
Home » Posts tagged 'organic chemistry' (Page 49)
Tag Archives: organic chemistry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
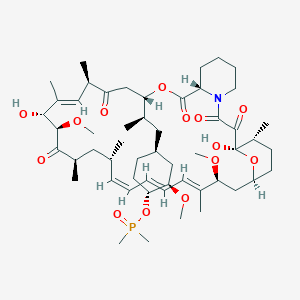
Phase 3 , breast cancer, Ridaforolimus (MK-8669; AP23573; formerly Deforolimus) Merck, license,Ariad Pharmaceuticals

Ridaforolimus
572924-54-0
(1R,2R,4S)-4-[(2R)-2-[(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28Z,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxycyclohexyl dimethylphosphinate
Dimethyl-phosphinic Acid C-43 Rapamycin Ester
42-(dimethylphosphinate) Rapamycin
- AP 23573
- AP23573
- Deforolimus
- MK 8669
- MK-8669
- MK8669
- Ridaforolimus
- Taltorvic
- UNII-48Z35KB15K
An mTOR inhibitor for the treatment of cancer.
Ridaforolimus (MK-8669; AP23573; formerly Deforolimus)
Merck, under exclusive worldwide license agreement with Ariad Pharmaceuticals
Method of Action: Oral inhibitor of mammalian target of rapamycin inhibitor (mTOR)
Indications/Phase of Trial: Maintenance therapy for metastatic soft-tissue sarcoma and bone sarcomas after at least four chemotherapy cycles (under review after receiving Complete Response letter from FDA in June; NME)
Ridaforolimus is an investigational small-molecule inhibitor of the protein mTOR, a protein that acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival, integrating signals from proteins, such as PI3K, AKT and PTEN, known to be important to malignancy.
TARGET- mTOR
Ridaforolimus (also known as AP23573 and MK-8669; formerly known as Deforolimus[1]) is an investigational targeted and small-molecule inhibitor of the protein mTOR, a protein that acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival, integrating signals from proteins, such as PI3K, AKT and PTEN known to be important to malignancy. Blocking mTOR creates a starvation-like effect in cancer cells by interfering with cell growth, division, metabolism, and angiogenesis.
It has had promising results in a clinical trial for advanced soft tissue and bone sarcoma.
RIDAFOROLIMUS
NMR….http://file.selleckchem.com/downloads/nmr/S102201-Deforolimus-HNMR-Selleck.pdf
HPLC . http://file.selleckchem.com/downloads/hplc/S102201-Deforolimus-HPLC-Selleck.pdf
MSDS..http://www.selleckchem.com/msds/Deforolimus-MSDS.html
| Commercial arrangements |
Ridaforolimus is being co-developed by Merck and ARIAD Pharmaceuticals. On May 5, 2010, Ariad Pharmaceuticals and Merck & Company announced a clinical development and marketing agreement. With this agreement, Ariad received $125 million in upfront payments from Merck and $53 million in milestone payments. Future payments are triggered upon acceptance of the NDA by the FDA with another payment when the drug receives marketing approval. There are similar milestones for acceptance and approval in both Europe and Japan. Other milestone payments are tied to revenue goals for the drug.[2] ARIAD has opted to co-promote ridaforolimus in the U.S. Merck plans to submit a New Drug Application (NDA) for ridaforolimus to the U.S. Food and Drug Administration (FDA) and a marketing application in the European Union in 2011.[3]
Clinical trials
Phase III SUCCEED
On June 6, 2011, Ariad and Merck announced detailed results from the largest randomized study ever in the soft tissue and bone sarcoma population, the Phase III SUCCEED clinical trial. SUCCEED evaluated oral ridaforolimus, in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. In this patient population, ridaforolimus improved progression-free survival (PFS) compared to placebo, the primary endpoint of the study. The complete study results were presented by Sant P. Chawla, M.D., director, Sarcoma Oncology Center, Santa Monica, CA, during the 2011 American Society of Clinical Oncology (ASCO) annual meeting.
The SUCCEED (Sarcoma Multi-Center Clinical Evaluation of the Efficacy of Ridaforolimus) trial was a randomized (1:1), placebo-controlled, double-blind study of oral ridaforolimus administered at 40 mg/day (five of seven days per week) in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. Oral ridaforolimus was granted a Special Protocol Assessment (SPA) by the FDA for the SUCCEED trial.
Based on 552 progression-free survival (PFS) events in 711 patients, (ridaforolimus (N=347), placebo (N=364) determined by an independent radiological review committee, the study achieved its primary endpoint of improvement in PFS, with a statistically significant (p=0.0001) 28 percent reduction in the risk of progression or death observed in those treated with ridaforolimus compared to placebo (hazard ratio=0.72).
Median PFS was 17.7 weeks for those treated with ridaforolimus compared to 14.6 weeks in the placebo group. Furthermore, based on the full analysis of PFS determined by investigator assessment, there was a statistically significant (p<0.0001) 31 percent reduction by ridaforolimus in the risk of progression or death compared to placebo (hazard ratio=0.69). In the investigator assessment analysis, median PFS was 22.4 weeks for those treated with ridaforolimus compared to 14.7 weeks in the placebo group [4
EU WITHDRAWAL IN NOV 2012
Merck, known as MSD outside the U.S. and Canada, announced today that it has formally notified the European Medicines Agency (EMA) of Merck’s decision to withdraw the Marketing Authorisation Application (MAA) for ridaforolimus.
The application for Marketing Authorisation for ridaforolimus was accepted by the EMA in August 2011. At the time of the withdrawal it was under review by the Agency’s Committee for Medicinal Products for Human Use (CHMP). In its letter to the EMA, Merck said that the withdrawal of ridaforolimus was based on the provisional view of the CHMP that the data available to date and provided in the Marketing Authorisation Application were not sufficient to permit licensure of ridaforolimus in the European Union for the maintenance treatment of patients with soft tissue sarcoma or primary malignant bone tumor.
Although the application for these uses was withdrawn, Merck is studying ridaforolimus in combination with other drugs in other tumor types. The withdrawal of the European application of ridaforolimus for the maintenance treatment of patients with soft tissue sarcoma or primary malignant bone tumor does not change Merck’s commitment to the ongoing clinical trials with ridaforolimus.
Description
42-(dimethylphosphinate) Rapamycin (Ridaforolimus) represented by the following formula I:

2. Description of RelatedArt
The mammalian target of Rapamycin (mTOR) is known as a mechanistic target of Rapamycin (H), which is found in the studies of Rapamycin. On the other hand, 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I) is a derivative of Rapamycin (II), which is also a kind of mTOR inhibitor. Ridaforolimus (I) can inhibit cell division and possibly lead to tumor cell death. Hence, there are many studies related to solid tumor treatments and blood cancer treatments using Ridaforolimus (I). In addition, in 2011, Merck also applied a certification of this compound against soft tissue and bone cancer.
U.S. Pat. No. 7,091,213 discloses a process for preparing 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I), and the process thereof is shown in the following Scheme I.

In this process, a solution of Rapamycin (II) in dichloromethane (DCM) was respectively added with 2,6-di-tert-butyl-4-methylpyridine or 3,5-lutidine as a base, and followed by the addition of a solution of dimethylphosphinic chloride (DMP-Cl) to perform a phosphorylation reaction at 0° C., under a stream of N2(g). The crude product was purified by flash chromatography (eluted with MeOH/DCM/EtOAc/hexane=1:10:3:3) to provide 42-(dimethyl- phosphinate) Rapamycin (Ridaforolimus) (I), which is a phosphorylated compound at 42-hydroxyl position of Rapamycin (II). In addition, this patent also disclosed a side product of 31,42-bis(dimethyl phosphinate) Rapamycin (III), which is a phosphorylated compound at both 31- hydroxyl position and 42- hydroxyl position of Rapamycin (II).
…………………..
SYNTHESIS
Some additional transformations of potential interest to the practitioner are shown below, including the preparation of reagents for generating the described C-43 phosphorus-containing rapalogs:
Preparation of Diakyl/diaryl Chlorophoshates

Preparation of Alkyl Halide Phosphonates

Illustrative routes for using the foregoing sorts of reagents to prepare certain rapalogs of this invention are shown below.

The synthesis of compounds of this invention often involves preparation of an activated form of the desired moiety “J”, such as a phosphoryl chloride as shown above (e.g. (R)(RO)P—Cl or RR′P(═O)—Cl, etc), and reaction of that reagent with rapamycin (or the appropriate rapalog) under conditions yielding the desired product, which may then be recovered from residual reactants and any undesired side products. Protecting groups may be chosen, added and removed as appropriate using conventional methods and materials.
Purification of Compounds of the Invention
A variety of materials and methods for purifying rapamycin and various rapalogs have been reported in the scientific and patent literatures and may be adapted to purification of the rapalogs disclosed herein. Flash chromatography using a BIOTAGE prepacked cartridge system has been particularly effective. A typical protocol is disclosed in the Examples which follow.
Physicochemical Characterization of Compounds of the Invention
The identity, purity and chemical/physical properties of the rapalogs may be determined or confirmed using known methods and materials, including HPLC, mass spectral analysis, X ray crystallography and NMR spectroscopy. High resolution 1D 1H and 31P NMR spectra acquired using a typical relaxation delay of 3 seconds have proved useful, as has reverse phase HPLC analysis (analytical column, 3 micron particle size, 120 ansgstrom pore size, thermostatted to 50° C. with a mobile phase of 50% acetonitrile, 5% methanol and 45% water (all % s by volume), for example, in an isocratic elution system, with elution of product and impurity peaks followed by UV detection at 280 nanometers). Normal phase HPLC may also be used, especially to evaluate the level of residual rapamycin or rapalog by-products. The presence of residual solvent, heavy metals, moisture and bioburden may be assessed using conventional methods.
Example 9
Dimethyl-phosphinic Acid C-43 Rapamycin Ester

Dimethyl-phosphinic Acid C-43 Rapamycin Ester
To a cooled (0° C.) solution of rapamycin (0.1 g, 0.109 mmol) in 1.8 mL of dichloromethane was added 0.168 g (0.82 mmol) of 2,6-di-t-butyl-4-methyl pyridine, under a stream of N2, followed immediately by a solution of dimethylphosphinic chloride (0.062 g, 0.547 mmol) in 0.2 mL of dichloromethane. The slightly yellow reaction solution was stirred at 0° C., under an atmosphere of N2, for 3.5 h (reaction monitored by TLC). The cold (0° C.) reaction solution was diluted with ˜20 mL EtOAc then transferred to a separatory funnel containing EtOAc (150 mL) and saturated NaHCO3 (100 mL). Upon removing the aqueous layer, the organic layer was washed successively with ice cold 1N HCl (1×100 mL), saturated NaHCO3 (1×100 mL), and brine (1×100 mL), then dried over MgSO4 and concentrated. The crude product was purified by silica gel flash chromatography (eluted with 1:10:3:3 MeOH/DCM/EtOAc/hexane) to provide 0.092 g of a white solid:
1H NMR (300 MHz, CDCl3) d 4.18 (m, 1H), 4.10 (m, 1H), 3.05 (m, 1H), 1.51 (m, 6H);
31P NMR (121 MHz, CDCl3) d 53.6; 1013 m/z (M+Na).
Example 9
Alternative Synthesis
Rapamycin and dichloromethane are charged into a nitrogen-purged reaction flask. The stirred solution is cooled to approximately 0° C. (an external temperature of −5±5° C. is maintained throughout the reaction). A solution of dimethylphosphinic chloride (2.0 molar equivalents) in dichloromethane is then added over a period of approximately 8–13 minutes.
This is followed immediately by the addition of a solution of 3,5-lutidine (2.2 molar equivalents) in dichloromethane over a period of approximately 15–20 minutes. Throughout both additions, the internal temperature of the reaction sssstays below 0° C. The cooled reaction solution is stirred for 1 hour and then transferred, while still cold, to an extractor containing saturated aqueous NaHCO3 and methyl-t-butyl ether (MTBE), ethyl acetate or diethyl ether. In-process samples are removed at 30 and 60 minute time points.
Samples are prepared in a similar fashion to that described for the reaction workup. Reaction progress is monitored by TLC (1:10:3:3 MeOH/DCM/EtOAc/hexanes) and reverse-phase HPLC analyses. The isolated organic layer is successively washed with ice cold 1N HCl, saturated aqueous NaHCO3 (2×), saturated aqueous NaCl, and dried over sodium sulfate. Upon filtration and solvent removal, the residue undergoes solvent exchange with acetone followed by concentration in vacuo to provide crude product, which may be analyzed for purity by normal- and reversed-phase HPLC.
…………………….
SYNTHESIS
The process of the present invention is shown in the following Scheme II.


- “ARIAD Reports First Quarter 2009 Development Progress and Financial Results- Ridaforolimus New USAN Name to Replace Deforolimus”. ARIAD Pharmaceuticals. 2009. Retrieved 2009-05-07.
- “ARIAD – News release”. Phx.corporate-ir.net. Retrieved 2012-10-07.
- “ARIAD – News release”. Phx.corporate-ir.net. 2011-03-17. Retrieved 2012-10-07.
- “ARIAD – News release”. Phx.corporate-ir.net. 2011-06-06. Retrieved 2012-10-07.
|
7-11-2012
|
FULLY HUMAN ANTI-VEGF ANTIBODIES AND METHODS OF USING
|
|
|
10-28-2011
|
METHODS OF TREATMENT
|
|
|
1-21-2011
|
ANTI-IGF1R
|
|
|
1-5-2007
|
Methods for treating neurofibromatosis 1
|
|
|
4-16-2004
|
Phosphorus-containing compounds and uses thereof
|

Generic approval FDA- Travatan ZTravoprost Ophthalmic Solution/Drops 0.004% – Par Pharmaceutical, Inc.

Travoprost is a synthetic prostaglandin F analogue.
Its chemical name is [1R-[lα(Z),2β(lE,3R*),3α,5α]]-7-[3,5-Dihydroxy-2-[3-hydroxy-4-[3-(trifluoromethyl)phenoxy]-1 -butenyl]cyclopentyl]-5-heptenoic acid, 1 -methylethylester.
It has a molecular formula of C26H35F3O6 and a molecular weight of 500.55.
The chemical structure of travoprost is:
Travoprost is a clear, colorless to slightly yellow oil that is very soluble in acetonitrile, methanol, octanol, and chloroform. It is practically insoluble in water.
TRAVATAN® (travoprost ophthalmic solution) 0.004% is supplied as sterile, buffered aqueous solution of travoprost with a pH of approximately 6.0 and an osmolality of approximately 290 mOsmol/kg.
TRAVATAN® contains Active: travoprost 0.04 mg/mL; Preservative: benzalkonium chloride 0.15 mg/mL; Inactives: polyoxyl 40 hydrogenated castor oil, tromethamine, boric acid, mannitol, edetate disodium, sodium hydroxide and/or hydrochloric acid (to adjust pH) and purified water.
GENERIC APPROVAL MARCH 2013
- Travoprost Ophthalmic Solution/Drops 0.004%
Approved: March 1, 2013 – Par Pharmaceutical, Inc.
Generic for: Travatan Z
Travoprost ophthalmic solution is a topical medication used for controlling the progression of glaucoma or ocular hypertension, by reducing intraocular pressure. It is a synthetic prostaglandin analog (or more specifically, an analog of prostaglandin F2α)[1][2] that works by increasing the outflow of aqueous fluid from the eyes.[3] It is also known by the brand names of Travatan and Travatan Z, manufactured by Alcon, and Travo-Z, manufactured by Micro Labs.
- Alcon Laboratories, Inc. (September 2011). “TRAVATAN – travoprost solution”. DailyMed. Bethesda, MD: U.S. National Library of Medicine. Retrieved 2011-09-30.
- Alcon Laboratories, Inc. (September 2011). “TRAVATAN Z (travoprost) solution”. DailyMed. Bethesda, MD: U.S. National Library of Medicine. Retrieved 2011-09-30.
- AHFS Consumer Medication Information (2011-01-01).“Travoprost Ophthalmic”. MedlinePlus. Bethesda, MD: U.S. National Library of Medicine. Retrieved 2011-09-30.
Phase3 Rindopepimut ,CDX-110 Celldex Therapeutics’ brain cancer vaccine

http://clinicaltrials.gov/ct2/show/NCT01480479
MAR 2013
Rindopepimut
Immunotherapeutic vaccine called Rindopepimut showed positive results in prolonging survival in patients with newly diagnosed EGFRvIII-positive glioblastoma (GB), one of the most aggressive forms of brain cancer
Celldex Therapeutics’ brain cancer vaccine, rindopepimut, also known as CDX-110, targets EGFRvIII, an activated mutation of the epidermal growth factor receptor (EGFR). This mutation is found in about 31% of cases of glioblastoma multiforme, a form of fast-growing brain cancer and the most common type of primary brain tumor. It can contribute to tumor growth, and is linked with poor long-term survival. It is not seen in normal tissue.
In the ACT III Phase II trial, which involved people with newly diagnosed EGFRvIII-positive glioblastoma, 65 patients were given rindopepimut in combination with standard-of-care treatment (temozolomide), after having undergone surgery and standard chemotherapy and radiation therapy.
focus on is Rindopepimut, an immunotherapy treatment that targets EGFRvIII. As it’s not found at significant levels in normal tissues but expressed in 30ish% of primary glioblastoma, it’s an ideal target that has produced promising results to date. The drug candidate has shown consistent benefit for patients across three phase 2 studies- that’s no fluke! It’s currently in a global phase 3 trial in patients with newly diagnosed glioblastoma with results due in a couple years
| Phase 3 Study of Rindopepimut in Patients With Newly Diagnosed Glioblastoma (ACT IV) | |
 |
|
| Design: | Phase 3, double-blind, study of rindopepimut compared with KLH control |
| Status: | Currently enrolling at multiple centers in the US; additional centers outside the US planned to be activated in 2012 |
ABOUT THE CLINICAL TRIAL
This 2-arm, randomized, Phase 3 study will investigate the efficacy and safety of the addition of rindopepimut to the current standard of care, temozolomide, in patients with recently diagnosed EGFRvIII positive glioblastoma. All patients will be administered temozolomide. Half the patients will be randomly assigned to receive rindopepimut (given along with GM-CSF as a vaccine adjuvant) and half the patients will be randomly assigned to receive a keyhole limpet hemocyanin (KLH). Patients will be treated in a blinded fashion (neither the patient nor the doctor will know which arm of the study the patient is on). Patients will be treated until disease progression or intolerance to therapy and all patients will be followed for survival.
All patients enrolled in the study will be closely monitored to determine if their cancer is responding to treatment and for any side effects that may occur.
Dainippon Sumitomo receives approval for Surepost, Repaglinide in combination with biguanides and thiazolidenediones

Repaglinide is an antidiabetic drug in the class of medications known as meglitinides, and was invented in 1983. It is sold by Novo Nordisk under the name of Prandin in theU.S., GlucoNorm in Canada, Surepost in Japan,Repaglinide in Egypt by EIPICO, andNovoNorm elsewhere. In Japan it is produced by Dainippon Sumitomo Pharma.
Repaglinide lowers blood glucose by stimulating the release of insulin from the pancreas. It achieves this by closing ATP-dependent potassium channels in the membrane of the beta cells. This depolarizes the beta cells, opening the cells’ calcium channels, and the resulting calcium influx induces insulin secretion.
Precursor drugs to repaglinide were invented in late 1983 by scientists at Dr Karl Thomae GmbH, a German drug manufacturer located at Biberach an der Riß in southern Germany which was acquired by Boehringer Ingelheim in 1990. The drug that became repaglinide was later licensed by Boehringer to Novo Nordisk, which filed an Investigational New Drugapplication for the compound with the Food and Drug Administration (FDA) in April 1992. Novo Nordisk filed its New Drug Application (NDA) for Prandin in July 1997 and it was quickly approved, gaining FDA approval in December 1997. The drug was the first of the meglitinide class. It was branded Prandin because its quick onset and short duration of action concentrates its effect around meal time (the prandium was the Roman meal which is comparable to the modern lunch).
After several attempts to file for U.S. patent protection, a filing was made in March 1990 which eventually became U.S. Patents 5,216,167 (June 1993), 5,312,924 (May 1994) and 6,143,769 (November 2000). After filing its NDA for repaglinide in 1997, Novo Nordisk applied for patent extension under the Hatch-Waxman Act. This process, called patent term restoration, allows drug patents to be extended based on the time that a drug spent in clinical trials and in the approval process. Previously it had been decided by the U.S. Patent and Trademark Office that the expiration date of U.S. Patents 5,216,167 and 5,312,924 would be 5 September 2006. In February 2001 Prandin’s patent life was extended to 14 March 2009 in response to Novo Nordisk’s patent term restoration application, with U.S. Patent 5,216,167 having been reissued as RE37035.[1]
Prior to the end of repaglinide’s patent term, Novo Nordisk obtained a new patent, U.S. Patent 6,677,358 (January 2004), covering the combination therapy of repaglinide together with the generic anti-diabetic drug metformin. This new patent was due to expire June 2018. In January 2011, a federal court ruled Novo Nordisk’s new patent invalid on the grounds of obviousness, and unenforceable on the grounds of inequitable conduct on the part of Novo Nordisk’s patent attorneys.[2]
- “Details for Patent: RE37035”.
- Frankel, Alison (2011-01-27). “Judge Finds Novo Nordisk Diabetes Drug Patent Invalid and Unenforceable, Questions In-House Patent Lawyer’s Conduct”. Retrieved 2011-02-05.
KYTHERA Biopharmaceuticals, Inc. Announces Positive Interim Results from Open-Label Study of ATX-101 in the Reduction of Unwanted Submental Fat (SMF) or “Double Chin”
http://clinicaltrials.gov/ct2/show/NCT01426373
synthesis………..https://newdrugapprovals.org/2014/07/14/some-thing-for-your-chin-fda-accepts-kytheras-atx-101-new-drug-application/
The drug is sodium deoxycholate for injection, code-named ATX-101 was developed for the treatment of lipomas – benign tumors of subcutaneous adipose tissue, as well as other unwanted fatty growths, such as a double chin. This substance, which is a salt of one of the bile acids, emulsifies fats, destroying their excess deposits


ATX-101 (a first-in-class injectable drug being studied for the reduction of localized fat. ATX-101 is a proprietary formulation of deoxycholate a well-studied endogenous compound that is present in the body), a facial injectable drug for the reduction of unwanted fat under the chin, or submental fat. V. Leroy Young, MD, FACS, presented the initial results at the American Society for Aesthetic Plastic Surgery (ASAPS) 45th Annual Aesthetic Meeting in Vancouver, British Columbia, on May 4, 2012.
In August 2010 Bayer Consumer Care AG signed a licensing and development collaboration agreement with KYTHERA, thereby obtaining commercialization rights to ATX-101 outside the US and Canada. KYTHERA and Bayer are collaborating on the development of ATX-101 in Europe.
KYTHERA Biopharmaceuticals Inc. 02 MAR 3013, announced positive interim results from a Phase IIIb multi-center open-label study (ATX-101-11-26) to evaluate the safety and efficacy of ATX-101 an investigational injectable drug for the reduction of unwanted submental fat (SMF) commonly known as double chin. The results presented at the Late Breaking Research Symposium at the 71st American Academy of Dermatology (AAD) Annual Meeting in Miami Beach Fla. found that ATX-101 is well-tolerated and may be effective in reducing SMF by both clinician and patient reported outcome measures. The ATX-101 global clinical development program has enrolled more than 2500 total patients of which more than 1500 have been treated with ATX-101.
“In my practice patients often request a non-surgical way to treat their submental fat or undesirable double chin” said investigator Susan Weinkle MD FAAD a board certified dermatologist and affiliate clinical professor at the University of South Florida. “For these patients double chin is often resistant to diet and exercise. The results of this study suggest that microinjections of ATX-101 can reduce submental fat without worsening skin laxity.”
ATX-101 is a proprietary synthetically-derived formulation of deoxycholic acid (DCA) a naturally-occurring molecule found in the body that aids in fat metabolism. In this open-label Phase IIIb study interim results three months after the last ATX-101 treatment found:
- Reduction of submental fat
- 87 percent of patients achieved at least a one-grade improvement from baseline on the Clinician-Reported Submental Fat Rating Scale (CR-SMFRS)
- Similarly 83 percent of patients achieved at least a one-grade improvement on the Patient-Reported Submental Fat Rating Scale (PR-SMFRS)
- 96 percent of patients had unchanged or improved skin laxity based on the clinician rated Submental Skin Laxity Grading Scale (SMSLG)
- 95 percent of patients were satisfied with treatment based on the Global Post Treatment Satisfaction Scale
- Adverse events were of mild to moderate intensity transient and primarily associated with the treatment area

Topline results from this study were announced in November 2012. As previously announced 71.3 percent of subjects had at least a one-grade improvement on the CR-SMFRS / PR-SMFRS composite and 14.0 percent had at least a two-grade improvement on the same composite measure.
These results are based on a multicenter 12-month open-label Phase IIIb study conducted at 21 sites across the United States evaluating 165 adults who received injections of ATX-101 for up to six treatments at four-week intervals. Patients received ATX-101 (2 mg/cm2) by subcutaneous microinjections directly into their SMF and were evaluated three months after their last treatment. The study population includes females (77.6 percent) and males (22.4 percent) with a mean age of 47 who report at least moderate SMF and dissatisfaction with the appearance of their chin. All Fitzpatrick Skin Types an industry standard scale to categorize skin tone are represented.
“We are pleased with these ATX-101 study results” said Patricia S. Walker M.D. Ph.D. chief medical officer KYTHERA Biopharmaceuticals Inc. “These results along with efficacy analyses in double-blind placebo-controlled studies support ATX-101 entering the market as potentially the first medical aesthetic drug approved for the reduction of submental fat.”
About ATX-101
ATX-101 is a potential first-in-class injectable drug candidate under clinical investigation for the reduction of unwanted submental fat. ATX-101 is a proprietary formulation of synthetic deoxycholic acid a well-characterized endogenous compound that is present in the body to promote the natural breakdown of dietary fat. ATX-101 is designed to be a locally-injected drug that causes proximal preferential destruction of adipocytes or fat cells with minimal effect on surrounding tissue. Based on clinical trials conducted to date ATX-101 has exhibited significant meaningful and durable results in the reduction of submental fat which commonly presents as an undesirable “double chin.” These results correspond with subject satisfaction measures demonstrating meaningful improvement in perceived chin appearance.
In August 2010 Bayer signed a licensing and collaboration development agreement with KYTHERA thereby obtaining development and commercialization rights to ATX-101 outside of the U.S. and Canada. Bayer recently completed two pivotal Phase III trials of ATX-101 in Europe for the reduction of submental fat. Topline results from these trials were reported in the second quarter of 2012. KYTHERA completed enrollment in its pivotal Phase III clinical program for ATX-101 in more than 1000 subjects randomized to ATX-101 or placebo in 70 centers across the United States and Canada in August 2012. The Company expects to release topline results in mid-2013.
About KYTHERA Biopharmaceuticals Inc.
KYTHERA Biopharmaceuticals Inc. is a clinical-stage biopharmaceutical company focused on the discovery development and commercialization of novel prescription products for the aesthetic medicine market. KYTHERA initiated its pivotal Phase III clinical program for ATX-101 in March 2012 and completed enrollment of more than 1000 patients randomized to ATX-101 or placebo in 70 centers across the U.S. and Canada in August 2012. KYTHERA also maintains an active research interest in hair and fat biology. Find more information at www.kytherabiopharma.com.
Medical Imaging Drugs Advisory Committee Recommends Approval of Guerbet NDA for Dotarem (gadoterate meglumine)

| Cas No. | 98059-18-8 |
| Name | 2-[4,7-bis(carboxylatomethyl)-10-(carboxymethyl)-1,4,7, 10-tetrazacyclododec-1-yl]acetate; gadolinium(3+); (2R,3R,4R,5S)-6-(methylamino)hexane-1,2,3,4,5-pentol |
Dotarem (gadoterate meglumine)
Company: Guerbet
Treatment for: Diagnostic
Dotarem (gadoterate meglumine) is a gadolinium-based contrast agent under review for use in magnetic resonance imaging (MRI).

VILLEPINTE, France, Feb. 14, 2013 Guerbet, the contrast agent specialist for medical imaging, today announced that the Medical Imaging Drugs Advisory Committee to US Food and Drug Administration (FDA) has voted unanimously by votes of 17 to 0 to recommend that FDA approve the New Drug Application (NDA) for Dotarem (gadoterate meglumine) for adults, and for pediatric use for children two years of age and older. The Committee voted 10 to 6 (with one member abstaining) not to recommend at this time approval of the indication for children under two years of age.
Dotarem is the only macrocyclic and ionic gadolinium-based contrast agent (GBCA) for the intravenous use with magnetic resonance imaging (MRI) in the brain (intracranial), spine and associated tissues in adults and pediatric patients to detect and visualize areas with disruption of the blood-brain barrier (BBB) and/or abnormal vascularity. The Guerbet NDA recommended dose is 0.1 mmol Gd/kg.

Gadoteric acid
Gadoteric acid (trade names Artirem, Dotarem) is a macrocycle-structured gadolinium-based MRI contrast agent. It consists of the organic acid DOTA as a chelating agent, and gadolinium (Gd3+), and is used in form of the meglumine salt.[1] The drug is approved and used in a number of countries worldwide.[2]
References
- Herborn, C. U.; Honold, E.; Wolf, M.; Kemper, J.; Kinner, S.; Adam, G.; Barkhausen, J. (2007). “Clinical Safety and Diagnostic Value of the Gadolinium Chelate Gadoterate Meglumine (Gd-DOTA)”. Investigative Radiology 42 (1): 58–62. doi:10.1097/01.rli.0000248893.01067.e5. PMID 17213750. edit
- Drugs.com: Gadoteric Acid

A gadolinium chelate paramagnetic contrast agent. When placed in a magnetic field, gadoterate meglumine produces a large magnetic moment and so a large local magnetic field, which can enhance the relaxation rate of nearby protons; as a result, the signal intensity of tissue images observed with magnetic resonance imaging (MRI) may be enhanced. Because this agent is preferentially taken up by normal functioning hepatocytes, normal hepatic tissue is enhanced with MRI while tumor tissue is unenhanced. In addition, because gadobenate dimeglumine is excreted in the bile, it may be used to visualize the biliary system using MRI.
FDA has approved a new use of Avastin® (bevacizumab) in combination with fluoropyrimidine-based irinotecan or oxaliplatin chemotherapy for people with metastatic colorectal cancer (mCRC).
Bevacizumab, CAS NO 216974-75-3
A MONOCLONAL ANTIBODY
January 23, 2013
Avastin (bevacizumab) is a recombinant humanized monoclonal IgG1 antibody that binds to and inhibits the biologic activity of human vascular endothelial growth factor (VEGF) in in vitro and in vivo assay systems. Bevacizumab contains human framework regions and the complementarity-determining regions of a murine antibody that binds to VEGF. Avastin has an approximate molecular weight of 149 kD. Bevacizumab is produced in a mammalian cell (Chinese Hamster Ovary) expression system in a nutrient medium containing the antibiotic gentamicin. Gentamicin is not detectable in the final product.
FDA has approved a new use of Avastin® (bevacizumab) in combination with fluoropyrimidine-based irinotecan or oxaliplatin chemotherapy for people with metastatic colorectal cancer (mCRC).
On January 23, 2013, the FDA has approved a new use of Avastin® (bevacizumab) in combination with fluoropyrimidine-based irinotecan or oxaliplatin chemotherapy for people with metastatic colorectal cancer (mCRC). The new indication will allow people who received Avastin plus an irinotecan or oxaliplatin containing chemotherapy as an initial treatment (first-line) for mCRC to continue to receive Avastin plus a different irinotecan or oxaliplatin containing chemotherapy after their cancer worsens (second-line treatment).
People who start on Avastin for mCRC can now stay on Avastin after their cancer worsens.
Bevacizumab (trade name Avastin, Genentech/Roche) is an angiogenesis inhibitor, a drug that slows the growth of new blood vessels. It is licensed to treat various cancers, including colorectal, lung, breast (outside the USA), glioblastoma (USA only), kidney and ovarian.
Bevacizumab is a humanized monoclonal antibody that inhibits vascular endothelial growth factor A (VEGF-A).[1] VEGF-A is a chemical signal that stimulates angiogenesis in a variety of diseases, especially in cancer. Bevacizumab was the first clinically available angiogenesis inhibitor in the United States.[citation needed]
Bevacizumab was approved by the U.S. Food and Drug Administration (FDA) for certain metastatic cancers. It received its first approval in 2004, for combination use with standard chemotherapy for metastatic colon cancer.[2] It has since been approved for use in certain lung cancers, renal cancers, and glioblastoma multiforme of the brain.
At one point bevacizumab was approved for breast cancer by the FDA, but the approval was revoked on 18 November 2011.[3][4]
- Los, M.; Roodhart, J. M. L.; Voest, E. E. (2007). “Target Practice: Lessons from Phase III Trials with Bevacizumab and Vatalanib in the Treatment of Advanced Colorectal Cancer”. The Oncologist 12 (4): 443–50. doi:10.1634/theoncologist.12-4-443. PMID 17470687.
- http://www.gene.com/gene/products/information/pdf/avastin-prescribing.pdf
- Pollack, Andrew (18 November 2011). “F.D.A. Revokes Approval of Avastin for Breast Cancer”. New York Times.
- “Cancer drug Avastin loses US approval”. BBC. November 18, 2011.
SEQUENCE
>1bj1_H|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||VH-CH1 (VH(1-123)+CH1(124-215))|||||||231||||MW 24867.8|MW 24867.8| EVQLVESGGGLVQPGGSLRLSCAASGYTFTNYGMNWVRQAPGKGLEWVGWINTYTGEPTY AADFKRRFTFSLDTSKSTAYLQMNSLRAEDTAVYYCAKYPHYYGSSHWYFDVWGQGTLVT VSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVL QSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHT >1bj1_L|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||L-KAPPA (V-KAPPA(1-107)+C-KAPPA(108-213))|||||||214||||MW 23451.0|MW 23451.0| DIQMTQSPSSLSASVGDRVTITCSASQDISNYLNWYQQKPGKAPKVLIYFTSSLHSGVPS RFSGSGSGTDFTLTISSLQPEDFATYYCQQYSTVPWTFGQGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC >1bj1_J|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||L-KAPPA (V-KAPPA(1-107)+C-KAPPA(108-213))|||||||214||||MW 23451.0|MW 23451.0| DIQMTQSPSSLSASVGDRVTITCSASQDISNYLNWYQQKPGKAPKVLIYFTSSLHSGVPS RFSGSGSGTDFTLTISSLQPEDFATYYCQQYSTVPWTFGQGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC >1bj1_K|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||VH-CH1 (VH(1-123)+CH1(124-215))|||||||231||||MW 24867.8|MW 24867.8| EVQLVESGGGLVQPGGSLRLSCAASGYTFTNYGMNWVRQAPGKGLEWVGWINTYTGEPTY AADFKRRFTFSLDTSKSTAYLQMNSLRAEDTAVYYCAKYPHYYGSSHWYFDVWGQGTLVT VSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVL QSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHT
Chelsea encouraged by FDA talks on Northera(droxidopa)

(2R,3S)-2-amino-3-(3,4-dihydroxyphenyl)-3-hydroxypropanoic acid
L-DOPS (L-threo-dihydroxyphenylserine; Droxidopa; SM-5688) is a psychoactive drugand synthetic amino acid precursor which acts as a prodrug to the neurotransmittersnorepinephrine (noradrenaline) and epinephrine (adrenaline).[1] Unlike norepinephrine and epinephrine themselves, L-DOPS is capable of crossing the protective blood–brain barrier(BBB).[1]
L-DOPS was developed by Sumitomo Pharmaceuticals under the trade name Droxidopafor the treatment of hypotension, including NOH,[2] and NOH associated with variousdisorders such as MSA, FAP, and PD, as well as IDH. The drug has been used in Japanand some surrounding Asian areas for these indications since 1989. Following a merge with Dainippon Pharmaceuticals in 2006, Dainippon Sumitomo Pharma licensed L-DOPS to Chelsea Therapeutics to develop and market it worldwide except in Japan, Korea, China, and Taiwan.
Clinical trials
Though L-DOPS has been used in Japan and Southeast Asia already for some time, it is also currently in clinical trials at the phase IIIpoint in the United States (U.S.), Canada, Australia, and throughout Europe. Provided L-DOPS successfully completes clinical trials, it could be approved for the treatment of NOH as early as 2011.[4] Additionally, phase II clinical trials for IDH are also underway. Chelsea Therapeutics obtained orphan drug status (ODS) for L-DOPS in the U.S. for NOH, and that of which associated with PD, PAF, and MSA, and is the pharmaceutical company developing it in that country.
FEBRUARY 21, 2013
Shares in Chelsea Therapeutics International have leapt after the company said it will resubmit its previously-rejected treatment of neurogenic orthostatic hypotension, Northera, after helpful discussions with US regulators.
A year ago, the Food and Drug Administration issued Chelsea with a complete response letter asking for more data regarding its filing for Northera (droxidopa)for NOH. That came as something of a surprise given that the agency’s Cardiovascular and Renal Drugs Advisory Committee had earlier voted 7-4 in favour of the therapy.
Now Chelsea says that following a meeting, it has received written guidance from the Director of the Office of New Drugs at the FDA stating that an ongoing study has the potential to serve as the basis for a resubmission.
The guidance suggests that “data strongly demonstrating a short-term clinical benefit of droxidopa in patients with NOH would be adequate for approval, with a possible requirement to verify durable clinical benefit post-approval”.
Encouraged by this, Chelsea plans to refile Northera in the late second quarter of 2013. Chief executive Joseph Oliveto said the firm looks forward to submitting the totality of our clinical experience to date to the agency for review…we now have a regulatory path forward”.
Chelsea also intends to initiate a new clinical trial in the fourth quarter of 2013.
- Goldstein, DS (2006). “L-Dihydroxyphenylserine (L-DOPS): a norepinephrine prodrug”. Cardiovasc Drug Rev 24 (3-4): 189–203. doi:10.1111/j.1527-3466.2006.00189.x.PMID 17214596.
- Mathias, Christopher J (2008). “L-dihydroxyphenylserine (Droxidopa) in the treatment of orthostatic hypotension”. Clin Auton Res 18 (Supplement 1): 25–29. doi:10.1007/s10286-007-1005-z.
- Crofford, LJ (2008). “Pain management in fibromyalgia”. Curr Opin Rheumatol 20 (3): 246–250.doi:10.1097/BOR.0b013e3282fb0268. PMID 18388513.
- Search of: “Droxidopa” – List Results – ClinicalTrials.gov
- Robertson, David (2008). “The pathophysiology and diagnosis of orthostatic hypotension”. Clin Auton Res 18(Supplement 1): 2–7. doi:10.1007/s10286-007-1004-0.

ONO Pharmaceutcal files for approval of Additional Indication for Onoact® 50 injection, Short-Acting Selective β1 Blocker in Japan

disease. AF/AFL with LV dysfunction accompanying by persistent elevated heart rate would lead to further deterioration of cardiac performance. Swift rate control is inevitable to be restored from this detrimental condition, however, no drug on market can provide both the features of fast-acting and easy titratability for tachyarrhythmia (AF/AFL) with LV dysfunction.
Onoact® 50 for injection is the short-acting selective β1 blocker which reduces heart rate by selectively blocking β1 receptors located chiefly in the heart and this fast-acting drug can be easily titrated.
We expect that Onoact® 50 for injection can contribute to promptly reducing heart rate without causing deterioration of cardiac performance in treatment of tachyarrhythmia (AF/AFL) with LV dysfunction.
This short-acting selective β1 blocker drug is discovered and developed by ONO and has been widely used by many patients since its launch. The drug has firstly received approval for emergency treatment of intra-operative tachyarrhythmia (atrial fibrillation and flutter, and sinus tachycardia) in July 2002. Then, it had also been approved for additional indication of emergency treatment of post-operative tachyarrhythmia (atrial fibrillation and flutter, and sinus tachycardia) with monitoring of circulatory dynamics in October 2006.
| Identifiers | |
|---|---|
| CAS number | 133242-30-5 |
| Chemical data | |
| Formula | C25H39N3O8 |
| Mol. mass | 509.59 g/mol |
Landiolol (INN) is a drug which acts as a highly cardioselective, ultra short-acting beta blocker. It is used as an anti-arrhythmic agent.
- Yoshiya I (December 1998). “[Landiolol hydrochloride, a new sympathetic beta blocker]” (in Japanese). Masui 47 Suppl: S126–32. PMID 9921175.
- Ogata J, Okamoto T, Minami K (2003). “Landiolol for the treatment of tachyarrhythmia associated with atrial fibrillation”. Can J Anaesth 50 (7): 753. doi:10.1007/BF03018726. PMID 12944459
FDA approves UCLA IND application to commence embryonic stem cell-based trial
12 feb 2013
The USFDA has approved the investigator investigational new drug (IND) application of University of California, Los Angeles (UCLA), the clinical partner of Advanced Cell Technology (ACT), to commence a clinical trial using the human embryonic stem cells (hESCs)-derived cells to treat severe myopia.
Embryonic stem cell-based trial was designed to assess the hESC-derived ACT’s retinal pigment epithelial (RPE) cells in patients with severe myopia (nearsightedness).
ACT chairman and CEO Gary Rabin said, “We are pleased to be on track to broaden the scope of our RPE program with the initiation of the new Investigator IND.”
Human embryonic stem cells (hESCs) are pluripotent cells derived from the inner cell mass of the blastocyst. They have the ability to renew themselves and to differentiate into a variety of different cell types that are found in the body. Unlike somatic or ‘adult’ stem cells, hESCs proliferate indefinitely. This, together with their ability to differentiate into most adult cell types, has resulted in the preferred use of these cells for research and therapeutic applications, as they represent a potentially indefinite source of therapeutic cells. Any cell therapy derived from hESCs would be allogeneic by nature. Some current studies involve the potential therapeutic application of hESCs for spinal cord injury, age-related macular degeneration (AMD), cardiovascular diseases, and diabetes. Among the start up cell therapy companies, Geron and Advanced Cell Technologies have pioneered clinical trials using cells differentiated from hESCs.
