
Home » Posts tagged 'Monoclonal antibody' (Page 5)
Tag Archives: Monoclonal antibody
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Amgen-AstraZeneca Psoriasis Drug Brodalumab (AMG 827) Hits Phase 3 Endpoints

AstraZeneca and Amgen announced that the Phase 3 AMAGINE-1TM study evaluating brodalumab in patients with moderate-to-severe plaque psoriasis met all primary and secondary endpoints for both evaluated doses.
Brodalumab is a human monoclonal antibody designed for the treatment of inflammatory diseases.[1] It is being tested for the treatment of moderate to severe psoriasis[2] in Phase III clinical trials as of November 2013.[3][4]
Brodalumab was developed by Amgen, Inc.

Mechanism of action
Brodalumab binds to the interleukin-17 receptor and so prevents interleukin 17 (IL-17) from activating the receptor. This mechanism is similar to that of another anti-psoriasis antibody, ixekizumab, which however binds to IL-17 itself.[2]
At present, brodalumab is the only experimental drug in development that inhibits the IL-17 receptor, thus inhibiting several of the IL-17 ligands at once from transmitting signals to the body. Other agents currently in development seek to target the individual IL-17 ligands. By inhibiting the attachment of these ligands with the receptor, brodalumab stops the body from receiving signals that may otherwise cause inflammation and other ailments.
Researchers are currently investigating brodalumab for the treatment of psoriasis (Phase II and planned Phase III), asthma (Phase II), and psoriatic arthritis (Phase II).
Psoriasis is a chronic disease of the immune system that causes the skin cells to grow at a faster rate. Worldwide, the condition affects around 125 million individuals. Even though several types of psoriasis exist, around 80% of sufferers have plaque psoriasis. Plaque psoriasis can cause painful and itchy red, scaly patches to appear on the skin.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Interleukin 17 receptor A |
| Clinical data | |
| Legal status | Investigational |
| Identifiers | |
| CAS number | 1174395-19-7 |
| ATC code | None |
| KEGG | D10061 |
| Chemical data | |
| Formula | C6372H9840N1712O1988S52 |
| Mol. mass | 144.06 kDa |
About Brodalumab (AMG 827)
Brodalumab is a novel human monoclonal antibody that binds to the interleukin-17 (IL-17) receptor and inhibits inflammatory signaling by blocking the binding of several IL-17 ligands to the receptor. By stopping IL-17 ligands from activating the receptor, brodalumab prevents the body from receiving signals that may lead to inflammation. The IL-17 pathway plays a central role in inducing and promoting inflammatory disease processes. In addition to moderate-to-severe plaque psoriasis (Phase 3), brodalumab is currently being investigated for the treatment of psoriatic arthritis (Phase 3) and asthma (Phase 2).
About the Amgen and AstraZeneca Collaboration
In April 2012, Amgen and AstraZeneca formed a collaboration to jointly develop and commercialize five monoclonal antibodies from Amgen’s clinical inflammation portfolio. With oversight from joint governing bodies, Amgen leads clinical development and commercialization for brodalumab (Phase 3 for moderate-to-severe plaque psoriasis and psoriatic arthritis, Phase 2 for asthma) and AMG 557/MEDI5872 (Phase 1b for autoimmune diseases such as systemic lupus erythematosus). AstraZeneca, through its biologics arm MedImmune, leads clinical development and commercialization for MEDI7183/AMG 181 (Phase 2 for ulcerative colitis and Crohn’s disease), MEDI2070/AMG 139 (Phase 2 for Crohn’s disease) and MEDI9929/AMG 157 (Phase 2 for asthma).
About Amgen
Amgen is committed to unlocking the potential of biology for patients suffering from serious illnesses by discovering, developing, manufacturing and delivering innovative human therapeutics. This approach begins by using tools like advanced human genetics to unravel the complexities of disease and understand the fundamentals of human biology.
Amgen focuses on areas of high unmet medical need and leverages its biologics manufacturing expertise to strive for solutions that improve health outcomes and dramatically improve people’s lives. A biotechnology pioneer since 1980, Amgen has grown to be the world’s largest independent biotechnology company, has reached millions of patients around the world and is developing a pipeline of medicines with breakaway potential.
For more information, visit www.amgen.com and follow us on www.twitter.com/amgen.

About AstraZeneca
AstraZeneca is a global, innovation-driven biopharmaceutical business that focuses on the discovery, development and commercialisation of prescription medicines, primarily for the treatment of cardiovascular, metabolic, respiratory, inflammation, autoimmune, oncology, infection and neuroscience diseases. AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. For more information please visit: www.astrazeneca.com.
References
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Brodalumab”. American Medical Association.
- “Neue Antikörper in der Pipeline”. Pharmazeutische Zeitung (in German) (12). 2012.
- ClinicalTrials.gov NCT01708590 Study of Efficacy, Safety, and Withdrawal and Retreatment With Brodalumab in Moderate to Severe Plaque Psoriasis Subjects (AMAGINE-1)
- ClinicalTrials.gov NCT01708629 Study of Efficacy and Safety of Brodalumab Compared With Placebo and Ustekinumab in Moderate to Severe Plaque Psoriasis Subjects (AMAGINE-3)
http://ksclinic.exblog.jp/18270693/
学術面で最初の講演は、米国のJames Krueger教授による「Th1細胞,Th17細胞,Th22細胞が複雑なサイトカインネットワークによって、細胞レベル、分子レベルで乾癬を引き起こす」でした。その要約を示すスライドを幾枚か失敬します(Krueger先生、ごめんなさい)。
 乾 癬の原因究明、病態(病気の起こり方)解明の主役となった免疫学的研究の最先端を行くKrueger先生の、最新情報がコンパクトにまとまった素晴らしい 講演でした。生物学的製剤の治療根拠となるサイトカインネットワークは、現在TipDC – Th17経路によって、きわめて明快に説明されるようになり、Th17細胞が放出するIL17が表皮細胞(ケラチノサイト)の乾癬化を起こします。現在使 用されている抗TNFα製剤、抗IL12/23製剤が、より上流(免疫反応の根っこ)で免疫反応を抑制するのに比べ、IL17はより末梢における乾癬の原 因サイトカインであることから、IL17の抑制は、より乾癬をピンポイントで、そして副作用もミニマムにすることが期待される。
乾 癬の原因究明、病態(病気の起こり方)解明の主役となった免疫学的研究の最先端を行くKrueger先生の、最新情報がコンパクトにまとまった素晴らしい 講演でした。生物学的製剤の治療根拠となるサイトカインネットワークは、現在TipDC – Th17経路によって、きわめて明快に説明されるようになり、Th17細胞が放出するIL17が表皮細胞(ケラチノサイト)の乾癬化を起こします。現在使 用されている抗TNFα製剤、抗IL12/23製剤が、より上流(免疫反応の根っこ)で免疫反応を抑制するのに比べ、IL17はより末梢における乾癬の原 因サイトカインであることから、IL17の抑制は、より乾癬をピンポイントで、そして副作用もミニマムにすることが期待される。
 現在、3種類のIL17抑制薬剤が開発され、治療研究が進められている。
現在、3種類のIL17抑制薬剤が開発され、治療研究が進められている。
①IL17A抗体(Secukinumab Novartis社)
②IL17A抗体(Ixekizumab Lilly社)
③IL17A受容体抗体(Brodalumab Amgen社)


その一つ、Secukinumabの効果(PASI75)=すごく乾癬がよくなる)では、たった3回の注射で90%以上の患者がPASI75を達成する。


PASI90(=乾癬がほとんどなくなる)でみても、60%の患者で達成されている。

Secukinumabの臨床効果。上の段は「プラセボ(偽薬)」、下の段がSecukinumab。

Ixekizumabの効果(PASI90)。約80%の患者で達成されている。驚異的である。

Brodalumabの臨床効果
印象深かった講演をもう一つ、詳細に紹介いたします。
米 国のAnne Bowcock教授の”The genetics of psoriasis: Old risks, novel loci (乾癬の遺伝子研究:昔から言われていた異常、新しく見つかった場所)です。Bowcock教授は、乾癬の原因遺伝子について世界で最初に報告した研究者 です。ここでも少し講演スライドを拝借(Bowcock先生、ごめんなさい)。
 Bowcock 教授は1999年、乾癬家系の詳細な遺伝子調査から第17染色体に乾癬と関わり深い遺伝子異常があることをみつけ、科学雑誌Scienceに報告した。 21世紀を迎える直前のことであり、遠からず乾癬の原因遺伝子が確定し、完治治療を開発することも夢ではないと、当時期待したものでした。
Bowcock 教授は1999年、乾癬家系の詳細な遺伝子調査から第17染色体に乾癬と関わり深い遺伝子異常があることをみつけ、科学雑誌Scienceに報告した。 21世紀を迎える直前のことであり、遠からず乾癬の原因遺伝子が確定し、完治治療を開発することも夢ではないと、当時期待したものでした。
ところが、次々と関連遺伝子はみつけられるものの(現在は30種類以上)、肝心の原因遺伝子、特定のタンパク、メカニズムは不明のままでした。
 Bowcock 教授の息の長い研究は、第17染色体上にあるCARD14と呼ばれるタンパクの、その異常が直接乾癬を起こすことを説き明かしました。CARD14は細胞 膜上にあるタンパクで、細胞外で起こる炎症から生じる様々な刺激物質を、細胞の膜から細胞の中へ伝える役割を果たしています。その伝達経路はNFκBを介 しています(乾癬ではこの経路が活発に動いていることが、高知大学の佐野教授により解明されました)。
Bowcock 教授の息の長い研究は、第17染色体上にあるCARD14と呼ばれるタンパクの、その異常が直接乾癬を起こすことを説き明かしました。CARD14は細胞 膜上にあるタンパクで、細胞外で起こる炎症から生じる様々な刺激物質を、細胞の膜から細胞の中へ伝える役割を果たしています。その伝達経路はNFκBを介 しています(乾癬ではこの経路が活発に動いていることが、高知大学の佐野教授により解明されました)。

遺伝性膿疱性乾癬患者では、このCARD14遺伝子に点突然変異が起こっていることを発見しました。この点突然変異だけで、特殊タイプではありますが、乾癬の原因が特定されたのです。

点突然変異だけではなく、CARD14遺伝子に起こりやすい変異も、ほかの遺伝子異常(PSORS1、MHC遺伝子)、あるいは環境変化が加わると乾癬を引き起こすことも証明しました。
大変感銘深い講演でした。
会議の模様、IFPA代表者会議の報告は、また後日掲載いたします(『2012年9月教室抄録』をご覧ください)。
ブログ「PHOTO & ESSAY」もご覧ください。
FDA Approves Cyramza, ramucirumab (IMC-1121B) for Stomach Cancer
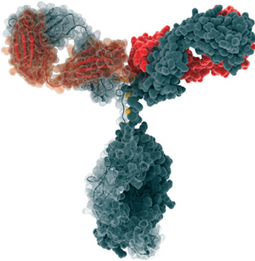
April 21, 2014 — The U.S. Food and Drug Administration today approved Cyramza (ramucirumab) to treat patients with advanced stomach cancer or gastroesophageal junction adenocarcinoma, a form of cancer located in the region where the esophagus joins the stomach.
Stomach cancer forms in the tissues lining the stomach and mostly affects older adults. According to the National Cancer Institute, an estimated 22,220 Americans will be diagnosed with stomach cancer and 10,990 will die from the disease, this year.
Cyramza is an angiogenesis inhibitor that blocks the blood supply to tumors. It is intended for patients whose cancer cannot be surgically removed (unresectable) or has spread (metastatic) after being treated with a fluoropyrimidine- or platinum-containing therapy.
“Although the rates of stomach cancer in the United States have decreased over the past 40 years, patients require new treatment options, particularly when they no longer respond to other therapies,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Cyramza is new treatment option that has demonstrated an ability to extend patients’ lives and slow tumor growth.”
Cyramza’s safety and effectiveness were evaluated in a clinical trial of 355 participants with unresectable or metastatic stomach or gastroesophageal junction cancer. Two-thirds of trial participants received Cyramza while the remaining participants received a placebo. The trial was designed to measure the length of time participants lived before death (overall survival).
Results showed participants treated with Cyramza experienced a median overall survival of 5.2 months compared to 3.8 months in participants receiving placebo. Additionally, participants who took Cyramza experienced a delay in tumor growth (progression-free survival) compared to participants who were given placebo. Results from a second clinical trial that evaluated the efficacy of Cyramza plus paclitaxel (another cancer drug) versus paclitaxel alone also showed an improvement in overall survival.
Common side effects experienced by Cyramza-treated participants during clinical testing include diarrhea and high blood pressure.
The FDA reviewed Cyramza under its priority review program, which provides an expedited review for drugs that have the potential, at the time the application was submitted, to be a significant improvement in safety or effectiveness in the treatment of a serious condition. Cyramza was also granted orphan product designation because it is intended to treat a rare disease or condition.
Cyramza is marketed by Indianapolis-based Eli Lilly.
Source: FDA
old article
Eli Lilly’s third-quarter earnings fell 9 percent compared with last year, when the maker of Cymbalta and Cialis booked a sizeable revenue-sharing payment from a former drug developer partner.
The Indianapolis company beat Wall Street expectations for the quarter and narrowed its earnings forecast for the year.
Lilly also said Wednesday that the U.S. Food and Drug Administration will give its stomach cancer treatment ramucirumab a priority review, which means the drugmaker will learn about its fate inside of eight months rather than a year, which is the norm.
read at
http://www.dddmag.com/news/2013/10/eli-lillys-profit-slides-gets-priority-review
cut paste old article
Eli Lilly and Co. announced that results from the Phase 3 REGARD trial of ramucirumab (IMC-1121B) as a single agent in patients with advanced gastric cancer who have had disease progression after initial chemotherapy were published today in The Lancet. REGARD is the first Phase 3 study with either a single-agent biologic or an anti-angiogenic therapy to show improved overall survival and progression-free survival in advanced gastric cancer patients.
READ ALL AT
Ramucirumab (IMC-1121B)[1] is a fully human monoclonal antibody (IgG1) being developed for the treatment of solid tumors. It is directed against the vascular endothelial growth factor receptor 2 (VEGFR2). By binding to VEGFR2 it works as a receptor antagonist blocking the binding of vascular endothelial growth factor (VEGF) to VEGFR2. VEGFR2 is known to mediate the majority of the downstream effects of VEGF inangiogenesis.
Ramucirumab is being tested in several phase III clinical trials for the treatment of metastatic gastric adenocarcinoma,[2] non-small cell lung cancer,[3] among other types of cancer. On September 26, 2013 Eli Lilly announced that its Phase III study for ramucirumab failed to hit its primary endpoint on progression-free survival among women with metastatic breast cancer.[4][5]
This drug was developed by ImClone Systems Inc. It was isolated from a native phage display library from Dyax.
- Statement On A Nonproprietary Name Adopted By The USAN Council – Ramucirumab, American Medical Association.
- ClinicalTrials.gov NCT01170663 A Study of Paclitaxel With or Without Ramucirumab in Metastatic Gastric Adenocarcinoma (RAINBOW)
- ClinicalTrials.gov NCT01168973 A Study in Second Line Non Small Cell Lung Cancer
- ClinicalTrials.gov NCT00703326 Phase III Study of Docetaxel + Ramucirumab or Placebo in Breast Cancer
- Fierce Biotech. “In another stinging setback, Eli Lilly’s ramucirumab fails PhIII breast cancer study”. Retrieved 27 September 2013.

Glenmark Pharmaceuticals Ltd. through its Swiss Subsidiary receives USD 5 Mn. as milestone fee payment from Sanofi

Glenmark Pharmaceuticals Ltd. through its Swiss Subsidiary receives USD 5 Mn. as milestone fee payment from Sanofi
Total Payment received for GBR 500 monoclonal antibody programme from Sanofi is USD 55 Mn
MUMBAI, April 15, 2014: Glenmark Pharmaceuticals Ltd. has informed the Stock Exchange today that the company through its Swiss subsidiary has received USD 5 million as
milestone payment from Sanofi on a collaboration of its VLA2 (alpha2-beta1) integrin monoclonal antibody. GBR 500 is a first-in-class therapeutic monoclonal antibody for chronicautoimmune disorders.
Glenmark has received from Sanofi already USD 50 Mn as an upfront payment in FY2011-12. Hence, the total amount received by Glenmark from Sanofi for its first in class VLA-2monoclonal antibody is USD 55 million
read at
http://www.moneycontrol.com/stocks/stock_market/corp_notices.php?autono=790416
(copy paste on browser)
MD and CEO Mr Glenn Saldanha
old updates
Glenmark GBR 500 enters into Phase II clinical development for ulcerative colitis
17 September 2012
Glenmark Pharmaceuticals, a wholly-owned subsidiary of Glenmark Pharmaceuticals, has commenced the Phase II study of GBR 500 for ulcerative colitis.
GBR 500, an antagonist of the VLA2 (alpha2-beta1) integrin, is a first-in-class therapeutic monoclonal antibody for chronic autoimmune disorders.
The randomised, double-blind, placebo-controlled study will investigate the efficacy and safety of GBR 500 in patients with moderate to severe ulcerative colitis (UC).
Glenmark Pharmaceuticals chief scientific officer Dr Michael Buschle said that UC represents an area of substantial unmet medical need, despite treatment advances in recent years.
“We’re pleased with the continued progress of our partnership with Sanofi and excited about the commencement of this trial,” Buschle said.
The trial, which will be conducted at multiple clinical sites in North America and Europe, is expected to involve approximately 84 patients.
Patients participating in the study will receive multiple doses of either GBR 500 or placebo, administered over a period of several weeks.
Glenmark has completed Phase I of GBR 500 in the US, won licensing rights to all therapeutic indications from Sanofi and is conducting the clinical development programme.
The trial is part of a strategic global collaboration between Glenmark and Sanofi to investigate GBR 500 for the treatment of chronic inflammatory disorders.
MUMBAI, India, May 16, 2011
Glenmark Pharmaceuticals Out-Licenses Novel Monoclonal Antibody, GBR 500, to Sanofi
Combined Upfront and Potential Development, Regulatory and Commercial Milestone Payments Could Total US$613 Mn
MUMBAI, India, May 16, 2011 /PRNewswire-FirstCall/ — Glenmark Pharmaceuticals S.A (GPSA), a wholly owned subsidiary of Glenmark Pharmaceuticals Limited India (GPL), announced today that it has entered into an agreement with Sanofi to grant Sanofi a license for the development and commercialization of GBR 500, a novel monoclonal antibody for the treatment of Crohn’s Disease and other inflammatory conditions. The transaction is expected to close in the coming month subject to customary closing conditions, including the expiration or early termination of the waiting period under the HSR Antitrust Improvements Act.
Under the terms of the agreement, Glenmark will receive an upfront payment of US$ 50 million, of which US$ 25 million will be paid upon closing of the transaction and US$ 25 million, which is contingent upon Sanofi’s positive assessment of certain data to be provided by Glenmark. In addition, Glenmark could receive potential success-based development, regulatory and commercial milestone payments. The total of these payments could reach US$613 Mn. In addition, Glenmark is eligible to receive tiered double-digit royalties on sales of products commercialized under the license.
GBR 500 is an antagonist of the VLA-2 (alpha2-beta1) integrin. It is a first-in-class therapeutic monoclonal antibody and has established proof of concept in animal models across a range of anti-inflammatory conditions. Glenmark has completed Phase I dosing of GBR 500 in the US and the drug has been well tolerated with a good pharmacokinetic profile. Plans are in place to initiate clinical proof of concept studies in Crohn’s Disease. Sanofi has licensed the rights to all therapeutic indications.
“There continues to be a strong medical need for safer and more efficacious products for the treatment of Inflammatory Diseases,” said Elias Zerhouni, M.D., President, Global Research & Development, Sanofi. “GBR500 brings an innovative approach to Sanofi’s Immuno-Inflammation portfolio, which we believe may address a significant gap in treating Inflammatory Diseases which would be of huge benefit to patients”.
Glenn Saldanha MD and CEO of GPL, “This collaboration on a novel first-in-class monoclonal antibody validates Glenmark’s world-class innovative R&D capabilities in the drug discovery arena. We are pleased to have this second licensing collaboration with Sanofi, one of the largest pharmaceutical companies in the world and the first one from Glenmark in the field of novel biologics”.
Amgen Drug Evolocumab Hits Endpoint of Cholesterol Reduction
 Amgen announced that the Phase 3 TESLA (Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities) trial evaluating evolocumab met its primary endpoint of the percent reduction from baseline at week 12 in low-density lipoprotein cholesterol (LDL-C). The percent reduction in LDL-C, or “bad” cholesterol, was clinically meaningful and statistically significant………….read at
Amgen announced that the Phase 3 TESLA (Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities) trial evaluating evolocumab met its primary endpoint of the percent reduction from baseline at week 12 in low-density lipoprotein cholesterol (LDL-C). The percent reduction in LDL-C, or “bad” cholesterol, was clinically meaningful and statistically significant………….read at
| Monoclonal antibody | |
|---|---|
| Source | Human |
| Target | PCSK9 |
| Clinical data | |
| Legal status | ? |
| Identifiers | |
| CAS number | 1256937-27-5 |
| ATC code | None |
| Chemical data | |
| Formula | C6242H9648N1668O1996S56 |
| Mol. mass | 141.8 kDa |
Evolocumab[1] is a monoclonal antibody designed for the treatment of hyperlipidemia.[2] Evolocumab is a fully human monoclonal antibody that inhibits proprotein convertase subtilisin/kexin type 9 (PCSK9).

PCSK9 is a protein that targets LDL receptors for degradation and thereby reduces the liver’s ability to remove LDL-C, or “bad” cholesterol, from the blood.
Evolocumab, being developed by Amgen scientists, is designed to bind to PCSK9 and inhibit PCSK9 from binding to LDL receptors on the liver surface. In the absence of PCSK9, there are more LDL receptors on the surface of the liver to remove LDL-C from binding to LDL receptors on the liver surface. In the absence of PCSK9, there are more LDL receptors on the surface of the liver to remove LDL-C from the blood.

On 23 January 2014 Amgen announced that the Phase 3 GAUSS-2 (Goal Achievement After Utilizing an Anti-PCSK9 Antibody in Statin Intolerant Subjects-2) trial evaluating evolocumab in patients with high cholesterol who cannot tolerate statins met its co-primary endpoints: the percent reduction from baseline in low-density lipoprotein cholesterol (LDL-C) at week 12 and the mean percent reduction from baseline in LDL-C at weeks 10 and 12. The mean percent reductions in LDL-C, or “bad” cholesterol, compared to ezetimibe were consistent with results observed in the Phase 2 GAUSS study.[3]
The GAUSS-2 trial evaluated safety, tolerability and efficacy of evolocumab in 307 patients with high cholesterol who could not tolerate effective doses of at least two different statins due to muscle-related side effects. Patients were randomized to one of four treatment groups: subcutaneous evolocumab 140 mg every two weeks and oral placebo daily; subcutaneous evolocumab 420 mg monthly and oral placebo daily; subcutaneous placebo every two weeks and oral ezetimibe 10 mg daily; or subcutaneous placebo monthly and oral ezetimibe 10 mg daily.
Safety was generally balanced across treatment groups. The most common adverse events (> 5 percent in evolocumab combined group) were headache (7.8 percent evolocumab; 8.8 percent ezetimibe), myalgia (7.8 percent evolocumab; 17.6 percent ezetimibe), pain in extremity (6.8 percent evolocumab; 1.0 percent ezetimibe), and muscle spasms (6.3 percent evolocumab; 3.9 percent ezetimibe).

Evolocumab, a PCSK9 inhibitor, was safe and effective at lowering low-density lipoprotein cholesterol (LDL-C) after one year of treatment, according to a study published online Nov. 19 inCirculation and presented simultaneously at the American Heart Association scientific session in Dallas.
The Open-Label Study of Long-term Evaluation Against LDL-C (OSLER) trial took place at 156 study centers around the world that participated in at least one of four phase 2 studies of between October 2011 and June 2012. Evolocumab is a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor made by Amgen.
Investigators led by Michael J. Koren, MD, of the Jacksonville Center for Clinical Research in Florida, randomized 1,104 participants in a 2:1 ratio to receive either evolocumab (420 mg every four weeks) plus standard-of-care therapy (based on guidelines for treatment of hypercholesterolemia) or evolocumab alone, which served as the control. After 12 weeks, lipid results were unblinded and investigators were able to adjust standard-of-care therapy in either group.
The main efficacy objective was to determine the effects of longer-term evolocumab therapy on cholesterol levels and the main safety endpoints included incidence of adverse events, serious adverse events and adverse events resulting in discontinuation of the drug.
Patients who received evolocumab for the first time in the OSLER study had an average LDL-C reduction of 52.3 percent at one year. Patients previously dosed with evolocumab in a prior trial and were in the evolocumab and standard-of-care group in OSLER had an average LDL-C reduction of 52.1 percent at the end of the study compared with 50.4 percent at baseline. Patients who terminated evolocumab when they entered OSLER had their LDL-C levels returned to around their baseline.
Adverse events occurred in 73.1 percent of the standard-of-care group and 81.4 percent of the evolocumab plus standard-of-care group. The researchers determined that 5.6 percent of adverse events were related to evolocumab. Serious adverse events occurred in 6.3 percent of the control group and 7.1 percent in the combination group.
The authors explained that their findings offer more insight into the use of this class of drugs to lower LDL-C in at-risk patients.
“Challenging patients such as those who fail to reach current lipid goals despite maximum doses of highly effective statin agents or those with well-documented statin intolerance are thus logical populations for treatment with PCSK9 inhibitors,” they concluded.

References
- World Health Organization (2012). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 108” (PDF). WHO Drug Information 26 (4).
- Statement On A Nonproprietary Name Adopted By The USAN Council – Evolocumab
- Estel Grace Masangkay, “Amgen Phase III GAUSS-2 Trial of Evolocumab (AMG 145) Meets Co-Primary Endpoints Of LDL Cholesterol Reduction”, Bioresearch Online (January 24 2014)


Late-stage success for Sanofi/Regeneron RA drug sarilumab

SARILUMAB
PRONUNCIATION sar il’ ue mab
THERAPEUTIC CLAIM Treatment of rheumatoid arthritis and
ankylosing spondylitis
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human interleukin 6 receptor α) (human REGN88 heavy
chain), disulfide with human REGN88 light chain, dimer
2. Immunoglobulin G1, anti-(human interleukin-6 receptor subunit alpha (IL-6RA,
membrane glycoprotein 80, CD126)); human monoclonal RGN88 γ1 heavy chain (219-
214′)-disulfide with human monoclonal RGN88 κ light chain dimer (225-225”:228-
228”)-bisdisulfide
MOLECULAR FORMULA C6388H9918N1718O1998S44
MOLECULAR WEIGHT 144.13 kDa
SPONSOR Regeneron Pharmaceuticals, Inc.
CODE DESIGNATION REGN88, SAR153191
CAS REGISTRY NUMBER 1189541-98-7
sarilumab
Sarilumab (REGN88/SAR153191) is a fully-human monoclonal antibody directed against the IL-6 receptor (IL-6R). Sarilumab is a subcutaneously delivered inhibitor of IL-6 signaling, which binds with high affinity to the IL-6 receptor. It blocks the binding of IL-6 to its receptor and interrupts the resultant cytokine-mediated inflammatory signaling.
Sanofi and Regeneron’s investigational rheumatoid arthritis drug sarilumab has succeeded in a late-stage trial.
The year-long Phase III study enrolled 1,200 patients with active, moderate-to-severe RA who were inadequate responders to methotrexate. Patients were randomised to one of three subcutaneous treatment groups, all in combination with MTX and dosed every other week – sarilumab 200mg, 150mg or placebo.http://www.pharmatimes.com/Article/13-11-22/Late-stage_success_for_Sanofi_Regeneron_RA_drug.aspx
Sarilumab is a human monoclonal antibody against the interleukin-6 receptor.
Regeneron and Sanofi are currently co-developing the drug for the treatment of rheumatoid arthritis, for which it is in phase III trials. Development inankylosing spondylitis has been suspended after the drug failed to show clinical benefit over methotrexate in a phase II trial.[1][2]
On May 15th, 2013, both companies announced that 2 new trials were starting (COMPARE and ASCERTAIN) and the first patients had already been enrolled.[3]
On November 22nd, 2013, both companies On May 15th, 2013, both companies announced positive phase 3 results for the RA-MOBILITY trial
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Sarilumab”. American Medical Association.
- http://investor.regeneron.com/releasedetail.cfm?releaseid=590869
- http://en.sanofi.com/Images/33027_20130515_sari_en.pdf
fully human monoclonal antibody directed against the interleukin-6 receptor (IL-6R) in combination with methotrexate (MTX) therapy improved disease signs and symptoms as well as physical functionw while inhibiting progression of joint damage in adults with RA who saw little improvement through MTX therapy alone.
Sarilumab met all three primary endpoints of the 52-week SARIL-RA-MOBILITY Phase III trial by demonstrating clinically relevant and statistically significant improvements compared to the placebo group in the two groups treated with the drug candidate. The trial enrolled about 1,200 patients with active, moderate-to-severe rheumatoid arthritis who were inadequate responders to MTX therapy.
Of patients treated with the 200 mg dose of sarilumab plus MTX, 66% saw improvement in signs and symptoms of RA at 24 weeks, as measured by the American College of Rheumatology score of at-least 20% improvement. The percentage dipped to 58% of sarilumab 150 mg dose patients, and 33% of placebo patients.
Sarilumab 200 mg patients showed the least progression of structural damage after 52 weeks, registering a 0.25 change in the modified Van der Heijde total Sharp score. That contrasts with scores of 0.90 in patients taking sarilumab 150 mg, and 2.78 in the placebo group.
In addition, sarilumab 200 mg patients showed improvement in physical function, as measured by change from baseline in the Health Assessment Question-Disability at week 16. However, the companies did not quantify those results in their announcement. Sanofi and Regeneron said additional analyses of efficacy and safety data from SARIL-RA-MOBILITY will be presented “at a future medical conference.”
“We are encouraged by these Phase III results and the impact sarilumab demonstrated on inhibition of progression of structural damage assessed radiographically in this study,” Tanya M. Momtahen, Sanofi’s sarilumab global project head, said in a statement.
Sarilumab—known as SAR153191 and REGN88—blocks the binding of IL-6 to its receptor and interrupts the resultant cytokine-mediated inflammatory signaling characteristic of RA. Sarilumab was developed using Regeneron’s VelocImmune® antibody technology.
The positive results continue what has been mostly strong success in clinical trials for the partners, whose development collaborations include alirocumab (REGN727), dupilumab (REGN668), and enoticumab (REGN421). Alirocumab is a PCSK9 antibody being evaluated for its ability to manage LDL cholesterol, including in people who do not get to their target LDL levels using statin medicines alone. Dupilumab is an antibody to the receptors for interleukin-4 and interleukin-13 under evaluation in atopic dermatitis and eosinophilic asthma. Enoticumab is a fully human monoclonal antibody to delta-like ligand-4 (Dll4) now in Phase I study for advanced malignancies.
On its own, however, Sanofi’s R&D efforts have shown more mixed results, with the pharma giant earlier this month ending development of cancer drug candidate fedratinib (SAR302503) after it was placed on clinical hold by the FDA following reports that some patients in clinical trials developed symptoms consistent with Wernicke’s encephalopathy. Another cancer compound, iniparib, had its development halted earlier this year after a disappointing Phase III trial.
Necitumumab

Necitumumab
Necitumumab is a fully human IgG1 monoclonal antibody designed to block the ligand binding site of the human epidermal growth factor receptor (EGFR), which is a target in several anti-cancer treatments because it sparks cancer progression, both by promoting angiogenesis, or the formation of new blood vessels for tumors, and by inhibiting apoptosis, or cell death. Recently approved therapies for non-squamous NSCLC, including afatinib and erlotinib, target specific EGFR mutations, but those drugs are used to treat patients with nonsquamous histology.Lilly did not provide specific data regarding the results of the trial, but the company announced that it plans to present that data at a scientific meeting next year, and to request a review of the drug by regulatory authorities before the end of 2014.
Necitumumabis one of three monoclonal antibodies in Phase III
development that targets EGFR, the target of the approved antibodies
cetuximab and panitumumab. However, necitumumab is a fully human
IgG1 antibody, distinguishing it from both the approved agents.
Necitumumab is directed against the ligand binding site of EGFR and is
being co-developed by Eli Lilly and Bristol-Myers Squibb in the United
States, Canada, and Japan, while Eli Lilly alone is developing it for other
markets. Necitumumabfirst entered clinical development in 2004 and
is now in Phase III development for the treatment of non–small-cell
lung cancer and Phase II for the treatment of colorectal cancer. The
primary indication chosen further distinguishes necitumumabfrom both
cetuximab and panitumumab, but it is an indication for which EGFR
kinase inhibitors such as erlotinib are approved.
In December 2009, Eli Lilly stressed the long half-life of necitumumab
(7–10 days, which permits dosing at 2–3 week intervals) and its potential
both for reduced hypersensitivity reactions (i.e., better tolerability) and
for induced host-mediated anticancer activity. In addition, it highlighted
that necitumumabdisplays similar or superior activity to cetuximab
in anticancer models. Preliminary data were presented from the Phase
II study in colorectal cancer showing antitumor activity in 73% of 44
patients treated with necitumumabplus FOLFOX.
Both Phase III studies in non–small-cell lung cancer are in stage IV
disease and in groups of 947 patients treated with necitumumabplus
cisplatin and a second agent. The INSPIRE study in non-squamous
disease began in November 2009 and uses pemetrexed as the second
drug, while the SQUIRE study commenced in January 2010 in
squamous disease and uses gemcitabine. Both studies have primary
completion dates in late 2011 and study completion dates of mid-2012,
which points to BLA submission in 2013.

A Phase I study in patients with solid tumors suggested that skin
toxicity was the dose-limiting toxicity and suggested that 800 mg of
necitumumab (at weekly or fortnightly intervals) be the maximum dose
(Kuenen et al. 2010).16 This dose was employed in the initial colorectal
cancer study, at 14-day intervals, which revealed a 60% partial response
(Taberno et al. 2008).17
The development strategy for necitumumab appears to have been
designed to establish it initially in a major indication where it will not
be competing with established antibody products, while seeking
to exploit the reported advantages over cetuximab appears to be
a secondary priority. While the reported Phase II data are very
encouraging, it will be some time before a better assessment of the
commercial prospects of necitumumab can be made. However, it does
appear to have significant potential.
Necitumumab (proposed INN) is a monoclonal antibody and an antineoplastic. It binds to the epidermal growth factor receptor(EGFR).[1] As of October 2009, two Phase III clinical trials are planned to investigate its effects on non-small cell lung carcinoma.[2][3]
- International Nonproprietary Names for Pharmaceutical Substances (INN, prepublication copy), World Health Organization.
- ClinicalTrials.gov NCT00981058 Squamous Non-Small Cell Lung Cancer (NSCLC) Treatment With the Inhibitor of Epidermal Growth Factor Receptor (EGFR) (SQUIRE)
- ClinicalTrials.gov NCT00982111 NonSquamous Non-Small Cell Lung Cancer Treatment With the Inhibitor of Epidermal Growth Factor Receptor (INSPIRE)
Possible Efficacy Of Lilly’s Necitumumab (IMC-11F8) In Lung Cancer Subset
18.4 2013
Eli Lilly announced yesterday their very preliminary and non-quantitative conclusions on the SQUIRE study, a 1093-patient Phase III trial of their anti-epidermal growth factor receptor (EGFR) antibody, necitumumab (IMC-11F8), against Stage IV squamous, non-small cell lung carcinoma (NSCLC).http://www.forbes.com/sites/davidkroll/2013/08/14/possible-efficacy-of-necitumumab-imc-11f8-in-squamous-nsclc-lung-cancer-subset/

Reslizumab
Reslizumab(CINQUIL) is a humanized monoclonal antibody
targeted against IL-5 that is being developed by Cephalon for the
treatment of eosinophilic asthma. In September 2010, Cephalon
indicated that it hopes to file a BLA in 2013, focusing on this subset of
severe asthmatics. Such patients are ca. 30% of the asthmatic population,
with the 750,000 patients in the United States suggested to offer the potential for peak market sales of $1 billion. However, previous
attempts to develop recombinant IL-5 antagonists for the treatment of asthma saw very disappointing clinical results with both mepolizumab
(GlaxoSmithKline) and reslizumab (Schering-Plough and Celltech).
Schering-Plough (now Merck) had been developing reslizumab in
partnership with Celltech (now UCB), utilizing the latter’s antibody
technology, but terminated development in 2002 after disappointing
clinical results. The rights were acquired by Ception Therapeutics in
2007, with development reinitiated for both pediatric eosinophilic
esophagitis and eosinophilic asthma. Cephalon acquired an option to
acquire Ception in January 2009 and exercised this option in April 2010
despite unpromising results in the Phase II/III study of reslizumab in
pediatric eosinophilic esophagitis patients.
In its November 2009 R&D presentation, Cephalon presented data
(from Schering-Plough) showing that reslizumab treatment of asthmatics
results in a sustained suppression of eosinophil levels; the protocols
employed in the Phase II/III study in pediatric eosinophilic esophagitis
and a Phase II study in eosinophilic asthma were described. The Phase
III study in asthmatics has yet to commence, but a 190-patient openlabel Phase III extension study in eosinophilic esophagitis is ongoing.
The Phase II/III study showed no discernable symptom improvement
despite suppression of eosinophil levels at all three doses tested (see
Walsh 2010).43 The outcome of the 106-patient Phase II study, in
February 2009, in asthmatics prompted Cephalon to acquire Ception.
Reslizumab treatment produced significant improvement in lung
function and reduced airway inflammation.
Reslizumab is currently the most advanced of three anti–IL-5monoclonal antibodies in development, but the 2013 submission date for a BLA seems optimistic given that Phase III studies have yet to start.
Mepolizumab is now in Phase II studies for the treatment of severe
asthma and nasal polyposis (having previously been filed for approval
in Europe for the treatment of hypereosinophilic syndrome), but the
filing was withdrawn and development for that indication discontinued
in late 2009. MedImmune and Kyowa Hakko Kirin’s benralizumab has
successfully completed a Phase IIa study in asthma with data presented
in September 2010, and a 108-patient study in asthma was completed
in October 2010. A similar-size Phase II study in COPD commenced in
November 2010.
Reslizumab is a humanized monoclonal antibody intended for the treatment of eosinophil-meditated inflammations of the airways, skin and gastrointestinal tract.[1] As of September 2009, the drug is undergoing Phase II/III clinical trials.[2]
- Walsh, GM (2009). “Reslizumab, a humanized anti-IL-5 mAb for the treatment of eosinophil-mediated inflammatory conditions”. Current opinion in molecular therapeutics11 (3): 329–36. PMID 19479666.
- ClinicalTrials.gov
Solanezumab, Eli Lilly’s anti-beta-amyloid monoclonal antibody for Alzheimer’s disease

- immunoglobulin G1-kappa, anti-[Homo sapiens amyloid-beta (Abeta)
peptide soluble monomer], humanized monoclonal antibody;
gamma1 heavy chain [humanized VH (Homo sapiens IGHV3-23*04
(87.60%) -(IGHD)-IGHJ4*01) [8.8.5] (1-112) -Homo sapiens
IGHG1*01, CH3 K130>del (113-441)], (215-219′)-disulfide with
kappa light chain (1’-219’) [humanized V-KAPPA (Homo sapiens
IGKV2-30*01 (90.00%) -IGKJ1*01) [11.3.9] (1′-112′) -Homo sapiens
IGKC*01 (113′-219′)]; (221-221″:224-224″)-bisdisulfide dimer
neuroprotective agent
C6396H9922N1712O1996S42 955085-14-0
Heavy chain / Chaîne lourde / Cadena pesada
EVQLVESGGG LVQPGGSLRL SCAASGFTFS RYSMSWVRQA PGKGLELVAQ 50
INSVGNSTYY PDTVKGRFTI SRDNAKNTLY LQMNSLRAED TAVYYCASGD 100
YWGQGTLVTV SSASTKGPSV FPLAPSSKST SGGTAALGCL VKDYFPEPVT 150
VSWNSGALTS GVHTFPAVLQ SSGLYSLSSV VTVPSSSLGT QTYICNVNHK 200
PSNTKVDKKV EPKSCDKTHT CPPCPAPELL GGPSVFLFPP KPKDTLMISR 250
TPEVTCVVVD VSHEDPEVKF NWYVDGVEVH NAKTKPREEQ YNSTYRVVSV 300
LTVLHQDWLN GKEYKCKVSN KALPAPIEKT ISKAKGQPRE PQVYTLPPSR 350
DELTKNQVSL TCLVKGFYPS DIAVEWESNG QPENNYKTTP PVLDSDGSFF 400
LYSKLTVDKS RWQQGNVFSC SVMHEALHNH YTQKSLSLSP G 441
Light chain / Chaîne légère / Cadena ligera
DVVMTQSPLS LPVTLGQPAS ISCRSSQSLI YSDGNAYLHW FLQKPGQSPR 50
LLIYKVSNRF SGVPDRFSGS GSGTDFTLKI SRVEAEDVGV YYCSQSTHVP 100
WTFGQGTKVE IKRTVAAPSV FIFPPSDEQL KSGTASVVCL LNNFYPREAK 150
VQWKVDNALQ SGNSQESVTE QDSKDSTYSL SSTLTLSKAD YEKHKVYACE 200
VTHQGLSSPV TKSFNRGEC 219
Disulfide bridges location / Position des ponts disulfure / Posiciones de los puentes disulfuro
Intra-H 22-96 139-195 256-316 362-420
22”-96” 139”-195” 256”-316” 362”-420”
Intra-L 23′-93′ 139′-199′
23”’-93”’ 139”’-199”’
Inter-H-L 215-219′ 215”-219”’
Inter-H-H 221-221” 224-224”
N-glycosylation sites / Sites de N-glycosylation / Posiciones de N-glicosilación
292, 292
Solanezumab, Eli Lilly’s anti-beta-amyloid monoclonal antibody for Alzheimer’s disease
The market for Alzheimer’s disease therapies is set to nearly triple between 2012 and 2022, despite increasing genericisation and the fact that few new product launches are expected during this time, according to new forecasts.
The key driver of growth in the AD market will be Eli Lilly’s anti-beta-amyloid monoclonal antibody solanezumab, the first potentially disease-modifying therapy (DMT) to launch for AD, according to the study, from Decision Resources. It reports that solanezumab is expected to launch in the seven major pharmaceutical markets – the US, France, Germany, Italy, Spain, the UK and Japan – starting in 2018 and that, by 2022, the drug is forecast to attain sales in excess of $5 billion in these markets.
More than 85% of solanezumab’s projected total use in 2022 will be in the mild AD market – the population in which the drug is currently being tested – followed by the pre-AD 1-2 years market segment, says the firm, which defines this latter population as those patients who will go on to develop overt AD within the next one to two years.
Solanezumab (proposed INN) is a monoclonal antibody being investigated by Eli Lilly as a neuroprotector[1] for patients withAlzheimer’s disease.[2][3]
It binds to the amyloid-β peptides that make up the protein plaques seen in the brains of people with the disease.
2012 results of the EXPEDITION 1 & 2 phase 3 clinical trials were only mildly encouraging.[4][5][6] but were said to be the “first evidence that targeting the amyloid cascade can slow the progression of disease.”[7]
- International Nonproprietary Names for Pharmaceutical Substances (INN, prepublication copy), World Health Organization.
- ClinicalTrials.gov NCT00749216 Solanezumab Safety Study in Japanese Patients With Alzheimer’s Disease
- ClinicalTrials.gov NCT00905372 Effect of LY2062430 on the Progression of Alzheimer’s Disease (EXPEDITION)
- “Lilly’s Solanezumab Slows Down Alzheimer’s Progression”. 9 Oct 2012.
- Solanezumab Did it actually work
- “Eli Lilly’s solanezumab faces grim prospects of attaining conditional FDA approval in mild Alzheimer’s”. 4 Sep 2012.
- “ALZHEIMER’S DRUG SLOWS MEMORY LOSS BY ONE THIRD”. 10 Oct 2012.

yellow coloured SOLANEZUMAB blocks beta amyloid from aa 16 to aa 25

Amyloid precursor protein (APP)
FDA approves Gazyva for chronic lymphocytic leukemia
Drug is first with breakthrough therapy designation to receive FDA approval
The U.S. Food and Drug Administration today approved Gazyva (obinutuzumab) for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia (CLL).
read all at
http://www.pharmalive.com/fda-approves-roche-s-gazyva
my old article cut paste
Roche’s new leukaemia drug, Obinutuzumab, superior to Rituxan in clinical trial
JULY 25, 2013 12:52 AM / 6 COMMENTS / EDIT
 July 24 2013 | By Márcio Barra
July 24 2013 | By Márcio Barra
Roche has announced that its experimental leukemia drug GA101, or obinutuzumab, used in combination with chemotherapy, was better than Rituxan at helping people with chronic lymphocytic leukemia live longer without their disease worsening, according to the results from the second phase of the clinical trial. Both drugs were tested and compared in combination with chlorambucil.
Roche’s Phase III leukemia drug Obinutuzumab (GA101) yields positive results
- GA101 is the first glycoengineered, type II anti-CD20 mAb.

Roche’s Phase III leukemia drug Obinutuzumab (GA101) yields positive results
Obinutuzumab (GA101)
| FORMULA | C6512H10060N1712O2020S44 |
|---|
GA101 is the first glycoengineered, type II anti-CD20 monoclonal antibody (mAb) that has been designed for increased antibody-dependent cellular cytotoxicity (ADCC) and Direct CellDeath.1 This agent is being investigated in collaboration with Biogen Idec.
Swiss pharmaceutical company Roche has announced that its early Phase III trial of Leukemia drug obinutuzumab (GA101) demonstrated significantly improved progression-free survival in people with chronic lymphocytic leukemia (CLL).
The positive results yield from stage 1 of a three-arm study called CLL11, designed to investigate the efficacy and safety profile of obinutuzumab (GA101) plus chlorambucil, a chemotherapy, compared with chlorambucil alone in people with previously untreated chronic lymphocytic leukemia (CLL).
This phase of the study met its primary endpoint and an improvement in progression-free survival was achieved; obinutuzumab plus chlorambucil significantly reduced the risk of disease worsening or death compared to chlorambucil alone.
Roche chief medical officer and global product development head Hal Barron said; “the improvement in progression-free survival seen with GA101 is encouraging for people with CLL, a chronic illness of older people for which new treatment options are needed.”
“GA101 demonstrates our ongoing commitment to the research and development of new medicines for this disease.”
Obinutuzumab is Roche’s most advanced drug in development for the treatment of hematological malignancies.
It has been specifically designed as the first glycoengineered, type 2 anti-CD20 monoclonal antibody in development for B cell malignancies.
Afutuzumab is a monoclonal antibody being developed by Hoffmann-La Roche Inc. for the treatment of lymphoma.[1] It acts as an immunomodulator.[2][3] It was renamed obinutuzumab in 2009.[4]
References
- Robak, T (2009). “GA-101, a third-generation, humanized and glyco-engineered anti-CD20 mAb for the treatment of B-cell lymphoid malignancies”. Current opinion in investigational drugs (London, England : 2000) 10 (6): 588–96. PMID 19513948.
- Statement On A Nonproprietary Name Adopted By The Usan Council – Afutuzumab,American Medical Association.
- International Nonproprietary Names for Pharmaceutical Substances (INN), World Health Organization.
- International Nonproprietary Names for Pharmaceutical Substances (INN), World Health Organization.
-
OBINUTUZUMAB ISMONOCLONAL ANTIBODY TYPE Whole antibody SOURCE Humanized (from mouse) TARGET CD20

{kind=link}