
Home » Posts tagged 'NEW PATENT' (Page 3)
Tag Archives: NEW PATENT
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
LUPIN LIMITED, WO 2016181313, NEW PATENT, SOFOSBUVIR
![]()
WO2016181313, A PROCESS FOR THE PREPARATION OF SOFOSBUVIR INTERMEDIATES & ITS POLYMORPH
| LUPIN LIMITED [IN/IN]; Kalpataru Inspire 3rd Floor, Off Western Express Highway Santacruz (East) Mumbai 400 055 (IN) |
SINGH, Girij, Pal; (IN).
SRIVASTAVA, Dhananjai; (IN).
MEHARE, Kishor, Gulabrao; (IN).
MALIK, Vineet; (IN).
DEOKAR, Sharad, Chandrabhan; (IN).
DANGE, Abhijeet, Avinash; (IN)


SUCCESS QUOTIENT: Lupin chairman DB Gupta (sitting) with managing director Kamal K Sharma (centre), directors Vinita Gupta (right) and Nilesh Gupta.
The present invention provides a novel process for preparation N-[(2,3,4,5,6- Pentafluorophenoxy)phenoxyphosphinyl]-L-alanine 1-methylethyl ester (formula 2) and resolving the formula 2 in the presence base to form N-[(S)-(2,3,4,5,6- Pentafluorophenoxy)phenoxyphosphinyl]-L-alanine 1-methylethyl ester (formula 2′).
Sofosbuvir is chemically named as (S)-isopropyl 2-((S)-(((2R,3R,4R,5R)-5-(2,4- dioxo3,4-dihydropyrimidin-l(2H)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran- 2yl)methoxy)-(phenoxy)phosphorylamino)propanoate and is represented by the following chemical structure:

Formula 1
PCT publications WO2011123645 and WO2010135569 describes process for preparation of compound of formula 2′ by reacting isopropyl (chloro(phenoxy)phosphoryl)-L-alaninate and pentaflurophenol in the presence of base.

Formula 2′
Example-1:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium hydride
10.2g of sodium hydride was dissolved in 100 ml anhydrous THF. This solution was slowly added to a solution of pentafluorophenol (50g) in THF (100ml), Reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained solid was dried under vacuum at 45-50°C (yield=55g, confirmed by IR)
Example-2:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium methoxide
2,3,4,5, 6-pentafluorophenol (lOg) was dissolved in methanol (100ml), solution was cooled to 5-10°C. To this was added a solution of sodium methoxide in methanol. The reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained residue was striped with toluene. Obtained solid was dried under vacuum at 45-50°C (yield=l lg)
Example 3:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium hydroxide
2,3,4,5, 6-pentafluorophenol (lOOg) dissolved in methanol (—ml), solution was cooled to 5-10°C. To this was added a solution of sodium hydroxide (— g) in methanol. The reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained residue was striped with dichloromethane. Obtained solid was dried under vacuum at 45-50°C (yield=— g)
Example 4:
Preparation of (2S)-isopropyl-2-((chloro(phenoxy)posphoryl)amino)propanoate:
phenyl phosphodichloridate (30.6g) was dissolved in dichloromethane , to this was added a solution of 1-alanine isopropyl ester free base (19.16g) in dichloromethane at-60°C under nitrogen. Solution of triethylamine (20.7ml) was added to above reaction mass. Reaction mass was stirredat -60°C for 30 min and then temperature was raised to 25 °C. Reaction mass was stirred at 20-25 °C for 60 min & filtered and washed with dichloromethane. Clear filtrate was distilled under reduced pressure obtained residue was stirred with diisopropyl ether & filtered. Clear filtrate was distilled under reduced pressure to get (2S)-isopropyl-2-((chloro(phenoxy)posphoryl)amino)propanoate compound.
Example 5:
Preparation of isopropyl ((perfluorophenoxy)(phenoxy)phosphoryl)-L-alaninate (formula 2):

(Formula 2)
Obtained (2S)-isopropyl-2-((chloro(phenoxy)phosphoryl)amino)propanoate (1.2 mol eq.) was dissolved in dichloromethane and cooled to 0-5°C under nitrogen atmosphere. To this was added solution of sodium 2,3,4,5,6-pentaflurophemolate (1 mol eq.) in tetrahydrofuran . Temperature of reaction mass was raised to 25°C and reaction mass was stirred for 3 hrs. After completion of reaction, reaction mass was distilled under reduced pressure & obtained residue was dissolved I ethyl acetate. Ethyl acetate layer was washed with water, dried over sodium sulfate & distilled off under reduced pressure. Diisopropyl ether was added to obtained residue and stirred for 60 min at 25 °C, obtained mass was filtered & washed with diisopropyl ether. Solid product was dried under vacuum at 40-45 °C .(yield=20g, enantiomer purity=93.45%)
Example 6:
Preparation of (S)-isopropyl 2-(((S)- (perfluorophenoxy)phenoxy)phosphoyl)amino)propanoate (Formula 2′):

Formula 2′
(2S)-isopropyl-2-((chloro(phenoxy)phosphoryl)amino)propanoate (1.2 mol eq.) was dissolved in tetrahydrofuran (3.5 volumes). The reaction mass was cooled to -10°C. Solution of sodium salt of pentafluorophenol (1 mol eq.) in tetrahydrofuran (3.5 volumes) was added dropwise to the reaction mass at -10°C. After completion of the reaction solvent was distilled off. Ethyl acetate and water were added to the reaction mass. Reaction mass was stirred, ethyl acetate layer was separated and washed with sodium bicarbonate solution and brine. Ethyl acetate layer was concentrated under reduced pressure. Reaction mass was stripped with n-hepatane to get crude product. Crude product was dissolved in Methyl tert-butyl ether and n-heptane (1 : 1 ratio). The pH of reaction mass was adjusted to pH 8 by using triethylamine. Reaction mass was stirred overnight. Solid mass was filtered and washed with a mixture of methyl tertiary-butyl ether: n-heptane (1 : 1). The obtained product was dissolved in ethyl-acetate and washed with water and 20% brine solution. Ethyl acetate layer was separated; solvent was distilled off under reduced pressure. Reaction mass was stripped with diisopropyl ether. Di-isopropyl ether was added to the reaction mass. Reaction mass was stirred at 45-50°C. Reaction mass was cooled to 5-10°C and stirred. The titled compound was isolated by filtration and washed with di-isopropyl ether. The titled compound was dried under reduced pressure at 40°C. Yield 66.81%.

Vinita Gupta, CEO, Lupin Pharmaceuticals Inc

Desh Bhandu Gupta- Founder and chairman of Lupin Limited
////////////LUPIN LIMITED, WO 2016181313, NEW PATENT, SOFOSBUVIR
WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED


WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED
LINK>>> (WO2016147197) A NOVEL PROCESS FOR PREPARING (2S,3R,4R,5S,6R)-2-[4-CHLORO-3-(4-ETHOXYBENZYL)PHENY 1] -6-(HY DROXY METHYL)TETRAHYDRO-2H-PY RAN-3,4,5-TRIOL AND ITS AMORPHOUS FORM
HARMAN FINOCHEM LIMITED [IN/IN]; 107, Vinay Bhavya Complex 159-A, C.S.T. Road Kalina, Mumbai 400098 Maharashtra (IN)
![]()
KADAM, Vijay Trimbak; (IN).
SAIKRISHNA; (IN).
CHOUDHARE, Tukaram Sarjerao; (IN).
MINHAS, Harpreet Singh; (IN).
MINHAS, Gurpreet Singh; (IN)

CHAIRMAN

HARPREET SINGH MINHAS
Owner, HARMAN FINOCHEM LIMITED

(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is sodium dependent glucose transporter (SGLT) which is currently under investigation for the treatment of type-2 diabetes. (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is marketed under the tradename Farxiga or Forxiga.
(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is also known as D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4ethoxyphenyl)methyl]phenyl]-, (I S). (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3, 4,5 -triol is a white to off-white powder with a molecular formula of C2iH25C106 and a molecular weight of 408.87

Formula-I
US 6,515,117 B2 discloses (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol and its pharmaceutically acceptable salts. US 6,515,117 B2 also describes process for preparation of (2S,3R,4R,5S,6R)-2-[4- chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol which comprises reaction of 5-bromo-2-chloro-4′-ethoxydiphenylmethane with 2,3,4,6-tetra-O-trimethylsilyl- -D-glucolactone in presence of THF/Toluene, methansulfonic acid to yield o-methylglucoside product which further reacts with Et3SiH, BF3Et20 in presence of MDC and acetonitrile to yield yellow solidified foam which is dissolved in MDC, pyridine and followed by acetylation with acetic anhydride, DMAP to yield tetra acetylated- β-C-glucoside as a white solid which is further deprotected with LiOH H20 in presence of THF/MeOH/H20 to get (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol.
The drawback of said prior art is having multiple process steps which makes the process very lengthy and tedious. Moreover the process discloses use of hazardous chemicals like pyridine which is not applicable to industry.
Process for preparation of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenylJ-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is disclosed in US 7,375,213 B2 and J.Med.Chem.2008, 51, 1145-1149. The preparation process is depicted in Scheme-I.

Scheme-1
Prior art US’213 describes reaction of 2-chloro-5-bromo-4′-ethoxy-diphenylmethane with 2,3,4,6-tetra-O-trimethylsilyl-D-gluconolactone, n-BuLi in presence of THF and Heptane. After basification with TEA, the oily residue of methyl- l-C-(2-chloro-4′- ethoxy-diphenylmethan-3-yl)-a-D-glucopyranose obtained as solid compound after workup. This compound reacts with acetic anhydride in presence of THF, DIPEA and DMAP to get oily residue of methyl-2,3,4,6 tetra-0-acetyl-l-C-(2-chloro-4′-ethoxydiphenylmethan-3-yl)-a-D-glucopyranose which further undergoes reduction reaction in presence of acetonitirle, t riethylsilane, boron trifluoride etherate to yield 2,3,4,6-tetra-0-acetyl-l-C-(2-chloro-4′-ethoxydi henylmethan-3-yl)-β-D-glucopyranose which is further deprotected by reacting with LiOH monohydrate in presence of THF/MeOH/H20 to get (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol.
The said prior art describes multiple, time consuming process steps which involves getting the intermediate products as oily residue at various stages of the process, which is difficult to purify and handle for further process step. More over the workup involves multiple evaporation of product which may result in decomposition. Another drawback of the process is that the process describes n-BuLi reaction with two pot reaction. It is very difficult to transfer the material from one reactor to second reactor at -78 °C at industrial level with highly moisture sensitive reaction mass. This makes process uneconomical, cumbersome and commercially not viable. Further when practically the said method followed, a-Isomer of the final product is formed in the range of 6-8% along ith Des-bromo impurity formed in the range of 7-9 %, which increases after addition of n-butyllithium and kept the mass for overnight reaction. Moreover lactone ring cleavage is also observed in the range of 3-4% after addition of Methanesulphonic Acid/Methanol and maintained overnight for reaction completion, the removal of which is difficult from the final product.
WO 2008002824 A 1 discloses crystalline forms of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol comprising (S)-propylene glycol (PG), (R)-PG, EtOH, ethylene glycol (EG), 1 :2 L-proline, 1 : 1 L-proline, 1 : 1 L-proline hemihydrate, 1 : 1 L-phenylalanine and its preparation process.
In the light of the above drawbacks, it is necessitated to provide economical, robust, safe and commercially viable process for preparing (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol.
Accordingly, it is an objective of the present invention to provide a commercially viable process for the preparation of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxyb.enzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, prepared via riovel intermediates which gives higher yield and purity and facilitates easy recovery of the final compound. The purification process does not involve any costly technique/equipment, however, carried out with solvents which are industrially feasible. More over the present invention discloses the n-BuLi insitu reaction that makes the present invention cost-effective over the teachings of prior art.



Scheme-II

Formula-Ill Formula-IV
Formula-V where R1= allyl, prop-2-ynyl,isopropyl
Scheme-Ill

where R = allyl, prop-2-ynyl
Scheme-IV

Scheme-V
Examples:
Example-1: Preparation of 3,4,5-Tris-trimethylsiIanyloxy-6-trimethylsiIanyloxymethyl-tetrahydro-pyran-2-one
To 750 cc of dry THF added 1.12 mole 3,4,5-Trihydroxy-6-hydroxymethyl-tetrahydro-pyran-2-one at ambient temperature and stirred for 20 min. To the reaction mass added 9.0 mole N-Methyl morpholine and stirred for another 30.0 min at ambient temperature. Reaction mass was cooled to -5 °C to 0 °C and stirred for 30.0 min. Added 18.0 mole Trimethyl sillyl chloride at the temp -5 °C to 0 °C and stirred for 30.0 min. Temperature was raised to 25 °C to 30 °C and maintained for 18-20hrs. After reaction complies by GC, the reaction mass was cooled to -5 deg to 0 deg. Added Sat.Sodium bicarbonate solution to obtain the pH 7-8 and stirred for 1 hr at 0 °C. Added 500 cc toluene and stirred for lhr. Reaction mass was settled down for 30.0 min and layers were separated. To the Aqueous layer added 250 cc of toluene and stirred for 30.0 min. Layers separated and both the organic layers mixed and back washed with sat.brine solution. Organic layer was distilled under reduced pressure at a temperature of about 40 – 48 deg. Unload the oily mass . Purity: 92-96 %
Example-2: Preparation of 2-Allyloxy-2-[4-chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyI-tetrahydro-pyran-3,4,5-triol
To the mixture of 10 cc THF and 10 cc Toluene added 0.138 mole 4-(5-bromo-2-chlorobenzyl)phenyl ethyl ether at ambient temperature and stirred for 15 min. Cooled to -70 to -80°C in dry ice /acetone bath and stirred for 15 min. Added a solution of 0.014 mole n-Butyl lithium (1.9M in hexanes) at -70 to -80°C. and stirred for lhr. Added solution of 3, 4, 5-Tris-trimethylsilanyloxy-6-trimethylsilanyloxymethyl-tetrahydro-pyran-2-one in 5 cc of Toluene at -70 to -80°C and stirred for 2 to 3hrs. After the compliance of the reaction, reaction mass was quenched with Methane sulphonic acid and Allyl alcohol mixture at -70 to -80°C. Temperature was raised to ambient temperature and stirred overnight. Reaction mass was quenched with 30 cc sat.sodiumbicarbonate solution to bring the pH neutral to alkaline and stirred for 30.0 min. Layers separated and aqueous layer was extracted with 10 cc of Toluene. Organic layer was combined and washed with 30cc water and 50 cc sat. brine solution. Organic layer was distilled under reduced pressure to recover toluene. Solid compound was dissolved in 50cc of toluene and quenched in n-Hexane to obtain 83 % the compound as crystalline solid.
HPLC purity: 88 – 91 %
I R data:
Anomeric C-0 stretching: 1242 cm“1
Allylic C- O stretching: 1 177 cm“1
Allylic C- H stretching: 3010 – 3120 cm“1
Aromatic C- CI stretching: 820 cm“1
Lactones O – H stretching: 3240 – 3380 cm“1
Lactones C – 0 stretching: 1045 – 1092 cm“1
Aromatic C=C stretching: 1510 , 1548 , 1603 , 1703 cm“1
Alkane C – H stretching: 2877,2866, 2956, 2958, 2962 cm“1
Aromatic C – H stretching: 3050 – 3090 cm“1
Dip-Mass
(M+Na) 487.19 m/z
(M+K) 503.17 m/z
Example 3: Preparation of 2-prop-2ynyl-2-[4-Chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
To the mixture of 10 cc THF and 10 cc Toluene added 0.138 mole 4-(5-bromo-2-chlorobenzyl)phenyl ethyl ether at ambient temperature and stirred for 15 min. Cooled to -70 to -80°C in dry ice /acetone bath and stirred for 15 min. Added a solution of 0.014 mole n-Butyl lithium (1.9M in hexanes) at -70 to -80°C. and stirred for lhr. Added solution of 3, 4, 5-Tris-trimethylsilanyloxy-6-trimethylsilanyloxymethyl-tetrahydro-pyran-2-one in 5 cc of Toluene at -70 to -80°C and stirred for 2 to 3hrs. After the compliance of the reaction, the reaction mass was quenched with Methane sulphonic acid and propargyl alcohol mixture at -70 to -80°C. Temperature was raised to ambient temperature and stirred overnight. Reaction mass was quenched with 30 cc sat.sodiumbicarbonate solution to bring the pH neutral to alkaline. Reaction mass stirred for 30.0 min. Layers separated and aqueous layer was extracted with 10 cc of Toluene. Organic layer were combined and washed with 30cc water and 50 cc sat. brine solution. Organic layer was distilled under reduced pressure to recover toluene. Solid compound dissolved in 50cc of toluene and quenched in n-Hexane to obtain 75 – 80 % the compound as crystalline solid.
HPLC purity: 88 – 93 %
IR data:
Anomeric C-0 stretching: 1242 cm“1
Propargyl ~c CH stretching: 2125 cm“1
Propargyl C- H stretching : 3010 – 3120 cm“1
Aromatic C- CI stretching: 820 cm“1
Lactones O – H stretching: 3240 – 3380 cm“1
Lactones C – 0 stretching: 1045 – 1092 cm“1
Aromatic C=C stretching: 1510 , 1548 , 1603 , 1703 cm“1
Alkane C – H stretching: 2877, 2866,2956,2958,2962 cm“1
Aromatic C – H stretching: 3050 – 3090 cm“1
Dip-Mass
(M+Na) 485.25 m/z
(M+K) 501.25 m/z
Example-4: Preparation of 2-[4-Chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyI-tetrahydro-pyran-3,4,5-trioI
To the mixture of 20 cc (1 : 1 MDC + ACN) added 0.1 1 mole 2-Allyloxy-2-[4-chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol under argon atmosphere, and stirred the reaction mass for 30.0 min. Cooled the reaction mass to -40 to -55°C in a dry ice/acetone bath under argon atmosphere. Charged 3 mole Triethylsilane at -40 to -55°C and stirred the reaction mass for 30.0 min at -50 to -55°C. Slowly added Borontrifloride in diethyl ether solution at -40 to -55°C and stirred the reaction mass for 2 hrs. Quenched the reaction mass with 50 cc sat. sodium bicarbonate solution at -40 to -55°C . and stirred the reaction mass for 30.0 min. Slowly raised the temperature to 25 to 30°C. Settled down the reaction mass and separated the layers, extracted the aqueous layer with 100 cc of MDC. Combined the organic layer and wash with 500 cc water. Washed the organic layer with 500 cc of sat. Brine solution. Distilled out the MDC under reduced pressure below 40°C. to get 85 %the light yellow solid.
HPLC purity: 92-95 %
Example 5: Preparation of 2-[4-Chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
To the mixture of 20 cc (1 :1 MDC + ACN) added 0.11 mole 2-prop-2-ynyl-2-[4-Chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol under argon
atmosphere. Stirred the reaction mass for 30.0 min. Cooled the reaction mass to -40 to -55°C in a dry ice/acetone bath under argon atmosphere. Charged 3 mole Triethylsilane at -40 to -55°C and stirred the reaction mass for 30.0 min at -50 to -55°C. Slowly added Borontrifloride in diethyl ether solution at -40 to -55°C and stirred the reaction mass for 2 hrs. Quenched the reaction mass with 50 cc sat. sodium bicarbonate solution at -40 to -55°C and Stirred the reaction mass for 30.0 min. Slowly raised the temperature to 25 to 30°C. Settled down the reaction mass and separated the layers, extracted the aqueous layer with 100 cc of MDC. Combined the organic layer and washed with 500 cc water. Washed the organic layer with 500 cc of sat. Brine solution. Distilled out the MDC under reduced pressure below 40°C. to get 85%the light yellow solid.
HPLC purity: 90%
Example 6: Preparation of amorphous form of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
To the solid obtained from example 4 charged 500cc of n-heptane and stirred for ½hrs at ambient temperature. Heated the reaction mass to 55-60°C and stirred it for 2-3 hrs.; cooled to room temperature and maintained for 4-5 hrs. Filtered the solid and washed the, cake with 100 cc n-heptane. Dried at 40-45°C under vacuum to get 85% amorphous form of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol.
HPLC purity: 91-93%
Example 7: Preparation of amorphous form of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
To the solid obtained from example 5 charged 500cc of n-heptane and stirred for ½ hrs at ambient temperature. Heated the reaction mass to 55-60°C and stirred it for 2-3 hrs., cooled to room temperature and maintained for 4-5 hrs. Filtered the solid and washed the cake with 100 cc n-heptane. Dried at 40-45 °C under vacuum to get 85-88% amorphous form of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol.
HPLC purity: 89-91%
Example 8: Preparation of L-proline – (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyI]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol co crystal
To the 10 cc of Ethyl acetate charged 1.0 mole (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol under argon atmosphere at ambient temperature and stirred for 30.0 min to get clear solution. Slowly heated the reaction mass to 60 – 65°C and stirred for 1 hr. Slowly added L-proline at 60 -65°C and maintained for 1 hr. Slowly added 15 cc n-Heptane to the reaction mass at 60 -65°C and stirred the mass for 2.5 hrs. Cooled the mass to ambient temperature for 3-4 hrs and maintained for 5 hrs. Filtered the mass under argon atmosphere. Washed the cake with 10 cc n-Heptane. Dried the cake at 50-55°C under reduced pressure to get 92% titled compound.
HPLC purity: 99%
Example 9: Preparation of L-proline – (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triolco crystal
To the 10 cc of acetone charged 1.0 mole (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol under argon atmosphere at ambient temperature and stirred for 30.0 min to get clear solution. Slowly heated the reaction mass to 60 – 65°C and stirred for 1 hr. Slowly added“ proline at 60 -65°C and maintained for 1 hr. Slowly added 15 cc n-Heptane to the reaction mass at 60 -65°C and stirred the mass for 2.5 hrs. Cooled the mass to ambient temperature for 3-4 hrs and maintained for 5 hrs. Filtered the mass under argon atmosphere. Washed the cake with 10 cc n-Heptane. Dried the cake at 50-55°C under reduced pressure to get 93-95% titled compound.
HPLC purity: 98-99%
Example 10: Preparation of amorphous form of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
To the 15 cc ethyl acetate added (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol at ambient temperature and stirred for 30.0 min. Slowly added 5- 8 cc sat. sodium bicarbonate solution at ambient temperature and stirred for 1.5 hr to get the clear solution. Settled down and separated layers. Extracted the aqueous layer with 25 cc ethyl acetate.
Combined the organic layers and washed the ethyl acetate layer with 50 cc sat. Sodium chloride solution. Distilled out ethyl acetate under reduced pressure at 40 – 45°C to get ■fluffy solid. Charged 50 cc n-Heptane and stirred for 5 hrs to get 70-78% the title compound as Amorphous soild.
HPLC purity: 99.8-99.95 %
Example 11: Preparation of 2-chloro -4′- ethoxydiphenylmethane (impurity)
To the 20 cc THF and 20 cc Toluene added 0.25 mole 2-ehloro-5-bromo-4′- ethoxydiphenylmethane under argon atmosphere. Cooled the reaction mass to – 78° C. Slowly added n-Butyl lithium (1.9 M in hexane) at – 78° C and stirred for 30 min. Slowly added 20 % Ammonium chloride solution to the reaction mass. Brought the reaction mass to ambient temperature and stirred for 30 min. Settled and separated layers. Extracted the aqueous layer with 50 cc toluene. Washed the combined organic layer with 500 cc brine solution. Distilled out the toluene and charged heptanes, stirred for 2 – 3 hrs at ambient temperature. Filtered the product and dried the product at 45 – 50°C under reduced pressure to get 93 % titled compound.
Mass: (m+1) 247 m/z found 247.1 1
HPLC purity: 96.33 %
SHENDRA AURANGABAD, MAHARASHTRA, INDIA

Bhupinder Singh Manhas


////////WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED
WO 2016147120, AZILSARTAN, NEW PATENT, SMILAX Laboratories Ltd
![]()
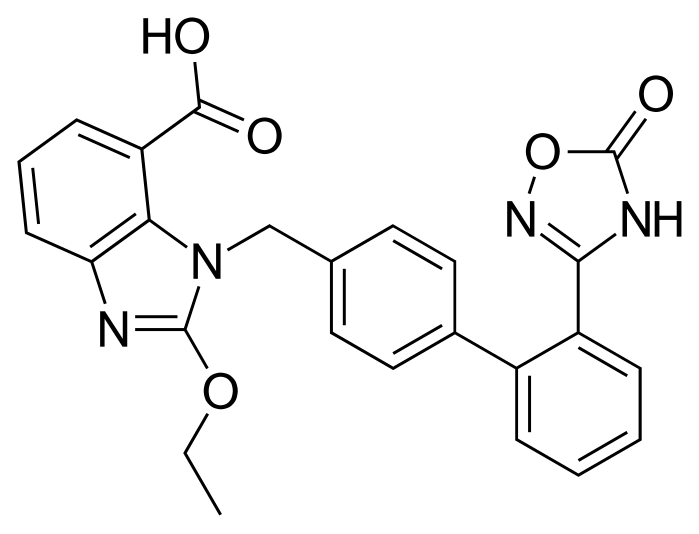
WO-2016147120, AZILSARTAN, NEW PATENT, SMILAX Laboratories Ltd
SMILAX LABORATORIES LIMITED [IN/IN]; Plot No. 12/A, Phase – III, I.D.A. Jeedimetla, Hyderabad 500 055 (IN).
YENUMULA, Raghavendra Rao; (IN).
BANDARI, Mohan; (IN).
SURYADEVARA, Murali Krishna; (IN)

(WO2016147120) AN IMPROVED PROCESS FOR THE PREPARATION OF SUBSTANTIALLY PURE AZILSARTAN
Azilsartan (I) is an angiotensin receptor II antagonist used in the treatment of hypertension. Angiotensin II causes vasoconstriction via an angiotensin II receptor on the cell membrane and elevates blood pressure.
Azilsartan medoxomil i.e. (5-methyl-2-oxo-l,3-dioxol-4-yl)methyl 2-ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid is developed by Takeda pharmaceuticals and is marketed under the trade name Edarbi. It was approved by USFDA on 25 Feb, 2011 and EMEA on 7 Dec 2011 for the treatment of high blood pressure in adults.
Azilsartan medoxomil and its salts thereof are imbibed with properties such as strong and long lasting angiotensin II antagonistic activity and hypotensive action which has an insulin sensitizing activity useful for the treatment of metabolic diseases such as diabetes and the like., and a useful agent for the prophylaxis or treatment of circulatory diseases such as hypertension, cardiac diseases, nephritis and stroke. Azilsartan medoxomil is the prodrug of 2-Ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan).
Methods of preparing benzimidazole derivative useful as an angiotensin II receptor antagonist such as Azilsartan Medoxomil and salts thereof are disclosed by Takeda in US 5,243,054 (herein after referred as US ‘054 patent). The US’054 patent describes several synthetic routes for preparing Azilsartan. According to one of the synthetic process, the compound of formula II is reacted with hydroxylamine hydrochloride in a conventional organic solvent and sodium methoxide in methanol to give the amidoxime compound of formula III which on further reaction with ethyl chloroformate in presence of triethylamine base in refluxing xylene undergoes cyclization to provide a compound of formula IV. Azilsartan was prepared by hydrolysis of compound of formula IV in presence of lithium hydroxide by adjusting the pH with HC1. The process is as depicted below in Scheme A:


However, the amidoxime compound of formula III obtained by the above process contains about 50% of amide imputiy along with desired product, owing to the strong reaction conditions which impairs the quality and loss of yield. The pH adjustment with HC1 in the hydrolysis step of compound IV results in the formation of an undesired desethyl impurity of formula V due to acid sensitive nature of the ether linkage in the benzimidazole moiety of Azilsartan.
Formula V

According to another method disclosed in US’054 for the preparation of Azilsartan comprises by reacting ethoxycarboimidoyl biphenyl benzimidazole derivative of compound with ethyl chloroformate to give N-methoxycarbonyl ethoxycarboimidoyl biphenyl benzimidazole derivative, which is further converted to compound of formula IV and then to Azilsartan of formula I by hydrolysis.
According to one another embodiment method for the preparation of Azilsartan disclosed in US ‘054, cyanobiphenyl aminobenzoate derivative compound reacts with hydroxylamine hydrochloride in presence of triethylamine subsequently followed by addition of ethyl chlorocarbonate results in the formation of compound of formula IV which is further hydrolyzed to obtain Azilsartan of formula I.
J. Med. Chem. Vol. 39, No. 26, 5230-5237 (1996) describes the use of triethylamine as base during the conversion of compound of formula II to amidoxime compound of formula III and use of 2-ethylhexylchloroformate instead of ethylchloroformate as cyclizing agent.
Processes for the preparation of Azilsartan medoxomil and its potassium salt are described in US 7,157,584 which comprises reacting Azilsartan with 4-hydroxymethyl-5-methyl-l,3-dioxol-2-one in presence of dimethylacetamide, p-toluoyl sulfonylchloride, 4-dimethylaminopyridine and potassium carbonate.
PCT publication WO 2012/107814 discloses process for the preparation of Azilsartan or its esters or salts by reacting amidoxime compound of formula III with carbonyl source such as carbodiimides, dialkyl carbonate and phosgene equivalents in presence of a suitable base to obtain compound of formula IV which is further converted to Azilsartan and its pharmaceutically acceptable salts. The process for the preparation of Azilsartan is as depicted in Scheme B:

Scheme – B
This publication also discloses that use of a carbonyl source reduces the formation of the content of desethyl impurity during cyclization.
Polymorphs of Azilsartan and its salts are disclosed in WO 2013/044816 and WO 2013/186792.
All the above prior art methods for the preparation of Azilsartan have inherent disadvantages such as the usage of unsafe reagents, high boiling solvents, extreme reaction conditions invariably resulting in the formation of low pure intermediates as well as Azilsartan having a considerably higher content of desethyl impurity. Accordingly, there remains a need for the industrial preparation of substantially pure Azilsartan which is free of impurities with high yield.

Examples
Example-1: Preparation of Methyl-2-ethoxy-l-[[2′- ((hydroxycarbamimidoyl)biphenyl)-4-yl]methyl]-lH-benzimidazole-7-carboxylate (Formula-Ill):
To a stirred solution of DMSO (1500.0 mL), Hydroxylamine hydrochloride ( 126.7g 1.83mol) and Dipotassium hydrogen phosphate (634.9g 3.65mol) was added Methyl l-[[2′-cyanobiphenyl-4-yl]methyl]-2-ethoxybenzimidazole-7-carboxylate (lOO.Og 0.243mol) at 25-30°C. The reaction mass temperature was raised to 80-85°C and maintained for 30-40 hours. Reaction completion was monitored by TLC. Upon completion of reaction, reaction mass was cooled to 10- 15°C, and was poured into water (3000.0 mL), stirred for 45min at 20-25°C. and was filtered. The filtered wet solid was washed with water and dried at 65°C to get crude Methyl-2-ethoxy-l-[[(2′-(hydroxycarbarmrmdoyl)biphenyl-4-yl]methyl]-lH-benzimidazole-7-carboxylate. The wet material was slurried in Acetone (optional) at reflux and filtered at room temperature to obtain pure compound.
Yield: 79.92 g, 74.0%; HPLC Purity: 97.78%; Desethyl impurity: 0.318%; Amide impurity: 1.42%.
Example-2: Preparation of Methyl 2-ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (Formula-IV) :
To the pre cooled solution of Methylene dichloride (375.0 mL) and Methyl-2-ethoxy-1 -[[2′ -((hydroxycarbamimidoyl)biphenyl)-4-yl] methyl]- lH-benzimidazole-7-carboxylate (75.0g, 0.168mol) was added ethyl chloroformate ( 18.3g 0.168mol)
followed by addition of triethylamine (18.75g 0.185mol). The reaction mass was maintained at 0-5 °C for about 1 hour. Upon completion of the reaction, reaction mass was poured into water (200.0 mL), organic layer was separated and washed with 5% NaHC03 solution (150.0 mL) and then with water (150.0 mL). The organic layer was dried over sodium sulfate and distilled to obtain the crude material (optionally be isolated using cyclohexane solvent). To this obtained crude material, ethyl acetate (750.0mL) and potassium carbonate (112.5g 0.814mol) were added and heated to reflux for 6 to 8 hours. The contents were cooled, filtered and wet solid was slurried in water. Wet material so obtained was slurried in ethyl acetate at reflux and filtered at room temperature and dried at 60-65°C to give Methyl 2-ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate.
Yield: 64.27 g, 81.0 %; HPLC Purity: 99.80%; Desethyl impurity: 0.085%.
Example-3: Preparation of 2-Ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan)
A mixture of 0.4N NaOH solution (395.8 mL) and Methyl 2-ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (25. Og) were stirred at 50-55°C for period of 60min. The reaction mass was cooled to room temperature and the product layer was washed with ethyl acetate (125.0mL). pH of the separated aqueous product layer was adjusted to 4.0 to 5.0 using dilute acetic acid at 0-5 °C. The obtained solid material was filtered and washed with water (lOO.OmL). This material was dried to obtain the title product.
Yield: 20.0 g, 82.47%; HPLC Purity : 99.80%; Desethyl impurity: 0.10%.
Example-4: Preparation of 2-Ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan)
A mixture of 0.4N NaOH solution (633.33 mL) and Methyl 2-ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (40.0g) were stirred at 50-55°C for period of 60min. The reaction mass was cooled to room temperature and the product layer was washed with ethyl acetate (200.0mL). pH of the separated aqueous product layer was adjusted to 4.0 to 4.5 using acetic acid at 10-15°C. The obtained solid material was filtered and washed with water (lOO.OmL). This material was dried to obtain the title product.
Yield: 32.35 g, 83.37%; HPLC Purity: 99.45%; Desethyl impurity: 0.12%.
Example-5: Preparation of 2-Ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan)
A mixture of 0.4N NaOH solution (791.66 mL) and Methyl 2-ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (50.0g) were stirred at 50-55°C for period of 60min. The reaction mass was cooled to room temperature and the product layer was washed with ethyl acetate (250.0mL). pH of the separated aqueous product layer was adjusted to 3.0 to 4.0 using citric acid at 10-15°C. The obtained solid material was filtered and washed with water (125.0mL). This material was dried to obtain the title product.
Yield: 37.0 g, 76.28%; HPLC Purity: 99.69%; Desethyl impurity: 0.083%.
Example-6: Preparation of 2-Ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan)
A mixture of 0.4N NaOH solution (395.83 mL) and Methyl 2-ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (25. Og) were stirred at 50-55°C for period of 60min. The reaction mass was cooled to room temperature and the product layer was washed with ethyl acetate (lOO.OmL). pH of the separated aqueous product layer was adjusted to 3.0 to 4.0
using hydrochloric acid at 10-15°C. The obtained solid material was filtered and washed with water (72.5 mL). This material was dried to obtain the title product. Yield: 20.22 g, 83.37%; HPLC Purity: 99.45%; Desethyl impurity: 0.217%.
Example-7: Purification of 2-Ethoxy-l-[[2′-(2,5-dihydro-5-oxo-l,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (Azilsartan)
Charged 2-Ethoxy- 1 – [[2′ -(2,5-dihydro-5-oxo- 1 ,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid (lOO.Og), methanol (600.0ml) and methylene dichloride (600.0ml) and were stirred for 10 min at 25-30°C to get a clear solution. Above solution was treated with Activated carbon (lO.Og) and stirred for 10.0 min at 25-30°C. Reaction mixture was passed through a hyflow bed and washed with a mixture of (1: 1) ratio of 200.0ml methanol and methylene dichloride. The solvent mixture was distilled out at below 50°C till the solid formation was observed. Reaction mixture is stirred for 30.0min at 30°C, then the solid was filtered and washed with 200.0ml of methylene dichloride. To the obtained solid, methanol (450.0 ml) was charged at 25-30°C, heated to 45°C, stirred for 30 min at 45°C and then cooled to 30°C. After cooling, the solid was filtered and washed with methanol (90.0ml) which was further dried at 50-55°C for 12 hours.
Yield: 80.0 g, 80.0%; HPLC Purity: 99.96%; Desethyl impurity : 0.012%.


Smilax Managing Director, S. Murali Krishna received the award from Hon’ble Chief Minister of Andhra Pradesh Shri. N. Kiran Kumar Reddy.
Nacubactam, A diazabicyclooctane beta-lactamase inhibitor, for treating bacterial infection


Nacubactam
RG-6080, FPI-1459, OP-0595, WK ?, WK-?, WK?
(2S,5R)-N-(2-amino ethoxy)-6-(sulfooxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
Beta lactamase inhibitor
Roche, under license from Meiji Seika Pharma and Fedora Pharmaceuticals is developing nacubactam hydrate
Meiji Seika Pharma Co., Ltd., Meiji Seikaファルマ株式会社
A diazabicyclooctane beta-lactamase inhibitor, for treating bacterial infection. In July 2016, nacubactam was reported to be in phase 1 clinical development
PATENTS , IN2015MU287, WO2016116878, WO 2016120752, INDICATE INTEREST FROM WOCKHARDT


Sulfuric acid, mono[(1R,2S,5R)-2-[[(2-aminoethoxy)amino]carbonyl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
A β-lactamase inhibitor potentially for the treatment of bacterial infections.
![]()
RG-6080; FPI-1459; OP-0595
CAS No. 1452458-86-4
| Molecular Formula | C9 H16 N4 O7 S |
| Formula Weight | 324.31 |
- Originator Fedora Pharmaceuticals
- Developer Meiji Seika Pharma
- Class Antibacterials; Azabicyclo compounds
- Mechanism of Action Beta lactamase inhibitors
- Phase I Bacterial infections
Most Recent Events
- 13 Jan 2015 OP 0595 licensed to Roche worldwide, except Japan ,
- 30 Nov 2014 Meiji Seika Pharma completes a phase I trial in Healthy volunteers in Australia (NCT02134834)
- 01 May 2014 Phase-I clinical trials in Bacterial infections (in volunteers) in Australia (IV)

Bacterial infections continue to remain one of the major causes contributing towards human diseases. One of the key challenges in treatment of bacterial infections is the ability of bacteria to develop resistance to one or more antibacterial agents over time. Examples of such bacteria that have developed resistance to typical antibacterial agents include: Penicillin-resistant Streptococcus pneumoniae, Vancomycin-resistant Enterococci, and Methicillin-resistant Staphylococcus aureus. The problem of emerging drug-resistance in bacteria is often tackled by switching to newer antibacterial agents, which can be more expensive and sometimes more toxic. Additionally, this may not be a permanent solution as the bacteria often develop resistance to the newer antibacterial agents as well in due course. In general, bacteria are particularly efficient in developing resistance, because of their ability to multiply very rapidly and pass on the resistance genes as they replicate.
The persistent exposure of bacterial strains to a multitude of beta- lactam antibacterial agents has led to overproduction and mutation of beta-lactamases. These new extended spectrum beta-lactamases (ESBL) are capable of hydrolyzing penicillins, cephalosporins, monobactams and even carbapenems. Such a wide spread resistance to many of the existing beta-lactam antibacterial agents, either used alone or in combination with other agents, is posing challenges in treating serious bacterial infections.
Due to various reasons, the oral therapeutic options for treating bacterial infections (including those caused by ESBL strains) are limited. For example, a combination of amoxicillin and clavulanic acid is effective against Class A ESBLs producing bacteria. However, the usefulness of this combination is compromised against bacteria producing multiple or mixed beta-lactamase enzymes (such as, for example, bacteria producing Class A and Class C ESBLs concurrently), and Klebsiella pneumoniae carbapenemases (KPCs). Therefore, oral antibacterial agents or combinations with activity against a range of bacterial strains (including those producing multiple ESBLs and KPCs) are urgently desired.
Cephalosporin antibacterial agents are known for treatment for various bacterial infections. Surprisingly, it has been found that pharmaceutical compositions comprising a cephalosporin antibacterial agent and certain nitrogen containing bicyclic compound (disclosed in PCT/IB2013/053092, PCT/JP2013/064971 and PCT/IB 2012/002675) exhibit unexpectedly synergistic antibacterial activity, even against highly resistant bacterial strains.
SYNTHESIS
WO 2015046207,
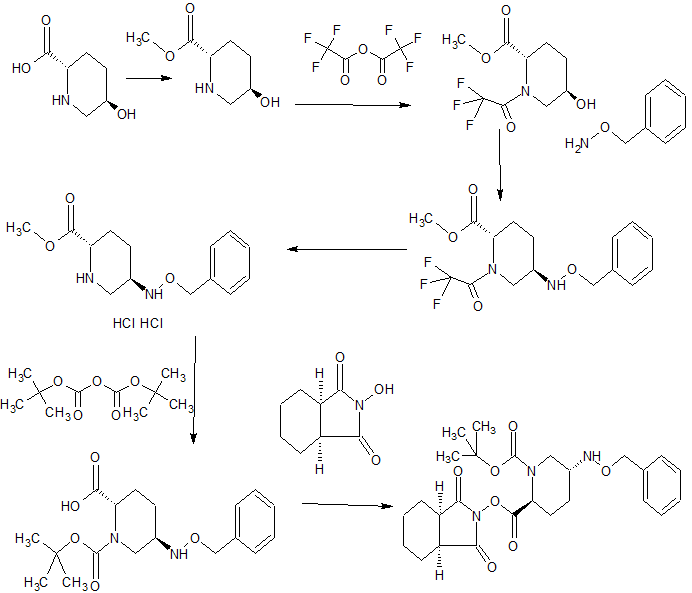
CONTD…………………..
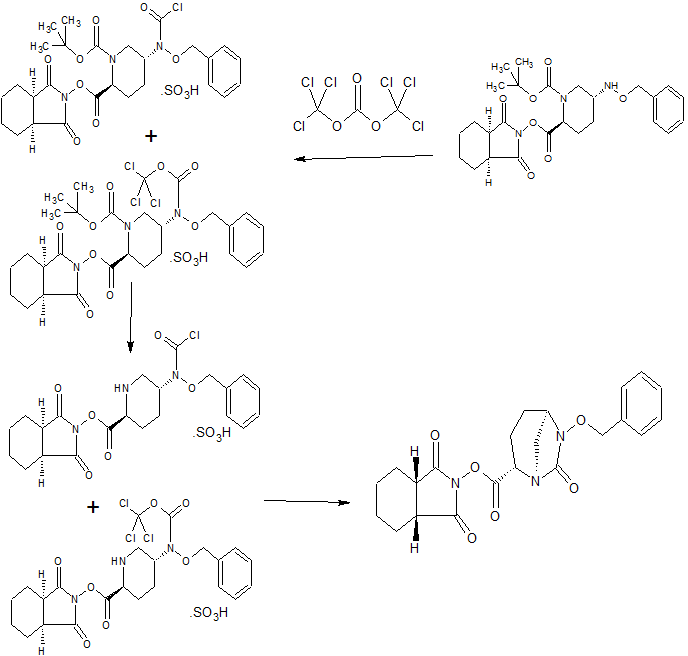
CONTD………………………………..
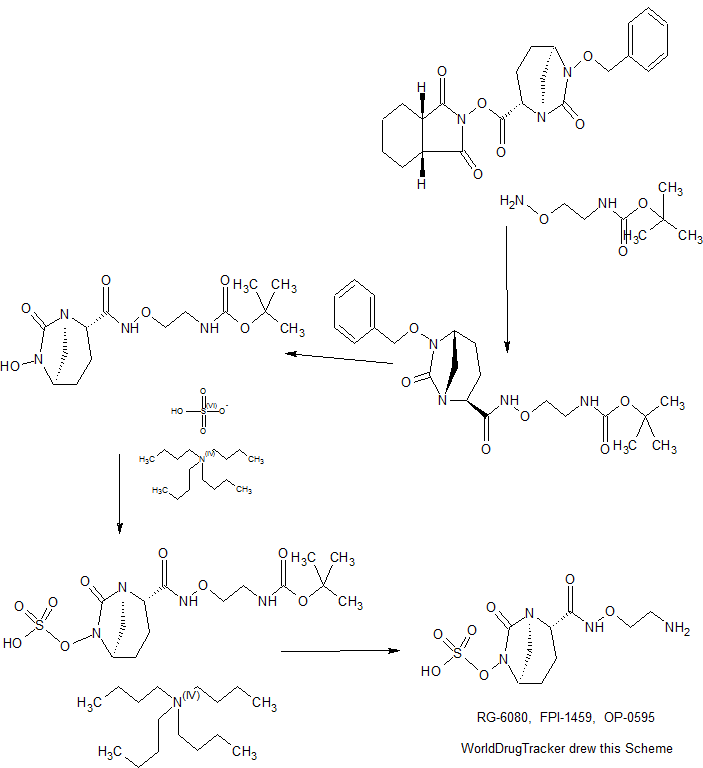
Patent
The novel heterocyclic compound in Japanese Patent 4515704 (Patent Document 1), preparation and shown for their pharmaceutical use, sodium trans-7-oxo-6- (sulfooxy) as a representative compound 1,6-diazabicyclo [3 .2.1] discloses an octane-2-carboxamide (NXL104). Preparation in regard to certain piperidine derivatives which are intermediates Patent 2010-138206 (Patent Document 2) and JP-T 2010-539147 (Patent Document 3) are shown at further WO2011 / 042560 (Patent Document 4) NXL104 to disclose a method for producing the crystals.
In Patent 5038509 (Patent Document 5) (2S, 5R) -7- oxo -N- (piperidin-4-yl) -6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane – 2- carboxamide (MK7655) is shown, discloses the preparation of certain piperidine derivatives with MK7655 at Patent 2011-207900 (Patent Document 6) and WO2010 / 126820 (Patent Document 7).
The present inventors also disclose the novel diazabicyclooctane derivative represented by the following formula (VII) in Japanese Patent Application 2012-122603 (Patent Document 8).
Patent Document 1: Japanese Patent No. 4515704 Pat
Patent Document 2: Japanese Patent Publication 2010-138206 Pat
Patent Document 3: Japanese patent publication 2010-539147 Pat
Patent Document 4: International Publication No. WO2011 / 042560 Patent
Patent Document 5: Japanese Patent No. 5038509 Pat
Patent Document 6: Japanese Patent Publication 2011-207900 Pat
Patent Document 7: International Publication No. WO2010 / 126820 Patent
Patent Document 8: Japanese Patent application 2012-122603 Pat.
[Chemical formula 1] (In the formula, R 3 are the same as those described below)
Reference Example
5 of 5 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
Formula 43]
step 1 tert-butyl {2 – [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate (IV-1)(2S, 5R)-6-(benzyloxy) -7-oxo-1,6-diazabicyclo [3.2.1] octane-2-carboxylic acid (4 .30g, dehydrated ethyl acetate (47mL) solution of 15.56mmol) was cooled to -30 ℃, isobutyl chloroformate (2.17g, washing included dehydration ethyl acetate 1mL), triethylamine (1.61g, washing included dehydration ethyl acetate 1 mL), successively added dropwise, and the mixture was stirred 1 hour at -30 ° C.. To the reaction solution tert- butyl 2-dehydration of ethyl acetate (amino-oxy) ethyl carbamate (3.21g) (4mL) was added (washing included dehydration ethyl acetate 1mL), raising the temperature over a period of 1.5 hours to 0 ℃, It was further stirred overnight. The mixture of 8% aqueous citric acid (56 mL), saturated aqueous sodium bicarbonate solution (40 mL), sequentially washed with saturated brine (40 mL), dried over anhydrous magnesium sulfate, filtered, concentrated to 5 mL, up to 6mL further with ethanol (10 mL) It was replaced concentrated. Ethanol to the resulting solution (3mL), hexane the (8mL) in addition to ice-cooling, and the mixture was stirred inoculated for 15 minutes. The mixture was stirred overnight dropwise over 2 hours hexane (75 mL) to. Collected by filtration the precipitated crystals, washing with hexane to give the title compound 5.49g and dried in vacuo (net 4.98 g, 74% yield). HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 50/50, 1.0 mL / min, UV 210 nm, Retweeted 4.4 min; 1 H NMR (400 MHz, CDCl 3 ) [delta] 1.44 (s, 9H), 1.56-1.70 (m, 1H), 1.90-2.09 (m, 2H), 2.25-2.38 (m, 1H), 2.76 (d, J = 11.6 Hz, 1H), 3.03 (br.d., J = 11.6 Hz , 1H), 3.24-3.47 (m, 3H), 3.84-4.01 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 5.44 (br. . s, 1H), 7.34-7.48 (yd, 5H), 9.37 (Br.S., 1H); MS yd / z 435 [M + H] + .
Step 2
tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate
(V-1) tert-butyl {2 – [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl ] carbonyl} amino) oxy] ethyl} carbamate (3.91 g, to a methanol solution (80 mL) of 9.01mmol), 10% palladium on carbon catalyst (50% water, 803 mg) was added, under hydrogen atmosphere and stirred for 45 minutes . The reaction mixture was filtered through Celite, after concentrated under reduced pressure to give 3.11g of the title compound (quantitative).
HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 75/25, 1.0 mL / min, UV 210 nm, Retweeted 3.9 from min; 1 H NMR (400 MHz, CD 3 OD) [delta] 1.44 (s, 9H) , 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 (br.dd., J = 15.0, 7.0 Hz, 1H), 3.03 (d, J= 12.0 Hz, 1H), 3.12 (br.d., J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS M / Z 345 [M Tasu H] Tasu .
Step 3
Tetrabutylammonium tert- butyl {2 – [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate
(VI-1) tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct 2-yl] carbonyl} amino) oxy] ethyl} carbamate (3.09g, in dichloromethane (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide – pyridine complex (3 .58g) was added, and the mixture was stirred overnight at room temperature. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, washed the aqueous layer with chloroform, tetrabutylammonium hydrogen sulfate to the aqueous layer and (3.47 g) chloroform (30 mL) was added and stirred for 10 minutes. The aqueous layer was extracted with chloroform, drying the obtained organic layer with anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the title compound 5.46g (91% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 80/20, 1.0ML / Min, UV210nm, RT 2.0 Min; 1 H NMR (400 MHz, CDCl 3 ) Deruta 1.01 (T, J = 7.4 Hz, 12H), 1.37-1.54 (m , 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30- 2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85-3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s , 1H), 9.44 (Br.S., 1H); MS yd / z 425 [M-Bu 4 N + 2H] + .
Step 4 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
tetra butylammonium tert- butyl {2 – [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (5.20g, 7.82mmol) in dichloromethane (25mL) solution of ice-cold under trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed the resulting residue with diethyl ether, adjusted to pH7 with aqueous sodium bicarbonate, subjected to an octadecyl silica gel column chromatography (water), after freeze drying, 1.44 g of the title compound obtained (57% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 99/1, 1.0ML / Min, UV210nm, RT 3.1 Min; 1 H NMR (400 MHz, D 2O) Deruta 1.66-1.76 (M, 1H), 1.76-1.88 (m, 1H ), 1.91-2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz , 2H), 3.18 (br d , J = 12.0 Hz, 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4 3.2 Hz &, 1H); MS yd / z 325 [M + H] + .
PATENT
Example
64 tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy ] ethyl} carbamate (V-1)
[of 124]
tert- butyl {2 – [({[(2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl } carbamate (example 63q, net 156.42g, 360mmol) in methanol solution (2.4L) of 10% palladium carbon catalyst (50% water, 15.64g) was added, under an atmosphere of hydrogen, stirred for 1.5 hours did. The catalyst was filtered through celite, filtrate was concentrated under reduced pressure until 450mL, concentrated to 450mL by adding acetonitrile (1.5 L), the mixture was stirred ice-cooled for 30 minutes, collected by filtration the precipitated crystals, washing with acetonitrile, and vacuum dried to obtain 118.26g of the title compound (net 117.90g, 95% yield). Equipment data of the crystals were the same as those of the step 2 of Reference Example 3.
Example
65 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VI-1)
tert- butyl {2 – [({[(2S, 5R) -1,6- -6- hydroxy-7-oxo-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (example 64,537.61g, 1.561mol) in acetonitrile (7.8L) solution of 2,6-lutidine (512.08g), sulfur trioxide – pyridine complex (810.3g) was added, at room temperature in the mixture was stirred overnight. Remove insolubles and the mixture was filtered, the filtrate concentrated to 2.5 L, diluted with ethyl acetate (15.1L). The mixture was extracted with 20% phosphoric acid 2 hydrogencarbonate aqueous solution (7.8L), the resulting aqueous layer into ethyl acetate (15.1L), added tetrabutylammonium hydrogen sulfate (567.87g), was stirred for 20 min. The organic layer was separated layers, dried over anhydrous magnesium sulfate (425 g), after filtration, concentration under reduced pressure, substituted concentrated tetrabutylammonium tert- butyl with dichloromethane (3.1L) {2 – [({[(2S, 5R ) -7-oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate was obtained 758g (net 586.27g, Osamu rate 84%).
The tetra-butyl ammonium salt 719g (net 437.1g, 0.656mol) in dichloromethane (874mL) solution was cooled to -20 ℃, dropping trifluoroacetic acid (874mL) at 15 minutes, 1 the temperature was raised to 0 ℃ It was stirred time. The reaction was cooled to -20 ° C. was added dropwise diisopropyl ether (3.25L), and the mixture was stirred for 1 hour the temperature was raised to 0 ° C.. The precipitate is filtered, washed with diisopropyl ether to give the title compound 335.36g of crude and vacuum dried (net 222.35g, 99% yield).
The title compound of crude were obtained (212.99g, net 133.33g) and ice-cold 0.2M phosphate buffer solution of pH5.3 mix a little at a time, alternating between the (pH6.5,4.8L). The solution was concentrated under reduced pressure to 3.6L, it was adjusted to pH5.5 at again 0.2M phosphate buffer (pH6.5,910mL). The solution resin purification (Mitsubishi Kasei, SP207, water ~ 10% IPA solution) is subjected to, and concentrated to collect active fractions, after lyophilization, to give the title compound 128.3 g (96% yield). Equipment data of the crystals were the same as those of step 3 of Reference Example 3.
PATENT
US 20140288051
WO 2014091268
WO 2013180197
US 20130225554
PATENT
IN2015MU287
PATENT
Example 59
(2S, 5R) -N- (2- aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (II-059)

Step 1
tert- butyl {2 – [({[(2S, 5R) -6- Benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl } carbamate

Acid of Example 9 or 16 (6b, 1.34g, 4.87mmol) in methylene chloride (35mL) solution of triethylamine (2.71mL), N- ethyl -N ‘- (3- dimethylaminopropyl) carbodiimide hydrochloride (1.41g), 1- hydroxybenzotriazole monohydrate (1.15g), were added tert- butyl of Reference Example 9, wherein 2- (amino-oxy) ethyl carbamate (1.12g), room temperature It was stirred overnight Te.Water was added to the reaction solution to a residue obtained by concentration under reduced pressure, and extracted with ethyl acetate. The resulting organic layer with 0.1M hydrochloric acid, saturated aqueous sodium bicarbonate solution, washed with saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered and concentrated.The resulting residue was purified by silica gel column and purified by chromatography (hexane / ethyl acetate = 8 / 2-0 / 10) to give the title compound 1.77g (84% yield).
[Α] D 20 -0.08 ° (c 0.29, CHCl 3); 1 H NMR (400 MHz, CDCl 3), δ: 1.44 (s, 9H), 1.56-1.70 (m, 1H), 1.90-2.09 (m , 2H), 2.25-2.38 (m, 1H), 2.76 (d, J = 11.6 Hz, 1H), 3.03 (br d, J = 11.6 Hz, 1H), 3.24-3.47 (m, 3H), 3.84-4.01 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 5.44 (br s, 1H), 7.34-7.48 (m, 5H), 9.37 (br s, 1H); MS m / z 435 [M + H] +; enantiomeric excess of 99.9% or higher ee (CHIRALPAK AD-H, 4.6x150mm, hexane / ethanol = 2/1, UV210nm, flow rate 1mL / min, retention time 4.95min (2R, 5S), 6.70min (2S, 5R).
Step 2
tert- butyl {2 – [({[(2S, 5R) -1,6- -6- hydroxy-7-oxo-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate

Compound of the above Step 1 (3.91g, 9.01mmol) in methanol (80mL), 10% palladium on carbon catalyst (50% water, 803mg) was added, under hydrogen atmosphere and stirred for 45 minutes. The reaction mixture was filtered through Celite, then concentrated under reduced pressure, to give 3.11g of the title compound (quantitative).
1 H NMR (400 MHz, CD 3 OD), δ: 1.44 (s, 9H), 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 ( br dd, J = 15.0, 7.0 Hz, 1H), 3.03 (d, J = 12.0 Hz, 1H), 3.12 (br d, J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS m / z 345 [M + H] +.
Step 3
(2S, 5R) -N- (2- aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide The above step 2 compound (3. 09g, in methylene chloride (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide – was added pyridine complex (3.58g), and stirred at room temperature overnight. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, and washed the aqueous layer with chloroform, and tetrabutylammonium hydrogen sulfate (3.47g) and chloroform (30mL) was added to the aqueous layer and stirred for 10 minutes. After extracting the aqueous layer with chloroform, drying the resulting organic layer over anhydrous sodium sulfate, filtered, concentrated under reduced pressure tetrabutylammonium tert- butyl {2 – [({[(2S, 5R) -7- oxo – 6- (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate was obtained 5.46g (91% yield).
1 H NMR (400 MHz, CDCl 3), δ: 1.01 (t, J = 7.4 Hz, 12H), 1.37-1.54 (m, 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30-2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85- 3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s, 1H), 9.44 (br s, 1H); MS m / z 425 [M-Bu 4 N + 2H] +.
The tetrabutyl ammonium salt (5.20g, 7.82mmol) in methylene chloride (25mL) solution of under ice-cooling trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed resulting residue with diethyl ether, at aqueous sodium bicarbonate was adjusted to pH7, it performs an octadecyl silica gel column chromatography (water), after freeze-drying, 1.44g of the title compound The obtained (57% yield).
[Α] D 24 -63.5 ° (c 0.83, H 2 O); 1 H NMR (400 MHz, D 2 O), δ: 1.66-1.76 (m, 1H), 1.76-1.88 (m, 1H), 1.91 -2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz, 2H), 3.18 (br d, J = 12.0 Hz , 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4, 3.2 Hz, 1H); MS m / z 325 [ M + H] +.
PATENT

ANTIBACTERIAL COMPOSITIONS OF A BETA-LACTAMASE INHIBITOR WITH A CEPHALOSPORINAbstract:
PATENT
WO 2016120752, WOCKHARDT, NEW PATENT, Nacubactam
![]()
Formula (I), chemically known as (25, 5i?)-N-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide has antibacterial properties and is disclosed in PCT International Patent Application No. PCT/IB2013/053092, PCT/JP2013/064971 and PCT/IB2012/002675. The present invention discloses a process for preparation of a compound of Formula (I).
Formula (I)

(VII) (VIII) (IX)
Scheme 2
Example 1
Synthesis of fert-butyl-r2-(aminooxy) ethyllcarbamate (III)
Preparation of fert-butyl-2-hydroxy ethylcarbamate (VIII):
Formula (VIII)

To a stirred solution of ethanolamine (50.0 g, 0.8186 mol) in dichloromethane (1000 ml), was added triethylamine (124 g, 1.228 mol) at 0°C. After 10 minutes, di-teri-butyl dicarbonate (VII, 214.15 g, 0.9823 mol) was added drop wise at 0°C under continuous stirring. Then reaction mass was allowed to warm to 25°C and stirred further for 3 hours. After completion of reaction, the resulting reaction mixture was poured into water (250 ml) and the organic layer was separated and dried over anhydrous sodium sulfate. The dried organic layer was concentrated under reduced pressure to obtain 130 g of the titled product as colorless oil in 98% yield.
Analysis:
Mass: 162 (M+l); for Molecular Weight of 161.2 and Molecular Formula of C7H15NO3.
1H NMR (400MHz, CDC13): δ 4.92(br s,lH), 3.72-3.68(q,2H), 3.30-3.26(q,2H), 2.33(br s,lH), 1.44(s,9H).
Preparation of A7-Boc-2-(2-aminoethoxy)isoindoline-l,3-dione (IX):

To a stirred solution of teri;butyl-2-hydroxy-ethylcarbamate (VIII, 50 g, 0.3106 mol) in tetrahydrofuran (500 ml), was added triphenylphosphine (89.5 g, 0.3416 mol) at 25°C. After stirring for 10 minutes, a solution of N-hydroxyphthalimide (50.66 g, 0.3106 mol) in dichloromethane (250 ml) was added to the reaction mass at 25 °C over a period of 10 minutes. After stirring for further 10 minutes, diisopropyl azodicarboxylate (69.1 g, 0.3416 mol) was added to the reaction mass in small portions (exothermic reaction was observed up to 34°C). The resulting reaction mass was stirred further at 25°C. After 16 hours, the reaction mass was concentrated under reduced pressure to obtain colorless oily material. The oily residue was diluted with diisopropyl ether (200 ml) and stirred for 30 minutes. The separated solid was filtered under suction. The filtrate was evaporated under reduced pressure and the residue subjected to di-isopropyl ether treatment (200 ml). This procedure was repeated once again. The filtrate was concentrated to obtain a solid product. The obtained solid was washed with diisopropyl ether (50 ml) and dried under reduced pressure. This solid contains small amount of triphenylphosphine oxide, along with the product. This was used as such for the next reaction without further purification.
Analysis:
Mass: 307.2 (M+l); for Molecular Weight of 306.3 and Molecular Formula of Ci5Hi8N205; 1H NMR of purified material (400MHz, CDC13): 7.85-7.25 (m,4H), 5.62(br s,lH), 4.26-4.23(t,2H), 3.46-3.42(q,2H), 1.46(s,9H).
Step 3: Preparation of fert-butyl-[ -(aminooxy) ethyl]carbamate (III):
Formula (III)

To a stirred solution of N-Boc-2-(2-aminoethoxy)isoindoline-l ,3-dione (IX, 97 g, 0.3167 mol) in dichloromethane (970 ml) was added hydrazine hydrate (31.7 g, 0.6334 mol) , at 0°C, drop wise, over a period of 45 minutes and the stirring continued further. After 2 hours, the reaction mass was filtered under suction. Filtrate was washed with water (485 ml), and the organic layer was diluted with an aq. solution of 10% potassium hydrogen sulfate (485 ml) and stirred for 15 minutes. The aqueous layer was separated, neutralized with solid sodium hydrogen carbonate and extracted with dichloromethane (2 x 485 ml). The organic layer was separated, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain colorless oil, this was used as such for further reaction immediately (28g, overall yield of step II and step III was 60%)
Analysis:
Mass: 177.2 (M+l) for Molecular Weight of 176.2 and Molecular Formula of C7H16N2O3.
Example 2
Synthesis of (25,5R)-jV-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l,6-diaza-bicvclor3.2.11octane-2- carboxamide (I)
Step 1: Preparation of (25,5R)-iV-(2-Boc-aminoethoxy)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (IV):

To a clear solution of sodium (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II, 42.67 g, 0.143 mol; prepared according to the procedure disclosed in Indian Patent Application No. 699/MUM/2013) in water (426 ml) was added EDC.HC1 (67.1 g, 0.349 mol) at 15°C
under stirring. After 10 minutes, a solution of teri-butyl-[2-(aminooxy) ethyl]carbamate (III, 28.0g, 0.159 mol; prepared as per the literature procedure depicted in Scheme 2) in dimethylformamide (56 ml) was added drop wise at 10°C under continuous stirring. The temperature of the reaction mass was allowed to warm to 25°C and then HOBt (21.5g, 0.159 mol) was added in small portions over a period of 15 minutes and the resulting mixture was further stirred at room temperature for 16 hours. The reaction was continuously monitored using thin layer chromatography using mixture of acetone and hexane (35 :65) as solvent system. After completion of reaction, the resulting mixture was filtered and the residue was washed with water (130 ml). The obtained white residue was suspended in water (130 ml) and the mixture stirred at 50°C for 3 hours. The resulting suspension was filtered, the residue dried under reduced pressure to obtain 51 g of (2S,5R)-N-(2-Boc-aminoethoxy)-6-(benzyloxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (IV) as off white solid in 73% yield.
Analysis:
Mass: 433.4 (M-l ); for Molecular Weight of 434.5 and Molecular Formula of C21H30N4O6;
1H-NMR (400MHz, CDC13): δ 9.32 (br s, 1H), 7.41 -7.26(m,5H), 5.41(br s, 1H), 5.06-4.88(dd, 2H), 3.98-3.96(d,lH), 3.91-3.90(m,2H), 3.39(m, 1H), 3.31-3.26(m, 2H), 3.04-3.01(d,lH), 2.77-2.74(d, 1H), 2.33-2.28(m, 1H), 2.03-1.93(m, 2H), 1.67-1.64(m, 1H), 1.44(s, 9H);
Purity as determined by HPLC: 99.4%.
Step 2: Preparation of (2S,5R)-iV-(2-Boc-aminoethoxy)-6-(hydroxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (V):

A solution of (25,5i?)-N-(2-Boc-aminoethoxy)-6-(benzyloxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1] octane-2-carboxamide (IV, 38 g, 0.0875 mol) in a mixture of dimethylformamide and dichloromethane (2: 8, 76 ml: 304 ml), containing 10% Pd/C (7.6 g, 50% wet) was hydrogenated at 50 psi hydrogen atmosphere at 25°C for 3 hours. The resulting mixture was filtered through a celite pad. The residue was washed with dichloromethane (75 ml). The solvent from the combined filtrate was evaporated
under reduced pressure to obtain 30 g (25,5i?)-N-(2-Boc-aminoethoxy)-6-(hydroxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide (V) as an oil, which was used as such for the next reaction without further purification.
Analysis:
Mass: 343.3 (M-l ) for Molecular Weight of 344.3 and Molecular Formula of C14H24N4O6.
Step 3: Preparation of (25,5R)-iV-(2-Boc-aminoethoxy)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide,tetrabutyl ammonium salt (VI):

To a stirred solution of (25,5i?)-N-(2-Boc-aminoethoxy)-6-(hydroxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide (V, 30.0 g, 0.0875 mol) in dimethylformamide (150 ml) was added sulphur trioxide dimethylformamide complex (16.06 g, 0.105 mol) in one portion, at 10°C. The reaction mass was stirred at the same temperature for 30 minutes and then allowed to warm to room temperature. After 2 hours, a solution of tetrabutylammonium acetate (31.6 g, 0.105 mol) in water (95 ml) was slowly added to the reaction mixture and stirred for another 2 hours. The solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2 x 60 ml) to obtain thick mass. This mass was partitioned between 1 : 1 mixture of dichloromethane (300 ml) and water (300 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (150 ml). The combined organic extracts were washed with water (3 x 150 ml) and dried over anhydrous sodium sulphate. The solvent was evaporated under reduced pressure and the resulting oily mass was triturated with ether (3 x 60 ml). Each time the ether layer was decanted and the residue was finally concentrated under reduced pressure to obtain the sticky mass. The so obtained material was purified by column chromatography over silica gel using mixture of methanol and dichloromethane as elution solvent. The solvent from the combined fractions was evaporated to obtain 47.5 g of (25,5i?)-N-(2-Boc-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide,tetrabutyl ammonium salt as white foam in 70% yield.
Analysis:
Mass: 423.4 (M-l) as free sulphonic acid; for Molecular Weight of 665.9 and Molecular Formula of C30H59N5O9 S;
1H- NMR (400MHz, CDC13): δ 9.52(br s, 1H), 5.53(br s, 1H), 4.33(s, 1H), 3.95-3.92(m,3H), 3.37-3.27(m, 1 1H), 2.87-2.84(d, 1H), 2.35-2.30(m, 1H), 2.17(m, 1H), 1.96-1.88(m, 2H), 1.74-1.60(m,8 H), 1.47-1.40(m, 17H), 1.02-0.98(m, 12H).
Step 4: Preparation of (2S R)-iV-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (I):
Formula (I)

To a stirred solution of (2S,5i?)-N-(2-Boc-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide, tetrabutyl ammonium salt (VI, 17 g, 0.0225 mol) in dichloromethane (85 ml) was added trifluoroacetic acid (85 ml) drop wise at -10°C over a period of 45 minutes. The resulting mass was further stirred at same temperature for 1 hour. The resulting reaction mixture was poured into cyclohexane (850 ml), stirred well for 30 minutes and the separated oily layer was collected. This procedure was repeated one more time and finally the separated oily layer was added to tert-butyl methyl ether (170 ml) under vigorous stirring at 25°C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with tert-butyl methyl ether (2 x 170 ml). The solid thus obtained was stirred with fresh dichloromethane (170 ml) for 30 minutes and filtered. The residual solid was dried at 45°C under reduced pressure to yield 7.3g of the titled compound in crude form. The obtained solid was further dissolved in water, (7.3 ml) and to this solution was added basic resin (Amberlyst A-26 -OH ion exchange resin, 4.4 g) under stirring. After 0.5 hour, the resin was filtered and to the filtrate isopropanol (51 ml) was added slowly at 25°C. The solution was further stirred for 12 hours. The separated solid was filtered and washed with additional isopropanol (7.5 ml) and dried under reduced pressure to obtain 4.3 g of (2S ,5R)-N-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide as off-white solid in 52 % yield.
Analysis:
Mass: 323.1 (M-l); for Molecular Weight of 324.31 and Molecular Formula of C9H16N4O7S; 1H-NMR (400MHz, D20): δ 4.07-4.06(d, 1H), 4.05-4.03(t, 2H), 3.96-3.94(d, 1H), 3.20(br s, 1H), 3.16-3.13(t, 2H), 3.02-2.99(d, 1H), 2.04-1.68(m, 4H);
Purity as determined by HPLC: 94.88%.

REF
| WO2015110969A3 * | Jan 21, 2015 | Nov 26, 2015 | Wockhardt Limited | Nitrogen containing compounds and their use as antibacterial agents |
| WO2015150941A1 * | Mar 12, 2015 | Oct 8, 2015 | Wockhardt Limited | A process for preparation of sodium (2s, 5r)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate |
| WO2016088863A1 * | Dec 4, 2015 | Jun 9, 2016 | Meiji Seikaファルマ株式会社 | Method for producing crystals of diazabicyclooctane derivative and stable lyophilized preparation |
| EP2931723A4 * | Dec 11, 2012 | Jun 1, 2016 | Fedora Pharmaceuticals Inc | New bicyclic compounds and their use as antibacterial agents and -lactamase inhibitors |
| US8933232 | Mar 29, 2013 | Jan 13, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole beta-lactamase inhibitors |
| US8933233 | Mar 29, 2013 | Jan 13, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole β-lactamase inhibitors |
| US8940897 | Mar 29, 2013 | Jan 27, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole β-lactamase inhibitors |
| US8962843 | Mar 29, 2013 | Feb 24, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole beta-lactamase inhibitors |
| US8962844 | Mar 29, 2013 | Feb 24, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole β-lactamase inhibitors |
| US9120795 | Mar 14, 2014 | Sep 1, 2015 | Cubist Pharmaceuticals, Inc. | Crystalline form of a β-lactamase inhibitor |
| US9120796 | Oct 2, 2014 | Sep 1, 2015 | Cubist Pharmaceuticals, Inc. | B-lactamase inhibitor picoline salt |
| US9309245 | Apr 2, 2013 | Apr 12, 2016 | Entasis Therapeutics Limited | Beta-lactamase inhibitor compounds |
| US9393239 | Apr 15, 2014 | Jul 19, 2016 | Fedora Pharmaceuticals Inc. | Bicyclic compounds and their use as antibacterial agents and betalactamase inhibitors |
/////////////IN2015MU287, WO-2016120752, nacubactam, WOCKHARDT, NEW PATENT, WK ?, WK-?, WK?, CAS 1452458-86-4, C9 H16 N4 O7 S, 324.31, Beta lactamase inhibitor, Roche, Meiji Seika Pharma, Fedora Pharmaceuticals, nacubactam hydrate , PHASE 1, A diazabicyclooctane beta-lactamase inhibitor, bacterial infection, July 2016, phase 1 clinical development, RG-6080, 1452458-86-4, FPI-1459, OP-0595, Phase I , β-lactamase inhibitor, bacterial infections, Fedora parmaceuticals, Meiji Seika Pharma
NCCONC(=O)[C@@H]2CC[C@@H]1C[N@]2C(=O)N1OS(=O)(=O)O
WO 2016113415, Sandoz, Riociguat, New Patent

WO 2016113415, Sandoz, Riociguat, New Patent
STEFINOVIC, Marijan; (AT).
RICHTER, Frank; (AT).
GRIESSER, Ulrich; (AT).
LANGES, Christoph; (AT)
SANDOZ AG [CH/CH]; Lichtstrasse 35 4056 Basel (CH)
Novel method for purifying riociguat, useful for treating chronic thromboembolic pulmonary hypertension, pulmonary arterial hypertension, systemic sclerosis and Raynaud’s phenomenon. Also claims novel crystalline solvates of riociguat (eg ethyl acetate or butan-2-one solvate), useful as intermediates in the purification of riociguat. Bayer and licensee Merck have developed and launched riociguat.
The present filing appears to be the first filing from Sandoz on riociguat; however see WO2015095515, assigned to Novartis, parent company of Sandoz, claiming an ophthalmic composition comprising a soluble guanylate cyclase activator (eg riociguat).
Riociguat (BAY 63-2521 ), having the chemical name N-[4,6-Diamino-2-[1-(2-fluorobenzyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]pyrimidin-5-yl]-N-methylcarbamic acid methyl ester, or sometimes also called or also sometimes called Methyl-(4,6-diamino-2-(1-(2-fluorobenzyl)-1 H-pyrazolo[3, 4-b]pyridin-3-yl)-5-pyrimidinyl)(methyl)carbamate is a stimulator of the soluble guanylate cyclase.
Riociguat has been approved for the treatment of inoperable, or persistent, recurrent chronic thromboembolic pulmonary hypertension (CTEPH) after surgery in adult patients and for the treatment of pulmonary arterial hypertension and is in development for the treatment of systemic sclerosis and Raynaud’s phenomenon.

(I)
The preparation of the compound of formula (I) and its purification are known. According to the experimental procedure of Example 8 of WO 03/095451 (comparable description in Chem. Med. Chem 2009, 4, 853-865), iodomethane is used as an alkylating agent in a late step and the purification of the crude riociguat either comprised preparatory HPLC steps or several steps of extracting, precipitating, suspending, washing, redissolving and reprecipitating riociguat, resulting in a long and tedious workup procedure with moderate yield.
In WO 201 1/064171 a potential genotoxic azo compound of formula III is used as a key intermediate, which under catalytic hydrogenation forms a compound of formula VIII.

The compound of formula VIII is further reacted with a methyl chloroformate or with a dimethyl carbonate derivative to form a compound of formula VI. The compound of formula VI is then methylated to form crude riociguat of formula (I).

Crude riociguat of formula (I) is then purified by a process comprising the intermediate isolation of a riociguat DMSO solvate of formula (II).
For the pharmaceutical use of riociguat, the solvent DMSO has to be removed. To that end, the compound of formula (II) is boiled in pharmaceutically acceptable solvents such as ketones, esters, ethers or alcohols. However, the riociguat obtained in this manner contains detectable amounts of DMSO.
These processes for the preparation of riociguat and their laborious purification protocols have a number of disadvantages which are unfavorable for industrial realization on a large scale.
On the one hand, the purification process according to WO 03/095451 require the repeated isolation of solid intermediates or preparatory HPLC, which ultimately results in a reduced yield of pure riociguat of formula (I) of pharmaceutical grade. Yet, traces of compound of formula (III) remain.
It is therefore one of the objects of the present invention to provide a process for the preparation of pure riociguat – compound of the formula (I) – which yields riociguat free from any genotoxic impurity and/or mutagenic impurity.
On the other hand, the process for the preparation of riociguat described in WO 201 1/064171 has a different serious drawback. It comprises the use of a DMSO solvate.
DMSO is an active pharmaceutical ingredient by itself. It is used as an active pharmaceutical ingredient in the treatment of interstitial cystitis. DMSO removal is difficult to achieve by the published processes. It is thus a further object of the invention to provide riociguat essentially free from DMSO and suitable for pharmaceutical use.
WO 2014/128109 discloses forms of riociguat, such as polymorphs and solvates, and describes a ¼ ethyl acetate solvate of riociguat in example 6. The X-ray powder
diffractogram in Tab.3 and figure 4 comprises reflexes at °2Theta positions of 9.1 and 25.6.
Thus, there is a need in the art for a process, which allows the preparation of pure riociguat free from any genotoxic impurity and/or mutagenic impurity which at the same time does not comprise residual DMSO.
Surprisingly, we have now identified a process for the purification of crude riociguat which yields riociguat which is essentially free from genotoxic impurities and DMSO. In particular, this novel process differs from the processes known to date in that the isolation of intermediates prior to the formation of riociguat is not required. This process allows to overcome the disadvantages of the processes known to date and to obtain riociguat in high yield and high purity and pharmaceutical acceptable quality essentially free of genotoxic impurities.
Examples
Preparative example
Preparation of crude riociguat
Riociguat was prepared as disclosed in example 7 of WO 201 1/064171 and had a chemical purity of 91.7% by the area of the riociguat peak in the HPLC-UV elution profile.
Comparative Example 1
Preparation of DMSO solvate
An amount of 4.505 g (0.0107 moles) of crude riociguat was dissolved in 8 ml DMSO at 100 °C. The obtained brownish, turbid solution was then cooled to 75 °C within 16 minutes. After that 55 ml of ethylacetate were added and the mixture was cooled to 25 °C (30 minutes). After 22 h the obtained precipiate was filtered off, washed with 14 ml EtOAc and dried for 4 hours at 50 °C at reduced pressure (50 mbar). The precipitate was analysed with XRPD, confirming that riociguat DMSO was obtained. The product was also analyzed by HPLC-UV-MS. Purity was calculated based on UV detection at 254nm. The so obtained riociguat DMSO solvate was 91 .92% pure.
Comparative Example 2
Preparation of riociguat form I from riociguat DMSO solvate
The entire product prepared in comparative example 1 (4.283 g = 0.009 moles) was reflux heated in 77 ml of ethylacetate at 78 °C for 1 h and then cooled to 25 °C. The white solid was filtered off with suction, washed with a total of 18 ml of ethyl acetate and dried at 50 °C under reduced pressure (50 mbar) for 5 hours. The dried product was then analyzed by XRPD, confirming identity of riociguat form I unequivocally.
Yield (dry): 3.224 g (0.0076 moles) = 75% for comparative example 2 and 72% overall (C.ex. 1 and 2). Total organic volatile impurity is higher than 1000 ppm and total DMSO content is higher than 100 ppm.
Example 1 ; Preparation of Riociguat ethylacetate solvate
Crude Riociguat (500 mg; Form I; 91 .7% percentage area purity) was dissolved in 2 ml DMF and heated to 100 °C to obtain a slightly turbid solution. After filtration through a 0.44 micron filter, 20 ml EtOAc were added to the hot solution (water bath 70°C) and allowed to stand. The temperature was slowly decreased to ambient temperature. Crystallization started after
10min. The yellowish, fine powder was filtered off and dried at ambient conditions. The PXRD indicated the formation of a new ethylacetate solvate. Yield 71 %, 97.8% purity.
Example 2; Preparation of the Methyl ethyl ketone (butan-2-one) solvate of Riociguat.
Crude Riociguat (500 mg; Form I; 91 .7% percentage area purity) was dissolved in 2 ml DMF at 100 °C to obtain a clear solution. After filtration through a 0.44 micron filter, 20 ml MEK were added. The hot solution (water bath 70 °C) was allowed stand. The temperature was then slowly decreased to ambient temperature. After 30 minutes yellowish, square-shaped crystals appeared, which were analyzed. Analysis confirmed that they were a new crystalline MEK-solvate. Yield 43%, 97.2% purity.
Example 3 ; Conversion of Solvated forms to Form I
Both the solvates from examples 1 and 2 can be converted to riociguat Form I by heating the material to 150°C under vacuum for an appropriate amount of time.
Example 4; Direct preparation of riociguat form I from crude riociguat using DMF-Acetone Crude Riociguat (200 mg; Form I; 91 .7% percentage area purity) was dissolved in 1.0 ml DMF at 100 °C to obtain a clear solution. After filtration through a 0.44 micron filter, 5 ml acetone was added. The hot solution (water bath 70 °C) was allowed to stand. Crystallisation occurred while the temperature was slowly decreased to ambient temperature. After 24 hours the precipitate was filtered off and dried at ambient conditions to obtain form I. Yield 78% ; 97.6% purity
///////////WO 2016113415, Sandoz, Riociguat, New Patent
Carbotegravir, Dolutegravir, New Patent, WO 2016113372, Lek Pharmaceutical and Chemical Co DD
![]()
Carbotegravir, New Patent, WO 2016113372, Lek Pharmaceutical and Chemical Co DD
LEK PHARMACEUTICALS D.D. [SI/SI]; Verovskova 57 1526 Ljubljana (SI)
MARAS, Nenad; (SI).
SELIC, Lovro; (SI).
CUSAK, Anja; (SI)
ViiV Healthcare is developing cabotegravir (first disclosed in WO2006088173), which in July 2016, was reported to be in phase 2 clinical development.
WO-2016113372
Process for preparing integrase inhibitors such as dolutegravir and cabotegravir and their analogs, useful for treating viral infections eg HIV infection. Also claims a process for preparing intermediates of dolutegravir and cabotegravir.
(4R, 12aS)-N-[(2,4-Difluorophenyl)methyl]-3 ,4,6,8, 12, 12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1-b][1 ,3]oxazine-9-carboxamide (Formula A):

Formula A
known by the INN name dolutegravir, is a new efficient antiviral agent from the group of HIV integrase inhibitors which is used in combination with some other antiviral agents for treatment of HIV infections, such as AIDS. The compound, which belongs to condensed polycyclic pyridines and was first disclosed in WO2006/1 16764, is marketed.
Another compound disclosed in WO2006/1 16764 is (3S, 1 1 aR)-N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 1 1 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide (Formula

Formula C
known by the INN name cabotegravir.
The complex structures of dolutegravir and cabotegravir present a synthetic challenge. The first description of the synthesis in WO2006/1 16764 shows a 16-steps synthesis (see Scheme A), which is industrially impractical due to its length and low overall yield.

Scheme A
WO 2010/068253 and WO 2006/1 16764 describe an alternative synthesis. The 1 1 -step synthesis, shown in Scheme B1 and Scheme B2, is based on bromination of the 9-position for further introduction of the carboxylic group. The synthesis relies on the use of expensive palladium catalysts and toxic selenium compounds. Furthermore, some variations of these approaches involve pyrone intermediates in several steps. In some cases pyrones are liquids which can complicate purification, while further reactions form complex mixtures.
![]()

 doiutegravir
doiutegravir
Scheme B2
In further alternative syntheses, acetoacetates were used as starting materials. Such an approach is challenging in terms of introducing the hydroxy group in the 7-position. The variation in Scheme C1 , described in WO2012/018065, starts from 4-benzyloxyacetoacetate. The procedure requires 9 steps, but use expensive reagents like palladium catalysts. Moreover, there is described a possibility of formation a co-crystal between an intermediate and hydroquinone, wherein however the additional step may diminish yields and make the process longer and time consuming.

Scheme C1
The variation in Scheme C2, described in WO2012/018065, starts from 4-chloroacetoacetate. The process is not optimal because of problems in steps which include pyrones and because of problems with conversion of 7-chloro to 7-hydroxy group which includes a disadvantageous use of silanolates with low yield (25%).

Scheme C2
The variation in Scheme C3, described in WO201 1/1 19566, starts from unsubstituted acetoacetate. For the introduction of the 7-hydroxy group, bromination is used and substitution of bromo with hydroxy is performed by a use of silanolates. The substitution of the bromine is achieved in a 43% yield.

Scheme C3
The variation in Scheme C4, described in WO201 1/1 19566, starts from 4-methoxyacetoacetate aiming at preparing dolutegravir or cabotegravir. The process uses lithium bases to affect a difficult to control selective monohydrolysis of a diester.

The object of the present invention is to provide short, simple, cost-effective, environmentally friendly and industrially suitable processes for beneficially providing dolutegravir and analogues thereof and cabotegravir and analogues thereof, in particular dolutegravir.
Scheme 1

According to an embodiment of the process of the invention the building block 3-aminobutanol can suitably be substituted with other aminoalcohols to give dolutegravir analogues. For example, using (S)-alaninol gives cabotegravir as the final product. Similarly, using amines other than 2,4-difluorobenzylamine in the amidation step results in the synthesis of other dolutegravir analogues.
According to the another preferred embodiment cabotegravir or a pharmaceutically acceptable salt thereof is prepared by the analogue process, which comprises providing a compound of formula (5c)

5c
converting the compound of formula (5c) to a compound of formula (6c)

6c
by carrying out a chlorination reaction, and converting the compound of formula (6c) to cabotegravir and/or a pharmaceutically acceptable salt thereof.
The compound of formula (5c) can preferably be provided by converting a compound of formula (3) to a compound of formula (4c)

Scheme 2

1. ) EtOCOCI, Et3N / Me2CO
2. ) 2,4-difiuorobenzylamine

Scheme 3

Analogous compound of formula 7c is a useful intermediate in the synthesis of cabotegravir. Scheme 3a

Scheme 4

Examples
The following examples are merely illustrative of the present invention and they should not be considered as limiting the scope of the invention in any way. The examples and modifications or other equivalents thereof will become apparent to those versed in the art in the light of the present entire disclosure. Particularly, all Examples related to the preparation of dolutegravir and intermediates thereof can be used by the analogy for the preparation of cabotegravir and intermediates thereof.
Example 1 :

Methyl acetoacetate (1 , 25.22 g) and dimethylformamide dimethyl acetal (DMFDMA, 35 mL) was heated at 50-55°C for 2 h, then methanol (60 mL), aminoacetaldehyde dimethyl acetal (24 mL) and acetic acid (4 mL) was added an the mixture was heated under reflux for one hour, then concentrated. MTBE (100 mL) was added and the mixture was kept at 5 °C overnight to crystallize. Upon filtration 46 g (92%) of product 2 was recovered.
1H NMR (DMSO-d6): δ 2.31 (s, 3H), 3.30 (s, 6H), 3.49 (m, 2H), 3.61 (s, 3H), 4.43 (m, 1 H), 8.02 (d, 1 H), 10.8 (bs, 1 H). 13C NMR (DMSO-d6): δ 30.52, 35.48, 50.53, 54.23, 98.99, 102.47, 160.70, 166.92, 197.21 .
Example 2:

Compound 2 (5.00 g) was dissolved in 2-propanol, dimethyl oxalate (7.02 g) was added and heated to 40 °C. Sodium methylate (25% in methanol; 20 mL) was slowly (10 min) added, the mixture was then heated to 50-55 °C and stirred at that temperature for 2-2.5 h. The mixture was cooled to ambient temperature, then sodium hydroxide solution (1 M, 65 mL) was added to the mixture and stirred for another 2 h, followed by addition of concentrated hydrochloric acid (1 1 mL) and stirred for another 2 h. The precipitate was filtered and dried to give 8.08 g (NMR assay 47%; 65% yield) of compound 3.
1H NMR (DMSO-d6): δ 2.50 (m, 2H), 3.30 (s. 6H), 4.49 (m, 1 H), 7.06 (s, 1 H); 8.70 (s, 1 H). 13C NMR (DMSO-d6): δ 55.23, 55.37, 102.34, 1 15.47, 120.24, 145.17, 162.71 , 165.22, 178.55.
Example 3:

Compound 2 (158.37 g) was dissolved in methanol (548 mL), followed by the addition of dimethyl oxalate (202.2 g). While keeping the temperature below 30°C, potassium ferf-butoxide (192.1 g) was added and reaction mixture was heated at 50 °C overnight. The suspension was then filtered and the filter cake washed with methanol. The filtrate was concentrated (approximately to 680 mL), then water (680 mL) was added, followed by addition of lithium hydroxide hydrate (143.7 g) while keeping the temperature below 40 °C. The suspension was then stirred at ambient temperature overnight and filtered. To the obtained filtrate, concentrated hydrochloric acid (339 mL) was added while keeping the temperature below 30 °C. The suspension was aged for 2 h and filtered to give 4 as a white powder (95.6 g, NMR assay 100%; 52% yield).
Example 4:

Compound 2 (5.00 g) was dissolved in 2-propanol, dimethyl oxalate (7.02 g) was added and heated to 40 °C. Sodium methylate (25% in methanol; 15 mL) was slowly (10 min) added then the mixture was heated to 50-55 °C and stirred at that temperature for 72 h. The mixture was concentrated and components were separated by flash column chromatography (ethyl acetate/methanol 9:1 to 6:4). Early fractions gave compound 22 upon concentration, late fractions gave compound 23.
Compound 22: 1H NMR (DMSO-d6): δ 2.49 (m, 2H), 3.28 (s, 6H), 3.73 (s, 3H), 3.85 (s, 3H), 4.41 (m, 1 H), 4.50 (m, 1 H), 6.65 (s, 1 H), 8.36 (s, 1 H). 13C NMR (DMSO-d6): δ 51.63, 53.36, 54.25, 55.47, 102.71 , 1 18.24, 123.60, 140.81 , 150.21 , 162.44, 164.49, 173.43.
Compound 23: 1H NMR (DMSO-d6): δ 2.49 (m, 2H), 3.26 (s, 6H); 3.70 (s, 3H); 4.33 (d, 1 H); 4.60 (m, 1 H), 6.19 (s, 1 H), 8.12 (s, 1 H). 13C NMR (DMSO-d6): δ 50.03, 51.34, 54.59, 54.85, 102.91 , 1 16.04, 1 18.19, 148.32, 152.12, 163.46, 165.24, 174.99
Example 5:

Compound 3 (5.5 g; assay 53%) was suspended in acetonitrile, acetic acid (6 mL) and methanesulfonic acid (2.5 mL) were added followed by the heating of mixture to 70 °C for 4 h. The suspension was filtered and filtrate cooled to ambient temperature. Triethylamine (6.6 mL) and (R)-3-amino-butan-1 -ol (1.24 mL) was added followed by heating the mixture at reflux temperature for 20-24 h. The mixture was filtered, filtrate concentrated and 1 M HCI (100 mL) was added, followed by extraction with dichloromethane (3 x 50 mL). Combined organic fractions were concentrated, 2-propanol was added (10 mL) and suspension was stirred at 70-80 °C for 10 min, left to cool to ambient temperature then filtered to give 2.19 g of compound 4 (73%).
1H NMR (DMSO-de): δ 1.31 (d, 3H), 1.52 (m, 1 H), 1 .97 (m, 1 H), 3.89 (m, 1 H), 4.01 (m, 1 H), 4.46 (m, 1 H), 4.64 (m, 1 H), 4.78 (m, 1 H), 5.50 (m, 1 H), 7.29 (s, 1 H), 8.88 (s, 1 H), 15.83 (s, 1 H). 13C NMR (DMSO-d6): δ 15.22, 29.14, 45.26, 51.13, 62.09, 76.03, 1 16.31 , 1 18.79, 140.53, 146.79, 155.36, 165.24, 178.75.
Example 6:

Compound 3 (14.55 g; assay 49%) was suspended in acetonitrile (125 mL), acetic acid (15 mL) and methanesulfonic acid (6.25 mL) were added followed by the heating of mixture to 70 °C for 4 h. The suspension was filtered and filtrate cooled to ambient temperature. Triethylamine (16.5 mL) and (S)-2-aminopropanol (2.45 mL) was added followed by heating the mixture at reflux temperature for 24 h. The insoluble product was filtered, washed with 2-propanol (20 mL) and dried to give (3S, 1 1 aR)-3-methyl-5,7-dioxo-2,3,5,7, 1 1 ,1 1 a-hexahydrooxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxylic acid (5.2 g, 75%).
1H NMR (DMSO-d6): δ 1.31 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.8 Hz, 1 H), 4.13 (dd, J = 1 1.7, 10.3 Hz, 1 H), 4.28 (m, 1 H), 4.39 (dd, J = 8.6, 6.8 Hz, 1 H), 4.92 (dd, J = 12.3, 4.2 Hz, 1 H), 5.45 (dd, J = 10.2, 4.1 Hz, 1 H), 7.16 (s, 1 H), 8.84 (s, 1 H), 15.74 (s, 1 H).
Example 7:

Compound 4 (0.63 g) was dissolved in dichloromethane (15 mL), cooled to 5°C, then triethylamine (0.31 mL) was added, followed by ethyl chloroformate (0.26 mL), followed by slow (30 min) addition of 2,4-difluorobenzylamine. The mixture was then stirred at ambient temperature for 24 h. Water (10 mL) was added, organic phase was separated and washed with 1 M HCI (15 mL) and water (15 mL), concentrated and treated with 2-propanol to give the product 5 in a quantitative yield.
1H NMR (CDCI3): δ 1.39 (d, 3H), 1.52 (s, 1 H), 2.19 (m, 1 H), 4.00 (m, 2H), 4.16 (m, 1 H), 4.31 (m, 1 H), 4.62 (d, 2H), 5.00 (m, 1 H), 5.27 (m, 1 H), 6.80 (m 2H), 7.33 (m, 2H), 8.49 (s, 1 H), 10.48 (s, 1 H). 13C NMR (CDCI3): 15.50, 29.22, 36.43, 45.19, 51.83, 62.79, 103.71 , 103.91 , 1 1 1 .0, 1 1 1 .18, 120.59, 123.04, 130.40, 137.41 , 144.58, 156.27, 163. 87, 177.83.
Example 8:

To a suspension of 4 (2.84 g, 10 mmol) in a mixture of triethylamine (2.24 mL, 16 mmol) and acetone (50 mL) stirring on an ice bath was added ethyl chloroformate (1 .20 mL, 12 mmol). After stirring for 10 min, 2,4-difluorobenzylamine (1.21 mL, 10 mmol) was added and the mixture left stirring at room temperature for 1 h. The product was isolated by slowly diluting the reaction mixture with water (50 mL), partial concentration, filtration, washing with water (2 50 mL) and drying. There was obtained 5 as a white powder (3.48 g, 86%): mp 181.0-184.7 °C. 1H NMR (DMSO-d6): δ 1.29 (d, J = 7.0 Hz, 3H), 1 .56 (dd, J = 13.9, 2.0 Hz, 1 H), 1 .93-2.06 (m, 1 H), 3.90 (ddd, J = 1 1.6, 5.0, 2.1 Hz, 1 H), 3.98 (td, J = 12.0, 2.2 Hz, 1 H), 4.45 (dd, J = 13.6, 6.6 Hz, 1 H), 4.72 (dd, J = 13.6, 3.8 Hz, 1 H), 4.74-4.81 (m, 1 H), 5.44 (dd, J = 6.6, 3.8 Hz, 1 H), 8.93 (s, 1 H), 15.14 (s, 1 H). 13C NMR (DMSO-d6): δ 15.78, 29.13, 44.89, 52.88, 61 .63, 75.61 , 1 13.54, 128.49, 136.42, 145.64, 154.62, 164.58, 174.58
Example 9:

To a suspension of 4 (1 1.36 g, 40 mmol) in acetonitrile (80 mL) stirring at room temperature was added TCCA (9.29 g, 38 mmol) and DABCO (0.23 g, 5 mol%). After stirring at room temperature for 1 h, the reaction was quenched with a mixture of DMSO (5.26 mL) and water (1.33 mL). The insoluble cyanuric acid was removed by filtration and the filtrate evaporated under reduced pressure to give viscous oil. This was triturated in methanol (20 mL) to induce crystallization. The product was filtered, washed with cold methanol (10 mL) and dried to give 7 as a yellowish powder (5.13 g, 41 %): mp 191 .3-198.7 °C.
Example 10:

Attempted chlorination of 23: Compound 23 (0.54g) was suspended in acetonitrile (10 mL) and trichlorocyanuric acid (0.44 g) was added and the solution was stirred at ambient temperature overnight. Precipitate was filtered. Only traces of a product corresponding to the compound 26 could be detected in the reaction mixture by LC-MS analysis. Conversion did not improve with time.
Example 11 :

Attempted chlorination of 3: Compound 3 (0.30 g) was suspended in acetonitrile (5 mL) and trichlorocyanuric acid (0.13 g) was added. The suspension was stirred at ambient temperature overnight. Only traces of a product corresponding to the compound 24 could be detected in the reaction mixture by LC-MS analysis.
Example 12:

9 10
Trichloroisocyanuric acid (0.23 g) was added in a single portion to a stirred solution of the diethyl 1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (9, 0.66 g) in dry acetonitrile (4 mL) at room temperature. The resulting suspension was stirred at room temperature for ca. 24 h. The reaction mixture was diluted with dichloromethane and filtrated. The filtrate was then concentrated in vacuo to afford crude oil (0.86 g). Purification by flash chromatography (eluting ethyl acetate/cyclohexane) furnished diethyl 3-chloro-1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate, 10 as a yellow semi-solid (0.38 g). 1H NMR (CDCI3): δ 1.28 (t, J=7A Hz, 3H), 1 .37 (t, J=7.2 Hz, 3H), 3.35 (s, 6H), 3.89 (d, J=5.0 Hz, 2H), 4.27 (q, J=l A Hz, 2H), 4.43 (q, J=l A Hz, 2H), 4.48 (t, J=4.9 Hz, 1 H), 8.15 (s, 1 H). 13C NMR (CDCI3): δ 13.83, 14.13, 55.82, 57.09, 61.41 , 63.72, 102.52, 1 17.35, 126.90, 140.22, 146.92, 160.67, 164.13, 168.95.
Example 13:

Diethyl 1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (9, 0.64 g) was dissolved in anhydrous acetonitrile (6 mL) and treated sequentially with acetic acid (560 μί) and methanesulfonic acid (40 μί). The resulting mixture was heated to 62 °C and stirred for 4 h and more methanesulfonic acid (40 μΙ_) was added. After additional 2 h, more methanesulfonic acid (80 μΙ_) was added. This was repeated after additional 2 h, when more methanesulfonic acid (80 μΙ_) was added. The reaction mixture was stirred additional 17 h at 62 °C then was treated with a mixture of (R)-3-aminobutanol (0.22 g), triethylamine (0.5 mL) and acetonitrile (0.7 mL). The reaction mixture was stirred additional 22 h at 62 °C and then concentrated in vacuo. The crude material was partitioned between dichloromethane and 1 M HCI solution (15 mL). The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford the crude (4R, 12aS)-ethyl 4-methyl-6,8-dioxo-3,4,6,8, 12,12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1 -b][1 ,3]oxazine-9-carboxylate (11 ) as a brownish oil (0.61 g).
1H NMR (CD3OD): δ 8.44 (s, 1 H), 7.16 (m, 1 H), 5.48 (t, J=4.8 Hz, 1 H), 4.86 (m, 1 H), 4.49 (dd, J=13.6, 4.0 Hz, 1 H), 4.30-4.25 (m, 3H), 4.09 (dt, J=12.1 , 2.3 Hz, 1 H), 3.96 (ddd, J=1 1.7, 5.0, 2.1 Hz, 1 H), 2.18-2.10 (m, 1 H), 1.60-1 .56 (m, 1 H) 1 .39 (d, J=7A Hz, 3H), 1.33 (t, J=7A Hz, 3H). 13C NMR (CDCI3): δ 8.45, 14.08, 15.39, 29.17, 45.04, 45.72, 51 .56, 60.86, 62.61 , 76.33, 1 19.54, 123.72, 136.96, 145.67, 156.26, 163.68, 175.43
Example 14:

10
Diethyl 3-chloro-1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (10, 1.23 g) was dissolved in 85% formic acid (25 mL) at room temperature. The mixture was warmed to 40 °C and stirred for 23 h. The reaction mixture was concentrated in vacuo, and then partitioned between dichloromethane and aqueous NaHC03 solution. The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford brownish oil (0.49 g). The crude oil was dissolved in anhydrous toluene (5 mL) and treated sequentially with (R)-3-aminobutanol (0.19 g), methanol (0.2 mL) and acetic acid (96 μί). The resulting mixture was heated to 90 °C and stirred for 20 h. The reaction mixture was cooled to room temperature and then partitioned between dichloromethane and aqueous NaHC03 solution. The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford the crude (4R,12aS)-Ethyl 7-chloro-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5] pyrazino [2, 1-b][1 ,3]oxazine-9-carboxylate (12) as a brownish oil (0.24 g).
Example 15:

To a solution of 4 (5.68 g, 20 mmol) in dichloromethane (50 mL) stirring in an ice bath was added triethylamine (5.6 mL, 40 mmol), followed by ethyl chloroformate (2.61 mL, 26 mmol). After 20 min, ethanol (50 mL) was added. The mixture was then left stirring 24 h at room temperature and concentrated under reduced pressure. The residue was triturated in acetone (80 mL). The insoluble salt (triethylamine hydrochloride) was removed by filtration. The filtrate was evaporated under reduced pressure to give 11 as an amorphous solid in a quantitative yield (6.1 g).
Example 16:

To a stirring solution of 11 (0.94 g, 3.0 mmol) in acetonitrile (8 mL) heated at 40 °C was added TCCA in portions during 1 h (0.44 g, 1 .8 mmol). After an additional 1 h, the reaction mixture was diluted with a solution of NaHS03 (0.60 g) in water (60 mL), extracted with dichloromethane (50 mL) and the extract evaporated under reduced pressure to give a crude product which was purified by flash chromatography (CH2CI2 : MeOH, from 98 : 2 to 80 : 20) to give 12 (0.45 g, 44%).
1H NMR (CDCI3): δ 1.37 (t, J = 7.1 Hz, 3H), 1.38 (d, J = 7.0 Hz, 3H), 1 .56 (dq, J = 13.9, 2.2 Hz, 1 H), 2.21 (m, 1 H), 3.99 (d, J = 2.3 Hz, 1 H), 4.00 (t, J = 1.8 Hz, 1 H), 4.10 (dd, J = 13.2, 6.6 Hz, 1 H), 4.37-4.27 (m, 3H), 4.98 (m, 1 H), 5.35 (dd, J = 6.6, 3.8 Hz, 1 H), 8.07 (s, 1 H).
13C NMR (CDCI3): δ 14.20, 16.09, 29.34, 44.87, 53.73, 61.49, 62.29, 76.01 , 1 16.22, 133.1 1 , 134.18, 144.52, 155.48, 163.88, 169.98.
Example 17:

To a mixture of 7 (3.89 g, 12.2 mmol) in methanol (12 mL) was added sodium methylate (22.3 mL, 97.6 mmol). The reaction mixture was stirred for 24 h at 30 °C and then quenched with a slow addition of 3M hydrochloric acid (35 mL) while stirring in an ice bath. The mixture was concentrated under reduced pressure to remove most of the methanol, then extracted with dichloromethane (2 30 mL), the combined extracts washed with water (30 mL) and evaporated under reduced pressure. Methanol (20 mL) was added to the obtained amorphous residue and removed under reduced pressure to yield the solid 8 (3.69 g, 98%).
1H NMR (CDCI3): δ 15.04 (s, 1 H), 8.42 (s, 1 H), 5.29 (dd, J=5.6, 3.9 Hz, 1 H), 5.01 -4.96 (m, 1 H), 4.42 (dd, J=13.6, 3.6 Hz, 1 H), 4.25 (dd, J=13.6, 6.0 Hz, 1 H), 4.05 (s, 3H), 4.00-3.97 (m, 2H), 2.21 -2-14 (m, 1 H), 1.53 (dd, J=14.1 , 1.9 Hz, 1 H), 1.36 (d, J=7 Hz, 3H). 13C NMR (CDCI3): δ 176.35, 165.94, 155.03, 153.70, 143.08, 130.90, 1 15.94, 76.05, 62.65, 61.45, 53.86, 44.96, 29.43, 16.06.
Example 18:

To a suspension of 7 (2.55 g, 8.0 mmol) in a mixture of triethylamine (1 .46 mL, 10.4 mmol) and acetone (32 mL) stirring on an ice bath was added ethyl chloroformate (0.88 mL, 8.8 mmol). After stirring for 10 min, 2,4-difluorobenzylamine (1.07 mL, 8.8 mmol) was added and the mixture left stirring at room temperature for 1 h. The product was isolated by slowly diluting the reaction mixture with water (40 mL), filtration, washing with water (2 30 mL) and drying. There was obtained 2.91 g of 6 as a white powder (83%).
1H NMR (CDCI3): δ 1.30 (d, J = 7.0 Hz, 3H), 1 .49 (dd, J = 14.0, 2.2 Hz, 1 H), 2.14 (ddd, J = 14.6, 1 1.1 , 6.4 Hz, 1 H), 3.89-3.95 (m, 2H), 4.09-4.15 (m, 1 H), 4.26 (dd, J = 13.4, 3.8 Hz, 1 H), 4.55 (d, J = 5.8 Hz, 2H), 4.89-4.98 (m, 1 H), 5.18 (dd, J = 6.2, 3.8 Hz, 1 H), 6.68-6.79 (m, 2H), 7.23-7.31 (m, 1 H), 8.41 (s, 1 H), 10.24 (t, J = 5.8 Hz, 1 H). 13C NMR (CDCI3): δ 16.09, 26.95, 29.30, 36.79, 45.1 1 , 45.28, 53.86, 62.47, 75.93, 103.87 (t, J = 25.4 Hz), 1 1 1 .21 (dd, J = 21 .0, 3.4 Hz), 1 17.32, 130.58 (dd, J = 9.3, 5.8 Hz), 133.40, 143.54, 155.34, 163.16, 163.25, 163.35, 172.88.
Example 19:

To a suspension of 5 (1 .67 g, 4 mmol) in acetonitrile (20 mL) was added DABCO (23 mg, 5 mol%) and TCCA (0.62 g, 2.52 mmol). The mixture was stirred 18 h at 40 °C protected from light and then quenched with a mixture of DMSO (0.48 mL) and water (0.12 mL). The insoluble cyanuric acid was removed by filtration and washed with acetonitrile (5 mL). The filtrate was evaporated under reduced pressure to give viscous oil that was crystallized from a mixture of methanol (6 mL) and water (3 mL), by slowly cooling the solution from 60 °C to room
temperature. The product 6 was filtered, washed with cold methanol (5 mL) and dried to give an off-white powder (1.07 g, 61 %).
1H NMR (CDCI3): δ 1.30 (d, J = 7.0 Hz, 3H), 1 .49 (dd, J = 14.0, 2.2 Hz, 1 H), 2.14 (ddd, J = 14.6, 1 1.1 , 6.4 Hz, 1 H), 3.89-3.95 (m, 2H), 4.09-4.15 (m, 1 H), 4.26 (dd, J = 13.4, 3.8 Hz, 1 H), 4.55 (d, J = 5.8 Hz, 2H), 4.89-4.98 (m, 1 H), 5.18 (dd, J = 6.2, 3.8 Hz, 1 H), 6.68-6.79 (m, 2H), 7.23-7.31 (m, 1 H), 8.41 (s, 1 H), 10.24 (t, J = 5.8 Hz, 1 H). 13C NMR (CDCI3): δ 16.09, 26.95, 29.30, 36.79, 45.1 1 , 45.28, 53.86, 62.47, 75.93, 103.87 (t, J = 25.4 Hz), 1 1 1 .21 (dd, J = 21.0, 3.4 Hz), 1 17.32, 130.58 (dd, J = 9.3, 5.8 Hz), 133.40, 143.54, 155.34, 163.16, 163.25, 163.35, 172.88.
Example 20:

To a suspension of 6 (0.44 g) in anhydrous methanol (1 mL) was added a 25% methanolic solution of sodium methylate (1 .14 mL) and the mixture stirred for 4 h at 40 °C. The reaction was quenched with acetic acid (0.4 mL), diluted with water (8 mL), extracted with 2-methyltetrahydrofuran (12 mL), the extract washed with 1 M NaOH(aq) (8 mL), water (8 mL) and evaporated under reduced pressure. To the oily residue was added methanol (8 mL) and evaporated under reduced pressure to give 27 as a white solid (0.38 g, 88%).
Example 21 :

The suspension of (4R, 12aS)-7-chloro-N-(2,4-difluorobenzyl)-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1 -b][1 ,3]oxazine-9-carboxamide (6, 0.44 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2 mL) was stirred at room temperature for 24 h. The reaction was quenched with 2M H2S04 (1 .18 mL) and left stirring for 2 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with water (2 x 2 mL). The obtained white precipitate (0.38 g) was suspended in THF-water (1 :1 , 4.5 mL) and stirred at room temperature for ca. 2 h. The reaction mixture was filtered through fitted funnel rinsing with water (2 χ 1 mL) and dried in vacuo at 40°C to afford pure dolutegravir as a white solid (0.33 g, HPLC purity: 99.38%).
1H NMR (DMSO-d6): δ 12.51 (s, 1 H), 10.36 (t, J=5.9 Hz, 1 H), 8.50 (s, 1 H), 7.41-7.36 (m, 1 H), 7.26-7.21 (m, 1 H), 7.07-7.03 (m, 1 H), 5.45 (dd, J=5.4, 4.3 Hz, 1 H), 4.81 -4.76 (m, 1 H), 4.59-4.53 (m, 3H), 4.36 (dd, J=13.8, 5.8 Hz, 1 H), 4.05-4.00 (m, 1 H), 3.91-3.88 (m, 1 H), 2.05-1 .97 (m, 1 H), 1.55-1.52 (m, 1 H), 1 .33 (d, J=7.1 Hz, 3H). 13C NMR (DMSO-d6): δ 170.27, 163.68, 162.29, 161 .78 (dd), 159.82 (dd), 154.61 , 140.64, 130.74 (d), 130.67 (d), 122.37 (d), 1 16.73, 1 15.38, 1 1 1 .33 (d), 103.80 (t), 62.01 , 51 .16, 44.69, 35.74, 29.13, 15.21.
Example 22:

A suspension of dolutegravir (0.31 g) in methanol (4 mL) was cooled to 0 °C.25% Solution of sodium methoxide in methanol was added to the mixture and the resulting suspension was stirred at 0 °C for 2 h, then at room temperature for 23 h. The reaction mixture was then filtered through fitted funnel rinsing with methanol (3 x 10 mL). The white precipitate was dried overnight at room temperature to afford pure dolutegravir sodium as a white solid (0.26 g, HPLC purity: 99.84%).
1H NMR (DMSO-d6): δ 10.70 (t, J=5.8, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J=6.4Hz, 1H), 4.51 (d, J=5.5Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J=14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J=12.9 Hz, 1H), 1.24 (d, J=7.0Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 23:

The suspension of 6 (0.44 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2 mL) was stirred at room temperature for 24 h. The reaction was diluted with absolute ethanol (10 mL) and left stirring for ca. 30 min at room temperature. The reaction mixture was filtered through fitted funnel rinsing with absolute ethanol (3 x 10 mL) and dried in vacuo at room temperature to afford dolutegravir sodium as a pale yellow solid (0.43 g, HPLC purity: 98.80%). 1H NMR (DMSO-d6): δ 10.70 (t, J = 5.8 Hz, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J = 6.4 Hz, 1H), 4.51 (d, J = 5.5 Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J= 14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J = 12.9 Hz, 1H), 1.24 (d, J = 7.0 Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 24:

The suspension of (4R,12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-6][1,3]oxazine-9-carboxamide (27, 0.43 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2.5 mL) was stirred at room temperature for ca.24 h. The reaction was diluted with mixture of water/ethanol (5 mL, 1:1) and left stirring for ca. 1.5 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with mixture of water/ethanol (3 x 5 mL, 1:1) and dried in vacuo at room temperature to afford 15 as a pale yellow solid (0.41 g, HPLC purity: 98.87%).
1H NMR (DMSO-de): δ 10.70 (t, J = 5.8 Hz, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J = 6.4 Hz, 1H), 4.51 (d, J = 5.5 Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J = 14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J = 12.9 Hz, 1H), 1.24 (d, J = 7.0 Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 25:

The suspension of {4R, 12aS)-7-chloro-4-methyl-6,8-dioxo-3,4, 6,8, 12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-6][1,3]oxazine-9-carboxylic acid (7, 0.31 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2.5 mL) was stirred at 50 °C for 3 days. The reaction was quenched with 2M H2S04 (1.2 mL) and left stirring for 7 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with water (3×5 mL) and ethanol (5 mL) dried in vacuo at 40°C to afford 28 as a pale yellow solid (0.17 g).
1H NMR (DMSO-d6): δ 15.37 (s, 1H), 12.76 (s, 1H), 8.66 (s, 1H), 5.51-5.49 (m, 1H), 4.80-4.78 (m, 1H), 4.65 (dd, J=13.8, 3.7 Hz, 1H), 4.43 (dd, J=13.8, 5.9 Hz, 1H), 4.05 (t, J^^.b Hz, 1H), 3.91 (dd, J=11.4, 3.1 Hz, 1H), 2.07-2.00 (m, 1H), 1.56 (d, J=13.8 Hz, 1H), 1.34 (d, J=7.0 Hz, 3H).13C NMR (DMSO-de): δ 172.21, 165.39, 161.73, 153.61, 141.11, 118.66, 112.99, 75.95, 62.03, 51.50, 44.90, 29.08, 15.18.
Example 26:

The suspension of (4R,12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8, 12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1 ,3]oxazine-9-carboxamide (27, 0.88 g) and solid sodium hydroxide (0.24 g) in absolute ethanol (20 mL) was stirred at 30 °C for 1.5 h. The reaction was quenched with 2M H2S04 (1 .5 mL) and left stirring for 3 hours at room temperature. The reaction mixture was filtered through fritted funnel and rinsed with water (3 x 2 mL) and ethanol (4 mL), and dried in vacuo at 40 °C to afford O-ethyl dolutegravir (29) as a pale yellow solid (0.25 g). The filtrate was extracted with ethyl acetate (3 x 5 mL). The combined organic layers were dried over MgS04, filtered and concentrated, then dried in vacuo at 40 °C to afford more 29 as a pale yellow solid (0.27 g).
1H NMR (CDCI3): δ 10.37 (t, J = 5.8 Hz, 1 H), 8.36 (s, 1 H), 7.37-7.32 (m, 1 H), 6.83-6.77 (m, 2H), 5.19 (dd, J = 5.9, 3.8 Hz, 1 H), 5.04-4.98 (m, 1 H), 4.61 (d, J = 6Hz, 2H), 4.26-4.22 (m, 3H), 4.1 1 (dd, J = 13.4, 5.9 Hz, 1 H), 3.97 (t, J = 2.4 Hz, 1 H), 3.96 (d, J = 2.4 Hz, 1 H), 2.21-2.14 (m, 1 H), 1.51 (dq, J = 14.0, 2.3 Hz, 1 H), 1 .47 (t, J = 7.0 Hz, 3H), 1 .35 (d, J = 7.1 Hz, 3H).
13C NMR (CDCI3): δ 174.78, 164.17, 162.49 (dd), 160.51 (dd), 155.72, 154.08, 142.32, 130.60 (dd), 129.33, 121 .51 (dd), 1 18.67, 1 1 1 .23 (dd), 103.78 (t), 76.15, 69.74, 62.58, 53.42, 44.58, 36.50 (d), 29.44, 16.04, 15.64.
Example 27:

The suspension of (4R, 12aS)-7-(benzyloxy)-4-methyl-3,4, 12,12a-tetrahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1-b][1 ,3]oxazine-6,8-dione (30, 0.68 g, prepared according to prior art) and solid sodium hydroxide (0.40 g) in absolute ethanol (5 mL) was stirred at 50 °C for 14 h. The reaction was quenched with formic acid (0.35 mL), water (2 mL) was added and mixture was left stirring for additional 1 h at room temperature. The reaction mixture was extracted with ethyl acetate (3 x 5 mL) and the combined organic layers concentrated to afford a crude oil. Purification by flash chromatography (eluting with CH2CI2/methanol) afforded 32 as an orange solid (0.26 g, 52 %).
The above procedure if done at room temperature in same time period, affords 31 as orange oil (0.24 g, 43 %).
Compound 32: 1H NMR (DMSO-d6): δ 7.64 (d, J = 7.4 Hz, 1 H), 6.20 (d, J = 7.3 Hz, 1 H), 5.40 (dd, J = 5.1 , 4.2 Hz, 1 H), 4.83-4.78 (m, 1 H), 4.35 (dd, J = 13.6, 3.9 Hz, 1 H), 4.13 (dd, J = 13.6, 5.4 Hz, 1 H), 4.05-4.00 (m, 1 H), 3.90-3.85 (m, 1 H), 2.03-1.95 (m, 1 H), 1.52 (dd, J = 13.9, 1 .9 Hz, 1 H), 1.33 (d, J = 7.1 Hz, 3H). 13C NMR (DMSO-d6): δ 170.96, 163.01 , 153.48, 137.96, 1 16.83, 1 13.52, 76.18, 62.05, 50.39, 44.53, 29.21 , 15.28.
Compound 31 : 1H NMR (DMSO-d6): δ 7.67 (d, J = 7.4 Hz, 1 H), 6.28 (d, J = 7.4 Hz, 1 H), 5.29 (dd, J = 5.4, 3.8 Hz, 1 H), 4.82-4.75 (m, 1 H), 4.32 (dd, J = 13.6, 3.6 Hz, 1 H), 4.10 (dd, J = 13.5, 5.6 Hz, 1 H), 4.03-3.93 (m, 3H), 3.85 (ddd, J = 1 1 .6, 5.0, 2.2 Hz, 1 H), 1.97-1 .89 (m, 1 H), 1 .48 (dd, J = 13.8, 2.1 Hz, 1 H), 1.27 (d, J = 7.1 Hz, 3H), 1.26 (d, J = 7.0 Hz, 3H). 13C NMR (DMSO-d6): δ 174.38, 156.1 1 , 150.82, 139.48, 1 16.39, 1 13.52, 75.92, 67.31 , 61 .80, 51 .36, 44.22, 29.29, 15.76, 15.36.
Exa

The transformation of 6 to dolutegravir with sodium hydroxide in ethanol was monitored for the interconversion of intermediates. The suspension of 6 (0.44 g) and solid sodium hydroxide (0.20 g) in ethanol (3.33 ml.) was stirred at 22 °C. Samples of the reaction mixture were taken after 3, 8 and 24 h for UPLC analysis. After 24 h, the reaction mixture was quenched with 2 M H2S04 (5 ml_), and left stirring at room temperature. The reaction mixture was filtered through fritted funnel, the product rinsed with water (30 ml.) and dried in vacuo at 50 °C overnight to afford dolutegravir as a white solid (0.27 g, 64 %).
The results of reaction monitoring:
Time UPLC analysis (area%)
Entry
(h) compound 6 compound 29 dolutegravir
1 3 h 37.50 20.63 39.99
2 8 h 0.78 15.46 80.32
3 24h 0.31 8.56 88.21
Example 29:

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 27 (0.86 g) in MeOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2 ml.) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring demethylation of 27 in MeOH:

Example 30:

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 6 (0.88 g) in EtOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2 mL) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring of the transformations of 6 in ethanol with NaOH:

dol. = dolutegravir
Exa

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 27 (0.88 g) in EtOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2ml_) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring of the transformations of 27 in ethanol with NaOH:

dol. = dolutegravir
Example 32:

Compound 3 (30 g, 1 10 mmol; assay 99%) was suspended in acetonitrile (450 mL), acetic acid (73 mL) and methanesulfonic acid (25 mL) were added. The reaction mixture was stirred 4 h at 70 °C. The clear red solution was cooled to 25 °C. Triethylamine (77 mL) and (S)-2-aminopropanol (17 mL) were added and the mixture was stirred at reflux temperature for 20 h. The reaction mixture was cooled to 25 °C and the insoluble product filtered, washed with 1 M HCI(aq) (60 mL), water (3 * 60 mL) and dried to give 4c (19.49 g, 67%): mp = 313-315 °C; 1H NMR (DMSO-d6): δ 1.31 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.8 Hz, 1 H), 4.13 (dd, J = 1 1.7, 10.3 Hz, 1 H), 4.28 (m, 1 H), 4.39 (dd, J = 8.6, 6.8 Hz, 1 H), 4.92 (dd, J = 12.3, 4.2 Hz, 1 H), 5.45 (dd, J = 10.2, 4.1 Hz, 1 H), 7.16 (s, 1 H), 8.84 (s, 1 H), 15.74 (s, 1 H); 13C NMR (DMSO-d6) 16.5, 51.6, 52.9, 72.4, 81.6, 1 15.8, 1 18.1 , 141.5, 147.6, 153.4, 165.3, 179.0.
Example 33

Compound 4c (2.78 g) was suspended in dimethylformamide (40 mL), cooled to 0 °C, then triethylamine (3.52 mL) was added, followed by ethyl chloroformate (1 .31 mL). After 10 min there was added 2,4-difluorobenzylamine (1 .57 mL). The mixture was then stirred at 25 °C for 1 h. Water (150 mL) was added and the mixture extracted with dichloromethane (50 mL). The organic phase was separated, washed with water (2 χ 50 mL), dried over sodium sulfate and evaporated under reduced pressure. The residue (4.31 g) was treated with boiling 2-propanol (40 mL), the suspension cooled, the product filtered and dried to give the product 5c as a white powder (2.70 g, 69%): 99.80 area% by HPLC at 258 nm; mp = 222-223 °C; MS (ESI) m/z = 390 [MH]+; 1H NMR (DMSO-d6): δ 1 .30 (d, J = 6.3 Hz, 3H), 3.63 (dd, J = 8.6, 6.8 Hz, 1 H), 4.02 (m, 1 H), 4.26 (m, 1 H), 4.37 (dd, J = 8.6, 6.8 Hz, 1 H), 4.53 (d, J = 6.0 Hz, 2H), 4.84 (dd, J = 12.2, 4.2 Hz, 1 H), 5.40 (dd, J = 12.2, 4.2 Hz, 1 H), 6.91 (s, 1 H), 7.05 (m, 1 H), 7.24 (m, 1 H), 7.38 (m, 1 H), 8.62 (s, 1 H), 10.43 (t, J = 6.0 Hz, 1 H).

To a suspension of 5c (2.70 g, 6.9 mmol) in acetonitrile (32 mL) was added DABCO (39 mg, 5 mol%) and TCCA (1.01 g, 4.3 mmol). The mixture was stirred 20 h at 40 °C protected from light and then quenched with a mixture of DMSO (0.81 mL) and water (0.20 mL). The insoluble cyanuric acid was removed by filtration and washed with acetonitrile (10 mL). The filtrate was evaporated under reduced pressure to give viscous oil that was crystallized from a mixture of methanol (10 mL) and water (5 mL), by slowly cooling the solution from 60 °C to room temperature. The product 6c was filtered, washed with cold methanol (8 mL) and dried to give an off-white powder (1 .20 g, 41 %): mp = 225-227 °C; MS (ESI) m/z = 424 [MH]+; 1H NMR
(DMSO-d6): δ 1.28 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.9 Hz, 1 H), 4.09 (m, 1 H), 4.26 (m, 1 H), 4.35 (dd, J = 8.6, 6.6 Hz, 1 H), 4.54 (d, J = 5.9 Hz, 2H), 4.85 (dd, J = 12.3, 3.8 Hz, 1 H), 5.42 (dd, J = 10.1 , 3.8 Hz, 1 H), 7.06 (m, 1 H), 7.24 (m, 1 H), 7.40 (m, 1 H), 8.67 (s, 1 H), 10.24 (t, J = 6.0 Hz, 1 H).
Example 35

cabotegravir
The suspension of 6c (1.00 g, 2.4 mmol) and sodium hydroxide (0.57 g, 14.2 mmol) in absolute ethanol (7 mL) was stirred at 40 °C for 16 h. The reaction was quenched with 0.5M H2S04 (15 mL), extracted with dichloromethane (20 mL), the extract washed with water (20 mL) and evaporated under reduced pressure. The residue was triturated in MTBE (10 mL), the product filtered, washed with MTBE (10 mL) and dried to give cabotegravir as an off-white solid (0.74 g, 77%): MS (ESI) m/z = 405 [MH]+.
Lek, a Sandoz company, opens the first production facility in Slovenia for drug substances for innovative medicines at its Mengeš site
Vojmir Urlep, president of Lek Board of Management

Lek, a Sandoz company, awarded for cooperation in practical training of students of the Faculty of Chemistry and Chemical Technology
At a ceremony held on 22 January 2015 at the Faculty of Chemistry and Chemical Technology, University of Ljubljana, the Maks Samec awards and recognitions for 2014 were presented for the best doctoral thesis in the field of chemistry, the best doctoral thesis in the field of chemical engineering and chemical technology and for services and merits to the Faculty in the year 2014. On this occasion, the Faculty also wanted to thank all the companies and individuals who shared their knowledge and resources to help the Faculty on its education and research path.
Lek, a Sandoz company, received a plaque for taking part in the implementation of practical training, which was collected, on behalf of the company, by Samo Roš, Head of Human Resources and a Member of the Lek Board of Management. By doing so, the Faculty of Chemistry and Chemical Technology thanked all the mentors who directly transfer their expertise and valuable experience onto students, teaching them specific skills, encouraging their development, guiding them through the work process and ensuring that students become socialized in the workplace.
Lek, a Sandoz company, is one of key pillars of the second-largest generic pharmaceutical company globally. Its role within Sandoz is to act as: a leading global development center for technologically demanding products and technologies; a global manufacturing center for active pharmaceutical ingredients and medicines; a competence center for the development of vertically integrated products; a Sandoz competence center in the field of development and manufacturing of biosimilar products; and, a supply center for the markets of Central and Eastern Europe (CEE), South East Europe (SEE) and Commonwealth of Independent States (CIS), and it is responsible for sales on the Slovenian market. For further information please visit http://www.lek.si/en.
Sandoz, the generic pharmaceuticals division of Novartis, is a global leader in the generic pharmaceutical sector. Sandoz employs over 26,400 employees and its products are available in more than 160 countries, offering a broad range of high-quality, affordable products that are no longer protected by patents. With USD 9.6 billion in sales in 2014, Sandoz has a portfolio of approximately 1,100 molecules, and holds the #1 position globally in biosimilars as well as in generic injectables, ophthalmics, dermatology and antibiotics, complemented by leading positions in the cardiovascular, metabolism, central nervous system, pain, gastrointestinal, respiratory, and hormonal therapeutic areas. Sandoz develops, produces, and markets these medicines, as well as active pharmaceutical and biotechnological substances. Nearly half of Sandoz’s portfolio is in differentiated products, which are defined as products that are more difficult to scientifically develop and manufacture than standard generics. In addition to strong organic growth since consolidating its generics businesses under the Sandoz brand name in 2003, Sandoz has benefitted from strong growth of its acquisitions, which include Lek (Slovenia), Sabex (Canada), Hexal (Germany), Eon Labs (US), EBEWE Pharma (Austria), Oriel Therapeutics (US), and Fougera Pharmaceuticals (US).
Sandoz is on Twitter. Sign up to follow @Sandoz_global at http://twitter.com/Sandoz_Global.
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, eye care, cost-saving generic pharmaceuticals, preventive vaccines and over-the-counter products. Novartis is the only global company with leading positions in these areas. In 2014, the Group achieved net sales of USD 58.0 billion, while R&D throughout the Group amounted to approximately USD 9.9 billion (USD 9.6 billion excluding impairment and amortization charges). Novartis Group companies employ approximately 130,000 full-time-equivalent associates. Novartis products are available in more than 180 countries around the world. For more information, please visit www.novartis.com
////////////Carbotegravir, Dolutegravir, New Patent, WO 2016113372, Lek Pharmaceutical and Chemical Co DD
New Patent, WO 2016110874, Artemisinin , IPCA Laboratories Ltd
![]()

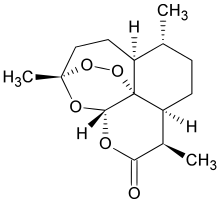
New Patent, WO 2016110874, Artemisinin , IPCA Laboratories Ltd
FOR Cancer; Parasitic infection; Plasmodium falciparum infection; Viral infection
KUMAR, Ashok; (IN).
SINGH, Dharmendra; (IN).
MAURYA, Ghanshyam; (IN).
WAKCHAURE, Yogesh; (IN)

Dr. Ashok Kumar, President – Research and Development (Chemical) at IPCA LABORATORIES LTD
IPCA LABORATORIES LIMITED [IN/IN]; 48, Kandivli Industrial Estate, Charkop, Kandivali (West), Mumbai 400067 (IN)

Novel process for preparing artemisinin or its derivatives such as dihydroartemisinin, artemether, arteether and artesunate. Also claims novel intermediates of artemesinin such as artemisinic acid or dihydroartemisinic acid. Discloses the use of artemisinin or its derivatives, for treating malaria, cancer, viral and parasitic infections.
In July 2016, Newport Premium™ reported that IPCA was capable of producing commercial quantities of artemether, arteether and artesunate; and holds an inactive US DMF for artemether since February 2009. In July 2016, IPCA’s website lists artemether, arteether and artesunate under its products and also lists artemether and artesunate as having EDMF and WHO certificates. The assignee also has Canada HPFB certificate for artemether.
The Central Drug Research Institute (CDRI) in collaboration with IPCA is developing CDRI-97/78 (1,2,4 trioxane derivative), a synthetic artemisinin substitute for treating drug resistant Plasmodium falciparum infection. In July 2016, CDRI-97/78 was reported to be in phase 1 clinical development. IPCA in collaboration with CDRI was also investigating CDRI-99/411, a synthetic artemisinin substitute for treating malaria; but its development had been presumed to have been discontinued; however, this application’s publication would suggest otherwise.

Writeup
Artemisinin is an active phytoconstituent of Chinese medicinal herb Artemisia annua, useful for the treatment of malaria. Generally, artemisinin/artemisinic acid is obtained by extraction of the plant, Artemisia annua. The plant Artemisia annua was first mentioned in an ancient Chinese medicine book written on silk in the West Han Dynasty at around 200 B.C. The plant’s anti-malarial application was first described in a Chinese pharmacopeia, titled “Chinese Handbook of Prescriptions for Emergency Treatments,” written at around 340 A.D.
Artemisinin being poorly bioavailable limits its effectiveness. Therefore semisynthetic derivatives of artemisinin such as artesunate, dihydroartemisinin, artelinate, artemether, arteether have been developed to improve the bioavailability of Artemisinin.
Artemisinin and its derivatives – dihydroartemisinin, artemether, arteether, and artesunate being a class of antimalarials compounds used for the treatment of uncomplicated, severe complicated/cerebral and multi drug resistant malaria. Additionally, there are research findings that artemisinin and its derivatives show anti-parasite, anti-cancer, and anti-viral activities.


Dihydroartemisinin Artesunate
The content of Artemisinin in the plant Artemisia annua varies significantly according to the climate and region/geographical area where it is cultivated. Further, the extraction methods provide artemisinin or artemisinic acid from the plant in very poor yields and therefore not sufficient to accommodate the ever-growing need for this important drug. Consequently, widespread use of these valuable drugs has been hampered due to the low availability of this natural product. Therefore, research has focused on the syntheses of this valuable drug in a larger scale to meet the increasing global demand and accordingly ample literature is available on the synthesis of artemisinin or its derivatives, but no commercial success being reported / known till date.
Artemisinin can be prepared synthetically from its precursors such as artemisinic acid or dihydroartemisinic acid according to literature methods known to skilled artisans. For example, dihydroartemisinic acid can be converted to artemisinin by a combination of photooxidation and air-oxidation processes as described in U.S. Patent No. 4,992,561.
Amorphadiene is an early starting material for synthesis of Artemisinic acid or dihydroartemisinic acid, which is an important intermediate for producing Artemisinin commercially, and WO2006128126 reported a preparation method as mentioned in scheme- 1.

acid
In accordance with the scheme 1, the amorphadiene is treated with di(cyclohexyl)borane ( δΗι ΒΗ followed by reaction with H2O2 in presence of NaOH to obtain the amorph-4-ene 12-ol which is further oxidized to dihydroartemisinic acid using CrCb/ifcSC^. The formation of amorph-4-ene 12-ol is taking place via epoxidation of the exocyclic double bond. However, the reported yields of this synthesis are very low, making it unviable to produce artemisinic acid at a cheaper cost than natural extraction, for commercial use.

Amorpha -4, 11-diene
A similar method is published in, WO2009088404, for synthesis of dihydroartemisinic acid through preparation of amorph-4-ene-12-ol via epoxide formation, albeit, predominantly at exo position by reacting the amorpha-4,11-diene with H2O2 in presence of porphyrin catalyst (TDCPPMnCl). During reaction, epoxidation also occurred at endo position leading to formation of Amorphadiene- 4,5- epoxide that remain as impurity. The formed exo epoxide (amorphadiene – 11, 12 – epoxide) is further reduced to get amorph- 4-ene 12-ol and then converted to dihydroartemisinic acid and finally converted into artemisinin.

Amorphadiene-11,12-epoxide
This process involves expensive & industry unfriendly reagents. Moreover, desired stereo isomers were obtained only in poor yields, because several purification steps were needed to get desired stereo isomers leading to escalated production/operational costs.
Therefore there remains a need in the art to improve the yield of Dihydroartemisinic acid, which could potentially reduce the cost of production of Artemisinin and/or its derivatives. Consequently it is the need of the hour to provide a synthetic and economically viable process to meet the growing worldwide demand by improving the process for Artemisinin and/or its derivatives to obtain them in substantially higher yields with good purity by plant friendly operations like crystallization/extractions rather than column chromatography/other cost constraint procedures.
Therefore, the object of the invention is to prepare Artemisinic acid of formula-II, Dihydroartemisinic acid of formula-IIa, Artemisinin and its derivatives through Amorphadiene- 4,5- epoxide.


DHAA methyl ester
Scheme 2
Method 4 (From compound of formula IV (R = CI)):
In the 4-neck round bottom flask was charged Diphenyl sulfoxide (23.8 g), NaHC03 (32.96 g) and DMSO (80 ml) at 30°C. Further a solution of compound of formula IV (R = CI) (10 g) in DMSO (20 ml) was charged to the reaction mass at 30°C followed by heating and maintaining the temperature for 40 hours at 80°C till completion. DMSO was distilled out under vacuum. The reaction mass was cooled followed by charging water
(100 ml) and toluene (100 ml) to the reaction mass with stirring for 30 minutes at 28°C. The layers were separated out and aqueous layer was back extracted with toluene (2 X 100 ml). The organic layer was washed with water (100 ml) and saturated brine solution (100 ml). Solvent was distilled out under vacuum at 50°C, and the crude mass degassed under vacuum at 50-55°C. IPA (40 ml) was charged to the mass. Simultaneous addition of hydrazine hydrate (65% in aqueous solution) (3.8 g) and hydrogen peroxide (50% in aqueous solution) (2.5 ml) was done at 30-32°C over a period of 3.25 hours. After completion, reaction mass was cooled up to 5-10°C and water (100ml) was added to the reaction mass. The pH of the reaction mass was adjusted to 3.8 with dilute 8% aqueous HC1 (24 ml) at 10°C. Ethyl acetate (60 ml) was added to the reaction mass at 10°C and stirred for 15 minutes at 15-20°C. The layers were separated. Aqueous layer was back extracted with ethyl acetate (2 X 20 ml). The combined organic layer was washed with 10%) sodium metabisulfite solution (50 ml), water (50 ml) and saturated brine solution (50 ml). The organic layer was distilled out under vacuum at 45°C and the obtained crude mass was degassed at 50-55°C. To this was added DME (40 ml), Biphenyl (0.9 g) and Li-metal (1.63 g) and the reaction mass was maintained for 10 hours at 80-85°C till reaction completion. The reaction mass was cooled up to 0-5°C followed by drop wise addition of water within one hour, and the reaction stirred for two hours at 20-25°C. Toluene (35 ml) was charged with stirring and layers were separated. The aqueous layer was washed with toluene (35 ml) and the combined toluene layer was washed with water (20 ml). The combined aqueous layer was again washed with toluene (20 ml). The aqueous layer was cooled to 10-15°C and pH adjusted to 3.5-4 with dilute 16% aqueous HC1. MDC (50 ml) was charged and stirred 30 minutes at 20-25°C followed by separation of layers. The aqueous layer extracted with MDC (25 ml) and the combined MDC layer was washed with water (50 ml), then with saturated NaCl solution (25 ml). The solvent was distilled out under vacuum at 40-45°C and the crude mass (Purity: 70-80%>) was degassed at 65-70°C. The crude product (10 g) was dissolved in ethyl acetate (200 ml). 10%> aqueous NaOH (100 ml) was charged to the reaction mass and stirred one hour at 20°C followed by layer separation. Again 10%> aqueous NaOH (100ml) was added to the organic layer, stirred for 30 minutes and layers were separated out. The pH of the combined NaOH solution wash was adjusted to 4.0 with dilute 16%> aqueous HC1 at 5-10°C under stirring. Ethyl acetate (850 ml) was charged to aqueous acidic mass, stirred 30 minutes and layers were separated out. The aqueous layer was back extracted with ethyl acetate (2 X 30 ml) and the combined organic layer was washed with water (100 ml) and saturated brine (50 ml). The organic layer was dried over sodium chloride, solvent was distilled out under vacuum and the purified mass was degassed under vacuum at 50-55°C to obtain Dihydroartemisinic acid (Purity: 90-95%).
b) Methyl ester of Dihydroartemisinic acid:
To a clear solution of Dihydroartemisinic acid (40 g) dissolved in MDC (120 ml) was added thionyl chloride (SOCh) (14.85 ml) at 10±2°C and reaction mass was heated to reflux temperature 40±2°C. After the completion of reaction, solvent was distilled out and excess SOCh was removed under reduced pressure. The resulting concentrated mass of acid chloride was dissolved in MDC (200 ml). In another RBF was taken triethylamine (30.6 ml) and methanol (120 ml). To this solution was added above acid chloride solution at 30±2°C and maintained till completion of reaction. To the reaction mass was added water (400 ml) and organic layer was separated. The aqueous layer was washed with MDC and mixed with main organic layer and the combined organic layer was back washed with water till neutral pH. Then organic layer was concentrated to give methyl ester of Dihydroartemisinic acid as a brown color oily mass.
Weight: 41.88 gm
Yield = 98%
c) Artemisinin:
Methyl ester of dihydroartemisinic acid (67.7 g) was dissolved in methanol (338 ml). To this solution was added Sodium molybdate (29.5 g), 50% hydrogen peroxide (147.3 g) was added at 30±2°C and reaction was maintained for 3-4 hours. After completion of reaction was added water (300 ml) and MDC (300 ml) to the reaction mass. The organic layer was separated and aqueous layer washed with MDC (100 ml). The combined organic layer was concentrated to 475 ml containing hydroperoxide intermediate and directly used for next stage reaction. In another RBF containing MDC (475 ml) was added benzene sulfonic acid (1.27 g) and Indion resin (6.7 g). This heterogeneous solution was saturated with oxygen by passing O2 gas for 10 min at 0±2°C. To this was added previous stage hydroperoxide solution at same temperature with continuous 02 gas purging within 30-40 minutes. The oxygen gas was passed at same temp for 4 hours and temperature raised to 15±2°C with continued passing of oxygen for 5 hours. The
mixture was stirred at 25-30°C for 8-10 hours followed by filtration of resin. The filtrate was washed with water (200 ml X 3) and the combined aqueous layer back washed with MDC (50 ml). The combined organic layer was concentrated to give crude Artemisinin. Weight: 54 gm
Yield= 70.7%
Purification of Artemisinin:
Crude Artemisinin (10 g) was dissolved in ethyl acetate (25 ml) at 45-50°C. The solution was cooled to 30-35°C followed by addition of n-Hexane (100 ml). The material was isolated, stirred for 2 hours, filtered and vacuum dried at 45°C.
Weight: 4 gm
Yield: 40%

THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India
////////New Patent, WO 2016110874, Artemisinin , IPCA Laboratories Ltd, malaria, Cancer, Parasitic infection, Plasmodium falciparum infection, Viral infection, artemether artemisinin, artemotil, artenimol, artesunate,
WO 2016110798, Piramal Enterprises Ltd, New Patent, Lurasidone

Lurasidone – it having been developed and launched by Sumitomo Dainippon Pharma. Lurasidone was launched for schizophrenia in the US by Sumitomo’s US subsidiary Sunovion Pharmaceuticals.
WO 2016110798, Piramal Enterprises Ltd, New Patent, Lurasidone
An improved process for the preparation of lurasidone and its intermediate
PIRAMAL ENTERPRISES LIMITED [IN/IN]; Piramal Tower Ganpatrao Kadam Marg, Lower Parel Mumbai 400013 (IN)
GHARPURE, Milind; (IN).
TIWARI, Shashi Kant; (IN).
WAGH, Ganesh; (IN).
REVANAPPA, Galge; (IN).
WARPE, Manikrao; (IN).
ZALTE, Yogesh; (IN).

From left: Anand Piramal, executive director, Piramal Group; Swati Piramal, vice-chairperson, Piramal Group; Ajay Piramal, chairman, Piramal Group; Nandini Piramal, executive director, Piramal Enterprises; and Peter DeYoung, president, Piramal Enterprises
Improved process for preparing pure (3aR,7aR)-4′-(benzo[d]isothiazol-3-yl)octahydrospiro[isoindole-2,1′-piperazin]-1′-ium methanesulfonate, useful as a key intermediate in the synthesis of lurasidone. Also claims a process for purifying lurasidone hydrochloride, useful for treating schizophrenia and bipolar disorders. In July 2016, Newport Premium™ reported that Piramal Enterprises was capable of producing commercial quantities of lurasidone hydrochloride and holds an active US DMF for the drug since March 2015.
Lurasidone (the Compound-I), is an atypical antipsychotic used in the treatment of schizophrenia and bipolar disorders.The drug is marketed as hydrochloride salt (the compound-I.HCl) by Sunovion Pharms Inc.under the tradename”LATUDA”, in the form of oral tablets. Latuda is indicated for the treatment of patients with schizophrenia. Lurasidone hydrochloride has the chemical name ((3aR,4S,7R,7aS)-2-[((lR,2R)-2-{ [4-(l,2-benzisothiazol-3-yl)-piperazin-l-yl]methyl}cyclohexyl)-methyl]hexahydro-lH-4,7-methanisoindol-l,3-dione hydrochloride, and is structurally represented as follows;

Compound-I.HCl
Lurasidone being an important antipsychotic agent; a number of processes for its preparation as well as for its intermediates are known in the art.
US Patent No. 5,532,372 describe a process for the synthesis of Lurasidone, which is illustrated below in Scheme-I. In the process, the compound, cyclohexane- l,2-diylbis(methylene) dimethanesulfonate(referred to as the compound-Ill) is reacted with 3-(l-piperazinyl-l,2-benzisothiazole(referred to as the compound-IV) in acetonitrile, and in the presence of sodium carbonate to provide corresponding quaternary ammonium salt as 4′-(benzo[d]isothiazol-3-yl)octahydrospiro[isoindole-2, r-piperazin]-l’-ium methanesulfonate (the compound-II). The compound-II is further treated with bicyclo[2.2.1]heptane-2-exo-3-exo-dicarboximide in xylene, in the presence of potassium carbonate and dibenzo-18-crown-6-ether to provide lurasidone.

Scheme-I
US Published Patent Application 2011/0263848 describes a process for the preparation of the quaternary ammonium salt (the compound-II) which comprises reacting 4-(l,2-benzisothiazol-3-yl)piperazine with (lR,2R)-l,2-bis(methanesulfonyloxymethyl)- cyclohexane in a solvent such as toluene in the presence of a phosphate salt.
Indian Published Patent Application 2306/MUM/2014 (” the IN’2306 Application”) describes a process for the synthesis of lurasidone and the intermediates thereof, comprising reacting (R,R) trans l,2-bis(methane sulphonyl methyl)cyclohexane with 3-(Piperazine-l-yl)benzo[d]isothiazole in presence of a mixture of two or more polar aprotic solvents selected from acetonitrile, N,N-dimethyl formamide (DMF) and/or Ν,Ν-dimethyl acetamide (DMAc), and a base at reflux temperature to obtain the quaternary ammonium salt (the compound II), which is then converted to lurasidone. The IN’2306 application demonstrated preparation of the compound II using the solvent combination such as acetonitrile-DMF and acetonitrile-DMAc.
US Published Patent Application 2011/0263847 describes a process for the preparation of the quaternary ammonium salt (the compound-II) comprising reacting 4-(l,2-benzisothiazol-3-yl)piperazine with (lR,2R)-l,2-bis(methanesulfonyloxymethyl)cyclohexane in a solvent such as toluene, wherein the piperazine compound is used in an excess amount i.e. 1.8 to 15 moles with respect to ( 1R,2R)- 1 ,2-bis(methanesulfonyloxymethyl)cyclohexane.
Chinese Published Patent Application 102731512 describes a process for the preparation of the quaternary ammonium salt (the compound-II) comprises reaction of 4-(l,2-benzisothiazol-3-yl)piperazine with (lR,2R)-l,2-bis(methanesulfonyloxymethyl)cyclohexane in a solvent such as toluene in the presence of a phase transfer catalyst.
In addition to the afore discussed patent documents, there are a number of patent documents that describe a process for the preparation of the quaternary ammonium salt (the compound-II), the key intermediate for the synthesis of lurasidone. For instance, Published PCT application WO2012/131606 A 1, Indian Published patent application 217/MUM/2013, Chinese published patent applications 102863437, 103864774 and 102827157 describe a process for the preparation of the quaternary ammonium salt (compound-II) comprises reaction of 4-(l,2-benzisothiazol-3-yl)piperazine with (lR,2R)-l,2-bis(methanesulfonyloxymethyl)cyclohexane in a solvent or a solvent mixture such as acetonitrile, acetonitrile : water solvent mixture, toluene or DMF, in the presence of a base.
It is evident from the discussion of the processes for the preparation of the quaternary ammonium salt (the compound-II), described in the afore cited patent documents that the reported processes primarily involve use of acetonitrile either as the single solvent or in a mixture of solvents. Acetonitrile is a relatively toxic, and not an environment friendly solvent. Due to its toxic nature, it can cause adverse health effects also. Acetonitrile is covered under Class 2 solvents i.e. solvents to be limited, and residual solvent limit of acetonitrile is 410 ppm in a drug substance as per the ICH (International Conference on Harmonisation) guidelines for residual solvents. Moreover, acetonitrile is a costlier solvent, which renders the process costlier and hence, is not an industrially feasible solvent.
It is also evident from the discussion of the processes described in afore cited patent documents that some of the reported processes involve use of high boiling solvents such as toluene and dimethylformamide as reaction solvent, which subsequently require high reaction temperatures, and this in turn leads to tedious workup procedures. In view of these drawbacks, there is a need to develop an industrially viable commercial process for the preparation of lurasidone and its intermediates; which is simple, efficient and cost-effective process and provides the desired compounds in improved yield and purity.
Inventors of the present invention have developed an improved process that addresses the problems associated with the processes reported in the prior art. The process of the present invention does not involve use of any toxic and/or costly solvents. Moreover, the process does not require additional purification steps and critical workup procedure. Accordingly, the present invention provides a process for the preparation of lurasidone and its intermediates, which is simple, efficient, cost effective, environmentally friendly and commercially scalable for large scale operations.

Scheme-II

Scheme-Ill
EXAMPLES
Example-1: Preparation of (3aR,7aR)-4′-(benzo[d]isothiazol-3-yl)octahydrospiro[isoindole-2,l’-piperazin]-l’-ium methanesulfonate(the compound II)
Charged 150.0 mL (3v) of isopropyl alcohol (IPA) in a flask followed by the addition of the compound-Ill (50.0 g) , 3-(l-Piperazinyl)-l, 2-Benzisothiazole (32.84 g), sodium carbonate granular (10.79 g) and water 50 mL (lv). The reaction mixture was heated at a temperature of 82-85 °C for 24 to 25 h. Cooled the reaction mixture to room temperature, filtered on Buchner funnel and the filtrate was collected.
The filtrate was evaporated under vacuum at 55-65°C till visible solid appears in the reaction mass. The solid was stirred in 75 mL of toluene at room temperature and the solid was filtered. The wet cake was transferred to a flask and added 125 mL of acetone to it; followed by stirring at room temperature. The resulting solid was filtered to yield the pure title compound (II).
Yield: 63.4 g (90 %)
Purity (by HPLC): 99.79 %
Unreacted compound-IV as impurity in 0.05 % .
Example-2: Preparation of Lurasidone free base.
Charged 150.0 mL of Ν,Ν-dimethylformamide (DMF) in a flask followed by the addition of 50.0 g of the compound-II (as obtained in the above example-1), 19.5 g (3aR,4S,7R,7aS)-4,7-methano-lH-isoindole-l,3(2H)-dione and 19.5 g of potassium carbonate. The reaction mixture was heated at a temperature of about 125 °C for 24 h. The reaction mixture was cooled to room temperature and 400 mL of water was added to it. The reaction mixture was stirred, and the precipitated product was filtered. The wet cake was washed with IPA and Lurasidone free base is obtained as the pure product. [Yield: 46.52 g (80 %)]
Example-3: Purification of Lurasidone hydrochloride.
Charged water (200 ml) and IPA (200 ml) in flask followed by the addition of Lurasidone hydrochloride (50 gm, residual acetone: 5769 ppm). The reaction mixture was heated at a temperature of 75-80 °C for about 30 min. The reaction mixture was cooled to 20-30 °C and stirred for about 2 hours. The precipitated solid was filtered and isolated as pure Lurasidone hydrochloride (residual acetone: 2 ppm)
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India
///////////////WO 2016110798, Piramal Enterprises Ltd, New Patent, Lurasidone
NEW PATENT, WO 2016108172, OSPEMIFENE AND FISPEMIFENE, OLON S.P.A.

Ospemifene is useful for treating menopause-induced vulvar and vaginal atrophy; while fispemifene is useful for treating symptoms related with male androgen deficiency and male neurological disorders.
In July 2016, Newport Premium™ reported that Olon was potentially interested in ospemifene and holds an active US DMF for ospemifene since September 2015. Olon’s website also lists ospemifene under R&D APIs portfolio.
PROCESS FOR THE PREPARATION OF OSPEMIFENE AND FISPEMIFENE
![]()
OLON S.P.A. [IT/IT]; Strada Rivoltana, Km. 6/7 20090 Rodano (MI) (IT)
| CRISTIANO, Tania; (IT). ALPEGIANI, Marco; (IT) |
Process for preparing ospemifene or fispemifene, by reacting a phenol with an alkylating agent.
Ospemifene, the chemical name of which is 2-{4-[(lZ)-4-chloro-l,2-diphenyl-l-buten-l-yl]phenoxy}ethanol (Figure), is a non-steroidal selective oestrogen-receptor modulator (SERM) which is the active ingredient of a medicament recently approved for the treatment of menopause-induced vulvar and vaginal atrophy.
The preparation of ospemifene, which is disclosed in WO96/07402 and WO97/32574, involves the reaction sequence reported in Scheme 1 :

Ospemifene
Scheme 1
The first step involves alkylation of 1 with benzyl-(2-bromoethyl)ether under phase-transfer conditions. The resulting product 2 is reacted with triphenylphosphine and carbon tetrachloride to give chloro-derivative 3, from which the benzyl protecting group is removed by hydrogenolysis to give ospemifene.
A more direct method of preparing ospemifene is disclosed in WO2008/099059 and illustrated in Scheme 2.

Ospemifene
Scheme 2
Intermediate 5 (PG = protecting group) is obtained by alkylating 4 with a compound X-CH2-CH2-O-PG, wherein PG is a hydroxy protecting group and X is a leaving group (specifically chlorine, bromine, iodine, mesyloxy or tosyloxy), and then converted to ospemifene by removing the protecting group.
Alternatively (WO2008/099059), phenol 4 is alkylated with a compound of formula X-CH2-COO-R wherein X is a leaving group and R is an alkyl, to give a compound of formula 6, the ester group of which is then reduced to give ospemifene (Scheme 3)

Ospemifene
Scheme 3
Processes for the synthesis of ospemifene not correlated with those reported in schemes 2 and 3 are also disclosed in the following documents: CN104030896, WO2014/060640, WO2014/060639, CN103242142 and WO201 1/089385.
Fispemifene, the chemical name of which is (Z)-2-[2-[4-(4-chloro-l,2-diphenylbut-l-enyl)phenoxy]ethoxy]ethanol (Figure) is a non-steroidal selective oestrogen-receptor modulator (SERM), initially disclosed in WOO 1/36360. Publications WO2004/108645 and WO2006/024689 suggest the use of the product in the treatment and prevention of symptoms related with male androgen
deficiency. The product is at the clinical trial stage for the treatment of male neurological disorders.
According to an evaluation of the synthesis routes for ospemifene and fispemifene described in the literature, those which use compound 4 (Schemes 2 and 3) are particularly interesting, as 4 is also a key intermediate in the synthesis of toremifene, an oestrogen-receptor antagonist (ITMI20050278).
Leaving group X of the compound of formula 7 is preferably a halogen, such as chlorine, bromine or iodine, or an alkyl or arylsulphonate such as mesyloxy or tosyloxy.
In one embodiment of the invention, in the compound of formula 7, X is a leavmg group as defined above and Y is -(OCH2CH2)nOH wherein n is zero, and the reaction of 7 with 4 provides ospemifene, as reported in Scheme 4.

Scheme 4
In another embodiment of the invention, in the compound of formula 7, X and Y, taken together, represent an oxygen atom, the compound of formula 7 is ethylene oxide, and the reaction of 7 with 4 provides ospemifene, as reported in Scheme 5.

Scheme 5
In another embodiment of the invention, X is a leaving group as defined above and n is 1, and the reaction of 7 with 4 provides fispemifene, as reported in Scheme 6.

Scheme 6
The reaction between phenol 4 and alkylating reagent 7, wherein X is a leaving group as defined above and Y is the -(OCHbCEh^OH group as defined above, can be effected in an aprotic solvent preferably selected from ethers such as tetrahydrofuran, dioxane, dimethoxyethane, tert-butyl methyl ether, amides such as N,N-dimethylformamide, Ν,Ν-dimethylacetamide and N-methylpyrrolidone, nitriles such as acetonitrile, and hydrocarbons such as toluene and xylene, in the presence of a base preferably selected from alkoxides, amides, carbonates, oxides or hydrides of an alkali or alkaline-earth metal, such as potassium tert-butoxide, lithium bis-trimethylsilylamide, caesium and potassium carbonate, calcium oxide and sodium hydride.
The reaction can involve the formation in situ of an alkali or alkaline earth salt of phenol 4, or said salt can be isolated and then reacted with alkylating reagent 7. Examples of phenol 4 salts which can be conveniently isolated are the sodium salt and the potassium salt. Said salts can be prepared by known methods, for example by treatment with the corresponding hydroxides (see preparation of the potassium salt of phenol 4 by treatment with aqueous potassium hydroxide as described in document ITMI20050278), or from the corresponding alkoxides, such as sodium methylate in methanol for the preparation of the sodium salt of phenol 4, as described in the examples of the present application.
Example 1
Sodium hydride (4.2 g) is loaded in portions into a solution of 4-(4-chloro-l,2-diphenyl-buten-l-yl)phenol (10 g) in tetrahydrofuran (120 ml) in an inert gas environment, and the mixture is maintained under stirring at room temperature for 1 h. 2-Iodoethanol (11 ml) is added dropwise, and the reaction mixture is refluxed for about 9 h. Water is added, and the mixture is concentrated and extracted with ethyl acetate. The organic phase is washed with sodium carbonate aqueous solution and then with water, and then concentrated under vacuum. After crystallisation of the residue from methanol-water (about 5: 1), 9.9 g of crude ospemifene is obtained.
Example 2
A solution of sodium methylate in methanol (6.25 ml) is added to a solution of 4-(4-chloro-l,2-diphenyl-buten-l-yl)phenol (10 g) in methanol (100 ml) in an inert gas environment, and maintained under stirring at room temperature for 1 h. The mixture is concentrated under vacuum and taken up with tetrahydrofuran (100 ml). A solution of 2-iodoethanol (3.5 ml) in tetrahydrofuran (30 ml) is added dropwise, and the reaction mixture is refluxed for about 3 h. Water is added, and the mixture is concentrated and extracted with ethyl acetate. The organic phase is washed with a saturated sodium hydrogen carbonate aqueous solution, and finally with water. The resulting solution is then concentrated under vacuum and crystallised from methanol-water to obtain 5.8 g of crude ospemifene.
Example 3
Potassium tert-butylate (2.0 g) is added to a solution of 4-(4-chloro-l,2-diphenyl-buten-l-yl)phenol (5 g) in tert-butanol (75 ml) in an inert gas environment, and maintained under stirring at room temperature for 1 h. The solvents are concentrated under vacuum, and the concentrate is taken up with tetrahydrofuran (50 ml). A solution of 2-iodoethanol (1.7 ml) in tetrahydrofuran (15 ml) is added in about 30 minutes, and the reaction mixture is then refluxed for about 2 h. The process then continues as described in Example 1, and 2.9 g of crude ospemifene is obtained.
Example 4
A 50% potassium hydroxide aqueous solution (4.4 ml) is added to a solution of 4-(4-chloro-l,2-diphenyl-buten-l-yl)phenol (2 g) in toluene (20 ml) in an inert gas environment, and maintained under stirring at room temperature for 15
minutes. 2-Iodoethanol (2.2 ml) is added in about 30 minutes, and the reaction mixture is refluxed and maintained at that temperature for about 7 h. After the addition of water, the phases are separated. The organic phase is washed with a saturated sodium hydrogen carbonate aqueous solution, and finally with water. The organic phase is then concentrated under vacuum. After crystallisation of the residue from methanol-water (about 5:1), 0.85 g of crude ospemifene is obtained.
//////NEW PATENT, WO 2016108172, OSPEMIFENE, FISPEMIFENE, OLON S.P.A.
EP 03031800, New patent, Miglustat, Navinta LLC

MIGLUSTAT
![]()
Gauchers disease type I; Niemann Pick disease type C
EP-03031800, Process for the preparation of high purity miglustat
Navinta, LLC ; Shah, Shrenik K. ; Kharatkar, Raju Mahadev ; Bhatt, Chiragkumar Anilkumar ; Kevat, Jitendra Bhagwandas
The present invention provides a process for the preparation and isolation of crystalline miglustat without the use of a column chromatography or ion exchange purification. The crystalline miglustat has a high purity and a melting point of 128 °C and an endothermic peak is 133 °C.
Process for preparing and isolating crystalline form of miglustat with a high purity is claimed. Represents a first PCT filing from the inventors on miglustat. Actelion, under license from Oxford GlycoSciences (OGS; then Celltech, now UCB), which licensed the compound from GD Searle & Co, has developed and launched miglustat.
Product patent WO9426714, will expire in the US in 2018.
Kharatkar is affiliated with Sterling Biotech, Bhatt is affiliated with Intas and Kevat is affiliated with Orchid Chemicals & Pharmaceuticals.
INVENTORS Shah, Shrenik K.; Kharatkar, Raju Mahadev; Bhatt, Chiragkumar Anilkumar; Kevat, Jitendra Bhagwandas
About Navinta
Navinta, LLC in Ewing, N.J. is a technology driven Pharmaceutical Company that focuses on novel routes of synthesis of new and existing drug molecules, complex pharmaceutical ingredients, novel formulations of liquid dosage form, novel oral dosage form, novel injectable dosage form and implantable drug delivery devices. Navinta has currently at least fifteen (15) patents granted or pending with the United States Patent and Trademark Office.
![]()
EP-03031800 LINK EMBEDDED
|
Miglustat is a potent inhibitor of glycosyltransferase. It is primarily used in the treatment of Gaucher’s disease. Miglustat is chemically known as N-butyl-1,5-dideoxy-1,5-imino-D-glucitol of formula (I) and is sometimes referred as N-butyl-1-deoxynojirimycin. Miglustat is a white to off-white crystalline solid with a melting point of 125-126° C. Its empirical formula is C10H21NO4 and has a molecular weight of 219.28 g/mol.
|



Example 1
Synthesis of 2, 3, 4, 6-tetra-O-benzyl-N-butyl-1-deoxynojirimycin hydrochloride of Formula (VI)
To a solution of 2, 3, 4, 6-tetra-O-benzyl-1-deoxynojirimycin hydrochloride (V) (prepared as in Organic Process Research & Development, 2008, 12, 414-423) (45 g, 0.08 mol) in 1575 mL of methanol, n-butyraldehyde (21.6 g, 0.24 mol) and sodium cyanoborohydride (25.2 g, 0.4 mol) were added and stirred. The reaction was maintained under stirring at a temperature from about 25.degree. C. to about 30.degree. C. After the completion of the reaction, the reaction was quenched by adding 765 ml of 10% HCl in methanol, while keeping the temperature between 25.degree. C. to 30.degree. C. The reaction mass was cooled to 0.degree. C. to 5.degree. C. and the resulting precipitate solids were filtered. The filtrate was treated with aqueous HCl and the solid formed was filtered, suspended in 1 N HCl, stirred for 1 hour and filtered. The collected solid was washed with diisopropylether and dried under vacuum to furnish 46.2 g of compound (IV) (46.2 g, 0.075 mol, 94% yield) of high chemical purity based on HPLC analysis (>99.0%).
Example 2
Synthesis of Miglustat Hydrochloride of Formula (III)
A solution of 2, 3, 4, 6-tetra-O-benzyl-N-butyl-1-deoxynojirimycin hydrochloride (VI) (100 g, 0.16 mol) in methanol (1000 mL), 10% HCl solution in methanol (100 mL), and 10% Pd/C (50% wet) (10 g) were mixed and stirred under hydrogen atmosphere at a temperature of about 25.degree. C. to about 30.degree. C. until completion of the reaction. The reaction mass was filtered and the solvent was removed from the filtrate by rotary evaporation. Ethyl acetate (1000 mL) was added to the residue from the rotary evaporation to precipitate the solid. The solid was filtered and dried to isolate Miglustat hydrochloride (III) (42 g, 0.16 mol, 100% yield) of >99.5% purity as measured by HPLC analysis. The DSC thermogram of this product is provided as FIG. 3, and the FTIR spectrum of this product is provided as FIG. 4.
Example 3
Synthesis of Miglustat of Formula (I)
Miglustat hydrochloride (III) (42 g, 0.16 mol) obtained from Example 2 was dissolved in 420 mL of methanol and DBU (1,8-diazabicycloundec-7-ene) (34.1 mL) was added. The reaction mass was warmed slightly and stirred for about 2 hours. The reaction was concentrated by removal of methanol. Dichloromethane (900 mL) was added to the residue. The resulting solid was filtered and dried to obtain crystalline miglustat (I) (27 g, 0.12 mol, 75% yield) of >99.5% purity as measured by HPLC analysis. The melting point of the crystalline miglustat (I) is 128.degree. C. The DSC thermogram and FTIR spectrum of the product are provided as FIG. 1 and FIG. 2, respectively. The crystalline miglustat (I) contained <0.05% of the 5R isomer (IV) as measured by HPLC.

{kind=link}