D-gTrp-CHO was purified by preparative HPLC (Waters, delta pak, C18, 40×100 mm, 5 μm, 100 A).
Yield=52%.
C26H30N6O3, 474 g.mol−1.
1H NMR (400 MHZ, DMSO-d6)+1H/1H correlation: δ 1.21 (s, 3H, CH3 (Aib)); 1.43 (s, 3H, CH3 (Aib)); 2.97 (m, 2H, (CH2)β); 3.1 (m, 2H, (CH2)β′); 4.62 (m, 1H, (CH)αA&B); 5.32 (q, 0.4H, (CH)α′B); 5.71 (q, 0.6H, (CH)α′A); 7.3 (m, 4H5 and H6 (2 indoles)); 7.06-7.2 (4d, 2H, H2A et H2B (2 indoles)); 7.3 (m, 2H, H4 or H7 (2 indoles)); 7.6-7.8 (4d, 2H, H4A and H4B or H7A et H7B); 7.97 (s, 3H, NH2 (Aib) and CHO (Formyl));8.2 (d, 0.4H, NH1B (diamino)); 8.3 (m,1H, NHA&B); 8.5 (d, 0.6H, NH1A (diamino)); 8.69 (d, 0.6H, NH2A (diamino)); 8.96 (d, 0.4H, NH2B (diamino)); 10.8 (s, 0.6H, N1H1A (indole)); 10.82 (s, 0.4H, N1H1B (indole)); 10.86 (s, 0.6H, N1H2A (indole)); 10.91 (s, 0.4, N1H2B (indole)).
Mass Spectrometry (Electrospray), m/z 475 [M+H]+, 949 [2M+H]+.
………………………………
UPDATED INFO AS ON JAN 6 2014
Quebec City, Canada, January 6, 2014 – Aeterna Zentaris Inc. (NASDAQ: AEZS) (TSX: AEZS) (the “Company”) today announced that the U.S. Food and Drug Administration (“FDA”) has accepted for filing the Company’s New Drug Application (“NDA”) for its ghrelin agonist, macimorelin acetate, in Adult Growth Hormone Deficiency (“AGHD”). The acceptance for filing of the NDA indicates the FDA has determined that the application is sufficiently complete to permit a substantive review.
The Company’s NDA, submitted on November 5, 2013, seeks approval for the commercialization of macimorelin acetate as the first orally-administered product that induces growth hormone release to evaluate AGHD. Phase 3 data have demonstrated the compound to be well tolerated, with accuracy comparable to available intravenous and intramuscular testing procedures. The application will be subject to a standard review and will have a Prescription Drug User Fee Act (“PDUFA”) date of November 5, 2014. The PDUFA date is the goal date for the FDA to complete its review of the NDA.
David Dodd, President and CEO of Aeterna Zentaris, commented, “The FDA’s acceptance of this NDA submission is another significant milestone in our strategy to commercialize macimorelin acetate as the first approved oral product for AGHD evaluation. We are finalizing our commercial plan for this exciting new product. We are also looking to broaden the commercial application of macimorelin acetate in AGHD for use related to traumatic brain injury victims and other developmental areas, which would represent significant benefit to the evaluation of growth hormone deficiency, while presenting further potential revenue growth opportunities for the Company.”
Macimorelin acetate, a ghrelin agonist, is a novel orally-active small molecule that stimulates the secretion of growth hormone. The Company has completed a Phase 3 trial for use in evaluating AGHD, and has filed an NDA to the FDA in this indication. Macimorelin acetate has been granted orphan drug designation by the FDA for use in AGHD. Furthermore, macimorelin acetate is in a Phase 2 trial as a treatment for cancer-induced cachexia. Aeterna Zentaris owns the worldwide rights to this novel patented compound.
AGHD affects about 75,000 adults across the U.S., Canada and Europe. Growth hormone not only plays an important role in growth from childhood to adulthood, but also helps promote a hormonally-balanced health status. AGHD mostly results from damage to the pituitary gland. It is usually characterized by a reduction in bone mineral density, lean mass, exercise capacity, and overall quality of life.
Aeterna Zentaris is a specialty biopharmaceutical company engaged in developing novel treatments in oncology and endocrinology. The Company’s pipeline encompasses compounds from drug discovery to regulatory approval.
back to home for more updates
DR ANTHONY MELVIN CRASTO Ph.D
GT Biologics, a developer of live biotherapeutics for the treatment of autoimmune diseases, has received orphan drug designation from the US Food and Drug Administration (FDA) for its lead product candidate, Thetanix.
read all at
read all on
http://microbewiki.kenyon.edu/index.php/Bacteroides_thetaiotaomicron

MACITENTAN
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide,
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl] -N’-propylsulfamide
CAS NO 441798-33-0
ACT-064992, Opsumit,UNII-Z9K9Y9WMVL
Mechanism of Action: Endothelin receptor antagonist (ERA)
Date of Approval: October 18, 2013(US)
Indication: Pulmonary Hypertension (PAH)
Company: Actelion Pharmaceuticals Ltd
PCT patent application: WO2002053557
FDA N204410, MACITENTANTABLET; ORAL10MG, OPSUMIT, ACTELION PHARMS LTD
Macitentan is achiral
Macitentan is a crystalline powder that is insoluble in water. In the solid state macitentan is very stable, is not hygroscopic, and is not light sensitive.
Macitentan (Opsumit® )is a novel dual endothelin receptor antagonist that resulted from a tailored drug discovery process. Macitentan has a number of potentially key beneficial characteristics – i.e., increased in vivo preclinical efficacy vs. existing ERAs resulting from sustained receptor binding and tissue penetration properties. A clinical pharmacology program indicated a low propensity of macitentan for drug-drug interactions.
Macitentan (ACT-064992) is a tissue-targeting dual ET(A)/ET(B) endothelin (ET) receptor antagonist designed for tissue targeting. Macitentan inhibited ET-1-induced contractions in isolated endothelium-denuded rat aorta (ET(A) receptors) and sarafotoxin S6c-induced contractions in isolated rat trachea (ET(B) receptors). In diabetic rats, chronic administration of macitentan decreased blood pressure and proteinuria and prevented end-organ damage. Treatment with macitentan enhanced the cytotoxicity mediated by paclitaxel as measured by the degree of apoptosis in tumor cells and tumor-associated endothelial cells. A Phase III clinical trial of macitentan was successfully completed in 2012.


Macitentan is an investigational drug being studied for the treatment of pulmonary arterial hypertension. It acts as a dualendothelin receptor antagonist and is being developed by Actelion.[1] A Phase III clinical trial was successfully completed in 2012.[2]
on 22 October 2012 – Actelion (SIX: ATLN) announced that it has submitted a New Drug Application (NDA) to the US Food and Drug Administration (FDA) seeking approval for macitentan (Opsumit®) for the treatment of patients with pulmonary arterial hypertension
Actelion’s experimental lung drug macitentan prolonged overall survival by more than a third according to detailed study data, which the company hopes will convince investors it has a viable follow-up product to secure its commercial future.
Europe’s largest standalone biotech company wants the drug, which treats pulmonary arterial hypertension — a disease that causes high blood pressure in the arteries of the lungs — to replace blockbuster Tracleer.
Tracleer currently makes up 87 percent of sales but loses patent protection in 2015 and has also seen its market share eroded by Gilead’s Letairis.

Macitentan has an active metabolite, ACT-132577, which is an oxidative depropylation product. Both macitentan and ACT-132577 are mainly excreted in form of hydrolysis products via urine (about 2/3 of all metabolites) and faeces (1/3).[3]
Co-administration of ciclosporin has only a slight effect on the concentrations of macitentan and its active metabolite, whilerifampicin decreases the area under the curve (AUC) of the drug’s blood plasma concentration by 79%, and ketoconazoleapproximately doubles it. This corresponds to the finding that macitentan is mainly metabolised via the liver enzyme CYP3A4.[4]

SYNTHESIS
The synthesis begins with the reaction of chlorosulfonyl isocyanate (1) (dissolved in dichloromethane at 0 ° C) with one equivalent of tert-butanol. This produces a by BOC protected Aminosulfonylchlorid (2). With one equivalent of n-propylamine (dissolved in 3 eq. Of triethylamine, dichloromethane, at 0 ° C, RT 16 h) is produced by a hydrochloric acid elimination BOC-protected sulfamide (3). This is dissolved in 5 M HCl and dioxane (4-8 h), the BOC protecting group is cleaved. The sulfamide formed (4) is potassium tert-butoxide-(dissolved in MeOH, 3h) is converted to the potassium salt (5). Tert-butoxide potassium acts as a very strong base for deprotonation. This sulfamide potassium salt reacts with the nucleophilic substituents on the heteroaromatic Dichlorpyrimidinderivat (6) (dissolved in dimethyl sulfoxide, at room temperature, RT 42-72 h) under KCl-cleavage to a Monochlorpyrimidin intermediate (7). By treatment with ethylene glycol (dissolved in dimethyl ether, potassium-tert-butoxide,), the ethylene glycol side chain is generated (8). With 2-chloro-5-bromo-pyrimidine (dissolved in tetrahydrofuran, close, at 60-75 ° C) is formed under elimination of HCl in an S N 1 reaction Macitentan (9)…………Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 .

Synthesis of Macitentan
…………………………………………….
SYNTHESIS


YOU CAN READ AT YAOPHA.COM, lovely site to see for drugs


如果您有关于yaopha.com的问题或建议请与我们联系,具体方式如下:
E-mail:contactus@yaopha.com
………………………….
SYNTHESIS
(WO2006/051502A2, JMC2012, 7849). Chlorosulfonyl isocyanate ( 1 ) reaction with tert-butyl alcohol 2 , which is then reacted with n-propylamine 3 . 3 de-boc protected through the acid after reaction with potassium t-butoxide 4 . Another compound 5 with NaH after acidic protons off with dimethyl carbonate ( 6 ) to obtain 7 . 7 and formamidine hydrochloride ( 8 ) to ring chlorinated later POCl3 9 . 9 and 4 SNAr reaction occurs 10 . 10under basic conditions with ethylene glycol SNAr reaction occurs again in alkaline conditions with11 SNAr reaction occurs MACITENTAN.

………………………
http://www.google.com/patents/WO2014155304A1?cl=en
LC-MS (Agilent MS detector G1956B with Agilent 1200 Binary Pump and DAD).
Parameters of the LC-MS method:
Injection volume: 2 |jL
Column: Kinetex C18, 2.6 μιη, 2.1 x 50 mm
Column flow rate: 1 mL/min
Eluents: Eluent A: water + 0.08% TFA
Eluent B: MeCN + 0.012% TFA
Gradient: 2.0 min 95% B
2.8 min 95% B
3.0 min 5% B
Temperature: 40°C Detector wavelength 210 nm

Preparation B: N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]- 4-pyrimidinyl] -N’-propylsulfamide (macitentan):
N-(5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl)propane- 1-sulfamide (200 g; 0.46 mol; see Example 2 or 3) and 5-bromo-2-chloropyrimidine (117 g; 0.60 mol; 1.3 eq) were dissolved in toluene (3 L) and DMF (400 mL). The reaction mixture was warmed up to 50°C and toluene (approx. 400 mL) was distilled our under reduced pressure. The mixture was cooled to 0 °C and tBuOK (156 g, 3 eq, 1.38 mol) was added portionwise. It was stirred at 20 °C for 1 h. Water (1 L) was added and the pH of the solution was adjusted to 3-5 using 33% aq. HC1. The mixture was heated to 50°C and the layers were separated. The org. phase was treated with charcoal at 50°C and filtered over Celite. The filter cake was rinsed with toluene. At 50°C, water (1 L) was added to the org. layer. The layers were separated. The org. layer was concentrated under reduced pressure to a total volume of 1 L and cooled to 0°C. The solid obtained was filtered off. It was rinsed with toluene and MeOH. The crude material was suspended in EA (1 L) and heated to 50°C. 300 mL of EA were distilled out and MeOH (400 mL) was added. The suspension was cooled down to 0°C. The solid was filtered off, rinsed with MeOH and dried under reduced pressure to afford the title compound as a white solid (225 g; 83% yield).

……………………
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm3009103

Starting from the structure of bosentan (1), we embarked on a medicinal chemistry program aiming at the identification of novel potent dual endothelin receptor antagonists with high oral efficacy. This led to the discovery of a novel series of alkyl sulfamide substituted pyrimidines. Among these, compound 17 (macitentan, ACT-064992) emerged as particularly interesting as it is a potent inhibitor of ETA with significant affinity for the ETB receptor and shows excellent pharmacokinetic properties and high in vivo efficacy in hypertensive Dahl salt-sensitive rats. Compound 17 successfully completed a long-term phase III clinical trial for pulmonary arterial hypertension
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (17)
……………
WO 2015004265 click
Example 3 : N-(5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl)pr opane- 1- sulfamide (reaction in and work-up with MIBK):
EG (124 mL, 3.7 mol, 6.0 eq.) was added to a warm (40-50°C) suspension of the compound of Preparation A (150 g, 0.37 mol) in MIBK (600 mL). Solid KOtBu (114 g, 1.11 mol, 3.0 eq.) was added portionwise so that IT < 60°C. The mixture was stirred for
2- 3 h at 100-105°C. After completion of the reaction (LC-MS control), it was cooled to 50 °C. A 40%) aq. solution of citric acid monohydrate (300 mL) was added until pH 4 was reached. The layers were separated. The org. phase was washed with water (450 mL) and the layers were separated. Water (450 mL) was added and the mixture was warmed to 50°C. It was stirred at 50°C for 5 min. The layers were separated. The org. phase was concentrated under vacuum at 50°C until 200 mL of MIBK were removed. Hept (800 mL) was added dropwise at 70-75°C until turbidity was observed. The mixture was seeded with an analytically pure sample of N-(5-(4-bromophenyl)-6-(2 hydroxy ethoxy)pyrimidin-4-yl)propane-l-sulfamide and stirred at 60-65°C for 30 min. It was allowed to cool to 5°C within 5 h. It was filtered off, rinsed with a cold MIBK/Hept mixture (300 mL, 1 : 1) and dried under vacuum at 50°C to yield the title compound as a white solid (121 g; 76% yield).
The product had NMR data equivalent to those reported in Bolli et al, J. Med. Chem. (2012), 55, 7849-7861. [M+H]+ = 430 and 432. LC-MS: tR = 1.46 min; purity: 98.4% a/a. Residual ethylene glycol (GC-FID): 530 ppm.
…….
CN 104447572 click
(l) Martin H. Bolli et al. Reported the synthesis of Marcy cefotetan follows:

[0008] The method W 5- (4- desert phenyl) -4,6-dichloro-chewing clever as a starting material, N- propyl amine Lai ugly bell in DMS0 as a reaction solvent, an alcohol bell as t a base under substitution reaction conditions, the reaction temperature needs of 24-7 to give


The intermediate compound 15, compound 15 in hexylene glycol dimethyl off as the reaction solvent, a tertiary alcohol under conditions with a strong base clock as hexanediol substitution reaction, l〇 (TC Reaction of 18-2 to give compound 17, Compound 17 was then reacted with 5-chloro-chewing desert -2 clever substitution reaction at tetraammine Qiao Nan as a reaction solvent, ammoniated axis as the alkali conditions, the reaction to give the final product of Marcy cefotetan The route every step the higher the yield, the experimental use of N- propyl amine Lai ugly bell hygroscopic, unstable and a long time before the two-step reaction, the reaction at the second step requires l〇 (TC high temperature 18-2 technology is not suitable for industrial production.
[0009] International Patent W02002 / 053557 discloses some preparation methods and other Massey cefotetan column derivative method at each step of the preparation of the reaction times are longer, some reactions up to 4 days, and the resulting intermediate are purified by column chromatography method is not suitable for industrial production.



[00 pairs (3) N- [5- (4- desert) -6-mouth – [(5-desert -2- chew clever-yl) oxy] hexyl oxy] -4-chewing clever yl] -N ‘- Lai ugly propyl amine (Formula I) Synthesis
[0036] Weigh 20gN-5- (4- desert) -6- (2-2- light hexyl group -) 4- chew clever group -N ‘- Lai ugly propyl amine, 200ml dried DMS0 added to 1L H jar, add 20g of alcohol t-clock was added in portions, then add 17. 7g5- desert – dichloro chew clever, 30-4 (TC reduction reaction, the reaction and the reaction solution. a 10% sample skillfully acid to adjust PH value 3 to 4, the reaction mixture was added to 1000ml water, olive mix, suction. suction Massey cefotetan get wet crude product 42g, 450ml of methanol was added at room temperature and then beating 20min, filtration and dried 45C to give white solid was dried under vacuum to give 23.2 Marcy cefotetan yield;.. 85%
[0037] The compound (Formula I) relating to the physical and chemical properties, spectroscopic data are as follows:
[0038] branded point; 135-136 ° C; we NMR (300MHz, DMS0) 5 (egg m):… 9 8 (s, lH), 8 7 (s, 2H), 8 5 (s, l H,) 7. 5 (s, 2H), 7. 2 (s, IH), 7. 1 (s, 2H,) 4. 7 (s, 2H), 4. 6 (s, 2H,) 2. 8 (s, 2H,), 1. 5 (m, 2H,), 0. 81 (m, 3H), MS Qiaoqiao m / z 589 ([M + Tin +).
…………
see
WO 2002053557
http://www.google.com/patents/WO2002053557A1?cl=en
………..


Assignment of the signals mentioned in the text of the H-NMR spectrum of the drug Macitentan
Solvent: CDCl 3
δ 8.51 (s, 2H, CH) 11 , 8.49 (s, 1 H, CH) 10 , 7.58 to 7.63 (m, 2H, CH) 9 , 7.16 to 7.21 ( m, 2H, CH) 8 , 6.88 (s, 1H, NH) 7 , 5.61 (t, J = 6.2 Hz, 1H, NH) 6 , 4.72 to 4.76 (m, 2H , CH 2 ) 5 , 4.62 to 4.66 (m, 2H, CH 2 ) 4 , 2.99 (q, J = 6.8 Hz, 2H, CH 2 ) 3 , 1.61 (h, J = 7.3 Hz, 2H, CH2 ) 2 , 0.97 (t, J = 7.4 Hz, 3H, CH 3 ) 1 . [Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 .]

Solvent: CDCl 3
δ 11.6, 22.7, 46.1, 65.3, 65.9, 104.8, 112.4, 123.7, 128.0, 131.7, 133.0, 155.7, 156 , 4, 159.7, 163.5, 166.3. [ Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 . ]
NMR PREDICT BY ME
1H NMR PREDICT

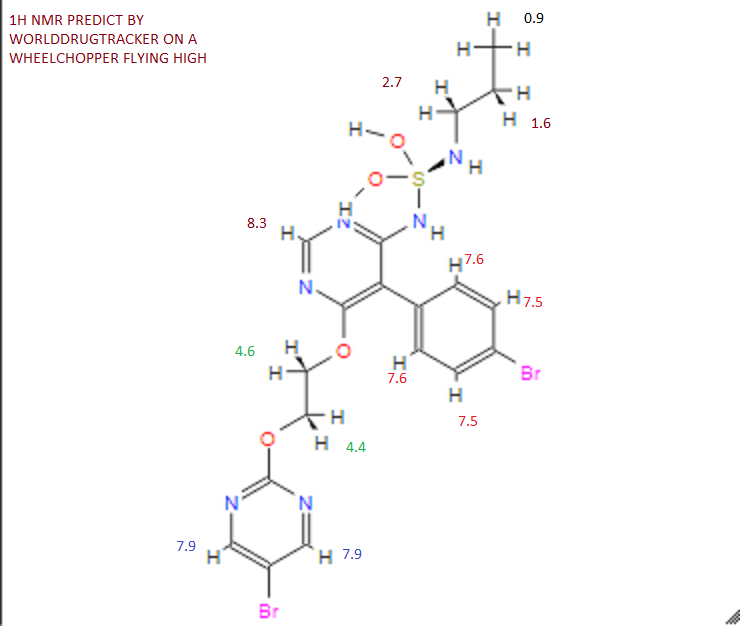
13C NMR PREDICT BY ME


COSY PREDICT BY ME, WORLDDRUGTRACKER ON A WHEELCHOPPER SCALING NEW HEIGHTS
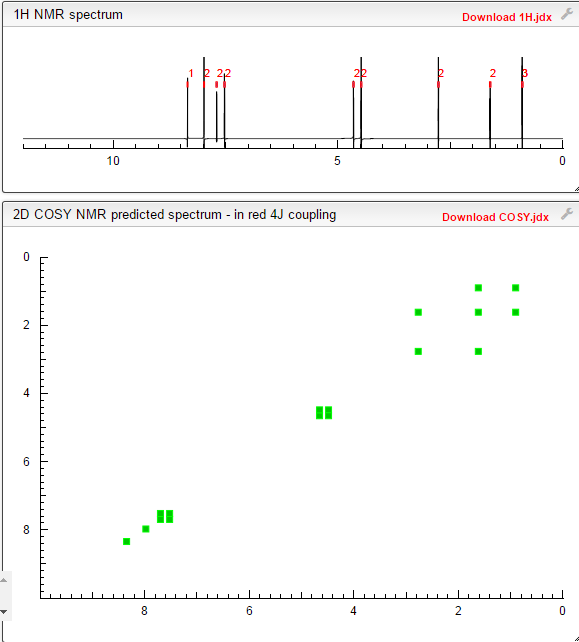
REFERENCES
Actelion Ltd
Actelion Ltd is a biopharmaceutical company with its corporate headquarters in Allschwil/Basel, Switzerland. Actelion’s first drug Tracleer®, an orally available dual endothelin receptor antagonist, has been approved as a therapy for pulmonary arterial hypertension. Actelion markets Tracleer through its own subsidiaries in key markets worldwide, including the United States (based in South San Francisco), the European Union, Japan, Canada, Australia and Switzerland. Actelion, founded in late 1997, is a leading player in innovative science related to the endothelium – the single layer of cells separating every blood vessel from the blood stream. Actelion’s over 2,400 employees focus on the discovery, development and marketing of innovative drugs for significant unmet medical needs. Actelion shares are traded on the SIX Swiss Exchange (ticker symbol: ATLN) as part of the Swiss blue-chip index SMI (Swiss Market Index SMI®).
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide
|
|
| Clinical data | |
| Trade names | Opsumit |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Metabolism | Hydrolysis, oxidation (CYP3A4) |
| Excretion | 2/3 urine, 1/3 faeces |
| Identifiers | |
| CAS Registry Number | 441798-33-0 |
| ATC code | C02KX04 |
| PubChem | CID: 16004692 |
| ChemSpider | 13134960 |
| ChEBI | CHEBI:76607 |
| Synonyms | ACT-064992 |
| Chemical data | |
| Formula | C19H20Br2N6O4S |
| Molecular mass | 588.273 g/mol |
| Patent | Submitted | Granted |
|---|---|---|
| Sulfamides and their use as endothelin receptor antagonists [US7094781] | 2004-04-22 | 2006-08-22 |
| Sulfamides and their use as endothelin receptor antagonists [US7285549] | 2006-08-10 | 2007-10-23 |
| Stable Pharmaceutical Compositions Comprising a Pyrimidine – Sulfamide [US2008233188] | 2008-09-25 | |
| Combination Comprising Paclitaxel for Treating Ovarian Cancer [US2010311774] | 2010-12-09 | |
| Stable pharmaceutical compositions comprising a pyrimidine-sulfamide [US2010004274] | 2010-01-07 | |
| SULFONYLUREA MODULATORS OF ENDOTHELIN RECEPTOR [US2011082151] | 2011-04-07 | |
| ENDOTHELIN RECEPTOR ANTAGONISTS FOR EARLY STAGE IDIOPATHIC PULMONARY FIBROSIS [US2010022568] | 2007-04-12 | 2010-01-28 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2011136818] | 2011-06-09 | |
| Therapeutic Compositions Comprising a Specific Endothelin Receptor Antagonist and a PDE5 Inhibitor [US2009318459] | 2009-12-24 |
Patent and Exclusivity
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
|
|---|---|---|---|---|---|---|---|
| N204410 | 001 | US7094781 | Oct 12, 2022 | Y | Y | ||
| N204410 | 001 | US8268847 | Apr 18, 2029 | U – 1446 | |||
| N204410 | 001 | US8367685 | Oct 4, 2028 | Y | U – 1445 |
| Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N204410 | 001 | ODE | Oct 18, 2020 |
| N204410 | 001 | NCE | Oct 18, 2018 |
U1446 METHOD OF TREATING PULMONARY HYPERTENSION COMPRISING ADMINISTERING MACITENTAN IN COMBINATION WITH A COMPOUND HAVING PHOSPHODIESTERASE-5 INHIBITORY PROPERTIES
U1445 METHOD OF TREATING PULMONARY ARTERIAL HYPERTENSION BY ADMINISTERING A PHARMACEUTICAL COMPOSITION COMPRISING MACITENTAN AND A POLYSORBATE, WHERIN THE POLYSORBATE REPRESENTS 0.1 TO 1% OF THE WEIGHT OF SAID PHARMACEUTICAL COMPOSITION
OPSUMIT (macitentan) is an endothelin receptor antagonist. The chemical name of macitentan is N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide. It has a molecular formula of C19H20Br2N6O4S and a molecular weight of 588.27. Macitentan is achiral and has the following structural formula:
 |
Macitentan is a crystalline powder that is insoluble in water. In the solid state macitentan is very stable, is not hygroscopic, and is not light sensitive.
OPSUMIT is available as a 10 mg film-coated tablet for once daily oral administration. The tablets include the following inactive ingredients: lactose monohydrate, magnesium stearate, microcrystalline cellulose, polysorbate 80, povidone, and sodium starch glycolate Type A. The tablets are film-coated with a coating material containing polyvinyl alcohol, soya lecithin, talc, titanium dioxide, and xanthan gum.
//////

Droxidropa
FDA Deems Resubmission a Complete Response; PDUFA Date Set as
February 14, 2014
CHARLOTTE, N.C., Sept. 4, 2013 (GLOBE NEWSWIRE) — Chelsea Therapeutics International, Ltd. (Nasdaq:CHTP) today announced that the U.S. Food and Drug Administration (FDA) has acknowledged receipt of the New Drug Application (NDA) resubmission seeking approval to market NORTHERA(TM) (droxidopa), an orally active synthetic precursor of norepinephrine
read all at
http://www.pharmalive.com/chelsea-therapeutics-announces-fda-acceptance-of-northera-nda-resubmission
UPDATE………………….

DROXIDOPA
ORPHAN DRUG,
CAS 23651-95-8, 3916-18-5
ROTATION –FORM
|
(2S,3R)-2-amino-3-(3,4-dihydroxyphenyl)-3-hydroxypropanoic acid
|


Droxidopa (INN; trade name Northera; also known as L-DOPS, L–threo-dihydroxyphenylserine, L-threo-DOPS and SM-5688) is a synthetic amino acid precursor which acts as a prodrug to the neurotransmitter norepinephrine (noradrenaline).[1] Unlike norepinephrine, droxidopa is capable of crossing the protective blood–brain barrier (BBB).[1]
CLIP




REF http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/203202Orig1s000ChemR.pdf
Distribution
Droxidopa exhibits plasma protein binding of 75% at 100 ng/mL and 26% at 10,000 ng/mL with an apparent volume of distribution of about 200 L.
Droxidopa starting dose is 100mg three times daily (which can be titrated to a maximum of 600 mg three times daily). One dose should be taken in late afternoon at least 3 hours prior to bedtime to reduce the potential for supine hypertension during sleep.
Droxidopa was developed by Sumitomo Pharmaceuticals for the treatment of hypotension, including NOH,[2] and NOH associated with various disorders such as MSA, FAP, and PD, as well as IDH. The drug has been used in Japan and some surrounding Asianareas for these indications since 1989. Following a merge with Dainippon Pharmaceuticals in 2006, Dainippon Sumitomo Pharmalicensed droxidopa to Chelsea Therapeutics to develop and market it worldwide except in Japan, Korea, China, and Taiwan. In February 2014, the Food and Drug Administration approved droxidopa for the treatment of symptomatic neurogenic orthostatic hypotension.[3]
Chelsea Therapeutics obtained orphan drug status (ODS) for droxidopa in the U.S. for NOH, and that of which associated with PD, PAF, and MSA. In 2014, Chelsea Therapeutics was acquired by Lundbeck along with the rights to droxidopa which was launched in the US in Sept 2014.[4]
REGULATORY
CLICK ON IMAGE TO VIEW
FDA agreement on overall development program (Sep 2007)
• FDA agreement on Study 301 design under a Special Protocol Assessment (Feb 2008) – Included agreement: length of patient exposure was adequate for the safety evaluation
• FDA agreement on changing primary endpoint of Study 301 while it was ongoing and prior to any unblinding (Nov 2009) – From dizziness to the OHQ – SPA remained intact
• FDA agrees to NDA package (Dec 2010) – Studies 301, 302, 303, 304 and 305 – Renal safety study conducted post-marketing
• FDA accepts droxidopa NDA and grants priority review (Sep 2011)
Droxidopa is a prodrug of norepinephrine used to increase the concentrations of these neurotransmitters in the body and brain.[1][What, if any, are the other neurotransmitters droxidopa increases concentrations of? “These neurotransmitters” implies multiple(see above)] It ismetabolized by aromatic L-amino acid decarboxylase (AAAD), also known as DOPA decarboxylase (DDC). Patients with NOH have depleted levels of norepinephrine which leads to decreased blood pressure or hypotension upon orthostatic challenge.[5] Droxidopa works by increasing the levels of norepinephrine in the peripheral nervous system (PNS), thus enabling the body to maintain blood flow upon and while standing.
Droxidopa can also cross the blood–brain barrier (BBB) where it is converted to norepinephrine from within the brain.[1] Increased levels of norepinephrine in the central nervous system (CNS) may be beneficial to patients in a wide range of indications. Droxidopa can be coupled with a peripheral aromatic L-amino acid decarboxylase inhibitor (AAADI) orDOPA decarboxylase inhibitor (DDC) such as carbidopa (Lodosyn) to increase central norepinephrine concentrations while minimizing increases of peripheral levels.
With over 20 years on the market, droxidopa has proven to have few side effects of which most are mild. The most common side effects reported in clinical trials include headache, dizziness nausea, hypertension and fatigue.[6][7][8][8]
L-threo-dihydroxyphenylserine, also known as droxidopa, L-threo-DOPS, or L-DOPS, is an orally active synthetic precursor of norepinephrine. Droxidopa thus replenishes depleted norepinephrine, allowing for re-uptake of norepinephrine into peripheral nervous system neurons. This reuptake, in turn, stimulates receptors for vasoconstriction, providing physiological improvement in symptomatic neurogenic orthostatic hypotension patients. It has also shown efficacy in other diseases, such as Parkinson’s disease and depression.
Droxidopa has been used in Japan for many years for the treatment of orthostatic hypotension. It was originally approved in 1989 for the treatment of frozen gait or dizziness associated with Parkinson’s disease and for the treatment of orthostatic hypotension, syncope or dizziness associated with Shy-Drager syndrome and Familial Amyloidotic Polyneuropathy.
Marketing approval was later expanded to include treatment of vertigo, dizziness and weakness associated with orthostatic hypotension in hemodialysis patients.
The preparation of droxidopa generally involves a multi-step synthesis. Typically, one or more of the necessary steps in the synthesis requires that reactive sites other than that site targeted for reaction are temporarily protected. Thus, the synthesis of droxidopa typically comprises at least one protecting and associated deprotecting step. For example, the catechol moiety, the amine moiety, and/or the carboyxyl moiety may require protection and subsequent deprotection, depending upon the synthetic route and the reagents used in the preparation of droxidopa.
U.S. Patent Nos. 4,319,040 and 4,480,109 to Ohashi et al. describe processes for the preparation of optically active D- and L- threo-DOPS by optically resolving a racemic mixture of threo-2-(3,4-methylenedioxyphenyl)-N-carbobenzyloxyserine or threo-2-(3,4-dibenzyloxy-phenyl)- N-carbobenzyloxyserine, respectively. Following optical resolution of these racemic mixtures to give the desired L-enantiomer, the methylene or benzyl groups must be removed from the catechol moiety and the carbobenzyloxy (CBZ) group must be removed from the amine group to give droxidopa. The methylene group can be readily removed by reaction with a Lewis acid {e.g., aluminum chloride). The CBZ group (and the benzyl catechol protecting groups, where applicable) is removed from the amine by hydrogenolysis to give the desired compound. The hydrogenolysis step is noted to be carried out by treating the optically resolved salt with hydrogen in the presence of a catalyst, e.g., palladium, platinum, nickel, or the like.
However, for large-scale production of pharmaceutical compounds, hydrogenolysis may not be desirable. For example, hydrogenolysis requires expensive, specialized equipment, which represents a large capital investment. Labor costs are also high, as the process requires careful handling and disposal of certain compounds (e.g., the pyrophoric catalyst). Further, due to the hazards associated with both the reagents and the high pressure system required for hydrogenolysis, it is desirable to avoid synthetic methods that require hydrogenolysis.
In an alternative method for the production of droxidopa, taught by U.S. Patent No.
4,562,263 to Ohashi et al, hydrogenation is not required. In this process, the amine group is protected via a phthaloyl group. Following optical resolution, the phthaloyl group is removed from the droxidopa precursor by hydrazine.
However, hydrazine is known to be genotoxic and has been classified by the EPA as a
Group B2 probable human carcinogen. Thus, it is desirable to remove even trace amounts of hydrazine from pharmaceutical compounds. In practice, the method described in the ‘263 patent suffers from the inability to remove 100% of the hydrazine from the final product. Thus, there is some level of contamination by hydrazine using this method. The Food and Drug Administration has established a maximum genotoxic impurity level of 1.5 micrograms per day. Therefore, based on the maximum daily dose of droxidopa (1.8 g), the maximum allowable hydrazine level therein is 0.8 ppm. Accordingly, it would be advantageous to find a new synthetic route for the preparation of droxidopa that avoids the use of hydrogenolysis and also avoids the use of hydrazine
https://www.google.ch/patents/WO2013142093A1?cl=en
The synthetic route for the preparation of droxidopa comprises the following steps: a) converting piperonal to 2-amino-3-(benzo-l,3-diox-5-yl)-3-hydroxypropanoic acid

c) optical resolution and separation of the desired isomer



Experimental Section
Example 1 : Screening of Deprotection Strategies for Phthaloyl Group

Example 2: Exemplary Synthesis of Droxidopa
The synthesis of droxidopa according to the methods provided herein can be conducted as a continuous process or can be conducted in a series of individual steps. Both processes are intended to be encompassed by the present disclosure.
Synthesis of N-carbomethoxy phthalimide

3-Methoxy phthalimide 1 (120 kg) is added to a vessel containing dimethylformamide (420 L) and stirred (95 ± 10 RPM) at 25 – 30 °C for 30 min. The contents are cooled to 18 – 20 °C and triethylamine (124 L) is added. The contents are further cooled to -10 °C to -5 °C and
methylchloroformate 2 (85 kg) is added. The reaction temperature is maintained in the range of -10 °C to 0 °C to control the exothermicity during the addition of methylchloroformate. The temperature of the mixture is maintained at 0 – 5 °C for 1 h after the addition of
methylchloroformate .
The reaction mixture is then heated to 25 – 30 °C for 1 h. An in-process sample is taken to confirm a phthalimide content limit <2.5%. The mixture is sampled again to confirm a phthalimide content <0.5%. The mixture is transferred to another reactor, cooled to 0 – 5 °C, and the reaction is quenched with the addition of demineralized water (1260 ± 10 L) at a temperature of 10 ± 5 °C. The mixture is then heated at 25 – 30 °C for 1 h.
The material is centrifuged for 2 h and the wet cake is washed three times with
demineralized water (360 L). The wet cake is dried at a temperature of 55 – 60 °C and a sample is taken after 12 h of drying to confirm water content <1.0% w/w.
Expected yield of N-carbomethoxy phthalimide (3): 144-158 kg. This material is not isolated and is used directly in the next step.
Synthesis of 2-amino-3-(benzo-l ,3-dioxol-5-yl)-3-hydroxypropanoic acid

Piperonal 4 (229 ± 1 kg) is added to toluene (310 ± 5 L) in a reactor and the mixture is stirred (85 – 95 RPM) until a clear solution is obtained (approximately 30 min). The piperonal solution is transferred to a vessel for later use. Methanol (415 ± 5 L) is added to the reactor followed by the addition of potassium hydroxide (85 kg). The mixture is stirred for approximately 30 min at 25 – 30 °C to provide a clear solution. The potassium hydroxide solution is cooled to 20 – 25 °C, and then glycine 5 (52 ± 1 kg) and toluene (310 ± 5 L) are added while stirring at 20 – 25 °C. The contents of the reactor are cooled to 15 – 20 °C. The solution of piperonal in toluene is slowly added to the reactor while maintaining the temperature at 15 – 20 °C. The reactor temperature is increased to 20 – 25 °C and maintained for 18 h. An in-process sample is taken to determine glycine content by TLC (limit <5.0%).
The reaction mass is transferred to another reactor, the temperature is increased to 40 °C, and the solvents (toluene and methanol) are distilled off under vacuum until the mixture becomes thick. Additional toluene (210 ± 5L) is added to the reaction mass three times and distilled out for complete removal of methanol and toluene. The reaction mixture is kept under vacuum at 40 °C. After 3 h, the reaction mixture is cooled to 18 – 22 °C and a dilute hydrochloric acid solution (230 ± 5L hydrochloric acid and 1145 ± 10 L demineralized water) is added and mixed for 30 min.
The mixture is allowed to settle for 30 min to separate into organic and aqueous layers. The aqueous layer is washed with toluene (310 ± 5 L) and separated. Glacial acetic acid (218 ± 2 kg) is added to the washed aqueous layer at 20-25 °C. Caustic solution (580 ± 5 L DM Water and 200 ± 1 kg caustic flakes) is slowly added into the reaction mass to bring the pH 5.0 to 5.1 while maintaining the temperature at 25 – 30 °C. The pH of the mixture is brought to 5.45 – 5.50 at 25 – 30 °C, while stirring for 30 min. The mixture is centrifuged for 8 h 30 min to 9 h and the resulting wet cake is washed with demineralized water (50 ± 5 L). The cake is dried at 50 – 55 °C under vacuum, and a sample is taken after 12 h to confirm that water content is <10% w/w. The purity is analyzed by HPLC (limit < 10%).
Expected yield of 2-amino-3-(benzo-l ,3-dioxol-5-yl)-3-hydroxypropanoic acid (6):
135 – 145 kg.
Synthesis of 2-phthalimido-3-hydroxy-3-(3,4-methylenedioxyphenvnpropionic acid

Intermediate 6 (140 kg) is added to a reactor containing demineralized water (1 120 L) and stirred (85-95 RPM) for 10 min at 20-25 °C. The contents are cooled to 15-20 °C and compound 3 (140 kg) is added followed by a sodium carbonate solution (63.5-68.3 kg sodium carbonate in 189-203 L demineralized water) within 45-60 min. The mixture is heated to 30-35 °C and held for 90 min. An in-process sample is taken to measure for Stage II (<2.5%) and Stage-I intermediate (<2.5 %). After acceptance criteria are met, the mixture is cooled to 15-20 °C. A dilute sulfuric acid solution (134 kg sulfuric acid in 1120 L demineralized water) at 15-20 °C is added to the mixture to bring the pH to 1.0-2.0. The mixture is maintained at this temperature and pH for 30 min, and then the mixture is heated to 20-25 °C for 2 h.
The mixture is centrifuged for 9 h and the resulting wet cake is washed twice with 518 L of demineralized water. The wet cake is removed from the centrifuge, washed in a reactor containing demineralized water (2590 L), and stirred for 1 h at 25-30 °C. The material is centrifuged for 9 h and the wet cake is washed twice with demineralized water (518 L). The final wet cake is dried at 45-50 °C under vacuum until water content is <1.0% w/w. Intermediate (6) output is considered as standard input and a mean of 140 kg is taken for all inputs.
Expected yield of 2-Phthalimido-3-hydroxy-3-(3,4-methylenedioxyphenyl)propionic acid (7): 187 – 208 kg.
Synthesis of L-threo (N-phthaloyl-3-(3,4-methylenedioxyphenyl)serine) norephedrine salt

L-Norephedrine 8 (89 kg) is added to a reactor containing methanol (296 L) and stirring (45-50 RPM) is started. The mixture is maintained at 25 – 30 °C for 15 – 20 min, and then transferred into a vessel for later use.
2-Phthalimido-3-hydroxy-3-(3,4-methylenedioxyphenyl)propionic acid 7 (197.5 kg) is added to a reactor containing methanol (395 L). The material is stirred for 15 – 20 min at 25 – 30 °C. The L-norephedrine solution is added and mixed for 3 h. If precipitation is not observed within 30 min of adding the L-norephedrine solution, it is seeded with L-threo(N-phthaloyl-3-(3,4- methylenedioxyphenyl) serine (approximately 50 g). After 3 h of mixing, the mixture is cooled to 10 – 15 °C and maintained for 1 h. An in-process sample is taken to check for purity by HPLC (>99.0% a/a). The mixture is centrifuged for 1 h to 1 h 30 min and the wet cake is washed with methanol (49 L) followed by isopropyl alcohol (197.5 L). The wet cake is checked for purity. If purity is <99% a/a, the wet cake is washed with a prechilled solution of methanol (197.5 L) followed by isopropyl alcohol (99 L). After achieving the required purity level, as measured by HPLC, the wet cake is removed from the centrifuge. The cake is dried at 45 – 50 °C until loss on drying <1.0% w/w.
Expected yield of L-threo (N-phthalpyl-3-(3,4-methylenedioxyphenyl)serine) norephedrine (9) salt: 85-99 kg.
Synthesis of L-threo flSi-phthaloyl-S-CS^-methylenedioxyphenvDserine)

Demineralized water (552 L) is added to a reactor and cooled to 10 – 15 °C. Sulfuric acid (20 kg) is added while maintaining the temperature below 30 °C and stirring for 15 – 20 min. The solution is cooled to 15 – 20 °C and 9 (92 kg) is slowly added while stirring and maintaining temperature. The solution is heated to 45 – 50 °C for 6 h, cooled to 25 – 30 °C, and held for 1 h. The pH is checked to confirm the solution is <2.0.
The mixture is centrifuged for 1 h and the wet cake is washed two times with demineralized water (138 L). The wet cake is removed and added to a reactor containing demineralized water (460 L). The temperature is maintained at 25 – 30 °C and stirred for 1 h. The material is centrifuged for 30 min and the wet cake is washed two times with demineralized water (138 L). The material is collected and placed into preweighed containers.
Expected yield of L-threo(N-phthaloyl-3-(3,4-methylenedioxyphenyl)serine) (10): 60-64 kg.
Synthesis of L-threo(N-phthaloyl-3-(3 ,4-dihydroxyphenyl serine)

Compound 10 (62 kg) wet cake is added to a reactor containing methylene chloride (1240 L) and stirred for 10 min. The mixture is heated to remove methylene chloride and water under azeotropic reflux. After methylene chloride (1550 L) is removed and no water remains in the distillate, the mixture is cooled to 25 – 30 °C. An in-process sample is taken to determine water content (limit <0.1 %).
Methylene chloride (186 L) is added to another reactor at 25-30 °C. An in-process sample is taken to check for water content (limit <0.2% w/w). Aluminum chloride (81 kg) is added and the contents are stirred at 25 – 30 °C for 10 – 15 min. The mixture is cooled to 10 – 15 °C and octanethiol (78 kg) is added. The mixture is cooled to -20 to -10 °C.
The slurry of 10 in methylene chloride controlled at -20 to -7 °C is added to the stirred mixture that is temperature controlled at -15 to -10 °C for 20 – 30 min. The mixture is heated to 10-15 °C for 1.5-2.5 h. An in-process sample is taken to determine 10 content (limit <3.5%). The mixture is further cooled to -20 to -10 °C and then transferred to another reactor containing oxalic acid (62 kg), methylene chloride (186 L), and demineralized water (744 L) while maintaining the temperature below -3 °C to quench the reaction. The quenched material is slowly heated to 25 – 30 °C and maintained at this temperature for 12 h. Methylene chloride is distilled out at 25 – 30 °C under vacuum until the mixture volume is reduced to 1364 L.
The mixture is centrifuged for 3 h and the wet cake is washed with demineralized water (62 L). The wet cake is added to a reactor containing oxalic acid (2.5 kg) and demineralized water (248 L) and the contents are stirred at 25-30 °C for 2 h to obtain a clear solution. The material is centrifuged for 1 h 15 min to 1 h 30 min and the wet cake is washed twice with demineralized water (186 L). The wet cake is added to a reactor containing demineralized water (248 L) at 25 – 30 °C and the contents are stirred for 2 h. The material is centrifuged for 1 h 30 min to 2 h and the wet cake is washed twice with demineralized water (186 L). The material is collected and placed into preweighed containers.
Expected yield of L-threo(N-phthaloyl-3-(3,4-dihydroxyphenyl)serine (11): 40-50 kg. Synthesis of L-threo (3,4-dihydroxyphenvP)serine

Methanol (360 L) is added to a reactor and cooled to 20 – 25 °C. Compound 11 (45 kg) is added to the reactor while stirring at 25 – 30 °C for 15 – 20 min. Demineralized water (225 L) and sodium bicarbonate (17 kg) are added to another reactor and cooled to 20 – 25 °C. Hydroxylamine hydrochloride (14 kg) is added and mixed for 15 – 20 min at 20 – 25 °C to obtain a clear solution. The solution of 11 in methanol is transferred through a sparkler filter into a reactor. The hydroxylamine and sodium bicarbonate solution is added to the reactor while maintaining the temperature at 25 – 30 °C. The reaction mixture is heated to 65 – 70 °C and refluxed for 16 h. An in-process sample is taken to determine 11 content (limit <3%). The material is cooled to 25-30 °C with mixing for 2 h.
The material is centrifuged for 1 h and the wet cake is washed three times with methanol (23 L). The wet cake is dried at 40 – 45 °C until water content is <1.0% w/w.
Expected yield of L-threo(3,4-dihydroxyphenyl)serine (12): 20-24 kg. Synthesis of L-threo(3,4-dihvdroxyphenyl)serine hydrochloride

L-threo (3,4-dihydroxyphenyl)serine 12 (22 kg) material is added to a reactor containing demineralized water (55 L) and stirred for 15-30 min. The material is cooled to 20-25 °C and concentrated hydrochloric acid (13 L) is added to form L-threo(3,4-dihydroxyphenyl)serine hydrochloride) (13). The mixture is stirred for 30^15 min until a white thick suspension is observed. The mixture is stirred for an additional 2.0 h ± 15 min. Isopropyl alcohol (132 L) is slowly added and the mixture is stirred for 5 hr ± 15 min. The mixture is cooled to 15-20 °C and stirred for 30^5 min. The mixture is centrifuged for 30 min and the wet cake is washed twice with chilled isopropyl alcohol (22 L) at 15-20 °C. The material is unloaded from the centrifuge and a sample is taken to check the individual impurity by HPLC (limit <0.05%) and purity by HPLC (limit >99.0%).
Reprocessing: If the individual impurity by HPLC does not meet the limit <0.05%, compound 13 is reprocessed by adding the material to a reactor containing demineralized water (28 L) and stirring for 15 – 30 min. Concentrated hydrochloric acid (3 L) is added at 20-25 °C and mixing is continued for 15 – 30 min. Continue mixing for 2 h ± 15 min at the same temperature. Isopropyl alcohol (74 L) is added over a period of 2 – 3 h at 25 – 30 °C. Mixing is continued at 25 – 30 °C for 5 h ± 15 min followed by cooling to 15 – 20 °C and mixing for 30 – 45 min. The mixture is centrifuged for 30 min and washed twice with chilled isopropyl alcohol (22 L) and checked for the individual impurity by HPLC (limit <0.05%).
Expected yield of L-threo(3,4-dihydroxyphenyl)serine hydrochloride) (13): 19-20 kg. Synthesis of L-threo(3,4-dihvdroxyphenyl)serine

Compound 13 (19.5 kg) is added to a reactor containing demineralized water (195 L) while stirring at 25 – 30 °C. Concentrated hydrochloric acid (6 L) is added and mixed for 25 – 30 min. For complete dissolution, the contents can be mixed for another 15 – 20 min. Activated carbon (1 kg) and celite (0.2 kg) are added and mixed for 30 – 40 min. The mixture is filtered through a sparkler filter and the filter is washed with demineralized water (1 X L). The filtrate is transferred to another reactor. A solution containing triethylamine (14 kg) and methanol (41 L) is slowly added to the reaction mass (reactor) while mixing. The pH of the filtrate is adjusted to 7.0 – 7.25 over a period of 3 h at 25 – 30 °C. The contents are stirred for 20-30 min. An in-process sample is taken to confirm the pH is 7.0 – 7.25. The mixture is stirred for 3 h. The mixture is centrifuged for 1 h and the wet cake is washed twice with demineralized water (19.5 L). The wet cake is removed from the centrifuge and kept for a slurry wash. The wet cake is added to a reactor containing methanol (58.5 L) while stirring at 25 – 30 °C for 30 – 40 min. The material is centrifuged for 1 h and the wet cake is washed with methanol (19.5 L). The wet cake is unloaded from the centrifuge and retained for water washing.
The wet cake is added to a reactor containing demineralized water (39 L) while stirring for 30 – 40 min. The material is centrifuged for 10 min and the wet cake is washed twice with methanol (19.5 L). The wet cake is unloaded and a sample is taken to check the chloride content (<200 ppm). The wet cake is dried at 40 – 45 °C until the water content is <0.1 % w/w. A sample is taken after 16 h of drying to confirm loss on drying is <0.1% w/w. The dry material is sieved through a sifter (400 micron) and packed. A sample is taken for quality control testing.
Expected yield of L-threo(3,4-dihydroxyphenyl)serine: 14-15 kg.
Scheme 1: Overview of Droxidopa Synthesis
a) converting piperonal to 2-amino-3-(benzo-l,3-diox-5-yl)-3-hydroxypropanoic acid

d) removal of the catechol protecting group


https://www.google.com/patents/CN103086906A?cl=en
droxidopa, the English name Droxidopa, chemical name (_) _ (2S, 3R) _2_ amino _3_ hydroxy-3- (3,4-hydroxyphenyl) propionic acid, the formula is as follows:

· It is a synthetic (-) _ norepinephrine precursor amino acids by intestinal absorption and metabolism of norepinephrine, in patients with Parkinson’s disease is mainly used to improve gait stiffness and postural dizziness, and for Treatment of orthostatic hypotension drugs.
JP 09-031038 discloses droxidopa preparation, preparation process is as follows:

That structural formula I obtained in the presence of a copper catalyst structure formulas II, and then the open-loop, elimination of R and X groups of structure III of droxidopa.
JP05 – 239025 also provides a droxidopa preparation, the preparation process is as follows.

JP 59-055861 discloses a method of droxidopa preparation of optically active substances: Xi Qu racemic 多巴加 heat in water to form a saturated solution, the first addition to control the amount of optical after cooling Activity droxidopa as a seed, heat after the second cooling crystallize. JP 64-022849 discloses a method for purifying droxidopa: A mixture of alcohol solvents crude droxidopa was dissolved in water and inorganic acid, and then with an organic or inorganic base to neutralize, to precipitate crystals obtained purification. JP 59-055861 claims to obtain optically active by the method droxidopa, and JP64-022849 purification process is not considered to be heated, to avoid caused by heating droxidopa degradation.
Example 1
Preparation L- threo-3- (3,4-dihydroxyphenyl) serine 1: 300kg methanol successively and LN- carbonyl benzyloxy-3- (3,4-benzyloxyphenyl) serine Add 30kg 500L hydrogenation reactor, added dropwise 3mol / L hydrochloric acid to dissolve the solid, was added 5% palladium carbon 8kg, introducing hydrogen pressure was maintained at 0.02Mpa, the reaction temperature is controlled at 40 ± 5 ° C, 6 hours After discharge, the addition of concentrated hydrochloric acid 7kg and 0.3kg activated carbon and stirred for 20 minutes, filtration, the filtrate with 30% NaOH aqueous solution adjusted to pH 6-7, filtered crystallization two hours, that was (reaction formula below).

L- Su _3_ (3,4-light-phenyl) Preparation of silk atmosphere acid 2:
Dad was added to the reaction in ethanol 100kg, was added under stirring L- threo-3- (3,4-light-phenyl) -3-light-2 phthalamide imino acid, stirred at room temperature until The solid was dissolved clear, liquid solution of hydrazine hydrate at 40-45 ° C, after completion of the addition of hydrazine hydrate, the reaction was refluxed for 16 hours, cooled to below 30 ° C, concentrated hydrochloric acid was added dropwise 30kg, maintaining 30 ° C under stirring for I hour, Rejection filter cake washed once with aqueous hydrochloric acid, and the filtrate with 30% NaOH solution pH was adjusted to 6_7, filtered crystallization two hours, that was (reaction formula below).


The condensation of 3,4-dimethoxybenzaldehyde (I ) with glycine (II) by means of KOH in hot methanol gives racemic threo-3- (3,4-dimethoxyphenyl) serine (III), which is acylated with N – (ethoxycarbonyl) phthalimide (IV) by means of Na2CO3 in water yielding the corresponding N-phthaloyl derivative (V) The reaction of (V) with AlCl3 and ethyl mercaptan in dichloromethane affords N-phthaloyl-3- (3,4-. dihydroxyphenyl) serine (VI), which is deprotected with hydrazine in refluxing ethanol to racemic threo-3- (3,4-dihydroxyphenyl) serine (VII). The resolution of the racemic form (VII) is performed through its benzyloxy derivative.
| CN101657193A * | Mar 7, 2008 | Feb 24, 2010 | 切尔西治疗公司 | Droxidopa and pharmaceutical composition thereof for the treatment of fibromyalgia |
| EP0141613A2 * | Oct 24, 1984 | May 15, 1985 | Zaidan Hojin Biseibutsu Kagaku Kenkyu Kai | A process for producing an optically active 3-(3,4-dihydroxphenyl) serine and a proteced derivative thereof |
| JPS6422849A * | Title not available | |||
| JPS60160895A * | Title not available | |||
| WO2005085178A1 * | Mar 4, 2005 | Sep 15, 2005 | Estechpharma Co., Ltd. | Method of preparing optically active serine derivative |
| WO2006123678A1 * | May 17, 2006 | Nov 23, 2006 | Dainippon Sumitomo Pharma Co., Ltd. | Stable tablet containing droxidopa |
| WO2011001976A1 * | 29. Juni 2010 | 6. Jan. 2011 | Dainippon Sumitomo Pharma Co., Ltd. | Method for producing threo-3-(3,4-dihydroxyphenyl)-l-serine |
| US4319040 | 18. Juli 1980 | 9. März 1982 | Sumitomo Chemical Company, Limited | Process for the production of optically active threo-3-(3,4-dihydroxyphenyl)serine |
| US4480109 | 3. Jan. 1983 | 30. Okt. 1984 | Sumitomo Chemical Company, Limited | Process for producing threo-3-(3,4-dihydroxyphenyl)serine |
| US4562263 | 25. Mai 1984 | 31. Dez. 1985 | Sumitomo Chemical Company, Limited | Process for producing 3-(3,4-dihydroxyphenyl) serine |
| US20080015181 | 28. Juni 2007 | 17. Jan. 2008 | Chelsea Therapeutics, Inc. | Pharmaceutical Compositions Comprising Droxidopa |
| US20080221170 | 7. März 2008 | 11. Sept. 2008 | Chelsea Therapeutics, Inc. | Droxidopa and pharmaceutical composition thereof for the treatment of fibromyalgia |
| US20080227830 | 12. März 2008 | 18. Sept. 2008 | Chelsea Therapeutics, Inc. | Droxidopa and pharmaceutical composition thereof for the treatment of neurally mediated hypotension |
| US20090023705 | 7. Mai 2008 | 22. Jan. 2009 | Chelsea Therapeutics, Inc. | Droxidopa and pharmaceutical composition thereof for the treatment of mood disorders, sleep disorders or attention deficit disorders |
| Referenz | ||
|---|---|---|
| 1 | * | “Protection for the amino group” In: PETER G M WUTS; THEODORA W GREENE: “GREENE’S PROTECTIVE GROUPS IN ORGANIC SYNTHESIS,“, 2007, WILEY-Interscience,, HOBOKEN, NJ, USA, XP002685963, ISBN: 978-0-471-69754-1 page 696-700, 790-793, 799-802, the whole document |
| 2 | * | A. ARIFFIN ET AL.: “Suggested Improved Method for the Ing-Manske and Related Reactions for the Second Step of Gabriel Synthesis of Primary Amines“, SYNTHETIC COMMUNICATIONS: AN INTERNATIONAL JOURNAL FOR RAPID COMMUNICATION OF SYNTHETIC ORGANIC CHEMISTRY, vol. 34, no. 24, 2004, pages 4439-4445, XP002695538, Taylor & Francis Inc. ISSN: 0039-7911 |
| 3 | T. W. GREEN; P. G. M. WUTS: ‘Protective Groups in Organic Synthesis‘, vol. 583-584, 1999, WILEY-INTERSCIENCE pages 744 – 747 | |
For Immediate Release: Feb. 18, 2014
FDA approves Northera to treat neurogenic orthostatic hypotension
The U.S. Food and Drug Administration today approved Northera capsules (droxidopa) for the treatment of neurogenic orthostatic hypotension (NOH). NOH is a rare, chronic and often debilitating drop in blood pressure upon standing that is associated with Parkinson’s disease, multiple-system atrophy, and pure autonomic failure.
Symptoms of NOH include dizziness, lightheadedness, blurred vision, fatigue and fainting when a person stands.
“People with neurogenic orthostatic hypotension are often severely limited in their ability to perform routine daily activities that require walking or standing,” said Norman Stockbridge, M.D., Ph.D, director of the Division of Cardiovascular and Renal Drugs in the FDA’s Center for Drug Evaluation and Research. “There are limited treatment options for people with NOH and we are committed to helping make safe and effective treatments available.”
The FDA is approving Northera under the accelerated approval program, which allows for approval of a drug to treat a serious disease based on clinical data showing that the drug has an effect on an intermediate clinical measure (in this case, short-term relief of dizziness) that is reasonably likely to predict the outcome of ultimate interest (relief of dizziness during chronic treatment). This program provides patient access to promising drugs while the company conducts post-approval clinical trials to verify the drug’s clinical benefit, which for this approval is a long-term effect on patient symptoms in NOH, a chronic disease.
Northera has a boxed warning to alert health care professionals and patients about the risk of increased blood pressure while lying down (supine hypertension), a common problem that affects people with primary autonomic failure and can cause stroke. It is essential that patients be reminded that they must sleep with their head and upper body elevated. Supine blood pressure should be monitored prior to and during treatment and more frequently when increasing doses.
The most common adverse events reported by clinical trial participants taking Northera were headache, dizziness, nausea, high blood pressure (hypertension) and fatigue.
The effectiveness of Northera was shown through two-weeks in two clinical trials in people with NOH. People taking Northera reported a decrease in dizziness, lightheadedness, feeling faint, or feeling as if they might black out compared to those taking an inactive pill (placebo). Durability of the improvement in patient symptoms beyond two weeks has not been demonstrated.
Northera received orphan-product designation from the FDA because it is intended to treat a rare disease or condition.
Northera is made by Charlotte-based Chelsea Therapeutics Inc.
For more information:
FDA: Approved Drugs
FDA: Drug Innovation
National Institute of Neurological Disorders and Stroke: Orthostatic Hypotension
|
|
| Systematic (IUPAC) name | |
|---|---|
|
(2S,3R)-2-Amino-3-(3,4-dihydroxyphenyl)-3-hydroxypropanoic acid
|
|
| Clinical data | |
| Trade names | Northera |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 90% |
| Metabolism | Hepatic |
| Biological half-life | 1.5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | 23651-95-8 |
| ATC code | C01CA27 (WHO) |
| PubChem | CID 6989215 |
| ChemSpider | 83927 |
| UNII | J7A92W69L7 |
| ChEBI | CHEBI:31524 |
| ChEMBL | CHEMBL2103827 |
| Synonyms | β,3-Dihydroxytyrosine |
| Chemical data | |
| Formula | C9H11NO5 |
| Molar mass | 213.18734 g/mol |
NORTHERA capsules contain droxidopa, which is a synthetic amino acidprecursor of norepinephrine, for oral administration. Chemically, droxidopa is (–)-threo-3-(3,4- Dihydroxyphenyl)-L-serine. It has the following structural formula:
 |
Droxidopa is an odorless, tasteless, white to off-white crystals or crystalline powder. It is slightly soluble in water, and practically insoluble in methanol, glacial acetic acid, ethanol, acetone, ether, and chloroform. It is soluble in dilute hydrochloric acid. It has a molecular weight of 213.19 and a molecular formula of C9H11NO5.
NORTHERA capsules also contain the following inactive ingredients: mannitol, corn starch, and magnesium stearate. The capsule shell is printed with black ink. The black inks contain shellac glaze, ethanol, iron oxide black, isopropyl alcohol, n-butyl alcohol, propylene glycol, and ammonium hydroxide. The capsule shell contains the following inactive ingredients: 100 mg – gelatin, titanium dioxide, FD&C Blue No. 2, black and red iron oxide; 200 mg – gelatin, titanium dioxide, FD&C Blue No. 2, black and yellow iron oxide; 300 mg – gelatin, titanium dioxide, FD&C Blue No. 1, FD&C Yellow No. 5 (tartrazine), and FD&C Red No. 40. NORTHERA capsules differ in size and color by strength
CLIP
//////////DROXIDOPA, Chelsea Therapeutics, orphan drug status, FDA 2015, 3916-18-5, NORTHERA, SUMITOMO, Antiparkinsonian
THESIS
http://ncl.csircentral.net/668/1/th1739.pdf
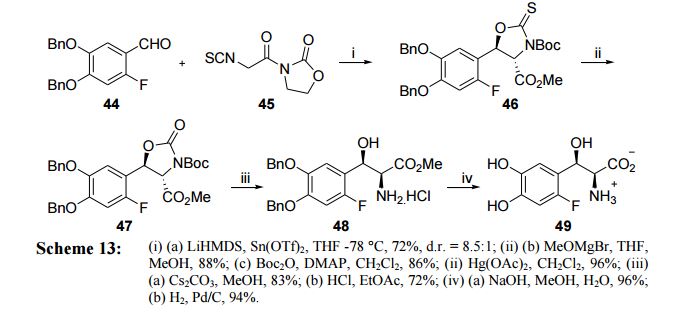
Review of Literature Literature search showed that there are only few reports available,42-44 which describe the synthesis of L-threo-DOPS (43) as detailed below. Kirk’s approach (2001)43 Kirk et. al have reported the synthesis of fluorinated analogue of L-threo-DOPS (49) starting from reaction of aldehyde 44 with isothiocyanate 45 in presence of LHMDS and Sn(OTf)2 to give ester 46 with d.r = 8.5:1. Subsequently, removal of chiral auxiliary with methoxymagnesium bromide followed by Boc protection of the thiocarbamate nitrogen was carried out. Thiocarbamate 46 was then converted to oxygen analogue 47 in 96% yield by treating it with Hg(OAc)2. This was followed by subsequent cleavage of 47 using Cs2CO3 and its Boc depotection gave the ester 48. Saponification of ester 48 followed by hydrogenation afforded fluoro analogue of L-threo-DOPS (49) (Scheme 13).
Sang-Ho’s approach (2007)44 Sang-Ho et. al have achieved an enzyme-catalyzed synthesis of L-threo-DOPS (43) by reacting in one-pot glycine, 3,4-dihydrobenzaldehyde, 2-mercaptoethanol, pyridoxal-5- phosphate solution, sodium sulfite and Triton X-100 in presence of E.coli at 15 °C

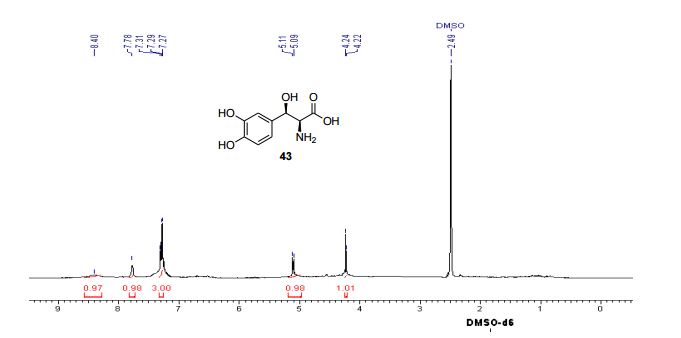
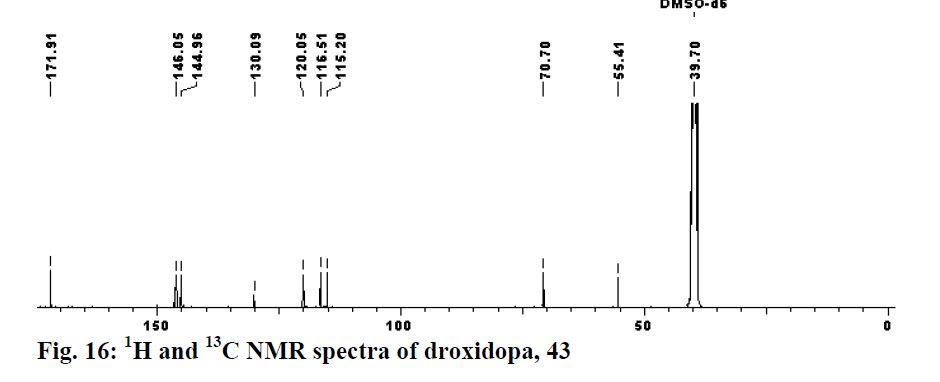
Yield: 94%; mp: 230-233 ºC; (lit.42 mp: 232-235 ºC); [α] 25 D: -38.7 (c 0.4, 1N aq. HCl); {lit.42 [α] 25 D: -39 (c 1, 1N HCl)}; IR (CHCl3, cm-1 ): 3018, 2399, 2366, 2345, 1652, 1519, 1215, 1018, 929, 756, 669; 1H NMR (200 MHz, DMSO-d6): δ 4.23 (d, J = 4.3 Hz, 1H), Droxidopa Chapter IV 226 5.10 (d, J = 3.8 Hz, 1H), 7.27-7.31 (m, 3H), 7.78 (br s, 1H), 8.40 (br s, 1H); 13C NMR (100 MHz, DMSO-d6): δ 55.41, 70.70, 115.21, 116.51, 120.06, 130.09, 144.96, 146.06, 171.91; Analysis: C9H11NO5 requires C, 50.70; H, 5.20; N, 6.57%; found: C, 50.99; H, 5.01; N, 6.33%
42 Hegedus, V. B.; Krasso, A. F.; Noack, K.; Zeller, P. Helv. Chim. Acta 1975, 58, 147.
ncl.csircentral.net/668/1/th1739.pdf
I here by declare that the thesis entitled “Enantioselective synthesis of bioactive molecules …. Section III Enantioselective synthesis of L-threo-DOPS (droxidopa).

CAMBRIDGE, Mass., Aug 14, 2013 (AP) — The Food and Drug Administration has granted an “orphan drug designation” to a potential hemophilia treatment from Alnylam Pharmaceuticals Inc.

Orphan drug status is awarded to drugs that could treat diseases that affect fewer than 200,000 people in the United States. It comes with some added marketing exclusivity.
The Cambridge, Mass., company said Wednesday that the agency gave the designation to a drug labeled ALN-AT3 for the treatment of hemophilia B. Alnylam has tested the drug in mice and plans to start studies involving humans early next year.
for the treatment of hemophilia B. Alnylam has tested the drug in mice and plans to start studies involving humans early next year.
http://www.pharmalive.com/alnylam-hemophilia-drug-garners-orphan-drug-status

