
Home » Posts tagged 'MGCD516'
Tag Archives: MGCD516
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sitravatinib
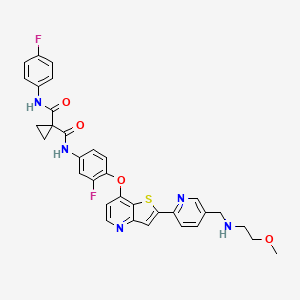

Sitravatinib
1-N‘-[3-fluoro-4-[2-[5-[(2-methoxyethylamino)methyl]pyridin-2-yl]thieno[3,2-b]pyridin-7-yl]oxyphenyl]-1-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide
| シトラバチニブ; ситраватиниб , سيترافاتينيب , 司曲替尼 , |
| Formula | C33H29F2N5O4S |
|---|---|
| Cas | 1123837-84-2 |
| Mol weight | 629.6763 |
MGCD516
Antineoplastic, Receptor tyrosine kinase inhibitor
Sitravatinib (MGCD516) is an experimental drug for the treatment of cancer. It is a small molecule inhibitor of multiple tyrosine kinases.
Sitravatinib is being developed by Mirati Therapeutics.[1]
Ongoing phase II trials include a trial for liposcarcoma,[2] a combination trial for non-small cell lung cancer,[3] and a combination trial with nivolumab for renal cell carcinoma.[4]
Mirati Therapeutics and licensee BeiGene are developing sitravatinib, an oral multitargeted kinase inhibitor which inhibits Eph, Ret, c-Met and VEGF-1, -2 and -3, DDR, Trk, Axl kinases, CHR4q12, TYRO3 and Casitas B-lineage, in combination with immune checkpoint inhibitors, for treating advanced solid tumors.
In March 2021, sitravatinib was reported to be in phase 3 clinical development.

PDT PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009026717
WO2009026717 , in which sitravatinib was first disclosed, claiming heterocyclic compounds as multi kinase inhibitors.
Scheme 10
Example 52
N-(3-Fluoro-4-(2-(5-((2-methoxyethylamino)methyl)pyridin-2-yl)thieno[3,2-b]pyridin-7- yloxy)phenyl)-N-(4-fluorophenyl)cyclopropane- 1 , 1 -dicarboxamide
Step 1 : tert-Butyl (6-(7-(2-Fluoro-4-(1-(4-fluorophenylcarbamoyl)-cyclopropanecarboxamido)phenoxy)thieno [3 ,2-b]pyridin-2-yl)pyridin-3 -y l)methyl(2-methoxyethyl)carbamate (146)
To aniline 126 (0.58 g, 1.1 mmol) and DIPEA (0.58 mL, 0.43 g, 3.3 mmol) in dry DMF
(20 mL) was added 1-(4-fluorophenylcarbamoyl)cyclopropanecarbpxylic acid (0.35 g, 1.5 mmol) and HATU (0.72 g, 1.9 mmol) and the mixture was stirred at r.t. for 18 h. It was then partitioned between ethyl acetate and water, the organic phase was washed with water, IM NaOH, brine, dried (MgSO4), filtered, and concentrated. Silica gel chromatography (ethyl acetate) afforded title compound Ϊ46 (0.60 g, 74 % yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.40 (s, 1H), 10.01 (s, 1H), 8.52-8.49 (m, 2H), 8.33 (s, 1H), 8.27-8.24 (m, 1H), 7.92-7.88 (m, 1H), 7.78 (dd, J = 8.2, 2.1 Hz, 1H) 7.65-7.60 (m, 2H), 7.52-7.42 (m, 2H), 7.14 (t, J = 8.8 Hz, 2H), 6.65 (d, J = 5.1 Hz 1H), 4.47 (s, 2H), 3.42-3.30 (m, 4H), 3.22 (s, 3H), 1.46-1.30 (m, 13H). MS (m/z): 730.1 (M+H).
Step 2. N-(3-Fluoro-4-(2-(5-((2-methoxyethylamino)methyl)pyridin-2-yl)thieno[3,2-blpyridin-7-yloxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (147)
To the compound 146 (0.59 g, 0.81 mmol) in dichloromethane (50 mL) was added TFA (3 mL). The solution was stirred for 18 h then concentrated. The residue was partitioned between dichloromethane and 1 M NaOH, and filtered to remove insolubles. The organic phase was collected, washed with IM NaOH, brine, dried (MgSO4), filtered, and concentrated to afford title compound 147 (0.35 g, 69 % yield).
1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.40 (s, 1H), 10.01 (s, 1H), 8.55 (d, J = 1.6 Hz, 1H), 8.51 (d, J = 5.3 Hz, 1H), 8.31 (s, 1H), 8.22 (d, J = 8.0 Hz, 1H), 7.92-7.87 (m, 2H), 7.65-7.61 (m, 2H), 7.52-7.43 (m, 2H), 7.17-7.12 (m, 2H), 6.64 (d, J = 5.5 Hz, 1H), 3.77 (s, 2H), 3.40 (t, J = 5.7 Hz, 2H), 3.23 (s, 3H), 2.64 (t, J = 5.7 Hz, 2H), 1.46 (br s, 4H). MS (m/z): 630.1 (M+H).
PATENT
WO 2009026720
https://patents.google.com/patent/WO2009026720A1
PATENT
WO-2021050580
Novel, stable crystalline polymorphic forms (form D) of sitravatinib , useful for treating a multi tyrosine kinase-associated cancer eg sarcoma, glioma, non-small cell lung, bladder, kidney, ovarian, gastric, breast or liver cancer.
International publication No. W02009/026717A disclosed compounds with the inhibition activities of multiple protein tyrosine kinases, for example, the inhibition activities of VEGF receptor kinase and HGF receptor kinase. In particular, disclosed N-(3-fluoro-4-((2-(5-(((2-methoxyethyl)amino)methyl)pyridin-2-yl)thieno[3,2-b]pyridin-7-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane- 1,1 -di carboxamide (Compound 1) is a multi-tyrosine kinase inhibitor with demonstrated potent inhibition of a closely related spectrum of tyrosine kinases, including RET, CBL, CHR4ql2, DDR and Trk, which are key regulators of signaling pathways that lead to cell growth, survival and tumor progression.
[003]
Compound 1
[004] Compound 1 shows tumor regression in multiple human xenograft tumor models in mice, and is presently in human clinical trials as a monotherapy as well as in combination for
treating a wide range of solid tumors. Compound 1 is presently in Phase 1 clinical trial for patients with advanced cancer, in Phase 2 studies for patients with advanced liposarcoma and non-small cell lung cancer (NSCLC).
[005] The small scale chemical synthesis of the amorphous Compound 1 had been disclosed in the Example 52 (compound 147) of W02009/026717A, however, in order to prepare the API of Compound 1 with high quality and in large quantity, crystalline forms of Compound 1 would be normally needed so the process impurities could be purged out by recrystallization.
Practically, it is difficult to predict with confidence which crystalline form of a particular compound will be stable, reproducible, and suitable for phamaceutical processing. It is even more difficult to predict whether or not a particular crystalline solid state form will be produced with the desired physical properties for pharmaceutical formulations.
[006] For all the foregoing reasons, there is a great need to produce crystalline forms of Compound 1 that provide manufacturing improvements of the pharmaceutical composition.
The present invention advantageously addresses one or more of these needs.
EXAMPLE 1
Preparation of N-(3-fluoro-4-((2-(5-(((2-methoxyethyl)amino)methyl)pyridin-2- yl)thieno[3,2-b]pyridin-7-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-l,l- dicarboxamide (Compound 1)
[0085] This Example illustrates the preparation ofN-(3-fluoro-4-((2-(5-(((2-methoxyethyl)amino)methyl)pyridin-2-yl)thieno[3,2-b]pyridin-7-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane- 1,1 -di carboxamide (Compound 1).
[0086] Step 1: N-(Y6-bromopyridin-3-vDmethvD-2-methoxyethan-l-amine (Compound 1A)
Compound 1A
[0087] To a stirred solution of 2-Methoxyethylamine (3.0 eq) in dichloromethane (DCM) (12 vol) was added Molecular sieves (0.3 w/w) and stirred for 2 hours at 25±5°C under nitrogen atmosphere. The reaction mass water content was monitored by Karl Fischer analysis until the water content limit reached 0.5 % w/w. Once the water content limit was reached, the reaction mass cooled to 5±5°C and 6-bromonicotinaldehyde (1.0 eq) was added lot wise over period of 30 minutes to the above reaction mass at 5±5°C. The reaction mass was stirred for 30±5 minutes at 5±5°C and acetic acid (1.05 eq) was added drop wise at 5±5°C. After completion of the addition, the mass was slowly warmed to 25±5°C and stirred for 8 h to afford Compound 1 A. The imine formation was monitored by HPLC.
[0088] Step 2: tert-butyl (Y6-brom opyri din-3 -vQmethvO(2-m ethoxy ethvDcarbamate (Compound
IB)
Compound 1B
[0089] Charged Compoud 1A (1.0 eq) in THF (5.0 vol) was added and the reaction mass was stirred for 30 minutes at 25±5°C under nitrogen atmosphere. The reaction mass was cooled to temperature of about 10±5°C. Di-tert- butyl dicarbonate (1.2 eq) was added to the reaction mass at 10±5°C under nitrogen atmosphere and the reaction mass temperature was raised to 25±5°C and the reaction mass for about 2 hours. The progress of the reaction was monitored by HPLC. After IPC completion, a prepared solution of Taurine (1.5 eq) in 2M aq NaOH (3.1 vol) was charged and stirred at 10±5°C for 16 h to 18 h. The reaction mass was further diluted with 1M aq.NaOH solution (3.7 vol) and the layers were separated. The aqueous layer was extracted with DCM (2 x 4.7vol) and the extract combined with the organic layer. The combined organic layers were washed with 1M aq.NaOH solution (3.94 vol), followed by water (2×4.4 vol), and dried over sodium sulfate (2.0 w/w) . The filtrate was concentrated under reduced pressure below 40° C until no distillate was observed. Tetrahydrofuran (THF) was sequentially added (1×4 vol and lx 6vol) and concentrated under reduced pressure below 40°C until no distillate was observed to obtained Compound IB as light yellow colored syrup liquid.
[0090] Step 3: tert-butyl 7-chlorothieno[3.2-b1pyridin-2-yl)pyridin-3-yl )methyl)(2-
methoxyethvDcarbamate (Compound 1C)
Compound 1C
[0091] To a stirred solution of 7-chlorothieno[3,2-b]pyridine (1.05 eq) in tetrahydrofuran (7 vol) was added n-butyl lithium (2.5 M in hexane) drop wise at -15±10°C and stirred for 90 minutes at same temperature under nitrogen atmosphere. Zinc chloride (1.05 eq) was added to the reaction mass at -15±10°C. The reaction mass was slowly warmed to 25±5°C and stirred for 45 minutes under nitrogen atmosphere to afford Compound 1C. The progress of the reaction was monitored by HPLC.
[0092] Step 4: tert-butyl (Y6-(7-(4-amino-2-fluorophenoxy)thieno[3.2-b1pyridin-2-v0pyridin-3-vDmethvD(2-methoxyethvDcarbamate (Compound ID)
Compound 1D
[0093] 3-fluoro-4-hydroxybenzenaminium chloride (1.2 eq) in DMSO (3.9 vol) at 25±5°C was charged under nitrogen atmosphere and the reaction mass was stirred until observance of a clear solution at 25±5°C. t-BuOK was added lot wise under nitrogen atmosphere at 25±10°C. The reaction mass temperature was raised to 45±5°C and maintained for 30 minutes under nitrogen atmosphere. Compound 1C was charged lot-wise under nitrogen atmosphere at 45±5°C and stirred for 10 minutes at 45± 5°C.The reaction mixture was heated to 100± 5°C and stirred for 2 hrs. The reaction mass is monitored by HPLC.
[0094] After reaction completion, the reaction mass was cooled to 10± 5°C and quenched with chilled water (20 vol) at 10±5°C. The mass temperature was raised to 25± 5°C and stirred for 7-8 h. The resulting Compound ID crude was collected by filtration and washed with 2 vol of water. Crude Compound ID material taken in water (10 vol) and stirred for up to 20 minutes at 25±5°C. The reaction mass was heated to 45±5°C and stirred for 2-3 h at 45±5°C, filtered and vacuum-dried.
[0095] Crude Compound ID was taken in MTBE (5 vol) at 25±5°C and stirred for about 20 minutes at 25±5°C. The reaction mass temperature was raised to 45±5°C, stirred for 3-4 h at 45±5°C and then cooled to 20±5°C. The reaction mass was stirred for about 20 minutes at 20±5°C, filtered, followed by bed wash with water (0. 5 vol) and vacuum-dried.
[0096] The crude material was dissolved in acetone (10 vol) at 25±5°C and stirred for about 2h at 25±5°C. The reaction mass was filtered through a celite bed and washed with acetone (2.5 vol). The filtrate was slowly diluted with water (15 vol) at 25±5°C. The reaction mass was stirred for 2-3 h at 25±5°C, filtered and bed washed with water (2 vol) & vacuum-dried to afford Compound ID as brown solid.
[0097] Step 5 : 1 -((4-((2-(5-(((tert-butoxycarbonv0(2-methoxy ethvOaminolmethvOpyri din-2 -yl )thieno[3.2-b]pyridin-7-yl )oxy)-3 -fluorophenyl icarbamoyl level opropane-1 -carboxylic acid (Compound IE)
Compound 1E
[0098] To a solution of Compound ID (1.0 eq.) in tetrahydrofuran (7 vol.), aqueous potassium carbonate (1.0 eq.) in water (8 vol.) was added. The solution was cooled to 5±5°C, and stirred for about 60 min. While stirring, separately triethylamine (2.0 eq.) was added to a solution of 1,1-cyclopropanedicarboxylic acid (2.0 eq.) in tetrahydrofuran (8 vol.), at 5±5°C, followed by thionyl chloride (2.0 eq.) and stirred for about 60 min. The acid chloride mass was slowly added to the Compound ID solution at 5±5°C. The temperature was raised to 25±5°C and stirred for 3.0 h. The reaction was monitored by HPLC analysis.
[0099] After reaction completion, the mass was diluted with ethyl acetate (5.8 vol.), water (5.1 vol.), 10% (w/w) aqueous hydrochloric acid solution (0.8 vol.) and 25% (w/w) aqueous sodium chloride solution (2 vol.). The aqueous layer was separated and extracted with ethyl acetate (2 x 5 vol.). The combined organic layers were washed with a 0.5M aqueous sodium bicarbonate solution (7.5 vol.). The organic layer was treated with Darco activated charcoal (0.5 w/w) and sodium sulfate (0.3 w/w) at 25±5°C for 1.0 h. The organic layer was filtered through celite and washed with tetrahydofuran (5.0 vol.). The filtrate was concentrated under vacuum below 50°C to about 3 vol and co-distilled with ethyl acetate (2 x 5 vol.) under vacuum below 50°C up to ~ 3.0 vol. The organic layer was cooled to 15±5°C, stirred for about 60 min., filtered, and the solid was washed with ethyl acetate (2.0 vol.). The material was dried under vacuum at 40±5°C until water content was less than 1% to afford Compound IE as brown solid.
[00100] Step 6: tert-butyl (Y6-(7-(2-fluoro-4-(T-(Y4-fluorophenvDcarbamovDcvclopropane-l-carboxamido)phenoxy)thieno[3.2-b]pyridin-2-v0pyri din-3 – (2-
methoxyethvDcarbamate (Compound IF)
[00101] Pyridine (1.1 eq.) was added to a suspension of Compound IE (1.0 eq.) in tetrahydrofuran (10 vol.) and cooled to 5±5°C. Thionyl chloride (2.0 eq.) was added and stirred for about 60 min. The resulting acid chloride formation was confirmed by HPLC analysis after quenching the sample in methanol. Separately, aqueous potassium carbonate (2.5 eq.) solution (7.0 vol. of water) was added to a solution of 4-fluoroaniline (3.5 eq.) in tetrahydrofuran (10 vol.), cooled to 5±5°C, and stirred for about 60 min. The temperature of the acid chloride mass at 5±5°C was raised to a temperature of about 25±5°C and stirred for 3 h. The reaction monitored by HPLC analysis.
[00102] After completion of the reaction, the solution was diluted with ethyl acetate (25 vol.), the organic layer was separated and washed with a 1M aqueous sodium hydroxide solution (7.5 vol.), a 1M aqueous hydrochloric acid solution (7.5 vol.), and a 25% (w/w) aqueous sodium chloride solution (7.5 vol.). The organic layer was dried and and filtered with sodium sulfate (1.0 w/w). The filtrate was concentrated ~ 3 vol under vacuum below 50°C and co-distilled with ethyl acetate (3 x 5 vol.) under vacuum below 50°C to ~ 3.0 vol. Ethyl acetate (5 vol.) and MTBE (10 vol.) were charged, heated up to 50±5°C and stirred for 30-60 min. The mixture was cooled to 15±5°C, stirred for about 30 min., filtered, and the solid was washed with ethyl acetate (2.0 vol.). MGB3 content was analyzed by HPLC analysis. The material was dried under vacuum at 40±5°C until the water content reached about 3.0% to afford Compound IF as brown solid.
[00103] Step 7 : N-(3-fluoro-4-((2-(5-(((2-methoxyethv0amino)methv0pyridin-2-yl )thieno[3.2-b]pyridin-7-yl )oxy)phenyl)-N-(4-fluorophenyl level opropane-1. 1 -dicarboxamide (Compound 1)
Compound 1
[0100] To a mixture of Compound IF in glacial acetic acid (3.5 vol.) concentrated hydrochloric acid (0.5 vol.) was added and stirred at 25±5°C for 1.0 h. The reaction was monitored by HPLC analysis.
[0101] After reaction completion, the mass was added to water (11 vol.) and stirred for 20±5°C for 30 min. The pH was adjusted to 3.0 ± 0.5 using 10% (w/w) aqueous sodium bicarbonate solution and stirred for 20±5°C for approximately 3.0 h.. The mass was filtered, washed with water (4 x 5.0 vol.) and the pH of filtrate was checked after every wash. The material was dried under vacuum at 50±5°C until water content was about 10%.
[0102] Crude Compound 1 was taken in ethyl acetate (30 vol.), heated to 70±10°C, stirred for 1.0 h., cooled to 25±5°C, filtered, and washed with ethyl acetate (2 vol.). The material was dries under vacuum at 45±5°C for 6.0 h.
[0103] Crude Compound 1 was taken in polish filtered tetrahydrofuran (30 vol.) and pre washed Amberlyst A-21 Ion exchange resin and stirred at 25±5°C until the solution became clear. After getting the clear solution, the resin was filtered and washed with polish filtered tetrahydrofuran (15 vol.). The filtrate was concentrated by -50% under vacuum below 50°C and co-distilled with polish filtered IPA (3 x 15.0 vol.) and concentrated up to -50% under vacuum below 50°C. Charged polish filtered IPA (15 vol.) was added and the solution concentrated under vacuum below 50°C to – 20 vol. The reaction mass was heated to 80±5°C, stirred for 60 min. and cooled to 25±5°C. The resultant reaction mass was stirred for about 20 hours at 25±5°C. The reaction mass was cooled to 0±5°C, stirred for 4-5 hours, filtered, and washed with polish filtered IPA (2 vol.). The material was dried under vacuum at 45±5°C, until the water content was about 2%, to obtain the desired product Compound 1. ¾-NMR (400 MHz, DMSO- d): 510.40 (s, 1H), 10.01 (s, 1H), 8.59 – 8.55 (m, 1H), 8.53 (d, J= 5.6 Hz, 1H), 8.32 (s, 1H), 8.23 (d, J= 8.0 Hz, 1H), 7.96 – 7.86 (m, 2H), 7.70 – 7.60 (m, 2H), 7.56 – 7.43 (m, 2H), 7.20 – 7.11 (m, 2H), 6.66 (d, J= 5.6 Hz, 1H), 3.78 (s, 2H), 3.41 (t, J= 5.6 Hz, 2H), 3.25 (s, 3H), 2.66 (t, J= 5.6 Hz, 2H), 1.48 (s, 4H)ppm. MS: M/e 630 (M+l)+.
EXAMPLE 2
Preparation of Crystalline Form D of N-(3-fluoro-4-((2-(5-(((2- methoxyethyl)amino)methyl)pyridin-2-yl)thieno[3,2-b]pyridin-7-yl)oxy)phenyl)-N-(4- fluorophenyl)cyclopropane-l, 1-dicarboxamide
EXAMPLE 2A: Preparation of Compound 1 Crystalline Form D
[0104] To a 50 L reactor, 7.15 Kg of Compound 1, 40 g of Form D as crystal seed and 21 L acetone (>99%) were added. The mixture was heated to reflux ( ~56 °C) for 1~2 h. The mixture was agitated with an internal temperature of 20±5 °C for at least 24 h. Then, the suspension was filtered and washed the filter cake with 7 L acetone. The wet cake was dried under vacuum at <45 °C, to obtain 5.33 kg of Compound 1 of desired Form D
[0105] X-Ray Powder Diffraction (XRPD)
The XRPD patterns were collected with a PAN alytical X’ Pert PRO MPD diffractometer using auincident beam of Cu radiation produced using au Optix long, fine-focus source. An elliptically graded multilayer mirror was used to focus Cu Ka X -rays through the specimens and onto the detector. Prior to the analysis, a silicon specimen (NIST SRM 640e) was analyzed to verify the observed position of the Si Ill peak is consistent with the NIST-certified position. A specimen of each sample was sandwiched between 3 -pm -thick films and analyzed in transmission geometly. A beam-stop, short autiscatter extension, and an autiscatter knife edge were used to minimize the background generated by air. Sober slits for the incident aud diffracted beauls were used to minimize broadening from axial divergence. The diffraction patterns were collected using a scanning position-sensitive detector (X’Celerator) located 240 mm from the specimens and Data Collector software v. 2.2b. Pattern Match v2.3.6 was used to create XRPD patterns.
[0106] The X-ray powder diffraction (XRPD) pattern was used to characterize the Compound 1 obtained, which showed that the Compound 1 was in Crystalline Form D of Compound 1 (Compound 1 Form D), see Figure 1A. The XRPD pattern yielded is substantially the same as that shown in Figure 3C.
[0107] Differential Scanning Calorimetry (DSC)
[0108] DSC was performed using a Mettler-Toledo DSC3+ differential scanning calorimeter. Temperature calibration was performed using octane, phenyl salicylate, indium, tin, and zinc. The TAWN sensitivity was 11.9. The samples were placed into aluminum DSC pans, covered with lids, and the weights were accurately recorded. A weighed aluminum pan configured as the sample pan was placed on the reference side of the cell. The pan lids were pierced prior to sample analyses. The method name on the thermograms is an abbreviation for the start and end temperature as well as the heating rate; e.g., -30-250-10 means “from ambient to 250°C, at 10°C/min.” The nitrogen flow rate was 50.0 mL/min. This instrument does not provide gas pressure value as required by USP because it is the same as atmospheric pressure.
[0109] A broad small endotherm with a peak maximum at approximately 57°C to 62°C (onset ~20°C to 22°C) followed by a sharp endotherm with a peak maximum at approximately 180°C (onset ~178°C) were observed. These events could be due to the loss of volatiles and a melt, respectively (see Figure IB).
[0110] In an alternative embodiment Form D was prepared as follows. Designated Material O was suspended in 600 pL of acetone. Initial dissolution was observed followed by re precipitation. The amount of suspended solids was not measured because the target of the experiment was to get a suspension with enough solids to slurry isolate and collect XRPD data. Based on the solubility of Form D in acetone a very rough estimate for the scale of the experiment is about 80-100mg. The suspension was stirred at ambient temperature for approximately 2 5 weeks after which the solids were isolated by centrifugation with filtration. XRPD data appeared to be consistent with Form D The sample was then dried in vacuum oven at ~40 °C for ~2 5 hours. The XRPD pattern of the final solids was consistent with Form D EXAMPLE 2B: Preparation of Compound 1 Form D
[0111] 427.0 mg of Compound 1 was dissolved in 5 mL of THF to obtain a clear brown solution. The resulting solution was filtered, and the filtrate evaporated under flow of nitrogen. A sticky solid was obtained, which was dried under vacuum in room temperature for ~5 min, still a sticky brown solid obtained. It was dissolved in 0.2 mL of EtOAc and sonicated to dissolve. The solution obtained was stirred at room temperature for 15 min and a solid precipitated. The resulting solid was added 0.4 mL of EtOAc and stirred in room temperature for 21 h 40 min to ontian a suspension. The solid was spparated from mother liquor by centrifugation, then the resulting solid was resuspended the in 0.6 mL of EtOAc and stirred in room temperature for 2 days. The solid was isolated by centrifugation, to obtain Compound 1 of desired Form D.
[0112] The X-ray powder diffraction (XRPD) pattern was used to characterize the Compound 1 obtained, which showed that the Compound 1 was in Crystalline Form D of Compound 1 (Compound 1 Form D).
EXAMPLE 2C: Preparation of Compound 1 Form D
[0113] Single crystal X-ray diffraction data of Compound 1 was collected at 180 K on a Rigaku XtaLAB PRO 007HF(Mo) diffractometer, with Mo Ka radiation (l = 0.71073 A). Data reduction and empirical absorption correction were performed using the CrysAlisPro program. The structure was solved by a dual-space algorithm using SHELXT program. All non-hydrogen atoms could be located directly from the difference Fourier maps. Framework hydrogen atoms were placed geometrically and constrained using the riding model to the parent atoms. Final structure refinement was done using the SHELXL program by minimizing the sum of squared deviations of F2 using a full-matrix technique.
Preparation of Compound 1 Form D ( a Single Crystal )
[0114] Compound 1 Form D was dissolved in a mixture of acetone/ ACN (1/2) with the concentration of Compound 1 at ~7 mg/mL. A block single crystal was obtained, which was a single crystal.
[0115] The XRPD pattern was used to characterize the single crystal of Compound 1 Form D obtained, see Figure 2A. The crystal structural data are summarized in Table IB. The refined single crystal structure were shown in Figure 2B. The single crystal structure of Compound 1 Form D is in the P-1 space group and the triclinic crystal system. The terminal long alkyl chain is found to have large ellipsoids, indicating high mobility with disordered atoms.
[0116] The theoretical XRPD calculated from the single crystal structure and experimental XRPD are essentially similar (Figure 2A). A few small peaks are absent or shift because of orientation preference, disorder and tested temperature (180 K for single crystal data and 293 K for experimental one).
[0117] Table IB. Crystal Data and Structure Refinement for Compound 1 Form D (a Single Crystal)
References
- ^ http://www.mirati.com/go/mgcd516/
- ^ “MGCD516 in Advanced Liposarcoma and Other Soft Tissue Sarcomas – Full Text View – ClinicalTrials.gov”.
- ^ “Phase 2 Study of Glesatinib, Sitravatinib or Mocetinostat in Combination With Nivolumab in Non-Small Cell Lung Cancer – Full Text View – ClinicalTrials.gov”.
- ^ “MGCD516 Combined With Nivolumab in Renal Cell Cancer (RCC) – Full Text View – ClinicalTrials.gov”.
| Identifiers | |
|---|---|
| showIUPAC name | |
| CAS Number | 1123837-84-2 |
| ChemSpider | 52083477 |
| UNII | CWG62Q1VTB |
| KEGG | D11140 |
| Chemical and physical data | |
| Formula | C33H29F2N5O4S |
| Molar mass | 629.68 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCOCCNCc1ccc(nc1)c2cc3c(s2)c(ccn3)Oc4ccc(cc4F)NC(=O)C5(CC5)C(=O)Nc6ccc(cc6)F | |
| hideInChIInChI=1S/C33H29F2N5O4S/c1-43-15-14-36-18-20-2-8-25(38-19-20)29-17-26-30(45-29)28(10-13-37-26)44-27-9-7-23(16-24(27)35)40-32(42)33(11-12-33)31(41)39-22-5-3-21(34)4-6-22/h2-10,13,16-17,19,36H,11-12,14-15,18H2,1H3,(H,39,41)(H,40,42)Key:WLAVZAAODLTUSW-UHFFFAOYSA-N |
///////////// sitravatinib, phase 3, シトラバチニブ , MGCD516, MG-516, Sitravatinib (MGCD516), UNII-CWG62Q1VTB, CWG62Q1VTB, MGCD-516, ситраватиниб , سيترافاتينيب , 司曲替尼 , Antineoplastic, MGCD 516
#sitravatinib, #phase 3, #シトラバチニブ , #MGCD516, #MG-516#Sitravatinib (MGCD516), #UNII-#CWG62Q1VTB, #CWG62Q1VTB, #MGCD-516, ситраватиниб , سيترافاتينيب , 司曲替尼 , #Antineoplastic, #MGCD516
COCCNCC1=CN=C(C=C1)C2=CC3=NC=CC(=C3S2)OC4=C(C=C(C=C4)NC(=O)C5(CC5)C(=O)NC6=CC=C(C=C6)F)F

{kind=link}